Клиническая фармакология.: учебник для вузов / Под ред. В.Г. Кукеса.- 4-е издание., перераб. и доп., - 2009. - 1056 с.
|
|
|
|
ГЛАВА 7. КЛИНИЧЕСКАЯ ФАРМАКОГЕНЕТИКА
Фармакогенетика - наука, изучающая роль генетических факторов в формировании фармакологического ответа организма человека на ЛС. Фармакогенетика возникла на стыке фармакологии и генетики. Хотя роль наследственности в формировании индивидуального ответа на ЛС была известна давно, понимание механизмов влияния генетических факторов на эффективность и безопасность фармакотерапии стало возможным лишь в связи с развитием методов молекулярной биологии и реализацией международной программы «Геном человека». Фармакокинетические и фармакодинамические процессы, протекающие с участием различных белков организма человека (ферментов, ионных каналов, молекул-переносчиков, рецепторов и т.д.), находятся под генетическим контролем.
Различные наследуемые изменения (мутации) в генах, кодирующих эти белки, могут приводить к изменению фармакокинетики и/или фармакодинамики ЛС, в результате чего изменяется фармакологический ответ. Такие мутации, передаваясь из поколения в поколение, могут распространяться в популяции. Явление, когда в популяции существуют различные аллельные варианты одного и того же гена, носит название генетического полиморфизма. Гены, для которых известен множественный аллелизм, называются полиморфными маркёрами. В случае наличия закономерности между определённым аллельным вариантом такого маркёра и каким-либо фенотипическим признаком говорят об ассоциации полиморфного маркёра с данным явлением. Полиморфизм соответствующих генов может быть причиной различий в фармакологическом ответе на ЛС. В последние два десятилетия после разработки метода ПЦР стало возможным диагностировать такие маркёры у пациентов. Этот метод получил название генотипирования.
Методы генотипирования позволяют прогнозировать фармакологический ответ на ЛС, а значит, повысить эффективность и безопасность применения ЛС. При выявлении соответствующего аллельного варианта у больного необходима коррекция терапии (доза, путь введения и его кратность, замена ЛС и т.д.). Этот подход лежит в основе так называемой персонализированной медицины. Изучение генов, ответственных за фармакокинетику и фармакодинамику ЛС, широко внедряется в клиническую практику во всех развитых странах.
В настоящее время проводится разработка и внедрение генетических микрочипов (microarray-technology) для обнаружения мутантных аллелей, ответственных за изменение фармакологического ответа. Данная задача стала приоритетной в новом направлении клинической фармакологии - фармакогеномике. Следует отметить, что некоторые полиморфные маркёры, ассоциированные с изменением фармакокинетики и фармакодинамики ЛС, часто приводят к развитию также некоторых заболеваний (онкопатология, болезнь Альцгеймера, болезнь Паркинсона, атеросклероз и др.). По этой причине фармакогенетические исследования способствуют более полному пониманию этиологии и патогенеза этих заболеваний.
С клинических позиций генетически детерминированные изменения фармакологического ответа можно классифицировать следующим образом:
• ЛС, приводящие к возникновению побочных эффектов (например, дефицит глюкозо-6-фосфатдегидрогеназы), применение ЛС в этом случае противопоказано;
• ЛС, приводящие к нежелательным реакциям, не относящимся к серьёзным (например, носительство «медленных» аллельных вариантов гена CYP2D6, приводящее к фенотипу «медленного метаболизатора»); ЛС назначают в низкой дозе;
• неэффективные ЛС или ЛС с низкой эффективностью (например, дупликация функциональных аллелей гена CYP2D6, приводящая к фенотипу «быстрого метаболизатора»), ЛС используют в высокой дозе.
Генетические факторы, влияющие на фармакокинетику лекарственных средств
Все этапы фармакокинетики ЛС (всасывание, распределение, биотрансформация, или метаболизм, выведение) находятся под контролем соответствующих генов, кодирующих ферменты биотрансформации ЛС и транспортёры ЛС.
Генетический полиморфизм характерен для генов, кодирующих ферменты как первой (изоферменты цитохрома Р-450, дигидропиримидин дигидрогеназа, бутирилхолинэстераза, параоксоназа), так и второй фазы метаболизма (N-ацетилтрансфераза, тиопурин S-метил- трансфераза, эпоксид гидролаза). Генетический полиморфизм может приводить к синтезу ферментов с изменённой активностью, что может влиять на скорость биотрансформации ЛС (замедление или ускорение). Скорость биотрансформации ЛС можно оценить по отношению концентрации ЛС-субтрата к концентрации его метаболита в плазме крови или в моче (так называемое метаболическое отношение). Гене-
тически детерминированные межиндивидуальные различия по этому показателю позволяют выделить группы индивидуумов, различающиеся по активности того или иного фермента биотрансформации.
• «Экстенсивные» метаболизаторы (extensive metabolism, ЕМ) - лица с нормальной скоростью биотрансформации определённых ЛС; как правило, гомозиготы по «дикому» аллелю гена соответствующего фермента. К «экстенсивным» метаболизаторам принадлежит большинство населения.
• «Медленные» метаболизаторы (poor metabolism, РМ) - лица со сниженной скоростью биотрансформации определённых ЛС; как правило, гомозиготы или гетерозиготы по «медленному» аллелю гена соответствующего фермента. Иногда выделяют и «промежуточных» метаболизаторов (intermedium metabolism, IM), к которым относят гетерозигот по «медленному» аллелю (при аутосомно-рецессивном типе наследования). У этих индивидуумов происходит синтез «дефектного» фермента либо вообще отсутствует синтез фермента биотрансформации. В результате ферментативная активность снижается или отсутствует. У этой категории лиц регистрируются высокие значения отношения концентрации ЛС к концентрации его метаболита. При этом у «медленных» метаболизаторов ЛС накапливается в организме в высоких концентрациях, что приводит к появлению выраженных нежелательных лекарственных реакций, вплоть до интоксикации. В связи с этим для «медленных» метаболизаторов должен быть осуществлён тщательный подбор дозы ЛС: она должна быть меньше по сравнению с назначаемой «экстенсивным» метаболизаторам.
• «Сверхактивные», или «быстрые», метаболизаторы (ultraextensivc metabolism, UM) - лица с повышенной скоростью биотрансформации определённых ЛС. Это, как правило, гомозиготы (при аутосомно-рецессивном типе наследования) или гетерозиготы (при аутосомно-доминантном типе наследования) по «быстрому» аллелю гена соответствующего фермента или, что встречается чаще, несущие копии функциональных аллелей. У этой категории лиц регистрируют низкие значения отношения концентрации ЛС к концентрации его метаболита. В результате концентрация ЛС в крови недостаточна для достижения терапевтического эффекта. Для «сверхактивных» метаболизаторов доза ЛС должна быть выше, чем такая для активных метаболизаторов.
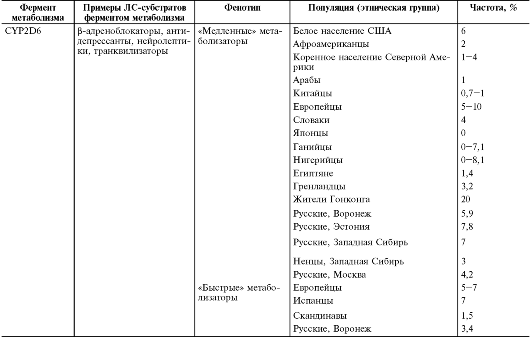
Распространённость генотипов «медленных» и «быстрых» метаболизаторов по отдельным ферментам биотрансформации ЛС в различных популяциях (этнических группах) представлена в табл. 7-1.
Таблица 7-1. Распространённость «медленных» и «быстрых» метаболизатров по отдельным ферментам метаболизма в различных популяциях
 Продолжение табл. 7-1
Продолжение табл. 7-1
 Окончание табл. 7-1
Окончание табл. 7-1
 Исследования
подобного рода весьма актуальны. Они могут определять целесообразность
внедрения методов генотипирования по ферментам метаболизма ЛС в
регионах, в которых проживают определён- ные этнические группы.
Исследования
подобного рода весьма актуальны. Они могут определять целесообразность
внедрения методов генотипирования по ферментам метаболизма ЛС в
регионах, в которых проживают определён- ные этнические группы.
Генетический контроль всасывания, распределения и выведения ЛС изучен недостаточно хорошо, его клиническое значение требует уточнения. Однако есть данные о роли генетического полиморфизма гена гликопротеина Р (белок-переносчик ЛС в энтероцитах) в изменении фармакокинетики некоторых ЛС (дигоксин и др.). Ниже описаны наиболее клинически значимые генетические полиморфизмы, влияющие на фармакокинетику ЛС.
Генетический полиморфизм изофермента цитохрома Р-450 2D6 (CYP2D6)
Изофермент цитохрома Р-450 2D6 метаболизирует около 20% всех известных ЛС, в том числе антипсихотические ЛС, антидепрессанты, β-адреноблокаторы (см. главу 1 «Клиническая фармакокинетика»). В конце 70-х гг. XX в. были выявлены отличия гипотензивного эффекта у больных артериальной гипертензией, применявших дебризохин*, ЛС из группы α-адреноблокаторов. Тогда же было сформулировано предположение о различии в скорости метаболизма (гидроксилирования) дебризохина у разных индивидуумов. У «медленных» метаболизаторов дебризохина гипотензивный эффект этого ЛС был наиболее выражен. Позднее было показано, что у «медленных» метаболизаторов дебризохина замедлен метаболизм и некоторых других ЛС, в том числе нортриптилина, фенформина®, L-спартеина сульфата*, энкаинида*, пропранолола, амитриптилина , метопролола . Это становится причиной изменения фармакологического ответа при применении ЛС-субстратов CYP2D6 у «медленных» метаболизаторов по CYP2D6. Например, у «медленных» метаболизаторов по CYP2D6 при применении кардиоселективного β-адреноблокатора метопролола гораздо чаще наблюдается такой побочный эффект, как бронхоспазм. Это связано с тем, что у «медленных» метаболизаторов по CYP2D6 метопролол накапливается в крови в таких высоких концентрациях, при которых кардиоселективность этого препарата исчезает.
В качестве ещё одного примера изменения фармакологического ответа можно привести более выраженный β-адреноблокирующий эффект антиаритмика пропафенона у «медленных» метаболизаторов по CYP2D6. Это связано с тем, что пропафенон метаболизируется до 5-гидроксипропафенона, который и обладает β-адреноблокирующим эффектом. 5-Гидроксипропафенон метаболизируется CYP2D6, поэтому у «медленных» метаболизаторов по CYP2D6 концентрация 5-гид- роксипропафенона будет повышена.
Риск возникновения побочных эффектов нейролептиков (экстрапирамидные расстройства) и трициклических антидепрессантов (гипотония, ажитация, сонливость) также выше у пациентов «медленных» метаболизаторов по CYP2D6. В качестве примера можно привести метаболизм трициклического антидепрессанта имипрамина у «медленных» метаболизаторов по CYP2D6. Имипрамин сначала подвергается N-деметилированию за счёт изофермента CYP2C19 до активных метаболитов: дезипрамина и нортриптилина. Дезипрамин и нортриптилин, в свою очередь, метаболизируются изоферментом CYP2D6 путём 4-гидроксилирования до неактивных метаболитов. Было показано, что у «медленных» метаболизаторов по CYP2D6 почти всегда отмечаются такие выраженные побочные эффекты имипрамина, как гипотензия, седативный эффект, тремор, кардиотоксичность. Это связано с наличием в крови высоких концентраций имипрамина, а также его активных метаболитов дезипрамина и нортриптилина. Как правило, «медленным» метаболизаторам по CYP2D6 для предотвращения нежелательных лекарственных реакций и интоксикации необходимо назначение ЛС-субстратов CYP2D6 в меньших дозах. Фармакологический ответ на кодеин у «медленных» метаболизаторов по CYP2D6 также изменён: у «медленных» метаболизаторов по CYP2D6 менее выражен анальгетический эффект кодеина. Этот феномен объясняется снижением О-деметилирования кодеина, при котором образуется морфин. Аналогичным образом анальгетический эффект трамадола также менее выражен у «медленных» метаболизаторов по CYP2D6.
Есть примеры, когда у «медленных» метаболизаторов по CYP2D6 могут реже возникать побочные эффекты при применении некоторых ЛС, что связано с замедлением образования метаболитов, вызывающих эти реакции. Так, при длительном применении антиаритмического ЛС прокаинамида у «медленных» метаболизаторов по CYP2D6 гораздо реже наблюдается такой побочный эффект, как волчаночноподобный синдром. Это, по-видимому, обусловлено тем, что за развитие волчаночноподобного синдрома ответственен метаболит прокаинамида N-гидроксипрокаинамид, образующийся в результате N-гидроксили- рования прокаинамида под действием CYP2D6.
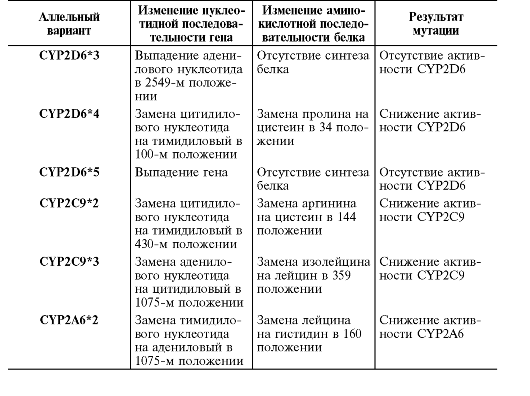
Генетические исследования показали, что «медленные» метаболизаторы по CYP2D6 являются носителями (гомозиготами) мутантных аллелей гена цитохрома 2D6. Результатом этих мутаций может быть отсутствие синтеза CYP2D6, синтез дефектного белка, не обладающего активностью или со сниженной активностью. На сегодняшний день таких мутантных аллелей выявлено более 30, однако 95% всех «медленных» метаболизаторов по CYP2D6 оказываются носителями трёх «медленных» аллельных вариантов: CYP2D6*3A, CYP2D6*4A,
CYP2D6*5 (см. табл. 7.2). Все эти аллельные варианты наследуются по аутосомно-рецессивному типу. Распространённость «медленных» метаболизаторов среди населения сильно колеблется: от 1% среди арабского населения до 30% среди жителей Гонконга. Распространённость «медленных» метаболизаторов в Европе в среднем составляет 5-10%, среди афроамериканцев - 1,8%, китайцев - 1%, среди японского населения «медленные» метаболизаторы по CYP2D6 практически не встречаются (см. табл. 7-1). Кроме того, есть данные о том, что у «медленных» метаболизаторов по CYP2D6 чаще развиваются некоторые злокачественные новообразования: рак мочевого пузыря, желудка, глотки, лёгких (в особенности у курильщиков), первичный рак печени. Предполагают, что причиной более частого возникновения рака лёгкого у курящих «медленных» метаболизаторов по CYP2D6 является их неспособность метаболизировать никотин.
Кроме «медленных» метаболизаторов по CYP2D6, существуют и «быстрые» метаболизаторы, распространённость которых в Европе достигает 20% (см. табл. 7-1). У «быстрых» метаболизаторов выявляется «дупликация» гена CYP2D6. Эта мутация наследуется по аутосомнорецессивному типу. У «быстрых» метаболизаторов по CYP2D6 при применении ЛС-субстратов CYP2D6 отмечается снижение их терапевтической эффективности за счёт ускорения метаболизма ЛС-субстратов CYP2D6, что приводит к снижению их концентрации в плазме крови. Например, противорвотное ЛС ондансетрон у больных, являющихся «быстрыми» метаболизаторами по CYP2D6, не предотвращает рвоту при проведении химиотерапии злокачественных опухолей. Поэтому «быстрым» метаболизаторам требуется назначение ЛС-субстратов по CYP2D в больших дозах. В настоящее время уже разработаны алгоритмы выбора нейролептиков и антидепрессантов и их режимов дозирования в зависимости от генотипов по CYP2D6.
Генетический полиморфизм изофермента цитохрома Р-450 2С9 (CYP209)
CYP2С9 метаболизирует 18,5% всех известных ЛС, в том числе многие НПВС, фенитоин, гипогликемические средства для приёма внутрь (производные сульфонилмочевины), S-варфарин (см. главу 1 «Клиническая фармакокинетика»). При применении ЛС-субстратов CYP2С9 выяснилось, что у части людей («медленные» метаболизаторы) снижен клиренс этих ЛС, и соответственно у них чаще наблюдались побочные эффекты. У «медленных» метаболизаторов по CYP2С9 чаще наблюдается гипогликемия при применении пероральных гипогликемических средств (толбутамида, глипизида, глибенкламида), геморрагические осложнения при применении непрямых антикоагулянтов (варфарина, аценокумарола). Генетические методы показали, что «медленные»
метаболизаторы несут аллельные варианты CYP2C9*2 и CYP2C9*3 (табл. 7.2). Аллельный вариант CYP2C9*2 представляет собой замену в нуклеотидной последовательности в 430-м положении цитидилового нуклеотида на тимидиловый, в результате чего в аминокислотной последовательности CYP2С9 в 144-м положении аргинин заменяется на цистеин. При этом образуется CYP2С9.2 со сниженной активностью (15% активности нормального CYP2С9).
В случае аллельного варианта CYP2C9*3 в нуклеотидной последовательности в 1075-м положении адениловый нуклеотид заменяется на цитидиловый, что приводит к замене в аминокислотной последовательности CYP2С9 в 359-м положении изолейцина на лейцин. При этом образуется CYP2С9.3 со сниженной активностью (5% активности нормального CYP2С9). Распространённость «медленных» метаболизаторов по CYP2С9 следующая: в США - 0,06%, среди афроамериканцев - 0,005%, в Китае - 0,026%, среди европейского населения - 1- 3% (см. табл. 7.1). «Медленные» метаболизаторы по CYP2С9 требуют назначение в меньших дозах таких ЛС, которые метаболизируются
Таблица 7-2. Наиболее распространённые клинически значимые «медленные» аллельные варианты изоферментов цитохрома Р-450
 CYP2С9.
Это относится, прежде всего, к ЛС с узкой терапевтической широтой:
пероральным гипогликемическим средствам (толбутамид, глипизид,
глибенкламид) и непрямым антикоагулянтам (варфарин, аценокумарол). В
настоящее время разработан алгоритм выбора начальной дозы варфарина в
зависимости от генотипов по CYP2C9 (www.warfarindosing.org).
CYP2С9.
Это относится, прежде всего, к ЛС с узкой терапевтической широтой:
пероральным гипогликемическим средствам (толбутамид, глипизид,
глибенкламид) и непрямым антикоагулянтам (варфарин, аценокумарол). В
настоящее время разработан алгоритм выбора начальной дозы варфарина в
зависимости от генотипов по CYP2C9 (www.warfarindosing.org).
Генетический полиморфизм изофермента цитохрома Р-450 2С19 (CYP2d9)
CYP2C19 метаболизирует 8,3% всех известных ЛС, в том числе имипрамин, диазепам, барбитураты, вальпроевую кислоту, противомалярийные препараты (см. главу 1 «Клиническая фармакокинетика»). «Медленные» метаболизаторы по CYP2C19 оказываются носителями ряда мутантных аллелей (более десяти). Распространённость «медленных» метаболизаторов по CYP2C19 среди европейского населения составляет 2-5%, среди азиатского населения 15-20% (см. табл. 7.1). Применение у «медленных» метаболизаторов по CYP2C19 ЛС-субстратов CYP2C19 приводит к более частому возникновению побочных эффектов, в особенности при применении ЛС с узкой терапевтической широтой, таких, как трициклические антидепрессанты, диазепам, некоторые барбитураты. С другой стороны, у «медленных» метаболизаторов по CYP2C19 ингибиторы протонного насоса (омепразол, пантопразол, лансопразол) вызывают более выраженный антисекреторный эффект. Есть данные о более высоком риске злокачественных новообразований головы и шеи у «медленных» метаболизаторов по CYP2C19.
Генетический полиморфизм дигидропиримидин дигидрогеназы
Физиологическая функция фермента дигидропиримидин дигидрогеназы (ДПДГ) - восстановление урацила и тимидина. Это первая реакция трёхэтапного метаболизма этих соединений до β-аланина. Кроме того, ДПДГ является основным ферментом, который метаболизирует фторурацил (5-фторурацил*). Он широко применяется в составе комбинированной химиотерапии рака молочной железы, яичников, пищевода, желудка, толстой и прямой кишки, печени, шейки матки, вульвы, мочевого пузыря, предстательной железы, опухолях головы, шеи, слюнных желёз, надпочечников, поджелудочной железы.
С середины 80-х годов появились сообщения о серьёзных осложнениях, возникающих при применении фторурацила, связанных с низкой активностью ДПДГ. Позже было показано, что низкая активность ДПДГ наследуется по аутосомно-рецессивному типу. У пациентов с низкой активностью ДПДГ отмечается удлинённый период полувыведения фторурацила (до 160 мин, при нормальном периоде полувыве-
дения 8-22 мин). Есть чёткая закономерность: чем ниже активность ДПДГ, тем тяжелее побочные эффекты (нейротоксичность, кардиотоксичность) фторурацила.
Генетические исследования последних лет, проводимые с помощью метода ПЦР, позволили выявить ряд мутаций гена ДПДГ, ответственных за сниженную активность этого фермента, а следовательно, и за повышенную чувствительность к фторурацилу. Наиболее рас- пространёнными мутациями оказались делеция в 165-м положении, замена гуанилового нуклеотида на адениловый в 14-м положении и сочетание этих двух мутаций. На сегодняшний день распространён- ность гомозигот по мутантным аллелям гена ДПДГ известна только среди японцев и составляет 1 на 10 000. Однако следует отметить, что повышенная чувствительность к фторурацилу, отмечается не только у гомозигот, но и у гетерозигот по мутантным аллелям гена ДПДГ. ДПДГ можно считать ферментом, обладающим генетическим полиморфизмом. В будущем, по-видимому, ожидается внедрение методов фенотипирования и генотипирования ДПДГ в онкологическую практику для обеспечения безопасности химиотерапии фторурацилом.
Генетический полиморфизм бутирилхолинэстеразы (псевдохолинэстеразы)
Физиологическая функциия бутирилхолинэстеразы - гидролиз ацетилхолина. Кроме того, бутирилхолинэстераза катализирует реакцию гидролиза деполяризующего миорелаксанта суксаметония. Суксаметония йодид (дитилин*, сукцинилдихолин*) широко применяется в анестезиологии. С начала 50-х годов появились сообщения о повышенной чувствительности к суксаметонию, которая обусловлена сниженной активностью бутирилхолинэстеразы. Бутирилхолинэстеразу со сниженной активностью в литературе часто называют атипичной псевдохолинэстеразой. Ещё в 50-е годы XX в. были описаны случаи продолжительной остановки дыхания (апноэ) при применении суксаметония: вместо 2-3 мин апноэ у лиц с парадоксальной реакцией продолжалось два часа и более.
В начале 70-х годов ХХ в. предположили возможность аутосомнорецессивного моногенного наследования низкой активности бутирилхолинэстеразы. Генетические исследования последних лет, проведён- ные с помощью ПЦР, позволили выявить ряд мутаций гена бутирилхолинэстеразы. Повышенная чувствительность к суксаметонию наблюдается только у гомозигот. Наиболее распространённой мутацией, приводящей к синтезу бутирилхолинэстеразы со сниженной активностью, оказалась замена в нуклеотидной последовательности в 209-м положении аденилового нуклеотида на гуаниловый. В результате синтезируется фермент, у которого в 70-м положении аспарагинат заменён
на глицин. Его обычно называют атипичной бутирилхолинэстеразой 1. Гомозиготы по этому мутантному аллелю проявляют повышенную чувствительность к суксаметонию. Распространённость гомозигот среди белого североамериканского населения составляет 1:3000.
Другая мутация, приводящая к синтезу бутирилхолинэстеразы с резко сниженной активностью, - «вставка» в 117-м положении нуклеотидной последовательности. В результате синтезируется белок, длина которого - 22% «длины» нормальной бутирилхолинэстеразы. Его в литературе называют «тихой» бутирилхолинэстеразой. Гомозиготы по этой мутантной аллели также проявляют повышенную чувствительность к суксаметонию. Распространённость гомозигот среди белого североамериканского населения 1:100 000.
Замена в 243-м положении треонина на метионин - ещё одна мутация, приводящая к синтезу фермента со сниженной активностью. В результате синтезируется бутирилхолинэстераза, которую в литературе называют фторрезистентной бутирилхолинэстеразой 1. Гомозиготы по этому мутантному аллелю также проявляют повышенную чувствительность к суксаметонию, однако период апноэ у них длится меньше, около 30 мин. Распространённость гомозигот среди белого североамериканского населения 1:150 000. Частота гетерозигот и гомозигот по всем мутантным аллелям, которые определяют сниженную активность бутирилхолинэстеразы среди европейского населения, составляет соответственно 2-4% и 1:2500.
Фенотипирование бутирилхолинэстеразы для определения её сниженной активности осуществляется с помощью так называемого дибукаинового теста, основанного на подавлении активности бутирилхолинэстеразы дибукаином в стандартных условиях. Результат теста представляется в виде «дибукаинового числа», которое является степенью подавления фермента, выраженной в процентах. У людей с нормальной активностью бутирилхолинэстеразы дибукаиновое число равно 80%. У гомозигот по мутантным аллелям, которые определяют сниженную активность бутирилхолинэстеразы, дибукаиновое число равно 20%, а у гетерозигот по этим аллелям - 60%. Таким образом, дибукаиновый тест позволяет не только выявлять лиц с повышенной чувствительностью к суксаметонию, но и гетерозигот. Это важно для профилактики осложнений при применении суксаметония у потомства. Генотипирование бутирилхолинэстеразы с помощью метода ПЦР используется пока только в научных исследованиях, однако внедрение этого метода в клиническую практику позволит более точно выявлять лиц с повышенной чувствительностью к суксаметонию и обеспечить высокую безопасность применения суксаметония.
Генетический полиморфизм параоксоназы (ароматической эстеразы)
Параоксоназа - фермент из группы арилэстераз. Свое название фермент приобрёл из-за способности метаболизировать параоксон, антихолинэстеразный препарат, применяемый местно при глаукоме. Кроме параоксона, параоксоназа инактивирует путём эфирного гидролиза такие ксенобиотики, как фосфорорганические соединения, органофосфаты, карбаматы, эфиры уксусной кислоты. Эти соединения широко применяются в сельском хозяйстве и промышленности, используются в качестве лекарственных препаратов [антихолинэстераз- ные средства параоксон, индигокармин (армин*)], а некоторые из них являются боевыми отравляющими веществами (зарин, зоман).
С начала 70-х гг. появляются сообщения о наличии различий в активности параоксоназы среди населения. Лишь в 90-е годы были идентифицированы мутации, приводящие к изменению активности параоксоназы. Таким образом был доказан генетический полиморфизм параоксоназы. Наиболее распространённой мутацией, обуславливающей изменение активности параоксоназы, оказалась замена в ферменте в 192-м положении глутамина на аргинин (мутация Gln192Arg). Эта мутация наследуется по аутосомно-рецессивному типу. Носители мутации параоксоназы Gln192Arg, особенно гомозиготы, более чувствительны в отношении фосфорорганических соединений. По данным этих же авторов, распространённость гомозигот по этой мутации среди испанского населения составляет 16%, среди североевропейского населения - 9%. Наибольшая распространённость этой мутации зафиксирована в Японии и составляет 41,4%. Именно это обстоятельство явилось причиной больших жертв при применении зарина во время террористического акта в Токийском метро в марте 1995 г.
Генетический полиморфизм N-ацетилтрансферазы
N-ацетилтрансфераза катализирует реакцию ацетилирования ряда ЛС, в том числе изониазида, сульфаниламидов, прокаинамида, гидралазина и др. Выделено два изофермента N-ацетилтрансферазы: N-ацетилтрансфераза 1 (NAT1) и N-ацетилтрансфераза 2 (NAT2). Изофермент NAT1 ацетилирует небольшое количество ариламинов и не обладает генетическим полиморфизмом. Таким образом, основной фермент ацетилирования - изофермент NAT2.
Впервые полиморфизм ацетилирования был описан в 1960 г., при этом были выделены «медленные» и «быстрые» ацетиляторы изониазида. Тогда было отмечено, что у «медленных» ацетиляторов в связи с накоплением (кумуляцией) изониазида чаще наблюдаются полиневриты. Так, у «медленных» ацетиляторов период полувыведения изониазида составляет 3 ч, в то время как у «быстрых» ацетиляторов 1,5 ч.
Появление полиневритов связано с тем, что изониазид тормозит переход пиридоксина (витамина В6) в активный кофермент дипиридоксинфосфат, который необходим для синтеза миелина. Индивидуальная скорость ацетилирования существенно не влияет на режимы дозирования ЛС при ежедневном приёме, но может уменьшать эффективность терапии при прерывистом применении изониазида.
При применении изониазида для лечения туберкулёза в составе комбинированной терапии у «медленных» ацетиляторов закрытие полостей в лёгких идёт быстрее. «Медленные» ацетиляторы являются гомозиготами по «медленной» аллели NAT2, а быстрые метаболизаторы - гомозиготами либо гетерозиготами по «быстрой» аллели NAT2. Позднее было показано, что полиморфизм ацетилирования характерен не только для изониазида, но и для гидралазина и сульфаниламидов. Позже было обнаружено, что в этот список входят несколько десятков ЛС. Применение прокаинамида и гидралазина у «медленных» ацетиляторов гораздо чаще вызывает поражение печени (гепатотоксичность). Получены данные о том, что фенотип «быстрого» ацетилирования чаще встречается у светлоглазых и светловолосых людей. Также описан феномен снижения частоты встречаемости «медленных» ацетиляторов с севера на юг.
Распространённость «медленных» ацетиляторов широко варьирует от 10-15% среди монголоидов до 50% среди представителей европеоидной расы (см. табл. 7.1). Только с конца 80-х гг. начали идентифицировать мутации гена NAT2, приводящие к «медленному» ацетилированию. На сегодняшний день известно около 15 мутантных аллелей гена NAT2. Все эти мутации наследуются по аутосомно-рецессивному типу. Тип ацетилирования определяют как методами фенотипирования, так и генотипированием NAT2. В качестве маркёрных субстратов ацетилирования широко используются дапсон и сульфадимидин (сульфадимезин*). Отношение концентрации моноацетилдапсона к концентрации дапсона в плазме крови через шесть часов после введения препарата менее 0,35 характерно для медленных ацетиляторов, а более 0,35 - для быстрых ацетиляторов. В случае если в качестве маркёрного субстрата используется сульфадимидин, наличие менее 25% сульфадимидина в плазме через шесть часов и менее 70% в моче, собранной через 5-6 ч после введения препарата, говорит о фенотипе медленного ацетилирования.
Генетический полиморфизм тиопурин S-метилтрансферазы
Тиопурин S-метилтрансфераза (ТРМТ) - фермент, который катализирует реакцию S-метилирования производных тиопурина. Это основной путь метаболизма цитостатиков из группы антагонистов
пурина: меркаптопурина (6-меркаптопурин*), тиогуанина (6-тиог- уанин*), азатиоприна. Меркаптопурин (6-меркаптопурин*) широко используется в составе комбинированной химиотерапии миелобластного и лимфобластного лейкозов, хронического миелолейкоза, лимфосаркомы, саркомы мягких тканей. 6-тиогуанин применяется в основном при острых лейкозах. Активность ТРМТ имеет значительные различия: 88,6% людей имеют высокую активность ТРМТ, 11,1% промежуточную, а у 0,3% активность ТРМТ весьма низкая или вовсе отсутствует. При этом известно, что у людей с низкой активностью ТРМТ выявляют повышенную чувствительность к меркаптопурину, тиогуанину и азатиоприну, которая проявляется опасными для жизни гематотоксическим (лейкопения, тромбоцитопения, анемия) и гепатотоксическими эффектами. В условиях низкой активности ТРМТ метаболизм меркаптопурина идёт по альтернативному пути, до высокотоксичного соединения 6-тиогуанина нуклеотида (6TGN). Чем меньше активность ТРМТ, тем больше концентрации 6TGN в плазме крови и тем более выражены побочные эффекты меркаптопурина. Низкая активность ТРМТ наследуется по аутосомно-рецессивному типу, при- чём гомозиготы проявляют низкую активность ТРМТ, а гетерозиготы - промежуточную.
Генетические исследования последних лет, осуществлённые с помощью ПЦР, позволили выявить ряд мутаций гена ТРМТ, определяющих её низкую активность. Распространённость гомозигот по мутантным аллелям, определяющим низкую активность ТРМТ, среди европейского населения составляет 3,7%, среди афроамериканцев 4,6%. Повышенная чувствительность к тиопуринам отмечается не только у гомозигот, но и у гетерозигот по мутантным аллелям гена ТРМТ. Таким образом, для обеспечения безопасности проводимой химиотерапии перед назначением тиопуринов необходимо определять активность ТРМТ в эритроцитах пациента (фенотипирование ТРМТ) или определять генотип пациента с помощью ПЦР (генотипирование ТРМТ). Фенотипирование и генотипирование ТРМТ уже используется в клиниках Европы и США. Разработана коррекция дозировок меркаптопурина в зависимости от активности ТРМТ или генотипа этого фермента, при этом безопасные дозы для пациентов с низкой активностью ТРМТ должны быть в 10-15 раз ниже среднетерапевтических.
Генетический полиморфизм гликопротеина Р
Как указывалось в главе 1, генетический фактор может существенно влиять на активность гликопротеина Р. Наиболее распространённый полиморфный маркёр гена MDR1, кодирующего гликопротеин Р, - заме-
на в нуклеотидной последовательности в положении 3435 цитидилового нуклеотида на тимидиловый (полиморфный маркёр С3435Т). Это приводит к снижению экспрессии гена MDR1 и к снижению количества гликопротеина Р. У больных с генотипом ТТ по полиморфному маркёру С3435Т гена MDR1 наблюдаются высокие концентрации дигоксина в плазме крови и, следовательно, у них чаще развивается дигиталисная интоксикация. У этой группы пациентов также определяются более высокие концентрации циклоспорина, что сопровождается повышением риска его побочных эффектов (нефротоксичность, нейротоксичность). Распространённость генотипа ТТ по полиморфному маркёру С3435Т гена MDR1 в европейской популяции высока и составляет 24%. У больных с генотипом ТТ по полиморфному маркё- ру С3435Т гена MDR1 терапия дигоксином и циклоспорином должна обязательно проводится наряду с контролем концентрации данных ЛС в плазме крови (терапевтический лекарственный мониторинг). В будущем ожидается внедрение в клиническую практику фармакогенетических исследований гликопротеина Р для повышения эффективности и безопасности терапии дигоксином, циклоспорином и другими ЛС - субстратами гликопротеина Р.
Генетический полиморфизм транспортёра органических анионов ОАТР-С
В настоящее время обнаружено, что полиморфизм гена, кодирующего транспортёр органических анионов ОАТР-С, может влиять на эффективность и безопасность ингибиторов ГМГ-КоА-редуктазы (статинов). Из-за угнетения «захвата» гепатоцитами статинов у пациентов, несущих аллели С и G по полиморфным маркёрам Т521С и T628G соответственно, при их применении отмечается ослабление гиполипидемического эффекта и одновременно возрастает риск поражения поперечно-полосатой мускулатуры (рабдомиолиза).
Генетические факторы, влияющие на фармакодинамику лекарственных средств
Причиной изменения фармакодинамики ЛС могут быть мутации генов белков, являющихся фармакологическими мишенями для ЛС (рецепторы, ферменты, ионные каналы и др.). Примерами генетического полиморфизма фармакологических мишеней могут служить полиморфизм генов, кодирующих β1- и β2-адренорецепторы, В2-брадикининовые рецепторы, ионные каналы и полиморфизм генов, ответственных за синтез компонентов ренин-ангиотензин-альдостероновой системы (РААС), АПФ и ангиотензиногена. К этой же группе фармакогенетических феноменов относится развитие гемолиза при применении некоторых ЛС у лиц с недостаточностью глюкозо-6-фос-
фатдегидрогеназы и так называемая злокачественная гипертермия при применении средств для наркоза и миорелеаксантов.
Генетический полиморфизмβ2-адренорецептора
Хорошо изучена мутация гена β2-адренорецептора, в результате которой в аминокислотной последовательности рецептора в 16-м положении аргинин заменяется на глицин (мутация Arg16Gly). У гомозигот по этой мутации в пять, а у гетерозигот в два раза чаще отсутствует бронхолитический эффект при применении короткодействующих агонистов β2-адренорецепторов (альбутерол®, сальбутамол) для устранения бронхоспазма по сравнению с лицами, не имеющими этой мутации. Данное явление можно объяснить наличием у носителей этой мутации предрасположенности к снижению плотности β2-адренорецепторов в бронхах на фоне применения короткодействующих агонистов β2-адренорецепторов («dowm-регуляции). Распространённость гомозигот по этой мутации высока и достигает в европейской популяции 40%, поэтому терапия бронхообструктивного синдрома у таких пациентов становится серьёзной проблемой. Показано, что препаратами выбора у таких больных должны быть длительно действующие (пролонгированные) агонисты β2-адренорецепторов (салметерол , формотерол), на бронхолитический эффект которых носительство мутации гена β2-адренорецептора Arg16Gly не влияет.
Генетический полиморфизм β1-адренорецептора
Полиморфизм гена, кодирующегоβ1-адренорецепторов (ADRB1), способен влиять непосредственно на фармакодинамику β-адреноблокаторов. В настоящее время подобного рода исследования выполнены у пациентов с артериальной гипертензией и ХСН. Существуют две несинонимичные замены в кодирующем регионе гена ADRB1:
• замена в нуклеотидной последовательности гена ADRB1 аденина на гуанин в положении 145, приводящая к замене в аминокислотной последовательности β1-адренорецептора глицина на серин в положении 49 (полиморфный маркёр Gly49Ser);
• замена в нуклеотидной последовательности гена ADRB1 гуанина на цитозин в положении 1165, приводящая к замене в аминокислотной последовательности β1-адренорецептора глицина на аргинин в положении 389 (полиморфный маркёр Gly389Arg).
Полиморфный маркёр Gly49Ser локализован во внеклеточной части β1-адренорецептора, а полиморфный маркёр Gly389Arg во внутриклеточной части, в центре связывания с G-белком. Частота аллеля 49Gly приблизительно равна 15%, без расовых отличий (европеоидная
и негроидная), тогда как аллель 389Gly чаще встречается у европеоидов (42%), чем у представителей негроидной расы (27%).
Активно изучается влияние носительства полиморфного маркёра Gly389Arg на гипотензивное действие β-адреноблокаторов у больных с артериальной гипертензией. У пациентов, несущих аллель Arg389, отмечается более интенсивное снижение систолического и диастолического АД как при однократном, так и при длительном применении β-адреноблокаторов. У больных с ХСН, являющихся носителями аллеля 389Arg, β-адреноблокатор метопролол в большей степени повышает фракцию выброса левого желудочка, снижает смертность больных по сравнению с лицами, не несущими этот аллель.
Генетический полиморфизм АПФ
Полиморфизм гена АПФ связан с наличием (вставка, insertion, I) или отсутствием (выпадение, deletion, D) 287-й пары нуклеотидных оснований. Он получил название I/D полиморфизма. Наибольшая активность АПФ в плазме крови отмечается у лиц с DD-генотипом, наименьшая - у лиц с II-генотипом. Лица с ID-генотипом занимают промежуточное положение. Данные о влияние I/D полиморфизма на антигипертензивное действие ингибиторов АПФ и блокаторов ангиотензиновых рецепторов противоречивы. Также противоречивы и данные о влиянии I/D полиморфизма на эффективность ингибиторов АПФ у больных с ХСН. Есть данные о том, что ингибиторы АПФ не оказывают положительного влияния на функцию почек (нефропротективный эффект) при недиабетических заболеваниях почек у больных с DD-генотипом, но эффективны у больных с II-генотипом и ID-генотипом.
Также получены данные о влиянии I/D полиморфизма на эффективность ЛС из других групп. Обнаружено, что достоверное увеличение фракции выброса левого желудочка, а также снижение конечного систолического и диастолического объёмов у больных ХСН на фоне длительной терапии с включением спиронолактона наблюдалась только у пациентов с ХСН с генотипами II и ID, но не DD. В другом исследовании было показано, что в группе больных ХСН, не принимающих β-адреноблокаторы, смертность была выше у лиц с генотипом DD. В группе больных ХСН, принимающих β-адреноблокаторы, смертность была ниже по сравнению с группой, не принимающих эти препараты, и не различалась в зависимости от генотипа АПФ. У больных ИБС с генотипами II и ID флувастатин достоверно лучше вызывал регрессию коронарографических изменений по сравнению с пациентами с генотипом DD. У больных с эректильной дисфункцией с генотипом DD эффективность силденафила достоверно ниже, чем у больных с гено-
типами ID и II. Однако окончательное значение I/D полиморфизма для фармакотерапии требует уточнения.
Генетический полиморфизм В2-брадикининовых рецепторов
Сухой кашель является специфической нежелательной лекарственной реакцией ингибиторов АПФ, возникающий у 10% пациентов. Сухой кашель связан с накоплением брадикинина в слизистой оболочке трахеи и крупных бронхов, который, в свою очередь, способствует активации провоспалительных пептидов (субстанции Р, фосфолипазы С или А2, простагландинов, нейропептида Y), а также местному высвобождению гистамина. Данная нежелательная лекарственная реакция чаще встречается у женщин, чем у мужчин, и проходит через несколько дней после отмены ЛС (максимум через четыре недели). Через В2-брадикининовые рецепторы реализуются большинство «воспалительных» эффектов брадикинина, в том числе сухой кашель, индуцированный ингибиторами АПФ. В2-брадикининовые рецепторы относят к рецепторам, сопряжённым с G-белками, они состоят из семи трансмембранных доменов. Генетический полиморфизм в промоторной области -58Т/С может влиять на развитие сухого кашля при применении ингибиторов АПФ. Было показано, что частота СС генотипа и С аллеля выше у пациентов с артериальной гипертензией. В то же время генотип ТТ и Т аллель встречались достоверно чаще у пациентов, у которых возник сухой кашель при применении ингибиторов. Частота Т аллеля у пациентов с кашлем составляет 67%, а у пациентов без кашля только 38%. Эта тенденция больше выражена у женщин. I/D полиморфизм АПФ, полиморфизм химазы, а также структурные полиморфизмы В2-брадикининовых рецепторов не влияют на частоту возникновения сухого кашля при применении ингибиторов АПФ.
Недостаточность (дефицит) глюкозо-6-фосфатдегидрогеназы
Причиной изменения фармакодинамики ЛС могут быть мутации генов ферментов, ответственных за защиту от окисления сульфгидрильных групп белков клеточных мембран под действием некоторых ЛС, в частности глюкозо-6-фосфатдегидрогеназы (Г-6-ФД). При этом у носителей подобных мутаций из-за дефицита Г-6-ФД возникает гемолиз эритроцитов при применении ряда ЛС (табл. 7.3). Для понимания этого явления необходимо представлять физиологическую роль Г-6-ФД. Этот фермент катализирует переход глюкозо-6-фосфата в фосфоглюконат, при этом коферментом этой реакции является НАДФ, который восстанавливается до НАДФН. НАДФН является важным донором электронов в реакции, где окисленный глутатион превращается в восстановленный под действием глутатионредуктазы. Образующийся
Таблица 7-3. Лекарственные средства, провоцирующие гемолиз эритроцитов при недостаточности Г-6-ФД
 восстановленный глутатион - активный антиоксидант, он защищает белки клеточных мембран от окисления.
восстановленный глутатион - активный антиоксидант, он защищает белки клеточных мембран от окисления.
В условиях недостаточности Г-6-ФД уменьшается образование НАДФН, и, следовательно, наблюдается дефицит восстановленного глутатиона. В связи с этим при применении ЛС, обладающих окислительными свойствами (см. табл. 7.3), возможен гемолиз эритроцитов. Это происходит из-за отсутствия защиты от окисления сульфгидрильных групп белков их клеточных мембран. У лиц с недостаточностью Г-6-ФД гемолиз эритроцитов возникает не только при применении ЛС, но и при употреблении некоторых продуктов питания, в частности конских бобов (Vicia faba). По этой причине данное заболевание часто называют фавизмом.
Наследование мутаций гена Г-6-ФД, обуславливающих её недостаточность, сцеплено с полом. Подобных мутаций выявлено более 50, однако можно выделить две формы недостаточности Г-6-ФД: «негроидную» и «средиземноморскую». «Негроидная» форма характеризуется ускоренным разрушением Г-6-ФД, поэтому гемолизу подвергаются только «старые» эритроциты (старше 55 дней). При этом острый гемолиз отмечается только при первом применении ЛС и длится несколько дней. При продолжении применения ЛС имеет место лишь хронический слабо выраженный гемолиз эритроцитов. «Средиземноморская» форма характеризуется наличием дефектной Г-6-ФД со сниженной активностью, поэтому гемолизу подвергаются как «молодые», так и «старые» эритроциты. При этой форме отмечается выраженный гемолиз эритроцитов, возникающий при первом применении ЛС и продолжающийся в течение всего периода назначения ЛС. Распространённость недостаточности Г-6-ФД варьирует в различных популяциях от 1-15%,
в некоторых регионах достигает 30-40% (Азербайджан). В европейской популяции недостаточность Г-6-ФД встречается крайне редко.
Злокачественная гипертермия при применении лекарственных средств
Злокачественная гипертермия представляет собой заболевание, возникающее при применении местных анестетиков, препаратов для ингаляционного наркоза, сукцинилхолина*. Для злокачественной гипертермии характерен аутосомно-доминантный тип наследования. Симптоматика злокачественной гипертермии складывается из лихорадочного синдрома, сопровождающегося нарушениями ритма сердца, ОПН, а также некротическими изменениями в поперечно-полосатой мускулатуре.
В основе патогенеза злокачественной гипертермии лежит увеличение концентрации внутриклеточного кальция, которое индуцировано вышеперечисленными ЛС. В последнее время выяснилось, что возникновение злокачественной гипертермии обусловлено носительством ряда аллельных вариантов гена, кодирующего рианодиновые рецепторы 1 типа (RYR1), расположенного в локусе 19q13.1. На сегодняшний день выявлено более 40 аллельных вариантов гена RYR1, ответственных за развитие злокачественной гипертермии. Обсуждается вопрос о целесообразности идентификации этих мутаций у всех пациентов, которым предполагается использовать местные анестетики, препараты для ингаляционного наркоза или сукцинилхолин.
Изменение фармакологического ответа при генетических (наследственных) заболеваниях
Ещё одной задачей клинической фармакогенетики стало изучение изменений фармакологического ответа при генетических (наследственных) заболеваниях. Характерными примерами подобных заболеваний являются порфирия, врождённые метгемоглобинемии.
Изменение фармакологического ответа при порфирии
Порфирия - наследственная патология обмена гема, в основе которой лежит повышение активности синтетазы δ-аминолевуленовой кислоты, что сопровождается избыточной продукцией δ-аминолевуленовой кислоты и порфобилиногена. Различают три формы порфирий, которые наследуется по аутосомно-доминантному типу. Клиническая картина обострения заболевания складывается из резких абдоминальных болей, полиневрита, психических нарушений, эпилептических припадков. Некоторые ЛС могут провоцировать обострение порфирии (табл. 7-4). Механизм этого феномена, по-видимому, связан с повышением активности синтетазы δ-аминолевуленовой кислоты под
действием некоторых ЛС (см. табл. 7-4), таких, как барбитураты, сульфаниламиды, эстрогены, гризеофульвин. Поэтому фармакотерапия больных порфирией должна проводится с особой осторожностью.
Таблица 7-4. Опасные и безопасные лекарственные средства у больных с порфирией
Опасные ЛС | Потенциально опасные ЛС | Относительно безопасные ЛС | Безопасные ЛС |
Антипирин* (МНН - феназол) Аминопирин Аминоглутетимид Барбитураты Блокаторы медленных кальциевых каналов Вальпроевая кислота Гризеофульвин Даназол Дапсон Диклофенак Карбамазепин Кетоконазол Ламотриджин Метоклопрамид Мефенитоин Нефезадон Нифедипин Препараты спорыньи Примидон Прогестерон (прогестины) Рифампин® (МНН - рифампицин) Сульфониламиды Фенилбутазон Хлорпопамид Эналаприл Этосуксимид | 5-фторурацил* (МНН - фторурацил) Алкилирующие цитостатики Бензодиазепины Бусульфан Гидролазин Диазепам Дилтиазем Золота препараты Ифосфамид Каптоприл Кетамин Лизиноприл Мефенамиовая кислота Мифепристон Метилдопа Налидиксовая кислота Нитразепам Нитрофурантоин Пентазоцин® Пиразинамид Прокарбозин Спиронолактон Теофиллин Трамадол Трициклические антидепрессанты Троглитазон Цефалоспорины Эстрогены синтетические | Адреналин* (МНН - эпинефрил) Азатиоприн Азота закись* (МНН - динитрогена оксид) Аценокумарол Амитриптилин Витамин В Витамин С* (МНН - аскорбиновая кислота) Даунорубицин, Дигоксин Доксазозин Ибупрофен Индомерацин Колхицин Лабеталол® Литий Лозартан Напроксен Неостигмин Нотриптилин® Пеницилламин Резерпин Тироксин* (МНН - левотироксин натрия) Тубокурарин*® (МНН - тубокурарина хлорид) Хлорамфеникол Хлорохин Цизаприд® Циклоспорин Цитарабин Эстрогены природные | Аллопуринол Амилорид® Атропин Ацетаминофен* (МНН - парацетамол) Ацетазоламид Ацетилсалициловая кислота Бромиды Буметанид Гентамицин Глюкокортикоиды Инсулин Наркотические анальгетики Кумарины* (МНН - амми большой плодов фурокумарины) Офлоксацин Пенициллины Пропранолол Стрептомицин Сукцинилхолин Тетрациклин Фенотиазины Флуоксетин Хлоралгидрат Циметидин |
Изменение фармакологического ответа при наследственной метгемоглобинемии
Причиной наследственной метгемоглобинемии могут быть либо аномалия молекулы гемоглобина (наследуется по аутосомно-доминантному типу), либо снижение активности диафоразы I, участвующей в окислении гемоглобина (наследуется по аутосомно-рецессивному типу). При этом резко повышена концентрация окисленного производного гемоглобина, метгемоглобина, неспособного переносить кислород. Применение даже в терапевтических дозах некоторых ЛС (сульфаниламиды) у таких больных может вызвать ещё большее увеличение концентрации метгемоглобина. Возникающий при этом выраженный цианоз иногда требует медикаментозного лечения (метиленовый синий и аскорбиновая кислота). В связи с этим необходимо избегать применение перечисленных ЛС у таких больных.