Клиническая генетика. Геномика и протеомика наследственной патологии : учеб. пособие. - 3-е изд., перераб. и доп. - Мутовин Г.Р. 2010. - 832 с. : ил
|
|
|
|
ГЛАВА 7 ОБМЕН ВЕЩЕСТВ В КЛЕТКЕ И ОРГАНИЗМЕ
Метаболизм как превращение энергии
С молекулярных позиций, жизнь многоклеточного организма - это сложно организованные генетические и биохимические реакции, упорядоченно протекающие на клеточном, межклеточном, тканевом, органном и системном уровнях организации и отражающие обмен веществ или взаимодействие между молекулами и атомами различных химических соединений (биологических материалов).
Реакции, происходящие в ходе обмена веществ, называются метаболизмом или метаболическими реакциями. Как сказано в предыдущей главе, в числе главных химических соединений клетки значатся: вода (70-90% объема), определяющая свойства биологических материалов; растворенные в воде соли Na, Ca, K, Mg, Cl и другие микроэлементы; органические соединения (10-30% объема), являющиеся ценным видом биологического топлива. Разнообразие органических молекул создается в ходе соединения атомов углерода с атомами других химических элементов.
Каждый химический элемент характеризуется валентностью или способностью образовывать определенное количество ковалентных связей. Именно так образуются простые молекулы: спирты (алкоголь), включающие углеродную цепь и гидроксильную группу (НО), амины (аминогруппа - NH2), кислоты (карбоксильная группа - СООН) и др. Из разных сочетаний простых молекул образуются сложные молекулы, в том числе наиболее важные для организма молекулы нуклеиновых кислот, белков, жиров и углеводов. Все эти молекулы образуются в результате метаболических реакций. Известны два типа метаболических реакций: анаболические или синтез необходимых для жизни молекул (анаболизм) и катаболические или распад молекул (катаболизм). Участник метаболической реакции - это метаболит, результат метаболической реакции - это продукт. Если продукт метаболической реакции служит исходным веществом для следующей реакции - это субстрат.
Цепь из таких последовательных реакций - это метаболический путь. Метаболические пути сложны и взаимосвязаны. Каждый путь
должен мгновенно подстраиваться к текущей ситуации в отдельной клетке и во всем организме, исходя из информации, поступающей от других путей или разных звеньев одного пути. Любой метаболический путь основан на превращениях энергии, которые определяют, какой путь возможен, а какой нет.
Превращения энергии (ее преобразования) - это с одной стороны согласование производства биологического топлива с постоянно меняющимися потребностями в нем; с другой стороны это подчинение скорости и направлений преобразования энергии в отдельных клетках нуждам и жизненному ритму всего организма, который периодически принимает пищу и голодает, работает и отдыхает, вынашивает, кормит, растит и обучает свое потомство опыту взаимодействия с окружающей средой. Кроме того, в ходе своего функционирования любой организм кратковременно или длительно, редко или часто болеет, затрачивая в период болезни запасы имеющейся энергии.
Таким образом, превращения энергии в клетках, тканях, органах и системах организма происходят постоянно в течение всей жизни индивида. Однако условно можно считать, что они начинаются с приема пищи, когда молекулы пищевых веществ «разбираются» клетками для получения энергии (биологического топлива), которая затем используется для метаболизма и синтеза необходимых организму веществ.
Координация и интеграция всех энергетических превращений обеспечивается главными регуляторными системами организма: нервной и эндокринной, а также иммунной системой, которая проявляет свое регулирующее действие либо опосредованно через нейроэндокринную регуляцию, либо через свои лимфоидные органы, обладающие эндокринной функцией (см. главу 14). В целом контроль за превращениями энергии в организме обеспечивается совместным действием нейромедиаторов, гормонов, регуляторных факторов роста, а также множеством сигнальных молекул-посредников энергетического метаболизма (см. главу 8).
Говоря о роли отдельных клеток и тканей в распределении и потреблении энергии, поступающей в организм в виде продуктов питания, а также о роли воды (см. главу 6), следует выделить наиболее энергоемкие и энергозатратные клетки печени, мышц, мозга, жировые клетки и эритроциты. Например, клетки печени при нормальном питании запасают глюкозу в виде гликогена, а при голодании освобождают ее до тех пор, пока не исчерпан весь его запас. Если запас
гликогена иссяк, печень превращает в глюкозу аминокислоты (глюконеогенез), а жиры (жирные кислоты) сначала превращает в кетоновые тела, а потом (через окисление) синтезирует из них триглицериды, являющиеся переносчиками энергии. Триглицериды поступают в кровь и разносятся во все ткани и органы, включая мозг, который не имеет собственных энергетических запасов. Поэтому транспорт глюкозы в нейроны носит пассивный характер, не требующий затрат энергии извне. В системе пассивного транспорта участвуют регуляторные поры, каналы межклеточных соединений и клеточных мембран (см. главу 6). Эти каналы контролируют прохождение различных молекул и потоков ионов, и их пропускная способность (например, для ионов Са2+, К+ и Na+) зависит от внешних для клетки сигналов, поступающих к воротам каналов, в которых расположены распознающие их рецепторы.
В свою очередь, транспорт глюкозы в эритроциты носит активный характер, так как происходит за счет концентрационного градиента или облегченной диффузии благодаря двустороннему встречному движению анионов Cl- и HCO- через плазматическую мембрану эритроцита. При этом активный транспорт совершается за счет внешнего источника энергии, выделяемой при гидролизе АТР, и идет (благодаря такому перемещению) против концентрационного градиента.
Разнообразными источниками энергии для мышц служат глюкоза и гликоген, жирные кислоты, кетоновые тела и аминокислоты. Особое место во внутриклеточном энергетическом обмене занимают митохондрии, ответственные за «тканевое дыхание» или энергетический обмен за счет процессов окисления и фосфорилирования, а также синтеза АТР для «критически зависимых» тканей и органов, функционирование которых полностью зависит от своевременного пополнения запасов АТР. В числе таких «критически зависимых» морфофункциональных структур находятся клетки и ткани головного мозга, миокарда, скелетных мышц, сетчатки глаз, островки Лангерганса в поджелудочной железе и др. Происходящие в них нарушения энергетического обмена вносят значительный вклад в спектр и объем наследственной патологии человека.
При этом часто «страдают» и сами митохондрии. Так, к настоящему времени идентифицирован большой класс митохондриальных болезней (свыше 200 нозологий), проявляющихся инвалидизацией больных за счет тяжелой нейродегенеративной симптоматики на фоне значительного снижения энергетических запасов (см. главу 26).
В частности, нарушения структуры и функции митохондрий выявлены при болезнях Альцгеймера и Паркинсона (см. главу 28), кардиомиопатическом синдроме, сахарном диабете и другой патологии.
Трофическое обеспечение
Трофическое обеспечение или трофика, - это совокупность метаболических реакций, определяющих сохранение структуры и функционирования клеток, тканей, органов и систем организма, их увеличение в процессе избыточной функциональной нагрузки (состояние гипертрофии) или уменьшение в процессе функциональной бездеятельности (состояние гипотрофии). Трофическое обеспечение всех структур организма происходит с помощью нервных волокон и сигнальных молекул, поступающих к рецепторам клеточных мембран в клетках-мишенях. Последние не только информируют нейроны о своем состоянии, но и оказывают на них стимулирующее воздействие, инициируя в нейронах адекватные функциональные изменения и, следовательно, опосредуя обратное воздействие на клеткимишени. При этом сама нервная система обеспечивает собственную трофику исключительно за счет сигнальных молекул, поступающих в нейроны от клеток-мишеней, которые они иннервируют. Все метаболические реакции, происходящие в многоклеточном организме, катализируются регуляторными белками-ферментами и протекают благодаря связыванию ферментов с субстратами. Такого рода реакции называются ферментативными.
Ферменты и ферментативные реакции
Молекула фермента (энзима) способна к образованию активного центра или «кармана», в который попадает молекула субстрата и в котором она «атакуется» разными функционально активными группами.
Согласно закономерностям классической генетики, одна биохимическая реакция катализируется одним ферментом. В ее основе лежит формула, предложенная в 1941 году Дж. Бидлом и Э. Тейтемом: «Один ген - один фермент», которая потом трансформировалась в формулу: «Один ген - одна полипептидная цепь», длительное время считавшуюся центральной догмой молекулярной биологии (см. главу 1).
Необходимо отметить, что этот общий для энзимологии принцип и теперь достаточно часто соблюдается для многофункциональных ферментов и многоферментных систем (комплексов).
Протекающие в организме ферментативные реакции основаны на модели Михаэлиса-Ментен, которая учитывает все известные формулы генной экспрессии:
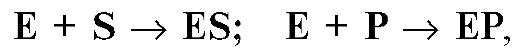 где E - фермент; S - субстрат; ES иЕР - комплексы фермента с субстратом S и фермента с продуктом Р.
где E - фермент; S - субстрат; ES иЕР - комплексы фермента с субстратом S и фермента с продуктом Р.
Таким образом, для одной ферментативной реакции необходимы как минимум субстрат и фермент. При этом субстратом могут быть молекулы ДНК, РНК, белка или другие молекулы, а ферментом - молекулы регуляторных белков. Ферментативные реакции сопровождают все альтернативные процессы в клетке и организме: прогресс и регресс, синтез и распад, развитие и инволюция (старение), возбуждение и торможение, сон и бодрствование и вообще любые другие молекулярные процессы (физикохимические, генетические и биохимические, морфологические, физиологические и патофизиологические), связанные с онтогенезом (см. главу 12).
Функции клетки и организма, обеспечиваемые с помощью ферментов
Как известно, ферменты обеспечивают многочисленные функции клетки и организма. Перечислим наиболее важные из ферментативных реакций. Это:
• экспрессия генов, производящих структурные и функциональные белки для клеток, тканей, органов и систем организма (реакции ДНК-мРНК-белок);
• защитные реакции организма: врожденный и приобретенный иммунитет, свертывание крови, действие цитохрома Р450 и др. (реакции фермент-белок, фермент-субстрат);
• узнавание молекул при их трансмембранном переносе (включая контроль потоков ионов), взаимодействии гормонов и других сигнальных молекул с рецепторами, генерации и проведении нервных импульсов и др. (те же реакции);
• умственная деятельность, работа мышечных клеток и др. (те же реакции). Кроме того, это также ферментативные реакции, происходящие между нуклеиновыми кислотами, начиная с этапа оплодотворения гамет. Например, в реакции ДНК-ДНК участвуют ферменты цитоплазмы яйцеклетки и ядерные факторы транскрипции, содержащиеся в яйцеклетке и сперматозоиде. Другие примеры - это реакции ДНК-мРНК в ходе транскрипции, реакции мРНК-рРНК, мРНК-тРНК в ходе трансляции, а также реакции
сайт-специфического связывания между аминокислотными остатками ферментов и нуклеотидными последовательностями ДНК.
ОСНОВНЫЕ СОБЫТИЯ ВНУТРИКЛЕТОЧНОГО
МЕТАБОЛИЗМА
Среди множества разных по биологической значимости событий внутриклеточного метаболизма в первую очередь следует рассмотреть основные «технологические» процессы, происходящие на молекулярном уровне, - это метаболизм пуриновых и пиримидиновых нуклеотидов, заменимых и незаменимых аминокислот, деградация ДНК и белков.
Метаболизм нуклеотидов
Нуклеотид - это соединение трех элементов: фосфат-сахар- основание. В этом соединении с образованием энергии связан фосфат. При этом центральную роль играет главный энергетический субстрат - АТР, хотя другие энергетические соединения (GTP, CTP, UTP) также принимают участие. Углеводные компоненты нуклеотида представлены дезоксирибозой или рибозой. С ними через атом азота (N) в девятом положении пурина и первом положении пиримидина связаны азотистые основания, обеспечивающие передачу наследственной информации.
Азотистые основания нуклеотида - это пурины А и G и пиримидины С, Т и U.
К ДНК-нуклеотидам (дезоксирибонуклеотидам) относятся: аденин (dАМР), гуанин (dGMP), цитозин (dСМР) и тимин (dТМР).
К РНК-нуклеотидам (рибонуклеотидам) относятся: адениловая (АМР), гуаниловая (GMP), цидиловая (СМР) и уридиловая (UMP) кислоты.
Синтез пуриновых нуклеотидов
Большинство клеток синтезируют пурины de novo из предшественников, имеющих небольшую молекулярную массу. Источниками свободных пуринов служат нуклеиновые кислоты, распадающиеся в лизосомах клетки при деградации молекулы ДНК, а местом их синтеза является печень.
Пуриновый цикл - это реакция риботилирования или присоединения комплектующих фрагментов к рибозо-5-фосфату. Этот
механизм характерен для синтеза пуринов de novo, свободных пуринов и пиримидиновых нуклеотидов при участии 5-фосфорибозил- 1-пирувата (активная форма рибозо-5-фосфата) или фосфорибозил- 1-пирофосфата (ФРПФ). Образование ФРПФ происходит по пентозофосфатному пути при переносе пирофосфатной группы (РР) с АТР глутамина, катализируемого с помощью ФРПФ-синтетазы. Продукт этой реакции - 5-фосфорибозиламин. После его образования происходит ряд последовательных реакций (их всего 9), завершающихся сборкой первого пуринового нуклеотида, включающего гипоксантин, - это инозиновая кислота (IMP) .
Образование IMP служит своеобразным метаболическим перекрестком: из этой кислоты образуются аденин или гуанин.
В ходе указанных преобразований из NH10-формилтетрагидрофолата или тетрагидрофолата (FH4) также образуется формильная группа (FH) - это реакция формилирования. Участник реакции формилирования - тетрагидрофолат (FH4) является коферментом витамина F или фолиевой (птероилглутаминовой) кислоты. FH4 восстанавливается из гидрофолатредуктазы (FH2) при участии NADPH и формилтрансферазы.
Формильная группа также поступает от серина, который в присутствии серингидроксиметилазы переносит гидроксиметильную группу (СН2ОН) на FH4, и в результате образуются глицин, вода и N5N10-метилен-FH4. Вместе с тем, последнее соединение еще не готово к участию в формилировании, так как метиленовая группа (СН2) более восстановлена, чем формильная группа (FH). Поэтому СН2 окисляется ферментом NADP+ до метильного производного, которое после гидролиза превращается в N10-формил-FH4, или донор формильной группы, необходимый для синтеза пуриновых нуклеотидов. Здесь уместно заметить, что синтез пуринов de novo не дает свободных пуринов, так как новые пурины сразу превращаются в пуриновые нуклеотиды. Известен также путь синтеза пуриновых нуклеотидов, в ходе которого в них превращаются свободные пурины, получаемые при распаде нуклеотидов и сохраняющиеся после взаимодействия с ФРПФ. В этом пути участвуют две фосфорибозилтрансферазы: одна катализирует образование пуриновых нуклеотидов из аденина, другая - из гипоксантина и гуанина. Причем во втором случае (синтез пуриновых нуклеотидов из гипоксантина или рибонуклеотида АМР, а синтез гуанина из рибонуклеотида GMP) принимает участие гипоксантин-гуанинфосфорибозилтрансфераза
(ГГФРТ), которая, взаимодействуя с ФРПФ, образует IMP и неорганический фосфор (Р). Для клеток человека сбережение аденина, по-видимому, менее важно, чем сбережение гипоксантина и гуанина. Свободный гипоксантин образуется из АМР при удалении фосфатной группы (РН) с помощью 5-нуклеотидазы и при этом превращается в аденозин, из которого удаляется NH2-группа с помощью фермента аденозиндезаминазы (АДА) . В итоге аденозин превращается в инозин, из которого с помощью другого фермента - нуклеозидфосфорилазы - образуются гипоксантин и рибозо-1-фосфат.
Следует также отметить, что при отсутствии в этом метаболическом пути АДА развивается аутосомно-рецессивная болезнь лимфоцитов (20q13.11), проявляющаяся тяжелым комбинированным иммунодефицитом (ТКИД). Предложенный в США в 1990 г. способ лечения ТКИД стал первым случаем применения в медицине метода генной терапии, суть которого заключалась во введении в стволовые клетки костного мозга in vitro нормального гена АДА и последующей аутотрансплантации этих клеток in vivo (см. главу 20).
В заключение необходимо подчеркнуть, что синтез пуриновых нуклеотидов требует от клетки больших энергетических затрат, и поэтому механизм реутилизации свободных пуринов более выгоден для нее, ибо позволяет клетке ограничить синтез de novo.
Кроме того, в организме имеются уникальные клетки (эритроциты), которые не способны синтезировать пурины de novo, и поэтому используют лишь готовые пуриновые основания. Значение механизма реутилизации свободных пуринов можно продемонстрировать на примере Х-сцепленного рецессивного синдрома Леша-Найана (Xq26-27), связанного с умственной отсталостью, нарушениями координации и аутоагрессией (из-за отсутствия ГГФРТ). У таких больных в клетках печени резко увеличен синтез пуриновых нуклеозидов de novo (есть сахар и основание, но нет фосфата), что ведет к увеличению уровня ФРПФ, образованию большого количества мочевой кислоты (в печени) и отложению кристаллов уратов (в почках).
Схожая симптоматика наблюдается при подагре, однако в этом случае у больных нет неврологических расстройств (по неизвестным пока причинам). По-видимому, подагра - это фенокопия синдрома Леша-Найана. При этом важно отметить, что мочевая кислота, образующаяся в печени из гипоксантина и гуанина, ингибируется аллопурином. Этот препарат применяется для лечения подагры, он вызывает преимущественное накопление не уратов, а гуанина
и гипоксантина, которые растворимы в воде и поэтому легко выводятся из организма.
Путь синтеза пуриновых нуклеотидов de novo - это пример аллостерического ингибирования по принципу обратной связи. При этом местом контроля ингибирования служит первая (обратимая) реакция синтеза пуринов. Ее катализирует ФРПФ-синтетаза, ингибируемая рибонуклеотидами AMP, ADP,GMP и GDP.
Синтез пиримидиновых нуклеотидов
Большинство клеток синтезирует пиримидиновые нуклеотиды de novo. Вместе с тем, известен путь реутилизации свободных пиримидинов, который менее ярок, чем у пуринов.
Синтез свободных пиримидинов начинается с аспарагиновой кислоты и ведет к образованию оротовой кислоты (соединение с циклической структурой), которая в присутствии ФРПФ и под действием киназ преобразуется в урацил (UМР).
В ходе сбалансированного производства дезоксинуклеотидтрифосфатов (так же, как пуринов) осуществляется аллостерическая регуляция по принципу обратной связи (см. главу 8). Их восстановление идет на уровне дифосфатов при помощи NADPH (переносит электроны на редуктазу). Только после этого они в несколько стадий превращаются в трифосфаты (в результате фосфорилирования киназами с участием АТР). Сначала не участвующий в синтезе dUTP (он содержит тимин) гидролизуется в dUMP с образованием РР. Затем dUMP метилируется в dTMP.
Метилирование - это перенос обладающей высокой активностью метильной группы (СН3) от донора-метионина на молекулы других соединений, включая ДНК (см. ниже). При этом нуклеозид-тимидин ресинтезируется до ТМП под действием тимидинкиназы.
Метилирование dUMP происходит также под действием фермента тимидилатсинтетазы, коферментом которой является N5N10-метилен- FH4 или метилентетрагидрофолат (FH4). Далее dTMP фосфорилируется в dTTP (метилированный урацил), который превращается в dTTP.
В случае синтеза пуринов метиленовая группа (СН2) окисляется с образованием формильной группы (см. выше), а в случае синтеза тимидилата она восстанавливается и переносится к метильной группе (СН3) тимина в присутствии тимидилатсинтетазы. При этом FH4 превращается в FH2. Для обратной восстановительной реакции необходим фермент дегидрофолатредуктаза, с дефицитом которого связано появление одной из генокопий фенилкетонурии (см. ниже).
Другой метаболический путь - это превращение FH4 в метилен- FH4 в реакции с серином, но для этого FH2 восстановливается до FH4 под действием дегидрофолатредуктазы.
Показано, что антифолатами или структурными аналогами фолата, ингибирующими FH2-редуктазу, служат антилейкемические препараты: аметоптерин и аминоптерин, которые подавляют образование dTMP, крайне необходимого лейкозным клеткам. Причем образование dTMP идет по пути ингибирования FH2-редуктазы и торможения процесса превращения FH2 в FH4. Далее фермент серингидроксиметилаза способствует образованию метилен-FH (из фолиевой кислоты), а затем в присутствии dUMP образуется dTMP.
Кроме этих реакций, известна реакция, в ходе которой N5N10- метилен-FH4 восстанавливается до N5-метилен-FH4, который поставляет метильные группы для превращения гомоцистеина в метионин в присутствии метионинсинтетазы.
Метионинсинтетазе необходим кофактор - витамин В12, при полном отсутствии которого развивается аутосомно-рецессивная пернициозная анемия (6р12-р21.2). При этом заболевании в организме не образуется желудочный гликопротеин, необходимый для восстановления в кишечнике витамина В12, хотя с пищей его поступает много. В связи с этим запасы FH4 становятся недоступными для синтеза пуринов, и тетрагидрофолат превращается в метил- FH4, вызывая у больных неврологические расстройства, связанные с метилмалоновым ацидозом. При дефиците витамина В12 развивается аутосомно-рецессивная врожденная мальабсорбция фолата (мегалобластная анемия). Один из ее генов-кандидатов картирован на 11q13.3-q14.1.
Известны две реакции, зависимые от витамина В12. Они катализируются разными ферментами: метилмалонил-СоА-мутазой (6р12- р21.2) и метионинсинтазой (ген не картирован). При недостатке первого фермента развивается смертельный ацидоз, тогда как недостаток второго фермента вызывает только раннюю задержку психомоторного развития, сопровождающуюся неврологической симптоматикой, обусловленной токсическим действием гомоцистеина.
Метилирование последовательностей ДНК
Метилирование последовательностей ДНК (например, остатков цитозина в положении 5) происходит с образованием 5-метилцитозина (5-mC) при действии ряда ферментов, получивших общее название: цитозин-ДНК-метилтрансфераз или М-таз.
М-таза - это «поддерживающий» фермент, узнающий и метилирующий только наполовину метилированные последовательности ДНК, формирующиеся в ходе репликации, когда вновь синтезированная дочерняя цепь еще не метилирована. Известны четыре таких фермента (Dnmt 1, Dnmt 2, Dnmt 3а и Dnmt 3b). Наиболее изучен Dnmt 1 или белок с молекулярной массой около 190 кДа, имеющий 2 домена: каталитический (расположен в С-концевой части фермента), структурно близкий к бактериальным цитозиновым М-тазам, и регуляторный (расположен в N-концевой части), содержащий сигнальную последовательность, направляющую фермент в активные репликативные комплексы в делящихся клетках.
Ферментативная активность Dnmt 1 резко возрастает с началом синтеза ДНК. Возможно, что промотор гена этого фермента активируется продуктом гена H-ras, участвующим в переносе митогенного сигнала.
Показано, что остатки цитозина в основном метилируются в составе динуклетидов CpG, или CpG-островков (см. главы 1 и 25). Всего в геноме эукариот метилируется около 70% CpG-островков и 6-7% остатков цитозина. Такое поддерживающее метилирование отражено на рис. 32: в результате репликации метилированные динуклеотиды CpG присутствуют в материнской нити ДНК. ДНК-метилтрансфераза распознает в ней метилированные CpG и воссоздает тот же рисунок метилирования в дочерней нити. Следует отметить, что М-тазы обладают лишь ограниченной способностью метилировать последовательности ДНК de novo в полностью неметилированных участках и метилировать олигонуклеотиды, содержащие ошибочно спаренные основания (см. главу 10).
В настоящее время клонированы гены, продукты которых проявляют высокую способность к метилированию ДНК de novo и, возможно, ответственны за этот процесс.
Метилирование остатков цитозина влияет на структурные характеристики ДНК, что проявляется в облегчении перехода ее метилированных участков из В-формы в Z-форму, увеличении шага спирали ДНК и изменении кинетики образования крестообразных структур. При этом метильная группа 5-mC выступает на поверхности большой бороздки ДНК, находящейся в В-форме, что увеличивает ее гидрофобность и в ряде случаев становится решающим фактором взаимодействия ферментов с соответствующими участками молекулы ДНК. Кроме того, показано метилирование других последовательностей ДНК, например CpNpG, а также опи-
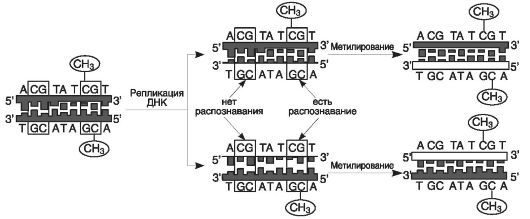 Рис. 32. Поддерживающее метилирование в геноме (по: Herman et al., 1999; http//www.kletca.ru/stem-cells/glossary/)
Рис. 32. Поддерживающее метилирование в геноме (по: Herman et al., 1999; http//www.kletca.ru/stem-cells/glossary/)
сан механизм метилирования аденина и гуанина - это метилирование с помощью сульфониевого катиона S-аденизилметионина или SАМ. В частности, механизм метилирования гуанина в положении 7 (N7-метилгуаниновая группа), а также в положении 2 (группа ОН второго и иногда третьего нуклеотида) играет важную роль в кэпировании мРНК с помощью РНК-полимеразы II или модификации мРНК с 5'-конца. В этом месте в составе первого нуклеотида имеется трифосфатная группа, и ее концевой фосфат удаляется с заменой на остаток GMP. Благодаря этому кэпированная мРНК достраивается до функционально активной мРНК. Кроме метилирования аденина и гуанина, для SАМ показано метилирование аминокислот и других веществ: креатина, ФХ и адреналина (относится к катехоламинам).
Деградация ДНК
Деградация ДНК - это универсальная для большинства клеток замена в ходе репликации старых молекул на новые. Процесс деградации ДНК считается необратимой терминальной стадией апоптоза, которая контролируется белками семейства Вс1-2 (см. главу 11).
Деградация мРНК
Выделено 2 механизма деградации мРНК (процессы NMD и SMD), относящихся к восстановительным механизмам клетки (см. главу 10).
На рис. 33 приведена схема механизма - NMD у млекопитающих: процесс NMD происходит во время трансляции, и с его помощью разрушается мРНК, содержащая преждевременные стоп-кодоны (ПСК), что прерывает трансляцию на расстоянии 50-55 нуклеотидов вперед по ходу считывания экзон-экзонной последовательности, возникшей в результате сплайсинга.
Предшественник мРНК (пре-мРНК) в ядре связан с гетеродимером СВР80 - СВР20 главного ядерного кэп-связывающего белка (CBP).
После формирования З'-конца пре-мРНК соединяется с ядерным поли (А)-связывающим Upf3a-белком или РАВР?-белком (PABP). Затем пре-мРНК подвергается сплайсингу и превращается в мРНК,
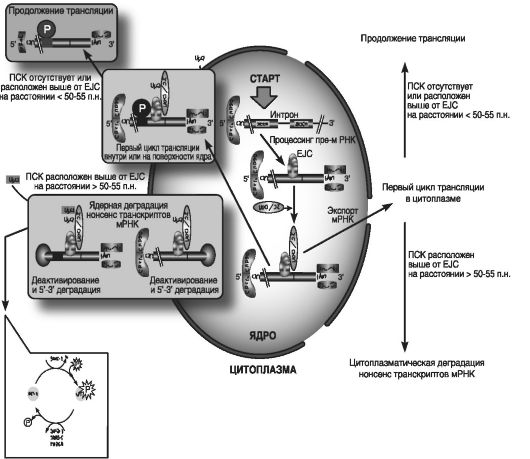 Рис. 33. Схема NMD у млекопитающих (по Maquat L., 2005)
Рис. 33. Схема NMD у млекопитающих (по Maquat L., 2005)
которая связывается с комплексом, включающим СВР80-СВР20, РАВРN1 и цитоплазматический белок PABPC, и после этого присоединяется к белкам экзон-экзонного комплекса или EJC, находящегося на расстоянии 20-24 нуклеотида вперед. Компонентами EJC служат ряд белков, включая:
• белки сплайсинга пре-мРНК (Pnn/DRS, RNPS1, SRm160, UAP56);
• белки, участвующие в экспорте мРНК (REF/Aly, Y14, Magoh);
• белки, функции которых полностью не выяснены (PYM, eIF4AIII и Barentsz/MLN512).
К комплексу EJC также могут присоединяться дополнительные белки:
• факторы NMD (Upf3 или Upf3a, Upf3X или Upf3b, Upf2);
• белок Upf1 (присоединяется, по-видимому, транзиторно).
Как полагают, белки Upf3/Upf3X, имеющие преимущественно ядерную локализацию, способны перемещаться в цитоплазму и взаимодействовать с белком Upf2, который сконцентрирован вдоль цитоплазматического края ядерной оболочки. При их перемещении образуется инициирующий комплекс первичной трансляции или мРНП. Этот комплекс проходит первичный цикл трансляции либо в ассоциации с ядром, либо с цитоплазмой, так как мРНК служит ассоциированным с ними субстратом для NMD.
Процесс NMD происходит после распознавания ПСК во время первого цикла трансляции. Если трансляция прерывается в ПСК, который находится на расстоянии более чем 50-55 нуклеотидов выше от экзон-экзонного соединения, то Upf1 инициирует процесс NMD путем взаимодействия с белком Upf2, ассоциированным c EJC.
Непосредственная деградация нонсенс-транскриптов в клетках млекопитающих происходит как в направлении 5' - 3', так и 3' - 5', включая соответственно декэпирование и действие 5' - 3' экзонуклеотических факторов или же деаденилирование и действие 3' - 5' экзосомальных факторов.
Участие EJC-комплексов в процессе NMD подтверждается данными о том, что мРНК, содержащие ПСК, и мРНК, происходящие из генов, не содержащих интроны, не подвергаются NMD. Мишенью для NMD служат только вновь синтезированные мРНК, тогда как стабильные мРНК не подвергаются деградации.
Для NMD показано разрушение ряда нонсенс-транскриптов, включая:
• мРНК, относящуюся к продуктам альтернативного сплайсинга;
• мРНК, необходимую для селенопротеинов;
• мРНК с открытой рамкой считывания;
• мРНК, содержащую интрон в 3'-нетранслируемой области гена. В случае цитоплазматической деградации пока не ясно, происходит
ли NMD в комплексе с рибосомами, транслирующими белки, или цитоплазматическими внерибосомными участками деградации мРНК.
Эффективность NMD, как правило, не зависит от локализации ПСК и увеличения количества EJC-комплексов. Однако эффективность NMD может быть повышена за счет других механизмов, например за счет моделирования (замены) различных последовательностей генов.
В настоящее время продолжает изучаться роль NMD в других клеточных процессах. Например, один из его факторов - SMG1 - принимает участие в распознавании и/или репарации повреждений ДНК, другой фактор - Upfl - участвует в нонсенс-опосредованном альтернативном сплайсинге, а также в недавно открытом новом пути деградации мРНК - так называемой опосредованной деградации мРНК или SMD. В случае SMD-механизма деградации РНКсвязывающий белок напрямую взаимодействует с белком Upfl, вызывая деградацию мРНК на достаточно удаленном расстоянии от ПСК, включающем нормальный стоп-кодон.
Вместе с тем, для SMD-механизма окончательно не определено функциональное назначение указанных факторов.
Метаболизм аминокислот и его нарушения
Аминокислоты образуются из кетокислот и аммиака (аминогруппы). Они используются для синтеза различных белков, в том числе компонентов клеточных мембран, нейромедиаторов (например, 5-гидрокситриптамина и гамма-аминобутирата или GАВА), гормонов (например, тироксина), гема и других веществ. Источниками аминокислот служат пищевые продукты и продукты клеточного метаболизма.
Основные аминокислоты делятся на:
• незаменимые (поступают исключительно извне), их 10;
• заменимые (поступают извне и синтезируются в организме), их также 10.
Только одна аминокислота - аргинин - необходима организму в течение ограниченного периода развития (исключительно во время его роста), тогда как остальные 19 аминокислот необходимы всегда.
Абсолютно необходимыми считаются лизин, фенилаланин и триптофан. Цистеин можно получить из фенилаланина, а тирозин - из метионина. Основные аминокислоты либо кетогенны (образуют ацетил-СоА, который превращается в кетоновые тела), либо гликогенны (увеличивают уровень глюкозы в крови, а у диабетиков в моче). Вместе с тем, лейцин и лизин относятся и к тем и к другим (кетогенным и гликогенным).
На рис. 34 приведена общая схема метаболизма аминокислот. Как показано на схеме, избыток аминокислот формируется за счет аминокислотных остатков, не использованных в биосинтезе белков или на другие нужды клетки.
Сформировавшийся избыток аминокислот в виде метаболического фонда используется (при их расщеплении) для производства энергии и создания энергетических запасов (жиров и гликогена), а аминный азот выводится с мочой в виде мочевины.
В случае необходимости резервами для производства аминокислот могут стать функциональные мышечные белки (их в организме больше всего). У человека выявлен целый ряд наследственных болезней, связанных с нарушениями метаболизма аминокислот (см. главу 21). Для больных с такими нарушениями характерен либо дефицит, либо избыток какой-либо определенной аминокислоты, что ведет к пло-
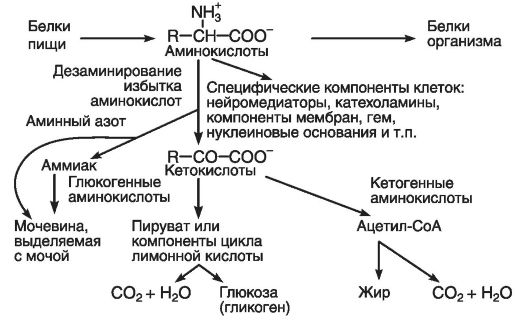 Рис. 34. Общая схема метаболизма аминокислот (по Эллиот В., Эллиот Д., 2002)
Рис. 34. Общая схема метаболизма аминокислот (по Эллиот В., Эллиот Д., 2002)
хому перевариванию и всасыванию пищи, истощению организма, задержке психомоторного и физического развития, отекам тканей, неврологической и другой симптоматике, базирующейся на синтезе неполноценных белков.
Наиболее характерным примером этих заболеваний служит аутосомно-рецессивная фенилкетонурия (ФКУ), развивающаяся в результате дефицита фермента фенилаланин-4-гидроксилазы
(12q24.2).
ФКУ имеет ряд генокопий (чаще всего злокачественных), развивающихся в результате дефицита 6-пирувоилтетрагидроптерин- синтазы (11q22.3-q23.3), цитозольной дигидроптеридинредуктазы (4р15.31), цитозольной гуанозин-циклогидролазы (ген не картирован) или тетрагидробиоптерина - кофактора ВН4 гидролаз ароматических аминокислот: фенилаланина, тирозина и триптофана (ген также не картирован).
Сюда же относится генокопия ФКУ вследствие дефицита дегидрофолатредуктазы.
Прежде чем рассмотреть механизмы патогенеза генокопий фенилкетонурии, отметим, что в нормально функционирующем организме фенилаланин не подвергается дезаминированию, а превращается в тирозин (под действием фенилаланин-4-гидроксилазы).
При ФКУ синтез тирозина затруднен либо полностью заблокирован, и фенилаланин «вынужденно» подвергается дезаминированию в фенилпируват (кетокислота), который выводится с мочой.
В связи с этими особенностями обмена фенилаланина в организме и наличием генокопий ФКУ был проведен анализ состава свободных аминокислот и результатов метаболизма фенилаланина в плазме крови небеременных и беременных женщин - носителей гена ФКУ (Васильева О.В., 1999). Состав свободных аминокислот в сыворотке крови у небеременных (первая группа) характеризовался соотношением заменимых и незаменимых аминокислот 38 и 62% соответственно, а также отношением гидрофобных и нейтральных к другим аминокислотам, составляющим 71 к 29%. Отмечались наибольшие концентрации аланина, треонина, лизина и аргинина; наименьшие - аспарагиновой и глутаминовой кислот.
При анализе корреляционных связей между количественными характеристиками аминокислотного спектра были выделены три степени сопряженности количественных показателей взаимно корре-
лируемых систем: высокие, средние и низкие уровни объединения аминокислот.
Соответственно, высокие уровни - это лизин, фенилаланин и тирозин; средние уровни - это гистидин, цистеин и валин; низкие уровни - это лейцин, изолейцин, метионин, аланин и аспарагиновая кислота. Кроме этих трех уровней, были выделены уровни корреляционных связей, характерные для аргинина и глутаминовой кислоты, а также серина и треонина. Полученные данные были объяснены автором структурно-функциональными особенностями аминокислот:
• фенилаланин является предшественником тирозина;
• фенилаланин, тирозин и лизин участвуют в синтезе ацетил-СоА без промежуточного образования пирувата;
• аргинин, гистидин и валин могут участвовать в синтезе глутаминовой кислоты;
• лейцин, изолейцин и метионин относятся к гидрофобным аминокислотам;
• аланин образуется при переаминировании пирувата, который может стать основой для синтеза глицина; донором аминогруппы служит аспартат.
Физиологическая беременность приводила к изменениям фонда свободных аминокислот за счет снижения содержания в первом триместре-глицина, валина, лейцина; во втором триместрегистидина, фенилаланина и цистеина. Если по данным массового (пилотного) скрининга беременных уровень фенилаланина в плазме крови соответствовал 1,2 мг%, то это оценивалось как критерий отбора женщин в группу «потенциальных гетерозигот» по гену ФКУ.
Если экспрессия скрытого в материнском организме гена ФКУ приводила в первом триместре беременности к возрастанию уровня фенилаланина в плазме крови выше 10 мг%, то это оценивалось как причина нарушений развития плода.
Облигатное гетерозиготное носительство гена ФКУ у матерей, родивших детей с ФКУ (вторая группа), в сравнении с женщинами без гена ФКУ (третья группа) проявлялось нарушениями метаболического фонда аминокислот в виде высокой концентрации глутаминовой и аспарагиновой кислот, треонина и глицина.
Отмечая большое теоретическое и практическое значение работы О.В. Васильевой, можно сделать вывод, что имеющиеся сегодня представления о спектре и механизмах проявления различных генокопий
ФКУ (а возможно, и генокопий при других наследственных болезнях обмена - НБО аминокислот) должны быть значительно расширены. Это может быть сделано не только за счет анализа фонда всех аминокислот, объединенных в отдельные группы в зависимости от степени сопряженности с их структурно-функциональными характеристиками, но и на основе анализа роли белков-ферментов, участвующих в метаболизме этих аминокислот. В пользу такого вывода свидетельствует также пример аутосомно-рецессивного лейциноза или болезни мочи «кленового сиропа», при которой выделены три генокопии, обусловленные дефицитом фермента: дегидрогеназы альфа-кетокислот с разными боковыми цепями, тип I A (19q13.1-13.2), тип I B (6p21-p22) и тип II (1p31). Лейциноз развивается в результате нарушения окислительного декарбоксилирования альфа-кетокислот, сопутствующих образованию алифатических аминокислот: лейцина, изолейцина и валина.
Завершая рассмотрение данных о значении метаболизма отдельных аминокислот, важно отметить, что наряду с дефицитом может наблюдаться их избыток, и тогда у больных диагностируются, например, такие заболевания, как:
• алкаптонурия (3q2) - результат нарушенного расщепления тирозина из-за избытка гомогентизиновой кислоты, являющейся его продуктом дифенолом; в этом случае дифенол соединяется с кислородом воздуха и образует пигмент, благодаря которому моча окрашивается в темный цвет;
• цистатионурия (16q) - это избыток цистатиона (см. ниже).
Синтез аминокислот
Предшественниками основных аминокислот являются 5 химических соединений: альфа-кетоглутарат, 3-фосфоглицерат, оксалоацетат (R= СН2СОО), фосфоенол-пируват, пируват (R=СН3) и два моносахарида пентозофосфатного пути. Рассмотрим механизмы их преобразования в аминокислоты.
Аминокислоты как продукты метилирования
Аминокислоты могут быть продуктами метилирования или переноса метильной группы от донора-метионина на различные соединения (см. выше). Когда метионин реагирует с АТР, то его NH3+-группа активируется с образованием сульфониевого катиона, или S-аденизилметионина (SАМ). Перенос метильной группы катализируется трансметилазами.
В ходе этой реакции три фосфатные группы АТР превращаются в пирофосфат (РР) и неорганический фосфор (Р), затем РР расщепляется до двух молекул Р.
Сначала SАМ превращается в S-аденозилгомоцистеин, который превращается в гомоцистеин-метионин (у него вместо группы S-CH3 имеется SH-группа, или тиольная группа). Далее тиольная группа с гомоцистеина переносится на серин с образованием цистеина. Промежуточным соединением в этой реакции является цистатион, избыток которого выводится с мочой.
Продуктами метилирования SАМ также являются: креатин, фосфолипид - ФХ и катехоламин - адреналин.
Аминокислоты как продукты трансаминирования
Аминокислоты могут быть продуктами трансаминирования или дезаминирования. Например, глутаминовая кислота синтезируется с помощью глутаматдегидрогеназы, кофакторами которой являются NAD+ и NADР+. Эта реакция обратима.
Донорами глутаминовой кислоты служат аспарагиновая кислота и аланин, образующиеся при трансаминировании оксалоацетата и пирувата. Глутаминовая кислота дезаминируется путем отщепления двух атомов водорода в присутствии глутаматдегидрогеназы, использующей в качестве окислителя NAD+или NADP+. Этот фермент аллостерически ингибируется ATP и GTP (они указывают на большие запасы энергии), но активируется ADP и GDP (они указывают на недостаток энергии).
После дезаминирования глутаминовой кислоты образуется альфакетоглутарат, который участвует в цикле Кребса (цикл лимонной кислоты), что делает возможным окисление глутаминовой кислоты
до Н2О и СО2.
Поскольку альфа-кетоглутарат превращается в оксалоацетат, он может участвовать в синтезе глюкозы, т.е. глутаминовая кислота - это гликогенная аминокислота.
Для других аминокислот (кроме глутаминовой) нет соответствующих дегидрогеназ. Поэтому их дезаминирование идет не в одну, а в две стадии: первая стадия - переаминирование, вторая стадия - дезаминирование. В целом этот общий для всех аминокислот метаболический путь называется трансаминированием или дезаминированием.
Трансаминирование катализируется аминотрансферазами (трансаминазами), которые специфичны для разных аминокислот.
В активных центрах трансаминаз находится кофермент пиридоксаль-5-фосфат (ПФ), выступающий в роли электрофильного интермедиатора, который сначала принимает аминогруппу
(служит ее акцептором), а затем (как донор) передает ее кетокислоте.
Рабочей группой ПФ является альдегидная группа (СНО) .
ПФ включает три производных витамина В6: пиридоксаль, пиридоксин и пиридоксамин.
Механизм трансаминирования можно продемонстрировать на примере аланина, являющегося транспортной формой аминного азота в крови.
В аланине сосредоточено около 30% аминного азота, поступающего в печень после расщепления мышечных белков и образующегося из пирувата при трансаминировании других аминокислот.
При взаимодействии аланина и альфа-кетоглутарата возникает пируват, который вступает в реакцию с глутаматом, - это первая стадия. Во второй стадии глутамат присоединяет NAD+ и воду, образуя кетоглутарат, который взаимодействует с NaРН+ и NH4. В печени аланин дезаминируется. Образующийся при этом аммиак идет на синтез мочевины, а пируват - на синтез глюкозы, которая возвращается с кровью в мышцы, замыкая глюкозо-аланиновый цикл переноса аммиака. Этот цикл приобретает особое значение при голодании, когда в ходе глюконеогенеза в печени используются аминокислоты, образующиеся при расщеплении мышечных белков.
Синтез серина и глицина
Серин синтезируется в три стадии из гликозила-3-фосфатглицерина, который сначала окисляется в кетокислоту (трифосфатгидроксипируват); она трансаминируется глутаминовой кислотой и превращается в 3-фосфат-серин, гидролизующийся до серина и неорганического фосфора.
Глицин синтезируется путем удаления из серина гидроксиметильной группы (см. выше). Реакция идет с участием тетрагидрофолиевой кислоты, являющейся переносчиком моноуглеродных групп.
Такого рода перенос важен для синтеза нуклеотидов.
Синтез других аминокислот
Глутамин (так же как аланин) служит транспортной формой аммиака в крови, который образуется при дезаминировании аминокислот.
Аммиак токсичен и поэтому в печень поступает не в свободном виде, а в соединении с глутаминовой кислотой, образуя при участии фермента глутаминсинтетазы амид глутаминовой кислоты, или
глутамин. В качестве промежуточного продукта образуется гаммаглутамилфосфат (ангидрит глутаминовой и фосфорной кислот) - это макроэргическое соединение, способное взаимодействовать с ионами аммония при участии синтетазы (см. главу 8).
Источником глутамина служит альфа-кетоглутарат из цикла Кребса, подвергающийся трансаминированию другими аминокислотами.
Глутамин переносится кровью в печень, где гидролизуется глутаминазой, а высвобождающийся при этом аммиак используется для синтеза мочевины.
Фенилаланин - это ароматическая аминокислота, избыток которой в нормально функционирующем организме превращается в тирозин при участии фенилаланин-4-гидроксилазы, поставляющей 2 атома водорода от кофермента - тетрагидробиоптерина (см. выше).
Лейцин, изолейцин и валин - это алифатические аминокислоты, промежуточные продукты которых накапливаются в виде кетокислот при лейцинозе (см. выше).
Метаболизм других соединений из аминокислот
Кроме белков, из аминокислот образуются: амины (в результате декарбоксилирования); катехоламины или гормоны, сходные по структуре с катехолом (например, 1,2-дегидроксибензол), включая дофамин, адреналин и норадреналин; нейромедиаторы (GABA и 5-окситриптамин), а также гормон - тироксин.
Аминогруппы после их удаления из аминокислот выводятся с мочой в виде мочевины - это инертное водорастворимое нетоксическое соединение.
Мочевина образуется в печени при отщеплении гуанидиновой группы от аргинина. При этом попутно образуется аминокислота - орнитин, не входящая в состав основных белков организма.
Для превращения орнитина обратно в аргинин используются атом углерода, получаемого из углекислого газа, и аминный азот, выделяемый в ходе метаболизма любой из основных аминокислот.
Образование аргинина из орнитина идет в несколько стадий. В качестве промежуточного продукта образуется аминокислота - цитруллин, также не входящая в состав основных белков организма; она стимулирует синтез мочевины в печени (как орнинтин и аргинин).
Деградация белков
Деградация белков - это замена старых белковых молекул на новые молекулы. Она происходит во всех клетках и тканях организма в ходе метаболизма. Белки имеют различную продолжительность жизни. К белкам-долгожителям относятся структурные белки и гемоглобин. Несколько дней живут белки печени.
Многие белки имеют продолжительность жизни не более 20 ч, а часть из них живут не более десяти или даже двух минут.
В связи с разной продолжительностью жизни белков их деградация характеризуется высокой селективностью.
В ходе синтеза аминокислот и последующего производства из них структурных и регуляторных белков их абсолютная структурная (и функциональная) точность не всегда соблюдается. Поэтому в клетках неизбежно образуются ошибочные аминокислоты, что влечет за собой неправильный фолдинг белков (см. главу 3), и такие белки уничтожаются клеткой, т.е. подвергаются процессу деструкции. Для селективной деструкции важен белок - убиквитин. Это небольшой белок, участвующий в АТР-зависимой реакции, при которой его концевая карбоксильная группа связывается с аминогруппой боковой цепи белка-мишени (остатками лизина), который должен подвергнуться деструкции, т.е. он как бы «метится» для нее.
Выбор убиквитином белков для уничтожения, по-видимому, определяется их N-концевыми аминокислотными остатками. В мечении также могут участвовать белки - молекулярные шапероны (см. главу 3).
В деградации долгоживущих белков принимают участие лизосомы цитоплазмы, ответственные за аутолитическую деструкцию клеток в ходе их регресса. Деградация клеточных белков жизненно необходима при голодании организма, когда распадаются мышечные белки, за счет которых может происходить глюконеогенез аминокислот.