Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 10 ФОРМЫ ГИБЕЛИ КЛЕТКИ
Общие данные
Формы гибели клетки определяются характером и степенью ее повреждения. Они зависят от типа клетки, энергетического статуса (уровень АТР) и иммунного статуса (активность Т-киллеров).
Известны следующие основные формы гибели клетки или прекращения ее биологической активности с последующим фагоцитозом остатков - это апоптоз (программированная гибель), некроз (гибель при повреждении) и смешанные формы.
В частности, при необратимой гибели клеток почечной ткани, индуцированной токсинами или острой почечной недостаточностью, некроз может включать элементы апоптоза.
В ацинарных клетках поджелудочной железы наблюдается форма апонекроза (некрозоподобный фенотип).
В исследованиях in vitro на культуре клеток 293Т выделена форма параапоптоза, неапоптотического типа программированной гибели с частично апоптозной морфологией, набуханием митохондрий, но без фрагментации ядра клетки.
Кроме того, высказано предположение о существовании апоптозонекрозного континуума, определяемого перекрестным взаимодействием процессов гибели клетки и процессов сохранения гомеостаза (cross-talk).
Все указанные формы гибели клетки отличаются по своим морфологическим, молекулярно-биохимическим и клиническим критериям.
Морфологические критерии апоптоза и некроза
Морфологические критерии апоптоза и некроза приведены на рис. 47.
Для апоптоза характерны: конденсация хроматина, целостность плазматической и внутриклеточных мембран, набухание митохондриальных мембран.
При апоптозе клеточное содержимое не попадает в межклеточное пространство и не вызывает воспалительной реакции, так как остатки клетки фагоцитируются макрофагами или соседними клетками.
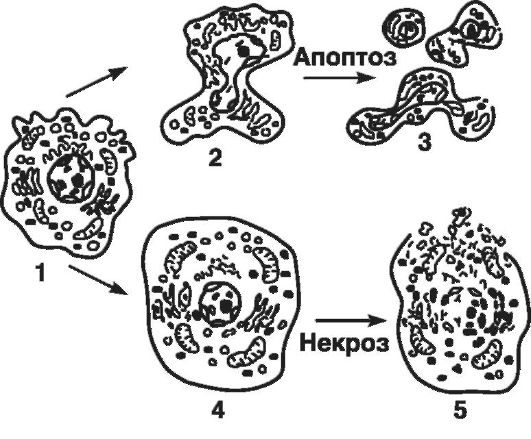 Рис. 47. Морфологические изменения клетки при апоптозе и некрозе (по Самуилову В.Ф., 2001):
Рис. 47. Морфологические изменения клетки при апоптозе и некрозе (по Самуилову В.Ф., 2001):
1 - нормальная клетка; 2 - апоптическое сморщивание с образованием пузырчатых выростов; 3 - фрагментация мембраны с образованием апоптотических телец; 4 - набухание и некротическая дезинтеграция клетки
Для некроза характерны: набухание всей клетки и всех ее органелл, разрыв плазматической и внутриклеточных мембран, активация лизосомных ферментов и воспалительная реакция (в результате попадания внутриклеточного содержимого во внеклеточную среду).
ХАРАКТЕРИСТИКА АПОПТОЗА
В здоровом организме процесс апоптоза сбалансирован процессом физиологической регенерации клетки. Апоптоз - это генетически контролируемый молекулярный (биохимический) механизм, ответственный за поддержание постоянной численности нормальных клеток, выбраковку и удаление дефектных клеток и, следовательно, выживание, старение и смерть клеток и тканей организма. Как правило, апоптоз - это результат опосредованного (через рецепторные системы клетки) неспецифического или нефизиологического действия внешних факторов, выступающих как индукторы, способные при интенсивном воздействии вызвать гибель клетки. Вместе с тем, апоптоз связан с внеклеточными и внутриклеточными сигналами, выступающими в роли мощных физиологических факторов.
Прежде всего, к таким факторам относятся гормоны (например, половые гормоны), которые либо индуцируют, либо ингибируют гибель клетки в зависимости от стадии ее дифференцировки (см. главы 9 и 14).
Другими физиологическими факторами являются цитокины или продуцируемые клетками сигнальные (полифункциональные) пептиды, связывающие специфические рецепторы на клетках-мишенях. В зависимости от функции цитокины делятся на факторы роста, факторы семейства TNF, интерлейкины и интерфероны (см. главу 8). Действие цитокинов на клетки неоднозначно (так же как у гормонов): для одних клеток они являются индукторами, для других - ингибиторами апоптоза, что связано с типом клетки, стадией дифференцировки, функциональным состоянием. Следовательно, программированная гибель клетки зависит от соотношения специфических и неспецифических регуляторов (физиологических и нефизиологических).
Молекулярно-биохимические критерии апоптоза
К молекулярно-биохимическим критериям апоптоза относятся:
• энергозависимость; определяется самой клеткой благодаря наличию АТР (соотношение в клетке АТР/АДР);
• расщепление ядерной ДНК на фрагменты, кратные 180-200 н.п., что приводит к формированию на электрофореграмме ДНКовой лестницы;
• уменьшение объема клетки; клетка сморщивается в течение нескольких минут, теряя до 1/3 своего объема;
• активация цистеиновых протеаз (каспаз), вызывающих деградацию белков;
• экстернализация или появление ФС на наружной поверхности плазматической мембраны; связана с формированием из мембраны апоптотических телец (фрагментов), способствующих их узнаванию макрофагами и последующему фагоцитозу.
Фазы апоптоза
Выделены четыре фазы апоптоза: инициация апоптотического сигнала (первая), трансдукция или проведение апоптотического сигнала (вторая), активация каспаз (третья), деградация ДНК (четвертая).
Инициация, трансдукция и проведение апоптотического сигнала
В зависимости от характера стимула, инициирующего апоптоз, выделяют два сигнальных каскада (пути): рецепторный и митохондриальный.
Рецепторный путь опосредован специфическими «рецепторами смерти», расположенными на плазматической мембране - это факторы TNF .
Наиболее изучен рецептор Fas (Fas-R), или CD95. Его цитоплазматический домен обогащен цистеином, а интегральная (мембранная) часть содержит «домен смерти», или DD, вовлекаемый в белокбелковое взаимодействие с генерацией «сигнала смерти». Лиганд этого рецептора (Fas-L) относится к цитокинам - пептидам из семейства TNF; он экспрессируется на активированных Т-лимфоцитах и естественных киллерах. Связывание лиганда Fas-L с рецептором Fas-R приводит к олигомеризации последнего и формированию сигнального комплекса, инициирующего каскад реакций апоптоза.
В олигомеризации Fas-R участвуют: цитоплазматический DD-домен рецептора, адапторный пептид FADD (Fas- ассоциированный DD, содержащий DED-эффекторный «домен смерти») и прокаспазы-8. Образование этих белков активируют каспазы-8 (см. ниже), что вызывает каскад апоптотических реакций.
В последние годы установлено, что мутации в генах Fas и Fas-L обусловливают развитие ряда аутоиммунных заболеваний, а в случае появления опухолевых клеток активность каспазы-8 может ингибироваться специфическим белком FLIP, препятствующим апоптозу.
Также описан апоптоз, инициированный связыванием рецепторов семейства TNF, содержащих домены DD (TNF-R), с лигандом TRAIL. Их олигомеризация аналогична Fas-R / Fas-L и осуществляется с участием FADD и сходного с ним пептида TRADD (TNF- R-ассоциированный DD). Все указанные примеры характеризуют рецепторный путь апоптоза.
Активация каспаз
Каспазы - это специфические цистеиновые протеазы, катализирующие расщепление белков. Они считаются ключевыми ферментами апоптоза, расщепляющими пептидную связь по карбоксильному концу остатка аспарагиновой кислоты, что ведет к активации или инактивации белка. Мишенями для каспаз служат белки, деградация которых вызывает развитие характерных для апоптоза необратимых процессов - это мембранные белки, белки ядра и цитоплазмы клетки.
Выделено 14 типов каспаз, синтезируемых в виде зимогенов или прокаспаз (имеют большой молекулярный вес), которые затем активируются путем протеолитического процессинга.
В зависимости от функции семейство каспаз делится на 2 подсемейства.
• Первое подсемейство - это каспазы-1, -4 и -5, участвующие в созревании цитокинов. Сюда также относятся интерлейкин-1 бета и интерлейкин-18, усиливающие противовоспалительные свойства цитокинов.
• Второе подсемейство - это каспазы, участвующие в ферментативном каскаде реакций апоптоза. Среди них выделяют эффекторы (гидролизуют структурные белки) и индукторы (принимают и передают апоптотические сигналы).
В неповрежденной клетке активность каспаз регулируется путем:
• взаимодействия индуктора апоптоза со специфическими рецепторами, например, активация каспазы-8 в Fas/Fas-L системе;
• образования гетеродимеров каспаз со специфическими регуляторными белками семейства Вс1-2 (см. ниже);
• взаимодействия каспаз с сериновой протеазой или гранзимом В (GrB) при воздействии цитотоксических Т-лимфоцитов.
Высказаны предположения, что действие каспаз на белки цитоскелета клетки приводит к изменению ее формы и что ингибиторы каспаз не полностью блокируют сморщивание клетки.
В апоптозе может участвовать цитозольная фосфолипаза, которая служит субстратом для каспаз 2 и 8 (см. главы 6 и 10). Возможно, что инактивация цитозольной фосфолипазы ведет к снижению синтеза противовоспалительных простагландинов, образующихся из арахидоновой кислоты, которая является продуктом гидролиза фосфолипидов.
Для TNF-индуцированного апоптоза также показана активация каспазой-3 цитозольной фосфолипазы.
Участие фосфолипаз, вероятно, усиливает повреждение лизосом, что связано с наличием «лизосомо-митохондриальной оси» апоптоза, определяющей эффекты каспаз, лизосомных гидролаз и генерацию митохондриями АФК. На рис. 48 показаны некоторые пути передачи апоптозного сигнала в клетке, включая митохондриальный путь, при котором действие индукторов апоптоза ведет к резкому снижению величины электрохимического потенциала митохондрий и сопровождается выходом в цитоплазму цитохрома-с, который инициирует активацию каспаз.
Наряду с выходом в цитоплазму цитохрома-с возможно высвобождение митохондриями других медиаторов: прокаспаз-2, -3 и -9, AIF, эндонуклеазы G, что, по-видимому, контролируется белками
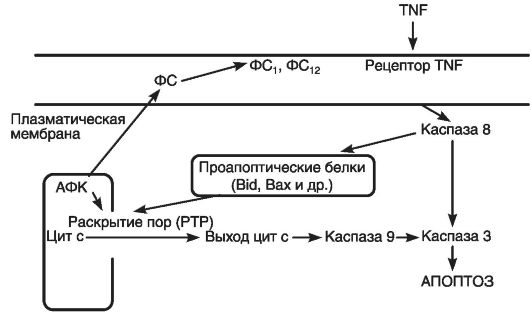 Рис. 48. Некоторые пути передачи апоптозного сигнала в клетке (по Скулачеву В.П., 2001)
Рис. 48. Некоторые пути передачи апоптозного сигнала в клетке (по Скулачеву В.П., 2001)
семейства Вс 1-2 (см. выше), которые кодируются одноименными генами и относятся к наиболее известным внутриклеточным регуляторам апоптоза (для них характерны гомологичные домены или домены ВН 1-4).
В зависимости от функции среди этих регуляторов апоптоза выделяют: антиапоптотические белки (Вс1-2, Вс1-3, Вс1-х, Мс1-1) с доменами ВН 1, 2, 4 и проапоптотические белки (Bid, Bad, Bax, Bim) c доменом BH 3. Например, белок Bid (tBid) взаимодействует с митохондриальными липидами, индуцируя высвобождение эндонуклеазы G и внедрение лизофосфолипидов (особенно лизоФХ) в мембрану митохондрий с последующим ее разрывом. В этом случае эффект разрыва мембраны, вероятно, связан с «заякореванием» С-концевых доменов проапоптотических белков Вс1-2, что способствует их воздействию на мембраны эндоплазматического ретикулума и митохондрий с возможным формированием в них гигантских транзитных пор (РТР) или мегаканалов.
Считается, что соотношение белков этого семейства определяет предрасположенность или устойчивость клеток к апоптозу. Важной общей чертой для митохондриального и рецепторного путей апоптоза
является деполяризация (дезинтеграция) митохондрий, сопровождающаяся глубокими изменениями проницаемости их мембран с последующим образованием мегаканалов. Одним из них служит неспецифический канал, образующийся при окислении SH-группы Cys-56 в аденин-нуклеотидтранслоказе - это белок внутренней мембраны митохондрий, осуществляющий антипорт АТФ/ΑΌΦ (см. главу 6). Этот мегаканал (РТР-комплекс) содержит множество мишеней для внешнего воздействия и регулируется такими эндогенными факторами, как АФК и оксид азота, либо изменениями электрохимического потенциала митохондрий и концентрации ионов Са2+ и Mg2+, а также изменениями соотношений АТФ и ΑΌΦ, липидов и белков, белков Вс 1-2 и др.
Считается, что гигантская пора интегрирует ответные реакции клетки на стрессовое воздействие, и ее раскрытие происходит практически при всех процессах клеточной гибели, что изменяет проницаемость митохондрий.
Деградация ДНК
Если начальные стадии апоптоза различаются в зависимости от типа клеток и индуцирующих сигналов, то фаза деградации ДНК универсальна для большинства клеток. Это необратимая терминальная стадия апоптоза, контролируемая белками семейства Вс1-2.
К хорошо изученным ядерным белкам-регуляторам апоптоза относится белок р53 (мол. масса 53 кДа) или фактор транскрипции, запускающий апоптоз и блокирующий процесс деления клетки с поврежденной ДНК - это первая особенность апоптоза. Так, на ранних стадиях повреждения ДНК экспрессия фактора р53 повышается, что блокирует клеточный цикл в фазах G1 и G2 (см. главу 9) и предоставляет клетке возможность репарировать поврежденную ДНК фосфолипазой PLA2, предотвращая тем самым появление мутаций.
Если активность клеточных систем репарации недостаточная (см. главу 11) и повреждения в ДНК сохраняются, то в таких клетках индуцируется апоптоз.
Накопление в клетке фактора р53 возможно при обоих путях апоптоза.
Вместе с тем, почти у 50% типов изученных опухолевых клеток ген р53 был инактивирован, и связанная с ним регуляция клеточного гомеостаза была нарушена, что приводило к накоплению дефектных клеток.
Вторая особенность апоптоза - это концентрация в цитоплазме свободного ионизированного кальция. Повышение его уровня способствует запуску апоптозного митохондриального каскада сигналов, инициирующего образование транзитных пор и выход в цитоплазму цитохрома-с и эндонуклеазы G.
В освобождении кальция из клеточных депо принимают участие вторичные мессенджеры: диглицерид и инозитол-1,4,5-трифосфат (IP3), генерация которых в клетке связана с активацией разных изоформ фосфолипазы С (см. выше).
С нарушением нормального гомеостаза кальция связана инициация апоптоза некоторыми токсинами. Однако для этого необходимо сохранение основного пула АТР, иначе клетка может перейти к другому механизму гибели - некрозу (см. ниже).
Роль фосфолипидов и других соединений в апоптозе
Завершение апоптоза - это фаза фрагментации погибшей клетки на «апоптотические тельца«. В этой фазе наиболее существенные процессы протекают в плазматической мембране, где происходит перераспределение фосфолипидов, а во внешнем монослое мембраны наблюдается экстернализация ФС, его присоединение к ней (транслокация) и последующее окисление (см. главу 6).
Транслокация ФС осуществляется с участием Са2+- активированной скрамблазы и КЛ митохондрий (в случае свободнорадикального окисления). Эта скрамблаза - сигнал к поглощению «рецептора внешнего ФС», который экспрессируется в разных клетках и тканях, и благодаря этому «апоптотические тельца» захватываются и перевариваются не только фагоцитами, но и соседними клетками. При этом рецепторы к ФС оказывают противовоспалительное действие, подавляя иммунную реакцию фагоцитирующей клетки. Кроме того, в апоптотической клетке возрастает генерация лизоФХ, который служит хемотаксическим фактором - источником сигнала «seak me», стимулирующего приток фагоцитов к данной области.
В апоптоз вовлечены в качестве сигнальных вторичных мессенджеров такие производные фосфолипидов, как церамиды, сфингозин и SIP, способные к регуляции как процессов выживания, так и гибели клетки.
Например, церамид регулирует ключевые факторы апоптоза: полярное перераспределение и олигомеризацию рецептора Fas с формированием суперкаталитического домена, дестабилизацию митохондриальных мембран (путем участия Вах-индуцированных неспецифических
церамидных пор и повышения проницаемости мембран), а также высвобождает апоптогенные факторы, например, цитохром-с. В свою очередь, сфингозин (производное церамида) усиливает апоптоз под действием церамидаз, ингибируя протеинкиназу С (см. главу 6).
Из церамида же образуется SIP или сигнальный липид, обладающий антиапоптотическими свойствами. Его эффекты реализуются с помощью четырех механизмов:
• активации антиапоптотических сигнальных путей (NFkB, ERK- киназа);
• снижения в лимфоцитах экспрессии проаптотических белков Вс1-2 (Вах), блокирующих апоптоз (см. выше);
• антиапоптотического действия по предотвращению процессов, индуцированных стрессом в митохондриях (блокирование выхода цитохрома-с и активации каспаз);
• блокирования апоптоза в эндотелиальных клетках путем повышения уровня оксида азота.
В последние годы была также продемонстрирована специфическая роль в апоптозе мембранных рафтов, что связано с регуляцией ионных каналов и сборкой сигнальных комплексов (см. главу 8). При этом индукторы апоптоза вызывали реорганизацию рафтов и их укрупнение в большие «мембранные платформы», обогащенные церамидами.
Завершая рассмотрение механизмов апоптоза, следует отметить, что повреждение клеточной мембраны не является его первопричиной, хотя и служит важной составляющей механизма апоптоза (здесь происходят олигомеризация и наружная ориентация апоптозных рецепторов и локализуются многие апоптотические сигналы, включая экстернализацию ФС).
ХАРАКТЕРИСТИКА НЕКРОЗА
Некроз - это форма пассивной гибели клетки под действием внешних факторов, на которые клетка не может или не успевает отреагировать с помощью собственных защитных систем. В результате происходит нарушение клеточных функций (в первую очередь повреждаются клеточные мембраны).
В зависимости от своей природы факторы, вызывающие некроз, делятся на физические, химические и биологические (биогенные). Устойчивых к ним клеток в живой природе нет.
Молекулярно-биохимические механизмы некроза
Биохимические механизмы некротической гибели приведены на рис. 49.
В этих механизмах ключевую роль играют три сопряженных процесса:
• резкое повышение концентрации в цитозоле ионов Са2+;
• разобщение дыхания и окислительного фосфорилирования в митохондриях с истощением запасов клеточного АТФ;
• увеличение проницаемости плазматической мембраны и ее повреждение.
Все сопряженные процессы относятся к деструктивным. Они протекают одновременно и взаимосвязанно, и каждый процесс усиливается генерацией АФК и нарастающим окислительным стрессом. Вместе с тем процесс увеличения проницаемости плазматической мембраны и ее повреждение - это еще обратимый этап некроза. Поэтому важной основой для эффективного терапевтического воздействия на погибающие в ходе некроза клетки является направленное торможение или блокирование всех трех сопряженных процессов, если оно сочетается с репарацией поврежденных мембран.
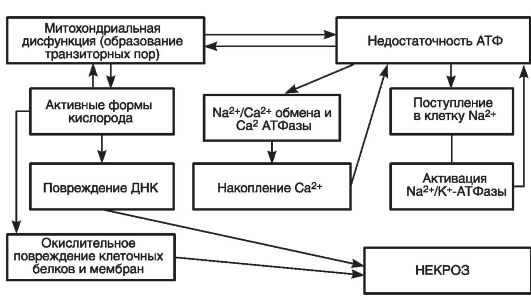 Рис. 49. Биохимические процессы при некротическом повреждении клетки (по Bhatia M., 2004)
Рис. 49. Биохимические процессы при некротическом повреждении клетки (по Bhatia M., 2004)
Механизмы некроза зависят от характера патогенного воздействия, структурных и функциональных особенностей поврежденной ткани, а также конституциональных особенностей организма.
Типы некроза
В зависимости от механизмов развития различают два типа некроза: первый тип - это прямое или непосредственное травматическое (токсическое) воздействие; второй тип - это непрямое или опосредованное воздействие через нейроэндокринную или сердечнососудистую системы организма. Например, при первом типе некроза происходит резкое энергетическое «истощение» клеток: разобщаются митохондриальные процессы окислительного фосфорилирования, существенно снижается синтез АТР, нарушается работа АТР-зависимых ионных каналов, утрачивается регуляция ионного гомеостаза и разрушаются белки цитоскелета клетки. При этом плазматическая мембрана теряет способность регулировать клеточный объем и ионную проницаемость. В результате усиливается транспорт ионов, нарушается осмотическое равновесие, увеличивается концентрация ионов Na+ в цитозоле.
В свою очередь, реакции резкого истощения активируют Na+-, К+-АТРазу, что еще больше истощает запасы АТФ, способствуя набуханию митохондрий и развитию отека клетки.
В самом начале повреждения ионный гомеостаз еще поддерживается одновременным оттоком ионов К+, однако в дальнейшем ионные насосы блокируются, и приток в клетки ионов Na+ и воды увеличивается.
В последние годы на различных клеточных культурах была показана роль некоторых ионных насосов и трансмембранных каналов в развитии некротического процесса. Среди них: Na+ / К+-насос; Na+/ Н+- и Na+/ Са2+-ионные каналы; Na+-, K+-, 2С1--котранспортер.
Например, неспособность клеточных мембран поддерживать низкую концентрацию Са2+ в цитоплазме и регулировать гомеостаз митохондриального Са2+ лежит в основе необратимости молекулярнобиохимических нарушений при ишемии мозга и миокарда, ряде аутоиммунных заболеваний и других болезней, а также при цитотоксическом воздействии многих химических соединений, включая лекарства.
Высокие концентрации Са2+ в цитоплазме в условиях окислительного стресса раскрывают в митохондриях транзитные поры (как при
апоптозе), что снижает митохондриальный трансмембранный потенциал, приводит к деградации ?+,К+-АТРазы и еще больше усиливает повреждение мембраны.
Наряду со снижением мембранного потенциала нарушается осмотический баланс между матриксом и межмембранным пространством митохондрий, после чего в матрикс поступает вода, вызывая его набухание и разрыв наружной мембраны. В результате в поврежденных митохондриях резко снижается генерация АТР и наступает некроз клетки.
Раскрытие пор и увеличение проницаемости митохондриальных мембран контролируют специфические вторичные месседжеры (и проаптотические белки семейства Вс1-2 - см. выше), что характерно для атеросклероза, сахарного диабета, нейродегенеративных заболеваний, процессов старения и цитолитического гепатита.
РАЗЛИЧИЯ И ОБЩНОСТЬ АПОПТОЗА И НЕКРОЗА
Некроз отличается от апоптоза следующим. При некрозе:
• наблюдается аутолиз клетки, и ее содержимое изливается в межклеточное пространство, где развивается процесс воспаления;
• отсутствует зависимость от энергетического статуса клетки;
• отсутствует генетический контроль с помощью специфических белков-регуляторов;
• в процесс одновременно вовлечена большая группа соседних клеток и тканей, подвергшихся общему интенсивному воздействию повреждающих факторов;
• структурно-функциональные изменения в клетке подчинены сопряженным друг с другом общим молекулярно-биохимическим закономерностям, первопричиной которых служит нарушение ионного гомеостаза.
Несмотря на разную природу повреждающих факторов, при некрозе выделен ряд общих с апоптозом биохимических каскадов. Среди них:
• нарушение окислительного фосфорилирования (синтеза АТР) и гликолиза;
• нарушение внутриклеточного баланса Са2+ (его накопление);
• увеличение проницаемости плазматической мембраны и мембран внутриклеточных органелл;
• генерация АФК и свободных радикалов;
• необратимое разрушение митохондрий;
• повреждение ядра клетки с активацией эндонуклеаз и деполимеризацией (дезинтеграцией) ДНК и РНК.
Рассмотрим на примере разрушения митохондрий значение некоторых из этих каскадов биохимических реакций. Именно степень разрушения митохондрий определяет возможность выживания клетки и путь ее гибели (апоптоз или некроз).
Известно, что митохондрии нормальной клетки служат источником АФК, допустимый уровень которых поддерживается защитной ферментной системой, включающей цитохромоксидазу, супероксиддисмутазу, глутатионпероксидазу (см. главу 11).
В поврежденной клетке концентрация АФК нарастает, что ведет к открытию транзитных пор с выходом в цитоплазму цитохрома-с и АФК, что сдерживает их дальнейшую генерацию.
Если транзитные поры остаются открытыми (и при этом сохраняется избыток АФК), то наступает гибель митохондрий или так называемый митоптоз, ведущий либо к апоптозу (за счет цитохрома-с и AIF), либо к некрозу (при нехватке АТР).
Клинические проявления некроза отличаются выраженным полиморфизмом, в основе которого лежат «местная смерть» группы соседних клеток и выключение из функционирования жизненно важных клеточных зон. Таковы, например, причины инфарктов миокарда, ишемии головного мозга, некрозов коркового вещества почек, прогрессирующего некроза печени и острого панкреатита, осложненного панкреонекрозом.
Следовательно, некроз, являясь результатом непрограммированных, но биохимически, морфологически и клинически максимально выраженных деструктивных процессов в клетке, вносит существенный вклад в патогенез многих заболеваний.
Обратимость и необратимость повреждений при некрозе
Повреждение митохондрий при некрозе может быть обратимым в случае нерезко выраженного радиационного воздействия (включая УФО). При этом обратимым считается само явление формирования транзитных пор.
Возможна обратимость повреждений целых клеток, но только на начальных этапах.
Обратимая стадия - это, во-первых, неспецифическая реакция в ответ на любое раздражение клетки. Для нее характерны:
• увеличение проницаемости мембран для ионов Са2+ и последующая активизация гидролаз с развитием метаболических нарушений;
• изменение состава липидов и микровязкости фосфолипидного бислоя мембран;
• возрастание доли холестерина при атеросклерозе и старении;
• частично компенсируемые процессы ПОЛ при сахарном диабете (см. главу 9).
Во-вторых, обратимость повреждений наблюдается при ишемии миокарда с последующей реперфузией. Всего два фактора обусловливают необратимость повреждений при ишемии - это дисфункция митохондрий клетки с резким дефицитом АТФ и дисфункция клеточных мембран.
Вместе с тем, в настоящее время не определены критерии перехода поврежденной клетки из обратимого состояния к необратимому.