Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 27 ЭКСПАНСИЯ НУКЛЕОТИДНЫХ ПОВТОРОВ ДЕЗОКСИРИБОНУКЛЕИНОВОЙ КСИЛОТЫ И БОЛЕЗНИ ЭКСПАНСИИ
Общие данные
В хромосомной ДНК выделяют два класса последовательностей нуклеотидов (см. главу 2): уникальные последовательности или экзоны (они реплицируются и транскрибируются), и повторяющиеся последовательности или интроны (они реплицируются, но, как правило, не транскрибируются).
Последовательности ДНК разнообразны по спектру: ди-, три-, тетра-, пента-, гекса- и полинуклеотидные. При этом ди-, тетра- и пентануклеотидные последовательности редко обнаруживаются в экзонах, тогда как три- и гексануклеотидные последовательности в них выявляются часто, что объясняется способностью первых и неспособностью вторых к сдвигу рамки считывания в ходе транскрипции. Установлена прямая связь между длиной (размером) последовательности, ее стабильностью и изменчивостью в популяции. При этом размер последовательности важный, но не единственный фактор, влияющий на ее стабильность.
В свою очередь, стабильность экзонов и интронов связана со степенью их «чистоты». Например, информативность последовательности со вставками нуклеотидов или маркерами, нарушающими ее непрерывность, значительно ниже, чем у последовательностей без таких вставок. Потеря вставок в экзонах - это, как правило, результат формирования делеции или однонуклеотидной замены (например, аденина на цитозин), что ведет к образованию неканонической (не «уотсон-криковской») пары азотистых оснований.
Наряду с информативностью последовательностей ДНК показана ассоциативная связь между их нестабильностью и степенью совершенства. Так, нормальные аллели гена SCA1 (ген спиноцеребеллярной атаксии, тип I) имеют большую длину повтора CAG, содержат 1-3 вставки повтора САТ, тогда как аллели с экспансией копий повторов характеризуются непрерывными трактами СAG.
Вместе с тем, было показано, что наряду с уникальными последовательностями транскрипционной активностью обладает небольшая часть повторяющихся последовательностей нуклеотидов (их не менее 1%) - они оказались способными кодировать рРНК, тРНК и даже гистоны (см. главу 1). Например, такая способность наблюдалась у ряда кодирующих и некодирующих последовательностей ДНК, если в них сверх нормы возрастало количество копий (одинаковых повторов), состоящих из три-, тетрануклеотидов ДНК и более, которые экспрессировались в протяженные цепи-тракты полипептидов, сформированных из одинаковых последовательностей аминокислот: аланина, глицина, глутаминовой кислоты, лейцина, пролина или серина. В совокупности такие тракты занимают 75% общего объема всех аминокислотных трактов, представляющих собой последовательности из пяти и более идентичных аминокислотных остатков, которые определяются в клетках и тканях как белки с общей длиной около 400 аминокислот - это одна из универсальных характеристик феномена экспансии (см. ниже). Впервые такие протяженные аминокислотные тракты-повторы были описаны V. Verkerk и соавт. в 1991 г. при синдроме Мартина-Белл.
Для молекулярной медицины особенно значимыми оказались тринуклеотидные повторы ДНК, экспрессирующиеся в полиглутаминовые и полиаланиновые тракты аминокислотных остатков, обусловливающие развитие 18 нозологий, а также микросателлитные повторы, экспрессирующиеся в аминокислотные последовательности полипептидов, обусловливающих развитие еще 9 нозологий (всего 27 нозологий).
Результатом анализа этих необычных данных стало открытие нового биологического феномена - экспансии нуклеотидных повторов, роста числа их копий в геноме человека, а также нового класса болезней экспансии.
ФЕНОМЕН ЭКСПАНСИИ НУКЛЕОТИДНЫХ
ПОВТОРОВ И ЕГО ОСОБЕННОСТИ
Феномен экспансии нуклеотидных повторов имеет ряд характерных особенностей.
Универсальные характеристики
1. Кодирующие и некодирующие нуклеотидные повторы обнаруживаются в генах нормальных индивидов, они полиморфны по числу содержащихся в них копий повторов, что объясняет приобретение токсических функций молекулами агрегатов моноаминокислотных трактов, сформировавшихся в ходе экспансии.
2. Нуклеотидные повторы располагаются в разных областях генов, в том числе в промоторной области (гены FMR1 и FMR2), транскрибируемой, но нетранслируемой части гена (ген DMPK), экзонах (гены нейродегенеративных заболеваний) и интронах (ген FRDA);
3. Отмечается наличие порога предрасположенности к экспансии, после которого начинается генетическая и фенотипическая нестабильность (до порога - норма или состояние премутации, после порога - полная или динамическая мутация).
4. В случае превышения порога предрасположенности к экспансии наблюдается (одномоментное, скачкообразное) появление дополнительных копий повтора.
5. Не все нуклеотидные повторы предрасположены к нестабильности, которая зависит от нуклеотидного состава повторов, состояния кариотипа (хромосомного окружения) и биохимического статуса клетки. Зависимость стабильности повторов от их нуклеотидного состава связана со способностью повторов к образованию вторичных структур (повторы типа CNGn, где n - число повторов), которые образуют «несовершенные шпильки», стабилизированные взаимодействием как канонических пар оснований (АТ- и ГЦ-пары), так и неканонических пар оснований (другие сочетания двух азотистых оснований).
Если шпильки не образуются, то предрасположенность к экспансии резко снижается. По-видимому, здесь играет роль некий энергетический барьер, необходимый для образования шпилек. Вероятно, что этот энергетический барьер тесно связан с функцией тканевого (митохондриального) дыхания (см. главу 26).
В связи с такой характеристикой феномена экспансии был сделан вывод, что стабильность структур, образованных разными повторами, хотя и различается, но соответствует определенной шкале. В случае повтора CGG такая шкала выглядит как уравнение:
CGG > CCG = CTG > CAG
В этом уравнении первый повтор обладает наибольшим потенциалом для образования шпилек и демонстрирует наименьший для
всех известных тринуклеотидных повторов уровень репликативного скольжения (см. ниже).
6. Последовательность из 5 идентичных аминокислотных остатков и более определяется как полиаминокислотный тракт или белок длиной 400 аминокислот.
В протеоме человека содержатся 20% белков, в составе которых обнаруживается по крайней мере один полиаминокислотный тракт. В таких трактах-повторах почти не встречаются аминокислоты аргинин, валин, изолейцин, метионин, тирозин, фенилаланин и цистеин. В то же время лейцин встречается в 19% белков с одним полиаминокислотным трактом. При этом исключительным правилом для человека является вхождение полилейциновых повторов в сигнальные пептиды (см. главу 8).
7. Процесс экспансии копий нуклеотидных повторов связан с нарушениями систем репарации ДНК. Однако механизмы, приводящие к нестабильности повторов, и в том и в другом случае различаются.
8. Экспансия копий кодирующих и некодирующих нуклеотидных повторов сопровождается феноменом антиципации - это увеличение тяжести течения болезни по мере роста числа копий повторов. Одновременно наблюдается прямая корреляция между ростом числа копий повторов и временем манифестации заболевания.
Уникальные характеристики
1. Наличие одинаковых нуклеотидных повторов при разных нозологиях, проявляющихся разными патологическими фенотипами, обусловлено мутациями в разных генах.
Всего 9 болезней экспансии кодирующих повторов связаны с полиглутаминовыми трактами, образовавшимися в результате экспансии повтора CAG (кодирует глутамин в транслируемых областях генов). Включение таких трактов в структуру белков обусловливает их неправильное функционирование в ходе ДНК-белковых и белокбелковых взаимодействий и развитие патологических фенотипов.
Две болезни полиглутаминовых трактов обусловлены экспансией копий повтора СGG (гены FraXA и FraXE).
В свою очередь, из 9 болезней экспансии, связанных с образованием полиаланиновых трактов, четыре заболевания обусловлены полиморфизмом длин повторов GСА и GCT (гены: РАВРМ, RUNX2, ZIC2 и HOXA13). В редких случаях такие полиморфизмы связаны с дупликациями или делециями целых блоков смешанных повторов (GСN)n.
2. Кодирующие и некодирующие нуклеотидные повторы в случае болезней экспансии наследуются, как правило, по доминантному типу. Чтобы вызвать патологический фенотип, достаточно мутации в одном аллеле гена. Исключением из этого правила является прогрессирующая миоклонус-эпилепсия, обусловленная некодирующим повтором и наследуемая по рецессивному типу (см. MERRF-синдром в главе 26).
3. При болезнях экспансии, обусловленных кодирующими повторами ДНК, их копии в ряде случаев являются функционально полноценными кодонами генетического кода. Вместе с тем, они могут кодировать аминокислоты, из которых могут быть сформированы моноаминокислотные тракты. При этом изменение числа копий таких кодонов не ведет к сдвигу рамки считывания в ходе транскрипции.
4. Мутантные аллели при отдельных болезнях экспансии сцеплены с ломкими сайтами хромосом, называемыми маркерными сайтами, например, при синдроме Мартина-Белл - это сайты FraXF, Fra16A, Fra16B и Fra11. Такая особенность свидетельствует в пользу «эффекта родоначальника» при возникновении и сохранении мутаций в популяции.
5. По мере роста числа повторов мутантные аллели проявляют митотическую и мейотическую нестабильность.
6. Митотическая нестабильность проявляется тканевым (соматическим) мозаицизмом. Она обусловлена мутантными аллелями с разным числом копий повторов, проявляющими свое действие в одной или сразу нескольких тканях, например, при миотонической дистрофии и синдромах FraXA и FraXE. В частности, в первом случае соматический мозаицизм обнаружен у 13-16-недельных плодов, что выразилось у них в разной длине повторов, сохранявшейся в течение всей жизни.
7. Мейотическая нестабильность зависит от пути передачи мутантного аллеля по отцовской или материнской линии. В первом случае она проявляется быстрее (например, при нейродегенеративных заболеваниях), чем во втором случае (например, при миотонической дистрофии).
Молекулярные механизмы и модели экспансии
В последние годы предложен ряд моделей, характеризующих молекулярные механизмы феномена экспансии: репликативная, комбинативная и рекомбинационная модели.
Репликативная модель
Репликативная модель приведена на рис. 73.
Эта модель основана на модели репликации ДНК с участием фрагментов Оказаки (см. главу 2). Она свидетельствует, что динамическая мутация может быть результатом нарушения функционирования ДНК-полимеразы во время репликации, предшествующей митозу и мейозу. Фрагменты Оказаки имеют среднюю длину 150-200 н.п. Они участвуют в синтезе отстающей (запаздывающей) цепи ДНК, тогда как на основной (лидирующей) цепи синтез идет непрерывно. В данном случае при наличии на З'-конце протяженной монотонной последовательности будет наблюдаться скольжение вновь синтезированной нити относительно матричной нити ДНК.
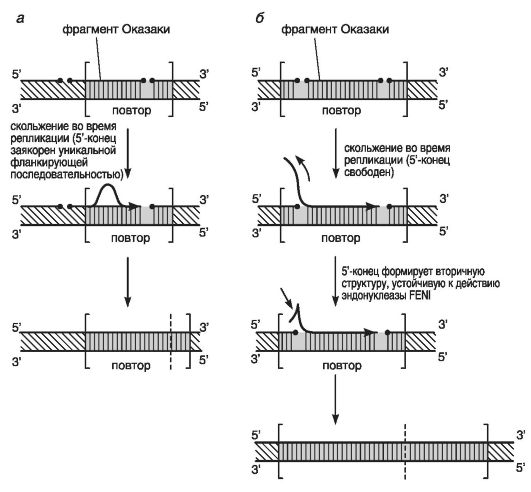
Рис. 73. Модель экспансии повторов на запаздывающей цепи ДНК (по Eichler Е.Е. et al., 1994)
При этом возможны 2 варианта: либо часть фрагмента Оказаки (например, только З'-конец), либо весь фрагмент целиком находятся в пределах повтора. Первый вариант: 5'-конец фрагмента Оказаки фиксирован уникальной последовательностью ДНК или AGG-вставками, другой З'-конец фрагмента Оказаки свободно скользит вдоль комплементарной цепи, образуя вторичные структуры.
Второй вариант: совершенный или «чистый» повтор длиной 70 триплетов не фиксирован по своим краям уникальными последовательностями и также свободно скользит вдоль комплементарной цепи, образуя шпильки.
Высказано предположение, что при обоих вариантах уменьшенный уровень репликативного скольжения лежит в основе мутационного процесса при экспансии числа копий повторов.
Таким образом, репликативная модель экспансии у человека предполагает, что динамическая мутация может быть результатом нарушения функционирования ДНК-полимеразы во время репликации, предшествующей митозу и мейозу.
Комбинативная модель
Изменение длин тандемных повторов может быть результатом неравного кроссинговера (см. главу 5). Однако такая экспансия, во-первых, не сопровождается обменом фланкирующих последовательностей ДНК и, во-вторых, нельзя исключить неравный кроссинговер между сестринскими хроматидами, при котором в одинаковой степени может произойти как экспансия, так и сокращение длин повторов, что экспериментально не подтверждено.
Рекомбинационная модель
Наиболее обоснованной считается модель экспансии в результате рекомбинации или генной конверсии (см. главу 5).
Генная конверсия - это нереципрокный обмен между сестринскими хроматидами одной хромосомы или отклонение фактического обмена от ожидаемого теоретического обмена. Она может быть вызвана двухнитевыми разрывами вблизи нуклеотидных повторов
ДНК.
Не исключено, что генная конверсия может индуцироваться аномальной репликацией повторов и сопровождаться кроссинговером. В результате генной конверсии образуются как удлиненные, так и укороченные повторы.
Однако в отличие от неравного кроссинговера при генной конверсии это равновесие сдвигается в сторону преобладания нуклеотидных последовательностей с экспансией повторов.
На рис. 74 приведены модели генной конверсии, вызванной двухнитевыми разрывами ДНК.
Проблемы и пути определения механизмов патогенеза экспансии
В решении проблем патогенеза экспансии копий нуклеотидных повторов нет полной ясности. Экспансия реально существует при наличии в клетках стабильных систем репарации ДНК. Поэтому речь может идти о несостоятельности локальной репарации ДНК, зависящей от различий в генетическом и биохимическом микроокружении нуклеотидных повторов. В пользу такого предположения свидетельствуют две сходные по клинике, но разные по этиологии нозологии: одна - это прогрессирующая миоклонус-эпилепсия (если считать, что нет дефектов репарации), а другая - это митохондриальная миоклонус-эпилепсии с РКМВ, сопровождающаяся дефектами репарации или MERRF-синдром.
В пользу перспективности этого пути указывают данные о том, что все 27 нозологий болезней экспансии относятся к МБ человека. Как известно, в последние годы были показаны ранее неизвестные механизмы патогенеза многих МБ (см. главы 26, 28 и 29).
ОБЩАЯ ХАРАКТЕРИСТИКА БОЛЕЗНЕЙ ЭКСПАНСИИ
БЭ обусловлены ростом числа копий кодирующих и некодирующих повторов ДНК. В основном это тринуклеотидные повторы. Однако имеются заболевания, обусловленные тетра-, пента- и двенадцатинуклеотидными повторами (см. ниже).
Выделены четыре типа классификаций болезней экспансии: А,
Б, В и Г.
• А. Классификация в зависимости от эффекта мутантного гена.
В основу классификации положено деление БЭ на два класса.
- Первый класс - болезни, обусловленные мутациями с эффектом «минус». Такие мутации приводят к нарушениям экспрессии генов: при атаксии Фридрейха (ген Х25), миотонической дистрофии (гены DMPK и ZNF9), прогрессирующей миоклонус-эпилепсии (ген CSIB) и УО, синдроме Мартина-
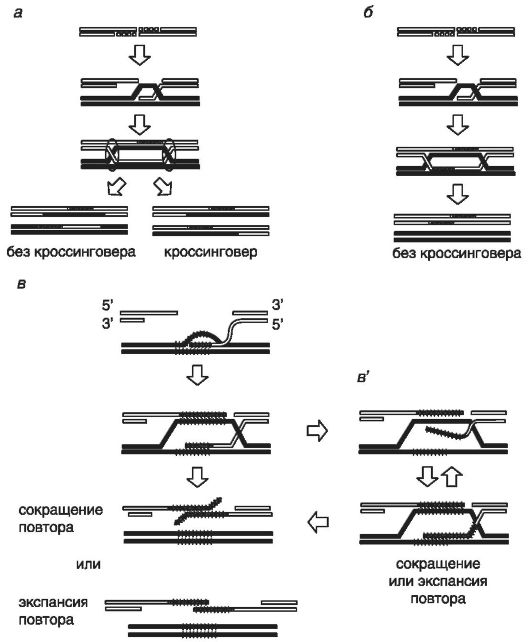 Рис. 74. Модели генной конверсии, инициированной двухнитевыми разрывами ДНК (по Cummings C.J. and Zoghbi H.Y., 2000).
Рис. 74. Модели генной конверсии, инициированной двухнитевыми разрывами ДНК (по Cummings C.J. and Zoghbi H.Y., 2000).
Стрелки указывают направление синтеза ДНК в рамках репарации двухнитевых разрывов. Поврежденная молекула ДНК выделена белым цветом. Матричная цепь ДНК выделена черным цветом. Дочерняя вновь синтезированная цепь ДНК имеет серый цвет. Области тандемных повторов обозначены вертикальными линиями.
Модель a демонстрирует разрешение двух четырехнитевых структур (структуры Холидея) - они обведены кругами. Модель a ведет к генной конверсии, ассоциированной с кроссинговером в 50% случаев. Модель б - синтеззависимый отжиг цепей ДНК. Модель в - перестройки тандемных повторов по модели б. Модель в' - отделение вновь синтезированной цепи и ее повторный ошибочный отжиг на матрице
Белл (ген FRAXA) или УО, сцепленной с ломкой Х-хромосомой (ген FRAXE). Патогенез этих заболеваний связан либо с выключением экспрессии гена в результате инактивации его промоторной области (гены FMR1 и FMR2), либо с нарушением (блокированием) транскрипции гена (гены DMPK и FRDA).
- Второй класс - болезни, обусловленные мутациями с эффектом «плюс». Такие мутации приводят к появлению токсических белков, повреждающих подкорковые структуры мозга - это все остальные болезни, не вошедшие в класс болезней с эффектом «минус». Их патогенез обусловлен увеличением длины полиглутаминового тракта (9 болезней, связанных с триплетом CAG, кодирующим глутаминовую кислоту) или увеличением длины полиаланинового тракта (одна болезнь). Следует отметить, что белки полиглутаминового тракта приобретают характеристики, напоминающие характеристики прионных белков (см. главу 29): они вызывают токсический эффект в нейронах подкорковых отделов мозга, что связано с апоптозом.
• Б. Классификация в зависимости от кодирующих и некодирующих повторов. В ее основу также положено деление БЭ на два класса.
- Первый класс - болезни, связанные с кодирующими повторами. Среди них 18 заболеваний, включая 9 нозологий, обусловленных полиглутаминовыми трактами: болезнь Кеннеди (Xq12), денторубропаллидольюисова атрофия (12р13.31), СЦА 6 типов: 1 (6р23), 2 (12q24.1), 3 (14q32.1), 6 (19р13), 7 (3р14.1) и 17 (6q27), хорея Гентингтона (4р16.3); а также 9 нозологий, обусловленных полиаланиновыми трактами: блефарофимоз и эпикант (3q23), врожденный синдром центральной гиповентиляции (4р12), клайдокраниальная дисплазия (6р21), семейная алобарная голопрозэнцефалия (13q32), околофарингеальная мышечная дистрофия (14q11), рука-нога-генитальный синдром (17р15), синполидактилия (2q31), Х-сцепленная УО в сочетании с дефицитом гормона роста (Xq26.3), Х-сцепленная УО в сочетании с эпилепсией (Хр22.13);
- Второй класс - болезни, связанные с некодирующими (микросателлитными) повторами. Среди них 9 заболеваний: атаксия Фридрейха (9q13), миотоническая дистрофия, тип 1 (19q13.3) и тип 2 (3q21), синдром Мартина-Белл или УО FRAXA (Xq27.3),
УМО FRAXE (Xq28), прогрессирующая миоклонус-эпилепсия (21q22.3), СЦА трех типов: 8 (13q21), 10 (22q13) и 12 (5q31-33).
• В. Классификация в зависимости от состава моноаминокислотного тракта. Эта классификация входит составной частью в первый класс из 18 болезней классификации Б, включая 9 болезней полиглутаминового и 9 болезней полиаланинового трактов (нозологии приведены выше).
• Г. Классификация в зависимости от локализации копий повторов в транслируемой и нетранслируемой областях генов. В первом случае экспансия колеблется в пределах 40-80 копий повторов, нарушения транскрипции и трансляции мутантных генов отсутствуют, и БЭ возникают в результате неправильного функционирования протяженных трактов. Для таких болезней характерна поздняя манифестация с неуклонным прогрессированием (например, хорея Гентингтона и разные типы СЦА). Во втором случае экспансия достигает больших значений и колеблется от нескольких сотен до нескольких тысяч копий повторов. При таких болезнях характер клинических проявлений и темп прогрессирования тесно связаны с неправильным функционированием протяженных трактов, ибо большое число копий нуклеотидных повторов делает ген нестабильным, приводя к феномену антиципации в семейных родословных.
К этому классу принадлежат: атаксия Фридрейха, миотоническая дистрофия и синдром Мартина-Белл.
БОЛЕЗНИ ЭКСПАНСИИ КОДИРУЮЩИХ ПОВТОРОВ
В табл. 17 приведены 18 таких заболеваний, относящихся к классификации Б.
Таблица 17. Характеристика болезней экспансии кодирующих повторов
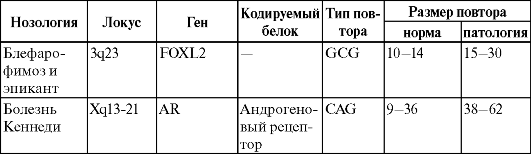
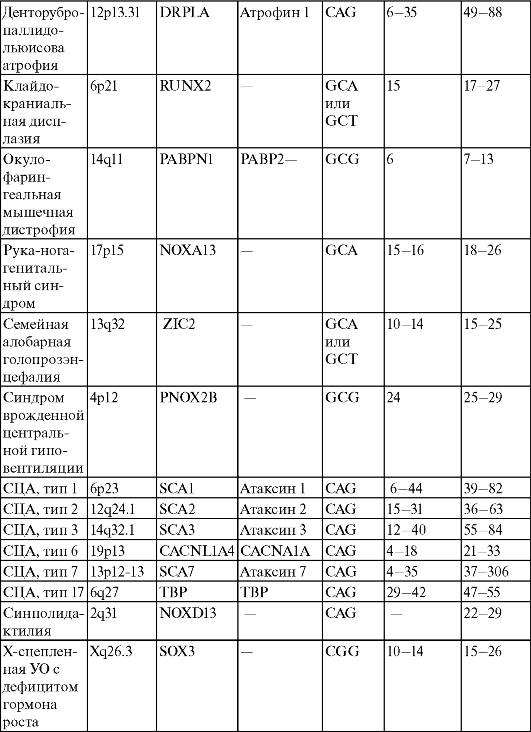
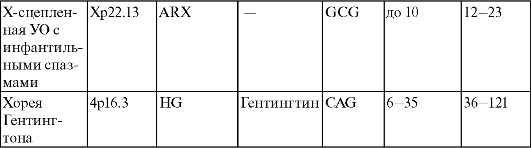 Примечание. Знак «-» обозначает отсутствие данных.
Примечание. Знак «-» обозначает отсутствие данных.
Таким образом, приведенные в этой таблице типы тринуклеотидных повторов кодируют либо белки увеличенного полиглутаминового тракта (САG-повтор экспрессирует белки: гентингтин, атрофин, ТВР и САСNAIA, белки андрогенового рецептора и белки атаксинов 1-3 и 7), либо белки увеличенного полиаланинового тракта (повторы GCG, GCA и GCT).
При этом размеры кодирующего повтора САG меньше, чем размеры некодирующего микросателлитного повтора GCG.
Причинно-следственные связи
В случае БЭ, связанных с полиглутаминовыми трактами, гомология между белками, содержащими такие тракты, отсутствует, хотя они объединены общим механизмом экспансии на основе кодирующего повтора CAG. Для этих заболеваний характерна прогрессирующая дисфункция нейронов, развивающаяся у больных обычно в зрелом возрасте и приводящая к выраженной дегенерации нервных клеток. И хотя при БЭ поражаются практически все ткани организма, дегенерации подвержена лишь небольшая группа нейронов, специфическая для каждой нозологии.
По-видимому, патогенез болезней, связанных с полиглутаминовым и полиаланиновым трактами, заключается в приобретении белками с протяженными трактами функций цитотоксичности при их агрегации между собой или другими белками - именно этим определяется общность внутриклеточных эффектов мутантных белков (см. выше).
Например, в случае полиглутаминовых трактов основной патогенетический механизм заключается в том, что трансглутаминаза связывает полиглутаминовый тракт с полипептидами, содержащими лизиловые группы, что ведет к формированию кополимеров с цитотоксическим действием.
С другой стороны, антипараллельные бета-цепи полиглутаминовых повторов удерживаются вместе водородными связями, что ведет к их мультимеризации и последующей агрегации молекул. Иными словами, образование токсических агрегатов не является необходимым условием для развития БЭ, что нашло подтверждение у больных с синдромом Кеннеди, СЦА типов 2 и 7 и хореей Гентингтона при обнаружении у них белковых агрегатов не только в селективно поврежденных нейронах, но и в неповрежденных клетках других тканей. Эта причина цитотоксичности была подтверждена для всех БЭ.
События, сопутствующие механизму экспансии
Причины и механизмы формирования протяженных белковых агрегатов и их токсического действия можно представить как многозвеньевую цепочку метаболических событий в клетке, в которой первое звено - это экспансия полиглутаминового или полиаланинового трактов; второе звено - это изменение пространственной организации патологических белковых молекул; третье звено - это устойчивость патологических белковых молекул к протеосомной утилизации; четвертое звено - это увеличение селективной дегенерации клеток, содержащих такие молекулы.
Следует отметить, что для третьего звена данной цепочки было показано, что патологические молекулы белка - атаксина 1 (92-глу) более устойчивы к убиквитин-опосредованной деградации in vitro, чем нормальные молекулы.
Помимо основных событий механизма экспансии, в поврежденной клетке наблюдается скопление протеосом и белков - молекулярных шаперонов, которые регулируют сборку пространственной структуры патологического белка и уменьшают его цитотоксичность, контролируя степень агрегации и скорость обмена с другими белками.
В качестве одного из патогенетических механизмов следует упомянуть внутриядерное накопление белковых агрегатов, которые изменяют экспрессию генов, аномально взаимодействуя с регуляторами транскрипции. Например, был выделен ТАТА-связывающий белок (ТВР), служащий причиной СЦА 17-го типа (см. ниже).
При хорее Гентингтона выделен белок - гентингтин, который в норме (когда нет молекул экспансии) не взаимодействует с репрессором транскрипции (N-COR). При появлении молекул экспансии гентингтин связывается с ними, повреждая аппарат транскрипции.
В нейронах у таких больных выявлена эктопическая цитоплазматическая локализация N-CoR и одного из его корепрессоров - Sin3a.
Кроме этих белков, при БЭ обнаружены транскрипционные факторы: СВР, TAFII130 (один из ТВР-ассоциированных факторов). Все это указывает на серьезные повреждения транскрипции вследствие экспансии патологических белковых трактов.
Наконец, следует отметить участие каспаз, ибо при БЭ, связанных с полиглутаминовыми трактами, им принадлежит особая роль - эти ферменты (цистеиновые протеазы) участвуют в апоптозе (см. главу 11).
Болезни экспансии, связанные с полиглутаминовыми трактами
Следует отметить, что полный клинический спектр последствий экспансии полиглутаминового тракта в специфических нейробелках остается пока неизвестным.
Болезнь Кеннеди
Болезнь Кеннеди или рецессивная сцепленная с полом спинобульбарная мышечная атрофия, манифестирует после 40 лет. Характерны поражение бульбарной группы черепных нервов и нисходящие параличи. Периферические парезы начинаются с проксимальных отделов верхних конечностей и мускулатуры надплечий. Снижение мышечной силы сопровождается атрофиями мышц и выраженными фасцикуляциями. Вначале угнетаются рефлексы с двуглавой и трехглавой мышц, затем появляется тремор пальцев. В дальнейшем присоединяется слабость в проксимальных группах мышц нижних конечностей с угнетением коленных и ахилловых рефлексов. Страдают мимическая мускулатура и мышцы, иннервируемые бульбарной группой черепных нервов, что ведет к дисфагии и дизартрии. Описаны эндокринные нарушения в виде гинекомастии.
Ген болезни локализован в Х-хромосоме (Xq13-21), кодирует андрогеновый рецептор AR.
Болезнь проявляется при количестве копий повтора CAG, равном 38-62. При позднем дебюте и мягких клинических проявлениях число копий этого повтора обычно не превышает 40.
Денторубропаллидольюисова атрофия
Денторубропаллидольюисова атрофия впервые описана в 1982 г. в Японии в пяти семьях. Распространенность заболевания в Японии
составляет 4 случая на 1 млн человек. В других странах описано 7 семей неяпонского происхождения с подобным заболеванием. Болезнь дебютирует после 20 лет. Смерть наступает после 40 лет.
Симптоматика характеризуется миоклонус-эпилепсией, атаксией, хореоатетозом и деменцией. При морфологических исследованиях обнаруживается дегенерация денторубральной и паллидольюисовой систем. Феномен антиципации наблюдается только при получении мутантного аллеля от отца. У всех больных наблюдается четкая корреляция между возрастом начала болезни и величиной экспансии повтора CAG.
Пациенты с более ранним дебютом и быстро прогрессирующей миоклонус-эпилепсией имеют большую протяженность области повтора. Характерным является расширение этой области в среднем на 4 повтора при прохождении мутантного аллеля через сперматогенез и сохранение или даже уменьшение длины повтора при получении мутации от матери.
Доказана аллельная природа этой болезни и сходного с ней синдрома Хау-Ривера, описанного в 1989 г. в нескольких афроамериканских семьях. Оба заболевания манифестируют в возрасте 15-30 лет с проявления атаксии, эпилептических припадков, хореиформных гиперкинезов и прогрессирующей деменции. Часто развивается шизофреноподобная симптоматика.
При морфологическом исследовании выявляется дегенерация нейронов зубчатого ядра, микрокальцификация бледного шара, нейроаксональная дистрофия nucleus gracilis и демиелинизация centrum semiovale.
Ген болезни DRPLA локализован в хромосоме 12 (12р13.31), он кодирует белок - атрофин 1. Болезнь проявляется при наличии 40-88 копий повтора CAG.
Спиноцеребеллярные атаксии
В основе этой группы заболеваний лежат прогрессирующие дегенеративные изменения в нейронах мозжечка, головного мозга и спиноцеребеллярном тракте. Характеризуются атаксией, дизартрией, офтальмоплегией. СЦА дебютируют с изменения походки, затем появляются изменения в руках (дизметрия, интенционный тремор). Постепенно нарастает мышечная слабость (сначала в ногах), которая сопровождается повышением тонуса и глубоких рефлексов. Появляются мозжечковая дизартрия, офтальмопарез, птоз и тотальная офтальмоплегия (с отсутствием зрачковых реакций на свет). Часто выявляются нарушение памяти и снижение критики.
Спиноцеребеллярная атаксия, типы 1 и 2
СЦА, тип 1 (оливопонтоцеребеллярная атаксия), или болезнь Менцеля, а также СЦА, тип 2, имеют сходную симптоматику.
Оба заболевания начинаются в тертьем-четвертом десятилетии жизни с появления легкого нарушения походки и неловкости при быстрой ходьбе и беге. По мере прогрессирования болезни развиваются мозжечковая атактическая походка, интенционный тремор, неустойчивость в позе Ромберга, асинергия мимической мускулатуры с мозжечковым гримасничаньем.
Расстройства речи возникают рано и носят сложный мозжечководизартрический характер.
Характерны экстрапирамидные нарушения в виде разнообразных гиперкинезов: статокинетический тремор конечностей, туловища и головы, миоклонии, кривошея, хореиформные, атетоидные, дистонические гипердвижения. Реже встречается синдром паркинсонизма.
На первом-втором годах от начала заболевания наблюдаются: глазодвигательные расстройства, нистагм, нарушения глотания и фонации, расстройства сфинктеров, пирамидная симптоматика разной степени выраженности, спинальные симптомы (амиотрофии, фасцикуляции, арефлексия, нарушения глубокой чувствительности), деменция. В отдельных семьях отмечены атрофия зрительных нервов, дегенерация сетчатки, катаракта.
Иногда зрительные нарушения могут стать началом манифестации болезни и на много лет опередить появление неврологической симптоматики.
Продолжительность жизни больных от момента манифестации не превышает 10-15 лет. Причина смерти - инфекционные осложнения.
Основные критерии клинического диагноза: дебют в возрасте 30-40 лет; мозжечковая атаксия, дизартрия, экстрапирамидные симптомы (тремор, миоклонии, торсионно-дистонические гиперкинезы), глазодвигательные нарушения, дисфагия и дисфония, расстройства сфинктеров, деменция; неуклонное прогрессирование в течение всей болезни.
При КТ мозга наблюдается истончение средней ножки мозжечка, расширение субарахноидального пространства полушарий и червя мозжечка, расширение большой цистерны, 4-го желудочка, боковых желудочков и субарахноидального пространства больших полушарий мозга. При МРТ дополнительно визуализируются атрофия моста и
продолговатого мозга. В настоящее время возможна прямая ДНКдиагностика, основанная на идентификации:
• гена болезни SCA1 (кодирует белок - атаксин 1) в локусе хромосомы 6 (6р22-23); болезнь проявляется при экспансии повтора CAG, равной 39-82 копиям;
• гена болезни SCA2 (кодирует белок - атаксин 2) в локусе хромосомы 12 (12q24.1); болезнь проявляется при экспансии повтора CAG, равной 36-64 копиям.
Спиноцеребеллярная атаксия, тип 3
СЦА, тип 3, или болезнь Мачадо-Джозефа, впервые описана в 70 годах XX в. в семьях португальского происхождения.
Дебютирует в возрасте от 20 до 60 лет. Выделяют четыре патологических фенотипа: первый проявляется преимущественно экстрапирамидными и пирамидными симптомами; второй характеризуется сочетанием мозжечковой атаксии, пирамидных и экстрапирамидных симптомов; третий - сочетание мозжечковой симптоматики и амиотрофии; четвертый - сочетание синдрома паркинсонизма с легкой мозжечковой симптоматикой, дистальной мотосенсорной невропатией и амиотрофией.
Выделяют основные и дополнительные (малые) диагностические критерии. К основным критериям относятся: мозжечковая атаксия, пирамидная симптоматика разной степени выраженности, периферические амиотрофии, экстрапирамидные расстройства (мышечная дистония, синдром паркинсонизма). К малым критериям относятся: прогрессирующая наружная офтальмоплегия, подергивание мышц лица и языка при целенаправленных движениях (напоминают крупные фасцикуляции), «выпученные глаза» или широко раскрытые глазные щели с неподвижными, фиксированными глазными яблоками (обусловлено офтальмоплегией и ретракцией верхних век).
Продолжительность заболевания около 20 лет. Больные умирают от инфекционных осложнений.
При КТ и МРТ мозга наблюдается выраженное расширение 4-го желудочка и относительная сохранность коры полушарий, червя мозжечка и варолиева моста (даже при многолетнем течении болезни).
Ген болезни локализован на хромосоме 14 (14q32.1), кодирует белок-атаксин. Болезнь проявляется при наличии 55-84 копий повтора CAG.
Спиноцеребеллярная атаксия, тип 6
СЦА, тип 6, или наследственная пароксизмальная церебелляр-
ная атаксия, в одних случаях сочетается с эпизодической атаксией, а в других случаях - с семейной гемиплегической мигренью. Заболевание характеризуется поздним дебютом, мягким течением и медленным прогрессированием (длится 20-30 лет).
Клиническая симптоматика включает: мозжечковую атаксию, нистагм, скандированную речь, легкое снижение вибрационной и проприоцептивной чувствительности. Постепенно пациенты утрачивают способность к самостоятельному передвижению, позднее у них развивается парез гортани.
При МРТ мозга обнаруживается атрофия мозжечка и спинного мозга. При морфологическом исследовании - значительная потеря клеток Пуркинье и (в меньшей степени) гранулярных клеток, нейронов зубчатого ядра и нижних олив.
При этом заболевании наблюдается достоверно значимая обратная корреляция между возрастом начала болезни и величиной экспансии повтора CAG. Вместе с тем, в отличие от других БЭ здесь нет прямой связи между дополнительной экспансией мутантного аллеля и феноменом антиципации.
Ген болезни САСNL1А4 локализован в хромосоме 19 (19р13); он кодирует белок САСNА1А.
Болезнь проявляется при наличии 21-33 копий повтора CAG. В этой же области хромосомы 19 (19р13) картированы еще два гена, связанных с семейными нейродегенеративными нарушениями. Первый ген отвечает за развитие наследственной пароксизмальной церебеллярной атаксии (НРСА) или эпизодической атаксии (ЕА), характеризующейся нистагмом и периодическими приступами атаксии, продолжающимися от нескольких минут до нескольких часов (иногда дней).
Причем в перерывах между приступами сохраняется нормальная координация.
Частота и тяжесть приступов с возрастом уменьшаются. Приступы могут быть связаны с эмоциональным стрессом, приемом алкоголя, гипоксией и другими причинами.
При появлении симптомов атаксии часто развивается негемиплегическая мигрень.
При МРТ мозга обнаруживаются признаки церебеллярной атрофии.
Второй ген связан с семейной гемиплегической мигренью (FHM), при которой у пациентов с раннего детского возраста развиваются периодические приступы односторонней пульсирующей головной
боли. В некоторых семьях наблюдаются дегенеративные изменения мозжечка, клинически проявляющиеся атаксией, нистагмом, вестибулярными нарушениями, сходными с таковыми при НРСА/ ЕА. Перекрывание симптомов этих двух заболеваний позволило высказать предположение об их аллельной природе, а периодичность приступов указывала на возможность дефекта гена, кодирующего один из ионных каналов клетки.
В некоторых семьях у родственников больных наблюдалось перекрывание симптомов, характерных для всех трех приведенных выше форм заболеваний. Например, в одной из семей с диагнозом «эпизодическая атаксия» симптомы заболевания у разных пациентов варьировали от изолированного нистагма до тяжелой прогрессирующей церебеллярной атаксии. При этом обнаружилась экспансия повтора CAG в мутантном аллеле гена, где было 23 копии триплета, однако точковые мутации в кодирующей области гена САСNL1А4 отсутствовали. У другой семьи мутантный аллель CAG25 оказался связан с прогрессирующей церебеллярной атаксией, а аллель CAG20 ассоциирован с эпизодической атаксией.
В настоящее время считается, что небольшие экспансии CAG- повтора в гене САСNL1А4 являются мейотически нестабильными, и их мутации могут объяснить варьирующий полиморфизм признаков болезни у родственников из одной и той же семейной родословной.
Спиноцеребеллярная атаксия, тип 7
СЦА, тип 7 характеризуется сочетанием мозжечковой атаксии и прогрессирующей пигментной дегенерации сетчатки. В дальнейшем возможно развитие дисфагии вследствие констрикторных расстройств. Возраст начала болезни варьирует от 1,5 до 70 лет. Характерна антиципация, особенно выраженная при получении мутантного аллеля от отца. В этом случае болезнь дебютирует на 20 лет раньше в каждом последующем поколении. Но анализ характера наследования этого заболевания часто осложняется получением больными мутантного аллеля от матери, что указывает на возможность селективного отбора, направленного против аллелей, проявляющихся большой экспансией, которая происходит на уровне зародышевых клеток или ранних зародышей.
При длине CAG-повторов более 59 копий болезнь дебютирует с ухудшения зрения, тогда как при меньших значениях количества копий CAG-повторов первым симптомом является атаксия.
Спиноцеребеллярная атаксия, тип 17
СЦА, тип 17 - это аутосомно-доминантная форма с хорошо
изученным патогенезом. В ее основе лежит механизм аномального взаимодействия проникших в клеточное ядро молекул, содержащих протяженные полиглутаминовые тракты, с регуляторами транскрипции, что нарушает экспрессию генов. Причина - белок ТВР, являющийся субъединицей транскрипционного фактора D, входящего в состав белкового комплекса РНК-полимеразы II (TFIID), который необходим для экспрессии большинства генов.
Как известно, РНК-полимераза II участвует в синтезе РНК на матрице ДНК, но самостоятельно она не способна узнавать промоторные области генов. Для этого существуют факторы транскрипции, последовательно связывающиеся с промотором и образующие ОИК, кэп-сайт (см. главу 3). При данном заболевании первым в этом комплексе стоит TFIID, сформированный из белка ТВР. В N-концевой области молекулы данного белка расположен протяженный полиглутаминовый повтор, определяющий ДНК-связывающую активность С-концевой области молекулы.
ТВР-белок в комплексе с ТАТА-боксом приобретает специфическую пространственную структуру (седловидную форму), в которой вогнутая внутренняя часть контактирует с ДНК, а внешняя выпуклая часть - с субъединицами комплекса TFIID, которые являются ТВР-ассоциированными факторами транскрипции. Нарушения нормальной пространственной структуры ТВР изменяют аффинность ТВР-ассоциированных факторов, что ведет к нарушениям экспрессии генов и клеточной гибели.
Хорея Гентингтона
Хорея Гентингтона известна с 1872 г. Она относится к тяжелой нейродегенеративной патологии, наследуемой по аутосомнодоминантному типу с полной пенетрантностью. Ген болезни локализован в хромосоме 4 (4р16.3). Он кодирует белок - гентингтин, аномально взаимодействующий с транскрипционными факторами при его проникновении в клеточное ядро. В норме молекулы гентингтина не вступают во взаимодействие с репрессором транскрипции N-CoR, в то время как молекулы белка с экспансией полиглутаминового тракта связывают этот репрессор. Болезнь проявляется при количестве копий повтора CAG, равном 36-121. Клинические признаки включают триаду симптомов: гиперкинезы, деменция, психические расстройства. Выделяют гиперкинетический (90% случаев) и кинетикоригидный (10%) варианты болезни. Первый вариант характеризуется нарастающими гиперкинезами, манифестацией
заболевания в четвертом-пятом десятилетиях жизни и заканчивается смертью через 15-20 лет после появления первых признаков. Второй вариант (болезнь Вестфаля) возникает в первом-втором десятилетиях жизни, проявляется мышечной ригидностью, контрактурами, тремором рук, атаксией, миоклонией, пирамидными симптомами, но гиперкинезов при этом нет. Наследственная передача в этом случае идет исключительно по линии отец-сын.
Болезни экспансии, связанные с полиаланиновыми трактами
Блефарофимоз и эпикант
Блефарофимоз и эпикант в сочетании с другими глазными аномалиями (короткая и узкая глазная щель, неправильный рост ресниц, микрокорнеа, миопия высокой степени и иногда - микрофтальмия, ювенильная катаракта) - это редкое заболевание с аутосомнорецессивным типом наследования. Ген болезни FOXL2 локализован в хромосоме 3(3q23). Он обусловливает развитие патологического фенотипа при количестве копий повтора GCG, равном 15-30.
Клайдокраниальная дисплазия
Клайдокраниальная дисплазия (черепно-ключичный дизостоз) известна с 1897 г.
Минимальные диагностические признаки: аплазия ключицы или ее части, поздние: закрытие родничков, оссификация черепных швов и прорезывание зубов.
Клинически синдром представляет собой генерализованную скелетную дисплазию. Это длительно незакрывающиеся (открытые) черепные швы и роднички, что ведет к чрезмерному развитию лобных, височных и затылочных бугров, наличию вормиевых костей.
Созревание скелета замедлено. Отмечается отставание в росте. Характерны: укорочение средних фаланг 5 пальца кисти, дополнительные эпифизы 2 и 5 метакарпальных костей, широкое лонное сочленение, аномалии позвонков и ребер, сколиоз, узкая грудная клетка, повышенная ломкость костей. Патология зубов включает гипоплазию эмали и кариес.
Описаны глухота, расщелина нёба и сирингомиелия.
Ген болезни RUNX2 локализован в хромосоме 6 (6р21). Болезнь проявляется при 17-27 копиях повторов GCA и GCT.
Окулофарингеальная мышечная дистрофия
ОФМД обусловлена кодирующим повтором GCG c изменяю-
щейся длиной. Это легкая по течению форма заболевания. Она выявляется у гетерозигот, но у них, как правило, отсутствуют выраженные клинические проявления, что можно объяснить состоянием премутации, ибо количество GCG-повторов не превышает 6 (соответствует нормальному значению). В случаях, когда число повторов GCG увеличивается от 7 до 13 копий, наблюдается возрастание протяженности полиаланинового тракта, расположенного в N-конце поли (А)-связывающего белка РАВР2, что обусловливает развитие тяжелой доминатной формы заболевания, которая начинает проявляться уже при экспансии, равной семи повторам. Частота аллеля (GCG)7 в нормальной популяции не превышает 2%. У гетерозигот (GCG)6 / (GCG)7 нет признаков ОФМД, тогда как у гомозигот (GCG)7 / (GCG)7 развивается аутосомно-рецессивная форма болезни.
Таким образом, аллель GCG7 выступает в роли модификатора как доминантно, так и рецессивно наследуемых патологических фенотипов.
Рука-нога-генитальный синдром
Рука-нога-генитальный синдром развивается у женщин, наследуется по аутосомно-доминантному типу. Патологический фенотип включает комплекс врожденных нарушений развития: аномалии кистей (клинодактилия мизинца, укорочение метакарпальных костей, дислокация большого пальца; аномалии стоп (маленькие размеры, большой палец меньше других вследствие укорочения метатарзальной кости); аномалии внутренних половых органов (удвоение тела или шейки матки, перегородки влагалища).
Ген болезни NOXA13 локализован в хромосоме 17 (17р15). Он обусловливает развитие заболевания при количестве копий повтора GCA, равном 18-26.
Семейная алобарная голопрозэнцефалия
Семейная алобарная голопрозэнцефалия или аринэнцефалия встречается с частотой 1:16 тыс. человек, характеризуется аутосомно-рецессивным типом наследования. Дети нежизнеспособны. Наблюдаются неразделение конечного мозга на сферы, двусторонние расщелины губы и нёба, отсутствие фильтра, гипотелоризма. В тяжелых случаях выявляются цебоцефалия и циклопия. На рентгенограмме: отсутствие мозолистого тела, единая вентрикулярная полость, свободно сообщающаяся с субарахноидальным простран-
ством. Другие морфологические изменения: отсутствие обонятельных луковиц, обонятельного тракта и пластинок, гипоплазия гиппокампа; крупные извилины, деформация передней черепной ямки.
Ген болезни ZIC2 локализован в хромосоме 13 (13q32). Болезнь проявляется при 15-25 копиях повторов GCA и GCT.
Синдром врожденной центральной гиповентиляции
СВЦГ проявляется в виде альвеолярной гиповентиляция и врожденной гиповентиляции. Первая форма редко диагностируется, проявляется как апноэ во время сна, характеризуется снижением насыщения крови кислородом при сохранности двигательной (механической) функции легких. Объясняется нарушением чувствительности рецепторов к кислороду и углекислоте.
При этой форме выделяют первичный (идиопатический) тип и вторичный тип - результат неврологического поражения. Основными жалобами являются: частые пробуждения, ощущение невысыпания и головные боли по утрам. Лечение разработано недостаточно.
Вторая форма - еще более редкое состояние. Это наблюдаемые с первых месяцев жизни нарушения легочной вентиляции; проявляются во время сна. С возрастом у больных детей происходит постепенное «дозревание» хеморецепторных систем, и считается, что с 4-5 лет такие больные уже не требуют обязательной госпитализации в случае респираторной инфекции. Эта форма центральной гиповентиляции нередко сопровождает другие наследственные болезни (болезнь Гиршпрунга, нейробластома). Ген PNOX2B локализован в локусе 4р12. Болезнь проявляется при количестве копий повтора GCG, равном 25-29.
Синполидактилия
Синполидактилия - это синдром полидактилии (лишние пальцы), сочетающийся с синдактилией (сросшиеся пальцы). Встречается чаще других аномалий развития пальцев. Количество лишних пальцев - от одного (шестипалость) до трех (восьмипалость) и даже пяти (десятипалость). Добавочные пальцы имеют либо все элементы фаланг, либо только их часть; обычно они прикрепляются по краям кисти и стопы либо в области II-IV пальцев. У больных часто выявляются другие БАР и МАР.
Ген болезни NOXD13 локализован в хромосоме 2 (2q31). Развитие патологического фенотипа - при количестве копий повтора CAG, равном 22-29.
Х-сцепленная умственная отсталость в сочетании с дефицитом гормона роста
Х-сцепленная УО в сочетании с дефицитом гормона роста описана H. Renpenning в 1953 г. Болезнь характеризуется тяжелой УО, микроцефалией, скошенным лбом, маленькими яичками, пре- и постнатальной задержкой роста (вследствие дефицита СТГ).
Ген болезни SOX3 локализован в Х-хромосоме (Xq26.3), его патологическое действие проявляется при количестве копий повтора GGG, равном 15-26.
Х-сцепленная умственная отсталость в сочетании с инфантильными спазмами
Х-сцепленная УО в сочетании с инфантильными спазмами - синдром, описанный в 1891 г. Ч. Вестом. Это заболевание детского возраста встречается с частотой 1:4,5-10 тыс. человек. Болеют преимущественно мальчики (60% случаев). Характерны: кратковременные серии пароксизмов с вовлечением сгибательных групп мышц (аксиальных), прогрессирующее нарушение психоневрологических функций и резистентность к терапии. Могут выявляться как криптогенные, так и симптоматические инфантильные спазмы, в том числе при первом типе спазмов отсутствует четкая причина болезни, наблюдается нормальное нервное и психическое развитие ребенка; до начала манифестации спазмов нет других типов приступов; не обнаруживается симптоматика при КТ и МРТ мозга.
При втором типе спазмов этиология известна. Это могут быть: хромосомный синдром, ВПР мозга, дисгенезии мозга, нейрокожные синдромы, НБО, а также неспецифические пренатальные повреждения мозга, связанные с патологией беременности, перинатальные гипоксические (ишемические) поражения мозга, травмы, опухоли и постнатальные инфекции. Характерна задержка нервно-психического развития до начала приступов, нередки признаки поражения, выявляемые при КТ и МРТ мозга.
Ген болезни ARX локализован в локусе Хр22.13. Патологический фенотип проявляется при количестве повторов GCG, равном 12-23 копиям.
Следует отметить, что с геном ARX могут ассоциироваться и другие болезни: неспецифическая УО при инсерции GCG 2 и мутации гуанина G286S (856 G >A); УО при делеции: 429 del 24; лиссэнцефалия (420- 451 del 32); УО при синдромах Веста и Партингтона (429-452 del 24).
БОЛЕЗНИ ЭКСПАНСИИ НЕКОДИРУЮЩИХ ПОВТОРОВ
В таблице 18 приведены 9 БЭ некодирующих (микросателлитных) повторов.
Таблица 18. Характеристика болезней экспансии некодирующих (микросателлитных) повторов
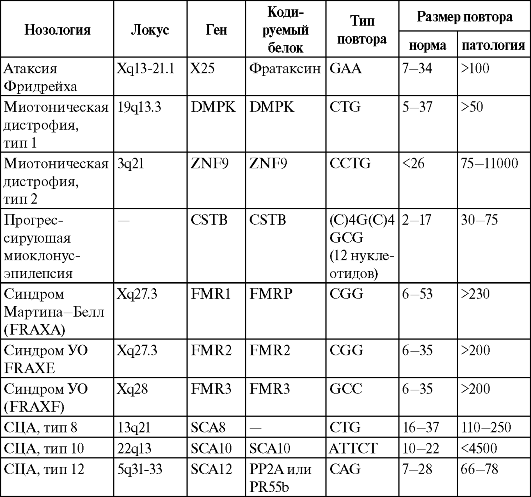 Примечание. Знак «-» обозначает отсутствие данных.
Примечание. Знак «-» обозначает отсутствие данных.
Все указанные в этой таблице болезни обусловлены значительным увеличением размеров микросателлитных повторов. Они, как большинство БЭ кодирующих повторов, сопровождаются дегенерацией и дисфункцией разных тканей и органов и выраженностью
клинических проявлений. При этом, несмотря на ряд общих свойств, некодирующие микросателлитные повторы различаются по нуклеотидному составу и расположению относительно кодирующих и регуляторных элементов генома, что обусловливает упомянутую выше уникальность патогенетических механизмов при каждом конкретном заболевании.
В последние годы было показано, что некодирующие микросателлитные повторы, как правило, происходят из небольшого пула повторов промежуточных размеров.
Атаксия Фридрейха
Атаксия Фридрейха - это редкая БЭ, наследуемая по аутосомнорецессивному типу, при которой нет феномена антиципации. Клиника: атаксия, дизартрия, снижение рефлексов, кардиомиопатия, сахарный диабет. Морфологически выявляется дегенерация нейронов спинного мозга, ганглиев дорсальных корешков и сенсорных ганглиев. Причина - экспансия повтора GAA (мутация в гене фратаксина), ведущая к снижению его экспрессии и появлению протяженного повтора, богатого АТ-парами, затрудняющими транскрипцию. В результате в митохондриях накапливаются ионы железа, истощается мтДНК (феномен деплеции), угнетаются клеточное дыхание и работа железосерозависимых ферментов, что вызывает повышенную чувствительность к окислительному стрессу (см. главу 26).
Миотоническая дистрофия
Миотоническая дистрофия, тип 1
МД, тип 1 - это аутосомно-доминантное заболевание с мультисистемными проявлениями: катаракта, миотония, мышечная дистрофия, нарушение проводимости миокарда, эндокринные расстройства. При врожденной форме МД 1 наблюдаются множественные ВПР, гипотония, респираторный дистресс-синдром и УО.
Болезнь обусловлена мутациями в гене, локализованном в 19q13, и развивается в результате экспансии копий повтора CTG, который может одновременно присутствовать в 3'-нетранслируемой области гена протеинкиназы DMPK и промоторной области гена на хромосоме 19. Экспансия повтора CTG воздействует на уровень фермента DMPK, нарушая транскрипцию гена, процессинг и трансляцию
мРНК.
Внутри ядра накапливается мРНК с протяженным повтором CUS; она не попадает в цитоплазму, и поэтому изменяется внутриклеточ-
ная концентрация фермента DMPK, что ведет к аномальному фосфорилированию субстрата для этого фермента.
В последние годы выделены два гена, фланкирущих ген DMPK. Предполагается, что экспансия повтора CTG влияет на структуру хроматина и изменяет экспрессию близлежащих двух генов: гена DMAHP (в пределах его промоторной области расположен повтор CTG) и гена DMWD (расположен проксимально от гена DMPK, но вблизи от него). Полагают, что изменение функционирования этих двух генов может быть причиной МД 1.
Еще один механизм - это приобретение новой функции первичным транскриптом, имеющим протяженный тракт CUS (см. выше), т.е. возможен трансдоминантный эффект, нарушающий процессинг разных типов РНК, что обусловливает блокирование РНКсвязывающих белков CUS-ВР и разнообразие симптоматики.
В целом, можно выделить единый механизм МД 1 - это воздействие экспансии копий повтора CTG на свойства мРНК, кодируемой геном DMPK, и на экспрессию генов, расположенных вблизи этого повтора.
Миотоническая дистрофия, тип 2
МД, тип 2 - это гетерогенная болезнь, обусловленная тетрануклеотидным повтором CCTG, который расположен в транскрибируемой, но не транслируемой области гена ZNF9. Этот повтор в норме содержит 26 нуклеотидов; при МД 2 его размеры составляют от 75 до 11 000 (среднее значение около 5000) нуклеотидов.
Экспансия указанного повтора вызывает (независимо от хромосомного окружения и функционирования генов в клетке) выраженные мультисистемные расстройства, т.е. она крайне патогенна. При этом нет никакой функциональной гомологии между белком ZNF9 и протеинкиназой DMPK. По-видимому, в случае МД 2 основной причиной служит способность молекул с экспансией повторов CCTG создавать вторичные структуры в виде двухнитевой РНК, препятствующей нормальному ферментному расщеплению молекул, их созреванию и выходу в цитоплазму, что обусловливает накопление РНК с экспансией и мультисистемные расстройства.
Прогрессирующая миоклонус-эпилепсия
Прогрессирующая миоклонус-эпилепсия - это аутосомнорецессивное заболевание с хроническим течением и сочетанием миоклонических гиперкинезов с эпилептическими приступами, что ведет к церебеллярной атаксии.
Большинство мутаций - это крупные двенадцатинуклеотидные повторы (C)4G(C)4GCG или CCCCGCCCCGCG, расположенные в промоторной области гена CSTB. Причина болезни - мутация в гене цистатина В (CSTB), кодирующем фермент - ингибитор цистеиновых протеаз, что ведет к нарушению пространственной организации промоторных элементов и гиперметилированию. При этом экспансия увеличивает расстояние от сайта связывания активатора транскрипции до точки ее инициации, ограничивая пространственное взаимодействие активатора с другими элементами транскрипционного комплекса.
Как следствие этих событий, происходит избыточное связывание рецепторов транскрипции, нарушение синтеза соответствующих мРНК и белка, изменяется структура хроматина.
Ген болезни CSTB не локализован; он кодирует одноименный белок.
Болезнь проявляется при количестве повторов, (С)4G(С)4 GСG, равном 30-75 копиям.
Синдром Мартина-Белл
Синдром Мартина-Белл (СМБ) - синдром наследственной УО. Синонимы: синдром FRAXA, синдром Х-ФРА, fra(X)-синдром. СМБ широко распространен в популяции: 0,3-1:1000 (мужчины); 0,2- 0,6:1000 (женщины); в среднем - 0,5-1:1000. Такая частота сопоставима только с частотой синдрома Дауна (1:550-650).
У нормальных индивидов в сегменте Xq 27 существуют микросателлитные последовательности ДНК (повторы), содержащие 2-5 пар оснований. В случае мутации в гене FMR1 происходит не привычная замена одного нуклеотида на другой, а увеличение числа копий этих повторов. Экспансия (свыше 230 копий) - это полная мутация, проявляющаяся на клиническом уровне симптоматикой СМБ, а на молекулярном уровне - аномальным метилированием CpG-островка промоторной области гена FMRI, ибо в его 5'-нетранслируемой области содержатся повторы CGG. Эти повторы препятствуют транскрипции, что ведет к отсутствию мРНК и белка FMRP, который относится к РНК-связывающим белкам, циркулирующим между ядром и цитоплазмой.
В цитоплазме белок FMRP формирует РНП-комплекс, связывающийся с рибосомами.
Возможно, в нейронах белок FMRP определяет внутриклеточное расположение и регулирует трансляцию специфических мРНК-
мишеней. По-видимому, полная мутация как причина наследственной УО - это нарушение синтеза белков, необходимых для образования и функционирования синаптических контактов.
Установлена генетическая гетерогенность данного синдрома. Помимо «классической» формы синдрома - FRAXA, обнаружена микроделеция гена FМR1 (без амплификации триплета CGG и аномального метилирования), выделены новые ломкие участки, обозначаемые как формы: FRAXE (тот же триплет CGG, локализация гена в том же сегменте Xq27.3, но дистальнее на 500 тысяч пар оснований от сайта FRAXA; состояние мутации - свыше 200 копий) и форма FRAXF (триплет GCC; локализация гена в сегменте Xq28 на 1-2 млн пар оснований дистальнее сайта FRAXE или ближе к теломере; состояние премутации - 12-26; состояние мутации - свыше 200 копий). Тип наследования СМБ не укладывается в критерии Х-сцепленного рецессивного наследования. Основные клинические признаки: умеренная или глубокая УО с аутистическими расстройствами, мышечной гипотонией и гиперактивностью.
Выявляется черепно-лицевой дисморфизм: большие оттопыренные ушные раковины, прямоугольное лицо, периорбитальная гиперпигментация, высокий и выступающий лоб, тонкий длинный нос, высокое нёбо (часто выявляются подслизистые расщелины нёба или язычка), массивный подбородок, широкие кисти и стопы, макроорхизм, гиперпигментация мошонки. Также встречаются: гинекомастия, гипоспадия, высокая эластичность соединительной ткани (гиперподвижность межфаланговых, коленных и голеностопных суставов), ожирение, пролапс митрального клапана, плоскостопие, сколиоз.
Комбинация всех перечисленных признаков характерна для 50-60% больных мужчин.
Остальные больные мужчины не имеют дисморфизма лица (10%), еще часть мужчин (также 10%) имеют только УО, но без остальных признаков, а оставшиеся 20-30% мужчин не имеют макроорхизма. Клиническая диагностика СМБ сложна из-за недостаточной специфичности и широкой вариабельности симптомов. Даже наиболее специфический из них - макроорхизм - имеет диагностическую ценность только в пубертатном и постпубертатном периодах, а УО может быть симптомом многих других заболеваний.
Примерно половина больных мужчин - это трансмиттеры, или носители (передатчики) неэкспрессируемого мутантного гена, кото-
рый становится экспрессируемым при передаче последующим поколениям в одной семейной родословной.
Наряду с этим описаны два типа женщин - гетерозиготных носителей гена синдрома, в том числе: дочери нормальных мужчинтрансмиттеров, не имеющие клинических признаков и генетических нарушений, а также родные сестры больных мужчин-трансмиттеров, имеющие клинические признаки болезни в 35% случаев. Как оказалось, вероятность развития УО зависит от положения индивида в родословной. При этом четко наблюдается феномен антиципации - более раннее и тяжелое проявление симптомов заболевания в последующих поколениях родословной, или парадокс Шермана.
Умственная отсталость, ассоциированная с ломким участком Х-хромосомы
Это второй вариант синдрома наследственной УМО, ассоциированной с ломким участком Х-хромосомы, или синдром FRAXE. Частота в популяции - 1:50 тыс. человек (всего описано около 30 случаев). Основной симптом - задержка интеллектуального развития, задержка речи, развития навыков чтения и письма, рассеянное внимание, гиперактивность; редко наблюдается аутизм. В отличие от классического СМБ здесь нет черепно-лицевого дисморфизма. Характерна экспансия повтора GCC, локализованного в гене FMR2, который экспрессируется в гиппокампе. Эта мутация ассоциируется с аномальным метилированием (гиперметилированием) промоторной области гена FMR2, что ведет к подавлению транскрипции и отсутствию мРНК и белка.
Спиноцеребеллярные атаксии Тип 8
СЦА, тип 8 - это прогрессирующая атаксия с атрофией мозжечка и «оживленными» рефлексами. Причина - экспансия повтора CTG в 3'-терминальном экзоне гена SCA8, экспрессирующемся в тканях головного мозга. Транскрипт этого гена не транслируется и, возможно, служит эндогенной антисмысловой РНК, регулирующей экспрессию других генов. Кроме такой РНК, была выделена РНК, транскрибируемая в ориентации, обратной гену SCA8. Этот первичный транскрипт гомологичен продукту гена KELCH у дрозофилы. Его значение пока не определено.
Тип 10
СПА, тип 10 - это аутосомно-доминантное заболевание, обусловленное экспансией пентануклеотидного повтора ATTCT в интроне 9 гена SCA10.
В норме наблюдается 10-22 нуклеотида, тогда как у больных их число достигает 4500. Патогенез пока не известен.
Тип 12
ЦСА, тип 12 - это редкое заболевание с экспансией повтора CAG в 5'-нетранслируемой области гена PP2А или гена PP55b, кодирующего специфическую для нервной ткани регуляторную субъединицу фермента - фосфатазы 2А (РР2А). Несмотря на локализацию повтора CAG вблизи консервативных промоторных элементов, влияние экспансии на транскрипцию гена PP2А не доказано. Патогенез не известен.
Подходы к диагностике
Клиническая и молекулярно-генетическая диагностика БЭ основана на особенностях феномена экспансии. Первое появление признаков болезни соответствует превышению порога экспансии нуклеотидных повторов, который можно выявить при прохождении клеток через мужской и женский мейоз. К основным диагностическим признакам (их должно быть не менее трех) относятся следующие.
• Первый признак - это усиление тяжести симптомов у больных родственников в каждом последующем поколении в пределах одной семейной родословной, т.е. собственно феномен антиципации (см. выше). Антиципация объясняется ростом числа копий повторов в циклах клеточных (митотических и мейотических) делений либо по отцовской линии (например, рост повторов в мужском мейозе при хорее Гентингтона), либо по материнской линии (например, рост повторов в женском мейозе при миотонической дистрофии). Следует отметить, что у потомков пораженных лиц прогрессирование симптомов будет приходиться на более ранний возраст, а тяжесть течения болезни - нарастать с большей скоростью.
• Второй признак - это корреляция тяжести симптомов с числом повторов у больных как в одной и той же семейной родословной, так и разных семейных родословных.
• Третий признак - это парадокс Шермана или увеличение количества больных лиц в каждом последующем поколении. Причем этот парадокс зависит от того, кто из родителей передает мутацию потомкам. Основа парадокса Шермана - это наличие в семейной родословной здоровых носителей премутации (состояние до превышения порога экспансии).
Например, при синдроме Мартина-Белл переход из состояния премутации в полную мутацию возникает только в женском мейозе. При таком переходе число копий повторов будет заметно выше у сыновей, чем у дочерей женщин-носительниц мутации.
После обнаружения всех трех диагностических признаков врач вправе поставить предварительный диагноз БЭ и после этого обязан направить пациента на уточняющую лабораторную диагностику, принципы которой приведены на рис. 75. Для конкретной БЭ имеется свой вариант ПЦР-анализа, связанный с генокопированием мутантных аллелей. Разработаны два подхода.
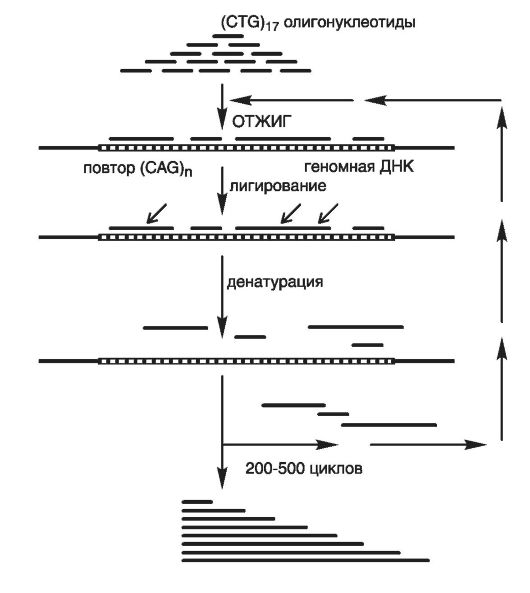 Рис. 75. Принцип метода RED (по Cummings C.J. and Zoghbi H.Y., 2000)
Рис. 75. Принцип метода RED (по Cummings C.J. and Zoghbi H.Y., 2000)
Первый подход - экспансия в пределах 150 повторов регистрируется только методом ПЦР-анализа полиморфных повторов с последующей визуализацией продуктов амплификации и только с праймеров, которые фланкируют область повтора. При этом подходе применяется метод RED, позволяющий проводить скрининг генома пробанда на наличие потенциальной экспансии (премутации). Метод RED (обнаружение повторов экспансии) основан на отжиге* олигонуклеотидов - это повторы длиной 15-20 триплетов и последующем лигировании соседних олигонуклеотидов с их денатурацией с матрицы. Циклы повторяются сотни раз до накопления необходимого конечного продукта (см. рис. 75).
Второй подход основан на том, что при количестве копий повтора более 200 диагностика с помощью ПЦР-анализа затруднена, и тогда определение экспансии проводится с помощью блот-гибридизации ДНК-зондами, например, с помощью зондов STB12.3 и Ox 055 при синдроме FraXA или зонда DNA25 при МД.
Кроме того, разработан простой и надежный метод диагностики синдрома фрагильной Х-хромосомы по образцам ДНК с метилированным (инактивированным) и неметилированным (функционально активным) промотором гена FMR1.
Подходы к лечению
Лечение БЭ традиционно симптоматическое.
В случае детского аутизма применяются нейролептики, антидепрессанты, психостимуляторы, а также поэтапная психологопедагогическая коррекция. В России разработан эффективный способ коррекции детского аутизма по методу И.А. Скворцова (1992-1997).
Сущность метода заключается в метамерном микроинъекционном или безыгольном введении микродоз препаратов, изготовленных из мозга животных (церебролизин, церебролизат, церебролизат-М) или других биологических тканей.
* Отжиг - это техника гибридизации или процесс охлаждения ДНК, приводящий к восстановлению водородных связей между парами оснований. Этот процесс противоположен плавлению ДНК или разделению ее цепей под действием температуры (около 95 °С).