Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 28 ИМПРИНТИНГ И БОЛЕЗНИ ИМПРИНТИНГА
Характеристика геномной памяти
Молекулярно-генетический процесс, в ходе которого модифицируются (маркируются) аллели родительских генов в локусах хромосом отцовского и материнского происхождения и обеспечивается моноаллельный характер их экспрессии, называется импринтом или эпигеномным процессом. Этот процесс также называют импринтингом или геномной памятью (см. главу 4), а сами маркированные гены называются импринтированными генами.
В настоящее время в геноме человека предполагается наличие от 100 до 200 генов, связанных с геномной памятью (уже выделено около 70 генов). Импринтированные гены локализованы в локусах большинства аутосомных хромосом (хромосомы 1-3, 5-7, 11, 13-15, 18, 19, 20 и 22) и локусах Х-хромосомы.
В указанных генных локусах в ходе их модификации возникают разные мутации: делеции (в том числе микроделеции, выявляемые при FISH-анализе), инверсии, транслокации и другие структурные изменения (см. ниже). Таким образом, механизмы геномной памяти связаны с эпигеномным воздействием на экспрессию импринтированных генов. В последние годы обсуждается ряд таких механизмов. Среди них:
• амплификация некодирующих микросателлитных последовательностей;
• инактивация крупных блоков структурного гетерохроматина;
• нарушение метилирования импринтированных генов;
• структурные нарушения регуляторных районов импринтированных генов;
• транспозиция повторяющихся нуклеотидных последовательностей.
ОБЩАЯ ХАРАКТЕРИСТИКА БОЛЕЗНЕЙ
ИМПРИНТИНГА
С нарушениями процессов геномной памяти связаны болезни импринтинга или болезни геномной памяти (БГП), объединяющие
24 нозологии, которые в зависимости от этиологической причины делятся на три класса:
• болезни генного импринтинга - 14 нозологий (58,3% всех БГП);
• болезни хромосомного импринтинга - 10 нозологий (31,7%);
• болезни ошибок импринтинга, обусловленные микроделециями в регуляторных областях импринтированных генов или центрах импринтинга (ЦИ) - две нозологии (входят в предыдущие два класса).
Болезни генного импринтинга
Этиология
При болезнях генного импринтинга наблюдается моноаллельная экспрессия в локусах хромосом одного из родителей. Причина - точковые мутации в генах, дифференцированно экспрессирующихся в зависимости от материнского и отцовского происхождения и приводящих к специфическому метилированию цитозиновых оснований в молекуле ДНК.
Эти мутации обусловливают развитие заболеваний, для которых большое значение имеют характер наследования и происхождение хромосом. К таким заболеваниям относятся:
• болезнь Гиршпрунга, обусловленная мутацией в гене RET (10q11.2); чаще всего наследуется по материнской линии;
• нейрофиброматоз Реклингаузена (тип 2) - мутация в гене SCH (22q12); наследуется по материнской линии;
• псориаз - проявляется тяжелее, если наследуется по отцовской линии;
• семейная гипертрофическая кардиомиопатия - наследуется по материнской линии;
• синдром Ангельмана (СА) - делеция критического района, находящегося в материнской хромосоме 15 (15q11.2-q13);
• синдром Вильямса - проявляется более выраженной задержкой физического и умственного развития и микроцефалией, если делеция затрагивает материнскую хромосому 7 (7q11.23);
• синдром «крика кошки» - проявляется более выраженно, если делеция захватывает отцовскую хромосому 5 (5р15.3);
• синдром Корнелии де Ланге (3q26) - проявляется более выраженно, если наследуется по материнской линии;
• синдром Прадера-Вилли (СПВ) - делеция критического района, находящегося в отцовской хромосоме 15 (15q11.2-q13);
• синдром Турета и поликистоз почек - проявляются раньше и тяжелее, если наследуются по материнской линии;
• тяжелая (злокачественная) шизофрения - проявляется более выраженно, если наследуется по отцовской линии;
• spina bifida - наследуется по материнской линии (в 2 раза чаще, чем по отцовской линии);
• эпилепсия - проявляется тяжелее, если наследуется по материнской линии.
Следует отметить, что в случае СА и СПВ особенности молекулярной организации критического района хромосомы 15 связаны с гомологической рекомбинацией, мейотической нестабильностью и объясняют высокую частоту хромосомных аберраций. В свою очередь, в семьях с повторными случаями СА и СПВ в геномах здоровых родственников обнаружены близко расположенные, но противоположно импринтированные гены, которые названы генамикандидатами этих заболеваний (см. ниже).
Кроме того, обнаружена потеря фрагментов хромосом в клетках злокачественных опухолей, расцениваемая как потеря гетерозиготности (см. главы 17 и 25). Например, при нейробластомах утрачивался локус 1р36 материнского происхождения или наблюдалась гиперамплификация гена N-Myc в локусе 2р24 отцовского происхождения.
Механизмы патогенеза
Известны два механизма патогенеза, касающиеся как самой молекулы ДНК, так и молекул белков, входящих в состав хроматина хромосом, подверженных импринтингу.
Первый механизм - это нарушение метилирования импринтированных генов. В настоящее время хорошо изучена эпигенетическая модификация или специфическое метилирование цитозиновых остатков ДНК по 5-му углеродному атому. Это единственная допустимая в физиологических условиях химическая модификация, стабильно сохраняющаяся в ряду клеточных поколений и прямо или косвенно влияющая на экспрессию генов. У человека дифференцированное метилирование родительских аллелей наблюдается, как правило, внутри или рядом с ГЦ-богатыми районами, содержащими разные типы нуклеотидных повторов, между которыми нет гомологии, а длина единицы повтора каждый раз другая, и возможно его любое расположение по отношению к импринтированному гену, промотору или регуляторному участку. По-видимому, такие повторы вовлекаются в установку процесса импринтинга (метилирования гена). Они
служат мишенями для маркирования определенного аллеля за счет организации уникальной для него вторичной структуры ДНК. Так, показано, что эти повторы создают свернутые структуры, узнаваемые гетерохроматинспецифическими белками. Например, метилирование CpG-районов изменяет структуру ДНК с образованием формы z-ДНК, что ведет к полной инактивации импринтированных генов.
Второй механизм связан с особенностями структурной организации и функционирования хроматина в локусах, в которых располагаются импринтированные гены. В пользу этого указывают результаты экспериментов по изучению времени репликации импринтированных хромосомных доменов в S-фазе митоза. В частности, до репликации в клеточном ядре наблюдаются два гибридизационных сигнала, соответствующих материнскому и отцовскому аллелям импринтированного гена, а после репликации такой сигнал приобретает сдвоенную структуру. Асинхронность вычисляется как соотношение сдвоенных и одиночных сигналов.
В случае СПВ и СА критический район соответственно на отцовской или материнской хромосоме 15 раньше реплицируется в S-фазе. При этом время репликации коррелирует с уровнем активности генов и зависит от конденсации хроматина в районах промоторов и примыкающих к нему районах.
Инактивация транскрипции сопровождается уплотнением хроматина (гетерохроматизацией), в результате чего ДНК становится менее доступной для РНК-полимеразы и факторов, необходимых для инициации транскрипции. Химическая природа модификаций гетерохроматина до сих пор не выяснена. Имеются данные о взаимосвязи процессов упаковки хроматина и метилирования ДНК. Например, показано, что транскрипционно активный хроматин имеет пониженное содержание линкерного гистона Н1, связывающего между собой нуклеосомы и упаковывающего их в фибриллы (см. главу 3). Этот гистоновый белок предпочтительно связывается с метилированными последовательностями ДНК.
Такое же сродство (аффинность) имеют негистоновые белки хроматина группы МеСР. При этом сила связывания определяется плотностью метилированных CG-динуклеотидов, а не конкретной нуклеотидной последовательностью. Некоторые из негистоновых белков хроматина подавляют транскрипцию непосредственно, например в белке МеСР2 для этого имеется специальный домен.
Таким образом, установка процессов метилирования ДНК про-
исходит только в импринтированных локусах на последующих этапах дифференцировки гамет. Основным ферментом, обеспечивающим метилирование de novo у млекопитающих, является ДНКметилтрансфераза, или Dnmt1 (см. главу 7). Вместе с тем, остается невыясненным механизм распознавания нуклеотидных последовательностей ДНК, которые должны быть по-разному метилированы в отцовском и материнском гаметогенезе. В этой связи были выделены два альтернативных способа сплайсинга 5'-экзонов гена Dnmt1, один из которых реализуется в оогенезе, другой - в сперматогенезе.
При прямом эпигеномном воздействии на экспрессию конкретного гена метилированию подвергается сам импринтированный ген. В этом процессе участвуют ДНК-связывающие белки, вызывающие гетерохроматизацию метилированного локуса. В результате доступ активаторов транскрипции к ДНК ограничивается, и экспрессия гена останавливается. При этом действие многочисленных факторов транскрипции зависит от характера метилирования ДНК. Среди этих факторов выделяют, с одной стороны, метилчувствительные активаторы и метилзависимые репрессоры (они опосредуют инактивацию метилированного гена), а с другой стороны - метилчувствительные репрессоры и метилзависимые активаторы (они обеспечивают экспрессию метилированного гена).
В случае косвенного влияния метилирования на экспрессию импринтированного гена предполагается модификация не самого гена, а другого гена - импринтора, находящегося на той же хромосоме в цис-положении. При этом функция гена-импринтора направлена на поддержание моноаллельной экспрессии одного или нескольких импринтированных локусов в пределах конкретного кластера генов.
Как оказалось, гены-импринторы часто (если не всегда) кодируют нетранслируемые РНК - это универсальный механизм конкурентной экспрессии, необходимый для поддержания в соматических клетках моноаллельной экспрессии всех импринтированных генов, включая гены, относящиеся к кластеру генов СПВ и СА.
Таким образом, общей особенностью импринтированных генов, находящихся в составе критических районов хромосом, является наличие генов, кодирующих нетранслируемую РНК. Например, для СПВ и СА обнаружено несколько таких генов (ZNF127AS, PAR5, PARSN, IPW, PAR1, C15orf2, PWCR1, UBE3A-AS). Некоторые из них синтезируются на антисмысловой цепи соответствующих генов (AS
означает «антисенс» - см. главу 20). В частности, на антисмысловой цепи белок-кодирующего гена ZNF127 транскрибируется в противоположном направлении нетранслируемая антисмысловая РНК, или ZNF127AS. Причем ген ZNF127 и его антисмысловой аналог ZNF127AS активны только на отцовской хромосоме.
Антисмысловая РНК также обнаружена для гена UBE3A, но его транскрипция (как и UBE3A-AS) происходит на разных родительских хромосомах. Так, UBE3A-AS экспрессируется на отцовской хромосоме и только в тех тканях мозга, в которых UBE3A подвержен импринтингу и активен только на материнской хромосоме. В остальных тканях, где UBE3A экспрессируется биаллельно, транскрипт UBE3A-AS не обнаруживается.
В целом можно заключить, что механизмы генного импринтинга остаются малоизученными.
Болезни хромосомного импринтинга
Этиология
Для болезней хромосомного импринтинга характерна однородительская дисомия (ОРД) или наличие двух копий хромосомы либо отцовского, либо материнского происхождения (см. главу 4). Термины: «однородительская дисомия» или «изодисомия» впервые предложены Э. Энжелом в 1980 г. для обозначения одного из типов анеуплоидии в гаметах млекопитающих. Эти термины указывают на наличие у диплоидного потомства двух локусов, полученных от одного и того же родителя, тогда как в норме наследуется только по одному локусу от каждого родителя. Примерами таких заболеваний служат:
• ОРД по материнской хромосоме 2, сопровождающаяся дизэмбриогенезом и задержкой развития;
• ОРД по длинному плечу отцовской хромосомы 6 (6q23-q24), сопровождающаяся неонатальным сахарным диабетом;
• ОРД по длинному плечу материнской хромосомы 7 при муковисцидозе;
• ОРД по короткому плечу материнской хромосомы 7 в области гена GRB10 при синдроме Сильвера-Рассела;
• частичная трисомия отцовской хромосомы 11 (11р15.5) или сбалансированная транслокация с точками разрывов в материнской хромосоме 11 при синдроме Беквитта-Видемана.
В свою очередь, ОРД по материнской хромосоме 14 обусловливает: акромикрию, гипотонию, задержку физического, моторного
и умственного развития, сколиоз, умеренно выраженный черепнолицевой дисморфизм, а ОРД по этой же хромосоме отцовского происхождения сопровождается скелетно-мышечными аномалиями и тяжелой УО.
Кроме того, материнская и отцовская ОРД по хромосоме 15 ведет к СПВ и СА соответственно (см. ниже). ОРД по длинному плечу отцовской хромосомы 20 в области гена GNAS1 ведет к псевдогипопаратиреоидизму. ОРД по Х-хромосоме отцовского происхождения сопровождается дисгенезией гонад и низким ростом (но это только единственное наблюдение).
Механизмы патогенеза
Механизмы хромосомного импринтинга - это формирование ОРД, наличие в кариотипе двух хромосом одной пары, полученных от одного из родителей: либо отца, либо матери, а не двух родителей, как это происходит в норме.
Известны три механизма формирования ОРД.
Первый механизм - это комплементация гамет, случайное слияние двух гамет с разнонаправленной анеуплоидией по одной и той же хромосоме. Например, оплодотворение диплоидной по хромосоме 15 яйцеклетки нуллисомным по хромосоме 15 сперматозоидом ведет
к СПВ.
Второй механизм - это коррекция трисомии до дисомии, т.е. оплодотворение одной дисомной гаметы одной гаплоидной (нормальной) гаметой и формирование трисомной зиготы с элиминацией из нее (в последующих делениях дробления) хромосомы, находившейся в нормальной гамете. Доказательством служат два случая трисомии по материнской хромосоме 15, выявленных при пренатальных исследованиях ворсин хориона. После рождения у обоих детей наблюдался СПВ, подтвержденный анализом ДНК материнского происхождения.
Третий механизм - это коррекция моносомии до дисомии при оплодотворении нормальной и нуллисомной гамет с формированием моносомной зиготы и последующей дупликацией в ней моносомной хромосомы.
На основе этих механизмов формируется ОРД двух типов.
1. Гетеродисомия или наследование двух разных гомологичных хромосом от одного из родителей. Этот тип ОРД обусловлен либо комплементацией гамет, либо коррекцией трисомии до дисомии и наблюдается у большинства больных с СПВ в результате нерасхожде-
ния материнских хромосом в первом делении мейоза. Вероятность нерасхождения, по-видимому, зависит от возраста матери, превышающего 30 лет на момент рождения ребенка.
2. Изодисомия или наследование двух копий одной хромосомы от одного из родителей.
Болезни ошибок импринтинга
Этиология
Болезни ошибок импринтинга - результат микроделеций в регуляторных областях импринтированных генов, или центрах импринтинга (ЦИ).
Первое сообщение о таких болезнях относится к 1993 г., когда у больных с СПВ и СА не было отмечено типичных делеций критического района хромосомы 15 (15q11.2-q13), но также (как и в случае ОРД) наблюдался одинаковый характер метилирования обеих родительских копий этого критического района. В дальнейшем выяснено, что в одном случае была одинаковой и соответствовала характеру метилирования на обеих родительских хромосомах экспрессия импринтированных генов, локализованных в критическом районе и характерных для СПВ (гены: SNRPN, ZNF127, PAR1, PAR5, IPW,
PWCR1, C15orf2).
Молекулярно-генетические исследования показали, что ошибки импринтинга часто сопровождаются микроделециями ЦИ. Сами делеции в ЦИ не связаны с клиническими проявлениями, но могут выявляться у здоровых родителей и других родственников больных пробандов. Анализ родословных в таких семьях свидетельствует, что делеции ЦИ наследуются (без клинических проявлений) в ряду поколений у лиц одного и того же пола. Таким образом, к заболеваниям этой группы относятся те же два синдрома (СПВ и СА), однако вызваны они другими генетическими причинами, т.е. это их генетические варианты.
Механизмы патогенеза
При генетических вариантах СПВ и СА показано, что на одной из родительских хромосом изменен на обратный или инвертирован характер метилирования (и экспрессии) генов. Это объясняется переключением (эпигеномной заменой) генов хромосомы 15 в гаметогенезе одного из родителей, возникшим в результате ошибок в ЦИ. Предложены две модели такой замены в гаметогенезе:
• первая модель - эпигеномное переключение только в той гомологичной хромосоме, которая получена от родителя противоположного пола, тогда как вторую гомологичную хромосому модификация не затрагивает;
• вторая модель - это предварительное «стирание» существующей геномной «памяти» на обеих родительских хромосомах с последующим установлением «памяти», соответствующей данному полу; эта модель наиболее предпочтительна.
ОТДЕЛЬНЫЕ НОЗОЛОГИИ
Синдром Прадера-Вилли
СПВ впервые описан в 1956 г. Встречается с частотой 1:10-20 тыс. Как правило, возникает спорадически, хотя известны семейные формы.
Основные признаки: черепно-лицевой дисморфизм (долихоцефалия, миндалевидный разрез глазных щелей, гипертелоризм, эпикант, микрогнатия, «рыбий рот», высокое нёбо, диспластичные ушные раковины, акромикрия, нарушения дерматоглифики), низкий рост, ожирение, мышечная гипотония, гипогонадизм, гипопигментация, задержка умственного развития.
Продолжительность жизни больных не превышает 25-30 лет.
Этиология и механизмы патогенеза СПВ и «альтернативного» ему по генетической природе СА были изучены в последние годы. Выделены три генетические причины СПВ
Первая (основная) причина - это делеция критического района отцовской хромосомы 15 (15q11.2-q13) - 95% случаев генного импринтинга. Столь высокая частота этой мутации обусловлена мейотической нестабильностью указанного хромосомного района. Показано: делеции (как и в случае СА) могут возникать в гаметогенезе в результате внутриили межхромосомных перестроек. Причем внутрихромосомные делеции происходят путем образования большой петли, имеющей 3-4 млн н.п.
Рассмотрим молекулярную организацию критического района хромосомы 15. На рис. 76 приведена молекулярная структура этого критического района хромосомы 15.
На рис. 76 обозначены два кластера точек разрывов при делециях, картированных проксимальнее гена ZNF127, локализованного в этом критическом районе.
Здесь же расположены три белок-кодирующих гена (NDN, MAGEL2, SNRPN) и 7 генов, кодирущих нетранслируемые РНК (ZNF127AS, PAR5, PARSN, IPW, PAAR1, C15orf2, PWCR1), - все гены в этих локусах при СПВ экспрессируются моноаллельно на отцовской хромосоме. Для трех из указанных генов (гены NDN, ZNF127, SNRPN) показана тканеспецифическая экспрессия в нейронах головного мозга.
Неимпринтированная область хромосомы 15 находится в дистальной части критического района 15q11.2-q13. Она включает несколько биаллельно функционирующих генов, в том числе локус Р (отвечает за пигментацию кожи и глаз), гены GABRB3, GABRA3 и GABRG3 (кодируют субъединицы рецептора гамма-аминомасляной кислоты, или ГАМК-А), и ген HERC2. Последний ген филогенетически относится к самым древним генам: он считается родоначальником семейства END-псевдогенов, возникшего в результате нескольких последовательных дупликаций. Считается, что END-псевдогены способствуют процессам гомологической рекомбинации и неравного кроссинговера между повторяющимися последовательностями ДНК в критическом районе 15q11.2-q13 (см. главу 3).
К первопричинам СПВ, обусловливающим только 5% случаев генного импринтинга, относятся: инвертированная дупликация (тетрасомия), простая дупликация (трисомия), несбалансированная транслокация (моносомия), а также сбалансированная транслокация и инверсия критического района отцовской хромосомы 15 (15q11.2-q13).
Вторая причина СПВ - это ОРД по материнской хромосоме 15, обусловливающая 25% случаев болезни (см. выше).
• Третья причина СПВ - это ошибки импринтинга (см. выше). Генами-кандидатами СПВ являются следующие. Ген SNURF-SNRPN - состоит из 10 экзонов и имеет необычную бицистронную структуру. Его мРНК-транскрипт содержит две неперекрывающиеся рамки считывания для двух разных белков: первый белок SNURF (по-видимому, участвует в регуляции экспрессии импринтированных генов на отцовской хромосоме), второй белок N или SmN (является компонентом малых ядерных рибонуклеопротеинов - мяРНП). Анализ экспрессии двойного транскрипта гена SNURF-SNRPN показал его присутствие во всех тканях. Кроме двойного транскрипта, в мышечной и почечной тканях была обнаружена укороченная мРНК, соответствующая 1-34 экзонам этого гена.
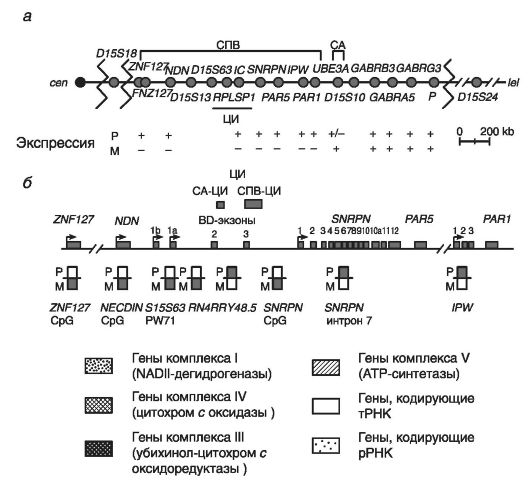 Рис. 76. Молекулярная структура критического хромосомного района 15q11.2- q13 (по Genomic Imprinting, 1997):
Рис. 76. Молекулярная структура критического хромосомного района 15q11.2- q13 (по Genomic Imprinting, 1997):
а - генетическая карта критического района 15(q11-q13), в которой положение генов обозначено кружками, точки разрывов хромосомы (при стандартных делециях) обозначены зигзагами; транскрипция отцовского (Р) и материнского (М) аллелей обозначена знаками: «+» - экспрессируется, «-» - не экспрессируется; б - характер аллельного метилирования локусов критического района 15 (q11-q13).
Экзоны генов показаны прямоугольниками, сайты инициации транскрипции каждого гена - стрелками. ЦИ имеет два альтернативных сайта инициации транскрипции. Отсутствие метилирования показано незакрашенными прямоугольниками, наличие метилирования - закрашенными прямоугольниками
• ZNF127 - в составе этого гена выделены две формы: собственно ZNF127 и антисенс ZNF127AS; обе формы транскрибируются в противоположных направлениях только на отцовской хромосоме. Ген содержит ряд мотивов типа «цинковые пальцы« (см. главу 8). Экспрессия гена ZNF127 показана во всех тканях с максимумом проявления в семенниках. Высокий уровень транскрипта ZNF127AS также показан в тканях мозга и легких; в других тканях его почти нет. Предполагается, что потеря экспрессии этих генов в ряде тканей организма обусловливает развитие таких симптомов, как бесплодие, гипогонадизм, неврологические и психические расстройства, ожирение.
• NDN - некдиновый ген. Он не содержит интронов и кодирует белок, угнетающий рост нейронов (подобно белку RBI при ретинобластоме, являющемуся супрессором опухолевого роста). Данный белок тормозит переход G1-cтадии клеточного цикла в стадию S. При этом некдин максимально экспрессируется в нейронах гипоталамуса, контролирующего процессы развития ожирения и гипогонадизма у больных.
• Локус Р - располагается в дистальной части критического района хромосомы 15, экспрессируется биаллельно, контролирует пигментацию кожи и радужки глаз. Потеря двух аллелей локуса Р ведет к альбинизму и гипопигментации кожи (у гетерозигот).
Синдром Ангельмана
СА впервые описан в 1965 г. под названием синдрома «счастливой куклы». Основные признаки: микробрахицефалия с уплощенным затылком, большая нижняя челюсть, открытый рот, выступающий язык, макростомия, редкие зубы, гипопигментация.
У больных детей наблюдается задержка психомоторного развития, невозможность обучения, явления атаксии, гиперкинезия, мышечная гипотония, судорожная готовность и гиперрефлексия. Постоянны приступы неконтролируемого смеха и хлопания в ладоши.
В основе синдрома лежат те же причины, что и при СПВ, однако затронута не отцовская, а материнская хромосома.
Первая причина - варианты делеции критического района материнской хромосомы 15 (15q11.2-q13), возникающие в гаметогенезе в результате внутри- и межхромосомных перестроек (см. выше).
Вторая причина - ОРД отцовской хромосомы 15, возникающая при нерасхождении хромосом в оогенезе и зависящая от возраста матери на момент рождения ребенка (см. выше).
Третья причина - ошибки в ЦИ. Генами-кандидатами СА являются следующие.
• Ген UBE3A - основной ген-кандидат, мутации в котором обусловливают развитие 20% случаев СА. Этот ген кодирует фермент У6-АР убиквитин-протеинлигазу, экспрессия которой обнаружена во всех тканях. Кодируемый белок служит коактиватором для рецепторов стероидных гормонов. В ряде структур головного мозга ген UBE3A активен только в материнской хромосоме, что может объяснить развитие атаксии и тремора дефицитом кодируемого геном фермента в клетках Пуркинье, а развитие эпилептических припадков и невозможность обучения ребенка - отсутствием экспрессии этого гена в нейронах гиппокампа. Ген UBE3A транскрибируется с нескольких промоторов и имеет 7 нетранслируемых экзонов со стороны 5'-конца. Среди мутаций данного гена преобладают мутации со сдвигом рамки считывания и нонсенсмутации. Описаны также инверсии с точками разрывов внутри
гена UBE3A.
• Ген АТР1 С - кодирует аминофосфолипидтранспортирующую АТРазу. Экспрессируется преимущественно с материнского аллеля в фибробластах кожи и в разных структурах мозга. Передает сигналы в ЦНС и поддерживает контакты между клеточными мембранами. Отсутствие продуктов экспрессии этого гена в мозге больных СА ведет к тяжелому детскому аутизму.
• Ген GABRB3 является одним из генов кластера ГАМКА- рецепторных генов, расположенных между геном UBE3A и локусом Р. Эти рецепторные гены попадают в область наиболее частых делеций критического района. Ген GABRB3 кодирует бета-3-субъединицу ГАМКА-рецептора. Поэтому симптоматика СА (моторные нарушения, нарушения памяти, неспособность к обучению, судорожные припадки) может быть обусловлена гаплонедостаточностью данного гена.
Синдром Беквитта-Видемана
Синдром Беквитта-Видемана (СБВ) относится к распространенным наследственным заболеваниям с частотой в популяции 1:10- 12 тыс. Тип наследования болезни аутосомно-доминантный с неполной пенетрантностью и варьирующей экспрессивностью.
Примерно 15% всех случаев СБВ расцениваются как семейные формы.
Основные клинические признаки СБВ: гигантизм (масса тела при рождении свыше 3900 г), черепно-лицевой дисморфизм (долихоцефалия, гипоплазия верхней челюсти и средней трети лица, прогнатизм, макроглоссия, вертикальные насечки на мочках и небольшие полулунные ямочки на задней поверхности завитков ушных раковин), гемангиомы на лбу, пигментные пятна на коже затылка и лица, пупочная грыжа. У всех больных наблюдается вицеромегалия внутренних органов или увеличение размеров гонад, надпочечников, печени, поджелудочной железы, предстательной железы, почек и селезенки. Предполагается, что вицеромегалия связана с сохранением паракринной и эндокринной функций частью эмбриональных клеток на пренатальном и постнатальном этапах онтогенеза. У 15% больных с СБВ выявляется УО. У многих больных наблюдается повышенная предрасположенность к развитию онкогенных заболеваний, например частота нефробластомы достигает 59%, карциномы надпочечников - 15%, гепатобластомы - 2%.
Важный диагностический признак - гемигипертрофия, наблюдаемая в 12,5% случаев СБВ, а у больных с опухолями этот признак проявляется в каждом втором случае.
Критической областью для СБВ является терминальная часть короткого плеча хромосомы 11 (11р15.5). В этой области расположен кластер импринтированных генов, перемешанных с неимпринтированными генами (рис. 77).
Критическая область включает в левой части (по отношению к центромере) 6 генов и ограничена геном NAP2; в правой части - 7 генов и ограничена геном L 23.
Кластер генов критической области хромосомы 11(11р15.5) начинается с первого гена - NAP2, биаллельно экспрессирующегося в эмбриональных и зрелых тканях.
Второй ген - IPL/TSSC3 - преимущественно экспрессируется с материнского аллеля и ограниченно - с отцовского аллеля; наиболее активен в плаценте, легких, миокарде, печени, поджелудочной железе и почках. В клетках мозга взрослых людей и лимфоцитах крови он экспрессируется биаллельно. При развитии опухолей головного мозга ген IPL/TSSC3, по-видимому, выполняет роль гена-супрессора материнского аллеля.
Третий ген - IMPTI/TSSC5 - кодирует белок мембранного переносчика, контролирующего полиспецифический транспорт и множественную лекарственную устойчивость.
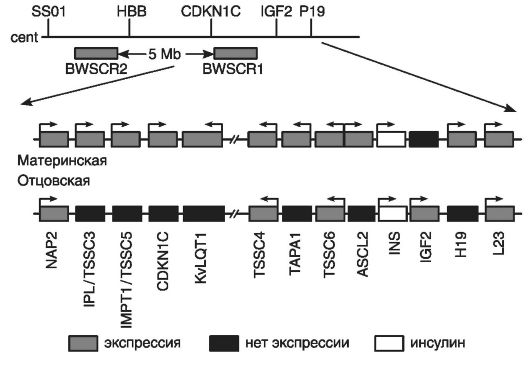 Рис. 77. Схема молекулярной организации критической области 11р15.5 (по Немцовой М.В. и Залетаеву Д.В., 2004)
Рис. 77. Схема молекулярной организации критической области 11р15.5 (по Немцовой М.В. и Залетаеву Д.В., 2004)
Кроме того, точковые мутации этого гена и потеря его материнской копии были идентифицированы при раке грудной железы, нефробластоме и рабдомиосаркоме.
С антисмысловой цепи ДНК транскрибируется другой ген этого локуса - ген IMPTI-AS, имеющий множество сайтов инициации транскрипции, но экспрессирующий только два транскрипта. Считается, что этот ген не транслируется и поэтому не относится к импринтированным генам.
Четвертый ген - CDKN1C - также известен как ген р57К1Р2. Он кодирует ингибитор циклинзависимой киназы, относящийся к CIP-зависимым регуляторам клеточного цикла. Его гиперэкспрессия останавливает клеточный цикл в предсинтетической стадии.
В гене обнаружены точковые мутации, в связи с чем он считается реальным геном-кандидатом синдрома СБВ. Мутации гена р57 KIP
встречаются в 40% семейных и 5% спорадических случаев СВБ. В самом гене суммарная частота мутаций составляет 10-15%.
В клетках таких опухолей, как карцинома надпочечников, нефробластома и рабдомиосаркома, выявляется гиперэкспрессия гена р57 KIP с материнского аллеля.
Роль пятого гена - KvLQT1 - в этиологии СБВ точно не определена, однако считается, что он участвует в процессе импринтинга при этом заболевании. Данный ген кодирует белок, формирующий заслонку калиевых каналов. Он экспрессируется биаллельно только в сердечной мышце; его точковые мутации сопровождаются доминантно наследуемым дефектом проводимости сердечной мышцы или LQT-синдромом (удлиненный Q-Т-интервал и желудочковая аритмия), а также аутосомно-рецессивным синдромом Джервила-Ланге-Нильсона, сочетающимся с аналогичными нарушениями проводимости миокарда и глухотой. Ген этого синдрома (KvLQT1) экспрессируется с материнского аллеля и обнаруживается в большинстве тканей организма, но он почти не экспрессируется в мозге и скелетных мышцах. В интроне гена KvLQT1 обнаружен ген LIT1 (см. ситуацию «ген в гене», глава 1), транскрибирующийся с антисмысловой цепи, но только с отцовской хромосомы.
При СБВ установлена биаллельная экспрессия этого гена, тогда как при нефробластоме наблюдается лишь моноаллельная его экспрессия.
Шестой ген TSSC4 и восьмой ген TSSC6 не относятся к импринтированным генам СБВ, но они кодируют белки с опухоль-подавляющей функцией, например белки с предполагаемой опухоль-подавляющей функцией в случае рабдомиосаркомы. Между этими генами расположен седьмой ген - ТАРА1 (CD81), кодирующий интегральный мембранный белок, обнаруженный в разных типах клеток. Этот ген участвует в развитии на ранних этапах Т-лимфоцитов. Он экспрессируется только с материнской хромосомы.
Девятый ген - ASL2, также экспрессирующийся с материнского аллеля. Его белковый продукт является фактором транскрипции, содержащим мотив: спираль-петля-спираль (см. главу 8). Этот белок участвует в развитии трофоэктодермы.
Десятый ген - INS (ген инсулина) имеет биаллельную экспрессию, а находящийся рядом с ним инсулиноподобный фактор роста IGF2 кодирует фетальный фактор роста.
Одиннадцатый ген или IGF2-фактор экспрессируется только с отцовского аллеля, может быть митогеном для всех типов клеток, но у человека он специфически модулирует рост и дифференцировку мышечных клеток. Нарушение экспрессии IGF2 обусловливает органомегалию при СВБ. Биаллельную экспрессию IGF2 обнаруживают в нефробластомах, карциномах коры надпочечников, рабдомиосаркомах, других эмбриональных опухолях, встречающихся при СБВ.
Двенадцатый ген Н19 кодирует нетранслируемую РНК с неустановленной функцией. Этот ген тесно сцеплен с предыдущим геном IGF2; их экспрессия регулируется общим энхансером, расположенным в 3'-конце гена Н19, который имеет противоположный импринтинг с материнской хромосомы.
Тринадцатый ген L23 кодирует рибосомоподобный белок, экспрессирующийся биаллельно в эмбриональных и зрелых тканях. Этим геном ограничена дистальная часть критической области 11р15.5.
К генетическим вариантам СБВ относятся:
• частичная трисомия дистальной части короткого плеча хромосомы 11 (результат аномальной сегрегации в мейозе структурных перестроек отцовского происхождения);
• спорадические дупликации критической области хромосомы 11 (11р15.5); их частота (совместно с частичной трисомией) - около 2%;
• однородительская отцовская дисомия (результат постзиготической митотической нестабильности - рекомбинации с высокой долей мозаицизма);
• сбалансированная транслокация между хромосомами 11 и 22, унаследованная от матери.
Помимо этих генетических вариантов, идентифицированы точковые мутации в одном из двух генов-кандидатов СБВ - гене CDKN1C, c которым связывают 10-15% всех мутаций.
Другой ген-кандидат СБВ - IGF2 не имел точковых мутаций, но его экспрессия у больных проходила либо только с материнской хромосомы, либо наблюдалась ОРД по отцовской хромосоме 11, т.е. в данном случае имела место потеря импринтинга (ПИ).
Описан больной с унаследованной от матери сбалансированной инверсией короткого плеча хромосомы 11 с точками разрывов в области СБВ-ХР2 (см. ниже), что также объяснялось ПИ при нормальном эпигенотипе Н19.
Таким образом, определенная часть спорадических случаев СВБ не связана с указанными выше генетическими вариантами, а объяс-
няется ПИ. В общей сложности, структурные аномалии хромосомы 11 выявляются у 2% больных с СБВ. Первым по частоте повреждаемости является район локализации гена KvLQT1, обозначаемый как СБВ-ХР1; два других района - СБВ-ХР2 и СБВ-ХР3 - расположены ближе к центромере на 5 и 7 млн н.п. соответственно. В районе СБВ-ХР2 выделены два гена, содержащих мотив «цинковые пальцы» (см. главу 8). Это гены ZNF214 и ZNF215, второй ген имеет пять альтернативных и один антисмысловой транскрипты, в норме экспрессирующиеся только с материнского аллеля. Структура хромосомного района СБВ-ХР3 пока не изучена.
Подходы к диагностике
Непосредственными причинами болезней импринтинга являются не только генетические нарушения (точковые, структурные и хромосомные мутации), но и функциональные нарушения, связанные с взаимодействием между импринтированными и неимпринтированными генами в критических областях хромосом.
В случае мутаций диагностическими тестами являются молекулярногенетические и цитогенетические методы исследования наследственного материала. Например, при СПВ и СА критическим районом служат локусы 15q11.2-q13, а при СБВ - локус 11р15.5. В случае функциональных нарушений с диагностической целью исследуется экспрессия импринтированных генов, и ее результаты строго соотносятся с характером метилирования. При СПВ и СА происходит инактивация генов за счет либо отцовского, либо материнского гиперметилирования двух локусов: D15S63 (ген PW71) и D16S9 (ген ZNF127), а также инактивация промоторной области гена SNRPN. При СБВ гиперметилирование касается импринтированных генов IGF2 и LIT1, которые моноаллельно экспрессируются (метилируются) с отцовского или материнского аллелей.
В основе тестов для диагностики СПВ, СА и СБВ лежит анализ аллельного метилирования критических областей хромосом. Применяются три метода.
• Анализ аллельного метилирования локусов импринтированных генов. Используя метод, учитывают, что при СПВ независимо от его причины присутствуют только метилированные аллели, а при СА - только неметилированные аллели локусов критических областей хромосом.
• Бисульфатная модификация ДНК и метилспецифическая ПЦР. Метод основан на модификации геномной ДНК бисульфитом
• натрия, позволяющим осуществить химическое превращение неметилированных остатков цитозина в урацил, в то время как 5-метилцитозин остается неизменным. По измененному нуклеотидному составу судят о состоянии метилирования каждого цитозинового остатка на исследуемом фрагменте ДНК. В частности, при СПВ образуется продукт той длины, которая соответствует метилированному материнскому аллелю, а при СА обнаруживается неметилированный отцовский аллель. Для анализа применяется набор из трех праймеров (общий, материнский и отцовский). Материнский и отцовский праймеры попарно с общим праймером дают продукты амплификации, равные 313 и 221 н.п. соответственно. Разновидностью этого метода является МС-ПЦР, используемая для скрининговой диагностики СПВ и СА, но без определения их генетических причин. Анализ микросателлитного полиморфизма локусов критического района 15q11.2-q13. Метод позволяет идентифицировать делеции и ОРД на основе анализа микросателлитного полиморфизма в локусах D15S11 (входит в область наименьшего перекрывания всех делеций при СПВ), D15S113 (расположен внутри области перекрывания всех делеций при СА) и GABRB3 (характеризуется большим количеством аллелей и гетерозиготностью). Обычно формируются делеции стандартной протяженности (4 млн н.п.), перекрывающие все три локуса. Анализ этих трех локусов можно проводить методом мультиплексной ПЦР с тремя парами праймеров. При отсутствии делеций и ОРД при диагнозе СПВ и СА, подтвержденном на основе анализа метилирования, можно предполагать наличие делеции или мутации в ЦИ. В случае СА (если результат анализа метилирования отрицательный) причиной наследственной природы заболевания может быть мутация в гене-кандидате UBE3A, что имеет большое значение для МГК семьи и прогноза рождения здорового потомства.
Подходы к лечению
Подходы к радикальной этиологической и патогенетической терапии болезней импринтинга не разработаны, поэтому их лечение только симптоматическое.