Клиническая фармакология.: учебник для вузов / Под ред. В.Г. Кукеса.- 4-е издание., перераб. и доп., - 2009. - 1056 с.
|
|
|
|
ГЛАВА 9. КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ. ДОКАЗАТЕЛЬНАЯ МЕДИЦИНА
Безопасность и эффективность новых ЛС должны быть установлены в клинических исследованиях. Клиническое исследование - любое исследование, проводимое с участием человека в качестве субъекта для выявления или подтверждения клинических и/или фармакологических эффектов исследуемых продуктов и/или выявления нежелательных реакций на исследуемые продукты, и/или изучения их всасывания, распределения, метаболизма и выведения с целью оценить их безопасность и/или эффективность. Однако до начала клинических испытаний потенциальное ЛС проходит сложный этап доклинических исследований.
ДОКЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
Доклинические исследования начинают вскоре после синтеза новой потенциально эффективной молекулы ЛС. Новое ЛС следует соответствующим образом испытать in vitro и на животных, прежде чем назначать его людям. Цель доклинических исследований - получение информации о фармакологических особенностях тестируемого соединения: фармакокинетике, фармакодинамике, потенциальной токсичности и безопасности препарата.
При фармакологическом исследовании потенциальных препаратов подробно изучают фармакодинамику веществ: их специфическую активность, длительность эффекта, механизм и локализацию действия. Для определения активности и селективности действия вещества используют различные скрининговые тесты, проводимые в сравнении с эталонным препаратом. Выбор и количество тестов зависят от задач исследования. Так, для изучения потенциальных антигипертензивных ЛС, действующих предположительно как антагонисты α-адренорецепторов сосудов, исследуют in vitro связывание с этими рецепторами. В дальнейшем изучают антигипертензивную активность соединения на моделях экспериментальной артериальной гипертензии животных, а также возможные побочные эффекты. Важный аспект исследования - изучение фармакокинетики веществ (всасывание, распреде-
ление, метаболизм, экскреция). Особое внимание уделяют изучению путей метаболизма самого вещества и его основных метаболитов. Сегодня существует альтернатива экспериментам на животных - это исследования на культурах клеток in vitro (микросомы, гепатоциты или образцы тканей), которые позволяют оценить важные фармакокинетические параметры. В результате таких исследований может возникнуть необходимость химической модификации молекулы вещества для достижения более желательных фармакокинетических или фармакодинамических свойств.
О безопасности нового соединения судят по результатам изучения его токсичности в экспериментах на моделях животных. Это исследования общетоксического действия (определение острой, субхронической и хронической токсичности). Параллельно с этим препараты исследуют на специфическую токсичность (мутагенность, репродуктивная токсичность, включая тератогенность и эмбриотоксичность, иммунотоксичность, аллергенность и канцерогенность с использованием различных режимов дозирования). Использование физиологических, фармакологических, биохимических, гематологических и других методов исследования на животных позволяет оценивать токсические свойства препарата и прогнозировать степень безопасности его применения в клинике. Однако следует учитывать, что полученные сведения нельзя в полной мере экстраполировать на человека, а редкие нежелательные реакции обычно выявляют лишь на этапе клинических испытаний. Общая продолжительность доклинических исследований оригинального препарата превышает 5-6 лет. В результате такой работы из 5-10 тыс. новых соединений отбирают около 250 потенциальных препаратов.
Заключительная задача доклинических исследований - выбор способа производства исследуемого препарата (например, химический синтез, генная инженерия). Обязательный компонент доклинической разработки ЛС - оценка его стабильности в лекарственной форме и разработка аналитических методов контроля ЛС.
КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
Влияние клинической фармакологии на процесс создания новых ЛС проявляется при проведении клинических исследований. Многие результаты фармакологических исследований на животных раньше автоматически переносили на человека. Когда была осознана необходимость проведения исследований на человеке, клинические испытания проводили на пациентах без их согласия. Известны случаи проведения
заведомо опасных исследований на социально незащищённых лицах (заключённые, душевнобольные и т.д.). Потребовалось длительное время, чтобы сравнительный дизайн исследования (наличие «опытной» группы и группы сравнения) стал общепринятым. Вероятно, именно ошибки в планировании исследований и анализе их результатов, а порой фальсификации последних стали причиной ряда гуманитарных катастроф, связанных с выпуском токсических препаратов, например раствора сульфаниламида в этиленгликоле (1937), а также талидомида (1961), который назначали в качестве противорвотного средства на ранних сроках беременности. В это время врачи не знали о способности талидомида тормозить ангиогенез, что привело к рождению более 10 000 детей с фокомелией (врождённой аномалией нижних конечностей). В 1962 г. талидомид запрещён для медицинского применения. В 1998 г. применение талидомида получило одобрение американского FDA (Управление по контролю качества пищевых продуктов, медикаментов и косметических средств в США, Food and Drug Administration) для использования при лечении лепры, а в настоящее время проводятся его клинические испытания для терапии множественной рефрактерной миеломы и глиомы. Первой государственной организацией, регулирующей вопросы клинических испытаний, стало FDA, предложившее в 1977 г. концепцию качественной клинической практики (Good Clinical Practice, GCP). Важнейший документ, определяющий права и обязанности участников клинических исследований, - Хельсинкская декларация Всемирной медицинской ассоциации (1964). После многочисленных доработок появился итоговый документ - Руководство по надлежащей клинической практике (Consolidated Guideline for Good Clinical Practice, GCP) Международной конференции по гармонизации технических требований к регистрации фармацевтических продуктов, предназначенных для применения человеком (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH). Положения ICH GCP согласуются с требованиями к проведению клинических испытаний ЛС в РФ и отражены в Федеральном законе «О лекарственных средствах» (? 86-ФЗ от 22.06.98 с изменениями от 02.01.2000). Основной официальный документ, регламентирущий проведение клинических испытаний в РФ, - национальный стандарт Российской Федерации «Надлежащая клиническая практика» (утверждён Приказом Федерального агентства по техническому регулированию и метрологии от 27 сентября 2005 г. ? 232-ст), который идентичен ICH GCP.
Согласно этому документу, Надлежащая клиническая практика (GCP) - международный этический и научный «стандарт планирования, выполнения, мониторинга, аудита и документального оформле-
ния клинических испытаний, а также обработки и представления их результатов; стандарт, который служит для общества гарантией достоверности и точности полученных данных и представленных результатов, а также защищённости прав, здоровья и анонимности субъектов исследования».
Выполнение принципов Надлежащей клинической практики обеспечивается соблюдением следующих основных условий: участием квалифицированных исследователей, распределением обязанностей между участниками исследования, научным подходом к планированию исследования, регистрацией данных и анализом представляемых результатов.
Выполнение клинических исследований на всех его этапах подвергается многостороннему контролю: со стороны спонсора исследования, органов государственного контроля и независимого этического комитета, а вся деятельность в целом осуществляется в соответствии с принципами Хельсинской декларации.
Цели клинического исследования
Цели клинического исследования - исследование фармакологического действия препарата на человека, установление терапевтической (лечебной) эффективности или подтверждение терапевтической эффективности в сравнении с другими ЛС, изучение безопасности и переносимости ЛС, а также определение терапевтического применения, то есть той «ниши», которую может занимать данный препарат в современной фармакотерапии.
Исследование может быть этапом подготовки препарата к регистрации, способствовать продвижению на рынок уже зарегистрированного препарата или служить инструментом решения научных проблем.
Этические и правовые нормы клинических исследований
Гарантии прав субъектов исследования и соблюдение этических норм - сложная проблема клинических испытаний. Они регламентированы вышеперечисленными документами, гарантом соблюдения прав пациентов служит Независимый этический комитет, одобрение которого необходимо получить до начала клинических испытаний. Основная задача Независимого этического комитета - защита прав и здоровья субъектов исследования, а также гарантия их безопасности. Независимый этический комитет рассматривает информацию о препарате, структуру протокола клинического испытания, содержание информированного согласия и биографии исследователей с последующей оценкой ожидаемых польза/риск для пациентов.
Субъект может участвовать в клинических испытаниях только при полном и сознательном добровольном согласии. Каждый участник исследования должен быть заранее информирован о целях, методах, ожидаемом риске и пользе, обеспечении его необходимой медицинской помощью в случае выявления нежелательных реакций в ходе испытания, страховании на случай причинения ущерба здоровью, связанного с участием в данном исследовании. Исследователь должен получить от субъекта подписанное и датированное информированное согласие на участие в исследовании. Каждый участник должен знать, что его участие в исследовании добровольное и что он может в любое время выйти из исследования. Принцип информированного согласия является краеугольным камнем этичности клинических исследований. Важная сторона защиты прав субъектов исследования - соблюдение конфиденциальности.
Участники клинического исследования
Первое звено клинических испытаний - спонсор (обычно фармацевтическая компания), второе - медицинское учреждение, на базе которого проводят клиническое исследование, третье - субъект исследования. Связующим звеном между спонсором и медицинским учреждением могут выступать контрактно-исследовательские организации, которые берут на себя задачи и обязанности спонсора и осуществляют контроль за данным исследованием.
Последовательность проведения исследования
Постановка исследовательского вопроса (например, действительно ли препарат Х достоверно снижает уровень АД или действительно ли препарат Х способен снижать уровень АД более эффективно, чем препарат Y?). Одно исследование может позволить ответить сразу на несколько вопросов.
Разработка протокола исследования.
• Дизайн исследования. В первом примере более уместно сравнительное плацебоконтролируемое исследование (препарат Х и плацебо), а во втором примере необходимо сравнивать препараты X и Y между собой.
• Объём выборки. В протоколе необходимо определить, какое именно число исследуемых потребуется для доказательства исходной гипотезы (величина объема выборки рассчитывается математически на основании законов статистики).
• Продолжительность исследования. Следует предусмотреть продолжительность исследования (например, антигипертензивный эффект клофелина будет зарегистрирован уже после однократно-
го приёма, а для исследования современных ингибиторов АПФ могут потребоваться более продолжительные сроки).
• Критерии включения и исключения больных. В данном примере исследование не даст достоверных результатов, если в качестве испытуемых окажутся лица с нормальным уровнем АД. С другой стороны, включая в исследования больных с артериальной гипертензией, исследователи должны позаботиться о том, чтобы больные имели примерно одинаковый уровень АД. Не следует включать в исследование лиц со злокачественной (неподдающейся никакому лечению) гипертензией, лиц с резко изменённым метаболизмом (печёночная недостаточность) и экскрецией (почечная недостаточность). Таким образом протокол исследования должен включать точные критерии, по которым будут проводить отбор больных, вместе с тем выбранная для исследования популяция должна соответствовать популяции больных, на которую рассчитан гипотетический препарат Х.
• Оценка эффективности. Исследователь должен выбрать индикаторы эффективности препарата (критерии исхода заболевания - «конечные точки»). В данном примере ему следует уточнить, как именно будет оценен гипотензивный эффект - путём однократного измерения АД; путём вычисления среднесуточной величины АД; или же эффективность лечения будут оценивать по влиянию на качество жизни больного или по способности ЛС предотвращать появления осложнений артериальной гипертензии.
• Оценка безопасности. В протоколе необходимо предусмотреть клинические и лабораторные методы выявления нежелательных явлений и методы их коррекции.
• Процедура статистической обработки полученных данных. Этот раздел протокола разрабатывают совместно со специалистами по медицинской статистике.
Предварительная работа по протоколу, его пересмотр, создание форм регистрации данных исследования.
Представления протокола исследования в органы государственного контроля и этический комитет.
Проведение исследования.
Анализ полученных данных.
Формулирование выводов и публикация результатов исследования.
Проведение клинических исследований
Надёжность результатов клинических испытаний полностью зависит от тщательности их планирования, проведения и анализа. Любое
клиническое испытание следует проводить по строго определённому плану (протоколу исследования), идентичному для всех медицинских центров, принимающих в нём участие.
Протокол исследования - основной документ исследования, который «описывает задачи, методологию, статистические аспекты и организацию исследования». На основании рассмотрения протокола выдаётся разрешение на проведение исследования. Внутренний (мониторинг) и внешний (аудит) контроль за проведением исследования прежде всего оценивает соответствие действий исследователей процедуре, описанной в протоколе.
Включение пациентов в исследования осуществляют сугубо добровольно. Обязательное условие включения - ознакомление пациента с возможным риском и пользой, которую он может извлечь из участия в исследовании, а также подписание им информированного согласия. Правила ICH GSP не допускают использования материальных стимулов для привлечения больных к участию в исследовании (исключение делается для здоровых волонтеров, привлекаемых для исследования фармакокинетики или биоэквивалентности ЛС). Больной должен соответствовать критериям включения/исключения.
Критерии включения должны чётко идентифицировать популяцию, которую предстоит исследовать.
Критерии исключения определяют тех пациентов, которые входят в группу повышенного риска развития нежелательных реакций (например, больных бронхиальной астмой при испытании новых β-адреноблокаторов, язвенной болезнью - новых НПВС).
Обычно не допускается участие в исследованиях беременных, кормящих, больных у которых может быть изменена фармакокинетика исследуемого препарата, больных алкоголизмом или наркоманией. Недопустимо включение в исследование недееспособных пациентов без согласия попечителей, военнослужащих, заключенных, лиц с аллергией на исследуемый препарат или больных, которые одновременно участвуют в другом исследовании. Больной вправе прекратить свое участие в исследовании в любой момент без объяснения причин.
Клинические испытания на несовершеннолетних пациентах проводят лишь в случаях, когда исследуемый препарат предназначен исключительно для лечения детских болезней или исследование необходимо для получения информации об оптимальной дозировке препарата у детей. Результаты изучения данного ЛС у взрослых служат основой для планирования исследований у детей. При исследовании фармакокинетических параметров ЛС следует помнить о том, что по мере роста функциональные показатели детского организма быстро меняются.
С определёнными проблемами сопряжено изучение действия ЛС у пациентов пожилого возраста в связи с наличием у них сопутствующих заболеваний, нуждающихся в фармакотерапии. При этом может возникать взаимодействие лекарственных препаратов. Следует учитывать, что нежелательные реакции у пожилых могут возникнуть раньше и при применении меньших доз, чем у больных среднего возраста (например, лишь после широкого применения НПВС беноксапрофена было обнаружено, что он токсичен для больных пожилого возраста в дозах, относительно безопасных для лиц среднего возраста).
Дизайн исследования
Клиническое исследование может иметь различный дизайн. Исследования, в котором все пациенты получают одинаковое лечение, в настоящее время практически не используют из-за низкой доказательности получаемых результатов. Наиболее распространено сравнительное исследование в параллельных группах (группа «вмешательство» и группа «контроля»). В качестве контроля может выступать плацебо (плацебоконтролируемое исследование) или другой активный препарат. Использование плацебо позволяет разграничить собственно фармакодинамические и суггестивные эффекты препарата, отличить эффект ЛС от спонтанных ремиссий в течение заболевания и влияния внешних факторов, избежать получения ложноотрицательных заключений (например, равная эффективность исследуемого препарата и плацебо может быть связана с применением недостаточно чувствительного метода оценки эффекта или низкой дозой ЛС). Исследования со сравнительным дизайном требуют проведения рандомизации - распределения испытуемых на опытную и контрольную группы случайным образом, что позволяет создать схожие исходные условия и свести к минимуму систематическую ошибку и предвзятость отбора больных. Процесс рандомизации, длительность лечения, последовательности периодов лечения и критерии прекращения испытания отражают в дизайне исследования. С проблемой рандомизации тесно связана проблема «слепоты» исследования. Цель слепого метода - устранение возможности влияния (осознанного или случайного) врача, исследователя, пациента на полученные результаты. Идеально испытание с использованием двойного слепого метода, когда ни пациент, ни врач не знают, какое лечение получает пациент.
Исследователь может получить доступ к информации о том, какое именно ЛС получает пациент (это может потребоваться при возникновении серьёзных нежелательных реакций), но в этом случае пациент должен быть исключён из исследования.
Индивидуальная регистрационная карта
Под индивидуальной регистрационной картой понимают «печатный, оптический или электронный документ, созданный для регистрации всей требуемой в протоколе информации о каждом субъекте исследования». ИРК служит связующим информационным звеном между исследователем и спонсором исследования. На основании индивидуальных регистрационных карт создают базу данных исследования для проведения статистической обработки результатов.
Регистрация нежелательных явлений
Проводят на всех этапах исследования. В протоколах с I по III фазу должны быть описаны методы мониторинга нежелательных явлений. При этом регистрируется любое изменение самочувствия или объективных показателей испытуемого, возникшее в период приёма препарата и после окончания лечения, даже в том случае, когда связь этого явления с приёмом препарата представляется более чем сомнительной.
Фазы клинического исследования
Производитель и общество заинтересованы в том, чтобы в ходе исследований, предшествующих регистрации нового ЛС, была получена как можно более точная и полная информация о клинической фармакологии, терапевтической эффективности и безопасности нового ЛС. Подготовка регистрационного досье невозможна без ответа на эти вопросы. Общий цикл исследований нового ЛС обычно превышает 10 лет (рис. 9-1). В связи с этим неудивительно, что разработка новых ЛС остаётся уделом только крупных фармацевтических компаний, а общая стоимость исследовательского проекта превышает 500 млн долларов США.
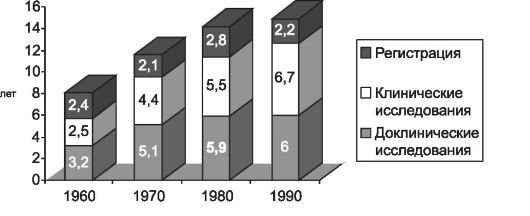 Рис. 9-1. Время, необходимое для разработки и внедрения нового ЛС.
Рис. 9-1. Время, необходимое для разработки и внедрения нового ЛС.
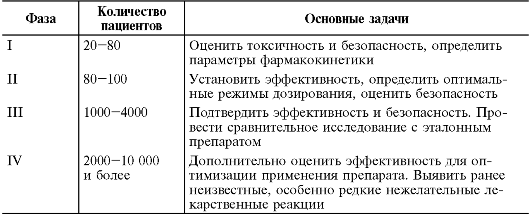
Клинические испытания нового ЛС - завершающая стадия длительного и трудоёмкого процесса их разработки. Клинические испытания ЛС перед их официальным разрешением к медицинскому применению проводят в 4 этапа, традиционно называемые «фазы клинических исследований» (табл. 9-1).
Таблица 9-1. Фазы клинических исследований ЛС
 Фаза
I - начальный этап клинических исследований, поисковый и особенно
тщательно контролируемый. Обычно клинические испытания I фазы проводят
на здоровых добровольцах мужского пола (18- 45 лет), однако при изучении
препаратов с высокой потенциальной токсичностью (например,
противоопухолевые, противоретровирусные препараты) может быть получено
разрешение на исследование у пациентов. Цель I фазы - получение
информации о максимальной безопасной дозе. Исследуемое соединение
назначают в низких дозах с постепенным их повышением до появления
признаков токсического действия, параллельно определяют концентрацию
препарата или его активных метаболитов в плазме крови, тщательно
контролируют клинические и лабораторные данные испытуемых для выявления
нежелательных лекарственных реакций. Начальную токсическую дозу
определяют в доклинических исследованиях, у человека она составляет 1/10
экспериментальной. Клинические испытания I фазы проводят в
специализированных клиниках, оснащённых оборудованием для оказания
экстренной медицинской помощи.
Фаза
I - начальный этап клинических исследований, поисковый и особенно
тщательно контролируемый. Обычно клинические испытания I фазы проводят
на здоровых добровольцах мужского пола (18- 45 лет), однако при изучении
препаратов с высокой потенциальной токсичностью (например,
противоопухолевые, противоретровирусные препараты) может быть получено
разрешение на исследование у пациентов. Цель I фазы - получение
информации о максимальной безопасной дозе. Исследуемое соединение
назначают в низких дозах с постепенным их повышением до появления
признаков токсического действия, параллельно определяют концентрацию
препарата или его активных метаболитов в плазме крови, тщательно
контролируют клинические и лабораторные данные испытуемых для выявления
нежелательных лекарственных реакций. Начальную токсическую дозу
определяют в доклинических исследованиях, у человека она составляет 1/10
экспериментальной. Клинические испытания I фазы проводят в
специализированных клиниках, оснащённых оборудованием для оказания
экстренной медицинской помощи.
Фаза II - ключевая, так как полученные сведения определяют целесообразность продолжения исследования нового ЛС. Цель - доказательство клинической эффективности и безопасности ЛС при испытании на чётко очерченных контингентах больных, установление оптимального режима дозирования. Сравнивают эффективность и безопасность изучаемого препарата с эталонным и плацебо. Испытания
Фазы II подразумевают наличие спланированного дизайна, чётких критериев включения/исключения, рандомизации, ослепления, процедур последующего контроля. Обычно эта фаза продолжается около 2 лет.
Фаза III - если препарат оказался эффективен и безопасен во II фазе, его исследуют в фазе III. Клинические испытания III фазы - контролируемые, многоцентровые исследования (исследования, проводимые по единому протоколу более чем в одном исследовательском центре), спланированные для определения безопасности и эффективности ЛС в условиях, приближённых к тем, в которых оно будет использовано в случае его разрешения к медицинскому применению. Полученные сведения уточняют эффективность лекарственного препарата у больных с учётом сопутствующих заболеваний, различных демографических характеристик и режима дозирования. Обычно исследования имеют сравнительный дизайн по отношению к существующей стандартной терапии. После завершения этой фазы и регистрации фармакологическое средство приобретает статус ЛС (процесса последовательных экспертных и административно-правовых действий) с занесением в Государственный реестр РФ и присвоением ему регистрационного номера.
Дженерики допускают в обращение после истечения срока патентной защиты оригинального препарата на основании оценки регистрационного досье сокращенного объёма и данных по биоэквивалентности.
Конкуренция с новыми препаратами заставляет продолжать исследования и после регистрации, для того чтобы подтвердить эффективность препарата и его место в фармакотерапии.
Фаза IV (постмаркетинговые исследования). Клинические испытания IV фазы проводят после того, как препарат разрешён для клинического применения по определённому показанию. Цель IV фазы - уточнение особенностей действия ЛС, дополнительная оценка его эффективности и безопасности у большого количества пациентов. Расширенные пострегистрационные клинические исследования характеризуются широким применением нового лекарственного препарата в медицинской практике. Их назначение - выявить ранее неизвестные, особенно редкие побочные эффекты, а также случаи лекарственного взаимодействия на большой и разнородной популяции пациентов, влияние отдаленных эффектов препарата на выживаемость (снижение или повышение уровня смертности). Полученные данные могут послужить основанием для внесения соответствующих изменений в инструкцию по медицинскому применению препарата. Несмотря на существенные затраты и строгую оценку эффективности лишь 1 из
каждых 10 новых зарегистрированных препаратов занимает лидирующее положение на рынке ЛС, принося производителю значительную прибыль. Другие 8 новых зарегистрированных ЛС примерно окупают расходы на своё создание, и ещё 1 препарат из 10 причиняет убытки своему производителю и/или снимается с производства.
ДОКАЗАТЕЛЬНАЯ МЕДИЦИНА
Предложенная в начале 90-х годов концепция доказательной медицины, или медицины, основанной на доказательствах (evidence-based medicine), подразумевает добросовестное, точное и осмысленное использование лучших результатов клинических исследований для выбора лечения конкретного больного. Подобный подход позволяет уменьшить количество врачебных ошибок, облегчить процесс принятия решения для практических врачей, администрации лечебных учреждений и юристов, а также снизить расходы на здравоохранение. В концепции доказательной медицины рассматривают методы корректной экстраполяции данных рандомизированных клинических исследований для решения практических вопросов, связанных с лечением конкретного больного. При этом доказательная медицина - концепция или метод принятия решений, она не претендует, чтобы её выводы полностью определяли выбор ЛС и другие аспекты лечебной работы.
Доказательная медицина призвана решать важные вопросы.
• Можно ли доверять результатам клинического исследования?
• Каковы эти результаты, насколько они важны?
• Можно ли использовать эти результаты для принятия решений при лечении конкретных больных?
Уровни (классы) доказательности
Удобным механизмом, позволяющим специалисту оценить качество любого клинического исследования и достоверность полученных данных, служит предложенная в начале 90-х годов рейтинговая система оценки клинических исследований. Обычно выделяют от 3 до 7 уровней доказательности, при этом с возрастанием порядкового номера уровня качество клинического исследования снижается, а результаты представляются менее достоверными или имеющими лишь ориентировочное значение. Рекомендации из исследований различного уровня принято обозначать латинскими буквами A, B, C, D.
Уровень I (А) - хорошо разработанные, крупные, рандомизированные, двойные слепые, плацебоконтролируемые исследования. К этому же уровню доказательности принято относить данные, получен-
ные в результате метаанализа нескольких рандомизированных контролируемых исследований.
Уровень II (В) - небольшие рандомизированные и контролируемые исследования (если статистически корректные результаты не получены из-за малого количества больных, включённых в исследование).
Уровень III (С) - исследования «случай-контроль», или когортные исследования (иногда их относят к уровню II).
Уровень IV (D) - сведения, содержащиеся в отчётах экспертных групп или консенсусах специалистов (иногда их относят к уровню III).
«Конечные точки» в клинических исследованиях
Для оценки эффективности нового ЛС по результатам клинических исследований могут быть использованы первичные, вторичные и третичные «конечные точки». Эти основные показатели оценивают в контролируемых сравнительных исследованиях по результатам лечения по крайней мере в двух группах: основной (больные получают новый способ лечения или новый препарат) и группе сравнения (больные не получают изучаемый препарат или принимают известный препарат сравнения). Например, при исследовании эффективности лечения и профилактики ИБС выделяют следующие «конечные точки».
• Первичные - основные показатели, связанные с возможностью увеличения продолжительности жизни больного. В клинических исследованиях к ним относят снижение общей смертности, смертности от сердечно-сосудистых заболеваний, в частности инфаркта миокарда и инсульта.
• Вторичные показатели отражают улучшение качества жизни либо вследствие снижения заболеваемости, либо облегчения симптомов заболевания (например, уменьшение частоты приступов стенокардии, увеличение толерантности к физической нагрузке).
• Третичные - показатели, связанные с возможностью профилактики заболевания (например, у больных с ИБС - стабилизация АД, нормализация содержания глюкозы в крови, снижение концентрации общего холестерина, ЛПНП и т.д.).
Метаанализ - метод поиска, оценки и объединения результатов нескольких контролируемых исследований. В результате метаанализа можно установить положительные или нежелательные эффекты лечения, которые не могут быть выявлены в отдельных клинических исследованиях. Необходимо, чтобы включённые в метаанализ исследования были тщательно рандомизированы, их результаты опубликованы с подробным протоколом исследования, указанием критериев отбора
и оценки, выбора конечных точек. Например, в двух метаанализах установлено благоприятное действие лидокаина при аритмии у больных с инфарктом миокарда, а в одном - увеличение количества смертельных случаев, что является наиболее важным показателем для оценки действия этого препарата. Целесообразность назначения низких доз аспирина для снижения смертности и развития сердечно-сосудистых осложнений у пациентов высокого риска была установлена на основании метаанализа 65 рандомизированных клинических исследований, в которые были включены около 60 000 пациентов.
Значение доказательной медицины в клинической практике
В настоящее время концепцию доказательной медицины широко используют при решении вопроса о выборе ЛС в конкретных клинических ситуациях. Современные руководства по клинической практике, предлагая те или иные рекомендации, снабжают их рейтингом доказательности. Существует также международная Кокрановская инициатива (библиотека Кокрана), объединяющая и систематизирующая все накопленные в этой области сведения. При выборе ЛС наряду с рекомендациями лекарственного формуляра используют международные или национальные руководства по клинической практике, то есть систематически разработанные документы, предназначенные для облегчения практикующему врачу, юристу и пациенту принятия решений в определённых клинических ситуациях. Однако исследования, проведённые в Великобритании, показали, что врачи общей практики далеко не всегда склонны применять национальные рекомендации в своей работе. Кроме того, создание чётких систем рекомендаций вызывает критику со стороны специалистов, полагающих, что их применение ограничивает свободу клинического мышления. С другой стороны, использование подобных руководств стимулировало отказ от рутинных и недостаточно эффективных методов диагностики и лечения и в конечном счёте повысило уровень медицинской помощи больным.
В заключение следует отметить, что результаты современных клинических исследований нельзя признать окончательными и абсолютно надёжными. Очевидно, что эволюционные скачки в изучении новых ЛС происходили и будут происходить, что приводит и будет приводить к принципиально новым клинико-фармакологическим представлениям, а следовательно, и к новым методическим подходам к изучению препаратов при проведении клинических испытаний.