Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 26 ПЕРОКСИСОМЫ И МИТОХОНДРИИ И ИХ БОЛЕЗНИ
Происхождение и наследование пероксисом и митохондрий
При образовании зиготы в ней объединяются все структурнофункциональные элементы ядра и цитоплазмы яйцеклетки и элементы ядра (головки) сперматозоида; шейка и хвост мужской гаметы в цитоплазму женской гаметы не проникают (см. главы 4, 6 и 12). На этой основе (два ядра и одна цитоплазма) начинается онтогенез многоклеточного организма, в ходе которого сначала идут стадии деления-дробления зиготы, затем последующего митоза соматических клеток, а во внутриутробном онтогенезе появляются еще и мейотические деления, которые затем останавливаются. Остановка мейоза продолжается до постнатального этапа половой зрелости организма, когда начинают формироваться функционально зрелые женские или мужские гаметы.
В свою очередь, митотические деления клеток продолжаются до завершающего этапа онтогенеза.
Говоря о материнском происхождении пероксисом и митохондрий, следует отметить, что они (так же, как аппарат Гольджи, лизосомы и ЭР) происходят из женской гаметы, которая после оплодотворения мужской гаметой становится зиготой, и далее ее структурнофункциональные элементы передаются каждой соматической клетке в ходе развития многоклеточного организма.
Передача цитоплазмы из клетки в клетку идет в три этапа:
• первый этап (митоз) - цитоплазма исходно делящейся соматической клетки поступает только в одну из двух образующихся дочерних клеток;
• второй этап (мейоз) - дочерняя клетка, получившая всю цитоплазму материнской клетки, в свою очередь делится на две дочерние клетки, в одну из которых передается вся цитоплазма; вторая дочерняя клетка цитоплазму не получает;
• третий этап - одна из четырех дочерних клеток, получившая цитоплазму, становится функционально активной, способной к
оплодотворению яйцеклеткой; три другие дочерние клетки цитоплазму не получают - это полярные тельца.
Иными словами, передача цитоплазмы (и ее структур) от исходной соматической клетки идет исключительно через женские гаметы по линии мать-дочь. Это материнское наследование.
Однако полное материнское наследование возможно только в случае, если в зиготу не попадут даже единичные отцовские митохондрии или их деление будет заблокировано. Данный факт имеет принципиальное значение для понимания механизмов формирования полиморфизма мтДНК, ибо при отсутствии митохондрий отцовского происхождения невозможен кроссинговер между родительскими гомологами мтДНК и образование новых комбинаций митохондриальных генов, т.е. мтДНК - всегда материнского происхождения. Как показали результаты выполнения международной программы «Геном человека», мтДНК, выделенная из костей черепа современного человека, по нуклеотидному составу соответствует мтДНК, выделенной из костей черепа ископаемого человека, проживавшего на севере африканского континента предположительно 150-200 тыс. лет тому назад. Данный факт был положен в основу гипотезы Г. Уилсона (1987) о происхождении вида: Homo sapiens (Человек разумный) произошел от этого ископаемого первобытного человека.
Вместе с тем, данной гипотезой воспользовались современные креационисты, посчитавшие, что на ее основе базируется теория божественного сотворения мира, согласно которой человек произошел от общих прародителей - Евы и Адама, живших в Библейском Раю (междуречье между Тигром и Ефратом), и именно этот географический район Земли оказался поблизости от севера африканского континента, где была обнаружена мтДНК, извлеченная из ископаемых останков первобытного человека (см. главу 1).
Однако вернемся к материнскому происхождению мтДНК. Как пример, следует привести наследственную атрофию Лебера, которая в одних случаях обусловлена дефектами самой мтДНК, а в других случаях - дефектами ядерной ДНК, т.е. болезнь передается из поколения в поколение либо по нетрадиционному варианту (материнское наследование), либо по традиционному варианту (моногенное наследование) соответственно. Во втором случае характерны два типа наследования: аутосомно-доминантный и аутосомно-рецессивный (см. главу 4).
ХАРАКТЕРИСТИКА ПЕРОКСИСОМ И ПЕРОКСИСОМНЫХ БОЛЕЗНЕЙ
Пероксисомы
Пероксисомы представляют собой круглые или овальные органеллы, находящиеся во всех клетках организма (кроме зрелых эритроцитов); их диаметр 0,2-1,0 микрон. Они имеют матрикс, окруженный одинарной мембраной.
Количество, размеры и форма пероксисом варьируют в разных тканях: больше всего пероксисом в клетках печени и почек, меньше (и меньшего размера) - в клетках кожи и мозга.
В пероксисомах нет своей ДНК (как в митохондриях), и поэтому они не производят, а «импортируют» составляющие их белки - пероксины, вовлеченные в биогенез органелл. Изучены функции 20 пероксинов, в том числе локализованы гены 9 белков.
Белки матрикса и мембраны пероксисом синтезируются на свободных полисомах в цитозоле и транспортируются пероксисомами, в которых содержатся многочисленные ферменты (не менее 50), представленные в основном оксидазами, использующими кислород для окисления разных субстратов клетки. При этом продуктом восстановления кислорода служит не вода, а перекись водорода, которая окисляет субстраты, например алкоголь. Пероксисомам приписываются анаболическая и катаболическая функции. Их работа (наряду с процессами окисления) направлена на защиту клетки с помощью каталаз от образующегося в ней (в основном в митохондриях) атомарного кислорода (см. главу 11).
Ферменты пероксисом также участвуют в биосинтезе эстерифицированных фосфолипидов, окислении глютаровой, L-пипеколиновой и фитановой кислот, D-аминокислот и некоторых фенолов, бетаокислении части длинноцепочечных и всех ОДЦЖК, которые не могут быть окислены в митохондриях (до укорочения длинных цепей), и метаболизме простагландинов и холестерина (см. главы 6 и 7). Кроме того, имеются все основания считать, что пероксисомы причастны к переработке холестерина в желчные кислоты (например, клофибрат, снижающий его уровень в крови, вызывает значительное увеличение числа пероксисом в печени).
Специфичность функционирования пероксисом проявляется в том, что в них:
• не окисляются короткие, среднецепочечные и большая часть длинноцепочечных жирных кислот - это функция митохондрий (см. ниже);
• ацил-Коа оксидаза пероксисом переносит электроны непосредственно на кислород, тогда как в митохондриях ацил-Коа дегидрогеназа переводит ацил-Коа в эноил-Коа, и электроны переносятся на ФАД+;
• ферменты пероксисом и митохондрий кодируются разными генами.
В пероксисомах протекают начальные этапы биосинтеза плазмалогенов (глицеролипидов), содержащих ненасыщенный спирт, соединенный простой эфирной связью с глицерином фосфолипида. Плазмалогены входят в состав 5-20% фосфолипидов клеточных мембран и формируют структуру миелина. Они непосредственно участвуют в активации тромбоцитов, удалении свободных радикалов, переработке холестерина в желчные кислоты и других реакциях (см. главы 6 и 13).
Нарушения биогенеза пероксисом сопровождается снижением их количества или полным отсутствием в клетках разных тканей организма, что связано с развитием пероксисомных болезней.
Пероксисомные болезни Общие данные
Пероксисомные болезни (ПерБ) обусловлены нарушениями структуры и функционирования пероксисом, т.е. связаны со сложными метаболическими реакциями, в том числе нарушениями транспорта белков через мембраны пероксисом и с работой мембранных рецепторов.
Популяционная частота ПерБ составляет 1:25-50 тыс.
Известно не менее 17 нозологий. Большинство из них наследуются по аутосомно-рецессивному типу (кроме Х-сцепленной адренолейкодистрофии), и практически все они проявляются в раннем детском возрасте (кроме гипероксалурии I типа и Х-сцепленной адренолейкодистрофии). Примерами наиболее распространенных форм ПерБ служат: адренолейкодистрофия, ризомиелическая точечная хондродистрофия, синдромы Рефсума и Цельвегера. Для этих заболеваний характерны генетическая гетерогенность и клинический полиморфизм. В частности, продемонстрировано развитие нескольких симптомокомплексов при мутациях в одном и том же гене и развитие одного симптомокомплекса при мутациях в разных генах. Причем
в первом случае различия обеспечивались аллельными сериями одного и того же гена или его плейотропным действием, а во втором случае - мутациями в разных генах (аллелях) и генокопированием сходной клинической картины (см. главу 4). Например, при ризомиелической точечной хондродисплазии выделены мутации в двух разных генах, хотя наблюдается один и тот же фенотип.
Широко известна морфофункциональная классификация ПерБ, основанная на двух критериях: количество пероксисом в клетках печени (морфологический критерий) и степень нарушения функций пероксисом (физиологический критерий). В соответствии с этой классификацией выделяют три группы ПерБ: первая группа отличается значительным снижением количества пероксисом в клетках печени и нарушением в них всех биохимических процессов; вторая группа характеризуется нормальным количеством пероксисом в клетках печени и нарушением в них только некоторых биохимических процессов; третья группа сопровождается полным подавлением функции пероксисом при нормальном их количестве в клетках печени.
В последние годы произошло уточнение этой классификации, и теперь среди всех ПерБ выделяют два основных класса. Первый класс болезней - это комплексные дефекты или генерализованное нарушение функций (пероксисомы отсутствуют или их число резко снижено). Примеры: болезнь Рефсума новорожденных, неонатальная адренолейкодистрофия, ризомиелическая точечная хондродисплазия, цереброгепаторенальный синдром Цельвегера и цельвегероподобный синдром. Второй класс болезней - это болезни, при которых структура пероксисом сохранена, но имеется мутация в гене, контролирующем единичный фермент (наблюдается его дефицит). Примеры: акаталазия, болезнь Рефсума взрослых, гипероксалурия (тип I), гиперпиколовая ацидемия, глютаровая ацидурия (тип III), дефицит би(три)-функционального белка, ди- и тригидроксихолестанемия, псевдонеонатальная адренолейкодистрофия и Х-сцепленная адренолейкодистрофия.
Механизмы патогенеза
Установлено, что развитие ПерБ связано с нарушениями:
• окисления ОДЦЖК, пристановой кислоты, ди- и тригидроксихолестановых кислот или окисления метаболитов жирных кислот;
• деградации фитановой и пипеколиновой кислот;
• транспорта белков-пироксинов через мембраны пероксисом и работой их рецепторов;
• синтеза плазмалогенов; при этом токсический эффект от накапливающихся в клетках метаболитов проявляется как атрофия коры надпочечников; демиелинизация белого вещества мозга и суданофильная лейкодистрофия с частичным периваскулярным накоплением лимфоцитов; фиброз печени.
Основные симптомы
При первом и втором классах ПерБ у больных наблюдается варьирующая экспрессивность симптомов, что связано с нозологией болезни. Например, тяжело протекает синдром Цельвегера, тогда как легкое течение отмечается при болезни Рефсума новорожденных, а средняя тяжесть болезни наблюдается в случае адренолейкодистрофии.
Клинические различия касаются времени манифестации, тяжести поражения нервной системы и продолжительности жизни.
Большинство ПерБ (15 из 17 нозологий) имеют выраженную неврологическую симптоматику. Основные симптомы: гепатомегалия, неврологические нарушения (задержка раннего психомоторного развития, мышечная гипотония, нейросенсорное снижение слуха), ретинопатия (дефекты пигментации сетчатки и побледнение дисков зрительных нервов) или катаракта, черепно-лицевой дисморфизм и (иногда) аномалии развития скелета (ризомиелический тип укорочения конечностей).
Отдельные нозологии пероксисомных болезней Синдром Цельвегера
Синдром Цельвегера (СЦ) - это гетерогенное заболевание, обусловленное мутациями в генах пероксинов 1-3, 5, 6 и 12 (гены РМР 70 и РМР35). Иногда выявляются делеция или инверсия в сегменте 7q11.3. Всего известны 11 групп комплементации СЦ.
Манифестация болезни начинается с рождения. У больных детей появляются судорожные припадки и геморрагический синдром. Характерны: внутриутробная гипотрофия (масса тела ниже 2500 г), черепно-лицевой дисморфизм: увеличенный в размерах лоб, монголоидный разрез глаз, периорбитальная полнота (припухлость) тканей, короткий вздернутый нос, микрогнатия. В первые месяцы жизни наблюдаются длительная желтуха и симптомы надпочечниковой недостаточности. Типичны резкая мышечная гипотония (вплоть до атонии) и поликистоз почек.
У всех больных выявляются пороки головного мозга (агенезия мозолистого тела, очаги демиелинизации в белом веществе мозга,
гидроцефалия, лизэнцефалия, полимикрогирия). Иногда наблюдаются врожденная катаракта и глаукома, пороки сердца и наружных половых органов. Во всех случаях характерна грубая задержка раннего психомоторного развития.
Продолжительность жизни больных резко снижена (обычно они умирают на первом году жизни).
При морфологическом исследовании биоптатов печени наблюдается резкое снижение или полное отсутствие пероксисом в клетках печени. Биохимические показатели - значительное снижение активности основных ферментов, участвующих в синтезе плазмалогенов (кожные фибробласты и эритроциты), метаболизме фитановой кислоты, бета-окислении жирных кислот, деградации пипеколоновой кислоты. Важный биохимический признак - повышение концентрации ОДЦЖК в моче.
Цельвегероподобный синдром
Цельвегероподобный синдром - это недостаточность пероксисомной 3-оксоацил-Коа-тиолазы. Симптоматика мало отличается от классического СЦ. Характерны те же клинические и биохимические признаки, но в клетках печени обнаруживаются нормальные пероксисомы, хотя их число уменьшено. Диагностика основана на выявлении сочетания основных симптомов (черепно-лицевой дисморфизм, мышечная гипотония, судороги, гепатомегалия с конъюгированной гипербилирубинемией, высокая концентрация ОДЦЖК и пристановой кислоты в плазме крови, изолированная недостаточность 3-оксоацил-Коа-тиолазы в фибробластах и биоптате печени).
Ген болезни картирован в сегменте 3р22-23. Выявлены мутации гена пероксисомной 3-оксоацил-Коа-тиолазы, приводящие к ее дефициту во всех тканях организма.
Неонатальная адренолейкодистрофия
Неонатальная адренолейкодистрофия - это гетерогенный синдром, сходный с СЦ. Описано 6 групп комплементации. На первый план выступает тяжелое течение болезни с признаками дегенерации ЦНС, проявляющейся демиелинизацией головного мозга.
Первичный молекулярно-генетический и биохимический дефекты не известны.
Болезнь Рефсума новорожденных
Инфантильная болезнь Рефсума - это гетерогенный синдром, сходный с СЦ, но с более легким течением и большей продолжительностью жизни. Первые 6 мес жизни выражены нарушения вскарм-
ливания, связанные с гепатомегалией, пролонгированной желтухой, гипохолестеринемией и гипотрофией. Поэтому кормить ребенка можно только через зонд.
Продолжительность жизни не превышает 1,5-3 лет.
Описаны две группы комплементации. Первичный молекулярногенетический и биохимический дефекты не известны.
Следует отметить, что с учетом особенностей течения указанных выше нозологий ПерБ можно выстроить схему снижения тяжести течения заболевания у больных детей: СЦ и цельвегероподобный синдром - неонатальная адренолейкодистрофия - болезнь Рефсума новорожденных.
Болезнь Рефсума взрослых
Болезнь Рефсума взрослых манифестирует в возрасте до 50 лет. Характеризуется накоплением фитановой кислоты в крови и клетках разных тканей вследствие недостаточности гидроксилазы фитановой кислоты. Проявляется высокой концентрацией белка в спинномозговой жидкости, мозжечковой атаксией, периферической полиневропатией, пигментным ретинитом. Не постоянны: пороки развития сердца и скелета, аносмия, ихтиоз, нейросенсорная глухота.
Первичный молекулярно-генетический и биохимический дефекты неизвестны.
Ризомиелическая точечная остеохондродисплазия
Ризомиелическая точечная остеохондродисплазия - это изолированная недостаточность дегидроксиацетонфосфатацилтрансферазы (алкилдегидроксиацетонфосфатсинтазы). Выделяют классическую форму (одна группа комплементации) и атипичную форму (две группы комплементации). Манифестация болезни происходит в неонатальном периоде, реже - на первом году жизни. Наблюдается острое течение. Большинство детей умирают на первом году жизни.
Фенотип: задержка роста, микроцефалия, плоское лицо с запавшей переносицей, антимонголоидный разрез глаз, катаракта, диспропорциональное укорочение конечностей (преимущественно в проксимальных отделах), множественные контрактуры суставов, ихтиозоформная дисплазия кожи и алопеция. Характерны: тяжелая задержка психомоторного развития, изменения на КТ и МРТ головного мозга.
При этом заболевании наблюдаются нарушения биосинтеза плазмалогенов (вследствие дефицита дегидроксиацетонфосфата и алкилирующей дегидроксиацетофосфатсинтетазы), окисления фитановой
кислоты, стимуляции зрелой формы тиолазы пероксисом (без накопления ОДЦЖК в плазме крови).
Морфологически выявляется уменьшенное число или полное отсутствие пероксисом, увеличенные их размеры в клетках печени при неизмененной их структуре в фибробластах.
На рентгенограмме - очаги точечной минерализации костей - кальцификаты или нарушение эндохондриального окостенения, оссификация вентральной и дорсальной части позвонков (исчезает после двухлетнего возраста), симметричное укорочение и деформация метафизов плечевой и бедренной костей.
Ген болезни не картирован. Предполагается биохимический дефект импорта пероксинов.
Х-сцепленная адренолейкодистрофия
Х-сцепленная адренолейкодистрофия относится к одной из наиболее распространенных форм НБО (частота - 1:20 тыс.). Характеризуется значительной вариабельностью возраста манифестации (от 2,5 до 10 лет), разной экспрессивностью признаков и прогредиентностью течения. Выделяют ряд форм. Первая форма встречается наиболее часто. Манифестация в детском возрасте (между 5 и 10 годами жизни). Характеризуется наиболее тяжелыми проявлениями: отсутствие зрения, речи и слуха, полная обездвиженность. Питание ребенка возможно только через зонд. В дальнейшем развиваются демиелинизация и воспалительные процессы в тканях мозга; появляются судороги. Смерть наступает через несколько лет. Вторая форма также часто встречается, начинается во взрослом возрасте. Характеризуется дисфункцией тазовых органов и прогрессирующим парапарезом вследствие поражения спинного мозга. При обеих формах может быть надпочечниковая недостаточность, проявляющаяся одновременно с неврологической симптоматикой.
Кроме этих форм выделяют: болезнь Адисона (без неврологических симптомов, но с характерной биохимической картиной Х-сцепленной адренолейкодистрофии), подростковую форму и церебральную форму взрослых. Все указанные пять форм могут встречаться в пределах одной семейной родословной.
Первичный молекулярно-генетический дефект - мутации структурного гена интегрального белка мембраны пероксисом (ген ALDP), обусловливающие дефицит этого белка в нейронах мозга, коре надпочечников, лейкоцитах, эритроцитах, фибробластах. Ген локализован в сегменте Хq28. Первичный биохимический дефект связан с
накоплением ОДЦЖК в плазме крови (вследствие нарушения бетаокисления в пероксисомах).
Псевдонеонатальная адренолейкодистрофия
Псевдонеонатальная адренолейкодистрофия - это недостаточность пероксисомной ацил-Коа оксидазы. Болезнь гетерогенна, имеет симптоматику, сходную с таковой неонатальной адренолейкодистрофии, но без признаков черепно-лицевого дисморфизма. Характеризуется ранним появлением судорог и выраженной задержкой психомоторного развития.
Ген болезни не локализован.
Дефицит би-(три-)функционального белка пероксисом
Дефицит би-(три-)функционального белка пероксисом имеет сходные признаки с неонатальной адренолейкодистрофией. Число и морфология пероксисом не изменены.
Накопление метаболитов в нервной ткани сопровождается интоксикацией, что морфологически проявляется демиелинизацией, микронодулярным циррозом, нарушениями цитоархитектоники и полимикрогирией головного мозга, фиброзом и холестазом печени.
Болезнь манифестирует на первом году жизни. Продолжительность жизни - до трех лет.
Ген не локализован. Предполагаются мутации би-(три-)функциональных белков во всех клетках, кроме эритроцитов.
Би- и тригидроксихолестановая ацидемия
Би- и тригидроксихолестановая ацидемия манифестирует в неонатальном периоде, проявляется гепатомегалией, тяжелой печеночной недостаточностью, отсутствием психомоторного развития и роста, черепно-лицевым дисморфизмом. Характеризуется накоплением токсических метаболитов ди- и тригидроксихолестановой и пристановой кислот. Смерть наступает в возрасте от 6 мес до 2 лет.
Ген не локализован. Предполагается первичный биохимический дефект, связанный с недостаточностью холестаноил-Коа оксидазы.
Глютаровая ацидурия, тип III
Описан единичный случай глютаровой ацидурии III типа у новорожденной девочки - с затруднениями при кормлении, рвотой после приема пищи, задержкой развития.
Отмечалась повышенная экскреция с мочой глютаровой кислоты. Ген не локализован. В качестве первичного биохимического дефекта предполагается дефицит глютарат-Коа оксидазы.
Гипероксалурия, тип I
Гипероксалурия, тип I, отличается выраженным клиническим полиморфизмом. Выделяют неонатальную, детскую и взрослую формы, различающиеся тяжестью проявления. Сопровождается нефролитиазом и нефрокальцинозом, приводящими к ранней почечной недостаточности (до 20 лет) вследствие системного оксалоза экстрапочечных тканей. Описаны артриты и атриовентрикулярная блокада. Больные умирают в течение одного года от уремии или других осложнений (гематурия, острая почечная колика).
Идентифицированы мутации структурного гена пероксисомной аланинглиоксилат-аминотрансферазы (АГТ), приводящие к ее дефициту в печени. Ген локализован в сегменте 2q36-37. Описана мутация гена, приводящая не к снижению активности, а к нарушению компартментализации фермента G630A в пероксисомах.
Гиперпиколовая ацидемия
Фенотип больного с гиперпиколовой ацидемией сходен с фенотипом при СЦ (см. выше). Гиперпиколовая ацидемия характеризуется высоким уровнем гиперпиколовой кислоты, сочетанием симптомов генерализованного поражения пероксисом и болезни Жуберта (дисплазия червя мозжечка, изменения печени, нарушения дыхания). Больные умирают в возрасте до трех лет. Первичный молекулярногенетический и биохимический дефекты не известны.
Акталаземия
Акталаземия, или болезнь Такахары, имеет доброкачественное течение, проявляющееся изъязвлением и воспалением слизистой оболочки полости рта. Частота распространения в Японии - 1:250 тыс. В 25-50% случаев болезнь осложняется грануломатозными псевдоопухолями в полости носа и придаточных пазухах. Ген болезни локализован в сегменте 11р13.
Биохимический дефект связан со снижением активности каталазы в эритроцитах.
Подходы к диагностике
Принцип диагностики ПерБ - это анализ сочетания данных клинического обследования и общих лабораторных данных. Основные дифференциально-диагностические признаки:
• морфологический и физиологический критерии: количество пероксисом и снижение их функции (см. выше);
• клинические данные: выраженная неврологическая симптоматика, надпочечниковая и печеночная недостаточность, в том
• числе неврологические расстройства, нарушения функций надпочечников и печени, снижение остроты зрения и слуха; часто манифестации болезни предшествуют: нарушения вскармливания, связанные с гепатомегалией и пролонгированной желтухой, задержка психомоторного развития; при осмотре пациентов в ряде случаев определяются арефлексия, гипорефлексия, мышечная (генерализованная) гипотония, нейросенсорная тугоухость, пигментная дегенерация сетчатки, судороги; лабораторные данные: увеличение концентрации в плазме крови ди- и тригидрокси-5-бета-холестановой кислоты (соответственно ДГХК и ТГХК), ОДЦЖК и фитановой кислоты, пристановой кислоты; определение гипохолестеринемии, повышения уровня трансаминаз, уровня связанного билирубина и некоторых факторов свертывания крови и др. К основным клинико-инструментальным и клиниколабораторным методам диагностики ПерБ относятся: аудиометрия, исследование глазного дна, электронная микроскопия биоптатов печени, ЭЭГ, КТ и МРТ головного мозга, УЗИ печени и почек.
Подходы к лечению
Методы радикальной терапии ПерБ не разработаны. В некоторых случаях - пересадка печени и почек (гипероксалурия, тип I). При Х-сцепленной адренолейкодистрофии высокоэффективна гормонотерапия надпочечниковой недостаточности.
В остальных случаях ПерБ назначается симптоматическая терапия: купирование судорог антиконвульсантами, мышечных болей и спазмов - балкофеном, нормализация сна и т.д.
ХАРАКТЕРИСТИКА МИТОХОНДРИЙ И МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Митохондрии
Митохондрии впервые описаны Р. Альтманом в 1894 г., однако окончательное название «митохондрии» им дал К. Бенда в 1897 г. Митохондрии состоят из четырех компонентов: матрикс, окруженный внутренней мембраной (кристы и трубочки), межмембранное пространство и наружная мембрана. В матриксе содержатся кольцевая митохондриальная ДНК (мтДНК), специфические мРНК, тРНК и
собственные рибосомы, которые по размерам меньше, чем рибосомы цитоплазмы клетки. Митохондрии присутствуют во всех клетках, кроме зрелых эритроцитов. Их число в одной клетке может достигать нескольких сотен. Много митохондрий в ооцитах и, наоборот, в сперматозоидах их всего четыре, и они, как сказано выше, не проникают в яйцеклетку при оплодотворении.
Основная функция митохондрий - физико-химические реакции, связанные с тканевым дыханием и синтезом АТФ (аденозинтрифосфат), происходящим благодаря сопряжению процессов окисления и фосфорилирования. АТФ запасает и хранит энергию для окисления. Его синтез происходит на внутренних мембранах митохондрий, площадь которых многократно больше площади внешней мембраны за счет крист (гребней), выступающих в пространство матрикса.
Кроме указанных процессов, у митохондрий имеются функции: превращение лимонной кислоты; синтез аминокислот, гема, витаминов, липидов и пиримидинов; окисление жирных кислот. Обеспечение такого многообразия функций требует присутствия в митохондриях большого количества полипептидов, синтезируемых самой соматической клеткой (около 10%).
Геном митохондрий полностью расшифрован (см. главу 2).
Хемиосмотическая теория
Важной вехой в развитии биоэнергетики митохондрий стала хемиосмотическая теория, предложенная П. Митчеллом в 1961 г. Согласно этой теории, формирование запасов энергии в митохондриях происходит в результате электрохимической активности протонов, их перемещения из матрикса через внутреннюю мембрану в межмембранное пространство с помощью протонного насоса, функционирование которого зависит от переноса электронов по дыхательной цепи митохондрий в ходе окисления.
В дыхательной цепи митохондрий участвуют пять комплексов субъединиц, кодируемых как ядерной ДНК, так и мтДНК. Их общее количество достигает 10-35 и 1-7 субъединиц соответственно. При этом небольшая по размерам молекула мтДНК кодирует 13 полипептидов, входящих в состав четырех из пяти ферментных комплексов. Из них 7 полипептидов включены в комплекс I (NADH-дегидрогеназа с коэнзимом Q оксидоредуктазы), 1 полипептид служит субъединицей комплекса III (коэнзим Q с цитохром-с-оксидоредуктазой); 3 полипептида являются субъединицами комплекса IV (цитохром-с-оксидазы) и 2 полипептида - субъединицами комплекса V (АТР-синтетазы).
В свою очередь, субъединицы белков комплекса II (в том числе сукцинатдегидрогеназа) кодируются исключительно генами ядерной ДНК. Все белки митохондрий, кодируемые ядерной ДНК, синтезируются в рибосомах цитоплазмы и транспортируются в одно из четырех структурных образований: внутреннюю и наружную мембраны, матрикс и межмембранное пространство. На рис. 72 приведена схема пространственного расположения пяти комплексов дыхательной цепи митохондрий.
Митохондриальные болезни Общие данные
МТБ - это обширная группа заболеваний, обусловленных разобщением процессов окисления и фосфорилирования в митохондриях. Поэтому их часто называют болезнями окислительного фосфорилирования или OXPHOS-болезнями.
Среди МТБ выделяют четыре класса, включая один класс ненаследственных (приобретенных) и три класса наследственных МТБ. Первый класс - приобретенные болезни, связанные с токсическим действием на митохондрии. Второй класс - болезни, обусловленные дефектами
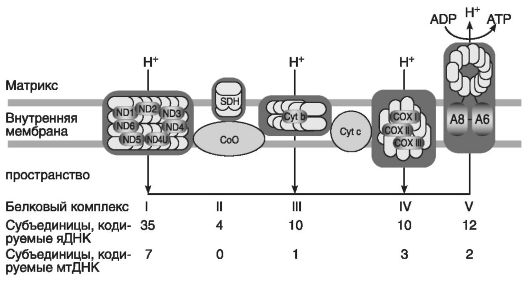
Рис. 72. Митохондриальная дыхательная цепь (по Васильеву В.Б., 2005). Число субъединиц указано под стрелками. Протоны Н+ перекачиваются из матрикса в межмембранное пространство с помощью комплексов I,II,IV. Возвращение в матрикс осуществляется комплексом V (АТР-синтетазы) и сопровождается образованием АТР. Коэнзим Q10 (СоО_) и цитохром с Cyt c) - протеины, кодируемые яДНК
ядерной ДНК: дефицит субстратов транспорта; дефицит субстратов утилизации; нарушение цикла Кребса; нарушение процессов окисления и фосфорилирования; дефекты дыхательных цепей; дефекты белков, импортируемых в митохондрии. Третий класс - болезни, обусловленные дефектами мтДНК: точковые мутации генов, кодирующих структурные белки; большие по размерам делеции генов структурных белков. Четвертый класс - болезни, связанные с интергеномными дефектами. Сюда относится одно аутосомно-доминантное заболевание, ассоциированное с множественными делециями мтДНК. Механизмы патогенеза
При OXPHOS-болезнях в митохондриях нарушаются один или сразу несколько биохимических процессов.
• Транспорт субстратов, контролируемый транслоказами, которые переносят глутамат, дикарбоновые кислоты, длинно- и среднецепочечные жирные кислоты, ионы кальция.
• Окисление субстратов мультиферментным пируватдегидрогеназным комплексом; дефицит этого комплекса развивается вследствие мутаций, обусловливающих накопление пирувата, переходящего в лактат и проявляющегося лактат-ацидозом.
• Цикл трикарбоновых кислот (цикл Кребса), центральный путь метаболизма углеродсодержащих молекул, ведущий к полному аэробному разложению пирувата.
• Работа дыхательной цепи - из-за появления изолированных и сочетанных ее дефектов. Известно, что дыхательная цепь митохондрий состоит из пяти мультиферментных комплексов, из которых четыре осуществляют транспорт электронов, а пятый катализирует АТР. Эта цепь находится под двойным контролем (со стороны ядерной и мтДНК). В ней участвуют пять цитохромов. При этом электроны транспортируются в дыхательную цепь, а освобождающаяся энергия прокачивает протоны через внутреннюю мембрану в межмембранное пространство митохондрий. Очевидно, что в любом из перечисленных звеньев дыхательной цепи митохондрий может возникнуть дефект.
• Сопряжение процессов окисления и фосфорилирования, процессов энергетического обмена в митохондриях. Показано, что нарушения процессов энергетического обмена в митохондриях обусловливают не только развитие OXPHOS-болезней (их 21 нозология), но и развитие многих МБ и БНП, симптоматика которых тесно связана с энергетической недостаточностью тканей и орга-
нов, сильно зависящих от запасов АТР. Среди таких органов и тканей: головной мозг, островки Лангерганса, сердечная мышца, скелетная мускулатура, сетчатка глаз и др. Дегенеративные изменения на фоне сниженного синтеза АТР также происходят в митохондриях пожилых людей при развитии болезней Альцгеймера и Паркинсона, кардиомиопатий и сахарного диабета. Кроме того, в последние годы отмечено, что к механизмам процессов старения и связанных с ними дегенеративных изменений относятся 4 события, сопровождающих нарушения окислительного фосфорилирования, в том числе:
• дестабилизация митохондрий как главного источника энергии для всех тканей и органов;
• существенное сокращение запасов АТР в митохондриях;
• появление и накопление мутаций в ядерной ДНК и мтДНК, участвующих в сопряжении процессов окисления и фосфорилирования в митохондриях;
• возрастное снижение интенсивности процессов окисления и фосфорилирования в митохондриях.
Характерной особенностью МТБ у детей является отсутствие какой-либо симптоматики в первые годы жизни, что объясняется гетероплазмией или одновременным присутствием в клетках растущего организма сотен митохондрий, содержащих как нормальную, так и мутантную мтДНК. С возрастом количество мутантной мтДНК в клетках нарастает, и когда оно достигает некоего «порогового уровня», энергетический метаболизм таких клеток уже не обеспечивает потребности энергоемких тканей и органов в АТР, что ведет к их дегенерации и появлению OXPHOS-болезней.
Следует отметить, что накопление мутантных копий в мтДНК объединяет эти болезни с болезнями экспансии нуклеотидных повторов, в основе которых лежит накопление кодирующих и некодирующих кодонов (три-, тетра- и пентаплетов) в ядерной ДНК, и в случае превышения их «порогового уровня» развивается полная или динамическая мутация (см. главу 27). Чем старше человек, тем OXPHOS-болезни, как правило, тяжелее протекают и заканчиваются летальным исходом.
Причины МТБ хорошо изучены: это точковые мутации и обширные делеции в мтДНК или ядерной ДНК, в которой возникают межгеномные сигнальные эффекты, прямо или косвенно приводящие к феномену деплеции или истощения митохондрий.
Основные симптомы
Основные симптомы МТБ связаны с поражением нервной системы, что обусловлено высокой потребностью в энергии нервной ткани и высокой чувствительностью нейронов к недостатку их энергоснабжения. Характерны раннее начало болезни на втором-третьем году жизни ребенка, прогрессирующее течение и вовлечение многих систем организма. Симптомы со стороны ЦНС: атаксия, задержка психомоторного развития, миоклонус-эпилепсия, мышечная гипотония или гипертония, а также деменция и инсультоподобные состояния. Часто поражаются: сердце (кардиомиопатия, нарушение проводимости миокарда) и органы зрения (атрофия зрительных нервов, катаракта, наружная офтальмоплегия, пигментная дегенерация сетчатки). Возможны: гепатомегалия, диарея, нанизм, непереносимость белков, поражение поджелудочной железы и других органов, тубулопатия.
Лабораторные данные: ацидоз, изменение соотношения пируват/ лактат с преобладанием пирувата (в норме оно равно 20). Отмечается повышение концентрации кетоновых тел.
Отдельные формы митохондриальных болезней ARP-синдром
ARP-синдром - это синдром атаксии, нейропатии и пигментного ретинита. Описан в 1990 г. Симптоматика проявляется с трехлетнего возраста. Характерны: атаксия, деменция, задержка развития, слабость в проксимальных отделах конечностей, сенсорная нейропатия, судороги, пигментный ретинит. Молекулярно-генетический дефект - точковая мутация мтДНК.
Атрофия Лебера
Наследственная оптическая нейроретинопатия Лебера (LHON) характеризуется быстро нарастающей слепотой у лиц в возрасте до 20 лет, вызванной двусторонней атрофией зрительного нерва. В дальнейшем развивается ретробульбарная нейропатия. При исследовании глазного дна выявляются отек диска зрительного нерва и поражение сосудов сетчатки (микроангиопатия). У больных наблюдаются двигательные, неврологические и психические расстройства: гиперрефлексия, дистония, нарушения координации, периферическая нейропатия, тремор. Преобладают больные мужского пола.
При КТ выявляется общая атрофия коры головного мозга. На ЭКГ - нарушения сердечной проводимости и сократимости. Молекулярно-генетический дефект - точковая мутация. Описаны
четыре типа мутаций мтДНК, наиболее частой из которых является замена гуанина на аденин в положении 11 778. Это основание входит в состав нуклеотидов гена, кодирующего четвертую субъединицу НАДН-дегидрогеназы. Такая замена развивается в ряде случаев у пациентов с гетероплазмией, имеющей тенденцию к гомоплазмии по мутантной мтДНК. При этом в ходе трансляции мутантной мтДНК аргинин в положении 340 заменяется на гистидин.
Также предполагается существование Х-сцепленного фактора, модифицирующего экспрессию дефекта мтДНК. Аналогичный дефект отмечался при миссенс-мутациях мтДНК, и всего были выделены три критерия, позволяющих считать такие мутации непосредственно связанными с атрофией Лебера.
В числе трех морфологических и биохимических признаков синдрома LHON значатся: гетероплазмия с нарастанием доли мутантной мтДНК; более высокая доля мутантной мтДНК у больных пациентов, чем в митохондриях здоровых лиц; замена эволюционно консервативной аминокислоты, постоянно присутствующей в нормальном белке.
При анализе семейных родословных у больных часто встречаются точковые мутации мтДНК, последовательно накапливающиеся в поколениях одной семьи, что, по-видимому, происходит в результате снижения эффективности переноса электронов в целом в дыхательной цепи митохондрий.
Болезнь Кирнс-Сэйра
Болезнь Кирнс-Сэйра известна с 1958 г. Большинство случаев спорадические. Характерно начало в первом десятилетии жизни. Основные симптомы: гиперпаратиреоидизм, мозжечковые нарушения (атаксия), нарушение сердечной проводимости, пигментная ретинопатия, прогрессирующая офтальмоплегия. Реже встречаются: гипопаратиреоз, изолированный дефицит гормона роста, низкий рост, сахарный диабет, снижение слуха, судороги, УО. Диагноз ставится при обнаружении лактат-ацидоза и определении в биоптате мышц РКМВ.
При ДНК-анализе выявляются крупные делеции мтДНК (предполагается, что это результат мутации в зиготе).
Болезнь Лея
Болезнь Лея - это подострая некротизирующая энцефаломиелопатия с атаксией, гипотензией, мышечной слабостью, нарушениями дыхания, недостаточной массой тела, пониженной остротой
зрения и слуха. Болезнь манифестирует в период новорожденности или на первом году жизни. Наблюдаются как бессимптомные, так и тяжелые формы. Выделяют неонатальную и позднюю формы. Неонатальная форма характеризуется респираторным дистресссиндромом, мышечной гипотонией, судорогами, трудностями вскармливания. Смерть наступает в первые недели или на первом году жизни ребенка.
Поздняя форма характеризуется апноэ, атрофией зрительных нервов и косоглазием, задержкой роста, мозжечковыми нарушениями, рвотой, судорогами, тетрапарезом.
Молекулярно-генетический дефект - делеция мтДНК, приводящая к дефициту пируватдегидрогеназного комплекса и цитохром С-оксидазы.
Доминантно наследуемая митохондриальная миопатия
Доминантно наследуемая митохондриальная миопатия наиболее часто встречается как форма МТБ с поздним началом. Она манифестирует на третьем десятилетии жизни с проявления офтальмоплегии и слабости мышц верхних и нижних конечностей.
В дальнейшем присоединяется двусторонняя катаракта.
MELAS-синдром
MELAS-синдром также называют синдромом митохондриальной миопатии, энцефалопатии, молочнокислого ацидоза (лактатацидоза) и инсультоподобных эпизодов. Описано более 70 случаев. Синдром развивается до 40-летнего возраста. Характерны: деменция, инсультоподобные приступы, мигренеподобные боли, судороги. Нередки: гемианопсия и гемипарезы, мышечная слабость в конечностях, низкий рост, снижение слуха. У всех больных выявляются лактат-ацидоз и РКМВ в биоптатах мышц. У каждого третьего больного обнаруживаются кальцификаты базальных ганглиев, а у каждого десятого - прогрессирующая наружная офтальмоплегия.
Молекулярно-генетический дефект - точковая мутация мтДНК.
MERRF-синдром
MERRF-синдром - это прогрессирующая миоклональная эпилепсия с РКМВ. Описано 25 случаев. Основные симптомы: деменция, мозжечковые нарушения (прогрессирующая атаксия), нарушение глубокой чувствительности, приступы генерализованных судорог, снижение слуха. Иногда выявляются атрофия зрительных нервов и низкий рост.
На аутопсии - губчатая дегенерация задних отделов головного мозга и спиноцеребеллярного тракта. В биоптате мышц - РКМВ. Молекулярно-генетический дефект - точковая мутация мтДНК (см. также главу 27).
MIMYCA-синдром
MIMYCA-синдром - это синдром кардиомиопатии или миопатии без вовлечения ЦНС. Молекулярно-генетический дефект - точковая мутация мтДНК.
Синдром Альпера
Синдром Альпера - это прогрессирующая детская полидистрофия. Характерны: деменция, дисфункция печени, слепота, спазмы, судороги, обусловленные специфической дегенерацией тканей головного мозга.
Молекулярно-генетический дефект не установлен. Синдром Вольфрама
Синдром Вольфрама характеризуется манифестацией в возрасте от одного года до второго десятилетия жизни. Характерны: атрофия зрительных нервов, задержка психомоторного развития, отставание в росте, пигментная дегенерация сетчатки, сахарный диабет.
Молекулярно-генетический дефект - делеция мтДНК.
Синдром Пирсона
Для синдрома Пирсона характерны: гипопластическая анемия и панцитопения раннего детского возраста в сочетании с атрофией селезенки и фиброзом поджелудочной железы. Болезнь обусловлена делецией мтДНК.
Хроническая прогрессирующая наружная офтальмоплегия Хроническая прогрессирующая наружная офтальмоплегия клинически напоминает синдром Кирнс-Сэйра. Сочетается с блефароптозом, параличом наружных мышц глазного яблока и расстройствами ЦНС. Молекулярно-генетический дефект не установлен.
Подходы к диагностике
Клиническое проявление отдельных форм МТБ может изменяться, например, в случаях «обменов» симптомами между разными нозологиями, что затрудняет их диагностику. Только в последние годы, когда были идентифицированы разные типы мутаций мтДНК, удалось систематизировать нозологии с учетом обусловливающих их развитие молекулярных механизмов, связанных с расстройствами процессов окисления и фосфорилирования в митохондриях, и каждый синдром стал определяться в зависимости от конкретной мутации. Молекулярно-
генетическая диагностика МТБ направлена на идентификацию мутаций мтДНК, включая выявление нуклеотидных замен в генах, кодирующих полипептиды (миссенс-мутации); нуклеотидных замен в генах, кодирующих тРНК; делеций и вставок в мтДНК; мутаций, изменяющих число копий мтДНК. Важными для диагностики являются морфологические изменения в мышечной ткани - РКМВ. Этот признак более свойственен состояниям, обусловленным крупными делециями мтДНК. Основными диагностическими тестами служат:
• обнаружение в биоптатах тканей гетероплазии или неравномерного распределения митохондрий с мутантной ДНК по тканям в эмбриональном периоде, что ведет к поражению мышц, печени и др.;
• выявление при ДНК-анализе в клетках разных тканей протяженных делеций или точковых мутаций мтДНК, которые реплицируются быстрее и поэтому быстро накапливаются;
• повышение концентрации метаболитов (в крови и мышечных биоптатах), являющихся биохимическими маркерами нарушений окислительного фосфорилирования;
• нарушение активности ферментов дыхательной цепи митохондрий в мышечном биоптате (дефицит дегидрогеназы, цитохромс-оксидазы).
Подходы к лечению
Лечение МТБ в основном симптоматическое. Оно направлено на снятие явлений ацидоза и выведение лактата. Применяемая терапия: жирная диета (без карбонгидратов), витамины С, К и У, кокарбоксилаза, коэнзим Q.
В последние годы был разработан патогенетический подход: стимуляция продукции АТР и снижение токсического воздействия патологических метаболитов.
Такое лечение ведется в соответствии с утвержденными протоколами, предусматривающими ряд обязательных назначений:
• купирование церебрального лактат-ацидоза дихлоацетатом (таблетки или внутривенные инъекции): соответственно 15 мг несколько раз или 200 мг один раз в день;
• прием коэнзима Q10 в дозе 100 мг (на три порции в день) с добавлением сукцината или прием идебенола (сходен с коэнзимом Q10);
• витаминотерапия аскорбатом (витамин С) в дозе 1 г в день, менадионом (витамин КЗ) в дозе 40-80 мг в день, тиамином (300 мг в день), рибофлавином (100 мг в день) и витамином Е (антиоксидант) по 500 мг 2 раза в день.