Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ЧАСТЬ 3. МОЛЕКУЛЯРНЫЕ БОЛЕЗНИ С ТРАДИЦИОННЫМ И НЕТРАДИЦИОННЫМ НАСЛЕДОВАНИЕМ. ОТДЕЛЬНЫЕ КЛАССЫ И НОЗОЛОГИИ. ПРОФИЛАКТИКА НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ ГЛАВА 21 МОНОГЕННЫЕ БОЛЕЗНИ
Общая характеристика
Моногенные болезни (МБ) подчиняются закономерностям менделевского наследования. Они обусловлены точковыми мутациями в единичных генах (см. главу 4).
По данным ВОЗ, среди новорожденных детей МБ встречаются с частотой 30 на 1000 (3%). В отдельных регионах России этот показатель достигает 42-65 больных детей - 4,2-6,5%. В структуре общей смертности детей до 5 лет включительно на долю МБ приходится 12-14%. Известно более 4500 нозологий МБ. Они широко представлены во всех областях медицины. Так, в дерматологии их более 400, неврологии - около 350, оториноларингологии и офтальмологии - по 200-250 нозологий и т.д.
Популяционный риск развития МБ связан с неравномерностью распределения патологических генов этих болезней. Раньше считалось, что МБ, встречающиеся с частотой 1:10 000 человек и более, - это частые болезни; если их частота менее 1:100 000 человек - это редкие болезни; МБ с частотой от 1:10 000 до 1:100 000 - широко распространенные МБ. Последнее определение постепенно вытеснило два первых, ибо в его пользу свидетельствуют данные о том, что, например, в популяциях Средней Азии доля относительно частых аутосомно-рецессивных форм МБ мала по сравнению с таковой в европейских популяциях, а спектр МБ чрезвычайно широк, хотя и представлен в основном редко встречающимися нозологиями.
В настоящее время список МБ, гены (аллели) или локусы которых картированы на хромосомах, насчитывает 1500 нозологий, тогда как генов самих МБ идентифицировано всего 1100. В этом несоответствии нет противоречий, ибо идентифицированы разные нозологии МБ, связанные с разными точковыми мутациями в одних и тех же генах, и при 400 нозологиях это мутации по типу аллельной серии (см. главу 5). Установлен первичный генный дефект (замена основания, внутригенная делеция и др.) или биохимический дефект (ошибка синтеза или модификации белковой молекулы) почти для
500 генов МБ.
ПРИНЦИПЫ КЛАССИФИКАЦИИ
К настоящему времени разработаны многочисленные классификации МБ. Однако ни одна из них не является исчерпывающей и полностью отражающей специфику этой патологии. Поэтому целесообразно рассмотреть только основные принципы классификаций.
Классификация по типу наследования
Тип наследования МБ связан с локализацией патологического гена в аутосоме или гоносоме и зависит от доминантности или рецессивности гена (см. главу 4). Мужчины и женщины поражаются с одинаковой частотой в случае аутосомного типа наследования и с разной частотой в случае сцепленного с полом или ограниченного полом наследования. На основе этого принципа выделяют:
• аутосомно-доминантные болезни (ахондроплазия, несовершенный остеогенез, нейрофиброматоз Реклингаузена, ретинобластома, семейная гиперхолестеринемия, синдромы Крузона, Марфана, Холт-Орама, хорея Гентингтона);
• аутосомно-рецессивные болезни (алкаптонурия, альбинизм, атаксия-телеангиэктазия, болезнь Тея-Сакса, врожденная гиперплазия коры надпочечников - АГС), галактоземия, МВ, недостаточность аденозиндезаминазы, пигментная ксеродерма, ФКУ);
• Х-сцепленные доминантные болезни (гипофосфатемия - витамин D-резистентный рахит, орофациодигитальный синдром, синдром недержания пигмента);
• Х-сцепленные рецессивные болезни (гемолитическая анемия, гемофилия АиВ, миодистрофия Дюшенна-Беккера, недостаточность глюкозо-6-фосфат-дегидрогеназы, синдромы Леша- Найяна, Лоу, Мартина-Белла, Менкеса).
Классификация по органному и системному типу
• Болезни желудочно-кишечного тракта (синдромы мальабсорбции, в том числе врожденный дефицит лактазы, врожденная хлоридная диарея, дефицит энтерокиназы, непереносимость белка коровьего молока и белка сои, первичная гипомагнеземия, целиакия).
• Болезни кожи и ее придатков (ангидротическая и гидротическая эктодермальная дисплазия, буллезный эпидермолиз, веретенообразная аплазия волос, врожденная аплазия кожи, вялая кожа, ихтиоз и ихтиозоформные дерматозы, ладонно-подошвенные
кератодермии (гиперкератозы), пигментная ксеродерма, синдром Блума, синдром недержания пигмента).
• Болезни нервной системы. Это заболевания нервной ткани, в том числе болезни пирамидной и экстрапирамидной систем, мозжечка, ангиоматозы Гиппеля-Линдау и Штурге-Вебера, атаксия-телеангиэктазия, нейрофиброматоз, туберозный склероз, миотонии (болезнь Томсена и др.), прогрессирующие мышечные дистрофии (например, болезнь Дюшенна-Беккера), спинальные мышечные атрофии (болезни Верднига-Гоффмана и Кугельбергера-Веландер).
• Болезни органов дыхания (альвеолярный микролитиаз, идиопатический диффузный фиброз легких, изолированный легочный гемосидероз, наследственный спонтанный пневмоторакс, первичная легочная гипертензия, синдром Картегенера, трахеобронхомегалия).
• Болезни почек и мочевыводящих путей (синдром Альпорта, болезнь де Тони-Дебре-Фанкони, глюкодиабет, гипофосфатемический рахит, наследственный нефрит, несахарный диабет, поликистозная болезнь - детский и взрослый типы, почечный канальцевый ацидоз).
• Болезни сердечно-сосудистой системы (семейные формы гиперхолестеринемии, гиперлипопротеинемии и гипертриглицеринемии, наследственный амилоидоз, недостаточность карнитина, семейная недостаточность липопротеиновой липазы).
• Болезни соединительной ткани и скелета (ахондроплазия, врожденная косолапость, метатропическая дисплазия, муколипидозы, мукополисахаридозы, несовершенный остеогенез, псевдоахондроплазия, спондилодисплазия и множественная эпифизарная дисплазия, синдромы Марфана, Элерса- Данлоса).
• Болезни эндокринных органов: АГС, врожденный гипотиреоз, гипофизарный нанизм).
• Болезни других органов и систем организма.
Классификация по типу обмена веществ
Благодаря данной классификации многие МБ называются НБО. Среди них выделяют:
• болезни аминокислот (алкаптонурия, альбинизм, гистидинемия, гомоцистинурия, лейциноз, ФКУ);
• болезни биосинтеза кортикостероидов (АГС, гипоальдостеронизм);
• болезни лимфоцитов и лейкоцитов (недостаточность аденозиндезаминазы, септический гранулематоз);
• болезни липидов (ганглиозидозы, гиперлипидемия, гиперхолестеринемия, муколипидозы, сфинголипидозы, цереброзидозы);
• болезни металлов (болезни Вильсона-Коновалова, Менкеса, семейный периодический паралич);
• болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглер-Найяра, Порфирии);
• болезни пуринов и пиримидинов (оротовая ацидурия, подагра, синдром Леша-Найяна);
• болезни транспорта систем почек (болезнь де Тони-Дебре- Фанкони, витамин D-резистентный рахит, тубулопатии);
• болезни углеводов (галактоземия, гликогенозы, мукополисахаридозы, непереносимость фруктозы);
• болезни эритрона: анемия Фанкони, гемолитические анемии, недостаточность глюкозо-6-фосфатдегидрогеназы.
Кроме того, в рамках НБО выделяют особую группу болезней накопления, тезаурисмозов (см. главу 4). Эта группа МБ объединяет НБО, связанные с дефицитом ферментов лизосом (наследуются с цитоплазмой по материнской линии). Такие НБО проявляются прогрессирующим накоплением предшественников метаболических реакций в клетках разных тканей, например отложением макромолекул гликогена, гликопротеинов, гликолипидов, мукополисахаридов, сфинголипидов, которые в условиях нормального обмена веществ «самоперевариваются» лизосомальными ферментами, что необходимо при очищении организма от продуктов жизнедеятельности и мертвых клеток.
Классификация по этиологии
• Болезни с установленным первичным молекулярным (биохимическим) дефектом. Число таких болезней продолжает неуклонно расти: если по каталогу В. Мак-Кьюсика в 1990 г. насчитывалось около 100 нозологий, то в конце 1993 г. их было уже 328, в 2001 г. - 700, а в 2006 г. - 1500 (33,3%).
• Болезни с неустановленным первичным молекулярным (биохимическим) дефектом. На эти заболевания приходится оставшаяся
часть МБ (66,7%).
В последние годы появилось множество подтверждений ведущей роли первичного молекулярного дефекта. Например, ранее труднообъяснимый факт, что мутации в одном и том же гене (локусе) приводят к разным клиническим проявлениям, теперь объясняется наличием каждый раз разных точковых мутаций в одном гене. Так, врачам-невропатологам давно известны рано начинающаяся тяжелая форма детской миодистрофии Дюшенна и более легкая форма юношеской миодистрофии Беккера, обусловленные дефектами одного и того же гена - дистрофина (см. главу 17). Аналогичный пример касается гена адренорецептора, сцепленного с Х-хромосомой: в одном случае конкретная точковая мутация одного гена является причиной болезни Кеннеди, а в другом случае уже другая точковая мутация этого же гена служит причиной синдрома Морриса или тестикулярной феминизации (см. главы 5
и 17).
Другой пример - ген МВ, в разных точках которого выделены около 1400 мутаций, приводящих к формированию сходных в целом фенотипов, но с разной тяжестью клинических проявлений.
Вообще составление классификаций МБ на основе выявления обусловливающих их первичных молекулярных (биохимических) дефектов - чрезвычайно трудное дело. Если еще учесть, что при некоторых МБ наблюдается высокий процент вновь возникших мутаций (например, при миодистрофии Дюшенна 30% мутаций de novo), то еще больше становится понятной ведущая роль первичного молекулярного дефекта именно на уровне гена. Следствием такого дефекта являются структурные и функциональные нарушения на другом уровне - уровне белка-дистрофина.
Более того, для осуществления простых или сложных биохимических реакций первоначально всегда требуется координированная экспрессия нескольких генов. Например, в случае ФКУ разными генами кодируются разные субъединицы олигомерного белка-фермента: фенилаланингидроксилазы, дефицит которой обусловливает развитие заболевания. При этом субъединицы олигомерного белка различаются между собой по молекулярной массе и изоэлектрическим точкам. Следовательно, при определении болезни на основе критерия первичного биохимического дефекта в рамках одной моногенной формы будут выявляться фенотипы, не только обусловленные разными мутациями в разных точках одного гена, но и связанные с мутациями в разных генах, т.е. речь идет о генокопировании сходной
клинической картины с разными особенностями течения болезни у разных больных (см. главу 5).
Таким образом, главную роль в возникновении и последующем фенотипическом проявлении первичного биохимического дефекта играет координированная генная экспрессия, которая зависит от вида клеток, периода онтогенеза, влияния генов-модификаторов и факторов среды. Подтверждением ведущей роли первичного молекулярного дефекта является также классификация МБ, опубликованная В.С. Барановым и др. (2005).
Классификации МБ на основе мутационных спектров и оптимальных алгоритмов ДНК-диагностики
Классификация базируется на двух принципах: это уникальность спектра мутационных изменений каждого гена (причины уникальности приведены в главе 5) и оптимальные алгоритмы для их ДНКдиагностики.
Авторами классификации предложено разделение МБ на два класса.
Первый класс - это болезни, вызванные немажорными мутациями, равномерно распределенными по всей нуклеотидной последовательности гена. В их число включены замены, делеции, дупликации и инсерции одного или нескольких нуклеотидов. Данный класс МБ представлен: аутосомно-доминантным врожденным поликистозом почек, гемофилией А и В, множественными эндокринными неоплазиями (МЭН), семейным раком грудной железы и раком яичников, семейным аденоматозно-полипозным раком толстого кишечника, CRASH-синдромом (болезнь LI), синдромом Смита-Лемли-Опица, Х-сцепленной формой синдрома Альпорта, Х-сцепленной гидроцефалией.
Для молекулярной диагностики этого класса МБ применяются методы ДНК-анализа для выявления неизвестных мутаций, но чаще других используется комбинация методов SSCP (анализ однонитевого конформационного полиморфизма) и методов анализа отдельных экзонов с последующим прямым сиквенированием тех фрагментов ДНК, где подозревают наличие мутаций (см. главу 19).
Второй класс - это болезни, вызванные мажорными мутациями, которые принципиально важны для ДНК-диагностики. Среди таких патологий: ахондроплазия, болезни Вильсона-Коновалова, Леша- Найяна и Ретта, болезни накопления, МВ, рецессивные формы
наследственной глухоты (несиндромальная несенсорная тугоухость), ФКУ. Для идентификации каждого типа мажорной мутации предложен оптимальный метод из общего арсенала методов ДНК-анализа, включающий мультиплексную ПЦР с анализом внутригенных полиморфных сайтов рестрикции и др. (см. главу 19).
Третий класс - это болезни, вызванные протяженными дупликациями и делециями, возникшими в результате неравного кроссинговера или неправильного спаривания гомологичных хромосом во время мейоза, которое происходит по фланкирующим повторам участка ДНК, содержащего ген. Такая мутация называется генной конверсией (см. главу 5), и если в участок нуклеотидной последовательности ДНК (область мутации) вовлечен, например, ген РМР22 (17р11.2), контролирующий синтез периферического миелинового белка 22, то возникнет аутосомно-доминантная моторно-сенсорная нейропатия Шарко-Мари-Тус.
Особенно много мутаций в результате генной конверсии возникает в псевдогенах. В этом случае происходит перенос фрагмента одного аллеля в другой аллель гена или перенос фрагмента псевдогена в нормальный ген. Например, такая мутация происходит при переносе последовательности псевдогена в ген 21-гидроксилазы (CYP21B) при
АГС.
К данному классу мутаций принадлежит мутация, обусловливающая развитие болезни Гоше - это самая частая аутосомнодоминантная болезнь накопления. В ее основе лежит мутация гена GBA (1q21), гена глюкоцереброзидазы (лизосомной гидроксилазы), рядом с которым локализован псевдоген psGBA.
В настоящее время выделены 115 точковых мутаций гена GBA. В их числе - дупликации, делеции и конверсии, возникающие в мейозе между геном и псевдогеном.
Типичной патологией этого класса МБ считается сцепленная с полом миодистрофия Дюшенна-Беккера (Хр21), обусловленная мутациями (протяженными делециями и дупликациями, микроделециями и нонсенс-мутациями) самого крупного гена человека, кодирующего белок - дистрофин, входящий в состав сарколеммы мышечного волокна. Кроме того, сюда относятся семейная мышечная атрофия трех типов, включающая болезнь Верднига-Гоффмана, болезнь Кугельбергера-Веландер и промежуточную форму. Все типы мышечной атрофии представляют собой аллельные варианты мутаций одного дуплицированного гена SMN1 (5q13).
Копия этого гена - ген SMN2 - вариант псевдогена, располагается ближе к центромере и отличается от гена SMN1 по 8 нуклеотидам, причем 5 из них находятся в интронах, а 3 - в экзонах (различия в гене и псевдогене выявляются методом SSCP и методом ПДРФанализа).
Четвертый класс - это болезни экспансии, в основе которых лежат динамические мутации (см. главу 27).
В последние годы была опубликована еще одна классификация, согласно которой все МБ делят на три группы в соответствии с видом наследственной изменчивости (см. главы 2 и 5):
• болезни, обусловленные мутационной изменчивостью генома; в каталоге В. Мак-Кьюсика (OMIM) перечислено 1100 мутаций идентифицированных, отчетливо проявляющихся в фенотипах больных при 1,5 тыс. МБ (см. выше);
• болезни, обусловленные вариационной (комбинативной) изменчивостью генома, в составе которого выделены не менее 50% повторяющихся последовательностей ДНК, относящихся к 5 классам: повторы, возникшие из транспозонов, неактивные перемещенные копии клеточных генов, простые повторяющиеся последовательности, сегментарные дупликации, блоки тандемноповторяющихся генов. Эти повторы ДНК перестраивают геном, вызывая эктопические нарушения, приводящие к развитию болезней генома;
• болезни, обусловленные эпигенетической (эпигеномной) изменчивостью, связанной не со структурными нарушениями генома, а с изменениями регуляции генной активности, которые, хотя и наследуются, но обратимы. К примерам эпигеномных нарушений относятся: инактивация Х-хромосомы, эффект положения теломеры, сайлесинг гена, тканевая специфичность и возрастзависимая модификация ДНК, парамутации, трансфекции, импринтинг, цитодукция и гомологзависимые процессы;
• болезни, обусловленные изменениями в геноме, в том числе гонадный мозаицизм, мейотический драйв, прионизация, цитоплазматическая наследственность, экспансия нуклеотидных повторов.
Завершая рассмотрение всех известных классификаций МБ, следует отметить: в последние три десятилетия выделены новые формы МБ, не вошедшие по разным причинам в указанные классификации, которые поэтому нельзя считать совершенными.
В частности, еще в 70-80-е годы прошлого века появились первые сообщения о митохондриальных и пероксисомных болезнях, которые (совместно с тезаурисмозами - см. выше) были сначала отнесены к болезням обмена клеточных органелл, а затем причислены к классу болезней материнского наследования. В свою очередь, в 90-е годы XX в. выделены болезни геномного импринтинга и болезни экспансии числа нуклеотидных повторов, которые, правда, частично вошли в классификацию МБ, учитывающую мутационные спектры и методы ДНК-диагностики. Наконец, совсем недавно был выделен класс прионных болезней (см. главы 3 и 28).
Примеры наиболее распространенных МБ
В странах Запада в родовспомогательных учреждениях (центрах планирования семьи и репродукции) проводится обязательный неонатальный скрининг новорожденных - это либо массовое (тотальное), либо селективное (выборочное) «просеивание» доступного биологического материала, взятого от новорожденных, с целью молекулярногенетической или биохимической диагностики наиболее распространенных МБ, в том числе наследственных болезней обмена веществ. Общее количество «просеиваемых» нозологий в странах «семерки» уже достигло 300 заболеваний (в России - только 5 нозологий). Ниже приводится выборочный, примерный список таких нозологий и указаны их частоты в популяции (в порядке снижения).
• Синдром Мартина-Белл (фрагильная Х-хромосома или сцепленная с Х-хромосомой УО). Ген заболевания (FMR1) локализован в сегменте Xq27 (хромосомный маркер - fra Xq27). Частота - 0,5: 1000.
• МВ. Ген CFTR локализован в сегменте 7q32 и кодирует белокрегулятор трансмембранной проводимости ионов хлора. Частота в странах Европы и Северной Америки - 1:2 тыс.; в азиатских странах встречается редко.
• Миодистрофия Дюшенна-Беккера (Хр21) - 1:3-3,5 тыс. у мужчин.
• Нейрофиброматоз Реклингаузена, тип 1 (17q11.2) и тип 2 (22q11.2) - 1:3-5 тыс.
• Врожденный гипотиреоз (8q24.3) - 1:4,7 тыс. При незобных формах гипотиреоза гены локализованы в сегментах 1р13 и 14q31.
• Миотоническая дистрофия (миотония Томсена). Ген локализован в сегменте 19q13.6. Частота - 1:10 тыс.; в некоторых популяциях
1:1000.
• ФКУ (12q24.1). При ФКУ, обусловленной дефицитом дегидроптеридинредуктазы, ген локализован в 4p15.1. Частота - 1:10 тыс.
• Гемофилия А (Xq28). При гемофилии В ген локализован в Xq25.3. Частота - 1:10 тыс. у мужчин.
• Опухоль Вилмса (11p14.2) При втором типе опухоли Вилмса ген локализован в 11p15.3. Частота - 1:10 тыс.
• Недостаточность альфа-1-антитрипсина (14q32) - 1:3-20 тыс.
• Хорея Гентингтона (4р16) - 1:12,5-25 тыс. и гораздо выше в популяциях-изолятах.
• Ретинобластома (13q14.2) - 1:14 тыс.
• Несовершенный остеогенез (7q21.3; 17q21.4) - 1:15 тыс.
• Гистидинемия (12q21.4) - 1:23 тыс.
• Атаксия-телеангиэктазия (11q23.1) - 1:30-50 тыс.
• Галактоземия (9р13.1) - 1:35-50 тыс.
• Лейциноз или болезнь с запахом мочи, напоминающим запах кленового сиропа (болезнь кленового сиропа). Это гетерогенная группа, дефектные гены локализованы в локусах 19q13.1, 1р31.2 - тип 2, 6р22.2 - тип 3. Частота - 1:90-120 тыс.
• Аргининянтарная ацидурия (7q11.2) - 1:300 тыс.
• Болезнь Тея-Сакса (15q22.4) - 1:360 тыс. в большинстве популяций и 1:3,6 тыс. у евреев-ашкенази.
• Тирозинемия, тип I (15q22.5). При тирозинемии II типа ген локализован в локусе 16q23.2. Частота 1:900 тыс.
• ТКИД или недостаточность АДА (20q13.1) - 1:1,5 млн.
• Недостаточность глюкозо-6-фосфатдегидрогеназы (Xq28) - редкий этнический вариант; в отдельных популяциях частота у мужчин составляет от 1:4 до 1:20.
• Серповидноклеточная анемия (11р15.3) - характерна для популяций американских негров и Экваториальной Африки - 1:400.
• Талассемия альфа (16р13.1) и бета (11p15.3) - встречается в эндемичных по малярии регионах мира.
Показания для лабораторной диагностики наследственных болезней обмена
А. Симптоматика, выявляемая на первом году жизни ребенка.
• Задержка психомоторного и физического развития в сочетании со следующими признаками:
- прогредиентно развившееся к концу первого года жизни отставание психоневрологических функций, эпилептические при-
падки в первые месяцы жизни;
- рвота, дегидратация, желтуха, мышечная гипотония или гипертония, нарушение дыхания, судороги, летаргия, кома, асцит;
- необычный цвет и запах мочи и/или запах немытого тела, в том числе «мышиный» запах, запах потных ног, солода, кленового сиропа;
- диарея, гипотрофия (исключить экзогенные причины);
- гепатомегалия и/или спленомегалия неясной этиологии.
• Выявляемые при лабораторном исследовании основных биологических жидкостей больного (кровь, моча) метаболический ацидоз, алкалоз, сахар, белок, ацетон в моче, лейкоцитопения и/или тромбоцитопения, изменение иммунологических показателей.
Б. Симптоматика, выявляемая на втором году и в последующие годы жизни ребенка.
• Прогредиентное развитие УО и неврологической симптоматики после периода нормального развития разной продолжительности.
• УО в сочетании со следующими признаками:
- задержка физического развития;
- гаргоилический дисморфизм, тугоподвижность суставов, искривление позвоночника;
- гидроцефалия, глухота, помутнение роговицы, катаракта, вывих (подвывих) хрусталика;
- изменение волос и ногтей;
- гепато- и/или спленомегалия, поражение почек, экзема;
- эпилептические припадки, интоксикация, летаргия, кома, рвота, диарея, алалия, дизлексия.
• УО неясной этиологии.
• Гипотрофия неясной этиологии.
• Непереносимость отдельных продуктов питания, проявляющаяся анорексией, диареей, задержкой развития.
• Нефролитиаз.
• Лабораторные данные (те же, что и для первого года жизни).
ХАРАКТЕРИСТИКА ОТДЕЛЬНЫХ НОЗОЛОГИЙ МОНОГЕННЫХ БОЛЕЗНЕЙ
Галактоземия
Галактоземия относится к моносахаридозам, НБО простых Сахаров. Впервые описана в 1908 г. А. Рессом. Это аутосомно-рецессивная форма, проявляющаяся с частотой 1:15 тыс. Известны три генетических варианта, связанных с недостаточностью ферментов, превращающих галактозу в глюкозу в клетках печени. На рис. 64 представлена схема развития метаболических блоков при галактоземии.
Как видно на схеме, на первом этапе метаболических превращений образовавшаяся в кишечнике пищевая лактоза фосфорилируется в печени под действием галактокиназы или галактозо- 1-фосфорилазы (ген локализован в сегменте 17р24). В результате образуется галактозо-1-фосфат, который под действием галактозо-1-
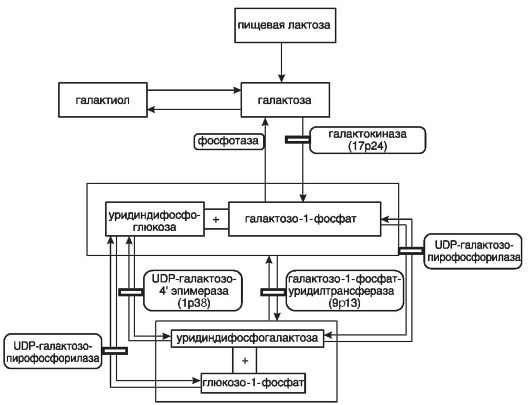 Рис. 64. Схема превращений галактозы и метаболические блоки, приводящие к галактоземии (по Иванову В.И., 2006)
Рис. 64. Схема превращений галактозы и метаболические блоки, приводящие к галактоземии (по Иванову В.И., 2006)
фосфатуридилтрансферазы или фермента Г1ФУТ (9р13) с участием в качестве кофермента NAD+ превращается в уридиндифосфогалактозу (UDP-галактоза) и глюкозо-1-фосфат. На следующем этапе UDP-галактоза под действием UDP-галактозо-4-эпимеразы (1р38) преобразуется в уридинфосфоглюкозу (UDP-глюкоза) и глюкозо-1- фосфат.
Все эти реакции обратимы, и глюкоза легко превращается в галактозу. Наиболее распространенным считается вариант галактоземии I типа, недостаточности фермента Г1ФУТ (9р13), обусловленной однонуклеотидными заменами (мажорная мутация - Cln188Arg). В основе патогенеза галактоземии лежит токсическое действие накапливающихся в результате метаболического блока галактозы и галактозо-1-фосфата на клетки и ткани мозга, печени, кишечника и почек. В этих тканях и органах ингибируется бактерицидная активность лейкоцитов и развитие септических проявлений. При значительном накоплении галактозы она метаболизируется по побочному пути с образованием спирта - галактиола, вызывающего разрыв эонулярных волокон хрусталика и развитие катаракт.
Манифестация первых признаков заболевания происходит в виде повторной рвоты, диареи и желтухи, развивающихся через несколько дней после первого кормления младенца материнским молоком. При отсутствии лечения в первые недели жизни формируется гепатомегалия и появляются гемолитические изменения.
В дальнейшем наблюдаются: задержка психомоторного развития, внутричерепная гипертензия, гипотрофия, почечная недостаточность и катаракты; возможна смерть от почечной или церебральной недостаточности. Генокопиями галактоземии служат другие генетические варианты, приводящие к метаболическим блокам.
Диагностика галактоземии проводится на доклинической стадии путем тотального биохимического скрининга новорожденных. Возможна пренатальная диагностика: исследование активности фермента Г1ФУТ в культуре клеток плода и определение присутствия галактизола в амниотической жидкости.
У детей старшего возраста диагноз ставится на основе характерной симптоматики, повышения уровня галактозы и галактозо-1-фосфата в плазме крови и моче, недостаточности Г1ФУТ в эритроцитах, лейкоцитах и фибробластах, а также с помощью ДНК-диагностики (идентификация мутаций в гене Г1ФУТ и других генах (см. выше).
Лечение галактоземии проводится на основе безлактозной диеты. Эффективность лечения снижена из-за перенасыщения глюкозой, образующейся в организме больного.
Гиперфенилаланинемии
ГФА - это гетерогенная группа аутосомно-рецессивных заболеваний, связанных с нарушениями метаболизма ФА - незаменимой аминокислоты, которая не синтезируется в организме, а поступает с пищей.
Под действием ФАГ основное количество поступившего с пищей ФА превращается в тирозин, являющийся субстратом для синтеза аминов и меланина, и лишь небольшое его количество используется для синтеза белка.
На рис. 65 приведена схема превращения ФА в тирозин и последующего превращения тирозина в другие продукты метаболизма с выделением в этих реакциях основных метаболических блоков в случае развития МБ (альбинизм, алкаптонурия, врожденный гипотиреоз и ФКУ).
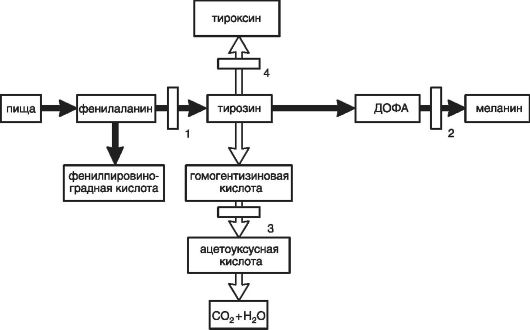 Рис. 65. Схема превращений фенилаланина и тирозина и основные метаболические блоки на их пути (по Иванову В.И., 2006)
Рис. 65. Схема превращений фенилаланина и тирозина и основные метаболические блоки на их пути (по Иванову В.И., 2006)
Прямоугольниками на стрелках указаны метаболические блоки: 1 - фенилкетонурия; 2 - альбинизм; 3 - алкаптонурия; 4 - врожденный гипотиреоз
ФАГ - это печеночная оксигеназа со смешанной функцией. У плода ФАГ состоит из двух белковых субъединиц: стабильной (в качестве кофактора она имеет птеридин) и лабильной. Во взрослом организме имеются три стабильные субъединицы: ФАГС, тетрагидробиоптерин (ВН4) и молекулярный кислород. При этом ВН4 служит восстановителем молекулярного кислорода при встраивании его в ряд субстратов: ФА, тирозин и триптофан.
Кроме того, от ВН4 зависят тирозиновая и триптофановая гидроксилазы.
На рис. 66 представлена схема ферментативных реакций с участием тетрагидробиоптерина в качестве кофактора, переходящего в дегидроформу или форму дегидробиоптерин-хинона. Образовавшийся тирозин тремя разными путями превращается либо в тироксин (основной путь), либо в ДОФА, либо в гомогентизиновую кислоту.
В случае возникновения мутаций в генах, кодирующих эти метаболические пути, развиваются указанные выше метаболические блоки. Один из таких блоков возникает в ходе основного пути гидроксилирования ФА в тирозин, что ведет к избыточному накоплению ФА и развитию гиперфенилаланинемий.
Известны три причины гиперфенилаланинемий:
• мутации в гене, кодирующем злокачественную (классическую) ФКУ (ген РАН); ген локализован на хромосоме 12 (12q22-q24.2);
• мутации в генах, кодирующих ферменты, контролирующие синтез биоптерина; один из этих генов - ген гуанозинтрифосфатциклогидролазы (GCH I), локализован на хромосоме 14 (14q22.1-q22.2); кроме того, известны три аллельные формы (тяжелая, средней тяжести и транзиторная) гена 6-пирувоилтетрагидробиоптерин- синтетазы (PTS), локализованного на хромосоме 11 (11q22.3-q23.3);
• мутации в генах, кодирующих ферменты, контролирующие дальнейшее превращение ФА: две аллельные формы (тяжелая и средней тяжести) гена дегидроптеридинредуктазы - ген QDPR, локализованный на хромосоме 4 (4р15.3), птеридин-4 альфакарбиноламиндегидротаза - ген PCBD, локализованный на хромосоме 10
(10q22).
Вместе с тем, имеются данные, согласно которым обратные превращения кофакторов ФАГ в нормальную форму не ведут к нарушениям метаболизма ФА (Дадали Е.Л. и Барышникова Н.В., 2006).
В настоящее время разработана общая классификация ГФА (три группы).
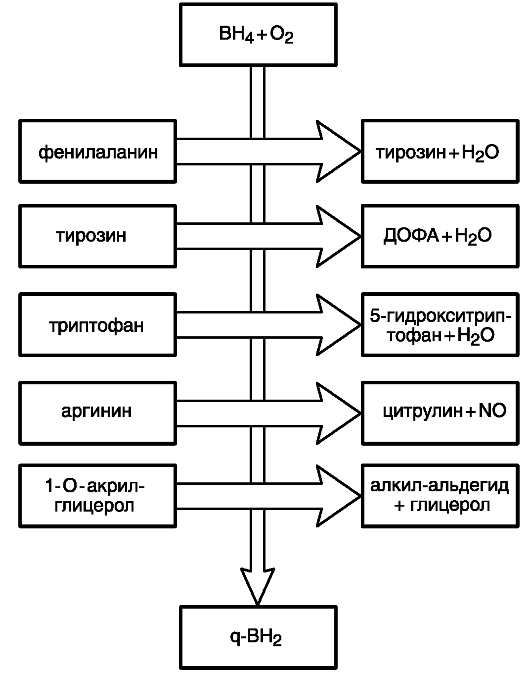 Рис. 66. Схема ферментативных реакций с участием биоптерина в качестве кофактора (по Иванову В.И., 2006)
Рис. 66. Схема ферментативных реакций с участием биоптерина в качестве кофактора (по Иванову В.И., 2006)
• ФАГ-зависимые ГФА (95% всех случаев). ФАГ-зависимые ГФА сопровождаются снижением активности ФАГ.
• Выделяют:
- при резко сниженной активности ФАГ - классическую форму злокачественной ФКУ;
- при умеренно сниженной активности ФАГ - атипичную форму
ФКУ.
• ФАГ-независимые ГФА (5%) сопровождаются нормальной активностью ФАГ.
Выделяют:
- биоптеринзависимые (нарушение синтеза и нарушение метаболизма); они реже встречаются, чем ФКУ, и отличаются от нее отсутствием эффекта от диетотерапии; наиболее распространены две формы: первая форма - недостаточность дегидроптеридинредуктазы, которая восстанавливает активную форму тетрагидробиоптерина (мутация в гене, локализованном на хромосоме 4 (4р15.3), частота 1:100 тыс.; при отсутствии лечения дети погибают в раннем возрасте; вторая форма более мягкая, обусловлена недостаточностью пирувоилтетрагидроптеринсинтетазы, преобразующей дегидроптеридинтрифосфат в тетрагидробиоптерин; частота - 1:30 тыс.;
- другие ГФА, включая тяжелые клинические формы, не связанные с установленными биохимическими дефектами, формы без клинических проявлений.
• Транзиторные формы ГФА. Эти формы обусловлены незрелостью ферментных систем у новорожденного с глубокой недоношенностью и функциональной несостоятельностью (вследствие незрелости).
Транзиторные формы ГФА вызывают временное увеличение концентрации ФА в сыворотке крови в результате тирозинемии, спровоцированной преждевременными родами и чрезмерным употреблением белковой пищи беременной (такая пища ингибирует собственные ферменты у плода). Такую тирозинемию следует отличать от тирозиноза у франкоязычного населения Канады.
Фенилкетонурия
ФКУ - это классическая форма аутосомно-рецессивной ФАГзависимой ГФА с резко сниженной активностью ФАГ.
ФКУ была впервые описана Феллингом в 1934 г. Теперь известно, что она вызвана мутацией гена РАН, локализованного на хромосоме 12 (12q22-q24.2). Частота в популяции - 1:10 тыс. человек.
Особенности ФКУ:
• высокая частота болезни в европейских странах (Ирландия - 1:4560, Белоруссия - 1:5,6 тыс., Германия - 1:6,7 тыс., Чехия - 1:8 тыс., Россия - 1: 6,35 тыс., г. Москва - 1:11,8 тыс.);
• тяжелый исход болезни (необратимая УО) в случае несвоевременной диагностики (до 6 мес жизни);
• высокая частота гетерозиготного носительства гена болезни - 1:50 человек.
Манифестация ФКУ происходит в первом полугодии жизни: обычно в возрасте 2-6 мес, иногда через 2-3 нед после рождения ребенка. Первые проявления болезни: вялость, отсутствие интереса к окружающему, повышенная возбудимость, беспокойство, частое срыгивание, мышечная гипотония или гипертония, судороги, признаки аллергического дерматита. Появляется специфический «мышиный» запах. Характерны гипопигментация кожи, волос и радужной оболочки глаз. Отчетливо формируется задержка психомоторного развития. При отсутствии лечения развивается необратимая УО.
В основе патогенеза ФКУ лежат аминоацидурия и ацидоз тканей, причинами которых являются следующие метаболические нарушения:
• внутриклеточное накопление (до метаболического блока) ФА и его производных (фенилпировиноградная, фенилмолочная и фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин);
• высокая концентрация ФА и его производных в крови и моче;
• нарушение реабсорбции и транспорта ФА и тирозина.
В результате этих нарушений наблюдается прямое токсическое действие продуктов метаболизма на ЦНС, нарушаются функции печени, обмен белков, липо- и гликопротеидов, метаболизм гормонов.
Диагностируют ФКУ на основе совокупности генеалогических данных (в том числе о ФКУ у сибсов), результатов клинического и биохимического обследования (превышение уровня ФА в крови более 900-1200 мкмоль/л); наличия положительной проба Феллинга.
Прямая диагностика ФКУ - идентификация наиболее частой мутации R408W (в Китае и Японии не встречается); она проводится с помощью ПЦР и последующей рестрикции.
Методом патогенетического лечения ФКУ является диетотерапия, ограничивающая поступление в организм белка и ФА до уровня минимальной возрастной потребности.
Для коррекции питания назначают белковые гидролизаты, лишенные ФА, но имеющие все другие необходимые аминокислоты (апонти, берлофен, нофелан). Дополнительно назначают витамины группы В, микроэлементы, минеральные вещества. Выбор тактики лечения (особенности диетотерапии) зависит от первичного биохимического дефекта.
Муковисцидоз
МВ (или кистофиброз) - одно из наиболее тяжелых заболеваний детского возраста. Относится к патологии экзокринных желез (бронхиальных, потовых, слезных, слюнных, ушных), а также поджелудочной железы и печени (имеют экзокринную и эндокринную функции).
Болезнь наследуется по аутосомно-рецессивному типу. Средняя продолжительность жизни больных МВ составляет 25-30 лет. Частота МВ колеблется от 1:2 тыс. (Америка, Англия, Россия, страны Северной Европы) до 1:40 тыс. (Финляндия) и 1:170 тыс. (Африка). В России ежегодно рождаются 700-750 детей с МВ.
Ген болезни локализован в сегменте 7q32.1. Он кодирует белокрегулятор трансмембранной проводимости ионов хлора (cftr - кистофиброзный трансмембранный регулятор), являющийся транспортным белком АТФазы. Чаще всего, развитие болезни связано с мажорной мутацией или делецией кодона в 508 положении гена - del F508 (70-85% всех мутаций этого гена).
Всего выделено около 1400 точковых мутаций гена МВ (предполагается до 5 тыс.).
При этом заболевании речь идет о значительной по объему и спектру полиморфной аллельной серии, представленной разными мутациями в разных точках одного гена.
Нарушения в ионном канале обусловлены вторичными дефектами в клеточном метаболизме. Изменения количества электролитов и белка в секрете экзокринных желез нарушают его физико-химические свойства, вызывая повышенную дегидратацию и вязкость, что сопровождается закупоркой протоков желез и развитием вторичных изменений, особенно выраженных в легких, поджелудочной железе и кишечнике. В результате плохо отделяемый секрет закупоривает мелкие респираторные (бронхиальные) пути и небольшие протоки поджелудочной железы. При этом изменяется среда эпителиальной поверхности, что приводит к росту бактериальных колоний, нейтрофильному воспалению и нагноению слизистой и обусловливает хронический легочный процесс вследствие недостаточности экзокринной функции поджелудочной железы.
Клиническая картина МВ зависит от типа точковой мутации и характеризуется полисистемным поражением. Наиболее тяжело протекают инфекции дыхательных путей. Быстро развиваются бронхиты, пневмонии, абсцессы и бронхоэктазы. У 27% больных развивается
астматический синдром. Процесс осложняется дыхательной недостаточностью и развитием легочного сердца. Возможны пневмоторакс, кровохарканье, кровотечение.
Тяжело протекает поражение желудочно-кишечного тракта. Кишечные симптомы связаны с нарушением активности ферментов поджелудочной железы. Гнилостные процессы в кишечнике приводят к вздутию живота, появлению обильного жирного стула с резко выраженным запахом (родители отмечают появление жирных пятен на белье).
При закупорке меконием (мекониальный илиус) развивается кишечная непроходимость (7-15% случаев). У 15-30% больных выявляется локальный или генерализованный билиарный цирроз печени.
Для больных МВ также характерны: выраженная гипотрофия (несмотря на повышенный аппетит), явления авитаминоза К (гематомы, кровотечения и кровоизлияния), авитаминозы А, Е и D.
Выделяют следующие клинические формы МВ:
• смешанная (70% случаев);
• преимущественно легочная (11%); при ней поражение органов пищеварения минимально или отсутствует;
• абортивная или стертая (11%);
• преимущественно кишечная (5%);
• мекониальная непроходимость (3%).
Это условное деление, так как одна форма практически всегда переходит в другую форму. Диагностические тесты
Потовый тест или стимуляция потовых желез пилокарпином с помощью ионофореза. После стимуляции потоотделения пот собирается на фильтровальную бумагу и проводится его химический анализ с определением концентрации натрия и хлора (положительным считается превышение уровня в 60 ммоль/л у детей и 70 ммоль/л у взрослых).
Бромидный тест или определение концентрации брома в поте и крови после нагрузки бромом (положительным считается повышение уровня брома до 50-100% при норме 20%).
Тест на протеолитическую активность кала или определение уровня ферментов поджелудочной железы в кале (отдельно анализируется содержание нейтрального жира, клетчатки, мышечных волокон и крахмала; проводится рентгенопленочный тест на отсутствие трипсина в кале).
Кроме того, определяется активность ферментов дуоденального содержимого.
Помимо перечисленных диагностических тестов, разработаны программы массового скрининга новорожденных на МВ, включающие два этапа: определение иммунореактивного трипсина в пятнах крови и альбумина в меконии, а также электролитов в поте.
Лечение муковисцидоза в основном симптоматическое. Применяются непрерывная или прерывистая антибиотикотерапия, сочетание аминогликозидов с цефалоспоринами, бронходилататоры, гормонотерапия, витамины, ферменты и диета.
Обязательны ЛФК, вибрационный массаж, постуральный дренаж. Прямой метод лечения - генотерапия с применением векторных систем (аденовирусы и липосомы) - находится в стадии клинических испытаний.
С 1980 г. в Англии, Франции и США осуществляются тысячи успешных трансплантаций легких, а с 1985 г. - сотни трансплантаций легких и сердца. В этих странах активно проводится пренатальная диагностика МВ. Например, в США подсчитано, что своевременная пренатальная диагностика МВ и прерывание беременности ежегодно экономят в одном штате 187,5 тыс. долларов.
В России имеется крайне ограниченное число центров по лечению детей с МВ, а потому МГК охвачены только 10-15% больных.
Болезни накопления
К болезням накопления относятся МБ, связанные с нарушениями структуры и функционирования лизосом цитоплазмы, основная функция которых заключается в ферментативном расщеплении и окислении разных макромолекул клетки (см. главу 6).
Наиболее известные лизосомные болезни - это гликогенозы, МЛ,
МПС и СФЛ.
Гликогенозы
Гликогенозы - группа НБО полисахаридов, развивающихся в результате нарушений синтеза или распада гликогена на простые сахара. При этом нормальный и аномальный гликоген одновременно накапливаются в цитоплазме клеток печени и других органов, что позволяет рассматривать гликогенозы, с одной стороны, как болезни накопления, а с другой стороны - как болезни материнского наследования (см. главы 4 и 6).
Известны 12 типов гликогенозов, наследуемых в основном по аутосомно-рецессивному типу (кроме IX типа - Х-сцепленный рецессивный тип).
Классификация гликогенозов основана на различиях в дефектах ферментов, лежащих в основе заболеваний. Выделяют следующие гликогенозы.
• Гликогеноз I типа или болезнь Гирке (дефект глюкозо-6- фосфатазы ЭР). Это наиболее частая форма (37%). Известны несколько генетических вариантов первый вариант обусловлен мутацией в гене, картированном на хромосоме 17(17q21); второй вариант связан с мутациями в гене Са2+-связывающего белка, стабилизирующего каталитическую активность глюкозо-6- фосфатазы (локус не картирован); третий вариант связан с мутациями гена Т1 (11q23), производящего белок, обеспечивающий микросомный транспорт глюкозо-6-фосфата.
• Гликогеноз II типа или болезнь Помпе. Развивается в результате дефицита лизосомного фермента альфа-D-глюкозидазы (у детей) или недостаточности кислой мальтазы (у взрослых). Такая недостаточность ведет к отложению негидролизованного гликогена в лизосомах мышцы сердца и скелетных мышц, где появляются «пенистые клетки», создающие ложную картину прогрессирующей мышечной дистрофии. Выделяют три формы болезни: младенческую (может вызвать «смерть в колыбели»), детскую и взрослую. По частоте проявления занимает 10% от всех гликогенозов. Ген локализован в сегменте 17q23.
• Гликогеноз III типа (лимитдекстриноз) или болезнь Форбса- Кори. При этом заболевании снижена или вообще отсутствует активность фермента амило-1,6-глюкозидазы. Ген болезни локализован на хромосоме 1 (1р21). Частота достигает 26% от всех гликогенозов.
• Гликогеноз IV типа (амилопектиноз) или болезнь Андерсен. Наблюдается дефицит амило-1,4,1,6-трансглюкозидазы. Ген картирован на хромосоме 3 (3р12).
• Гликогеноз V типа, или болезнь Мак-Ардля. Наблюдается дефицит мышечной фосфорилазы. Ген локализован на хромосоме
11 (11q13).
• Гликогеноз VI типа или болезнь Герса. Характерен дефицит фосфорилазы печени. Локализация гена предполагается на хромосоме 14.
• Гликогеноз VII типа или болезнь Таруи. Наблюдается снижение активности фосфофруктокиназы. Ген локализован на хромосоме 1 (1q32).
• Гликогенозы типов VIII, IX, Х, XI и 0 (нулевой тип) - встречаются редко.
Симптоматика гликогенозов проявляется на первом году жизни гипогликемией и лактат-ацидозом, сопровождающимися рвотой, судорогами, потерей сознания (вплоть до комы). Больные отстают в росте, у них отмечается локальное отложение жировой клетчатки на щеках («кукольное лицо»), груди, ягодицах, бедрах; могут возникать ксантомы на локтях и коленях.
Особенности течения болезни зависят от места депонирования гликогена: печень, почки, мышечная ткань. Соответственно выделяют цирроз печени, почечную форму и мышечную форму (включая сердечную форму).
Появление и нарастание гипогликемии может привести к развитию двух клинических синдромов: «внезапной смерти» или «смерти в колыбели», и «респираторного дистресс-синдрома». При развитии у больных легочной гипертензии и сердечной недостаточности смерть наступает в юношеском возрасте.
Лечение гликогенозов в основном симптоматическое. При отсутствии лечения продолжительность жизни резко снижена и не превышает одного-трех лет.
Муколипидозы
Муколипидозы (МЛ) - это группа заболеваний, отличающихся большим фенотипическим сходством с МПС и связанных с генокопированием одного и того же (сходного) фенотипа, однако при МЛ нет характерного для МПС клинико-биохимического показателя экскреции с мочой ГАГ, т.е. МЛ - это «мукополисахаридозы без мукополисахаридозурии».
В цитоплазме фибробластов кожи таких больных обнаружены множественные вакуоли (лизосомы), наполненные включениями в виде нерасщепленных фрагментов субклеточных мембран и соединений гликозаминопротеогликановой и гликолипидной природы. Наличие этих включений послужило основанием для выделения МЛ II типа или I-клеточной болезни (I - inclusion или включение), что связано с обнаружением в биоптатах тканей больных «раздутых» клеток, содержащих нерасщепленные продукты метаболизма. Ген I-клеточной болезни локализован в сегменте 4q21-23. Он контроли-
рует присоединение маннозо-6-фосфатной группировки, обеспечивающей транпорт гидролаз, которая «узнается» рецепторами мембран лизосом.
Выделены также МЛ I, III (псевдолиподистрофия Гурлер) и IV типов. Они различаются между собой специфическими ферментативными нарушениями, морфологическими признаками и тяжестью течения болезни. Гены МЛ II и III типов локализованы в сегменте
4q21.4.
Следует отметить, что при I-клеточной болезни и псевдолиподистрофии Гурлер их характерной особенностью служит недостаточность лизосомных гидролаз в фибробластах кожи и лейкоцитах крови (альфа-нейроминидаза, арилсульфатаза А, бета-галактозидаза, гексоаминидаза А и В, сфингомиелиназа). При этом в жидких средах организма наблюдается существенное повышение их активности, которое обусловливает увеличение активности гидролаз в сыворотке крови, что связано с отсутствием в молекулах упомянутой выше маркерной группировки (маннозо-6-фосфата). При всех четырех типах МЛ в фибробластах кожи повышается общее содержание сиаловых кислот, Gm3- и Gd3-ганглиозидов.
Клиническая картина МЛ (особенно I типа) во многом напоминает таковую гаргоилического дисморфизма: грубые черты лица, поражение опорно-двигательного аппарата (но не при IV типе), разные офтальмологические симптомы (особенно при IV типе), УО (но не при III типе), миоклонические судороги, иногда висцеромегалия.
МЛ диагностируется на основе генеалогических данных, фенотипических признаков, электронномикроскопического исследования биоптатов тканей и органов, показателей нормальной почечной экскреции ГАГ, олигосахаридурии, высокой активности лизосомных гидролаз в сыворотке крови, идентификации продуктов накопления и их выделения. Лечение МЛ во многом соответствует лечению
МПС.
Мукополисахаридозы
Одна из первых форм мукополисахаридозов (МПС) была описана Гурлер в 1917 г. С тех пор она называется болезнью Гурлер (см. ниже).
МПС - это гетерогенная группа заболеваний, отнесенных к НБО сложных сахаров и сопровождающихся нарушениями ферментативного катализа ГАГ, их избыточным накоплением в лизосомах
клеток разных тканей и органов, а также повышенной экскрецией с мочой. ГАГ - это кислые мукополисахариды, состоящие из глюкуроновой кислоты, сульфатированного гексоамина, аминосахароз и нейтральных сахаров, соединенных с белками. Существуют в форме протеогликанов: гепаран-, дерматан-, кератан-, хондроитин-4(6)- сульфатов, которые служат важными компонентами структурного белка-коллагена (белка соединительной ткани) и альфа-кератина (белка волос).
Большинство МПС характеризуется аутосомно-рецессивным типом наследования, кроме болезни Хантера (Х-сцепленный рецессивный тип). Манифестация МПС происходит до 7-летнего возраста (обычно в 2-3 года). Характерны: задержка роста (нанизм), карликовость, контрактуры суставов, кифосколиоз, массивный череп с глубоким и удлиненным турецким седлом, короткая шея, деформация грудной клетки, веслообразные ребра, укорочение трубчатых костей, грубые черты лица, помутнение роговицы, гепатоспленомегалия, задержка психического развития, кондуктивная глухота, паховые и пахово-мошоночные грыжи, пороки сердца, гликозоаминогликанурия (до 100-200 мг в сутки).
Этот симптомокомплекс получил название гаргоилического дисморфизма или патологического фенотипа, сходного с фенотипом «уродцев», некогда украшавших барельеф собора Парижской Богоматери (до пожара в XVII в.). Именно такой фенотип у звонаря Квазимодо, описанного в романе классика французской литературы Виктора Гюго.
Гетерогенность МПС
Всего выделено не менее 10 генокопий, включая 5, связанных с нарушением активности сульфатаз и гликозидаз (4) и дефицитом трансферазы (1). Среди генокопий следующие:
• МПС I типа или болезнь Гурлер; сопровождается сниженной активностью альфа-L-идуронидазы и накоплением в тканях дерматан- и гепаратансульфатов. Ген локализован на хромосоме 4
(4р16.3).
• МПС типа IS и МПС типа H/S . Оба типа рассматриваются как аллельные формы гена МПС I типа. Протекают более благоприятно.
• МПС II типа или болезнь Хантера (Xq28). Сопровождается снижением активности L-идуросульфатсульфатазы и отложением в тканях дерматан- и гепарансульфатов. Клинические признаки
менее выражены. Продолжительность жизни больше, чем при других типах.
• МПС III типа или болезнь Санфиллипо. В зависимости от первичного биохимическогодефекта выделяют4 варианта:А(дефицит гепарансульфатазы), В (дефицит фермента N-ацетил-альфа-глюкозаминидазы), С (дефицит фермента глюкозамин^-ацетилтрансферазы) и D (дефицит фермента ^ацетил-глюкозамин-6-сульфатсульфатазы). В клинической картине преобладают психические расстройства: агрессивность и деменция. Продолжительность жизни не превышает 20 лет. Ген варианта D локализован в сегменте 12q14; гены других вариантов не картированы.
• МПС IV типа или болезнь Моркио. Выделяют подтип А (дефицит галактозамин-6-сульфатсульфатазы) и подтип В (дефицит бета-галактозидазы). В тканях откладывается кератансульфат. Преобладают поражение скелета и непропорционально низкий рост. Ген подтипа А локализован в сегменте 16q24.3, подтипа В - в сегменте 22q13-qter.
• МПС V типа или болезнь Шейе. Относится к МПС I типа. Ген не картирован.
• МПС VI типа или болезнь Марото-Лами. Дефицит фермента N-ацетил-галактозамин-4-сульфатсульфатазы (арилсульфатазы В). В тканях накапливается дерматансульфат. Фенотипически напоминает МПС I типа, но интеллект не снижен. Ген локализован в сегменте 5q13-q14.
• МПС VII типа или болезнь Слая. Дефицит фермента бетаглюкуронидазы. В тканях накапливаются дерматан-, гепаран- и хондроитинсульфаты. Фенотипически напоминает МПС I типа, но имеет более доброкачественное течение. Ген локализован в сегменте 7q21.1-q22.
Диагноз МПС ставится на основе совокупных данных генеалогического анализа, клинических проявлений, типичных рентгенологических данных, экскреции с мочой оксипролина (снижение), ГАГ и их фракций (превышение в 5-10 раз).
Однако точная идентификация типов МПС возможна только с помощью определения активности соответствующих ферментов в лимфоцитах и лейкоцитах крови, культуре фибробластов кожи, биоптатах печени, а также моче.
Лечение МПС в основном симптоматическое. Оно заключается в назначении терапии, способствующей нормализации (стабилизации)
процесса в опорно-двигательном аппарате, ССС и ЦНС, паренхиматозных органах, органах зрения и слуха.
Перспективным считается плазмаферез, очищающий организм больного от чрезмерного количества ГАГ.
Сфинголипидозы
Среди болезней накопления выделяют гетерогенную группу СФЛ, обусловленных снижением активности ферментов, обеспечивающих деградацию сложных липидов - сфингомиелинов, к которым относятся галактозиды, цереброзиды и церамиды. Эти соединения являются производными сфингозина - аминоспирта, имеющего 18 углеродных остатков, к которым через аминовые и гидроксильные группы присоединяются другие компоненты (например, гексозы) и сиаловая кислота.
Разнообразие СФЛ определяет особенности распределения отдельных сфинголипидов, входящих в состав клеточных мембран головного мозга и других тканей, а также характер развивающихся метаболических блоков при нарушениях метаболизма. СФЛ наследуются, как правило, по аутосомно-рецессивному типу и проявляются крайне редко (1:360 тыс.), кроме болезни Тея-Сакса, частота которой в популяции евреев-ашкенази достигает 1:3600.
Среди СФЛ выделяют группы, подгруппы, отдельные генетические варианты (типы) и клинические формы, различающиеся по времени манифестации заболевания, выраженности симптоматики и тяжести течения. Клиническую вариабельность связывают с уровнем остаточной активности ферментов, специфичностью субстратов и их распределением в тканях.
СФЛ, сопровождающиеся поражением ЦНС, делятся на две группы:
• болезни с преимущественным нарушением обмена миелина, содержащегося в белом веществе мозга;
• болезни с преимущественным нарушением обмена цереброзидов, содержащихся в сером веществе мозга.
Следует отметить, что неврологическая симптоматика характерна для большинства СФЛ, за исключением некоторых форм: болезни Гоше, Нимана-Пика и Фабри (см. ниже).
Среди основных симптомов СФЛ выделены: двигательные расстройства, поражение опорно-двигательного аппарата (типичное также для МПС), изменения кожи, сетчатки глаз и внутренних органов (почки, печень, селезенка), прогрессирующее слабоумие.
Продолжительность жизни больных резко снижена. Наиболее известными формами СФЛ являются ганглиозидозы или болезни, связанные с генерализованным распадом ганглиозных клеток, развитием глиоза и вторичной демиелинизацией. Общепринято латинское обозначение ганглиозидозов: символ G обозначает ганглиозид; символы m1 (моно), m2 (ди), m3 (три) - количество остатков сиаловой кислоты. Среди них выделены три типа.
• Gm1 - ганглиозидоз или дефицит изоферментов А, В и С бетагалактозидазы с накоплением в тканях ганглиозида m1, асило- Gm1-ганглиозида и кератансульфата. Ген локализован в сегменте 3р21.2. Болезнь известна также под другими названиями: болезнь Тея-Сакса с висцеральной локализацией, псевдогурлер-синдром или нейровисцеральный ганглиозидоз. Выделяют две клинические формы: юношеская (болезнь Дерри) и инфантильная; первая отличается от второй более поздней манифестацией симптоматики.
• Gm2 - ганглиозидоз или мутации генов болезни Сандхоффа (тип II), ювенильного ганглиозидоза (тип III), типов IV-VII (крайне редки). Ген болезни Сандхоффа локализован в сегменте 5q13.2. На этой же хромосоме предполагается локализация гена АВ-варианта этой болезни. Ген ювенильной и взрослой форм ганглиозидоза Gm2 локализован в сегменте 15q22.4 (там же, где ген болезни Тея-Сакса). Иногда к III типу ганглиозидозов относят ювенильную форму болезни Сандхоффа.
• Gm3 - ганглиозидоз или блокада фермента галактозоаминотрансферазы.
Болезнь Тея-Сакса
Болезнь Тея-Сакса - это наиболее известная форма ганглиозидоза, связанная с недостаточностью лизосомного фермента гексоаминидазы А во всех тканях и органах. В них одновременно повышена активность гексоаминидазы В. Ген болезни Тея-Сакса локализован на хромосоме 15 (15q22.4). Вследствие недостаточности гексоаминидазы А в мозге больных накапливается Gm2-ганглиозид (в 100-300 раз выше нормы).
Заболевание проявляется в возрасте 5-6 мес безразличием ребенка к окружающему. Плач становится слабым и протяжным, появляется гиперакузия (вздрагивание от внезапного шума). Постепенно развивается атония мышц, снижается двигательная активность. Позднее в ответ на минимальный раздражитель развиваются судороги с опи-
стотонусом. Для больных характерны макроцефалия и «кукольное» лицо. В 90% случаев на глазном дне в области желтого пятна сетчатки обнаруживается вишнево-красное пятно - это патогномоничный симптом «вишневой косточки».
В терминальной стадии болезни Тея-Сакса наступает полная обездвиженность ребенка, развиваются бульбарные расстройства, глухота, слепота, трофические нарушения, кахексия, явления декортикации и смерть. Болезнь диагностируется на основе совокупности данных генеалогического анамнеза, клинических и лабораторных данных.
Болезнь Нимана-Пика
Аутосомно-рецессивная болезнь Нимана-Пика - это второй по частоте встречаемости СФЛ. Выделены два гена болезни: первый локализован в сегменте (11р15.3), он кодирует сфингомиелиназу и идентифицируется при I и II типах болезни. Второй ген локализован в сегменте (18q11-12), он кодирует мембраносвязанный стерочувствительный белок-фосфолипазу М и выявляется при III типе болезни.
Редкие сфинголипидозы
К числу редких СФЛ относятся следующие.
• Аутосомно-рецессивный сульфатидный липидоз или болезнь Шольца. Эта болезнь также называется метахроматической лейкодистрофией из-за накопления в белом веществе мозга метахроматических сульфатидов. Болезнь связана с мутациями в двух генах: арилсульфатазы А (22q13.31) и просапозина (10q21-22). Выделяют 3 клинические формы: поздняя инфантильная, ювенильная и взрослая.
• ТригексозилцерамидныйлипидозилиболезньФабри. Заболевание сцеплено с Х-хромосомой (Xq21.6) и сопровождается снижением активности лизосомальной альфа-галактозидазы А. В результате происходит отложение гликосфинголипидов в стенках мелких сосудов всех органов, эпителии роговицы, клубочках почек, миокарде, нейронах вегетативных ганглиев. Болезнь прогрессирует медленно.
• Глюкозилцереброзидный липидоз или болезнь Гоше. Для заболевания характерно накопление глюкозилцерамида (глюкоцереброзида), связанное со снижением или отсутствием активности бета-глюкозидазы. Ген локализован в сегменте 1q21.2. Выделяют три типа болезни Гоше: взрослая или хроническая (80% всех случаев), инфантильная (15%), ювенильная (5%).
• Церамидный липидоз (липогранулематоз) или болезнь Фарбера. Характеризуется блокадой синтеза лизосомного фермента - церамидазы. В результате во многих тканях и органах (в коже, гортани, клапанах сердца, плевре и вокруг суставов) накапливаются Gm3- ганглиозид и церамид. Характерно неуклонно прогрессирующее течение.
• Нейрональный цероидлипофусциноз или липогранулематоз. Выделяют три клинические формы: инфантильная, ювенильная (болезнь Баттена) и взрослая (болезнь Куфса). Характеризуются накоплением липофусцина в тканях головного мозга и внутренних органов. Локализация генов инфантильной и ювенильной форм связана с хромосомой 1 (1p32). Биохимические дефекты не установлены.
• Лейкодистрофия или болезнь Краббе. Дефектный фермент - бета-галактозидаза. Ген локализован на хромосоме 14 (14q21-31).
• Метахроматическая лейкодистрофия или генерализованный ксантоматоз (болезнь Вольмана). Дефектный фермент - просапозин (дефицит кислой липазы). В тканях накапливаются холестерин и его эфиры, а также триглицериды. Ген локализован на хромосоме 10 (10q21-22). Известен вариант болезни Вольмана - липидоз с накоплением только эфиров холестерина.
• Абеталипопротеинемия. Характеризуется отсутствием транспорта триглицеридов из тонкого кишечника и печени. В результате развивается дефицит ЛПНП в плазме крови. Ген локализован на хромосоме 2 (2р24).
• Болезнь танжьерских островов. Характеризуется дефицитом липопротеидов высокой плотности (см. главу 6). Это семейная форма болезни.
Лечение ганглиозидозов, сфингомиелиноза и других липидозов симптоматическое: оно направлено на улучшение деятельности центральной и периферической нервной системы и паренхиматозных органов.