Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 25 ОНКОГЕННЫЕ БОЛЕЗНИ
Общая характеристика
ОГБ - одна из наиболее проблемных областей теоретической и клинической медицины со времен XIX в. В последние годы эта проблема стала рассматриваться с молекулярно-генетических позиций.
Исторически первое упоминание о возможном наследственном характере семейной формы рака грудной железы относится к 1869 г., когда французский хирург Брока опубликовал результаты исследования родословной своей жены и обнаружил такую опухоль у 10 из 24 женщин - ее близких и дальних родственниц.
В настоящее время стало очевидным, что при проведении клинико-генеалогического обследования онкологических больных врачи-генетики почти не встречают семейных родословных, в которых бы не фиксировались случаи онкологической заболеваемости и смертности у родственников больного пробанда.
Более того, в последние 50-60 лет неуклонно выросла частота многих форм ОГБ, таких, как рак грудной железы, желудочнокишечного тракта, легких, яичников и др. Например, с 1950 г. частота меланомы увеличилась в 20 раз, а с 1980 г. частота ретинобластомы выросла в 3 раза (теперь - 1 случай на 10-15 тыс. человек); частота карциномы почки ежегодно увеличивается на 2%, а нефробластома и нейробластома встречаются с частотой 1:8-10 тыс. человек.
В настоящее время ОГБ встречаются в общей популяции с высокой частотой (детское население - 1:160 человек, взрослое население - 1:10 человек).
Рост частоты ОГБ объясняется не только значительно выросшими в молекулярной медицине возможностями для их диагностики, но и неблагоприятной в ряде случаев экологической обстановкой (см. главу 17). Так, было показано, что после аварии в апреле 1986 г. на Чернобыльской АЭС достоверно выросла частота рака щитовидной железы среди населения, проживающего в районах, подвергшихся радиоактивному заражению.
Окончательное выяснение главенствующей роли генетических и средовых факторов в развитии опухолей у человека произошло на
рубеже XX-XXI вв. благодаря достижениям международной программы «Геном человека» (см. главу 1). Сегодня уже не возникает сомнений, что рак - это полигенное заболевание с наследственной предрасположенностью (90% всех случаев) или моногенное заболевание (10%).
Именно на этой основе ОГБ следует рассматривать как одну из форм наследственной патологии. Какие данные свидетельствуют в пользу такого заключения?
Во-первых, подтверждение в качестве причин ОГБ:
• генных мутаций, как при МБ и ВПР (см. главы 21 и 23);
• хромосомных аберраций, как при XБ (см. главу 24);
• суммарного действия генов и факторов среды, как при МФЗ (см. главу 22);
• гиперэкспрессии и эктопической экспрессии генов, как при тератомах (см. ниже).
Во-вторых, в большинстве случаев механизмы канцерогенеза связаны с потерей гетерозиготности протоонкогенов и наличием гомозиготности генов-супрессоров опухолевого роста (см. главу 17).
В-третьих, гетерозиготные носители мутаций протоонкогенов имеют 100% риск развития опухоли, что прямо указывает на генетическую детерминированность опухолевого роста.
В-четвертых, имеет место идентификация в раковой клетке нарушений ее жизненного цикла, не связанных с апоптозом и некрозом. Это позволяет предположить, что происходящие в такой клетке молекулярные преобразования можно рассматривать как ее стремление к ускоренной смерти организма хозяина (см. главу 11).
Современный взгляд на проблемы канцерогенеза
Из главы 17 известно, что этиология и патогенез опухолевого роста связаны с:
• возникновением в протоонкогенах доминантных гетерозиготных мутаций, ведущих к нарушениям экспрессии генов и, как следствие, к образованию патологических белковых продуктов с агрессивной функцией по отношению к нормальным клеткам;
• эффектом генов-супрессоров опухолевого роста, находящихся в гомозиготном состоянии активирующих их мутаций.
Следует отметить, что в первом случае патологические белки проявляют способность к передаче сигналов, индуцирующих нормальные клетки к злокачественному делению при отсутствии внеш-
них регуляторных стимулов со стороны контролирующих систем организма.
По-видимому, такой же способностью обладают так называемые «слитые белки», являющиеся продуктами химерных транскрибируемых генов, образующихся при хромосомных перестройках (см. ниже). В частности, образованию таких белков предшествует слияние по интронным областям двух генов, расположенных в местах разрывов хромосом (см. эффект положения в главе 5). Причем эти два гена могут быть самые разные, в том числе не связанные с регуляцией роста клетки и дифференцировкой, т.е. «слитые гены» и их белковые продукты приобретают несвойственные им ранее функции по активации новых транскрипционных факторов.
Нарушение экспрессии протоонкогенов обычно связывают с их гиперэкспрессией, но иногда оно обусловлено эктопической экспрессией или работой в местах, не свойственных нормальному расположению (например, в случаях, связанных с тератомами), или находящихся на неподходящей для этого стадии клеточного цикла.
Вместе с тем, гиперэкспрессия клеточных онкогенов (включая протоонкогены) чаще всего является результатом избирательной амплификации. Типичный пример - гиперэкспрессия протоонкогенов, оказавшихся в результате хромосомной перестройки в районах расположения генов Т-клеточных рецепторов или генов иммуноглобулинов и попавших под контроль их мощных промоторов, что приводит к появлению химерных белков с новыми агрессивными функциями и, следовательно, к гиперэкспрессии протоонкогенов.
Как известно, для нормальной клетки характерно физиологическое состояние ритмичного перехода от одного клеточного цикла к другому, обеспечивающее ее пролиферацию, дифференцировку и восстановление поврежденных структур (см. главы 9 и 11). Если обеспечение этого состояния невозможно, клетка переходит к программированной гибели (см. главу 10) либо к неконтролируемому злокачественному росту. При этом внутриклеточные процессы по-прежнему обеспечиваются работой разных групп генов, своевременно включающихся и выключающихся в ходе клеточного цикла.
При переходе клетки к неконтролируемому росту происходит смена каскадов включения-выключения (переключения) групп экспрессирующихся генов, что отражается на общей регуляции клеточного цикла. Очевидно, что важную роль в таком переключении
каскадов играют межклеточные взаимодействия, медиаторами которых служат небольшие молекулы пептидов или факторы роста.
Компоненты пускового механизма опухолевого роста
В любой клетке, находящейся в G0 стадии жизненного цикла (см. главу 9), можно вызвать процесс деления при воздействии факторов роста на ее трансмембранные рецепторы (см. главу 6). Такое воздействие вызывает в этих рецепторах биохимические сигналы, активирующие или репрессирующие ядерные факторы транскрипции, определяющие наборы генов, работающих в клетке, и ее вхождение в жизненный цикл, т.е. речь идет о пусковой роли факторов роста и трансмембранных рецепторов.
Для запуска опухолевого роста в нормальных клетках, по-видимому, существуют некие узловые точки их жизненного цикла, при прохождении которых требуется определенное соотношение позитивных и негативных регуляторов клеточного роста. Преобладание негативных регуляторов нарушает переход нормальной клетки к следующей стадии жизненного цикла, и такая клетка либо вступает в апоптоз, либо начинает бесконтрольно делиться.
Возникает вопрос: «Что же происходит за пределами нормального клеточного цикла?»
Ответ на этот вопрос, по-видимому, следует искать в организации и функционировании сигнальных молекул, регулирующих клеточный цикл (см. главы 8 и 17). Как известно, передача биохимического сигнала осуществляется путем реакций фосфорилирования и связана с участием в них регуляторных белков и белков - вторичных мессенджеров, обладающих киназной активностью.
В последние годы для многих генов человека, кодирующих факторы роста, были обнаружены гомологи среди вирусных онкогенов и тем самым было показано, что факторы роста могут действовать как самостоятельные клеточные онкогены - это первый компонент пускового механизма опухолевого роста. Например, к таким онкогенам относится ген PDGE или ген тромбоцитарного фактора роста, у которого бета-цепь гомологична белку, кодируемому геном v-sis вируса саркомы обезьян. Причем в опухолевых тканях находят не мутантные формы гена этого фактора роста, а наблюдают амплификацию, гиперэкспрессию или нарушение тканеспецифической экспрессии.
К другим примерам можно отнести опухолевые ткани при раке желудка и грудной железы: наблюдается гиперэкспрессия генов
INT2 и NST, относящихся к семейству факторов роста фибробластов.
Второй компонент пускового механизма опухолевого роста - это
трансмембранные рецепторы факторов роста (ТРФР), с помощью которых осуществляется перевод внешнего (внеклеточного) сигнала во внутриклеточный сигнал роста. ТРФР состоят из трех доменов: внешний лигандсвязывающий, трансмембранный и внутренний (цитоплазматический) тирозинкиназный; все они обладают аутофосфорилирующей способностью, возникающей при димеризации молекулы рецептора в процессе его взаимодействия с лигандом.
Показана роль многих генов рецепторных киназ как протоонкогенов в случае возникновения доминантных мутаций, вызывающих постоянную активацию рецептора при отсутствии лиганда. Например, детально изучен онкогенный механизм действия мутаций в гене EGFR рецептора эпидермального фактора роста, цитоплазматический домен которого имеет 95% гомологию с онкогеном v-erb вируса эритробластоза у птиц.
Дополнительными свидетельствами в пользу такого пускового механизма онкогенного действия мутаций являются:
• миссенс-мутации гена NEU и мутации генов нерецепторных тирозинкиназ (семейство scr-белков - продукт вируса саркомы Рауса, для которой характерно наличие двух некаталитических последовательностей SH2 и SH3);
• мутации гена ABL (гомологичен онкогену v-abl вируса мышиной лейкемии Абельсона);
• мутации генов RAF и MOS серин-треонинкиназ, участвующих в формировании многих типов опухолей (как вторичные мессенджеры);
• мутации в генах гуанин-нуклеотидсвязывающих белков или G-белков.
В этих случаях были выделены две группы производимых мутантных белков: большие белки-гетеродимеры (с эндогенной ГТФазной активностью) и малые белки-мономеры.
Мутации больших G-белков обнаружены при ряде опухолей слизистых оболочек человека, что связано с увеличением продукции циклического АМФ или основного митогенного фактора в тканях слизистых оболочек, секретирующих факторы роста.
В свою очередь, малые G-белки являются важными регуляторами митогенной ативности. Среди них хорошо изучено семейство ras- белков (см. главу 8).
Двухударная модель канцерогенеза
Двухударная модель (теория) канцерогенеза была предложена в конце XX в. (Knudson A.G., 1986). Приводился пример гена RB1, описанного как первый ген-cупрeccор опухолевого роста. Согласно рассматриваемой модели, для перехода нормальной клетки в опухолевую необходимы два последовательных события (удара), или две мутации (рис. 70). Первая мутация (первый удар) - это мутантный аллель гена RB1, приводящий к образованию клетки с повышенным риском злокачественной трансформации или к развитию доброкачественной ретиномы сетчатки. Далее для образования спорадической злокачественной ретинобластомы необходимо появление в этой клетке второй мутации (второй удар), которая возникает в доброкачественной ретиноме сетчатки в 90% случаев.
При этом первая мутация может быть унаследована от одного из родителей или появиться во втором аллеле гена RB1, что приведет к злокачественной ретинобластоме.
В последующие годы двухударная модель развития ретинобластомы была подтверждена наличием гомозиготности по полиморфным
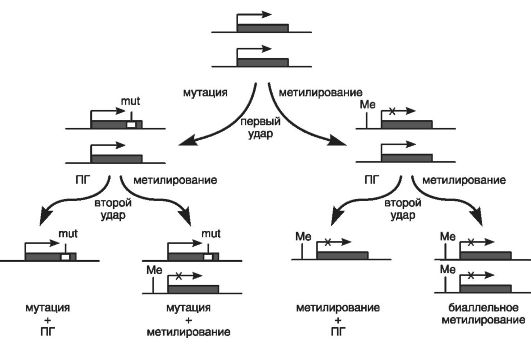 Рис. 70. Двухударная
модель канцерогенеза (по Залетаеву Д.В. и соавт., 2005); ПГ - потеря
гетерозиготности; Ме - метилирование; mut - мутация
Рис. 70. Двухударная
модель канцерогенеза (по Залетаеву Д.В. и соавт., 2005); ПГ - потеря
гетерозиготности; Ме - метилирование; mut - мутация
ДНК-маркерам, хотя в клетках крови таких больных сохранялась гетерозиготность по этим маркерам. Тем самым было отмечено состояние, связанное с потерей нормальной копии гена.
Следовательно, вторым важным событием стали мутация в неповрежденном аллеле гена и превращение гиперпластической клетки с повышенным риском злокачественной трансформации в раковую клетку, которая начала безостановочно делиться, вызывая рост опухоли.
Было также показано, что второй мутацией может быть не только независимое событие во втором аллеле гена RB1, но и полная потеря локуса хромосомы, содержащего данный ген, или потеря гетерозиготности.
Таким образом, согласно двухударной модели канцерогенеза, основными генетическими механизмами опухолевого роста служат:
• первая мутация в виде структурного нарушения гена-супрессора опухолевого роста, вызывающая гиперпластический, но еще не злокачественный процесс;
• последующая потеря неизмененного аллеля гена в клетках опухоли (потеря гетерозиготности), что служит маркером злокачественного роста.
Именно так считалось до недавнего времени. Однако при анализе экспрессии генов, обусловливающих злокачественный рост, было показано отсутствие экспрессии одних и наличие гиперэкспрессии других генов, поэтому механизмы происходящих в таких клетках изменений оставались невыясненными.
В дальнейшем обнаружено: наиболее важным событием опухолевого роста является аномальное метилирование СрG-островков (динуклеотидов), находящихся как в промоторных, так и внутригенных областях генов-супрессоров.
Оказалось, что метилирование СрG-островков и последующая инактивация генов-супрессоров могут возникнуть не только как результат первой и второй мутаций, но и как результат функциональной мутации (см. ниже), например делеции гена RB1. В связи с этими фактами стала очевидной необходимость определения и анализа профиля метилирования как известных, так и клонированных генов, являющихся кандидатами в гены опухолевого роста.
Как было сказано выше, одной из функций таких генов является их участие в регуляции клеточного цикла в опухолевой клетке.
Окончательным подтверждением такого участия стала идентификация целого ряда генов, вовлеченных в канцерогенез. В частности, выяснилось: большое значение имеет метилирование регуляторных (промоторных) областей, приводящее к инактивации геновсупрессоров в клетках опухоли.
Было также продемонстрировано, что не только генетические причины и механизмы могут определять развитие опухолевого роста - такого рода факторами могут стать эпигенетические (надгеномные) события, касающиеся и наследственных, и ненаследственных изменений экспрессии генов без вмешательства в них структурных нарушений нуклеотидных последовательностей ДНК.
Эпигенетическая регуляция экспрессии генов в клетках опухоли
Эпигенетическая регуляция экспрессии генов играет важную роль в опухолевом росте. Она базируется в том числе на аномальном метилировании СрG-островков (находятся в промоторных областях генов), приводящем к полной инактивации генов (см. выше). В геноме человека выделены два типа СрG-динуклеотидов: первый тип - это рассеянные по всему геному отдельные динуклеотиды (их 80%), они расположены в межгенных последовательностях (спейсерах) и редко - в транскрибируемых последовательностях ДНК; второй тип - это островки СрG-динуклеотидов или районы ДНК, обогащенные ими; они расположены вблизи 60% структурных генов, локализованных преимущественно в 5'-районах, где содержатся регуляторные последовательности промоторов. Размеры СрG-островков занимают от 0,5 до 1,5 тыс. н.п.; содержание в них ГЦ-пар превышает 60%, а соотношение СрG/GpC составляет не менее 0,6. За исключением импринтированных генов (см. главу 4), СрG-островки промоторных районов генов нормальных тканей не метилированы.
Тканеспецифические гены не имеют в своих промоторах таких островков, и поэтому в них обнаруживаются только единичные СрG- динуклеотиды, степень метилирования которых различна в разных клетках и тканях, что препятствует локальному связыванию факторов транскрипции.
Метилирование последовательностей ДНК
Метилирование последовательностей ДНК, ее обратимая ковалентная модификация касается только одного из четырех азотистых
оснований - цитозина. Метилирование сопровождается деметилированием. В ходе реакции метилирования-деметилирования образовавшийся в СрО-динуклеотиде цитозиновый остаток метилируется с помощью ДНК-метилтрансфераз, переносящих метильную группу S-аденозилметионина в позиции N5 пиримидинового кольца (служит мишенью для метилирования). Остатки S-аденозилметионина удаляются с помощью ДНК-деметилазы или гликозилазы.
Этот фермент деметилирует геном опухолевых клеток. Он способен узнавать метилированные СрО-динуклеотиды, трансформировать 5-метилцитозин в цитозин и тем самым деметилировать нуклеотидные последовательности ДНК на значительном участке, нарушая стабильность молекулы (рис. 71).
Таким образом, обнаружение связи между снижением активности гена (его выключением) и метилированием промоторной области
5-метилцитозин
NH2
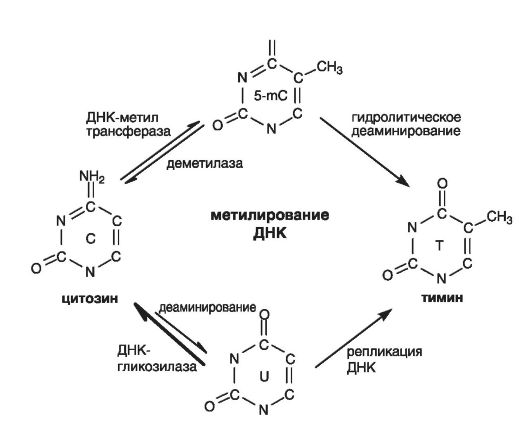
Рис. 71. Схема метилирования-деметилирования (по Залетаеву Д.В. и соавт., 2005)
гена является наиболее веским подтверждением эпигенетического механизма регуляции генной экспрессии.
Кроме того, метилирование последовательностей ДНК включает в себя другие механизмы, способствующие полному прекращению экспрессии гена. Например, метильные группы, выступая в большую бороздку молекулы ДНК, нарушают механизм взаимодействия в реакциях ДНК-белок, препятствуя связыванию специфических транскрипционных факторов. В свою очередь, метилированные районы ДНК могут связывать белки, служащие репрессорами транскрипции, что опосредованно модифицирует состояние хроматина.
Например, процесс деацетилирования гистонов (идет параллельно процессу метилирования) обусловливает более плотную упаковку нуклеосом (см. главу 2), чем вызывает полную инактивацию гена без нарушений его структуры.
Значение метилирования генов, вовлеченных в канцерогенез
Как сказано выше, реакции метилирования с последующей инактивацией клеточных онкогенов могут выступить не только в роли первой или второй мутации (механизм двойного удара), но и функциональной мутации (см. выше). Показана именно такая роль метилирования для многих генов (табл. 14).
Таблица 14. Механизмы инактивации и двойного удара с помощью метилирования одного или двух аллелей генов в опухолевой клетке
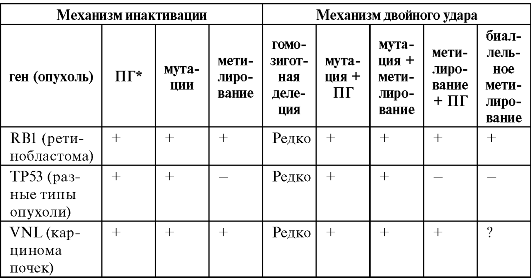
 Примечание. «*» - ПГ (промотор гена); «+» - наличие данных; «-» - отсутствие данных, «?» - разноречивые данные.
Примечание. «*» - ПГ (промотор гена); «+» - наличие данных; «-» - отсутствие данных, «?» - разноречивые данные.
В последние годы был идентифицирован ряд вовлеченных в канцерогенез генов-супрессоров, инактивация которых основана на метилировании их регуляторных областей. Эти гены приведены в табл. 15.
Таблица 15. Гены-супрессоры, СрО-островки которых метилируются при злокачественных опухолях
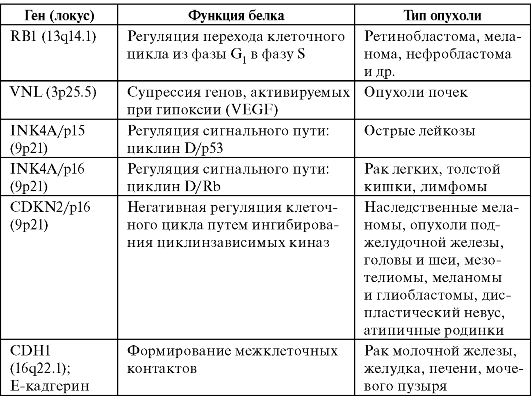
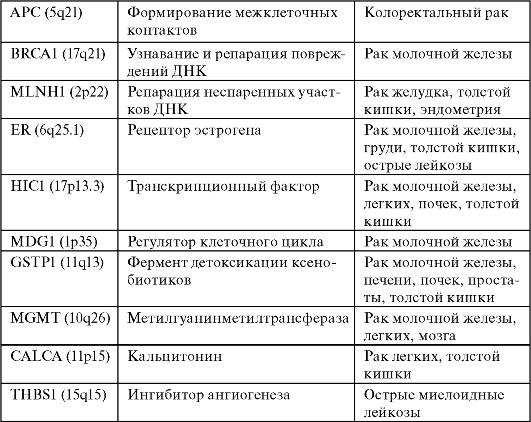 Следует
отметить, что анализ метилирования промоторных районов указанных
генов-супрессоров (как и ряда генов МБ) при эпигенетической регуляции их
экспрессии оказался в целом намного проще и экономически выгоднее (в
сравнении с традиционными молекулярно-генетическими технологиями) для
оценки вклада клеточных онкогенов в развитие как нормальных, так и
злокачественных клеток. Аномальное метилирование-деметилирование в
настоящее время стало современным диагностическим тестом и
прогностическим маркером при выявлении определенных типов опухолей.
Следует
отметить, что анализ метилирования промоторных районов указанных
генов-супрессоров (как и ряда генов МБ) при эпигенетической регуляции их
экспрессии оказался в целом намного проще и экономически выгоднее (в
сравнении с традиционными молекулярно-генетическими технологиями) для
оценки вклада клеточных онкогенов в развитие как нормальных, так и
злокачественных клеток. Аномальное метилирование-деметилирование в
настоящее время стало современным диагностическим тестом и
прогностическим маркером при выявлении определенных типов опухолей.
Характеристика профиля (паттерна) метилирования известных генов человека и вновь клонированных генов в разных тканях и типах опухолей вносит существенный вклад в понимание механизмов опухолевого роста. Именно это направление стало новым направлением молекулярной медицины, которое, как полагают ученые, приведет в ближайшие годы к разработке эффективных диагностических
технологий и методов терапии как разных типов рака, так и МБ (см. главы 4, 7, 17 и 21).
ДНК-диагностика рака
В настоящее время в ДНК-диагностике рака выделено пять направлений. Среди них диагностика наследственных форм рака; маркеров неблагоприятного прогноза рака; маркеров микрометастазов рака (минимальной остаточной болезни); маркеров начальных стадий рака; маркеров, увеличивающих риск определенного типа рака.
Первое направление включает идентификацию мутаций геновсупрессоров опухолевого роста, участвующих в регуляции клеточного цикла. В частности, ведется поиск мутаций одного из аллелей гена RB1, наследуемого по аутосомно-доминантному типу и проявляющегося варьирующей экспрессивностью и неполной пенетрантностью (90%) - это первый пример. В данном случае доброкачественная ретинома сетчатки глаза развивается в результате инактивации неповрежденного аллеля гена - это первая мутация (удар), возникающая, как правило, в раннем детском возрасте и гораздо реже - во внутриутробном периоде развития. Вторая мутация (удар) может возникнуть в сетчатке глаза (что вызовет злокачественную ретинобластому) или других соматических клетках (что вызовет вторичную опухоль). При этом в роли вторичной опухоли может выступить основное заболевание (лимфоклеточный лейкоз, рак легких или мелкоклеточный рак, рак грудной железы, рак половых органов и остеосаркома).
Второй пример - это идентификация мутаций при нефробластоме, которая относится к генетически гетерогенным заболеваниям, в том числе в каждом третьем случае она связана с инактивацией генасупрессора WT1. В ряде случаев нефробластома обусловлена делецией сегмента короткого плеча хромосомы 11 (11р13) и приводит к синдрому WAGR (опухоль Вилмса, аномалии мочеполовой системы и задержка физического и умственного развития). В более редких случаях нефробластома связана с точковыми мутациями гена транскрипционного фактора (миссенс-мутации) или белкового мотива «цинковые пальцы», обусловливающего развитие синдрома Дениса-Дрэша (см. главы 16 и 17), проявляющегося прогрессирующей нефропатией, псевдогермафродитизмом, нефробластомой и гонадобластомой.
Третий пример - это синдром фон Хиппель-Линдау, объединяющий четыре типа опухолей (гемангиобластома, карцинома почки, феохромоцитома и опухоль поджелудочной железы), обусловленных
мутациями гена VNL (3p25.5), к которым относятся: крупные делеции; мутации сдвига рамки считывания; миссенс-мутации экзонов 1 и 3; соматические мутации экзонов 1 и 2 (только в клетках изолированных непапиллярных карцином почки).
При этом 20% опухолей связаны с метилированием промоторной части гена.
Четвертый пример - семейный аденоматозный полипоз желудочно-кишечного тракта и его вариант - аутосомнодоминантный синдром Гарднера с преимущественным поражением толстого кишечника. При этом заболевании часто выявляются гепатобластомы, медуллобластомы, фибромы, рак щитовидной железы, которые обусловлены делециями и нонсенс-мутациями, ведущими к преждевременной остановке синтеза белка в результате повреждения двух кодонов (1061 и 1309). Причем мутации второго кодона вызывают быстрое появление полипов и их злокачественное перерождение, тогда как соматические мутации, как правило, затрагивают кодоны 1286 и 1513.
Пятый пример - синдром Горлина или синдром базальноклеточного невуса, связанный с мутацией в гене-супрессоре РТС1 (9р22.3). Этот синдром - редкое аутосомно-доминантное заболевание, проявляющееся множественными аномалиями развития, невусами и вторичными злокачественными опухолями (астроцитомы, базально-клеточные карциномы, карциномы и фибромы яичников, медулобластомы). При этих типах опухолей выявляется генетический (геномный) полиморфизм по промоторным областям гена - маркерам сегмента 9р22.3.
Другие примеры мутаций генов наследственных форм рака:
• гена-супрессора р53 или гена синдрома Ли-Фрамени; сопровождается опухолями мозга, саркомами мягких тканей, остеосаркомами, карциномой молочной железы, лейкемией;
• гена атаксии-телеангиэктазии или синдрома Луи-Бар (11q23.1); обусловливает развитие глиомы, В-клеточной лимфомы, медуллобластомы и Т-клеточной лейкемии;
• гена NBS1 или гена синдрома Ниймиген; обусловливает аномалии фенотипа, иммунодефицит, предрасположенность к развитию гемобластом, лимфобластной лейкемии, лимфом и нейробластом; в последнем случае: нейробластома - это одна из частых и хорошо изученных у детей солидных опухолей трех типов, развивающихся по разным генетическим причинам (потеря гете-
• розиготности 1р или 14q; варьирующая активность теломеразы; разная экспрессия гена TRK-A); генов рака грудной железы (BRCA1 и BRCA2, обусловливают развитие вторичного рака поджелудочной железы или яичников). Второе направление связано с поиском маркеров неблагоприятного прогноза рака.
Известно, что 90% случаев рака связаны с генами наследственной предрасположенности, сочетающейся с действием факторов среды, т.е. с мультифакториальными причинами. Такой рак нередко возникает спорадически, без отягощенного семейного анамнеза, обязательного при наследственных формах рака.
При этом всякий раз в геноме соматической клетки выявляются уникальные индивидуальные изменения, характеризующие процесс злокачественного перерождения и связанные с типом мутации, а также типом экспрессии (инактивация или амплификация) и другими особенностями.
Иными словами, всякий раз необходимо определение индивидуального молекулярного профиля опухолевого роста, что затрудняет определение прогноза и тактики лечения онкологического заболевания.
Следует отметить, что раннее обнаружение прогностических маркеров рака связано с решением ряда вопросов (поиск опухоли, анализ ее инвазивности и способности к метастазированию, восприимчивость или невосприимчивость к терапии, определение периода ремиссии и процента выживаемости больного).
В настоящее время накоплены данные о молекулярной структуре ряда опухолей, установлены характерные для каждой нозологии молекулярные маркеры, позволяющие поставить точный диагноз уже при анализе гистологического препарата.
В свою очередь, прогностически ценно (например, при раке грудной железы) установление факта экспрессии или гиперэкспрессии некоторых генов в клетках опухоли, таких, как гены онкогенов RNAMM и HER2/neu, кадгерина-11, сиалилтрансфераз ST3Gall III и ST6Gal I, стероидной сульфатазы, циклина CCND1, альфа- и бетаэстрогенных рецепторов, лекарственной резистентности MDr1 и фактора роста гепатоцитов HGF.
Одновременная экспрессия сразу нескольких генов может свидетельствовать об относительно благоприятном прогнозе, например, в случаях совместной экспрессии антиметастатического гена nm 23, гена ICAM-1 и гена простат-специфического антигена (PSA).
О благоприятном прогнозе будет также свидетельствовать потеря гетерозиготности по маркерам хромосомы 16 (16q23.2-q24.2), тогда как потеря гетерозиготности по короткому плечу хромосомы 17 (17р13.3), наоборот, будет указывать на неблагоприятный прогноз.
В свою очередь, при раке простаты прогноз неблагоприятен при гиперэкспрессии гена орнитиндекарбоксилазы, эктопической экспрессии TSPY и отсутствии экспрессии TGF-бета в тестикулах.
При раке легкого на неблагоприятный прогноз будет указывать снижение экспрессии белка-ингибитора клеточного цикла р27 и потеря аллелей гена-супрессора FNIT, тогда как благоприятный прогноз будет связан с экспрессией тромбоспондина TSP2, ингибирующего рост и метастазирование опухоли.
В случае колоректального рака прогноз неблагоприятен при выраженной экспрессии генов таких белков, как катепсин В, CD44, p53 и тимидилатсинтетаза, NRP1 рецептор витамина D, а также при отсутствии экспрессии гена (локализован на длинном плече хромосомы 18), кодирующего белок р21.
При агрессивных карциномах пищевода и желудка будет наблюдаться гиперэкспрессия генов ST3, BM-4-/SPARK, MET, uPA и потеря гетерозиготности по маркерам длинного плеча хромосомы 18.
Кроме того, прогноз в отношении других типов опухолей неблагоприятен при гиперэкспрессии генов: транскрипционного фактора Id2, каспазы-1, тимидинфосфорилазы, промоторов DPC1 на коротком плече хромосомы 1 и DCC1 на длинном плече хромосомы 18 (рак поджелудочной железы); генов опухоль-индуцированного белка-антигена GAGE и делеции длинного плеча хромосомы 10 (генсупрессор PTEN) - соответственно выявляются при глиомах и высокоинвазивных менингиомах; генов циклина А (карцинома печени) и D (саркома мягких тканей конечностей); гена Lerk-5 (агрессивная меланома) и гена кадгерина-6 (карцинома почки).
Третье направление связано с диагностикой минимальной остаточной болезни или поиском метастазов высокоинвазивных опухолей. Оно базируется на определении экспрессии ряда опухоль-специфических генов в образце биологического материала с помощью ПЦР с праймерами на обратно транскрибируемую мРНК или ОТ-мРНК. Такие праймеры получены для агрессивной меланомы (экспрессия тирозиназы; гликопротеина MUC18, участвующего в клеточной адгезии, и бета1-4-N-ацетил-галактозаминилтрансферазы); метастазирующей нейробластомы (экспрессия тирозингидроксилазы); рака грудной
железы (экспрессия маммоглобин-тканеспецифического маркера, цитокератина 19 или гена СК19, рецептора эпидермального фактора роста или гена EGFR); рака желудка (экспрессия гена СК19 и карциноэмбрионального антигена СЕА); рака эпителия желудочнокишечного тракта (экспрессия цитокератина 20); рака поджелудочной железы или легких (экспрессия гена СК19); рака простаты (экспрессия гена PSA); саркомы Юинга и рабдомиосаркомы.
Четвертое направление позволяет определить заболевание на ранних стадиях опухолевого процесса, т.е. это проспективная диагностика, проводимая при диспансеризации населения в группах лиц повышенного риска определенной онкопатологии. Такая диагностика предусматривает мониторинг молекулярных маркеров задолго до манифестации роста опухоли.
Методы диагностики этого направления основаны на исследованиях ДНК, выделенной из плазмы и клеток крови, желудочного сока, мокроты, слюны, мазков из половых путей, а также мочи и кала.
Особое место среди этих методов занимает высокоточная диагностика с помощью стандартных наборов ДНК-маркеров по клеткам и плазме крови, в которых определяют: аномальное метилирование генов RB1, p15 и p16 (гепатоклеточная карцинома, лейкозы и лимфомы), генов MLH1 и CDH1; микросателлитную нестабильность и потерю гетерозиготности в определенных районах хромосом; мутации генов р53 и K-ras.
Например, аномальное метилирование гена р16 определяется в гиперплазированных клетках бронхолегочного эпителия при раке легких (в мокроте) и для идентификации вирусов папилломы XVI и XVIII типов при ранних стадиях рака шейки матки.
Наличие вируса папилломы указывает на рак пищевода.
Аномальное метилирование гена MLH1 свидетельствует о начальных стадиях рака желудка и аденоме толстого кишечника (определяется в смыве ректального содержимого), а функциональная инактивация генов АРС, CDKN2A указывает на рак желудка.
Пятое направление связано с поиском полиморфизмов ДНК, вовлеченных в канцерогенез и обусловливающих усиление или ослабление экспрессии генов. Такие полиморфизмы могут быть локализованы как в кодирующих, так и в некодирующих районах генов и наиболее часто идентифицируются у пациентов групп высокого риска - лиц с моногенными и онкологическими заболеваниями, не восприимчивых к терапии некоторыми лекарственными препаратами.
В этом направлении диагностики важное значение имеют полиморфизмы цитохрома Р450 (см. главу 11), среди которых аллели нуклеотидного повтора ТТТА7 в интроне 4 гена CYP19, которые идентифицированы у больных раком грудной железы и гомозигот по этому аллелю, имеющих родственников, болеющих раком легких, толстого кишечника, яичников и др. В то же время другой аллель ТТТА12 в 5 раз чаще выявляется у здоровых лиц, чем у больных.
Кроме того, один из полиморфизмов по третьему экзону гена CYP19 (замена гуанина на аденин в кодоне 80, контролирующем валин) обнаружен у больных с аллелем ТТТА7, что связано с увеличением активности цитохрома Р450 и нечувствительностью к действию его ингибитора 4-гидроксиандростендиона.
Повышенный риск развития рака грудной железы может быть связан с другим цитохромом: Р450с17альфа (ген CYP17), участвующим в синтезе стероидных гормонов. В этом случае в позиции 27 в области 5'-конца гена выявлена замена тимина на цитозин, создающая сайт узнавания для фермента MspAI и транскрипционного фактора Sp1, который в 2,5 раза увеличивает экспрессию гена CYP17 и риск развития рака грудной железы. У таких больных также идентифицирована миссенс-мутация в гене MnSOD (замена тимина на цитозин), приводящая к замене валина на аланин и редко встречающаяся у здоровых женщин. Показано также, что у женщин репродуктивного возраста, являющихся гомозиготами по аланину (А/А), риск развития рака грудной железы в четыре раза выше, чем у женщин с одним или двумя аллелями по валину.
В настоящее время доказана роль гена андрогенового рецептора (AR) в развитии рака простаты, что связано со снижением его транскрипционной активности и увеличением числа тринуклеотидных повторов (САО)п. Причем укороченные по длине повторы (меньше 18 триплетов) чаще ассоциированы с раком простаты, чем повторы, содержащие 26 триплетов и более.
Кроме того, показана роль других генетических полиморфизмов, обусловливающих развитие разных типов рака. Так, например, замена тимина на цитозин в позиции 264 ниже участка полиаденилирования в гене CYP1A1 в 2,4 раза увеличивает риск развития сквамозной карциномы; замена аргинина на пролин (в результате замены гуанина на цитозин) в кодоне 72 гена р53 увеличивает риск развития рака легких у жителей острова Тайвань; замена изолейцина на валин в кодоне 104 гена GSTP1 ведет к увеличению риска рака
грудной железы, пищевода и тестикул; аллельный полиморфизм протоонкогена Hras1 свидетельствует о неблагоприятном течении рака легкого; аллель А4 намного чаще выявляется при агрессивном низкодифференцированном немелкоклеточном раке легкого.
В процесс канцерогенеза вовлечен ген PADPRP, принимающий участие в репарации, репликации и рекомбинации ДНК, а также его процессированный псевдоген, локализованный на хромосоме 13 (13q33), имеющий биаллельный полиморфизм по бета-аллелю (делеция 193 н.п.), а гомозиготный вариант по этому бета-аллелю повышает в 2,4 раза риск развития аденокарциномы у курильщиков и т.д.
Молекулярные механизмы опухолевого ангиогенеза
Данные о более интенсивной по сравнению с нормой васкуляризации опухолевой ткани накапливаются в онкологии более 100 лет. В 1971 г. И. Фолкман впервые предложил гипотезу зависимости опухолевого роста от ангиогенеза. Согласно этой гипотезе, эндотелиальные и опухолевые клетки, находящиеся внутри опухоли, формируют единую экосистему, в которой покоящиеся клетки эндотелия переводятся в фазу быстрого роста с помощью химических сигналов, исходящих от опухолевых клеток.
Но только в конце 80-х годов XX в. были получены экспериментальные и клинические доказательства зависимости процесса метастазирования от ангиогенеза. Чтобы опухоль метастазировала, опухолевые клетки должны получить доступ к сосудам первичной опухоли, выжить в кровотоке, выйти из микрососудов в микрососуды органа-мишени, закрепиться и пролиферировать в них, индуцировать ангиогенез. Только потом они приобретают способность отвечать на митогенный сигнал ростовых факторов. При этом количество опухолевых клеток, попавших в кровяное русло через пролиферирующие капилляры (они характеризуются текучестью из-за фрагментарности базальной мебраны), коррелирует с плотностью сосудов опухоли, а также количеством легочных метастазов, наблюдаемых позднее.
Далее опухоль секретирует ангиогенные факторы bFGF и VPF/ VEGF, индуцирует пролиферирующими эндотелиальными клетками секрецию активаторов плазминогена и эластазы, что вносит дополнительный вклад в деградацию базальной мембраны сосуда и способствует проникновению опухолевых клеток в кровяное русло.
В случае, когда метастазы лишены ангиогенной активности, они могут неопределенно долго сохраняться в виде микроскопических
опухолей в «спящем» состоянии, которому соответствует баланс пролиферации и апоптоза опухолевых клеток.
Иными словами, плотность кровеносных сосудов во многих типах опухолей является прогностическим фактором риска метастазирования.
Последовательность событий, приводящих к васкуляризации опухолей, служит отражением событий физиологического ангиогенеза (см. главу 12). Среди последних: активация эндотелиальных клеток около сосудов опухоли, индукция протеолитической активности (обусловливает деградацию базальной мембраны сосудапредшественника и прилежащего внеклеточного матрикса), миграция и пролиферация клеток эндотелия, формирование капиллярных структур и организация кровяного потока.
Вместе с тем, опухолевый ангиогенез имеет ряд особенностей:
а) в зону около опухоли привлекаются клетки, участвующие в воспалении (лимфоциты, макрофаги, тромбоциты, тучные клетки и др.); они усиливают ангиогенный сигнал, продуцируя ряд цитокинов, обладающих способностью к митогенной стимуляции эндотелия: интерлейкины 1, 2 и 8, TNF-альфа и др.;
б) имеет место повышенная экспрессия на клеточной поверхности тирозинкиназных рецепторов (Flk1, Flt1, Tie1,Tie2, ERB2/her и др.); они обеспечивают проведение митогенных сигналов от ангиогенных факторов (VEGF, ангиопоэтины 1 и 2, EOF и др.); при этом клетки эндотелия теряют чувствительность к антипролиферативным сигналам перицитов, интегрированных в базальную мембрану эндотелия сосудов и в норме секретирующих мощный ингибитор ангиогенеза - TOF-бета;
в) имеет место повышенная проницаемость сосудов, питающих опухоль; причина - высокая экспрессия вазодилататора - оксида азота на фоне гиперэкспрессии VEOF; действуя совместно, эти два фактора образуют широкие промежутки между клетками эндотелия, заполненные микротромбами, и базальная мембрана имеет обширные разрывы;
г) имеют место плазморрагии, гиалиноз стенок и внутристеночные кровоизлияния в предшествующих кровеносных сосудах, инфильтрирующих опухолевую ткань;
д) имеет место дальнейшая дополнительная дестабилизация кровеносных сосудов за счет дезинтеграции их базальной мембраны на основе действия протеиназ (гепариназа, плазмин, урокиназа и др.).
Особенно васкулиризованными опухолями являются: гемангиобластома, глиома, легочная карцинома, меланома и фибросаркома.
В таблице 16 представлены основные регуляторные ангиогенные факторы опухолевого ангиогенеза.
Таблица 16. Основные ангиогенные факторы, участвующие в регуляции опухолевого ангиогенеза (по Луценко С.В. и соавт., 2004)
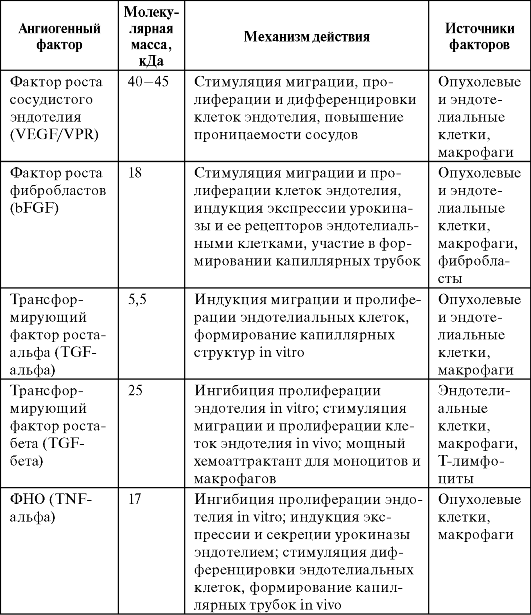
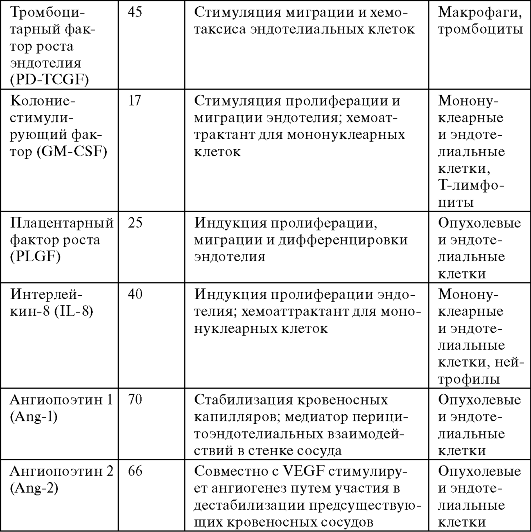 Как
следует из данных этой таблицы, одним из основных факторов,
экспрессируемых большинством васкуляризованных опухолей, является VEGF с
широким спектром действия (стимуляция пролиферации, миграции и
дифференцировки клеток эндотелия; регуляция их активности совместно с
неспецифическими факторами роста - bFGF, TGF-бета, PDGF). Этот фактор
является не только наиболее важным прогностическим показателем
интенсивности васкуляризации опухолей, но и одним из первых факторов,
для борьбы с которым стала разрабатываться антиангиогенная терапия.
Как
следует из данных этой таблицы, одним из основных факторов,
экспрессируемых большинством васкуляризованных опухолей, является VEGF с
широким спектром действия (стимуляция пролиферации, миграции и
дифференцировки клеток эндотелия; регуляция их активности совместно с
неспецифическими факторами роста - bFGF, TGF-бета, PDGF). Этот фактор
является не только наиболее важным прогностическим показателем
интенсивности васкуляризации опухолей, но и одним из первых факторов,
для борьбы с которым стала разрабатываться антиангиогенная терапия.
Проангиогенные болезни, подходы к их терапии и терапии рака
К проангиогенным болезням относятся: ангиофиброма, артрит, атеросклероз, гемангиома, гемофилическая артропатия, глаукома, гранулема, диабетическая ретинопатия, несросшийся перелом, псориаз, ретроленталевая фибропатия, синдромы Ослера-Вебера и Хиппеля-Линдау, склеродермия, солидные опухоли, трахома и некоторые другие.
В настоящее время разработан целый ряд антиангиогенных препаратов для лечения проангиогенных заболеваний. Такая терапия строится на основе ингибирования регуляторного сигнала, поступающего после формирования лигандрецепторного комплекса и связанного с блокированием процесса фосфорилирования внутриклеточного домена рецепторов тирозина, что полностью тормозит митогенное действие антиангиогенных факторов роста.
К некоторым из таких препаратов относятся: ангиозимы, каталитически активные нуклеиновые кислоты, блокирующие секрецию рецептора VEGF на стадии мРНК; антисмысловые олигонуклеотидные последовательности комплементарных мРНК, блокирующие экспрессию bFGF, VEGF и других факторов роста и их рецепторов; химиопрепараты, воздействующие на разные этапы каскада реакций внутриклеточной передачи регуляторного сигнала от фактора роста и др.