Детские болезни: учебник / Под ред. А.А. Баранова - 2-е изд., - 2009. - 1008 с.
|
|
|
|
ГЛАВА 13 РЕВМАТИЧЕСКИЕ БОЛЕЗНИ
РЕВМАТИЗМ (ОСТРАЯ РЕВМАТИЧЕСКАЯ ЛИХОРАДКА)
Ревматизм (болезнь Сокольского-Буйо) - системное воспалительное заболевание соединительной ткани с преимущественным поражением сердечно-сосудистой системы, развивающееся у предрасположенных к нему лиц молодого возраста (7-15 лет) после инфекции, вызванной β-гемолитическим стрептококком группы А. В англоязычной литературе ревматизм часто называют острой ревматической лихорадкой.
Заболеваемость ревматизмом в среднем составляет 5 на 100 000 населения Земли, но в развивающихся странах, по данным ВОЗ, она значительно выше (2,2 на 1000 детей). В Российской Федерации распространённость ревматизма в среднем 0,3 на 1000 детского населения. Первичная заболеваемость детей в Москве достаточно низкая
(0,03 на 1000).
Этиология
Острая ревматическая лихорадка развивается через 2-4 нед после перенесённой инфекции, вызванной β-гемолитическим стрептококком группы А. У стрептококка выделяют несколько Аг и токсинов, в частности М-протеины клеточной стенки (фактор вирулентности), стрептолизины S и О, стрептокиназу и гиалуронидазу. Подтверждением воздействия стрептококка на организм больного ревматизмом служит обнаружение у большинства больных противострептококковых АТ - антистрептолизина-О, антистрептогиалуронидазы, антистрептокиназы, антидезоксирибонуклеазы В, способных повреждать различные ткани и клетки организма.
В возникновении ревматизма важную роль играет наследственная предрасположенность больного. Ревматизмом заболевают только 0,3- 1% детей, перенёсших стрептококковую инфекцию. На «семейный» характер ревматизма в начале ХХ столетия указал известный россий-
ский педиатр А.А. Кисель. Значение роли генетических факторов в развитии ревматизма подтверждается более частым его развитием у родных братьев и сестёр и более высокой заболеваемостью среди монозиготных, чем среди гетерозиготных, близнецов. Аг D-8, D-17 B-лимфоцитов выявляют у 98% больных кардитом и полиартритом и у 75% пациентов с хореей ревматического генеза.
Патогенез
Развитие ревматической лихорадки определяют несколько механизмов. Определённую роль может играть прямое токсическое повреждение компонентов миокарда кардиотропными ферментами β-гемоли- тического стрептококка группы А. Однако ведущее значение придают особенностям клеточного и гуморального иммунного ответа на различные Аг стрептококка, приводящим к синтезу противострептококковых АТ, перекрёстно реагирующих с Аг миокарда (феномен молекулярной мимикрии), а также цитоплазматическими Аг нейрональной ткани, локализующимися в субталамической зоне и базальных ганглиях головного мозга. Кроме того, М-протеин обладает свойствами «суперантигена», т.е. способен вызывать активацию Т и В-лимфоцитов без предварительного процессинга Аг-представляющими клетками и взаимодействия с молекулами класса II главного комплекса гистосовместимости.
Патоморфология
Для ревматизма характерно преимущественное поражение соединительной ткани. Классически выделяют четыре стадии патологического процесса при ревматизме: мукоидное набухание, фибриноидные из- менения, пролиферативные реакции и склероз. В стадии мукоидного набухания возможно обратное развитие процесса. Пролиферативная стадия характеризуется формированием ревматической гранулёмы, состоящей из крупных базофильных клеток гистиоцитарного происхождения, гигантских многоядерных клеток, а также из лимфоидных, плазматических и тучных клеток. Типичные ревматические гранулё- мы выявляют только в сердце (в настоящее время довольно редко). В патологический процесс вовлекаются также сосуды микроциркуляторного русла, серозные оболочки, суставы и нервная система. В основе поражения нервной системы лежит ревматический васкулит, а при хорее - поражение клеток подкорковых ядер. Изменения кожи и подкожной клетчатки также обусловлены васкулитом и очаговой воспалительной инфильтрацией.
Клиническая картина
Ревматизм чаще всего возникает у детей школьного возраста и значительно реже у дошкольников. Заболевание характеризуется поли-
морфизмом симптомов, главными из которых являются полиартрит, кардит и поражение подкорковых ядер головного мозга. Поражения внутренних органов в виде ревматической пневмонии, нефрита, абдо- минального синдрома (ревматического перитонита) в настоящее время практически не наблюдают.
Ревматический полиартрит. Ревматический полиартрит возникает, по данным разных авторов, в 40-60% случаев заболевания. Для него характерно острое начало на фоне невысокого подъёма температуры тела, боли и припухлость преимущественно крупных, иногда средних суставов, летучесть и быстрое обратное развитие процесса. Ревматический артрит может быть отнесён в группу РеА, возникших вследствие перенесённой инфекции.
Ревматический кардит. Поражение сердца (ревмокардит) является ведущим в клинической картине болезни и определяет её течение и прогноз. В 70-85% случаев болезни возникает первичный ревмокардит. При ревмокардите могут поражаться все оболочки сердца - миокард, эндокард и перикард. Наиболее распространённым принято считать поражение миокарда - диффузный миокардит. Однако на ранних этапах болезни разграничить миокардит и эндокардит клинически часто бывает очень затруднительно, для этого необходимо комплексное клинико-инструментальное обследование.
Обычно больные ревмокардитом не предъявляют жалоб. Родители отмечают, что через 2-3 нед после перенесённой ангины у ребёнка продолжают сохраняться вялость, быстрая утомляемость, субфебрилитет. В этот период появляются клинические симптомы ревмокардита в виде тахикардии, реже брадикардии, расширения границ сердца, приглушённости тонов сердца. При аускультации выслушивают систолический шум. При ФКГ обнаруживают снижение амплитуды, деформацию, уширение и обеднение высокочастотными осцилляциями, преимущественно первого тона. На ЭКГ возможно выявление различных аритмий, миграции водителя ритма, замедления атриовентрикулярной проводимости (иногда вплоть до атриовентрикулярной диссоциации).
Значительные трудности представляет клиническая диагностика поражения клапанов сердца на ранних этапах заболевания, имеющая большое прогностическое значение. Большую роль в диагностике играет ЭхоКГ. Наиболее часто поражается митральный клапан. При этом на ЭхоКГ обнаруживают утолщение и «лохматость» эхосигнала от створок и хорд клапана, ограничение подвижности задней его створки. При рентгенографии у детей с поражением митрального клапана обнаруживают «митральную» конфигурацию сердца, увеличение размеров левых камер. При поражении аортального клапана на ЭхоКГ выявляют мелкоамплитудное диастолическое трепетание его створок.
На рентгенограммах видна аортальная конфигурация сердца с преимущественным увеличением левого желудочка.
Исход ревмокардита при длительности острого периода от 1,5 до 2 мес зависит от формирования порока сердца (в 20-25%). Чаще всего формируется недостаточность митрального клапана, реже - недо- статочность аортального клапана, митрально-аортальный порок, митральный стеноз.
Возвратный ревмокардит чаще развивается на фоне приобретённого порока сердца. Клинически он обычно проявляется нарастанием ранее существовавших или появлением новых шумов, развитием недостаточности кровообращения.
Малая хорея. Эта форма ревматизма возникает в 7-10% случаев, преимущественно у девочек школьного возраста. Основные симптомы заболевания обусловлены поражением подкорковых ядер головного мозга. Характерны эмоциональные расстройства (плаксивость, раздражительность, неустойчивость настроения), к которым присоединяются двигательные нарушения на фоне снижения мышечного тонуса. Гиперкинезы (беспорядочные, некоординируемые, насильственные движения отдельных групп мышц) приводят к невнятности речи, изменению почерка, неопрятности при еде, а иногда и к невозможности самообслуживания. Гиперкинезы усиливаются при волнении, чаще бывают двусторонними. Вызывая коленный рефлекс, можно выявить симптом Гордона (тоническое сокращение четырёхглавой мышцы). Гипотония мышц затрудняет обычный образ жизни. У таких больных бывает положительным симптом «дряблых плеч»: при поднятии стоящего больного за подмышечные впадины со стороны спины голова глубоко погружается в плечи. Возможна полная обездвиженность больного («мягкая» хорея). Течение хореи часто имеет затяжной и рецидивирующий характер. Обычно активная фаза длится до 2 мес.
Кольцевидная эритема. Кольцевидная эритема - сыпь в виде бледно-розовых колец на коже груди и живота. Сыпь не сопровождается зудом, не возвышается над поверхностью кожи, быстро исчезает без пигментации и шелушения.
Ревматические узелки. Ревматические узелки - округлые плотные образования размером до 0,5-1 см, определяемые в местах прикрепления сухожилий, в затылочной области. В настоящее время у детей появляются чрезвычайно редко.
Классификация
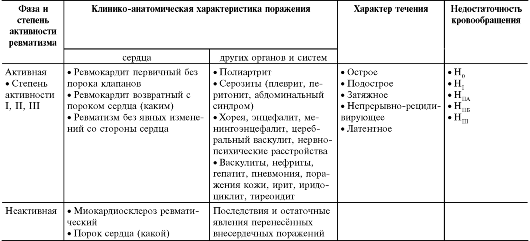
Общепринятой в настоящее время является классификация и номенклатура ревматизма А.И. Нестерова (табл. 13-1). Классификация составлена с учётом фазы болезни, клинико-анатомического пораже- ния органов, характера течения болезни и состояния кровообращения.
Таблица 13-1. Рабочая классификация и номенклатура ревматизма
Диагностика
Критерии диагностики ревматизма разработали А.А. Кисель (1940), Джонс (1944), дополнил А.И. Нестеров (1963). Основные проявления
1. Кардит.
2. Полиартрит.
3. Хорея.
4. Подкожные узелки.
5. Кольцевидная эритема.
6. «Ревматический» анамнез (связь с перенесённой носоглоточной стрептококковой инфекцией, наличие случаев ревматизма в семье).
7. Доказательство ex juvantibus - улучшение состояния больного после 2-3-недельного курса специфического лечения.
Дополнительные проявления А. Общие
1. Повышение температуры тела.
2. Адинамия, утомляемость, слабость.
3. Бледность кожных покровов.
4. Потливость.
5. Носовые кровотечения.
6. Абдоминальный синдром.
Б. Специальные (лабораторные показатели)
1. Лейкоцитоз (нейтрофильный).
2. Диспротеинемия: увеличение СОЭ, гиперфибриногенемия, появление С-реактивного белка, повышение концентрации α2 и γ-глобулинов, повышение концентрации сывороточных му- копротеинов.
3. Изменения серологических показателей: появление Аг стрептококков в крови, повышение титров антистрептолизина-О, антистрептокиназы, антистрептогиалуронидазы.
4. Повышение проницаемости капилляров.
В настоящее время наиболее распространены критерии ВОЗ (1989), разработанные Американской ревматологической ассоциацией (табл. 13-2).
Наличие у больного двух больших или одного большого и двух малых критериев свидетельствует о высокой вероятности острого ревматизма, особенно при подтверждённых данных о перенесённой инфекции, вызванной стрептококками группы А.
Таблица 13-2. Критерии диагностики ревматизма*

* Из: Насонова В.А. и соавт. Клиническая ревматология. М., 1989.
Дифференциальная диагностика
Ревматический полиартрит дифференцируют с РеА, дебютом ЮРА и ЮАС, СКВ, геморрагическим васкулитом. Ревмокардит следует отграничить от неревматического кардита, ПМК, ВПС, инфекционного эндокардита. Хорею дифференцируют с функциональными тиками, гиперкинезами при СКВ, тиреотоксикозе, опухолях мозга.
Комплексный метод терапии первичного ревматизма включает одновременное назначение небольших доз (0,5-0,7 мг/кг/сут) глюко- кортикоидов и НПВС.
• Дозы глюкокортикоидов выбирают в зависимости от тяжести патологического процесса и выраженности изменений в сердце. Начальную дозу постепенно снижают до полной отмены в среднем через 1,5 мес.
• Из НПВС наиболее часто назначают индометацин и диклофенак. НПВС комбинируют с глюкокортикоидами и одним из базисных препаратов, особенно при затяжном течении заболевания и формировании порока сердца.
• В качестве базисной терапии используют хинолиновые производные: хлорохин, гидроксихлорохин.
Учитывая стрептококковую природу ревматизма, в течение первых 10-14 дней терапии назначают бензилпенициллин или его аналоги. В комплексную терапию входит также санация очагов хронической инфекции, в частности хронического тонзиллита. При декомпенсиро-
Лечение
ванном хроническом тонзиллите необходима тонзиллэктомия. Через 6-8 мес после острого периода рекомендуют санаторно-курортное лечение.
Профилактика
Рекомендации ВОЗ (1989) по предупреждению ревматизма и его рецидивов включают следующие мероприятия.
1. Первичная профилактика - мероприятия, обеспечивающие правильное развитие ребёнка:
• закаливание с первых месяцев жизни;
• полноценное питание с достаточным содержанием витаминов;
• рациональная физическая культура и спорт;
• борьба с инфекцией, вызванной стрептококком группы А (ангина, скарлатина), включающая назначение препаратов пенициллина. Рекомендуемый препарат - феноксиметилпенициллин.
2. Вторичная профилактика направлена на предупреждение рецидивов и прогрессирования болезни. Наиболее оптимальна круглогодичная профилактика, проводимая ежемесячно в течение не менее 5 лет. Всем детям, перенёсшим ревматизм, назначают бензатина бензилпенициллин.
Прогноз
Прогноз в последние годы значительно улучшился благодаря мерам первичной и вторичной профилактики. Первичный ревмокардит приводит к формированию пороков сердца только у 20-25% больных. Реже наблюдают случаи тяжёлого течения ревматизма. Летальность снизилась с 11-12% до 0,4-0,1%.
ЮВЕНИЛЬНЫЙ РЕВМАТОИДНЫЙ АРТРИТ
Ювенильный ревматоидный артрит (ЮРА) - хроническое воспалительное заболевание суставов у детей до 16 лет с неизвестной этиологией и сложным патогенезом, характеризующееся неуклонно прогрессирующим течением, сопровождающееся у некоторых больных вовлечением внутренних органов, нередко заканчивающееся инвалидизацией.
Среди ревматических заболеваний детского возраста ЮРА занимает по распространённости первое место. Заболевание наблюдают в различных регионах земного шара с частотой от 0,05 до 0,6% в популяции. Первичная заболеваемость также колеблется в значительных пределах, составляя от 6 до 19 случаев на 100 000 детского населения.
ЭТИОЛОГИЯ
Этиология ЮРА до настоящего времени неизвестна. Среди его причин рассматривают совокупность различных факторов внешней среды (вирусная и бактериальная инфекции, травма сустава, переохлаждение организма, инсоляция, введение белковых препаратов и др.). В основе неадекватной ответной реакции у больных ЮРА лежит их «сверхчувс- твительность к различным факторам внешней среды» (Е.М. Тареев), в результате чего формируется сложный иммунный ответ, приводящий к развитию прогрессирующего заболевания. Определённую роль играет и семейно-наследственная предрасположенность к ревматическим заболеваниям. Исследования последних десятилетий выявили связь ЮРА с наличием у больных DR-локуса HLA с преобладанием DR4 у пациентов с системными формами болезни и DR5 - с преимущественно суставным вариантом болезни.
ПАТОГЕНЕЗ
Патологический процесс при ЮРА начинается в синовиальной оболочке сустава с нарушения микроциркуляции и поражения клеток, выстилающих синовиальную мембрану. В ответ на перечисленные выше изменения в организме больного образуются изменённые IgG, которые воспринимаются собственной иммунной системой как аутоантигены. Иммунокомпетентные клетки, в том числе плазматические клетки синовиальной оболочки сустава, в ответ вырабатывают АТ - анти-IgG. Эти АТ, названные ревматоидным фактором, в при- сутствии комплемента взаимодействуют с аутоантигеном, происходит формирование иммунных комплексов. ЦИК оказывают повреждающее воздействие как на эндотелий сосудов, так и на окружающие ткани. В первую очередь страдает синовиальная оболочка сустава, в результате чего развивается артрит. В синовиальной жидкости и тканях сустава при этом образуется избыточное количество цитокинов макрофагального происхождения - ИЛ1 и ИЛ6, фактора некроза опухолей-α (ФНОа). ИЛ1 индуцирует воспаление и разрушает хрящ. Этим же свойством обладает ФНОа. ИЛ6 способствует гиперпродукции белков острой фазы воспаления - С-реактивного белка и фибриногена. Происходит дальнейшая активизация ферментных систем, разрушающих хрящ. Усиление новообразования сосудов, или ангиогенез, вследствие действия на ткани цитокинов, также усиливает деструкцию хряща. В процессе воспаления в тканях сустава формируется большое число клеток, образующих так называемый паннус, или плащ, закрывающий поверхность суставного хряща, тем самым препятствуя нормальным процессам обмена и усиливая деструкцию костно-хрящевых образований.
ПАТОМОРФОЛОГИЯ
При биопсии синовиальной мембраны в начальном периоде болезни выявляют ворсинчатую гипертрофию и гиперплазию поверх- ностного слоя. Воспаление в суставе при ЮРА, как и у взрослых больных, приводит к образованию эрозий и деструкции хряща. Однако эти процессы у детей развиваются медленнее и в меньшем проценте случаев. Как правило, количество и глубина эрозий соответствуют глубине и тяжести процесса. Одновременно с патологическим процессом в суставах у детей наблюдают увеличение лим- фатических узлов, обусловленное неспецифической фолликулярной гиперплазией. У детей при ЮРА, как и у взрослых, во всех органах могут развиваться васкулиты, не имеющие специфических морфологических признаков. Наблюдают также атрофии мышечных волокон. Наиболее выражены изменения в мышцах, прилегающих к поражён- ным суставам.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина ЮРА разнообразна. Начало заболевания может быть острым или подострым. При остром начале обычно повыша- ется температура тела, появляется болезненность, а затем отёк в одном или нескольких суставах, чаще симметричных. Однако симметричность поражений иногда становится очевидной не сразу, а в течение нескольких дней или недель от начала болезни. Поражаются, как правило, крупные суставы - коленные, голеностопные, лучезапястные, но иногда с самого начала болезни страдают мелкие суставы рук и ног (плюснефаланговые, межфаланговые) (рис. 13-1 и 13-2 на вклейке). Типично для ЮРА поражение суставов шейного отдела позвоночника. Все суставы резко болезненны, отёчны, в редких случаях кожа вокруг них гиперемирована. Температура тела постепенно повышается и может достигать 38-39 ?C. При этом нередко на коже туловища и конечностей появляется полиморфная аллергическая сыпь, увеличиваются периферические лимфатические узлы, печень и селезёнка. В общем анализе крови выявляют анемию, часто нейтрофильный лейкоцитоз со сдвигом лейкоцитарной формулы влево, увеличение СОЭ до 40-60 мм/ч, повышение концентрации Ig, преимущественно
IgG.
Острое начало болезни обычно свойственно тяжёлым формам ЮРА - генерализованной суставной или суставно-висцеральной (системной) форме болезни с часто рецидивирующим течением и неблагоприятным прогнозом. Эту форму чаще наблюдают у детей дошкольного и младшего школьного возраста, но она может возникать и у подростков.
Подострое начало болезни характеризуется менее яркой симптоматикой. Артрит, как правило, начинается с одного сустава - коленного или голеностопного (рис. 13-3 на вклейке). Сустав распухает, нарушается его функция, иногда даже без выраженной болезненности. У ребёнка изменяется походка, а дети до 2 лет перестают ходить. Наблюдают так называемую утреннюю скованность в суставах, выражающуюся в том, что больной после ночного сна чувствует некоторое время затруднение при движениях в суставах и самообслуживании. Он с трудом встаёт, походка его замедленна. Утренняя скованность может продолжаться от нескольких минут до 1 ч и более. Процесс в течение длительного времени может ограничиться одним суставом (ревматоидный моноартрит). Такая форма заболевания, особенно у девочек дошкольного возраста, нередко сопровождается ревматоидным поражением глаз - ревматоидным увеитом, односторонним или двусторонним. При ревматоидном увеите затронуты все обо- лочки глаза, вследствие чего резко падает острота зрения вплоть до полной его потери, причём иногда в течение полугода. В редких случаях развитие ревматоидного увеита может предшествовать суставному процессу, что чрезвычайно затрудняет своевременную диагностику.
Подострое начало болезни может протекать и с вовлечением в процесс нескольких суставов - чаще 2-4. Такую форму болезни на- зывают олигоартикулярной. Боли в суставах могут быть умеренными, как и экссудативные изменения. В процесс могут вовлекаться, например, два голеностопных и один коленный сустав и наоборот. Температура тела не повышается, полиаденит умеренный. Эта форма ЮРА протекает более доброкачественно, с менее частыми обострениями.
В последующем, при прогрессировании болезни, возможны две основные формы - преимущественно суставная и суставно-висцераль- ная формы в соотношении 65-70% и 35-30% соответственно.
Суставно-висцеральная форма
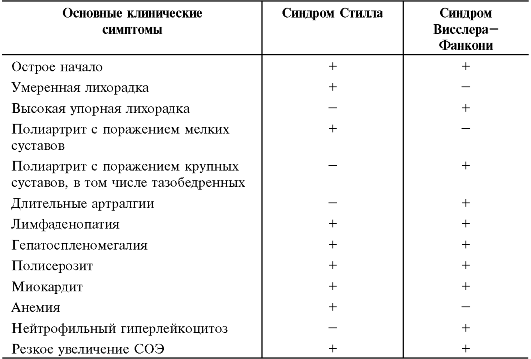
Суставно-висцеральная (системная форма) включает пять признаков: упорная высокая лихорадка, полиморфная аллергическая сыпь, лимфаденопатия, гепатолиенальный синдром, артралгии/ар- трит. Эта форма ЮРА имеет два основных варианта (табл. 13-3) - синдром Стилла, чаще развивающийся у детей дошкольного возраста, и синдром Висслера-Фанкони, обычно наблюдаемый у школьников.
Таблица 13-3. Варианты системной формы ювенильного ревматоидного артрита
Длительно рецидивирующее течение ЮРА может осложниться вторичным амилоидозом, чему способствует постоянная циркуляция в кровеносном русле иммунных комплексов. Амилоид откладывается в стенках сосудов, в почках, печени, миокарде, кишечнике, что приводит к нарушению их функций. Чаще всего амилоидоз поражает почки, о чем свидетельствует стойкая протеинурия с развитием в последующем ХПН.
Суставная форма
При суставной форме прогрессирование ЮРА приводит к стойкой деформации суставов с частичным или полным ограничением под- вижности в них. До 25% детей становятся инвалидами (рис. 13-4 и 13-5 на вклейке).
КЛАССИФИКАЦИЯ
Основные формы ЮРА представлены в рабочей классификации болезни (отечественный вариант, табл. 13-4). Кроме того, в настоящее время широко распространена англо-американская классификация
ЮРА (табл. 13-5).
Таблица 13-4. Рабочая классификация ювенильного ревматоидного артрита
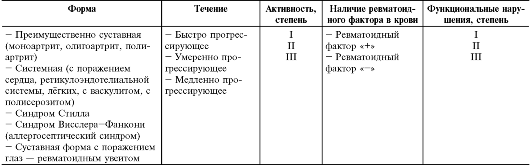
Таблица 13-5. Англо-американская классификация ювенильного ревматоидного артрита
_

ДИАГНОСТИКА
Диагностика ЮРА часто представляет трудности, особенно на ранних этапах болезни. В России приняты следующие диагностические критерии.
Клинические признаки
1. Артрит, продолжительностью 3 мес и более.
2. Артрит второго сустава, возникший через 3 мес и позже.
3. Симметричное поражение мелких суставов.
4. Контрактуры суставов.
5. Тендосиновит или бурсит.
6. Мышечная атрофия.
7. Утренняя скованность.
8. Ревматоидное поражение глаз.
9. Ревматоидные узелки.
10. Выпот в полость суставов. Рентгенологические признаки
11. Остеопороз, мелкокистозная перестройка костной структуры эпифиза.
12. Сужение суставных щелей, костные эрозии, анкилоз суставов (рис. 13-6 на вклейке).
13. Нарушение роста костей.
14. Поражение шейного отдела позвоночника. Лабораторные признаки
15. Положительный ревматоидный фактор.
16. Положительные данные биопсии синовиальной оболочки.
В зависимости от количества выявленных положительных признаков определяют степень вероятности наличия заболевания (при обязательном наличии артрита):
• 3 признака - вероятный ЮРА;
• 4 признака - определённый ЮРА;
• 8 признаков - классический ЮРА.
Используют также рентгенологические критерии изменений в суставах по классификации американского рентгенолога Штейнброккера, подразделяемые на четыре стадии.
I степень - остеопороз без деструктивных изменений.
II степень - незначительные разрушения хряща и кости, небольшое сужение суставной щели, единичные узуры костей.
III степень - значительные разрушения хряща и кости, выраженное сужение суставной щели, множественные узуры, подвывихи, локтевая девиация.
IV степень - симптомы степени III в сочетании с анкилозом.
В результате ревматоидного воспаления развиваются изменения в суставах, имеющие три степени в зависимости от характера и тяжести нарушения их функций. Степени нарушения функций суставов следующие.
I степень - умеренное ограничение профессиональной деятельности
(учёбы в школе), но полное сохранение самообслуживания.
II степень - лишение способности выполнять профессиональную деятельность (учёбу в школе) и умеренное ограничение самообслуживания.
III степень - утрата возможности самообслуживания и необходимость постороннего ухода.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
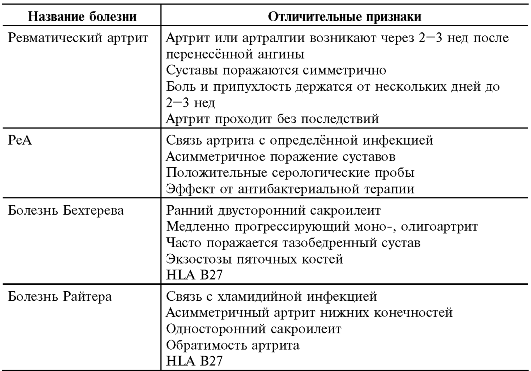
Дифференциальную диагностику ЮРА следует проводить с ревматическим артритом, РеА, анкилозирующим спондилоартритом, болезнью Райтера. В табл. 13-6 приведены отличительные признаки этих заболеваний и суставного синдрома в частности.
Таблица 13-6. Дифференциальная диагностика ювенильного ревматоид- ного артрита*
* Из: Насонова В.А. и соавт. Клиническая ревматология. М., 1989.
ЛЕЧЕНИЕ
Лечение при ЮРА необходимо проводить комплексно и этапно. В активном периоде болезни больные нуждаются в стационарном лечении, в неактивном - в амбулаторном наблюдении и санаторно- курортном лечении. Значительную часть времени больные лечатся в амбулаторных условиях в связи с длительностью болезни. В поликлинике дети продолжают получать сочетанную терапию, включающую медикаментозное лечение, ЛФК, курсы массажа и физиотерапии. Только длительное и непрерывное лечение под контролем врача и медицинской сестры может дать положительный эффект.
В период обострения лечение включает НПВС, в тяжёлых случаях в сочетании с глюкокортикоидами и иммунодепрессантами (хинолино- вые производные, пеницилламин, метотрексат, циклоспорин), а также с иммуноглобулином человеческим нормальным. Ниже приведены основные препараты, их дозы и длительность применения.
Основные лекарственные препараты, применяемые при лечении ювенильного ревматоидного артрита
I. Нестероидные противовоспалительные препараты: диклофенак, ацетилсалициловая кислота, индометацин, ибупрофен, напроксен.
II. Глюкокортикоиды: преднизолон - внутрь; метилпреднизолон - внутривенно, метилпреднизолон и бетаметазон - внутрисуставно.
III. Базисные препараты
• Хинолиновые: гидроксихлорохин и хлорохин.
• Метотрексат.
• Сульфасалазин.
• Циклоспорин.
Базисные препараты назначают на длительный срок, от одного до нескольких лет, в зависимости от клинической картины болезни. Проведение базисной терапии приводит к уменьшению потребности в НПВС и глюкокортикоидах (следовательно, снижает риск развития побочных эффектов, возникающих на фоне лечения этими препаратами), позволяет улучшить качество жизни, снизить инвалидность, улучшить отдалённый прогноз, увеличить продолжительность жизни.
IV. Иммунотерапия
Иммуноглобулин человеческий нормальный (например, «Пентаглобин», «Интраглобин», «Сандоглобулин»).
Местная терапия
Широко применяют местное лечение поражённого сустава - внутрисуставное введение препаратов, преимущественно глюкокортикои- дов, временную иммобилизацию сустава с помощью съёмной лангеты,
различные физиотерапевтические методы лечения, ЛФК, массаж. При наличии контрактур накладывают скелетное вытяжение, проводят механотерапию на специальной аппаратуре.
Осложнения терапии
Препараты, применяемые для лечения ЮРА, имеют много побочных эффектов. Так, НПВС и глюкокортикоиды при пероральном приёме повышают кислотообразующую функцию желудка и могут вызвать хронический гиперацидный гастрит с развитием эрозивноязвенного процесса, поэтому их необходимо принимать после еды и желательно запивать щелочным питьём. Если ребёнок, страдающий ЮРА и получающий лечение, жалуется на боли в животе, необходимо срочно показать его врачу, провести эндоскопическое исследование желудка, чтобы не пропустить тяжёлого осложнения, например прободения язвы.
ПРОГНОЗ
ЮРА - заболевание пожизненное, однако при правильно подобранной терапии и систематическом наблюдении ревматолога возможна длительная ремиссия с удовлетворительным качеством жизни (возможна учёба, приобретение среднего и высшего образования, работа по профессии). При часто рецидивирующем течении, системных проявлениях болезни прогноз более пессимистичен - рано происходит инвалидизация, активная жизнь ограничена.
ЮВЕНИЛЬНЫЙ АНКИЛОЗИРУЮЩИЙ СПОНДИЛОАРТРИТ
Ювенильный анкилозирующий спондилоартрит (ЮАС) - хроническое воспалительное заболевание периферических суставов и поз- воночника, преимущественно у лиц мужского пола.
Заболевание известно давно, однако как самостоятельная нозологическая единица было выделено В.М. Бехтеревым в
Эпидемиология
Точных данных о частоте ЮАС нет. Среди взрослых частота манифестного типа заболевания составляет 2 на 1000 и выше, из всех случаев анкилозирующего спондилоартрита 10-25% дебютируют в детском возрасте. Заболеваемость ЮАС в различных регионах мире варьирует, что связано с популяционной частотой HLA B27. По дан-
ным Минздрава России (2002) заболеваемость ЮАС составила 1,7 на 100 000 детского населения (среди детей до 18 лет). Мальчики болеют в 5-7 раз чаще, чем девочки. В то же время выявлено, что среди носителей HLA B27 частота рентгенологически определяемого сакроилеита у женщин примерно такая же, как и у мужчин, однако заболевание носит клинически менее выраженный характер.
Этиология и патогенез
ЮАС - сложное мультифакториальное заболевание с наследственной предрасположенностью. Роль наследственности подтверждается накоплением в семьях повторных случаев заболевания. У больных с анкилозирующим спондилоартритом частота Аг HLA B27 составляет 70-80%, а в общей популяции - 4-10%. Такая высокая ассоциация с иммуногенетическим маркёром позволила подтвердить роль наследственности в развитии заболевания и использовать его наличие в качестве диагностического критерия.
Большинство ревматологов признают роль инфекции как этиологического фактора анкилозирующего спондилоартрита. У части больных с РеА, особенно у носителей HLA B27 в дальнейшем развивается анкилозирующий спондилоартрит. Большинство исследователей предполагают этиологическую роль клебсиеллы и некоторых других энтеробактерий. Ряд исследований показал наличие перекрёстной серологической реактивности и структурное сходство Аг клебсиеллы и
HLA B27.
Существует несколько гипотез, объясняющих взаимодействие наследственных и инфекционных факторов в развитии анкилозирую- щего спондилоартрита. Гипотеза молекулярной мимикрии позволяет предположить, что в результате структурного сходства HLA B27 и ряда микроорганизмов АТ, вырабатываемые к инфекционным агентам, перекрёстно реагируют с собственными белковыми структурами (с Аг HLA B27), что вызывает развитие аутоиммунного процесса.
Существует гипотеза о влиянии Аг HLA на иммунный ответ организма, в частности доказано, что носители HLA B27 имеют сниженную фагоцитарную активность.
Клиническая картина и диагностика
• Симптоматика ЮАС обычно включают поражение периферических суставов, энтезиты, поражение осевого скелета, общие симптомы (обычно минимальны, может быть субфебрильная лихорадка), экстраартикулярные проявления (поражение глаз, сердца). Суставной синдром при ЮАС представлен моноили олигоартритом асимметричного характера, с преимущественным поражением суставов ног. Чаще в процесс вовлекаются коленные, реже - го-
леностопные и тазобедренные суставы. Возможно поражение суставов верхних конечностей (асимметричного характера), а также грудино-ключичных, грудино-рёберных, лобковых сочленений. Довольно типичны для ЮАС поражение суставов плюсны, плюснефаланговых суставов I пальца ноги. Поражение межфаланговых суставов пальцев стоп носит асимметричный характер, сопровождается воспалением околосуставных тканей с развитием так называ- емого дактилита. Поражение суставов свода стопы сопровождается воспалительными изменениями периартикулярных тканей, рентгенологически выявляют остеопороз костей свода стопы, возможны эрозии суставных поверхностей и даже анкилоз суставов стопы. Мелкие суставы кистей поражаются редко.
• Энтезиты - воспаление в местах прикрепления связок, сухожилий, фасций или капсул суставов. Энтезиты - наиболее ранний симптом ЮАС, в детском возрасте их наблюдают чаще, чем у взрослых пациентов. При спондилоартритах энтезиты возникают у 90% пациентов, с другой стороны, они возможны и при других ревматических заболеваниях (ЮРА, СКВ). Энтезиты проявляются всеми признаками воспаления - спонтанной болью, или болевой реакцией при пальпации в этой области, можно отметить припухание, реже - повышение местной температуры. Наиболее часто энтезопатии развиваются в области пяточных костей, в месте прикрепления ахиллова сухожилия. Для анкилозирующего спондилоартрита характерен ахиллобурсит. Типичны для ЮАС талалгии - боли в пятках, а также боли в области свода стопы, что затрудняет ходьбу, особенно на пятках или на носках. Энтезопатии также могут развиваться в местах прикрепления подошвенного апоневроза в области верхнего и нижнего полюса надколенников, в проекции гребней подвздошных костей, в области бугристости большеберцовых костей, в проекции большого вертела бедренной кости, по ходу пупартовых связок, в области ягодиц.
• Симптомы поражения позвоночника в дебюте ЮАС, в отличие от анкилозирующего спондилоартрита у взрослых, минимальны и развиваются по мере прогрессирования заболевания. Поражение позвоночника чаще начинается с пояснично-крестцового отдела, реже - с шейного. Боль в спине или иррадиирующая боль в ягодицах, в паховой области, в области бёдер носит ноющий характер, отмечается в покое, менее выражена при движениях. Боль возникает исподволь, сопровождается скованностью в утренние часы. Может исчезать самостоятельно или рецидивировать.
- Ригидность (нарушение подвижности) позвоночника позволяет подтвердить диагноз ЮАС. Для выявления ригидности позвоночника используют пробу Томайера - определение расстояние
между пальцами и полом при наклоне пациента. Это расстояние не должно превышать
- Со временем у пациентов появляется нарушение физиологических изгибов позвоночника, усиливаются или сглаживаются грудной кифоз, поясничный лордоз, иногда выявляют ограничение экскурсии грудной клетки.
- Один из первых и частых признаков вовлечения позвоночника - поражение крестцово-подвздошных сочленений, которое проявляется спонтанными болями либо болезненностью при физикальном обследовании, для чего предложен ряд тестов - прямое надавливание на область крестцово-подвздошных сочленений, сдавливание руками с двух сторон таза, надавливание на гребень подвздошной кости в положении больного на боку.
- Рентгенологические изменения при развитии сакроилеита проходят несколько стадий: I - явные ранние изменения: псевдорасширение суставных щелей крестцово-подвздошного сочленения; II - сужение и нечёткость суставной щели, эрозии, частичный анкилоз; III - полный костный анкилоз крестцово-подвздошного сочленения. В последние годы для диагностики сакроилеита рекомендуют проведение КТ или МРТ. В отличие от взрослых пациентов с анкилозирующим спондилоартритом, обнаружить рентгенологические изменения вышележащих отделов позвоночника у детей с ЮАС удаётся крайне редко. Межпозвонковые суставы поражаются у детей на более поздних сроках заболевания.
• Поражение глаз в виде иридоциклита, увеита более характерно для взрослых пациентов с анкилозирующим спондилоартритом, однако они возможны и при ЮАС (10-20% случаев). Увеит развивается остро, чаще в виде одностороннего процесса, проявляется покраснением, светобоязнью, слёзотечением.
• Поражение сердца - довольно редкое проявление ЮАС. Преимущественно в процесс вовлекается аорта с развитием недостаточности аортального клапана.
• Поражение почек при ЮАС также наблюдают редко. Причинами нефропатии у больных ЮАС может быть приём НПВС (лекарст-
венная нефропатия), болезнь Берже (IgA-нефропатия), а также развитие вторичного амилоидоза. Диагностические критерии
Диагноз анкилозирующего спондилоартрита у взрослых устанавливают на основании диагностических Римских и/или Нью-Йоркских критериев, однако они ориентированы на развёрнутые стадии заболе- вания и практически неприменимы в детской практике. Специально для диагностики ЮАС группой немецких ревматологов разработаны критерии Garmisch-Partenkirchen (GP) (табл. 13-7).
Согласно этим критериям, диагноз вероятного ЮАС устанавливают при наличии 2 основных критериев или 1-2 основных и 2 дополнительных. Определённому диагнозу ЮАС соответствует тот же набор признаков в сочетании с рентгенологическим подтверждением сакроилеита (двухсторонний сакроилеит II стадии или односторонний сакроилеит III стадии).
Таблица 13-7. Диагностические критерии ЮАС Garmisch-Partenkirchen (GP)

Дифференциальная диагностика
Поскольку в клинической картине ЮАС преобладает поражение периферических суставов, проведение дифференциальной диагностики с другими хроническими артритами у детей часто бывает затруд- нено.
• Олигоартрит с поражением глаз характерен для одного из вариантов ЮРА, однако последний преимущественно развивается у девочек раннего возраста. Увеит при этом варианте ЮРА чаще носит подострый характер, развивается исподволь, без ярких клинических проявлений. Для этой формы ЮРА характерно повышение уровня антинуклеарного фактора в 40-50% случаев, отсутствует связь с HLA B27 (возможны ассоциации с HLA А2, DR5).
• Наличие асимметричного олигоартрита, выявление связи с перенесённой кишечной или урогенительной инфекцией, наличие маркёров хламидийной, микоплазменной, кишечной инфекций позволяет диагностировать РеА или болезнь Райтера (при наличии артрита, уретрита, конъюнктивита). Несмотря на сходство клинической картины и схожесть патогенеза РеА и ЮАС, наличие энтезопатий и ригидности позвоночника для первого не характерно. Принципиально важный дифференциально-диагностический критерий - отсутствие рентгенологических признаков сакроилеита при РеА.
• Псориатический спондилоартрит предполагает наличие у пациента характерных кожных изменений и/или наличие в семейном анамнезе больных псориазом.
• Болезнь Крона и неспецифический язвенный колит также могут протекать со спондилоартритом. При этой патологии отмечают изменение характера стула, нарастающую дистрофию; эндоскопическое исследование кишечника позволяет поставить правильный диагноз.
• Принципиальное значение имеет дифференциальная диагностика с заболеваниями неревматической природы, обладающими схожей симптоматикой, - инфекционными артритами (туберкулёзным, бруцеллёзным и др.), а также патологией невоспалительного характера (дисплазии, остеохондропатии) и онкологической патологией.
Лечение
• С противовоспалительной целью для лечения ЮАС применяют НПВС. Наиболее эффективным противовоспалительным препаратом в лечении ЮАС является индометацин, который назначают перорально по 2-3 мг/кг/сут в 2-3 приёма. Тем не менее в связи с высокой частотой побочных эффектов индометацина, лечение рекомендуют начинать с напроксена (перорально 15-20 мг/кг/сут в 2 приёма). Реже применяют диклофенак, другие НПВС при ЮАС считают неэффективными.
• Глюкокортикоиды, как наиболее мощные противовоспалительные средства, используют в период обострения суставного синдрома. Их применение ограничивается преимущественно внутрисуставным введением. При необходимости можно воспользоваться коротким курсом пульс-терапии: быстрое (30-60 мин) внутривенное введение больших доз метилпреднизолона (5-15 мг/кг) в течение 3 дней. При наличии аортита, поражения почек и торпидного к лечению увеита возможно назначение преднизолона перорально в дозах не более 0,5 мг/кг/сут. Для лечения увеита также применяют местно противовоспалительные и глюкокортикоидные препараты.
• Иммуносупрессивная терапия: в качестве базисного препарата наиболее часто применяют сульфасалазин по 30-40 мг/кг/сут, также существуют данные об использовании метотрексата по 10 мг/м2 1 раз в неделю.
Прогноз
Прогноз для жизни благоприятен. При длительном течении ЮАС у больных нарастает тугоподвижность в позвоночнике. Причиной инвалидизации может быть поражение тазобедренных суставов, анкило- зирование межпозвоночных суставов. При адекватном наблюдении и лечении удаётся приостановить прогрессирование заболевания.
РЕАКТИВНЫЙ АРТРИТ
Реактивный артрит (РеА) - асептическое воспалительное заболевание суставов, развивающееся в ответ на внесуставную инфекцию, при котором предполагаемый первичный агент не может быть выделен из суставов на обычных искусственных питательных средах.
Термин «реактивный артрит» введён в литературу в
ЭПИДЕМИОЛОГИЯ
В
структуре ревматических заболеваний разных стран доля РеА составляет
от 8,6 до 41,1%. Такая вариабельность показателей объясняется
сложностью диагностики заболевания при слабой выраженности
предшествующей инфекции, разными диагностическими подходами, а также
наличием перекрёстной клинической симптоматики с другими артритами. В
последнее десятилетие в структуре ревматических заболеваний наблюдают
тенденцию к нарастанию частоты РеА. В России по данным отдела
медицинской статистики МЗ РФ распространён- ность РеА в
ляет 57,5%, у подростков - 41,8%. Представленные показатели доказывают, что проблема РеА в детском возрасте весьма актуальна.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
В настоящее время к РеА относят в основном заболевания, связанные с кишечной и мочеполовой инфекциями, которые ассоциируются с Аг гистосовместимости В27 (HLA B27). Выделяют две группы ар- тритов: урогенитальные и постэнтероколитические. Этиологические факторы урогенитальных РеА включают Chlamydia trachomatis (серовар D, K), уреаплазму. Этиологические факторы постентероколитических РеА - иерсинии (Yersinia enterocolitica, 03 и 09 серотип; Yersinia pseudotuberculosis), сальмонеллы (Salmonella enteritidis, Salmonella typhimurium и др.), шигеллы (Shigella flexneri II-IIa), кампилобактер (Campylobacter jejuni). Инфекции респираторного тракта, связанные с Mycoplasma pneumoniae и, особенно, Chlamydia pneumoniae также являются частыми причинами развития РеА.
Урогенитальные реактивные артриты
В настоящее время одна из наиболее частых причин развития РеА - хламидийная инфекция (до 80% случаев), что связано с пандемией хламидиоза в мире, особенностями путей передачи хламидийной инфекции, цикла развития хламидий и реакции на терапию.
Основная особенность хламидий - внутриклеточный паразитизм. Источник инфекции - люди, млекопитающие, птицы. Заражение человека Chlamydia pneumoniae и Chlamydia psittaci происходит воздушно-капельным и воздушно-пылевым путём, Chlamydia trachomatis пе- редаётся половым, вертикальным, контактно-бытовым путём и при прохождении плода через инфицированные родовые пути матери. В детском возрасте половой путь передачи не является приоритетным.
Перенесённая инфекция не обеспечивает пожизненного иммунитета, естественная резистентность отсутствует.
Хламидии имеют две клеточные формы: элементарное тельце (высокоинфекционная, не проявляющая метаболическую активность форма) и ретикулярное тельце (репродуктивная внутриклеточная форма). Жизненный цикл хламидий проходит несколько стадий. Внутриклеточный цикл развития хламидий продолжается 2-3 сут, однако он может задержаться в репродуктивной фазе на несколько дней и месяцев.
Хламидии также могут трансформироваться в L-формы. К этому предрасполагают аномальные реакции иммунной системы, а также применение неадекватных доз антибиотиков и антибиотиков, к которым хламидии не чувствительны. L-формы обладают очень слабой способностью к антигенному раздражению иммунокомпетентных клеток и могут длительно находиться внутри клеток, при делении они
передаются дочерним клеткам. Это приводит к длительной персистенции возбудителя и хроническому течению процесса.
У больных с хроническим хламидийным РеА, как правило, выявляют аномалии иммунного ответа - нарушение соотношения между Т-супрессорами и Т-хелперами (снижение количества последних), выраженное уменьшение относительного и абсолютного количества В-клеток, снижение количества NK-клеток. Все указанные изменения в иммунном ответе организма больных способствуют развитию хронизации процесса. В реализации генетически детерминированной предрасположенности индивидуума к развитию хронического РеА придают большое значение носительству HLA-B27.
Постэнтероколитические реактивные артриты
В происхождении РеА, связанных с кишечной инфекцией, основное значение также придают инфекции и генетической предрасположенности. Тем не менее истинный характер взаимоотношений микро- и макроорганизма всё ещё не ясен.
«Артритогенные» микроорганизмы проникают в слизистую оболочку кишечника и размножаются внутри полиморфноядерных лейкоци- тов и макрофагов. В дальнейшем происходит транспорт бактерий и продуктов их жизнедеятельности из первичного очага в органы мишени. По данным экспериментальных исследований, наиболее длительное время микроорганизмы персистируют в клетках, экспрессирующих HLA В27. Предполагают, что наличие HLA B27 обусловливает развитие аномальной иммунной реакции на патогенную кишечную и урогенитальную микрофлору. HLA-B27 даёт перекрёстные сероло- гические реакции с хламидиями и некоторыми грамотрицательными энтеробактериями, что обусловлено феноменом микробной антигенной мимикрии. В составе клеточной стенки ряда кишечных бактерий и хламидий присутствуют белки, которые содержат фрагменты, имеющие структурное сходство с отдельными участками молекулы HLA B27. Допускают, что перекрёстно реагирующие АТ способны оказывать повреждающее действие на собственные клетки организма, которые экспрессируют достаточное для этого количество молекул HLA B27. С другой стороны, считают, что такое перекрёстное реагирование препятствует осуществлению адекватного иммунного ответа против внутриклеточных паразитов и их эффективной элиминации, способствуя персистированию инфекции.
О значении генетических факторов в патогенезе РеА свидетельствует тесная ассоциация с HLA-B27, который выявляют при постэн- тероколитических артритах в 80-90% случаев и несколько реже при урогенитальных РеА (популяционная частота этого HLA D-27 составляет 7-10%).
КЛИНИЧЕСКАЯ КАРТИНА
Синдром Райтера (уретро-окуло-синовиальный синдром) впервые был описан в
Синдром Райтера чаще всего начинается с симптомов поражения урогенитального тракта через 2-4 нед после перенесённой кишечной инфекции или предполагаемого заражения хламидиозом. В последующем присоединяются симптомы поражения глаз и суставов.
• Поражение урогенитального тракта у детей характеризуется стёртостью клинической картины. У мальчиков может развиваться баланит, инфицированные синехии, фимоз. У девочек поражение урогенитального тракта может ограничиваться вульвитом, вульвовагинитом, лейкоцит- и микрогематурией, а также циститом. Поражение урогенитального тракта может на несколько месяцев предшествовать развитию суставного синдрома.
• Поражение глаз характеризуется развитием коньюнктивита чаще катарального, невыраженного, непродолжительного, но склонного к рецидивам. При иерсиниозном РеА конъюнктивит может быть гнойным, тяжёлым. У трети больных развивается острый иридоциклит, угрожающий слепотой. Поражение глаз также может на несколько месяцев или лет предшествовать развитию суставного синдрома.
• Поражение опорно-двигательного аппарата проявляется ограниченным асимметричным моно-, олиго-, реже полиартритом. Характерно преимущественное вовлечение в процесс суставов ног, с наиболее частым поражением коленных, голеностопных суставов, плюснефаланговых, проксимальных и дистальных межфаланговых суставов пальцев стоп. Артрит может начинаться остро, с выраженными экссудативными изменениями. У некоторых пациентов повышается температура тела, вплоть до фебрильных значений. Экссудативный артрит может протекать без боли, скованности, выраженного нарушения функции, с большим количеством синовиальной жидкости, непрерывно рецидивируя. Поражение суставов характеризуется длительным отсутствием деструктивных изменений, несмотря на рецидивирующий синовит. Типичным для РеА считают поражение первого пальца стопы, формирование «сосискообразной» деформации пальцев стоп (за счёт выраженного отёка и гиперемии пора- жённого пальца), развитие теносиновита и бурсита, ахиллобурсита.
У ряда больных отмечается развитие энтезита и энтезопатий (боли и болезненность при пальпации в местах прикрепления сухожилий к костям), частые боли в пятках, боли, скованность, ограничение подвижности в шейном и поясничном отделе позвоночника и илеосакральных сочленениях. Эти клинические симптомы характерны для мальчиков подросткового возраста и ассоциируются с HLA B27. В таких случаях высок риск формирования ЮАС. При затяжном (6-12 мес) или хроническом (более 12 мес) течении болезни характер суставного синдрома меняется. Увеличивается количество поражённых суставов, артрит становится более симметричным, чаще вовлекаются суставы верхних конечностей и позвоночник.
• Часто классические симптомы синдрома Райтера хронологически не связаны между собой, что затрудняет диагностику. Иногда даже при тщательном обследовании не удаётся выявить одного из симптомов (уретрита или конъюнктивита). В таких случаях заболевание расценивают как неполный синдром Райтера.
• Помимо классической триады симптомов при болезни Райтера нередко выявляют поражение кожи и слизистых оболочек. Они проявляются кератодермией ладоней и стоп, псориазоподобными высыпаниями, трофическими изменениями ногтей. У детей также могут развиваться эрозии слизистой оболочки полости рта по типу стоматита или глоссита, которые часто клинически не проявляются и остаются незамеченными. Из других внесуставных проявлений следует отметить лимфаденопатию, реже гепатоспленомегалию, миоперикардит, аортит.
• При лабораторном исследовании выявляют повышение СОЭ, концентрации C-реактивного белка, a2-глобулинов, фибриногена, серомукоида и других неспецифических показателей воспаления. Постэнтероколитический РеА протекает более остро, агрессивно,
чем РеА, ассоциированный с хламидийной инфекцией.
Вероятный реактивный артрит. В ряде случаев РеА протекает без от- чётливых внесуставных проявлений синдрома Райтера (конъюнктивита, уретрита, кератодермии). В таких случаях ведущим в клинической картине становится суставной синдром, который характеризуется преимущественным поражением суставов нижних конечностей асимметричного характера. В целом характер и течение артрита аналогичен таковому при синдроме Райтера. Вне зависимости от наличия внесуставных проявлений у этих детей существует риск формирования ЮАС.
В случае отсутствия полной клинической картины синдрома Райтера, даже при характерном суставном синдроме, диагноз РеА представляет значительные трудности. Наличие характерного моно-, олигоартрита, артрита с преимущественным поражением суставов ног,
выраженной экссудацией, связанного с перенесённой кишечной или урогенитальной инфекцией, или с наличием серологических признаков перенесённой инфекции позволяет отнести заболевание к «вероятному реактивному артриту».
ДИАГНОСТИКА
Диагноз синдрома Райтера или РеА основывается на информации о предшествующей инфекции, анализе особенностей клинической кар- тины, данных лабораторных (табл. 13-8) и инструментальных методов обследования, результатах этиологической диагностики.
Таблица 13-8. Лабораторные тесты для подтверждения «артритогенной» инфекции*
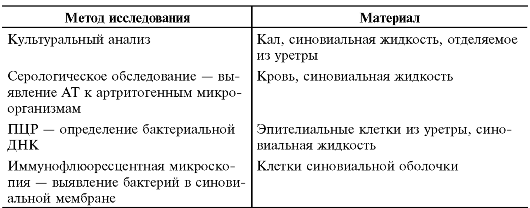
* Kingsly G., Sieper J. Third Internetional Workshop on Reactive Arthritis: an overview // Ann Rheum Dis. - 1996. - Vol. 55. - P. 564-570.
Этиологическая диагностика включает следующие тесты.
• Иммунологический метод.
- Выявление Аг хламидий в эпителиальных клетках, полученных в результате соскобов из уретры и конъюнктивы, а также в синовиальной жидкости (прямой иммунофлюоресцентный анализ и др.); выявление АТ к Аг хламидий в сыворотке крови и в синовиальной жидкости (реакция связывания комплемента, прямая и непрямая иммунофлюоресценция).
- Выявление АТ к бактериям кишечной группы в сыворотке крови
(РПГА, РСК).
• Морфологический метод: выявление морфологических структур возбудителя (окраска препаратов, иммунофлюоресцентный анализ).
• Культуральный метод: выделение хламидий (культура клеток, куриные эмбрионы, лабораторные животные).
• Молекулярно-биологические методы (ПЦР и др.): выявление ДНК возбудителя в крови и синовиальной жидкости.
• Бактериологическое исследование кала.
• Бактериологическое исследование мочи. Диагностические критерии
Диагноз
РеА устанавливают на основании диагностических критериев, принятых на
III Международном совещании по реактивным артритам в Берлине в
Таблица 13-9. Берлинские диагностические критерии РеА*
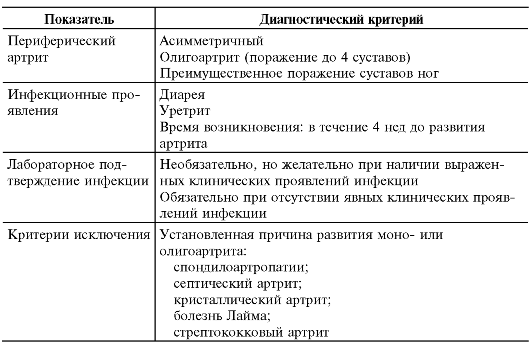
* Kingsly G., Sieper J. Third International Workshop on Reactive Arthritis: an overview // Ann Rheum Dis. - 1996. - Vol. 55. - P. 564-570.
Согласно данным критериям, диагноз «реактивный артрит» можно поставить лишь в том случае, если у больного имеет место типичный периферический артрит, протекающий по типу асимметричного оли- гоартрита с преимущественным поражением суставов нижних конечностей. При наличии клинических признаков инфекции (диареи или уретрита), перенесённой за 2-4 нед и до развития артрита, лабораторное подтверждение желательно, но не обязательно. При отсутствии клинических проявлений инфекции учитывают лабораторные данные, её подтверждающие.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Проведение дифференциальной диагностики РеА от других артритов у детей часто бывает затруднено. Наиболее часто РеА приходит-
ся дифференцировать с инфекционными артритами, заболеваниями инфекционной этиологии, сопровождающимися артритами, а также ортопедической патологией и некоторыми формами ЮАС.
• Вирусные артриты: клиническая картина чаще представлена артралгиями, чем артритами. Клинические симптомы продолжаются в течение 1-2 нед и исчезают без остаточных явлений. Диагноз вирусного артрита устанавливают на основании хронологической связи с перенесённой вирусной инфекцией или с вакцинацией, клинической картины острого артрита.
• Постстрептококковый артрит развивается после перенесённой стрептококковой инфекции. Диагностические критерии постстрептококкового артрита включают: появление артрита на фоне или спустя 1-2 нед после перенесённой носоглоточной инфекции стрептококковой этиологии; наличие повышенных титров постстрептококковых АТ; выявление хронических очагов инфекции в носоглотке (хронический тонзиллит, фарингит, гайморит); восстановление функции опорно-двигательного аппарата в результате лечения, включающего санацию хронических очагов инфекции.
• Болезнь Лайма проявляется поражением кожи (в виде мигрирующей эритемы), суставов, нервной системы. Развивается при инфицировании спирохетами Borrelia burgdorferi вследствие укуса клеща вида Ixodes. Диагностика болезни Лайма основана на наличии характерной клинической картины, пребывания пациента в эндемичной зоне, наличия в анамнезе факта укуса клеща. Подтверждают диагноз серологическими методами, которые выявляют АТ к Borrelia burgdorferi.
• Септический артрит: диагноз основывается на клинической картине (общие симптомы интоксикации, проявления генерализованной инфекции, преимущественно моно-, реже олигоартрит с резко выраженными симптомами воспаления), характере синовиальной жидкости, результатах посева синовиальной жидкости на флору (высевают преимущественно Staphylococcus aureus и Haemophilus influenzae) с определением чувствительности к антибиотикам, а также рентгенологических данных (в случае развития остеомиелита).
• Туберкулёзный артрит диагностируют на основе данных семейного анамнеза (контакт с больным туберкулёзом); сведений о вакцинации БЦЖ, данных реакции Манту и её динамики; общих симптомов туберкулёзной инфекции (интоксикация, субфебрильная лихорадка); рентгенологической картины; анализа синовиальной жидкости; биопсии синовиальной оболочки.
• Ювенильный ревматоидный артрит: диагноз основывается на прогрессирующем течении артрита, наличии иммунологических изменений, характерных иммуногенетических маркёров (HLA A2, DR-5,
DR-8), выявлении рентгенологических изменений в суставах, характерных для ЮРА.
• Ювенильный анкилозирующий спондилоартрит - возможный исход хронического течения РеА у предрасположенных лиц (HLA B27 носителей). Суставной синдром, как и при РеА, представлен асимметричным моно-, олигоартритом с преимущественным поражением суставов ног. Кардинальные признаки, позволяющие поставить диагноз ЮАС, - рентгенологические данные, свидетельствующие о наличии сакроилеита (одноили двустороннего), спондилоартрита.
• Ортопедические заболевания и болевые синдромы (болезни Пертеса, Кальве, Осгуд-Шлаттера, боли роста): диагноз основывается на клинико-рентгенологических особенностях, отсутствии признаков воспаления.
ЛЕЧЕНИЕ
В лечении РеА выделяют три вида терапии: этиотропную, патогенетическую и симптоматическую.
Этиотропная терапия
Реактивный артрит, ассоциированный с хламидийной инфекцией
Поскольку хламидии - внутриклеточные паразиты, выбор антибактериальных препаратов ограничивается только теми, которые способны накапливаться внутриклеточно. К таким антибиотикам относят макролиды, тетрациклины и фторхинолоны, однако последние две группы достаточно токсичны и обладают побочными эффектами, ограничивающими их применение в детской практике. В связи с этим наиболее часто для лечения хламидиоза у детей используются макролиды (азитромицин, рокситромицин). У подростков возможно использование доксициклина и фторхинолонов. Терапия антибиотиком более эффективна в острой стадии РеА. При хламидиозе следует воздержаться от назначения антибиотиков пенициллинового ряда, в связи с возможностью перехода хламидий в L-формы и развития хронической персистирующей инфекции.
Высокой антихламидийной активностью обладает азитромицин, обладающий бактерицидным действием в очаге воспаления, где со- здаются его высокие концентрации. Детям в первый день приёма доза препарата составляет 10 мг/кг, в последующие 5-7 дней - 5 мг/кг в один приём. Лучший эффект достигается при использовании антибиотика в течение 7-10 дней.
При
остром артрите, ассоциированном с хламидиями, также можно применять
другие макролиды: кларитромицин (дети старше 6 мес - 15 мг/кг/сут в 2
приёма), спирамицин (детям массой более
(5-8 мг/кг/сут), джозамицин (30-50 мг/кг/сут в 3 приёма). Курс лечения этими препаратами должен составлять 7-10 дней.
У детей подросткового возраста (старше 8-10 лет) допустимо применять антибиотик из группы тетрациклинов, высокоэффективный против хламидий, - доксициклин.
Реактивный артрит, ассоциированный с кишечной инфекцией В отношении РеА, связанных с кишечной инфекцией, однозначных рекомендаций по антибактериальной терапии не существует. Всем детям с РеА, у которых присутствуют АТ к бактериям кишечной группы в диагностических титрах или при бактериологическом обследовании кала обнаружены бактерии кишечной группы, рекомендуют проведение антибактериальной терапии. Препараты выбора - аминогликозиды: амикацин по 15-20 мг/кг/сут в 1-2 введения внутривенно или внутримышечно в течение 7-10 сут; гентамицин по 5-7 мг/кг/сут в 2 введения внутривенно или внутримышечно в течение 7-10 сут; также применяют фторхинолоны (у детей старше 12 лет).
Патогенетическая терапия
Монотерапия антибиотиками оказывает недостаточный эффект при затяжном и хроническом течении РеА, ассоциированного с персистирующей хламидийной инфекцией. В этот период, как правило, рецидивирует лишь суставной синдром, а не вся триада симптомов. Недостаточная эффективность антибиотиков связана с особенностями жизненного цикла хламидии и развитием персистирующей инфекции. В этом случае хламидии располагаются внутриклеточно и недосягаемы и для АТ, и для антибиотиков. У больных с хлами- дийной инфекцией иммунная система функционирует неадекватно и полноценный иммунный ответ не формируется, иммунопатологические реакции преобладают над защитными. Поэтому представляется целесообразным для лечения хронического хламидийного артрита использовать различные иммуномодулирующие средства.
Для лечения хронических РеА хламидийной этиологии разработаны и апробированы схемы терапии с использованием иммуномодуляторов - экстракта тимуса («Тактивин»), «Ликопида» (глюкозаминил- мурамилдипептид - синтетический иммуномодулятор, получаемый из клеточной стенки M. lysodeicticus), полиоксидония (производное ТчГ-окси-поли-1,4-этиленпиперазина - синтетический иммуномодулятор последнего поколения).
Для купирования островоспалительных изменений в суставах больным целесообразно применение НПВС в возрастных дозах, а также внутрисуставное введение глюкокортикоидов. Контроль эффективности проведённой патогенетической и этиотропной терапией целесообразно проводить не ранее чем через 1 мес, оптимально - через
3 мес после проведённого лечения. При неэффективности проведён- ного курса комбинированной терапии можно рекомендовать повторные курсы лечения со сменой иммуномодуляторов и антибиотиков. Важный фактор успешного лечения ребёнка с РеА, ассоциированным с хламидийной инфекцией, - диагностика и лечение членов семьи больного.
Симптоматическая терапия
Для купирования суставного синдрома при РеА применяют НПВС: диклофенак перорально по 2-3 мг/кг/сут в 2-3 приёма, индомета- цин перорально по 1-2 мг/кг/сут в 2-3 приёма (в связи с высокой частотой побочных эффектов использовать в детской практике не рекомендуют), напроксен перорально по 15-20 мг/кг/сут в 2 приёма, пироксикам перорально по 0,3-0,6 мг/кг/сут в 2 приёма, ибупрофен перорально по 35-40 мг/кг/сут в 2-4 приёма, нимесулид перорально по 5 мг/кг/сут в 2-3 приёма или мелоксикам перорально по 0,3- 0,5 мг/кг/сут в 1 приём.
Глюкокортикоиды, как наиболее мощные противовоспалительные средства, используют в период обострения суставного синдрома. Их применение ограничивается преимущественно внутрисуставным спо- собом введения. При необходимости можно воспользоваться коротким курсом пульс-терапии: быстрое (в течение 30-60 минут) внутривенное введение больших доз метилпреднизолона по 5-15 мг/кг в течение 3 дней.
Иммуносупрессивная терапия
При тяжёлом и торпидном течении заболевания, появлении признаков спондилоартрита, высокой клинической и лабораторной ак- тивности возможно применение иммуносупрессивных препаратов. Наиболее часто используется сульфасалазин (по 30-40 мг/кг/сут), реже метотрексат (по 10 мг/м2 в неделю).
ПРОГНОЗ
У большинства детей РеА заканчивается полным выздоровлением. Такой исход типичен в случае развития РеА, связанного с иерсиниозной и кампилобактерной инфекцией. У части больных эпизоды РеА рецидивируют и в дальнейшем появляются признаки спондилоартрита, особенно у HLA В27-позитивных больных. Существуют данные, что у позитивных по HLA B27 пациентов после перенесённого РеА, вызванного сальмонеллами, может развиться псориаз. По нашим данным у некоторых больных с РеА происходит трансформация в типичный ЮРА со всеми соответствующими клинико-рентгенологическими изменениями.
ДИФФУЗНЫЕ БОЛЕЗНИ СОЕДИНИТЕЛЬНОЙ ТКАНИ
Диффузные болезни соединительной ткани представляют собой группу заболеваний с системной прогрессирующей дезорганизацией соединительной ткани и поражением микроциркуляторного русла. Болезни этой группы развиваются при наличии генетической предрасположенности под воздействием неблагоприятных факторов окружающей среды.
Согласно рабочей классификации и номенклатуре ревматических заболеваний (ВНОР, 1985), в группу диффузных болезней соедини- тельной ткани включены СКВ, ССД, дерматомиозит/полимиозит, диффузный фасциит, болезнь Шёгрена, смешанное заболевание соединительной ткани, ревматическая полимиалгия, рецидивирующий панникулит, рецидивирующий полихондрит.
У детей наиболее часто развивается СКВ, несколько реже - ССД и ювенильный дерматомиозит, остальные заболевания представляют собой большую редкость. В соответствии со структурой Международной классификации болезней и причин смерти десятого пересмотра - МКБ-10 (ВОЗ, 1995) диффузные болезни соединительной ткани включены в блок «Системные поражения соединительной ткани». Диффузные болезни соединительной ткани развиваются у детей реже, чем у взрослых, но нередко протекают более тяжело, приводят к ранней инвалидизации, а при отсутствии правильного лечения - к неблагоприятному исходу.
Системная красная волчанка
Системная красная волчанка (СКВ) - системное аутоиммунное заболевание, вызванное генетически обусловленным нарушением им- мунной регуляции, приводящим к образованию аутоантител к широкому спектру антигенных компонентов клетки и развитию иммунного воспаления. СКВ - одно из наиболее тяжёлых и часто развивающихся заболеваний из группы диффузных болезней соединительной ткани, отличающееся клиническим полиморфизмом, хроническим вариабельным течением и, при отсутствии лечения, нередко неблаго- приятным прогнозом.
Распространённость СКВ составляет в различных регионах 4- 250:100 000 населения. Около 20% пациентов заболевают в возрасте до 16 лет. По данным J.A. Mills (1994), частота СКВ у детей до 15 лет составляет 1:100 000. В возрасте до 15 лет девочки болеют СКВ в 4,5 раза чаще мальчиков. Пик заболеваемости приходится на возраст
12-14 лет.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
СКВ считают заболеванием с многофакторным типом наследования, предрасположенность к которому формируется с участием как генетических, так и средовых факторов.
О роли наследственности свидетельствуют следующие факты.
• Наличие семейных случаев СКВ.
• Частое выявление у здоровых родственников первой степени родства больных СКВ антинуклеарных АТ, гипергаммаглобулинемии, ложноположительной реакции фон Вассермана и др.
• Значительно более высокий показатель конкордантности по СКВ (частота поражения обоих партнёров близнецовой пары) монозиготных близнецов по сравнению с дизиготными.
• Ассоциация заболевания с носительством определённых гаплотипов HLA класса II (в частности, с DRВ, DQА).
• Связь с генетически обусловленным дефицитом компонентов комплемента (C1q, C2, C4).
• Полиморфизм генов некоторых цитокинов (в частности, ФНОа и др.).
Существуют косвенные данные о возможной роли вирусной инфекции в качестве пускового фактора. Среди факторов внешней среды первостепенное значение имеет инсоляция, провоцирующая начало и последующие обострения СКВ. УФО стимулирует апоптоз клеток кожи, что приводит к экспрессии внутриклеточных Аг на их мембране, индуцируя развитие аутоиммунного процесса. Не подлежит сомнению большое значение гормональных факторов. Отмечено, что эстрогены способствуют иммунологической гиперреактивности за счёт поликло- нальной активации В-клеток и повышения синтеза АТ, а андрогены в целом оказывают супрессивное действие на иммунитет.
В основе патогенеза СКВ лежит нарушение регуляции иммунных процессов с развитием реакций против собственных Аг из-за утраты к ним толерантности. Полагают, что аутоиммунные нарушения при СКВ обусловлены взаимосвязанными процессами: поликлональной активацией В-лимфоцитов и Аг-специфической Т-зависимой стимуляцией синтеза аутоантител. Фундаментальное нарушение иммунной системы у больных СКВ заключается в дефекте апоптоза лимфоцитов. У больных СКВ выявляют увеличение количества В-клеток, коррели- рующее с наличием гипергаммаглобулинемии. Обнаружены разнообразные дефекты иммунорегуляторных субпопуляций Т-лимфоцитов, которые приводят к нарушению их супрессорной активности и способствуют В-клеточной гиперактивности.
Аутоантитела могут реагировать с чрезвычайно широким спектром Аг - компонентами ядра, цитоплазмы и мембран клеток, белками сыворотки и др., образуя иммунные комплексы. Развитие иммунно-
го воспаления в различных органах связано с отложением ЦИК на базальной мембране сосудов, с локальным образованием иммунных комплексов в тканях, а также с цитокинзависимым повреждением эндотелия, активацией лейкоцитов, системы комплемента и др.
ПАТОМОРФОЛОГИЯ
Для СКВ характерны следующие морфологические феномены: формирование богатого ядерным детритом и нуклеопротеидами фибри- ноида, ядерная патология (кариолизис, кариопикноз и кариорексис), образование гематоксилиновых телец и волчаночных клеток (LE-кле- ток). Гематоксилиновые тельца образуются в результате деструкции клеточных ядер и имеют вид овальных слабо базофильных образований, содержащих нуклеопротеиды и иммунные комплексы. LE-клет- ки представляют собой полиморфноядерные нейтрофилы (реже - эозинофилы или базофилы) с фагоцитированным ядром другой клетки или отдельными его фрагментами, опсонизированными антинуклеарными АТ.
Наиболее характерные для СКВ морфологические изменения развиваются в коже и некоторых внутренних органах.
• В области видимых кожных изменений и в участках неизменённой кожи обнаруживают патогномоничный для СКВ признак - пятнистые или сплошные линейные отложения Ig (IgG или IgM) и иммунных комплексов с компонентом комплемента C3 по ходу базальной мембраны эпидермиса.
• Изменения в почках при СКВ представлены различными вариантами иммунокомплексного гломерулонефрита. Специфичным для СКВ считают обнаружение феномена «проволочной петли», возникающего в результате массивного субэндотелиального отложения иммунных комплексов, компонентов комплемента и Ig по ходу базальной мембраны капилляров клубочков, что приводит к её утолщению и гомогенизации.
• Диагностически значим феномен «луковичной шелухи», представляющий собой периваскулярный склероз центральных и кисточковых артерий селезёнки с концентрическими наслоениями коллагеновых волокон.
• У всех пациентов выявляют признаки поражения вилочковой железы: интерстициальный тимит, увеличение внутридольковых периваскулярных пространств и атрофия собственно паренхимы железы. Патогномоничный для СКВ признак - обнаружение характерного периваскулярного склероза междольковых артерий вилочковой железы, аналогичного «луковичному» периартериальному склерозу селезёнки.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина СКВ чрезвычайно многообразна. В остром периоде у больных отмечают лихорадку неправильного типа, слабость, недомогание, похудание, алопецию (очаговое или гнёздное выпадение волос), признаки поражения различных органов и систем, сопровождающиеся изменениями в крови.
Кожа и слизистые оболочки
Наиболее характерное клиническое проявление СКВ - эритематозные высыпания на лице (преимущественно в области скуловых дуг и переносицы) в форме «бабочки» (рис. 13-7 на вклейке). Эритема представлена гиперемией кожи с чёткими границами, с инфильтрацией, фолликулярным гиперкератозом и последующей рубцовой атрофией. Кроме того, эритематозные элементы могут располагаться на коже верхней трети груди и спины - в области «декольте», над локтевыми и коленными суставами (рис. 13-8 на вклейке). В некоторых случаях кожные высыпания у больных могут быть представлены дискоидными эритематозными очагами.
Больным СКВ свойственна фотосенсибилизация (повышенная чувствительность к УФО): после длительного пребывания на солнце ухудшается общее состояние, появляются высыпания на коже или усиливается их яркость. В остром периоде СКВ на коже ладоней и подошвенной поверхности стоп у большинства больных можно увидеть признаки капиллярита - отёчные эритематозные высыпания с телеангиэктазиями, а иногда и с ишемическими некрозами кончиков пальцев (рис. 13-9 на вклейке). Для подострой кожной волчанки (субтип СКВ) характерны папулосквамозные и кольцевидные высыпания с гипопигментацией и телеангиэктазиями в центре. Типичный признак СКВ - люпус-хейлит - изменения красной каймы губ. При осмотре полости рта у боль- ных нередко выявляют афтозный стоматит (эрозивные или язвенные очаги с кератотическим ободком и интенсивной эритемой).
Суставы и мышцы
Суставной синдром при СКВ отличается мигрирующим характером течения и быстрым исчезновением после начала лечения глюкокортикоидами. Развиваются артралгии или артриты без нарушения функций, выраженных деформаций и изменений на рентгенограммах. Характерно множественное и преимущественно симметричное поражение проксимальных межфаланговых и пястно-фаланговых суставов II-V пальцев кистей, а также локтевых, коленных и голеностопных суставов. У детей значительно реже, чем у взрослых, развиваются асептические некрозы костей с костно-хрящевой секвестрацией и
вторичным остеосклерозом. Наиболее часто они локализуются в эпифизах головок бедренных костей.
У каждого второго ребёнка, больного СКВ, отмечают миалгии или полимиозит. В остром периоде возможно снижение мышечной силы, быстро исчезающее на фоне лечения.
Серозные оболочки
При СКВ у детей часто наблюдают полисерозит: плеврит, перикардит, реже - асептический перитонит, перигепатит, периспленит. Значительное скопление экссудата в полостях бывает редко. Обычно при УЗИ обнаруживают утолщение плевры и/или перикарда, плевроперикардиальные спайки.
Внутренние органы
При СКВ у детей наиболее часто поражается сердце. Чаще всего развивается миокардит, проявляющийся расширением границ сердца, систолическим шумом «мышечного характера», нарушениями сердечного ритма и проводимости, снижением сократительной способности миокарда. Реже обнаруживают вовлечение в процесс пристеночного и клапанного эндокарда (преимущественно митрального клапана, иногда клапанов аорты или трёхстворчатого). Обычно это вальвулит или уплотнение створок клапанов, однако в отличие от ревматизма пороки сердца при СКВ формируются крайне редко. Диагностическое значение имеет обнаружение абактериального веррукозного эндокардита Либмана-Сакса. Он представлен тромботическими бородавчатыми наложениями в участках изъязвлений эндокарда, мелкими перфорациями створок клапанов и разрывами хорд.
Поражение лёгких - пневмонит - выявляют у детей достаточно часто. Пневмонит включает поражение сосудов лёгких (васкулит и/ или склероз) и интерстициальной ткани (интерстициальная пневмония или пневмофиброз). При высокой активности СКВ он может проявляться симптомокомплексом, характерным для острой пневмонии. При этом рентгенологически выявляют очаговоподобные тени с неровными контурами, иногда - дисковидные ателектазы. У большинства больных (при отсутствии жалоб) обнаруживают усиление и нечёткость интерстициального рисунка лёгких, расширение просвета сосудов, высокое стояние диафрагмы.
Волчаночный нефрит наблюдают у 70% больных. Характер почечного процесса во многом определяет прогноз заболевания в целом. Согласно классификации ВОЗ (1982), выделяют шесть типов поражения почек при СКВ.
I тип - отсутствие светооптических изменений в биоптате, но наличие отложений иммунных комплексов по ходу базальных мем- бран капилляров клубочков.
II тип - мезангиальный гломерулонефрит.
III тип - очаговый пролиферативный гломерулонефрит.
IV тип - диффузный пролиферативный гломерулонефрит.
V тип - мембранозный гломерулонефрит.
VI тип - хронический гломерулосклероз. Клинически выделяют следующие формы нефрита.
• Нефротический нефрит с распространёнными отёками вплоть до анасарки, массивной протеинурией и гипопротеинемией.
• Нефрит выраженной формы без нефротического синдрома с умеренной протеинурией (1,5-3 г/сут) и значительной гематурией.
• Латентный нефрит (протеинурия менее 1,5 г/сут, гематурия с содержанием эритроцитов менее 20 в поле зрения).
Наиболее тяжёлый вариант поражения почек - быстропрогрессирующий волчаночный нефрит, характеризующийся быстрым и неуклонным падением функций почек, наличием нефротического синдрома и выраженной (иногда злокачественной) артериальной гипертензией.
Патология органов ЖКТ включает поражение слизистой оболочки пищевода, желудка и двенадцатиперстной кишки, иногда с образованием эрозий или язв. Патология кишечника может быть связана с поражением сосудов брыжейки (васкулит и/или тромбоз), приводящим к возникновению геморрагий, инфарктов, некрозов с возможной последующей перфорацией и развитием кишечного кровотечения или фибринозно-гнойного перитонита. В отдельных случаях развивается терминальный илеит, клинически проявляющийся симптомокомплексом болезни Крона.
Поражение нервной системы возникает у каждого второго ребён- ка, больного СКВ. В патологический процесс могут вовлекаться лю- бые её отделы. Клинические проявления чрезвычайно многообразны. Характерны появление психических расстройств, рецидивирующих эпилептиформных припадков, упорных головных болей, нарушение когнитивных функций (памяти, внимания, мышления) и эмоционально-личностные расстройства. Могут развиться хореические гиперкинезы, преходящие нарушения мозгового кровообращения (очень редко - инсульты), поперечный миелит. Поражение периферической нервной системы обычно протекает по типу симметричной дистальной, преимущественно сенсорной или сенсомоторной, полиневропатии, редко - множественной мононевропатии.
Антифосфолипидный синдром
Приблизительно у 1/3 детей, больных СКВ, диагностируют антифосфолипидный синдром (АФС). АФС - симптомокомплекс, вклю- чающий венозные и/или артериальные тромбозы, некоторые формы акушерской патологии и тромбоцитопению. Формирование АФС свя-
зывают с присутствием АТ к мембранным фосфолипидам и связанными с ними гликопротеинами.
Спектр антифосфолипидных АТ включает: АТ к кардиолипину; АТ, выявляемые с помощью фосфолипидзависимых коагуляционных тес- тов (волчаночный антикоагулянт, АТ к протромбину, факторам V и Х, Р2-гликопротеину 1 и др.); АТ, не выявляемые стандартными методами определения антифосфолипидных АТ (к белку С, к белку S, к тромбомодулину, к липопротеинам низкой плотности и др.).
В основе АФС лежат невоспалительная тромботическая васкулопатия, затрагивающая сосуды любого калибра и локализации, а также гематологические нарушения, что определяет спектр его клинических проявлений (табл. 13-10).
Таблица 13-10. Клинические и лабораторные проявления антифосфоли- пидного синдрома*
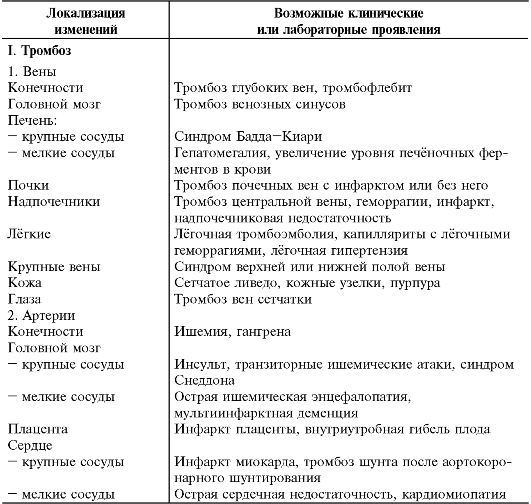
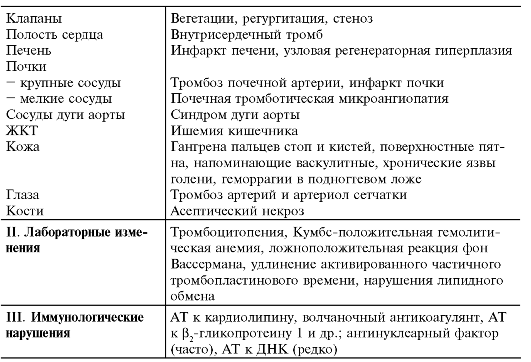
* По Е.Л. Насонову и соавт., 1999.
Один из ведущих клинических симптомов АФС при СКВ у детей - тромбозы, они могут развиться на разных этапах болезни. У детей преобладают тромбозы мелких сосудов кожи, проявляющиеся цианотическими пятнами, поверхностным некрозом кожи и сетчатым ливедо. Реже выявляют тромбоз мелких сосудов головного мозга, почек, печени и др.
Для диагностики АФС разработаны клинические критерии, дополненные лабораторными тестами (табл. 13-11).
Таблица 13-11. Классификационные критерии антифосфолипидного синд- рома*


ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ
При лабораторном исследовании в активном периоде СКВ обычно обнаруживают повышение СОЭ, лейкопению, реже тромбоцито- пению и аутоиммунную гемолитическую анемию с положительной реакцией Кумбса. Характерны гипергаммаглобулинемия, повышение IgM и IgG, а также ЦИК. У больных с волчаночным нефритом отмечают снижение общей гемолитической активности комплемента и его отдельных компонентов, коррелирующее с активностью почечного процесса. Большое диагностическое значение придают иммунологическим тестам.
• У 70% детей с СКВ обнаруживают LE-клетки (образуются при наличии АТ к комплексу ДНК-гистон).
• У 95% больных выявляют антинуклеарный фактор - гетерогенная группа АТ, реагирующих с различными компонентами ядра. Специфичность этого теста относительно невелика (антинуклеарный фактор обнаруживают у здоровых людей и больных другими ревматическими и неревматическими заболеваниями, при некоторых инфекциях, у больных, получающих некоторые лекарственные препараты).
• АТ к нативной (двухспиральной) ДНК относительно специфичны для СКВ и с большим постоянством обнаруживаются у больных с волчаночным нефритом, их концентрация коррелирует с активностью заболевания.
• АТ к гистонам более характерны для лекарственной волчанки.
• АТ к Sm-Аг высокоспецифичны для СКВ, но их обнаруживают лишь у 20-30% больных.
• АТ к SS-A/Ro- и SS-В/La-Аг менее специфичны для СКВ, их чаще обнаруживают при синдроме Шёгрена, подострой кожной и лекарственной волчанке, иногда у здоровых людей.
• Ревматоидный фактор (аутоантитела класса IgM, реагирующие с Fc-фрагментом IgG) при СКВ у детей выявляют довольно часто с помощью реакции латекс-агглютинации или реакции ВаалераРоуза.
ДИАГНОСТИКА
Для диагностики СКВ в настоящее время наиболее широко применяют критерии, разработанные в
При наличии у больного четырёх или более признаков в любом сочетании диагноз СКВ считают достоверным.
Таблица 13-12. Диагностические критерии системной красной волчанки


ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
В отсутствие типичных кожных изменений приходится дифференцировать СКВ с учётом доминирующих в клинической картине синдромов, наиболее часто - с другими ревматическими заболеваниями: при наличии суставного и кардиального синдромов - с ревматизмом, ЮРА, при наличии геморрагических высыпаний - с болезнью Шёнляйна-Геноха, идиопатической тромбоцитопенической пурпурой, а также с системными васкулитами, первичным АФС и др. Кроме того, нередко приходится исключать злокачественные лимфо- пролиферативные и инфекционные заболевания (туберкулёз, болезнь Лайма, бруцеллёз, иерсиниоз, гепатит В, С и др.).
ЛЕЧЕНИЕ
Лечить больных СКВ необходимо длительно, непрерывно, этапно, в соответствии с разработанными терапевтическими программами. Препаратами выбора служат глюкокортикоиды короткого действия - преднизолон или метилпреднизолон. Дозы глюкокортикоидов варьируют в зависимости от тяжести состояния больного, ведущих клинических синдромов заболевания, а также остроты течения и активности процесса.
• При высокой активности СКВ доза преднизолона обычно составляет 1-1,5 мг/кг/сут (не более 70 мг/сут), при умеренной активности - 0,5-1 мг/кг/сут. Препарат назначают преимущественно в утренние часы. Лечение глюкокортикоидами в максимальных дозах обычно продолжают в течение 4-6 нед до получения значимого клинического эффекта, а затем, по мере снижения активности заболевания и достижения ремиссии, дозу препарата медленно уменьшают в течение 6-12 мес до поддерживающей (в среднем 0,2 мг/кг/сут - 10-15 мг/сут). Лечение поддерживающими дозами глюкокортикоидов следует продолжать длительно и непрерывно для предупреждения рецидивов и поддержания ремиссии.
• Для лечения больных с полисиндромным поражением применяют пульс-терапию: внутривенно в течение трёх последовательных дней вводят сверхвысокие дозы метилпреднизолона из расчёта в среднем 10-30 мг/кг (не более 1000 мг/сут). Проведение пульс-терапии позволяет быстрее купировать многие проявления заболевания, сократить продолжительность активного периода и быстрее начать снижение пероральной дозы глюкокортикоидов.
Для лечения больных с наиболее тяжёлыми и прогностически неблагоприятными вариантами СКВ, особенно с волчаночным нефри- том или тяжёлым поражением ЦНС, при неэффективности глюкокортикоидов или наличии выраженных осложнений при их применении используют цитостатические средства.
• Препарат выбора при лечении волчаночного нефрита - циклофосфамид. Его вводят в мегадозах внутривенно в виде пульс-терапии 1 раз в месяц в дозе 0,5-0,75 г/м2 поверхности тела в течение 6- 12 мес, а затем - 1 раз в 3 мес ещё в течение 12-18 мес; в качестве альтернативного препарата для лечения волчаночного нефрита используют микофенолата мофетил в дозе 600 мг/м2 2 раза в день перорально длительно.
• При неэффективности циклофосфамида для купирования нефротического синдрома при мембранном типе нефрита в последние годы применяют циклоспорин в дозах до 5 мг/кг/сут.
• При волчаночном кризе (почечном или полиорганном) проводят плазмаферез синхронно с пульс-терапией метилпреднизолоном и циклофосфамидом.
• Больным с менее тяжёлыми вариантами СКВ при неэффективности или выраженных осложнениях лечения глюкокортикоидами, а также для поддержания ремиссии нефрита к лечению добавляют азатиоприн в дозе 1-2 мг/кг/сут внутрь.
Лечение АФС в первую очередь направлено на устранение гиперкоагуляции и профилакику тромбообразования. С этой целью используют прямые (нефракционированный гепарин, низкомолекулярные гепарины) и непрямые антикоагулянты, а также антиагреганты (ацетилсалициловая кислота).
Для лечения больных СКВ с низкой активностью применяют аминохинолиновые препараты (преимущественно гидроксихлорохин), обладающие, кроме того, антитромботическим и антигиперлипидемическим эффектами.
Наряду с глюкокортикоидами и цитостатическими средствами больным СКВ по показаниям назначают антигипертензивные препараты, антибиотики и симптоматические средства.
Рецидивы болезни могут возникнуть и после многолетней ремиссии, поэтому больным СКВ необходимо пожизненно находиться под диспансерным наблюдением врача.
ПРОГНОЗ
При раннем установлении диагноза и активном длительном лечении прогноз благоприятный. Выживаемость детей, больных СКВ, через 5 лет после начала заболевания составляет в среднем 95%, через 10 лет - 80-90%. Наиболее часто больные погибают в первые годы после начала заболевания в связи с развитием волчаночного криза (в первую очередь почечного или полиорганного) или тяжёлых инфекционных осложнений.
Ювенильный дерматомиозит
Ювенильный дерматомиозит - заболевание из группы диффузных болезней соединительной ткани с преимущественным поражением проксимальных скелетных мышц, развитием мышечной слабости, а также лиловой эритемы на коже. Поскольку этиология заболевания неясна, ювенильный дерматомиозит включают в состав гетерогенной группы идиопатических воспалительных миопатий с ведущим клиническим проявлением - поражением скелетной мускулатуры воспалительного генеза. В соответствии с классификацией R.L. Woltman (1994), кроме ювенильного дерматомиозита, в эту группу входят и другие миопатии (табл. 13-13).
Таблица 13-13. Классификация воспалительных миопатий
I. Идиопатические воспалительные миопатии
• первичный полимиозит;
• первичный дерматомиозит;
• ювенильный дерматомиозит;
• миозит, ассоциирующийся с другими диффузными болезнями соединительной ткани;
• миозит, ассоциирующийся с опухолями;
• миозит «с включениями»;
• миозит, ассоциирующийся с эозинофилией;
• оссифицирующий миозит;
• гигантоклеточный миозит;
• локализованный, или очаговый, миозит
II. Миопатии, вызванные инфекцией
III. Миопатии, вызванные лекарственными средствами и токсинами
У детей, кроме ювенильного дерматомиозита, другие типы миопатий наблюдают очень редко.
По распространённости ювенильный дерматомиозит занимает третье место среди диффузных болезней соединительной ткани и составляет 1,9:1 000 000 детей в возрасте до 16 лет. Заболеваемость
детей в среднем составляет 1/5-1/8 от числа заболевших взрослых. Ювенильный дерматомиозит чаще наблюдают у девочек, чем у мальчиков (соотношение 2:1). Отмечают два пика заболеваемости - в 3- 5 лет и 7-9 лет.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Среди возможных триггеров дерматомиозита обсуждают роль инфекции (вирусы Коксаки А и В, ECHO, ВИЧ, токсоплазмы, боррелии), вызывающей заболевание у генетически предрасположенных людей. Обсуждают три возможных пути воздействия вируса на организм.
• Прямое поражение мышечной ткани.
• Синтез АТ к вирусным Аг, находящимся на поверхности мышечных волокон.
• Синтез аутоантител к инфекционным Аг, перекрёстно реагирующих с аутоантигенами (антигенная мимикрия).
О роли генетических факторов свидетельствуют наличие семейных случаев дерматомиозита, развитие заболевания у близнецов, ассоциация дерматомиозита с HLA, DR3, ВК5, DR7. Так же как и другие системные заболевания соединительной ткани, ювенильный дерматомиозит развивается при участии средовых и эндокринных факторов.
Иммунные нарушения при ювенильном дерматомиозите выявляют как на клеточном, так и на гуморальном уровнях. Поражённые мышцы инфильтрированы активированными Т-лимфоцитами и В-лимфо- цитами, а также макрофагами, причём, как показали исследования, Т-клетки обладают цитотоксической активностью в отношении миофибрилл. При дерматомиозите развивается гуморальный иммунный ответ, приводящий к активации комплемента, что сопровождается поражением сосудов микроциркуляторного русла. Уточняют значение широкого спектра миозит-специфических АТ: к аминоацилсинтета- зам тРНК, к частицам сигнального распознавания, к белково-ядерному комплексу Mi-2 и др.
ПАТОМОРФОЛОГИЯ
При морфологическом исследовании кожи больных дерматомиозитом обнаруживают продуктивные и продуктивно-деструктивные васкулиты всех слоёв дермы, периваскулярную лимфоцитарную инфильтрацию, истончение эпидермиса, склерозирование дермы в поражённых участках. В биоптатах мышц также выявляют клеточные инфильтраты в перимизии и вокруг сосудов микроциркуляторного русла и вен, состоящие преимущественно из лимфоцитов, а также макрофагов, гистиоцитов и плазматических клеток. Диагностическое значение имеет обнаружение крупных макрофагов, внедрившихся в некротизированные мышечные волокна с признаками избыточного
фагоцитоза. Некробиотические процессы в миофибриллах сочетаются с их выраженной регенерацией. При длительном течении заболевания выявляют атрофию мышечных волокон, нарастание фиброза и склероза эндо- и перимизия.
КЛИНИЧЕСКАЯ КАРТИНА
У детей дерматомиозит чаще начинается остро или подостро, в дебюте заболевания нередко возникают лихорадка, слабость, недомогание, снижение массы тела, миалгии, артралгии, прогрессирующее снижение мышечной силы. Клиническая картина дерматомиозита обычно полисиндромна, но наиболее характерны изменения со стороны кожи и мышц.
Поражение кожи
Поражение кожи - характерный признак дерматомиозита. К кожным проявлениям дерматомиозита относят эритематозные высыпания с лиловым оттенком на лице в параорбитальной области (симптом «дерматомиозитных очков») (рис. 13-10 на вклейке), в области декольте, над пястно-фаланговыми и проксимальными межфаланговыми суставами кистей. Приподнимающиеся или плоские эритематозные шелушащиеся высыпания, локализирующиеся над суставами пальцев и над крупными суставами конечностей, в первую очередь локтевыми и коленными, получили название признак Готтрона. В остром периоде у больных нередко отмечают поверхностный некроз кожи в местах поражения, а в последующем развивается атрофия с участками депигментации. У некоторых больных наблюдают покраснение, шелушение и растрескивание кожи ладоней («рука механика»).
У детей, больных дерматомиозитом, обычно возникает яркое ливедо, особенно в области плечевого и тазового поясов, капиллярит ладоней и стоп, телеангиэктазии. Генерализованное поражение сосудов особенно характерно для детей дошкольного возраста.
При остром и подостром течении наблюдают выраженные трофические нарушения в виде ксеродермии, ломкости ногтей, алопеции.
Поражение подкожной клетчатки
Над поражёнными мышцами конечностей и на лице нередко появляется тестоватый или плотный отёк. Возможно развитие парци- альной липодистрофии лица и конечностей, обычно сочетающейся с мышечной атрофией.
Поражение мышц
Обычно в начале заболевания больные дерматомиозитом жалуются на быструю утомляемость при физической нагрузке, боли в мышцах,
возникающие спонтанно и усиливающиеся при пальпации и движениях. Для дерматомиозита характерно симметричное поражение в первую очередь проксимальных мышц конечностей, вследствие чего дети не могут носить портфель в руках, им трудно поднимать руки вверх и удерживать их в этом положении, они не могут самостоятельно причесаться («симптом расчёски»), одеться («симптом рубашки»), быстро устают при ходьбе, часто падают, не могут подниматься по лестнице, встать со стула, поднять и удерживать ноги. При тяжёлом поражении мышц шеи и спины больные не могут оторвать голову от подушки, повернуться и встать с кровати. В наиболее тяжёлых случаях развивается генерализованная мышечная слабость с акцентом на проксимальную группу, вследствие чего пациенты могут быть почти полностью обездвижены.
При поражении мышц гортани и глотки появляется гнусавость и охриплость голоса, а также нарушение глотания, что может приводить к аспирации пищи и слюны. При поражении мимических мышц отмечают маскообразность лица, а при поражении глазодвигательных мышц - диплопию и птоз век. Тяжёлые поражения диафрагмы и межрёберных мышц приводят к нарушению дыхания. В исходе полимиозита развивается гипотрофия мышц.
У детей, в отличие от взрослых, нередко формируются стойкие, иногда болезненные сухожильно-мышечные контрактуры, резко ограничивающие объём движений.
Поражение суставов
Поражение суставов наблюдают более чем у 75% больных. Развиваются артралгии или полиартрит. Наиболее часто поражаются мелкие суставы кистей (преимущественно проксимальные межфаланговые суставы), коленные и локтевые. Суставные изменения характеризуются умеренной дефигурацией и болезненностью при пальпации и движениях. В большинстве случаев суставной синдром быстро купируется на фоне лечения, лишь у 25% больных отмечают формирование контрактур, деформаций и подвывихов в межфаланговых суставах с некоторым ограничением функциональных возможностей.
Кальциноз
Кальциноз при дерматомиозите у детей возникает в 3-4 раза чаще, чем у взрослых. Он развивается почти у 40% больных преимущественно в сроки от 1 года до 5 лет после начала заболевания. Кальцинаты могут быть ограниченными в виде отдельных очагов или пластин и локализоваться подкожно или в соединительной ткани вокруг мышечных волокон, располагаться в зонах наибольшей травматизации - вокруг коленных или локтевых суставов, вдоль ахиллова сухожилия,
на бёдрах, ягодицах, плечах. У больных с дерматомиозитом непрерывно-рецидивирующего течения кальциноз обычно имеет диффузный характер.
Поражение внутренних органов
При дерматомиозите наиболее часто развивается миокардит, проявляющийся преимущественно нарушениями ритма и проводимости, снижением сократительной способности сердечной мышцы. У 25% больных развивается перикардит с нерезко выраженными признаками, быстро исчезающими после начала лечения глюкокортикоидами.
Поражение лёгких (пневмонит) связано с сосудисто-интерстициальными изменениями и клинически проявляется непродуктив- ным кашлем, одышкой, непостоянными хрипами при аускультации. Прогностически неблагоприятно развитие диффузного альвеолита с образованием альвеолярно-капиллярного блока, быстрым развитием лёгочной недостаточности и летальным исходом. Поражение лёгких при дерматомиозите может быть также обусловлено развитием аспирационных и банальных гипостатических пневмоний вследствие поражения мышц, участвующих в глотании и дыхании. Нередко у детей обнаруживают плеврит, при высокой степени активности процесса иногда сопровождающийся образованием экссудата.
Поражение почек выявляют редко. Почечный синдром бывает представлен преходящим мочевым синдромом, в отдельных случаях сопровождающимся нарушением функции почек вплоть до развития ОПН вследствие массивной миоглобинурии.
Нередко у детей при высокой активности процесса возникают эзофагит, гастродуоденит, энтероколит, возможно развитие эрозивно-язвенного процесса, осложняющегося перфорацией и кровотечением. Изредка наблюдают псевдоабдоминальный синдром, возникающий в результате поражения мышц передней брюшной стенки, с отёком, уплотнением и резкой болезненностью при дыхании и пальпации.
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ
При лабораторном исследовании у больных в активном периоде заболевания обычно выявляют повышение СОЭ, умеренную анемию, у некоторых пациентов - умеренный лейкоцитоз, гипергаммаглобулинемию.
Среди биохимических показателей к характерным изменениям, отражающим поражение скелетных мышц, следует отнести повышение активности креатинфосфокиназы, а также альдолазы. Кроме того, у больных нередко выявляют увеличение концентрации ЛДГ и аминотрансфераз в сыворотке крови. У ряда больных возникает миоглобинурия.
Выявление миозит-специфических АТ имеет важное значение в первую очередь для классификации, т.е. уточнения клинико-иммунологического подтипа дерматомиозита и полимиозита. У части больных выявляют АТ к аминоацилсинтетазам тРНК, в первую очередь АТ к гистидил-тРНК-синтетазе (Jo-1). При наличии указанных АТ в крови развивается антисинтетазный синдром, характеризующийся острым началом миозита, интерстициальным поражением лёгких, лихорадкой, симметричным артритом, синдромом Рейно, поражением кожи кистей по типу «руки механика», неполным ответом на применение глюкокортикоидов и частым развитием обострений на фоне снижения их дозы, дебютом заболевания преимущественно в весенний период.
ДИАГНОСТИКА
Разработаны следующие критерии диагностики дерматомиозита (Tanimoto К. et al., 1995).
1. Поражение кожи.
а. Гелиотропная сыпь - красно-фиолетовые эритематозные высыпания на веках.
б. Признак Готтрона - красно-фиолетовая шелушащаяся атрофическая эритема или пятна на разгибательной поверхности кистей над пястно-фаланговыми и проксимальными межфаланговыми суставами.
в. Эритема на разгибательной поверхности конечностей, над локтевыми и коленными суставами.
2. Слабость мышц проксимальных отделов конечностей и туловища.
3. Повышение активности креатинфосфокиназы и/или альдолазы в сыворотке крови.
4. Миалгии или болезненность мышц при пальпации.
5. Изменения на электромиографии (короткие полифазные потенциалы моторных единиц со спонтанными потенциалами фибрилляций).
6. Обнаружение АТ Jo-1.
7. Недеструктивный артрит или артралгии.
8. Признаки системного воспаления (лихорадка более
9. Гистологические изменения: воспалительные инфильтраты в скелетных мышцах с дегенерацией или некрозом мышечных фибрилл; активный фагоцитоз или признаки активной регенерации.
При наличии первого и любых четырёх из последующих критериев диагноз дерматомиозита считают достоверным. Диагностика дерматомиозита трудна в тех случаях, когда заболевание дебютирует с одного синдрома, особенно в том случае, когда ведущим синдромом становится миопатия, а кожные изменения неяркие.
При наличии типичного кожного и миопатического синдромов диагностика дерматомиозита обычно вызывает меньшие трудности, однако у детей раннего возраста ранняя диагностика затруднена в связи со сложностью выявления мышечной слабости и правильной трактовки появляющихся симптомов.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Дифференциальную диагностику дерматомиозита следует проводить с полиневропатиями, заболеваниями с поражением нервно-мы- шечных синапсов (миастения), прогрессирующими мышечными дистрофиями (миодистрофия Эрба, миодистрофия Ландузи-Дежерина), эндокринными миопатиями, инфекционными миозитами. Мышечную слабость следует отличать от общей слабости и мышечной усталости, возникающих при многих заболеваниях, в частности при анемии, у детей раннего возраста при рахите, при злокачественных новообразо- ваниях и др.
ЛЕЧЕНИЕ
Препаратами выбора для лечения дерматомиозита служат глюкокортикоиды короткого действия - преднизолон и метилпреднизолон. Их дозы определяют с учётом тяжести состояния больного, активности заболевания и эффективности предшествующей терапии. При высокой активности процесса первоначальная доза преднизолона для перорального приёма составляет 1-1,5 мг/кг/сут (не более 70 мг/сут), при умеренной активности - 0,5-0,9 мг/кг/сут. Клинический эффект при лечении дерматомиозита развивается у больных медленнее, чем при СКВ, поэтому лечение максимальными дозами глюкокортикоидов проводят длительнее (до 2 мес). В последующем по мере снижения активности заболевания и улучшения состояния пациентов дозы медленно снижают до поддерживающих (не менее 10-15 мг/сут - 0,2 мг/кг/сут). Лечение поддерживающими дозами следует продолжать в течение многих лет, что будет способствовать предупреждению рецидивов заболевания.
Для лечения больных с наиболее тяжёлыми формами дерматомиозита проводят пульс-терапию метилпреднизолоном: препарат вводят внутривенно в дозе 10-20 мг/кг/сут (не более 1000 мг/сут) в течение трёх последовательных дней или более, что позволяет в более короткие сроки подавить активность патологического процесса. Для лечения больных с миопатическим кризом и при торпидности к проводимой стандартной терапии применяют плазмаферез, синхронизируя его проведение с пульс-терапией глюкокортикоидами, что позволяет добиться положительных результатов, в том числе и у пациентов, резистентных к стероидной терапии.
В последние годы для лечения больных дерматомиозитом уже в ранние сроки с момента начала заболевания в комбинации с глюкокортикоидами широко применяют метотрексат. Препарат назначают перорально в дозе 10-12,5 мг/м2/нед (до 20 мг/нед с учётом переносимости), лечение проводят длительно. Комбинированная терапия позволяет быстрее добиться клинического эффекта и начать снижение дозы глюкокортикоидов, что уменьшает выраженность их побочных эффектов, предотвращает прогрессирование кальциноза. Для лечения кальциноза применяют этидроновую кислоту, имеются данные об эффективности дилтиазема.
ПРОГНОЗ
Прогноз при ювенильном дерматомиозите менее благоприятен, чем при дерматомиозите у взрослых. Летальные исходы отмечают преимущественно в первые годы после начала заболевания на фоне высокой активности процесса и кризового течения. Выживаемость больных через 5 лет после установления диагноза в среднем составляет более 90%. При раннем же установлении диагноза и активном длительном лечении у большинства больных удаётся добиться длительной ремиссии на много лет. Худший прогноз наблюдают у детей, заболевших в раннем возрасте, а также у больных с тяжёлым поражением ЖКТ, лёгких.
Системная склеродермия
Системная склеродермия (ССД) - полисиндромное заболевание из группы диффузных болезней соединительной ткани, характеризую- щееся прогрессирующими фиброзно-склеротическими изменениями кожи, опорно-двигательного аппарата, внутренних органов и вазоспастическими нарушениями по типу синдрома Рейно.
Заболеваемость ССД взрослых составляет 4,5-14:1 000 000. Больные в возрасте до 10 лет составляют менее 2%, а в возрасте до 16 лет - 3%, девочки заболевают чаще мальчиков (соотношение 3-4:1). ССД у детей чаще начинается в дошкольном и младшем школьном возрасте.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Этиология ССД изучена недостаточно. Определённое значение для её развития имеет генетическая предрасположенность, о чём свидетельствует наличие семейных случаев. Носительство специфических генетических маркёров - HLA А9, В8, Bw35, DR1, DR3, DR5, DR11, DR52 ассоциировано, как предполагают, не с риском развития заболевания в целом, а с синтезом определённых АТ.
Факторы внешней среды могут выступать в качестве триггерных, вызывая повреждение эндотелия сосудов с последующим развитием
воспалительных реакций и формированием фиброза. К ним относят вирусы (цитомегаловирус, ретровирусы), токсины бактерий, контакт с некоторыми химическими веществами (например, поливинилхлоридом), длительный контакт с кремниевой пылью или бензином, приём определённых ЛС (блеомицина, L-триптофана), употребление в пищу недоброкачественных продуктов питания («испанское токсическое масло»).
Патогенез ССД окончательно не установлен. Развивающиеся патологические изменения являются следствием нарушении функций иммунной системы, эндотелия и фибробластов. Нарушения клеточного иммунитета, вероятно, играют ведущую роль развитии ССД. Об этом свидетельствует формирование мононуклеарноклеточной инфильтрации в ранней стадии патологического процесса, нарушение функции Т-хелперов и естественных киллеров и высвобождение различных цитокинов, хемокинов и факторов роста, играющих важную роль в повреждении эндотелия сосудов, возникновении неконтролируемой пролиферации фибробластов и синтеза ими коллагена, что приводит к развитию фиброза. Возможно, определённое значение имеют и нарушения гуморального иммунитета, поскольку у больных ССД выявляют широкий спектр специфических антинуклеарных и антинуклеолярных аутоантител - антицентромерных, антитопоизомеразных, к РНК-полимеразе, к рибонуклеопротеину и др.
Особое значение в патогенезе, определяющее специфику заболевания, имеют процессы усиленного образования коллагена и фиброз. Выявлена устойчивая гиперактивность фибробластов, структурные и функциональные аномалии клеточных мембран, вследствие чего фибробласты становятся относительно автономными и приобретают способность к избыточной пролиферации и синтезу коллагена на фоне отсутствия дополнительных стимулов, избегая гомеостатического контроля.
Существенное звено патогенеза - нарушение микроциркуляции с пролиферацией и деструкцией эндотелия, утолщением стенки и сужением просвета микрососудов, вазоспазмом, агрегацией форменных элементов, стазом, деформацией и редукцией капиллярной сети.
ПАТОМОРФОЛОГИЯ
При ССД наблюдают атрофию эпидермиса с вакуолизацией клеток базального слоя и накоплением меланина, сужением сосочкового слоя. Наряду с утолщением дермы выявляют мукоидное набухание и фибриноидные изменения, участки склероза и гиалиноза, отмечают кариопикноз и кариорексис. В начальном периоде отмечают продуктивные васкулиты, обнаруживают лаброциты в периваскулярных про-
странствах, спастическое сокращение субэпидермальных артериол, мукоидное набухание внутренней оболочки сосудов дермы с сужением и даже с полной обтурацией просвета. В поздних стадиях отмечают редукцию и склероз сосудистого русла. При ССД обнаружено усиление неофибриллогенеза с аномалией коллагеновых структур.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина ССД отличается полисиндромностью и полиморфностью.
Поражения кожи
Наиболее характерным проявлением заболевания считают поражение кожи, проходящее последовательно стадии плотного отёка, индурации (уплотнения) и атрофии. Кожа на поражённых участках становится плотной, плохо собирается в складку, нарушаются её эластичность и пигментация. При развитии атрофии кожа истончается. Выделяют несколько форм ССД.
• При диффузной форме отмечают генерализованное поражение кожи. При поражении кожи груди и спины у больного возникает ощущение надетого «корсета» или «панциря». При распространён- ном поражении кожи конечностей с вовлечением периартикулярных областей значительно ограничивается объём движений в суставах.
• При проксимальной форме отмечают поражение кожи туловища и проксимальных отделов конечностей выше метакарпальных и метатарзальных суставов.
• Очень характерен внешний вид больного при акросклеротическом варианте ССД: лицо маскообразное, амимичное, уши, губы и нос истончены («птичий нос»), открывание рта затруднено и сопровождается формированием вокруг него морщин («кисетный рот»), нарушено смыкание век. Кроме того, при акросклеротическом варианте поражаются дистальные отделы конечностей, вначале кисти рук и стопы, а затем - предплечья и голени. Вследствие отёка и индурации кожи пальцы трудно сжать в кулак, формируются контрактуры, кисти приобретают своеобразный вид «когтистой лапы».
• При гемисклеродермии отмечают поражение кожи и подлежащих тканей конечностей и туловища с односторонней локализацией, что нередко приводит к аномалии развития: постепенно происходит уменьшение объёма поражённых конечностей, нарушается их рост, появляется выраженная асимметрия (рис. 13-11 на вклейке).
• У детей часто возникает атипичная форма ССД с очаговым поражением кожи, скудной висцеральной патологией и преобладанием функциональных нарушений.
• Практически не развивается висцеральная форма с преобладанием в клинической картине поражений внутренних органов и сосудов и минимальными изменениями кожи.
• Исходом дистрофических изменений в тканях может быть подкожный кальциноз, локализующийся в местах повышенной травматизации (на пальцах, в области локтевых и коленных суставов). Подкожный кальциноз наблюдают при особой форме ССД - CREST-синдроме, объединяющем пять признаков: «С» - подкожный кальциноз, «R» - синдром Рейно, «E» - нарушение моторики пищевода, «S» - склеродактилию, «T» - телеангиэктазии.
Синдром Рейно
Типичный и ранний (нередко - первый) признак ССД, особенно часто развивающийся при диффузной и акросклеротической фор- мах, - синдром Рейно. При его периферической форме возникает внезапное побеление или посинение кистей, реже стоп, онемение языка. Из-за нарушения микроциркуляции дистальные отделы конечностей обычно холодные на ощупь. При нарастании микроциркуляторных расстройств у больных появляются поверхностные некрозы и язвочки, в тяжёлых случаях может развиться гангрена. Вследствие трофических нарушений происходит истончение концевых фаланг пальцев, развивается остеолиз концевых фаланг. При генерализованных вазоспастических реакциях отмечают повышение АД, кардиалгии, головные боли, нарушения зрения.
Поражения суставов и мышц
При ССД отмечают полиартралгии, артриты без эрозивных изменений суставного хряща и периартриты с фиброзно-склеротическими изменениями периартикулярных тканей, формированием контрактур и деформаций. Склеротические изменения также развиваются в мышцах. При фиброзирующем миозите происходит постепенное замещение мышечных волокон соединительной тканью, что сопровождается их атрофией и снижением мышечной силы.
Поражения внутренних органов
Для ССД характерно поражение сердца. В начальном периоде ССД у детей возникает отёк, уплотнение и нечёткость структур миокарда, в более поздние сроки развивается кардиосклероз, формируется малое ригидное сердце. Поражение эндокарда у детей чаще всего поверхностное, в связи с чем пороки формируются относительно редко. При рентгенологическом исследовании и УЗИ часто выявляют утолщение перикарда и плевроперикардиальные спайки.
В лёгких при ССД развивается пневмосклероз, в первую очередь в базальных отделах. При инструментальном исследовании выявляют утолщение плевры.
Поражение почек у детей обычно выявляют в поздние сроки. Симптоматика, как правило, скудная, в виде минимальной протеи- нурии или минимального мочевого синдрома. Иногда отмечают нефритоподобные проявления, сопровождающиеся нарушением функций почек и повышением АД. У детей редко развивается истинная склеродермическая почка, обусловленная поражением сосудов с множественными кортикальными некрозами, клинически проявляющаяся быстрым нарастанием протеинурии, повышением АД, азотемией и олигурией.
Для ССД очень характерно поражение ЖКТ. Классический признак ССД - гипотония пищевода с расширением его в верхних отделах и сужением в нижней трети. При поражении пищевода нарушается глотание, больной запивает сухую пищу водой, появляется отрыжка. Нарушение моторики подтверждает задержка пассажа бариевой взвеси, выявляемая при рентгенологическом исследовании. В отдельных случаях формируются стриктуры и укорочение пищевода, фиброзирование кардиального сфинктера с нарушением прохождения пищевого комка. Из-за поражения слизистых оболочек нарушается переваривание пищи в желудке и всасывание её в кишечнике. При поражении кишечника у больных ССД обычно отмечают ослабление перистальтики, характерны упорные запоры.
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ
Среди лабораторных показателей отмечают увеличение СОЭ, серомукоида, повышение концентрации Ig (особенно IgG), титров антинуклеарных АТ и ревматоидного фактора, реже - АТ к ДНК. Диагностическое значение имеет обнаружение у больных специфических аутоантител.
• АТ Scl-70 (АТ к топоизомеразе I) обнаруживают у 20-30% детей с
ССД.
• АТ к центромере обнаруживают у 7% детей с ССД преимущественно с CREST-синдромом.
ДИАГНОСТИКА
Для установления диагноза ССД применяют следующие критерии
(табл. 13-14).
Таблица 13-14. Предварительные диагностические критерии ювенильной ССД (Pediatric Rheumatology European Society, 2004)

ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
В первую очередь проводят с другими заболеваниями склеродермической группы: диффузным эозинофильным фасциитом, склередемой Бушке, мультифокальным фиброзом, псевдосклеродермией (мета- болической, наследственной и др.), индуцированной склеродермией (вибрационной, иммунологической, паранеопластической); други-
ми диффузными болезнями соединительной ткани; ЮРА, болезнью Рейно, различными кожными заболеваниями.
ЛЕЧЕНИЕ
Наиболее важный препарат патогенетической терапии ССД - пеницилламин. Он подавляет синтез и усиливает распад коллагена, тормозит избыточное образование фиброзной ткани. Терапевтическую дозу подбирают с учётом переносимости препарата (5-8 мг/кг/сут - 125-500 мг/сут), назначают на длительный срок.
При наличии клинических и лабораторных признаков, свидетельствующих о высокой активности патологического процесса, для ликвидации системного или локального иммунного воспаления больным назначают глюкокортикоиды короткого действия (преднизолон или метилпреднизолон) в средних дозах (15-25 мг/сут) с последующим их снижением при достижении терапевтического эффекта вплоть до полной отмены. Глюкокортикоиды неэффективны в поздних стадиях заболевания при наличии выраженных фиброзно- склеротических изменений. В последние годы для лечения больных с быстропрогрессирующими формами ССД при наличии высокой иммунологической активности применяют метотрексат, циклофосфамид, циклоспорин.
Большое значение в лечении ССД придают улучшению микроциркуляции. Больным назначают вазодилататоры (блокаторы кальциевых каналов, препараты никотиновой кислоты), пентоксифиллин, дезаг- реганты (дипиридамол, пирикарбат), при необходимости - антикоагулянты (гепарин натрий), ингибиторы АПФ, препараты, укрепляющие сосудистую стенку (эсцин+тиамин, троксерутин, рутозид). В последние годы для лечения выраженных ишемических осложнений у больных ССД используют Пг (алпростадил).
Местное лечение включает аппликации раствора диметилсульфоксида с добавлением сосудорасширяющих средств на поражённые участки кожи, тепловые процедуры (парафиновые аппликации), фонофорез с различными лекарственными средствами, грязевые и озокеритовые аппликации и т.д. Применяют мази («Хондроксид», «Гепатромбин», гепариновую мазь, троксевазиновую и др.), кремы и гели.
ПРОГНОЗ
Прогноз в отношении жизни благоприятен, в то же время при ССД у детей могут сформироваться выраженная асимметрия лица или конечностей, контрактуры, что приводит к инвалидизации. Чем раньше возникает, острее протекает и позже диагностируется ССД, тем тяжелее её исход.
СИСТЕМНЫЕ ВАСКУЛИТЫ
Васкулит (ангиит) - первичное или вторичное воспаление стенок кровеносных сосудов. Первичные системные васкулиты - группа заболеваний, характеризующихся поражением стенки сосудов различного калибра (от микроциркуляторного русла до аорты и её ветвей) по типу очагового воспаления и некроза с последующим вовлечением в патологический процесс органов и тканей в зоне сосудистого повреждения. Вторичные васкулиты развиваются при инфекционных, аутоиммунных, ревматических, онкогематологических и других заболеваниях.
Распространённость заболеваний этой группы у детей неизвестна. Болезнь Шенляйна-Геноха (геморрагический васкулит), ювенильный полиартериит, болезнь Кавасаки, артериит Такаясу (неспецифический аортоартериит) наблюдают преимущественно в детском возрасте. В отличие от взрослых, системные васкулиты у детей хотя и развиваются реже, но отличаются остротой течения, яркими манифестными симптомами и в то же время - более оптимистичным прогнозом при условии ранней и адекватной терапии.
Этиология и патогенез
Этиология большинства первичных васкулитов неизвестна. Полагают, что их возникновению у ребёнка могут способствовать час- тые острые инфекционные болезни, очаги хронической инфекции, лекарственная аллергия, наследственная предрасположенность к сосудистым или ревматическим заболеваниям. Бактериальные или вирусные инфекции (стрептококковая, герпесвирусная, парвовирусная, гепатит В или С), с одной стороны, аллергия или отягощённый аллергологический анамнез, с другой стороны, по мнению специалис- тов, служат двумя равновеликими факторами, формирующими фон для гиперсенсибилизации организма, или выступают как триггерные, провоцирующие моменты.
Большое значение в развитии васкулитов имеют формирование, циркуляция и осаждение на стенках сосудов иммунных комплексов, антинейтрофильных цитоплазматических АТ, иммунное пролиферативно-деструктивное воспаление стенки артерий различного калибра, повреждение эндотелия сосудов, повышение сосудистой проницаемости, гиперкоагуляция с нарушением кровотока, ишемия в зоне повреждения сосудов.
Клиническая картина
В начальном периоде васкулитов наблюдают общие проявления неспецифического воспалительного синдрома: субфебрильная или
фебрильная лихорадка, артралгии, похудание, симптомы периферических и висцеральных сосудистых расстройств, умеренная анемия, лейкоцитоз в периферической крови, увеличение СОЭ, признаки ги- перкоагуляции, диспротеинемия, повышение уровня С-реактивного белка, IgA, ЦИК и криоглобулинов. Однако наряду с общими признаками каждому заболеванию свойственны собственные клинические синдромы (табл. 13-15). Своеобразие клинических проявлений обусловлено локализацией васкулита, калибром поражённых сосудов, распространённостью патологического процесса, особенностями мор- фологических изменений (преобладание деструктивных или пролиферативных процессов), степенью расстройств гемодинамики и ишемии органов и тканей.
Таблица 13-15. Клинические синдромы ювенильных форм системных вас- кулитов
Наименование заболевания | Характерные клинические синдромы |
Болезнь Шенляйна-Геноха Болезнь Кавасаки Узелковый полиартериит Болезнь Такаясу | Кожный геморрагический, суставной, абдоминальный, почечный Лихорадочный, поражения слизистых оболочек (конъюнктивит, фарингит), кожный, лимфонодулярный, сердечный с возможным формированием аневризм коронарных артерий Абдоминальный, почечный, артериальной гипертензии, множественный асимметричный мононевритический, кожный, тромбангиитический Асимметрии или отсутствия пульса, быстрой утомляемости, перемежающейся хромоты, цереброваскулярный, абдоминальный, артериальной и лёгочной гипертензии |
Диагностика
Диагноз каждого из системных васкулитов базируется на характерных клинических синдромах. Лабораторные показатели отражают лишь воспалительную активность. По показаниям проводят инструментальные исследования, позволяющие выявить уровень сосудистого поражения и характер деформации сосудов, признаки нарушения вне- и внутриорганного кровообращения (УЗИ, допплерография, КТ, МРТ, РЭГ, реовазография, аорто- и коронарография), а также диагностическую биопсию.
Общие принципы лечения
Выбор методов лечения болезни предполагает воздействие на возможную причину и основные механизмы развития болезни.
• Подавление иммунного воспаления путём назначения препаратов противовоспалительного и иммунодепрессивного действия: глюкокортикоидов (преднизолон, метилпреднизолон), цитостатиков (циклофосфамид, метотрексат).
• Удаление Аг, ЦИК: внутривенное введение Ig, плазмаферез синхронно с пульс-терапией глюкокортикоидами и/или цитостатиками.
• Коррекция гемостаза: назначение антикоагулянтов, антиагрегантов.
• Симптоматическая терапия. Лечение назначают с учётом нозологического диагноза, фазы болезни и её клинических особенностей. Эффект лечения оценивают по динамике клинических синдромов и лабораторных показателей. Лечение в острой фазе заболевания проводят в стационаре, затем продолжают амбулаторно при обязательном диспансерном наблюдении и контроле.
БОЛЕЗНЬ ШЕНЛЯЙНА-ГЕНОХА
Болезнь Шенляйна-Геноха (геморрагический васкулит) - распространённое системное заболевание с преимущественным поражением микроциркуляторного русла. Заболевание обычно возникает в возрасте 7-12 лет. Частота составляет 13,5:100 000 детей.
Клиническая картина
Заболевание начинается обычно остро с субфебрильной, реже - фебрильной лихорадки (иногда без температурной реакции), с одного или нескольких характерных синдромов, в зависимости от чего выделяют простую и смешанную формы болезни.
Кожный синдром. Поражение кожи в виде пурпуры возникает у всех больных. Чаще в самом начале болезни, иногда - вслед за абдоминальным или другим характерным синдромом на коже появляется мелкопятнистая или пятнисто-папулёзная геморрагическая сыпь. Сыпь симметричная, локализуется преимущественно на коже нижних конечностей, на ягодицах, вокруг крупных суставов, реже - на верхних конечностях, туловище, лице. Интенсивность сыпи различна - от единичных элементов до обильной сливной, иногда в сочетании с отёком Квинке. Высыпания волнообразно рецидивируют, сгущаются в дистальных отделах («чулочная болезнь»). Геморрагические элементы оставляют после себя пигментацию, проходящую бесследно.
Суставной синдром. Это второй по частоте признак болезни. Степень поражения суставов варьирует от артралгий до обратимых артритов, преимущественно крупных суставов.
Абдоминальный синдром. Он развивается почти у 70% детей в результате отёка и геморрагий в стенке кишки, брыжейке или брюшине.
Самый частый признак - схваткообразная боль в животе. Болевые приступы могут повторяться многократно в течение дня и сопровождаться диспепсическими расстройствами в виде тошноты, рвоты, жид- кого стула с примесью крови.
Почечный синдром. Поражение почек возникает реже и проявляется гематурией различной степени выраженности, иногда - развитием гломерулонефрита. Нефротический синдром и почечная недостаточность развиваются редко. Именно развитие гломерулонефрита может лишить ребёнка шанса на выздоровление.
Течение болезни чаще острое циклическое, с выздоровлением в течение 2 мес, но может быть затяжным, рецидивирующим и продолжаться 6 мес, редко год и более. Хроническое течение свойственно вариантам с развитием гломерулонефрита или с изолированным, непрерывно рецидивирующим кожным геморрагическим синдромом.
Осложнения. При развитии абдоминального синдрома возможны хирургические осложнения (инвагинация, кишечная непроходимость, перфорация кишки с развитием перитонита). Гломерулонефрит может осложниться ОПН или ХПН.
Диагностика
Диагноз устанавливают по характеру остро возникшего кожного синдрома, прежде всего по наличию симметрично расположенной мелкопятнистой геморрагической сыпи на ногах. Трудности возникают, если первым проявлением болезни служат боли в суставах, животе или изменения в анализах мочи. В этих случаях диагноз возможно поставить лишь при последующем появлении типичной сыпи.
Лечение
В острый период обязательны госпитализация, постельный режим, гипоаллергенная диета, базисная терапия гепарином и антиагрегантами. Дозу гепарина натрия подбирают индивидуально в зависимости от степени тяжести заболевания. Лечение проводят в течение 4-6-8 нед с постепенной отменой препарата. Из дезагрегантов назначают дипиридамол, пентоксифиллин, из антиагрегантов 3-го поколения - индобуфен, тиклопидин. С целью активации фибринолиза применяют никотиновую кислоту, ксантинола никотинат. При тяжёлом течении заболевания в острый период применяют преднизолон в суточной дозе 0,5-1 мг/кг (коротким курсом), внутривенное введение декстрана [мол. масса 30 000-40 000] («Реополиглюкина»), глюкозо-новокаиновой смеси (в соотношении 3:1), по показаниям проводят сеан- сы плазмафереза синхронно с пульс-терапией метилпреднизолоном. Антибиотики назначают только в случае наличия очаговой инфекции.
Прогноз
60-65% больных выздоравливают, но возможно рецидивирующее течение. В случае развития гломерулонефрита ребёнок нуждается в наблюдении у нефролога, так как исходом может быть ХПН.
БОЛЕЗНЬ КАВАСАКИ
Болезнь Кавасаки (слизисто-кожно-лимфатический синдром) - острый системный некротизирующий васкулит с поражением крупных, средних и мелких артерий, сочетающийся с кожно-слизисто-железистым синдромом.
Болеют дети младше 8 лет (до 80%), чаще - мальчики. Распространённость болезни значительно превышает частоту всех остальных форм васкулитов и ревматической лихорадки. Частота болезни Кавасаки в Японии, Китае и Корее составляет 100-110:100 000 детей до 5 лет, в США - 10-22:100 000, в Германии - 9:100 000, заболеваемость в России неизвестна. Наличие сезонной изменчивости и цикличности заболевания позволяет предположить инфекционную его природу, но до настоящего времени подтвердить это предположение не удалось.
Клиническая картина
Заболевание начинается остро, повышается температура тела, появляется гиперемия конъюнктив, сухость и гиперемия губ, слизистой оболочки полости рта. У 50-70% больных увеличиваются шейные лимфатические узлы с одной или с обеих сторон. В последующие дни присоединяются интенсивная эритема пальцев рук и ног, полиморфные или скарлатиноподобные высыпания на туловище, конечностях и в паховых областях, плотный отёк кистей и стоп. Могут также появляться артралгии, кардиомегалия, приглушённость сердечных тонов, систолический шум, увеличение размеров печени, диарея. Возможно формирование аневризм коронарных сосудов. Высокая лихорадка продолжается от 12 до 36 дней. На второй неделе сыпь, конъюнктивит, увеличение лимфатических узлов исчезают, язык становится «малиновым», появляется пластинчатое шелушение пальцев рук и ног.
Осложнения. Инфаркт миокарда, разрыв коронарной артерии.
Лабораторное исследование
При лабораторном исследовании обнаруживают лейкоцитоз и тромбоцитоз в периферической крови, увеличение СОЭ, анемию. При поражении коронарных сосудов на ЭКГ выявляют признаки ишемии миокарда, а при помощи допплерографии и коронарографии - аневризмы.
К 6-10-й неделе все клинические и лабораторные симптомы исчезают, наступает выздоровление. В эти же сроки возможен и внезапный смертельный исход.
Диагностика
Диагноз болезни Кавасаки устанавливают при наличии пяти из шести основных клинических критериев или четырёх основных и ко- ронарита (см. ниже). Основные критерии болезни Кавасаки следующие.
• Повышение температуры тела, продолжающееся не менее 5 дней.
• Гиперемия конъюнктив.
• Воспалительные изменения слизистой оболочки губ и полости рта.
• Ладонная и подошвенная эритема с отёком и последующим шелушением кожи пальцев.
• Полиморфная сыпь.
• Негнойное увеличение одного или нескольких шейных лимфатических узлов (более
Лечение
Лечение антибиотиками и глюкокортикоидами неэффективно. Предупредить формирование коронарных аневризм и их осложнений помогает только лечение ацетилсалициловой кислотой и высокими дозами внутривенно вводимого Ig. Ig назначают в дозе 2 г/кг массы тела ребёнка в сочетании с ацетилсалициловой кислотой (30-80 мг/ кг/сут) на время лихорадочного периода). В последующем в течение 3 мес назначают поддерживающую дозу ацетилсалициловой кислоты (2-5 мг/кг/сут). Аневризмы обнаруживают в 8% случаев при лечении Ig в течение первых 10 дней от начала лихорадки и в 29% - при более позднем начале лечения. Без лечения аневризмы коронарных артерий формировались более чем у 60% больных.
Прогноз
Прогноз чаще благоприятный. Большинство пациентов выздоравливают. При задержке или отсутствии лечения самый высокий риск развития аневризм отмечен у детей в возрасте до 1 года. Летальность составляет 0,1-0,5%. Смерть чаще всего является результатом разрыва аневризмы коронарной артерии или инфаркта миокарда.
УЗЕЛКОВЫЙ ПОЛИАРТЕРИИТ
Узелковый полиартериит - острое, подострое или хроническое заболевание, в основе которого лежит поражение периферических и висцеральных артерий, преимущественно мелкого и среднего калибра. В МКБ-10 выделены три варианта узелкового полиартериита.
• Узелковый полиартериит (классический).
• Полиартериит с поражением лёгких (синдром Черджа-Стросс).
• Ювенильный полиартериит.
В настоящее время болезнь выявляют редко, особенно её классический вариант, ассоциированный с вирусом гепатита В. Распространённость неизвестна. Возникает у детей всех возрастов, ювенильный полиартериит - чаще у девочек.
Клиническая картина
Заболевание начинается в большинстве случаев остро. Появляются высокая ремиттирующая лихорадка, профузные поты, сильные боли в мышцах, крупных суставах, животе, истощение. При лабораторном исследовании обнаруживают нейтрофильный гиперлейкоцитоз, значительное увеличение СОЭ, гипергаммаглобулинемию, маркёры вируса гепатита В (при отсутствии симптоматики гепатита). Через несколько недель, а при постепенном начале - месяцев появляется несколько характерных клинических признаков: узелки, древовидное ливедо (синевато-фиолетовая сосудистая сеть на коже в виде ветвей дерева), множественный мононеврит, церебральные сосудистые кризы, боли в животе, коронарит, артериальная гипертензия. У детей в восемь раз чаще, чем у взрослых, развивается тромбангиитический синдром - некрозы кожи, слизистых оболочек, дистальная гангрена. Особенность полиартериита с поражением лёгких - обязательно предшествующий и сопутствующий системному васкулиту синдром гиперэозинофильной бронхиальной астмы.
Клиническая картина узелкового полиартериита различается в зависимости от варианта, преимущественной локализации и выраженности васкулита (табл. 13-16).
Осложнения. Инсульт, отёк мозга, инфаркт миокарда, почечная не- достаточность, перитонит, дыхательная недостаточность, инфицирование очагов некроза.
Диагностика
Диагноз устанавливают при наличии у больного характерных клинических симптомов. Из лабораторных показателей учитывают вы- сокую степень нейтрофильного лейкоцитоза и увеличения СОЭ, свойственные ювенильному полиартерииту, положительные маркёры вируса гепатита В, характерные для классического полиартериита, и повышение эозинофилов в периферической крови выше 20-30% при синдроме Черджа-Стросс. В сложных случаях производят биопсию кожи и подкожной клетчатки, редко - почки, а также - аортографию (нефрографию).
Таблица 13-16. Типичные симптомы различных вариантов узелкового по- лиартериита
Клинический вариант | Симптоматика острой фазы |
Все варианты Классический Ювенильный Синдром ЧерджаСтросс | Лихорадка, боль в суставах, мышцах, гиперестезия, похудание Синдром артериальной гипертензии ренального генеза, множественный мононеврит, церебральные сосудистые кризы, коронарит, некроз кишечника Узелки, ливедо, локальные отёки, мононеврит, очаги некроза кожи, слизистых оболочек, дистальная гангрена Бронхиальная астма или рецидивирующий обструктивный синдром в анамнезе, гиперэозинофильный синдром в сочетании с любыми из обозначенных выше симптомов активного полиартериита |
Лечение
В активном периоде лечение проводят в стационаре, в тяжёлых случаях - в условиях палаты интенсивной терапии. Всем больным назначают преднизолон в суточной дозе 1-2 мг/кг. При высокой ак- тивности процесса и выраженных висцеритах назначают циклофосфамид в суточной дозе 2-3 мг/кг ежедневно или в виде пульс-терапии (10-15 мг/кг 1 раз в месяц на протяжении не менее года). При выраженной артериальной гипертензии применяют только циклофосфамид или его комбинацию с коротким курсом преднизолона в низких дозах (менее 0,5 мг/кг). Для усиления противовоспалительного эффекта назначают фенилбутазон.
Через 4-6 нед лечения (при достижении положительных результатов) дозу глюкокортикоидов начинают снижать до поддерживающей. Лечение гормонами продолжают не менее 2 лет. Для улучшения кро- вообращения применяют антикоагулянты, антиагреганты, ангиопротекторы. При выраженных болях назначают болеутоляющие средства.
Прогноз
Ювенильный полиартериит характеризуется хроническим рецидивирующим течением, но по мере взросления опасность формирования гангрены становится минимальной. При классическом варианте воз- можна многолетняя ремиссия, но в острый период (обычно в первый год болезни) высок риск (до 40%) смертельного исхода от осложнений злокачественной артериальной гипертензии. При полиартериите с поражением лёгких удаётся достичь стойкой ремиссии васкулита, но бронхиальная астма может стать гормонозависимой.
НЕСПЕЦИФИЧЕСКИЙ АОРТОАРТЕРИИТ
Неспецифический аортоартериит (болезнь Такаясу) - деструктивно-продуктивный сегментарный аортит и субаортальный панартериит, характеризующийся образованием аневризм и/или стенозов аорты и её ветвей вплоть до сегментарной артериальной окклюзии, что клинически проявляется ишемическими расстройствами и синдромом асимметрии и отсутствия пульса. У лиц женского пола выявляют в 5-7 раз чаще, чем у мужчин. В подавляющем большинстве случаев болезнь начинается в 10-20 лет, у детей дошкольного возраста диагностируют редко. Заболеваемость составляет 1,2-6,3 случая на 1 млн населения.
Клиническая картина
Заболевание имеет две фазы развития: острую (от нескольких недель до нескольких месяцев и лет) и хроническую, с волнами обострения или без них.
Начало болезни у большинства детей острое. Первые симптомы неспецифичны - лихорадка, анорексия, артралгии и миалгии, геморрагические или нодозные высыпания. По мере развития болезни на фоне стойко увеличенной СОЭ (до 40-60 мм/ч) возникают боли в мышцах рук (ног) или усталость при физической нагрузке, синдром перемежающейся хромоты, онемение пальцев, тахикардия, расширение границ сердца, сосудистые шумы при аускультации. Нередки головная боль, головокружение, ухудшение зрения и другие симптомы локального дефицита кровообращения в зависимости от уровня поражения артерий. Выделяют четыре варианта (типа) локализации процесса (табл. 13-17).
Таблица 13-17. Типы неспецифического аортоартериита*
Тип | Локализация |
I II III IV | Дуга аорты и отходящие от неё артерии Нисходящий, брюшной отделы аорты, чревная, почечная, бедренная и др. артерии Смешанный вариант (распространённое поражение сосудов области дуги и других отделов аорты) Поражение лёгочных артерий, сочетающееся с любым из трёх типов заболевания |
* По E.Lupi-Herrera et al., 1977.
Динамика клинических проявлений острой фазы и некоторые синдромы, сохраняющиеся при хронической фазе, в немалой степени обусловлены местом поражения и степенью дефицита кровообращения (табл. 13-18).
Таблица 13-18. Связь клинических синдромов с локализацией аортоарте- риита
Поражённые артерии | Проявления |
Подключичные, плечевые, бедренные, подколенные артерии Сонные артерии Лёгочные артерии Почечные артерии Чревная, мезентериальная артерии | Синдром перемежающейся хромоты, синдром асимметрии или отсутствия пульса Головная боль, ухудшение зрения, нару- шение мозгового кровообращения Лёгочная гипертензия Артериальная гипертензия почечного происхождения Боль в животе, рвота, диарея |
Активный период при этом заболевании может продолжаться месяцами и даже годами, несмотря на клиническое улучшение на фоне лечения.
Осложнения развиваются при распространённом поражении аорты, почечных, сонных артерий. Могут возникнуть расслоение стенки аневризмы аорты, разрыв аневризмы, инсульт, ХПН.
Диагностика
Диагноз чаще устанавливают лишь при выявлении синдрома асимметрии или отсутствия пульса, обычно к концу 2-го года болезни. Возможна более ранняя диагностика при условии тщательного поиска причины локального снижения кровообращения у пациента (чаще девочки-подростка) с повышенной СОЭ неустановленной этиологии.
Внимательный осмотр, пальпация пульса и измерение АД на обеих руках и ногах, аускультация сонных, подключичных, бедренных артерий, аорты с целью выявления патологических сосудистых шумов, инструментальные исследования позволяют установить диагноз неспецифического аортоартериита.
Лабораторные и инструментальные исследования помогают выявить признаки активности процесса. В анализе крови определяют значительно увеличенную СОЭ, анемию, увеличение концентрации C-реактивного белка, IgA. С помощью допплерографии, дуплексного сканирования, МРТ, аортографии возможно выявить и проследить в динамике деформацию сосудистого русла, изменение линейной скорости кровотока, утолщение стенок аорты или устья крупных артерий, артериальную окклюзию.
Лечение
В острой фазе назначают преднизолон по 0,5-1 мг/кг/сут со снижением дозы через 1-2 мес до поддерживающей, метотрексат не ме-
нее 10 мг/м2 поверхности тела 1 раз в неделю, препараты, улучшающие коллатеральное кровообращение (пентоксифиллин, винпоцетин и др.). С наступлением хронической фазы в течение 1-2 лет продолжают поддерживающее лечение преднизолоном и метотрексатом. По показаниям производят оперативное вмешательство (сосудистую пластику, иссечение аневризмы и др.).
Прогноз
Большинство больных, перенёсших острую фазу неспецифического аортоартериита или несколько обострений, длительное время остаются трудоспособными. При поражении почечных артерий и артериальной гипертензии возможно развитие ХПН.