Детская неврология : учебник : в двух томах / А. С. Петрухин. - Т. 2. - 560 с. : ил.
|
|
|
|
ГЛАВА 4. НАСЛЕДСТВЕННЫЕ МЕТАБОЛИЧЕСКИЕ ЗАБОЛЕВАНИЯ НЕРВНОЙ СИСТЕМЫ У ДЕТЕЙ
Наследственные метаболические заболевания (НМЗ) - огромный класс моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные белки. На сегодняшний день насчитывается более 500 нозологических форм, и это число постоянно увеличивается. Ранняя диагностика НМЗ дает врачу возможность применять эффективные методы лечения, которые являются малоили безуспешными на более поздних стадиях патологического процесса. Кроме того, правильный окончательный диагноз необходим для проведения грамотного медико-генетического консультирования семьи. Все НМЗ подразделяют на 22 подкласса в зависимости от ведущего биохимического и/или молекулярно-генетического дефекта. Наиболее распространенными и хорошо изученными среди них являются:
• лизосомные болезни накопления;
• митохондриальные энцефаломиопатии;
• пероксисомные болезни;
• нарушения обмена аминокислот/органических кислот;
• нарушения обмена углеводов.
4.1. Лизосомные болезни накопления
Лизосомные болезни накопления характеризуются нарушением расщепления макромолекул в лизосомах. Лизосомы - это органеллы, окруженные одной мембраной и содержащие около 50 различных ферментов. При мутациях в генах, кодирующих лизосомные ферменты или транспортные белки, нерасщепленные субстраты накапливаются в лизосомах, что вызывает нарушение их функции и приводит к гибели клеток. В зависимости от природы накапливаемых в лизосомах макромолекул выделяют: мукополисахаридозы, гликопротеинозы, сфинголипидозы, нейрональные цероидные липофусцинозы. Частота встречаемости ЛБН составляет 1 на 7000-8000 живых новорожденных.
Болезнь Краббе (БКр глобоидно-клеточная лейкодистрофия, недостаточность галактоцереброзидазы). В зависимости от возраста начала заболевания выделяют 4 клинические формы: инфантильную, позднюю инфантильную, ювенильную и взрослую. Болезнь Краббе - аутосомнорецессивное прогрессирующее заболевание. Ген картирован на хромосоме 14q21-q31. В результате снижения активности лизосомного фермента
галактоцереброзидазы происходит накопление предшественника галактоцереброзида - галактозилцерамида (психозина) в бимолекулярном слое миелина. Психозин токсичен и вызывает гибель олигодендро- цитов, распад миелинового волокна. В глиальной ткани образуются характерные включения - «глобоидные клетки».
Клиника. Наиболее часто встречается инфантильная (классическая) форма (85-90% случаев). Она начинается в первые 6 мес жизни и проходит несколько стадий. Для 1-й стадии характерны немотивированные подъемы температуры, гипервозбудимость (беспричинный крик, нарушение сна), диспепсия (отказ от еды, срыгивание, рвота), спастический гипертонус. К 6-8-му мес жизни становится заметным отставание в психомоторном развитии, в некоторых случаях присоединяются судороги. На 2-й стадии заболевания утрачиваются ранее приобретенные навыки, нарастает мышечный тонус до опистотонуса; характерны сухожильная гиперрефлексия, прогрессирующая деменция, атрофия зрительных нервов со снижением реакции зрачков на свет, гипотрофия вплоть до кахексии. У 80-90% больных отмечаются судороги (миоклонические, генерализованные тонико-клонические), резистентные к антиэпилептической терапии. Для терминальной стадии характерно отсутствие контакта с окружающим, поза децеребрационной или декортикационной ригидности, бульбарнопсевдобульбарный синдром, сухожильная гипо- и арефлексия.
При поздней инфантильной и ювенильной формах начинается в возрасте от 2 до 6 лет, как правило, с нарушения зрения в виде зрительной агнозии, гемианопсии. Позднее присоединяются нарушения походки, которые обусловлены спастическими парезами и параличами, прогрессирующей мозжечковой атаксией и невропатией. По мере прогрессирования болезни зрение снижается практически до амавроза, развиваются грубый спастический тетрапарез, бульбарнопсевдобульбарный синдром, органическая деменция.
Диагностика. На начальных стадиях заболевания на КТ и МРТ головного мозга выявляют поражение белого вещества мозжечка, подкорковых структур (зубчатого ядра, базальных ганглиев и/или таламуса), пирамидных трактов. Позднее возникают атрофия большого мозга, поражение мозолистого тела (задних отделов ствола и/или валика) и теменно-затылочных отделов белого вещества головного мозга.
Диагноз подтверждают определением активности фермента галактоцереброзидазы с использованием радиоактивного меченого или искусственного субстрата в лейкоцитах крови или культуре клеток кожных фибробластов, активность которого составляет 0-5% нормы.
Профилактика и лечение. Специфическая терапия в настоящее время не разработана. На доклинической стадии заболевания, особенно при поздних формах, применяют трансплантацию костного мозга. Пренатальная диагностика проводится путем измерения активности галактоцереброзидазы в амниоцитах и клетках ворсин хориона или методами ДНК-диагностики, если генотип пробанда известен.
Метахроматическая лейкодистрофия (МЛД, сульфатидный липидоз, диффузный склероз мозга, метахроматическая лейкоэнцефалопатия). МЛД является аутосомно-рецессивным прогрессирующим заболеванием, возникающим при снижении активности лизосомного фермента арилсульфатазы А (АСА), которая участвует в гидролизе цереброзидсульфатов. Происходит накопление сульфатидов и снижение уровня цереброзидов в белом веществе центральной и периферической нервной системы. Миелиновая оболочка становится нестабильной и разрушается. Ген арилсульфатазы А картирован на хромосоме 22q13.31-qter. Частота встречаемости заболевания - 1 на 40 000. Выделяют несколько форм МЛД в зависимости от возраста дебюта: врожденную, позднюю инфантильную, ювенильную и взрослую. Наиболее часто встречается поздняя инфантильная (классическая) форма.
Возраст начала болезни варьирует от 6 мес до 4 лет. Для 1-й стадии характерна прогрессирующая полиневропатия, проявляющаяся гипотонией мышц конечностей, снижением или отсутствием сухо- жильных рефлексов, нарушением походки. На 2-й стадии становятся заметными нарушения психомоторного и речевого развития. Типичны спастическая нижняя параплегия, туловищная атаксия, периодические боли в конечностях, атрофия зрительных нервов, нистагм. Во время 3-й стадии продолжается утрата ранее приобретенных навыков, развивается спастический тетрапарез, однако сохраняется способность дифференцировать окружающих, эмоционально реагировать на обращенную речь. Примерно у 25% больных появляются эпилептические приступы. Для терминальной стадии болезни характерны отсутствие контакта с окружающими, поза децеребрационной или декортикационной ригидности, бульбарно- псевдобульбарный синдром, учащение судорог, амавроз, снижение слуха до полной глухоты, нарушения речи вплоть до мутизма.
При ювенильной и взрослой формах первыми симптомами заболевания обычно являются нарушения интеллекта или/и поведения, которые часто расценивается как шизофреноподобный синдром. В дальнейшем присоединяются пирамидные (центральные парезы и параличи конечностей), экстрапирамидные (тремор, ригидность,
гиперкинезы) и мозжечковые (атаксия, дизартрия) расстройства, развивается органическая деменция, снижаются зрение и слух.
Диагностика. В большинстве случаев при МРТ головного мозга выявляют гиперинтенсивные в Т2-режиме очаговые изменения белого вещества. На ранних стадиях болезни на ЭНМГ снижены скорости проведения импульса по периферическим нервам. На поздних стадиях заболевания развивается атрофия зрительных нервов.
Основным методом подтверждения диагноза является определение активности АСА в лейкоцитах крови и культуре клеток кожных фибробластов. При МЛД активность фермента составляет 0-20% нормы.
Профилактика и лечение. В настоящее время эффективное лечение не разработано, поэтому применяют симптоматическую терапию. На стадии разработки находится фермент-заместительная терапия - препарат «метазим» (фирмы Zymenex), предназначенный для лечения поздней инфантильной формы заболевания. При поздней инфантильной и ювенильной формах проводится ТКМ. Пренатальная диагностика осуществляется путем измерения активности АСА в амниоцитах и клетках ворсин хориона или методами ДНК-диагностики, если генотип пробанда известен.
4.2. Гликогенозы (болезни накопления гликогена)
В расщеплении гликогена участвуют несколько ферментов, в том числе и лизосомный фермент - глюкозидаза. Поэтому гликогеноз 2-го типа (болезнь Помпе), связанный с нарушениями активности гликозидазы, относят как к ЛБН, так и к нарушениям обмена гликогена. Частота встречаемости заболевания - 1 на 40 000 живых новорожденных.
Болезнь Помпе (гликогеноз 2-го типа). Лизосомный фермент глюкозидаза (кислая мальтаза) участвует в расщеплении гликогена в лизосомах. Ген глюкозидазы картирован на длинном плече хромосомы 17q25. Мутации в гене вызывают снижение активности фермента глюкозидазы, что приводит к отложению гликогена в разных тканях, преимущественно в печени, сердце и скелетных мышцах.
Классическая инфантильная форма. Дебют заболевания приходится на первое полугодие жизни. Манифестными симптомами являются мышечная гипотония и диспепсия. В дальнейшем развивается быстро прогрессирующая гипертрофическая кардиомиопатия: тоны сердца глухие, сердечные аритмии, часто в виде ритма галопа, на рентгенограмме грудной клетки размеры сердца увеличены. У больных наблюдается
симптомокомплекс «вялого ребенка»: снижение спонтанной двигательной активности, диффузная мышечная гипотония, при тракции за руки происходит запрокидывание головы назад, сухожильные рефлексы снижены. Скелетные мышцы часто увеличены в объеме и плотные на ощупь. При ЭНМГ регистрируют первично-мышечные нарушения. Печень обычно не увеличена, но очень плотная при пальпации.
Ювенильная и взрослая формы. Заболевание характеризуется поздним дебютом и медленным прогрессированием основных клинических симптомов, ведущим из которых является миопатический синдром. На начальных этапах болезни появляются снижение мышечной силы и атрофии проксимальных отделов нижних конечностей, в дальнейшем поражаются диафрагма и другие группы мышц, участвующие в акте дыхания, что приводит к легочной недостаточности и затруднению/ нарушению дыхания во сне.
Диагностика. Основными методами подтверждения диагноза являются определение активности альфа-глюкозидазы (в фибробластах, мышечной ткани, лейкоцитах крови), а также методы ДНК-анализа.
Профилактика и лечение. Пренатальную диагностику проводят путем определения активности лизосомной альфа-глюкозидазы в биоптате хориона. В настоящее время создан препарат для ферментзаместительной терапии (препарат «Миозим» фирмы Genzyme).
Мукополисахаридозы - группа лизосомных болезней накопления, обусловленная нарушениями обмена гликозаминогликанов. Выделяют 10 форм мукополисахаридозов, различающихся по типу накапливаемых гликозаминогликанов, типу наследования и дефекту ферментов. Клиническими особенностями этой группы являются сочетания поражения нервной системы, скелета и внутренних органов. Наиболее хорошо изучены мукополисахаридоз I, II, III типов.
Мукополисахаридоз I типа - аутосомно-рецессивное заболева-ние, возникающее в результате снижения активности альфа-L-иду-ронидазы, которая участвует в метаболизме двух гликозаминогликанов - дерматансульфата и гепарансульфата. Поскольку идуроновая кислота входит в состав дерматансульфата и гепарансульфата, происходит нарушение внутрилизосомного распада этих гликозамингликанов и накопления их в лизосомах. Ген альфа-L-идуронидазы картирован на хромосоме 14р16.3. Частота встречаемости заболевания -1 на 144 000 живых новорожденных. Гликозоаминогликаны накапливаются повсеместно: в хрящах, сухожилиях, надкостнице, эндокарде и сосудистой стенке, печени, селезенке и нервной ткани. В зависимости от выраженности
клинических симптомов заболевания различают 3 формы заболевания: синдромы Гурлер, Гурлер-Шейе и Шейе.
Синдром Гурлер дебютирует на первом году жизни задержкой пси- хомоторного развития, грыжами (пупочной и пахово-мошоночной), частыми респираторными инфекциями. С возрастом постепенно формируются характерные черты лица - «гаргоилизм», нарушения со стороны опорно-двигательного аппарата. Типичный вид больного: большая голова, череп гидроцефальной формы, выступающие швы черепа и лобные бугры, большой лоб, широкая переносица, короткие носовые ходы с вывернутыми кнаружи ноздрями, полуоткрытый рот, большой высунутый язык, толстые губы, гиперплазия десен, неправильный рост зубов, короткая шея, тотальная платиспондилия и/или кифоз поясничного отдела (поясничный «гибус»), бочкообразная или колоколообразная грудная клетка. Отмечаются задержка роста, шумное дыхание, тугоподвижность мелких и крупных суставов. Также на первом году жизни может отмечаться помутнение роговицы, которое носит прогрессирующий характер; позднее может присоединиться глаукома. Для всех больных характерны задержка психомоторного развития, гепатоспленомегалия, прогрессирующая нейросенсорная тугоухость.
Диагностика. При МРТ головного мозга обнаруживают множественные кисты в проекции перивентрикулярного белого вещества, мозолистого тела, реже - базальных ганглиев. Диагноз подтверждают при выявлении повышенной экскреции дерматансульфата и гепарансульфата с мочой и определении активности альфа-L- идуронидазы (в фибробластах, лейкоцитах крови), а также с помощью ДНК-анализа.
Лечение и профилактика. Лечение симптоматическое. ТКМ может кардинально изменить течение заболевания и улучшить его прогноз, однако эта процедура имеет много противопоказаний. В настоящее время создан препарат для фермент-заместительной терапии - препарат «альдуразим» фирмы Genzyme.
Синдром Гурлер-Шейе дебютирует на первом десятилетии жизни, чаще в возрасте 3-8 лет. Характерны стертые гарголоидные черты лица, умеренная тугоподвижность суставов, нейросенсорная глухота, помутнение роговицы. Сердечно-сосудистые нарушения разной степени также наблюдаются практически у всех больных и включают поражение клапанов, утолщение миокарда, постепенное развитие сердечной недостаточности. У многих больных развиваются гидроцефалия и миелопатия шейного отдела позвоночника. Интеллект, как правило, не страдает.
Синдром Шейе дебютирует в конце первого десятилетия жизни. Основные симптомы заболевания: тугоподвижность суставов, туннельный синдром карпальной области (симптом «когтистой кисти»), черты лица по типу гаргоилизма, катаракта, глаукома, пигментная дегенерация сетчатки. Развиваются обструкция дыхательных путей, сопровождающаяся ночными апноэ; порок сердца в виде стеноза аорты. Миелопатия шейного отдела спинного мозга встречается реже, чем при синдроме Гурлер-Шейе.
Мукополисахаридоз II типа (синдром Хантера) - Х-сцепленное рецессивное заболевание, возникающее в результате снижения активности лизосомного фермента идуронатсульфатазы, которая участвует в метаболизме дерматансульфата и гепарансульфата. Ген картирован на длинном плече Х-хромосомы - Xq27-28. Частота встречаемости заболевания - 1 на 110 000 живых новорожденных. Патогенез син- дрома Хантера аналогичен патогенезу при синдроме Гурлер. По степени тяжести различают тяжелую и легкую формы заболевания.
Основные клинические симптомы сходны с синдромом Гурлер, но медленнее прогрессируют. Отмечаются хроническая диарея, эпилептические приступы; помутнение роговицы нехарактерно. У большинства больных отмечаются изменение лица по типу гаргоилизма, туннельный синдром карпальной области, паховые и пупочные грыжи. Реже выявляют локальные изменения на коже в виде образований, напоминающих «морскую гальку цвета слоновой кости», располагающиеся обычно на спине, плечах, боковой поверхности бедер. Тугоподвижность суставов, нейросенсорная тугоухость, комбинированные пороки сердца, обструкция дыхательных путей в виде ночных апноэ могут развиться на 2-3-м десятилетии жизни. Интеллект, как правило, не страдает.
Диагностика. Основными методами подтверждения диагноза является выявление повышенной экскреции дерматансульфата и гепарансульфата с мочой и определение активности фермента идуронатсульфатазы (в фибробластах, лейкоцитах крови), а также методы ДНК-анализа.
Лечение симптоматическое. Разработан препарат «Elaprasetm» (фирма Shire) для фермент-заместительной терапии.
Мукополисахаридоз III типа (синдром Санфилиппо) - аутосомнорецессивное прогрессирующее и генетически гетерогенное заболевание. К заболеванию приводят мутации четырех разных генов: лизосомной α-Ν-ацетилглюкозаминидазы (ген картирован на 17q21), ацетил-КоА а-глюкозаминид-N-ацетилтрансферазы (ген картирован на 8р11-q13), лизосомной N-ацетилглюкозамин-6-сульфатазы (ген картирован на
12q14), сульфамидазы (ген картирован на хромосоме 5q32-q33.3). Все ферменты участвуют в метаболизме гепарансульфата.
По степени выраженности клинических проявлений и первичному биохимическому дефекту различают 4 нозологические формы (типы A, B, C и D). Обычно заболевание манифестирует на третьем году жизни. Первыми и ведущими симптомами заболевания являются прогрессирующие психоневрологические расстройства: задержка психоречевого развития, нарушения поведения (агрессивность, гиперактивность, утрата навыков опрятности); нарушения сна. Изменения черт лица по типу гаргоилизма и скелетные нарушения слабо выражены по сравнению с другими мукополисахаридозами. У большинства больных наблюдаются склонность к хронической диарее, грыжи и легкий гепатолиенальный синдром. По мере течения заболевания задержка психического развития сменяется прогрессирующей умственной отсталостью и потерей ранее приобретенных навыков, развивается спастический тетрапарез, бульбарно-псевдобульбарный синдром.
Диагностика. При МРТ головного мозга обнаруживают кортикальную и субкортикальную атрофию, множественные кистозные изменения в перивентрикулярном белом веществе. Диагноз подтверждают выявлением повышенной экскреции гепарансульфата с мочой и определением активности ферментов (в фибробластах, лейкоцитах крови), а также при помощи ДНК-анализа.
Профилактика и лечение. Эффективных методов не разработано. Проводится симптоматическая терапия. Возможна пренатальная диагностика биохимическими или молекулярно-генетическими методами.
Ганглиозидозы - заболевания, при которых накопление липидных включений происходит преимущественно в ядрах нейронов.
GMj-ганглиозидоз - аутосомно-рецессивное прогрессирующее заболевание, возникающее в результате снижения активности фермента β-галактозидазы. Недостаточность фермента приводит к нарушению распада в лизосомах ганлиозида Gm1 (важного компонента мембран нейронов) и кератансульфата (компонента гликозаминогли- канов экстраклеточного матрикса хрящевой ткани). Ген картирован на хромосоме 3р21-р14.2.
На основании клинических проявлений, биохимических и молекулярно-генетических данных заболевание подразделяют на 3 клинических формы. Различают острую инфантильную (тип 1), позднюю инфантильную/ювенильную (тип 2) и взрослую/хроническую (тип 3) формы. Течение заболевания - неуклонно прогрессирующее.
Инфантильная форма. В большинстве случаев после короткого, относительно нормального периода развития, обычно в 3-6 мес (реже - сразу после рождения) возникают первые симптомы заболевания. Манифестными симптомами в неонатальный период являются диспепсия (отказ от еды, слабость сосания, отрицательная весовая кривая, срыгивания), генерализованный или локальный отек конечностей, гипертрихоз, диффузная мышечная гипотония. В дальнейшем происходит регресс пси- хомоторного развития, диффузная мышечная гипотония трансформируется в мышечную ригидность, также выявляются сухожильная гиперрефлексия, гипотрофия, патологические рефлексы, высокие старт-рефлексы на звуковые раздражители, судороги, бульбарно-псевдобульбарный синдром, корковая глухота и слепота. В первые месяцы жизни формируются дизморфические черты лица (западение спинки носа, удлиненный фильтр, гирсутизм в области лба и шеи, гипертрофия десен, макроглоссия, глубоко посаженные глаза) и скелетные дисплазии по типу множественного дизостоза, а также атрофия межкостных мышц. Перечисленные нарушения имеют разную степень выраженности. На втором полугодии жизни, как правило, развивается гепатоспленомегалия, иногда - асцит. В 50% случаев отмечается макулодистрофия по типу «вишневой косточки».
Поздняя инфантильная/ювенильная форма заболевания начинается на первом году жизни или в возрасте от 2 до 6 лет с задержки психомоторного развития. Болезнь медленно прогрессирует. На втором году жизни появляются миоклонические и генерализованные тонико-клонические судороги, нарастает мышечный тонус вплоть до децеребрационной/декортикационной ригидности, возникает атрофия зрительных нервов. Краниофациальные дизморфии, «множественный дизостоз», гепатоспленомегалия и симптом «вишневой косточки» на глазном дне не выявляются.
Хроническая (взрослая) форма дебютирует на первом десятилетии жизни (3-8 лет) чаще всего мозжечковой симптоматикой (динамическая атаксия, дизартрия, атипичная спиноцеребелярная дегенерациия). В дальнейшем присоединяются экстрапирамидные (торсионно-дистонические гиперкинезы, тремор, дистония) и пирамидные расстройства (центральные парезы и параличи).
Диагностика. При МРТ головного мозга выявляют диффузную гипомиелинизацию, повышение интенсивности сигнала в области базальных ганглиев. При исследовании биоптата костного мозга, печени выявляются «пенистые клетки» - гистиоциты с большим количеством вакуолей. При биохимических исследованиях в лей-
коцитах крови, культуре клеток кожных фибробластов, аутоптатах органов определяется значительное снижение активности фермента β-галактозидазы - менее 10% нормы.
Профилактика и лечение. Методов эффективной терапии не разработано; лечение симптоматическое. Пренатальная диагностика возможна путем определения активности лизосомной β-галактозидазы в культуре амниоцитов, биоптате хориона.
GM2-ганглиозидозы. Клиническая картина достаточно разнообразна. GM2-ганглиозидозы - группа заболеваний, вызванная чрезмерным накоплением GM2 -ганглиозида и некоторых свободных гликолипидов в лизосомах преимущественно нервной системы.
В настоящее время создано несколько классификаций GM2-ганглиозидозов, основанных на различных критериях: клинических, биохимических, молекулярно-генетических. Зандгофф в 1971 г. предложил разделять их на основании биохимических критериев на три группы: вариант В - наличие активности гекзозаминидазы В и отсутствие активности гексозаминидазы А, вариант О - отсутствие активности обеих гесозаминидаз, вариант АВ - дефекты активаторного белка. В зависимости от возраста начала и течения выделяют следующие формы: инфантильную/острую, позднюю инфантильную/подострую и взрослую/хроническую. Разделение на клинические формы отражает уровень активности фермента и никак не коррелирует с лежащим в основе первичным биохимическим дефектом. Так инфантильная (острая) форма GM2-ганглиозидозов практически не различается по клинической картине ни при варианте В, О и АВ. Наибольшее распространение на сегодняшний имеет классификация, основанная на молекулярно-генетических критериях.
Причиной GM2 -ганглиозидозов являются мутации трех различных генов: α-субъединицы GM2 А (картирован на 15q23-q24), мутации в котором проводят к болезни Тея-Сакса, β-субъединицы GM2 АиВ (картирован на 15q13) - болезнь Зиндгоффа, и белка активатора GM2 А (картирован на 5q31.3-q33.1) - вариант АВ GM2-ганглиозидоза.
На основании клинических проявлений GM2-ганглиозидоз подразделяют на три клинических формы: острые или инфантильные (клас- сическая болезнь Тея-Сакса и Зандгоффа), позднеинфантильные/ ювенильные или подострые и взрослую или хроническую форму.
Инфантильная форма. В большинстве случаев после короткого периода относительно нормального развития, обычно на 3- 5-м мес жизни, возникают первые симптомы заболевания: задержка моторного развития и роста, мышечная гипотония, снижение
ответа на внешние раздражители. С течением заболевания продолжается утрата приобретенных к тому времени навыков; нарастают мышечная гипотония и слабость. Иногда начальным симптомом является снижение зрения с сохранной реакцией зрачков на свет, при офтальмоскопии обнаруживают симптом «вишневой косточки». К 8-10 мес жизни происходит практически полная утрата интереса к окружающему миру. В большинстве случаев в конце первого года наблюдаются судороги (генерализованные тонико-клонические, фокальные приступы), трудно поддающиеся АЭП. Также отмечается прогрессирующий прирост окружности головы (макроцефалия). На втором году жизни, как правило, развиваются децеребрационная ригидность, бульбарно-псевдобульбарный синдром, учащаются судороги.
Поздняя инфантильная/ювенильная форма обычно начинается на первом десятилетии жизни (от 2 до 10 лет). Первый симптом - незаметно появляющаяся неустойчивость, неловкость при быстрой ходьбе и беге. Наряду с нарушением ходьбы и неустойчивостью в позе Ромберга отмечаются интенционный тремор и дискоординация в руках. Расстройства речи возникают рано и носят сложный мозжечково-дизартрический характер. К другим типичным проявлениям GM2-ганлиозидоза относятся экстрапирамидные расстройства в виде разнообразных гиперкинезов: миоклонических, хореиформных, атетоидных, дистонических и других. Различные виды эпилептических приступов (генерализованные тонико-клонические, миоклониче- ские) появляются в конце первой декады жизни. У половины больных встречаются поведенческие нарушения: неадекватное поведение, повышенная аффективная возбудимость, ажитация, психотическая депрессия, психозы, галлюцинации, делирий, что часто расценивают как шизофреноподобный синдром. На поздних стадиях заболевания возникают снижение зрения и изменения на глазном дне (частичная атрофия зрительных нервов, дегенерация макулы по типу «вишневой косточки», пигментная дегенерация сетчатки). По мере прогрессирования заболевания развиваются децеребрационная или декортикационная ригидность, органическая деменция, учащаются судороги.
Диагностика. На начальных стадиях заболевания инфантильной формы GM2-ганглиозидоза в базальных ганглиях и белом веществе больших полушарий мозга по данным КТ выявляют снижение плотности, а по данным МРТ - гиперинтенсивные очаги (в Т2-режиме). На второй и третьей стадии болезни происходит дальнейший распад белого вещества, визуализируется грубая корково-подкорковая
атрофия. Основными методами подтверждения диагноза являются измерение активности ферментов Нех А и Нех В в культуре клеток фибробластов, лейкоцитах крови и молекулярно-генетические исследования.
Профилактика и лечение. В настоящее время эффективных методов лечения не разработано, показано проведение симптоматической терапии. Заместительная ферментативная терапия неэффективна. ТКМ рассматривается как возможный способ лечения при поздней инфантильной/ювенильной и взрослой формах GM2-ганглиозидоза.
4.3. Митохондриальные болезни
Молекулярно-генетическая классификация митохондриальных болезней:
1. Дефекты митохондриальной ДНК:
• точковые мутации;
• единичные делеции;
• дупликации или дупликации/делеции.
2. Дефекты ядерной ДНК:
• дефекты в генах яДНК, кодирующих КДЦМ;
• дефекты в генах яДНК, ответственных за сборку КДЦМ.
3. Дефекты митохондриальной ДНК, вызванные нарушениями ядерной ДНК:
• тканеспецифичные делеции и дупликации митохондриальной
ДНК.
Болезни дыхательной цепи митохондрий, обусловленные точковыми мутациями мтДНК. В настоящее время известно 5 клинических симптомокомплексов, связанных с точковыми мутациями мтДНК, которые наследуются по материнской линии. Названия синдромов представляют собой латинский акроним основных клинических составляющих. Это синдромы NARP (невропатия, атаксия, пигментная дегенерация сетчатки), MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные состояния), MERRF (миоклонус-эпилепсия, наличие «рваных красных волокон» в мышечном биоптате).
Синдром NARP (невропатия, атаксия, пигментная дегенерация сетчатки) манифестирует, как правило, на 1-5-м году жизни, но иногда и гораздо позже - на втором десятилетии жизни. Основные клинические симптомы: мышечная слабость, атаксия, пигментная дегенерация сетчатки. В отягощенных родословных у пораженных членов семьи встречаются также снижение интеллекта, судороги, сенсорная невропатия.
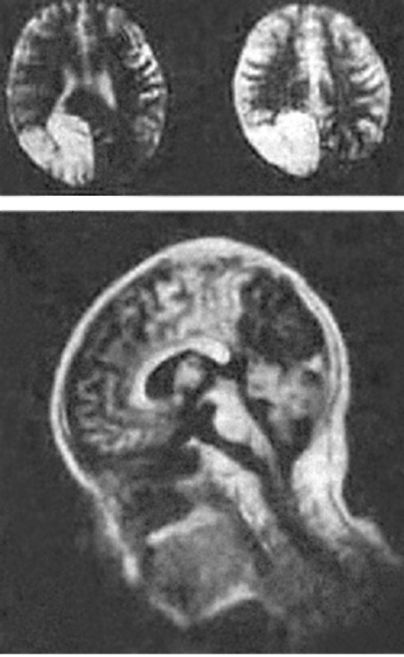
Рис. 4.1. MELAS, МРТ головного мозга: участок ишемического инфаркта в правой затылочной доле
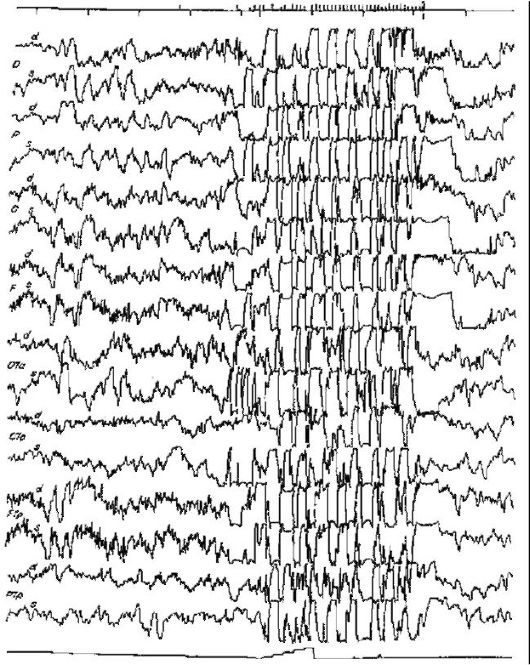
Рис. 4.2. ЭЭГ при MELAS-синдроме - генерализованная эпилептиформная активность
К числу редких симптомов относятся атрофия зрительных нервов и нейросенсорная тугоухость.
Синдром MERRF (миоклонусэпилепсия, наличие «рваных красных волокон» в мышечном биоптате). Клинический фенотип часто представлен неполными клиническими формами и характеризуется выраженным внутрисемейным клиническим полиморфизмом. Заболевание манифестирует в любом возрасте. Основной симптомокомплекс включает различные виды эпилептических приступов (миоклонические, генерализованные тонико-клонические и другие), мозжечковую атаксию и прогрес- сирующую мышечную слабость. Менее частыми симптомами являются нейросенсорная тугоухость, полиневропатический синдром, снижение интеллекта, атрофия зрительных нервов, спастические парезы/параличи.
Синдром MELAS (митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные состояния) характеризуется прогрессирующей митохондриальной энцефаломиопатией; пароксизмальными состояниями, напоминающими инсульты; лактат-ацидозом (рис. 4.1-4.3). Заболевание начинается в возрасте 5-35 лет либо инсультоподобными состояниями (кортикальный или субкортикальный инфаркт), либо злокачественной мигренью. Типичная
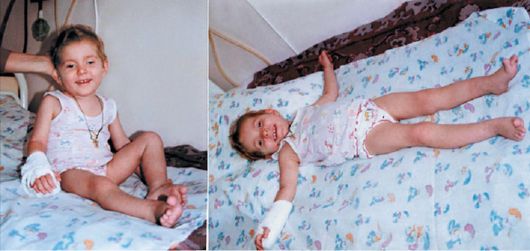
Рис. 4.3. Больная, 3 года, с MELAS-синдромом
локализация инсультоподобных очагов, выявляемая при проведении КТ/ МРТ головного мозга, - височная, теменная или затылочная область. В большинстве случаев инсультоподобные эпизоды сопровождаются гемипарезами и гемианопсией и имеют тенденцию к относительно быстрому восстановлению. Другими неврологическими симптомами являются судороги, мозжечковые расстройства, миоклонус-эпилепсия, корковая агнозия, мигренеподобные головные боли и подкорковые нарушения (мышечная дистония, различные виды гиперкинезов). Периферическая невропатия наблюдается редко. Мышечная слабость, нейросенсорная тугоухость являются типичными симптомами заболевания.
Болезни дыхательной цепи митохондрий, обусловленные крупными перестройками митохондриальной ДНК. Выделяют несколько синдромов, связанных с делециями мтДНК. Самыми распространенными являются синдромы Кернса-Сейра и прогрессирующая наружная офтальмоплегия.
Синдром KSS (синдром Кернса-Сейра) дебютирует в детском возрасте, наиболее часто - повышенной утомляемостью, мышечной слабостью, птозом, отставанием в росте, которые обычно остаются незамеченными. Классическая клиническая картина включает триаду симптомов: 1) дебют до 20 лет; 2) прогрессирующая наружная офтальмоплегия; 3) пигментная дегенерация сетчатки. К основной триаде присоединяются следующие симптомы: высокое содержание белка в ЦСЖ, блокада сердечной проводимости и мозжечковая симптоматика в виде атаксии, дизартрии.
Дефекты ядерной ДНК: из митохондриальных заболеваний, связанных с нарушениями яДНК, наиболее распространенной формой является синдром Ли (Leigh syndrome).
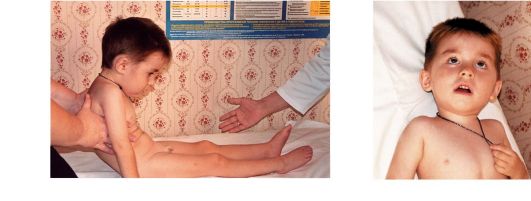
Рис. 4.4. Больной 2 лет с синдромом Лея:
а - резкая мышечная гипотония, невозможность самостоятельных движений; б - эпилептический приступ с заведением глаз
Синдром Ли - один из самых генетически гетерогенных и частых синдромов среди болезней дыхательной цепи митохондрий, дебютирующих в раннем детском возрасте. Он может быть обусловлен мутациями мтДНК (гены, кодирующие субъединицы АТФ-азы или тРНК), мутациями ядерных генов, кодирующих полипептиды полипептиды дыхательной цепи митохондрий (NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, SDHA), мутациями ядерных генов, контролирующих сборку КДЦМ на митохондриальной мембране (SURF1, COX10, COX15, SCO2, BCS1L).
Дебют заболевания - в первые месяцы жизни. С рождения отмечаются диспепсия (слабое сосание, нарушения глотания, снижение аппетита и повторная рвота), признаки легочной недостаточности, миокло- нические судороги, мышечная диффузная гипотония или спастическая диплегия, отсутствует фиксация взгляда, резко снижена реакция на яркий свет и звуковые раздражители. Заболевание быстро прогрессирует, приводя к смерти через несколько недель или даже дней, чаще вследствие паралича дыхательного центра (рис. 4.4, 4.5).
При манифестации болезни в первые 3 мес жизни наблюдаются эпизоды угнетения сознания вплоть до комы, тонические и миокло- нические судороги, диспепсия, снижение остроты зрения и прогрессирующая мышечная слабость. По мере прогрессирования заболевания усугубляются неврологические расстройства: мышечная диффузная гипотония трансформируется в спастический тетрапарез, учащаются эпилептические приступы, развивается бульбарно-псевдобульбарный паралич. У многих детей отмечаются птоз и наружная офтальмоплегия, атрофия зрительных нервов и иногда пигментная дегенерация
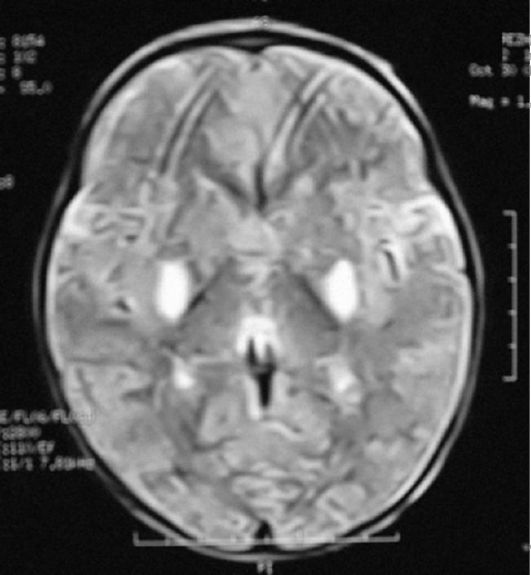
Рис. 4.5. МРТ головного мозга больного с синдромом Лея
сетчатки. Если заболевание начинается в старшем и подростковом возрасте, в клинике преобладают экстрапирамидно-мозжечковые расстройства, а затем присоединяются пирамидные нарушения, судороги.
Диагностика. Характерно повышение уровня лактата в крови или ЦСЖ. При МРТ головного мозга обнаруживают повышение интенсивности сигнала в перивентрикулярном белом веществе, в области базальных ганглиев и таламусе.
Профилактика и лечение. При обосновании тактики лечения митохондриальных болезней необходимо учитывать мультиси-
стемный характер патологии. Целесообразна терапия, направленная на коррекцию нарушений витальных функций у больных в острых, угрожающих жизни состояниях (тяжелый лакта-тацидоз, кома, инсульто- подобные состояния, судороги, дыхательная недостаточность), а также подключение к терапии кофакторов различных комплексов дыхательной цепи митохондрий: коэнзим Q10, филлоквинон (витамин К), аскорбат, сукцинат, тиамин, рибофлавин, витамин Е, препараты, улучшающие метаболизм в мышцах, - карнитин и никотинамид.
Пренатальная диагностика проводится при заболеваниях, связанных с мутациями ядерных генов. Проведение дородовой диагностики синдромов, обусловленных мутациями мтДНК, проблематично из-за феномена гетероплазмии и неравномерного распределения мутантной мтДНК по разным тканям.
4.4. Пероксисомные болезни
Пероксисомные болезни - обширная группа наследственных прогрессирующих заболеваний, возникающих в результате наруше- ния функции пероксисом.
Пероксисомные заболевания подразделяются на две большие группы:
1) нарушение биогенеза пероксисом (их полное отсутствие или нарушение их функциональной активности): синдром Целлвегера,
неонатальная адренолейкодистрофия, инфантильная болезнь Рефсума; 2) нарушение единичных белков пероксисом: акаталаземия, X-сцепленная адренолейкодистрофия. Нарушение единичных белков пероксисом
Х-сцепленная адренолейкодистрофия (Х-АЛД) - Х-сцепленное рецессивное заболевание, возникающее в результате нарушения работы мембрансвязанного белка пероксисом ALDP, основной зада-
чей которого является транспорт жирных кислот. Происходит накопление жирных кислот с очень длинной цепью (ОДЦЖК) в различных тканях организма: надпочечниках, нервной системе, яичках. Ген Х-АЛД находится на хромосоме Хд28. Частота встречаемости заболевания - 1 на 20 000 живых новорожденных лиц мужского пола.
На основании клинических проявлений, возраста дебюта, скорости нарастания неврологических симптомов заболевание подразде- лили на 7 фенотипов (детская, юношеская и взрослая церебральные, адреномиелоневропатия, изолираванная надпочечниковая недостаточность, асимптомная у гетерозиготных носительниц).
Детская и ювенильная формы Х-АЛД дебютируют в возрасте 7,2? 1,7 лет. В большинстве случаев (86%) неврологические и психические расстройства предшествуют клиническим и лабораторным признакам надпочечниковой недостаточности. Классическая детская форма характеризуется поведенческими, интеллектуальными и двигательными нарушениями (рис. 4.6, 4.7). Реже наблюдаются нарушения зрения (гомонимная гемианопсия, зрительная агнозия, острая потеря зрения, атрофия зрительных нервов) и слуха; признаки надпочечниковой недостаточности (гиперпигментация кожных покровов, общая слабость, периодически возникающая рвота и тошнота).
Ювенильная форма манифестирует в возрасте 10-21 лет и по клиническим проявлениям сходна с детской. Характерны прогрессирующая деменция, спастический тетрапарез, снижение остроты зрения и слуха, судороги.
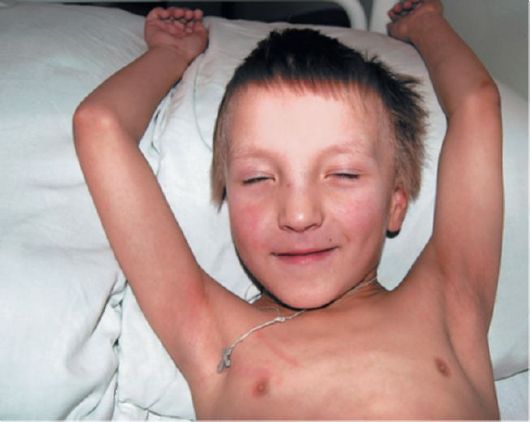
Рис. 4.6. Больной 6 лет с Х-сцепленной адренолейкодистрофией, правосторонним гемипарезом
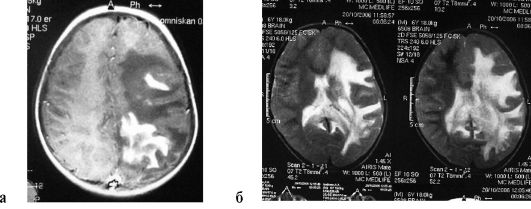
Рис. 4.7. МРТ головного мозга больного с Х-сцепленной адренолейкодистрофией (см. рис. 4.6). Массивная лейкодистрофия в левом полушарии (а, б)
Адреномиелоневропатия является наиболее частой формой Х-АЛД у взрослых. В детском возрасте она иногда сочетается с надпочечниковой недостаточностью. Преобладают неврологические нарушения в виде прогрессирующей миелопатии (спастический тетрапарез, нарушение всех видов чувствительности и функции тазовых органов).
Диагностика. При детской форме Х-АЛД белок в ЦСЖ часто повышен. Регистрируют нарушения при исследовании зрительных, слуховых, соматосенсорных вызванных потенциалов. Изменения на ЭНМГ при адреномиелоневропатии сходны с таковыми при аксональной, сенсомоторной полиневропатии. При МРТ головного мозга в Т2-режиме на начальных стадиях заболевания выявляется гиперинтенсивный сигнал в области мозолистого тела, кортико-спинальных и кортико-понтинных трактов, который по мере прогрессирования быстро распространяется в затылочные и заднетеменные отделы.
Методом биохимического подтверждения диагноза при Х-АЛД является выявление в плазме крови, эритроцитах, лейкоцитах, культуре клеток кожных фибробластов повышения уровня жирных кислот с очень длинной цепью, особенно тетраказаноиковой (С24:0) и гексазаноиковой (С26:0) кислот и их соотношений С24:0/С22:0 и С26:0/С22:0.
Профилактика и лечение. Вопрос о превентивной терапии Х-АЛД стоит особенно остро, так как диагноз можно поставить за многие годы до появления клинических симптомов заболевания. В экспериментах на культуре кожных фибробластов удалось получить снижение синтеза дексадеканоевой кислоты при добавлении олеиновой кислоты. На этом основании прежде для лечения больных с Х-АЛД использовали глицеротриолеатное масло, при применении которого
отмечалось снижение уровня жирных кислот с очень длиннойй цепью на 30-40%. Следующим этапом стало назначение смеси эруковой (С22-1) и олеиновой кислот в соотношении 1:4. Эта смесь известна под названием масла Лоренцо. Масло Лоренцо апробировано во многих центрах, но его эффективность оценить достаточно сложно. При назначении диеты был получен положительный эффект на доклинической стадии. По мнению ведущего исследователя болезней обмена Х. Мозера, у пациентов с выраженными неврологическими нарушениями такая диета неэффективна. В настоящее время основным методом лечения Х-АЛД является трансплантация костного мозга. Однако высокая степень риска тяжелых инфекционных осложнений в раннем посттрансплантационном периоде препятствует широкому внедрению этого метода. Пренатальная диагностика Х-АЛД проводится с использованием как биохимических (определение уровня жирных кислот с очень длинной цепью), так и молекулярно-генетических методов.
Заболевания, сопровождающиеся нарушением биогенеза пероксисом, - чрезвычайно гетерогенная группа болезней, которые манифестируют преимущественно в неонатальном периоде.
Синдром Целлвегера - редкое аутосомно-рецессивное заболевание, возникающее в результате нарушения лизисной функции пероксисом и сопровождающееся накоплением токсических метаболитов во всех тканях: нервной системе, печени, почках. Частота встречаемости синдрома Целльвегера - 1 на 100 000.
Дебют заболевания - с рождения. Для данного синдрома характерны краниофациальные дизморфии: высокий лоб, гипертелоризм, эпикант, гипоплазия орбит и надбровных дуг, готическое нёбо, запавшая, широкая переносица, микрогнатия, плоский затылок, расхождение швов черепа, увеличение переднего и заднего родничка, деформации ушной раковины, избыточные кожные складки на шее. Отмечаются вялое сосание (дети кормятся через зонд), гипотрофия, задержка роста, гепатомегалия с дисфункцией печени, крипторхизм, гипоспадия, эписпадия. Наблюдается симптомокомплекс «вялого ребенка»: выраженная диффузная мышечная гипотония, сухожильная гипорефлексия (ареф- лексия), грубая задержка психомоторного развития, а также отсутствие реакции на окружающее, снижение слуха, судороги. Изменения со стороны органов зрения включают помутнение роговицы, пятна Брушфильда, глаукому, катаракту, пигментную дегенерацию сетчатки, атрофию зрительного нерва. Реже встречаются точечная хондродисплазия, врожденные пороки сердца.
Диагностика. При МРТ головного мозга выявляют задержку миелини- зации и/или МР-признаки лейкодистрофии в сочетании с диффузными нарушениями серого вещества, полимикрогирией, расширением желудочков, кистами, гипоплазией мозолистого тела, нарушениями миграции нейронов. Основным методом подтверждения диагноза является определение уровня ОДЦЖК в крови, применяется также ДНК-анализ.
Профилактика и лечение. Эффективных методов лечения пока не разработано. Проводится симптоматическая терапия. При установленном молекулярно-генетическом дефекте возможно проведение пренатальной диагностики.
4.5. Нарушение обмена органических кислот/аминокислот
Органические ацидурии/аминоацидопатии - один из обширных классов наследственных метаболических заболеваний. К таким забо- леваниям относятся глутаровая ацидурия, тип 1, фенилкетонурия, болезнь с запахом «кленового сиропа мочи», множественная карбоксилазная недостаточность и другие.
Глутаровая ацидурия, тип 1 (ГА1) - аутосомно-рецессивное заболевание, обусловленное мутациями в гене, кодирующем фермент глутарилКоА дегидрогеназу (GCDH). Дефицит данного фермента приводит к накоплению в биологических жидкостях и тканях глутаровой и 3-гидроксиглутаровой кислот, оказывающих нейротоксическое действие. Частота встречаемости заболевания составляет в среднем 1 на 50 000 живых новорожденных в странах Западной Европы. Ген GCDH картирован на хромосоме 19p13.2.
ГА1 обычно дебютирует в раннем детском возрасте - от 6 до 18 мес. В 75% случаев наблюдается энцефалитоподобный вариант заболевания: внезапно возникают неукротимая рвота, срыгивания, судороги. Развивается диффузная мышечная гипотония, которая быстро трансформируется в мышечную ригидность/спастичность, появляются различные виды гиперкинезов (орофациальные, хореиформные, хореоатетоидные, баллистические). Происходит угнетение сознания до сопора и комы. Заболевание протекает волнообразно. После перенесенных метаболических кризов происходит медленное, но неполное восста- новление неврологических нарушений (рис. 4.8). В 25% случаев заболевание имеет менее острое, доброкачественное течение. На первом году жизни у детей наблюдается задержка психомоторного развития, а в дальнейшем происходит постепенная утрата ранее приобретенных навыков, присоединяются различные виды гиперкинезов (рис. 4.9).
Практически у всех пациентов интеллект не страдает.
Диагностика. Наиболее частыми нейрорадиологическими признаками при ГА1 являются: лобно-теменная гипоплазия/ атрофия, вентрикуломегалия, суб- дуральные гематомы, задержка миелинизации/ демиелинизация и некроз базальных ганглиев (рис. 4.10). Основными методами подтверждающей диагностики глутаровой ацидурии типа 1 являются биохимические - в биологических жидкостях определяют концентрацию органических кислот и/или ацилкарнитинов (глутарилкарнитина).
Лечение и профилактика. Всем больным с установленным диагнозом глутаровой ацидурии типа 1 назначают низкобелковую диету (1,5-2,0 г/кг/день) с низким содержанием триптофана (17-20 мг/кг/день) и лизина (80-100 мг/кг/ день). Ограничение поступления лизина является главным принципом диетотерапии. В настоящее время разработаны специальные смеси аминокислот («Глутаридин», фирма «Нутриция»). Основным методом метаболической коррекции является прием L-карнитина в дозе от 50-100 мг в день длительно, постоянно. Наряду со специфической терапией про- водится симптоматическое лечение: назначают препараты для коррекции мышечного тонуса, гиперкинетических расстройств, судорог.
Лейкодистрофия Канавана-Ван Богарта-Бертранда наследуется по аутосомно-рецессивному типу. Частота встречаемости в общей популяции не установлена ввиду редкости заболевания. Болезнь встречается во всех этнических группах, но с наибольшей частотой - у евреев ашкенази, у которых распространенность болезни Канавана составляет 1 на 5000 новорожденных.
Болезнь Канавана обусловлена недостаточностью фермента аспартоацилазы, которая приводит к накоплению N-ацетиласпартата
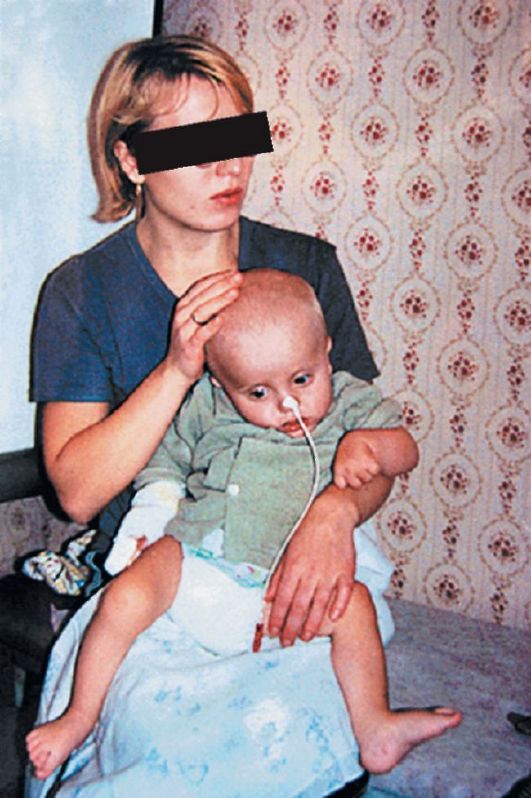
Рис. 4.8. Больной 1 года глутаровой ацидурией, период криза с утратой всех навыков. Макрокрания
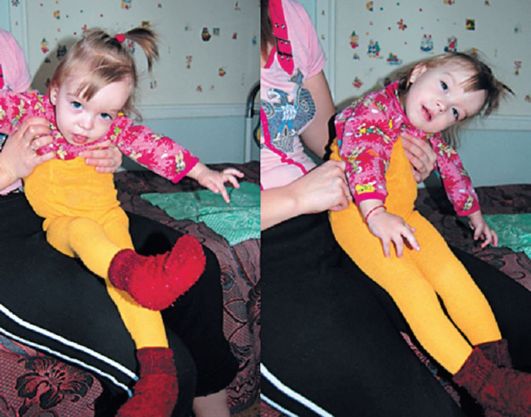
Рис. 4.9. Больная 3 лет с глутаровой ацидурией. Гиперкинезы
в мозге, спинномозговой жидкости, плазме и моче. Молекулярные механизмы патогенеза БК неясны. Предполагается токсическое действие N-ацетиласпартата или его метаболитов на ткань мозга, что способствует, по всей вероятности, возникновению хронического отека головного мозга.
Адачи и соавт. описали 3 клинические формы болезни Канавана в зависимости от возраста дебюта: врожденную, инфантильную, ювенильную. Инфантильная форма заболевания наиболее хорошо изучена клинически. На первом месяце у детей часто отмечаются: нарушение фиксации взора, повышенная возбудимость, вялое сосание, слабый зрительный контакт «глаза в глаза», судороги, диффузная мышечная гипотония. Основные клинические признаки болезни Канавана становятся очевидными к 3 мес жизни: выраженная мышечная гипотония, неспособность удерживать голову в вертикальном положении, в даль- нейшем трансформация диффузной мышечной гипотонии в спастичность. Макроцефалия является характерным симптомом заболевания, но в раннем возрасте окружность головы может оставаться в пределах нормы. По данным Дж. Гаскона, мышечная гипотония, сопровождающаяся снижением двигательной активности, в последующем сменяется спастическим гипертонусом, указывая на вовлечение в патологический процесс пирамидной системы. Наблюдаются повышение сухожильных рефлексов, появление патологических кистевых и стопных рефлек-
сов. По мере прогрессирования болезни развивается децеребрационная или декортикационная ригидность; нарастает нарушение психомоторного развития. Кроме того, у 50% больных присоединяются генерализованные тоникоклонические судороги. Атрофия зрительных нервов обычно развивается на втором году жизни.
Диагностика. У пациентов с болезнью Канавана на КТ и МРТ головного мозга обнаруживают диффузную дегенерацию белого вещества с вовлечением в пато- логический процесс полушарий мозга, в меньшей степени поражаются мозжечок и ствол мозга. При офтальмоскопии выявляют атрофию дисков зрительных нервов. Определение концентрации N-ацетиласпартата в моче и измерение активности аспартоацилазы в культуре кожных фибробластов являются надежными биохимическими тестами, применяемыми для установления диагноза болезни Канавана. При ней отмечается повышение уровня N-ацетиласпартата в моче - более 1000 нмоль/моль креатинина.
Профилактика и лечение. Эффективных методов лечения заболевания не разработано. Высокий уровень носительства в популяции евреевашкенази требует проведения простых скрининг-программ, например, как при болезни Тея-Сакса. В семейной паре, где оба родителя по данным ДНК-исследования являются носителями патологического гена, диагноз плоду можно поставить на основании ДНК анализа ворсин хориона.
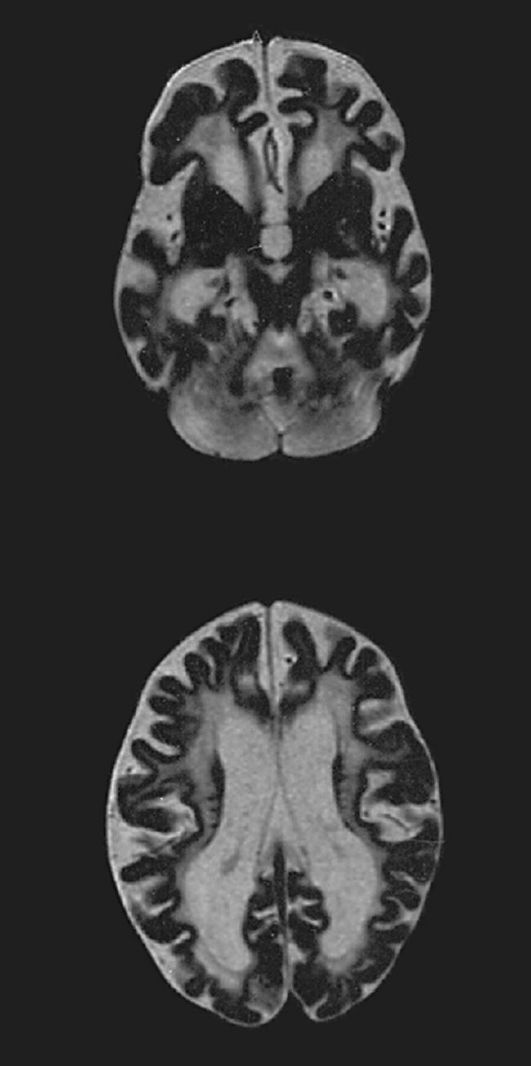
Рис. 4.10. МРТ больной с глутаровой ацидурией. Атрофия полушарий, вентрикуломегалия
4.6. Нарушения обмена углеводов
Галактоземия - одно из наиболее часто встречающихся нарушений обмена углеводов. Галактоземия I типа или классическая галактоземия (недостаточность галактозо-1-фосфатуридилтрансферазы) относится к группе наследственных заболеваний, обусловленных недостаточностью ферментов, участвующих в метаболизме галактозы. Частота встречаемости заболевания составляет 1 на 40-70 000 живых новорожденных. Ген галактозо-1-фосфатуридиотрансферазы картирован на 9p13. Следствием недостаточности фермента является накопление галактозы, галактозо-1-фосфата. Эти метаболиты токсически действуют на метаболизм многих тканей: мозг, печень, почки, кишечник.
Клинические проявления. Дебют заболевания - в первый месяц жизни с появления рвоты, диареи, отмечается недостаточная прибавка массы тела. К 2-недельному возрасту развивается гепатоспленомегалия, у 30% больных - катаракта, мышечная гипотония. У нелеченных больных постепенно формируется печеночная недо- статочность, которая может привести к летальному исходу. У ряда больных галактоземия протекает менее тяжело, и первые симптомы манифестируют к 3-6 мес, а в дальнейшем появляются задержка психоречевого развития, судороги и эндокринные нарушения.
Диагностика. У некоторых больных выявляют изменения при МРТ: корково-подкорковую атрофию, гипомиелинизацию, атрофию мозжечка, ствола головного мозга. Основными методами подтверждения диагноза галактоземии являются биохимические тесты, основанные на определении концентрации галактозы, галактозо-1- фосфата в крови и/или на измерении уровня активности фермента галактозо-1-фосфатуридиотрансферазы в эритроцитах.
Профилактика и лечение. Основным методом лечения галактоземии типа I является диетотерапия. Назначение безлактозных смесей приводит к быстрому купированию основных клинических симптомов и восстановлению функции печени. Однако, как показывает обширный клинический опыт, у большого числа пациентов, находящихся на строгой диете, возникают неврологические (снижение уровня интеллекта, атаксия, мышечная гипотония, интенционный тремор) и эндокринологические (задержка полового развития, первичная или вторичная аменорея) осложнения. В настоящее время галактоземия включена в программу массового скрининга новорожденных во многих странах, в том числе и в России.
Рекомендуемая литература
1. Бадалян Л.О., Таболин В.А., Вельтищев Ю.Е. Наследственные болезни у детей. - М.: Медицина, 1971. - 367 с.
2. Краснопольская К.Д. Наследственные болезни обмена веществ: справочное пособие для врачей. - М.: РОО «Центр социальной адаптации и реабилитации детей «Фохат», 2005. - 364 с.
3. Петрухин А.С. Неврология детского возраста. - М.: Медицина,
2004. - 784 с.
4. Темин П.А., Казанцева Л.З. Наследственные нарушения нервнопсихического развития детей. - М.: Медицина, 2001. - 428 с.
5. Aicardi Jean. Diseases of the Nervous System in Childhood: 2nd ed. -
Cambridge, 1998. - 897 р.
6. Menkes J.H.: textbook of Child Neurology: 3nd ed. - Philadelphia Lea & Febiger, 1985. - 827 p.