Клиническая фармакология.: учебник для вузов / Под ред. В.Г. Кукеса.- 4-е издание., перераб. и доп., - 2009. - 1056 с.
|
|
|
|
ГЛАВА 1. КЛИНИЧЕСКАЯ ФАРМАКОКИНЕТИКА
Клиническая фармакокинетика - раздел клинической фармакологии, изучающий пути введения, биотрансформацию, связь с белками крови, распределение ЛС и выведение их из организма человека, в частности больного. Главная задача клинической фармакокинетики - повышение эффективности и безопасности (снижение побочных эффектов) лекарственной терапии. К основным фармакокинетическим процессам относят всасывание, метаболизм (биотрансформация), распределение и выведение. Эти процессы проходят с участием различных белков: транспортных, белков плазмы крови, ферментов биотрансформации. От их функционирования зависит концентрация ЛС в плазме крови, а влияние на них различных факторов может в конечном счёте изменить фармакологический ответ. Следовательно в настоящее время современные знания в области клинической фармакокинетики позволяют врачу «управлять судьбой ЛС в организме больного», осуществляя максимально эффективную и безопасную терапию.
1.1. РОЛЬ ТРАНСПОРТЁРОВ ЛЕКАРСТВЕННЫХ СРЕДСТВ В ФАРМАКОКИНЕТИЧЕСКИХ ПРОЦЕССАХ
В настоящее время стало ясно, что в процессах всасывания, распределения и выведения ключевую роль играют транспортные белки - «транспортёры», система которых сформировалась в процессе эволюции, для переноса нутриентов, эндогенных соединений и ксенобиотиков. Большое количество ЛС - субстраты данных транспор- тёров, поэтому их ещё называют транспортёрами ЛС. В фармакокинетике ЛС транспортёры выполняют следующие функции в зависимости от расположения.
• При локализации в энтероцитах:
- «выброс» ЛС в просвет кишечника: гликопротеин Р (P-gp, MDR1), протеин резистентности рака груди (ВСRP), протеин 2, ассоциированный со множественной лекарственной устойчивостью (MRP2);
- всасывание ЛС: транспортёр I олигопептидов (РЕРТ1), полипептид В, транспортирующий органические анионы (ОАТР-В или ОАТР2В1).
• При локализации в гепатоцитах:
- захват ЛС из крови: полипептиды А, В и С, транспортирующие органические анионы (ОАТР-А или ОАТР1В3, ОАТР-В или ОАТР2В1, ОАТР-С или ОАТР1В1), протеины 1, 3 и 4, ассоциированные со множественной лекарственной устойчивостью
(MRP1, MRP3, MRP4);
- активная секреция ЛС в жёлчь: гликопротеин Р (MDR1), протеин 2, ассоциированный со множественной лекарственной устойчивостью (MRP2), протеин резистентности рака груди
• При локализации в эпителиоцитах проксимальных почечных канальцев:
- захват ЛС из крови: транспортёры органических анионов 1, 2, и 3 (ОАТ1, ОАТ2 и ОАТ3);
- активная секреция в мочу: гликопротеин Р (MDR1), транспор- тёр органических анионов 4 (ОАТ4), протеины 2 и 4, ассоциированные со множественной лекарственной устойчивостью
(MRP2, MRP4);
- реабсорбция ЛС: транспортёры 1 и 2 олигопептидов (РЕРТ1, РЕРТ2), протеин 1, ассоциированный со множественной лекарственной устойчивостью (MRP1).
• При локализации в эндотелиоцитах гематоэнцефалического барьера (ГЭБ) транспортёры осуществляют «выброс» ЛС в просвет сосуда, не допуская их проникновения в центральную нервную систему (ЦНС), - гликопротеин Р (MDR1).
На активность транспортёров ЛС могут влиять различные факторы, что приводит к изменению фармакокинетики и, следовательно, фармакодинамики ЛС. В конечном счёте это может привести к развитию нежелательных лекарственных реакций или недостаточной эффективности ЛС. Рассмотрим наиболее важные транспортёры, участвующие в фармакокинетике ЛС.
ГЛИКОПРОТЕИН Р
Гликопротеин Р - продукт гена MDR1 - представляет собой АТФзависимый насос, локализованный на цитоплазматических мембранах различных клеток. Осуществляет выброс во внеклеточное пространство различных ксенобиотиков, в том числе и ЛС (рис. 1-1), общее свойство которых - липофильность. Активность гликопротеина Р изначально изучали в опухолевых клетках, чтобы выяснить механизм резистентности опухолей к цитостатикам. Однако позже доказали, что гликопротеин Р обнаруживают и в нормальных тканях организма человека: в энтероцитах, гепатоцитах, клетках проксимальных почечных канальцев, эндотелиоцитах гистогематических барьеров (гематоэнце-
фалического, гематоовариального, гематотестикулярного и гематоплацентарного). В ЖКТ гликопротеин Р выполняет роль своеобразного насоса, «выкачивающего» ЛС из клетки в просвет кишки. Гликопротеин Р способствует выведению гепатоцитами ксенобиотиков в жёлчь, в эпителии почечных канальцев участвует в активной секреции ксенобиотиков в мочу. Гликопротеин Р эндотелиоцитов гистогематических барьеров препятствует проникновению ксенобиотиков в ЦНС, яичники, яички, через плаценту. Таким образом, гли-
копротеин Р обеспечивает адаптационный механизм, возникший в процессе эволюции для защиты организма от ксенобиотиков, он препятствует всасыванию ксенобиотиков и способствует скорейшему их выведению. Следует отметить, что содержание гликопротеина Р значительно различается у мужчин и женщин: у мужчин количество гликопротеина Р в 2,4 раза больше, чем у женщин. Именно этот феномен лежит в основе половых различий в фармакокинетике ряда ЛС. Например, равновесная концентрация дигоксина (при одинаковой дозе) выше у женщин по сравнению с мужчинами, поэтому гликозидная интоксикация встречается у женщин. Многие широко применяемые ЛС - субстраты гликопротеина Р: сердечные глкикозиды, блокаторы медленных кальциевых каналов, ингибиторы ГМГ-КоА-редуктазы (статины), блокаторы Н1-рецепторов гистамина, макролиды, некоторые цитостатики, противоретровирусные препараты и другие (прил. 1.1. Многие ЛС - субстраты не только гликопротеина Р, но и одновременно изофермента цитохрома Р-450 (CYP3A4).
На активность гликопротеина Р могут влиять следующие факторы:
• применение ЛС, относящихся к индукторам и ингибиторам гликопротеина Р (табл. 1.1; см. главу 5 «Взаимодействие лекарственных средств»);
• использование фитопрепаратов и продуктов питания, компоненты которых также индуцируют или ингибируют гликопротеин Р (см. главу 5 «Взаимодействие лекарственных средств»);
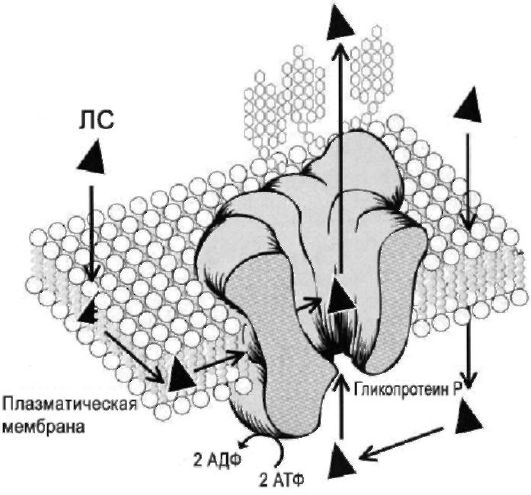 Рис. 1-1. Механизм функционирования гликопротеина Р (MDR1): АТФ - аденозинтрифосфат, АДФ - аденозиндифосфат (по Marzolini и соавт., 2004).
Рис. 1-1. Механизм функционирования гликопротеина Р (MDR1): АТФ - аденозинтрифосфат, АДФ - аденозиндифосфат (по Marzolini и соавт., 2004).
• генетический полиморфизм гликопротеина Р: носительство генотипов по определённым полиморфным маркёрам гена MDR1; может изменять фармакокинетику ЛС - субстратов гликопротеина Р (см. главу 7 «Клиническая фармакогенетика»).
ТРАНСПОРТЁРЫ ОРГАНИЧЕСКИХ АНИОНОВ И КАТИОНОВ
Транспортёры органических анионов и катионов - трансмембранные белки, ответственные за перенос через мембрану эндогенных веществ и ксенобиотиков различной химической структуры, в том числе ЛС и их метаболитов, общее свойство которых - гидрофильность. Транспортёры органических анионов формируют суперсемейство натрийнезависимых транспортных систем, производящих транспорт через мембрану ряда ЛС и их метаболитов. Они подразделяются на два семейства: OAT (organic anion-transporters) и OATP (organic aniontransporting polypeptides). Суперсемейство транспортёров органических катионов представлено одним семейством - ОСТ. ОАТ, ОАТР, ОСТ обнаруживают в печени, почках, головном мозге и кишечнике, что позволяет им играть важную роль во всасывании, распределении и выведении ЛС. ОАТ и ОСТ имеют важное значение в активной секреции гидрофильных ЛС в проксимальных почечных канальцах в мочу, а ОАТР - в гепатоцитах в жёлчь. К субстратам транспортёров органических анионов и катионов относят некоторые широко применяемые ЛС, включая β-лактамные антибиотики, диуретики, нестероидные противовоспалительные средства (НПВС), противовирусные и противоопухолевые средства, ингибиторы ГМГ-КоА-редуктазы (статины) (прил. 1.2).
Как и в случае с гликопротеином Р, на активность транспортёров органических анионов и катионов могут влиять следующие факторы:
• применение ЛС - индукторов и ингибиторов транспортёров органических анионов и катионов (прил. 1.2; см. главу 5 «Взаимодействие лекарственных средств»);
• использование фитопрепаратов и продуктов питания, компоненты которых также могут быть индукторами или ингибиторами транспортёров органических анионов и катионов (см. главу 5 «Взаимодействие лекарственных средств»);
• генетический полиморфизм транспортёров органических анионов и катионов: носительство генотипов по определённым полиморфным маркёрам генов, кодирующих транспортёры органических анионов и катионов, может изменять фармакокинетику ЛС-субстратов данных транспортёров (см. главу 7 «Клиническая фармакогенетика»).
1.2. ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Всасывание (абсорбция) - процесс поступления ЛС в кровеносную или лимфатическую систему из места его введения. Всасывание зависит от пути введения, растворимости ЛС в тканях в месте его поступления и кровотока в них, лекарственной формы и физико-химических свойств ЛС. От пути введения зависят скорость развития эффекта, его выраженность и продолжительность, а в отдельных случаях и характер действия ЛС. Всасывание при энтеральном и парентеральном пути введения ЛС различно. При внутривенном и внутриартериальном введении о всасывании говорить не приходится - ЛС сразу и полностью попадает в кровоток.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ЭНТЕРАЛЬНОМ ВВЕДЕНИИ
Самый распространённый путь введения ЛС - через рот (per os). Это наиболее доступный, удобный и простой путь введения.
Существуют четыре механизма всасывания ЛС при энтеральном введении (рис. 1-2):
• пассивная диффузия;
• активный транспорт;
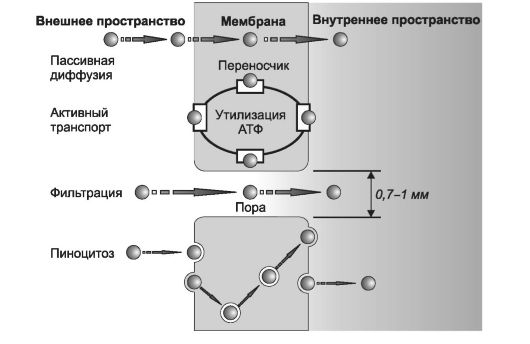 Q - молекула лекарственного вещества
Q - молекула лекарственного вещества
Рис. 1-2. Механизмы всасывания лекарственных средств при энтеральном введении.
• фильтрация через поры;
• пиноцитоз.
При всасывании ЛС обычно преобладает один из перечисленных механизмов в зависимости от пути введения и физико-химических свойств ЛС. Так, в ротовой полости, в желудке, ободочной и прямой кишке, а также с поверхности кожи всасывание ЛС происходит в основном путём пассивной диффузии и, в меньшей степени, фильтрации.
Пассивная диффузия - наиболее важный процесс. Для всасывания ЛС не требуется затрат энергии, и объём всосавшегося вещества прямо пропорционален градиенту концентрации и коэффициенту распределения ЛС в средах «липиды-вода». Жирорастворимые ЛС всасываются быстрее, чем водорастворимые, и между двумя ЛС сходного химического состава не будет наблюдаться конкуренции за всасывание. Вначале ЛС должно поступить в водный раствор на поверхности клеточной мембраны, затем раствориться в липидном слое мембраны и наконец перейти в водную фазу по другую сторону мембраны. Всасывание ЛС будет зависеть от физико-химических свойств вещества, в особенности от степени его ионизации в просвете ЖКТ. Диффузии подвергаются электролиты, находящиеся в недиссоциированном состоянии. Растворимость и степень ионизации ЛС зависят от рН желудка и двенадцатиперстной кишки. Показано, что при снижении рН лучше всасываются слабые кислоты, так как в кислой среде они находятся в менее ионизированной форме. Напротив, повышение рН (например, при ахлоргидрии) облегчает всасывание слабых оснований и задерживает всасывание слабых кислот.
Ионизированные формы ЛС обладают несущественной растворимостью в жирах, поэтому теоретически кислоты должны лучше всасываться при низком значении рН в желудке (тогда они находятся в менее ионизированном состоянии), чем при более высоком значении рН в кишечнике. Однако короткий срок пребывания ЛС в желудке и ограниченная по сравнению с кишечником площадь поверхности сводят на нет значение фактора рН.
Стоит заметить, что ЛС, абсорбированные путём пассивной диффузии, хорошо всасываются не только в тонкой, но и в толстой и прямой кишке. Этот факт - основа для разработки многих ЛС с замедленным выделением действующего вещества и ЛС для ректального применения.
Термин «активный транспорт» подразумевает энергетические затраты для перемещения ЛС через клеточную мембрану, часто против градиента концентрации. Этот высокоспецифичный механизм обусловливает транспорт таких природных веществ, как аминокислоты, са-
хара и некоторые витамины. Однако активный транспорт может быть вовлечён в процесс всасывания ЛС, имеющих структурное сходство с природными веществами. Активный транспорт обеспечивает всасывание гидрофильных полярных молекул, некоторых неорганических ионов, пиримидинов. Например, метилдопа, леводопа всасываются путём активного транспорта с участием аминокислот-переносчиков. В настоящее время хорошо изучено всасывание по механизму активного транспорта в кишечнике ЛС, в химической структуре которых имеются «остатки» аминокислот. Всасывание данных ЛС [ингиби- торы ангиотензинпревращающего фермента (АПФ), валацикловир] осуществляется путём работы транспортёров - олигопептидов PEPT1 и PEPT2 (табл. 1-1), что обеспечивает высокую биодоступность ЛС. Некоторые препараты были специально разработаны как субстраты PEPT1, в частности валацикловир, который был получен путём присоединения к молекуле ацикловира молекулы валина, что привело к увеличению биодоступности с 20 до 54%. Степень всасывания активно транспортированных ЛС зависит от введённой дозы, потому что в процесс вовлекаются механизмы, связанные с феноменом насыщения переносчиков.
Таблица 1-1. Локализация, функция и субстраты транспортёров олигопептидов
 В
течение долгого времени считали, что поры между клетками так малы, что
путём фильтрации через них могут быть абсорбированы только ЛС с
молекулярной массой менее 100 Д. Однако новейшие исследования указывают
на то, что данный путь всасывания имеет гораздо большее значение. ЛС,
использующие этот путь, часто обладают так называемым «окном всасывания»
в тонкой кишке. В этот период скорость всасывания ЛС резко падает, что
ведёт к неполному всасыванию. К таким ЛС относят фуросемид и атенолол.
Эти ЛС не подходят для выпуска в виде форм с замедленным высвобождением.
В
течение долгого времени считали, что поры между клетками так малы, что
путём фильтрации через них могут быть абсорбированы только ЛС с
молекулярной массой менее 100 Д. Однако новейшие исследования указывают
на то, что данный путь всасывания имеет гораздо большее значение. ЛС,
использующие этот путь, часто обладают так называемым «окном всасывания»
в тонкой кишке. В этот период скорость всасывания ЛС резко падает, что
ведёт к неполному всасыванию. К таким ЛС относят фуросемид и атенолол.
Эти ЛС не подходят для выпуска в виде форм с замедленным высвобождением.
Пиноцитоз - механизм всасывания, в процессе которого микроскопические частицы поглощаются клеточной мембраной, не играет серьёзной роли во всасывании ЛС, хотя может иметь определённое отношение к захвату макромолекул.
ФАКТОРЫ, ВЛИЯЮЩИЕ НА ВСАСЫВАНИЕ
Ниже перечислены некоторые известные факторы, влияющие на всасывание ЛС.
• Форма и характеристики ЛС:
- время расщепления таблетки/капсулы;
- время растворения;
- присутствие эксципиентов (высушивающих веществ) в таблетке или капсуле;
- стабильность в ЖКТ;
- физико-химические свойства ЛС (жирорастворимость, гидрофильность, pKa).
• Характеристики пациента:
- рН просвета ЖКТ;
- скорость опорожнения желудка;
- время прохождения содержимого по тонкой кишке;
- площадь абсорбирующей поверхности ЖКТ;
- наличие заболеваний ЖКТ;
- кровоток в брыжеечных сосудах;
- активность ферментов.
• Присутствие в ЖКТ других веществ:
- взаимодействие с другими ЛС, ионами;
- взаимодействие с пищей.
• Фармакокинетические характеристики ЛС:
- метаболизм в стенке кишки;
- метаболизм под действием кишечной микрофлоры.
Формы выпуска ЛС могут оказывать существенное влияние на его растворимость и дальнейшее всасывание. Присутствие эксципиентов (высушивающих веществ), считавшихся инертными, также может изменить всасывание ЛС. Бентонит - компонент некоторых гранулированных форм парааминобензойной кислоты (ПАСК) - способствует значительному ухудшению всасывания рифампицина при одновременном приёме. Причиной этого считают адсорбцию рифампицина на бентоните.
Скорость опорожнения желудка и перистальтика определяют быстроту попадания ЛС в тонкую кишку, где происходит всасывание большинства ЛС. Факторы, замедляющие опорожнение желудка, способствуют снижению скорости всасывания большинства ЛС (и наоборот).
Однако для некоторых ЛС, например плохо растворимых, неравномерно всасывающихся или усваиваемых в ЖКТ, степень всасывания может быть увеличена при замедлении опорожнения желудка или перистальтики тонкой кишки.
Нарушение всасывания некоторых ЛС может быть результатом синдрома недостаточности всасывания. Он обусловлен нарушением всасывания в слизистой оболочке тонкой кишки одного или нескольких питательных веществ в связи с расстройствами транспортных механизмов с последующим возникновением нарушений обменных процессов. Выделяют первичный (наследственно обусловленный) и вторичный (приобретённый) синдром нарушенного всасывания.
Влияние патологии ЖКТ на всасывание ЛС показано в табл. 1-2.
Таблица 1-2. Влияние патологии ЖКТ на всасывание лекарственных средств
 На всасывание ЛС могут влиять другие ЛС, а также пищевые продукты (см. главу 5 «Взаимодействие лекарственных средств»).
На всасывание ЛС могут влиять другие ЛС, а также пищевые продукты (см. главу 5 «Взаимодействие лекарственных средств»).
ВЛИЯНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ НА ВСАСЫВАНИЕ НУТРИЕНТОВ
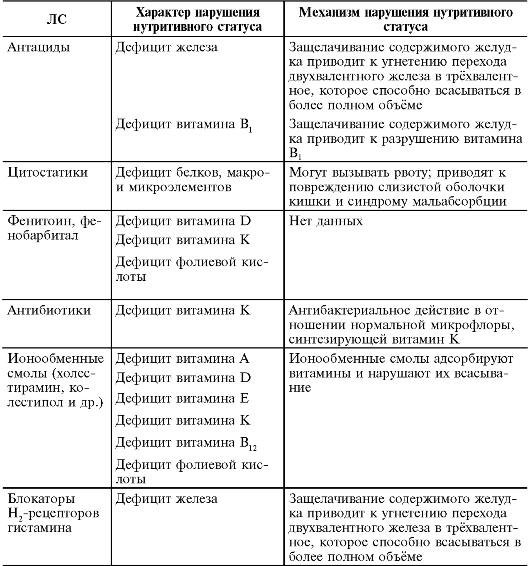
Многие ЛС могут оказывать влияние на всасывание нутриентов (белки, жиры, углеводы, витамины, микроэлементы и др.). Длительное применение подобных ЛС может приводить к дефициту тех или иных нутриентов (табл. 1-3).
Существуют ЛС, уменьшающие всасывание углеводов. Это бигуаниды и акарбоза. Бигуаниды также усиливают утилизацию глюкозы в периферических тканях, уменьшают глюконеогенез и снижают повышенное содержание инсулина у больных сахарным диабетом 2-го
Таблица 1-3. Влияние лекарственных средств на нутритивный статус
 типа
и страдающих ожирением. Акарбоза представляет собой группу ингибиторов
α-глюкозидаз, снижает абсорбцию большинства углеводов, таких, как
крахмал (60% всех углеводов, поступающих с пищей), декстрины, мальтоза и
сахароза. Акарбоза - аналог олигосахаридов (псевдотетрасахарид),
который имеет высокую аффинность (более чем в 1000 раз) к моно- и
дисахаридам кишки. Это конкурентное угнетение α-глюкозидаз снижает
постпрандиальное повышение глюкозы в крови. Таким образом, замедляются
процессы ферментирования и всасывания углеводов по всему кишечнику, при
этом само ЛС не вса-
типа
и страдающих ожирением. Акарбоза представляет собой группу ингибиторов
α-глюкозидаз, снижает абсорбцию большинства углеводов, таких, как
крахмал (60% всех углеводов, поступающих с пищей), декстрины, мальтоза и
сахароза. Акарбоза - аналог олигосахаридов (псевдотетрасахарид),
который имеет высокую аффинность (более чем в 1000 раз) к моно- и
дисахаридам кишки. Это конкурентное угнетение α-глюкозидаз снижает
постпрандиальное повышение глюкозы в крови. Таким образом, замедляются
процессы ферментирования и всасывания углеводов по всему кишечнику, при
этом само ЛС не вса-
сывается. Снижают абсорбцию глюкозы и такие ЛС, как сердечные гликозиды, поскольку они транспортируются тем же путём, что и активно резорбируемые сахара.
Существует ЛС, уменьшающее всасывание жиров (орлистат). Орлистат - ингибитор желудочно-кишечных липаз. Его механизм действия обусловлен образованием ковалентной связи с активным сериновым участком желудочной и панкреатической липаз в просвете желудка и тонкой кишки. Фермент при этом теряет способность расщеплять жиры, поступающие с пищей в виде триглицеридов, уменьшает всасывание свободных жирных кислот и моноглицеридов. Данное ЛС практически не всасывается в системный кровоток.
ПЕРОРАЛЬНЫЕ СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ С КОНТРОЛИРУЕМОЙ СКОРОСТЬЮ ВЫСВОБОЖДЕНИЯ
Хотя назначение per os - самый частый путь приёма ЛС, стандартные формы выпуска могут вызывать определённые проблемы. В некоторых случаях ЛС может иметь очень короткий период полуэлиминации и поэтому должно приниматься очень часто для поддержания устойчивой концентрации (например, прокаинамид). В других случаях высокая максимальная концентрация ЛС в плазме, вызванная быстрым высвобождением активного вещества из препарата, принятого per os, может вызвать побочные эффекты (индометацин, карбамазепин). Основной подход к решению этих проблем - разработка разнообразных лекарственных форм с замедленным высвобождением для снижения скорости всасывания.
Действие некоторых из имеющихся систем контролируемого высвобождения ЛС зависит от осмоса (рис. 1.3).
Основа действия этой системы - полупроницаемая мембрана, окружающая осмотически активную сердцевину ЛС. В каждой капсуле с помощью лазерных технологий очень точно просверлено по одному отверстию. Когда капсула или устройство проглатывается, вода из тонкой кишки поступает в ядро капсулы через полупроницаемую мембрану, растворяя ЛС на поверхности ядра. Таким образом, внутри устройства вырабатывается стабильное осмотическое давление, которое выталкивает наружу раствор ЛС через отверстие. Скорость доставки ЛС регулируется размером отверстия в устройстве. Быстрота высвобождения будет оставаться постоянной до тех пор, пока содержимое капсулы или устройства не растворится полностью, а всё увеличивающееся разбавление затем приведёт к постепенному снижению скорости доставки ЛС. Такая система впервые широко использовалась для индометацина, однако впоследствии с её помощью стали применять такие ЛС, как β-адреноблокаторы.
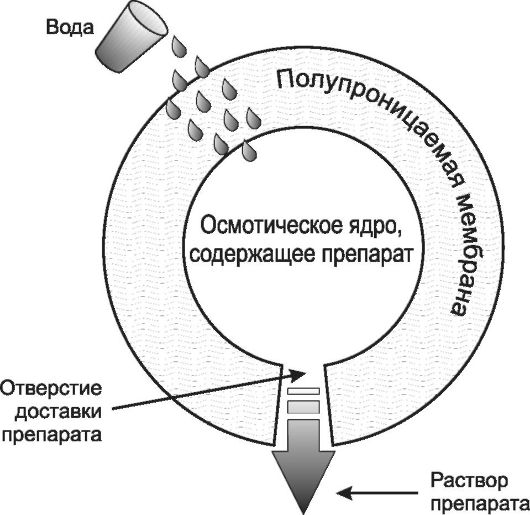 Рис. 1-3. Схематическое изображение типичной осмотической системы доставки лекарственного средства в разрезе.
Рис. 1-3. Схематическое изображение типичной осмотической системы доставки лекарственного средства в разрезе.
Разработаны различные системы контролируемого высвобождения ЛС. У этих систем две основные цели:
• доставить пациенту необходимое количество ЛС, не подвергая его действию ЛС в концентрациях ниже или выше терапевтических;
• обеспечить хороший контроль лекарственной терапии без вытекающих из этого дополнительных расходов.
Разработаны системы контролируемого высвобождения гормональных контрацептивов, которые могут быть имплантированы под кожу с помощью обычной иглы и способны доставлять ЛС на необходимой скорости в течение нескольких лет.
МЕСТНОЕ ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
ИЗ ПОЛОСТИ РТА И НОСА
В настоящее время увеличивается частота использования доставки ЛС в область полости рта или носа с терапевтическими целями. Хотя этот способ назначения обладает большим количеством потенциальных преимуществ, существуют и некоторые неудобства. Буккальное и сублингвальное введение способствует быстрому всасыванию ЛС и позволяет избежать эффекта первого прохождения, который наступает
при всасывании из тонкой кишки. Недостатки такого способа введения включают неприятный вкус ЛС и необходимость держать ЛС во рту, не разжёвывая и не проглатывая его. Этим способом традиционно применяют нитраты, однако в последнее время его всё чаще используют для приёма и других ЛС (каптоприла, пентазоцина и др.). Сублингвальный приём бупренорфина и морфина способствует более быстрому облегчению боли, чем применение per os в соответствующих дозах.
Интраназальный путь доставки ЛС условно используют для введения некоторых пептидов, таких, как десмопрессин, гонадотропинрилизинг-фактор и его аналоги. Несмотря на всасывание менее 1% дозы, такой метод доставки всё ещё считается эффективным. Кроме того, он позволяет избежать затруднений, встречающихся ввиду негативного отношения пациентов к лечению при инъекционном способе введения. Интраназально применяют и другие пептиды, например кальцитонин и соматорелин (соматотропин-рилизинг-фактор), а в настоящее время проводят испытания интраназальных форм пропранолола и контрацептивных препаратов. Многие пептиды всасываются лучше из полости носа, если в состав ЛС включены «промотеры». Эти вещества могут изменять свойства мукозного слоя слизистой полости носа или ослаблять тесные межклеточные связи эпителия.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ВВЕДЕНИИ В ПРЯМУЮ КИШКУ
Ректальный способ введения ЛС может быть выбран для того, чтобы избежать раздражения слизистой желудка. Всасывание из прямой кишки происходит теми же способами, что и в других отделах ЖКТ. Площадь поверхности слизистой меньше, и поэтому всасывание при этом способе не столь быстрое и полное, как при пероральном введении. Назначение per rectum - хорошая альтернатива парентеральному введению ЛС в случае тошноты или рвоты.
При применении ЛС per rectum можно частично избежать эффекта первого прохождения через печень, и это может объяснить лучшую биодоступность (по сравнению с пероральным приёмом) таких ЛС, как метопролол, метоклопрамид и морфин. Нижние и средние ректальные вены непосредственно впадают в нижнюю полую вену, поэтому кровь попадает в системный кровоток, минуя печень. На ректальное всасывание влияют те же факторы, которые обсуждались выше в отношении всасывания ЛС при приёме их внутрь. Липофильные ЛС (барбитураты, бензодиазепины) - идеальные средства для ректального применения, так как они находятся в основном в неионизированной форме и легко проникают через клеточную мембрану. Ректальное
введение тиопентала натрия или диазепама может быть использовано для быстрой анестезии при невозможности внутривенных инфузий или при наличии судорожного приступа.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ИНГАЛЯЦИОННОМ
ВВЕДЕНИИ
Многие ЛС хорошо всасываются из лёгких путём пассивной диффузии. В особенности это относится к вдыханию газообразных средств для общего наркоза, но также и к ЛС в виде аэрозолей или ингаляций.
Большинство ЛС, выпускаемых в виде аэрозоля для прямого проникновения в лёгкие (β-адреномиметики, ингаляционные глюкокортикостероиды и др.), всасываются именно путём пассивной диффузии. Чем меньше размер частицы, тем вероятнее, что ЛС хорошо абсорбируется. Частицы размером более 20 мкм могут откладываться в эпителии бронхиол, и в дальнейшем дыхательные реснички могут проталкивать частицы вещества обратно в гортань, где они затем проглатываются. Частицы размером 2 мкм могут достичь самых мелких бронхиол.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ВНУТРИМЫШЕЧНОМ
ВВЕДЕНИИ
Существует несколько основных причин, по которым ЛС могут назначаться парентеральным путём:
• ЛС разрушаются в кислой среде желудка (бензилпенициллин);
• в связи с тем, что они подвергаются активному метаболизму при первом прохождении через печень (лидокаин);
• для обеспечения быстрого начала действия ЛС.
К парентеральным путям введения относят подкожный, внутримышечный, внутривенный и некоторые другие. При внутримышечном введении препарата обеспечивается относительно быстрое наступления эффекта (10-30 мин).
Хотя растворимость в жирах способствует всасыванию ЛС при их внутримышечном введении, главный фактор, определяющий скорость и полноту всасывания ЛС, - растворимость в воде. ЛС должно быть достаточно водорастворимым при физиологическом значении рН организма для того, чтобы находиться в растворённом виде в интерстициальной жидкости мышечной ткани до того, как произойдёт его всасывание. Липофильность ЛС способствует диффузии его в капилляры. Водорастворимость ЛС может снижаться при физиологических значениях рН за счёт преципитации в месте инъекции. ЛС, плохо растворимые в воде (диазепам) или растворимые в воде только при нефизиологических значениях рН (фенитоин, хлордиазепоксид), скорее всего, будут обладать низкой биодоступностью после внутримы-
шечного введения. Такие ЛС оседают в месте введения, в то время как безводный растворитель постепенно уходит из места введения или происходит его промежуточное преобразование (буферизация). Всасывание фенитоина и дигоксина может быть очень медленным и непостоянным, поэтому внутримышечные инъекции не являются надёжным средством введения этих ЛС и его следует избегать. Хлордиазепоксид и диазепам всасываются медленно после внутримышечного введения, а диазепам может абсорбироваться не полностью. Более быстрый и надёжный эффект от этих ЛС может быть достигнут при их пероральном или внутривенном назначении. В то же время фенобарбитал и бензилпенициллин хорошо всасываются при внутримышечном введении. Оба ЛС являются слабыми кислотами и не образуют преципитатов в месте инъекции.
Внутримышечно можно вводить ЛС, оказывающие умеренное раздражающее действие, а также депо-препараты. Объём вводимого вещества не должен превышать 10 мл. Внутримышечный путь введения ЛС в малорастворимой форме (в масле или других основах) используют для пролонгирования фармакотерапевтического эффекта.
На всасывание ЛС после внутримышечной инъекции также оказывает влияние локальный кровоток. Например, кровоснабжение скелетных мышц может быть снижено из-за нарушения перфузии, связанной с уменьшением сердечного выброса при сердечной недостаточности или респираторном дистресс-синдроме. Морфин может всасываться медленнее при внутримышечном введении при остром инфаркте миокарда. Неодинаковую скорость всасывания также можно объяснить различиями в кровоснабжении отдельных мышечных групп. Например, всасывание лидокаина при его введении в дельтовидную мышцу происходит быстрее, чем при его введении в латеральную широкую мышцу спины или большую ягодичную мышцу.
Внутримышечный путь введения ЛС может стать причиной таких осложнений, как местная болезненность и постинъекционные абсцессы. Следует избегать инъекций вблизи нервных стволов, так как раздражающие вещества могут повреждать нервные волокна, в результате чего появляются сильные боли, а иногда парез мышц. Фармакокинетика ЛС может измениться при случайном попадании иглы в сосуд, а в некоторых случаях (при введении масляных растворов) такая оплошность бывает крайне опасной.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ТРАНСДЕРМАЛЬНОМ
ВВЕДЕНИИ
Большинство ЛС всасывается через кожу лишь до некоторой степени, и всасывание увеличивается, если поверхность кожи закрыть
полиэтиленом. У человека на всасывание ЛС через кожу влияет множество факторов. Например, важны толщина кожи и её регулярный контакт с атмосферным воздухом. Чрескожное всасывание обратно пропорционально толщине зернистого слоя и прямо пропорционально гидрофильности кожи. Вообще всасывание ЛС происходит лучше либо через тонкую кожу (например, за ушной раковиной), либо через те её участки, которые не постоянно находятся в контакте с окружающей средой. Существуют также немаловажные расовые различия состава кожи и реакции разных типов кожи на различные химические стимулы. ЛС несколько лучше всасываются у светлокожих, чем у темнокожих, хотя этот факт имеет сомнительное клиническое значение. Считается, что ЛС лучше всасываются через повреждённый кожный покров, однако это не всегда так.
ЛС можно использовать в виде трансдермальных форм как для однократного применения, так и для длительной терапии. Некоторые преимущества трансдермальной доставки ЛС по сравнению с привычным пероральным их приёмом перечислены ниже:
• избежание эффекта первого прохождения в печени или ЖКТ;
• предотвращение резких пиков и падений кривой, отражающей зависимость концентрации ЛС в плазме от времени;
• возможность быстро прервать поступление ЛС;
• уменьшение индивидуальной вариабельности всасывания, наблюдаемой обычно при пероральном приёме;
• длительность действия.
Пример трансдермальной лекарственной формы для однократного применения - скополамин, который был первым среди подобных лекарственных форм и успешно использовался в лечении как морской болезни, так и послеоперационной тошноты. Другие ЛС для однократного применения - азатидин и фентанил.
Большое количество ЛС сейчас применяют для длительной трансдермальной терапии; среди них - клонидин, эстрадиол, нитроглицерин, тимолол и тестостерон. Развитие системы контроля за скоростью высвобождения значительно продвинуло вперёд подобный вид терапии. Типичный пример строения трансдермальной системы показан на рис. 1-4.
Используемые в настоящее время системы в большинстве своём весьма эффективны, однако они не решают всех проблем терапии. Например, при использовании трансдермальной формы нитроглицерина нередко развивается толерантность, с которой можно справиться, лишь снимая систему с кожи на определённый отрезок времени ежедневно.
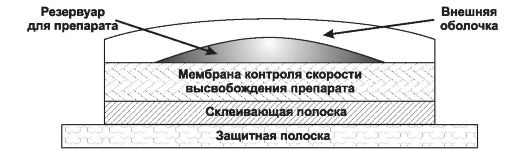 Рис. 1-4. Схематическое изображение типичной трансдермальной системы доставки лекарственного вещества в разрезе.
Рис. 1-4. Схематическое изображение типичной трансдермальной системы доставки лекарственного вещества в разрезе.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ЧЕРЕЗ КОНЪЮНКТИВУ
Конъюнктива функционирует в качестве специализированной покровной ткани, поэтому через неё, как и через кожу, могут проникать ЛС, предназначенные для локального и системного действия. Уже достигнуты определённые успехи при использовании применяемой местно формы пилокарпина с замедленным высвобождением.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ АЛЬТЕРНАТИВНЫХ СПОСОБАХ ИХ ВВЕДЕНИЯ
В настоящее время активно изучают другие способы доставки ЛС к месту их действия. В течение последних 10 лет исследуют применение моноклональных антител и липосом, однако продвижение по пути прогресса в данном случае идёт не так быстро, как можно рассчитывать.
Липосомы способны переносить широкий спектр ЛС к месту их действия и в процессе доставки уменьшать риск возникновения побочных эффектов. В настоящее время проходят исследования липосомных форм таких ЛС, как амфотерицин B , доксорубицин и гентамицин. В последнем десятилетии были значительно усовершенствованы технологии обеспечения захвата ЛС и поддержания стабильности липосом, и, скорее всего, липосомные системы будут иметь большое клиническое значение в ближайшем будущем.
1.3. РАСПРЕДЕЛЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ В ОРГАНИЗМЕ
Распределение подавляющего большинства ЛС в организме происходит неравномерно. Одни проходят через эндотелий капилляров и не способны проникать через другие мембраны, поэтому их накопление отмечают только в межклеточной жидкости. Другие свободно
проходят через мембраны и бывают распределены по всему организму. Основным результатом процессов распределения с точки зрения клинической фармакологии считают попадание ЛС в место его действия (в биофазу), где оно взаимодействует со специфическими активными центрами, определяющими его эффект. Чтобы достичь биофазы, небольшим молекулам ЛС достаточно обладать способностью вступать во взаимодействие с водой окружающей среды. Для более крупных молекул основным условием абсорбции и распределения в тканях считают растворимость в жирах.
Нерастворимые в воде и жирах молекулы организм усваивает лишь тогда, когда они способны проходить внутрь клетки через поры клеточных мембран или посредством транспортных систем. Содержание ЛС в той или иной ткани в произвольный момент определяют по алгебраической сумме потоков вещества из крови в ткань и из ткани в кровь. Поток ЛС из крови в ткань может составлять любую часть от общего количества вещества, содержащегося в артериальной крови. Поток зависит от соотношения скоростей поступления ЛС с кровью и его диффузии в клетки. Если препарат не проникает в ткани, то соответствующая доля общего потока вещества, поступающего в клетки, равна нулю. Если же потенциально возможная скорость поступления ЛС в клетку выше скорости потока крови через данную ткань, то реальный поток вещества в клетки будет равен скорости потока крови через ткань. Таким образом, распределение зависит от того, что будет лимитирующим фактором: клеточный транспорт или приток ЛС с кровью. Также распределение определяет величина фракции ЛС, не связанного с белками крови.
В месте действия молекулы ЛС могут быть включены в различные кинетические процессы.
• ЛС взаимодействуют со специфическими рецепторами, что и определяет клинический эффект данного вещества.
• Взаимодействуют с неспецифическими, неактивными участками, обычно с белками тканей.
• Могут оставаться в свободной растворённой форме.
• Попадают обратно в плазму крови в неизменённом виде.
• Проходят биотрансформацию.
• Выделяются в неизменённом виде.
Скорость движения ЛС из клеток во внешнюю среду зависит от градиента концентрации вещества между внутри- и внеклеточным пространством. Любые факторы, увеличивающие этот градиент, будут способствовать снижению внутриклеточного уровня вещества, а факторы, снижающие этот градиент, - приводить к накоплению ЛС в клетках. Следовательно, если процесс распределения детерминирован
по мембранному транспорту, то связывание с белками крови снижает интенсивность распределения, а связывание с внутриклеточными компонентами увеличивает её. Если же распределение ограничено током крови, то концентрация ЛС в тканях близка к его количеству в крови. В любом случае уменьшение концентрации свободной формы препарата в плазме крови ниже его уровня в ткани изменяет направление процесса распределения ЛС, т.е. вещество переходит из тканей в кровь.
Состояние гемодинамики считают определяющим фактором в распределении ЛС. Нарушения гемодинамики могут существенно изменять кинетику процесса. Так, при геморрагическом шоке или застойной сердечной недостаточности перфузия большинства органов снижена. Кровообращение в головном мозге и миокарде страдает в меньшей степени. Кроме того, нарушения клубочковой фильтрации и печёночного кровотока ведут к снижению почечного и печёночно- го клиренса соответственно. В результате концентрация ЛС в плазме крови, особенно после внутривенного введения, будет возрастать (например, продолжительность действия тиопентала натрия при шоке возрастает).
Связывание с белками крови. ЛС, попадая в кровеносное русло или лимфатические протоки, как и большинство инородных соединений (ксенобиотиков), в той или иной мере взаимодействуют с белками крови. Это оказывает существенное влияние на фармакокинетику и фармакологический эффект препарата, поскольку связанное с белками крови ЛС не взаимодействует с рецепторами, ферментами и теряет способность проникать через мембраны.
Белки крови имеют специфическую структуру и могут связывать различные вещества за счёт своих активных центров (табл. 1-4). Скорость и прочность связывания зависят от конформации и степени комплементарности (соответствия) этих центров и молекул веществ, а также от характера возникающих при взаимодействии физико-химических связей. По убыванию «прочности» физико-химические связи можно расположить в следующем порядке: ковалентные, ионные, водородные, ван-дер-ваальсовы.
Связывание ЛС с биологическими макромолекулами происходит, как правило, в результате одновременной реализации нескольких механизмов. Так, ван-дер-ваальсовы силы стабилизируют ионную связь и делают её более прочной. Катионы первичных, вторичных и третичных аминов образуют с анионами карбоновых кислот как ионные, так и водородные связи. При взаимодействии ароматических групп белка и ЛС гидрофобные взаимодействия дополняет комплексообразование с переносом заряда и т.д.
Таблица 1-4. Основные белки крови, связывающие лекарственные средства
 Прочность
комплекса, образованного несколькими комплементарно расположенными
центрами белка и ЛС, по-видимому, выше, чем просто сумма участвующих в
комплексообразовании отдельных связей. Естественно, что полную
комплементарность всех потенциальных центров, связывание ЛС с
биомакромолекулой определяет их максимальная специфичность. Примером
этого считают взаимодействие антител с антигенами или гаптенами.
Установлено, например, что антитело несёт электрический заряд,
противоположный по знаку и близкий по величине заряду гаптеновой группы.
Высокая специфичность антител обусловлена наличием у них участка,
структура которого строго комплементарна, соответствует структуре
антигена и размеру всего гаптена с точностью до 0,1 нм, что меньше
диаметра атома. Связывающий участок антитела имеет жёсткую конформацию,
которая не соответствует структуре гаптена.
Прочность
комплекса, образованного несколькими комплементарно расположенными
центрами белка и ЛС, по-видимому, выше, чем просто сумма участвующих в
комплексообразовании отдельных связей. Естественно, что полную
комплементарность всех потенциальных центров, связывание ЛС с
биомакромолекулой определяет их максимальная специфичность. Примером
этого считают взаимодействие антител с антигенами или гаптенами.
Установлено, например, что антитело несёт электрический заряд,
противоположный по знаку и близкий по величине заряду гаптеновой группы.
Высокая специфичность антител обусловлена наличием у них участка,
структура которого строго комплементарна, соответствует структуре
антигена и размеру всего гаптена с точностью до 0,1 нм, что меньше
диаметра атома. Связывающий участок антитела имеет жёсткую конформацию,
которая не соответствует структуре гаптена.
Взаимодействие между ЛС и белками крови считают обратимым процессом. Он подчинён закону действия масс. Эта реакция протекает очень быстро, Т1/2 составляет около 20 мс и не лимитирует удаление вещества из плазмы крови. Только несвязанные вещества могут диффундировать в ткани, поскольку комплекс белок + ЛС не способен пройти через мембрану клетки. Равновесие между фазами ЛС наступает при его распределении тогда, когда вводимое количество вещества эквивалентно его выведению. Проходя через печень, лёгкие, почки, мозг, ЛС может связываться с белками. Степень диссоциации в этом случае не всегда бывает равной комплексу альбумин + ЛС. В связи с этим наблюдают накопление ряда веществ в тканях и наоборот. Необходимо учитывать, что препарат может взаимодействовать с различными белками крови, имеющими несколько участков связывания. Например, альбумин имеет 10 мест для связи с основаниями.
Малое сродство участков связывания к этим лигандам обусловливает непрочность взаимодействия. В молекуле альбумина обнаружено только два места, в которых возникают прочные связи с кислыми ЛС. Липопротеины и кислый α1-гликопротеин взаимодействуют с основаниями (хинидин, хлорпромазин, имипрамин).
В качестве связывающих веществ могут выступать практически все белки, а также форменные элементы крови. Набор связывающих компонентов в тканях ещё более велик, ЛС могут взаимодействовать с одним или несколькими белками: альбуминами, глобулинами, липопротеинами, с кислым α1-гликопротеином. Например, тетрациклин в организме на 14% связан с альбуминами, на 38% - с различными липопротеинами, на 8% - с другими белками крови. В значительной степени морфин и кодеин взаимодействуют с глобулинами; хлорпромазин (аминазин*), имипрамин (имизин*) - с липопротеинами; пропранолол и верапамил - с кислымиα1-гликопротеинами. Когда рёчь идёт о связывании препарата с белками крови, имеют в виду суммарное связывание данного ЛС с белками и другими компонентами крови.
Поскольку существует равновесие между свободным и связанным с белком ЛС, то при выведении первого из организма происходит диссоциация комплекса белок + ЛС. При отщеплении ЛС от комплекса оно переходит в свободное активное состояние. Обратимость процессов образования и разрушения комплекса ЛС + белок неодинакова для различных классов фармакологических средств.
В большинстве случаев белок играет роль депо, регулирующего баланс между связанным препаратом и его активной формой. Обратимость взаимодействия лекарственных веществ с белками крови приводит к тому, что каждая удалённая из циркуляции молекула активного ЛС может быть замещена за счёт диссоциации белкового комплекса. Это положение правомочно только для тех ЛС, которые имеют одинаковое сродство к белкам крови и тканей. Если же сродство вещества к белкам и жирам тканей выше, то концентрация его в плазме низкая, а в тканях высокая. Например, тиопентал натрия связан с белками крови на 75%, но, попадая в головной мозг или жировую ткань, данное ЛС активнее взаимодействует с жировой тканью, а восполнение активной формы препарата происходит за счёт диссоциации от комплекса с белками крови. В связи с этим может наступить период, когда в плазме крови ЛС нет, а всё его количество сосредоточено в головном мозге или жировой ткани.
Ряд тканевых структур активно связывает определённые химические вещества. Например, ткань щитовидной железы накапливает соединения йода и меди, костная ткань - тетрациклины и т.д.
Необходимо учитывать, что насыщение белков крови в большинстве случаев происходит при концентрации ЛС, достигаемой при использовании значительно больших доз, чем разрешено в клинической практике. Исключение составляют малотоксичные ЛС, которые можно применять в больших дозах. Например, бензилпенициллин при тяжёлых септических состояниях вводят по 50-100 млн ЕД в сутки, при этом содержание бензилпенциллина превышает предел насыщения белков крови.
Обнаружены генотипические индивидуальные особенности взаимодействия отдельных ЛС с белками крови. Определённые нарушения связи ЛС с белками крови отмечают у новорождённых. К 3 мес эти различия выражены уже незначительно. Снижение способности белков крови связывать ЛС отмечено у пожилых людей, что объясняют уменьшением количества альбумина в сыворотке крови у этой группы пациентов.
В ряде случаев наблюдают изменение связывающей способности белков крови при приёме пищи с высоким содержанием жиров. Например, при алиментарной гиперлипидемии, вызванной употреблением 50 г масла, через 4 ч возрастает способность сывороточного альбумина связывать сульфаниламиды, и только через 6-7 ч она приходит к нормальному уровню.
Большое влияние на взаимодействие белков крови с ЛС оказывают хронические заболевания почек и печени, что в первую очередь связано с качественными изменениями альбуминов и, в ряде случаев, глобулинов (табл. 1-5). Повышение уровня кислых α1-гликопротеинов в сыворотке крови (при болезнях почек, ревматоидном полиартрите) усиливает связывание многих ЛС, относимых к основаниям, что приводит к уменьшению их эффективности (хлорпромазин, пропранолол). Концентрация α1-гликопротеина возрастает при физических стрессах, инфаркте миокарда, болезни Крона и других воспалительных заболеваниях, поэтому при этих патологиях свободная фракция ЛС бывает снижена (табл. 1-6).
Связывание хинидина с белками крови при декомпенсации ХСН снижено с 86 до 82%, повышено при хронической лёгочной недостаточности с 84 до 93% или в послеоперационном периоде с 78,5 до 87,5%. Это обусловлено, по-видимому, конформационными изменениями белков. У пациентов с нарушениями работы почек процент связанных с альбумином кислых ЛС [фенитоин (дифенин*), фенилбутазон (бутадион*), барбитураты, салицилаты, сульфаниламиды] меньше, чем у здоровых людей.
Уменьшение количества связывающих ЛС белков крови на 10-15% наблюдают при старении. Эти изменения наряду со снижением кро-
Таблица 1-5. Изменение связывания лекарственных средств с белками крови при заболеваниях почек или печени
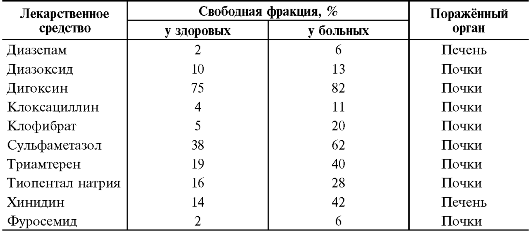 Таблица 1-6. Некоторые патологические состояния, при которых изменено содержание белков крови
Таблица 1-6. Некоторые патологические состояния, при которых изменено содержание белков крови
 вотока
через элиминирующие органы и ухудшением работы последних приводят к
замедлению выведения веществ. Особое значение это имеет для тех ЛС, у
которых лимитирующим фактором будет величина органного кровотока. Таким
образом, у пожилых лиц при назначении им стандартных доз препаратов
наблюдают увеличение концентрации ЛС в плазме крови и развитие побочных
эффектов.
вотока
через элиминирующие органы и ухудшением работы последних приводят к
замедлению выведения веществ. Особое значение это имеет для тех ЛС, у
которых лимитирующим фактором будет величина органного кровотока. Таким
образом, у пожилых лиц при назначении им стандартных доз препаратов
наблюдают увеличение концентрации ЛС в плазме крови и развитие побочных
эффектов.
Изменения взаимодействия ЛС с белками крови наблюдают при их конкуренции за связь с белками, когда более активные ЛС вытесняют вещества с меньшим сродством. Более подробно эти вопросы рассмотрены в главе «Взаимодействие лекарственных средств».
Клинический эффект в наибольшей степени зависит от концентрации свободной фракции ЛС. В результате снижения связи препарата с белками крови при заболеваниях печени и почек концентрация несвя-
занной формы ЛС может возрастать, поэтому иногда бывает необходимо уменьшать дозу или кратность введения. Конкурентное действие на связывание оказывают эндогенные субстраты, накопление которых происходит при различных заболеваниях (жирные кислоты, билирубин).
Поскольку с рецепторами взаимодействует только несвязанная форма ЛС, фармакологический эффект зависит не от общей концентрации препарата в биофазе, а от содержания свободных молекул. Это имеет практическое значение, если ЛС связано с белками крови более чем на 85%. Например, снижение связанного с белками количества ЛС с 98 до 96% приводит к увеличению концентрации в крови его свободной фракции в два раза, что, естественно, изменяет фармакологический эффект. При большем сродстве ЛС к тканевым белкам его концентрация в крови ниже, чем в тканях. Например, многие противовоспалительные препараты: диклофенак (ортофен*), фенилбутазон - имеют высокое сродство к белкам синовиальной жидкости, поэтому уже через 12 ч их в плазме крови практически нет, в то время как содержание их в воспалённом суставе остаётся высоким.
Гентамицин, тобрамицин , ампициллин взаимодействуют с белками крови незначительно и хорошо проникают в интерстициальную жидкость, в то время как клоксациллин и диклоксациллин, имеющие высокое сродство к белкам крови, плохо проходят через гистогематические барьеры. Причиной того, что асцитическая жидкость может содержать большое количество цефалоспоринов, считают высокую степень связывания этих препаратов с экстраваскулярными белками.
Проникновение ЛС через гистогематические барьеры. Гидрофильные ЛС и метаболиты не проникают через гистогематические барьеры. Липофильные вещества могут легко проходить через них, однако гликопротеин Р, локализованный на цитополазматической мембране эндотелиоцитов гистогематических барьеров, «выбрасывает» некоторые ЛС в просвет сосуда. Функционирование этого транспортёра имеет важное значение для работы ГЭБ и гематоплацентарного барьера. Однако некоторые ЛС - ингибиторы гликопротеина Р способны снижать его активность и повышать проницаемость гистогематических барьеров для ЛС-субстратов гликопротина Р. Кроме того, носительство генотипов по определённым полиморфным маркёрам гена MDR1, кодирующего гликопротеин Р, также может обусловливать повышенную проницаемость гистогематических барьеров для определённых ЛС.
1.4. МЕТАБОЛИЗМ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Метаболизм, или биологическая трансформация веществ, - общее понятие, включающее все химические изменения, происходящие с ЛС в организме. В результате метаболизма ЛС, с одной стороны, уменьшается растворимость препаратов в жирах (уменьшение липофильности) и повышается их растворимость в воде (увеличение гидрофильности), а с другой - изменяется фармакологическая активность данных веществ.
Некоторые ЛС выводятся почками в неизменённом виде. Чаще всего данные вещества представляют собой «малые молекулы», но иногда такие ЛС обладают способностью находиться в ионизированном состоянии при физиологических значениях рН. Однако большинство ЛС не имеют вышеуказанных физико-химических свойств. Фармакологически активные органические молекулы, находясь в неионизированном состоянии при физиологических значениях рН, как правило, липофильны. Данные ЛС обычно связаны с белками крови, плохо фильтруются в почечных клубочках и одновременно легко реабсорбируются в почечных канальцах. Система метаболизма (или биотрансформации) направлена на повышение растворимости молекулы ЛС (увеличение гидрофильности); это способствует выведению вещества из организма с мочой. Иными словами, липофильные ЛС превращаются в гидрофильные, а следовательно, более легко экскретируемые соединения (рис. 1-5).
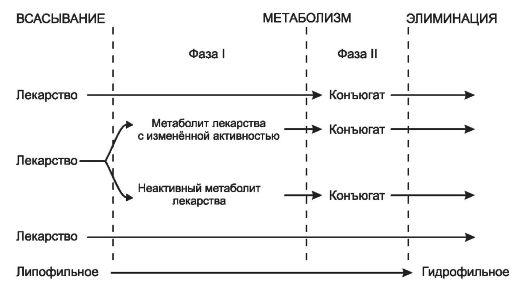 Рис. 1-5. Фазы метаболизма лекарственных средств [Katzung 1998].
Рис. 1-5. Фазы метаболизма лекарственных средств [Katzung 1998].
ПУТИ ИЗМЕНЕНИЯ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТИ ЛЕКАРСТВЕННЫХ СРЕДСТВ В РЕЗУЛЬТАТЕ МЕТАБОЛИЗМА
Фармакологически активное вещество превращается в фармакологически неактивное (процесс характерен для большинства ЛС).
Фармакологически активное вещество вначале трансформируется в другое фармакологически активное вещество (табл. 1-7), т.е. имеет активные метаболиты.
Таблица 1-7. Лекарственные средства и их метаболиты, сохраняющие фармакологическую активность
ЛС | Активный метаболит |
Аллопуринол Амитриптилин Ацетилсалициловая кислота Ацетогексамид Верапамил Глутетимид Диазепам Дилтиазем Имипрамин Кодеин Лидокаин Меперидин (тримеперидин) Миноксидил Морфин Прокаинамид Пропранолол Спиронолактон Фенилбутазон Флуразепам* Хлорохин | Аллоксантин Нортриптилин* Салициловая кислота Гидроксигексамид Норверапамил 4-Гидроксиглютетимид Оксазепам Миноксидила сульфат Дезимипрамин Морфин Моноэтилглицинксилидид Нормеперидин Миноксидила сульфат Морфин-6-глюкуронид N-ацетилпрокаинамид 4-Гидроксипропранолол Канкренон Оксифенилбутазон Дезэтилфлуразепам Гидроксихлорохин |
Неактивные лекарственные вещества, превращающиеся в организме в фармакологически активные средства, называют пролекарствами
(табл. 1-8).
Таблица 1-8. Лекарственные средства - пролекарства
Пролекарство | Активный метаболит |
Азатиоприн Квинаприл Ловастатин Набуметон* Озетамивир Периндоприл Преднизон Примидон Рамиприл Сулиндак* Трандолаприл Фенацетин*1 Хлоралгидрат Циклофосфан Эналаприл | Меркаптопурин Квинаприлат β-Гидроксикислотный метаболит 6-Метокси-2-нафтилуксусная кислота Озелтамивира карбоксилат Периндоприлат Преднизолон Фенобарбитал Рамиприлат Сулиндака сульфид Трандолаприлат Ацетаминофен* Трихлорэтанол 4-Кетоциклофосфан Эналаприлат |
1 Фенацетин в настоящее время не производят вследствие развития выраженных побочных эффектов, в частности нефротоксичности (препарат вызывает «фенацетиновый нефрит»).
Одна из главных целей создания пролекарств - улучшение фармакокинетических свойств препаратов, при этом отмечают ускорение метаболизма, а также увеличение количества всосавшегося вещества. Так были разработаны сложные эфиры ампициллина: пивампицин, талампицин и бикампицин - ЛС, в отличие от ампициллина практически полностью всасывающиеся при приёме внутрь (на 98-99%). В печени при участии ферментов (например, карбоксиэстераз) происходит гидролиз данных ЛС до ампициллина (обладает антибактериальной активностью). Ещё один пример - ингибиторы АПФ, содержащие карбоксильную группу (эналаприл, периндоприл, рамиприл, трандолаприл, квинаприл и некоторые другие). Эналаприл всасывается на 60% при приёме внутрь, превращаясь в печени под влиянием карбоксиэстераз до активного вещества - эналаприлата. Необходимо отметить, что эналаприлат при введении внутрь всасывается лишь на 10%.
Ещё одна цель создания пролекарств - увеличение избирательности действия ЛС для повышения их эффективности и безопасности. Дофамин* назначают для усиления почечного кровотока при ОПН, однако препарат вызывает ряд таких нежелательных побочных действий, как повышение артериального давления (АД), тахикардия и аритмии. Присоединение к дофамину* остатка глутаминовой кислоты позволило создать лекарственное вещество глутамил-дофа. Он гидролизуется
до дофамина* только в почках под влиянием глутамилтранспептидазы и декарбоксилазы L-ароматических аминокислот и, таким образом, практически не оказывает влияния на центральную гемодинамику.
Клиренс лекарственных веществ
Клиренс - объём плазмы крови, полностью очищаемый почками от лекарственного вещества в единицу времени. Существуют ЛС с высоким и низким печёночным клиренсом. Метаболизм большинства препаратов происходит в печени.
ЛС, метаболизирующиеся в печени, подразделяют на две подгруппы.
• ЛС, обладающие высоким печёночным клиренсом.
Для них характерна высокая степень извлечения (экстракции) из крови, обусловленная значительной активностью (ёмкостью) ферментных систем, которые отвечают за метаболизм данных веществ. Поскольку биологическая трансформация данных препаратов в печени происходит очень быстро и легко, то определяют печёночный клиренс таких ЛС величина и скорость кровотока.
• ЛС с низким печёночным клиренсом.
Печёночный клиренс зависит не от скорости печёночного кровотока, а от активности ферментов и степени связывания препарата с белками крови. Лекарственные средства, обладающие высоким печёночным клиренсом
• Алпренолол.
• Амитриптилин.
• Верапамил.
• Гидралазин.
• Дезипрамин*.
• Дигидроэрготамин*.
• Дилтиазем.
• Доксорубицин.
• Имипрамин*.
• Изопреналин.
• Изосорбида динитрат.
• Кетамин.
• Лабеталол*.
• 6-Меркаптопурин.
• Метопролол.
• Морфин.
• Налоксон.
• Налтрексон.
• Неостигмин.
• Никардипин*.
• Никотинамид.
• Нифедипин.
• Нитроглицерин.
• Папаверин.
• Пентазоцин*.
• Пентоксифиллин.
• Пропранолол.
• Скополамин.
• Тестостерон.
• Хлорпромазин.
• Цитарабин.
• Эстрадиол.
При одинаковой ёмкости ферментных систем ЛС, в значительной степени связанные с белками (например, дифенин, хинидин, толбутамид), имеют низкий клиренс по сравнению с препаратами, слабо связанными с белками (теофиллин, парацетамол*). Ёмкость ферментных систем - не постоянная величина. При увеличении дозы ЛС (вследствие насыщения ферментов) ёмкость системы уменьшается; это может привести к увеличению биодоступности вещества.
При приёме внутрь ЛС, обладающих высоким печёночным клиренсом, они всасываются в тонкой кишке и через систему воротной вены поступают в печень, где подвергаются активному метаболизму (примерно на 50-80%) ещё до поступления в системное кровообращение. Это явление называют пресистемной элиминацией, или эффектом «первого прохождения» («first-pass effect»). В результате (при приёме внутрь) данные ЛС имеют низкую биодоступность, однако их абсорбция может составлять почти 100%. Эффект «первого прохождения» наблюдают у таких ЛС, как хлорпромазин, ацетилсалициловая кислота и верапамил, гидралазин и изопреналин, имипрамин* и кортизон. Лабеталол*, лидокаин и морфин, метопролол, метилтестостерон, метоклопрамид и нортриптилин*, окспренолол* и органические нитраты, пропранолол, резерпин и салициламид, этмозин* и некоторые другие лекарства также активно метаболизируются до поступления в системный кровоток. Следует отметить, что незначительный метаболизм ЛС может проходить и в других органах (например, в просвете и стенке кишечника, в лёгких, плазме крови и почках).
Фазы метаболизма лекарственных средств
В целом, все реакции биологической трансформации ЛС относят к одной из двух категорий, их обозначают как фазы метаболизма I и II (см. рис. 1-5).
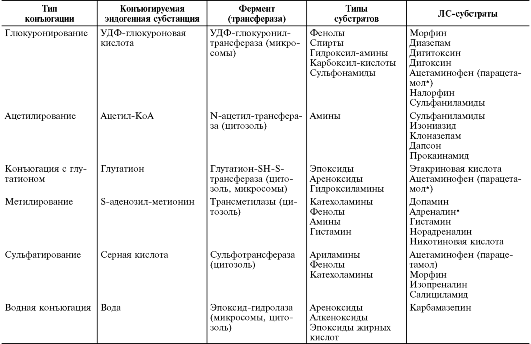
• Реакции I фазы (несинтетические реакции). В процессе несинтетических реакций ЛС трансформируются в более полярные и лучше растворимые в воде (гидрофильные) соединения по сравнению с исходным веществом. Процесс происходит за счёт присоединения или освобождения таких активных функциональных групп, как, например, гидроксильная (-ОН), сульфгидрильная (-SH) или аминогруппа (-NH2). Основные реакции I фазы - реакции окисления. Наиболее распространённой реакцией окисления, происходящей в организме, считают реакцию гидроксилирования - присоединения гидроксильного радикала (-ОН). Таким образом, можно считать, что в I фазе метаболизма происходит «взлом» молекулы ЛС (табл. 1-9). Катализаторы данных реакций - ферменты-оксидазы со смешанной функцией. Субстратная специфичность этих ферментов очень мала, поэтому они окисляют различные ЛС. К другим, происходящим значительно реже, реакциям I фазы относят процессы восстановления и гидролиза.
• Реакции II фазы (синтетические реакции). В результате реакций II фазы метаболизма или синтетических реакций происходит соединение (конъюгация) ЛС или его метаболитов с эндогенными веществами; при этом образуются полярные, хорошо растворимые в воде конъюгаты, легко выводимые через почки или с жёл- чью. Для вступления в синтетическую реакцию молекула должна обладать химически активным радикалом (группировкой), к которому может присоединиться конъюгирующая молекула. Если активные радикалы присутствуют в молекуле лекарственного вещества изначально, то реакция соединения может осуществляться, минуя превращения I фазы. В противном случае молекула ЛС приобретает активные радикалы в ходе реакций присоединения, происходящих в течение I фазы (табл. 1-10).
ЛС в процессе метаболизма могут превращаться только за счёт реакций I фазы либо исключительно при вступлении в реакции II фазы. Иногда часть лекарственного вещества трансформируется путём реакций I фазы, а другая его часть - в процессе реакций II фазы. Кроме того, существует возможность последовательного превращения ЛС при прохождении реакций I и II фазы.
Микросомальная система оксидаз со смешанной функцией
Многие ферменты, осуществляющие реакции превращения ЛС, располагаются на мембранах эндоплазматического ретикулума (ЭПР) печени и других тканей. При изоляции мембран ЭПР путём гомогенизации и фракционирования клетки они преобразуются в везику-
Таблица 1-9. Реакции I фазы (по B. Katzung,1998; с дополнениями)
 Таблица 1-10. Реакции II фазы (по В. Katzung, 1998; с дополнениями)
Таблица 1-10. Реакции II фазы (по В. Katzung, 1998; с дополнениями)
 лы,
называемые микросомами. Микросомы сохраняют большинство морфологических
и функциональных характеристик интактных мембран ЭПР, включая
шероховатость или гладкость поверхности. Микросомы, образованные из
шероховатой (рибосомальной) или гладкой (нерибосомальной)
эндоплазматической сети, обладают свойствами ЭПР-предшественника.
Основная функция шероховатых микросом - синтез белка. Гладкие микросомы
содержат относительно большое количество ферментов, ответственных за
окислительный метаболизм ЛС. В частности, в гладких микросомах
обнаруживают особый класс ферментов, называемых оксидазами. Эти ферменты
- цитохромы Р-450 - осуществляют различные (смешанные) функции.
лы,
называемые микросомами. Микросомы сохраняют большинство морфологических
и функциональных характеристик интактных мембран ЭПР, включая
шероховатость или гладкость поверхности. Микросомы, образованные из
шероховатой (рибосомальной) или гладкой (нерибосомальной)
эндоплазматической сети, обладают свойствами ЭПР-предшественника.
Основная функция шероховатых микросом - синтез белка. Гладкие микросомы
содержат относительно большое количество ферментов, ответственных за
окислительный метаболизм ЛС. В частности, в гладких микросомах
обнаруживают особый класс ферментов, называемых оксидазами. Эти ферменты
- цитохромы Р-450 - осуществляют различные (смешанные) функции.
Активность оксидаз требует присутствия как восстанавливающего агента (НАДФ-Н), так и молекулярного кислорода. При типичной реакции на молекулу субстрата расходуется (восстанавливается) одна молекула кислорода; при этом один кислородный атом включается в продукт реакции, а другой образует молекулу воды.
В окислительно-восстановительном процессе ключевую роль выполняют два микросомальных фермента - цитохром С-редуктаза и цитохром Р-450.
• Флавопротеин НАДФ-Н-цитохром Р-450-редуктаза. Один моль этого фермента содержит по одному молю флавинмононуклеотида (ФМН) и флавинадениндинуклеотида (ФАД). Поскольку цитохром С может служить акцептором электрона, данный фермент часто обозначают как НАДФ-цитохром С-редуктаза.
• Гемопротеин или цитохром Р-450 выполняет функцию конечной оксидазы. В действительности микросомальная мембрана содержит множество форм гемопротеина, и такая множественность возрастает при повторном введении ксенобиотиков. Относительное изобилие цитохрома Р-450 по сравнению с редуктазой печени делает реакцию восстановления гема, содержащегося в цитохроме Р-450, лимитирующей стадией в процессе окисления ЛС в печени.
Процесс микросомального окисления ЛС требует участия цитохрома Р-450, цитохрома Р-450-редуктазы, НАДФ-Н и присутствия молекулярного кислорода. Упрощённая схема окислительного цикла представлена на рис. 1-6. Окисленный (с помощью Fe3+) цитохром Р-450 соединяется с ЛС, при этом образуется бинарный комплекс. НАДФ- Н - донор электрона для флавопротеинредуктазы, которая, в свою очередь, восстанавливает окисленный комплекс цитохром Р-450-ле- карство. Второй электрон (переходит от НАДФ-Н через ту же флавопротеинредуктазу, восстанавливающую молекулярный кислород) формирует комплекс, состоящий из «активированного кислорода»,
цитохрома Р-450 и субстрата. Этот комплекс транспортирует «активированный кислород» к лекарственному субстрату с образованием окисленного продукта.
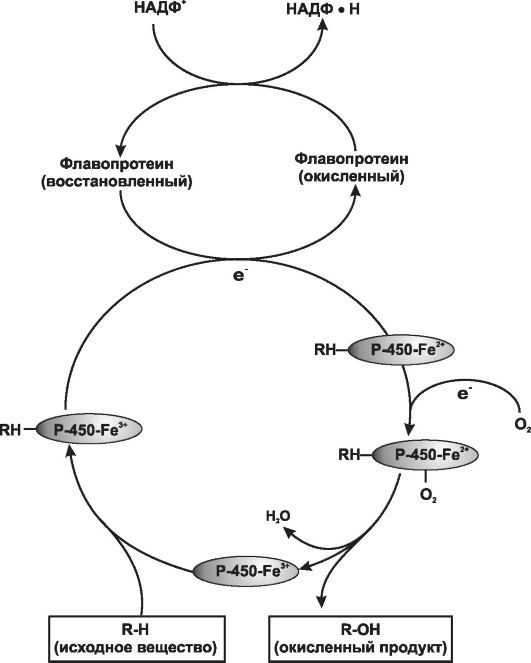 Рис. 1-6. Схема функционирования системы оксидаз со смешанной функцией (по B. Katzung, 1998).
Рис. 1-6. Схема функционирования системы оксидаз со смешанной функцией (по B. Katzung, 1998).
ЦИТОХРОМ Р-450
Цитохром Р-450 (CYP) представляет группу ферментов, осуществляющих не только метаболизм ЛС и других ксенобиотиков, но и участвующих в синтезе глюкокортикостероидных гормонов, холестерина, жёлчных кислот, простаноидов (тромбоксан А2 и простациклин). Как показали филогенетические исследования, цитохромы Р- 450 появились в живых организмах более трёх миллиардов лет назад. Цитохром Р-450 - гемопротеин - белок, содержащий гем. Название цитохрома Р-450 связано с особыми свойствами данного гемопротеина. В восстановленной форме белок связывает монооксид углерода, при этом происходит образование комплекса с максимальным поглощением света при длине волны 450 нм. Гем цитохрома Р-450 содержит железо, связанное не только с атомами азота четырёх лигандов (образуют порфириновое кольцо), но также существуют пятый и шестой лиганды, расположенные сверху и снизу кольца гема. Эти лиганды представляют собой атомы азота гистидина и серы цистеина, входящих в состав полипептидной цепи белковой части цитохрома Р-450. Данный факт объясняет наличие особых свойств у гемопротеина.
Наибольшее количество цитохрома Р-450 определяют в гепатоцитах. Однако цитохром Р-450 обнаруживают и в других органах, например в кишечнике и почках, лёгких и надпочечниках, головном мозге и коже, а также в плаценте и миокарде. Важнейшим свойством цитохрома Р-450 считают его способность подвергать биологической трансформации практически все известные химические соединения. Метаболизм, обусловленный действием гемопротеина, происходит, как правило, посредством реакции гидроксилирования. Цитохромы Р-450 включают один атом кислорода в субстрат (окисляя его), а другой - в молекулу воды, поэтому данные ферменты называют также монооксигеназами.
Цитохром Р-450 имеет множество изоферментов. На данный момент выделено более 1000 изоформ гемопротеина. Изоферменты цитохрома Р-450 по идентичности аминокислотного состава разделяют на семейства (Nebert, 1987). Каждое семейство, в свою очередь, содержит подсемейства. Изоферменты цитохрома Р-450, обладающие довольно высоким сходством аминокислотного состава (более 40%), объединяют в семейства (существует семнадцать семейств). Изоферменты цитохрома Р-450 объединяют в подсемейства, если их состав идентичен на 55% и более. В настоящее время выделяют 39 подсемейств. Семейства гемопротеинов принято обозначать римскими цифрами, подсемейства - латинскими буквами. Пример отнесения к классификации отдельного изофермента: арабская цифра (расположена на первом месте) обозначает семейство, латинская буква - подсемейство;
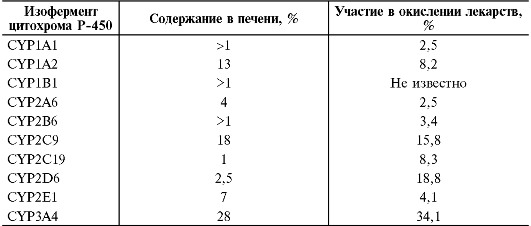
в конце указывают арабскую цифру, соответствующую изоферменту. Изоферменты цитохрома Р-450 - представители различных семейств и подсемейств; их отличают субстратная специфичность и регуляторы активности (ингибиторы и индукторы). В метаболизме ЛС принимают участие изоферменты I, II и III семейств. Изоферменты CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP3A4, - наиболее важные и хорошо изученные изоформы цитохрома Р-450, участвующие в метаболизме ЛС. Информация о содержании различных изоферментов цитохрома Р-450 в печени человека, а также об их влиянии на окисление ЛС представлена в табл. 1-11.
Таблица 1-11. Содержание изоферментов цитохрома Р-450 в печени человека и их участие в окислении лекарственных средств [Lewis et al., 1999]
 Субстратная
специфичность определённых изоферментов цитохрома Р-450 позволила
разработать методы их фенотипирования. При этом активность различных
ферментов метаболизма исследуют по фармакокинетике «маркёрного»
субстрата путём измерения концентрации неизменённого вещества и
содержания его метаболита в сыворотке или плазме крови (табл. 1-12).
Субстратная
специфичность определённых изоферментов цитохрома Р-450 позволила
разработать методы их фенотипирования. При этом активность различных
ферментов метаболизма исследуют по фармакокинетике «маркёрного»
субстрата путём измерения концентрации неизменённого вещества и
содержания его метаболита в сыворотке или плазме крови (табл. 1-12).
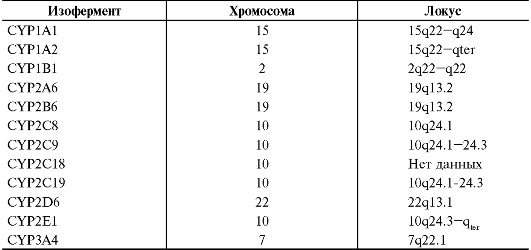
Каждый изофермент цитохрома Р-450 кодируется определённым геном. Гены изоферментов цитохрома Р-450 локализованы в разных хромосомах и занимают в них разные локусы. Расположение генов изоферментов цитохрома Р-450, участвующих в метаболизме ЛС, представлено в табл. 1-13. В настоящее время известны и изучены 53 гена изоферментов цитохрома Р-450. Определение изоформ цитохрома Р- 450 путём идентификации генов соответствующих изоферментов с помощью полимеразной цепной реакции (ПЦР) называют генотипированием изоферментов цитохрома Р-450 (см. главу 7 «Клиническая фармакогенетика»).
Таблица 1-12. Лекарственные средства, используемые в качестве «маркерных» субстратов для фенотипирования некоторых изоферментов цитохрома Р-450
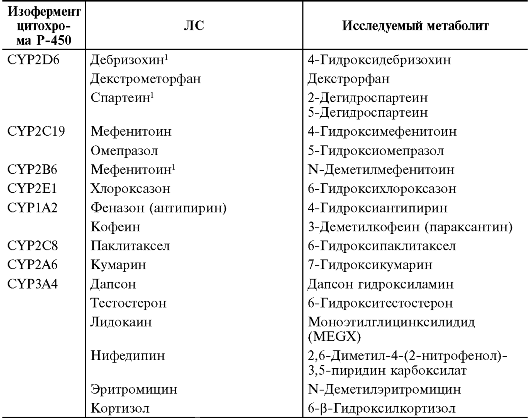 1 В России не зарегистрирован.
1 В России не зарегистрирован.
Таблица 1-13. Локализация генов изоферментов цитохрома Р-450, участвующих в метаболизме лекарственных средств
 Ферменты,
осуществляющие реакции I и II фаз метаболизма обладают генетическим
полиморфизмом (см. главу 7 «Клиническая фармакогенетика»), а также
способностью к индукции или ингибированию под действием различных
веществ (см. главу 5 «Взаимодействие лекарственных средств»).
Ферменты,
осуществляющие реакции I и II фаз метаболизма обладают генетическим
полиморфизмом (см. главу 7 «Клиническая фармакогенетика»), а также
способностью к индукции или ингибированию под действием различных
веществ (см. главу 5 «Взаимодействие лекарственных средств»).
Главные изоферменты цитохрома P-450, влияющие на биологическую трансформацию лекарственных веществ
Семейство CYPI
CYPI - семейство изоферментов цитохрома Р-450. Эндогенные субстраты для изоферментов семейства CYPI не определены до сих пор. Ферменты данного семейства метаболизируют ксенобиотики и полициклические ароматические углеводороды (ПАУ) - основные компоненты табачного дыма (также входят в состав продуктов сжигания органического топлива). Отличительная особенность изоферментов семейства CYPI - их способность индуцироваться под действием ПAУ, в том числе диоксина и 2,3,7,8-тетрахлордибензо-р-диоксина. Поэтому семейство CYPI в литературе называют цитохромом, индуцибельным ПAУ, диоксининдуцибельным цитохромом или TCDD-ин- дуцибельным цитохромом. В организме человека присутствуют изоферменты семейства CYPI, входящие в состав подсемейств IA и IB. К подсемейству IA относят изоферменты CYP1A1 и CYP1A2. CYPB1 - единственный представитель подсемейства CYPIB, обнаруженный у человека.
• CYP1A1 (IA1) - изофермент цитохрома Р-450, содержащийся в основном в лёгких, в меньшей степени - в лимфоцитах и плаценте. CYP1A1 не участвует в метаболизме однако в лёгких фермент активно участвует в биологической трансформации ПAУ, при этом некоторые ПАУ, например бензпирен или нитрозамины, превращаются в канцерогенные соединения, способные спровоцировать развитие злокачественных новообразований, в первую очередь рака лёгких. Такой процесс получил название биологической активации канцерогенов. ПAУ индуцируют CYP1A1 так же, как и другие изоферменты семейства CYPI. Механизм индукции CYP1A1 при действии ПАУ заключается в следующем: полициклические ароматические углеводороды, проникая в клетку, соединяются с Ah-рецептором (белок - регулятор транскрипции). Образовавшийся комплекс (ПAУ-Ah-рецептор) проникает в ядро с помощью другого белка (ARNT) и стимулирует экспрессию гена CYP1A1, связываясь со специфическим диоксинчувствительным участком (сайтом) гена. Таким образом, у курящих людей индукция CYP1A1 происходит наиболее
интенсивно, вызывая биологическую активацию канцерогенов. Именно этим объясняют высокий риск развития рака лёгких у курильщиков.
• CYP1A2 (1A2) - изофермент цитохрома Р-450 (обнаруживают преимущественно в печени). В отличие от CYP1A1, CYP1A2 метаболизирует не только ПАУ, но и некоторые лекарственные вещества (прил. 1.3). В качестве «маркёрных субстратов» для фенотипирования CYP1A2 используют фенацетин*, кофеин и антипирин*. При этом фенацетин* подвергают О-деметилированию, кофеин - 3-деметилированию, а антипирин* - 4-гидро- ксилированию.
Оценка клиренса кофеина - один из основных диагностических тестов, выполняемых для определения функционального состояния печени. Главный метаболизирующий фермент кофеина - CYP1A2, следовательно, при проведении теста изучают активность данного изофермента. Пациенту предлагают принять внутрь препарат кофеина, меченный радиоактивным изотопом углерода (C13-кофеин). Затем выдыхаемый пациентом в течение часа воздух собирают в мешок (используют специальный прибор) и анализируют. По соотношению объёмов радиоактивного и меченого углекислого газа (13CО2/12CО2) в выдыхаемом воздухе (измеряют с помощью масс-спектроскопии) определяют клиренс кофеина. Cуществует модификация теста: методом жидкостной высокоэффективной хроматографии исследуют концентрацию кофеина и его метаболитов в плазме крови, моче и слюне, взятых натощак. В этом случае определённый вклад в метаболизм кофеина вносят CYP3A4 и CYP2D6. Оценка клиренса кофеина - надёж- ный тест, используемый для диагностики функционального состояния печени, особенно при тяжёлых заболеваниях (например, цирроз печени). Однако при умеренном поражении печени, нередко получают ложноположительные результаты (метод недостаточно чувствителен). На результат теста влияют курение (индукция CYP1A2); возраст; совместное применение изменяющих активность изоферментов цитохрома Р-450 (его ингибиторов или индукторов).
Cедует отметить, что CYP1A2 - главный фермент, осуществляющий биологическую трансформацию теофиллина (рис. 1-7).
Семейство CYPII
CYPIIA - группа изоферментов, входящих в состав подсемейства цитохрома Р-450. Из всех известных изоферментов подсемейства CYPIIA наиболее важную роль в метаболизме ЛC играет изофермент CYP2A6 цитохрома Р-450. Cпособность к индукции под действием фенобарбитала - общее свойство изоферментов подсемейства
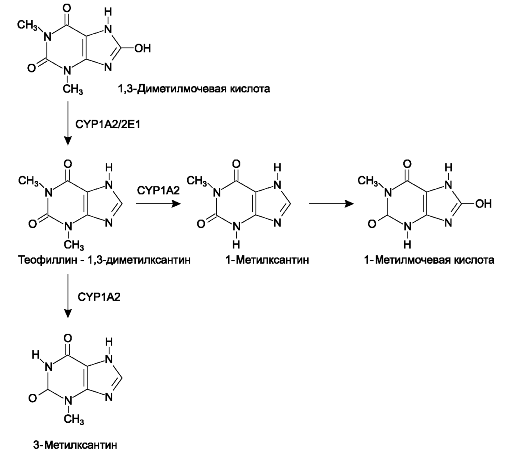 Рис. 1-7. Метаболизм теофиллина.
Рис. 1-7. Метаболизм теофиллина.
CYPIIA (их также называют фенобарбиталиндуцибельными цитохромами).
• IIА6 (CYP2A6) - изофермент цитохрома Р-450, обнаруживаемый, как правило, в печени. CYP2A6 метаболизирует небольшое число ЛС. Изофермент индуцирует превращение никотина в кетинин, 7-гидроксилирование кумарина, 7-гидроксилирование циклофосфана, а также вносит определённый «вклад» в биологическую трансформацию ритонавира (прил. 1.3). CYP2A6 принимает участие в биологической активации компонентов табачного дыма - нитрозоаминов - канцерогенов, вызывающих рак лёгких. CYP2A6 способствует активации 6-амино(х)ризена и 2-амино-3-метилмидазо(4,5-f)кванолина (мощные мутагены).
CYPIIC - подсемейство цитохрома Р-450. Из всех изоферментов подсемейства CYPIIC наиболее важную роль в метаболизме ЛC отводят изоферментам CYP2C9 и CYP2C19. Общее свойство цитохромов подсемейства CYPIIC - 4-гидроксилазная активность в отношении мефенитоина1 (противосудорожное ЛC) - маркёрного субстрата изоферментов подсемейства CYPIIC. Поэтому изоферменты подсемейства CYPIIC называют мефенитоин-4-гидроксилазами. CYP2C9 - изофермент цитохрома Р-450, содержащийся в основном в печени.
• CYP2C9 метаболизирует некоторые лекарственные вещества, в том числе НПВС фенитоин и S-варфарин.
• Изофермент 2C19 (CYP2C19) цитохрома P-450 также принимает участие в метаболизме ЛC (прил. 1.3).
Изоформы CYP2C9 и CYP2C19 обладают генетическим полиморфизмом (см. главу 7 «Клиническая фармакогенетика»).
CYPIID (IID) - ещё одно подсемейство цитохрома Р-450. В состав подсемейства CYPIID входит один изофермент - CYP2D6.
• IID6 (CYP2D6) - изофермент цитохрома Р-450, локализующийся преимущественно в печени. CYP2D6 метаболизирует около 20% всех известных ЛС Изофермент обеспечивает биологическую трансформацию нейролептиков, антидепрессантов, транквилизаторов, β-адреноблокаторов (прил. 1.3). Дебризохин, декстрометорфан и спартеин - маркёрные субстраты, используемые для фенотипирования цитохрома CYP2D6. В отличие от других изоферментов цитохрома Р-450, индукторов CYP2D6 не существует. Изофермент обладает генетическим полиморфизмом (см. главу 7 «Клиническая фармакогенетика»).
Подсемейство CYPIIE цитохрома Р-450. Из изоферментов подсемейства IIE наиболее важное (для метаболизма значение имеет изофермент CYP2E1. Cпособность к индукции под влиянием этанола - общее свойство изоферментов, входящих в состав подсемейства CYPIIE (их также называют этанолиндуцибельными цитохромами).
• CYP2E1 - изофермент цитохрома Р-450 - обнаруживают в печени, причём у взрослых людей количество CYP2E1 составляет примерно 7% всех изоферментов цитохрома Р-450. Cубстраты CYP2E1 - ЛC (прил. 1.3) и некоторые ксенобиотики: этанол, нитрозоамины; «небольшие» ароматические углеводороды (например, бензол и анилин); алифатические хлоруглеводороды. CYP2E1 катализирует превращение дапсона в гидроксиламиндапсон, N1-деметилирование и N7-деметилирование кофеина,
Препарат в России не зарегистрирован.
дегалогенизацию хлорфторуглеводородов и средств, используемых для ингаляционного наркоза (галотан), а также ряд других реакций. CYP2E1 и CYP1A2 совместно катализируют реакцию превращения ацетаминофена (парацетамола*) в N-ацетилбензо- хинонимин, оказывающий мощное гепатотоксическое действие. Есть данные об участии цитохрома CYP2E1 в атерогенезе. Как показали многочисленные исследования, CYP2E1 - наиболее важный изофермент цитохрома Р-450, окисляющий липопротеиды низкой плотности (ЛПНП). В процессе окисления ЛПНП принимают участие и другие изоферменты цитохрома Р-450, а также 15-липооксигеназа и НAДФ-Н-оксидазы. Продукты окисления ЛПНП - 7α-гидроксихолестерол, 7β-гидроксихолестерол, 5β-6β-эпоксихолестерол, 5α6β-эпоксихолестерол, 7-кетохолесте- рол, 26-гидроксихолестерол. Процесс окисления ЛПНП происходит в эндотелиоцитах, гладкой мускулатуре кровеносных сосудов и макрофагах. Окисленные ЛПНП стимулируют образование пенистых клеток, способствуя формированию атеросклеротических бляшек.
Семейство CYPIII
• CYPIIIA - подсемейство цитохрома Р-450. В состав подсемейства IIIA цитохрома Р-450 входят четыре изофермента: CYP3A3, CYP3A4, CYP3A5 и CYP3A7. Цитохромы подсемейства IIIA составляют 30% всех изоферментов цитохрома Р-450, содержащихся в печени, и 70% всех изоферментов стенки ЖКТ. При этом в печени, как правило, обнаруживают цитохризофермент CYP3A4, в стенке желудка и кишечника - изоферменты CYP3A3 и CYP3A5. Изофермент CYP3A7 содержится только в печени плода. Из изоферментов данного подсемейства наиболее важную роль в метаболизме ЛC отводят CYP3A4. CYP3A4 - изофермент, участвующий в биологической трансформации более чем 60% всех известных лекарственных веществ (прил. 1.3), в том числе блокаторов медленных кальциевых каналов, макролидных антибиотиков, некоторых антиаритмических препаратов. CYP3A4 катализирует реакцию 6β-гидроксилирования эндогенных глюкокортикостероидов (например, тестостерона, прогестерона и кортизола). Дапсон, эритромицин, нифедипин, лидокаин, тестостерон и кортизол - маркёрные субстраты, применяемые для определения активности CYP3A4. Метаболизм лидокаина протекает в гепатоцитах, где происходят оксидативное N-деэтилирование CYP3A4 и синтез моноэтилглицинксилидида.
Определение активности CYP3A4 по моноэтилглицинксилидиду - метаболиту лидокаина считают наиболее чувствительным и специ-
фичным тестом, характеризующим функциональное состояние печени при острых и хронических её заболеваниях, а также при синдроме системного воспалительного ответа (сепсисе). При циррозе печени от концентрации моноэтилглицинксилидида напрямую зависят прогноз и исход заболевания.
Cуществуют данные о внутривидовой вариабельности метаболизма ЛC происходящего при участии CYP3A4. Однако в последнее время причиной этого феномена считают не существование генетического полиморфизма CYP3A4, а нарушение экспрессии факторов транскрипции гена CYP3A4.
Связь реакций первой и второй фаз метаболизма
Реакции II фазы метаболизма или синтетические реакции представляют соединение (конъюгацию) ЛC или его метаболитов с эндогенными веществами - в результате происходит образование полярных, хорошо растворимых в воде комплексов, легко выводимых через почки или с жёлчью. Для вступления в реакцию второй фазы молекула должна содержать химически активный радикал (группировку) - область присоединения конъюгирующей молекулы. Иногда активные радикалы присутствуют в молекуле ЛC изначально; в этом случае соединение с эндогенным веществом осуществляется, минуя реакции первой фазы. Нередко молекулы ЛC присоединяют активные радикалы в процессе реакций I фазы. Глюкуронирование, ацетилирование, метилирование, сульфатирование и водная конъюгация - наиболее распространённые реакции II фазы метаболизма. Тип реакции конъюгации зависит прежде всего от химической структуры субстрата (лекарственного вещества). Так, глюкуронированию подвергаются соединения, содержащие гидроксильные, карбоксильные, карбамоильные, тиоловые, карбонильные группы, а также нитрогруппы. Cубстраты ацетилирования - ЛC и метаболиты, содержащие нитрогруппу. Реакцию сульфатирования, как правило, наблюдают у соединений, обладающих фенольной структурой. В большинстве случаев в результате реакций второй фазы ксенобиотики полностью утрачивают биологическую активность. Однако иногда отмечают образование активных метаболитов и канцерогенов в процессе реакций конъюгации. Таким образом, реакции первой и второй фазы - отдельные этапы единого процесса метаболизма (биологической трансформации) ксенобиотиков.
Cвязь и взаимодействие реакций различных фаз биологической трансформации на примере метаболизма ацетаминофена (парацетамола*) и изониазида.
Метаболизм ацетаминофена
Aцетаминофен (парацетамол*) - широко используемое антипиретическое ЛС Как показали исследования, в результате реакции соединения ацетаминофена (примерно 80-90%) с глюкуроновой (глюкуронирование) и серной (сульфатирование) кислотами происходит формирование конъюгированных метаболитов, нетоксичных и легко выводимых почками. Небольшое количество ацетаминофена (10-17%) подвергается окислению посредством CYP2E1 и CYP1A2 с образованием N-ацетилбензохинонимина (рис. 1-8), который, соединяясь с глутатионом, превращается в неактивное соединение (выделение происходит через почки). При увеличении дозы ацетаминофена возрастает количество N-ацетилбензохинонимина, возникает дефицит глутатиона, а N-ацетилбензохинонимин оказывает гепатотоксический эффект. Соединение N-ацетилбензохинонимина с нуклеофильными группами белков гепатоцитов приводит к развитию некроза печёночной ткани. Исследование механизма гепатотоксического действия ацетаминофена помогло разработать эффективный метод (а также внедрить его в клиническую практику) лечения интоксикации данным препаратом. Метод заключается в применении N-ацетилцистеина (восполняет запасы глутатиона в печени). Положительный эффект отмечают уже
 Рис. 1-8. Метаболизм ацетаминофена (парацетамола).
Рис. 1-8. Метаболизм ацетаминофена (парацетамола).
через 10-12 ч после первого применения ацетилцистеина. Гепатотоксическое действие ацетаминофена усиливается на фоне хронического злоупотребления алкоголем: с одной стороны, этанол истощает запасы глутатиона в печени, а с другой, провоцирует индукцию CYP2E1. Метаболизм изониазида
Как известно, изониазид - противотуберкулёзное ЛС, входящее в состав большинства современных схем комбинированной химиотерапии заболевания. CYP2E1 окисляет примерно 60-70% изониазида (в течение реакции первой фазы). В результате реакции окисления образуется нетоксичный и неактивный метаболит - изоникотиновая кислота (легко выводится почками). Остальная часть поступившего в организм изониазида (30-40%) вступает в реакцию ацетилирования (реакция второй фазы). Продукт реакции - ацетилизониазид (почки частично экскретируют вещество). Небольшое количество ацетилизониазида вследствие амидного гидролиза (реакция I фазы) преобразуется в ацетилгидразин. Aцетилгидразин - токсичное соединение, приводящее к формированию тяжёлых нарушений печени (оказывает гепатотоксическое действие).
Глюкуронирование
Глюкуронирование - одна из основных реакций второй фазы метаболизма ЛС. Глюкуронирование - присоединение (конъюгация) УДФ-глюкуроновой кислоты к субстрату. Надсемейство ферментов, называемых УДФ-глюкуронилтрансферазами, катализирует данную реакцию. В состав надсемейства УДФ-глюкуронилтрансфераз входят два семейства и более двадцати изоферментов, локализованных в эндоплазматической системе клеток (табл. 1-14). Они ускоряют глюкуронирование ксенобиотиков (включая ЛС и их метаболиты), пестицидов и канцерогенов. К соединениям, подвергающимся глюкуронированию, относят простые и сложные эфиры; вещества, содержащие карбоксильные, карбамоильные, тиольные и карбонильные группы, а также нитрогруппы. Глюкуронирование приводит к увеличению полярности химических соединений, облегчает их растворимость в воде и элиминацию из организма. УДФ-глюкуронилтрансферазы обнаруживают у всех позвоночных животных (рептилий, амфибий и млекопитающих). У новорождённых отмечают сниженную активность ферментов. Однако в течение первых трёх месяцев жизни происходит увеличение активности УДФ-глюкуронилтрансфераз до значений, соответствующих активности фермента у взрослых.
Таблица 1-14. Состав семейств УДФ-глюкуронилтрансферазы человека, локализация генов и маркерные субстраты изоферментов
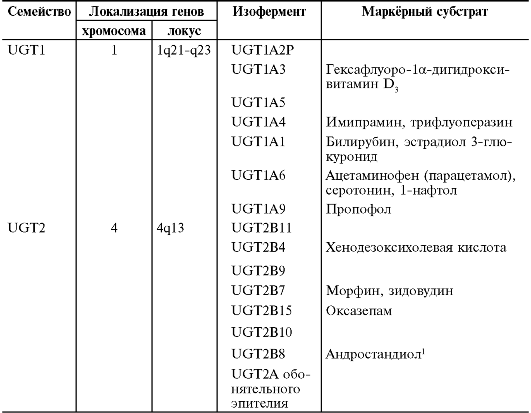 1Препарат снят с производства.
1Препарат снят с производства.
УДФ-глюкуронилтрансферазы обнаруживают в печени, кишечнике, лёгких, головном мозге, обонятельном эпителии и почках. Однако реакция соединения с глюкуроновой кислотой происходит главным образом в печени. Как показали исследования, экспрессия изоферментов УДФ-глюкуронилтрансферазы в различных органах неодинакова. Так, образование основного количества изофермента УДФ-глюкуронилтрансферазы, катализирующего реакцию глюкуронирования билирубина, происходит в печени; в других органах, например в почках, - значительно меньше. Экспрессия изоферментов УДФ-глюкуронилтрансферазы, ускоряющих соединение фенола и глюкуроновой кислоты, одинакова и в печени, и в почках. По идентичности аминокислотного состава надсемейство УДФ-глюкуронилтрансфераз подразделяют на два семейства - UGTI и UGTII. Сходство аминокислотного состава изоферментов семейства UGTI составляет 62-80%, а изоферментов семейства UGTII - 57-93%. Изоферменты, входящие в состав семейств УДФ-глюкуронилтрансферазы человека, а также ло-
кализация генов и маркёрные субстраты изоферментов, применяемые для фенотипирования, представлены в табл. 1-15.
Физиологическая функция УДФ-глюкуронилтрансфераз - глюкуронирование эндогенных соединений. Билирубин - продукт, образующийся в процессе катаболизма гема, - наиболее хорошо изученный эндогенный субстрат УДФ-глюкуронилтрансферазы. Соединение билирубина с глюкуроновой кислотой предотвращает накопление токсичного свободного билирубина. При этом билирубин выделяется с жёлчью в виде моноглюкуронидов и диглюкуронидов.
Участие в метаболизме гормонов - ещё одна важная физиологическая функция ферментов. Так, глюкуронирование тироксина и трийодтиронина происходит в печени, затем гормоны (глюкурониды) выводятся с жёлчью. УДФ-глюкуронилтрансферазы также участвуют в метаболизме глюкокортикостероидных гормонов, жёлчных кислот, ретиноидов, однако в настоящее время эти реакции изучены недостаточно.
Среди ЛС, подвергающихся глюкуронированию, присутствуют вещества, относящиеся к разным классам (многие из них, например, морфин и хлорамфеникол, имеют узкий терапевтический диапазон).
Таблица 1-15. Лекарственные средства и ксенобиотики, подвергающиеся глюкуронированию различными изоферментами УДФ-глюкуронилтрансферазы
 Представители различных химических групп, подвергающиеся глюкуронированию:
Представители различных химических групп, подвергающиеся глюкуронированию:
• фенолы:
- пропофол,
- ацетаминофен,
- налоксон;
• спирты:
- хлорамфеникол,
- кодеин,
- оксазепам;
• алифатические амины:
- ламотриджин,
- амитриптилин;
• карбоновые кислоты:
- фенилбутазон,
- сульфинпиразон* ;
• карбоксильные кислоты:
- напроксен,
- кетопрофен.
Таким образом, в реакциях глюкуронирования участвуют соединения, содержащие разные функциональные группы, - акцепторы для УДФ-глюкуроновой кислоты. B процессе реакции образуются полярные неактивные метаболиты, легко выводящиеся из организма. Однако при глюкуронировании морфина происходит его трансформация в активный метаболит - морфин-6-глюкуронид. Это практически единственный пример образования активного вещества в результате глюкуронирования. Морфин-6-глюкуронид оказывает значительный анальгетический эффект и обладает менее выраженными побочными действиями (реже, чем морфин, вызывает тошноту и рвоту). Нередко глюкуронирование приводит к биологической активации канцерогенов. К канцерогенным глюкуронидам относят N-глюкуронид 4-ами- нобифенила, N-глюкуронид N-ацетилбензидина, О-глюкуронид 4- [(гидрокисметил) нитрозоамино]-1-(3-пиридил)-1-бутанона.
Ацетилирование
Aцетилирование - один из самых ранних механизмов адаптации организмов в процессе эволюции. Данная реакция необходима для синтеза жирных кислот, глюкокортикостероидов, а также для полноценного функционирования цикла Кребса. Метаболизм или биологическая трансформация ксенобиотиков (ЛС, бытовых и промышленных ядов) - главная функция ацетилирования. Aцетилирование протекает при участии особого фермента - N-ацетилтрансферазы и кофермен-
та A. Контроль интенсивности ацетилирования в организме человека происходит при участии β2-адренорецепторов, а также за счёт метаболических резервов (пантотеновая кислота, пиридоксин, тиамин, липоевая кислота) и особенностей генотипа. Кроме того, ацетилирование зависит от функционального состояния печени и других органов, содержащих N-ацетилтрансферазу. Однако, как показали исследования, заболевания печени практически не влияют на интенсивность и качество ацетилирования (как и других реакций второй фазы). Между тем, ацетилирование ЛС и других ксенобиотиков происходит преимущественно в печени. Bыделяют два изофермента N-ацетилтрансфера- зы (NAT) - NAT1 и NAT2. NAT1 ацетилирует небольшое количество ариламинов и не обладает генетическим полиморфизмом. Таким образом, NAT2 - основной фермент реакции ацетилирования. Локализация гена NAT2: 8-я хромосома, локус 8р23.1-р21.3. NAT2 ацетилирует некоторые ЛС, например изониазид и сульфанилнамиды (табл. 1-16).
Таблица 1-16. Лекарственные средства, подвергающиеся ацетилированию
 * ПАБК - парааминобензойная кислота.
* ПАБК - парааминобензойная кислота.
Генетический полиморфизм (см. главу 7 «Клиническая фармакогенетика») - основное свойство NAT2.
Значение кишечника для метаболизма лекарственных средств
Кишечник - важный для биологической трансформации ЛС орган (второй по значимости после печени). B стенке кишечника протекают реакции как I фазы, так и II фазы метаболизма. Биологическая трансформация ЛС в стенке кишечника имеет значение при рассмотрении эффекта первого прохождения (пресистемного метаболизма). Уже доказана значительная роль метаболизма в стенке кишечника при «первом прохождении» таких ЛС, как циклоспорин A , нифедипин, мидазолам и верапамил. B основном в стенке кишечника обнаруживают изоферменты цитохрома Р-450 (ферменты первой фазы метаболизма). Среднее содержание цитохрома Р-450 в стенке кишечника человека составляет 20 пмоль на 1 мг микросомального белка (в печени - 300 пмоль на 1 мг микросомального белка). При прове-
дении исследований установили чёткую закономерность: по мере продвижения по кишечнику (от проксимальных отделов к дистальным) содержание изоферментов цитохрома Р-450 уменьшается. Кроме того, максимальное количество изоферментов цитохрома Р-450 определяют на вершине ворсинок кишечника, а минимальное - в криптах. CYP3A4 - основной изофермент цитохрома Р-450, обнаруживаемый в кишечнике. Содержание CYP3A4 составляет 70% всех изоферментов цитохрома Р-450 кишечника.
УДФ-глюкуронилтрансфераза и сульфотрансфераза - наиболее хорошо изученные ферменты, осуществляющие II фазу биологической трансформации лекарственных веществ в стенке кишечника. Распределение этих изоферментов в кишечнике аналогично содержанию изоферментов цитохрома Р-450. Основные фазы метаболизма некоторых ЛС происходят в кишечнике. Например, сульфатирование β2-адреномиметиков (тербуталина* и изопреналина) осуществляется только в стенке кишечника.
Однако, несмотря на определённое участие в биологической трансформации ЛС, стенка кишечника по своей метаболической способности значительно уступает печени.
1.5. ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ИЗ ОРГАНИЗМА
От процессов экскреции, как и от метаболизма и распределения в тканях, зависят продолжительность действия и скорость элиминации лекарственного вещества. Основные органы, участвующие в процессе выделения, - почки, гепатобилиарная система, лёгкие и кишечник (табл. 1-17).
ПОЧЕЧНАЯ ЭКСКРЕЦИЯ
Почечную экскрецию определяют три основных процесса:
• клубочковая фильтрация;
• канальцевая секреция;
• реабсорбция (преимущественно путём пассивной диффузии).
Клубочковая фильтрация носит избирательный характер, что предотвращает фильтрацию белков плазмы (в том числе альбумина) и способствует поддержанию осмотического градиента плазмы. Форма и размер молекулы, её электрический заряд относят к факторам, ограничивающим клубочковую фильтрацию. Приблизительно 130 мл плазмы ежеминутно (190 л/сут) подвергается клубочковой фильтрации, что и определяет основополагающую роль почек в процессе выведения ЛС. Любые лекарственные вещества, не связанные с белка-
Таблица 1-17. Основные пути выведения лекарственных средств из организма
 ми
плазмы или форменными элементами, подвергаются клубочковой фильтрации.
Молекулы радиусом более 2 нм могут иметь ограничения в процессе
ультрафильтрации, крупные молекулы с радиусом более 4,2 нм не будут
подвергаться клубочковой фильтрации. Скорость клубочковой фильтрации
заряженных молекул обычно ниже, чем нейтральных, даже при сопоставимой
молекулярной массе. Наибольшие ограничения в процессе фильтрации имеют
отрицательно заряженные молекулы (анионы), что, возможно, обусловлено
электростатическим взаимодействием между молекулами фильтрата и
отрицательно заряженными стенками клубочковых капилляров.
Преимущественно путём клубочковой фильтрации происходит удаление из
организма таких ЛС, как дигоксин, гентамицин, прокаинамид (новокаинамид*),
метотрексат. Скорость клубочковой фильтрации оценивают по величине
клиренса креатинина. Клиренс (CL) препаратов, которые элиминируются из
организма только благодаря клубочковой фильтрации, равен произведению
скорости клубочковой фильтрации (СКФ) на долю препарата, которая
присутствует в плазме в несвязанном виде (f):
ми
плазмы или форменными элементами, подвергаются клубочковой фильтрации.
Молекулы радиусом более 2 нм могут иметь ограничения в процессе
ультрафильтрации, крупные молекулы с радиусом более 4,2 нм не будут
подвергаться клубочковой фильтрации. Скорость клубочковой фильтрации
заряженных молекул обычно ниже, чем нейтральных, даже при сопоставимой
молекулярной массе. Наибольшие ограничения в процессе фильтрации имеют
отрицательно заряженные молекулы (анионы), что, возможно, обусловлено
электростатическим взаимодействием между молекулами фильтрата и
отрицательно заряженными стенками клубочковых капилляров.
Преимущественно путём клубочковой фильтрации происходит удаление из
организма таких ЛС, как дигоксин, гентамицин, прокаинамид (новокаинамид*),
метотрексат. Скорость клубочковой фильтрации оценивают по величине
клиренса креатинина. Клиренс (CL) препаратов, которые элиминируются из
организма только благодаря клубочковой фильтрации, равен произведению
скорости клубочковой фильтрации (СКФ) на долю препарата, которая
присутствует в плазме в несвязанном виде (f):
CСL = f *СКФ.
Факторы, определяющие скорость клубочковой фильтрации, могут изменять клиренс лекарственных веществ. К примеру, воспаление клубочковых капилляров может ускорять процесс ультрафильтрации
и, таким образом, увеличивать клиренс лекарств. Поскольку большинство лекарственных препаратов хотя бы частично связано с белками плазмы, скорость их ультрафильтрации ниже теоретической скорости клубочковой фильтрации. Чем прочнее связь лекарственного вещества с белками плазмы, тем значительнее удлинение периода полувыведения.
Пассивная диффузия. Процесс обратной диффузии через канальцевые мембраны в циркуляторное русло - важное звено почечной экскреции лекарств (особенно слабых электролитов). B целом, движение ЛС обратно в кровь определяет градиент их концентрации. pH мочи может значительно влиять на скорость пассивной обратной диффузии (табл. 1-18). Процесс обратной диффузии происходит в основном в дистальных канальцах и собирательных трубочках, где осуществляется подкисление мочи. Так как неионизированные молекулы лекарственных веществ диффундируют из просвета канальцев обратно в кровь, то процесс подкисления будет повышать реабсорбцию (снижать элиминацию) слабых кислот, таких, как салицилаты, и снижать реабсорбцию (повышать элиминацию) слабых оснований, таких, как амфетамины. Однако изменение pH мочи практически не влияет на элиминацию неионизированных форм лекарств, нерастворимых в жирах.
Bозможность изменения pH мочи и влияния на скорость элиминации ЛС используют в медицинской практике при лечении отравлений. К примеру, выведение барбитуратов (слабых кислот) ускоряется при назначении бикарбонатов пациенту. Экскрецию оснований можно ускорить, подкисляя мочу при использовании кислых солей (хлорида аммония).
Таблица 1-18. Лекарственные средства, выводимые почками в зависимости от pH мочи
Скорость экскреции выше в щелочной среде | Скорость экскреции выше в кислой среде |
Aминокислоты Пробеницид*9 Aцетазоламид Стрептомицин Барбитураты Салицилаты Сульфаниламиды Нитрофураны Налидиксовая кислота Фенилбутазон | Имипрамин Хлорохин Кодеин Хинидин Мексилетин Фенфлурамин*9 Морфин Aмфетамин Прокаин (новокаин*) Прокаинамид |
Активная канальцевая секреция. Большинство лекарств могут служить субстратами для двух активных систем секреции в клетках проксимальных канальцев. Эти системы, обеспечивающие транспорт лекарств из крови в просвет канальцев, независимы друг от друга: одна секретирует органические анионы, другая - органические катионы. Одно лекарственное вещество может конкурировать с другим одно- имённо заряженным веществом, что, в свою очередь, может снижать общую скорость элиминации каждого субстрата. Секреторная способность обеих систем может увеличиваться при высоких концентрациях ЛС в плазме крови. Каждый лекарственный препарат имеет свой собственный максимальный уровень секреции (транспортный максимум). Скорость работы транспортёров определяет общую скорость почечной или печёночной элиминации, общий клиренс лекарственного вещества. Транспортёры также регулируют фармакологические и токсические эффекты лекарств в силу своей способности лимитировать их распределение в тканях и, как следствие, развитие фармакологических и побочных эффектов того или иного лекарственного вещества.
Транспортёры в основном вовлечены в процессы канальцевой секреции и реабсорбции. Описано несколько активных транспортных систем, вовлечённых в процесс секреции, в проксимальных канальцах. Реабсорбция в ряде случаев идёт при помощи белков-транспортёров, однако большинство лекарственных веществ реабсорбируется путём пассивной диффузии на основании градиента концентраций между плазмой и содержимым нефрона. Субстраты для почечных транспор- тёров представлены в табл. 1-19. Данная таблица демонстрирует лекарственные вещества, подвергающиеся транспортёропосредованной почечной экскреции в неизменённом виде.
Таблица 1-19. Лекарственные средства, выделяемые путём активного транспорта в канальцах
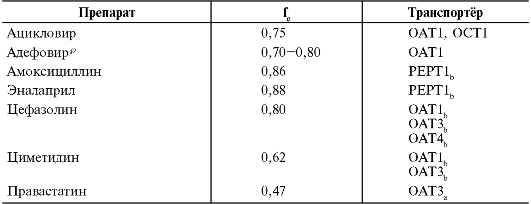 Примечания: fe
- фракция препарата, выделяющаяся с мочой в неизменённом виде; a -
лекарственный препарат - субстрат для данного белка-транспортёра; b -
лекарственный препарат - ингибитор данного белка-транспортёра.
Примечания: fe
- фракция препарата, выделяющаяся с мочой в неизменённом виде; a -
лекарственный препарат - субстрат для данного белка-транспортёра; b -
лекарственный препарат - ингибитор данного белка-транспортёра.
Детальные механизмы транспорта не изучены у большинства ЛС. Такие транспортёры, как гликопротеин P и ассоциированный с развитием лекарственной резистентности протеин 2-го типа, расположенные на апикальной поверхности канальцевого эпителия, отвечают за секрецию части конъюгированных метаболитов молекул лекарственных веществ (глюкурониды, сульфаты, конъюгаты глутатиона). Поскольку конъюгаты, как правило, не обладают фармакологической активностью, увеличение скорости их элиминации путём активной секреции не будет оказывать выраженного эффекта на общую продолжительность их действия. Эти системы активной секреции играют особенно важную роль в экскреции анионов и катионов, имеющих сильную связь с белками плазмы, так как данные комплексы не подвергаются процессу клубочковой ультрафильтрации. Однако, поскольку связь с белком обычно бывает обратимой, системы активной секреции могут быстро и эффективно удалять лекарственные вещества с белками из плазмы крови и транспортировать их в канальцевый секрет (табл. 1-20). Снижение секреторной активности приводит процесс экскреции к уровню, определяемому скоростью клубочковой фильтрации.
Таблица 1-20. Лекарственные средства, выделяемые путём активного транспорта в канальцах
Слабые кислоты | Слабые основания |
Ацетазоламид Этакриновая кислота Фуросемид Спиронолактон (верошпирон*) Гидрохлоротиазид Ацетилсалициловая кислота Фенилбутазон Индометацин Пробенецид*9 Клофибрат*9 Метотрексат Пенициллины Сульфаниламиды Фенобарбитал Хлорпропамид Налидиксовая кислота Хлорпромазин (аминазин*) | Трициклические антидепрессанты Амфетамин Гистамин Морфин Хинидин |
Активная канальцевая реабсорбция. Часть молекул, прошедших процесс клубочковой фильтрации, подвергается реабсорбции в проксимальных канальцах. Процесс активной реабсорбции особенно важен для эндогенных веществ, таких, как глюкоза и аминокислоты. Лишь небольшое количество ЛС подлежит активной реабсорбции. Большинство лекарственных препаратов замедляет процессы активного транспорта. Так, препараты, способствующие уменьшению концентрации мочевой кислоты (пробенецид * и т.п.) ингибируют активную реабсорбцию уратов, в то время как пиразинамид, замедляющий экскрецию уратов, может блокировать активную секрецию мочевой кислоты. Некоторые лекарственные вещества обладают способностью ингибировать процесс активной реабсорбции в одних дозах и активную секрецию в других, часто меньших, дозах. К примеру, салицилаты в малых количествах могут снижать выведение уратов, а в больших дозах, напротив, обладают урикозурическим эффектом. Данный пример объясняет парадоксальное влияние различных доз лекарственных веществ на общую скорость элиминации компонентов, выделяющихся путём активного транспорта.
Клиническое значение почечного пути выведения
Скорость ренальной экскреции лекарственных веществ зависит от объёма распределения лекарственного вещества, степени связывания с белками плазмы, и её определяют следующие факторы:
• скорость клубочковой фильтрации;
• pH канальцевой жидкости;
• величина обратной диффузии неионизированных форм;
• величина активной канальцевой секреции компонентов;
• величина активной канальцевой реабсорбции.
Нарушения одного из этих процессов могут привести к существенным изменениям клинических эффектов того или иного лекарственного вещества. В конечном счёте, количество лекарственного вещества, содержащегося в моче, представляет собой баланс между ультрафильтрацией, реабсорбцией (пассивной или активной) и секрецией. Продолжительность действия и выраженность фармакологического эффекта большинства лекарственных препаратов определяет функциональный статус почек, так как именно они играют ключевую роль в процессе выведение лекарственных веществ. Нарушение функционирования почек влечет за собой корректировку режима дозирования ЛС, с учётом степени почечной дисфункции.
Большое влияние на скорость почечной экскреции оказывает возраст больного. Так, количество ЛС, активно выделяемых в канальцевом аппарате почек, у пожилых лиц заметно меньше по сравнению с
молодыми. Также у них замедляется скорость клубочковой фильтрации ряда ЛС: прокаинамида , клофибрата*, пенициллинов, метилдопы, сердечных гликозидов. У детей, особенно на первом году жизни, выведение ЛС почками также уменьшено по сравнению со взрослыми людьми. Эти особенности необходимо учитывать при выборе ЛС и режима его дозирования.
Большое значение придают применению ЛС при ухудшении функции почек. У больных с почечной недостаточностью дозы многих ЛС необходимо снижать по следующим причинам:
• снижение клубочковой фильтрации, нарушение процессов секреции и реабсорбции приводят к задержке выведения ЛС и их метаболитов и к повышению их концентрации в плазме крови;
• токсичность ЛС или их метаболитов возрастает у больных с почечной недостаточностью в связи с нарушением функционального состояния основных систем;
• при почечной недостаточности может нарушаться метаболизм ЛС, что также ведёт к замедлению скорости элиминации;
• у этой группы больных связь ЛС с белками плазмы ослаблена, что приводит к увеличению их свободной фракции.
ВЫВЕДЕНИЕ С ЖЁЛЧЬЮ
Прохождение большинства чужеродных молекул из крови в печень не имеет серьёзных ограничений, так как эндотелий печёночного синуса - пористая мембрана. Следовательно, лекарственные вещества с молекулярной массой, меньшей, чем у белков плазмы, быстро выделяются в печёночные протоки. Все молекулы, выделяемые в жёлчь, можно подразделить на три группы:
• группа А - вещества, концентрация которых в жёлчи и крови идентична, отношение жёлчь/плазма равно 1 (глюкоза, ионы Na+ и K+);
• группа В - вещества, для которых соотношение жёлчь/плазма более 1, обычно от 10 до 1000 (соли жёлчи, прокаинамид);
• группа С - вещества, для которых соотношение жёлчь/плазма менее 1 (инсулин, белки).
Лекарственные вещества могут принадлежать к любой из этих категорий. Экскреция с жёлчью играет важную роль (5-95% полученной дозы) в выведении ряда анионов, катионов и неионизированных молекул, таких, как сердечные гликозиды. Печёночный путь выведения играет ключевую роль в выведении солей тяжёлых металлов. Сердечные гликозиды, анионы и катионы транспортируются из печени в жёлчь трёмя независимыми системами активного транспорта, сходными с системами канальцевой секреции в почках. Лекарственные вещест-
ва, связанные с белками плазмы, полностью доступны для активного транспорта в печени. Большинство лекарств секретируется печенью в жёлчь и затем попадает в тонкую кишку, откуда большая их часть обратно всасывается через кишечную стенку в кровь (энтерогепатическая рециркуляция). Этот процесс может идти до тех пор, пока лекарственное вещество, претерпев метаболические изменения в печени, не будет выведено из организма с мочой или жёлчью (табл. 1-21).
Таблица 1-21. Лекарственные средства, подвергающиеся энтерогепатической рециркуляции
Доксирубицин (адриамицин*) Амфетамин
1,25-Дигидроксивитамин D3
Эстрадиол
Индометацин
Метронидазол Морфин Фенитоин Сулиндак*
Полярные конъюгаты глюкуроновой
Назначение энтеросорбентов может прервать цикл энтерогепатической циркуляции и удержать лекарственный препарат в просвете кишки. Большая часть чужеродных веществ (в том числе лекарственных препаратов) полностью или частично проходит метаболизм в печени. Конъюгация способствует увеличению билиарной секреции молекул путём образования полярного (анионного) центра и увеличения молекулярного веса. Конъюгированные лекарства не могут быть реабсорбированы из гастроинтестинального тракта, пока не будут гидролизованы кишечными ферментами (3-глюкуронидаза). Хлорамфеникол, к примеру, секретируется в жёлчь, затем гидролизуется кишечной флорой и снова практически полностью реабсорбируется. Данный путь рециркуляции ЛС - причина его токсичности. Нарушение функции печени может привести к изменению секреции жёлчи и, как следствие, к аккумулированию ряда лекарственных веществ в плазме (пробеницид*, дигоксин, диэтилстильбэстрол). Усиление печёночной экскреции также может быть, к примеру, при длительном назначении фенобарбитала или спиронолактона. В этом случае наблюдают снижение концентрации лекарств в крови за счёт ускорения процесса печёночной элиминации.
В целом почки и печень способны активно транспортировать одни и те же органические анионы. Однако данные органы имеют определён- ные количественные различия, касающиеся аффинитета лекарств к системам транспортёров. В настоящее время известно несколько систем транспортёров, связанных с транспортом молекул лекарственных веществ.
Суммарный клиренс (CLtot) представляет сумму печёночного (CLh) и почечного (CL) клиренсов.
кислоты
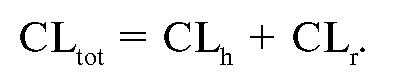 Данное
уравнение демонстрирует баланс между почечным и пе- чёночным путями
выведения. Кроме того, почечный и печёночный клиренс регулируют степень
аффинитета лекарственного вещества к тому или иному белку-транспортёру,
локализованному в печени или почках. К примеру, 41-47% общей дозы
правастатина выводится с мочой в неизменённом виде. Это подтверждает тот
факт, что правастатин - субстрат почечных транспортёров, в частности
органический анион-3, который расположен на базолатеральной мембране
проксимальных канальцев.
Данное
уравнение демонстрирует баланс между почечным и пе- чёночным путями
выведения. Кроме того, почечный и печёночный клиренс регулируют степень
аффинитета лекарственного вещества к тому или иному белку-транспортёру,
локализованному в печени или почках. К примеру, 41-47% общей дозы
правастатина выводится с мочой в неизменённом виде. Это подтверждает тот
факт, что правастатин - субстрат почечных транспортёров, в частности
органический анион-3, который расположен на базолатеральной мембране
проксимальных канальцев.
ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ЧЕРЕЗ ЛЁГКИЕ
Выведение ЛС с выдыхаемым воздухом через лёгкие зависит от его физико-химических свойств, растворимости в крови и тканях, а также от лёгочной вентиляции и лёгочного кровотока. Этот путь выведения основной только для некоторых ЛС. Около 90% этанола окисляется в печени, но часть его выводится через лёгкие. Содержание этанола в выдыхаемом воздухе легко определить, оно хорошо коррелирует с его концентрацией в крови, что имеет важное практическое значение.
ЛС, выделяемые через лёгкие:
• средства для ингаляционного наркоза;
• аммония хлорид;
• йодиды;
• камфора;
• непрямые антикоагулянты, производные 4-оксикумарина;
• эфирные масла;
• этанол.
Факторы, влияющие на элиминацию ЛС через лёгкие:
• физико-химические свойства;
• лёгочная вентиляция;
• лёгочный кровоток;
• растворимость ЛС в крови и тканях.
ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ С МОЛОКОМ МАТЕРИ
Этот путь экскреции не имеет существенного значения с точки зрения фармакокинетики ЛС, однако его необходимо всегда иметь в виду при проведении терапии у кормящих матерей. Концентрация ЛС в молоке бывает равной уровню ЛС в крови матери или даже большей (табл. 1-22).
Таблица 1-22. Параметры выведения с грудным молоком некоторых лекарственных средств
 Концентрация
ЛС в грудном молоке, а следовательно, и общее количество этого ЛС и его
метаболитов, попадающих в организм но- ворождённого, зависят от
физико-химических свойств ЛС, связывания его с белками крови, режима
применения (доза, частота и путь введения), режима кормления и др. ЛС,
хорошо связывающиеся с белком, остаются в плазме крови матери, а хорошо
связывающиеся с жиром - концентрируются в молоке (например,
барбитураты). Теофиллин возможно определить в молоке на уровне 70%
концентрации ЛС в плазме крови, хлорамфеникол (левомицетин*) - 50%,
пенициллины и цефалоспорины - до 20%. Хорошо проникают в молоко
эритромицин, тетрациклин, изониазид, сульфаниламиды, соли лития,
мепробамат, ацетилсалициловая кислота, тербуталин*. Следует также
учитывать, что даже малые концентрации ЛС в молоке могут вызывать
разнообразные аллергические реакции у грудных детей.
Концентрация
ЛС в грудном молоке, а следовательно, и общее количество этого ЛС и его
метаболитов, попадающих в организм но- ворождённого, зависят от
физико-химических свойств ЛС, связывания его с белками крови, режима
применения (доза, частота и путь введения), режима кормления и др. ЛС,
хорошо связывающиеся с белком, остаются в плазме крови матери, а хорошо
связывающиеся с жиром - концентрируются в молоке (например,
барбитураты). Теофиллин возможно определить в молоке на уровне 70%
концентрации ЛС в плазме крови, хлорамфеникол (левомицетин*) - 50%,
пенициллины и цефалоспорины - до 20%. Хорошо проникают в молоко
эритромицин, тетрациклин, изониазид, сульфаниламиды, соли лития,
мепробамат, ацетилсалициловая кислота, тербуталин*. Следует также
учитывать, что даже малые концентрации ЛС в молоке могут вызывать
разнообразные аллергические реакции у грудных детей.
Большинство ЛС при их поступлении в организм новорождённого с молоком матери относительно безопасны (исключая аллергизирующее действие). Однако существуют ЛС, необходимость приёма которых требует прекращения грудного вскармливания. К ним относят соли лития, противоопухолевые ЛС, изониазид, хлорамфеникол, цитостатики, радиоактивные диагностические средства. Следует также избегать контакта с инсектицидами (ДДТ, гексахлоран), обладающими высокой жирорастворимостью и поступающими в значительных количествах в организм новорождённого с молоком матери.
ЭКСКРЕЦИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ СО СЛЮНОЙ
Выведение ЛС со слюной имеет значение, поскольку даёт возможность определить фармакокинетические параметры, если концентрация ЛС в слюне коррелирует с его концентрацией в крови (табл. 1-23).
Основной транспортный механизм экскреции ЛС со слюной - пассивная диффузия. Скорость выведения зависит от липофильности и pKa ЛС, его связывания с белками плазмы крови, а также - от pH слюны. Значительная вариабельность этого показателя резко ог-
Таблица 1-23. Выведение лекарственных средств со слюной
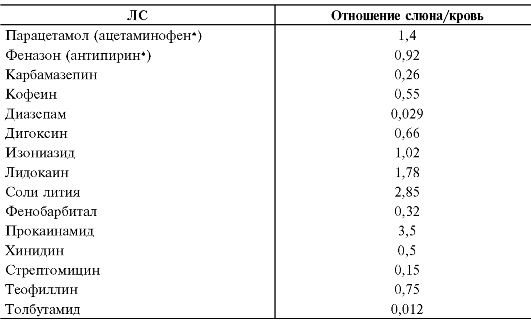 раничивает
возможности использования слюны для оценки индивидуальных
фармакокинетических параметров. Практически это можно осуществить лишь
для ЛС с pKa около 7 (т.е. неионизированных при физиологическом pH), так
как для них зависимость скорости экскреции от pH слюны минимальна. К
числу таких ЛС относят феназон (антипирин*), параметры фармакокинетики которого используют для оценки общей активности смешанных оксидаз печени.
раничивает
возможности использования слюны для оценки индивидуальных
фармакокинетических параметров. Практически это можно осуществить лишь
для ЛС с pKa около 7 (т.е. неионизированных при физиологическом pH), так
как для них зависимость скорости экскреции от pH слюны минимальна. К
числу таких ЛС относят феназон (антипирин*), параметры фармакокинетики которого используют для оценки общей активности смешанных оксидаз печени.
ДРУГИЕ ПУТИ ЭКСКРЕЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Экскреция ЛС с потом, слёзной жидкостью, вагинальным секретом и др. не имеет существенного практического значения. Усиление выведения ЛС с потом отмечают при выраженном потоотделении, при гипертензии, повышении тонуса симпатической нервной системы, внезапном падении АД, но даже в этих случаях количество выводимых ЛС незначительно и не имеет практического значения в оценке фармакокинетических параметров.
1.6. ОСНОВНЫЕ ФАРМАКОКИНЕТИЧЕСКИЕ ПАРАМЕТРЫ
Одним из основных показателей, определяющих фармакологический эффект, считают концентрацию ЛС на уровне рецептора, однако в условиях целостного организма установить её невозможно. В эксперименте было доказано, что в большинстве случаев между концен-
трацией препарата в крови и его содержанием в области рецептора существует корреляция.
В связи с этим для определения фармакокинетических параметров изучают содержание ЛС в крови. Для того чтобы получить соответствующее представление о поступлении препарата в кровь и выведении его из организма, отслеживают изменения концентрации ЛС в плазме крови на протяжении длительного времени. Содержание препаратов в плазме крови определяют методами жидкостной или газожидкостной хроматографии, с помощью радиоиммунного или иммуноферментного анализа и другими способами. На основании полученных данных строят график. На оси абсцисс отмечают время от начала исследования, а на оси ординат - концентрацию ЛС в плазме крови (в соответствующих единицах). Такой график носит название фармакокинетической кривой (рис. 1-9).
Описать все детали процесса распределения препарата в организме, во всех органах и тканях крайне сложно. Математические модели позволяют представить организм в виде одной или нескольких разделён- ных проницаемой мембраной частей (камер), в которых происходят распределение ЛС и их переходы из одного пространства в другое. Камеры выделяют условно, они не соответствуют какому-либо анатомически ограниченному пространству. Такое моделирование называют камерным. За центральную часть обычно принимают плазму крови, составляющие её элементы и хорошо перфузируемые органы (сердце, лёгкие, печень, почки, эндокринные железы), за периферическую - плохо перфузируемые органы и ткани (мышцы, кожу, жировую ткань). В этих камерах распределение ЛС происходит с разной скоростью: быстрее - в центральной и медленнее - в периферической. Самой простой считают однокамерную модель. В этом случае предполагают, что после введения препарата его концентрация убывает по моноэкспоненциальному закону. В соответствии с законами линейной кинетики скорость изменения количества препарата в камере пропорциональна его количеству в этом пространстве.
Основные фармакокинетические параметры (табл. 1-24):
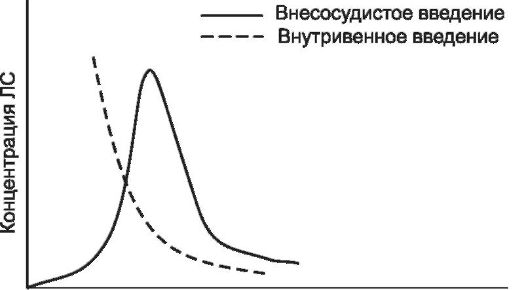 Время после введения ЛС Рис. 1-9. Фармакокинетическая кривая.
Время после введения ЛС Рис. 1-9. Фармакокинетическая кривая.
• максимальная концентрация (Сmах);
• время достижения максимальной концентрации (Tmax);
• общий клиренс (CL);
• константа элиминации (kel);
• период полувыведения (Т1/2);
• объём распределения (Vd);
• площадь под фармакокинетической кривой (AUC);
• биодоступность.
Общий клиренс (CL), измеряемый в мл/мин или л/ч, - это объ- ём плазмы или крови, который полностью очищается от препарата за единицу времени. В рамках линейной модели:
CL=Vd x kel, CL=Доза/АUС.
В связи с тем что основными путями выведения считают почки и печень, общий клиренс представляет собой, главным образом, сумму почечного и печёночного клиренсов. Под печёночным подразумевают метаболический клиренс в печени и выведение препарата с жёлчью. Например, почечный клиренс циметидина составляет около 600 мл/ мин; метаболический - 200 мл/мин и жёлчный - 10 мл/мин; следовательно, общий клиренс равен 810 мл/мин. Другие пути выведения
Таблица 1-24. Клиническое значение основных фармакокинетических параметров
 или
внепечёночный метаболизм не имеют большого практического значения, и
при расчёте общего клиренса их во внимание обычно не принимают.
или
внепечёночный метаболизм не имеют большого практического значения, и
при расчёте общего клиренса их во внимание обычно не принимают.
Основными физиологическими факторами, определяющими клиренс, считают функциональное состояние физиологических систем организма, объём притока крови и скорость кровотока в органе. Печё- ночный клиренс зависит от скорости печёночного кровотока или функциональной активности метаболизирующих ферментов. Например, клиренс лидокаина, который интенсивно метаболизируют ферменты печени, зависит прежде всего от скорости его доставки к печени, т.е. от объёма притекающей крови, скорости кровотока. В связи с этим при уменьшении печёночного кровотока в результате застойной сердечной недостаточности наблюдают снижение клиренса лидокаина.
В то же время клиренс фенотиазинов зависит в основном от функциональной активности метаболизирующих ферментов, поэтому при поражении гепатоцитов отмечают резкое снижение клиренса данной группы препаратов и, естественно, значительное повышение их концентрации в крови.
Объём распределения (Vd), измеряемый в л или л/кг, - гипотетический объём жидкости организма, необходимый для равномерного распределения всего количества ЛС (введённой дозы) в концентрации, аналогичной концентрации в плазме крови. При внутривенном введении
Vd=Доза/C0,
где С0 - начальная концентрация в крови.
Высокие значения объёма распределения свидетельствуют о том, что препарат активно проникает в биологические жидкости и ткани. При этом, если препарат активно связывается, например, жировой тканью, его концентрация в крови может практически мгновенно стать очень низкой, а объём распределения достигнет нескольких сотен литров, превысив реальное количество жидкостей организма. В связи с этим данную величину ещё называют «кажущимся объёмом распределения». Объём распределения зависит от различных факторов. Физико-химические свойства препарата (молекулярная масса, уровень ионизации и полярности, растворимость в воде и жирах) влияют на его прохождение через мембраны, а следовательно, и на объём распределения. Значение этого параметра зависит и от физиологических факторов: возраста, пола, общего количества жира в организме. Например, у пожилых людей и новорождённых объём распределения уменьшен. Кроме того, он может быть изменён при некоторых па-
тологических состояниях, особенно при заболеваниях печени, почек, сердечно-сосудистой системы и т.д.
Значение объёма распределения используют при подборе режима дозирования для расчёта нагрузочной дозы, требуемой для достижения необходимой концентрации препарата в крови:
Доза(нагрузочная)=Vd x C,
где С - эффективная концентрация ЛС в крови.
Константа скорости элиминации (kel), измеряемая в ч-1, характеризует снижение концентрации вещества за единицу времени (показывает долю препарата, выводимую из организма за единицу времени).
Общий клиренс, объём распределения и константа элиминации связаны между собой уравнением:
CL=Vd x kel,.
Период полувыведения (T1/2) - время, необходимое для снижения концентрации препарата в плазме крови на 50%:
T1/2=0,693/kel.
Практически за один период полувыведения организм элиминирует 50% ЛС, за два периода - 75%, за 3 периода - чуть меньше 90% и т.д.
Зависимость между периодом полуэлиминации и константой скорости элиминации важна для выбора интервала между введениями, а также определения промежутка времени, необходимого для достижения равновесной концентрации при многократном введении ЛС (обычно 5-7 Т1/2).
Если препарат вводят одинаковыми дозами через фиксированные интервалы времени, меньшие, чем время элиминации ЛС, то его концентрация в крови возрастает, а затем наступает период, когда в каждом интервале между приёмом очередных доз количество всасывающегося препарата равно количеству элиминируемого. Это состояние называют стационарным, или Steady state. Концентрация, достигаемая при этом, также носит название стационарной (реже равновесной) (Css). В результате наблюдают колебания концентрации ЛС в пределах средней величины с определёнными максимальными (Cssmax) и минимальными значениями (Cssmin).
Именно при установлении стационарной концентрации отмечают проявление клинического эффекта препарата в полном объёме. Чем короче период полувыведения соединения, тем быстрее достигают равновесной концентрации и тем более выражены её колебания. Например, прокаинамид (новокаинамид*) имеет период полувыведения
около 2-3 ч и для его стационарной концентрации характерен большой разброс значений при назначении препарата через каждые 6 ч. В связи с этим для предупреждения и уменьшения указанных колебаний стационарной концентрации в крови в последнее время всё шире используют препараты с замедленным высвобождением.
На практике равновесную концентрацию ЛС можно вычислить по концентрации данного препарата после однократного введения:
Сss=(1,44 x F xДоза xT1/2)/(Vd x τ),
где τ - интервал времени между введениями, F - биодоступность.
Для расчёта дозы, необходимой для поддержания нужной концентрации препарата в крови, т.е. поддерживающей дозы, используют значение клиренса:
Доза(поддерживающая )=CL x С 55.
При внесосудистом введении препарата по естественным причинам не всё его количество достигает системного кровотока.
Биодоступность (F), измеряемая в процентах, - часть препарата, достигшая системного кровотока после внесосудистого введения ЛС.
Биодоступность бывает абсолютной и относительной. Её вычисляют по площади под фармакокинетической кривой (AUC). Когда данные о концентрации препарата при внесосудистом введении сравнивают с данными при его внутривенном введении, получают абсолютную биодоступность. При введении одинаковых доз:
F=AUC (внутрь)/АUС (внутривенно).
При введении разных доз (D):
F=AUC (внутрь) χ D (внутривенно)/АUС (внутривенно) χ D (внутрь).
В случае, когда сравнивают два внесосудистых пути введения, говорят об относительной биодоступности. Если сравниваемые препараты аналогичны (действующее вещество, доза, лекарственная форма), но их выпускают на разных производствах, то такие ЛС называют дженериками. В этом случае говорят об их биоэквивалентности:
F=AUCЛС испытываемое /АUСЛС сравнения .
Два лекарственных препарата считают биоэквивалентными, если они обеспечивают одинаковую биодоступность действующего вещества. Таким образом, биоэквивалентность - это сравнительная биодоступность.