Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 9 НОРМАЛЬНОЕ ФУНКЦИОНИРОВАНИЕ, АДАПТАЦИЯ И ПОВРЕЖДЕНИЕ КЛЕТКИ
Параметры нормального функционирования клетки
На основе молекулярного мониторинга были изучены клеточные эффекты в нормальных клетках и клетках с измененными функциями. Показано, что условия нормального (физиологического) функционирования клетки определяют три главных фактора:
• наличие стабильных структурообразующих родительских геномов (отцовская и материнская молекулы ДНК);
• определенный диапазон функциональных характеристик структурообразующих молекул индивида;
• параметры окружающей среды: t = 37 °С и pH = 7,35 (для плазмы крови соответственно 36-37 °С и pH 6,5-7,5).
Клетка как структурно-функциональная единица организма постоянно зависит от поддержания баланса между ферментными системами, продуцирующими активный кислород (оксидантные системы), и антиоксидантными системами. Их соотношение формирует клеточный гомеостаз (и гомеостаз организма в целом), в котором общая сумма антиоксидантов создает «буферную систему», обладающую определенной защитной емкостью против реакций избыточного окисления.
Следовательно, в клетке постоянно поддерживается оптимальное равновесие между реакциями окисления и восстановления. Это равновесие также зависит от дополнительных параметров нормального функционирования. Они следующие:
• стабильная структура ядерных и цитоплазматических компонентов;
• целостность клеточных мембран и ферментных систем, включая восстановительные системы;
• доступность поступающих к клетке метаболических (энергетических и трофических) субстратов;
• эффективность собственного метаболизма;
• своевременность дифференциации и специализации собственных функций в окружении соседних клеток и др.
Адаптация, повреждение и выживаемость клетки
При воздействии на клетку факторов окружающей среды ею достигается новое устойчивое (приспособленное к ним) временное или стационарное состояние, называемое адаптацией.
В случаях, когда адаптационные возможности клетки ограничены, в ней развивается цепь событий, называемых повреждением, которое до какого-то определенного момента (некий пороговый уровень) может быть обратимым, и клетка сохраняет свою выживаемость (жизнеспособность), зависящую от защитных или восстановительных свойств самой клетки и целого организма.
Иными словами, превышение клеткой допустимого порогового уровня функционирования зависит от эффективности работы как ее защитных (восстановительных) ферментных систем, так и систем целого организма.
Жизненный цикл клетки
В организме два типа клеток: соматические и герминативные (гаметы).
Жизненный цикл соматической клетки или митотический цикл, включает время подготовки клетки к делению (изначально исходная клетка-зигота находится на стадии бластулы) и время, которое занимает непосредственно сам митоз. Такой цикл длится у человека от 12 до 36 ч (в среднем 24 ч) и включает ряд последовательных периодов (фаз).
• Go-период (фаза покоя) . В связи с тем, что постоянно «покоящихся» клеток не существует вообще, эта фаза считается условной и служит точкой отсчета.
• G1-период или предсинтетическая фаза , В это время производятся пуриновые и пиримидиновые основания и другие компоненты, необходимые для синтеза ДНК. Продолжительность этого периода занимает 40% времени всего митотического цикла (примерно 10 ч).
• S-период (фаза) синтеза ДНК или репликации (удвоения молекулы). В это время происходит полуконсервативная «сборка» на каждой из двух нитей старой молекулы двух новых молекул ДНК; продолжительность - 39% общего времени цикла (примерно 9 ч).
• G2-период (постсинтетическая фаза) . В это время синтезируются мономеры белка-тубулина и другие необходимые для митоза компоненты - 19% времени (примерно 4 ч).
• М-митоз, или равномерное разделение клетки (удвоенной молекулы ДНК) на две дочерние клетки - 2% времени (около 1 ч).
Если учесть, что три фазы жизненного цикла (G1, S и G2) объединены в интерфазу, то общее время подготовки клетки к митозу составит 23 ч (время интерфазы). К этому времени следует добавить еще 1 ч (время митоза), и, следовательно, общая продолжительность цикла составит 24 ч. Иными словами, в течение 23 ч клетка готовится к делению и в течение одного часа делится.
Именно эта закономерность считается «золотым правилом биологии«, согласно которому «работающая (готовящаяся к делению) клетка не делится, делящаяся клетка не работает».
Продолжительность S-периода контролируется теломерами хромосом или «органичителями» репликации ДНК. Эти концевые участки хромосом не несут генетической информации, но зато защищают хромосомы от потерь ДНК в ходе репликации и действия эндонуклеаз. Проблема репликации самих теломер связана с ДНК-полимеразами, осуществляющими синтез ДНК только с использованием РНКпраймеров. Когда праймеры удаляются, то 5'-концы укорачиваются на длину праймера, т.е. на 10-30 нуклеотидов, что должно приводить к потере генов в теломерных участках хромосом.
Теломеры хромосом человека содержат многократно повторяющуюся последовательность, состоящую из оснований - TTAGGG (кроме цитозина). Во время каждого деления теломеры теряют 5-20 таких последовательностей, и такое укорочение продолжается до определенной критической длины, что служит сигналом к прекращению деления, после чего клетка гибнет.
Для соматических клеток есть лимит на число клеточных делений. Так, при культивировании клетки новорожденных могут делиться 80-90 раз, тогда как клетки 70-летнего человека делятся только 20-30 раз, т.е. существует предельное число клеточных делений, или число Хейфлика. В свою очередь, жизненный цикл мейотической клетки связан с процессом оплодотворения, точнее нацелен на него.
После овуляции созревшая яйцеклетка сохраняет свою активность в течение 12-24 ч, тогда как активность зрелого сперматозоида в женских половых путях сохраняется всего несколько часов (см. ниже).
Митоз и его особенности
Митоз или деление соматической клетки, включает четыре фазы: профазу, метафазу, анафазу и телофазу. В зависимости от фазы мито-
за происходящие в клетке изменения затрагивают все структуры ядра и цитоплазмы клетки (рис. 45). В результате митоза из одной материнской клетки образуются две дочерние клетки, они имеют диплоидный набор хромосом (2n) и двойное количество ДНК (2с).
Мейоз и его особенности
Мейоз или деление половой клетки также называют гаметогенезом. В этом случае происходит следующее деление двух возникших в ходе митоза дочерних клеток на две новые клетки. В результате двух последовательных делений образуются 4 гаметы (рис. 46). В сравнении с митозом - это более сложный процесс, хотя между ними много общего.
• Мейоз включает первое и второе деления; каждое деление разделено на такие же по названиям фазы, как при митозе, но с символами I и II соответственно. Все изменения затрагивают структуру ядра и цитоплазмы.
• два деления мейоза разграничены интеркинезом. Это название напоминает интерфазу при митозе, но в данном случае нет репликации ДНК.
• В результате первого деления мейоза из исходной материнской клетки образуются две дочерние. Все они имеют диплоидный набор хромосом (2n) и двойное количество ДНК (2с).
• В результате второго деления мейоза каждая из двух образовавшихся после первого деления дочерних клеток снова делится на две. Каждая из четырех дочерних клеток имеет одинарный (гаплоидный) набор хромосом (n) и одинарное количество ДНК (с).
• Профаза первого деления мейоза имеет ряд последовательных стадий: пролептотена (премейотический митоз), лептотена, зиготена, пахитена, диплотена и диакинез.
• В результате одного сперматогенеза (мейоз у мужчин) образуются 4 функционально активные (способные к оплодотворению) гаметы. Сперматогенез начинается в период полового созревания, следует непосредственно за серией митотических делений и завершается образованием зрелых сперматозоидов.
• В результате одного оогенеза (мейоз у женщин) образуется одна функционально активная яйцеклетка (она получает всю цитоплазму исходной материнской клетки) и 3 полярных тельца (вообще не получают цитоплазму). В этой закономерности заключен эволюционный смысл «биологического
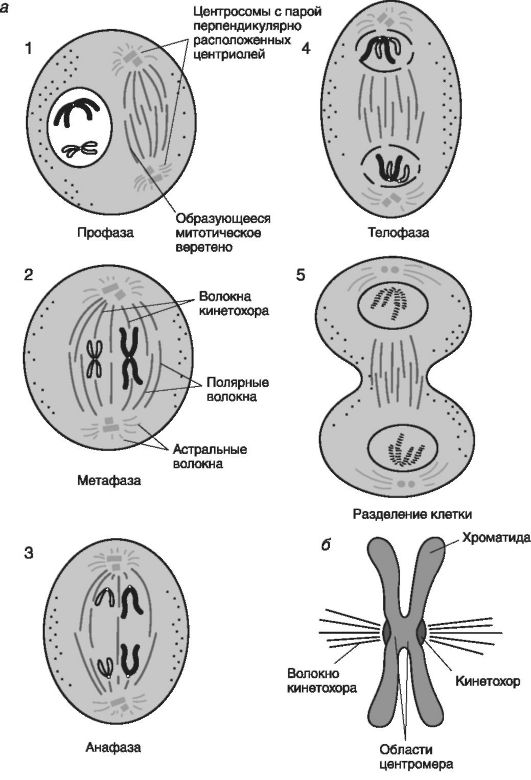 Рис. 45. Упрощенное
изображение митоза (по Эллиот В., Эллиот Д., 2002): а - стадии (фазы)
митоза: 1 - профаза, 2 - метафаза, 3 - анафаза, 4 - телофаза, 5 -
разделение цитоплазмы. Внутририсуночные обозначения волокон, являющихся
микротрубочками: астральные волокна - расположены в области полюсов
клетки; волокна кинетохора - расположены (прикреплены) в области
центромера (по ним перемещаются хромосомы к полюсам клетки); полярные
волокна - по ним полюса клетки расходятся в разные стороны; митотическое
веретено - веретено деления клетки; центросомы - каждая содержит пару
перпендикулярных центриолей; б - морфология хромосомы
Рис. 45. Упрощенное
изображение митоза (по Эллиот В., Эллиот Д., 2002): а - стадии (фазы)
митоза: 1 - профаза, 2 - метафаза, 3 - анафаза, 4 - телофаза, 5 -
разделение цитоплазмы. Внутририсуночные обозначения волокон, являющихся
микротрубочками: астральные волокна - расположены в области полюсов
клетки; волокна кинетохора - расположены (прикреплены) в области
центромера (по ним перемещаются хромосомы к полюсам клетки); полярные
волокна - по ним полюса клетки расходятся в разные стороны; митотическое
веретено - веретено деления клетки; центросомы - каждая содержит пару
перпендикулярных центриолей; б - морфология хромосомы
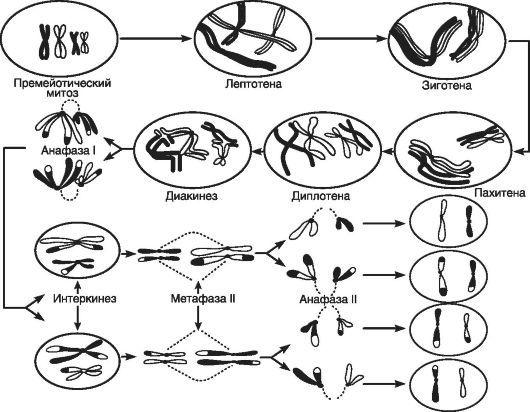 Рис. 46. Упрощенное изображение мейоза (по Эллиот В., Эллиот Д., 2002)
Рис. 46. Упрощенное изображение мейоза (по Эллиот В., Эллиот Д., 2002)
матриархата»: «цитоплазма материнской клетки передается всегда по линии мать-дочь и никогда по линиям мать-сын, отец-дочь, отец-сын» (см. главу 4). Оогенез начинается в эмбриогенезе, продолжается в фетогенезе, когда появляются первые пахитены мейоза (примерно 7-месячный плод). Затем оогенез прерывается на длительный период (до половой зрелости) и продолжается весь репродуктивный период, останавливаясь на время беременности, после которой снова продолжается и окончательно завершается в конце репродуктивного периода. Мало изученной особенностью женского мейоза является формирование во внутриутробном периоде развития организма девочки и сохранение до и после ее рождения резерва клетокродоначальниц для будущих ооцитов (их около 2 тыс). Этот резерв сохраняется до наступления половой зрелости женского организма
во время репродуктивного периода уменьшается на 300-400 яйцеклеток (одна яйцеклетка созревает каждые 28 дней)*.
Повреждения структурных компонентов клетки при патологии
Любой патологический процесс в организме начинается на уровне клеточных, молекулярных, субмолекулярных (или даже аттомолярных) структур: генетических, биохимических, физико-химических. При их повреждении происходит дезорганизация клеточных функций.
Нарушения первичной структуры ДНК
Причинами нарушений первичной структуры ДНК является повреждающее действие ультрафиолетового света, рентгеновских лучей, химических соединений и других факторов окружающей среды.
Как сказано в главах 5 и 7, такого рода повреждения чаще всего ведут к генным и гораздо реже - к хромосомным мутациям, включая:
• ошибки спаривания оснований в матричной цепи ДНК, например встраивание аденина вместо цитозина с образованием АГ-пары;
• спонтанное отщепление оснований от цепи ДНК, например депуринизация и последующее отщепление пуринов;
• дезаминирование цитозина и превращение его в урацил;
• присоединение метильных или этильных групп к основаниям, что изменяет их свойства и ведет к ошибкам спаривания оснований; например, при действии УФ-света в цепи ДНК между собой сшиваются соседние тиминовые основания и образуются тиминовые димеры, препятствующие репликации.
При рентгеновском облучении образуются однонитевые разрывы ДНК, а более жесткие виды излучения (например, гамма-частицы) ведут к двухнитевым разрывам.
При действии ряда химических соединений (митомицин С, иприты, псоралены) образуются сшивки двух цепей ДНК.
В случае алкилирования азотистых оснований наиболее часты два типа нарушений: первый тип - алкилирование аминогруппы
* Известно, что в антенатальном периоде развития как женского, так и мужского организма создается также резерв нейронов (150 млрд), тогда как в течение жизни их расходуется не более 3%, или 5 млрд (см. главу 13); эта проблема также мало изучена.
гуанина, в результате чего образуется метилгуанин, связывающийся с тимином вместо цитозина, что ведет к замене ГЦ-пары на АТ-пару в следующем цикле репликации; второй тип - дезаминирование 5-метилцитозина, также приводящее к замене оснований.
Кроме метильных и этильных групп, к азотистым основаниям могут присоединяться моноаддукты (более крупные химические группы), препятствующие нормальной репликации и транскрипции
ДНК.
Нарушения мембран и ферментных систем клетки
Мембраны и ферментные системы клетки - это первая мишень при различных ее повреждениях. При этом не всегда ясно, как результаты повреждений связаны с этиологией и патогенезом конкретного патологического признака или фенотипа, являются ли они его причиной или следствием, так как возможны и та, и другая ситуации.
Например, при сахарном диабете основной причиной повреждения мембранных ферментов может быть свободнорадикальное окисление или гликилирование их молекул, сопровождаемое нарушением регуляции экспрессии генов и увеличением количества ферментов в мембранах.
Кроме повреждения ферментов, большое значение имеет повреждение других структурообразующих компонентов мембран: фосфолипидов, ПНЖК, углеводов и т.д. В частности, при сахарном диабете в мембранах эритроцитов были обнаружены продукты окисления фосфолипидов (9-оксилинолевая кислота).
При болезни Альцгеймера в головном мозге снижается активность фосфолипаз - это необходимая компенсация в ответ на снижение скорости потери фосфолипидов. Наблюдаемые при этом рост количества глицерофосфорилхолина и увеличение активности лизофосфолипид-ацетилтрансферазы тесно связаны с изменением метаболизма холин-содержащих фосфолипидов и участием в этих реакциях амилоидных пептидов.
Другой пример - больные шизофренией и их потомки: в одних случаях у них наблюдается снижение уровня фосфомоноэфиров - метаболитов синтеза фосфолипидов (это важный показатель риска развития заболевания), а в других случаях - изменяется состав ПНЖК в фосфолипидах мозга.
Кроме того, описано множество случаев, когда у больных с дисфункцией нейронов коры головного мозга первостепенное значение
приобретают жирнокислотный состав мембранных фосфолипидов и активность фосфолипаз.
При ишемической болезни сердца и циррозе печени в мембранах эритроцитов растет соотношение холестерин / фосфолипиды, сопровождаемое резким снижением активности Ка+,К+-АТФазы, ростом уровня СМ, ФС, ФЭ и лизоФХ (при соответствующем снижении уровня ФХ) и деформацией эритроцитов (см. главу 6).
У больных с гиперинсулинемией без диабета (имеют избыточную массу тела), количество мембранного СМ коррелирует с уровнем инсулина плазмы крови и вязкостью мембран, а также изменением степени экспонирования фосфолипидов на поверхности эритроцитов, что обусловливает нарушение асимметрии мембран, повышает их адгезивность и приводит к воспалению.
Изменения фосфолипидов обнаружены в бронхоальвеолярной жидкости у детей с дефицитом легочного сурфактанта.
У взрослых больных с бронхиальной астмой также наблюдали снижение количества сурфактанта, что коррелировало с ослаблением легочной функции.
Все эти примеры свидетельствуют: связь характера повреждений структурных компонентов мембран и ферментных систем клетки с этиологией и патогенезом конкретного заболевания каждый раз уникальна, хотя спектр самих повреждений довольно широкий.
Причины нарушений
Среди причин нарушений (повреждений) мембран и ферментных систем клетки выделены:
• мутации в генах и хромосомах;
• физические факторы: травма, резкое изменение температуры, скачки атмосферного давления, радиация, электрошок;
• химические факторы: гипертонические растворы соли, глюкоза, гормоны, ионы тяжелых металлов, кислоты, лекарства, растворители, яды;
• биологические факторы: бактерии, микроорганизмы, вирусы, вакцины и сыворотки;
• анафилактический шок и иммунные нарушения;
• дефекты обмена аминокислот и витаминов, связанные с нарушением трофики;
• избыток активных форм кислорода (АФК), которые вырабатываются во многих ферментативных и неферментативных процессах; с их гиперпродукцией связывают: адаптационные свойства кле-
ток и апоптоз, клеточные эффекты при артритах, атеросклерозе и кардиомиопатиях, нейродегенеративных заболеваниях и психических расстройствах;
• дефицит кислорода; наблюдается при ишемии, дыхательной недостаточности, анемии и отравлениях окисью углерода;
• процессы канцерогенеза и старения.
Механизмы нарушений
К механизмам нарушений клеточных мембран и ферментных систем относятся: перекисное окисление липидов (ПОЛ), активация лизосомальных гидролаз и протеиназ, эффекты продуктов расщепления фосфолипидов и др. Рассмотрим эти и сопряженные с ними механизмы.
Перекисное окисление липидов
ПОЛ (свободно-радикальное) сопровождается окислительной модификацией мембранных белков и изменением сигнальных процессов в клетке (см. главу 10). При этом образуются перекисные липидные соединения (LOOH) и интермедиаторы или промежуточные соединения, родственные LOOH.
Инициаторами ПОЛ служат молекулы АФК, в том числе:
• реакционноактивная и менее реакционноактивная формы синглетного кислорода (1О2'-); обе формы являются побочными продуктами распада органических гидроперекисей - это так называемая дисмутация супероксидных анионов и пероксидных радикалов; кроме того, эти формы образуются при взаимодействиях Н2О2 и ОС1-, катализируемых ферментом-хлорпероксидазой;
• перекись водорода (Н2О2);
• гидроксильный (НО') и пероксидный (НО'2) реакционноактивные радикалы;
• первичные супероксидные радикалы (О2'-);
• пероксинитрит (ОNOО) и перокинитрокислота (О]ЧЮОН), являются продуктами взаимодействия пероксидных радикалов с ионами металлов переменной валентности (например, Fe2+) или со слабо окисляющимся оксидом азота (NO').
Воздействие АФК на клеточные мембраны служит универсальным механизмом повреждения их белковых и небелковых компонентов. При этом одними из мощных цитотоксических оксидантов белков являются первичные супероксидные радикалы (О2'-), образующиеся при окислении гистидина, метионина, тирозина, триптофана и цистина.
В результате происходит инактивация белков и их агрегация с образованием поперечных сшивок. В свою очередь, образование синглетного кислорода происходит при перекисном окислении микросомальных липидов и биосинтезе простагландинов. Этот радикал разобщает сигнальные процессы в клетке, вызывая защитный эффект с индукцией МАРК (см. главу 6).
Эффекты повреждения клеток АФК суммируются с эффектами, зависящими от физико-химических свойств ферментов: сложности строения молекул, места локализации (наружная или внутренняя мембрана), характера катализируемой реакции.
Как известно, у олигомерных ферментов имеется мембранная липидная матрица, которая выполняет роль «организатора» субъединиц (протомеров или мономеров) этих белков.
Повреждение липидной матрицы нарушает пространственное расположение и координацию отдельных звеньев ферментативных путей. Например, микросомальная глюкозо-6-фосфатаза состоит из каталитического и транспортного белков, для проявления активности которых необходимы два фосфолипида (соответственно ФХ и ФЭ), и их гидролиз под действием фосфолипаз инактивирует этот фермент.
Другими примерами липидзависимых мембранных ферментов служат транспортные К+-, Na+-, Са2+-, 1У^2+-АТРазы, липидное окружение которых контролирует амфифильность и микровязкость мембран. Эти ферменты высоко чувствительны к ПОЛ. Они имеют как водорастворимые, так и трансмембранные протомеры, разобщающиеся при повреждениях мембран, что ведет к инактивации ферментов.
Механизмы повреждения клетки, связанные с АТРазами, можно условно разделить на две группы: повреждение самого фермента и повреждение его липидного микроокружения, от которого также зависит активность фермента. Например, Са2+-АТРаза ведет к замедлению «оттока» ионов кальция из клетки, выравниванию их концентрации во внутри- и межклеточном пространстве, тогда как при повреждении этого фермента нарушаются гомеостаз и стабильность мембран клетки.
Кроме того, нарушение структурной организации липидного бислоя мембран может прямо воздействовать на фермент (изменяет его активный центр), что затруднит доступность субстрата, находящегося в водной фазе, и будет препятствовать его взаимодействию с ферментом.
Следовательно, разрушительное действие АФК на клеточные мембраны инициируется реакциями цепного окисления мембранных фосфолипидов. Эти реакции начинаются с внедрения в мембраны свободных радикалов (в основном НО'), которые вступают во взаимодействие с ПНЖК фосфолипидов, разрывают в них СН-связи около атома углерода, соседнего с двойной связью (-СН=СН-СН2-), и формируют липидные радикалы или гидроокислы.
Затем липидные радикалы обусловливают появление широкого спектра продуктов деградации фосфолипидов и изменяют свойства мембран.
В ходе окисления фосфолипидов также происходит образование мембраноактивных лизофосфолипидов, которые (как и их гидроокислы) дезорганизуют липидный бислой, увеличивая толщину мембраны, фрагментацию везикул, вытесняя белки из гидрофобной зоны в поверхностную зону мембраны. Эти нарушения изменяют заряд мембраны и конформацию липопротеиновых комплексов, обусловливают появление гидрофильных участков в гидрофобном слое и как следствие нарушают проницаемость и стабильность мембраны (вплоть до ее разрыва).
В свою очередь, перекисному окислению с образованием собственных гидроперекисей подвергается мембранный холестерин.
К сопряженным с ПОЛ реакциям относится изменение роли NO' в свободно-радикальных процессах. Известно, что NO' служит регулятором образования АФК и «тканевого (митохондриального) дыхания«. Эта маленькая молекула связывается с цитохромоксидазой и ингибирует аэробный метаболизм, контролируя скорость входа кислорода в дыхательную цепь митохондрий, что способствует накоплению оксиданта-пероксинитрита.
Кроме того, гидроокислы фосфолипидов могут независимо от АФК инициировать повреждение мембранных белков, что еще больше усугубляет повреждение мембран. В этих случаях пусковым механизмом служат две реакции: взаимодействие сульфгидрильных групп (SH) цистеина со свободными радикалами; образование дисульфитов и сульфоновой кислоты. Пример такого повреждения - это формирование белковых агрегатов в хрусталике глаза при катаракте.
Кроме цистеина, окислению в ходе ПОЛ подвергаются аргинин, гистидин, метионин, пролин, серин, тирозин, фенилаланин (включая их производные), а также гемсодержащие белки. Деструкция этих аминокислот делает их доступными для гидролизации под действием
протеаз, для которых окисленные аминокислотные остатки служат маркерами.
Образование комплексов белков с липидами также происходит в виде двух реакций: формирования ковалентной связи между свободными SH-группами аминокислот и альдегидами (карбоксильные группы окисленных липидов); образования полимерных продуктов за счет формирования поперечных сшивок в белковых молекулах.
При действии АФК возможно их участие в трансдукции сигналов от мембранных рецепторов. В ходе такой трансдукции АФК генерируются совместно с NO' при участии НАДФН-оксидазной системы и NOS-системы. Благодаря такому совместному действию АФК и NO' формируется механизм так называемой редокс-регуляции, нарушение которой повреждает сигнальные белки в клеточных мембранах (см. главу 10).
Активация лизосомальных гидролаз и протеиназ
Происходящая при повреждениях цитоплазмы клетки активация лизосом связана с освобождением из них гидролаз и протеиназ, которые разрушают мембраны, запуская события некротической гибели клетки (см. главу 11). Такой механизм включается при интоксикации гепатотропными ядами, к которым относятся: афлатоксин, галактозамин, змеиный яд, хлорпромазин, четыреххлористый углерод (СС14) и др. Эти яды освобождают в печени многие ферменты: ДНКазы и РНКазы, гидролазы и протеиназы (гамма-глюкуронидаза и кислая протеиназа). Изменение активности лизосом может быть вызвано действием ионов Са2+, Mg2+, К+, витаминов А и Е, фосфолипаз, прогестерона, продуктов ПОЛ и других соединений. При освобождении из лизосом кислых липаз (фосфолипаз) происходит ферментативный гидролиз структурообразующих компонентов (нуклеопротеины) и энергосберегающих веществ (гликоген) клетки.
В свою очередь, активация лизосомальных ферментов при холестазе связана с разрушением мембран лизосом под действием желчных кислот.
Эффекты продуктов расщепления фосфолипидов
При гидролизе фосфолипидов, сопровождающемся активацией фосфолипаз, растворимые в воде PLA2 оказывают мембранолитическое действие, вызывая появление дырок и последующий разрыв мембран.
В зависимости от нуклеотидной последовательности генов, кодирующих PLA2, были выделены 10 групп фосфолипаз (I-X), включая
изоформы секреторной (sPLA2), цитозольной Са2+-зависимой фосфолипазы (cPLA2) и цитозольной Са2+-независимой фосфолипазы (iPLA2). Для этих групп показано их участие в:
• апоптозе, некрозе, транслокации к внутриклеточным мембранам (фосфолипаза cPLA2);
• функциях, связанных с аллельными формами генов, приводящими к разной экспрессии (фосфолипаза iPLA2);
• процессе воспаления (фосфолипаза sPLA2);
• фракциях фосфолипидов в липопротеинах плазмы крови (фосфолипаза sPLA2).
В последних двух случаях субстратами служили: фактор активации тромбоцитов (PAF) или продукты окислительного и энзиматического расщепления фосфолипидов.
Показана также способность всех групп фосфолипаз освобождать арахидоновую кислоту (субстрат для простагландин-Н-синтетазы) при воспалении и ишемии (реперфузии).
Арахидоновая кислота выступает в роли модулятора повреждения клеток при действии лекарств, оксидантов и токсинов, гидролизующих фосфолипиды, повышающие проницаемость мембран и вызывающие лизис клеток. Кроме того, арахидоновая кислота служит субстратом для циклооксигеназы, участвующей в образовании эйкозаноидов (простагландины, простациклины и тромбоксаны), а под влиянием 5-липоксигеназы превращается в лейкотриены, которые регулируют проницаемость сосудистой стенки и являются химиотрактантами для нейтрофилов.
Механизмы повреждения структурных компонентов клеток печени
В настоящее время доказана способность нескольких миллионов химических соединений и более чем 300 лекарственных препаратов повреждать клетки печени - главного органа, в котором происходит метаболизм ксенобиотиков (чужеродных веществ), поступающих в организм с пищей, вдыхаемым воздухом, кровотоком (при переливании крови и ее заменителей) или парентерально. При этом пусковым механизмом является повреждение клеточных мембран гепатоцитов за счет либо прямого действия вирусов, ядов или токсинов, либо опосредованного действия продуктов окислительного стресса и других эндогенных факторов.
К механизмам повреждения структурных компонентов клеток печени также относятся: повреждение лекарственными препаратами, механическое повреждение цитоскелета и его отсоединение
от мембраны (способствует растяжению и разрыву), воздействие на мембранные белки или их комплексы ионами тяжелых металлов, бактериальными токсинами, ядами и другими соединениями.
Клетки печени особенно чувствительны к гипоксии и низкому содержанию гликогена. Так, если гликогена в гепатоцитах мало, в них угнетается синтез АТР, блокируются АТР-зависимые метаболические пути (синтез жирных кислот и гликонеогенез). В результате разрушается мтДНК и снижается функционирование митохондрий, что вызывает некроз клетки*.
В зависимости от механизма патогенеза при поражениях печени выделено четыре основных патологических фенотипа.
• Цитолиз (некроз гепатоцитов). Связан с нарушением проницаемости плазматической и внутриклеточных мембран, выходом в кровь и межклеточное пространство внутриклеточного содержимого, что ведет к увеличению концентрации билирубина, витамина В12 и Fe2+ и в конечном итоге повышает активность ферментов, участвующих в некрозе.
• Холестаз или нарушение желчевыводящей функции печени. Связан с повреждением цитоскелета гепатоцитов и приводит к обратному оттоку желчи в синусоиды печени.
• Синдром печеночно-клеточной недостаточности . Этот синдром приводит к нарушению синтетической и детоксицирующей функций печени, развитию гиперазотемии, падению активности холинэстеразы в крови.
• Иммуновоспалительный синдром. Этот синдром связан с сенсибилизацией иммунокомпетентных клеток и активацией клеток ретикулоэндотелиальной системы.
Рассмотрим более подробно механизмы лекарственного повреждения клеток печени. Согласно данным W. Lee (2003), при действии лекарств наиболее частыми механизмами повреждения клеток печени являются:
• метаболические преобразования лекарства с участием цитохрома Р450 и образованием аддуктов (ковалентное присоединение к молекуле цитохрома Р450 (см. главу 10);
• миграция образовавшихся аддуктов (лекарство и цитохром Р450) к клеточной поверхности и инициация на ней иммунных реакций,
* В настоящее время не вызывает сомнений, что молекулярные повреждения гепатоцитов человека индуцируют не только их некроз, но и апоптоз (см. главу 10).
обеспечивающих образование антител и гаптенов, прямое действие NK-клеток и Т-клеток;
• образование конъюгатов лекарства с внутриклеточными белками, что обусловливает дисфункцию органелл, нарушает ионный градиент, снижает уровень АТР, вызывает деградацию белка-актина, набухание, лизис и некроз гепатоцитов;
• воздействие на транспортные белки каналикулярной мембраны и инициация холестаза с последующей стимуляцией апоптоза гепатоцитов;
• вторичный цитокиновый ответ, инициирующий воспаление и каскад иммуноопосредованных реакций, ведущих к апоптозу гепатоцитов с участием факторов Fas и TNF;
• действие лекарства на митохондрии, что ведет к разобщению реакций окислительного фосфорилирования, ингибированию синтеза АТР и окислению жирных кислот с последующим развитием ацидоза и внутриклеточным накоплением микровезикул с триглицеридами.
Приведенный перечень механизмов лекарственного повреждения печени, по-видимому, не совсем полный. Это подтверждается отсутствием удовлетворительного ответа на вопрос: почему лекарство, являющееся безопасным для клеток печени у большинства больных с заболеваниями разной природы, вдруг оказывается гепатотоксичным для кого-то одного из них. Исчерпывающий ответ на этот вопрос, скорее всего, базируется на данных, указывающих на генетические вариации белков, способствующих образованию токсичных продуктов и соответствующих конформационным вариациям изоферментов, участвующих в образовании аддуктов с цитохромом Р450.
Поэтому причину идиосинкразии на вводимый лекарственный препарат, вероятно, можно объяснить (хотя бы частично) генетическими вариациями молекулы цитохрома Р450, обладающего разной каталитической активностью (его у человека не менее 50 форм). Следует также указать на связь механизмов лекарственного повреждения гепатоцитов с ПОЛ плазматических мембран. В частности, ПОЛ не только индуцирует в мембранах характерные нарушения, но и активирует действие фосфолипаз и протеаз на микрофиламенты клетки, нарушая их фиксацию в мембранах или реструктурируя актин, вызывающий некроз.
По такому механизму действуют, например, лекарства, метаболизируемые этанолиндуцируемой формой цитохрома Р450 (GYP 2E1).
Этот фермент вызывает развитие окислительного стресса через генерацию АФК. При этом погибшие под действием этанола гепатоциты и купферовские клетки в дальнейшем служат источниками эндотоксинов.
Кроме того, повреждение мембран ЭР в печени может сопровождаться инактивацией цитохрома Р450 и нарушением процессов гидроксилирования лекарств, пищевых добавок, токсических веществ, различных ядов и канцерогенов (см. главу 10). К таким веществам, например, относятся: алкилирующие агенты, гелиотрин, тиоацетамид, CC14 и эндогенные субстраты (жирные кислоты, простагландины, стероиды и холестерин).