Клиническая фармакокинетика: теоретические, прикладные и аналитические аспекты: руководство / Под ред. В.Г. Кукеса. - 2009. - 432 с
|
|
|
|
ГЛАВА 19 ПРИКЛАДНЫЕ АСПЕКТЫ ФАРМАКОКИНЕТИКИ НЕКОТОРЫХ АНТИАРИТМИЧЕСКИХ ПРЕПАРАТОВ
А.К. Стародубцев, А.Б. Кузнецов
Антиаритмические препараты - достаточно многочисленный класс ЛС. Современные рекомендации по лечению аритмий поддерживают назначение лишь весьма ограниченного круга антиаритмических препаратов.
Хинидин
Фармакологические свойства
Согласно классификации, предложенной E. VaughanWilliams (1969), хинидин относят к препаратам класса IA. Его электрофизиологические эффекты обусловлены блокадой натриевых (замедляет скорость деполяризации и проводимость в клетках миокарда) и калиевых каналов (удлинение потенциала действия и эффективного рефрактерного периода). Хинидин снижает
возбудимость миокарда, автоматизм и проводимость в предсердиях, атриовентрикулярном узле, пучке Гиса, волокнах Пуркинье и в желудочковом миокарде. Дополнительные фармакологические эффекты включают блокаду холинэргических рецепторов и α-адренорецепторов, что вызывает вазодилатацию (снижение АД) и рефлекторную тахикардию.
Клиническое применение
В настоящее время хинидин применяют для восстановления синусового ритма при мерцательной аритмии, для поддержания синусового ритма у пациентов с рецидивирующей мерцательной аритмией без органических заболеваний сердца, а также у пациентов с артериальной гипертензией и ИБС. Хинидин рассматривают как препарат второй или третьей линии, применяемый при неэффективности других, более активных и безопасных препаратов.
Базовые фармакокинетические параметры
Хорошо всасывается при приёме внутрь (кишечная абсорбция достигает 95%). Биодоступность - около 90%. Связывание с плазменными белками достигает 70-80%, при этом уровень свободного хинидина зависит от генетического полиморфизма плазменного орозомукоида. Объём распределения составляет 3±0,5 л/кг. Время достижения максимальной концентрации хинидина глюконата в плазме - 3-4 ч, хинидина сульфата - 1-1,5 ч. Время достижения равновесной концентрации - около двух дней. Терапевтическая концентрация препарата в сыворотке крови составляет 2-6 мкг/мл; токсические эффекты возникают при превышении концентрации 6-8 мкг/мл. Характерен эффект «первого прохождения через печень». Метаболическая трансформация хинидина происходит преимущественно в печени с участием CYP3A4. Основные метаболиты (3-гидроксихинидин, хинидин-10,11-дигидродиол, хинидин-N-оксид, О-десметилхинидин) демонстрируют антиаритмическую активность в эксперименте на животных. Наиболее значимый метаболит - 3-гид- роксихинидин, который обнаруживают в достаточной концентрации в плазме у человека; возможно, он вносит вклад в антиаритмическое действие при лечении хинидином. Выведение препарата осуществляют почки (до 10-50% - в неизменном виде). Период полувыведения составляет около 4,4-9 ч.
Влияние различных заболеваний (состояний) на фармакокинетические параметры
Изменения базовых фармакокинетических параметров отмечают у пожилых пациентов, а также у больных циррозом печени и сердечной недостаточностью.
У пациентов старше 65 лет обнаружено снижение клиренса хинидина, удлинение периода полувыведения (около 10 ч) при практически неизменном объёме распределения. Нельзя исключить, что сопутствующие заболевания способствуют изменению фармакокинетики препарата у пожилых пациентов. У больных с гепатитом или циррозом печени обнаружено увеличение общего клиренса, объё- ма распределения и времени полувыведения хинидина. Основной фактор, ответственный за изменение фармакокинетики препарата, - снижение синтеза плазменных протеинов (альбумина и кислых α-гликопротеинов), приводящее к повышению плазменной концентрации несвязанного хинидина, увеличению объёма распределения и повышению клиренса в результате увеличенного поступления свободного хинидина в печень.
У пациентов с сердечной недостаточностью обнаружено изменение фармакокинетики хинидина: снижение клиренса, связанное с уменьшением кровотока в печени; снижение объёма распределения (за счёт усиления связывания хинидина с протеинами плазмы крови) и уменьшение содержания свободной фракции препарата. Снижение клиренса и объёма распределения приводит к тому, что время полувыведения хинидина сходно с таковым у пациентов без сердечной недостаточности. Влияние сердечной недостаточности на фармакокинетику характеризуется высокой вариабельностью, что ранее приводило к существенным проблемам при определении режима дозирования. В настоящее время такой проблемы не существует, поскольку для лечения аритмий (желудочковых и наджелудочковых) у пациентов с сердечной недостаточностью рекомендовано использовать в основном β-адреноблокаторы и амиодарон.
Фармакокинетические взаимодействия
Фармакокинетические взаимодействия хинидина связаны с двумя факторами:
• препараты, угнетающие или стимулирующие активность системы CYP3A, могут существенно изменять метаболическую трансформацию хинидина;
• хинидин способен угнетать метаболическую трансформацию субстратов CYP2D6.
Сок грейпфрута увеличивает время достижения максимальной плазменной концентрации хинидина, способствует уменьшению площади под кривой «концентрация-время» (ПККВ) для 3-гидрок- сихинидина, но не влияет на ПККВ для хинидина и на отношение ПККВ 3-гидроксихинидина к ПККВ хинидина. Таким образом, сок грейпфрута ингибирует кишечную абсорбцию препарата и его метаболическую трансформацию в 3-гидроксихинидин.
Циметидин увеличивает ПККВ и пиковую плазменную концентрацию хинидина, вызывает снижение общего клиренса, биодоступности и увеличение времени его полувыведения. Наиболее вероятный механизм влияния цимедитина на кинетику хинидина - угнетение печёночного метаболизма последнего за счёт ингибирования CYP3A4 и снижения его ренального клиренса.
Интраконазол увеличивает плазменную концентрацию хинидина за счёт ингибирования CYP3A4, торможения метаболической трансформации и почечной секреции препарата, опосредованной Р-гликопротеином. Способностью ингибировать метаболизм хинидина и увеличивать ПККВ обладают и другие азоловые противогрибковые препараты (флуконазол и миконазол).
Эритромицин способствует увеличению плазменной концентрации и снижению клиренса хинидина за счёт ингибирования CYP3A4 и замедления метаболической трансформации препарата.
Рифампицин индуцирует существенное снижение плазменной концентрации (в три раза) и уменьшение времени полувыведения, ускорение печёночной трансформации и существенное возрастание клиренса хинидина за счёт 3-гидроксилирования (в девять раз) и N-оксидации (в шесть раз) в результате индукции CYP3A4. Другие индукторы CYP3A4 (фенитоин, фенобарбитал) оказывают сходные эффекты на кинетику препарата.
Вещества, подщелачивающие мочу (антациды, ингибиторы карбоангидразы, цитраты, натрия гидрокарбонат) увеличивают сывороточную концентрацию хинидина, способствуя усилению его почечной реабсобции.
Хинидин влияет на фармакокинетику некоторых β-адреноблокаторов, в метаболизме которых участвует CYP2D6. Например, он увеличивает ПККВ пропранолола за счёт снижения печёночного клиренса и ингибирования его метаболической трансформации (преимущественно 4- и 5-гидроксидирования), носящего стереозависимый
характер. Кроме того, он тормозит метаболическую трансформацию метопролола и тимолола малеата.
Хинидин снижает общий клиренс мексилетина и двух его основных метаболитов (гидроксиметилмексилетина и мексилетин-N-глю- куронида) за счёт снижения печёночной трансформации, по-видимому, в результате ингибирования CYP2D6.
Влияние хинидина на фармакокинетику пропафенона зависит от генетически детерминированной интенсивности гидроксилирования в печени. Он вызывает увеличение равновесной плазменной концентрации, снижение общего клиренса пропафенона и концентрации его основного метаболита - 5-гидрокипропафенона (только у «быстрых» метаболизаторов). У «медленных» метаболизаторов значимого влияния хинидина на кинетику препарата не отмечено. Хинидин также индуцирует увеличение выраженности эффектов блокады β-адренорецепторов пропафеноном у «быстрых» метаболизаторов.
За счёт модуляции кишечного Р-гликопротеина или других хинидинчувствительных транспортёров, влияющих на абсорбцию и биодоступность фентанила, хинидин способствует повышению плазменной концентрации и увеличению ПККВ последнего.
Хинидин увеличивает плазменную концентрацию, время полувыведения и ПККВ дигоксина (за счёт снижения его общего и ренального клиренса). У пациентов с нарушенной функцией почек отмечено снижение преимущественно неренального клиренса дигоксина. В эксперименте обнаружена способность хинидина ингибировать гепатобилиарный транспорт, увеличивать кишечную абсорбцию и биодоступность дигоксина, а также, ингибируя почечный и кишечный Р-гликопротеин, приводить к снижению его почечной и кишечной экскреции.
Совместное применение непрямых антикоагулянтов (производных кумарина) и хинидина приводит к выраженной гипопротромбинемии в результате снижения синтеза или усиления катаболизма факторов свёртывания крови под влиянием последнего, что требует коррекции дозы антикоагулянтов.
Режимы дозирования
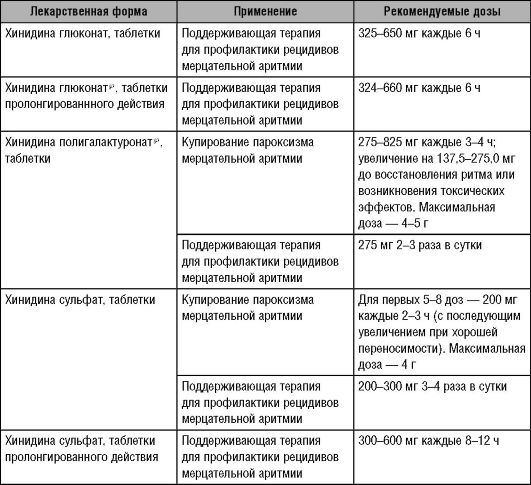
Существует несколько солей хинидина и их лекарственных форм. Рекомендуемые стандартные схемы дозирования каждой лекарственной формы хинидина для взрослых приведены в табл. 19-1.
Таблица 19-1. Рекомендуемые стандартные дозировки хинидина
 Для
индивидуализации лечения хинидином и достижения оптимальной
терапевтической концентрации используют методы, основанные на
фармакокинетических моделях и определении плазменной концентрации
препарата.
Для
индивидуализации лечения хинидином и достижения оптимальной
терапевтической концентрации используют методы, основанные на
фармакокинетических моделях и определении плазменной концентрации
препарата.
Метод расчёта дозы и кратности приёма, основанный на известных фармакокинетических параметрах, применяют, в том числе и при наличии тех или иных заболеваний (состояний), изменяющих фармакокинетику хинидина. Расчёт оптимальной дозы препарата для приёма внутрь предполагает:
• оценку периода полувыведения и константы скорости элиминации. Для взрослых пациентов с различными заболеваниями
(состояниями) принимают следующий период полувыведения (T1/2): при нормальной функции печени - 7 ч, при заболеваниях печени (цирроз, активный гепатит) - 9 ч, при сердечной недостаточности - 7 ч. Расчёт константы скорости элиминации производят по формуле:
•  вычисление объёма распределения (Vdr):
вычисление объёма распределения (Vdr):
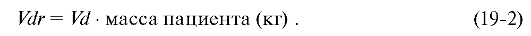 Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vd, нормализованного
по весу: при нормальной функции печени - 2,4 л/кг, при заболеваниях
печени (цирроз, активный гепатит) - 3,8 л/кг, при сердечной
недостаточности - 1,7 л/кг;
Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vd, нормализованного
по весу: при нормальной функции печени - 2,4 л/кг, при заболеваниях
печени (цирроз, активный гепатит) - 3,8 л/кг, при сердечной
недостаточности - 1,7 л/кг;
• определение клиренса хинидина:
•  где k - ранее рассчитанная константа скорости элиминации, Vdr - объём распределения у пациента; определение НД:
где k - ранее рассчитанная константа скорости элиминации, Vdr - объём распределения у пациента; определение НД:
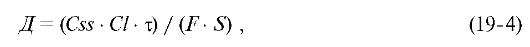 где Css - целевая равновесная концентрация хинидина (обычно в пределах 2-6 мкг/мл); Cl - ранее рассчитанный клиренс; τ - интервал между приёмами хинидина; F - биодоступность (0,70 - для большинства доступных пероральных форм хинидина); S -
относительное содержание активного хинидина в его солях (0,83 - для
хинидина сульфата в обычных таблетках и медленновысвобождающихся формах;
0,62 - для хинидина глюконата в медленновысвобождающейся форме; 0,60 -
для хинидина глюконата в обычных таблетках).
где Css - целевая равновесная концентрация хинидина (обычно в пределах 2-6 мкг/мл); Cl - ранее рассчитанный клиренс; τ - интервал между приёмами хинидина; F - биодоступность (0,70 - для большинства доступных пероральных форм хинидина); S -
относительное содержание активного хинидина в его солях (0,83 - для
хинидина сульфата в обычных таблетках и медленновысвобождающихся формах;
0,62 - для хинидина глюконата в медленновысвобождающейся форме; 0,60 -
для хинидина глюконата в обычных таблетках).
Для коррекции дозы хинидина при рецидивах аритмий или появлении симптомов токсичности рекомендуется определение плазменной концентрации хинидина и коррекция дозы для достижения новой, более эффективной или безопасной концентрации. Расчёт
новой поддерживающей дозы (ПД) определяется на основании уравнения линейной кинетики:
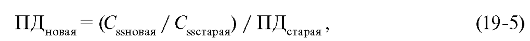 где Сssновая - равновесная концентрация, которую необходимо достигнуть в результае коррекции дозы, Cssстарая - измеренная плазменная
где Сssновая - равновесная концентрация, которую необходимо достигнуть в результае коррекции дозы, Cssстарая - измеренная плазменная
концентрация хинидина, ПДстарая - используемая доза.
Для коррекции дозы хинидина при рецидивах аритмий или возникновении симптомов интоксикации рекомендовано определение его плазменной концентрации и коррекция дозы для достижения новой, более эффективной или безопасной концентрации. Расчёт новой скорости поддерживающей инфузии определяют на основании уравнения линейной кинетики:
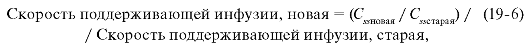 где Сssновая- равновесная концентрация, которую необходимо достигнуть в результате коррекции дозы; Cssстарая - измеренная плазменная
где Сssновая- равновесная концентрация, которую необходимо достигнуть в результате коррекции дозы; Cssстарая - измеренная плазменная
концентрация хинидина; Скорость поддерживающей инфузии, старая - используемая доза.
Клинический мониторинг
Современные рекомендации по лечению аритмий допускают применение хинидина для купирования пароксизмов мерцательной аритмии либо для поддержания синусового ритма у пациентов без органической патологии сердца и выраженных структурных изменений левого желудочка. Критерий эффективности лечения - восстановление и поддержание синусового ритма. Определение плазменной концентрации хинидина целесообразно проводить при рецидивах мерцательной аритмии, для коррекции поддерживающей дозы, а также для оценки комплаентности.
Неблагоприятные лекарственные реакции при лечении хинидином возникают при превышении плазменной концентрации 6-8 мкг/мл и включают желудочковую экстрасистолию, желудочковую тахикардию/фибрилляцию желудочков, полиморфную желудочковую тахикардию, синусовую брадикардию, асистолию, гипотонию, нарушения со стороны желудочно-кишечного тракта, головную боль,
спутанность сознания, судороги, нарушения зрения, гемолитическую анемию, тромбоцитопению, кожную сыпь, волчаночноподобный синдром .
Для мониторирования безопасности терапии хинидином рекомендована регулярная регистрация ЭКГ с целью определения длительности комплекса QRS и интервала Q-T (уширение комплекса QRS более чем на 50% свидетельствует о кардиотоксическом действии препарата); измерение АД; оценка нежелательных лекарственных реакций.
Существенное значение для обеспечения безопасности имеет определение плазменной концентрации хинидина при длительном лечении дозами, превышающими 2 г в сутки или при возникновении характерных нежелательных лекарственных реакций (особенно при расширении QRS более чем на 50%).
Прокаинамид Фармакологические свойства
Согласно общепринятой классификации, предложенной E. Vaughan-Williams (1969), прокаинамид - препарат класса IA. Электрофизиологические свойства прокаинамида сходны с таковыми хинидина. Наиболее значимый метаболит прокаинамида - ]-ацетилпрокаинамид - способен блокировать калиевые каналы и потенцировать удлинение интервала Q-T, индуцированное прокаинамидом. Дополнительные фармакологические эффекты последнего включают умеренную М-холиноблокирующую активность и вазодилатирующий эффект (преимущественно в отношении артериол).
Клиническое применение
Прокаинамид применяют для купирования желудочковых и наджелудочковых аритмий.
У пациентов с острым инфарктом миокарда (нестабильной стенокардией) прокаинамид можно применять в виде внутривенного болюса для лечения резистентной к электроимпульсной терапии желудочковой тахикардии/фибрилляции желудочков. Возможно его использование в качестве препарата второго ряда при неэффективности амиодарона; в качестве препарата резерва - для купирования устойчивой желудочковой тахикардии без гемодинамических нарушений у пациентов с ост-
рыми формами ИБС. При рецидивирующей желудочковой тахикардии у пациентов с острыми формами ИБС его применяют в качестве препарата первого ряда в сочетании с β-адреноблокаторами. Прокаинамид достаточно эффективен при лечении мономорфной устойчивой или рецидивирующей желудочковой тахикардии, не связанной с острыми формами ИБС, гемодинамически стабильной фокальной предсердной тахикардии, мерцательной аритмии. Кроме того, он рекомендован в качестве высокоэффективного препарата первого ряда для купирования суправентрикулярной тахикардии, а также тахикардии неясной локализации с широкими QRS-комплексами. Однако прокаинамид рассматривают как препарат, по эффективности уступающий антиаритмическим препаратам класса IC и III.
Базовые фармакокинетические параметры
Прокаинамид при приёме внутрь всасывается в полном объёме; абсорбция при приёме внутрь составляет 75-100%. Биодоступность различных лекарственных форм препарата достигает 79-85%; связывание с плазменными белками - 15-20%. Объём распределения прокаинамида составляет от 1,2 до 5,7 л/кг при приёме внутрь и 1,41 л/кг при внутривенном введении. При внутривенном введении максимальную концентрацию препарата в плазме обнаруживают немедленно, при приёме внутрь - через 1-2 ч. Терапевтическая концентрация прокаинамида в плазме крови составляет 4-10 мкг/мл.
Для прокаинамида характерен эффект «первого прохождения через печень». Метаболическая трансформация препарата (деэтилирование и ]-окисление) происходит в печени с участием CYP2D6. Основные метаболиты: N-ацетилпрокаинамид, дезэтилпрокаинамид, ]-ацетилдезэтилпрокаинамид, парааминобензойная кислота и её N-ацетилированныедериваты (гидроксиламин и нитропрокаинамид). Ацетилаторный клиренс прокаинамида составляет 10,1±1,7 л/мин (для пациента массой около 70 кг). N-ацетилпрокаинамид обладает способностью блокировать калиевые каналы, увеличивает интервал Q-T, влияет на фармакокинетику прокаинамида, увеличивая его Т1/2. До 25% введённой дозы препарата подвергается метаболической трансформации, у «быстрых ацетилаторов» - до 40%. Кроме того, у них отмечено уменьшение Т1/2 прокаинамида и несколько более высокая плазменная концентрация и почечная экскреция N-ацетил- прокаинамид. Гидроксиламинпрокаинамид и нитрозопрокаинамид, возможно, индуцируют развитие характерных для прокаинамида
нежелательных лекарственных реакций. Гидроксиламинпрокаинамид, кроме того, обладает цитотоксичностью. Метаболическую трансформацию прокаинамида в лейкоцитах (с образованием гидроксиламинпрокаинамида) и моноцитах (с образованием гидроксиламин- и нитрозометаболитов) рассматривают как возможный механизм развития агранулоцитоза и волчаночноподобного синдрома, индуцированного препаратом.
Прокаинамид выводят преимущественно почки (до 50-60% - в неизменённом виде). Основные механизмы - гломерулярная фильтрация и канальцевая секреция. Т1/2 прокаинамида зависит от пути введения и типа метаболизма. При приёме внутрь Т1/2 прокаинамида составляет 4-8 ч, при внутривенном введении - 2,4-4,4 ч. У «быстрых ацетилаторов» Т1/2 короче, чем у «медленных».
Влияние различных заболеваний (состояний) на фармакокинетические параметры
Возраст оказывает определённое влияние на кинетику препарата. Показано, что плазменная равновесная концентрации прокаинамида и N-ацетилпрокаинамида повышается с увеличением возраста пациента, что, возможно, связано с ухудшением функций почек и снижением скорости канальцевой секреции. Эти изменения требуют коррекции назначаемых доз и тщательного клинического мониторинга у пожилых пациентов.
У больных с ХПН отмечают более низкий уровень неренального клиренса и увеличение Т1/2 прокаинамида, более высокую плазменную концентрацию и увеличение Т1/2 N-ацетилпрокаинамида, что может играть существенную роль в развитии терапевтического и токсического эффектов препарата.
Фармакокинетика прокаинамида у пациентов с заболеваниями печени изучена недостаточно. При циррозе печени возможно снижение его клиренса, при этом клиренс N-ацетилпрокаинамида существенно не изменён. Существуют рекомендации о целесообразности уменьшения стартовых доз прокаинамида у пациентов с заболеваниями печени.
Фармакокинетические взаимодействия
Ранитидин увеличивает ПККВ, но не изменяет Т1/2 прокаинамида и N-ацетилпрокаинамида. В качестве основного механизма изменений
кинетики рассматривают снижение ренального клиренса препарата и его метаболита, которое обусловлено ингибированием канальцевой секреции. При этом у пациентов с более низким исходным почечным клиренсом ранитидин увеличивает почечный, но уменьшает метаболический клиренс, и наоборот, у пациентов с более высоким исходным почечным клиренсом ранитидин снижает почечный, но увеличивает метаболический клиренс. Кроме того, влияние ранитидина на кинетику прокаинамида носит дозозависимый характер.
Циметидин индуцирует увеличение плазменной концентрации прокаинамида и N-ацетилпрокаинамида у пожилых за счёт снижения общего и почечного клиренса прокаинамида, а поскольку циметидин - ингибитор CYP2D6, то можно предполагать также ингибирование печёночной трансформации препарата.
По данным исследований, триметоприм увеличивал плазменную концентрацию и ПККВ прокаинамида и N-ацетилпрокаинамида у здоровых добровольцев, что сопровождалось увеличением интервала Q-T. Влияние триметоприма на кинетику последнего обусловлено снижением почечного клиренса препарата и его метаболитов в связи с угнетением канальцевой экскреции. Кроме того, триметоприм, возможно, снижает ацетилаторный клиренс прокаинамида.
При совместном применении амиодарона и прокаинамида отмечают снижение плазменного клиренса и увеличение Т1/2 прокаинамида, при этом объём распределения последнего существенно не изменён. Наблюдают выраженное увеличение длительности комплекса QRS, интервала Q-T, рефрактерного периода в правом желудочке и продолжительности цикла индуцируемой тахикардии (по сравнению с монотерапией). Это свидетельствует об аддитивном электрофизиологическом действии.
На фоне лечения кордароном целесообразно уменьшение дозы внутривенновводимого прокаинамида на 20-30%.
Офлоксацин вызывает увеличение плазменной концентрации и ПККВ прокаинамида и снижение плазменного клиренса, практически не влияя на фармакокинетические параметры N-ацетилпрокаинамида.
Левофлоксацин увеличивает ПККВ и снижает почечный клиренс прокаинамида и N-ацетилпрокаинамида.
Ципрофлоксацин способствует снижению почечного клиренса прокаинамида и N-ацетилпрокаинамида. Взаимодействие фторхинолонов и прокаинамида, по-видимому, осуществляется на уровне почечной системы транспорта органических катионов.
Режимы дозирования
Современные рекомендации предусматривают применение прокаинамида только для купирования пароксизмальных желудочковых тахикардий и наджелудочковых аритмий.
Стандартные режимы дозирования предусматривают внутривенную инфузию препарата со скоростью 50 мг/мин до купирования аритмии или достижения максимальной дозы 1000 мг. Для профилактики рецидивов аритмии рекомендована инфузия со скоростью 2-6 мг/мин.
Для индивидуализации терапии прокаинамидом и достижения оптимальной терапевтической концентрации используют методы, основанные на фармакокинетических моделях и определении плазменной концентрации препарата.
• Методы расчёта НД и скорости поддерживающей инфузии основаны на известных фармакокинетических параметрах прокаинамида, в том числе при наличии тех или иных заболеваний (состояний), изменяющих его фармакокинетику. Расчёт оптимальной дозы предполагает: оценку периода полувыведения и константы скорости элиминации. Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие Т1/2: при нормальной функции печени и почек - 3,3 ч, при почечной недостаточности - 14 ч, при сердечной недостаточности - 5,5 ч. Расчёт константы скорости элиминации производят по формуле:
•  определение объёма распределения у конкретного пациента (Vdr). Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vd, нормализованного
по массе: при нормальной функции печени и почек - 2,7 л/кг, при
почечной недостаточности - j,7 л/кг, при сердечной недостаточности - j,6
л/кг. Для расчёта объёма распределения используют соотношение:
определение объёма распределения у конкретного пациента (Vdr). Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vd, нормализованного
по массе: при нормальной функции печени и почек - 2,7 л/кг, при
почечной недостаточности - j,7 л/кг, при сердечной недостаточности - j,6
л/кг. Для расчёта объёма распределения используют соотношение:
• 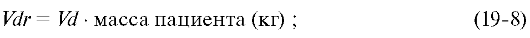 расчёт клиренса (Cl) прокаинамида:
расчёт клиренса (Cl) прокаинамида:
•  где k - ранее рассчитанная константа скорости элиминации; Vdr - общий объём распределения; расчёт НД:
где k - ранее рассчитанная константа скорости элиминации; Vdr - общий объём распределения; расчёт НД:
•  где Css - равновесная концентрация прокаинамида, которую необходимо получить после введения (обычно в пределах 4-10 мкг/мл); Vdr - ранее рассчитанный объём распределения; расчёт скорости поддерживающей инфузии:
где Css - равновесная концентрация прокаинамида, которую необходимо получить после введения (обычно в пределах 4-10 мкг/мл); Vdr - ранее рассчитанный объём распределения; расчёт скорости поддерживающей инфузии:
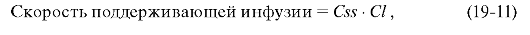 где Css - равновесная концентрация прокаинамида; Cl - ранее рассчитанный клиренс.
где Css - равновесная концентрация прокаинамида; Cl - ранее рассчитанный клиренс.
При рецидивах аритмий или возникновении симптомов токсичности рекомендовано определение плазменной концентрации прокаинамида и коррекция дозы для достижения новой, более эффективной или безопасной концентрации. Расчёт новой скорости поддерживающей инфузии определяют на основании уравнения линейной кинетики:
 где Сssновая - равновесная концентрация, которую необходимо достигнуть в результате коррекции дозы; Сssстарая - измеренная плазменная
где Сssновая - равновесная концентрация, которую необходимо достигнуть в результате коррекции дозы; Сssстарая - измеренная плазменная
концентрация прокаинамида; Скорость поддерживающей инфузии старая - используемая скорость поддерживающей инфузии.
Клинический мониторинг
Терапевтический уровень прокаинамида составляет 4-10 мкг/мл. Эффективность, частота и выраженность нежелательных лекарственных реакций в большинстве случаев зависят от концентрации препарата. При концентрации прокаинамида, соответствующей верхней границе терапевтического диапазона (≥8 мкг/мл) можно отметить умеренно выраженные нежелательные лекарственные реакции: желудочно-кишечные нарушения, слабость, недомогание, умеренную гипотонию, изменение некоторых показателей ЭКГ (удлинение интервалов P-R, QRS и Q-T на 30% и менее). При превышении плазменной концен-
трации j2 мкг/мл можно обнаружить удлинение интервалов P-R, QRS и Q-T на 30% и более, внутрижелудочковые блокады, желудочковые проаритмии (в том числе полиморфную желудочковую тахикардию), остановку сердца. Некоторые нежелательные лекарственные реакции (кожная сыпь, агранулоцитоз и волчаночноподобный синдром) не зависят от плазменной концентрации прокаинамида. В ряде случаев не обнаружено чёткой связи плазменной концентрации препарата с антиаритмическим эффектом и развитием нежелательных лекарственных реакций. Для получения клинического эффекта у некоторых пациентов необходимо достижение плазменной концентрации прокаинамида, составляющей 20 мкг/мл, при этом не регистрируют существенных нежелательных лекарственных реакций.
Методы клинического мониторирования лечения прокаинамидом:
• регистрация ЭКГ и оценка длительности интервалов P-R, QRS и Q-T;
• обнаружение нарушений внутрижелудочковой проводимости и «новых» желудочковых аритмий;
• контроль АД;
• контроль за возникновением некоторых клинических симптомов (желудочно-кишечные нарушения, слабость, недомогание, сыпь, волчаночноподобный синдром);
• определение титра антиядерных антител;
• исследование крови для обнаружения агранулоцитоза. Существенное значение для оптимизации дозирования имеет
определение сывороточной концентрации прокаинамида и N-аце- тилпрокаинамида при рецидивах аритмий на фоне лечения, при наличии нежелательных лекарственных реакций, характерных для препарата, заболеваний (состояний) или при лечении ЛС, влияющими на фармакокинетику прокаинамида.
Лидокаин
Фармакологические свойства
Согласно общепринятой классификации, предложенной E. Vaughan-Williams (j969), лидокаин - препарат класса IB. Он способен блокировать натриевые каналы, ингибируя входящий деполяризующий натриевый ток в клетках миокарда. Для него характерна
«быстрая» кинетика связывания с натриевыми каналами. Лидокаин не влияет на скорость деполяризации в нормальных условиях и при нормальной частоте ритма. Его сродство с натриевыми каналами существенно возрастает при высокой частоте ритма, ишемии, гипокалиемии и ацидозе, что приводит к замедлению скорости деполяризации и проведения импульса в миокарде. Кроме того, лидокаин укорачивает длительность потенциала действия и рефрактерный период в пучке Гиса, волокнах Пуркинье и желудочковом миокарде; снижает автоматизм и возбудимость желудочков. В терапевтических концентрациях препарат не оказывает существенного влияния на гемодинамику. Кроме того, лидокаин обладает свойствами местного анестетика.
Клиническое применение
Применение лидокаина в настоящее время ограничено купированием желудочковой тахикардии. При этом его рассматривают как препарат резерва, эффективность (безопасность) которого уступает препаратам первого ряда, в качестве которых рассматривают амиодарон и β-адреноблокаторы. Применение лидокаина возможно для лечения следующих аритмий: желудочковой тахикардии без гемодинамических нарушений или полиморфной желудочковой тахикардии у пациентов с острым инфарктом миокарда (нестабильной стенокардией); устойчивой мономорфной желудочковой тахикардии, не ассоциированной с острыми формами ИБС; желудочковой тахикардии типа «пируэт», при тахикардии с широкими комплексами QRS неизвестного генеза.
Базовые фармакокинетические параметры
В связи с низкой кишечной абсорбцией лидокаин применяют только парентерально, чаще в виде внутривенного болюса или инфузии. Связывание с плазменными белками составляет 40-80% и зависит от концентрации препарата. Диспозиционная кинетика характеризуется быстрым поступлением препарата в органы с высоким уровнем кровоснабжения (сердце, лёгкие, мозг, печень), а затем - в жировую и мышечную ткань. Объём распределения лидокаина - 2,3±0,6 л/кг. При внутривенном введении максимальной концентрации в плазме достигают практически немедленно. Терапевтическая концентрация в сыворотке крови составляет 1,5-5 мкг/мл; при концентрации выше
5 мкг/мл могут возникать токсические эффекты. Метаболизм лидокаина осуществляется в печени путём дезалкилирования аминогруппы и разрыва амидной связи с образованием фармакологически активных метаболитов - моноэтилглицинэксилидида и глицинэксилидида. Моноэтилглицинэксилидид - основной метаболит, который обнаруживают в крови в концентрации, составляющей 80% от таковой лидокаина. В отличие от последнего, моноэтилглицинэксилидид незначительно связывается с плазменными белками и, возможно, обладает собственной антиаритмической активностью. Вероятно, другие метаболиты также смогут вносить вклад в развитие антиаритмических или токсических эффектов препарата.
CYPjA2 - изофермент, ответственный за метаболическую трансформацию лидокаина. CYP3A4 также частично принимает участие в метаболической трансформации (деэтилировании) препарата. Основной конечный метаболит лидокаина - 2,6-ксилидин, образующийся в печени из лидокаина или моноэтилглицинэксилидида. Фермент, ответственный за его образование, локализован в лабильной субклеточной фракции гепатоцитов. Выведение лидокаина осуществляют преимущественно почки (до j0% - в неизменённом виде). Т1/2 лидокаина - j,8-4 ч; длительная инфузия способствует увеличению Т1/2.
Влияние различных заболеваний (состояний) на фармакокинетические параметры
У пациентов с серьёзным нарушением функции почек, не подвергающихся гемодиализу, отмечено снижение общего клиренса лидокаина и увеличение его Т1/2. Объём распределения и концентрация моноэтилглицинэксилидида не зависят от степени нарушения функции почек, однако концентрация глицинэксилидида у пациентов с ХПН существенно увеличена. Кроме того, у таких больных не отмечено ингибирования метаболической трансформации лидокаина.
Кинетика препарата нарушена у пациентов с заболеваниями печени, в частности, циррозом. Отмечают снижение метаболического клиренса лидокаина, связанное с уменьшением содержания CYPjA2 в печени. Кроме того, определённую роль в ингибировании печёноч- ной трансформации может играть нарушение кровоснабжения печени у больных циррозом вследствие развития патологических изменений в артериальной и портокавальной системе.
Гемодинамические нарушения - шок, гипотензия и сердечная недостаточность - снижают клиренс лидокаина и могут способствовать увеличению его плазменной концентрации.
Изменения кинетики препарата обнаружены у пациентов с острым инфарктом миокарда. Важный параметр, влияющий на кинетику, - функция левого желудочка. При её снижении у пациентов с острым инфарктом миокарда может меняться объём распределения и снижаться плазменный клиренс (как результат снижения кровоснабжения печени и уменьшения метаболической трансформации). В то же время у пациентов с острым инфарктом миокарда повышается плазменная концентрация кислого α-гликопротеина, что приводит к снижению концентрации несвязанного лидокаина без изменения его клиренса.
Фармакокинетические взаимодействия
Эритромицин индуцирует умеренное снижение клиренса лидокаина и увеличение Т1/2 и ПККВ у здоровых добровольцев. Он существенно меняет кинетику лидокаина только у пациентов с выраженным поражением печени (при сравнении со здоровыми добровольцами), при этом клиренс препарата снижен, а объём распределения и Т1/2 увеличены.
Флувоксамин способствует существенному снижению клиренса, повышению ПККВ и пиковой концентрации лидокаина, а также изменяет кинетику его основных метаболитов (снижает концентрацию моноэтилглицинкэксилидида) за счёт ингибирования CYP1A2. У пациентов с выраженным нарушением функций печени флувоксамин практически не влияет на кинетику препарата, по-видимому, в связи с исходно низкой концентрацией CYP1A2 в печени.
Ципрофлоксацин вызывает умеренное увеличение плазменной концентрации и ПККВ лидокаина и умеренно ингибирует его плазменный клиренс. Кроме того, он умеренно снижает ПККВ основных метаболитов - моноэтилглицинкэксилидида и 3-гидроксилидока- ина. Влияние ципрофлоксацина на кинетику лидокаина связано с ингибированием CYP1A2 и торможением метаболической трансформации препарата.
Заместительная гормональная терапия эстрадиолом и прогестероном у женщин в менопаузе способствует умеренному снижению ПККВ и укорочению Т1/2 лидокаина. В качестве основного механизма
изменения его кинетики рассматривают взаимодействие гормонов с CYPjA2 и CYP3A4.
Совместное применение с мексилетином приводит к увеличению плазменной концентрации лидокаина. При этом концентрация основных метаболитов существенно не изменяется, что позволяет предполагать неметаболический механизм взаимодействия. В качестве основного механизма увеличения концентрации лидокаина рассматривают конкуренцию между мексилетином и лидокаином на уровне тканевого связывания.
Совместное применение с амиодароном приводит к увеличению ПККВ лидокаина, к уменьшению ПККВ его метаболитов и снижению системного клиренса препарата; объём распределения и Т1/2 существенно не изменяются. Амиодарон и его основной метаболит (десэтиламиодарон) ингибируют CYP3A4 и тормозят метаболическую трансформацию лидокаина.
Пропафенон незначительно увеличивает ПККВ лидокаина и снижает его системный клиренс, но практически не влияет на объём распределения и Tj. Их совместное применение способствует увеличению выраженности и продолжительности нежелательных лекарственных реакций со стороны ЦНС. Пропафенон незначительно ингибирует метаболизм лидокаина с помощью механизмов, отличных от характерного для пропафенона ингибирования CYP2D6.
Режимы дозирования
Современные рекомендации предполагают внутривенное применение лидокаина для купирования желудочковой тахикардии: введение НД j мг/кг (50-j00 мг), повторное введение через 5 мин, после чего - поддерживающая инфузия со скоростью j-4 мг/мин. В настоящее время длительность инфузии рекомендовано ограничивать, поскольку она не влияет на частоту рецидивов аритмий. Кроме того, для профилактики рецидивов желудочковой тахикардии более эффективны и безопасны другие антиаритмические препараты, в частности, β-адреноблокаторы и амиодарон.
Для индивидуализации терапии лидокаином и достижения оптимальной терапевтической концентрации используют методы, основанные на фармакокинетических моделях и определении плазменной концентрации препарата.
Методы расчёта НД и скорости поддерживающей инфузии основаны на известных фармакокинетических параметрах лидокаина, в том числе при наличии тех или иных заболеваний (состояний), изменяющих фармакокинетику препарата. Расчёт оптимальной нагрузочной и поддерживающей дозы предполагает:
• оценку периода полувыведения и константы скорости элиминации. Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие 71/2: при нормальной функции печени - 1,5 ч, при заболеваниях печени (цирроз, активный гепатит) - 5 ч, при сердечной недостаточности - 2 ч; для пациентов, перенёсших инфаркт миокарда, - 4 ч. Расчёт константы скорости элиминации производят по формуле:
•  расчёт центрального объёма распределения (Vdc). Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vd: при
нормальной функции печени - 0,5 л/кг, при заболеваниях печени (цирроз,
активный гепатит) - 0,6 л/кг, при сердечной недостаточности - 0,3 л/кг;
для пациентов, перенёсших инфаркт миокарда, - 0,5 л/кг. Для расчёта
центрального объёма распределения используют соотношение:
расчёт центрального объёма распределения (Vdc). Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vd: при
нормальной функции печени - 0,5 л/кг, при заболеваниях печени (цирроз,
активный гепатит) - 0,6 л/кг, при сердечной недостаточности - 0,3 л/кг;
для пациентов, перенёсших инфаркт миокарда, - 0,5 л/кг. Для расчёта
центрального объёма распределения используют соотношение:
• 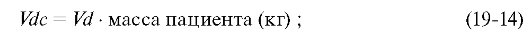 расчёт клиренса (Cl) лидокаина производят по формуле:
расчёт клиренса (Cl) лидокаина производят по формуле:
•  где k - ранее рассчитанная константа скорости элиминации; Vdt - общий объём распределения. Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vdt:
при нормальной функции печени - 1,5 л/кг; при заболеваниях печени
(цирроз, активный гепатит) - 2,6 л/кг; при сердечной недостаточности -
1,0 л/кг; для пациентов, перенёсших инфаркт миокарда, - 1,5 л/кг; расчёт
НД основан на двухкомпартментой модели и описан уравнением:
где k - ранее рассчитанная константа скорости элиминации; Vdt - общий объём распределения. Для взрослых пациентов с различными заболеваниями (состояниями) приняты следующие значения Vdt:
при нормальной функции печени - 1,5 л/кг; при заболеваниях печени
(цирроз, активный гепатит) - 2,6 л/кг; при сердечной недостаточности -
1,0 л/кг; для пациентов, перенёсших инфаркт миокарда, - 1,5 л/кг; расчёт
НД основан на двухкомпартментой модели и описан уравнением:
•  где - равновесная концентрация лидокаина, которую необходимо получить после его введения (обычно в пределах 1,5-5 мкг/мл); Vdc - ранее рассчитанный центральный объём распределения; расчёт скорости поддерживающей инфузии:
где - равновесная концентрация лидокаина, которую необходимо получить после его введения (обычно в пределах 1,5-5 мкг/мл); Vdc - ранее рассчитанный центральный объём распределения; расчёт скорости поддерживающей инфузии:
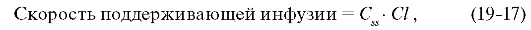 где Сss - равновесная концентрация лидокаина; Cl - ранее рассчитанный клиренс.
где Сss - равновесная концентрация лидокаина; Cl - ранее рассчитанный клиренс.
При рецидивах аритмий или возникновении симптомов токсичности рекомендовано определение плазменной концентрации лидокаина и коррекция дозы для достижения новой, более эффективной или безопасной концентрации. Расчёт новой Скорости поддерживающей инфузии основан на уравнении линейной кинетики:
 где Сssновая - равновесная концентрация, которую необходимо достигнуть в результате коррекции дозы; Сssстарая
- измеренная плазменная концентрация лидокаина; Скорость поддерживающей
инфузиистарая - используемая скорость поддерживающей инфузии.
где Сssновая - равновесная концентрация, которую необходимо достигнуть в результате коррекции дозы; Сssстарая
- измеренная плазменная концентрация лидокаина; Скорость поддерживающей
инфузиистарая - используемая скорость поддерживающей инфузии.
Клинический мониторинг
Симптомы токсичности лидокаина, как правило, связаны с передозировкой и возникают при превышении плазменной концентрации 8 мкг/мл. К нежелательным лекарственным реакциям относят изменения со стороны ЦНС (головокружение, сонливость, спутанность сознания, судороги и остановка дыхания) и сердечно-сосудистой системы (прогрессирующая внутрижелудочковая блокада, асистолия, угнетение сократимости миокарда, гипотензия). В то же время есть сообщения об отсутствии симптомов токсичности при превышении концентрации лидокаина, составляющей 8 мкг/мл, и наличии таковых при концентрации менее 8 мкг/мл.
Плазменная концентрация препарата коррелирует с антиаритмической активностью и токсическими эффектами. Её рассматривают как важную конечную точку клинического мониторинга. Определение плазменной концентрации лидокаина целесообразно при заболеваниях (состояниях), изменяющих метаболизм препарата.
Амиодарон
Фармакологические свойства
Согласно общепринятой классификации, предложенной E. Vaughan-Williams (1969), амиодарон - препарат класса III. Он способен активно блокировать калиевые каналы, существенно увеличивает продолжительность потенциала действия в клетках миокарда. Основной электрофизиологический эффект - удлинение потенциала действия и увеличение периода рефрактерности в миокарде предсердий, желудочков, системе Гиса-Пуркинье. Амиодарон способен подавлять сердечный автоматизм; незначительно блокирует натриевые и медленные кальциевые каналы; обладает умеренно выраженными свойствами блокатора α- и β-адренорецепторов и периферического вазодилататора.
Клиническое применение
Амиодарон целесообразно применять в условиях строгого клинического мониторирования у пациентов, для которых доказана эффективность амиодарона. Современные рекомендации по лечению аритмий рассматривают его как высокоэффективный препарат первого ряда. Препарат рекомендован для экстренного лечения:
• желудоковой тахикардии/фибрилляции желудочков, резистентной к электроимпульсной терапии;
• устойчивой желудочковой тахикардии без гемодинамических нарушений;
• непрерывно рецидивирующей желудочковой тахикардии (в комбинации с β-адреноблокаторами) у пациентов с острым инфарктом миокарда или нестабильной стенокардией;
• желудоковой тахикардии/фибрилляции желудочков в дополнение к электроимпульсной терапии;
• устойчивой желудочковой тахикардии без гемодинамических нарушений;
• непрерывно рецидивирующей желудочковой тахикардии и полиморфной желудочковой тахикардии, не связанных с острым коронарным синдромом.
Амиодарон эффективен для первичной профилактики внезапной сердечной смерти; профилактики и лечения желудочковой тахикардии, рецидивируюшей на фоне применения β-адреноблокаторов; у
пациентов, перенёсших инфаркт миокарда. Кроме того, он рекомендован для лечения желудочковой тахикардии у пациентов с гипертрофической кардиомиопатией и аритмогенной дисплазией правого желудочка при невозможности имплантации дефибриллятора-кардиовертера; для купирования тахикардии неясного происхождения (особенно у пациентов с дисфункцией левого желудочка), синоатриальной реципрокной тахикардии; для лечения и профилактики атриовентрикулярной реципрокной тахикардии, фокальной предсердной тахикардии и фокальной тахикардии атриовентрикулярного соединения; для купирования пароксизмов мерцательной аритмии и поддержания синусового ритма у пациентов с хронической сердечной недостаточностью или выраженной гипертрофией левого желудочка.
Базовые фармакокинетические параметры
Абсорбция при приёме внутрь медленная и вариабельная; всасывается 20-86% принятой дозы. Характерна интенсивная кишечно-печё- ночная рециркуляция. Биодоступность амиодарона характеризуется высокой вариабельностью и составляет 20-88%; связывание с плазменными белками - 96-99,99%; терапевтическая концентрация в сыворотке крови - 0,5-3,5 мкг/мл. Максимальная концентрация после однократного приёма препарата зависит от дозы (0,55±0,20 мкг/мл после приёма 400 мг амиодарона и 1,17±0,3 мкг/мл после приёма 600 мг). Период достижения максимальной концентрации составляет 2-7 ч. Фармакокинетический анализ стандартных режимов дозирования амиодарона для приёма внутрь показывает, что средние равновесные концентрации препарата расположены в терапевтическом диапазоне. Время достижения равновесной концентрации - около 30 дней. В процессе лечения отмечают нарастание плазменной концентрации основного метаболита амиодарона - десэтиламиодарона -и уменьшение соотношения плазменных концентраций амиодарон/десэти- ламиодарон c 9,2±5,0 (при введении начальной дозы) до 2,0±0,6 (после терапии в течение 1 мес). Объём распределения препарата в основном зависит от длительности лечения и достижения равновесной концентрации. После однократного приёма внутрь он составляет 7-21 л/кг. Объём распределения препарата в стадии достижения равновесной концентрации характеризуется высокой вариабельностью (от 22 до
110 л/кг). После внутривенного введения амиодарона снижение плазменной концентрации описывает двухфазная кривая, соответствующая двухкомпартментной модели. Объём распределения в первой фазе составляет 17 л, во второй фазе - 77 л, что может отражать распределение препарата в тканях.
Диспозиционная кинетика амиодарона достаточно сложна и вариабельна. Время полураспределения препарата из плазмы (центральный компартмент) в периферические ткани (периферический компартмент) составляет около 4 ч. Распределение амиодарона и десэтиламиодарона имеет определённую тканевую специфичность. Самую высокую концентрацию препарата определяют в печени, лёг- ких, жировой ткани и поджелудочной железе, несколько меньше - в почках, левом желудочке сердца и щитовидной железе; самые низкие концентрации обнаруживают в скелетных мышцах и коже. Самую высокую концентрацию десэтиламиодарона определяют в печени и лёгких, несколько меньше концентрации - в поджелудочной железе, жировой ткани, почках, левом желудочке сердца, щитовидной железе и головном мозге. В ряде органов и тканей концентрация десэтиламиодарона превышает концентрацию амиодарона (печени, лёгких, сердце, щитовидной железе и головном мозге). В жировой ткани, поджелудочной железе и плазме крови концентрация препарата выше, чем десэтиламиодарона.
Антиаритмическую активность амиодарона и десэтиламиодарона, по-видимому, определяет их концентрация в миокарде. При длительном лечении средний уровень амиодарона в различных областях миокарда составляет от 15 до 48 мкг/г, десэтиламиодарона - от 48 до 71 мкг/г, при этом максимальную концентрацию отмечают в субэпикардиальном миокарде. В исследовании R. Candinas и соавт. (1998) показано, что концентрация амиодарона и десэтиламиодарона составляла 13,2 и 28,3 мкг/г в предсердиях и 13,0 и 40,8 мкг/г - в желудочках. Средняя концентрация препарата в миокарде пациентов, получавших длительное лечение, колебались от 4±1,0 до 29±17,2 мкг/г, десэтиламиодарона - от 22±8,8 до 141±102,5 мкг/г. Концентрация десэтиламиодарона во всех зонах миокарда превышала концентрацию амиодарона. Среднее соотношение концентрации в сердце и в плазме составляло 12-35 для амиодарона и 35-61 - для десэтиламиодарона, что характеризует интенсивное поглощение препарата и метаболита тканями сердца. У пациентов, получав-
ших длительное лечение до трансплантации, амиодарон способен к перераспределению в миокард трансплантированного сердца. Содержание препарата в миокарде достигало пика на второй неделе после операции (87,6±80,7 мкг/г) и в течение 12 недель амиодарон сохранялся в нём в определяемых концентрациях. Препарат быстро поглощается миокардом при внутривенном введении (в течение 3-5 мин). Максимальную концентрацию в плазме отмечают через 2-5 мин. Быстрое распределение амиодарона в миокард ведёт к скорому развитию острых противоаритмических эффектов при внутривенном введении препарата.
Метаболическая трансформация амиодарона происходит в печени. Изоферменты цитохрома Р-450 (CYP2C8 и CYP3A4) принимают участие в печёночном N-деэтилировании препарата. Изоферменты CYP1A2, CYP2C19 и CYP2B6 вносят существенно меньший вклад в метаболическую трансформацию амиодарона. В то же время существует выраженная межиндивидуальная вариабельность вклада отдельных изоферментов цитохрома Р-450 в процесс N-деэтилирования препарата. Значение отдельных изоферментов цитохрома Р-450 зависит от их содержания в печени, а также от печёночной концентрации амиодарона. Основной его метаболит - десэтиламиодарон - способен увеличивать продолжительность потенциала действия кардиомиоцитов и рефрактерность миокарда и, по-видимому, обладает собственной антиаритмической активностью. Концентрация десэтиламиодарона тесно коррелирует с логарифмом кумулятивной дозы амиодарона и нарастает при увеличении продолжительности лечения. Период полувыведения десэтиламиодарона - 33-110 дней. В процессе метаболической трансформации амиодарона также происходит образование йодсодержащих метаболитов. В равновесной стадии до 64% йода содержат йодсодержащие метаболиты амиодарона и только 36% - амиодарон и десэтиламиодарон. Период полувыведения йодсодержащих метаболитов препарата превышает таковой для амиодарона и десэтиламиодарона.
Выведение амиодарона происходит в основном с жёлчью (до 85-95%), около 1% выводят почки. Общий клиренс препарата характеризуют как достаточно низкий; в фазу равновесной кинетики он составляет 90-220 мл/ч/кг. Клиренс препарата выше у женщин, чем у мужчин. Периоду его полувыведения свойственна высокая вариабельность. При длительном приёме в фазу равновесной кинетики Т1/2 составляет 9-120 дней.
Влияние различных заболеваний (состояний) на фармакокинетические параметры
Пол, возраст, рост, уровень креатинина и фракция выброса не влияют на фармакокинетические параметры амиодарона. Состояние сократительной функции миокарда не воздействует на фармакокинетику препарата, вводимого внутривенно. У пациентов с умеренно и существенно нарушенной функцией почек фармакокинетические параметры амиодарона и десэтиламиодарона существенно не отличаются от таковых у пациентов с нормальной функцией почек. Почечная недостаточность, в том числе выраженная, не требует коррекции доз амиодарона.
Фармакокинетические взаимодействия
Препаратов, модифицирующих фармакокинетику амиодарона, относительно немного. Совместное применение с орлистатом способствует снижению ПККВ и пиковой концентрации амиодарона, по-видимому, за счёт ингибирования кишечной абсорбции. Сок грейпфрута оказывает существенное влияние на кинетику препарата: отмечено увеличение ПККВ (на 50%) и максимальной концентрации (на 80%). Также он существенно (почти полностью) ингибирует образование N-десэтиламиодарона, что ведёт к уменьшению изменений интервалов P-R и Q-T, вызванных терапией амиодароном. Существенное снижение образования N-десэтиламиодарона обусловлено ингибированием CYP3A4 соком грейпфрута.
В то же время амиодарон модифицирует фармакокинетические параметры целого ряда препаратов, что обусловлено его ингибирующим влиянием на изофермент цитохрома Р-450 (CYP3A4) и воздействием десэтиламиодарона на целый ряд изоферментов: CYP1A1, CYP1A2, CYP2B6 и CYP2D6.
Амиодарон изменяет фармакокинетические параметры фенитоина: при совместном применении увеличивается ПККВ, максимальная и 24-часовая концентрация фенитоина и Т1/2: снижается его общий клиренс. Объём распределения и концентрация несвязанного фенитоина существенно не изменяются; снижается концентрация метаболитов препарата и уровень их почечной элиминации, что позволяет предполагать ингибирующее влияние амиодарона на метаболизм фенитоина.
Совместное применение амиодарона и дигоксина увеличивает плазменную концентрацию и Т1/2 последнего; объём его распре-
деления практически не изменяется. Амиодарон снижает системный, ренальный и экстраренальный клиренс дигоксина, увеличивает пиковую плазменную концентрацию препарата, принятого внутрь, и уменьшает время достижения пиковой концентрации и ПККВ. Изменений ренального клиренса дигоксина не обнаружено. Повидимому, амиодарон увеличивает биодоступность дигоксина за счёт механизма, независящего от элиминации.
Амиодарон изменяет фармакокинетику циклоспорина при лечении аритмий у пациентов с трансплантированным сердцем, существенно снижает общий клиренс последнего. Основной механизм, ответственный за изменение его кинетики, - ингибирование амиодароном печёночного метаболизма циклоспорина, по-видимому, за счёт угнетения активности изоферментов цитохрома Р-450.
Амиодарон влияет на кинетику симвастатина: увеличивает ПККВ, пиковую плазменную концентрацию и Т1/2. Взаимодействие препаратов может привести к выраженной манифестации нежелательных лекарственных реакций симвастатина: рабдомиолизу, нарушению функции почек (вплоть до развития острой почечной недостаточности), гепатотоксичности. Механизм взаимодействия обусловлен ингибированием CYP3A4 и торможением метаболической трансформации симвастатина. В то же время достоверного влияния амиодарона на кинетику правастатина не обнаружено. Наименьший риск нежелательных лекарственных реакций при совместном применении статинов и ингибиторов CYP3A4 отмечен для правастатина, розувастатина и аторвастатина, в то время как применение симвастина и ловастатина ассоциировано с более высоким риском нежелательных лекарственных реакций.
Амиодарон увеличивает плазменную концентрацию метопролола (почти в два раза), однако индивидуальная выраженность этого эффекта зависит от генотипа CYP2D6. Механизм взаимодействия определяется ингибирующим влиянием десэтиламиодарона на CYP2D6 и торможением метаболической трансформации метопролола.
При совместном применении с амиодароном отмечена тенденция к увеличению плазменной концентрации карведилола, достоверное увеличение концентрации S-карведилола (без существенного изменения концентрации R-карведилола), а также увеличение отношения плазменных концентраций S-/R-карведилола. Десэтиламиодарон - ингибитор CYP2C9, который преимущественно принимает участие в метаболической трансформации S-карведилола. Поскольку
S-карведилол определяет β-блокирующую активность, при совместном приёме карведилола с амиодароном может возрастать выраженность симптомов β-адреноблокады.
При совместном применении амиодарона и варфарина можно отметить увеличение выраженности антикоагулянтного эффекта последнего. Амиодарон уменьшает общий клиренс R- и S-варфарина. Препарат и его метаболиты ингибируют метаболизм варфарина, в частности, оксидативную трансформацию R- и S-варфарина в фенольные производные. Амиодарон вызывает более выраженное ингибирование метаболизма S-варфарина, чем R-варфарина, ингибирует изофермент CYP2C9, ответственный за трансформацию S-варфарина в S-7-гид- роксиварфарин, что приводит к усилению антикоагуляции.
При совместном применении амиодарона и лидокаина отмечают увеличение ПККВ последнего и снижение ПККВ монодесэтилидокаина. Отмечают снижение системного клиренса лидокаина без изменения объёма распределения и периода полувыведения. Ингибируя CYP3A4, амиодарон и десэтиламиодарон блокируют метаболическую трансформацию лидокаина.
Режимы дозирования
Рекомендуемые режимы дозирования амнодарона для приёма внутрь при лечении желудочковых аритмий предусматривают НД 800-1600 мг/сут в течение 7-21 дня до наступления терапевтического эффекта или развития нежелательных лекарственных реакций, затем - 600-800 мг/сут в течение 30 дней с последующим переходом на поддерживающую дозу 200-400 мг. Для лечения суправентрикулярных аритмий НД составляет 600-800 мг/сут в течение семи дней, затем - 400 мг/сут в течение 21 дня с последующим переходом на поддерживающую дозу 200-400 мг/сут. Фармакокинетический анализ режимов дозирования амиодарона для приёма внутрь (по 1600 мг/сут в течение двух дней; по 1200 мг/сут в течение пяти дней; по 800 мг/сут в течение семи дней; по 600 мг/сут в течение семи дней; по 400 мг/сут в течение 62 дней с последующей поддерживающей дозой 343-400 мг/сут) показал, что они позволяют достигнуть равновесной концентрации в пределах терапевтических значений у 90% пациентов.
Режимы внутривенного дозирования для купирования желудочковой тахикардии предусматривают болюсные введения амиодарона по 150 мг до достижения кумулятивной суточной дозы 2200 мг.
Клинический мониторинг
Диспозиционная кинетика препарата достаточно сложна. Амиодарону свойственна вариабельная биодоступность, распределение во многие органы и ткани, экстремально высокая липофильность, биотрансформация с образованием фармакологически активного метаболита, очень медленная элиминация препарата и метаболита. Это создает серьёзные проблемы при индивидуальном дозировании амиодарона. Его плазменная концентрация достаточно тесно коррелирует с дозой. В то же время тканевая концентрация и эффекты препарата не зависят от таковой в плазме. Тканевую концентрацию амиодарона и десэтиламиодарона определяет скорее общая (кумулятивная) доза, чем плазменная концентрация. Определение плазменной концентрации амиодарона и десэтиламиодарона не позволяет предсказать их содержание в миокарде. Минимальная эффективная доза амиодарона чётко не установлена, поскольку уникальная фармакокинетика препарата существенно затрудняет оценку зависимости эффекта препарата от дозы.
Тем не менее, применение стандартных режимов дозирования амиодарона для приёма внутрь, предусматривающих НД и период поддерживающей терапии, позволяет достигнуть его равновесной концентрации более 0,5 мкг/мл и клинического эффекта у большинства пациентов.
Для оценки кинетико-динамических отношений и прогнозирования эффекта при лечении амиодароном используют три клинических параметра: ЧСС, длительность интервала Q-T и обнаружение корнеальных микродепозитов (отмечают у большинства пациентов). У пациентов без снижения ЧСС и уменьшения содержания корнеальных микродепозитов обнаруживают крайне низкую концентрацию амиодарона, что может свидетельствовать об отсутствии клинического эффекта.
Нежелательные лекарственные реакции, как правило, возникают при применении высоких доз амиодарона и включают корнеальную микродепозицию (>90%), кожные изменения (4-9%), фоточувствительность (25-75%), нарушения функции щитовидной железы - гипо- (около 6%) и гипертиреоидизм (0,9-2,0%), поражения лёгких (1-7%), периферическую нейропатию (0,3%), желудочно-кишечные расстройства и гепатотоксичность (повышение активности ферментов - 15-30%, гепатит и цирроз - 0,6%).
Высокая частота и выраженность нежелательных лекарственных реакций связана с более высокой концентрацией амиодарона в плазме и подкожной жировой ткани. Определение препарата в плазме позволяет прогнозировать нежелательные лекарственные реакции. При плазменной концентрации менее 1,5 мг/л риск гепатотоксичности амиодарона минимален, при превышении концентрации 2,5 мг/л риск поражения печени существенно возрастает. Анализ взаимосвязи концентрации амиодарона с активностью аланинаминотрансферазы позволяет рекомендовать определение активности фермента в начале лечения, через 1, 3 и 6 мес, и затем каждые полгода для мониторирования и прогнозирования гепатотоксичности.
Таким образом, клиническое мониторирование лечения должно включать анализ ЧСС, длительность интервала Q-T и обнаружение корнеальных микродепозитов для прогнозирования эффективности препарата. Определение концентрации амиодарона в плазме и подкожно-жировой клетчатке, а также серийное определение активности аланинаминотрансферазы может быть полезным для оценки и прогнозирования нежелательных лекарственных реакций.