Детская неврология : учебник : в двух томах / А. С. Петрухин. - Т. 2. - 560 с. : ил.
|
|
|
|
ГЛАВА 5. НАСЛЕДСТВЕННО-ДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ
Наследственно-дегенеративные заболевания клинически совершенно разнородны, но характеризуются сходным течением. У здорового человека (ребенка или взрослого) спонтанно или после провоци- рующих факторов появляются патологические симптомы поражения не только ЦНС, но и других органов и систем. Постепенно клиническая выраженность этих симптомов усиливается, а состояние пациента неуклонно ухудшается. Скорость прогрессирования болезни вариабельна. Наследственно-дегенеративные заболевания приводят к утрате некоторых функций (движения, речи, мыслительных процессов, зрения, слуха и т.д.) и иногда заканчиваются летально. Причиной наследственно-дегенеративных заболеваний является патологический ген (или несколько генов). Поэтому возраст дебюта болезни зависит от времени экспрессии этого гена, а степень тяжести - от его пенетрантности: чем более выражен патологический признак, тем тяжелее течение болезни.
Выдающиеся неврологи XIX-XX вв. описали наследственнодегенеративные заболевания, но причины их возникновения долгое время оставались неизвестными. Новая эра в неврологии началась благодаря достижениям молекулярной генетики: были открыты гены и биохимические дефекты, отвечающие за развитие симптомов этих заболеваний. Согласно сложившейся традиции, они носят эпонимные названия, и это является данью уважения ученым, которые первыми описали данные заболевания.
Топически наследственно-дегенеративные заболевания подразделяют в зависимости от уровня поражения нервной системы на болезни с преимущественным поражением: 1) коры большого мозга; 2) базальных ганглиев; 3) ствола и мозжечка; 4) спинного мозга.
5.1. Наследственно-дегенеративные заболевания базальных ганглиев
Болезнь Гентингтона - наследственное медленно прогрессирующее заболевание нервной системы с аутосомно-доминантным типом наследования, характеризующееся хореическими гиперкинезами, психическими нарушениями и прогрессирующей деменцией. Частота встречаемости в популяции колеблется и составляет в среднем 3-7 на
100 000.
Историческая справка. Дж. Гентингтон был потомственным врачом. Под наблюдением его дедушки находилось несколько пациентов с наследственной формой хореи. Восьмилетний Джордж впервые увидел и зарисовал их движения. В 1872 г. Гентингтон впервые охарактеризовал это заболевание, впоследствии названное в его честь.
Молекулярная генетика и патогенез. Ген болезни Гентингтона картирован на хромосоме 4p16.3. Он кодирует белок гентингтин. Причиной болезни Гентингтона является увеличение числа тринуклеотидных цитозин-аденин-гуанин (САG)-повторов, расположенных в первом экзоне гена. В генах здоровых людей содержится от 10 до 35 повторов. При хорее Гентингтона наблюдается увеличение их числа (от 36 до 121). После того, как число тринуклеотидных повторов превысит 36, наблюдается накопление зоны повторов в последующих поколениях, что коррелирует с увеличением тяжести заболевания. Это явление получило название антиципации, и болезнь Гентингтона является лучшим его примером: чем раньше проявилось заболевание в ряду поколений, тем тяжелее оно протекает.
Триплет CAG кодирует аминокислоту глутамин, поэтому в белке образуется удлиненный полиглутаминовый участок, который при- водит к апоптозу. При болезни Гентингтона также нарушается функция митохондрий в нейронах полосатого тела. Эти изменения, вероятно, обусловлены накоплением свободных перекисных радикалов.
Патоморфология. При аутопсии головного мозга при болезни Гентингтона обнаруживают атрофию и глиоз хвостатых ядер и скорлупы (рис. 5.1). Уменьшено количество нейронов в бледном шаре, в коре лобных долей и субкортикальных отделах полушарий. Специфических гистологических маркёров не описано. В непо- врежденных нейронах и астроцитах накапливается липофусцин, в клетках бледного шара - железо, в периваскулярном пространстве - сидерофаги. В основном повреждаются нейроны хвостатых ядер, ответственные за секрецию тормозящего нейромедиатора - γ-аминомасляной кислоты.
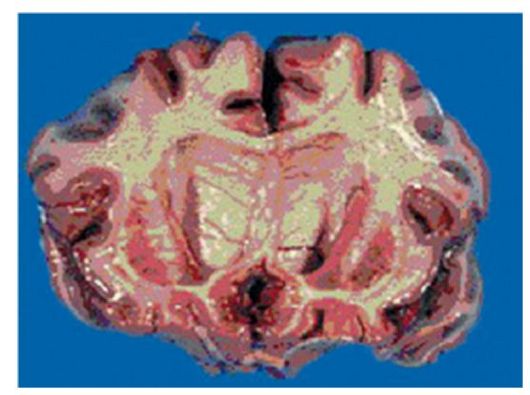
Рис. 5.1. Атрофия головного мозга, преимущественно хвостатого ядра, при болезни Гентингтона (макропрепарат)
Большие пирамидные клетки III, V и VI слоев коры большого мозга сморщиваются, приобретая неправильную форму. В начале болезни гибель клеток коры компенсируется за счет активного ветвления ден- дритов оставшихся пирамидных клеток.
Клинические проявления. Заболевание начинается в любом возрасте, чаще - в период с 20 до 60 лет (в среднем - в 40 лет). На ювенильную форму приходится около 10% всех случаев хореи Гентингтона. Самый ранний дебют заболевания описан в 3 года.
В начальной стадии заболевания непроизвольные движения в виде хореи возникают утром или при нервном напряжении. Хореические гиперкинезы в лицевой мускулатуре проявляются выразительными гримасами с высовыванием языка, подергиванием щек, поочередным подниманием и нахмуриванием бровей. Иногда отмечаются эпизоды шумного, глубокого дыхания. Хорея в руках выглядит как быстрое сгибание и разгибание пальцев, в ногах - как поочередное скрещивание и разведение ног, сгибание и разгибание пальцев стоп. Наряду с хореей в мышцах туловища и проксимальных отделах конечностей можно отметить атетоз. Гиперкинезы обычно симметричны, усиливаются при физической нагрузке или волнении и прекращаются во сне. По мере развития болезни они усиливаются, появляется грубая дистония, переходящая в ригидность.
Иногда заболевание начинается с дистонии: больные не могут длительно находиться в одной позе, отмечается торсия шеи, туловища и конечностей. При ювенильной форме в 50% случаев начальными сим- птомами являются брадикинезия, ригидность и паркинсонический тремор.
Судороги у взрослых с болезнью Гентингтона бывают редко, а у детей встречаются в 30-50% случаев. Наблюдаются различные типы приступов: фокальные, генерализованные тонико-клонические, абсансы, диалептические, миоклонические, обычно резистентные к противосудорожным препаратам. Изменения на ЭЭГ характеризуются генерализованной эпилептической активностью с частотой 2- 2,5 Гц и нерегулярными пик-волнами.
У больных прогрессируют расстройства речевых функций. На начальных стадиях хореи Гентингтона возникают нарушения, свя- занные со звукопроизношением (дизартрия). Постепенно изменяются скорость и ритм речи, она становится медленной и невнятной. Нарушения глотания обычно появляются в терминальной стадии. Частой причиной смерти является аспирационный синдром.
У 90% детей выявляют повышение сухожильных рефлексов и спастический гипертонус. Аксиальные рефлексы (хоботковый, соса- тельный, дистанс-оральный), как правило, возникают при грубых интеллектуальных нарушениях.
Глазодвигательные нарушения встречаются у большинства пациентов. Больные не могут плавно и точно следить за предметом, часто моргают. Характерен нистагм.
Часто болезнь Гентингтона в детском возрасте начинается с изменений поведения: снижаются успеваемость в школе и концентрация внимания, замедляется мышление, нарушается кратковременная память, появляется неусидчивость.
Редко в подростковом возрасте заболевание дебютирует с психозов, шизотипического расстройства. Для начальной стадии характерны снижение настроения (депрессия), тревога, раздражительность, эмо- циональная лабильность, апатия. Возникают суицидальные мысли.
Течение заболевания у детей характеризуется быстрым прогрессированием, что связано с феноменом антиципации.
Диагностика. Диагноз подтверждается при молекулярно-генетическом анализе. С помощью полимеразной цепной реакции определяют число САG-повторов в пораженном гене. При взрослой форме заболевания число повторов превышает 36, при ювенильной - 50.
На МРТ головного мозга видна атрофия головок хвостатых ядер, в меньшей степени - бледных шаров и гипоталамуса, лобных отде- лов коры. Однофотонная эмиссионная компьютерная томография (SPECT) выявляет низкий метаболизм глюкозы в хвостатых ядрах еще на доклинической стадии.
Дифференциальный диагноз проводят с другими заболеваниями детского возраста, проявляющимися хореей: доброкачественной непрогрессирующей семейной хореей, идиопатической торсионной дистонией, болезнью Галлервордена-Шпатца, болезнью Вильсона- Коновалова, ювенильной формой болезни Паркинсона, нейроакантоцитозом.
Пренатальная диагностика проводится молекулярно-генетическим методом.
Лечение. В настоящее время эффективного лечения не разрабо- тано, проводят симптоматическую терапию. Для уменьшения выраженности хореи показаны нейролептики. При ригидности назначают препараты леводопа, бромокриптин, амантадин, при возникновении судорог - антиэпилептическую терапию.
Гепатолентикулярная дегенерация (болезнь Вильсона, болезнь Вильсона-Коновалова) - это аутосомно-рецессивное заболевание, возникающее при нарушении обмена меди. Для него характерно сочетание поражения внутренних органов и головного мозга, в основном печени и чечевицеобразных ядер. Распространенность заболевания составляет 2-3 случая на 100 000 населения. Для помощи страдающим болезнью Вильсона-Коновалова разработана эффективная патогенетическая терапия, без которой заболевание быстро прогрессирует и заканчивается летальным исходом. Своевременно начатое систематическое лечение предотвращает развитие болезни или приводит к частичному регрессу симптомов.
Первое классическое описание болезни с ее типичными морфологическими изменениями в виде цирроза печени опубликовал английский невролог С. Вильсон в 1912 г. Основными клиническими симптомами он назвал непроизвольные движения в конечностях и туловище, тремор, мышечную ригидность, дисфагию и дизартрию, аффективные вспышки, иногда психические расстройства. Необходимо отметить, что при этой болезни, несмотря на то что она называется «гепатолентикулярная дегенерация», поражаются не только печень и чечевицеобразные ядра. Выдающийся отечественный невролог Н.В. Коновалов значительно расширил представления о патофизиологии, патогенезе и клинике болезни и создал ее классификацию.
Ген болезни Вильсона расположен на длинном плече хромосомы 13 (13q14.3). Он кодирует медь-транспортирующую АТФазу, участвующую в синтезе церуллоплазмина. Заболевание развивается только у гомозигот. Для гетерозигот (при одном нормальном гене и одном патологическом) характерно субклиническое течение.
В основе заболевания лежит нарушение метаболизма меди. Ион меди входит в состав ферментов дыхательной цепи (цитохромоксидазы и лизилоксидазы). Ежедневно человек употребляет с пищей от 1 до 5 мг меди, из которых усваивается около 40%. Всосавшиеся в проксимальных отделах ЖКТ ионы меди образуют прочное соединение с металлопротеином, транспортируются в клетки, участвуют во внутриклеточном обмене и экскретируются. При болезни Вильсона- Коновалова нарушается выведение меди из печени в составе церуллоплазмина. Медь накапливается в гепатоцитах, развивается гепатоз, а в дальнейшем - нодулярный цирроз печени. Непосредственное токсическое воздействие меди вызывает гемолитическую анемию.
Свободно циркулирующая медь откладывается в органах и тканях, в первую очередь в головном мозге и роговице. Формируются патологические изменения в базальных ядрах и кольцо Кайзера-Флейшера в роговице. Хроническая интоксикация приводит к поражению ЦНС. Летальный исход наступает от печеночной комы.
Патологическая анатомия. При вскрытии печень уменьшена вследствие атрофического цирроза, под микроскопом участки нормальной ткани чередуются с участками некроза и островками регенерации. При электронной микроскопии включения меди расположены диффузно в цитоплазме гепатоцитов. В почках выявляется дегенерация эпителия канальцев, цитоплазма также содержит включения меди. Селезенка обычно увеличена. Базальные ядра головного мозга выглядят коричнево-красными; чечевицеобразные ядра, особенно скорлупа, размягчены, содержат мелкие кисты и сморщены. Страдают также хвостатое тело, бледный шар, глубокие слои коры, зубчатые ядра мозжечка, субталамические ядра. Число нейронов уменьшено, аксоны их разрушены. Характерно появление глии Альцгеймера, которая образуется из обычных астроцитов: крупные, лишенные цитоплазмы, «голые» ядра и клетки с очень большим телом, порой со сморщенным ядром. Чем позднее начинается заболевание, тем медленнее оно течет, тем более диффузными становятся изменения мозга.
Отложение меди в роговице приводит к образованию колец Кайзера-Флейшера, цвет которых варьирует между желтым, зеленым и коричневым. Для клинической картины характерен полиморфизм неврологических и соматических симптомов. В соответствии с классификацией Н.В. Коновалова выделяют 5 форм заболевания.
Брюшная форма - тяжелое заболевание печени, приводящее к смерти до появления симптомов со стороны нервной системы; заболевают дети дошкольного возраста.
Ригидно-аритмо-гиперкинетическая, или ранняя, форма отличается быстрым течением (2-3 года), начинается также в детском возрасте. В клинической картине заболевания преобладают мышечная ригидность,приводящая к контрактурам, бедность и замедленность движений, хореоатетоз или дистонические гиперкинезы; лицо амимично, часто искажено застывшей гримасой. Обычны расстройства речи (дизартрия) и глотания (дисфагия), судорожные смех и плач, нередки судороги, аффективные расстройства и умеренное снижение интеллекта.
Дрожательно-ригидная форма встречается чаще других. Начинается в юношеском возрасте, течет несколько медленнее (в среднем 5-6 лет),
порой сопровождаясь ремиссиями и внезапными ухудшениями. Типичны грубая ригидность и ритмичный тремор (2-8 дрожаний в 1 с), который резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает; захватывает конечности, голову и туловище. Иногда к тремору присоединяются атетоз и хорея, наблюдаются также дисфагия и дизартрия.
Дрожательная форма начинается в возрасте 20-30 лет, течет довольно медленно (10 лет и больше); в клинике преобладает тремор, ригидность появляется в конце болезни. Нередки гипотония, амимия, медленная, монотонная речь (брадилалия), брадикинезия, изменения психики, аффективные вспышки. Наблюдаются эпилептические приступы.
Экстрапирамидно-корковая форма встречается реже других форм, длится 6-8 лет; начинается как одна из вышеописанных форм. Типичные экстрапирамидные нарушения в дальнейшем осложняются остро развивающимися парезами, судорогами и слабоумием, которые связаны с образованием обширных очагов в коре больших полушарий.
Диагностика
1. Роговичное кольцо Кайзера-Флейшера при офтальмологическом исследовании со щелевой лампой.
2. Исследование концентрации церуллоплазмина в крови (нижняя граница нормы - 20 мг/дл).
3. Повышение экскреции меди в суточной моче (более 80 мкг/сут).
4. Биопсия печени - увеличение содержания меди в сухом веществе.
5. При КТ, МРТ обнаруживается атрофия большого мозга и мозжечка, базальных ядер, расширение желудочков и субарахноидальных пространств.
6. Окончательно подтверждает диагноз генетический анализ. Дифференциальный диагноз проводится с другими наследственно-
дегенеративными заболеваниями, протекающими с гиперкинезами.
Лечение. Д-пеницилламин (купримин, депен) образует с медью прочное соединение, экскретирующееся почками. Препарат назначается в дозе 1-1,5 г/сут. В первые 3-6 мес происходит транзиторное ухудшение. Затем состояние больных начинает улучшаться: значительно уменьшаются неврологические симптомы, улучшаются бытовые навыки. Восстанавливается функция печени. После достижения устойчивого терапевтического эффекта дозу препарата можно
несколько уменьшить. Лечение проводится в течение всей жизни. Иногда развиваются побочные явления. Менее токсичным медь- связывающим препаратом является триентин (триен).
Диета не играет большой роли в лечении гепатолентикулярной дегенерации, тем не менее ее обычно рекомендуют (исключение из рациона пищи, богатой медью: какао, шоколада, грибов, орехов).
Торсионная дистония (G 24.1) и дистонические синдромы
Дистонии относятся к тем неврологическим состояниям, которые трудно диагностировать и лечить даже современному неврологу. С. Марсден, создатель современной классификации дистоний, отметил, что до 1970-х гг. многие пациенты с дистонией наблюдались и лечились психиатрами из-за стойкого мнения, что «столь курьезные движения могут возникнуть только у человека с нездоровой психикой». До сих пор точно не установлена распространенность дистоний; между тем они занимают второе место после тиков среди двигательных расстройств у детей.
Дистония впервые описана В. Швальбе в 1908 г. под названием «особой тонической формы спазма с симптомами истерии». В самостоятельную нозологическую форму - «dystonia musculorum deformans» - ее выделил Г. Оппенгейм в 1911 г., отметив прогрессирующее течение при отсутствии мышечной атрофии, парезов, атаксии и чувствительных расстройств.
Дистония - клинический синдром, характеризующийся неритмич- ными, медленными насильственными движениями в различных частях тела, своеобразными изменениями мышечного тонуса и патологическими позами. При дистонии непроизвольное сокращение мышц вызывает повторные выкручивающие движения. При длительном мышечном сокращении может возникать тремор.
Дистония, по-видимому, связана с нарушением плавности перехода позы в движение и наоборот. Дополнительной характеристикой дистонии является крайняя чувствительность к внешним раздражителям разных модальностей, особенно тактильных. Жесты и прикосновения усиливают или уменьшают дистонию, чем больные часто пользуются («корригирующие жесты»). Например, прикосновение к напряженным мышцам при кривошее вызывает появление дистонических поз в других частях тела, а прикладывание пальца к подбородку уменьшает кривошею.
Дистония действия часто возникает при специфических движениях. Например, писчий спазм - дистония мышц кисти и пальцев - возникает
исключительно при письме и никогда при шитье или других действиях. Дистония руки при ходьбе вперед полностью исчезает при ходьбе назад; гемидистония при ходьбе исчезает при беге.
Постуральная дистония (дистония покоя) выражается формированием длительно существующих патологических поз, которые исчезают во сне.
Дистонию классифицируют по возрасту дебюта, этиологии и распространенности гиперкинеза. По распространенности
• Фокальная дистония - с поражением одной части тела. Может быть шейная дистония, характеризующаяся патологическим положением головы (кривошеей) и спазмом мышц шеи. Краниальная дистония в круговой мышце глаза выглядит как редкое насильственное моргание. Оромандибулярная дистония проявляется дистонией жевательных мышц (тризм). Писчий спазм возникает в дистальных отделах доминантной руки, причем не только при письме (рис.5.2), но и при других действиях, например игре на пианино, гитаре, печатании на клавиатуре и т.д.
• Сегментарная дистония - с поражением двух смежных частей тела.
• Мультифокальная дистония - с поражением нескольких несмежных частей тела.
• Гемидистония - с поражением половины тела.
• Генерализованная дистония (рис. 5.3).
Выделяют 4 степени тяжести дистонии (Э. Фернанде-Альварес,
Ж. Айкарди, 2001):
• 1-я степень - дистония возникает только при специфических движениях;
• 2-я степень - постоянная дистония, иногда возможно расслабление;
• 3-я степень - постоянная дистоническая поза, не поддающаяся коррекции;
• 4-я степень - генерализованная постоянная дистония.
По этиологии дистонии делят на первичные (идиопатические) и вторичные (симптоматические).

Рис. 5.2. Торсионная дистония руки (писчий спазм)

Рис. 5.3. Торсионная дистония, генерализованная форма
Первичная дистония (G 24.1, синонимы: генерализованная дистония, торсионная дистония). Встречается с частотой 3-4 на 100 000 населения и включает 13 генетических форм. Самая частая первичная дистония - DYT1 - обнаруживается в 90% случаев этого заболевания у детей в популяции евреевашкенази и в 40-60% - в общей популяции населения земного шара.
При первичной дистонии не бывает морфологических изменений в мозге. Биохимический дефект локализуется в базальных ганглиях и связан с патологией нейромедиаторов. Применение ПЭТ (позитронноэмиссионной томографии) и фМРТ (функциональной МРТ) выявило, что при дистонии нарушается функциональная активность многих отделов: моторной коры, мозжечка, базальных ганглиев (в основном бледного шара). Электрофизиологические исследования показывают, что при дистонии нарушается центральное торможение рефлекторного напряжения мышц под действием тактильных раздражителей. Ведущая роль в патогенезе отводится дофамину и его метаболи- там. Нарушается корковый контроль планирования и выполнения
движений, таламус не подавляет рефлекторную активность ствола и спинного мозга. В результате возникают длительные патологические сокращения группы мышц агонистов и антагонистов.
Большинство случаев первичной дистонии дебютируют до15 лет. Дистония вначале поражает одну из конечностей - изменяется походка или нарушается почерк. Парадоксальные феномены (когда ребенок не может писать на листе бумаги, но пишет на доске) ошибочно принимаются за истерию, тем более что во сне дистония полностью исчезает. Течение первичной дистонии вариабельное и непредсказуемое. Часто локальная дистония переходит в генерализованную. Например, локальная дистония руки в дальнейшем сопровождается появлением дистонии в ноге. Тяжесть дистонии в конечностях может быть разной. Туловищная дистония с постоянными патологическими позами приводит к тяжелым скелетным деформациям (сколиоз, кифоз, лордоз). Постепенно дистония становится фиксированной, развиваются мышечные ретракции и контрактуры. Дистонии с ранним дебютом неблагоприятны. Тяжелые мышечные спазмы могут приводить к нарушениям функций внутренних органов, некрозу мышц, миоглобинурии и почечной недостаточности. Дистония сопровождается появлением других гиперкинезов, чаще миоклонуса и тремора. Интеллект детей не страдает.
Отечественные неврологи выделяют две основные формы первичной дистонии: ригидную и дистонически-гиперкинетическую (Иванова- Смоленская И.А., Маркова Е.Д.).
Первичная ригидная дистония (торсионная дистония) характеризуется повышением мышечного тонуса и развитием фиксированных патологических поз, чаще в ногах, но иногда в руках, шее, туловище. Заболевание начинается в возрасте от 4 до 16 лет. Течение относительно доброкачественное. Наиболее часто первые дистонические движения появляются в стопе, приводя к нарушениям походки. Вначале симптомы носят интермиттирующий характер и усиливаются под влиянием стресса, но через некоторое время становятся постоянными. К патологическим позам постепенно присоединяются легкие паркинсоноподобные симптомы: замедленность движений, «дистонический» тремор. Дистония охватывает мышцы рук, появляются выраженные торсионные спазмы шеи и туловища. Эти дистонические движения возникают в ответ на активность мышц в любой другой части тела, могут появляться спонтанно. В результате патологическая поза фиксируется, в конечностях наблюдаются периодические атетоидные гиперкинезы.
Первичная дистонически-гиперкинетическая дистония (миоклоническая дистония, дистония плюс) начинается в детском возрасте, характеризуется мягким течением и медленным прогрессированием. Торсионная дистония сочетается с миоклонусом, преимущественно мышц шеи, туловища и дистальных отделов рук. Миоклонические подергивания уменьшаются в покое, при беге, быстрой ходьбе; провоцируются испугом и волнением. Во сне миоклонии исчезают.
При первичной дистонии отмечается снижение уровня ГВК (гомовинилиновой кислоты), тетрабиоптерина - кофактора тиро- зингидроксилазы, превращающей L-тирозин в L-дофу; нарушен синтез дофамина. Немедленный и выраженный эффект леводопы является основным диагностическим критерием дофа-зависимой дистонии. На фоне приема леводопы (не более 500-1500 мг/сут) происходит быстрая нормализация состояния с полным регрессом симптоматики.
Диагноз дистонии основывается на клинических симптомах. Не существует параклинических методов для подтверждения первичной дистонии, кроме генетического исследования. Если заболевание начинается до 24 лет, следует провести генетический анализ на DYT1. Другие генетические анализы не проводятся из-за их технической сложности и высокой стоимости.
Вторичные (симптоматические) варианты проявляются дистонией в сочетании с симптомами со стороны других отделов нервной системы и внутренних органов. Вторичные дистонии характеризуются более тяжелым течением, чем первичные; быстрее возникают фиксированные контрактуры и скелетные деформации. При дистонии у детей обязательно следует исключать гепатолентикулярную дегенерацию, учитывая разработанную патогенетическую терапию этого заболевания.
Дистония в результате перинатального поражения головного мозга появляется у детей с патологией перинатального периода до трех лет жизни, по мере развития у ребенка гиперкинетической формы ДЦП.
Болезнь Галлервордена-Шпатца обычно дебютирует с дистонии, и лишь спустя 1-2 года у ребенка могут появиться типичные для этого заболевания спастические парезы и характерная МР-картина (рис. 5.4)- «глаза тигра».
Болезнь Фара (наследственная кальцификация базальных ганглиев на фоне патологии щитовидной и паращитовидных желез), ювенильная форма хореи Гентингтона и такие наследственные болезни
обмена, как глутаровая ацидурия, синдром Леша-Нихана, гомоцистинурия, синдром Ли у детей сопровождаются дистониями.
Гемидистония всегда имеет симптоматическую вторичную природу и указывает на органическое поражение контралатерального полушария. Она относится к вторичным дистониям, возникает вследствие энцефалитов, рассеянного склероза, черепно-мозговой травмы, опухоли.
Дистония в результате медикаментозной интоксикации (G 24.0) особенно часто возникает при применении фенитоина, карбамазепина, фенотиазина, бутирофенона, бензамина, трициклических антидепрессантов, антигистаминных препаратов, кетамина, лития и церукала.
Хронический нейролептический синдром возникает при длительном приеме высоких доз нейролептиков. Для него характерна орофациальная дистония, особенно примечательно стереотипное высовывание языка, смыкание губ и жевание («синдром кролика»). Для коррекции и предотвращения нейролептического синдрома назначается антихолинэргический препарат (циклодол).
Острый нейролептический синдром - неврологическое расстройство, которое возникает сразу после назначения нейролептиков, бло- кирующих дофаминовые рецепторы. Основные клинические особенности: лихорадка, тахикардия, артериальная гипертония, ригидность мышц затылка, судороги, спутанность сознания. Это состояние угрожает жизни больного и требует проведения неотложной помощи. Антидотом является дантролен в дозе 1,5 мг/кг и агонист рецепторов дофамина Д2 - бромокриптин. Злокачественный нейролептический синдром представляет собой тяжелый вариант острого нейролептического синдрома, как правило, приводящий к смерти вследствие быстрой декомпенсации функций жизненно важных органов.
Дистония в результате травмы периферической нервной системы может развиться только у детей старшего возраста и взрослых. Механизмы возникновения данной дистонии неясны.
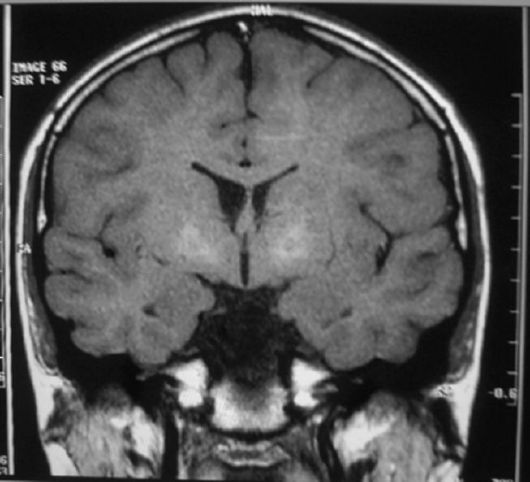
Рис. 5.4. МРТ при болезни Галлер- вордена-Шпатца - «глаза тигра», отложение железа в бледные шары
МРТ при вторичной дистонии всегда выявляет поражение базальных ганглиев, особенно часто - ограды, и кортико-стриарных связей. Однако следует сказать, что на ранних стадиях, например болезни Галлервордена-Шпатца, головной мозг по данным МРТ может быть интактен; поэтому при наличии в клинике дистонии необходимо провести динамическое магнитно-резонансное исследование.
Дифференциальный диагноз. Дистонии дифференцируют между собой, а также исключают другие заболевания, при которых дистония является симптоматической (например, ДЦП, болезнь Галлервордена-Шпатца и др.)
Лечение дистоний
Медикаментозное: для лечения дистонии разными авторами рекомендуются многие препараты [леводопа, нейролептики, баклофен, клоназепам, миорелаксанты (сирдалуд, мидокалм), карбамазепин], однако их применение ограничено большим количеством побочных эффектов и узким терапевтическим диапазоном.
Хемоденервация: применение инъекции ботулотоксина А в мышцы. Взаимодействие ботулотоксина с ацетилхолиновыми рецепторами расслабляет мышцы. Применяют при фокальной дистонии. Длительность действия - до 6 мес, после чего необходимо повторное введение.
Хирургическое: селективная денервация, ризотомия, миомектомия, билатеральная таламотомия, паллидотомия. Глубокая стимуляция бледного шара включена в протокол лечения дистонии в США в 2004 г. Криоталамэктомия проводится при локальных формах дистоний. Эта процедура может привести к клинической ремиссии в большинстве случаев гемидистонии и спастической кривошеи. Эффективность хирургического лечения детей с генерализованной формой мала, так как высоки вероятность возврата клинических симптомов и риск осложнений (нарушения речи, гемипарез, атаксия и эпилепсия).
Семейный (эссенциальный) тремор Минора
Наследственный (семейный) тремор, проявляющийся дрожанием рук при движениях, описал отечественный невролог Л.С. Минор. Распространенность в популяции высокая и составляет 5 на 1000. Заболевание передается по аутосомно-доминантному типу, возможны и спорадические случаи. На настоящий момент картированы два гена - на хромосоме 2р22-р25 и на хромосоме 3q13. Патогенез семейного тремора до сих пор неясен, морфологических изменений в мозге не находят.
Физиологический эссенциальный тремор с частотой от 8 до 12 Гц зависит от периферических рефлексов. Этот вид тремора возникает абсолютно у всех здоровых людей при стрессе, резко усиливается при высокой адренергической активности: утомлении, холоде, тревоге, гипогликемии, а также при приеме лекарственных средств: кофеина, тиреоидных гормонов, антидепрессантов и фенотиазинов. Во сне тремор полностью исчезает. Влияние тремора на активные движения различно. У некоторых больных сохраняется способность выполнять даже тонкие виды работ, другие вынуждены менять профессию.
Патологический эссенциальный тремор с частотой от 4до7 Гц возникает вследствие нарушения взаимодействия денторубрального пути и сегментарной двигательной иннервации (нарушение в спинальном аппарате гамма-альфа-сопряжения). Внутривенное введение пропранолола не влияет на патологический тремор.
Клинические проявления. Симптомы обычно становятся выраженными в пубертатный период, но известны случаи заболевания в 5-летнем возрасте и ранее. Чаще встречается у лиц мужского пола. Во сне тремор прекращается; значительно уменьшается или исчезает после приема небольшой дозы алкоголя. Так, один из наблюдавшихся С.Н. Давиденковым музыкант, страдавший эссенциальным тремором, мог выступать на сцене только после приема 100 г водки (1960). В нижних конечностях тремора не бывает. Дети с эссенциальным тремором могут прекрасно рисовать, вышивать, играть, собирать и склеивать игрушки. Речь и интеллект не нарушаются, не изменяются походка и мышечная сила. Вначале симптомы прогрессируют, но у взрослых клиническая картина стабилизируется, и тремор не влияет на повседневную деятельность. В более позднем возрасте состояние может внезапно ухудшиться, и эссенциальный тремор переходит в сенильный.
Диагноз эссенциального тремора клинический; основан на исключении других заболеваний базальных ганглиев и влияния лекарственных средств. Дрожание рук заметно при осмотре, для выявления незначительного тремора больному предлагают вытянуть руки, написать несколько слов, провести прямую линию и т.п. Легкое дрожание головы можно ощутить, положив руки на голову больного.
Диагностика
1. Продолжительность более 1 года.
2. Отсутствие пирамидных, мозжечковых, чувствительных нарушений и поражения периферических нервов.
3. Нормальный интеллект.
4. Тремор не связан с приемом лекарственных препаратов.
5. Отсутствие системных заболеваний (например, патологии щитовидной железы).
6. Нормальный результат МРТ.
Дополнительно подтверждает диагноз эссенциального тремора положительный семейный анамнез. Следует заметить, что локализация и выраженность тремора у членов одной семьи может отличаться.
Лечение. В большинстве случаев тремор не требует лечения. Тем не менее уменьшает проявление семейного тремора пропраналол, антагонист бета-адренорецепторов.
Ювенильная болезнь Паркинсона (G 20)
Симптомы ювенильной болезни Паркинсона появляются до 20-летнего возраста. В этом состоит различие между ювенильной болезнью Паркинсона и болезнью Паркинсона с ранним дебютом (появление симптомов в возрасте от 20 до 40 лет). Заболевание наследуется по аутосомно-доминантному и аутосомно-рецессивному типу. Ген аутосомно-доминантной формы картирован на хромосоме 4q21-23, аутосомно-доминантной формы с ранним дебютом - на хромосоме 2р13. Ген аутосомно-рецессивной формы ювенильной болезни Паркинсона картирован на хромосоме 6q15.2-27. Он кодирует белок паркин, который в избытке встречается во всех отделах мозга, включая черное вещество.
При морфологическом исследовании обнаруживаются гибель нейронов и глиоз в компактной части черного вещества.
Клиника. Первые симптомы аутосомно-рецессивной формы появ- ляются после 15 лет. Нарушается походка, появляются ретропульсия, тремор, гиперрефлексия и дистоническая установка стоп. Все симптомы уменьшаются во сне. Интеллектуально-мнестических расстройств нет. На МРТ изменения не определяются.
Дифференциальный диагноз. Ювенильную болезнь Паркинсона дифференцируют с болезнью Вильсона-Коновалова, дофа-зависимой дистонией и оливопонтоцеребеллярной атрофией.
Лечение. Заместительная терапия препаратами леводопы.
5.2. Наследственно-дегенеративные заболевания ствола, мозжечка и спинного мозга
Наследственно-дегенеративные заболевания ствола, мозжечка и спинного мозга характеризуются медленно прогрессирующим
Рис. 5.5. Атактическая походка

Рис. 5.6. Атаксия в положении стоя

Рис. 5.7. Больная с Атаксией Фридрейха
течением с распадом функций, которые регулируются этими мозговыми структурами. Дебют заболеваний - в детском и юно- шеском возрасте.
Этиология в большинстве случаев наследственная, заболевания передаются по аутосомнодоминантному или аутосомнорецессивному типу.
Патогенез. Прогрессирующее течение обусловлено атрофией нервной ткани в пределах пораженной области.
Классификация этих расстройств основана на генетических, клинических и патоморфологических данных.
Атаксия Фридрейха (G 11.1)
Атаксия Фридрейха (АФ) описана Н. Фридрейхом в 1863 г. Это наследственное заболевание, характеризующееся медленно прогрессирующей атаксией вследствие склеротического перерождения задних и боковых столбов спинного мозга, гипоплазии мозжечка и спинного мозга (рис. 5.5-5.7). Для него характерны атаксия, нистагм, кифосколиоз, деформация стопы. Больные отличаются особым дизморфическим статусом, имеют множество скелетных аномалий, часть из которых сформирована с рождения. Прибли- зительно у трех из четырех пациентов имеются высокий свод стопы (полая стопа), пальцы в
виде барабанных палочек, атрофированы мелкие мышцы стопы. Кифосколиоз наблюдается в 75-90% случаев.
Распространенность в популяции вариабельна - максимально до 10 случаев на 100 000 с высокой частотой гетерозиготного носительства мутантного гена - 1 на 120 человек.
Генетика. Заболевание передается аутосомно-рецессивным путем; ген картирован на хромосоме 9q13. Он кодирует митохондриальный белок фратаксин, расположенный на внутренней поверхности мембраны митохондрий и участвующий в обмене железа. В интроне патологического гена увеличена последовательность повторов ГАА (гуанинаденин-аденин). Количество ГАА-повторов находится в диапазоне от 6 до 29 у здоровых людей и от 120 до 1700 - у больных, причем размер повторов коррелирует с возрастом дебюта и тяжестью болезни. Патологически удлиненный аллель генетически нестабилен и способен к дальнейшей экспансии при его передаче в следующее поколение. В результате мутации снижается уровень нормального фратаксина, железо откладывается внутри митохондрий, происходит необратимое повреждение функции митохондрий и нарушение окислительного фосфорилирования. В результате гибнут клетки энергозависимых мишеней (мозга, сердца, поджелудочной железы, почек, печени).
Таким образом, атаксия Фридрейха - это митохондриальное заболевание, связанное с мутацией ядерного генома. У гетерозигот неврологических симптомов не наблюдается.
Патогенез связан с дегенерацией длинных проводников спинного мозга. Наряду с периферическими нервами также могут поражаться продолговатый мозг и, реже, мозжечок. В этих областях выявляются аксональная дегенерация, демиелинизация и компенсаторный глиоз. Дегенеративные изменения наиболее выражены в столбах Кларка и зубчатых ядрах мозжечка, но поражаются также ядра продолговатого мозга и клетки Пуркинье. Апоптоз нейронов и глиоз отмечаются в вестибулярных и слуховых ядрах. В миелиновой оболочке проводников снижен уровень протеолипидов. Возможна патология со стороны внутренних органов: кардиомегалия с гипертрофией миоцитов, а в поджелудочной железе - хронический интерстициальный фиброз и воспалительная инфильтрация. Нередко выявляется сахарный диабет.
Патоморфология. Выявляется гибель клеток столбов Кларка и начинающихся от них спиноцеребеллярных трактов, а также (в поздней стадии болезни) дегенерация ядер III, V, IX-X, XII пар черепных нервов, клеток Пуркинье, зубчатого ядра и верхней ножки мозжечка.
В указанных областях выявляются аксональная дегенерация, демиелинизация и компенсаторный глиоз.
Клинические проявления. Возраст дебюта вариабелен, однако в одной семье заболевание начинается в одном возрасте. Первые симптомы могут отмечаться уже в 2-летнем возрасте, средний возраст дебюта - 10 лет. Течение характеризуется появлением новых симптомов, относительно быстрым прогрессированием процесса и сочетанием типичных неврологических и экстраневральных нарушений.
Дети начинают ходить после года, часто падают. При более позднем дебюте возникает пошатывание, нарушена ходьба в темноте (признак заднестолбовой атаксии). Вскоре к атаксии при ходьбе при- соединяются дискоординация рук, изменение почерка, слабость в ногах.
Со стороны черепных нервов обнаруживаются нарушения остроты зрения из-за атрофии зрительных нервов, нистагм (в 20-40% случаев), а также снижение слуха. Кроме того, могут наблюдаться подергивания глазных яблок (миоклонии). Атрофия зрительных нервов может быть врожденной или быстро нарастает на первом году жизни. У 40% больных нарушено восприятие цветов.
Вестибулярные расстройства возникают рано, на поздних этапах болезни встречаются приблизительно у 50% пациентов. Также типич- на глухота, вызванная дегенерацией слуховых нейронов. Наиболее примечательным симптомом является комбинированная мозжечковосенситивная атаксия, вызванная поражением мозжечка и задних столбов с их чувствительными проводниками. Она более выражена в ногах, чем в руках, и выявляется при исследовании походки и статики ребенка. Можно выявить отсутствие вибрационной и проприоцетивной чувствительности, в далеко зашедших случаях в дистальных отделах конечностей нарушены другие виды чувствительности.
При неврологическом обследовании выявляется арефлексия коленных и ахилловых рефлексов. Возникают слабость дистальных мышц нижних конечностей и атрофия мелких мышц рук и ног. Часты жалобы на боли, судороги и парестезии в конечностях.
В развернутой клинической стадии нарушения координации нарастают, к ним присоединяется слабость и атрофия мышц ног, а затем и рук, вплоть до тетрапареза. Речь становится раскатистой в результате несогласованности дыхания и фонации. По поводу деменции мнения противоречивы: для детей умственная отсталость и деменция нехарактерны.
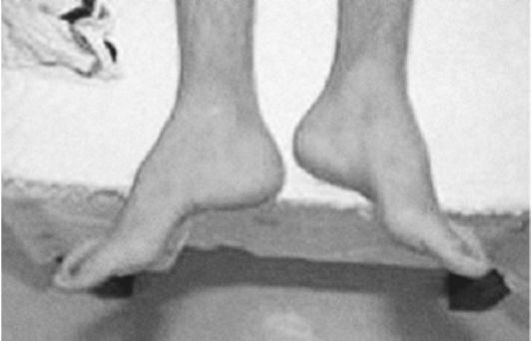
Рис. 5.8. Деформация стоп по типу Фридрейха
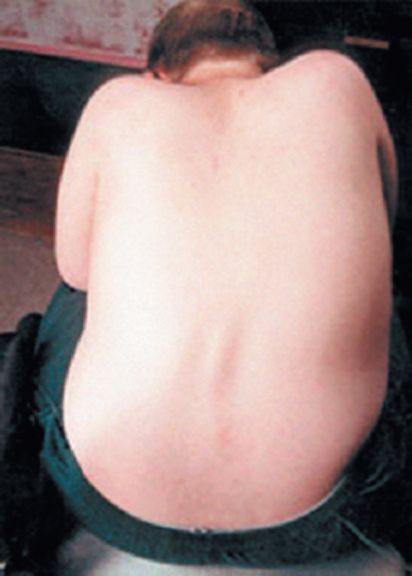
Рис. 5.9. Сколиоз при атаксии Фридрейха
Расстройства функций тазовых органов характерны для финальной стадии болезни, а ранним симптомом могут быть внезапные позывы к мочеиспусканию.
Среди экстраневральных проявлений болезни Фридрейха необходимо выделить поражение сердца, которое встречается более чем у 90% больных. Характерна прогрессирующая гипертрофическая или дилатационная кардиомиопатия. Она проявляется болями в области сердца, сердцебиением, одышкой при физической нагрузке, систолическим шумом и другими симптомами. Более чем у половины больных кардиомиопатия является непосредственной причиной смерти.
Деформации стоп - «стопа Фридрейха» - не патогномонична для болезни Фридрейха (рис. 5.8) и встречается при некоторых других дегенеративных заболеваниях нервной системы, например при невральной амиотрофии Шарко-Мари, спастической параплегии Штрюмпеля и др. Нередок также сколиоз (рис. 5.9). К экстраневральным проявлениям болезни Фридрейха относятся эндокринные расстройства (сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников).
Неврологические симптомы прогрессируют медленно, с продолжительностью заболевания до 20 лет, хотя возможно более быстрое течение болезни. Иногда наблюдаются периоды стабилизации состояния. Сопутствующие инфекции ухудшают течение заболевания и способствуют появлению новых симптомов. Больной с далеко зашедшей болезнью прикован к постели, страдает дисфагией и другими бульбарными симптомами. Смерть наступает от истощения или, чаще, от миокардита с тяжелой сердечной недостаточностью. При хорошем уходе пациенты могут доживать до 40-50 лет.
Дополнительные методы исследования. При исследовании зрительных вызванных потенциалов выявляются генерализованное снижение амплитуды потенциалов и удлинение времени их появления. Уменьшение амплитуды, вероятно, является последствием распада волокон зрительных путей.
Соматосенсорные вызванные потенциалы, регистрируемые от надключичных отведений, отличаются от нормальных уже на самых ранних стадиях болезни, но они не сопровождаются снижением проводимости по периферическому нерву.
МРТ может выявить расширение IV желудочка и атрофию верхнего червя, ствола и спинного мозга.
При проведении ЭКГ и Эхо-КГ признаки миокардита выявляются в 80-90% случаев. Особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки.
При цитохимическом исследовании ферментов-дегидрогеназ лимфоцитов выявляется достоверное снижение сукцинатдегидрогеназы (СДГ), α-глицерофосфадегидрогеназы (ГФДГ), глутаматдегидрогеназы (ГДГ), лактатдегидрогеназы (ЛДГ), малатдегидрогеназы (МДГ) и др.
Необходимо иметь в виду, что при молекулярно-генетическом обследовании пациентов с клинически типичными проявлениями не у всех обнаруживается увеличение тринуклеотида ГАА, расширение аллеля. Возможна точечная мутация или делеция в гене на обеих хромосомах. Описана аутосомно-рецессивная форма мозжечковой атаксии, при которой сухожильные рефлексы сохранены и нет атрофии зрительных нервов, диабета и нарушений со стороны сердца. Симптомы появляются в возрасте от 18 мес до 20 лет, течение медленнее, чем при классической форме. Почти у половины пациентов с такой клинической картиной можно найти увеличение повторов ГАА.
Диагноз. В типичном случае клинический диагноз ставится на основании имеющихся с раннего детства прогрессирующей атаксии, скелетных деформаций, нарушений зрительных вызванных потенциалов и кардиопатии. Диагноз подтверждается генетически (определение размера повторов ГАА).
Дифференциальный диагноз в первую очередь должен проводиться со второй по частоте встречаемости прогрессирующей атаксией с началом в детском возрасте - атаксией-телеангиоэктазией (болезнью Луи-Бар). Клинически она отличается наличием на коже телеангиоэктазий (чрезмерного локального расширения мелких сосудов, преимущественно пре-
капилляров и капилляров), отсутствием скелетных аномалий, частыми и тяжело протекающими инфекциями дыхательных путей, отсутствием или крайне низким уровнем IgA, высоким уровнем альфа-фетопротеина. На МРТ выявляется гипоплазия мозжечка, чаще его червя.
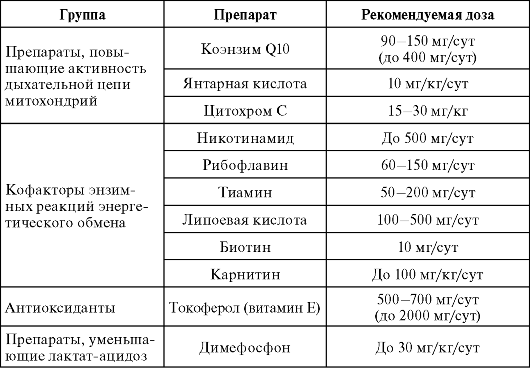
Лечение атаксии Фридрейха не разработано. Применяют препараты, поддерживающие функцию митохондрий (табл. 10). Рекомендуется одновременное назначение препаратов, повышающих активность дыхательной цепи митохондрий, кофакторов энзимных реакций энергетического обмена, антиоксидантов. Пациенты чувствуют себя лучше при ограничении количества углеводов в пище до 10 г/кг, поскольку они являются своеобразной «провокацией», усиливающей дефект энергетического обмена.
Дети с АФ могут оставаться активными максимально долго, занимаясь лечебной физкультурой, выполняя комплексы корректирующих упражнений, направленных на укрепление силы мышц и нормализацию баланса. При такой программе упражнений кардиомиопатия не развивается.
Ортопедическое хирургическое лечение скелетных деформаций, особенно прогрессирующего сколиоза, показано, если неэффективен ортопедический корсет.
Таблица 10. Медикаментозные препараты, применяемые для лечения БФ
Прогноз. Болезнь Фридрейха характеризуется неуклонно про- грессирующим течением, длительность болезни может варьировать в широких пределах, но чаще не превышает 20 лет. Непосредственными причинами смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения.
Спиноцеребеллярные атаксии [оливопонтоцеребеллярная дегенерация]. Оливопонтоцеребеллярная дегенерация - генетически и клинически гетерогенные состояния. Для них характерны прогрессирующая мозжечковая атаксия, тремор, головокружение, дизартрия, снижение глубокой чувствительности, глазодвигательные нарушения и пирамидные симптомы. Реже наблюдаются гиперкинезы, симптомы периферического паралича и тазовые нарушения. Патологический процесс поражает нейроны коры мозжечка, ядра варолиева моста и нижних олив, а также в той или иной степени спинной мозг и базальные ядра. Степень тяжести обусловлена характером мутации и длиной патологического гена. В результате молекулярно-генетических исследований в настоящее время выделено более 10 типов атаксий, которые получили название спиноцеребеллярных атрофий (СЦА). Но даже при молекулярно-генетическом исследовании приблизительно у половины семей с аутосомно-доминантной мозжечковой атаксией не обнаруживается ни одна из известных мутаций. Тем не менее диагноз аутосомнодоминантной мозжечковой атаксии базируется на идентификации генетической мутации.
Средний возраст дебюта этих заболеваний приходится на четвертое десятилетие жизни, однако ряд состояний встречается у детей.
Этиология. Гены картированы на хромосомах: СЦА1 - на 6р22-23, СЦА2 - на 12q24.1, СЦАЗ - на 14q32.1, СЦА4 - на 16q21, СЦА5 - на 11q13, СЦА7 - на 3р12-13, СЦА8 - на 13q 21, СЦА10 - на 22q13.
Ген формы СЦА6 картирован на хромосоме 19р13. И только при этой форме установлен механизм работы гена, который кодирует альфа-1-субъединицу вольтаж-зависимых кальциевых каналов.
Механизм мутаций при СЦА заключается в патологическом увеличении числа тринуклеотидных повторов. Длина повторов нарастает из поколения в поколение, поэтому чем длиннее повтор, тем раньше дебютирует заболевание и тем тяжелее оно протекает (антиципация). Такой характер повреждения гена и проявления болезни характерен для болезни Гентингтона, миотонической дистрофии, спинальнобульбарной амиотрофии Кеннеди и многих других неврологических заболеваний. Распространенность отдельных генетических форм СЦА
варьирует в различных популяциях. В Северной Америке преобладающей формой является СЦА3, в России чаще всего встречается СЦА1. При этой форме увеличенная полиглутаминовая последовательность провоцирует нейрональную дегенерацию. Обычно клиническая картина дебютирует в возрасте до 15 лет, причем у мальчиков раньше, так как повторы в большей степени удлиняются при наследовании по отцовской линии. Характерны атаксия, офтальмоплегия, пирамидные и экстрапирамидные симптомы.
Морфологически выявляется атрофия мозжечка и его ножек, а также основания моста. Наиболее сильно страдают клетки Пуркинье и нейроны зубчатого ядра, а также базальные ядра, спинной мозг, сетчатка глаза и периферическая нервная система.
Диагноз и дифференциальный диагноз основывается на времени дебюта, характерном сочетании симптомов и скорости их развития у детей, чьи родители страдают прогрессирующей атаксией.
Семейная спастическая параплегия. Заболевание передается аутосомно-доминантным, аутосомно-рецессивным или Х-сцепленным путем.
Патогенез. Основные изменения происходят в спинном мозге. Аксональная дегенерация пирамидальных путей всегда максимально выражена в дистальных отделах. В пораженных проводниках разрушаются осевой цилиндр и миелиновая оболочка. Поражаются также восходящие пути, в особенности задние столбы, спиноцеребеллярные волокна и клетки спинномозговых узлов, которые дегенерируют на фоне пролиферации глии. Признаков первичной демиелинизации не обнаруживается. При биопсии мышц можно обнаружить рваные красные волокна.
Клиническая картина при всех формах сходна. При рецессивных вариантах болезни средний возраст развития полной клинической картины - 11,5 года, а при доминантных - 20 лет. Однако у 40% пациентов первые симптомы появляются до 5-летнего возраста. Дети начинают позднее ходить, выявляются шаткость и неуклюжесть, перекрещивание ног в виде ножниц. Мышечный тонус в ногах и сухо- жильные рефлексы повышены, выявляются патологические стопные симптомы. Это заболевание часто протекает под маской детского церебрального паралича. Следует заметить, что при ССП не наблюдается атрофии мышц и, несмотря на поражение задних столбов, вибрационная чувствительность не нарушена.
Как правило, течение болезни очень медленное, причем быстрее прогрессирует рецессивная форма. Если ребенок страдает той или
иной доминантной формой, его состояние относительно стабильно до 30 лет. Верхние конечности часто остаются интактными вплоть до терминальной стадии. Соматические нарушения на ранних стадиях болезни не наблюдаются. В некоторых семейных случаях спастическая параплегия сочетается с деменцией, судорогами, гиперкинезами, невритом зрительного нерва, патологией сердца, гипопигментацией кожи.
Диагноз. При отсутствии семейного анамнеза диагноз наследствен- ной параплегии ставится методом исключения. Время проведения по двигательным и чувствительным нервам не нарушено, соматосенсорные вызванные потенциалы снижены, причем не только у больных, но и у клинически здоровых членов семьи. Прогрессирующее развитие симптомов опровергает диагноз ДЦП. Чувствительные расстройства и нарушение функций сфинктеров, обычно характерные для опухоли спинного мозга, редко встречаются на ранних стадиях болезни. Однако при отсутствии убедительного семейного анамнеза требуется проведение МРТ для исключения новообразований спинного мозга.
Лечение. Ввиду медленного прогрессирования болезни должна применяться активная программа физиотерапии и лечебной физкультуры для предотвращения контрактур.