Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 4 НАСЛЕДОВАНИЕ ГЕНОВ И ПРИЗНАКОВ
Общие данные
Результаты взаимодействия генов двух родительских геномов в зиготе и развившемся из нее многоклеточном организме проявляются в контролируемых ими признаках, которые в той или иной мере передаются из поколения в поколение. Такое наследование зависит от многих причин.
В классической генетике длительное время считалось, что вклад обоих родителей в геном потомства примерно одинаковый как по материнской, так и по отцовской линии семейных родословных. На этой основе сформулировали одно из первых правил наследования - равнозначность и взаимосвязанность функций двух разных по происхождению аллелей одних и тех же генов (или эквивалентность реципрокных скрещиваний). Выделили два варианта и ряд типов наследования с учетом количества генов, их происхождения (материнское или отцовское), локализации в аллельных или неаллельных локусах аутосом и половых хромосом, характера проявления (доминантность или рецессивность), а также особенностей (механизмов) взаимодействия между генами.
Однако уже во второй половине XX в. установили: во многих случаях вклад одного из родителей значительно отличается от вклада другого родителя. Было показано: функции родительских генов на протяжении всего онтогенеза могут изменяться вплоть до дифференциального отключения материнских либо отцовских аллелей. В основе этого явления лежит эпигеномный процесс или маркирование локусов хромосом одного из родителей, приводящее к выключению экспрессии расположенных в них аллелей. Данное явление получило название импринтинга. Термин впервые применен во втором десятилетии XX в. австрийским зоологом Конрадом Лоренцом. Наблюдая за поведением утят, только что вылупившихся из яичной скорлупы, он обратил внимание: они ищут взглядом свою маму-утку. Если в поле их зрения попадает хозяин или хозяйка утки либо пробегающая мимо собака или кошка - за ними, как за своей матерью, утята следуют всю дальнейшую жизнь.
Таким образом, речь идет о формировании до рождения ряда функций организма, запечатленных в геноме («геномная память»).
Позднее термин «импринтинг» стали применять невропатологи. Импринтингом, считают они, обусловлено происхождение ряда врожденных феноменов у новорожденных младенцев, например таких, как поиск соска материнской грудной железы или базисные функции (глотание, дыхание, жевание, кровообращение, пищеварение и сосание), которые сформировались во внутриутробной жизни.
Затем этот термин стали использовать генетики для объяснения необычного поведения половых хромосом (элиминация хромосом отцовского происхождения) у насекомых рода Sciara Coprophila (Кроуз Г., 1960). Было показано: отцовские Х-хромосомы каким-то образом маркируются (импринтируются) перед слиянием гамет в соответствии со своим родительским происхождением.
В настоящее время убедительно доказано общебиологическое значение импринтинга как эпигеномного процесса, связанного с регуляторными (функциональными) изменениями активности генома и не связанного с его структурными повреждениями (см. главу 28).
У человека в результате импринтинга, например, экспрессируется материнский ген (аллель), тогда как отцовский аллель блокируется или наоборот. Следовательно, у индивида имеет место только моноаллельная экспрессия или неэквивалентный вклад в геном одного из его родителей, а значит, налицо отклонение от менделевских законов наследования, основанных на диаллельной модели (см. главу 2).
Прежде чем рассмотреть такие отклонения, остановимся на закономерностях наследования генов и признаков, впервые описанных Г. Менделем на основе результатов его экспериментов по скрещиванию семян садового гороха. В дальнейшем эти закономерности стали называть законами наследственности и распространили на человека.
Законы наследственности
В современной редакции законы наследственности формулируются следующим образом.
• Первый закон - закон доминирования (или единообразия) признака у потомков первого поколения. В первом поколении у потомка проявляется действие доминантного гена (доминантный
признак), но не проявляется действие рецессивного гена (рецессивный признак). В последующих поколениях у потомка проявляется действие как доминантного, так и рецессивного гена.
• Второй закон - закон расщепления генов у потомка или закон «чистоты гамет». У потомка происходит альтернативное расщепление (распределение) генов в гаметах: одна половина гамет несет доминантные гены (А), а другая половина - рецессивные гены (а). Оба типа генов присутствуют в соматических клетках, не смешиваясь и не заменяя друг друга.
• Третий закон - закон независимого наследования неаллельных генов или случайных сочетаний наследственных задатков у потомков. Наследование двух генов (двух пар признаков) называется дигибридным, более двух генов (двух пар признаков) - полигибридным. Формула такого наследования соответствует биноминальному ряду: (3 + 1)п, где η - число генов (пар признаков). Для выведения формулы используется решетка Р. Пеннета. С ее помощью рассчитываются генотипы организмов, их количество и зависимость от типов гамет, содержащих доминантные и рецессивные гены (табл. 3).
Таблица 3. Решетка Р. Пеннета
Первый родитель | Второй родитель | |||
А | А | В | b | |
А' | А'А | А'а | А'В | А'Ь |
а' | а'А | а'а | а'В | а'Ь |
B' | В'А | В'а | В'В | B'b |
b' | Ь'А | Ь'а | Ь'В | b'b |
Примечание. А, В, А' и В' - доминантные гены; а, b, а'и b' - рецессивные гены; А'А, В'В, А'В и В'А - доминантные гомозиготные организмы; а'а, а'Ь, Ь'а, b'b - рецессивные гомозиготные организмы; А'а, а'Ь, а'А, а'В, В'а, В'Ь, Ь'а и Ь'В - доминантные гетерозиготные организмы.
Помимо трех законов наследственности, наследование генов и признаков определяют основные положения хромосомной теории наследственности.
Хромосомная теория наследственности
Во втором десятилетии XX в. Томас Хент Морган (1866-1945) и его ученики (К. Бриджес, Г. Меллер и А. Стертевант) сформулировали основные положения хромосомной теории наследственности (все
авторы стали нобелевскими лауреатами). С тех пор их формулировки почти не изменились, хотя некоторые уточнения и были сделаны.
Хромосомная теория наследственности в современной редакции включает следующие положения.
• Гены расположены в хромосомах; число генов в хромосоме пропорционально длине хромосомы. В дальнейшем оказалось: разные гены различны по длине, поэтому не всегда соблюдается указанная пропорция. Например, на хромосомах 5 и 9 генов идентифицировано больше, чем на самых длинных хромосомах 1 и 2.
• Гены расположены по длине хромосомы в линейном порядке. В дальнейшем выделили мобильные генетические элементы или «прыгающие гены» (транспозоны), которые перемещаются по геному с одной хромосомы на другую, нарушая линейный порядок расположения.
• Аллельные гены занимают идентичные локусы гомологичных хромосом. Из двух гомологичных хромосом по происхождению одна - отцовская, другая - материнская. Их локусы идентичны друг другу. В них находятся аллели одного и того же гена или аллельные гены. Каждый из двух аллелей одного и того же гена представляет собой отцовскую и материнскую копии; в норме это, как правило, диаллельная модель организационной структуры гена (см. рис. 12). В случае наследственной патологии у индивида может быть только один генный локус, и соответственно в нем будет находиться один аллель (либо отцовский, либо материнский); это состояние моноаллельности по данному аллелю отмечается, например, при синдроме Шерешевского-Тернера (кариотип: 45,ХО). Другой патологический вариант - наличие у индивида одновременно трех аллелей и более; это состояние полиаллельности по данному аллелю - например, три идентичных аллеля одного и того же гена трех хромосом 21 при синдроме Дауна (кариотип: 47,ХХ,+21); три аллеля трех хромосом 13 при синдроме Патау (кариотип: 47, XY+13) либо наличие в кариотипе от 4 до 11 Х-хромосом (4-11 аллелей одного и того же гена - 49, ХХХХ).
• Гены одной хромосомы образуют группу сцепления, обеспечивая совместное наследование контролируемых ими признаков. Следует отметить: сцепление генов с хромосомой постоянно нарушается в ходе кроссинговера - процесса гомологичной рекомбинации или обмена одинаковыми участками (генами и их фраг-
ментами) между гомологичными хромосомами в первом делении мейоза. Частота кроссинговера прямо пропорциональна расстоянию между генами. Взаимный обмен аллелями отцовского и материнского происхождения происходит между всеми парами гомологичных хромосом, за исключением Х- и Y-хромосом. Открытие кроссинговера также принадлежит школе Т.Х. Моргана. Теперь рассмотрим варианты и типы наследования генов и признаков.
ВАРИАНТЫ И ТИПЫ НАСЛЕДОВАНИЯ ГЕНОВ И ПРИЗНАКОВ
В настоящее время выделяют три варианта наследования генов и признаков: моногенный и полигенный при традиционном (классическом) наследовании и вариант неклассического или нетрадиционного наследования.
Моногенное наследование основано на первом и втором законах наследственности. Оно подразумевает наследование одного гена (одной пары признаков) и относится к аллельным генам.
Полигенное наследование основано на третьем законе наследственности. Оно подразумевает наследование двух генов (пар признаков) и более и относится к неаллельным генам.
Нетрадиционное наследование есть наследование генов и признаков, выходящее за рамки моногенного и полигенного вариантов.
Моногенное наследование
Моногенное наследование нередко называют простым менделевским наследованием.
На основе представлений о диаллельной модели структуры гена наследование механизмов взаимодействия между отцовским и материнским геномами рассматривается отдельно для каждой аллельной и неаллельной пары.
Типы моногенного наследования
В рамках моногенного наследования выделяют:
• аутосомно-доминантный тип (на одной из двух аутосом расположен доминантный ген);
• аутосомно-рецессивный тип (на одной из двух аутосом расположен рецессивный ген);
• Х-сцепленный доминантный тип (на Х-хромосоме расположен доминантный ген);
• Х-сцепленный рецессивный тип (на Х-хромосоме расположен рецессивный ген);
• Y-сцепленный тип или голандрическое наследование (ген расположен на Y-хромосоме).
Согласно каталогу В. Маккьюсика «Наследование менделевских признаков у человека» (Интернет-версия: online - http:www.ncbi.nlm. nih.gov/Omim), идентифицировано более 12,5 тыс. таких фенотипов. Среди них около 12 тыс. - фенотипы, наследуемые (или предположительно наследуемые) аутосомно-доминантно или аутосомнорецессивно (в том числе 9 тыс. фенотипов с установленным типом наследования).
В последние годы получен ряд данных о наследовании, сцепленном с Х- и Y-хромосомами, на которых локализованы соответственно
300 и 92 гена.
На рис. 20 приведена карта X-хромосомы и ряд сцепленных с ее локусами доминантных и рецессивных заболеваний.
В качестве примеров Х-сцепленных доминантных фенотипов следует привести редко встречающиеся заболевания: витамин D-резистентный рахит или гипофосфатемия (Хр22.2); синдром недержания пигмента, тип I (Xp11.1) и тип II (Xq28).
Примеры наиболее распространенных заболеваний, наследуемых по Х-сцепленному рецессивному типу: гемолитическая анемия (Xq21.2 или Xq28), гемофилия А (Xq28.2) и В (Xq27.2), миодистрофия Дюшенна-Беккера (Xp21.2), синдромы Леша-Найяна (Xq26.2),
Лоу (Xq25.2), Менкеса (Xp11.1), Мартина-Белл (Xq27.2 или Xq27.3),
точечная хондродисплазия Конради-Хюнерманна (Хр22.2), пигментный ретинит (Xp21.2-21.3; Xp22).
На рис. 21 приведено схематическое изображение локусов Y-хромосомы, в которых расположены гены, формирующие мужской пол, и гены, обусловливающие мужское бесплодие.
В качестве примеров заболеваний, сцепленных с Y-хромосомой, следует привести нарушения дифференцировки пола, формы мужского бесплодия в виде азооспермии (Yp - фактор 2; Yq11 - фактор 1), гонадобластому и др. Всего на схеме выделены гены, ответственные за семь таких заболеваний.
Кроме того, моногенное наследование может быть не только сцеплено с полом (Х- или Y-хромосомой), но и ограничено полом.
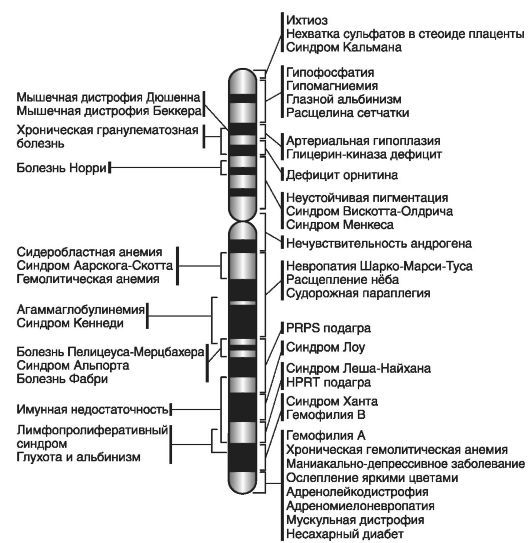 Рис. 20. Карта X-хромосомы человека (по мультимедийному учебнику «Биология» Copyright (c), 2007)
Рис. 20. Карта X-хромосомы человека (по мультимедийному учебнику «Биология» Copyright (c), 2007)
Например, ген плешивости проявляется у мужчин (доминантный эффект) и почти не проявляется у женщин (рецессивный эффект).
Критерии моногенного наследования
Аутосомно-доминантный тип:
• заболевание регулярно передается из поколения в поколение без пропусков, т.е. прослеживается в родословной по вертикали, кроме случаев мутаций de novo (см. главу 5);
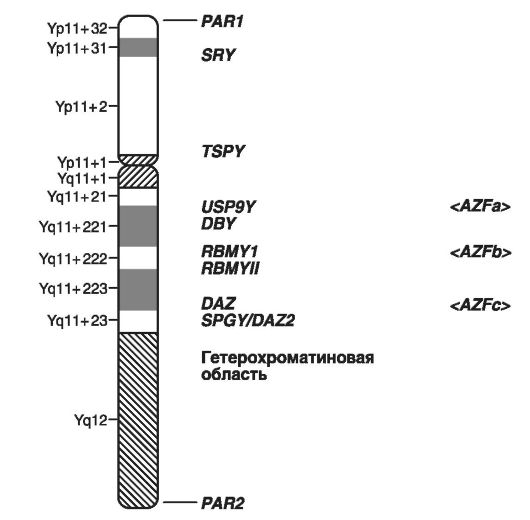 Рис. 21. Схематическое изображение Y-хромосомы (по GenBank, 2003)
Рис. 21. Схематическое изображение Y-хромосомы (по GenBank, 2003)
• риск рождения больного ребенка, если болен один из родителей, составляет 50%;
• здоровые индивиды имеют здоровых потомков;
• у больного индивида болен один из родителей, кроме случаев новой мутации;
• оба пола поражаются с одинаковой частотой. Аутосомно-рецессивный тип:
• родители больного пробанда (лицо, обратившееся за консультацией к врачу-генетику) здоровы, но аналогичное заболевание обнаруживается у родных, двоюродных и троюродных сибсов пробанда (его братья и сестры), т.е. прослеживается в родословной по горизонтали (в одном поколении);
• у больного родителя рождаются здоровые дети;
• риск рождения больного ребенка равен 25% (соотношение больных и здоровых лиц составляет 1:4);
• в случае кровнородственных браков между родителями больного пробанда наблюдается увеличение числа больных родственников в родословной.
Х-сцепленный доминантный тип:
• у больного пробанда обязательно болен один из родителей;
• у больного отца все дочери больны, а сыновья здоровы;
• у больной матери рождение больной дочери и больного сына одинаково вероятно;
• у здоровых родителей все дети будут здоровы;
• больных женщин в 2 раза больше, чем больных мужчин. Х-сцепленный рецессивный тип:
• заболевание наблюдается у мужчин - родственников больного пробанда по материнской линии;
• сыновья не наследуют заболевание отца;
• у больного отца все дочери здоровы и являются гетерозиготными носителями гена болезни отца;
• если женщина - гетерозиготный носитель гена болезни, то половина ее сыновей будут больны, а все дочери здоровы; причем половина дочерей также станут гетерозиготными носителями гена болезни.
Критерии голандрического наследования пока не разработаны. В последние годы у человека выделен ряд сложно наследуемых моногенных и полигенных болезней:
• дигенные болезни - рак грудной железы (две генокопии: гены ВRС1 и ВRС2);
• тригенные болезни - синдром Барде-Бидля (3 генокопии: гены ВВS1, ВВS2 и ВВS30);
• четырехгенные - пятигенные болезни: болезнь Альцгеймера (обусловлена четырьмя генокопиями: PS1, PS2, PS3 и PS4, а также геном прионного белка, см. главу 29);
• полигенные, или мультифакториальные болезни (МФБ), обусловленные «генными сетями» - артериальная гипертензия (170 генов, включая 17 главных генов), бронхиальная астма (около 20 генов), остеопороз (13 генов), эндометриоз (11 генов) и др.
Полигенное наследование
Полигенное наследование нередко называют мультигенным или мультифакториальным, имея в виду наследование одновременно не одного, а нескольких определенных генов, проявляющих свое
действие в специфических условиях окружающей среды, при наличии провоцирующих внешних факторов, как правило, усиливающих индивидуальное действие генов, эффект которых суммируется (аддитивное действие).
Критерии полигенного наследования (всего 5) систематизированы в 1969 г. К. Картером, а спустя 20 лет их дополнили (еще два) Ф. Фогель и А. Мотульски (1989).
Критерии полигенного наследования
Риск развития мультифакториального признака (заболевания) определяют следующие факторы.
• Наследуемость признака или болезни. Чем выше наследуемость признака или заболевания (чем больше унаследовано генов, за него ответственных), тем выше риск его развития у здоровых родственников. Наследуемость признака (заболевания) - это степень влияния на формирование данного признака (заболевания) наследственных факторов в сравнении с таковой факторов среды. Наследуемость выражается в абсолютных цифрах (от нуля до единицы) или процентах при помощи коэффициента h2 или Кн, который рассчитывается по формуле: Кн = G/Е x 100%, где Кн - коэффициент наследования, G - наследственные факторы, Е - факторы окружающей среды. В таблице 4 приведены значения ряда коэффициентов наследования мультифакториальных признаков и заболеваний.
Таблица 4. Коэффициенты наследования мультифакториальных признаков и заболеваний
Название патологии | Кн % |
Шизофрения: | |
злокачественная форма | 85 |
вялотекущая форма | 74 |
Бронхиальная астма | 80 |
Расщелина губы и нёба | 76 |
Сердечно-сосудистые заболевания | 65 |
Артериальная гипертензия | 62 |
Дефекты невральной трубки | 60 |
Изолированный врожденный порок сердца | 35 |
• Степень выраженности признака или тяжесть течения болезни у пробанда. Чем сильнее выражен признак или тяжелее протекает
заболевание у больного родственника, тем выше риск его развития у здоровых родственников.
• Общность генов у пробанда и его родственников (или близкая степень родства с больным родственником). Чем больше общих генов у больного и его родственников, тем выше риск развития у последних признака или заболевания. Например, популяционная частота псориаза составляет 0,75%. У родственников I степени родства частота его развития - 5,6%, у родственников II степени родства - 3,0-3,5%, у родственников III степени родства - 1,75%, у родственников IV степени родства - 0,75%.
• Редко поражаемый пол. Мультифакториальный признак или заболевание проявляется чаще у лиц редко поражаемого пола (критерий, названный эффектом Картера). Например, врожденный пилоростеноз у мальчиков встречается в 2-5 раз чаще, чем у девочек, т.е. в данном случае женский пол - редко поражаемый пол. Однако частота этой болезни у будущих детей пораженных пилоростенозом девочек достигнет 10-20%, тогда как у будущих детей пораженных пилоростенозом мальчиков - только 2-6%. Другой пример - язвенная болезнь желудка и двенадцатиперстной кишки, как правило, проявляющаяся у лиц мужского пола и гораздо реже - у лиц женского пола. Однако ее частота у детей больной женщины выше, чем у детей больного мужчины.
• Число больных родственников. Чем больше в родословной родственников, имеющих мультифакториальный признак или заболевание, тем выше риск его развития у потомков (табл. 5).
Таблица 5. Риск развития мультифакториального признака или заболевания у пробанда в зависимости от числа его больных родственников
Название патологии | Число больных и риск болезни | ||
1 | 2 | 3 | |
Врожденный порок сердца: | |||
дефект межжелудочковой перегородки | 1:20 | 1:7 | 1:2 |
тетрада Фалло | 1:40 | 1:12 | 1:4 |
Дефекты невральной трубки | 1:40 | 1:12 | 1:4 |
Эпилепсия | 1:30 | 1:10 | 1:3 |
Бронхиальная астма | 1:20 | 1:7 | 1:2 |
Недифференцированная УМО | 1:20 | 1:7 | 1:2 |
Шизофрения | 1:20 | 1:4 | 1:2 |
Паховая грыжа | 1:10 | 1:4 | 1:2 |
Инсулинзависимый сахарный диабет | 1:20 | 1:3 | 1:2 |
Общий риск для детей, если их родители здоровы, составляет 5-10%; если болен один из родителей - 10-20%; если больны оба родителя - до 40%;
Дополнительные критерии
• Близнецовый критерий . Если конкордантность (сходство) монозиготных близнецов по какому-то признаку или заболеванию в 4 раза выше конкордантности у дизиготных близнецов, то этот признак или заболевание наследуется по полигенному варианту.
• Критерий сегрегационного отношения пораженных сибсов в семьях с одним больным или двумя здоровыми родителями. Если доля больных сибсов в семьях с одним больным родителем выше в 2,5 раза (и более), чем доля больных сибсов в семьях с двумя здоровыми родителями, то особенно вероятен полигенный вариант наследования. Если указанное соотношение меньше 2,5, то полигенное наследование также не исключено.
Однако классические варианты наследования генов и признаков имеют место лишь в меньшей части всех случаев такого наследования, тогда как почти 2/3 из них - нетрадиционные варианты.
Нетрадиционное наследование
Нетрадиционное наследование отмечается при многих наследственных болезнях человека. В последние годы к ним отнесены ранее плохо изученные или вообще неизвестные заболевания 6 классов: болезни накопления (44 нозологии; см. главу 21); пероксисомные болезни (17 нозологий; см. главу 26); митохондриальные болезни (выделено свыше 200 нозологий, предполагается наличие еще около 1150; см. главу 26); болезни импринтинга (выделены 24 нозологии; см. главу 28); болезни экспансии числа нуклеотидных повторов (27 нозологий; см. главу 27); прионные болезни (9 нозологий; см. главу 29).
В целом на долю болезней нетрадиционного наследования приходится 321 нозологическая единица (без учета 1150 митохондриальных болезней). При наследовании заболеваний первых трех классов речь идет о материнском наследовании, т.к. лизосомы, митохондрии и пероксисомы, являясь цитоплазматическими структурами соматических клеток, наследуются исключительно по линии мать-дочь и никогда не передаются по линиям мать-сын, отец-сын или отец-дочь, что обусловлено биологическим матриархатом (см. главы 5 и 12).
Вместе с тем, при ряде митохондриальных болезней, помимо чисто материнского наследования, может наблюдаться смешанное с материнским моногенное наследование. Оно связано с нарушениями взаимодействия между митохондриальным (мтДНК) и ядерным (ДНК) геномами. В частности, гены, кодирующие митохондриальные белки, находятся либо в митохондриях (гены двух типов рРНК, 22 типов тРНК, 13 типов полипептидов), либо в ядре клетки (их примерно 1150). Ядерные гены транскрибируются в ядре, транслируются в цитоплазме, а результаты их экспрессии «импортируются» митохондриями.
Обычно все копии мтДНК идентичны между собой (гомоплазия). В случаях, когда в мтДНК возникают мутации (их частота в 10 раз чаще, чем в ядерной ДНК), возможно наличие в одной клетке двух типов мтДНК (гетероплазия), и тогда процесс распространения мутантной и нормальной мтДНК называется репликативной сегрегацией (см. главу 2).
В случае болезней импринтинга или эпигеномного выключения из экспрессии локусов хромосом одного из родителей (см. выше) фенотипические проявления действия гена могут меняться в результате трех причин: делеция гена (генетический импринтинг), однородительская изодисомия (хромосомный импринтинг) и нарушение генной экспрессии в центре импринтинга.
Генетический импринтинг основан на механизме специфического метилирования цитозиновых оснований молекулы ДНК, выключающем транскрипцию гена (см. главу 28).
В настоящее время предполагается существование около 200 генов, подверженных такому импринтингу и имеющих тканеспецифическую моноаллельную экспрессию, а также обнаружены три кластера генов, локализованных в критических областях хромосом (7q32, 11р15 и 15q11.2-q13) и оказывающих прямое влияние на развитие наследственной патологии по такому механизму, в том числе опухолей.
Геномный импринтинг основан на однородительской изодисомии по конкретной хромосоме либо материнского, либо отцовского происхождения. Он наблюдается при ряде наследственных и врожденных заболеваний, таких, как: синдромы Ангельмана (причина - однородительская изодисомия по отцовской хромосоме 15) и Прадера-Вилли (изодисомия по материнской хромосоме 15); транзиторный неонатальный сахарный диабет (отцовская хромосома 6); синдром Сильвера-Рассела (материнская хромосома 7); синдром
Беквитта-Видемана (отцовская хромосома 11); задержка физического и моторного развития, гипотония и преждевременное половое развитие (материнская хромосома 14); задержка внутриутробного развития (ЗВУР) плода (материнская хромосома 7), а также ЗВУР с ограниченным плацентарным мозаицизмом (материнская хромосома 16).
Эффект геномного импринтинга имеет место также при пузырном заносе. Отмечены 4 типа нарушений:
• андрогенез или двойной набор хромосом (2n): два сперматозоида с Х-хромосомами и яйцеклетка без ядра;
• гиногенез или двойной набор хромосом (2n): яйцеклетка с двойным набором хромосом, сперматозоиды не участвуют в оплодотворении;
• андроид или тройной набор хромосом (3n): 2 отцовских и 1 материнская;
• гиноид или тройной набор хромосом (3n): 2 материнские и 1 отцовская.
Импринтинг в результате ошибок генной экспрессии в центре импринтинга
происходит также при синдромах Ангельмана и Прадера-Вилли. Он обусловлен тем, что оба родителя передают больному потомку гены, несущие их специфические свойства, т.е. гены отца и матери активированы или супрессированы у потомка по-разному (так называемые импринтированные гены). Причем по линии как матери, так и отца наследуются именно родительские нарушения генной экспрессии в центре импринтинга.
В настоящее время выделено не менее 60 импринтированных генов (или их транскриптов) на 1, 5-7, 11, 13, 15, 19 и 20 аутосомах и Х-хромосоме.
Следует отметить, что при синдромах Ангельмана и Прадера- Вилли также обнаружены близко расположенные, но противоположно импринтированные гены. Их назвали генами-кандидатами заболеваний (в семьях с повторными случаями этих синдромов). Причем геныкандидаты синдрома Ангельмана экспрессировались исключительно на материнской хромосоме, но репрессировались на отцовской хромосоме, тогда как гены-кандидаты синдрома Прадера-Вилли экспрессировались на отцовской хромосоме, но репрессировались на материнской хромосоме (см. главу 28).
Таким образом, импринтинг есть результат качественных, а не количественных изменений наследственного материала.
В конце XX в. выделили также ранее неизвестный класс болезней с нетрадиционным наследованием - болезней экспансии нуклео-
тидных повторов, связанных с увеличением числа кодирующих и некодирующих последовательностей нуклеотидов в результате динамических мутаций. Динамическая мутация «движется» от состояния фенотипически не проявляющейся премутации к состоянию фенотипически проявляющейся полной мутации. Такие мутации лежат в основе ряда тяжелых наследственных нейродегенеративных заболеваний (27 нозологий; см. главу 27). Например, повтор ЦЦГ характерен для синдрома Мартина-Белл (Xq23), повтор ГГЦ - для второго варианта этого синдрома (Xq28); повтор ЦТГ - для спинобульбарной мышечной атрофии (Xq11-12); ЦАГ - для миотонической дистрофии (19q13.3); ЦТГ - для хореи Гентингтона (4р16.3); ЦТГ - для спиномозжечковой атаксии, тип I (6р21.3); ЦТГ - для болезни Мачадо-Джозефа (14q32.1); ТТЦ - для атаксии Фридрейха (9р13). Во всех случаях происходит постепенное накопление (экспансия) критического числа нуклеотидных повторов.
Параллельно с экспансией в каждом последующем поколении нарастает тяжесть течения заболевания - антиципация.
ПОКАЗАТЕЛИ ВЗАИМОДЕЙСТВИЯ ГЕНОВ КАК ДОПОЛНИТЕЛЬНЫЕ ПОНЯТИЯ ГЕНОМИКИ И ПРОТЕОМИКИ
Согласно закономерностям генетики, отцовский и материнский геномы, объединившиеся в генотипе одной клетки и целого организма, вступают между собой во взаимодействие на протяжении всего онтогенеза, создавая индивидуальную внешнюю и внутреннюю характеристику, фенотип организма.
Выделяют три группы механизмов взаимодействия между родительскими генами и соответственно три группы показателей, относящихся к дополнительным понятиям геномики и протеомики. Это взаимодействия между:
• аллельными генами; их характеризуют: доминирование, рецессирование, кодоминирование, неполное и условное доминирование, сверхдоминирование;
• неаллельными генами; их характеризуют: эпистаз, комплементарность, полимерия;
• отдельным геном и генотипом (как системой генов); их характеризуют: экспрессивность, пенетрантность, эффект положения,
генокопирование, эффект плейотропии, аллельные серии и фенокопирование.
Рассмотрим эти механизмы как дополнительные понятия геномики и протеомики.
Механизмы взаимодействия между аллельными генами
Доминирование и рецессирование
Если функциональное состояние одного аллеля (например, материнский аллель) не зависит от состояния другого аллеля (отцовский аллель), то у потомка проявится признак, контролируемый материнским аллелем. Такой ген и контролируемый им признак называются доминатными.
Первое описание эффекта доминирования относится к 1905 г., когда в родословной семьи больного с брахидактилией была отмечена короткопалость. Другими примерами служат: белый локон, «куриная слепота», габсбургская губа, полидактилия (многопалость), синдактилия (сращение мягких или костных тканей фаланг), арахнодактилия («паучьи пальцы»), хондродистрофия, а также многочисленные формы аутосомно-доминантных заболеваний, например хорея Гентингтона (4р16.3).
Если функциональное состояние одного аллеля (например, материнский аллель) зависит от состояния другого аллеля (отцовский аллель), то у потомка проявится признак, контролируемый и материнским, и отцовским аллелями одновременно. При этом и сам ген, и контролируемый им признак называются рецессивными.
Примеры наследования рецессивных признаков и фенотипов: альбинизм, мягкие прямые волосы, курносый нос, светлые глаза, резусотрицательная I группа крови, неспособность ощущать вкус фенилтиокарбамида; многочисленные формы аутосомно-рецессивных заболеваний, например генокопии фенилкетонурии (12q24.2; 4р15.1).
В некоторых случаях доминантность и рецессивность генов плохо соотносятся с доминантностью и рецессивностью признаков. Например, эпикант у монголоидов контролируется доминантным геном, а у бушменов и готтентотов - рецессивным геном.
Другой пример - упомянутые выше ген и признак плешивости, проявляющиеся у мужчин как доминантные, а у женщин как рецессивные. Как оказалось, такой механизм обусловлен действием гормонов, т.е. имеет место зависимое от пола (контролируемое полом) наследование.
Неполное доминирование
О неполном доминировании, или промежуточном действии генов (проявлении признаков), говорят при ослаблении действия доминантного гена в присутствии рецессивного гена, т.е. у гетерозигот. Однако четкую границу между промежуточным действием и доминантностью с одной стороны, а также промежуточным действием и рецессивностью с другой стороны, провести нельзя. Например, пигментация кожи у человека варьирует от белого цвета у альбиносов до черного цвета у негров. От браков между белыми и неграми рождаются мулаты, имеющие промежуточный цвет кожи.
Другой пример неполного доминирования - различия по 6 типам певческого голоса, которые контролируются одной аллельной парой. В частности, баритон и меццо-сопрано наблюдаются только у гетерозигот, тогда как тенор и бас, альт и сопрано характерны для гомозигот.
При дальнейших исследованиях, правда, обнаружилось: пигментация кожи и тип певческого голоса определяются не только этим механизмом взаимодействия, но и независимыми друг от друга факторами: влиянием половых гормонов, эффектом полимерии, сцепленным с полом или зависимым от пола наследованием.
Условное (неустойчивое) доминирование
При неустойчивом или условном, доминировании проявление признака у гетерозигот зависит от генотипа и внешних условий (модифицирующее воздействие генотипа на главный ген, пенетрантность гена, местоположение гена в составе хромосомы, воздействие температуры).
Кодоминирование
Если аллельные гены активны в одинаковой мере (обладают одинаковым доминантным действием), то это кодоминирование. Классический его пример - наследование IV группы крови (по системе АВО), определяемой тремя аллелями, расположенными в 9-й хромосоме (множественность аллелей). Среди них - два доминантных аллеля (IA и !В) и один рецессивный аллель (I0). Попарное сочетание этих аллелей дает 4 группы крови:
- первая группа - наличие двух одинаковых рецессивных аллелей - I0I0 (гомозигота), обусловливающих присутствие в сыворотке крови альфа- и бета-антител;
- вторая группа - наличие двух одинаковых доминантных аллелей IAIA (гомозигота) или двух разных аллелей IAI0 (гетерозигота), обусловливающих присутствие в сыворотке крови бета-антител;
- третья группа - наличие двух одинаковых доминантных аллелей МВ (гомозигота) или двух разных аллелей М0 (гетерозигота); в сыворотке крови присутствуют альфа-антитела;
- четвертая группа - наличие двух разных доминантных аллелей !А!В (гетерозигота); в сыворотке крови нет антител, оба аллеля взаимодействуют с одинаковой силой, нейтрализуя друг друга.
Еще один пример кодоминантности - наследование серповидноклеточной анемии, являющейся аутосомно-рецессивным заболеванием (11р15). В данном случае наблюдается гомозиготность (2 патологических аллеля одного гена, контролирующего синтез дефектного гемоглобина). Такие гомозиготы имеют характерную симптоматику, но они невосприимчивы к малярии, ибо малярийный плазмодий не воспроизводится на дефектном гемоглобине.
Вместе с тем, в гетерозиготном организме одновременно присутствуют нормальный и дефектный аллели одного и того же гена. Причем оба аллеля дают одинаковый доминантный эффект, и поэтому в клетках одновременно синтезируются два вида гемоглобина (нормальный и аномальный). У таких гетерозиготных носителей патологического гена симптомов серповидноклеточной анемии нет либо она проявляется в легкой форме и только в условиях кислородной недостаточности.
Сверхдоминирование
В ряде случаев аллели, находящиеся в гетерозиготном состоянии, фенотипически проявляются сильнее, чем аллели, находящиеся в гомозиготном состоянии (эффект сверхдоминирования). Такое их проявление напоминает эффект гетерозиса у растений (гибридная мощность или сила). Так, в случае браков между представителями разных рас показатели здоровья их потомков превосходят таковое самих родителей: дети отличаются более высокими жизнеспособностью, продолжительностью жизни и др.
Механизмы взаимодействия между неаллельными генами
Эпистаз
Эпистаз - подавление действия гена, находящегося в одной неаллельной паре, действием гена из другой неаллельной пары, например подавление геном А гена В, т.е. A >B или A >bb. Выделяют доминантный и рецессивный эпистаз.
Доминантный эпистаз: доминантный аллель одной неаллельной пары, находящийся в гомозиготном (АА) или гетерозиготном (Аа) состоянии, подавляет проявление неаллельного к нему доминантного аллеля другой аллельной пары, находящейся в состоянии АА или Аа. Гены, дающие доминантный эффект, называются эпистатическими генами или супрессорами (ингибиторами). 0ни могут быть как доминантными, так и рецессивными. Подавляемые гены именуются гипостатическими генами.
Если гены, находящиеся в других неаллельных парах, усиливают доминантное действие эпистатических генов, то они называются генами-модификаторами (интенсификаторами).
Такой тип взаимодействия характерен для неаллельных генов, участвующих в регуляции онтогенеза, например генов иммунного ответа (генная сеть - 2190 генов; см. главу 15) или генов эритропоэза (генная сеть - 200 генов).
Возможны два варианта доминантного эпистаза:
• гомозиготы с рецессивными аллелями (аа) отличаются по фенотипу от гомозигот с доминантными аллелями (АА);
• гомозиготы по доминантным аллелям (АА) не отличаются по фенотипу от гомозигот по рецессивным аллелям (аа).
Рецессивный эпистаз проявляется в том, что рецессивный аллель одного гена подавляет действие неаллельного ему доминантного гена (аа>В), а между доминантными генами наблюдается комплементарность (см. ниже). Примером рецессивного эпистаза у человека служит «бомбейский феномен», связанный с рождением детей с I (I0I0) и IV (IАIВ) группами крови от родителей с I (I0I0) и II (IAI°) группами крови, тогда как теоретически от таких родителей должны рождаться дети с I (I0I0) или II (IAI0) группами крови. Феномен можно объяснить либо наличием не распознанного у одного из родителей редкого гетерозиготного варианта III группы крови (IBI0), либо наличием в генотипе ребенка с IV группой крови (IАIВ) рецессивных генов-модификаторов, которые в гомозиготном состоянии подавляют экспрессию антигенов, находящихся на поверхности эритроцитов, т.е. дают непредсказуемый фенотипический эффект.
Кроме рецессивного эпистаза, выделен двойной рецессивный эпистаз; при нем у рецессивных генов собственное фенотипическое проявление, а в двойных гомозиготах рецессивные аллели подавляют друг друга: аа >bb, bb >аа.
Комплементарность
Комплементарность - тип взаимодействия не менее чем двух доминантных неаллельных генов из нескольких пар с разным сочетанием доминантных и рецессивных аллелей, обусловливающих развитие нового признака, отличного от родительских вариантов.
Известно три типа комплементарности:
• доминантные аллели (АВ) различаются по фенотипическому проявлению;
• доминантные аллели (АВ) сходны по фенотипическому проявлению;
• у доминантных (А) и рецессивных (а) аллелей из нескольких неаллельных пар - самостоятельное фенотипическое проявление.
Например, у человека нормальный слух обусловлен взаимодействием нескольких пар неаллельных генов, но в парах должен находиться как минимум один доминантный аллель. Если же человек окажется рецессивной гомозиготой (хотя бы по одной паре неаллельных генов), то он будет глухим.
Другими примерами комплементарности служат фенотипы онкологических больных с ретинобластомой и нефробластомой (см. главу 25).
Полимерия и формование количественных признаков
Полимерия - это обусловленность признака или фенотипа взаимодействием генов, локализованных в нескольких неаллельных парах и дающих одинаковый эффект. Такие гены называются полимерными генами или полигенами. Степень проявления признака (фенотипа) зависит как от числа доминантных генов в неаллельных парах, так и от числа неаллельных пар.
Такие признаки называются количественными признаками. 0ни существенно отличаются от качественных признаков.
Если количественные признаки контролируются генами, наследуемыми полигенно, и проявляются во многих состояниях как переходные формы, то качественные признаки контролируются генами, наследуемыми моногенно, и проявляются только в альтернативных состояниях без переходных форм.
Примерами количественных признаков являются: упоминавшиеся выше варьирующая пигментация кожи у человека и наличие промежуточного цвета кожи у мулатов.
Другие примеры количественных признаков: уровень (состояние) здоровья человека, продолжительность жизни, интеллектуальные способности, масса и длина тела.
В последние годы выделен феномен взаимодействия между многочисленными неаллельными генами - кумулятивная полимерия. В данном случае речь идет об аддитивном (суммирующем) действии генов, каждый из которых оказывает свое (часто небольшое) влияние на признак. Именно кумулятивная полимерия формирует упомянутые выше генные сети, контролирующие значительную часть количественных признаков организма.
Среди генов, влияющих на количественный признак, могут оказаться один главный ген и ряд более слабых по сравнению с ним генов (полигены). Действие главного гена порой значительно превосходит таковое других генов, и контролируемый главным геном признак наследуется как менделевский (моногенный вариант наследования), а признаки, контролируемые полигенами, наследуются по полигенному варианту. Пример - наследование карликовости, обусловленной главным геном в случае ахондроплазии, тогда как в нормальной популяции рост человека определяется аддитивным действием полигенов.
МЕХАНИЗМЫ ВЗАИМОДЕЙСТВИЯ МЕЖДУ
ОТДЕЛЬНЫМ ГЕНОМ И ГЕНОТИПОМ
Экспрессивность и пенетрантность
Эти понятия впервые введены в 1926 г. Н.В. ТимофеевымРессовским и 0. Фогтом для описания варьирующего проявления признаков и контролирующих их генов. Экспрессивность есть степень выраженности (варьирования) одного и того же признака у разных лиц, имеющих ген, контролирующий данный признак. Наблюдается низкая и высокая экспрессивность. Рассмотрим, например, разную выраженность ринита (насморка) у трех разных больных (А, Б и С) с одним и тем же диагнозом 0РВИ. У больного А ринит выражен в легкой степени («шмыгание носом»), позволяющей в течение дня обходиться одним носовым платком; у больного Б ринит выражен в средней степени (ежедневно 2-3 носовых платка); у больного С - высокая степень выраженности ринита (5-6 носовых платков). Когда говорят об экспрессивности не отдельно взятого признака, а заболевания в целом, врачи часто оценивают состояние больного как удовлетворительное или средней степени тяжести, или как тяжелое,
т.е. в данном случае понятие экспрессивности аналогично понятию «тяжесть течения болезни».
Пенетрантность - это вероятность проявления одного и того же признака у разных лиц, имеющих ген, контролирующий данный признак. Пенетрантность измеряется в проценте лиц с определенным признаком от общего числа лиц, являющихся носителями гена, контролирующего данный признак. 0на бывает неполной или полной.
Примером заболевания с неполной пенетрантностью служит все тот же ринит при 0РВИ. Так, можно считать, что у больного А нет ринита (но есть другие признаки заболевания), тогда как у больных В и С ринит есть. Поэтому в данном случае пенетрантность ринита составляет 66,6%.
Пример заболевания с полной пенетрантностью - аутосомнодоминантная хорея Гентингтона (4р16). 0на манифестирует преимущественно у лиц в возрасте 31-55 лет (77% случаев), у остальных же больных - в другом возрасте: как в первые годы жизни, так и в 65, 75 лет и более. Важно подчеркнуть: если ген этой болезни передан потомку от одного из родителей, то болезнь проявится обязательно, в чем заключается полная пенетрантность. Правда, пациент не всегда доживает до манифестации хореи Гентингтона, умирая от другой причины.
Эффект положения
Другой тип зависимости действия гена от генотипа - эффект положения. 0ткрыл его А. Стертевант (1925). Суть эффекта - в изменении экспрессии гена при изменении занимаемого им положения (позиции) в хромосоме (в ряду нуклеотидных последовательностей).
По современным представлениям, эффект положения не связан с нарушением структуры гена: он и его промоторная область сохраняются как единица транскрипции. Следовательно, эффект положения - эпигеномное событие, определяемое тремя условиями:
• в промоторе происходит инициация транскрипции;
• регуляторные элементы (энхансеры и сайленсеры), содержащие сайты связывания факторов транскрипции, увеличивают специфичность транскрипционного комплекса на промоторе;
• организация хроматина в районе локуса способствует повышенной чувствительности к действию нуклеаз.
Уровень экспрессии зависит от местоположения гена в геноме: либо в районах конденсированного гетерохроматина, либо в районах
недеконденсированного хроматина (эухроматина), который деконденсирован в интерфазе, содержит большинство генов и реплицируется в начале S-фазы. В свою очередь, гетерохроматин конденсирован в течение всего клеточного цикла, реплицируется в конце S-фазы и содержит в основном повторяющиеся последовательности. Центромерные области хромосом состоят из структурного гетерохроматина или плотно конденсированного хроматина, содержащего повторяющие последовательности.
В связи с этими особенностями при формировании структурных перестроек хромосом их разрывы ведут к изменению положения генов, что сопровождается изменением их экспрессии (рис. 22). Доминантный ген А, оказавшийся вблизи места разрыва хромосомы, в большей степени утрачивает свое влияние, чем ген В, расположенный дальше от места разрыва. Иными словами, ослабление эффекта доминантного гена пропорционально расстоянию между ним и точкой разрыва хромосомы.
В других случаях хромосомная перестройка может:
• отделить транскрипционную единицу от регуляторного района, полностью нейтрализовав его влияние на ген (отсутствие энхансера снижает или устраняет транскрипцию в соответствующей ткани); в свою очередь, разделение гена и его сайленсера приведет к аномальному увеличению экспрессии;
• переместить ген в район энхансера другого гена, что вызовет неадекватную экспрессию (например, при лимфоме Беркитта транслокация перемещает ген c-myc под контроль энхансера иммуноглобина);
• переместить ген и его регуляторные последовательности в район другого гена, что снижает уровень экспрессии первого гена;
• вызвать мозаичный эффект положения (например, красный и белый цвет глаз у дрозофилы).
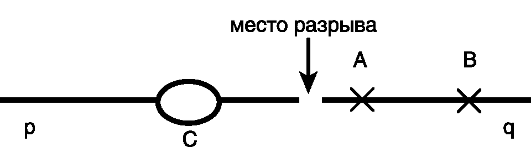 Рис. 22. Эффект положения в случае разрыва хромосомы:
Рис. 22. Эффект положения в случае разрыва хромосомы:
С - центромера; р и q - короткое и длинное плечи хромосомы; Х - генный
локус; А и В - доминантные гены
Другой пример проявления эффекта положения - индивиды с группами крови по системе Rh. В частности, лица с генным комплексом CDE/cDe имеют те же гены, что и лица с генным комплексом cDe/CDE (где заглавная буква - доминантный ген, а прописная буква - рецессивный ген). Однако у первых много антигена Е и мало антигена С, а у вторых - наоборот, что объясняется неодинаковой локализацией генов С и Е - на одной хромосоме или на разных хромосомах соответственно.
Еще один пример - аутосомно-доминантная лице-лопаточноплечевая миодистрофия типа 1А, обусловленная делецией вариабельного участка в субтеломерной части длинного плеча хромосомы (расположен в сегменте 4q35), который не содержит генов, но имеет нуклеотидные последовательности размерами 20-250 кб. Делеция этих последовательностей приводит к изменению структуры гетерохроматина в данной области хромосомы и опосредованному ингибированию транскрипции близлежащего гена (FRG1), расположенного проксимально от точки разрыва хромосомы.
Назовем еще ряд наследственных заболеваний, связанных с эффектом положения. Среди них:
• аниридия в результате делеции гена, расположенного в локусе
11р13;
• голопрозэнцефалия - мутация в гене SSN (7q36);
• краниосиностоз Сатре-Котцен - мутация в гене TWIST (7p21);
• цефалополисиндактилия Грейга в результате мутации генаСLIЗ
(7p13);
• синдром Ригера - ген PITX2(4q25-q27);
• XY-реверсия пола - мутация в гене SRY (Xq).
Генокопирование и его причины
Один и тот же признак (группа признаков) бывает обусловлен разными генетическими причинами (или гетерогенностью). Такой эффект, по предложению немецкого генетика Х. Нахтхайма, получил в середине 40-х годов XX в. название генокопирования. Известны три группы причин генокопирования.
Причины первой группы объединяет гетерогенность за счет полилокусности, или действия разных генов, расположенных в разных локусах на разных хромосомах. Например, среди наследственных болезней обмена сложных сахаров - глюкозоаминогликанов выделены 19 типов (подтипов) мукополисахаридозов. Все типы харак-
теризуются дефектами разных ферментов, но проявляются одной и той же (либо сходной) симптоматикой гаргоилического дисморфизма или фенотипа звонаря Квазимодо - главного героя романа «Собор Парижской Богоматери» классика французской литературы Виктора Гюго. Схожий фенотип нередко наблюдается и при муколипидозах (нарушениях обмена липидов).
Другой пример полилокусности - фенилкетонурия. Сейчас выделены не только ее классический тип, обусловленный дефицитом фенилаланин-4-гидроксилазы (12q24.2), но и три атипичные формы: одна вызвана дефицитом дигидроптеридинредуктазы (4р15.1), а еще две - дефицитом ферментов пирувоилтетрагидроптерин-синтетазы и тетрагидробиоптерина (соответствующие гены пока не определены).
Дополнительные примеры полилокусности: гликогенозы (10 генокопий), синдром Эллерса-Данлоса (8), нейрофибраматоз Реклингаузена (6), врожденный гипотиреоз (5), гемолитическая анемия (5), болезнь Альцгеймера (5), синдром Барде-Бидля (3), рак грудной железы (2).
Причины второй группы объединяет внутрилокусная гетерогенность. Она обусловлена либо множественным аллелизмом (см. главу 2), либо наличием генетических компаундов, или двойных гетерозигот, имеющих два одинаковых патологических аллеля в идентичных локусах гомологичных хромосом. Пример последнего - гетерозиготная бета-талассемия (11р15.5), формирующаяся в результате делеций двух генов, кодирующих бета-цепи глобинов, что ведет к повышенному содержанию гемоглобина HbA2 и повышенному (или нормальному) уровню гемоглобина HbF.
Причины третьей группы объединяет гетерогенность за счет мутаций в разных точках одного и того же гена. Пример - муковисцидоз (7q31-q32), развивающийся из-за наличия почти 1000 точковых мутаций в гене, отвечающем за болезнь. При общей длине гена муковисцидоза (250 тыс. н.п.) в нем предполагается обнаружить до 5000 таких мутаций. Данный ген кодирует белок, ответственный за трансмембранный перенос ионов хлора, что ведет к увеличению вязкости секрета экзокринных желез (потовых, слюнных, подъязычных и др.) и закупорке их протоков.
Другой пример - классическая фенилкетонурия, обусловленная наличием 50 точковых мутаций в гене, кодирующем фенилаланин-4- гидроксилазу (12q24.2); всего при этой болезни предполагается обнаружить более 500 точковых мутаций гена. Большинство их возникает
из-за полиморфизма по длине рестрикционных фрагментов (RFLP) или по числу тандемных повторов (VNTP). Установлено: главная мутация гена фенилкетонурии в славянских популяциях - R408 W/
NP2/VNTR3.
Эффект плейотропии
Вышеупомянутая неоднозначность характера связей между генами и признаками выражается также в эффекте плейотропии или плейотропного действия, когда один ген вызывает формирование целого ряда признаков.
Например, ген аутосомно-рецессивной атаксии-телеангиэктазии, или синдрома Луи-Бар (11q23.2) ответственен за одновременное поражение не менее шести систем организма (нервная и иммунная системы, кожные покровы, слизистые оболочки органов дыхания и желудочно-кишечного тракта, а также конъюнктива глаз).
Другие примеры: ген синдрома Барде-Бидля (16q21) обусловливает слабоумие, полидактилию, ожирение, пигментную дегенерацию сетчатки; ген анемии Фанкони (20q13.2-13.3), контролирующий активность топоизомеразы I, вызывает анемию, тромбоцитопению, лейкопению, микроцефалию, аплазию лучевой кости, гипоплазию пястной кости I пальца, пороки развития сердца и почек, гипоспадию, пигментные пятна кожи, повышенную ломкость хромосом.
Выделяют первичную и вторичную плейотропию. Первичная плейотропия обусловлена биохимическими механизмами действия мутантного белка-фермента (например, недостаточностью фенилаланин-4-гидроксилазы при фенилкетонурии).
Вторичная плейотропия обусловлена осложнениями патологического процесса, развившегося в результате первичной плейотропии. Например, за счет усиленного кроветворения и гемосидероза паренхиматозных органов у больного с талассемией возникают утолщение костей черепа и гепатолиенальный синдром.
Аллельные серии
Точковые мутации одного гена могут обусловить развитие не только одного, но и разных симптомокомплексов. Во втором случае играют роль аллельные серии одного и того же гена. Например, мутации гена адренорецептора, сцепленного с Х-хромосомой, становятся причиной болезни Кеннеди, если они захватывают область тринуклеотидных повторов в экзоне 1, но способны привести к син-
дрому Морриса или тестикулярной феминизации (фенотипически - девочка, генотипически - мальчик, т.е. наличие женского фенотипа при мужском кариотипе), если захватывают другие нуклеотидные последовательности данного гена (мутация в андрогенсвязывающем домене).
Другим примером гетерогенности по типу аллельной серии служат точковые мутации гена рецептора тирозинкиназы - RET, вызывающие четыре наследственные болезни: семейную медуллярную карциному щитовидной железы, болезнь Гиршпрунга, множественную эндокринную неоплазию двух типов - 2А (МЭН2А) и 2В (МЭН2В).
Хотя описываемый феномен открыт относительно недавно, уже известно более 100 моногенных болезней, обусловленных мутациями одного гена по типу аллельной серии. Их причинами служат:
• разные последовательности нуклеотидов (разные точки гена), контролирующие функционально разные домены белка;
• наличие в одном и том же гене модифицирующего мутантного аллеля (аллельный полиморфизм);
• воздействие генетического окружения на проявление мутантного аллеля (в том числе взаимодействие мутантного аллеля с аллелями одного или нескольких генов-модификаторов).
Фенокопирование
Фенокопирование - это копирование симптомокомплекса ненаследственной болезни (сформировавшейся под действием факторов среды) как симптомокомплекса наследственной генетической болезни.
Например, симптоматика эндемического зоба соответствует симптоматике врожденного гипотиреоза, но в первом случае причина - дефицит (либо полное отсутствие) неорганического йода в питьевой воде и продуктах питания (т.е. имеет место фенокопирование), а во втором - различные генетические факторы (т.е. имеет место генокопирование).
Другой пример генокопирования и фенокопирования симптоматики - слепота. Известно, что зрение контролируется группой генов (генная сеть), взаимодействующих друг с другом в ходе онтогенеза и обеспечивающих развитие и поддержание функционирования глаз и головного мозга. Разные причины нарушения целостности этой системы способны обусловить один и тот же результат (слепоту).
Так, вследствие генокопирования слепота может развиться из-за дефектов хрусталика, определяемых одними генами, дефектов сетчатки, определяемых другими генами, и дефектов роговицы, определяемых третьими генами.
Вследствие же фенокопирования за счет экзогенного влияния среды слепота может развиться из-за несоблюдения нормативов освещенности жилых и производственных помещений или в результате глазного травматизма.