Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 5 ИЗМЕНЧИВОСТЬ ОРГАНИЗМА
Общие данные
Изменчивость организма есть изменчивость его генома, обусловливающая генотипические и фенотипические различия человека и вызывающая эволюционное разнообразие его генотипов и фенотипов (см. главы 2 и 3).
Внутриутробное развитие зародыша, эмбриона, плода, дальнейшее постнатальное развитие организма человека (младенчество, детство, отрочество, юность, взрослая жизнь, старение и смерть) осуществляются в соответствии с генетической программой онтогенеза, сформировавшейся при слиянии материнского и отцовского геномов (см. главы 2 и 12).
В ходе онтогенеза геном организма индивида и закодированная в нем информация подвергаются непрерывным преобразованиям под действием факторов окружающей среды. Возникшие в геноме изменения могут передаваться из поколения в поколение, обусловливая изменчивость признаков и фенотипа организма у потомков.
В начале XX в. немецкий зоолог В. Хэкер выделил направление генетики, посвященное изучению связей и взаимоотношений между генотипами и фенотипами и анализу их изменчивости, и назвал его феногенетикой.
В настоящее время феногенетики выделяют два класса изменчивости: ненаследственную (или модификационную), которая не передается из поколения в поколение, и наследственную, которая передается из поколения в поколение.
В свою очередь, наследственная изменчивость также бывает двух классов: комбинативная (рекомбинационная) и мутационная. Изменчивость первого класса определяют три механизма: случайные встречи гамет при оплодотворении; кроссинговер, или мейотическая рекомбинация (обмен равными участками между гомологичными хромосомами в профазе первого деления мейоза); независимое расхождение гомологичных хромосом к полюсам деления при образовании дочерних клеток в ходе митоза и мейоза. Изменчивость второго
класса обусловлена точковыми, хромосомными и геномными мутациями (см. ниже).
Последовательно рассмотрим различные классы и типы изменчивости организма на разных этапах его индивидуального развития.
Изменчивость при оплодотворении гамет и начало функционирования генома зародившегося организма
Материнский и отцовский геномы не могут функционировать отдельно друг от друга.
Только двух родительских генома, объединившиеся в зиготе, обеспечивают зарождение молекулярной жизни, появление нового качественного состояния - одного из свойств биологической материи.
На рис. 23 отражены результаты взаимодействия двух родительских геномов при оплодотворении гамет.
Согласно формуле оплодотворения: зигота = яйцеклетка + сперматозоид, начало развития зиготы - это момент формирования двойного (диплоидного) при встрече двух гаплоидных наборов родительских гамет. Именно тогда зарождается молекулярная жизнь и запускается цепь последовательных реакций на основе сначала экспрессии генов генотипа зиготы, а затем генотипов появившихся из нее дочерних соматических клеток. Отдельные гены и группы генов в составе генотипов всех клеток организма начинают «включаться» и «выключаться» в ходе реализации генетической программы онтогенеза.
Ведущая роль в происходящих событиях принадлежит яйцеклетке, имеющей в ядре и цитоплазме все необходимые для зарожде-
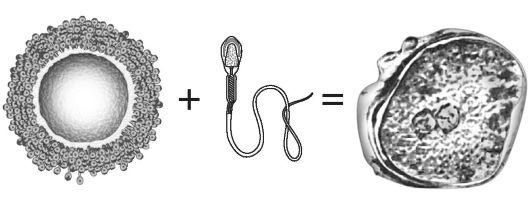 Рис. 23. Результаты
взаимодействия двух родительских геномов при оплодотворении гамет
(рисунки по www.bio.1september.ru; www.bio.fizteh.ru; www.
vetfac.nsau.edu.ru соответственно)
Рис. 23. Результаты
взаимодействия двух родительских геномов при оплодотворении гамет
(рисунки по www.bio.1september.ru; www.bio.fizteh.ru; www.
vetfac.nsau.edu.ru соответственно)
ния и продолжения жизни структурные и функциональные компоненты ядра и цитоплазмы (суть биологического матриархата). Сперматозоид же содержит ДНК и не содержит компонентов цитоплазмы. Проникнув в яйцеклетку, ДНК сперматозоида вступает в контакт с ее ДНК, и тем самым в зиготе «включается» функционирующий в течение всей жизни организма главный молекулярный механизм: ДНК-ДНК взаимодействие двух родительских геномов. Строго говоря, активизируется генотип, представленный примерно равными частями нуклеотидных последовательностей ДНК материнского и отцовского происхождения (без учета мтДНК цитоплазмы). Упростим сказанное: начало молекулярной жизни в зиготе - нарушение постоянства внутренней среды яйцеклетки (ее гомеостаза), а вся последующая молекулярная жизнь многоклеточного организма - стремление восстановить подверженный действию факторов среды гомеостаз или баланс между двумя противоположными состояниями: стабильностью с одной стороны и изменчивостью с другой. Таковы причинно-следственные связи, определяющие возникновение и непрерывность молекулярной жизни организма в ходе онтогенеза.
Теперь обратим внимание на результаты и значение изменчивости генома организма как продукта эволюции. Сначала рассмотрим вопрос об уникальности генотипа зиготы или клетки-родоначальницы всех клеток, тканей, органов и систем организма.
Само оплодотворение происходит случайно: одну женскую гамету оплодотворяет только одна мужская гамета из 200-300 млн сперматозоидов, содержащихся в эякуляте мужчины. Очевидно, что каждую яйцеклетку и каждый сперматозоид отличают друг от друга многие генотипические и фенотипические признаки: наличие измененных или неизмененных по составу и комбинациям генов (результаты комбинативной изменчивости), разные сиквенсы нуклеотидных последовательностей ДНК, разные размеры, форма, функциональная активность (подвижность), зрелость гамет и др. Именно эти отличия позволяют говорить об уникальности генома любой гаметы и, следовательно, генотипа зиготы и всего организма: случайность оплодотворения гамет обеспечивает появление на свет генетически уникального организма индивида.
Иными словами, молекулярная жизнь человека (как и жизнь биологического существа вообще) - «дар судьбы» или, если угодно, «божественный дар», ибо вместо данного индивида с одинаковой
вероятностью могли родиться генетически иные - его родные братья и сестры.
Теперь продолжим наши рассуждения о балансе между стабильностью и изменчивостью наследственного материала. В широком смысле, поддержание такого баланса - это одновременное сохранение и изменение (преобразование) стабильности наследственного материала под действием внутренних (гомеостаз) и внешних средовых факторов (норма реакции). Гомеостаз зависит от генотипа, обусловленного слиянием двух геномов (см. рис. 23). Норма реакции определяется взаимодействием генотипа с факторами окружающей среды.
Норма и диапазон реакции
Специфический способ реакции организма в ответ на действие факторов окружающей среды называется нормой реакции. Именно гены и генотип ответственны за развитие и диапазон модификаций отдельных признаков и фенотипа всего организма. Вместе с тем, в фенотипе реализуются далеко не все возможности генотипа, т.е. фенотип - частный (для индивида) случай реализации генотипа в конкретных условиях окружающей среды. Поэтому, например, между монозиготными близнецами, имеющими полностью идентичные генотипы (100% общих генов), выявляются заметные фенотипические различия, если близнецы растут в разных условиях окружающей среды.
Норма реакции бывает узкой или широкой. В первом случае стабильность отдельного признака (фенотипа) сохраняется практически вне зависимости от влияния окружающей среды. Примерами генов с узкой нормой реакции или непластичных генов служат гены, кодирующие синтез антигенов групп крови, окраску глаз, курчавость волос и др. Их действие одинаково при любых (совместимых с жизнью) внешних условиях. Во втором случае стабильность отдельного признака (фенотипа) изменяется в зависимости от влияния окружающей среды. Пример генов с широкой нормой реакции или пластичных генов - гены, контролирующие количество эритроцитов крови (разное у лиц, поднимающихся в гору, и лиц, спускающихся с горы). Другой пример широкой нормы реакции - изменение окраски кожных покровов (загар), связанный с интенсивностью и временем воздействия на организм ультрафиолетового облучения.
Говоря о диапазоне реакции, следует иметь в виду фенотипические различия, проявляющиеся у индивида (его генотипа) в зависимости от
«обедненных» или «обогащенных» условий окружающей среды, в которых находится организм. Согласно определению И.И. Шмальгаузена (1946), «наследуются не признаки, как таковые, а норма их реакции на изменения условий существования организмов».
Таким образом, норма и диапазон реакции - это пределы генотипической и фенотипической изменчивости организма при изменении условий окружающей среды.
Следует также отметить, что из внутренних факторов, оказывающих влияние на фенотипическое проявление генов и генотипа, определенное значение имеют пол и возраст индивида.
Внешние и внутренние факторы, определяющие развитие признаков и фенотипов, входят в указанные в главе три группы основных факторов, среди которых гены и генотип, механизмы межмолекулярных (ДНК-ДНКовых) и межгенных взаимодействий между родительскими геномами и факторы окружающей среды.
Безусловно, основой приспособления организма к условиям окружающей среды (основой онтогенеза) является его генотип. В частности, индивиды с генотипами, не обеспечивающими подавление отрицательного действия патологических генов и факторов среды, оставляют меньше потомков, чем те индивиды, у которых нежелательные эффекты подавляются.
Вероятно, что в генотипы более жизнеспособных организмов включены специальные гены (гены-модификаторы), подавляющие действие «вредных» генов таким образом, что вместо них доминантными становятся аллели нормального типа.
НЕНАСЛЕДСТВЕННАЯ ИЗМЕНЧИВОСТЬ
Говоря о ненаследственной изменчивости генетического материала, снова рассмотрим пример широкой нормы реакции - изменение окраски кожных покровов под действием ультрафиолетового излучения. «Загар» ведь не передается из поколения в поколение, т.е. не наследуется, хотя в его возникновении участвуют пластичные гены.
Точно так же не наследуются результаты травм, рубцовые изменения тканей и слизистых оболочек при ожоговой болезни, обморожениях, отравлениях и многие другие признаки, вызванные действием исключительно факторов среды. Вместе с тем, следует подчеркнуть: ненаследственные изменения или модификации связаны с наслед-
ственными свойствами данного организма, ибо образуются на фоне конкретного генотипа в конкретных условиях окружающей среды.
Наследственная комбинативная изменчивость
Как сказано в начале главы, кроме механизма случайных встреч гамет при оплодотворении, комбинативная изменчивость включает механизмы кроссинговера в первом делении мейоза и независимого расхождения хромосом к полюсам деления при образовании дочерних клеток в ходе митоза и мейоза (см. главу 9).
Кроссинговер в первом делении мейоза
За счет механизма кроссинговера сцепление генов с хромосомой регулярно нарушается в профазе первого деления мейоза в результате перемешивания между собой (обмена) генов отцовского и материнского происхождения (рис. 24).
В начале XX в. при открытии кроссинговера Т.Х. Морган и его ученики предположили: кроссинговер между двумя генами может происходить не только в одной, но и в двух, трех (соответственно двойной и тройной кроссинговер) и большем количестве точек. Отмечалось подавление кроссинговера в участках, непосредственно примыкающих к точкам обмена; такое подавление назвали интерференцией.
В конечном итоге подсчитали: на один мужской мейоз приходится от 39 до 64 хиазм или рекомбинаций, а на один женский мейоз - до 100 хиазм.
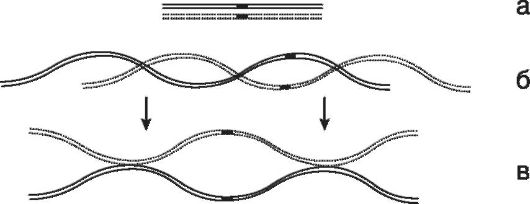 Рис. 24. Схема кроссинговера в первом делении мейоза (по Шевченко В.А. и соавт., 2004):
Рис. 24. Схема кроссинговера в первом делении мейоза (по Шевченко В.А. и соавт., 2004):
a - сестринские хроматиды гомологичных хромосом до начала мейоза; б - они же во время пахитены (видна их спирализация); в - они же во время диплотены и диакинеза (стрелки указывают на места кроссинговера-хиазмы, или участки обмена)
В результате сделали вывод: сцепление генов с хромосомами постоянно нарушается в ходе кроссинговера.
Факторы, влияющие на кроссинговер
Кроссинговер - один из регулярных генетических процессов в организме, контролируемый многими генами как непосредственно, так и через физиологическое состояние клеток в ходе мейоза и даже митоза.
К факторам, влияющим на кроссинговер, относятся:
• гомо- и гетерогаметный пол (речь идет о митотическом кроссинговере у самцов и самок таких эукариот, как дрозофила и тутовый шелкопряд); так, у дрозофилы кроссинговер протекает нормально; у тутового шелкопряда - либо тоже нормально, либо отсутствует; у человека следует обратить внимание на смешанный («третий») пол и конкретно на роль кроссинговера при аномалиях развития пола при мужском и женском гермафродитизме (см. главу 16);
• структура хроматина; на частоту кроссинговера в разных участках хромосом влияет распределение гетерохроматиновых (прицентромерные и теломерные участки) и эухроматиновых районов; в частности, в прицентромерных и теломерных участках частота кроссинговера снижена, и расстояние между генами, определяемое по частоте кроссинговера, может не соответствовать фактическому;
• функциональное состояние организма; по мере увеличения возраста меняется степень спирализации хромосом и скорость клеточного деления;
• генотип; в его составе выделены гены, увеличивающие или уменьшающие частоту кроссинговера; «запиратели» последнего - хромосомные перестройки (инверсии и транслокации), затрудняющие нормальную конъюгацию хромосом в зиготене;
• экзогенные факторы: воздействие температуры, ионизирующей радиации и концентрированных растворов солей, химические мутагены, лекарства и гормоны, как правило, повышающие частоту кроссинговера.
По частоте мейотического и митотического кроссинговера и СХО порой судят о мутагенном действии лекарств, канцерогенов, антибиотиков и других химических соединений.
Неравный кроссинговер
В редких случаях в ходе кроссинговера наблюдаются разрывы в несимметричных точках сестринских хроматид, и они обменива-
ются между собой неравными участками - это неравный кроссинговер.
Вместе с тем, описаны случаи, когда в ходе митоза наблюдается митотическая конъюгация (неправильное спаривание) гомологичных хромосом и рекомбинация происходит между несестринскими хроматидами. Такое явление получило название генной конверсии.
Значение данного механизма трудно переоценить. Например, в результате неправильного спаривания гомологичных хромосом по фланкирующим повторам может произойти удвоение (дупликация) или утрата (делеция) участка хромосомы, содержащего ген РМР22, что обусловит развитие наследственной аутосомно-доминантной моторно-сенсорной нейропатии Шарко-Мари-Тус.
Неравный кроссинговер - один из механизмов возникновения мутаций. Например, периферический белок миелин кодируется геном РМР22, расположенным в хромосоме 17 и имеющим длину около 1,5 млн н.п. Этот ген фланкируется двумя гомологичными повторами длиной около 30 тыс. н.п. (повторы расположены на флангах гена).
Особенно много мутаций в результате неравного кроссинговера происходит в псевдогенах. Тогда либо фрагмент одного аллеля переносится в другой аллель, либо фрагмент псевдогена - в ген. Например, подобная мутация отмечается при переносе последовательности псевдогена в ген 21-гидроксилазы (CYP21B) при адреногенитальном синдроме или врожденной гиперплазии коры надпочечников (см. главы 14 и 22).
Кроме того, за счет рекомбинаций в ходе неравного кроссинговера могут образовываться множественные аллельные формы генов, кодирующих антигены HLA класса I.
Независимое расхождение гомологических хромосом к полюсам деления при образовании дочерних клеток в ходе митоза и мейоза
Благодаря процессу репликации, предшествующему митозу соматической клетки, общее количество нуклеотидных последовательностей ДНК увеличивается вдвое. Формирование одной пары гомологичных хромосом происходит из двух отцовских и двух материнских хромосом. При распределении этих четырех хромосом в две дочерние клетки каждая из клеток получит одну отцовскую и одну материнскую хромосомы (для каждой пары хромосомного набора), однако какую именно из двух, первую или вторую, неизвестно. Имеет место
случайный характер распределения гомологичных хромосом. Легко подсчитать: за счет различных комбинаций 23 пар хромосом общее количество дочерних клеток составит 223, или более 8 млн (8 χ 106) вариантов комбинаций хромосом и расположенных на них генов. Следовательно, при случайном характере распределения хромосом в дочерние клетки каждая из них будет иметь свой уникальный кариотип и генотип (свой вариант комбинации хромосом и сцепленных с ними генов соответственно). Следует отметить и возможность патологического варианта распределения хромосом в дочерние клетки. Например, попадание в одну из двух дочерних клеток только одной (отцовской или материнской по происхождению) Х-хромосомы приведет к моносомии (синдром Шерешевского-Тернера, кариотип 45, ХО), попадание трех одинаковых аутосом приведет к трисомии (синдромы Дауна, 47,XY,+21; Патау, 47,ХХ,+13 и Эдвадса, 47,ХХ,+18; см. также главу 2).
Как отмечено в главе 5, в одну дочернюю клетку могут одновременно попасть две отцовские или две материнские по происхождению хромосомы - это однородительская изодисомия по конкретной паре хромосом: синдромы Сильвера-Рассела (две материнские хромосомы 7), Беквитта-Видемана (две отцовские хромосомы 11), Ангельмана (две отцовские хромосомы 15), Прадера-Вилли (две материнские хромосомы 15). В целом объем нарушений распределения хромосом достигает 1% всех хромосомных нарушений у человека. Эти нарушения имеют большое эволюционное значение, ибо создают популяционное разнообразие кариотипов, генотипов и фенотипов человека. Причем каждый патологический вариант является уникальным продуктом эволюции.
В случае мейоза также легко подсчитать: после первого деления общее число возможных типов комбинаций хромосом в дочерних клетках составит 223 или более 8 млн генотипов.
В результате второго мейотического деления образуются 4 дочерние клетки. В каждую из них отойдет по одной либо материнской, либо отцовской хромосоме из всех 23 хромосом.
Чтобы избежать возможных ошибок в наших дальнейших расчетах, примем за правило: в результате второго мейотического деления также образуется 8 млн вариантов мужских гамет и 8 млн вариантов женских гамет. Тогда ответ на вопрос, каков общий объем вариантов комбинаций хромосом и расположенных на них генов при встрече двух гамет, следующий: 246 или 64 χ 1012, т.е. 64 триллиона.
Образование такого (теоретически возможного) количества генотипов при встрече двух гамет наглядно объясняет смысл гетерогенности генотипов.
Значение комбинативной изменчивости
Комбинативная изменчивость важна не только для гетерогенности и уникальности наследственного материала, но и для восстановления (репарации) стабильности молекулы ДНК при повреждении ее обеих нитей. Примером служит образование одноцепочечной бреши ДНК напротив нерепарированного повреждения. Появившаяся брешь не может быть безошибочно исправлена без привлечения к репарации нормальной нити ДНК.
Мутационная изменчивость
Наряду с уникальностью и гетерогенностью генотипов и фенотипов в результате комбинативной изменчивости огромный вклад в вариабельность генома и фенома человека вносит наследственная мутационная изменчивость и обусловленная ею генетическая гетерогенность.
Вариации нуклеотидных последовательностей ДНК чисто условно можно разделить на мутации и генетический полиморфизм (см. главу 2). Вместе с тем, если гетерогенность генотипов - это постоянные (нормальные) характеристики вариабельности генома, то мутационная изменчивость - это, как правило, его патология.
В пользу патологической вариабельности генома свидетельствуют, например, неравный кроссинговер, неправильное расхождение хромосом к полюсам деления при образовании дочерних клеток, наличие генетических компаундов и аллельных серий. Иными словами, наследственная комбинативная и мутационная изменчивость проявляется у человека значительным генотипическим и фенотипическим разнообразием.
Уточним терминологию и рассмотрим общие вопросы теории мутаций.
ОБЩИЕ ВОПРОСЫ ТЕОРИИ МУТАЦИЙ
Мутация есть изменение структурной организации, количества и/или функционирования наследственного материала и синтезируемых им белков. Это понятие впервые предложил Гуго де Фриз
в 1901-1903 гг. в своей работе «Мутационная теория», где описал основные свойства мутаций. Они:
• возникают внезапно;
• передаются из поколения в поколение;
• наследуются по доминантному типу (проявляются у гетерозигот и гомозигот) и рецессивному типу (проявляются у гомозигот);
• не имеют направленности («мутирует» любой локус, вызывая незначительные изменения или затрагивая жизненно важные признаки);
• по фенотипическому проявлению бывают вредными (большинство мутаций), полезными (крайне редко) или безразличными;
• возникают в соматических и половых клетках.
Кроме того, одни и те же мутации могут возникнуть повторно.
Мутационный процесс или мутагенез, есть непрерывно идущий процесс формирования мутаций под действием мутагенов - факторов среды, повреждающих наследственный материал.
Впервые теория непрерывно идущего мутагенеза предложена в 1889 г. русским ученым из Петербургского университета С.И. Коржинским в его книге «Гетерогенезис и эволюция».
Как принято считать в настоящее время, мутации способны проявиться спонтанно, без видимых внешних причин, но под влиянием внутренних условий в клетке и организме - это спонтанные мутации или спонтанный мутагенез.
Мутации, вызванные искусственно путем воздействия внешних факторов физической, химической или биологической природы, - это индуцированные мутации, или индуцированный мутагенез.
Наиболее часто встречающиеся мутации называются мажорными мутациями (например, мутации в генах миодистрофии Дюшенна- Беккера, муковисцидоза, серповидноклеточной анемии, фенилкетонурии и др.). Сейчас созданы коммерческие наборы, позволяющие выявлять в автоматическом режиме наиболее важные из них.
Вновь возникшие мутации называются новыми мутациями или мутациями de novo. Например, к ним относятся мутации, лежащие в основе ряда аутосомно-доминантных болезней, таких, как ахондроплазия (10% случаев заболевания - семейные формы), нейрофиброматоз Реклингаузена I типа (50-70% - семейные формы), болезнь Альцгеймера, хорея Гентингтона.
Мутации от нормального состояния гена (признака) к патологическому состоянию называются прямыми.
Мутации от патологического состояния гена (признака) к нормальному состоянию называются обратными или реверсиями.
Впервые способность к реверсии установлена в 1935 г. Н.В. Тимофеевым-Рессовским.
Последующие мутации в гене, подавляющие первичный мутантный фенотип, называются супрессорными. Супрессия может быть внутригенной (восстанавливает функциональную активность белка; аминокислота не соответствует исходной, т.е. истинной обратимости нет) и внегенной (изменяется структура тРНК, в результате чего мутантная тРНК включает в полипептид другую аминокислоту вместо кодируемой дефектным триплетом).
Мутации в соматических клетках называются соматическими мутациями. Они формируют патологические клеточные клоны (совокупность патологических клеток) и в случае одновременного присутствия в организме нормальных и патологических клеток приводят к клеточному мозаицизму (например, при наследственной остеодистрофии Олбрайта экспрессивность заболевания зависит от количества аномальных клеток).
Соматические мутации могут быть как семейными, так и спорадическими (несемейными). Они лежат в основе развития злокачественных новообразований и процессов преждевременного старения.
Ранее считалось аксиомой, что соматические мутации не наследуются. В последние же годы была доказана передача из поколения в поколение наследственной предрасположенности 90% мультифакториальных форм и 10% моногенных форм рака, проявляющихся мутациями в соматических клетках.
Мутации в половых клетках называются герминативными мутациями. Считается, что они встречаются реже соматических мутаций, лежат в основе всех наследственных и некоторых врожденных болезней, передаются из поколения в поколение и также могут быть семейными и спорадическими. Наиболее изученная область общего мутагенеза - физический и, в частности, радиационный мутагенез. Любые источники ионизирующей радиации пагубны для здоровья человека, они, как правило, оказывают мощное мутагенное, тератогенное и канцерогенное воздействие. Мутагенный эффект однократной дозы облучения гораздо выше, чем при хроническом облучении; доза облучения в 10 рад удваивает частоту мутаций у человека. Доказано: ионизирующее излучение способно вызвать мутации, приводящие
к наследственным (врожденным) и онкологическим болезням, а ультрафиолетовое - индуцировать ошибки репликации ДНК.
Большую опасность представляет химический мутагенез. В мире существует около 7 млн химических соединений. В народном хозяйстве, на производстве и в быту постоянно применяются примерно 50-60 тыс. химических веществ. Ежегодно внедряются в практику около одной тысячи новых соединений. Из них 10% в состоянии индуцировать мутации. Таковы гербициды и пестициды (доля мутагенов среди них достигает 50%), а также ряд лекарственных препаратов (некоторые антибиотики, синтетические гормоны, цитостатики и др.).
Существует еще биологический мутагенез. К биологическим мутагенам относятся: чужеродные белки вакцин и сывороток, вирусы (ветряная оспа, коревая краснуха, полиомиелит, простой герпес, СПИД, энцефалит) и ДНК, экзогенные факторы (неполноценное белковое питание), соединения гистамина и его производные, стероидные гормоны (эндогенные факторы). Усиливают действие внешних мутагенов комутагены (токсины).
В истории генетики немало примеров значения связей между генами и признаками. Один из них - классификация мутаций в зависимости от их фенотипического эффекта.
Классификация мутаций в зависимости от их фенотипического эффекта
Такую классификацию мутаций впервые предложил в 1932 г. Г. Мёллер. Согласно классификации были выделены:
• аморфные мутации. Это состояние, при котором признак, контролируемый патологическим аллелем, не проявляется, так как патологический аллель не активен по сравнению с нормальным аллелем. К таким мутациям относятся ген альбинизма (11q14.1) и около 3000 аутосомно-рецессивных заболеваний;
• антиморфные мутации. В этом случае значение признака, контролируемого патологическим аллелем, противоположно значению признака, контролируемого нормальным аллелем. К таким мутациям относятся гены около 5-6 тыс. аутосомно-доминантных заболеваний;
• гиперморфные мутации. В случае такой мутации признак, контролируемый патологическим аллелем, выражен сильнее признака, контролируемого нормальным аллелем. Пример - гете-
розиготные носители генов болезней нестабильности генома (см. главу 10). Их число составляет около 3% населения Земли (почти 195 млн человек), а количество самих заболеваний достигает 100 нозологий. Среди этих заболеваний: анемия Фанкони, атаксиятелеангиэктазия, пигментная ксеродерма, синдром Блума, прогероидные синдромы, многие формы рака и др. При этом частота рака у гетерозиготных носителей генов этих заболеваний в 3-5 раз выше, чем в норме, а у самих больных (гомозигот по этим генам) частота рака в десятки раз выше, чем в норме.
• Гипоморфные мутации. Это состояние, при котором проявление признака, контролируемого патологическим аллелем, ослаблено по сравнению с признаком, контролируемым нормальным аллелем. К таким мутациям относятся мутации генов синтеза пигментов (1q31; 6p21.2; 7p15-q13; 8q12.1; 17p13.3; 17q25; 19q13; Xp21.2; Xp21.3; Xp22), а также более 3000 форм аутосомно-рецессивных заболеваний.
• Неоморфные мутации. О такой мутации говорят, когда признак, контролируемый патологическим аллелем, будет иного (нового) качества по сравнению с признаком, контролируемым нормальным аллелем. Пример: синтез новых иммуноглобулинов в ответ на проникновение в организм чужеродных антигенов.
Говоря о непреходящем значении классификации Г. Мёллера, следует отметить, что спустя 60 лет после ее публикации фенотипические эффекты точковых мутаций были разделены на разные классы в зависимости от оказываемого ими воздействия на структуру белкового продукта гена и/или уровень его экспрессии.
В частности, нобелевский лауреат Виктор Маккьюсик (1992) выделил мутации, изменяющие последовательность аминокислот в белке. Оказалось, что именно они отвечают за проявление 50-60% случаев моногенных болезней, а остальные мутации (40-50% случаев) приходятся на долю мутаций, затрагивающих экспрессию генов.
Изменение аминокислотного состава белка проявляется в патологическом фенотипе, например, в случаях метгемоглобинемии или серповидноклеточной анемии, обусловленной мутациями бетаглобинового гена. В свою очередь, были выделены мутации, затрагивающие нормальную экспрессию гена. Они приводят к изменению количества генного продукта и проявляются фенотипами, связанными с недостаточностью того или иного белка, например,
в случаях гемолитической анемии, обусловленной мутациями генов, локализованных на аутосомах: 9q34.3 (дефицит аденилаткиназы); 12p13.1 (дефицит триозофосфатизомеразы); 21q22.2 (дефицит фосфофруктокиназы).
Классификация мутаций В. Маккьюсика (1992) - это, безусловно, новое поколение классификаций. Вместе с тем, накануне ее публикации широкое признание получила классификация мутаций в зависимости от уровня организации наследственного материала.
Классификация мутаций в зависимости от уровня организации наследственного материала
Классификация включает следующее.
Точковые мутации (нарушение структуры гена в разных его точках).
Строго говоря, к точковым мутациям относятся изменения нуклеотидов (оснований) одного гена, ведущие к изменению количества и качества синтезируемых ими белковых продуктов. Изменения оснований - это их замены, вставки, перемещения или выпадения, которые можно объяснить мутациями в регуляторных областях генов (промотор, сайт полиаденилирования), а также в кодирующих и некодирующих областях генов (экзоны и интроны, сайты сплайсинга). Замены оснований ведут к появлению трех типов мутантных кодонов: миссенс-мутации, нейтральные мутации и нонсенс-мутации.
Точковые мутации наследуются как простые менделевские признаки. Они часто встречаются: 1 случай на 200-2000 рождений - это первичный гемохроматоз, неполипозный рак толстой кишки, синдром Мартина-Белл и муковисцидоз.
Точковые мутации, встречающиеся крайне редко (1:1 500 000), - это тяжелый комбинированный иммунодефицит (ТКИД) в результате дефицита аденозиндезаминазы. Иногда точковые мутации формируются не при воздействии мутагенов, а как ошибки репликации ДНК. При этом их частота не превышает 1:105-1:1010, так как они исправляются с помощью репарационных систем клетки почти на
99%.
Структурные мутации или аберрации хромосом (нарушают структуру хромосом и приводят к формированию новых групп сцепления генов). Это делеции (утраты), дупликации (удвоения), транслокации (перемещения), инверсии (поворот на 180°) или инсерции (вставки) наследственного материала. Такие мутации характерны для сомати-
ческих клеток (включая стволовые клетки). Их частота составляет 1 на 1700 клеточных делений.
Известен ряд синдромов, обусловленных структурными мутациями. Наиболее известные примеры: синдром «кошачьего крика» (кариотип: 46,ХХ,5р-), синдром Вольфа-Хиршхорна (46,ХХ, 4р-), транслокационная форма синдрома Дауна (кариотип: 47, ХУ, t (14;21)).
Другой пример - это лейкемии. При них происходит нарушение экспрессии гена в результате так называемого разделения (транслокация между структурной частью гена и его промоторной областью), и, следовательно, нарушается синтез белка.
Геномные (численные) мутации - нарушение числа хромосом или их частей (ведут к появлению новых геномов или их частей путем добавления или утраты целых хромосом или их частей). Происхождение этих мутаций обусловлено нерасхождением хромосом в митозе или мейозе.
В первом случае - это анеуплоиды, тетраплоиды с неразделенной цитоплазмой, полиплоиды, имеющие по 6, 8, 10 пар хромосом и более.
Во втором случае - это неразделение парных хромосом, участвующих в формировании гамет (моносомии, трисомии) или оплодотворение одной яйцеклетки двумя сперматозоидами (диспермия или триплоидный зародыш).
Их типичные примеры уже не раз приводились - это синдром Шерешевского-Тернера (45,ХО), синдром Клайнфельтера (47,ХХУ), регулярная трисомия при синдроме Дауна (47,ХХ, +21).
Геномные мутации являются наиболее частыми из всех классов мутаций, особенно синдром Дауна, частота которого составляет 1 случай на 550-650 человек.
Именно частота синдрома Дауна служит показателем общей частоты хромосомных и геномных мутаций в популяции человека. Существует правило, согласно которому среди 100 больных с разными хромосомными синдромами 95 больных (95%) будут иметь нарушения числа хромосом (включая 75% больных с синдромом Дауна) и только 5 больных (5%) - структурные нарушения хромосом.
В настоящее время в структурной геномике большое распространение получает классификация точковых мутаций в зависимости от размеров фрагментов ДНК, подвергающихся изменениям в ходе мутагенеза.
Причины уникальности мутаций и классификация точковых мутаций
Несмотря на наличие общих закономерностей мутационного процесса, структура и спектр мутационных повреждений каждого гена уникальны.
Причины уникальности мутаций скрыты в особенностях строения ДНК, включая:
• размеры гена;
• обогащенность гена ГЦ-парами; она предрасполагает к образованию точковых мутаций типа транцизии или замены цитозина на тимин в процессе метилирования-деметилирования;
• наличие в гене прямых и обратных (обращенных) повторов;
• наличие в генах последовательностей ДНК, гомологичных внегенным участкам, что может вызвать нарушение рекомбинации в мейозе;
• инсерции транспозонов (в Alu-, LINE- и SINE- последовательностях);
• динамические мутации транскрибируемых и нетранскрибируемых повторов.
Важно отметить, что точковые мутации каждого гена по своей локализации диаллельны: встречаются в двух вариантах в зависимости от отцовского или материнского происхождения (см. главу 2). Они могут затронуть любое звено на всем пути прохождения наследственной информации от молекулы ДНК к молекуле полипептида.
Согласно указанной выше классификации В. Маккьюсика (1992), среди точковых мутаций выделены мутации первого и второго типов.
Первый тип - это мутации транскрипции. Среди них:
• мутации в области промотора; примером служат два гена, характерных для синдромов Мартина-Белл и умственной отсталости, сцепленных с фрагильной Х-хромосомой, - это гены FraXA и FraXE; другой пример - это мутация фактора IX (положение 20 от старта транскрипции) при гемофилии В;
• мутации сплайсинга мРНК, в том числе мутации в 5'-донорскоми 3'-акцепторном сайтах интронов, а также мутации, приводящие к появлению новых сайтов сплайсинга из-за неправильного вырезания интрона или экзона; пример - мутация при муковисцидозе (621GT);
• мутации полиаденилирования;
• мутации кэп-сайта;
• мутации (делеции) энхансеров.
Второй тип мутаций - это мутации трансляции. Среди них выделяют следующие.
• Мутации кодона инициации транскрипции или индикаторного кодона (АУГ), а также мутации вблизи этого кодона.
• Мутации в терминирующих кодонах - ТАА, ТАГ и ТГА (приводит к синтезу длинных белков).
• Мутации сдвига рамки считывания или фреймшифт-мутации (приводят к делеции или инсерции участка молекулы ДНК, размеры которого не кратны трем нуклеотидам, и в результате нарушается нормальный отсчет кодирующих триплетов, возникают преждевременные стоп-кодоны (ПСК) и наблюдается синтез неполноценных белков. Эти мутации встречаются при муковисцидозе (3905 insT) и лежат в основе многих нервномышечных заболеваний. Например, протяженные делеции, охватывающие весь ген или значительную его область, обнаружены при Х-сцепленной рецессивной миодистрофии Дюшенна-Бекера и аутосомно-рецессивных спинальных мышечных атрофиях.
• Миссенс-мутации или замена одного аминокислотного остатка на другой в молекуле белка. Эти мутации приводят к нарушению экспрессии гена и, следовательно, к образованию новой структуры белка, которая может оказаться нестабильной; так, например, миссенс-мутация в кодоне 553 гена FAC ведет к замене лейцина на пролин, что делает продуцируемый геном белок неспособным компенсировать функциональный дефект в клетках больных с анемией Фанкони.
• Молчащая (нейтральная) мутация; такая мутация не сопровождается регистрируемым изменением признака и фенотипа; например, не всякая замена аминокислоты отражается на функциональной активности белка, т.е. она может оказаться нейтральной.
• Нонсенс-мутации или замены нуклеотидов, приводящие к замене информационно значимого кодона на стоп-кодон и, следовательно, к остановке бисинтеза белка или образованию укороченного белка. Эти мутации в большинстве случаев оказывают значительное повреждающее действие, и из-за преждевременного окончания трансляции они не способны к модификации, часто не защищены от действия протеолитических ферментов и быстро
деградируют. Однако на их долю приходится примерно одна треть всех моногенных заболеваний.
Примером служит нонсенс-мутация в третьем (последнем) экзоне гена бета-глобина, которая не вызывает процесс NMD (см. главу 7) в связи с отсутствием экзон-экзонного соединения и ведет к синтезу укороченного бета-глобина (он не может нормально связываться с альфа-глобином), что обусловливает доминантную форму талассемии. При нонсенс-мутации, приводящей к появлению преждевременного стоп-кодона в первом или втором экзонах, возникает рецессивная форма талассемии.
Нонсенс-мутации с появлением преждевременного стоп-кодона и мутация со сдвигом рамки считывания показаны соответственно для генов альфа-1-антитрипсина и аполипопротеина (АроВ1).
Другой пример - это нонсенс-мутация при муковисцидозе, приводящая к блоку синтеза полноразмерного белка - G542X.
• Нулевая мутация, приводящая к отсутствию синтеза функционально значимого генного продукта.
• Регуляторная мутация, затрагивающая регуляторные последовательности гена и нарушающая его экспрессию (см. главу 28).