Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 17 ЗДОРОВЬЕ, ГЕНЕТИЧЕСКИЙ ГРУЗ И НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ
ПОКАЗАТЕЛИ ЗДОРОВЬЯ И ГЕНЕТИЧЕСКИЙ ГРУЗ
ЧЕЛОВЕКА
Показателями здоровья человека как биологического вида являются: состояние генофонда и репродуктивной функции (см. главы 2 и 16).
Генофонд человека - это совокупность всех его генов (генотипов) в общей популяции, а общая популяция - это совокупность организмов всех людей, населяющих Землю (6,5-7,0 млрд человек). Для генофонда характерны:
• генетическая целостность;
• дифференцированность и неоднородность генотипов при сохраняющейся общей совокупности генов;
• зависимость генофонда современного человека от генофонда предков;
• наличие генетического груза или груза наследственной патологии, объем которого растет параллельно с прогрессом человечества и за всю историю медицины, начиная со времен Гиппократа, достиг 8,5-10,0%.
Генетический груз - это меньшая часть общей популяции, имеющая измененную наследственность, определяющую появление организмов с наследственной патологией, менее приспособленных к выживанию в окружающей среде и поэтому подвергающихся избирательной гибели в ходе естественного отбора.
Наследственная патология включает следующее.
• Генные или моногенные болезни (ГБ или МБ). Их количество достигло 4,5 тыс. нозологий, а частота в популяции составляет 3% или 30 детей на 1000 родившихся. В разных регионах России частота МБ составляет 4,2-6,5%.
• Сложно наследуемые (полигенные) болезни или мультифакториальные заболевания (МФЗ). На их долю приходится 94-96% хронической неинфекционной патологии; частота - 1,5%.
• Хромосомные болезни (ХБ). Их более 100 нозологий + 900 типов хромосомных нарушений; частота - до 1%.
• Врожденные болезни, большие и малые аномалии развития (БАР и МАР); их частота составляет 3%.
В последние годы были расшифрованы генетические причины и механизмы онкологических заболеваний. Поэтому эти болезни стали называть онкогенными (ОГБ) и относить к одной из форм наследственной патологии, причем около 90% ОГБ относятся к классу МФЗ и 10% - к классу МБ (см. главу 25). Частота ОГБ среди населения пожилого и старческого возраста достигает 10%, а среди детского населения - 1 случай на 160 детей (до 1%).
В общий объем генетического груза человека входят также гетерозиготные носители генов, или гетерозиготы. Считается, что в общей популяции количество гетерозигот не менее 3-5%. Гетерозиготы - это практически здоровые люди, в генотипах которых наряду с одной копией нормального аллеля гена присутствует вторая копия патологического аллеля этого же гена, полученная от одного из родителей. «Плохое» для гетерозигот начинается после их вступления в брак, когда речь заходит о прогнозе здоровья потомства. В таких семьях риск развития аутосомно-рецессивных и аутосомно-доминантных болезней составляет 25% и 50% соответственно.
Наиболее известными примерами таких заболеваний служат: АГС, анемия Фанкони, атаксия-телеангиэктазия, ВГ, муковисцидоз, пигментная ксеродерма, прогерия Вернера и Хатчинсона-Гилфорда, ФКУ, хорея Гентингтона и ряд других.
В таблице 11 приведена структура общей заболеваемости населения.
Таблица 11. Структура общей заболеваемости населения
Форма патологии | Частота,% |
Генетические болезни, в том числе: | 48,9-64,1 |
МБ | 3,9-6,5 |
МФЗ | 38,2-56,6 |
ХБ | 0,8-1,0 |
ВПР | 0,6-10,0 |
Негенетические болезни | 25,4 - 53,2 |
ВПР | 0,8 |
Основные показатели здоровья населения России в конце XX в. резко ухудшились. Так, средняя продолжительность жизни мужчин
в период с 1990 до 2005 г. уменьшилась на 15-16 лет, составив 74 года (1990) и 58,6 года (2005). Вместе с тем, у женщин продолжительность жизни изменилась мало (74 и 72 года соответственно). При этом рождаемость населения в России в расчете на 1000 человек уменьшилась в 2,5 раза и составила 8,6-10%, а младенческая смертность наоборот выросла до 19,9-23,4% против 9-11,0% в развитых странах. Из общего числа российских детей практически здоровы только 15-18% (каждый шестой ребенок), тогда как дети с хроническими болезнями составляют 32-35% (каждый третий ребенок), а дети с отклонениями физического и умственного развития - 47-53% (каждый второй ребенок). В структуре причин общей заболеваемости и смертности детского населения России значительный удельный вес стали занимать БАР и МАР, которые, например, по такому показателю, как потерянные и ухудшенные годы жизни, выдвинулись на одно из первых мест, оставив позади сердечно-сосудистые, онкологические и другие заболевания.
Неблагоприятная ситуация с генетической патологией у детей наблюдается во всех развитых странах мира, где такие дети постоянно госпитализируются в педиатрические клиники и занимают в них 30-40% коечного фонда, а в специализированных клиниках это каждые две койки из трех. При этом один из 15-ти детей имеет моногенное или врожденное заболевание, а с учетом хромосомных синдромов и МФЗ - это каждый четвертый ребенок (в специализированных стационарах - каждый второй-третий ребенок). Смертность детей в возрасте до 5 лет включительно при МФЗ достигла 35-42%, МБ - 10-14%, ХБ и ВПР - по 2-4%, т.е. генетические причины вызывают гибель 48-60% таких детей (каждый второй-третий из них)*.
ПРИЗНАКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Разный характер наследования
Известно, что патологический ген или хромосомное нарушение сохраняются у индивида в течение всей жизни. При этом патологический ген - это либо новая генная мутация, либо (что гораздо чаще) мутация, переданная от предыдущих поколений, а хромосомное
* Безусловно, эти данные заслуживают особого внимания, так как именно они являются предвестником приближающейся угрозы для человечества - его депопуляции: пока это только 10% генетического груза, а что будет дальше?
нарушение - это, как правило, новая хромосомная или геномная мутация.
Для МБ характерно менделевское наследование (см. главу 4). В случае МФЗ характерно полигенное наследование, и обусловливающие их развитие генные сети прямо передаются больному потомку как гены, общие с родителями и больными родственниками. При этом количество передаваемых потомку генов зависит от степени родства с предками (общности генов) и их проявление связано с действием факторов внешней среды, т.е. будет наблюдаться суммарный или аддитивный эффект действия генетических и средовых факторов.
Для ВПР характерно наличие или отсутствие наследования. Например, выделяют доминантно и рецессивно наследуемые моногенные пороки, пороки при хромосомных синдромах, пороки мультифакториальной и экзогенной природы.
Для ХБ наследование, как правило, нехарактерно, кроме случаев семейного носительства родителями больного потомка редких сбалансированных (фенотипически не проявляющихся) транслокаций, инверсий и инсерций.
Сегрегация в семейных родословных
Наследственная патология накапливается (сегрегирует) в семейных родословных. При этом сегрегация касается либо самой болезни, либо ее отдельных признаков, что обусловлено наличием генов, общих для потомка и его родителей и родственников. Выделяют семейные формы, характерные для всех без исключения форм наследственной патологии:
• МФБ - бронхиальная астма, гипертоническая болезнь, ишемическая болезнь сердца, психические расстройства, сахарный диабет, язвенная болезнь желудка и двенадцатиперстной кишки, псориаз и др.;
• МБ - атаксия Мари, атаксия Фридрейха, семейная гиперхолестеринемия, спастический паралич Штрюмпеля, эссенциальный тремор Минора и др.;
• ХБ - носительство редких сбалансированных транслокаций, инверсий и инсерций;
• ВПР - атрезия или стеноз ануса и прямой кишки, болезнь Гиршпрунга, врожденный вывих бедра, гипертрофический пилоростеноз, гипоспадия, симфалангия (отмечена у потомков 14-ти поколений английского полководца Джона Тальбота) и др.
Разное время манифистации и связь с возрастом
Для МБ и МФЗ характерно разное время манифестации, что связано с дифференцированной генной экспрессией, зависящей от типа клеток в составе тканей, периода онтогенеза, механизма взаимодействия отцовских и материнских (по происхождению) аллелей генов, их разной экспрессивности и пенетрантности под действием факторов среды. По данным канадских генетиков, манифестация большинства МБ (90%) и МФЗ (90%) приходится на детский и пожилой возраст соответственно.
В обзорной статье, опубликованной G. Jimenez-Sanchez et al. в журнале Nature в 2001 г., суммированы последние данные о времени манифестации в ходе онтогенеза почти 1000 патологических генов, лежащих в основе МБ. Эти данные отличаются от ранее опубликованных, хотя и в них преобладает детский возраст манифестации. Так, наибольшее количество МБ (33%) манифестирует у детей; эти болезни обусловлены мутациями белков со специальными функциями (белков терминальной дифференцировки). Мутации в генах, продуцирующих такие белки, обычно проявляются в первые годы жизни. Наличие многих дефектных белков, например, белков при НБО (см. ниже), отмечается уже в неонатальном периоде развития, других - в раннем подростковом периоде, и только некоторые из них появляются у взрослых, например белки при болезнях экспансии числа нуклеотидных повторов (см. главу 28).
Примерно 15% МБ обусловлены мутациями в генах, контролирующих процессинг мРНК , стабильность и функционирование белков процессинга, белков-транспортеров, белков-рецепторов и других белков. Мутации в генах, экспрессирующих эти белки, проявляются позже, и пик их манифестации приходится на подростковый и юношеский возраст. Так, большинство генов рецепторных белков манифестируют в период полового созревания. На третьем месте по частоте проявления находятся МБ, обусловленные мутациями в генах, контролирующих факторы транскрипции, - таких болезней около 10%. Гены болезней этой группы - филогенетически очень древние гены, производящие низкомолекулярные ядерные белки, выполняющие регуляторные функции по отношению к другим генам генотипа: они включают (выключают) и контролируют их работу. В связи с особыми функциями этих генов их белковые продукты могут стать критическими для клетки и организма, способными переключать программу онтогенеза. Например, к таким продуктам относятся белковые
факторы, продуцируемые геном SRY, которые в раннем эмбриогенезе (5-7 нед развития) включают программу, определяющую развитие организма по мужскому типу. Многочисленные примеры нарушений развития по мужскому типу были приведены в главе 16.
Таким образом, почти 60% МБ проявляются в детском возрасте. Манифестация оставшейся части МБ (около 40%) приходится на взрослый возраст. При этом МБ связаны с мутациями в генах, контролирующих синтез рецепторных белков (около 9%), белков внутриклеточного и внеклеточного матрикса (по 5% для каждого типа), белков-переносчиков (5%), гормонов (3%), иммуноглобулинов (2%).
Вместе с тем известны МБ с варьирующим временем манифестации. В частности, тяжелое нейродегенеративное аутосомно-доминантное заболевание с полной пенетрантностью хорея Гентингтона (ХГ) манифестирует в возрасте от 1 года до 75 лет. Причем начало болезни у 20% больных приходится на возраст 41-45 лет, у 77% больных - 31-55 лет. Со временем манифестации ХГ коррелирует ее прогредиентность (например, развитие хореотического гиперкинеза и деменции). Что касается манифестации МФЗ, то в детском возрасте проявляется только 10% этих заболеваний, а основная их часть манифестирует в пожилом возрасте.
В свою очередь, для большинства хромосомных аутосомных синдромов и врожденных аномалий развития характерна манифестация с момента рождения ребенка, тогда как ХБ, связанные с нарушением половых хромосом, проявляются только в период половой зрелости; до этого периода они протекают бессимптомно и поэтому диагностируются случайно при массовых врачебных осмотрах, например, юношей-допризывников или спортсменов.
Множественность поражения
При МБ и ХБ, а также в случае ВПР, в поражение вовлекается не менее двух-трех систем организма, что объясняется плейотропным действием мутантного гена (в случае МБ), генным или хромосомным дисбалансом (в случае ХБ и множественных ВПР). Для МФЗ, как правило, характерно поражение одной системы организма.
Прогредиентный или постоянный характер течения МБ и МФЗ
Отличительными чертами большинства МБ и МФЗ являются патокинез (движение) патологического процесса и его прогредиентность (прогрессирование) - см. главу 3.
Согласно И.В. Давыдовскому (1961), при данных болезнях имеет место: наличие латентного бессимптомного периода (продрома), периода появления (манифестации) первых симптомов, периода постепенного нарастания степени их выраженности и тяжести течения болезни. Исход заболевания разный (выздоровление, переход в хроническое течение, летальный исход).
В случае МБ, особенно НБО, в первые двое-трое суток жизни младенца симптоматики нет. Дети, как правило, рождаются внешне здоровыми и после нормально протекавшей беременности. Их состояние ухудшается внезапно по типу нейро-дистресс-синдрома. Ребенок отказывается от кормления, у него появляется и нарастает неврологическая симптоматика, задерживается психомоторное развитие, развиваются гипорефлексия, мышечная гипоили гипертония, рвота. Могут развиться судороги, остановка дыхания, кардиомиопатия, сосудистая недостаточность и коллапс (вплоть до клинического синдрома внезапной смерти - см. главу 3), летаргия, кома, почечная недостаточность. Постепенно нарастают дегидратация, ацидоз, кетоз, гипераммониемия. В дальнейшем такой ребенок отстает в физическом развитии.
Указанная симптоматика обусловлена постепенным накоплением в организме токсических продуктов метаболизма до метаболического (ферментативного) блока: кетокислот, метилмалоновой, пропионовой кислот, аммиака и других соединений, а также истощением запасов энергетических субстратов или невозможностью их использования вследствие такого блока.
Конституциональность, врожденность и непрогредиентность ХБ и ВПР
Клиническая картина при ХБ и ВПР остается постоянной с рождения до смерти индивида. Их главные отличительные особенности - это постоянство патологического фенотипа
Большие и малые врожденные аномалии развития
Большие и малые врожденные аномалии развития встречаются при всех формах наследственной патологии в виде отдельных множественных нарушений развития организма. Они могут рассматриваться не только как характерная особенность при разных формах наследственной патологии, но и как самостоятельное направление генетики - тератология (дисморфология).
Полиморфизм признаков
Полиморфизм признаков (разнообразие) характерен для всех форм наследственной патологии. В его основе лежит множество генетических причин (гетерогенность) - см. главу 2. Генетические причины полиморфизма признаков наследственной патологии - это варианты патологической изменчивости наследственного материала: множественный аллелизм и аллельные серии, влияние генов модификаторов, генокопирование, плейотропное действие генов, импринтинг, экспансия нуклеотидных повторов, эффект положения (см. главу 5).
В свою очередь негенетические (экзогенные) причины полиморфизма признаков наследственной патологии - это результат действия факторов окружающей среды при фенокопиях или их сочетанное действие с генетическими причинами.
Задержка физического и психомоторного (психического) развития
Задержка физического и психомоторного (психического) развития, олигофрения и выраженная неврологическая симптоматика характерны для большинства МБ и ХБ. При МФЗ и ВПР, как правило, этот признак не наблюдается.
Нарушение полового развития и функции репродукции
Нарушение полового развития и функции репродукции (включая мужское и женское бесплодие) характерно для многих МБ и гоносомных ХБ (см. главу 16). При остальных формах наследственной патологии эти признаки почти не выражены.
Снижение продолжительности жизни и повышенная смертность больных
Снижение продолжительности жизни и повышенная смертность больных характерны для многих форм наследственной патологии.
Резистентность к терапии
Только этиологическая и патогенетическая терапия направлена на полное устранение или блокирование причин и механизмов наследственной патологии (см. главу 30). Поэтому резистентность (невосприимчивость) к терапии наследственной патологии - это результат отсутствия радикальных методов лечения и причина нигилизма врачей и самих больных, считающих такой диагноз «пожизненным приговором». Вместе с тем, в последние годы благодаря внедрению
в молекулярную медицину радикальных методов генотерапии, клеточной и тканевой терапии многих форм наследственных болезней появились надежды на благоприятную перспективу (см. главу 20).
Характеристика молекулярных причин и механизмов наследственной патологии
Концепция о происхождении молекулярных болезней обсуждается в медицине с 80-х годов XIX в. Она предусматривает, что в основе наследственной патологии лежат нарушения физико-химических процессов, затрагивающих структурные и функциональные компоненты клетки в ходе ее метаболизма. Иными словами, молекулярные болезни обусловлены расстройствами действия генов (генотипа), влиянием факторов окружающей среды или тем и другим одновременно. Эти группы причин получили название основных факторов формирования нормальных и патологических признаков и фенотипов (см. главы 2 и 3).
Основные модели для изучения причин и механизмов наследственной патологии
Анализ причин и механизмов формирования признаков и фенотипов наследственной патологии позволяет выделить две особенности.
Первая особенность - эволюция человека «предоставила» три модели для изучения причин и механизмов наследственной патологии:
• модель, основанная на действии только патологических генов, - это МБ, меньшая часть ОГБ (около 10%) и врожденные аномалии развития (0,6-10%), обусловленные генными и хромосомными мутациями;
• модель, основанная на действии только патологических факторов среды (проявляющихся на фоне действия генов), - это экзогенные болезни, обусловленные ненаследственной (модификационной) изменчивостью (см. главу 5), и врожденные аномалии развития экзогенной природы (0,8%);
• модель, основанная на совместном действии патологических генов и факторов среды, - это полигенные (мультифакториальные) болезни, большая часть ОГБ (около 90%), ХБ и врожденные аномалии развития мультифакториальной природы.
Вторая особенность - врожденные аномалии развития и ОГБ можно изучать на основе каждой из указанных моделей в отдельности или сразу всех трех, включая две монофакторные модели (действие
гена, действие фактора среды) и одну многофакторную модель (суммарное действие генов и факторов среды).
Причины и патогенез моногенных болезней
Причины и патогенез МБ связаны с первичными молекулярными нарушениями структуры генов и кодируемых ими белков, нарушениями активации ферментов и метаболическими блоками. При этом экспрессия патологических генов не только ведет к синтезу патологических белков, но и (через промежуточные продукты метаболизма) изменяет физико-химические характеристики и количество белковых и небелковых компонентов клетки. Иными словами, действие патологического гена (белка) ведет к нарушениям обмена веществ в клетке и организме в целом (см. главы 7 и 8).
Подавляющая часть этих нарушений вызвана врожденной недостаточностью ферментов, или ферментопатиями. В основе примерно 75% ферментопатий лежит замена оснований в молекуле ДНК, ведущая к замене одной аминокислоты на другую в полипептиде, что изменяет последовательность аминокислот в структуре активного центра фермента или районе связывания апофермента с коферментом (чаще всего витамином или металлом). В результате выпадает каталитическая функция фермента и развивается метаболический блок, останавливающий ход метаболической реакции.
Вследствие метаболического блока накапливаются промежуточные метаболиты, образование которых предшествует блоку, и развивается дефицит конечных продуктов реакции.
Кроме того, метаболический блок вызывает нарушение транспорта участвующих в реакции химических соединений, которые накапливаются до блока и потому становятся токсичными. Например, при ФКУ в результате дефицита фенилаланингидроксилазы или кофермента-тетрагидробиоптерина в крови и тканях накапливается фенилаланин, который подвергается дезаминированию в фенилпируват, токсически (и необратимо) действующий на нервную ткань.
К токсическому поражению ЦНС ведет другое генетическое заболевание - лейциноз или болезнь с запахом мочи, напоминающим запах кленового сиропа. В этом случае накапливаются кетокислоты - продукты трех алифатических аминокислот: лейцина, изолейцина и валина, что сопряжено с нарушениями биохимических превращений в мозге.
К другим примерам МБ, обусловленных дефицитом конечных продуктов метаболизма (вследствие метаболического блока), относятся: АГС, альбинизм, врожденный гипотиреоз и галактоземия.
Как установлено, ферментопатии могут быть связаны с нарушениями клеточных рецепторов, мембранного транспорта, витаминной недостаточностью и дефицитом микроэлементов. Общее количество ферментопатий огромно (например, у человека не менее 180 ферментов являются металлопротеинами, и для каждого из них имеется свой вариант ферментопатии).
Таким образом, концепция о молекулярных болезнях стала фундаментом в исследованиях этиологии и механизмов патогенеза МБ, что со временем привело к значительным результатам. Бурное развитие геномики и протеомики позволило обосновать, разработать и внедрить в молекулярную медицину наряду с методами «прямой генетики» (путь от признака к гену) методы позиционного клонирования, выделения и изучения первичной структуры генов без предварительного знания их белковых продуктов, т.е. методы «обратной генетики» (путь от гена к признаку). На хромосомах человека картированы свыше 11 тыс. генов (включая 1100 генов наследственных болезней) и установлена их тесная связь с производимыми ими белками (см. главу 3).
Вместе с тем, возможности концепции о молекулярных болезнях оказались недостаточными для расшифровки причин и механизмов патогенеза ряда МБ и других форм наследственной патологии. Эти сложности обусловлены следующими причинами:
• во-первых, малоизученными особенностями формирования признаков МБ на протяжении всего многозвеньевого пути от гена к признаку, ибо каждое звено этого пути зависит от дифференцированной экспрессии генов, участвующих в формировании разных тканей, и модифицирующего влияния на экспрессирующие гены других генов генотипа и факторов окружающей среды;
• во-вторых, неоднозначным характером связей между мутацией в гене и степенью нарушения активности производимого им белка; такая неоднозначность наблюдается при миодистрофиях Дюшенна и Беккера, обусловленных мутацией одного и того же гена, но эта мутация проявляется у них по-разному: при миодистрофии Дюшенна болезнь развивается в раннем детском возрасте и протекает с глубокой инвалидизацией, а при миодистрофии Беккера - в юношеском возрасте и протекает более доброкаче-
ственно, не приводя к инвалидности (в дальнейшем эти различия были объяснены разными точковыми мутациями одного и того же гена по типу аллельной серии);
• в-третьих, регистрацией новых феноменов: импринтинга, прионизации, экспансии нуклеотидных повторов и антиципации (см. главы 27-29);
• в-четвертых, выделением новых механизмов нетрадиционного варианта наследования генов и признаков, обусловленных разным влиянием материнского и отцовского геномов на развитие митохондриальных болезней, болезней импринтинга и прионных болезней.
Так, при изучении митохондриальных болезней на примере атрофии Лебера выделили механизм наследования, смешанного с материнским (цитоплазматическим), и показали, что эта болезнь может передаваться всем потомкам по материнской линии в случае нарушения функций ферментов, синтезируемых мтДНК. Вместе с тем обнаружилось наличие аутосомно-доминантного и аутосомнорецессивного типов наследования данного заболевания, связанных с нарушениями структуры и функций ферментов, синтезируемых ядерной ДНК.
В дальнейшем описали три типа мутаций мтДНК (точковые мутации двух типов и крупные делеции) и три механизма патогенеза, связанные с мтДНК, ядерной ДНК или межгеномными сигнальными эффектами (см. главы 8 и 26).
Следовательно, кроме митохондриальных генов, процессы окислительного фосфорилирования (митохондриального дыхания) контролируют ядерные гены, экспрессирующие свыше 70 специфических белков.
Было также показано, что мутации ядерных генов ведут к изменению количества копий мтДНК, что обусловливает развитие феномена деплеции или истощения митохондрий. Причем такие мутации не нарушают наследование генов по моногенному варианту.
Другой пример - исследование механизмов эпигеномной регуляции генной активности. Роль геномной памяти в нетрадиционном наследовании генов и признаков (см. главы 4 и 28) была продемонстрирована на разных моделях (синдромы Ангельмана и Прадера- Вилли - первая модель, синдром Беквитта-Видемана - вторая модель и др.). Так, из болезней, связанных с геномной памятью, описаны: неонатальный сахарный диабет, синдром скелетной дисплазии с карлико-
востью и синдром Рассела-Сильвера. Всего проявление импринтинга было продемонстрировано для пяти материнских по происхождению хромосом (хромосомы 2, 7, 14, 15 и 16) и четыре отцовских по происхождению хромосом (хромосомы 6, 11, 14 и 15). Кроме того, были объяснены причины и механизмы патогенеза прионных болезней, ранее известных как спонгиоформные энцефалопатии, объединенные на основе сходства морфологических дефектов (см. главу 29). Завершая рассмотрение причин и механизмов патогенеза МБ, следует выделить наиболее тяжелые их формы, связанные с НБО:
• хорошо известные НБО, например приведенные выше ферментопатии;
• малоизвестные НБО - болезни клеточных рецепторов, сигнальной трансдукции, каналопатии (нарушения калиевых, кальциевых, натриевых и хлорных ионных каналов в мембранах мышечных, нервных и глиальных клеток) и коллагенопатии (нарушения молекулы коллагена).
Как заключительный пример рассмотрим коллагенопатии, при которых имеет место гетерогенность молекул коллагеновых белков, кодируемых разными генами, расположенными в разных локусах на разных хромосомах. Например, в основе несовершенного остеогенеза лежит экспрессия гена COL1A1 (17q21.31-22), синдрома Эллерса-Данлоса 1,2,4 и 7 типов - экспрессия генов COL1A2 (2q31), 3A1 (7q21), 5A1 и 5A2 (9q34.2-34.3) соответственно, синдромов Стиклера и Книста - COL 11А1 (1p21) и COL2A1 (12q13.11-13.2), аутосомно-рецессивного синдрома Альпорта - COL4A3 (2q36-37), Х-сцепленного рецессивного синдрома Альпорта - COL4A5 (Xq22), миопатии Бетлема - COL6A1 (21q22.3), буллезного эпидермолиза - COL7A1 (3p21.3), множественной эпифизарной дисплазии - COL9A1 (6q13). Безусловно, объединение болезней в отдельные группы облегчает решение вопроса индивидуальной терапии у больных с разными коллагенопатиями, однако такой подход несовершенен, ибо одна и та же нозология входит в разные классы болезней, например миопатия Бетлема и буллезный эпидермолиз, как правило, рассматриваются в группах нервных (нервно-мышечных) и кожных болезней соответственно.
Причины и патогенез мультифакториальных заболеваний
В исследованиях причин и механизмов патогенеза МФЗ, как и в случае МБ, произошли большие изменения. Была доказана роль
генетических причин и механизмов в развитии таких болезней, как атеросклероз, артериальная гипертония, бронхиальная астма (БА), сахарный диабет, шизофрения и другие аффективные состояния, язвенная болезнь желудка и двенадцатиперстной кишки (ЯБЖиДК), многие формы рака. Этиология и патогенез этих и других МФЗ стали широко изучаться с помощью анализа их сцепления с генамикандидатами и ассоциации с полиморфными генетическими маркерами (см. главу 19).
Понятия «сцепление» и «ассоциация» следует разграничить. Сцепление относится к двум генам, расположенным в одной хромосоме на определяемом расстоянии друг от друга, например, ген-кандидат в гены болезни сцеплен с другим геном (локусом). Ассоциация - это самая высокая частота гена-маркера, выявляемого при МФЗ. Она не подразумевает, что ген болезни и маркерный ген расположены в одной хромосоме. Например, с HLA-локусами, расположенными в хромосоме 6, могут быть сцеплены гены разных МБ, находящиеся на разном расстоянии от НLA-комплекса (АГС, гемохроматоз, СЦА). При этом нет оснований считать, что эти заболевания и HLA-комплекс как-то связаны между собой физиологически. С другой стороны, заболевания, ассоциирующиеся с аллелями HLA-комплекса, обычно не являются моногенными, а имеют мультифакториальную природу. К таким заболеваниям относятся аутоиммунные или иммунноассоциированные болезни: болезнь Аддисона, детская спру, миастения гравис, множественный склероз, ревматоидный артрит, сахарный диабет I типа, тиреотоксикоз и хронический гепатит. Показанные для них ассоциации касаются D/DR-антигенов HLA-системы. Причем семейные исследования выявили их сегрегацию у родственников пробандов, хотя отсутствовало менделевское наследование. Тем самым D/DR-гены HLA-комплекса и гены аутоиммунных заболеваний, будучи ассоциированными, не являются сцепленными. Вместе с тем, слабую ассоциацию со многими МФЗ показали группы крови системы АВО, и наоборот, сильную ассоциацию с МФЗ показали антигены системы HLA. Описана также ассоциация полиморфизма альфа-1- антитрипсина с хронической эмфиземой легких у взрослых, патогенез которой обусловлен генетической гетерогенностью или серией множественных аллелей локуса PI (23 разных фенотипа), которые способны индуцировать механизмы разрушения легочной ткани при хронической эмфиземе легких в результате инфицирования.
МФЗ нередко называют болезнями с наследственной предрасположенностью, имея в виду преобладание в их патогенезе генетических причин. Наследственная предрасположенность зависит от формирования комбинаций аллелей разных патологических генов - это генные сети, которые вносят общий суммарный (аддитивный) вклад в проявление признаков болезни, включающий вклад каждого отдельного гена (см. главу 22).
Важную роль в развитии МФЗ играют главные гены, от которых зависит инициация болезни и которые выступают координаторами действия всех ее генов.
Кроме того, при МФЗ выделены ассоциации с моногенно наследуемыми формами наследственной патологии. Например, у каждого второго больного с гиперлипидемией, приводящей к ишемической болезни сердца (ИБС), определяется моногенно наследуемая форма семейной гиперхолестеринемии, обусловленная мутациями в гене рецептора ЛПНП - LDLR (19p13.2-p31.1) или диагностируется семейный дефект гена Аро-В-100 (2р23-24).
Наибольшее число моногенно наследуемых форм выделено при инсулинзависимом сахарном диабете I типа; в их число входят первоначально изученные 6 форм: MODY1 (ген NHF4A), MODY2 (ген глюкокиназы или ген GCK), MODY3 (ген ядерного фактора гепатоцитов), MODY4 (ген фактора-1 промотора инсулина), MODY5 (ген печеночного фактора транскрипции 2), MODY6 (ген NEURODY) - см. главу 21.
В свою очередь, моногенно наследуемые формы выделены при эссенциальной гипертензии (ЭГ), развивающейся в результате неравного кроссинговера между генами альдостерон-синтазы и 11-бета- гидроксилазы. Например, идентифицированы четыре моногенные формы, в том числе редкая, купируемая глюкортикоидами форма - синдром Лиддла или синдром псевдоальдостеронизма (гены эпителиального натриевого канала: SCNN2 и SCNN3), синдром Гордона , тип 2, или синдром псевдогипоальдостеронизма (ген не локализован), а также синдром избытка минералокортикоидов (ген почечной 11-бета-гидроксистероид-дегидрогеназы).
Следует отметить, что основные клинические проявления ЭГ - это тяжелые осложнения (инсульты, инфаркты, почечная недостаточность). В настоящее время локализовано около 100 генов, продукты экспрессии которых участвуют в развитии этих тяжелых проявлений. К таким продуктам относятся:
• компоненты ренин-ангиотензиновой и калликреин-кининовой систем;
• вещества, обеспечивающие поддержание сосудистого тонуса (компоненты кальциевых каналов, синтазы закиси азота, эндотелины) и целостность структуры сосудов (коллагенсвязывающий белок, фибриллин, эластин);
• рецепторы адренергической системы, например рецепторы дофамина;
• продукты метаболизма стероидных гормонов (11-бета- и 17-альфа- гидроксилазы);
• продукты водно-солевого гомеостаза (аквопорины, рецепторы вазопрессинов);
• ионные каналы.
Вместе с тем, основными генами-кандидатами ЭГ считаются 9 генов, включая два гена, кодирующих субъединицы эпителиального ?+-канала, или гены SCNN 1B и SCNN IG, локализованные в сегменте 16p13-p12; ген APNH или ген антипортера Na+ / H+ (1р36.1- р35); ген REN или ренина (1q25-q32); ген AGT или ангиотензина I (1q42-q43); ген AGE или ангиотензин I-конвертирующего фермента (17q22-q24); ген PLA2 или панкреатической фосфолипазы А2 (12q23- q24.1); ген SAN или ген гипертензии, связанный с геном, экспрессирующимся в почке (16p13.11); ген NOS3 или ген эндотелиальной синтазы закиси азота (7q35-q36).
Кроме того, показано, что ряд генов, формирующих предрасположенность к ЭГ, вносят вклад в этиологию атеросклероза, что свидетельствует об общности патогенетических механизмов этих заболеваний, обусловленных нарушениями функционирования ренин-ангиотензиновой и калликреин-кининовой систем.
Однако ведущая роль в патогенезе атеросклероза принадлежит повышению концентрации липидов в плазме крови, нарушению процессов свертывания крови и целостности сосудистой стенки. Поэтому в качестве основных генов-кандидатов атеросклероза рассматриваются гены, продукты которых обеспечивают липидный обмен. Эти гены расположены на хромосоме 16 в локусе СЕРТ, кодирующем транспортный белок эфиров холестерина, и в кластере генов ароА1/ароС3/ароА4 на хромосоме 11.
При другом широко распространенном МФЗ - БА - также наблюдается значительное число локусов, сцепленных с этим заболеванием. Продукты экспрессии генов-кандидатов БА, патологические белки
и другие компоненты участвуют в трех звеньях патогенеза болезни: иммунологическом, воспалительном и нейрогенном. При этом особая роль принадлежит генам семейства интерлейкинов, цитокинов и цитокиновых рецепторов (см. главу 8), обеспечивающих нормальное функционирование слизистых оболочек бронхов и тучных клеток, контролирующих уровень IgE в крови и детоксикацию ксенобиотиков.
С учетом этих особенностей большое значение придается выявлению ассоциаций БА с HLA-комплексом (см. главу 15). Наиболее вероятными кандидатами на роль главных генов предрасположенности к БА считаются гены интерлейкина-9 и интерлейкина-4, картированные на хромосоме 5q31-q33. Вместе с тем, в случае БА у детей, сопровождающейся высокой концентрацией в крови IqE, была обнаружена достоверно значимая ассоциация с локусом короткого плеча хромосомы 20 (20р13), в котором локализован ген мембранассоциированного белка клеточного сурфактанта из семейства цитокиновых рецепторов.
Кроме этих двух генов в формировании предрасположенности к БА важную роль играют еще ряд генов, расположенных в разных хромосомах: ген TNFA (6р21.3-р21.1), гены IGEL и FCER1b (11q12-q13), гены IDIF и NOSI (12q15-q24.1) и ген ESD (13q14.2-q14.3). Их продуктами являются разные белки-ферменты: бета-фактор некроза опухолей, белок-регулятор уровня общего IqE, альфа-иммуноглобулиновый рецептор тучных клеток, гамма-интерферон, невральная синтетаза окиси азота и эстераза D.
Следует также отметить, что при исследовании причин и механизмов патогенеза МФЗ выявлен ряд генов, сцепленных с остеопорозом, ожирением, рассеянным склерозом, ревматоидным артритом, эндометриозом и другими болезнями с наследственной предрасположенностью.
Причины и патогенез хромосомных болезней
Говоря о причинах и механизмах патогенеза ХБ, следует указать на прямую связь наблюдаемых при них признаков с нарушениями количества наследственного материала в случае хромосомных и геномных мутаций. Иными словами, в основе признаков ХБ лежит хромосомный (и генный) дисбаланс, нарушающий координированную экспрессию генов и приводящий к молекулярным нарушениям. Результаты исследований указывают на особую роль в этих наруше-
ниях общеклеточных генов, к которым относятся структурные гены (кодируют белки, различные типы РНК и другие макромолекулы клетки), и регуляторных генов. Например, ген, находящийся на одной хромосоме, регулирует гены другой хромосомы. Проследим это на примере синдрома Дауна, при котором хромосомная перестройка приводит к дисбалансу как структурных, так и регуляторных генов. В частности, если в хромосоме 21 при трисомной форме синдрома Дауна содержится регуляторный ген, функция которого состоит не в активации, а в умеренной репрессии других генов, то можно полагать, что из-за тройной дозы гена эта репрессия превратится в более жесткую, что приведет к инактивации других генов или к извращению их функции.
Далее представим себе, что дисбаланс структурных генов и регуляторных участков касается нескольких десятков генных локусов. Этого уже будет достаточно, чтобы вызвать глубокие изменения в клеточных системах, в том числе нарушения координации взаимодействия генов, конечным следствием которых будут грубые нарушения процессов развития. Следует заметить: эффект дозы гена может и не иметь значения в патогенезе анеуплоидии, что было показано на примере фермента - супероксиддисмутазы-I при синдроме Дауна (см. главу 11). Вполне вероятно, в основе патогенеза анеуплоидии могут лежать другие события, например эффект положения гена (см. главу 5). Так, при сбалансированных транслокациях количество наследственного материала не меняется, но часто наблюдаются фенотипические нарушения развития (дисплазия и УО), которые можно объяснить с помощью такого эффекта. При этом гены, расположенные вблизи точки разрыва хромосомы, изменяют свое проявление (как правило, ослабевает их доминирование). Если эффект положения касается действия плейотропного гена, то становится весьма вероятным одновременное развитие многих нарушений в разных системах организма.
С другой стороны, эффект положения гена зависит от генотипической среды (генного окружения). Это означает, что в одном генном окружении функции генов, оказавшихся вблизи точки разрыва в хромосоме, могут быть изменены, тогда как в другом генном окружении они сохранятся. Возможно, именно этим объясняется явление, когда родители - носители сбалансированных хромосомных перестроек - являются фенотипически здоровыми людьми, а некоторые их дети (но не все) имеют ВПР.
Также интересно рассмотреть зависимость продолжительности жизни больного с ХБ от типа имеющегося у него хромосомного нарушения. Большинство носителей аутосомных трисомий умирают в первые дни жизни. У больных с трисомиями по половым хромосомам жизнеспособность практически не снижена, и патологический фенотип у них проявляется в период полового созревания, когда начинают функционировать гены, определяющие такое развитие и формирование вторичных половых признаков.
Следует указать на возможную связь развития ХБ плода и новорожденного и эндогенного влияния материнского организма. Приведем два примера. Первый пример - это связь возраста материнского организма и частоты проявления полных аутосомных трисомий, в частности частоты синдрома Дауна. Известно, что частота рождения ребенка с болезнью Дауна в оптимальные для материнского организма сроки деторождения (возраст с 19 до 35 лет) соответствует популяционному значению - 1 случай на 600-650 родов. Если возраст матери 36-40 лет, то частота рождения больного ребенка увеличивается в 2 раза (1 случай на 300 родов); при возрасте 41-45 лет - в 6 раз (1 случай на 100 родов); при возрасте 46-50 лет - в 12-13 раз (1 случай на 50 родов). Второй пример связан с повышением частоты хромосомных нарушений при гиповитаминозах B6 и B12, недостатке фолиевой кислоты и некоторых аутоиммунных заболеваниях у родителей и их детей.
Дополнительным аргументом служит упоминавшийся выше пример эндогенного влияния семейной предрасположенности на частоту сбалансированных транслокаций хромосом. Вполне вероятно, что рождение молодой матерью (до 19 лет) ребенка с синдромом Шерешевского-Тернера (кариотип: 45,ХО) обусловлено расширением спектра и разнообразия мутагенов внешней среды, особенно в условиях больших городов, - это «экологическая модель» и связь уже экзогенного влияния факторов среды на повторное возникновение гамет с числовым дисбалансом хромосом, что находит свое подтверждение в исследованиях по экспериментальной генетике.
Причины и патогенез врожденных аномалий развития
В этой области знаний почти ничего не изменилось с тех пор, как в 1973 г. J.C. Wilson впервые систематизировал причины и клеточные механизмы тератогенеза. Среди них были: генные мутации,
хромосомные аберрации, нарушение митоза, нарушение функции нуклеиновых кислот, отсутствие субстратов для биосинтеза белков, нарушение энергетических процессов, ингибиция энзимов, изменение осмотического и внутриклеточного давления, нарушение мембранных характеристик. Было отмечено, что патогенез ВПР на уровне тканей характеризуется клеточной гибелью, замедлением распада и рассасывания клеток, отмирающих в ходе нормального эмбриогенеза, нарушением адгезии тканей, митотическими задержками, нарушением дифференцировки клеток, сосудистой недостаточностью, торможением клеточной миграции.
Как и ранее, выделяют два класса причин, обусловливающих развитие врожденных аномалий.
• Класс эндогенных причин - это изменения наследственных структур (мутации), эндокринные болезни и метаболические дефекты, «перезревание» половых клеток и возраст родителей.
• Класс экзогенных причин и три группы факторов: а) физические факторы - это ионизирующая радиация и механическое воздействие (травма, сдавление); б) химические факторы - это лекарственные препараты, химические вещества, применяемые в быту и промышленности, гипоксия; в) биологические факторы - это вакцины, сыворотки, гормоны, препараты гистаминового ряда, вирусы и протозойные инфекции. Большинство тератологов полагают, что одной из наиболее частых причин ВПР служат мутации. В случае точковых мутаций аномалии развития наследуются доминантно (например, брахидактилия, габсбургская губа, хондродистрофия) или рецессивно (например, альбинизм). В случае хромосомных и геномных мутаций - это комплексы множественных ВПР.
Следует отметить: гормональные и метаболические нарушения у беременных нередко приводят к самопроизвольным абортам в результате нарушений дифференцировки клеток, тканей и органов плода (тератогенные синдромы), что существенно влияет на антенатальную и раннюю детскую смертность.
Тератогенный эффект был показан при вирилизирующих опухолях, врожденном гипотиреозе, галактоземии, гистидинемии, сахарном диабете I типа и ФКУ, выявляемых у беременных. Например, диабетическая эмбриопатия проявляется аномалиями развития у новорожденных (37% - пороки костно-мышечной системы, 24% - пороки сердца и сосудов, 14% - пороки ЦНС).
Специфическим нарушением развития является синдром диабетической эмбриоили фетопатии (синдром каудальной дисплазии) у плода и новорожденного в виде аплазии или гипоплазии крестца и копчика, поясничных позвонков и бедренных костей. Этот синдром также проявляется большой массой тела при рождении (отложение жировой клетчатки), гиперплазией поджелудочной железы, жировой дистрофией печени, ангиопатиями и ретинопатиями.
Другой пример - фенилаланиновая фетопатия, которая проявляется у детей, рожденных от матерей, страдающих ФКУ или являющихся гетерозиготными носителями гена ФКУ (см. главу 21).
«Перезревание гамет» как причина аномалий развития признается многими врачами-генетиками. Под этим термином подразумеваются изменения в гаметах, произошедшие в период времени от момента их полного созревания до момента оплодотворения. Чем длиннее это время, тем ниже способность к физиологическому оплодотворению гамет и больше аномальных абортусов и плодов. По-видимому, в основе «перезревания» лежит десинхронизация процессов овуляции и оплодотворения, связанная как с интрафолликулярными причинами (например, гормональные расстройства в преклимактерическом периоде), так и с экстрафолликулярными причинами (например, плохая проходимость маточных труб, приводящая к задержке продвижения по ним яйцеклетки и сперматозоидов навстречу друг другу, или недостаточная подвижность самих гамет). Предполагается также, что результатом «перезревания» является нерасхождение хромосом, проявляющееся анеуплоидией.
Широко известна связь между возрастом матери и наличием пороков развития у детей (см. выше). Также установлена связь возраста отцов с частотой рождения детей с расщелинами губы и нёба, ахондроплазией и комплексами множественных ВПР. По-видимому, здесь также имеет значение увеличение частоты мутаций в сперматозоидах и их «перезревание».
Нередкими причинами врожденных аномалий являются ионизирующая радиация, воздействие химических соединений и лекарственных препаратов, среди которых: анальгетики, анестетики, антибиотики (стрептомицин, тетрациклин), гемотерапевтические препараты (антикоагулянты, включая варфариновый ряд), наркотические вещества (изотретиноин, кокаин, комазин), противоопухолевые препараты, цитостатики (дипин), противосудорожные агенты (вальпроат, карбамазепин, триметадион, фенитоин, фенобарбитал),
стероидные гормоны, транквилизаторы (талидомид), а также алкоголь. В частности, при хроническом алкоголизме у беременной рождаются дети с синдромом алкогольной фетопатии (врожденная гипоплазия, постнатальный дефицит массы и длины тела, задержка физического и психомоторного развития, микроили гидроцефалия, черепно-лицевые аномалии).
Выделена форма эмбриопатий, связанная с метаболизмом металлов. Например, цинкзависимая эмбриопатия развивается при безмясной диете или при связывании цинка лекарственными препаратами (ацетазоамин, салицилаты), а также при нарушении адсорбции цинка (при хронических колитах). Такая патология проявляется умеренной гидроцефалией, микроили анофтальмией, расщелиной нёба, искривлением позвоночника, грыжами, пороками сердца.
Наконец, следует отметить гипоксию как одну из причин дефектов развития эмбриона и плода. Так, гипоксия в преимплантационном периоде ведет к гибели зародышей, в период органогенеза тормозит его имплантацию и развитие, ведет к порокам развития или гибели плода. Часто гипоксии способствуют анемии и маточные кровотечения у беременных женщин, наличие у них некомпенсированных пороков сердца.
К биологическим тератогенам относятся: вирусы герпеса, ветряной оспы, гриппа, инфекционного гепатита, краснухи, цитомегалии, эпидемического паротита, полиомиелита, СПИДа; возбудители протозойных инфекций (токсоплазмоз, сифилис); половые стероиды.
Таким образом, можно заключить: если со знанием причин врожденных аномалий развития дело обстоит в основном благополучно, то механизмы их формирования по-прежнему объясняются только общими нарушениями внутриутробного развития организма, которые представляются тератологами как нарушения одного или сразу нескольких процессов (гаметогенез, оплодотворение, имплантация зародыша в матке, его морфогенез и гистогенез в эмбриональном и фетальном периодах, рождение и раннее постнатальное развитие).
Однако практически не изучены молекулярные механизмы этих нарушений развития на уровне экспрессии патологических генов и модификации патологических белков, хотя в последние годы получены определенные результаты, указывающие на то, что такого рода нарушения обусловлены мутациями в генах раннего развития, контролирующих белковые факторы транскрипции (см. главы 8 и 12).
Больше внимание стало уделяться механизму плейотропного действия генов в случае наследственных синдромов, сопровождающихся комплексами множественных ВПР (см. главу 5). В частности, выделены два варианта плейотропии: относительная и мозаичная.
В случае относительной плейотропии нарушения развития могут быть связаны с общей причиной, например с дефектами структуры коллагена - белка соединительной ткани. Показано: именно этот дефект лежит в основе подвывиха хрусталика, скелетных и сердечнососудистых аномалий при синдроме Марфана.
В случае мозаичной плейотропии нарушения развития также объясняются общей причиной - действием одного патологического фактора (или группы аддитивно действующих факторов) в разные периоды времени и на разных стадиях развития. Например, при синдроме Эллиса-ван-Кревельда так объясняется формирование врожденного порока сердца, полидактилии, скелетной дисплазии (дефект соединительной ткани) и множественных уздечек в ротовой полости.
Причины и патогенез онкогенных болезней
В настоящее время окончательно доказана ключевая роль повреждений молекулы ДНК в развитии опухолевого процесса. Об этом свидетельствуют многочисленные данные, наиболее значимые из которых следующие:
• идентификация большого количества генных мутаций и хромосомных перестроек (делеции, инсерции, транслокации) в опухолевых тканях и культивируемых in vitro раковых клетках, что проявляется нестабильностью их генома и считается общим свойством опухолевых клеток;
• выделение наследственных форм рака;
• онкогенное влияние некоторых вирусов, способных к взаимодействию с геномом клеток хозяина и встраиванию в молекулу
ДНК;
• практически полное соответствие между физическими и химическими мутагенами и онкогенами по мутагенному и канцерогенному эффектам.
Однако до понимания истинных причин и механизмов канцерогенеза пока довольно далеко: молекулярные закономерности функционирования генома раковой клетки еще слабо изучены. И хотя известно, что опухолевые клетки - это активно делящиеся клетки, не контролируемые внешними регуляторными сигналами, до сих пор
не определены свойства тканеспецифической регуляции экспрессии генов, обусловливающих не только развитие многоступенчатого процесса озлокачествления, но и работу нормальных генов, по каким-то причинам уступающих пролиферативное преимущество раковым клеткам. При этом речь идет о следующих изменениях: частичная или полная утрата нормальными клетками способностей к пролиферации и дифференцировке; иммортализация (неограниченный во времени клеточный рост или продление времени жизни раковой клетки и увеличения числа ее делений)*; способность клетки к росту без прикрепления к субстрату; снижение уровня восприимчивости к клеточным факторам роста, находящимся в плазме крови; потеря механизма контактного ингибирования; исчезновение ряда поверхностных макромолекул (включая рецепторы). Все эти изменения «делают» злокачественные клетки менее уязвимыми для действия иммунной системы организма.
Известно, что в ходе эволюции были выработаны мощные механизмы, направленные на сохранение генетической стабильности клетки и организма и поддержание равновесия между действием двух систем регуляции клеточного деления: позитивных и негативных генов. В настоящее время постоянно увеличивается количество картированных и клонированных генов, вовлеченных в процесс озлокачествления. Нарушение равновесия между позитивными и негативными генами лежит в основе молекулярных реакций, запускающих механизмы канцерогенеза.
В этой связи следует выделить две группы пусковых причин:
• первая группа - это нарушение взаимодействия генов, регулирующих клеточный цикл;
• вторая группа - это мутации в онкогенах, принадлежащих к двум разным семействам, контролирующим процессы жизнеобеспечения клетки: протоонкогенам, вызывающим опухолевый рост, и антионкогенам (или генам-супрессорам опухолевого роста).
Нарушение взаимодействия генов, регулирующих клеточный цикл
Процесс озлокачествления нормальной соматической клетки связан с накоплением мутаций в ее геноме. Особое место в этиологии опухоли занимает комплексное повреждение генов, контролирующих клеточную пролиферацию, дифференцировку и апоптоз.
* Термин «иммортализация» приведен из словаря терминов по стволовым клеткам (http :www.kletca .ru /stem-cells/glossary).
Приведем пример накопления мутаций в геноме соматической клетки: рассмотрим наиболее часто повреждаемый локус короткого плеча хромосомы 9 (9р21), в котором находятся гены, кодирующие негомологичные ядерные белки: p16INK4a, p15INK4b и p14/ARF. Эти белки появляются в ходе транскрипции как результат альтернативных рамок считывания и выполняют в клетке супрессорные функции. Особую роль играет белок р16INК4а, связывающий циклинзависимые киназы Cdk4 и Cdk6 и препятствующий образованию комплексов с циклинами D, которые фосфорилируют ген RB1 (или ген ретинобластомы). Этот ген находится в комплексе с белком, являющимся его собственным продуктом. При распаде комплекса освобождается фактор транскрипции E2F. Действие гена RB1 тесно связано с функционированием генов, относящихся к генам-супрессорам опухолевого роста и протоонкогенам, продукты которых влияют на активность как самого гена RB1, так и клеточных генов (в результате освобождения фактора E2F), чем инициируется переход клетки в S-фазу клеточного цикла. Активность ядерных белков p16INK4a и p14/ARF в нормальных клетках в ходе экспрессии протоонкогенов Myc, Ras, Raf предотвращает дальнейшее размножение тех клеток, в которых произошла активация одного из онкогенов. Такое действие объясняется наличием в локусе CDKN2A/ARF участков связывания с фактором транскрипции E2F, который может активироваться многими онкогенами. Способом негативной регуляции активности гена RB1 служит механизм инактивации циклинзависимых киназ с помощью белка p16INK4a.
На рис. 59 приведена схема взаимодействия этого гена, других генов и их генных продуктов в процессе канцерогенеза. Ядерный белок p14/ARF способен стабилизировать и активировать ген-супрессор р53 в ходе экспрессии некоторых вирусных и клеточных онкогенов. При этом совместное функционирование гена-супрессора р53 и гена RB1 регулирует процесс апоптоза (см. главу 11). Подтверждением этому служат мутации, идентифицированные в разных злокачественных опухолях (глиобластома, наследственные спорадические меланомы, острый лимфолейкоз, рак мочевого пузыря, пищевода и поджелудочной железы), а также процесс метилирования локуса CDKN2A/ARF. Именно эти события вызывают инактивацию одного из трех белковых продуктов данного локуса: р16, р15 или pARF.
Следует выделить другой путь сигнализации или влияния на активность гена RB1 через клеточный протоонкоген Ras, который
играет центральную роль в передаче внутриклеточного митогенного сигнала. В этом случае ген Ras запускает киназный каскад: Raf - Mek - Erk, обеспечивающий передачу сигнала пролиферации от мембраны в ядро клетки и распределяющий сигнальные импульсы по отдельным путям через эффекторные белки. Например, мишенью для Ras-белка служит ген Raf, стимулирующий МАР-киназы, или митогенактивные белки, участвующие в каскаде запуска циклинзависимых протеинкиназ, необходимых для фосфорилирования гена RB1 и фосфатидил-инозитол-3'-киназы (PI3K).
Еще один механизм, но уже опосредованно влияющий на функционирование системы CDK-RB1, - вовлечение гена-супрессора АРС. В этом случае фактором транскрипции служит белок бетакатенин, активность которого регулируется путем связывания с трансмембранным гликопротеидом или Е-кадгерином, который участвует в образовании межклеточных контактов.
Инактивация гена АРС или мутация в гене бета-кадгерина (CTBN1) уменьшают связывание последнего с Е-кадгерином, стимулируя образование комплекса: бета-катенин Tcf4. Мишенью для этого комплекса являются ген циклина D1 и протоонкоген Myc. В результате активируются циклинзависимые киназы (комплексы
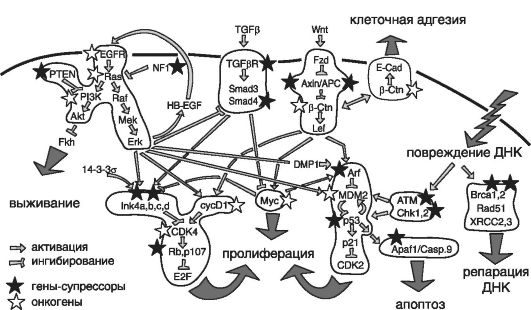 Рис. 59. Схема взаимодействия генов в процессе канцерогенеза (по Залетаеву Д.В. и соавт., 2005)
Рис. 59. Схема взаимодействия генов в процессе канцерогенеза (по Залетаеву Д.В. и соавт., 2005)
циклин D/cdk4,6 и циклин E/cdk2) и происходит фосфорилирование гена RB1, обусловливающее переход клетки в S-фазу.
Например, мутации в генах CDH1 (Е-кадгерин), бета-катенина и АРС были обнаружены в наследственных и спорадических опухолях желудочно-кишечного тракта, спорадических опухолях грудной железы и яичников. Предполагается, что онкогенный эффект мутаций гена Е-кадгерина связан с разобщением межклеточных сигналов, активирующих ген р53.
Кроме того, был выделен еще один путь регуляции клеточной дифференцировки: через взаимодействие клеточных матриксов между собой с помощью фактора роста TCF-бета и белков семейства Smad. В этом случае цитокин TCF-бета участвует в регуляции пролиферации нормальных эпителиальных, эндотелиальных и гемопоэтических клеток. В ходе такой регуляции активируются, например, рецепторы гемопоэтических клеток и фосфорилируются специфические сигнальные молекулы - белки Smad 2 и Smad 3, что вызывает их связывание с фактором транскрипции Smad 4. Образующиеся при этом комплексы переносятся из цитоплазмы в ядро клетки и там регулируют транскрипцию геновиндукторов циклинзависимых киназ р21/wafr1, p15/cdkn2a и p21/kip1. При активации этих генов блокируется фосфорилирование гена RB1 и происходит задержка перехода клетки из фазы G0 в фазу G1 клеточного цикла. Этот путь характерен для развития рака желчного пузыря, легких, поджелудочной железы и толстого кишечника.
Мутации в клеточных онкогенах
Протоонкогены
Идентифицировано свыше 100 протоонкогенов. Они считаются позитивными регуляторами клеточного роста: способны к индукции деления клетки и представляют собой группу генных семейств, играющих ключевую роль в пролиферации и дифференцировке клетки, функционировании ее рецепторов, репарации клеточной ДНК и формировании ответа клетки на внешние регуляторные воздействия. Продукты экспрессии протоонкогенов регулируют сигнальную трансдукцию, опосредованную механизмами:
• фосфорилирования серина, треонина и тирозина, содержащихся в остатках белков (при их деградации) путем отщепления фосфатной группы от АТР, что изменяет конфигурацию белковферментов и их активность;
• регуляции активности GTPазы, осуществляющей превращение GDP в GTP, играющего роль молекулярного медиатора для мембраноассоциированных тирозин-киназы и серин-треонинкиназы;
• регуляции репликации ДНК, экспрессии ядерных генов и апоптоза. Следует отметить, что значительная часть протоонкогенов была идентифицирована по их гомологии с вирусными онкогенами. Канцерогенный эффект протоонкогенов проявляется при наличии активирующей мутации в гетерозиготном состоянии.
Гены-супрессоры опухолевого роста
Гены-супрессоры опухолевого роста выступают как негативные регуляторы клеточной пролиферации и дифференцировки, препятствующие делению клетки. В нормальном состоянии они тормозят экспрессию протоонкогенов. Поэтому их иногда называют антионкогенами (по аналогии с антимутагенами - см. главу 5).
Канцерогенный эффект генов-супрессоров проявляется в гомозиготном состоянии активирующей их мутации (в отличие от доминантного эффекта мутации в гетерозиготном состоянии у протоонкогенов).
В настоящее время изучено 20 генов-супрессоров, включая:
• ген RB1 (13q14) или ген ретинобластомы; он связан с развитием остеосаркомы, рака грудной железы, легких, мочевого пузыря и простаты;
• ген WT1 (11p13), ген опухоли Вилмса (аниридия и аномалии развития мочеполовой системы), ген нефробластомы;
• ген опухолевого белка (tumor protein) - ТР53 или р53 (17р13.1), или общий ген, встречающийся при раке самых разных локализаций; его также называют геном синдрома Ли Фрамени, связанным с развитием опухолей любых тканей и внутренних органов (карцинома почек, остеосаркома, рабдомиосаркома, рак коры надпочечников, мозга, молочной железы, мочевого пузыря, поджелудочной железы, прямой кишки, лейкемия);
• ген VHL (3p25^26) или ген синдрома фон Хиппеля-Линдау; связан с гемангиобластомой, феохромоцитомой, почечно-клеточной карциномой и раком поджелудочной железы;
• ген BRCA, тип 1 (17q21), ген семейного рака грудной железы и яичников;
• ген BRCA, тип 2 (13q12-13), ген наследуемых (несемейных) форм рака грудной железы и яичников;
• ген АРС (5q21), ген семейного аденоматозного полипоза (синдром Гарднера); связан со злокачественными опухолями желудка, поджелудочной железы, прямой кишки;
• гены NF-1 (17q12-22) и NF-2 (22q) или гены нейрофиброматоза, типы 1 и 2 (болезнь Реклингаузена); связаны со шванномами и менингиомами центральной или периферической нервной системы;
• ген р16 (9р21), ген мезотелиомы, глиобластомы и семейной меланомы;
• ген MEN-1(11q) или синдрома множественной эндокринной неоплазии;
• ген рецептора тирозинкиназы, ген RET (10q21), ген синдрома множественной эндокринной неоплазии (типы МЕN-2А и MEN- 2В); связан с медуллярным (папиллярным) раком щитовидной железы и феохромоцитомой;
• ген PTEN (10q23.3), ген синдромов Банаяна-Зонана, Ковдена и Райли-Рувалка; связан с глиобластомами, неходжкинскими лимфомами, раком грудной, щитовидной и предстательной желез, раком эндометрия и яичников;
• гены Mut S или MSH2 (2p15-22) и Mut L MLH1 (3p21.3), гены наследственного неполипозного рака толстой кишки;
• ген CDH1 (16q22.1), ген рака желудка;
• гены TSC1 (9q34) и TSC2 (16p13.3), гены туберозного склероза; связаны с гемартромами мозга, глаз, кожи, почек, легких, сердца и костей.
Важно отметить, что как сами протоонкогены и гены-супрессоры, так и механизмы канцерогенеза, запускаемые с их участием, имеют существенные различия. В частности, мутации в протоонкогенах, обусловливающие их трансформацию в клеточные (целлюлярные) онкогены (c-onc), могут, с одной стороны, затронуть структурную часть гена и вызвать изменение его конечного продукта. В этом случае мутации изменяют конфигурацию белка, что активирует его аутофосфорилирование, увеличивает ферментативную активность, рост клетки.
Первый пример - это аутосомно-доминантные мутации, не нуждающиеся во второй аллельной копии онкогена для инициации злокачественной трансформации. К ним относятся мутации прото-
онкогенов семейства ras, например мутация кодона гена с-На-ras при карциноме мочевого пузыря. В этом случае в ДНК происходит замена гуанина на тимин, а в белке - замена глицина на валин. Второй пример - это точковая мутация гена с-N-ras при меланоме (замена лизина на глицин). Третий пример - это точковая мутация в двух разных точках одного кодона гена с-Ki-ras2 при остром миелоидном лейкозе и карциноме щитовидной железы, приводящая к мутациям в их белковых продуктах (замена глицина на валин и цистеин соответственно). Четвертый пример - это точковая мутация в гене erbA при эритробластозе (замена в белке Р75erb ряда аминокислот, находящихся в аминотерминальных положениях, на валин).
С другой стороны, мутации в протоонкогенах могут повысить уровень экспрессии генов за счет хромосомных перестроек или точковых мутаций в регуляторной части гена (например, в области промотора или энхансера) либо за счет амплификации или увеличения числа копий гена. В этом случае протоонкоген увеличивает свою транскрипцию. Первый пример - это протоонкогены с-mos (острый миелоидный лейкоз), c-myc (лимфома Беркитта) и c-abl (хронический миелоидный лейкоз), которые активируются транслокациями t(8;21), t(8;14) и t(9;22) соответственно. Их результатом может стать амплификация протоонкогена c-abl. Второй пример - это образовавшаяся в коротком плече хромосомы 11 делеция (например, рядом с областью расположения гена с-ras в сегменте 11p13), которая может индуцировать опухоль Вилмса. Третий пример - это инсерция, встроившаяся в регуляторные участки гена и представленная последовательностями некоторых ДНК-содержащих вирусов или провирусной ДНК ретровирусов, усиливающими экспрессию протоонкогенов и рост опухоли.