Детские болезни: учебник / Под ред. А.А. Баранова - 2-е изд., - 2009. - 1008 с.
|
|
|
|
ГЛАВА 18 ЭНДОКРИННЫЕ ЗАБОЛЕВАНИЯ
Врождённый гипотиреоз
Врождённый гипотиреоз - одно из наиболее распространённых врождённых заболеваний щитовидной железы у детей. Заболеваемость колеблется от 1 случая на 4000-5000 новорождённых в Европе и Северной Америке до 1 на 6000-7000 новорождённых в Японии. У девочек врождённый гипотиреоз регистрируют в 2-2,5 раза чаще, чем у мальчиков.
В
основе заболевания лежит полная или частичная недостаточность
тиреоидных гормонов, приводящая к задержке развития всех органов и
систем. В первую очередь у плода и новорождённого от недостатка
тиреоидных гормонов страдает ЦНС. Установлена тесная корреляция между
сроками начала заместительной терапии и индексом интеллектуального
развития ребёнка в дальнейшем. Благоприятного умственного развития
можно ожидать только при начале лечения в первый месяц жизни ребёнка. В
последние десятилетия появилась реальная возможность массового
обследования всех новорождённых на наличие у них врождённых
заболеваний, таких, как фенилкетонурия, галактоземия, врождённая
дисфункция коры надпочечников, болезнь кленового сиропа,
гомоцистинурия, а также врождённый гипотиреоз. Скрининг на
фенилкетонурию впервые был осуществлён в
Скрининг на врождённый гипотиреоз позволяет диагностировать заболевание в первый месяц жизни ребёнка. Наличие высоко- эффективного лекарственного средства для заместительной терапии (левотироксина натрия) позволяет проводить её в максимально физиологическом режиме и оптимальной дозировке. Врождённый ги-
потиреоз - заболевание, при котором своевременно начатое лечение предотвращает развитие умственной отсталости у ребёнка.
Этиология и патогенез
В последние годы в связи с развитием методов молекулярно-генетического анализа во многом изменились взгляды на этиологию врождённого гипотиреоза. Врождённый гипотиреоз достаточно гетерогенен по этиологии, обусловлен морфофункциональной незрелостью гипоталамо-гипофизарной системы, щитовидной железы или их анатомическим повреждением во внутриутробный период.
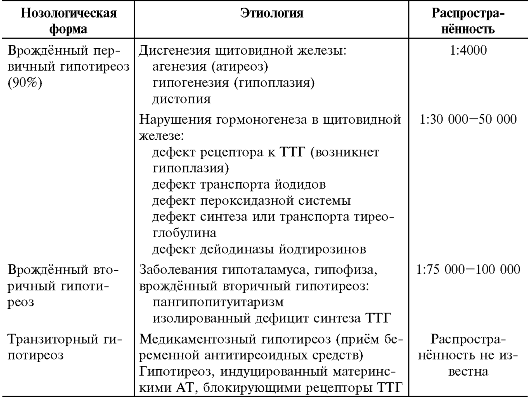
В подавляющем большинстве случаев (85-90%) диагностируют первичный врождённый гипотиреоз. Среди них приблизительно 85% являются спорадическими, а 15% - наследственными (табл. 18-1). Большинство спорадических случаев обусловлено дисгенезией щитовидной железы, причём случаи эктопии щитовидной железы наблюдают гораздо чаще, чем полное её отсутствие (агенезию) или гипоплазию. По данным различных авторов, в 22-42% регистрируют агенезию щитовидной железы, в 35-42% - эктопию, в 24-36% - гипоплазию.
Таблица 18-1. Этиология и распространённость основных форм врождён- ного гипотиреоза
Гипоталамо-гипофизарно-тиреоидная система плода развивается независимо от влияния организма беременной. К 10-12-й неделе внутриутробного развития щитовидная железа плода способна накапливать йод и синтезировать йодтиронины. К этому времени гипофиз плода может секретировать некоторое количество ТТГ.
Содержание T4 в сыворотке крови плода прогрессивно увеличивается с середины беременности к моменту родов. Концентрация T3 у плода до 20-й недели беременности относительно не высока, а затем значительно возрастает, достигая 60 нг/дл к концу беременности. Содержание ТТГ у плода постепенно возрастает с увеличением срока гестации и к моменту родов составляет около 10 мМЕ/л.
T4 беременной частично проникает через плаценту и влияет на развитие плода (особенно его головного мозга). Преимущественно это влияние осуществляется в первые недели беременности до того момента, когда начинается синтез собственных тиреоидных гормонов.
У плода, развивающегося в организме беременной с гипофункцией щитовидной железы, повышен риск развития патологии ЦНС. С другой стороны, при недостаточной выработке у плода тиреоидных гормонов проникающий через плаценту T4 беременной может до определённой степени предотвращать развитие у него гипотиреоза.
При рождении и в первые минуты жизни у новорождённого происходит значительный выброс ТТГ, концентрация которого к 30-й минуте после рождения достигает 70 мЕд/л (у доношенных новорож- дённых). Затем концентрация ТТГ постепенно снижается, достигая к концу 2-3-го дня 10 мЕд/л. Резкое повышение содержания ТТГ влечёт за собой значительное возрастание концентраций T4 и T3 в течение первых часов жизни ребёнка. T3 в значительной степени образуется на периферии благодаря процессам конверсии.
Клиническая картина
Типичную клиническую картину врождённого гипотиреоза у новорождённых, когда крайне важно поставить диагноз, наблюдают всего в 10-15% случаев. К наиболее типичным признакам заболевания в ранний постнатальный период относят:
• переношенную беременность (более 40 нед);
• избыток массы тела при рождении (более
• отёчные лицо, губы, веки, полуоткрытый рот с широким, «распластанным» языком;
• локализованные отёки в виде плотных «подушечек» в надключичных ямках, на тыльных поверхностях кистей, стоп;
• признаки незрелости при доношенной по сроку беременности;
• низкий, грубый голос при плаче, крике;
• позднее отхождение мекония;
• позднее отхождение пупочного канатика;
• плохую эпителизацию пупочной ранки;
• затянувшуюся желтуху.
В дальнейшем на 3-4-м месяце жизни, если не было начато лечение, возникают другие клинические симптомы заболевания:
• сниженный аппетит;
• затруднения при глотании;
• отставание в прибавке массы тела;
• метеоризм;
• запоры;
• сухость, бледность, шелушение кожных покровов;
• гипотермия (холодные кисти, стопы);
• ломкие, сухие, тусклые волосы;
• мышечная гипотония.
В поздние сроки, после 5-6-го месяца жизни, на первый план выступает нарастающая задержка психомоторного и физического развития ребёнка. Пропорции тела у детей с гипотиреозом приближаются к хондродистрофическим, отстаёт развитие лицевого скелета (широкая запавшая переносица, гипертелоризм, позднее закрытие родничков). Запаздывает прорезывание зубов, а затем их смена. Обращает на себя внимание кардиомегалия, глухость сердечных тонов, снижение АД, уменьшение пульсового давления, брадикардия (у детей первых месяцев частота пульса может быть нормальной). У детей с врождённым гипотиреозом низкий, грубый голос, часто наблюдают цианоз носогубного треугольника, стридорозное дыхание. При отсутствии адекватного лечения врождённого гипотиреоза в конечном итоге развивается кретинизм.
Лабораторные и инструментальные исследования
В качестве дополнительных методов обследования используют рентгенографию кистей (отмечают задержку появления ядер окостенения, их асимметрию, нарушение последовательности их возникновения, патогномоничным признаком служит эпифизарный дисгенез), общий анализ крови (анемия), биохимический анализ крови (гиперхолестеринемия), ЭКГ (снижение вольтажа, замедление проводимости, удлинение систолы, синусовая брадикардия).
Для подтверждения диагноза врождённого гипотиреоза необходимо исследовать содержание гормонов щитовидной железы (T3, T4) и ТТГ в сыворотке крови. При первичном гипотиреозе концентрации T4 и T3 снижены, а содержание ТТГ значительно превышает норму. При вторичном гипотиреозе концентрации тиреоидных гормонов снижены, а содержание ТТГ может быть сниженным или нормальным.
Диагностика и дифференциальная диагностика
До широкого внедрения в клиническую практику скрининга на врождённый гипотиреоз и радиоиммунологических методов определе- ния гормонов в сыворотке крови диагноз устанавливали на основании клинико-анамнестических данных, что приводило к позднему началу заместительной терапии.
Суммируя вышеперечисленные клинические признаки врождённого гипотиреоза, приводим шкалу Апгар, помогающую в раннем клиническом скрининге заболевания (табл. 18-2).
Таблица 18-2. Шкала Апгар для диагностики врождённого гипотиреоза у новорождённых*
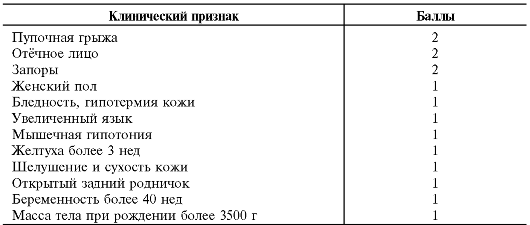
* Врождённый гипотиреоз следует заподозрить при оценке более 5 баллов.
Учитывая большую распространённость гипотиреоза, незначительную выраженность клинических признаков заболевания в первые дни, недели жизни, а также серьёзные последствия поздней диагностики заболевания, с середины 1970-х годов во многих развитых странах мира постепенно внедряют государственные системы неонатального скрининга на врождённый гипотиреоз.
Скрининг позволяет поставить диагноз в первые дни жизни ребён- ка, до развития клинической картины заболевания, и избежать таким образом тяжёлых последствий болезни, основными из которых являются задержка умственного и физического развития ребёнка. Экономически стоимость скрининга соотносится со стоимостью лечения ребёнка-инвалида в поздно диагностируемых случаях как 1:4.
Лечение
Сразу же после установления диагноза, а также в сомнительных случаях необходимо начинать заместительную терапию тиреоидными
препаратами. Препаратом выбора для лечения врождённого гипотиреоза служит левотироксин натрия. Он совершенно идентичен естественному гормону человека T4 - в этом его главное преимущество перед другими синтетическими препаратами. Кроме того, после при- ёма левотироксина натрия в крови создаётся «депо» этого препарата, которое расходуется по мере необходимости путём дейодирования T4 и превращения его в T3. Таким образом, удаётся избежать высоких, пиковых концентраций T3 в крови.
Всю суточную дозу необходимо принимать утром за 30 мин до завтрака, запивая небольшим количеством жидкости. Маленьким детям препарат следует давать во время первого утреннего кормления, в рас- толчённом виде.
Начальная доза левотироксина натрия составляет 12,5-25-50 мкг/сут или 10-15 мкг/кг/сут. Ориентировочные дозы левотироксина натрия, которые рекомендуют назначать детям для лечения врождённого гипотиреоза, указаны в табл. 18-3.
Таблица 18-3. Ориентировочные дозы левотироксина натрия для лечения детей с врождённым гипотиреозом

Самым надёжным показателем адекватности получаемого ребёнком лечения при гипотиреозе служит нормальное содержание ТТГ в сыворотке крови. Концентрация T4 нормализуется обычно через 1-2 нед после начала лечения, а концентрация ТТГ - через 3-4 нед.
При длительном лечении показателями адекватности применяемой дозы левотироксина натрия служат данные динамики роста, общее развитие ребёнка, показатели дифференцировки скелета.
В комплекс лечебных мероприятий при врождённом гипотиреозе следует включать симптоматическую терапию (антианемическую, антирахитическую, витаминотерапию), ЛФК, массаж, по показаниям - ноотропные препараты.
Прогноз
У всех детей с врождённым гипотиреозом при раннем и адекватном лечении можно достичь оптимального интеллектуального развития.
Диффузный токсический зоб (болезнь Грейвса)
Диффузный токсический зоб - аутоиммунное заболевание, характеризующееся диффузным увеличением щитовидной железы, наличием тиреотоксикоза и инфильтративной офтальмопатии.
Тиреотоксикоз - комплекс клинических и метаболических изменений, возникших в результате влияния на организм повышенного количества тиреоидных гормонов. Термин «гипертиреоз» используют в том случае, когда источником повышенной секреции тиреоидных гормонов является щитовидная железа.
ЭТИОЛОГИЯ
У детей, особенно младшего возраста, возможно развитие диффузного токсического зоба неиммунного генеза вследствие врождённой активирующей мутации гена рецептора ТТГ. У детей старшего воз- раста, как и у взрослых, тиреотоксикоз часто обусловлен токсической аденомой щитовидной железы. Редкая причина диффузного токсического зоба - аденома гипофиза (тиреотропинома).
ПАТОГЕНЕЗ
В щитовидной железе синтезируются тиреоидные гормоны - T4 и T3, при этом на долю первого приходится 90%. Большая часть циркулирующего в крови T3 поступает из периферических тканей, где под действием местных дейодиназ происходит его синтез из T4. Секрецию тиреоидных гормонов активирует ТТГ, а его секрецию, в свою очередь, увеличивает тиреотропин-рилизинг-гормон гипоталамуса. Рецепторы ТТГ расположены на поверхности фолликулярных клеток щитовидной железы.
Диффузный токсический зоб - аутоиммунное заболевание, в основе которого лежит образование АТ к рецептору ТТГ, так называемых тиреоидстимулирующих АТ. Последние, связываясь с рецептором ТТГ, оказывают эффект, подобный действию ТТГ. Происходит постоянная стимуляция секреции тиреоидных гормонов без участия ТТГ. Кроме того, повышенное содержание тиреоидных гормонов приводит к блокаде секреции ТТГ.
ПАТОМОРФОЛОГИЯ
Отмечают полиморфизм фолликулов, переход кубического эпителия в цилиндрический, гипертрофию и гиперплазию тиреоидного эпителия, иногда - сосочковые выросты в просвете фолликулов. Коллоид чаще жидкий с вакуолизацией. Между фолликулами наблюдают очаговую, диффузную или сочетанную лимфоидную инфильтрацию.
КЛАССИФИКАЦИЯ
По степени тяжести диффузный токсический зоб классифицируют следующим образом.
• Лёгкий тиреотоксикоз - ЧСС не превышает 100 в минуту, умерен-
ное снижение массы тела, отсутствуют признаки поражения других органов и систем.
• Тиреотоксикоз средней тяжести - ЧСС от 100 до 150 в минуту, выраженное снижение массы тела, снижение содержания холестерина, желудочно-кишечные расстройства, признаки надпочечниковой недостаточности (низкое диастолическое АД, гиперпигментация).
• Тяжёлый тиреотоксикоз - ЧСС более 150 в минуту, выраженная об-
щая дистрофия вплоть до кахексии, мерцательная аритмии, склонность к фибрилляции предсердий, при тяжёлом тиреотоксикозе часто развивается тиреотоксический криз.
КЛИНИЧЕСКАЯ КАРТИНА
Зоб
Увеличение щитовидной железы - наиболее характерный признак диффузного токсического зоба, наблюдаемый у 100% детей с этим заболеванием. При осмотре больного необходимо помнить о возможности загрудинного расположения щитовидной железы, когда её истинные размеры оценить невозможно и при первичном осмотре зоб может быть не диагностирован. Лёгкие и среднетяжёлые формы диффузного токсического зоба чаще сопровождаются небольшим увеличением щитовидной железы. Для тяжёлых форм, как правило, характерны большие размеры зоба, однако не всегда размеры зоба коррелируют с тяжестью тиреотоксикоза.
Кожные покровы
У детей с тиреотоксикозом кожные покровы, как правило, горячие и влажные. Это происходит в результате расширения кожных сосудов и повышенного потоотделения. Наиболее достоверно температуру и влаж- ность кожных покровов можно оценить на внутренней поверхности плеч и бёдер или на поверхности грудной клетки. Локти у больных с тиреотоксикозом гладкие и розовые. Кожа у этих больных легко краснеет. Ладони напоминают «печёночные», могут быть телеангиоэктазии. Иногда наблюдают гиперпигментацию, что свидетельствует о наличии надпочечниковой недостаточности. Ногти могут быть мягкими и ломкими.
Тиреотоксическая офтальмопатия
Изменения глаз у больных диффузным токсическим зобом могут быть как проявлением тиреотоксикоза, так и аутоиммунного процес-
са, локализованного в ретробульбарной клетчатке. Комплекс глазных симптомов, обусловленных тиреотоксикозом, называют тиреотоксической офтальмопатией, а аутоиммунное поражение ретробульбарной клетчатки и глазодвигательных мышц - инфильтративной офтальмопатией. Как правило, клинические признаки тиреотоксической офтальмопатии стихают по мере уменьшения выраженности симптомов тиреотоксикоза. Признаки аутоиммунной или инфильтративной офтальмопатии сохраняются после ликвидации симптомов тиреотоксикоза. Выраженность глазных симптомов, а также их динамика на фоне лечения диффузного токсического зоба - прогностические критерии эффективности консервативной терапии заболевания. Клинические признаки офтальмопатии наблюдают у 50-93% пациентов. Однако при проведении МРТ, УЗИ орбит признаки отёка глазодвигательных мышц, а также увеличение объёма ретробульбарной клетчатки находят у всех пациентов с диффузным токсическим зобов, независимо от наличия клинических симптомов офтальмопатии. Инфильтративная офтальмопатия может развиться и в отсутствии диффузного токсического зоба.
Один из характерных симптомов офтальмопатии при диффузном токсическом зобе у детей - экзофтальм (рис. 18-1 на вклейке). Тем не менее экзофтальм не считают обязательным признаком диффузного токсического зоба. По данным различных авторов, распространённость экзофтальма при диффузном токсическом зобе у детей колеблется от 52 до 93%. Экзофтальм, как правило, асимметричен. Односторонний экзофтальм у детей диагностируют крайне редко. Периорбитальный отёк может маскировать проявления экзофтальма. Иногда больные жалуются на чувство давления позади глазных яблок. Экзофтальм бывает настолько выраженным, что больные не могут закрыть глаза во время сна. Этот симптом называют лагофтальмом. К клиническим признакам тиреотоксической офтальмопатии также относят повышение светочувствительности, слезотечение, усиливающееся на ветру, инъекцию конъюнктивы. Достаточно часто отмечают нечёткость видения, диплопию, быстрое утомление глаз. Выраженная офтальмопатия часто сочетается с инфекционными поражениями конъюнктивы и изъязвлениями роговицы. Глазные симптомы диффузного токсического зоба приведены в табл. 18-4.
Степень
экзофтальма можно объективно оценить с помощью экзофтальмометра. Этот
прибор позволяет измерить расстояние между латеральным углом глаза и
наиболее выступающей точкой роговицы. В норме оно должно быть не более
Таблица 18-4. Глазные симптомы при диффузном токсическом зобе

Сердечно-сосудистая система
Нарушения сердечно-сосудистой системы считают самым важным признаком тиреотоксикоза как у взрослых, так и у детей. Наиболее частый кардиальный симптом тиреотоксикоза - тахикардия. Нередко она может предшествовать другим признакам заболевания. Часто дети с тиреотоксикозом жалуются на сердцебиение, возникающее спонтанно или на фоне минимальной психологической или физической нагрузки. При оценке частоты сердечных сокращений у детей необходимо помнить о возрастной норме. Тахикардия у детей с диффузным токсическим зобом носит постоянный характер и не проходит во сне и при эмоциональном переключении больного. При объективном осмотре можно диагностировать усиленный сердечный толчок. Тоны сердца чаще акцентированы, усилены. Тяжёлые формы заболевания могут сопровождаться приглушенными сердечными тонами. Довольно часто выслушивают неорганические функциональные шумы. Как правило, это систолические шумы на верхушке сердца, над лёгочной артерией, в точке Боткина-Эрба. Эхокардиография у некоторых больных позволяет определить признаки гипертрофии левого желудочка, которая носит функциональный характер и проходит на фоне компенсации основного заболевания. При исследовании метаболизма сердечной мышцы у больных с диффузным токсическим зобом определяют снижение энергетических ресурсов миокарда, уменьшение содержания гликогена, усиление катаболизма белка. При этом кровоток и утилизация кислорода в миокарде усиливаются. При проведении ЭКГ у детей диагностируют ускорение предсердно-желудочковой проводимости, повышение вольтажа зубцов P, QRS, T, синусовую
тахикардию. Кроме того, можно определить экстрасистолию. Такие симптомы, как мерцательная аритмия и пароксизмальная тахикардия, при диффузном токсическом зобе у детей наблюдают редко.
При диффузном токсическом зобе отмечают сниженное периферическое сопротивление и повышенный сердечный выброс. Это приводит к повышению систолического и снижению диастолического давления и, следовательно, к повышению пульсового давления.
Пищеварительная система
У пациентов с выраженным тиреотоксикозом часто отмечают повышение аппетита. Однако, несмотря на это, тяжёлые формы тиреотоксикоза у детей часто приводят к потере массы тела разной степени. Частота стула возрастает до нескольких раз в сутки. Диарею наблюдают редко. Анорексию, тошноту, рвоту и боли в животе относят к редким симптомам заболевания и отмечают их только в случае тяжёлых форм тиреотоксикоза. В связи с повышенной моторикой ЖКТ может возникать синдром мальабсорбции.
Нарушения функции печени отмечают только в случае тяжёлых форм тиреотоксикоза. Возможны гепатомегалия, желтушность, повы- шение активности печёночных ферментов. Гипоксия и повышенный уровень основного обмена приводят к некоторому снижению содержания гликогена. При тяжёлом тиреотоксикозе могут возникать жировая инфильтрация, локальный фиброз, лимфатическая инфильтрация и пролиферация эндотелия жёлчных протоков.
Нервная система
Расстройства нервной системы часто являются ведущими симптомами тиреотоксикоза у детей. Тиреотоксикоз приводит к нарушениям в психоэмоциональной и двигательной сферах. Часто первыми при- знаками заболевания у ребёнка становятся изменения в поведении, отмечаемые родителями и сверстниками. У детей возникают нервозность, эмоциональная лабильность, плаксивость, выраженная утомляемость, нарушения сна. Резкие перепады настроения, необоснованные вспышки гнева приводят к конфликтам с родителями, друзьями и преподавателями. Часто у детей страдает концентрация внимания и ухудшается память. Всё это негативно влияет на успеваемость ребён- ка и приводит к его социальной дезадаптации. Среди двигательных нарушений наиболее характерными считают гиперкинезы, напоминающие хорею: насильственные, быстрые, толчкообразные движения пальцев рук, головы, сокращения мимических мыщц и мускулатуры конечностей. При осмотре ребёнок не может сидеть на одном месте, барабанит по столу, поправляет волосы, застёгивает и расстёгивает
пуговицы. Движения быстрые, резкие, размашистые и часто бесцельные. При осмотре определяют мелкий тремор пальцев рук, языка и век. Однако у детей, в отличие от взрослых, тремор наблюдают реже и не относят к ранним симптомам заболевания. При проведении ЭЭГ диагностируют быстроволновую активность. Патогенетическая природа изменений нервной системы при тиреотоксикозе окончательно не выяснена. Предполагают, что неврологические нарушения могут быть обусловлены как повышенным тонусом симпатической нервной системы, так и непосредственным влиянием тиреоидных гормонов на нервную ткань, имеющую большое количество специфических рецепторов.
Костная система
Тиреотоксикоз характеризуется повышенным выведением кальция и фосфора через кишечник и почки. Кроме того, отмечают повышенное выведение продуктов распада коллагена с мочой. Всё это приводит к снижению плотности костной ткани. Однако патологические переломы при тиреотоксикозе у детей не возникают. В крови нередко определяют гиперкальциемию. Концентрации в сыворотке крови щелочной фосфатазы и остеокальцина также могут быть повышенными. Содержание ПТГ при этом чаще снижено или находится в пределах нормы.
Мочевыделительная система
Наиболее частым симптомом расстройства мочевыделения у детей бывает полиурия, развивающаяся вследствие усиления почечного кровотока и фильтрации. Полиурия и нарушения нервной системы нередко приводят к развитию у ребёнка чаще ночного, а иногда и дневного энуреза.
Система кроветворения
При тиреотоксикозе обычно наблюдают повышенную активность эритропоэза. В периферической крови увеличивается количество эритроцитов. Усиление эритропоэза происходит как в результате не- посредственного влияния тиреоидных гормонов на костный мозг, так и за счёт повышенной продукции эритропоэтина. Одновременно повышается объём плазмы, а гематокрит остаётся в пределах нормы. В периферической крови часто определяют лейкопению, обусловленную снижением абсолютного количества нейтрофилов, что приводит к относительному лимфоцитозу. Кроме того, могут быть абсолютный или относительный моноцитоз и эозинофилия. У 10% больных диагностируют спленомегалию. Нередко определяют генерализованную лимфаденопатию. Предполагают, что спленомегалия и лимфаденопатия обусловлены генерализованным аутоиммунным процессом, так
как для неаутоиммунных форм тиреотоксикоза эти изменения не характерны.
Эндокринная система
Влияние тиреотоксикоза на эндокринную систему наиболее выражено в отношении надпочечников. Ряд симптомов отражают надпо- чечниковую недостаточность, которая в разной степени выраженности развивается у всех детей с диффузным токсическим зобом. Общая слабость, утомляемость, пигментация кожных покровов, низкое диастолическое АД отражают недостаточность глюкокортикоидов. В результате усиления активности 11b-гидроксистероид дегидрогеназы повышается инактивация кортизола - преобразование 11-гидрок- сильной группы в кетогруппу. При этом секреция кортизола также возрастает, а концентрация его в крови не изменяется. Содержание свободного кортизола в суточной моче может быть несколько повы- шенно. Образование АКТГ гипофизом не изменяется. Активность ренина плазмы может быть повышена. Чувствительность к ангиотензину II снижена. Концентрации адреналина и норадреналина в крови находятся в пределах нормы.
Физическое развитие и костный возраст у детей с тиреотоксикозом, как правило, несколько ускорены, но ростовые показатели редко выходят за пределы нормы. Иногда может быть задержка полового развития.
Со стороны белкового обмена отмечают преобладание катаболических процессов. При этом повышается как синтез, так и распад и выведение белка из организма. В связи с этим наблюдают отрицательный азотистый баланс, снижение массы тела, мышечную слабость, может быть снижение концентрации альбумина.
У некоторых больных наблюдают патологию углеводного обмена - нарушение толерантности к глюкозе. Концентрация инсулина в плазме крови, как правило, повышена.
При диффузном токсическом зобе резко нарушается жировой обмен. Повышение липолитических процессов приводит к снижению в крови содержания холестерина и триглицеридов, а также к повышению концентраций свободных жирных кислот и глицерола. Потеря массы тела - один из ведущих и ранних симптомов диффузного токсического зоба у детей, хотя его наблюдают не во всех случаях. Иногда аппетит настолько повышен, что даже при значительном повышении уровня основного обмена больные не худеют.
Осложнения
Самое опасное для жизни больного осложнение тиреотоксикоза - тиреотоксический криз. Он может быть вызван различными причина-
ми, самой частой из которых считают проведение резекции щитовидной железы на фоне декомпенсированного тиреотоксикоза или в том случае, когда в пред- и послеоперационном периоде не использовали глюкокортикоиды. Кроме того, причинами криза могут быть отмена или неадекватное проведение антитиреоидной терапии, инфекционное заболевание, психическая травма, тяжёлая физическая нагрузка, оперативное вмешательство вне щитовидной железы. Патогенез тиреотоксического криза в первую очередь сводится к резкому повышению выделения в кровь тиреоидных гормонов. На этом фоне нарастают признаки относительной надпочечниковой недостаточности, так как тиреоидные гормоны увеличивают метаболизм кортизола. На фоне криза увеличивается потребность периферических тканей в глюкокортикоидах, что усиливает относительную надпочечниковую недостаточность. Криз, как правило, развивается в течение нескольких часов, реже - постепенно, в течение нескольких дней. Нарастают возбуждение и тахикардия, возникает аритмия, повышается температура тела, систолическое АД повышается, диастолическое - снижается. При дальнейшем развитии криза отмечают снижение как систо- лического, так и диастолического АД, нарастают признаки сердечной недостаточности. У больных могут быть бессонница, рвота, диарея, повышенная потливость, чувство страха, резкая головная боль, олигурия, гиперемия лица, частое дыхание, удушье, резкое двигательное беспокойство, беспорядочные движения, которые сменяются адинамией, апатией, сопором, переходящим в коматозное состояние.
ЛАБОРАТОРНЫЕ И ИНСТРУМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ УЗИ щитовидной железы позволяет оценить её размеры, а также структуру. Изменения структуры щитовидной железы при диффузном токсическом зобе сводятся к резкому снижению эхогенности (рис. 18-2 на вклейке) и появлению неравномерности структуры. Эти структурные изменения не относят к строго специфичным для диффузного токсического зоба, и поэтому их могут определять при других аутоиммунных заболеваниях щитовидной железы, например аутоиммунном тиреоидите.
Радиоизотопное исследование щитовидной железы у детей используют редко. Сцинтиграфию проводят в основном при подозрении на токсическую аденому щитовидной железы, она также позволяет выявить функционирующие метастазы высокодифференцированного рака щитовидной железы, которые могут быть причиной тиреотоксикоза.
Исследование иммунологического статуса позволяет определить повышение титра неспецифических, а также специфических АТ. Специфичны для диффузного токсического зоба АТ к рецептору ТТГ.
Связываясь с рецептором ТТГ, они активируют продукцию тиреоидных гормонов. Сохранение повышенных титров АТ к рецепторам ТТГ на фоне компенсации заболевания - прогностически неблагоприятный показатель рецидива. АТ к тиреоглобулину, микросомальному Аг не являются строго специфичными для диффузного токсического зоба. Повышенные титры этих АТ возможны и при других аутоиммунных заболеваниях щитовидной железы.
Исследование гормонального статуса. Определение концентраций ТТГ, T3 и T4 позволяет с высокой вероятностью подтвердить наличие гипертиреоза. Содержание ТТГ при диффузном токсическом зобе всегда снижено. Базальная концентрация T3 чаще повышена в большей степени, чем концентрация T4. Увеличение содержания ТТГ в сочетании с повышенными концентрациями тиреоидных гормонов позволяет заподозрить центральный тиреотоксикоз.
Биохимическое исследование крови указывает на снижение содержания холестерина, может быть гипергликемия. При проведении нагрузочного теста с глюкозой определяют нарушение толерантности к углеводам.
ДИАГНОСТИКА И ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА Визуально-пальпаторнаую оценку размеров щитовидной железы производят по классификации ВОЗ (1994):
0 степень - зоба нет;
1 степень - пальпируемый зоб;
II степень - видимый зоб (рис. 18-3 на вклейке).
При осмотре осуществляют оценку симптомов сдавления органов шеи. Дисфонию, парез голосовых связок относят к признакам сдавления возвратного нерва; кашель, икоту, брадикардию - блуждающего нерва. Затруднение глотания бывает при сдавлении пищевода, однако этот симптом можно наблюдать и при повышенной нервной возбудимости. Подтвердить сдавление пищевода помогает рентгеноконтрастное исследование с сульфатом бария. Синдром Бернара-Горнера (птоз, миоз, энофтальм), гиперемия половины лица, а также симпатоадреналовые кризы отмечают при сдавлении нервных ганглиев. Выраженное сдавление трахеи приводит к стридору. Начальные признаки сдавления трахеи легко диагностируют при УЗИ.
Наиболее часто в пубертатном возрасте диффузный токсический зоб приходится дифференцировать с СВД, тиреотоксической фазой аутоиммунного тиреоидита, реже с токсической аденомой щитовидной железы, ТТГ-секретирующей аденомой гипофиза, резистентностью к тиреоидным гормонам.
СВД - достаточно распространённое заболевание, особенно часто наблюдаемое в подростковом возрасте. При СВД могут быть тахикар-
дия, потливость, тремор, изменения АД - эти симптомы напоминают клиническую картину диффузного токсического зоба. Зоб в этих случаях не служит дифференциально-диагностическим признаком в связи с тем, что в йоддефицитных регионах его считают достаточно распространённым явлением. Однако клинические признаки СВД имеют свои особенности: тахикардия носит преходящий характер, прекращается во сне, при переключении внимания ребёнка в беседе часто сопровождается дыхательной аритмией. При диффузном токсическом зобе тахикардия постоянная, не зависит от сна и бодрствования, не изменяется при вдохе и выдохе. Потливость при СВД чаще регионарная (отмечают повышенную влажность ладоней, стоп, подмышечных областей), при диффузном токсическом зобе повышение влажности и потливости носит диффузный характер. Температура кожи у больных с СВД нормальная, ладони и стопы часто холодные и влажные. При диффузном токсическом зобе кожные покровы интенсивно тёплые или даже горячие. У больных с СВД часто отмечают тремор рук, однако тремор крупный, размашистый, непостоянный. При диффузном токсическом зобе тремор мелкий, постоянный, можно наблюдать так называемый симптом телеграфного столба. Окончательно подтвердить диагноз диффузного токсического зоба в этих случаях поможет гормональный профиль: уменьшение содержания ТТГ, повышение концентраций T4 и T3.
Автономную токсическую аденому щитовидной железы в
детском возрасте наблюдают чрезвычайно редко. Среди больных с
токсическими аденомами дети и подростки составляют всего от 2,2 до
8,6%. Во всех возрастных группах преобладают пациенты женского пола.
Патогенез токсической аденомы до конца остаётся не ясным. В последнее
время стало известно о мутациях в гене рецептора ТТГ, определяемых при
аденомах щитовидной железы. Размер большинства токсических аденом
превышает
Токсические аденомы иногда можно диагностировать при пальпации щитовидной железы. Наличие аденомы щитовидной железы подтверждают УЗИ и сканирование щитовидной железы с радиоактивным йодом. В случае наличия токсической аденомы показано оперативное лечение - аденомэктомия, а при определении карциномы объём операции расширяют до субтотальной или тотальной резекции щитовидной железы.
Тиреоидиты. Клинические признаки тиреотоксикоза также наблюдают при различных видах тиреоидитов: хроническом (хроническом аутоиммунном тиреоидите, зобе Хасимото), а также остром и подостром. У большинства пациентов с хроническим аутоиммунным тиреоидитом гормональная активность щитовидной железы может быть снижена или находится в пределах нормы. Всего у 5-10% детей и 6% взрослых с хроническим аутоиммунным тиреоидитом развивается клиническая картина тиреотоксикоза. Предполагают, что тиреотоксикоз в ранней фазе заболевания может быть вызван влиянием тиреоидстимулирующих АТ или лизисом клеток щитовидной железы и выходом тиреоидных гормонов в кровяное русло. Тиреотоксикоз, развивающийся при хроническом аутоиммунном тиреоидите, протекает нетяжело. В фазе тиреотоксикоза дифференциальная диагностика диффузного токсического зоба и хронического аутоиммунного тиреоидита очень сложна. При исследовании иммунологического статуса в обоих случаях определяют положитель- ные тиреоидстимулирующие АТ, АТ к микросомальному Аг и тиреоглобулину, поэтому эти показатели не могут служить дифференциально-диагностическим признаком. При обоих заболеваниях возможна офтальмопатия. УЗ-картина также неспецифична. Увеличение объёма, снижение эхогенности и неравномерность структуры могут быть и при хроническом аутоиммунном тиреоидите, и при диффузном токсическом зобе. Радиосцинтиграфия при хроническом аутоиммунном тиреоидите выявляет неравномерность захвата радиофармпрепарата, при диффузном токсическом зобе - признаки повышения функции щитовидной железы. Однако результаты этого исследования малоспецифичны, а само оно небезопасно, особенно у детей. Наиболее характерным дифференциально-диагностическим признаком служит динамика тиреотоксикоза: в случае хронического аутоиммунного тиреоидита тирео- токсическая фаза достаточно быстро (в течение нескольких месяцев) спонтанно переходит в гипотиреоз. Особенно быстро это происходит при назначении тиреостатических препаратов.
К классическим признакам острого тиреоидита относят озноб, гипертермию, покраснение кожи в области шеи, болезненность щи- товидной железы при пальпации и её асимметричное увеличение. Признаки тиреотоксикоза наблюдают у 2,5% детей.
Йодщдуцированный тиреотоксикоз возникает при повышенном потреблении йода, что приводит к увеличению синтеза T3 и T4. Он раз-
вивается у пациентов с автономией щитовидной железы, вызванной хронической йодной недостаточностью или автономными аденомами, возникшими на фоне диффузного токсического зоба. Предполагают, что поступление большого количества йода нарушает ауторегуляторные процессы в щитовидной железе. Хорошо изучен феномен Вольфа-Чайкова, происходящий в нормальной щитовидной железе, когда избыточное поступление йода приводит к блокаде синтеза тиреоидных гормонов. В случае йодиндуцированного тиреотоксикоза, наоборот, происходит усиление секреции и синтеза тиреоидных гормонов. Избыточное поступление йода может быть при использовании биологически активных пищевых добавок, местных антисептиков, ра- диофармпрепаратов, лекарственных средств (амиодарон), содержащих йод. При исследовании гормонального статуса определяют значительное повышение концентрации T4. При этом содержание T3 находится слегка повышено или в пределах нормы, отношение концентраций T4/T3 повышено. Лечение йодиндуцированного тиреотоксикоза в первую очередь требует прекращения поступления йода. Передозировка тиреоидных гормонов у детей в некоторых случаях может привести к клинически значимому тиреотоксикозу.
Тиреотоксикоз неаутоиммунного генеза - редкое заболевание, которое вызвано мутацией гена ТТГ. Рецептор ТТГ относят к семейству G-протеинсвязанных трансмембранных рецепторов. Структура гена ТТГ кодируется геном, расположенным на хромосоме 14. Проведённые исследования выявили у больных с врождённым неаутоиммунным тиреотоксикозом три активирующие мутации гена рецептора ТТГ. Выявленные мутации обусловливают конформационные изменения трансмембранных участков рецептора, что приводит к активации рецептора в отсутствии влияния ТТГ. Механизм спонтанной внутриклеточной активации приводит также к гиперплазии и гипертрофии тиреоцитов и, следовательно, к развитию зоба. У новорождённого с клиническими признаками тиреотоксикоза наличие активирующей мутации гена рецептора ТТГ можно предположить, если у матери отсутствуют заболевания щитовидной железы. При исследовании гормонального статуса отмечают повышение концентраций T3, T4, уменьшение содержания ТТГ, тиреоидстимулирующие АТ отсутствуют. Манифестация врождённого неаутоиммунного тиреотоксикоза во внутриутробном периоде может привести к тяжёлым последствиям, в связи с этим крайне важно пренатальное установление диагноза.
ЛЕЧЕНИЕ
Лечение начинают с применения тиреостатических препаратов и β-адреноблокаторов. Использование β-адреноблокаторов на первых этапах лечения болезни позволяет добиться быстрого клинического
эффекта: ребёнок становится спокойнее, уменьшаются ЧСС, потливость, тремор. Из группы β-адреноблокаторов применяют пропранолол в дозе 1 мг/кг/сут каждые 6-8 ч.
Антитиреоидные препараты назначают одновременно с β-адреноблокаторами. У детей из антитиреоидных препаратов применяют тиамазол и пропилтиоурацил. Эти препараты сходны по механизму действия и эффективности, а также по частоте и тяжести побочных явлений. Механизм действия антитиреоидных препаратов заключается в ингибировании присоединения йода к остаткам тирозина в тиреоглобулине, а также блокировании присоединения йодтирозиновых остатков к T4 и T3. Кроме того, пропилтиоурацил тормозит периферическую конверсию T4 в T3. Клинические данные в отношении иммуносупрессивного эффекта антитиреоидных препаратов противоречивы. Антитиреоидную терапию обычно начинают с максимальной дозы: 0,5-0,7 мг/кг/сут при применении тиамазола и 5-7 мг/кг/сут при ис- пользовании пропилтиоурацила. Обычно через 3-4 нед, а при тяжё- лом тиреотоксикозе через 6 нед β-адреноблокаторы отменяют и при стойком эутиреозе начинают снижать дозу тиреостатиков (тиамазола) по следующей схеме: один раз в неделю уменьшают суточную дозу на 5 мг; когда она составит 10 мг, её продолжают уменьшать на 2,5 мг в неделю. Поддерживающая доза тиамазола составляет 2,5-5 мг/сут и не изменяется в течение 2-3 лет терапии.
В тех случаях, когда применение тиреостатиков приводит к полной блокаде щитовидной железы, к терапии диффузного токсического зоба добавляют левотироксин натрия. К признакам переблокировки щитовидной железы относят увеличение объёма железы (по данным УЗИ) при отсутствии признаков рецидива тиреотоксикоза (зобогенный эффект); брадикардию, отёки, запоры (клинические признаки гипотиреоза), изолированное увеличение содержания ТТГ или в сочетании со снижением концентрации свободного T4. При этом, как правило, первым признаком переблокировки щитовидной железы служит на- растание концентрации ТТГ. В том случае, если удаётся диагностировать изолированное нарастание концентрации ТТГ, то можно начать с уменьшения дозы тиреостатиков.
Побочные эффекты антитиреоидных препаратов у детей наблюдают чаще, чем у взрослых. Наиболее часто диагностируют лейко- и тромбоцитопению, токсический гепатит, реже - лимфаденопатию, поли- невропатию, аллергические кожные реакции.
При развитии побочных эффектов производят замену препарата. В случаях, когда замена препарата не приводит к улучшению состояния, показано оперативное лечение.
Терапевтическая тактика нередко приводит к рецидивам тиреотоксикоза. Стойкой и длительной ремиссии удаётся достичь у 30-60%
детей, получающих консервативное лечение. Критериями полного выздоровления считают нормализацию размеров щитовидной железы, стойкий (в течение двух лет) клинический и гормональный эутиреоз, а также нормализацию титра антитиреоидных АТ.
Показания к оперативному вмешательству - отсутствие стабильной и длительной ремиссии тиреотоксикоза на фоне адекватно проводимой терапии, зоб больших размеров с признаками сдавления органов шеи, узлообразование, загрудинный зоб, невыполнение рекомендаций врача, невозможность избежать осложнений антитиреоидной терапии, манифестация диффузного токсического зоба в период беременности, выраженная офтальмопатия. Объём операции: субтотальная, субфасциальная резекция щитовидной железы.
ПРОФИЛАКТИКА
Учитывая аутоиммунную природу заболевания, необходима профилактика острых и хронических инфекционных заболеваний. Специфических мер профилактики не разработано.
ПРОГНОЗ
Консервативное лечение приводит к выздоровлению в 30-50% случаев. Более чем в половине случаев наблюдают рецидив заболевания. Правильно проведённое хирургическое лечение (тотальная тиреоидэктомия) приводит к ликвидации диффузного токсического зоба, но у ребёнка развивается гипотиреоз, требующий пожизненной заместительной терапии левотироксином натрия.
Надпочечниковая недостаточность
Надпочечниковая недостаточность (гипокортицизм) - симптомокомплекс, обусловленный сниженной выработкой гормонов коры надпочечников. По уровню поражения выделяют первичную надпо- чечниковую недостаточность, связанную с патологией самого надпочечника, вторичную, связанную со сниженной секрецией АКТГ и третичную, вызванную нарушением секреции кортикотропин-рилизинг гормона или других факторов, стимулирующих выработку АКТГ. Две последние формы называют ещё центральными.
Симптомы надпочечниковой недостаточности не специфичны и могут скрываться под маской различных заболеваний. Слабость, по- вышенная утомляемость, плохой аппетит, низкая прибавка массы тела у маленьких детей и потеря массы тела у детей старшего возраста характерны для многих заболеваний. Тошнота, многократная рвота, жидкий стул, боли в животе расценивают как проявления кишечных инфекций. У новорождённых и детей младшего возраста при-
знаком надпочечниковой недостаточности может быть гипогликемия. Выраженные гиперпигментации не всегда сопутствуют даже первичной надпочечниковой недостаточности. Учитывая отсутствие специ- фической клинической картины, надпочечниковую недостаточность редко диагностируют до развития сольтеряющих кризов, опасных для жизни больного. При своевременной диагностике надпочечниковую недостаточность успешно компенсируют заместительной терапией.
Этиология
Первичная надпочечниковая недостаточность
Ранее наиболее частой причиной первичного гипокортицизма считали туберкулёзное поражение надпочечников. До сих пор в некоторых регионах мира надпочечниковая недостаточность туберкулёзной этиологии занимает второе место после аутоиммунного поражения среди взрослых и детей старшего возраста. Гипокортицизм у детей младшего возраста чаще связан с отклонениями в развитии надпочечников и врождёнными нарушениями стероидогенеза. Выделяют 3 группы причин первичной надпочечниковой недостаточности.
• Врождённые нарушения развития надпочечников:
- врождённая гипоплазия надпочечников;
- дефект стероидогенного фактора 1;
- резистентность к АКТГ;
- семейная глюкокортикоидная недостаточность I и II типа;
- синдром Оллгрова (синдром 3А).
• Деструкция надпочечников:
- аутоиммунного генеза (аутоиммунные полигландулярные синдромы);
- адренолейкодистрофия;
- кровоизлияния в надпочечники;
- метастатическое поражение надпочечников;
- инфекционное поражение надпочечников (в том числе туберкулёзное);
- амилоидоз.
• Врождённые нарушения стероидогенеза:
- врождённая дисфункция коры надпочечников;
- митохондриальные болезни;
- дефицит ферментов метаболизма холестерина;
- синдром Смита-Лемли-Опица.
Относительная частота различных нозологических форм меняется в зависимости от пола и возраста пациентов.
• При рождении кровоизлияния в надпочечники вследствие гипоксии или сепсиса - самая частая причина развития острой надпочечниковой недостаточности.
• В период новорождённости и в младшем возрасте первое место среди причин гипокортицизма занимают различные формы врождён- ной дисфункции коры надпочечников у детей обоего пола и врож- дённая гипоплазия надпочечников у мальчиков.
• В старшей возрастной группе, как и у взрослых, наиболее распространены аутоиммунный полигландулярный синдром и адренолейкодистрофия. С возрастом растёт доля инфекционного и метастатического поражения надпочечников.
Вторичная и третичная надпочечниковая недостаточность Центральные формы гипокортицизма обусловлены дефицитом кортикотропин-рилизинг гормона и/или АКТГ. Все причины, вызывающие вторичную и третичную надпочечниковую недостаточность, можно разделить на 3 группы.
• Врождённый вторичный гипокортицизм:
- изолированный дефицит АКТГ;
- врождённый гипопитуитаризм.
• Деструкция гипоталамо-гипофизарных структур:
- опухоли ЦНС;
- черепно-мозговая травма;
- инфильтративные процессы;
- инфекционное поражение;
- хирургическое вмешательство;
- облучение головы.
• Ятрогенная супрессия гипоталамо-гипофизарно-надпочечниковой
системы экзогенными глюкокортикоидами.
Патогенез
Первичная надпочечниковая недостаточность
При первичной надпочечниковой недостаточности выпадает секреция всех 3 групп надпочечниковых гормонов: глюкокортикоидов, минералокортикоидов и андрогенов (в зависимости от формы). Поскольку глюко- и минералокортикоиды участвуют в поддержании гомеостаза, белкового, углеводного, жирового и водно-электролитного обмена, их недостаточность вызывает многочисленные расстройства.
• Кортизол активно участвует в углеводном обмене, действуя как антагонист инсулина. При дефиците кортизола снижаются синтез гликогена в печени, глюконеогенез и повышается чувствительность периферических тканей к инсулину. Все вышеперечисленное обусловливает развитие гипогликемии. Вероятность развития гипогликемии повышается при сопутствующем дефиците катехоламинов или недостаточной секреции СТГ, характерных для некоторых форм надпочечниковой недостаточности.
• Дефицит альдостерона приводит к выраженным сердечно-сосудистым нарушениям. Снижение реабсорбции натрия и повышение реабсорбции калия в почках приводят к гипонатриемии, гипокалиемии, уменьшению ОЦК и, как следствие, к развитию артериальной гипотензии вплоть до шока. Положение усугубляет гиперкалиемия, вызывающая нарушения сердечного ритма и миопатии. Дефицит глюкокортикоидов вносит свой вклад в развитие артериальной гипотензии, возможно, в результате снижения чувствительности сосудистой стенки к ангиотензину и норадреналину, а также в результате увеличения синтеза ПгI2. Снижение реабсорбции натрия в кишечнике вызывает диспептические расстройства, такие как боли в животе, нарушения всасывания.
• Дефицит надпочечниковых андрогенов способствует усилению катаболических процессов и ведёт к повышению уровня остаточного азота. В связи с недостаточной секрецией надпочечниковых андрогенов у детей отмечают задержанное адренархе.
При первичной надпочечниковой недостаточности низкое содержание кортизола по механизму отрицательной обратной связи приво- дит к повышению концентраций кортикотропин-рилизинг гормона, АКТГ и других производных проопиомеланокортина. В свою очередь, высокие концентрации АКТГ, меланоцит-стимулирующего гормона, воздействуя на рецепторы меланоцитов, вызывают повышение синтеза меланина, что проявляется клинической картиной гиперпигментации.
Вторичная и третичная надпочечниковая недостаточность
В отношении патогенеза к отличительным чертам центральных форм гипокортицизма относят отсутствие минералокортикоидной недостаточности и гиперпигментации. Поскольку регуляция синтеза альдостерона находится под контролем ренин-ангиотензиновой системы и лишь в малой степени зависит от содержания АКТГ, симптомов потери соли у пациентов с центральным гипокортицизмом не отмечают. Снижение концентрации АКТГ и других производных проопиомеланокортина объясняет отсутствие гиперпигментаций при вторичной и третичной надпочечниковой недостаточности.
Клиническая картина
Время возникновения первых симптомов, как и сами клинические симптомы надпочечниковой недостаточности, зависит от этиологи- ческого фактора.
При дисгенезе надпочечников, нарушении стероидогенеза и псевдогипоальдостеронизме признаки заболевания возникают вскоре после рождения и в первую очередь связаны с потерей соли, т.е. с дефици- том минералокортикоидов. У пациентов отмечают неукротимую рвоту
«фонтаном», нарастание гипотрофии, возникают признаки дегидратации вплоть до развития сосудистого коллапса. При отсутствии заместительной терапии такие пациенты погибают в неонатальном периоде.
У детей старшего возраста основной причиной гипокортицизма считают деструктивный процесс в надпочечниках. В этом случае клиническая картина развивается постепенно, по мере гибели клеток коры надпочечников. Первые клинические симптомы возникают только после деструкции более 90% всех клеток. В начале пациенты предъявляют жалобы на повышенную утомляемость, мышечную слабость, ухудшение аппетита, постуральные головокружения. Больные плохо переносят физические нагрузки, но их самочувствие улучшается в горизонтальном положении. При несвоевременной диагностике и отсутствии лечения у таких пациентов может развиться сольтеряющий криз. Первые симптомы острой надпочечниковой недостаточности - тошнота, рвота, жидкий стул, боли в животе, артериальная гипотензия. Довольно быстро в последующем развиваются дегидратация и шок.
У пациентов с диагностированной надпочечниковой недостаточностью, получающих лечение глюко- и минералокортикоидами, соль- теряющие кризы могут развиваться при присоединении инфекций, обширных травмах или проведении оперативных вмешательств без соответствующей коррекции терапии. У некоторых детей эмоциональный стресс может провоцировать развитие адреналового криза. Приём препаратов, ускоряющих метаболизм кортизола (левотироксина натрия, барбитуратов), также может привести к декомпенсации заболевания.
Характерным признаком первичной надпочечниковой недостаточности являются гиперпигментации, развивающиеся вследствие по- вышения концентрации АКТГ. Часто окружающие впервые замечают гиперпигментацию на открытых участках тела (лице, кистях рук). Максимальное развитие гиперпигментации наблюдают на коже наружных половых органов, подмышечных областей, коленей, локтей, также пигментируются соски, пупок, перианальная область и рубцы на месте повреждения кожи. Участки гиперпигментации могут быть на слизистых оболочках полости рта.
Первым признаком надпочечниковой недостаточности у детей может быть гипогликемия. Гипогликемия может сопровождаться кетозом, что служит поводом к диагностике кетотической гипогликемии. Многим детям назначают противосудорожную терапию, не распознав гипогликемического генеза судорог. Развитие гипогликемий особенно характерно для семейной глюкокортикоидной недостаточности и центральных форм гипокортицизма (вторичной и третичной надпочечниковой недостаточности).
При центральных формах гипокортицизма никогда не бывает гиперпигментаций, так как концентрация АКТГ всегда низкая. Для вто- ричной и третичной надпочечниковой недостаточности не характерен минералокортикоидный дефицит и, следовательно, симптомов потери соли у таких пациентов не будет. Остальные клинические признаки общие для первичной, вторичной и третичной надпочечниковой недостаточности.
Лабораторные и инструментальные исследования
Лабораторным подтверждением диагноза надпочечниковой недостаточности являются электролитные нарушения (гипонатриемия, гиперкалиемия) и данные гормональных исследований: снижение концентрации базального кортизола, альдостерона и повышение активности ренина плазмы крови. Необходимо уточнить, что исследование концентраций кортизола и альдостерона неинформативно при самой частой причине гипокортицизма в период новорождённости - врождённой дисфункции коры надпочечников (недостаточности 21-гидроксилазы). Высокое содержание предшественников кортизола за счёт перекрёстных реакций при используемых методах анализа может давать ложные высокие концентрации кортизола и альдостерона. При подозрении на врождённую дисфункцию коры надпочечников необходимо исследовать содержание промежуточных продуктов стероидогенеза, характерных для каждой из форм этого заболевания (см. раздел «Врождённая дисфункция коры надпочечников»).
Определение концентрации базального кортизола
При обследовании детей старшей возрастной группы с подозрением на гипокортицизм первым шагом должно быть определение концентрации базального кортизола в плазме крови. Это исследование не- обходимо проводить рано утром в 6.00-8.00 часов, что соответствует физиологическому пику секреции глюкокортикоидов. Концентрация кортизола в плазме крови менее 80 нмоль/л подтверждает наличие гипокортицизма, 80-150 нмоль/л - подозрительна на гипокортицизм, более 500 нмоль/л - исключает гипокортицизм.
Исследование содержания свободного кортизола в суточной моче может оказаться более информативным, так как в этом случае будут оценивать ещё и интегральную секрецию кортизола.
Одновременно с определением концентрации кортизола необходимо исследовать содержание электролитов крови и активность ренина плазмы, которые служат лабораторными критериями минералокортикоидной недостаточности.
Стимуляционный тест с АКТГ
При подозрении на надпочечниковую недостаточность следующим этапом диагностики должен стать стимуляционный тест с АКТГ.
Общепринятой признана короткая проба с АКТГ. Исходно берут кровь для определения концентрации кортизола, после чего внутри- венно вводят 250 мкг тетракозактида (синтетического аналога АКТГ) в 5 мл 0,9% раствора натрия хлорида, продолжительность инфузии составляет 2 мин. Затем через 30 и 60 мин берут кровь для повторного определения концентрации кортизола. В норме содержание кортизола на стимуляции превышает 500 нмоль/л. При первичной надпочечниковой недостаточности реакция на стимуляцию отсутствует или снижена, концентрация кортизола меньше 500 нмоль/л. При вторичной надпочечниковой недостаточности у большинства больных реакция на АКТГ отсутствует или снижена. Исключение составляют пациенты с недавно развившимся центральным гипокортицизмом.
При отсутствии препаратов АКТГ короткого действия возможно проведение аналогичной пробы с пролонгированными формами тет- ракозактида («Синактен-депо»). После внутримышечного введения 1 мл препарата кровь для определения концентрации кортизола берут через 10 и 24 ч. Результаты оценивают аналогично результатам теста с АКТГ короткого действия.
Вышеописанные методы исследования позволяют подтвердить наличие у пациента надпочечниковой недостаточности. Следующим шагом необходимо дифференцировать первичную надпочечниковую недостаточность от центральных форм. С этой целью показано исследование базальной концентрации АКТГ в плазме крови.
Определение базальной концентрации АКТГ
При первичной надпочечниковой недостаточности концентрация АКТГ превышает 100 пг/мл, тогда как при вторичной надпочечни- ковой недостаточности содержание АКТГ снижено или находится в пределах нормы.
Диагностика и дифференциальня диагностика
У новорождённых и детей младшего возраста первичная надпочечниковая недостаточность имеет яркие клинические признаки за счёт быстрого прогрессирования симптомов потери соли в сочетании с гиперпигментацией. Большую помощь в диагностике гипокортицизма у этой группы детей оказывает подробное выяснение семейного анамнеза и наличие сопутствующей патологии, характерной для различных нозологических форм (повторных случаев заболевания в семье, ранней смерти детей в младенчестве при сходной клинической картине).
Лечение
Заместительную терапию проводят глюко- и минералокортикоидами (в зависимости от формы).
Заместительная терапия глюкокортикоидами
Препаратом выбора у детей служит гидрокортизон - аналог природного гормона надпочечников кортизола. Физиологическая секреция кортизола у детей и подростков в среднем составляет 6-8 мг/м2/ сут. Доза заместительной терапии гидрокортизоном с учётом всасывания и метаболической биодоступности при пероральном приёме будет составлять 10-12 мг/м2/сут, равномерно разделённых на три приёма.
Индивидуальная потребность в этом препарате у различных пациентов колеблется от 5 до 20 мг/м2/сут. Подбор адекватной дозы глюкокортикоидов проводят в первую очередь на основании клинической картины. При недостаточной дозе гидрокортизона у больных отмечают слабость, гиперпигментацию, постуральную артериальную гипотензию, повышенную чувствительность к инфекционным заболеваниям и, кроме того, существует риск развития ночной гипогликемии. К признакам передозировки глюкокортикоидов относят торможение темпов роста и скелетного созревания, избыточную прибавку массы тела, появление стрий на коже, повышение АД и остеопороз. Многих побочных эффектов можно избежать, заменив короткоживущий гидрокортизон на препарат с длительным сроком действия (преднизолон, дексаметазон). Лабораторным критерием адекватности дозы глюкокортикоидов служит нормализация концентрации АКТГ в сыворотке крови.
Заместительная терапия минералокортикоидами
Терапию минералокортикоидами проводят больным с первичной надпочечниковой недостаточностью и изолированным гипоальдосте- ронизмом. Назначают препарат флудрокортизон в дозе 0,05-0,2 мг/сут. Ориентироваться в подборе дозы заместительной терапии, как и в случае с глюкокортикоидами, необходимо на клиническую картину. При недостатке минералокортикоидов у больных отмечают низкое АД, повышенную потребность в соли, тахикардию, у грудных детей - срыгивания. Лабораторными критериями минералокортикоидной недостаточности будут повышение концентрации ренина и склонность к гиперкалиемии. К симптомам передозировки препарата относят ар- териальную гипертензию, брадикардию, замедление скорости роста и подавление активности ренина плазмы крови. Повышение дозы флудрокортизона может потребоваться летом у пациентов, проживающих в условиях жаркого климата, в связи с тем, что недостаток альдостерона приводит к повышенному выведению натрия через потовые железы. Грудным детям в дополнение к минералокортикоидам обычно необходимо добавление к пище хлористого натрия или поваренной соли (1-2 г/сут).
Эндогенная секреция кортизола у здоровых людей повышается в случаях интеркурентных заболеваний и оперативных вмешательств.
Пациентам с надпочечниковой недостаточностью необходимо увеличивать дозу глюкокортикоидов в 2-3 раза равномерно в течение суток в случае инфекционного заболевания с лихорадкой или при проведении травматичных медицинских процедур (экстракции зуба, ФЭГДС, профилактических прививок). При невозможности перорального при- ёма препаратов необходимо внутримышечное или внутривенное введение гидрокортизона в увеличенной дозе. Дозу минералокортикоидов оставляют неизменённой.
Залогом успешной терапии надпочечниковой недостаточности является обучение пациента и его родителей правилам коррекции дозы препаратов. Каждый пациент должен иметь при себе идентификационную карту с указанием диагноза, получаемой терапии и телефона медицинского учреждения, где его наблюдают.
Терапия адреналового криза
При декомпенсации заболевания у пациентов с гипокортицизмом лечебные мероприятия необходимо начинать незамедлительно. В первую очередь усилия врача должны быть направлены на коррекцию водно-электролитных нарушений. Проводят массивную инфузионную терапию растворами натрия хлорида 0,9% и глюкозы 5-10% из расчёта 450 мл/м2 в течение первого часа, затем по 3000 мл/м2/сут. Одновременно внутривенно вводят гидрокортизон в дозе 2-5 мг/кг каждые 4 часа. После достижения стабильного состояния и коррекции электролитных нарушений больного переводят на пероральный приём гидрокортизона и добавляют к терапии флудрокортизон.
Прогноз
При своевременной диагностике и адекватном лечении возможно достижение нормальных продолжительности и качества жизни.
ОСТРАЯ НАДПОЧЕЧНИКОВАЯ НЕДОСТАТОЧНОСТЬ
Острая надпочечниковая недостаточность, или аддисонический криз, - неотложное, опасное для жизни состояние, характеризующе- еся гемодинамическими и метаболическими нарушениями на фоне резкого снижения концентрации гормонов коры надпочечников (глюко- и минералокортикоидов).
Этиология
Острая надпочечниковая недостаточность может быть обусловлена как первичным поражением надпочечников, так и нарушениями гипоталамо-гипофизарно-надпочечниковой регуляции (дефицит кор- тикотропин-рилизинг гормона или АКТГ после хирургических вмешательств в гипоталамо-гипофизарной области, при гипофизарном нанизме).
Нередко адреналовый криз относят к дебюту ранее не установленной хронической надпочечниковой недостаточности, проявившей себя в стрессовой для организма ситуации. У пациентов, страдающих хронической надпочечниковой недостаточностью любой этиологии, может возникать острая декомпенсация заболевания с развитием адреналового криза на фоне неадекватной заместительной терапии, острых интеркуррентных заболеваний, хирургических вмешательств, стресса.
Однако существуют также причины внезапного развития деструкции коры надпочечников, к которым относят тромбоэмболию, тромбоз надпочечниковых вен, геморрагический инфаркт, септический некроз, токсическое поражение. К факторам риска развития острой надпочечниковой недостаточности относят хирургические вмешательства, ожоговые травмы, сепсис любой этиологии, массивную антикоагулянтную терапию и коагулопатии различного генеза. Синдром Уотерхауса-Фридерихсена был описан как двустороннее кровоизлияние в надпочечники на фоне менингококцемии. В группу высокого риска входят пациенты с хроническими заболеваниями, сопровождающимися коагулопатией (СКВ, геморрагические васкулиты, АФС и др.).
У пациентов, получающих высокие лечебные дозы глюкокортикоидов, также может развиться острая надпочечниковая недостаточность при внезапной отмене или резком снижении дозы препаратов. К ятрогенным причинам адреналового криза относят осложнения лечения гиперкортицизма (болезни или синдрома Иценко-Кушинга): после адреналэктомии или аденомэктомии, а также на фоне лекарственной терапии блокаторами стероидогенеза (аминоглутетимид, кетоконазол, митотан).
Клиническая картина
Клиническая картина острой надпочечниковой недостаточности не зависит от причин её возникновения. Более того, её симптомы неспецифичны и присущи многим неотложным состояниям, что значительно затрудняет своевременную диагностику в условиях ургентной ситуации.
Гемодинамические нарушения характеризуются резкой артериальной гипотензией вплоть до сосудистого коллапса, тахикардией, акроцианозом, анурией. Повторная рвота, частый жидкий стул, боль в животе имитируют симптомы «острого живота» или ПТИ. Неврологические нарушения, включающие головную боль, менингеальные симптомы, судороги, острый психоз, супор и кому, могут быть обусловлены как интеркуррентным или фоновым заболеванием, так и гипогликемией в результате острого дефицита глюкокортикоидов.
Ни один из этих симптомов не является строго патогномоничным для острой надпочечниковой недостаточности. Они могут присутствовать в любом сочетании и выражаться в различной степени.
Диагностика
Клиническая диагностика острой надпочечниковой недостаточности при отсутствии указаний в анамнезе на хроническое заболевания надпочечников часто бывает затруднительна в критической ситуации, поэтому необходимо тщательно расспросить пациента или его близких о возможных факторах риска развития адреналового криза.
Лабораторная диагностика в условиях неотложного состояния, требующего незамедлительной коррекции, также весьма ограничена. К основным лабораторным маркёрам относят повышенное содержание калия, низкие концентрации натрия и глюкозы крови. Образцы крови для исследования содержания кортизола, АКТГ и активности ренина плазмы должны быть взяты, однако терапию необходимо начинать, не дожидаясь результатов.
Изменения на ЭКГ отражают метаболические нарушения, в частности гиперкалиемию, и представлены высоким зубцом Т, замедлением атриовентрикулярной проводимости с расширением зубца Р, удлинением интервала S-Т и расширением комплекса QRS.
Лечение
Лечение острой надпочечниковой недостаточности необходимо начинать незамедлительно при малейшем подозрении на её наличие. Учитывая высокую летальность в первые сутки от начала развития адреналового криза и обратимость возможных осложнений от массивной кратковременной терапии глюкокортикоидами, не стоит опасаться гипердиагностики.
Необходимо немедленно установить катетер в центральную вену, чтобы обеспечить условия для внутривенных вливаний. После забора образцов крови для биохимического и гормонального исследований тут же необходимо приступить к введению глюкокортикоидов и 0,9% раствора натрия хлорида с целью коррекции гемодинамики. Среди глюкокортикоидных препаратов предпочтение отдают гидрокортизону. Вначале внутривенно струйно вводят 100 мг гидрокортизона, затем под контролем содержания калия, натрия и АД следует продолжить капельное введение гидрокортизона. Доза гидрокортизона может доходить до 1000 мг в первые сутки лечения. Введение минералокортикоидов считают нецелесообразным ввиду минералокортикоидной активности гидрокортизона. Кроме того, масляный раствор дезоксикортона («Дезоксикортикостерона») реализует свой эффект лишь спустя несколько часов после внутримышечного введения. Терапию
глюкокортикоидами осуществляют на фоне массивной регидратации. За первые сутки может быть введено от 2 до
Учитывая высокую вероятность гипогликемии, в схему лечения включают введение 5-20% раствора глюкозы по возможности под контролем её содержания в плазме крови.
В зависимости от этиологии острой надпочечниковой недостаточности проводят лечение основного заболевания (антибиотикотерапию и др.).
Летальность в результате адреналового криза составляет около 50% и приходится на первые сутки заболевания.
После стабилизации состояния следует продолжить парентеральное введение глюкокортикоидов и 0,9% раствора натрия хлорида в течение нескольких дней под контролем гемодинамических показателей и содержания электролитов. Затем постепенно снижают дозу глюкокортикоидов и переходят к пероральному приёму препаратов, дополняя заместительной терапией минералокортикоидами (флудрокортизон) в заместительных дозах. Далее необходимо уточнить наличие хронической надпочечниковой недостаточности и перейти к постоянной адекватной заместительной терапии.
Профилактика
Основной задачей первичной профилактики аддисонического криза, а следовательно, смертности в группе пациентов с хронической надпочечниковой недостаточностью является своевременная адекватная терапия основного заболевания. Необходимо осуществлять регулярный медицинский контроль за адекватностью заместительной терапии. Огромную роль играет обучение пациентов и членов их семей самоконтролю, правилам поведения в различных ситуациях, потенциально провоцирующих развитие адреналового криза. При интеркуррентных заболеваниях, травмах, хирургических вмешательствах, стрессе дозу глюкокортикоидов следует увеличивать в 2 раза с последующим постепенным возвратом к прежней заместительной дозе. В мировой практике рекомендуют ношение специальных браслетов, на которых отражена информация о болезни пациента и основные пункты оказания скорой помощи: введение глюкокортикоидов, экстренная госпитализация для дальнейшей квалифицированной медицинской помощи.
ИЗОЛИРОВАННАЯ МИНЕРАЛОКОРТИКОИДНАЯ
НЕДОСТАТОЧНОСТЬ
Изолированный гипоальдостеронизм в отсутствии дефицита других гормонов коры надпочечников проявляется клинической картиной
потери соли. При данной патологии, в отличие от других форм надпочечниковой недостаточности, система гипоталамус-гипофиз-надпочечники интактна. Снижение концентрации альдостерона приводит к повышению активности только ренин-ангиотензиновой системы.
Выделяют 3 группы изолированной минералокортикоидной недостаточности:
• Врождённый первичный гипоальдостеронизм.
• Приобретённый вторичный дефицит альдостерона.
• Псевдогипоальдостеронизм.
Врождённый первичный гипоальдостеронизм
Врождённый первичный гипоальдостеронизм - редкая аутосомнорецессивная патология, характеризующаяся синдромом потери соли и задержкой физического развития. Данное заболевание обусловлено нарушением биосинтеза альдостерона вследствие дефицита фермента альдостеронсинтазы. В отличие от дефицита других ферментов стероидогенеза, дефицит альдостеронсинтазы не приводит к гиперплазии надпочечников, так как синтез кортизола при данном синдроме не нарушен. На рис. 18-4 представлены два последних этапа биосинтеза альдостерона, протекающие в клубочковой зоне коры надпочечников.
Известно, что синтез кортикостеронметилоксидазы (КМО) 1 и 2 типов происходит с одного гена CYP11B2, расположенного на хромосоме 8 (8q21). В зависимости от конкретной мутации поражается либо 18-гид- роксилазная активность фермента, либо альдегидсинтазная активность. Следовательно, выделяют 2 формы дефицита альдостеронсинтазы: недостаточность КМО-1 и КМО-2. Эти формы заболевания отличаются друг
от друга только гормональным профилем. При дефиците КМО-1 определяют низкие концентрации как альдостерона, так и 18-гид- роксикортикостерона, тогда как при дефиците КМО-2 значительно повышено содержание 18- гидроксикортикостерона, а концентрация альдостерона низкая. Дифференциально-диагностическим критерием этих состояний служит отношение 18-гидрокси- кортикостерона к альдостерону: при дефиците КМО-1 этот пока- затель меньше 10, а при дефиците КМО-1 - превышает 100.
Клинические признаки не зависят от формы заболевания.
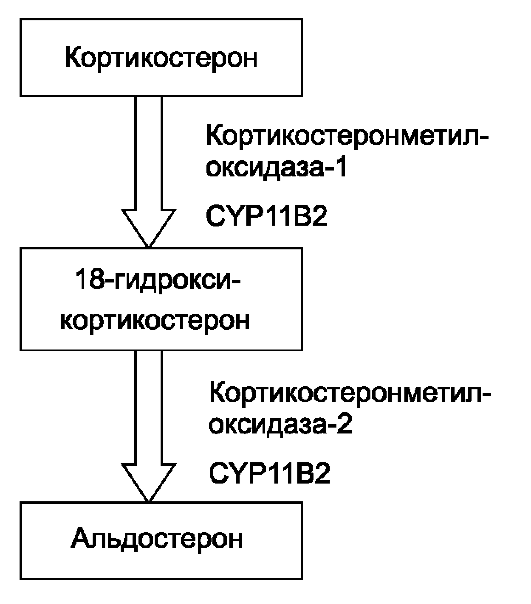
Рис. 18-4. Биосинтез альдостерона.
У новорождённых возникает рвота. Ребёнок отказывается от пищи, перестает прибавлять в массе тела - развивается дегидратация. По данным лабораторных исследований, отмечают гиперкалиемию, иногда гипонатриемию, высокую активность ренина плазмы крови. У данных пациентов с возрастом наблюдают положительную динамику относительно синдрома потери соли, однако отмечают задержку физического развития. При лечении пациентов с дефицитом альдостеронсинтазы используют добавление к пище хлористого натрия (поваренной соли) и минералокортикоидные препараты (флудрокортизон в дозе 0,05-0,1 мг/сут).
Приобретённый вторичный дефицит альдостерона
Приобретённый вторичный дефицит альдостерона обусловлен снижением биосинтеза ренина в почках. В результате отсутствия сти- муляции клубочковой зоны надпочечников ренин-ангиотензиновой системой снижается синтез альдостерона. Данное заболевание характеризуется хронической бессимптомной гиперкалиемией и некоторым снижением функций почек. Однако у части пациентов могут развиваться мышечная слабость и сердечные аритмии. Вторичный гипорениновый гипоальдостеронизм наблюдают у пациентов с СД, при СКВ, миеломной болезни, почечном амилоидозе, циррозе печени, серпо- видноклеточной анемии, вегетативной полиневропатии и СПИДе.
Причиной заболевания является поражение юкстагломерулярного аппарата почек, что приводит к сниженной секреции ренина. Существует несколько теорий, объясняющих гипоренинемию. Например, длительно существующая гиперволемия приводит к необратимому подавлению функции юкстагломерулярного аппарата. Другой причиной гипоренинемии может быть недостаточная активность вегетативной нервной системы, например у пациентов с СД. Третьим возможным механизмом может стать нарушение преобразования проренина в ренин вследствие дефицита калликреина или ПгI2.
Для пациентов с гипорениновым гипоальдостеронизмом характерна особая форма почечного тубулярного ацидоза. В развитии ацидоза играет роль не только дефицит минералокортикоидов, но и гиперкалиемия, которые снижают почечный аммониогенез, уменьшают секреторную активность дистального отдела нефрона для ионов Н+.
Диагноз вторичного гипоренинемического гипоальдостеронизма необходимо рассматривать у всех пациентов с хронической гипокалиемией. Клинический диагноз подтверждают низкое содержание альдостерона в крови в сочетании с низкой плазменной активностью ренина. Результаты стимуляционных диагностических тестов, вызывающих активацию ренин-ангиотензин-альдостероновой системы (ортостатическая проба, проба с фуросемидом), отрицательные.
Терапия гипоренинового гипоальдостеронизма направлена на коррекцию гиперкалиемии. У пациентов с умеренной гиперкалиемией без изменений на ЭКГ необходимо проводить мониторирование электролитов и ЭКГ. Таким пациентам нужно рекомендовать диету с ограничением калия и предостерегать их от приёма препаратов, вызывающих гиперкалиемию (β-адреноблокаторы, ингибиторы АПФ, гепарин натрия, калийсберегающие диуретики, ингибиторы циклооксигеназы). Терапию минералокортикоидами проводят пациентам с выраженной гиперкалиемией, без артериальной гипертензии и застойной сердечной недостаточности.
Псевдогипоальдостеронизм
Псевдогипоальдостеронизм - состояние, характеризующееся клинической картиной синдрома потери соли, но сопровождающееся высокими концентрациями альдостерона и ренина. Причиной дан- ной патологии считают нарушение механизма действия альдостерона. Выделяют псевдогипоальдостеронизм с аутосомно-рецессивным типом наследования, причиной которого является патология амилоридчувствительных натриевых каналов в дистальных отделах нефрона, что приводит к повышенной экскреции натрия из организма. Выявлены мутации в генах, кодирующих α- (SCNN1A), β- (SCNN1B) и γ-субъединицы (SCNN1G) амилорид-чувствительного натриевого канала, расположенные на хромосомах 12 (12p13) и 16 (16р13-р12).
При аутосомно-доминантных и спорадических формах заболевания причиной псевдогипоальдостеронизма является патология минералокортикоидного рецептора, ген которого расположен на коротком плече хромосомы 4 (4q31.1).
Отличительной особенностью клинической картины псевдогипоальдостеронизма, вызванного патологией натриевого канала, считают отсутствие поражения других минералокортикоидчувствительных тканей (потовые железы, кишечник). Лабораторно-диагностическими критериями данной патологии служит гиперкалиемия в сочетании с высоким содержанием альдостерона и ренина крови.
Минералокортикоиды в терапии псевдогипоальдостеронизма не эффективны, так как нарушен механизм самого действия альдостерона. Лечение таких пациентов сводится к возмещению потерь соли и воды.
Врождённая дисфункция коры надпочечников
Врождённая дисфункция коры надпочечников (адреногенитальный синдром, врождённая надпочечниковая гиперплазия) - группа заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из энзимов или транспортных белков,
принимающих участие в биосинтезе кортизола в коре надпочечников. Снижение биосинтеза кортизола по принципу обратной связи приводит к повышению секреции АКТГ и, как следствие, к развитию гиперплазии коры надпочечников и накоплению метаболитов, предшествующих дефектному этапу стероидогенеза. В основе ферментативных нарушений лежат дефекты генов, кодирующих тот или иной фермент биосинтеза стероидов.
В зависимости от того, какой фермент стероидогенеза выпадает, различают 5 основных форм этого заболевания. Наиболее тяжёлой формой считают липоидную гиперплазию надпочечников, обусловленную дефектом StAR-протеина. При этой форме заболевания практически отсутствует синтез всех гормонов коры надпочечников, и ранее считали, что это несовместимо с жизнью. Наиболее часто диагностируют заболевание, обусловленное недостаточностью фермента 21-гидроксилазы. На долю этой формы приходится 75% всех случаев заболевания. Более редко наблюдают дефект 3-гидроксистероиддегид- рогеназы, недостаточность 17а-гидроксилазы или 11b-гидроксилазы. На рис. 18-5 представлена схема стероидогенеза. Клиническая картина заболевания зависит от места блока синтеза стероидов. Понятно, что будут наблюдать дефицит стероидов ниже блока и, наоборот, избыток стероидов, которые синтезируются до блока.
Недостаточность 21-гидроксилазы - одно из самых частых врож- дённых ферментативных нарушений стероидогенеза. Заболеваемость классическими вариантами болезни в различных популяциях колеб- лется от 1:10 000 до 1:18 000 новорождённых. Чрезвычайно высокая заболеваемость определена в двух изолированных популяциях: у эскимосов западной Аляски -1:280 и у жителей острова Ла Руньон в Индийском океане - 1:2100. Заболевание наследуется по аутосомнорецессивному типу.
Этиология
Дефект 21-гидроксилазы обусловлен многочисленными мутациями гена, кодирующего этот фермент - CYP21. Ген расположен на коротком плече хромосомы 6.
Патогенез
21-гидроксилаза - микросомальный Р450-зависимый фермент, принимающий участие в биосинтезе кортизола и минералокортикоидов, трансформируя 17а-гидроксипрогестерон в 11-дезоксикортизол и прогестерон в дезоксикортикостерон. Недостаточность 21-гидрокси- лазы приводит к снижению продукции кортизола, что вызывает повы- шение секреции АКТГ и приводит к гиперплазии коры надпочечников. Надпочечники активно секретируют стероиды, предшествующие
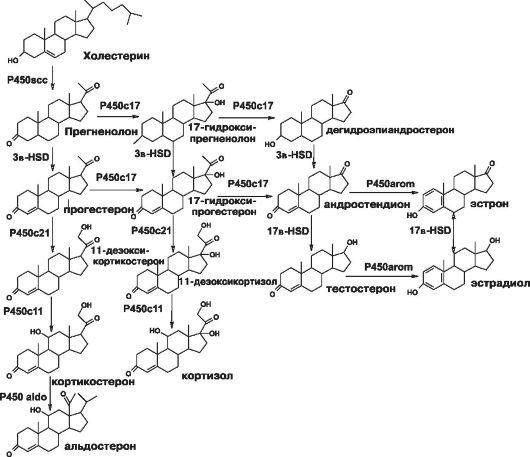
Рис. 18-5. Схема стероидогенеза.
ферментативному блоку: 17а-гидроксипрогестерон и андрогены, биосинтез которых не зависит от 21-гидроксилазы.
Минералокортикоидную недостаточность различной степени выраженности диагностируют у 75% детей с 21-гидроксилазным дефицитом. Снижение концентраций дезоксикортикостерона и альдостерона приво- дит к уменьшению реабсорбции натрия в почках. В связи с этим концентрация натрия в сыворотке крови падает и возрастает почечная реабсорбция калия. В результате этих нарушений развивается гипонатриемия, гиперкалиемия, ацидоз, обезвоживание. В ответ на снижение выработки минералокортикоидов возрастает рениновая активность плазмы.
Клиническая картина Пренатальная вирилизация
Классический вариант 21-гидроксилазной недостаточности приводит к выраженной гиперандрогении, формирующейся ещё внутриут-
робно. Предполагают, что активная вирилизация плода начинается с 20-25-й недели гестации, когда формируется влияние АКТГ на эмбриональный надпочечник и начинает синтезироваться кортизол. Внутриутробная гиперандрогения приводит к активной вирилизации наружных половых органов, что наиболее драматично выражено у девочек. К моменту рождения наружные половые органы девочки имеют бисексуальное строение: клитор гипертрофирован, отмечают сращение скротолабиального (мошоночного) шва различной степени выраженности. В некоторых случаях внутриутробная андрогенизация настолько выражена, что наружные половые органы практически соответствуют мужским, и девочку ошибочно регистрируют и воспитывают как мальчика (рис. 18-6 на вклейке).
Выделяют 5 степеней вирилизации наружных половых органов по Прадеру. Точкой отсчёта считают нормальные наружные половые органы девочки (вирилизация отсутствует). Максимальная степень вирилизации соответствует нормальному строению наружных половых органов мальчика.
I степень вирилизации - гипертрофия клитора и нормальный вход
во влагалище.
II степень вирилизации - гипертрофия клитора и частичное сращение больших половых губ (высокая задняя спайка).
III степень вирилизации - клитор гипертрофирован и сформирована его головка, сращение половых губ формирует урогенитальный синус (единое мочеполовое отверстие у основания клитора).
IV-V степени вирилизации - гипертрофированный клитор напоминает нормальный половой член, однако наблюдают его искривление (фиксацию к промежности), урогенитальный синус открывается на стволе или головке полового члена (пинеальная уретра). Степень выраженности вирилизации у детей с недостаточностью 21-гидроксилазы значительно варьирует и обусловлена в первую очередь характером мутации гена CYP21. У мальчиков при рождении наружные половые органы соответствуют полу ребёнка, может быть небольшое увеличение полового члена.
Постнатальная вирилизация
После рождения симптомы андрогенизации нарастают у детей обоего пола. У девочек увеличиваются размеры клитора, отмечают его напряжение. У мальчиков увеличиваются размеры полового члена, возникают эрекции. Следует отметить, что симптомы андрогенизации могут не проявляться в первые 1,5 года жизни ребёнка. К 1,5-2 годам у детей обоего пола формируется половое оволосение, acne vulgaris, грубеет голос, гипертрофируется мускулатура. В первые годы жизни линейный рост детей ускорен, однако степень костной дифференцировки опережает рост. Зоны роста закрываются к 9-10 годам.
Степень пре- и постнатальной андрогенизации у пациентов с недостаточностью 21-гидроксилазы может иметь значительные индивидуальные колебания даже у больных сибсов в одной семье с одинаковым генетическим дефектом. Это может быть связано с индивидуальными особенностями метаболизма предшественников андрогенов и различием в активности рецепторов андрогенов у конкретного пациента.
Сольтеряющий синдром (синдром потери соли)
Полная потеря активности 21-гидроксилазы, наблюдаемая у 75% детей с дефицитом Р450с21, приводит к снижению биосинтеза альдостерона. Альдостерон необходим для поддержания нормального натриевого гомеостаза, а его дефицит приводит к потере натрия через почки, кишечник и потовые железы. Наличие выраженного сольтеряющего компонента, связанного с минералокортикоидной недостаточностью, представляет серьёзную угрозу жизни ребёнка с первых дней жизни. Через 3-4 дня после рождения нарастает гиперкалиемия, а спустя несколько дней развиваются гипонатриемия и гипернатриурия. Потеря соли приводит к выраженному обезвоживанию и снижению массы тела. Дегидратация усугубляется частой массивной рвотой, вызванной гиперкалиемией. При отсутствии терапии может наступить смерть ребёнка в результате коллапса и кардиогенного шока.
Репродуктивная функция при классической форме недостаточности 21-гидроксилазы
Пубертатный период у нелеченных детей обоего пола наступает поздно. У девочек даже при крайней степени вирилизации могут развиваться молочные железы (не более II степени по Таннеру) и появляться менструальные выделения. Регулярный менструальный цикл возможен только на фоне адекватной глюкокортикоидной терапии. Яичники уменьшены, с признаками поликистоза. Причины нарушения менструальной функции прежде всего обусловлены избыточной концентрацией надпочечниковых андрогенов, которые подавляют циклическую секрецию гонадотропинов и непосредственно угнетают развитие фолликула, вызывая его преждевременную атрезию.
У мальчиков функция гонад более сохранна, чем у девочек. У взрослых нелеченных пациентов возможна олигоспермия.
У детей обоего пола при поздно начатом лечении препаратами глюкортикоидов возможна преждевременная активация гипоталамо-гипофизарно-гонадной системы - истинное преждевременное половое развитие. Как правило, этот феномен наблюдают у детей, чей костный возраст к началу лечения достигает пубертатного: 11,5-12 лет у девочек и 13,5-14 лет у мальчиков (развитие сесамовидной кости). У девочек начинают увеличиваться молочные железы, у мальчиков увеличивается объём яичек. Причина ранней активации истинного пубертата у этих детей окончательно не ясна. Возможно, избыток по-
ловых стероидов изменяет чувствительность гипоталамических центров и способствует их «созреванию». Быстрое снижение избыточной секреции надпочечниковых андрогенов при назначении глюкортикоидной терапии способствует активации секреции гонадотропин-рилизинг гормона гипоталамусом, стимулирующего гонадотропную и гонадную функции. Ранний истинный пубертат у детей с недостаточностью 21-гидроксилазы ухудшает ростовой прогноз и требует присоединения антигонадотропной терапии.
Неклассическая форма недостаточности 21-гидроксилазы
Распространённость неклассических вариантов дефицита 21-гид- роксилазы в общей популяции очень высока и составляет до 0,3%. В некоторых этнических группах неклассическую форму заболевания наблюдают ещё чаще: 1,6% в Югославии, 1,9% в Испании, 3,7% у евреев Западной Европы (Ashkenazi). При неклассических формах заболевания снижение активности фермента 21-гидроксилазы колеблется в достаточно широких пределах и может составлять 20-60% нормальных значений. В связи с этим клинические признаки гиперандрогении могут быть чрезвычайно вариабельны. Для детей с неклассической формой заболевания не характерны симптомы постнатальной вирилизации. При рождении наружные половые органы девочек сформированы по женскому типу. В редких случаях могут быть небольшое увеличение клитора и высокая задняя спайка на промежности, формирующая воронкообразный вход во влагалище. У детей обоего пола наиболее частым симптомом неклассической формы заболевания считают раннее появление лобкового и подмышечного оволосения (адренархе). Отмечают также небольшое увеличение скорости роста и костного созревания, однако конечный рост этих детей соответствует генетически ожидаемому.
У девочек пубертатного возраста и у взрослых женщин лёгкая недостаточность 21-гидроксилазы проявляется в виде гирсутизма. Возможно нарушение менструальной функции и формирование поликистозных яичников, что приводит к бесплодию. Однако у 50% женщин с неклассической формой недостаточности 21-гидроксилазы репродуктивная функция не нарушена.
Определение уровня 17а-гидроксипрогестерона показано всем новорождённым, имеющим аномальное строение наружных половых ор- ганов при отсутствии пальпируемых яичек.
Параллельно проводят кариотипирование. Определение кариотипа 46ХХ у ребёнка с бисексуальным строением наружных половых органов с 95% вероятностью свидетельствует о наличии 21-гидроксилаз- ной недостаточности. Высокая концентрация 17а-гидроксипрогесте- рона окончательно подтверждает диагноз.
У недоношенных и детей, перенёсших тяжёлую родовую травму или рождённых с дефицитом массы тела при нормальных сроках гестации, содержание 17а-гидроксипрогестерона может быть повышено при от- сутствии недостаточности 21-гидроксилазы. В этих случаях рекомендуют повторное проведение исследования (2-3-4 раза с интервалом 5-7 дней). Уменьшение содержания 17а-гидроксипрогестерона в динамике позволяет исключить 21-гидроксилазную недостаточность. Развитие сольтеряющего криза при недостаточности 21-гидроксилазы редко наблюдают у новорождённых и детей первых семи дней жизни. Однако до получения данных гормонального исследования, под- тверждающих или исключающих 21-гидроксилазную недостаточность, всем детям необходимо проводить мониторинг уровня электролитов в крови.
Нарастание концентрации калия и снижение содержания натрия в сыворотке крови у ребёнка с бисексуальным строением наружных половых органов, сопровождающиеся клиническими признаками сольтеряющего синдрома, следует рассматривать как проявление недостаточности 21-гидроксилазы и немедленно назначать терапию, не дожидаясь результатов гормонального анализа.
Наличие недостаточности 21-гидроксилазы у новорождённых мальчиков можно заподозрить лишь при наличии сольтеряющего синдрома. Всем новорождённым мальчикам с клиническими признаками гиперкалиемии, гипонатриемии и обезвоживания необходимо определять содержание 17а-гидроксипрогестерона.
Лабораторные и инструментальные исследования
Основной признак недостаточности 21-гидроксилазы - повышение уровня 17а-гидроксипрогестерона в сыворотке крови (в 10 раз и более) вследствие блока в синтезе кортизола. Необходимо исследо- вание уровня калия и натрия в сыворотке крови для оценки степени минералокортикоидной недостаточности.
Содержание ренина крови значительно повышается при минералокортикоидной недостаточности.
У всех детей с бисексуальным строением наружных гениталий необходимо исследовать кариотип.
Молекулярная диагностика, основанная на определении мутаций в гене CYP21, позволяет точно подтвердить или исключить наличие недостаточности 21-гидроксилазы. Для пренатальной диагностики молекулярно-генетический метод считают единственно достоверным способом выявления заболевания и его формы у плода.
Диагностика и дифференциальная диагностика
Неонатальный скрининг недостаточности 21-гидроксилазы
Диагностика
недостаточности 21-гидроксилазы основывается на клинических признаках
заболевания. В первые недели жизни ребёнка заподозрить заболевание
позволяет наличие вирилизации наружных половых органов у девочек и
развитие синдрома потери соли у детей обоего пола. Вирильную форму
заболевания у мальчиков диагностируют только к 4-5 годам жизни при
развитии симптомов преждевременного полового развития. Костный возраст
при этом уже значительно опережает хронологический. Подобный
клинический подход к диагностике недостаточности 21-гидроксилазы
приводит к большому количеству ошибок. До 30% девочек с тяжёлыми
признаками вирилизации наружных половых органов ошибочно регистрируют
как имеющих мужской пол. До 35% мальчиков с сольтеряющей формой
заболевания погибают в первые недели жизни, так как заболевание вовремя
не распознают. Избежать диагностических ошибок позволяет неонатальный
скрининг на выявление недостаточности 21-гидрокси- лазы. В основе
скрининга лежит определение содержания 17а-гид- роксипрогестерона в
сухом пятне крови на фильтровальной бумаге. К
Различные формы врождённой дисфункции коры надпочечников представлены в табл. 18-5.
Таблица 18-5. Дифференциальная диагностика различных форм врождён- ной дисфункции коры надпочечников
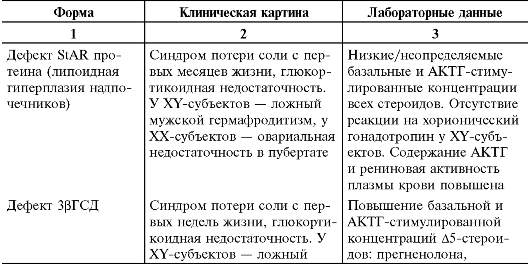

Синдром потери соли часто приходится дифференцировать с пилоростенозом, пилороспазмом и острыми заболеваниями, сопровождающимися рвотой. Неправильное строение наружных половых органов требует исключения истинного и ложного гермафродитизма. Синдром внутриутробной вирилизации у девочек иногда может быть обусловлен приёмом некоторых препаратов женщинами во время беременности. Гормональная и молекулярно-генетическая диагностика позволяет диагностировать и уточнять форму адреногенитального синдрома.
Лечение
Лечение врождённых нарушений надпочечникового стероидогенеза
Общие цели лечения всех форм врождённых нарушений надпочечникового стероидогенеза:
• заместить дефицит стероидов, секреция которых снижена в результате ферментативного дефекта;
• снизить концентрацию стероидов, секреция которых повышена в результате ферментативного дефекта;
• подавить избыточную секрецию АКТГ, используя минимально эф-
фективные дозы глюкокортикоидов;
• оптимизировать рост больных;
• предотвратить вирилизацию наружных половых органов;
• обеспечить нормальное половое созревание и фертильность.
Терапия недостаточности 21-гидроксилазы
Основным методом терапии недостаточности 21-гидроксилазы считают применение глюкокортикоидов, подавляющих гиперсекрецию АКТГ и нормализующих выработку андрогенов надпочечниками. Используются различные медикаментозные препараты, обладающие глюкокортикоидной активностью: преднизолон, кортизон, дексаметазон. Пролонгированные синтетические глюкортикоидные препараты (преднизолон, дексаметазон) оказывают негативное влияние на процессы роста. Их пролонгированный эффект может быстро привести к симптомам передозировки. Для детей с открытыми зонами роста, особенно младшего возраста, наиболее оптимальными препаратами следует считать таблетированные формы гидрокортизона (например, «Кортеф»). Первоначальная суточная доза гидрокортизона, необходимая для подавления АКТГ у детей первого года жизни, может достигать 20 мг/м2. Однако следует избегать длительного применения этих доз у ребёнка. Применение гидрокортизона в дозе 25 мг/м2 в течение года приводит к выраженной задержке или остановке роста! В случае длительной передозировки глюкокортикоидов в младшем возрасте отставание в росте ликвидировать невозможно, даже на фоне снижения дозы препаратов. В среднем суточная доза гидрокортизона должна составлять 10-15 мг/м2. Препарат назначают 3 раза в день в
равных дозах (в 7.00, 15.00, 22.00). Всем детям с сольтеряющей формой 21-гидроксилазной недостаточности необходимо дополнительное назначение минералокортикоидных препаратов.
К показаниям для назначения минералокортикоидной терапии относят:
• развитие клинических симптомов сольтеряющего криза;
• высокая концентрация калия в сыворотке крови при отсутствии
клинических симптомов потери соли;
• высокая рениновая активность плазмы крови при нормальной кон-
центрации калия и отсутствии клинических симптомов потери соли.
Доза флудрокортизона составляет 0,05-0,3 мг/сут. Такая терапия позволяет компенсировать недостаток минералокортикоидов, добиться более быстрого подавления избыточной секреции АКТГ при использовании менее высоких доз глюкокортикоидов. При наличии минералокортикоидной недостаточности потребность в минералокортикоидах максимальна у детей первого года жизни и составляет 0,1-0,3 мг/сут. Суточную дозу назначают в три приёма (в 7.00, 15.00, 23.00). Дополнительно вводят в рацион избыточное количество пова- ренной соли - до 2 г/сут.
В дальнейшем необходимость продолжения терапии минералокортикоидами основывается на показателях рениновой активности плазмы крови. У детей старшего возраста дозу минералокортикоидов снижают до 0,05-0,15 мг/сут. Суточную дозу назначают в два приёма (в 8.00 и 18.00).
Контроль адекватности терапии
Контроль адекватности терапии недостаточности 21-гидроксилазы основан на показателях физического развития и данных гормональ- ного исследования. Детей первых 2 лет жизни должен осматривать педиатр-эндокринолог каждые 3 мес. Детей старше 2 лет необходимо обследовать каждые 6-12 мес. При осмотре тщательно фиксируют показатели массы тела и роста. Определяют концентрации 17b-гидроксипрогестерона и электролитов. Проводят исследование рениновой активности плазмы крови. У детей старшего возраста ежегодно исследуют костный возраст, определяют содержание тестостерона.
Адекватная терапия глюкортикоидами обеспечивает нормальные темпы роста и костного созревания.
Терапия при ургентных состояниях
Родители пациентов с недостаточностью 21-гидроксилазы должны быть информированы об изменениях лекарственной терапии в неотложных и стрессовых ситуациях. Эту информацию необходимо выдать в виде письменных рекомендаций дополнительно к официальным медицинским выпискам. Дети старшего возраста и подростки должны
иметь при себе карту, в которую необходимо внести рекомендации о терапии в условиях неотложных ситуаций.
При интеркуррентных заболеваниях всем пациентам с высокой (>38,5 ?С) температурой тела, рвоте, травмах, хирургических вмешательствах необходимо дополнительное введение гидрокортизона. При стрессорных физических нагрузках (участие в соревнованиях) может возникнуть потребность в увеличении дозы глюкокортикоидов. Эмоциональные и психические стрессы (экзамены) не требуют дополнительного введения препаратов.
В период стресса дозу глюкортикоидов необходимо увеличить в 2-3 раза. В тех случаях, когда пероральный приём препаратов не возможен, следует обеспечить парентеральное введение гидрокорти- зона.
При тяжёлых интеркуррентных заболеваниях и оперативных вмешательствах следует применять внутримышечное введение гидрокор- тизона в дозе 3-5 мг/кг на инъекцию каждые 4-6 ч и дополнительное внутривенное капельное раствора натрия хлорида в количестве 150 мл/кг в сутки. При этом 25% указанного объёма жидкости необходимо ввести в первые 2 часа терапии.
Хирургическая стратегия коррекции наружных половых органов у девочек
Хирургическая коррекция наружных половых органов производят девочкам с симптомами внутриутробной вирилизации. Цель хирургической коррекции - добиться соответствия наружных половых органов избранному (женскому) полу, ликвидировать возможные анатомические препятствия, затрудняющие отток мочи, создать условия для нормальной репродуктивной функции.
Пренатальная диагностика и лечение недостаточности 21-гидрокси- лазы
Недостаточность 21-гидроксилазы в классическом и неклассическом варианте считают одним из самых распространённых заболеваний с аутосомно-рецессивным типом наследования. Разработка метода исследования содержания 17a-гидроксипрогестерона в капле крови, нанесённой на фильтровальную бумагу, позволила проводить массовый скрининг новорождённых в различных популяциях. В результате этих масштабных исследований было установлено, что средняя заболеваемость в мире составляет 1:13 500. В то же время в некоторых популяциях эта частота намного выше.
Профилактика
Как и при всех аутосомно-рецессивных наследственных заболеваниях, уменьшение частоты родственных браков сопровождается снижением заболеваемости.
Прогноз
Скрининг новорождённых и адекватная терапия с первых дней жизни значительно улучшают прогноз заболевания и социальную адаптацию детей. Пренатальная диагностика и лечение позволяют к моменту рождения избежать вирилизации наружных половых органов у девочек с недостаточностью 21-гидроксилазы.
Сахарный диабет
СД - группа заболеваний обмена веществ различной этиологии, характеризующихся хронической гипергликемией, возникающей в результате нарушения секреции или действия инсулина, либо обоих факторов одновременно (ВОЗ, 1999).
Исследовательская группа ВОЗ с учётом новых данных пересмотрела классификацию СД. Эта классификация включала клинические формы заболевания и классы статистического риска.
ЭТИОЛОГИЧЕСКАЯ КЛАССИФИКАЦИЯ САХАРНОГО ДИАБЕТА (ВОЗ, 1999)
• СД 1-го типа (деструкция β-клеток, приводящая обычно к абсолютной инсулиновой недостаточности):
- аутоиммунный;
- идиопатический.
• СД 2-го типа (с преимущественной инсулинорезистентностью и относительной инсулиновой недостаточностью или преимущественным дефектом секреции инсулина с инсулинорезистентностью или без неё).
• Гестационный СД.
• Другие специфические типы СД:
- генетические дефекты функции β-клеток;
- генетические дефекты в действии инсулина;
- болезни экзокринной части поджелудочной железы;
- эндокринопатии;
- СД, индуцированный лекарствами или другими химическими веществами;
- СД, индуцированный инфекциями;
- необычные формы иммуноопосредованного СД;
- другие генетические синдромы, иногда сочетающиеся с СД.
СД 1-го и 2-го типов - наиболее распространённые формы СД. Они различаются по клиническим, эпидемиологическим и иммунологическим характеристикам, характеру секреции инсулина, ассоциации с генетическими маркёрами.
СД 1-го типа наиболее часто наблюдают у детей и лиц молодого возраста, хотя это заболевание может манифестировать в любом возрасте. Аутоиммунный СД характеризуется деструкцией β-клеток, наличием аутоантител, абсолютной инсулиновой недостаточностью, полной инсулинозависимостью, тяжёлым течением со склонностью к кетоацидозу, ассоциацией с генами главного комплекса гистосовместимости HLA. Случаи заболевания идиопатическим СД регистрируют обычно у лиц, не относящихся к европейской расе, с деструкцией β- клеток, склонностью к кетозу, но неизвестным патогенезом.
СД 2-го типа среди взрослых является доминирующим. В детском возрасте его наблюдают крайне редко. СД 2-го типа в детском возрасте чаще протекает бессимптомно или с минимальной клинической симптоматикой. В то же время при инфекционных заболеваниях или сильных стрессах иногда может развиться кетоацидоз. В развитии заболевания у детей основное значение придают генетическому фактору. Монозиготные близнецы в 100% случаев конкордантны (сходны) по СД 2-го типа. У родителей также в большинстве случаев диагностируют СД 2-го типа, особенно при исследовании теста на толерантность к глюкозе. Большое значение в инициации заболевания имеют привычки поведения, такие как переедание и сниженная физическая активность. Внутриутробная задержка развития с дефицитом массы тела, а также недостаточное питание в раннем постнатальном периоде также могут способствовать развитию СД 2-го типа в детском возрасте из-за избыточного кормления ребёнка, приводящего к формированию ожирения, гиперинсулинизма, инсулинорезистентности.
Длительное время считали, что для детского возраста характерна лишь одна форма - СД 1-го типа. Однако исследования последнего десятилетия убедительно показали, что наряду с доминирующим СД 1-го типа, в детском возрасте диагностируют и более редкие сочетания СД с генетическими синдромами, СД 2-го типа, преобладающий у взрослых, а также MODY тип, считавшийся специфичным лишь для юношеского возраста. В настоящее время заболеваемость СД 1-го типа у детей в России составляет 9,24 на 100 000 детского населения.
ЭТИОЛОГИЯ
В основе развития СД 1-го типа лежит генетическая предрасположенность. Об этом свидетельствуют семейные случаи заболевания, а также высокая частота повторных случаев заболевания среди монозиготных близнецов.
Семейная концентрация СД 1-го типа (или частота повторных случаев заболевания в семьях больных) определяется следующими факторами:
• частотой СД в популяции;
• количеством больных и здоровых родственников, степенью их род-
ства;
• возрастом возникновения СД у пробанда, в некоторых случаях -
полом пробанда;
• возрастом обследуемых родственников, в некоторых случаях - их
полом.
В табл. 18-6 представлены эмпирически полученные показатели риска развития СД 1-го типа в различных группах родственников для популяций с высоким уровнем заболеваемости (0,4%).
Таблица 18-6. Эмпирический риск развития заболевания у родственников пациентов с сахарным диабетом 1-го типа (Эйзенбарт, 1994)

Как показали многочисленные исследования последних лет, СД 1-го типа - аутоиммунное заболевание у генетически предрасположенных лиц, при котором длительно текущий хронический лимфо- цитарный инсулит приводит к деструкции β-клеток с последующим развитием инсулиновой недостаточности.
Для начала аутоиммунного процесса необходим инициирующий или провоцирующий фактор внешней среды (триггер). На современном этапе не существует единого и несомненного взгляда на природу такого фактора. В настоящее время выделяют наиболее вероятные факторы, принимающие участие в запуске процессов разрушения островковых клеток.
• Вирусы: Коксаки B, краснухи, паротита, энтеровирусы, ротавирусы, цитомегаловирус, Эпстайна-Барр, ECHO и др.
• Факторы питания: коровье молоко и смешанное вскармливание на основе коровьего молока, продолжительность грудного вскармливания, нитраты.
• Воздействие токсинов.
ПАТОГЕНЕЗ
Механизмы повреждения β-клеток вирусами:
• прямое разрушение (лизис) β-клеток в результате инфицирования
вирусом;
• механизм молекулярной мимикрии, при котором иммунный ответ, направленный на вирусный Аг, сходный с собственным Аг β-клетки, повреждает и саму островковую клетку;
• нарушение функции и метаболизма β-клетки, в результате чего на
её поверхности экспрессируются аномальные Аг, что приводит к запуску аутоиммунной реакции;
• взаимодействие вируса с иммунной системой.
Стадии развития СД 1-го типа представлены на рис. 18-7.
Аутоантитела к различным структурам β-клеток рассматривают как иммунологические маркёры β-клеточной деструкции.
Инсулин - главный регулирующий обмен веществ гормон, конечным результатом действия которого является обеспечение энергети- ческих и пластических процессов в организме. К органам-мишеням действия инсулина относят печень, мышечную и жировую ткани. Инсулин может оказывать анаболическое и антикатаболическое действия. Анаболический эффект инсулина реализуется через стимуляцию синтеза гликогена и жирных кислот в печени, триглицеридов в жировой ткани, белка и гликогена в мышечной ткани. Антикатаболическое действие инсулина заключается в подавлении процессов гликогенолиза, глюконеогенеза (образование глюкозы из жиров и белков) и ке- тоногеза (образование кетоновых тел). В организме существуют и ин- сулиннезависимые ткани (почки, головной мозг, шванновские клетки периферических нервов, ткань хрусталика, артерии, сетчатка), в которых для переноса глюкозы внутрь клетки инсулин не требуется.
Механизм действия инсулина заключается в активации транспорта глюкозы через мембрану клетки, а также стимуляции различных ферментов, участвующих на разных стадиях в обменных процессах. Действие инсулина осуществляется при связывании со специфическими рецепторами на цитоплазматических мембранах.
Все клинические симптомы обусловлены недостатком выработки и действия инсулина. У детей это обусловлено преимущественно гибелью β-клеток поджелудочной железы, т.е. имеет место абсолютная недостаточность инсулина.
КЛИНИЧЕСКАЯ КАРТИНА
СД может развиться у ребёнка в любом возрасте. В течение первых месяцев жизни заболевание наблюдают редко. Риск увеличивается после 9 мес, достоверно нарастая после 5 лет и в пубертатном возрасте, и несколько снижается у взрослых.
Рис. 18-7. Стадии развития инсулинзависимого сахарного диабета (Зингер Α., Стендл Ε.): I стадия - генетическая предрасположенность, реализуемая менее чем у половины генетически идентичных близнецов и у 2-5% сибсов; II стадия - гипотетический пусковой момент, который вызывает развитие III стадии; III стадия - активный аутоиммунный процесс (первоначально лица даже с иммунными нарушениями имеют нормальную секрецию инсулина); IV стадия - при выраженных иммунных нарушениях отмечают снижение секреции инсулина в ответ на введение глюкозы, при этом уровень гликемии остаётся нормальным; V стадия - клиническая манифестация, которая развивается после гибели 80-90% β-клеток, при этом ещё сохраняется остаточная инсулиновая секреция; VI стадия - полная деструкция β-клеток.
У абсолютного большинства больных в детском возрасте развивается СД 1-го типа, который характеризуется выраженной инсулиновой недостаточностью и всегда является инсулинзависимым. Клинические признаки СД во многом зависят от возраста ребёнка и степени декомпенсации обменных процессов в момент его обследования. Для СД 1-го типа в детском возрасте характерно острое начало с быстрым нарастанием симптоматики вплоть до возникновения кетоацидоза, а при отсутствии своевременной диагностики возможно развитие кетоацидотической комы. У большинства детей время с момента появле- ния первых симптомов заболевания до возникновения коматозного состояния составляет от 3-4 нед до 2-3 мес.
У детей среднего и старшего возраста заподозрить наличие СД несложно. К основным симптомам СД относят:
• полиурию (повышенное мочеиспускание);
• полидипсию (жажду);
• сухость во рту;
• полифагию (повышенный аппетит);
• снижение массы тела;
• запах ацетона изо рта.
Полиурия - первый симптом глюкозурии, возникает при гипер- гликемии, превышающей почечный порог для глюкозы (в среднем 9 ммоль/л). Полиурия развивается в результате осмотического диуреза, обусловленного высокой концентрацией глюкозы в моче. Моча обычно бесцветная, имеет высокий удельный вес за счёт выделяющегося сахара. В дневное время этот симптом, особенно у детей школьного возраста, не привлекает внимания ни детей, ни взрослых. В то же время ночная полиурия и нередко сопутствующее ей недержание мочи, как правило, более заметны. Энурез характерен для тяжёлой полиурии и часто становится первым замечаемым симптомом СД. Нередко внимание родителей привлекает появление липкой мочи. Полиурия - компенсаторный процесс, так как способствует снижению гипергликемии и гиперосмолярности крови.
Полидипсия возникает на фоне полиурии вследствие обезвоживания организма и раздражения центра жажды головного мозга из-за гиперосмолярности крови. Так же, как и полиурия, жажда бывает более заметной в ночные часы, а также утром, до завтрака. Сухость во рту заставляет ребёнка в течение ночи несколько раз просыпаться и пить воду.
Полифагия (постоянное чувство голода) в сочетании со снижением массы тела является одним из характерных признаков СД. Они развиваются вследствие энергетического голодания клеток организма из-за нарушения утилизации глюкозы и потери её с мочой, с одной стороны, и усиления процессов липолиза и протеолиза в условиях
дефицита инсулина - с другой. Резкая потеря массы тела происходит также в связи с обезвоживанием организма. Родители не всегда характеризуют полифагию как патологический симптом и не фиксируют в числе жалоб, а нередко даже расценивают её как положительное явление в состоянии ребёнка. В большей степени родителей тревожит потеря массы тела ребёнка. Сочетание полифагии с потерей массы тела обычно заставляет обратиться к врачу. Однако нередко обследование ребёнка идёт в неправильном направлении (чаще всего по пути исключения глистной инвазии, желудочно-кишечного заболевания, хронической инфекции и др.), и, таком образом, больной остаётся без медицинской помощи. В дальнейшем у ребёнка развиваются общая и мышечная слабость, которые объясняют не только энергетическим голоданием клеток, но и нарастающими электролитными нарушениями.
Усиление липолиза в жировой ткани приводит к нарастанию в крови концентрации свободных жирных кислот, которые усиливают кетогенез из-за сниженной в условиях дефицита инсулина липосинтетической функции печени. Накопление кетоновых тел приводит к развитию диабетического кетоацидоза. У больных появляется запах ацетона изо рта, полифагия сменяется сниженным аппетитом, нарастает слабость, возникает одышка, вначале при физической нагрузке, а затем и в покое. Впоследствии к этим симптомам присоединяются анорексия, тошнота, рвота, сонливость. Это грозные предвестники развития коматозного состояния. Нередко в дебюте СД у детей можно наблюдать псевдоабдоминальный синдром. Боли в животе, тошноту, рвоту, возникающие при быстро развивающемся кетоацидозе, расценивают как симптомы хирургической патологии. Часто таких детей в связи с подозрением на острый живот ошибочно подвергают лапаротомии.
Кожные изменения часто регистрируют в дебюте СД. Сухость кожных покровов и слизистых оболочек вследствие обезвоживания организма становятся почти постоянным симптомом заболевания. На волосистой части головы может возникнуть сухая себорея, а на ладонях и подошвах - шелушение. Слизистая оболочка ротовой полости обычно яркокрасного цвета, сухая, в углах рта - раздражение, заеды. На слизистой оболочке ротовой полости могут развиваться молочница, стоматит. Диабетический румянец, захватывающий нередко, кроме щёк, лоб и подбородок, связан с парезом кожных капилляров при гипрегликемии и кетозе. Иногда обращает на себя внимание желтушное окрашивание кожи ладоней, подошв, носогубного треугольника (ксантоз). Оно вызвано отложением каротина в роговом слое кожи из-за нарушения его превращения в печени в витамин А. Тургор кожи, как правило,
снижен, особенно при резко выраженном обезвоживании. Чаще всего отмечают дефицит массы тела, иногда вплоть до кахексии.
Сердечно-сосудистые нарушения в дебюте заболевания наблюдают лишь при тяжёлой декомпенсации. К ним относят тахикардию, приглушение тонов сердца, наличие функциональных шумов. При ЭКГ исследовании регистрируют изменения метаболического характера.
Гепатомегалию у детей отмечают довольно часто. Её выраженность зависит от степени нарушения метаболизма. Увеличение печени при СД обычно связано с жировой инфильтрацией вследствие инсулиновой недостаточности. Назначение инсулина и компенсация углеводного обмена приводят к нормализации размеров печени.
Нарушения менструального цикла могут сопровождать дебют СД в пубертатном периоде у девочек. Возможны жалобы на зуд в области наружных половых органов и других частях тела.
Течение заболевания
Течение заболевания зависит от возраста ребёнка. У многих детей первых 5 лет жизни течение заболевания характеризуется крайней нестабильностью, склонностью к частым гипогликемическим состояниям, лёгкостью развития кетоза, повышенной чувствительностью к инсулину. Избежать гипогликемии у маленьких детей трудно изза неустойчивого аппетита и вариабельной физической нагрузки. Лабильное течение СД отмечают у детей как в препубертатный, так и пубертатный периоды. Оно обусловлено нестабильностью нейрогуморальной регуляции, напряжённостью обменных процессов в связи с интенсивным ростом и развитием. На всех стадиях пубертатного периода ярко выражена инсулинорезистентность. Следует особо учитывать эмоциональный фактор, который влияет на течение заболевания у подростков.
Осложнения
Диабетическая ретинопатия
Диабетическая ретинопатия является классическим примером сосудистых осложнений СД. Она занимает одно из первых мест среди причин, приводящих к снижению зрения и слепоте у лиц молодого возраста. Инвалидность по причине нарушения зрения наблюдают более чем у 10% больных СД. Слепота наступает в 25 раз чаще, чем в общей популяции.
Диабетическая ретинопатия - специфическое поражение сетчатой оболочки и сосудов сетчатки, характеризующееся развитием экссудативных очагов, ретинальных и преретинальных кровоизлияний, ростом новообразованных сосудов, а также развитием тракционной отслойки сетчатки, рубеозной глаукомы.
Диабетическая нефропатия
Диабетическая нефропатия - основная причина неблагоприятного прогноза у больных СД. Известно, что треть всех больных СД 1-го типа погибают от терминальной почечной недостаточности уже через 15-20 лет после начала заболевания. Наиболее неблагоприятный исход наблюдают у лиц, заболевших в детском возрасте.
Диабетическая невропатия
Диабетическая полиневропатия характеризуется возникновением болей в конечностях, снижением порога температурной и болевой чувствительности. Характерно развитие вегетативной полиневропатии, проявляющейся дисфункцией пищевода, гастропатией, диабетической диареей, запорами.
Риск развития сосудистых осложнений зависит от степени компенсации заболевания. При хорошей компенсации возможна нормальная по продолжительности и качеству жизнь. Чем хуже компенсация, тем быстрее развиваются и тяжелее протекают диабетические осложнения, продолжительность жизни сокращается на 15-20 лет, возникает ранняя инвалидизация.
Диабетическую катаракту, гепатоз и диабетическую хайропатию (ог- раничение подвижности суставов) также относят к типичным осложнениям СД.
При возникновении СД в раннем возрасте и плохой компенсации заболевания наблюдают задержку физического и полового развития. Крайняя степень выраженности этих симптомов (карликовость, от- сутствие вторичных половых признаков у подростков и гепатомегалия) носит название синдрома Мориака (рис. 18-8 на вклейке).
ЛАБОРАТОРНЫЕ И ИНСТРУМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
Диагноз подтверждают наличием гипергликемии, глюкозурии, у некоторых больных - кетоза или кетоацидоза. В норме содержание глюкозы в плазме крови натощак составляет 3,3-5,5 ммоль/л.
Глюкозурия служит важным диагностическим критерием СД. В норме у здорового человека глюкоза в моче отсутствует. Глюкозурия возникает тогда, когда концентрация глюкозы в плазме крови превышает 8,88 ммоль/л. Диагноз СД при обнаружении глюкозурии можно считать достоверным только после определения гипергликемии.
Кетонурия или ацетонурия. Наличие кетоновых тел (производных метаболизма липидов) в моче свидетельствует о тяжёлой декомпенсации СД, связанной с недостатком инсулина. Однако у детей кетонурию можно отмечать при инфекционных заболеваниях, протекающих с высокой температурой тела, при голодании, особенно у детей раннего возраста.
Определение содержания гликозилированного Hb (HbA1c) считают одним из современных методов диагностики нарушения углеводного
обмена. Кроме того, этот метод также используют для оценки степени компенсации углеводного обмена у больных СД, находящихся на лечении.
Содержание HbA1c зависит от концентрации глюкозы в плазме крови и служит интегральным показателем состояния углеводного обмена в течение последних 3 месяцев, учитывая, что «жизнь» эритроцита составляет 120 дней. Содержание HbA1c составляет 4-6% общего Hb в крови у здоровых лиц. Высокий уровень гликемии при СД способст- вует повышению процессов неферментативного гликозилирования белков Hb, поэтому у больных СД его содержание в 2-3 раза превышает норму. Для диагностики СД у детей этот показатель имеет большое значение.
Аутоантитела к Аг β-клеток (ICA, GADA, IAA, IAA) служат иммунологическими маркёрами инсулита, протекающего в поджелудочной железе. Их определение используют для ранней доклинической диагностики СД 1-го типа в группах высокого генетического риска либо для проведения дифференциальной диагностики между СД 1 и 2-го типов. При наличии у ребёнка классических симптомов СД определять аутоантитела к Аг β-клеток нет необходимости.
Определение содержания С-пептида в сыворотке крови даёт возможность оценить функциональное состояние β-клеток у лиц с высоким риском развития СД и, кроме того, помогает в дифференциальной диагностике СД 1 и 2-го типов. Базальная концентрация С-пептида у здоровых лиц составляет 0,28-1,32 пг/мл. При СД 1-го типа его со- держание снижено или не определяется. После стимуляции глюкозой, глюкагоном или сустакалом (питательной смесью с высоким содержанием кукурузного крахмала и сахарозы) концентрация С-пептида у больных СД 1-го типа не повышается, а у здоровых лиц - значительно возрастает. При наличии классических симптомов СД 1-го типа в дебюте заболевания у детей определение содержания С-пептида в сыворотке крови не имеет практической значимости.
ДИАГНОСТИКА И ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА Диагностику СД производят на основании классических симптомов заболевания. Дифференциальную диагностику проводят с почечным диабетом (глюкозурия без повышения концентрации глюкозы в плазме крови), несахарным диабетом (полиурия с низким удельным весом мочи и жажда при отсутствии глюкозурии и гипергликемии) и синдромом ацетонемической рвоты (кетонурия, ацидоз, запах ацетона изо рта при отсутствии гипергликемии).
До настоящего времени проблема дифференциальной диагностики СД в период его дебюта остаётся актуальной. У 88% детей СД диагностируют в состоянии кетоацидоза.
ЛЕЧЕНИЕ
СД 1-го типа - заболевание, в основе которого лежит абсолютная инсулиновая недостаточность, обусловленная аутоиммунным разру- шением инсулинпродуцирующих клеток поджелудочной железы, поэтому введение инсулина считают единственным на сегодняшний день патогенетическим методом его лечения. Кроме этого, к важным моментам терапии СД 1-го типа относят соблюдение диеты, правильный образ жизни, достаточную физическую нагрузку и самоконтроль.
Цели лечения детей и подростков с СД 1-го типа:
• достижение максимально близкого к нормальному состоянию уровня углеводного обмена;
• нормальное физическое и соматическое развитие ребёнка;
• нормальные психосоциальное состояние и адаптация ребёнка;
• развитие самостоятельности и мотивации к самоконтролю;
• профилактика специфических осложнений СД. Критерии компенсации СД:
• отсутствие жажды, полиурии, потери массы тела;
• гликемия натощак 4-7,6 ммоль/л;
• отсутствие глюкозурии;
• постпрандиальная гликемия менее 11 ммоль/л;
• концентрация глюкозы ночью не менее 3,6 ммоль/л;
• отсутствие тяжёлых гипогликемий (допускают наличие отдельных лёгких гипогликемий);
• содержание HbA1c менее 7,6%.
Больным СД необходимо ограничить потребление углеводов и жиров, при этом необходимо учитывать индивидуальные физиологичес- кие потребности в пищевых веществах и энергии. Рекомендуют соотношение белков, жиров и углеводов 1:0,8:3-3,5.
В настоящее время в России детям назначают только человеческие генно-инженерные препараты инсулина и их аналоги. Фармакокинетические характеристики этих препаратов представлены в табл. 18-7.
Таблица 18-7. Фармакокинетическая характеристика различных препара- тов инсулина
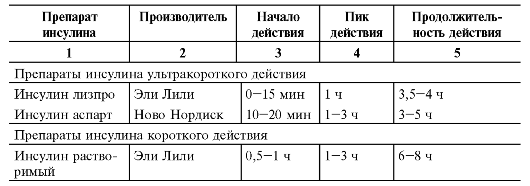

Кроме того, в настоящее время выпускают смешанные препараты инсулина, в состав которых входит инсулин средней продолжительности и короткого действия в различных соотношениях - от 90:10 до 50:50. Эти препараты более удобны, поскольку их применение позволяет уменьшить количество инъекций, проводимых с помощью шприц-ручек.
Сразу после установления диагноза СД у детей необходимо начинать инсулинотерапию. Начинают инсулинотерапию с дробного вве- дения препарата инсулина короткого действия 4-6 раз в сутки. Дозу подбирают индивидуально в зависимости от возраста ребёнка и степени гипергликемии. В среднем доза составляет 0,5-0,8 ЕД/кг/сут, и в дальнейшем её необходимо корректировать в зависимости от содержания глюкозы в плазме крови. Повторное исследование концентрации глюкозы в плазме крови проводят каждые 3-6 ч.
Наибольшее распространение в настоящее время получила интенсифицированная (или базис-болюсная) схема инсулинотерапии. Она заключается в том, что перед каждым основным приёмом пищи вводят препарат инсулина короткого действия, а препарат инсулина пролонгированного действия - 1-2 раза в сутки, чаще всего в вечерние и утренние часы. При этом с помощью препарата инсулина пролонгированного действия пытаются имитировать базальную секрецию, а
при использовании препарата инсулина короткого действия - посталиментарную секрецию.
В настоящее время «золотым стандартом» инсулинотерапии считают использование инсулиновой помпы. Инсулиновая помпа - современная технология, позволяющая эффективно компенсировать углеводный обмен. Помпа способна в непрерывном режиме вводить инсулин, имитируя базальную секрецию гормона β-клетками, а после еды - вводить инсулин в болюсном режиме, имитируя посталиментарную секрецию.
Осложнения инсулинотерапии
Гипогликемия - состояние, обусловленное низким содержанием глюкозы в плазме крови и являющееся одним из наиболее частых осложнений инсулинотерапии. По данным различных авторов, среди больных СД ярко выраженные и бессимптомные гипогликемии наблюдают в 25-58% случаев, а в 3-4% случаев причиной смерти больных СД становится гипогликемическая кома.
К симптомам гипогликемии относят снижение интеллектуальной деятельности, неуверенность в себе, вялость, заторможенность, плохую координацию движений, головную боль, чувство голода, слабость, повышенную потливость, сонливость, парастезии, головокружение, диплопию, «мушки» в глазах, раздражительность, ночные кошмары, неадекватное поведение, загруженность, гемиплегии, парезы, нарушение сознания и как крайнее проявление кому.
Самоконтроль
Накопленный клинический опыт показывает, что ни применение лучших препаратов инсулина, ни максимально подобранные доза и схема введения инсулина не могут решить проблему компенсации СД в детском и подростковом возрасте без проведения самоконтроля заболевания в домашних условиях.
Самоконтроль при СД считают одним из важнейших компонентов лечения. Для грамотного контроля СД больные должны хорошо понимать все аспекты своего заболевания. Проведение самоконтроля означает не только умение определять содержание глюкозы в плазме крови, но и правильно корректировать дозу инсулина в зависимости от уровня гликемии, изменений в питании, физических нагрузок и условий разнообразного спектра жизненных ситуаций. Для этого проводят обучение больных и их родителей в специально создаваемых школах самоконтроля.
НЕОТЛОЖНЫЕ СОСТОЯНИЯ
К острым неотложным состояниям при СД относят диабетический кетоацидоз и кетоацидотическую кому, а также гипогликемию и гипогликемическую кому. Гиперосмолярную некетоацидотическую кому и лактатацидоз в детском возрасте наблюдают крайне редко, хотя
состояние гиперосмолярности имеет большое значение в развитии кетоацидотической комы.
Диабетический кетоацидоз и кетоацидотическая кома
Диабетический кетоацидоз - тяжёлая метаболическая декомпенсация СД. При манифестации СД диабетический кетоацидоз развивается в 80% случаев, когда по тем или иным причинам задерживают диагностику заболевания, либо при уже установленном диагнозе откладывают назначение инсулина. Особенно быстро диабетический кетоацидоз развивается у маленьких детей.
У больных, уже получающих инсулинотерапию, причиной диабетического кетоацидоза и комы могут быть:
• неправильное лечение (назначение недостаточных доз инсулина);
• нарушение режима инсулинотерапии (пропуск инъекций, использо-
вание просроченных препаратов инсулина);
• грубые нарушения в питании (у девочек пубертатного возраста иногда сознательно с целью снижения массы тела);
• резкое возрастание потребности в инсулине, которое может возник-
нуть вследствие инфекционных заболеваний, стресса, хирургических вмешательств и др.
Тяжесть состояния при диабетическом кетоацидозе обусловлена резкой дегидратацией организма, декомпенсированным метаболическим ацидозом, выраженным дефицитом электролитов, гипоксией, в большинстве случаев гиперосмолярностью и нередко сопутствующими интеркуррентными заболеваниями.
К развитию комы при диабетическом кетоацидозе приводят следующие биохимические нарушения и патофизиологические процессы:
• резкое обезвоживание клеток головного мозга;
• гиперосмолярность вследствие гипергликемии;
• ацидоз;
• повышенное содержание азотистых шлаков вследствие распада бел-
ков и нарушения выделительной функции почек;
• тяжёлая гипоксия мозга из-за снижения мозгового кровотока, по-
вышения концентрации HbA1c, снижения содержания 2,3-дифосфо- глицерата в эритроцитах;
• недостаточность механизмов внутриклеточного энергетического
обеспечения;
• гипокалиемия;
• ДВС;
• общая интоксикация.
Лечение диабетического кетоацидоза включает 6 важнейших направлений.
• Введение жидкости для регидратации.
• Введение инсулина для прекращения катаболических процессов (кетоацидоза) и снижения гипергликемии.
• Коррекция электролитных нарушений.
• Купирование ацидоза с помощью бикарбонатов.
• Общие мероприятия.
• Лечение состояний, вызвавших диабетический кетоацидоз.
Отёк головного мозга - наиболее частая причина смертельных исходов. Причина развития отёка головного мозга во время лечения до конца не ясна, однако слишком быстрое снижение внутрисосудистой осмолярности может усиливать этот процесс. В связи с этим регидратацию у детей с диабетическим кетоацидозом необходимо проводить более медленно и осторожно, чем в других случаях дегидратации.
Объём вводимых растворов зависит от возраста ребёнка и составляет:
• в возрасте до 1 года - 1000 мл;
• 1-5 лет - 1500 мл;
• 5-10 лет - 2000 мл;
• 10-15 лет - 2000-3000 мл/сут.
При гликемии более 14 ммоль/л восполнение жидкости проводят 0,9% раствором натрия хлорида и раствором Рингера. При снижении гликемии ниже 14 ммоль/л в состав вводимых растворов подключают 5-10% раствор глюкозы для поддержания осмолярности и предотвращения быстрого снижения гликемии, так как при быстром падении осмолярности крови осмолярность ликвора остаётся намного выше (из-за медленно протекающих обменных процессов между ликвором и кровью). Устремляющаяся в ликвор по градиенту концентрации жидкость может стать причиной отёка головного мозга. Введение глюкозы также необходимо для устранения энергетического дефицита в организме, восстановления содержания гликогена в печени, снижения кетогенеза и глюконеогенеза.
Основные принципы инсулинотерапии при диабетическом кетоацидозе
• Начальная доза инсулина составляет 0,1 ЕД/кг/ч; у маленьких детей эта доза может составлять 0,05 ЕД/кг/ч, а при тяжёлой сопутствующей гнойной инфекции может увеличиваться до 0,2 ЕД/кг/ч.
• Снижение гликемии в первые часы должно составлять 4-5 ммоль/л/ч.
Если этого не происходит, дозу инсулина увеличивают.
При нормализации КЩС больного переводят на подкожное введение инсулина каждые 3-4 ч. При отсутствии кетоза на 2-3-и сутки ребёнка переводят на 5-6-разовое введение инсулина короткого действия, а затем на обычную комбинированную инсулинотерапию.
Купирование ацидоза
Постепенная нормализация кислотно-основного состояния развивается одновременно с лечением диабетического кетоацидоза благодаря регидратации и введению инсулина. Восполнение объёма жидкости
приводит к восстановлению буферных систем крови, а введение инсулина подавляет кетогенез.
Бикарбонаты используют только в крайних случаях:
• при снижении рН крови до уровня, подавляющего внешнее дыхание
(ниже 6,8), оказывающего отрицательное инотропное влияние на миокард, снижающего чувствительность кровеносных сосудов к катехоламинам, усиливающего инсулинорезистентность и продукцию лактата клетками печени;
• при нарушении сократимости миокарда в условиях персистирующе-
го шока (последний обычно развивается при неадекватных реанимационных мероприятиях и неадекватном действии инсулина при септических состояниях).
Обычно вводят 1-2 ммоль/кг
бикарбонатов (2,5 мл/кг 4% раствора гидрокарбоната натрия) внутривенно
капельно медленно (в течение 1 ч). Дополнительно вводят раствор калия
хлорида из расчёта 0,15-0,3 г сухого вещества на
Гипогликемия и гипогликемическая кома
Исходом тяжёлого гипогликемического состояния может стать гипогликемическая кома, если своевременно по разным причинам не будут предприняты меры к его купированию. Гипогликемическая кома - причина 3-4% случаев летальных исходов у больных СД. В последние годы в связи с расширением сети школ самоконтроля и повсеместным внедрением средств самоконтроля количество тяжёлых гипогликемических состояний, заканчивающихся коматозным состоянием, значительно сократилось.
Различают 3 степени тяжести гипогликемических состояний.
I степень - лёгкая. Ребёнок или подросток хорошо осознаёт своё со-
стояние и самостоятельно купирует гипогликемию (это не касается детей моложе 5-6 лет, поскольку в большинстве случаев они не способны сами себе помочь).
II степень - средняя. Дети или подростки не могут самостоятельно купировать гипогликемию и нуждаются в посторонней помощи, однако в состоянии принимать углеводы внутрь.
III степень - тяжёлая. Ребёнок или подросток находится в полубессознательном, бессознательном или коматозном состоянии, нередко в сочетании с судорогами, и нуждается в парентеральной терапии (внутримышечное введение глюкагона или внутривенное введение глюкозы).
К причинам возникновения гипогликемических состояний относят ошибки при введении инсулина, приём алкоголя, большую физическую нагрузку в дневные или вечерние часы, пропуски приёма пищи при введении инсулина.
В табл. 18-8 приведены основные различия кетоацидотической и гипогликемической комы.
Таблица 18-8. Дифференциальная диагностика гипогликемической и ке- тоацидотической комы
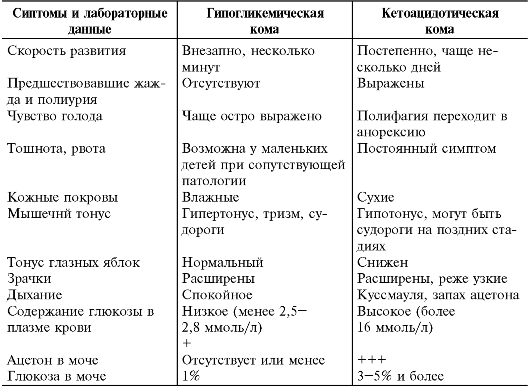
ПРОФИЛАКТИКА
В настоящее время возможна профилактика у близких родственников в семьях, где имеются больные СД 1 типа. На основании HLA-типирования рассчитывают риск заболевания и проводят регулярные иммунологические обследования на наличие АТ к островковым клеткам.
ПРОГНОЗ
Зависит от адекватности лечения. При поддержании близкого к норме показатель углеводного обмена (HbA1c не выше 7%) возможна нормальная по продолжительности и качеству жизнь. Средняя продолжительность жизни сокращается из-за развития специфических осложнений (диабетическая нефропатия и ХПН, ранний атеросклероз и ишемическая болезнь сердца, «диабетическая стопа» с развитием гангрены, поражение сосудов головного мозга с развитием инсульта). Эти осложнения напрямую связаны с плохой компенсацией СД.