Детская неврология : учебник : в двух томах / А. С. Петрухин. - Т. 2. - 560 с. : ил.
|
|
|
|
ГЛАВА 6. НЕРВНО-МЫШЕЧНЫЕ ЗАБОЛЕВАНИЯ
Дегенеративные заболевания с преимущественным поражением периферических нервов и мышц составляют значительную долю наследственной патологии человека. Диагностика нервно-мышечных заболеваний основана на молекулярно-генетических и электрофизиологических (ЭМГ) исследованиях.
Электронейромиография позволяет подтвердить диагноз и следить за динамикой заболевания. При нейрогенной мышечной патологии можно выявить признаки денервации: потенциалы фибрилляции, положительные острые волны, снижение амплитуды интерференционного потенциала, полифазные потенциалы. При первичной мышечной патологии ЭМГ-картина неспецифична и вариабельна; наиболее характерно снижение амплитуды потенциалов. Показатели скорости проведения импульса (СПИ) при аксонопатиях уменьшены незначительно или находятся на нижней границе нормы. При демиелинизирующих невропатиях СПИ значительно уменьшена. По изменению СПИ и амплитуды потенциалов действия (по чувствительным или смешанным нервам) можно диагностировать туннельные ней- ропатии, а также дифференцировать аксонопатии и миелинопатии. Увеличение латентного периода поздних ответов наблюдается при невропатиях и корешковом синдроме.
Значительную роль в диагностике играют морфологические, иммуногистохимические и электронно-микроскопические методы изучения биоптатов. Состояние мышечных волокон при световой биомикроскопии помогает дифференцировать первичную миогенную атрофию от вторичной денервационной (неврогенной или миелогенной) амиотрофии. Гистохимический анализ биоптатов необходим для обнаружения специфических метаболических дефектов в мышечной ткани. Электронная микроскопия открыла целый класс заболеваний, которые объединены понятием «структурная миопатия».
Лечение. Для многих заболеваний мышц, нервно-мышечных синапсов, периферических нервов и мотонейронов разработано этиологическое и патогенетическое лечение. В остальных случаях терапия направлена на замедление прогрессирования заболевания, продление периода ремиссии и повышение качества жизни больного. Лечение нервно-мышечных заболеваний требует совместных усилий неврологов и реабилитологов. Тактика лечения зависит от тяжести и скорости прогрессирования заболевания.
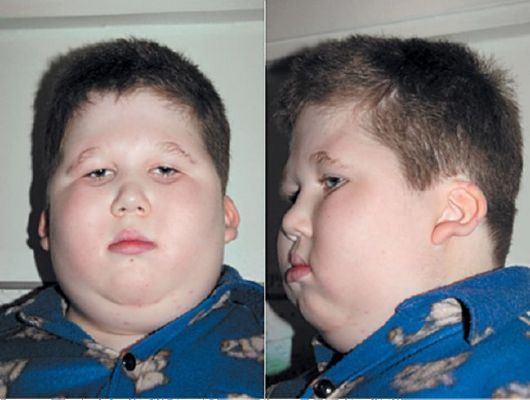
Рис. 6.1. Внешний вид ребенка 13 лет, получавшего длительную гормональную терапию. Кушингоид
Принципы длительной кортикостероидной терапии
Осложнения зависят от дозы и длительности лечения (рис. 6.1). Основные осложнения: синдром Кушинга, сахарный диабет, остеопороз, активизация туберкулеза, артериальная гипертония, психоз, подверженность инфекциям, язвенная болезнь.
При отмене кортикостероидов возможны 3 вида осложнений. 1. Осложнения, связанные с угнетением функции надпочечни-
ков. Оно развивается при дробном приеме преднизолона в дозе, превышающей 20-30 мг/сут в течение более одной недели. Полное восстановление занимает до одного года. При дозах, приближенных к физиологическим, функция надпочечников обычно остается сохранной, если длительность лечения не превышает 1 мес. После обычных доз кортикостероидов заместительной терапии не требуется.
2. Общие симптомы отмены (анорексия, тошнота, рвота, сонливость, головная боль, лихорадка, миалгия и артралгия, потеря массы) более вероятны после длительной терапии. Лечение симптоматическое, малыми дозами кортизона (10 мг/сут) в течение нескольких недель.
3. Обострение основного заболевания. Это одно из наиболее опасных осложнений при отмене кортикостероидов. Его риск уменьшается при постепенном снижении дозы. При нервно-мышечных заболеваниях чаще всего применяется преднизолон - препарат короткого действия для приема внутрь. Его можно назначать ежедневно (в несколько приемов или однократно утром) или через день (однократно утром). При коротком курсе (менее месяца) схема приема не имеет существенного значения. При длительном лечении дробный ежедневный прием способствует развитию синдрома Кушинга, угнетению функции надпочечников и снижает сопротивляемость инфекциям. При длительном курсе однократный утренний прием суточной дозы препарата короткого действия реже вызывает угнетение над-
почечников (хотя и не предотвращает возникновение синдрома Кушинга). При приеме через день удвоенной суточной дозы реже развиваются угнетение надпочечников, синдром Кушинга и снижение сопротивляемости инфекциям. Эта схема эффективна при большинстве нервно-мышечных заболеваний.
6.1. Прогрессирующие мышечные дистрофии
Термином «мышечные дистрофии» обозначают группу клинически полиморфных генетически детерминированных заболеваний, в основе которых лежат первичные прогрессирующие дегенеративные изменения в мышечных волокнах. Различные формы миодистрофий отличаются друг от друга своей генетической природой, типом наследования, сроком дебюта, топографическим своеобразием распределения мышечных атрофий. К характерному клиническому маркёру миодистрофий относится «утиная» походка, связанная со слабостью ягодичных мышц, которые фиксируют таз относительно бедренной кости. В результате во время ходьбы возникают наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). Кроме того, у больных можно наблюдать ходьбу на пальцах, частые падения, медленное двигательное развитие и специфические ограничения при поднимании рук вверх, подъеме по лестнице, вставании с пола.
Миодистрофии Дюшенна и Беккера. Форма Дюшенна широко распространена в мире и встречается с частотой 1 на 3500 новорожденных мальчиков, тогда как форма Беккера наблюдается примерно в 3-5 раз реже.
Этиология и патогенез. Миодистрофии Дюшенна и Беккера являются аллельными вариантами, наследуются по рецессивному сцепленному с Х-хромосомой типу и обусловлены либо полным отсутствием синтеза, либо синтезом дефектного высокомолекулярного цитоскелетного белка-дистрофина. Из-за отсутствия дистрофина миофибриллы утрачивают устойчивость к циклическим актам сокращения-расслабления и разрываются. Саркоплазматические мембраны становятся нестабильными, нарушается работа ионных каналов, в результате повышается концентрация свободного внутриклеточного ионизированного кальция, который оказывает некротизирующее влияние на мышечные волокна, вызывая их лизис (рис. 6.2).
Клиническая картина. Первые клинические симптомы у большинства мальчиков с миодистрофией Дюшенна возникают до 3-5 лет жизни: нарушается походка, дети начинают часто падать, утрачивают
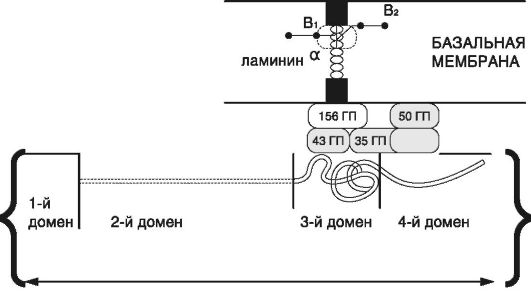
Рис. 6.2. Молекулярная организация дистрофина
Рис. 6.3. Больные, изображенные Г. Дюшенном
подвижность. Развивающаяся псевдогипертрофия икроножных мышц создает обманчивое впечатление о мышечной силе (рис. 6.3). Псевдогипертрофии могут развиваться также в ягодичных, дельтовидных мышцах, мышцах живота и языка. Наконец, мышечная слабость становится настолько явной, что ребенок с трудом встает с пола, ходит «утиной» походкой, использует миопатические приемы: «взбирание по себе», «подъем лесенкой» (симптомы Говерса).
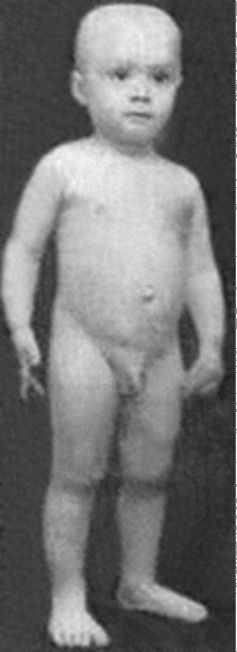
Рис. 6.4. Ребенок 1,5 года с болезнью Дюшенна
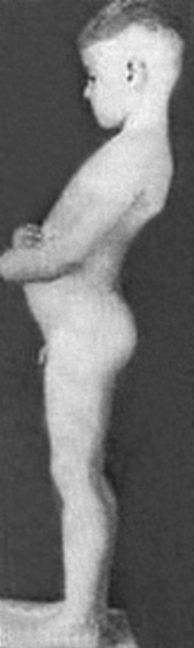
Рис. 6.5. Тот же ребенок в 5 лет. Псевдогипертрофии мышц, лордоз
Двигательные функции относительно стабилизируются между 3 и 6 годами жизни. В большинстве случаев возможность ходить и подниматься по лестнице сохраняется до 8-летнего возраста. От 3 до 8 лет происходит дальнейшее укорочение ахилловых сухожилий и формируются фиксированные сгибательные контрактуры в голеностопных суставах, развиваются компенсаторный поясничный гиперлордоз, кифосколиоз грудного отдела позвоночника, нарастают атрофии мышц бедра, тазового, а затем и плечевого пояса, спины и проксимальных отделов рук. Обращает на себя внимание наличие «свободных надплечий», «крыловидных лопаток», «осиной талии». Нередко атрофии мышц маскируются хорошо развитым подкожным жировым слоем. Часто развиваются деформации грудной клетки и стоп, диффузный остеопороз. Коленные, сгибательные и разгибательные локтевые рефлексы исчезают в первую очередь, тогда как ахилловы рефлексы могут сохраняться довольно долго. В возрасте 9 лет некоторые дети уже передвигаются с помощью кресла-каталки, но у большинства способность к самостоятельному передвижению сохраняется вплоть до 12-летнего, а возможность стоять - до 16-летнего возраста. Слабость дыхательной мускулатуры и диафрагмы обусловливает уменьшение жизненной емкости легких до 20 % нормы, что приводит к эпизодам ночной гиповентиляции (рис. 6.4-6.6).
У части больных обнаруживаются различные признаки эндокринопатии: адипозогенитальный синдром, низкорослость. В связи
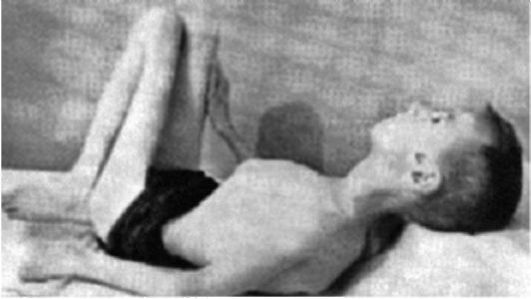
Рис. 6.6. Тот же ребенок в 14 лет. Выражены деформация позвоноч- ника, контрактуры сгибательного характера, атрофии мышц
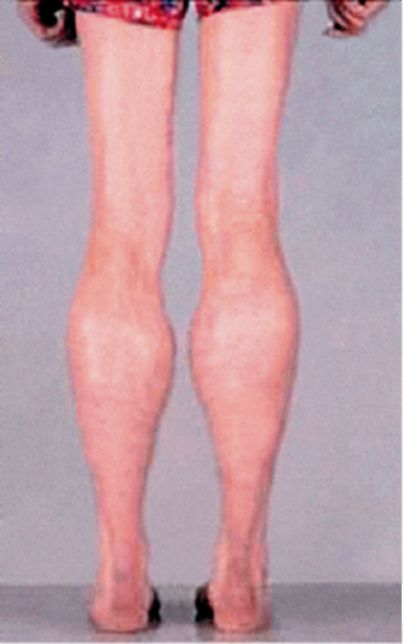
Рис. 6.7. Псевдогипертрофии мышц голеней при болезни Беккера
с дефицитом церебральных изоформ дистрофина - аподистрофинов у части больных с миодистрофией Дюшенна имеет место умственная отсталость различной степени. Тяжесть психических нарушений у детей не соотносится с выраженностью мышечного дефекта и стадией миодистрофического процесса. Облигатным признаком развернутой стадии миодистрофии Дюшенна является гипертрофическая, или дилатационная, кардиомиопатия, которая сопровожда- ется нарушениями ритма сердца, расширением его границ, явлениями сердечной недостаточности. Кардиомиопатия - наиболее частая причина летального исхода при миодистрофии Дюшенна. К летальности приводит также дыхательная недостаточность, которая провоцируется интеркуррентными инфекциями или аспирацией. Больные погибают на 2-3-м десятилетии жизни.
Миодистрофия Беккера (рис. 6.7) может развиваться после 15-
20 лет, протекает гораздо мягче. Больные с этой формой миодистрофии доживают до зрелого возраста. Нарушения интеллекта для нее нехарактерны, ретракции сухожилий и контрактуры менее выражены, чем при миодистрофии Дюшенна, кардиомиопатия может отсутствовать. Однако у некоторых больных нарушения деятельности сердца выступают на первый план и часто являются манифестным симптомом болезни. Кроме того, у части больных миодистрофией Беккера сохранена фертильность, поэтому взрослые больные через дочь могут передавать заболевание своим внукам («эффект деда»).
Диагностика. Для миодистрофии Дюшенна характерно значительное повышение уровня ферментов уже на ранних стадиях миодистро-
фического процесса. У больных до 5-летнего возраста уровень креатинфосфокиназы (КФК) может превышать верхнюю границу нормы в десятки и даже сотни раз. Затем концентрация фермента снижается приблизительно на 20% в год. Содержание в сыворотке альдолазы, лактат-дегидрогеназы, трансаминаз также повышено. Высокая активность КФК - практически облигатный признак болезни и, кроме миодистрофии Дюшенна, может встречаться при миодистрофии Беккера (как правило, не превышая 5000 ЕД/л), полимиозите, дерматомиозите, гипотиреозе, алкогольной миопатии и пароксизмальной миоглобинурии. ЭМГ выявляет признаки первично-мышечного поражения (низковольтажная кривая с обилием полифазных потенциалов, укорочение потенциалов действия двигательных единиц).
В настоящее время общепринятым «золотым стандартом» для диагностики миодистрофии Дюшенна и Беккера, выявления носи- тельства гена и пренатальной диагностики является мутационный анализ. Иммуногистохимическая реакция на дистрофин применяется при анализе процентного содержания дистрофина в мышцах и отличает формы Дюшенна и Беккера (при первой он отсутствует). У гетерозиготных носительниц (матерей и сестер больных) примерно в 70% случаев выявляются субклинические признаки патологии скелетных мышц: повышение КФК, первично-мышечные изменения на ЭМГ и при исследовании мышечных биоптатов. Изредка у носительниц отмечаются уплотнение и увеличение в объеме икроножных мышц, повышенная мышечная утомляемость при физической нагрузке, спазмы в мышцах после нагрузки (крампи).
Рентгенография костей помогает выявить атрофию диафизов длинных трубчатых костей, истончение кортикального слоя, сужение костно-суставного канала, диффузный остеопороз.
Поражение сердечно-сосудистой системы (кардиомиопатия) развивается у 73% больных детей. Недостаточность дистрофина в кар- диомиоцитах приводит к прогрессирующей атрофии кардиомиоцитов и замещению их фиброзной тканью. Кардиомиопатия впервые диагностируется в 6-7 лет, к 20 годам она имеется у 95% больных. Отмечаются также тахикардия, аритмия, лабильность пульса и АД, приглушение тонов, расширение границ сердца. На ЭКГ отмечаются нарушения сердечного ритма, желудочковые экстрасистолы, признаки гипертрофии левого желудочка (27%): глубокий зубец Q в отведениях II-III aVF и V6; высокий R в отведении V1, признаки ишемии миокарда (5%). На Эхо-КГ можно выявить гипертрофическую (55%) или дилатационную
(25%) кардиомиопатию, дефект межпредсердной перегородки, пролапс митрального клапана, миксому левого желудочка.
Биопсия сердечной мышцы обнаруживает атрофию мышечных волокон, интерстициальный фиброз, жировую инфильтрацию.
Дифференциальная диагностика миодистрофии Дюшенна и Беккера проводится с врожденной дисплазией тазобедренных суставов, витамин- D-резистентным рахитом, проксимальными типами спинальных амиотрофий, полимиозитом и дерматомиозитом, метаболическими и эндокринными миопатиями.
При наличии клинического фенотипа миодистрофии Дюшенна у девочек следует в первую очередь исключить наличие Х-аутосомных транслокаций или других хромосомных аберраций с заинтересован- ностью Х-хромосомы, а также некоторые другие редкие генетические варианты. Кроме того, требуется исключить синдром Шерешевского- Тернера (Х-моносомия). С этой целью проводят цитогенетическое исследование кариотипа.
Миодистрофия Эмери-Дрейфуса является медленно прогрессирующей формой миодистрофии с X-сцепленным рецессивным типом наследования, которая обусловлена мутацией в гене цитоскелетного мышечного белка - эмерина, продуцирующегося преимущественно в скелетных, гладких мышцах и кардиомиоцитах.
Клиническая картина (рис. 6.8). Заболевание начинается между 5 и 15 годами жизни. Самыми ранними и типичными симптомами являются нарастающие сгибательные контрактуры в локтевых суставах и разгибателях кистей, ретракции ахилловых сухожилий. Как правило, в 12-летнем возрасте у пациентов уже значительно выражены контрактуры в коленных, голеностопных и локтевых суставах. Затем возникают слабость и атрофия двуглавых и трехглавых мышц плеча, позже - дельтовидных и других мышц плечевого пояса. В некоторых случаях в качестве первого симптома отмечают ходьбу на пальцах и наружных краях стоп, которая возникает приблизительно в 5-летнем возрасте. До этого момента двигательное развитие детей обычно не нарушено. Мышечная слабость возникает незаметно и медленно прогрессирует. Примерно в 20-летнем возрасте наступает относительная стабилизация. Способность к ходьбе и подъему по лестнице сохраняется. Лицевая мускулатура не страдает. Слабость мышц представлена в руках (лопаточно-плечевая) и в ногах (перонеальная). Приемы Говерса и псевдогипертрофия икроножных мышц могут отсутствовать. Сухожильные рефлексы не вызываются. Часто укорочены заднешейные мышцы, отмечается ограничение
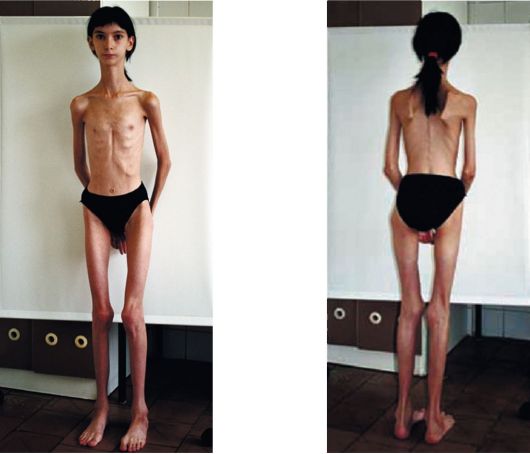
Рис. 6.8. Пациентка 12 лет с миодистрофией Эмери-Дрейфуса
движений в шейном отделе позвоночника (синдром ригидного позвоночника). Частыми и прогностически важными симптомами болезни являются нарушения сердечной проводимости и развивающаяся дила- тационная или гипертрофическая кардиомиопатия. Кардиомиопатия может осложняться развитием паралича предсердий вследствие фиброза пейсмекеров синусового узла. В этих случаях показана срочная имплантация искусственного водителя ритма.
Синкопальные состояния и приступы брадикардии в некоторых случаях могут предшествовать появлению мышечной слабости, но чаще возникают на 3-м десятилетии жизни. Изменения в проводящей системе сердца далеко не всегда обнаруживаются при стандартном ЭКГ-исследовании, но при мониторировании можно выявить атриовентрикулярные блокады и периоды Самойлова-Венкебаха. Аритмия, которую не удается скорригировать имплантацией искусственного водителя ритма, может привести к инсульту и смерти больного. Витальный прогноз при миодистрофии Эмери-Дрейфуса всецело зависит от степени поражения сердца.
Диагностика. Активность КФК повышена умеренно, лактатде- гидрогеназы и альдолазы - в меньшей степени. В пользу миодистрофии Эмери-Дрейфуса свидетельствует отсутствие иммунофлюоресцентной реакции на эмерин с 12 моноклональными антителами при биомикроскопии лейкоцитов, мышечных и кожных биоптатов. Для болезни характерны сочетанные ЭМГ-признаки первично-мышечного и неврогенного поражения с большой представленностью спонтанной денервационной активности.
Лице-лопаточно-плечевая миодистрофия (тип Ландузи-Дежерина). Заболевание наследуется по аутосомно-доминантному типу с высокой пенетрантностью и вариабельной экспрессивностью. Встречается с частотой 2,9 на 100 000 населения. Установлена генетическая гетерогенность лице-лопаточно-плечевой миодистрофии. Большинство случаев связано с мутацией длинного плеча хромосомы 4.
Клиническая картина. Заболевание обычно начинается на 2-м десятилетии жизни. Первоначально атрофии наблюдаются в плечевом поясе, позже распространяются на лицо. У больных обедняется мимика; речь становится неразборчивой. На высоте заболевания поражаются круговые мышцы рта и глаз, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая и трехглавая мышцы плеча. Отмечаются характерные симптомы в виде «поперечной улыбки» («улыбки Джоконды»), протрузии верхней губы («губы тапира»). Грудная клетка уплощается в переднезаднем направлении, плечевые суставы ротированы внутрь, лопатки приобретают крыловидную форму. Атрофии распространяются в нисходящем направлении. При вовлечении в процесс мышц ног слабость наиболее заметна в перонеальной группе мышц - «свисающая стопа». Характерна асимметрия атрофии. Может наблюдаться псевдогипертрофия мышц. Контрактуры и ретракции сухожилий выражены умеренно. Кардиомиопатия редка. Аномалии сосудов сетчатки при ангиоретинографии рассматривают как одно из фенотипических проявлений болезни. Тяжелые глазные симптомы сопровождаются теле- ангиэктазией, отеком и отслойкой сетчатки. Может наблюдаться снижение слуха. Телеангиэктазии ликвидируют коагуляцией, что предотвращает развитие слепоты. Течение болезни относительно благоприятно. Физические перегрузки, интенсивные спортивные занятия и нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни. Многие больные
сохраняют работоспособность, и качество их жизни не ухудшается. Других больных болезнь приковывает к креслу-каталке в зрелом возрасте.
Диагностика. Уровень КФК может повышаться в 5 раз. При ЭМГ регистрируются как миопатические двигательные единицы, так и денервационные потенциалы. Во многих мышцах конечностей гистологические изменения минимальны; в надлопаточных мышцах обнаруживают прогрессирующую дегенерацию и краевую денервацию. Необходимо исключить миастению и опухоль мозгового ствола.
Конечностно-поясные миодистрофии (КПМД) - случаи проксимальной мышечной слабости, которая начинает развиваться на 2-м или 3-м десятилетии жизни, медленно прогрессирует и приводит к глубокой инвалидизации лишь через 15-20 лет.
Этиология и патогенез. КПМД не является однородной в генетическом отношении; на сегодняшний день обнаружено около 10 различных генетических дефектов.
Клиническая картина. Первыми поражаются мышцы плечевого и тазового пояса. В развернутых стадиях значительно поражаются мышцы спины и живота, формируется поясничный гиперлордоз. Мышцы лица, как правило, не страдают. У больных обнаруживаются типичная «утиная» походка, миопатические приемы. Контрактуры и псевдогипертрофии мышц нехарактерны. Кардиомиопатии не развиваются; интеллект сохранен. Мужчины и женщины поражаются одинаково часто. Летальный исход может наступать от легочных осложнений.
Диагностика. Содержание КФК умеренно повышено. ЭМГ демонстрирует признаки первично-мышечного поражения. КПМД необходимо отличать от миопатии Беккера, ювенильной спинальной амиотрофии, миопатии с накоплением гликогена, эндокринных, токсических, лекарственных миопатий, полимиозита и миозита.
6.2. Врожденные структурные миопатии
Врожденные структурные миопатии (ВСМ) представляют собой генетически гетерогенную группу медленно прогрессирующих заболеваний скелетных мышц. Клиническая симптоматика различных ВСМ неспецифична. Основной клинический симптом - диффузная мышечная гипотония, которая может возникать еще внутриутробно и определять редкое шевеление плода. ВСМ принадлежит значительный удельный вес среди причин так называемого синдрома вялого ребенка. Гипотония превалирует в мышцах тазового пояса и прок-
симальных отделов ног. Мышцы плечевого пояса и рук поражаются в меньшей степени. Нередко выявляются врожденный вывих бедра, долихоцефалическая форма головы, готическое нёбо, конская стопа, кифосколиоз, гипоплазия мышц. Характерна задержка двигательного развития: дети поздно начинают держать голову, сидеть, вставать, ходить, часто падают при ходьбе, неспособны бегать. В дальнейшем они не могут выполнить простейших гимнастических упражнений, участвовать в подвижных играх. Сухожильные рефлексы у больных могут быть нормальными, сниженными или отсутствовать. Чрезвычайно важным критерием ВСМ является отсутствие прогрессирования или очень медленное нарастание мышечной слабости. При некоторых формах двигательные функции с возрастом могут несколько улучшаться.
Диагностика. Активность КФК в норме или чуть повышена. При ЭМГ регистрируются низкоамплитудные полифазные миопатические потенциалы двигательных единиц. Скорость проведения импульса по двигательным и чувствительным волокнам в норме. Диагноз достоверно устанавливается лишь при проведении мышечной биопсии с использованием световой и электронной микроскопии, кото- рая обнаруживает специфическое строение мышечного волокна. Исследование мышечных биоптатов у больных детей может выявить уникальные гистологические признаки, определившие целый ряд наименований: болезнь центрального стержня, миотубулярная миопатия, немалиновая миопатия, трехпластинчатая миопатия, миопатия с лизисом волокон I типа, миопатия со сферическими тельцами, миопатия с накоплением телец в виде «отпечатков пальцев», миопатия с цитоплазматическими включениями в виде редуцированных телец, миопатия с агрегацией трубочек и т.д.
Лечение мышечных дистрофий. Терапевтические возможности при миодистрофиях значительно ограничены. Этиологического и патогенетического лечения практически не существует. Симптоматическое лечение направлено на как можно более длительное поддержание имеющейся мышечной силы, снижение темпа развития атрофии и предотвращение формирования контрактур. Основная задача состоит в том, чтобы на максимально возможный срок продлить период активности.
Комплексное лечение состоит из медикаментозной терапии, физиотерапевтических процедур, лечебной гимнастики и массажа, ортопедической коррекции и соблюдения диеты. Важную роль играют психологическая поддержка, продолжение обучения, правильная профессиональная ориентация.
Физиотерапевтические процедуры включают электрофорез прозерина, хлорида кальция, синусоидально-модулированные или диадинамические токи различной проникающей способности, электромиостимуляцию, озокеритовые, парафиновые и грязевые аппликации, ванны (радоновые, хвойные, серные, сероводородные). Рекомендуется оксибаротерапия, поскольку кислород ингибирует процессы фиброзирования и коллагенообразования. Ортопедическая коррекция консервативного (специальные шины и укладки) и оперативного характера (ахиллотомия, миотомия) направлена на борьбу с контрактурами и формирующимися патологическими установками конечностей и также имеет цель сохранить способность больного к самостоятельному передвижению. В каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства. При развивающихся контрактурах после проведения тепловых процедур рекомендуется осторожное растяжение мышц до 20-30 раз в день с последующим наложением шины на время сна.
Больному рекомендуется диета, обогащенная белком, с ограничением жиров (особенно животного происхождения) и углеводов при оптимальном и сбалансированном содержании витаминов и микроэлементов. Необходимо избегать соленого, жареного, пряностей, маринадов, крепких мясных бульонов, кофе, шоколада, какао, пирожных, сдобного теста.
Медикаментозная терапия преследует цель компенсировать энергетический дефицит в мышечной ткани, улучшить тканевый метаболизм и кровообращение, стабилизировать мембраны мышечных волокон. Применяют никотиновую кислоту, витамины В6, В12, А и Е (аевит). Для улучшения белково-синтетических процессов используют аминокис- лотные препараты (церебролизин, глицин, метионин, глутаминовую, фолиевую кислоты). Назначают нестероидные анаболические средства (калия оротат), макроэргические препараты (фосфаден), кардиотрофики (рибоксин, карнитина хлорид, солкосерил), средства, улучшающие периферическое кровообращение (трентал, галидор, теоникол, оксибрал) и ноотропы [пантогам, пирацетам (ноотропил)]. Для улучшения энергетических процессов, происходящих в системе дыхательной цепи митохондрий, используют коэнзим Q10 (убихинон), лимантар, внутривенные инфузии цитохрома-С. Эффекты дезинтоксикации и улучшения реологических свойств крови, купирование слайдж-синдрома достигаются инфузиями вазоактивных препаратов, реополиглюкина, курсов плазмафереза. Относительной стабилизации клеточных мембран способствуют малые дозы преднизолона. Для коррекции
кардиомиопатии используют кардиотрофики (кроме больных с гипертрофической кардиомиопатией); при сердечной недостаточности - сердечные гликозиды, диуретики, каптоприл. При сердечных аритмиях назначают хинидин, β-адреноблокаторы, антагонисты кальция. При развитии полной атриовентрикулярной блокады актуальным становится вопрос о целесообразности имплантации искусственного электрокардиостимулятора.
Перспективы разработки методов генетической терапии при некоторых миодистрофиях (болезнях Дюшенна, Беккера) связаны с совершенствованием генетических технологий. Идет активный поиск генети- ческих носителей (векторов), способных встроить ген дистрофина или мини-гены в мышечные клетки больного реципиента. Исключительное значение придается медико-генетическому консультированию семьи, проведению пренатальной диагностики с исследованием ДНК плода.
6.3. Спинальные мышечные амиотрофии
Спинальные мышечные амиотрофии (СМА) - это гетерогенная группа наследственных заболеваний периферической нервной системы. Патогенез связан с прогрессирующей дегенерацией мотонейронов передних рогов спинного мозга (в некоторых случаях и двигательных ядер ствола мозга). Причиной этого является генетический дефект, вызывающий программируемую клеточную гибель - апоптоз клеток. Утрата мотонейронов приводит к развитию вялого паралича и денервационной атрофии поперечнополосатых мышц. В большинстве случаев наблюдается симметричное поражение проксимальных мышц конечностей; дистальные амиотрофии, поражение буль-
барной мускулатуры и асимметрия поражения развиваются реже. Центральный мотонейрон, как правило, интактен. Расстройств чувствительности не бывает.
Разные варианты СМА отличаются возрастом дебюта, характером течения, топографией поражения скелетной мускулатуры и типом наследования (рис. 6.9). Большинство форм наследуется по аутосомно-рецессивному типу. Несколько форм характеризуются
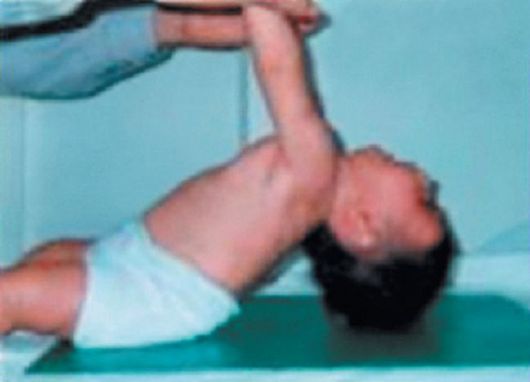
Рис. 6.9. Синдром вялого ребенка при СМА
аутосомно-доминантным и Х-сцепленным рецессивным типами наследования. При гистологическом исследовании мышечных биоптатов выявляют, что к группам волокон обычного размера прилежат мышечные волокна малого размера, пучки гипертрофированных и атрофичных мышечных волокон.
Если ЭМГ определяет бесспорные симптомы СМА, проведение мышечной биопсии необязательно. Принципы лечения и реабилитации СМА такие же, как при миодистрофиях. Этиотропное и патогенетическое лечение пока не разработано.
Проксимальные спинальные амиотрофии детского возраста наследуются по аутосомно-рецессивному типу. Выделяют три фенотипи- чески различных варианта, отличающихся возрастом клинической манифестации, течением и прогнозом:
• тип I, или острая злокачественная инфантильная СМА Верднига- Гоффмана;
• тип II, или хроническая инфантильная СМА (промежуточный тип);
• тип III, или ювенильная СМА Кугельберга-Веландера.
В их основе лежит единая генетическая мутация - делеция гена жизнеспособности моторного нейрона, расположенного на длинном плече хромосомы 5. Поиск мутации проводят при ДНК-диагностике, в том числе у плода при пренатальной диагностике, которая помогает избежать рождения больного ребенка.
Острая злокачественная инфантильная спинальная амиотрофия (болезнь Верднига-Гоффмана, или СМА I типа) встречается с частотой 1 на 25 500 новорожденных. Клинические симптомы отмечаются уже при рождении или появляются до 6 мес жизни. Еще внутриутробно отмечается вялое шевеление, свидетельствующее о снижении двигательной активности плода. У больного ребенка обнаруживают генерализованную слабость, преимущественно в проксимальных мышечных группах, гипотонию и арефлексию. В положении на спине наблюдается «поза лягушки» с разведением и наружной ротацией бедер. Мимическая мускулатура относительно сохранна, глазодвигательные мышцы не вовлечены. Дыхательная функция поначалу адекватная. Выявляются атрофия и фасцикуляции в языке, фасцикулярный тремор кистей. При развитии бульбарного синдрома исчезает глоточный рефлекс, значительно затрудняется кормление, что может привести к аспирационной пневмонии. Часто формируется деформация грудной клетки (рис. 6.10). Если мышечная слабость
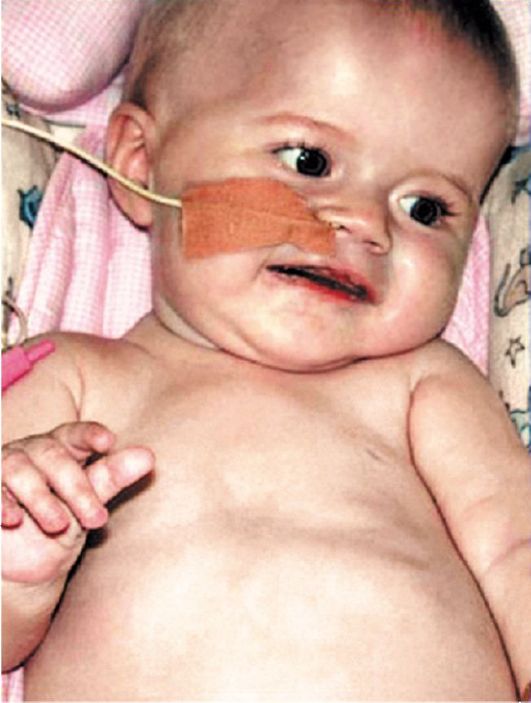
Рис. 6.10. Ребенок, 6 мес, с болезнью Верднига-Гоффмана
выявляется сразу после рождения, то смерть наступает приблизительно в 6-месячном возрасте, тогда как при появлении первых симптомов после 3 мес жизни срок выживаемости может составлять около 2 лет. Основная причина смерти - дыхательная недостаточность на фоне интер- куррентных респираторных заболеваний (рис. 6.11, 6.12).
Для диагностики выявляют мутацию гена путем молекулярно-генетического анализа. Концентрация КФК обычно в норме, но у детей с быстро прогрессирующей слабостью она может быть несколько повышена. ЭМГ обнаруживает потенциалы фибрилляций и фасцикуляции в покое и повышение средней амплитуды потенциалов двигательных единиц. Скорость проведения по двигательным аксонам периферических нервов, как правило, соответствует норме. СМА I типа необходимо дифференцировать от других состояний, вызывающих синдром «вялого ребенка». К ним относятся врожденные миодистрофии и невропатии, структурные миопатии, врожденная или неонатальная миастения, метаболические миопатии, внутриутробный полиомиелит, ботулизм, хромосомная патология, атоническая форма церебрального паралича, синдром Марфана.
Хроническая инфантильная спинальная амиотрофия (СМА II типа). Мышечная слабость, как правило, появляется между 6-м и 24-м мес жизни. Чем раньше дебютируют симптомы, тем злокачественнее течение. Первоначальные проявления слабости обычно симметричны и наблюдаются в проксимальных мышечных группах конечностей. Слабость мышц бедер - наиболее заметный симптом. В раннем периоде дистальная мышечная слабость минимальна или отсутствует. Сухожильные рефлексы с пораженных мышц резко снижены. Все больные способны сидеть, большинство - самостоятельно стоять, а некоторые даже могут ходить (рис. 6.13). Мимическая мускулатура
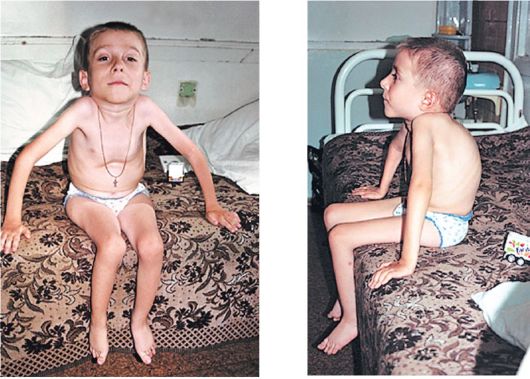
Рис. 6.11. Мальчик, 5 лет, с болезнью Верднига-Гоффмана
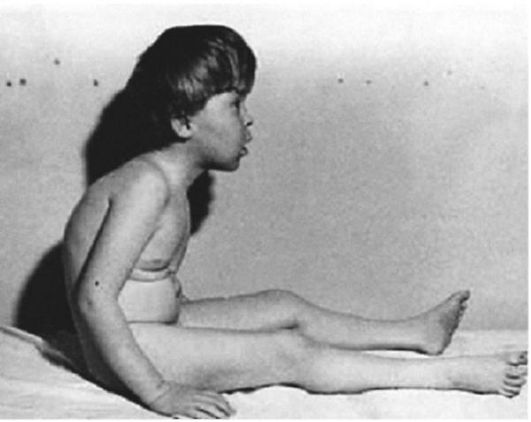
Рис. 6.12. Мальчик, 3 лет, с болезнью Верднига-Гоффмана
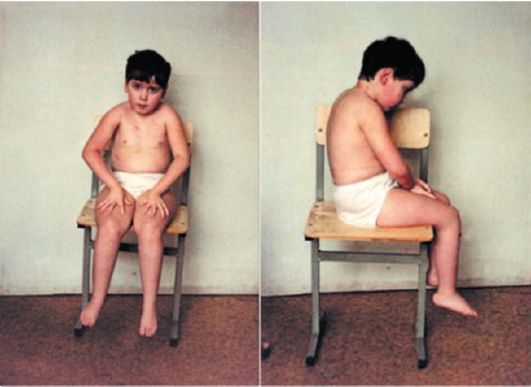
Рис. 6.13. Девочка, 9 лет, с болезнью Кугельберга-Веландера
и наружные мышцы глаза на ранних этапах болезни не поражаются. Мышечная слабость прогрессирует медленно. В отдельных случаях она остается стабильной многие годы, а затем прогрессирование воз- обновляется. Предполагается выживание больных вплоть до зрелого возраста, но даже в период относительной стабилизации ЭМГ выявляет
потенциалы фибрилляций и фасцикуляций. Формируются контрактуры, эквиноварусная деформация стоп. Уже в грудном возрасте у детей наблюдаются искривление позвоночника, деформации грудной клет- ки, дисплазия тазобедренных суставов.
Диагностика. Концентрация КФК в норме. Результаты генетического анализа и данные ЭМГ те же, что и при острой инфантильной форме.
Ювенильная спинальная амиотрофия (болезнь Кугельберга-Веландера, или СМА III типа) встречается в общей популяции с частотой 1,2 на 100 000. Двигательная активность во внутриутробном периоде достаточна; при рождении ребенок здоров. Дебют симптомов происходит между 2-м и 15-м годом жизни. Дети начинают неустойчиво ходить из-за нарастающей проксимальной мышечной слабости в ногах. Развиваются псевдогипертрофии икроножных мышц, что часто ведет к ошибочной диагностике миодистрофии Дюшенна. Заболевание течет доброкачественно, прогрессирует очень медленно. Кисти поражаются позже. Лицевые мышцы могут быть ослаблены, но движения глазных яблок всегда в полном объеме. Бульбарные нарушения нехарактерны. Примерно у половины больных могут развиваться костные деформации, изредка - сухожильные ретракции и контрактуры в суставах. Сухожильные рефлексы с ослабленных мышц отсутствуют или значительно угнетены. Часто регистрируется фасцикулярный тремор кистей.
Диагностика. Первостепенное значение имеет выявление генетической мутации. Концентрация КФК может превышать верхнюю границу нормы в 2-4 раза. У половины больных при ЭМГ регистрируется спонтанная активность (фасцикуляции, фибрилляции и положительные острые волны). При напряжении мышц отмечаются повышение амплитуды и полифазия, увеличение длительности и снижение числа потенциалов двигательных единиц. Проведение по чувствительным волокнам нервов всегда в норме. Скорость проведения по двигательным волокнам при длительном течении заболевания может уменьшаться. СМА III типа дифференцируют с конечностнопоясными миодистрофиями.
Бульбоспинальная амиотрофия Кеннеди - редкая Х-сцепленная рецессивная форма СМА, дебютирующая на 4-й декаде жизни; изредка отмечаются случаи дебюта симптомов в 12-15 лет. Ген картирован на длинном плече Х-хромосомы. Мутация затрагивает ген андрогенового рецептора, в том числе спинальных мотонейронов, что делает эти
рецепторы нечувствительными к влияниям мужских половых гормонов (андрогенов). Ядро клинической картины составляют слабость, атрофии и фасцикуляции в проксимальных мышечных группах конечностей, сухожильная арефлексия, лицевая слабость, атрофии и фасцикуляции в языке, периоральные фасцикуляции, дизартрия и дисфагия, тремор и болезненные мышечные спазмы (крампи). Изредка развивается аксональная невропатия. Бульбарные наруше- ния возникают, как правило, спустя 10 лет после начала болезни. Характерны эндокринные нарушения: гинекомастия, тестикулярная атрофия, снижение потенции и либидо, сахарный диабет, бесплодие, обусловленное азооспермией. Прогноз болезни в целом благоприятен: сохраняются способность к ходьбе и возможность самообслуживания. Продолжительность жизни не уменьшается, однако повышен риск возникновения злокачественных новообразований вследствие гормонального дисбаланса (в том числе рака грудной железы).
Диагностика. В настоящее время возможно проведение прямой ДНК-диагностики, установление гетерозиготного носительства и осуществление дородовой диагностики. ЭМГ выявляет признаки денервации. Уровень КФК может быть в норме. Заболевание необходимо отличать от бокового амиотрофического склероза.
6.4. Множественный врожденный артрогрипоз
Множественный врожденный артрогрипоз - синдром, основным проявлением которого является ограничение подвижности в суставах в сочетании с их деформациями. Обычно поражаются дистальные суставы (голеностопные, лучезапястные), реже - коленные и локтевые суставы. Мышечная слабость при артрогрипозе может носить как неврогенный, так и миогенный характер. Подавляющее большинство случаев относится к спорадическим, остальные случаи наследуются по аутосомно-рецессивному или X-сцепленному типу. При неврогенном артрогрипозе наиболее активная фаза болезни наблюдается во внутриутробном периоде и уже в периоде новорожденности нарушены дыхание и глотание; часть детей погибает от аспирации. В более легких случаях выживаемость выше, а мышечная слабость прогрессирует очень медленно или не прогрессирует вообще. Дыхательные расстройства и проблемы с кормлением в дальнейшем исчезают. Контрактуры представлены как в проксимальных, так и в дистальных суставах. У некоторых новорожденных определяются сопутствующие микрогнатия, высокое нёбо, лицевые аномалии, как при
синдроме Эдвардса (трисомии 18). У некоторых детей с неврогенным артрогрипозом обнаруживаются аномалии развития переднего мозга. Бывают сочетания с менингомиелоцеле, микроцефалией и задержкой психического развития. Синдром миогенного артрогрипоза может наблюдаться при миопатии с диспропорцией типов волокон, врожденных миодистрофиях, миотонической дистрофии, миастенических синдромах, дефиците фосфофруктокиназы.
Диагностика. Гистологическое исследование мышц выявляет характерные признаки денервации и реиннервации. Также выявляют проявления миопатии: повышение доли коллагеновых волокон и жировой ткани, хаотичность расположения волокон среднего размера, фиброз капсул мышечных веретен.
6.5. Воспалительные миопатии
Воспалительные миопатии представляют собой гетерогенную группу заболеваний, при которых мышечная слабость связана с воспалением мышц инфекционного или аутоиммунного характера. К воспалительным миопатиям относят полимиозит, дерматомиозит, миозит с внутриклеточными включениями, гранулематозный миозит, миозит при вирусных, бактериальных и паразитарных инфекциях, миопатию при саркоидозе и некоторые другие формы.
Дерматомиозит является системной иммунозависимой ангиопатией, при которой наблюдаются сосудистые окклюзии и инфаркты, приводящие к развитию всех характерных патологических изменений в мышцах, соединительной ткани, коже, желудочно-кишечном тракте и нервных волокнах. Патогенез связан с образованием антител и иммунных комплексов и активацией системы комплемента. В состав периваскулярного инфильтрата входят Т-лимфоциты, которые в подавляющей своей части являются Т-хелперами, В-лимфоциты и плазматические клетки.
Клиническая картина. Пик заболеваемости приходится на возраст 5-10 лет, но описаны случаи и более раннего начала (до 4-месячно- го возраста). Симптомы возникают постепенно или молниеносно. Скрытое начало характеризуется лихорадкой, недомоганием и утратой аппетита (анорексией). Мышечная слабость в это время может отсутствовать. Такие неспецифические симптомы сохраняются в течение недель и месяцев, что наводит на мысль о персистирующей инфекции. У большинства детей дерматит появляется раньше миозита. Сыпь вначале локализуется на верхних веках и имеет вид
эритемы с очагами нарушенной пигментации и отеком. Затем она распространяется вокруг глаз и на область щек. Эритема и отек на разгибательных поверхностях межфаланговых, локтевых и коленных суставов развиваются позже. Со временем кожа становится атрофичной и шелушащейся. Миопатические изменения включают проксимальную слабость, мышечную ригидность и боль. Слабость нарастает, быстро развиваются сгибательные контрактуры и деформации суставов. Сухожильные рефлексы снижаются, а затем исчезают. У 60% больных обнаруживаются кальцификаты в подкожной клетчатке, особенно под теми областями кожи, где нарушена пигментация. Множественные кальцификаты создают эффект «брони» при рентгенографии. У некоторых детей ведущим начальным симптомом является мышечная ригидность, а кожные и миопатические симптомы выражены не столь ярко. Инфаркты желудочно-кишечного тракта в терминальных стадиях болезни в прошлом приводили к смерти. Летальность при дерматомиозите в настоящее время снизилась и составляет менее 5%, что связано с совершенствованием методов лечения. Более чем у 30% взрослых с дерматомиозитом в дальнейшем выявляются злокачественные новообразования.
Диагностика. Сочетание лихорадки, сыпи, миалгии и слабости свидетельствует в пользу диагноза дерматомиозита. В начале болезни уровень КФК обычно повышен. Во время активного дерматомиозита ЭМГ покоя выявляет фибрилляции и положительные острые волны; при мышечном напряжении регистрируются укороченные низкоамплитудные полифазные потенциалы. Мышечная биопсия обнаруживает атрофию миофибрилл. Капиллярные некрозы вначале возникают по периферии мышечного пучка и вызывают ишемию прилежащих миофибрилл. Наиболее выражена атрофия в пучках, которые соприкасаются с большими фасциальными футлярами. Волокна I и II типов (тонические и фазические) поражаются в равной степени.
Лечение. Воспалительный процесс активен в течение 2 лет. Кортикостероиды снижают его активность, способствуя уменьшению симптоматики. Наилучшие результаты достигаются, когда кортикостероиды назначают на ранних стадиях болезни, в высоких дозах и применяют длительно. Препаратом выбора служит преднизолон. Начальная его доза дается из расчета 2 мг/кг в сутки, но не выше 100 мг/сут. Температура тела часто нормализуется в течение первых 48 ч от момента начала терапии. Иногда уровень КФК возвращается
к норме на 2-й нед лечения параллельно с заметным увеличением силы мышечного сокращения. В этом случае дальнейший прием преднизолона может осуществляться по схеме через день и в той дозе, которая позволит снизить тяжесть побочных эффектов стероидной терапии. Терапия преднизолоном одинаково эффективна при приеме препарата ежедневно или по схеме через день, но только в тех случаях, когда лечение не прерывается. Когда сила мышц нарастает, начальную дозу принимаемого через день преднизолона можно снижать на 10% в месяц в течение 5 мес. Дальнейшее уменьшение дозы преднизолона допустимо лишь на 5% в месяц. При решении вопроса о снижении дозы кортикостероидов недопустимо ориентироваться лишь на снижение активности КФК, поскольку заметное увеличение мышечной силы наступает только через 1-2 мес после уменьшения уровня фермента, т.е. ведущим критерием снижения дозы кортикостероидов служит позитивная клиническая динамика. У большинства больных поддерживающая доза преднизолона, принимаемого по схеме через день, которая необходима для нормализации силы мышечного сокращения и концентрации КФК, составляет 25% стартовой дозы.
При лечении преднизолоном у некоторых больных сыпь полностью исчезает, но у большинства остаются рубцовые изменения кожи. Длительная стероидная терапия требует контроля функции желудочно-кишечного тракта. Для защиты слизистой оболочки желудка назначают препараты хлорида калия и блокаторов Н2-рецепторов. Серьезным осложнением длительной терапии является развитие стероидной миопатии, которая может быть расценена как обострение основного заболевания. Отличить развивающуюся стероидную миопатию от обострения дерматомиозита по клиническим критериям довольно сложно. При стероидной миопатии, как правило, страдают проксимальные отделы конечностей, развиваются выраженные атрофии, активность КФК не увеличивается. У большинства детей с дерматомиозитом при лечении уже через 3 мес отмечается улучшение, но терапию преднизолоном необходимо продолжать в течение 2 лет. Если лечение прервано преждевременно, неизбежны рецидивы, развиваются кальциноз и контрактуры. Медикаментозное лечение дополняют физической реабилитацией, необходима дыхательная гимнастика. Массаж в активной фазе противопоказан. При правильном лечении благоприятный исход наблюдается у 80% детей с дермато- миозитом. При резистентности или непереносимости преднизолона
показано пероральное назначение цитостатиков: метотрексат в дозе от 10 до 20 мг/м2 поверхности тела 2 раза в неделю или азатиоприн в дозе 50-150 мг/сут. Во время терапии необходим регулярный контроль за функцией печени и клеточным составом крови. Комбинация кортикостероидов и цитостатиков позволяет избежать длительной терапии преднизолоном в высоких дозах. В случаях, когда прием кортикостероидов ограничен их побочным действием, применяют плазмаферез или курс внутривенных инфузий иммуноглобулина. В неактивной стадии обострений обычно не возникает.
Полимиозит. Этиология в большинстве случаев остается неиз- вестной. Предполагается, что в патогенезе играют роль клеточные и гуморальные механизмы, что подтверждается нередким развитием заболевания на фоне аутоиммунных процессов (системная красная волчанка, узелковый периартериит, ревматоидный артрит, склеродермия), а также хорошим эффектом применения кортикостероидов и иммуносупрессоров. Патогенез связан с клеточно-опосредованной цитотоксической реакцией, реализуемой Т-лимфоцитами, сенсибилизированными к поверхностным антигенам мышечных волокон.
Клиническая картина. Полимиозит обычно возникает в зрелом возрасте (45-55 лет), у детей и подростков встречается редко и не связан со злокачественными новообразованиями. Постепенно, исподволь нарастает симметричная проксимальная мышечная слабость, лихорадка и миалгии нетипичны. Часто развивается слабость сгибателей шеи («свисающая голова»). Для заболевания характерны дисфагия и приступы удушья. Постепенно слабость распространяется и на дистальные отделы конечностей. Выраженность парезов варьирует, а в тяжелых случаях развивается тетраплегия. Изредка слабость ограничивается дистальными группами мышц, мышцами глаза или лица. У больного могут наблюдаться периоды стабилизации и даже ремиссии, что может приводить к ошибочной диагностике конечностно- поясной миодистрофии. При хроническом течении болезни постепенно нарастают мышечные атрофии; возможно формирование контрактур. Сухожильные рефлексы вызываются на ранних стадиях болезни и снижаются при уменьшении мышечной массы, но полностью никогда не исчезают. Этот важнейший дифференциально- диагностический признак позволяет исключить полиневропатию. Иногда заболевание начинается остро с общего недомогания; резкая мышечная слабость развивается в течение нескольких дней, появляются боли в мышцах плечевого пояса. Атрофии мышц очень легкие
или отсутствуют. Часто при рентгенографии в мышцах обнаруживают кальцификаты. У взрослых типичны сердечно-легочные осложнения, нехарактерные для детской формы болезни.
Диагностика. Изменения КФК редки. ЭМГ-исследование практически всегда выявляет типичные признаки и миопатического, и неврогенного процессов. Мышечная биопсия обнаруживает различные патологические отклонения. Гистологически периваскулярная воспалительная инфильтрация наблюдается не всегда, поэтому отсутствие клеточных инфильтратов в образцах биоптатов не исключает диагноз полимиозита.
Для лечения полимиозита используется такая же схема, как при дерматомиозите. Больным, которые резистентны к кортикостерои- дам, показаны цитостатики (метотрексат). Плазмаферез и внутривенные введения иммуноглобулина являются оправданными альтернативными методами лечения при недостаточной эффективности общепринятой терапии.
Острый инфекционный миозит возникает после перенесенного гриппа или другой респираторно-вирусной инфекции. Симптомы вирусной инфекции сохраняются от 1 до 7 дней, а затем появляется интенсивная симметричная боль и слабость в мышцах. В тяжелых случаях за 1 сут больной становится обездвиженным. На фоне общей слабости проксимальные мышечные группы поражены тяжелее, чем дистальные. Болезненна пальпация мышц. Сухожильные рефлексы сохраняются. Уровень КФК обычно более чем в 10 раз превышает верхнюю границу нормы. Почти немедленно вслед за развитием миозита наблюдается его спонтанное обратное развитие. В худшем варианте для исчезновения болевого синдрома требуется от 2 до 7 дней постельного режима, после чего больной полностью выздоравливает.
Миотония. Феномен миотонии представляет собой замедленную реакцию расслабления (релаксации) мышцы после ее сокращения. Выделяют миотонию действия, перкуссионную или механическую миотонию и электромиографическую миотонию.
В патогенезе миотонии играет роль нестабильность мембраны мышечного волокна, что приводит к повторным сокращениям мышцы в ответ на одиночный раздражитель. Повторные миотонические импульсы возникают не спонтанно, а всегда при внешнем воздействии или в результате произвольного сокращения. Миотонию действия можно наблюдать у больного после интенсивного сокращения мышцы. Больного просят, например, сильно сжать кисть в
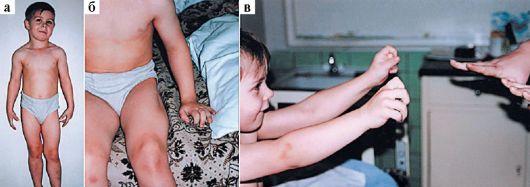
Рис. 6.14. Миотонические феномены у ребенка с миотонией Томсена:
а - псевдогипертрофия мыщц; б - мышечный валик при миатонической
реакции; в - невозможность расслабить кисти при повторных движениях
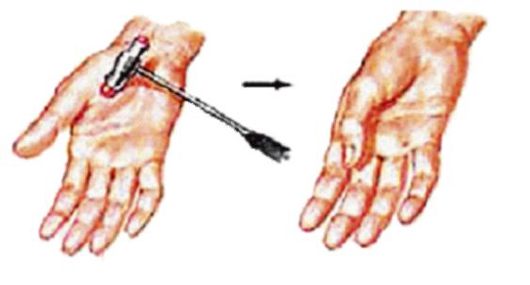
Рис. 6.15. Миотонические феномены у ребенка с миотонией Томсена
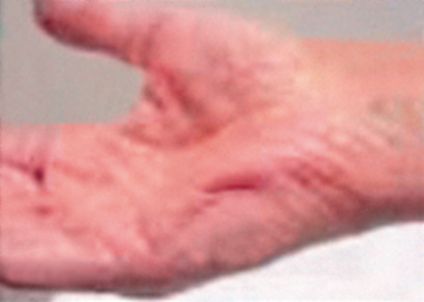
Рис. 6.16. Миотонические феномены у ребенка с миотонией Томсена
кулак и затем быстро его разжать (рис. 6.14-6.16). При этом возникает определенная временная задержка, прежде чем кисть полностью раскроется. При повторном выполнении такого же задания миотонический феномен с каждым разом уменьшается и в конечном итоге исчезает. При врожденной парамиотонии наблюдается обратное явление - нарастание миотонии при повторных движениях (парадоксальная миотония). Перкуссионная миотония проявляется мышечным сокращением после механической стимуляции (быстрый и энергичный удар молоточка по мышце). Этот феномен может наблюдаться в любой мышце, однако наиболее впечатляюще он выглядит при ударе по мышцам тенара: возникает быстрое сгибание и приведение к ладони большого пальца, которое продолжается несколько секунд. При перкуссии крупных мышц возникают симптомы «валика» и «ровика»; при поперечной перкуссии языка образуется «перетяжка» или «ямка» языка. Электромиографическая миотония регистрируется при введении в мышцу игольчатого
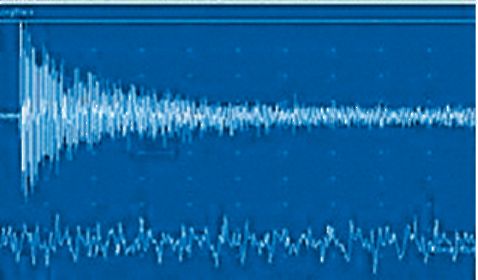
Рис. 6.17. ЭМГ при миотонии, «гул пикирующего бомбардировщика»
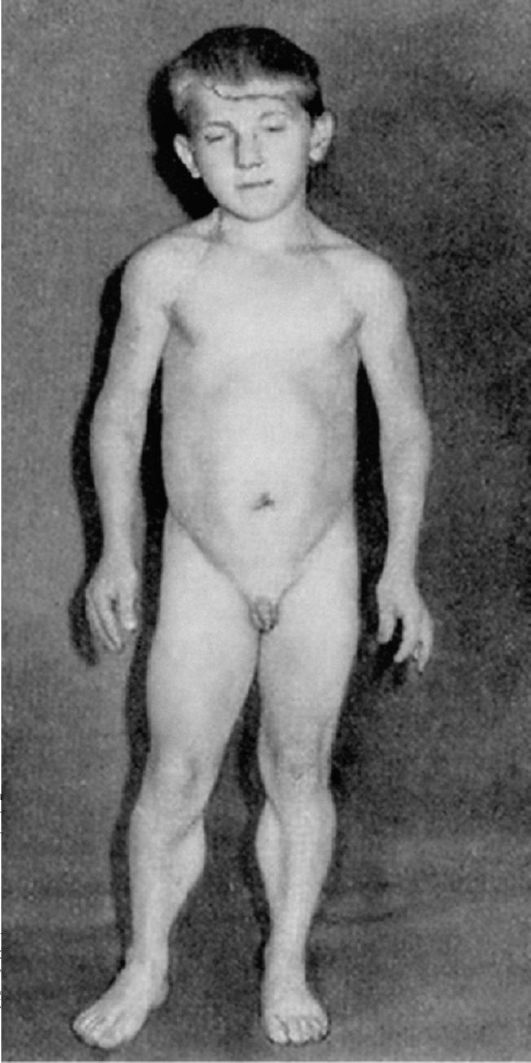
Рис. 6.18. Миотония Томсена у ребенка. «Геркулесовы мускулы»
электрода. Активное напряжение мышцы или ее перкуссия вызывает появление высокочастотных повторяющихся разрядов, которые вначале увеличиваются по частоте (от 100 до 150 Гц) и амплитуде, а затем уменьшаются. Общая продолжительность таких разрядов около 500 мс, а звуковой эквивалент напоминает гул пикирующего бомбардировщика (рис. 6.17).
Феномен миотонии является важнейшим симптомов нескольких гетерогенных наследственных заболеваний (рис. 6.18, 6.19).
Миотоническая дистрофия, или болезнь Россолимо-Куршмана- Штейнерта-Баттена, является мультисистемным заболеванием, которое наследуется по аутосомнодоминантному типу с вариабельной пенетрантностью патологического гена. Этиология болезни связана с нестабильностью участка ДНК хромосомы 19, что выражается в патологической его амплификации (повторяемости). В результате увеличивается число копий этого гена от 50 до нескольких тысяч. Миотоническая дистрофия с полным правом может быть отнесена к классу так называемых болезней экспансии нуклеотидных триплетов. Число повторов возрастает в последующих поколениях и коррелирует с более тяжелым течением заболевания (феномен антиципации). Количество повторов у ребенка
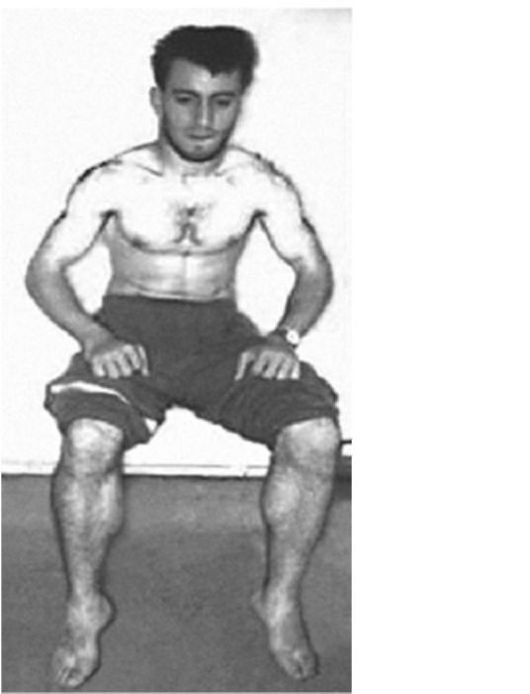
Рис. 6.19. Миотония Томсена у взрослого больного

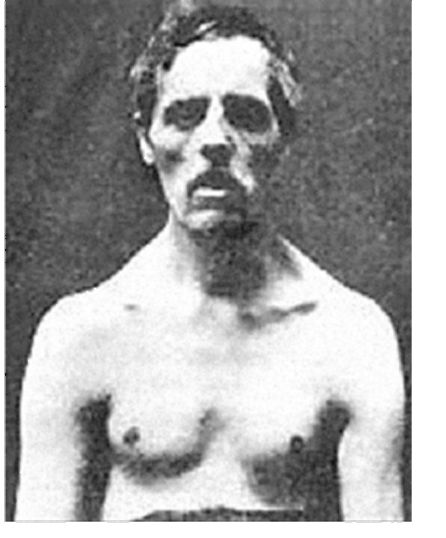
Рис. 6.20. Миотония Россолимо- Куршмана-Штейнерта-Баттена. Типичный вид больного
при наследовании болезни от матери возрастает в значительно большей степени, чем при наследовании от отца. У матери с 100 тринуклеотидными повторами риск рождения ребенка с 400 повторами выше 90%.
Болезнь представляет собой самый распространенный вид мышечной дистрофии, дебютирующей у взрослых. Встречаемость заболевания - 3-5 случаев на 100 000 населения. Оба пола поражаются с одинаковой частотой. Первые симптомы обычно появляются у подростков. В развернутых стадиях отмечаются миотония, слабость лицевой мускулатуры и дистальных отделов конечностей, катаракты, лобное облысение, множественная эндокринопатия. Атрофии мимических мышц настолько стереотипны по виду, что все больные выглядят похожими: лицо удлиненное и утонченное вследствие слабости височных и жевательных мышц; шея тонкая («лебединая») из-за атрофии грудиноключично-сосцевидных мышц; веки и углы рта опущены, нижняя половина лица провисает, что делает выражение лица печальным. Атрофии конечностей наиболее выражены в дистальных отделах: предплечьях и перонеальных мышцах (рис. 6.20, 6.21). Отмечается дисфагия, обусловленная поражением мышц глотки и гладких мышц пищевода. Сухожильные рефлексы снижаются и исчезают.
На поздних стадиях заболевания развиваются атрофии мелких мышц кистей. Больные жалуются на напряжение мускулатуры, затруднения при движениях из-за скованности. Миотония нарастает при охлаждении. В целом миотонические феномены не столь выражены, как при врожденной миотонии. Врач может выявить миотонический синдром при расспросе и подтвердить при осмотре. Например, при рукопожатии пациенту с миотонической дистрофией не удается сразу разжать кисть. Экстраневральные симптомы мио- тонической дистрофии - катаракта, лобная алопеция или эндокринные расстройства - возникают еще до клинически значимых симптомов миотонии. Часто регистрируются изменения на ЭКГ. В поздних стадиях может развиваться тяжелая кардиомиопатия с поперечной блокадой, приступами Адамса-Стокса-Морганьи и сердечной недостаточностью. Нарушается перистальтика кишечника, развивается мегаколон. Парез диафрагмы и межреберных мышц приводит к гиповентиляции и рецидивирующим бронхолегочным инфекциям. Эндокринные нарушения включают тестикулярную атрофию, бесплодие у женщин, гиперинсулинизм, сахарный диабет, атрофию надпочечников и нарушение секреции гормона роста. Нередко развиваются гиперсомния и обструктивные апноэ во сне, психические нарушения вплоть до выраженной деменции.
Диагностика базируется на характерных клинических проявлениях и семейном анамнезе. ЭМГ выявляет миотонические феномены, миопатические потенциалы и небольшие признаки денервации. Активность КФК чаще всего соответствует норме. Необходимости в мышечной биопсии для подтверждения диагноза нет. ДНК-анализ обнаруживает увеличение числа тринуклеотидных повторов; он может быть использован для выявления асимптомных пациентов и проведения пренатальной диагностики.
Лечение. Симптомы миотонии ослабевают при назначении препара- тов - мембраностабилизаторов: хинидина, новокаинамида, фенитоина
Рис. 6.21. Миотония Россолимо- Куршмана-Штейнерта-Баттена. «Ле- бединая» шея из-за атрофии грудиноключично-сосцевидной мышцы. Атрофия мышц разгибателей предплечий, перонеальных групп мышц, что приводит к появлению петушиной походки
(дифенина) и карбамазепина (финлепсина). Необходимо учитывать, что миотония сама по себе не инвалидизирует больного и не требует постоянной лекарственной терапии. К сожалению, лечение нарастающей мышечной слабости пока неэффективно. Больные часто относятся к лечению негативно; плохо переносят наркоз, который может осложниться развитием злокачественной гипертермии.
Врожденная миотоническая дистрофия. У матери с миотонической дистрофией вероятность рождения ребенка с врожденной формой болезни составляет 1:4, а если болен отец - 1:12. Основные признаки патологии внутриутробного периода при врожденной форме - снижение двигательной активности плода и многоводие. Преждевременно рождаются 50% детей. Роды могут быть затяжными из-за неадекватного сокращения матки, и часто возникает необходимость наложения акушерских щипцов. У некоторых новорожденных столь грубо страдает функция диафрагмы и межреберных мышц, что они вообще не способны к самостоятельному дыханию. В отсутствие немедленной интубации и ИВЛ многие из них сразу умирают. Наиболее заметные клинические симптомы у новорожденных: лицевая диплегия, при которой рот необычно заострен, а форма верхней губы напоминает перевернутую латинскую букву «V»; генерализованная мышечная гипотония; деформация суставов, варьирующая от двусторонней косолапости до распространенного артрогрипоза; дисфункция желудочно-кишечного тракта в виде пареза мышц желудка, нарушения глотания и аспирации. Слабость наиболее выражена в прок- симальных отделах конечностей. Сухожильные рефлексы отсутствуют. Миотонические феномены не вызываются перкуссией мышц и могут не определяться при ЭМГ. Неонатальная смертность достигает 16% и часто обусловлена кардиомиопатией. У выживших детей мышечная сила, как правило, нарастает, и процессы кормления и дыхания нормализуются в течение 1-го мес жизни.
Отдаленный прогноз неблагоприятен: у всех детей обнаруживаются задержка психического развития и выраженные клинические симптомы миотонической дистрофии. Диагностика требует постановки диагноза миотонической дистрофии у матери, у которой обычно находят множественные клинические признаки заболевания и миотонические ЭМГ-феномены.
Диагноз у матери и ребенка может быть уточнен после проведения амплификации участка ДНК хромосомы 19. Члены семьи входят в группу риска и в дальнейшем проходят генетическое тестирование для установления носительства.
Неотложная помощь новорожденному заключается в немедленной интубации и ИВЛ. Функция желудочно-кишечного тракта нормали- зуется при назначении церукала (метоклопрамида). Тугоподвижность суставов уменьшается при использовании физических методов терапии и иммобилизации.
Врожденная миотония - наследственное заболевание, характеризующееся скованностью и истинной гипертрофией мышц. В 19% семей прослеживается аутосомно-доминантное наследование (болезнь Томсена), реже - аутосомно-рецессивное наследование (болезнь Беккера). Большинство случаев носит спорадический характер. В целом у больных с аутосомно-рецессивной формой заболевание начинается позже и протекает с более тяжелыми миотоническими расстройствами, чем с аутосомно-доминантной. Однако симптомы обеих форм одинаковы, поэтому невозможно сделать вывод о типе наследования исключительно по клиническим критериям (см. рис. 6.18, 6.19).
Патологический ген и доминантной, и рецессивной форм врожденной миотонии картирован на длинном плече хромосомы 7, где расположен ген каналов ионов хлора.
Аутосомно-доминантная форма обычно дебютирует в грудном возрасте с изменения голоса при плаче; ребенок начинает задыхаться, а после плача лицо очень медленно расслабляется. Заболевание протекает легко. В зрелом возрасте миотония может приводить к генерализованной мышечной гипертрофии (атлетизму), но и в детстве мышцы имеют вид «геркулесовых мускулов». Иногда вовлекаются мышцы языка, лица и жевательные мышцы. Скованность мышц не сопровождается болью; она нарастает при пребывании больного на холоде. Выявляются перкуссионные миотонические симптомы. Мышечная масса, сила сокращений и сухожильные рефлексы - в норме. Сразу после отдыха мышцы остаются скованными, а движения - затрудненными. Однако после активизации скованность исчезает, восстанавливается обычный объем движений.
Диагностика. Диагноз подтверждается с помощью ЭМГ-исследования. Частота повторных мышечных осцилляций варьирует от 20 до 80 циклов в секунду от момента первоначального введения иглы в мышцу и до начала произвольного сокращения. Амплитуда и частота потенциалов прибывают и убывают, что сопровождается характерным звуком - «гулом пикирующего бомбардировщика». Признаки мышечной дистрофии отсутствуют. Уровень КФК в норме. В образцах мышечных биоптатов обнаруживается гипертрофия мышечных волокон.
Лечение. Миотония не всегда требует лечения, а лекарства недо- статочно эффективны. Иногда можно уменьшить скованность при назначении фенитоина (дифенина) или препаратов карбамазепина (финлепсина), которые дают в средних противосудорожных дозах. Новокаинамид назначают в начальной дозе 200 мг 2 раза в день, а затем ее постепенно повышают до 400 мг 3 раза в день. Препарат уменьшает мышечную скованность у детей с рецессивной формой болезни. Для некоторых больных эффективен диакарб (ацетазоламид). В тяжелых случаях показан короткий курс кортикостероидов. Полезны антагонисты кальция (нифедипин по 10-20 мг 3 раза в день), а также дизопирамид по 100-200 мг 3 раза в день. Необходимо учитывать, что сукцинилхолин, верошпирон, калий, антигиперлипидемические средства и β-адреноблокаторы способны усиливать миотонический синдром.
Ремиттирующая миотония (миотония, усиливающаяся при избытке калия) - аутосомно-доминантный синдром, связанный с мутацией гена натриевых каналов. Ген картирован на хромосоме 17. Клинические проявления схожи с врожденной миотонией. Дебют мышечной скованности обычно наблюдается после 10 лет и может быть спровоцирован общей анестезией. Миотонические феномены генерализованы, вовлекают туловище, конечности, глазодвигательные мышцы. Тяжесть миотонии день ото дня варьирует и уменьшается при согревании. Ухудшение состояния может наблюдаться после интенсивной физической нагрузки или приема большого количества калия с пищей.
Диагностика. ЭМГ-исследование выявляет миотонические феномены. В мышечных биоптатах патологии нет. Возможен ДНК-анализ мутантного гена, кодирующего α-субъединицу натриевого канала.
Лечение. Скованность при ремиттирующей миотонии может предот- вратить мексилетин - препарат, сходный по структуре с лидокаином; как и при других каналопатиях, может быть эффективен диакарб (ацетазоламид).
6.6. Периодические параличи
Периодические параличи, или пароксизмальная миоплегия, - объединяющий термин для группы каналопатий, редких наслед- ственных заболеваний, которые характеризуются приступами вялого паралича скелетных мышц вследствие патологии ионных каналов. Параличи подразделяют в зависимости от уровня калия в крови: гиперкалиемический (болезнь Гамсторп), гипокалиемический и нормокалиемический. Кроме того, периодический паралич может
быть первичным (генетически детерминированным) или вторичным. Вторичный гипокалиемический периодический паралич обусловлен потерей калия с мочой или его избыточным выведением из желудочнокишечного тракта. «Мочевые» потери калия связаны с первичным гиперальдостеронизмом, интоксикацией солодкой (лакричником), терапией амфотерицином Б и некоторыми почечными тубулярными дефектами. «Желудочно-кишечные» потери калия наиболее часто наблюдаются при тяжелой хронической диарее, длительном зондовом кормлении и гастрофистуле. Калий теряется у подростков с нервной анорексией, которые злоупотребляют диуретиками или искусственно вызывают у себя рвоту, чтобы «похудеть». Гипокалиемический периодический паралич осложняет тиреотоксикоз. Вторичный гиперкалиемический периодический паралич может быть обусловлен почечной или надпочечниковой недостаточностью.
Семейный гиперкалиемический паралич наследуется по аутосомнодоминантному типу с высокой пенетрантностью. Мутация расположена в гене натриевых каналов.
Клиническая картина. Дебют приступов мышечной слабости относится к раннему детскому и даже грудному возрасту. Приступы слабости возникают после интенсивной физической нагрузки. Перед приступом бывают чувствительные нарушения - парестезии в области лица, верхних и нижних конечностей, ощущение тяжести в спине. Изредка больной может замедлить развитие паралича ходьбой или переходом с места на место. У детей грудного и раннего возраста приступы выражаются внезапной потерей мышечного тонуса: они падают и не могут двигаться. У детей старшего возраста и взрослых могут наблюдаться как приступы средней тяжести (продолжаются менее часа и не приводят к глубокому параличу), так и тяжелые приступы (до нескольких часов). После нескольких тяжелых атак может оставаться некоторая остаточная мышечная слабость. Симптомы миотонии у больных с гиперкалиемическим параличом выражены умеренно и могут усиливаться при охлаждении. Характерна миотония век, языка, мышц предплечья и большого пальца.
Диагностика. Во время приступа содержание калия в крови обычно превышает 5 ммоль/л. Пероральный прием хлорида калия сразу после физической нагрузки немедленно провоцирует приступ слабости, на протяжении которого мышцы не реагируют на электрические стимулы.
Лечение. Острые приступы редко требуют лечения, поскольку они кратковременны. При развернутом приступе может помочь внутривен-
ное вливание 40% раствора глюкозы (до 40 мл) или 10% раствора глюконата кальция (до 20 мл). Ежедневный прием диакарба (ацетазоламида) предотвращает повторные приступы, механизм превентивного действия этого препарата при гиперкалиемическом и гипокалиемическом параличе неизвестен. Следует избегать приема пищи, богатой калием, увеличить в дневном рационе количество углеводов и поваренной соли.
Семейный гипокалиемический паралич наследуется по аутосомнодоминантному типу. Пенетрантность гена у женщин снижена. Мутация находится на длинном плече хромосомы 7, в гене кальциевых каналов. У 60% больных симптомы возникают до 16 лет, у остальных - до 20 лет жизни. Вначале приступы слабости нечастые, но затем бывают до нескольких раз в неделю. Приступы провоцируют: отдых после физической нагрузки (часто приступы наблюдаются ранним утром), обильный прием углеводистой пищи, избыток поваренной соли в рационе, эмоциональный стресс, прием алкоголя, переохлаждение; у женщин - менструации. До и во время приступа у больного могут отмечаться жажда и олигурия, болезненные ощущения в проксимальных группах мышц, затем развивается общая слабость. Иногда наблюдается тотальный паралич, при котором больной не в состоянии даже поднять голову. Слабость лицевых мышц бывает редко, движения глаз всегда сохранены. Дыхательная недостаточность не развивается. Большинство приступов длится от 6 до 12 ч, а некоторые - в течение всего дня (так называемый миоплегический статус). Мышечная сила быстро восстанавливается, но после нескольких тяжелых приступов могут отмечаться усталость, похудание, особенно проксимальных отделов конечностей, угнетение сухожильных рефлексов. Типичны вегетативные расстройства: гиперемия кожи, гипергидроз, лабильность пульса и артериального давления. Вне приступов мышечной слабости у больных отсутствуют симптомы нервно-мышечной патологии.
Диагностика. Во время приступа уровень калия в крови может снизиться до 1,5 ммоль/л, чему соответствуют изменения ЭКГ: брадикардия, уплощение зубца Т, увеличение интервалов Р-Q и Q-Т. Мышцы не сокращаются в ответ на электрические стимулы. С диагностической целью приступ можно спровоцировать приемом глюкозы в дозе 2 г/кг и одновременным подкожным введением 10-20 ЕД инсулина: приступ паралича развивается через 2-3 ч.
Лечение. Острые приступы у больных с адекватной функцией почек купируют повторными приемами калия в дозе от 5 до 10 г.
Такая же доза, принимаемая ежедневно, рекомендуется для профилактики их возникновения. У детей младшего возраста доза меньше. Ежедневный прием диакарба (ацетазоламида) оказывает благоприят- ное действие, предупреждая приступы во многих случаях. Он малотоксичен и обычно хорошо переносится даже при длительном применении. Следует снизить калорийность суточного рациона за счет углеводов и уменьшить количество поваренной соли. В то же время показаны продукты, богатые калием: сухофрукты, курага, чернослив, молочные продукты, картофель.
Семейный нормокалиемический паралич. В некоторых семьях наблюдаются случаи аутосомно-доминантного периодического паралича при нормальном уровне калия в крови. Это вариант гиперкалиемического периодического паралича с нарушением притока калия в кровь, когда невозможно оценить его истинное содержание в тканях. Миоплегия длится от нескольких дней до 2-3 нед. Темп нарастания и уменьшения мышечной слабости обычно медленный. Сухожильные рефлексы во время приступов исчезают. У части больных наблюдается гипертрофия отдельных мышечных групп. Приступы провоцируются отдыхом после интенсивной физической нагрузки, приемом алкоголя, охлаждением. Прием хлорида калия может спровоцировать приступ паралича, тогда как употребление 8-10 г поваренной соли ежедневно позволяет их избежать.
6.7. Миастения
Миастения (myastenia gravis) - аутоиммунное нервно-мышечное заболевание, клинически характеризующееся патологической слабостью и утомляемостью произвольной мускулатуры и связанное с повреждением ацетилхолиновых рецепторов (АХ-Р) постсинаптической мембраны поперечнополосатых мышц специфическими ком- плементфиксирующими антителами (АТ).
Распространенность миастении составляет 0,5-5 случаев на 100 000 населения во всех популяциях. Дети и подростки до 17 лет составляют 9-15% числа больных миастенией. Средний возраст начала болезни - 7,2 года. Дебют миастении возможен в любом возрасте. Описаны врожденные формы. Женщины болеют в 3 раза чаще, чем мужчины.
Этиология. Мультифакториальное заболевание, при котором определенное значение имеет наследственная предрасположенность, обусловленная иммунологическим дефектом и связанная с анти-
генами гистосовместимости B8 системы HLA. Причиной миастении могут быть вирусное поражение вилочковой железы, вследствие чего она начинает продуцировать Т-лимфоциты с измененными мембранными структурами; опухоль тимуса; в редких случаях - первичное поражение головного мозга различной этиологии.
Основой патогенеза миастении является аутоиммунная реакция на ацетилхолинэстеразные рецепторы (АХ-Р) скелетных мышц. Уровень антител к АХ-Р в крови больных коррелирует со степенью тяжести болезни. Антитела к АХ-Р блокируют нервно-мышечную проводимость, поскольку разрушают АХ, уменьшают скорость его восстановления, необратимо изменяя рецепторы постсинаптической мембраны.
Патологическая анатомия. Происходят дистрофические изменения терминалей аксонов, синаптических щелей и постсинаптических структур, в них откладываются иммуноглобулины и комплемент. В мышцах наблюдается умеренная дегенеративная атрофия, реже некроз волокон в сочетании с нерезко выраженной лимфоидной инфильтрацией и плазморрагией. У 70-90% больных выявляется патология вилочковой железы (гиперплазия зародышевых фолликулов, лимфоэпителиальные тимомы). В редких случаях отмечаются миокардит, тиреоидит, очаговые скопления лимфоцитов в различных органах.
Клиническая классификация миастении (по Б.М. Гехту).
1. Степень генерализации двигательных расстройств:
1) генерализованная;
2) локальная:
а) глазная,
б) бульбарная,
в) скелетная.
2. Степень тяжести двигательных нарушений:
1) легкая;
2) средняя;
3) тяжелая.
3. Течение миастенического процесса:
1) ремиттирующее (миастенические эпизоды);
2) непрогрессирующее (миастеническое состояние);
3) прогрессирующее;
4) злокачественное.
4. Степень компенсации двигательных расстройств под влиянием антихолинэстеразных препаратов:
1) полная (вплоть до восстановления работоспособности);
2) неполная (восстанавливается способность к самообслуживанию);
3) плохая (больные нуждаются в постороннем уходе). Клиническая картина. Миастения характеризуется патологической
утомляемостью и слабостью поперечнополосатых мышц. Больным трудно подниматься по лестнице, ходить, долго находиться в одной позе, носить тяжести.
Наиболее часто поражаются глазодвигательные, мимические, жевательные мышцы, а также мышцы глотки, гортани, языка. Поражение наружных мышц глаза во время первого осмотра выявляется у 40-50% больных, а по мере развития болезни - у 90-95%. Птоз может быть односторонним, причем возникает то с одной, то с другой стороны. Утром и после отдыха птоз меньше, нарастает при общей или зрительной нагрузке, к вечеру. При осмотре спровоцировать усиление птоза можно, попросив пациента несколько раз зажмуриться или присесть. Глазодвигательные нарушения асимметричны, изменчивы при нагрузке и не соответствуют зонам иннервации глазодвигательных нервов. За счет мышечной слабости возникает нистагм в крайних отведениях. Диплопия усиливается при зрительной и физической нагрузке, ярком свете, во второй половине дня (особенно при просмотре телевизора), более выражена при взгляде вдаль, уменьшается после отдыха с закрытыми глазами и по утрам (рис. 6.22).
Слабость жевательных и височных мышц приводит к утомлению при жевании иногда до отвисания нижней челюсти, больные во время еды поддерживают челюсть и помогают себе при жевании рукой. Важным сим- птомом является слабость мышц лица. Она более выражена в верхней половине лица (в круговых мышцах глаз), усиливается при повторном зажмуривании и общей физической нагрузке. Пациенту трудно надуть щеки, возникает «поперечная» улыбка вследствие слабости круговой мышцы рта. Также отмечается слабость жевательных и височных мышц.
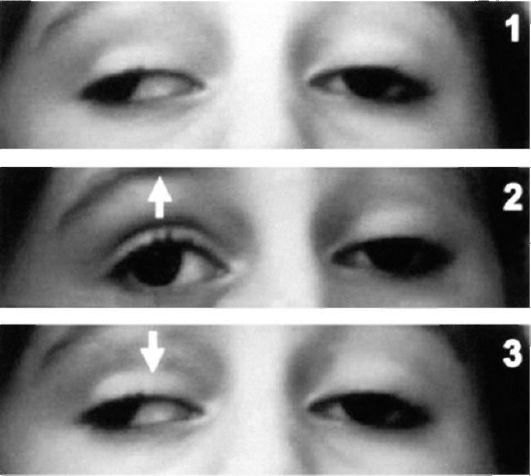
Рис. 6.22. Слабость глазных мышц при миастении
Поражение бульбарных мышц (мягкого нёба, глотки и верхних мышц пищевода), приводящее к дисфагии и дизартрии, развивает- ся у 40% больных. Оно усиливается при речевой, общей физической нагрузке, во время еды и уменьшается после отдыха. Нарушается глотание (пациент поперхивается при еде, жидкая пища попадает в носовые ходы). Речь становится гнусавой, может отмечаться охриплость голоса или нарушения модуляции, похожие на заикание. При тяжелой дизартрии больной не может глотать и говорить.
Слабость мышц шеи и туловища больше характерна для больных пожилого возраста. Слабость мышц спины проявляется нарушением осанки. За счет слабости задней группы мышц шеи возникает затруднение при подъеме головы в положении на спине или при разгибании шеи в вертикальном положении. Если миастения дебютирует со слабости мышц туловища, в дальнейшем развиваются бульбарные и дыхательные нарушения.
Жалобы на одышку при вдохе обусловлены слабостью диафрагмы или межреберных мышц. Ослабление кашлевого толчка приводит к скоплению густой мокроты, вязкой слюны, которую невозможно сплюнуть или проглотить.
Ослаблены мышцы конечностей, особенно проксимальных, шеи, туловища. При осмотре выявляются мышечные атрофии, снижение мышечного тонуса, лабильность сухожильных и надкостничных рефлексов. Слабость мышц конечностей может быть изолированной (без других симптомов миастении) или сочетаться со слабостью других групп мышц. Типична слабость проксимальных мышц-разгибателей. Наиболее часто поражаются дельтовидная мышца, трехглавая мышца плеча, подвздошная мышца.
Кроме двигательных расстройств, миастения сопровождается различными вегетативными и эндокринными нарушениями (гипо- и гипертиреоз, гипокортицизм и др.). Миастению характеризуют динамичность мышечной слабости в течение суток, усиление ее после нагрузки, обратимость или уменьшение слабости после отдыха. Ухудшение состояния провоцируют физическая нагрузка, отрицательные эмоции, менструация, инфекции, повышение температуры окружающей среды, а улучшают - ночной сон, отдых. Патогномонично уменьшение утомляемости после введения антихолинэстеразных препаратов (АХП).
Течение болезни чаще всего прогрессирующее, с ремиссиями или прогредиентное без ремиссий. При злокачественном течении буль- барные и дыхательные расстройства развиваются в течение первых недель заболевания. Миастения чаще дебютирует после ОРВИ или
стресса, одним симптомом (преходящий птоз, бульбарный парез и др.). Состояние больных с миастенией могут осложнять миастенические кризы или холинергические кризы.
Миастенический криз развивается вследствие декомпенсации миастении или недостаточной дозировки АХП; может провоцироваться бронхолегочной инфекцией. При этом происходит резкое ухудшение состояния с нарушением витальных функций. Дифференцировать миастенический криз от других тяжелых состояний, сопровождающихся респираторными расстройствами, можно по наличию асимметричного наружного офтальмопареза, птоза, бульбарного синдрома, гипомимии, слабости мышц конечностей и шеи, уменьшающихся в ответ на введение АХЭ препаратов (табл. 10).
Холинергический криз развивается при избыточной дозе АХЭ препаратов.
Таблица 10. Дифференциальная диагностика миастенического и холинергического кризов
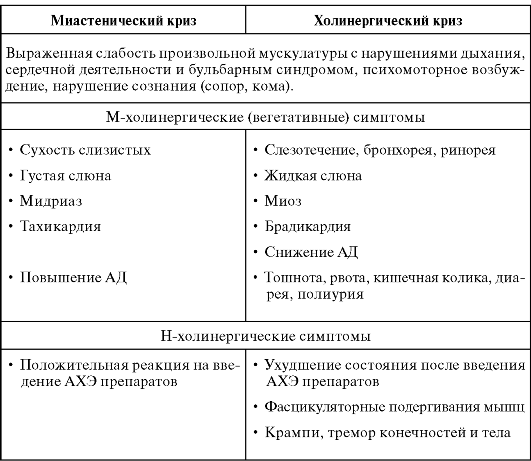
Смешанные (миастенический + холинергический) кризы встречаются у больных миастенией при неправильном приеме и/или изначально узком диапазоне терапевтических доз АХЭП, а также на фоне состояний, вызывающих общую или мышечную слабость различного генеза (интеркуррентные инфекции, соматические, гормональные нарушения, прием препаратов, влияющих на сократительную функцию произвольных мышц, и др.).
Прогноз зависит от клинической формы и проводимого лечения. Возможны практические выздоровления (примерно у 1/3 больных), значительное улучшение, инвалидность, летальные исходы, особенно при тимоме. Основными симптомами, угрожающими жизни больного, являются слабость мышц гортани и дыхательных мышц. Причины смерти при миастении: дыхательная недостаточность, аспирационная пневмония, побочные эффекты кортикостероидов и цитотоксических препаратов.
Диагностика включает сбор анамнеза, клинический осмотр, пробу с АХЭ-препаратами (прозерином, тензилоном, калимином), электромиографию, иммунологическое исследование, исследование вилочковой железы, морфологическое исследование мышечного биоптата, динамическое наблюдение.
Клинический осмотр включает исследование общего неврологического статуса и оценку силы произвольных мышц лица, шеи, туловища и конечностей до и после нагрузки. Мышечная сила оценивается от 0 до 5 баллов, где 0 - отсутствие силы, 5 - нормальная сила с учетом возраста и пола. Также выявляют синдром патологической мышечной утомляемости (нарастание симптомов после нагрузки) при отсутствии симптомов поражения центральной нервной системы.
Диагностические критерии
1. Птоз (односторонний, двусторонний, асимметричный, симметричный): появление или усиление птоза после долгого смотрения вверх или после быстрого многократного открывания или закрывания глаз.
2. Слабость жевательных мышц:
• недостаточное сопротивление насильственному закрытию нижней челюсти;
• пальпация височных мышц при жевании обнаруживает их слабое сокращение;
• больные не в состоянии плотно сомкнуть веки или оказать сопротивление пассивному открыванию глаз;
• больные не могут надуть щеки при надавливании на них.
3. Слабость мышц гортани и нёба выявляется, если:
• нёбо малоподвижно, рвотный рефлекс снижен или отсутствует;
• трудно глотать жидкую пищу.
4. Слабость мышц языка выявляется при надавливании языком на палец врача через щеку.
5. При выраженной слабости мышц шеи «голова свешивается».
6. Прозериновая проба с оценкой силы и утомляемости мышц проводится до подкожного введения 0,05% раствора прозерина в разовой возрастной дозировке и через 30-40 мин после него. Проба считается положительной, если мышечная сила нарастает. Различают:
++++ резкоположительную пробу, когда исчезают все миастенические симптомы;
+++ положительную пробу - остаются только отдельные симптомы;
++ слабоположительную пробу, при которой уменьшается выраженность миастенических симптомов;
+ сомнительную прозериновую пробу - степень выраженности проявлений миастении изменяется незначительно;
- отрицательную прозериновую пробу - клиническая симптоматика не изменяется после введения прозерина.
Подтверждением диагноза миастении считается наличие одного из первых трех вариантов прозериновой пробы.
Проводят ЭМГ наиболее ослабленных мышц для выявления особенностей нарушений нервно-мышечной передачи (мышца, отводящая мизинец, в двубрюшной мышце дна рта). Исследование проводят на фоне отмены АХЭП в течение суток, сразу после физической нагрузки и через 2 мин после нагрузки. Большое значение имеет обратимость ЭМГ-феноменов на фоне АХЭП - нарастание амплитуды М-ответа. При электромиографии отмечается уменьшение амплитуды второго потенциала действия мышц (в норме оба потенциала равны) в ответ на стимуляцию нерва спаренными импульсами с промежутком 0,1-0,7 с. При миастении снижение амплитуды потенциалов при постоянной стимуляции нерва сменяется фазой плато или повышением амплитуды, а при других заболеваниях происходит неуклонное снижение амплитуды ответа. При регистрации активности отдельных мышечных волокон часто выявляются характерные признаки поражения нервно-мышечных синапсов. В 95% случаев на ЭМГ обнаруживают патогномоничные изменения.
Для исключения опухоли или гиперплазии вилочковой железы, которая развивается у 75% больных с миастенией, проводят компьютерную томографию средостения, радионуклидное сканирование.
Иммунологическое исследование выявляет наличие антител к холинорецепторам у 50% больных с глазной формой миастении и у 80-90% больных с генерализованной формой. При тимоме также выявляются антигены к скелетным мышцам.
Иммунологическое исследование (ИФА, РИА) - количественный метод определения антител к АХР в сыворотке крови больных миастенией, который позволяет с вероятностью до 80% подтвердить диагноз.
Дифференциальный диагноз проводят с состояниями, ведущим симптомом которых является мышечная слабость:
• миастенические синдромы (ботулизм, отравление антибиотиками из группы аминогликозидов, болезнь Иценко-Кушинга, болезнь Аддисона, гипо- и гипертиреоз, полиомиозит);
• рассеянный склероз, нейроинфекции (энцефалит, полиневропатии, энцефаломиелополирадикулоневриты): у больных офтальмопарез сопровождается гипорефлексией, атаксией, нарушением чувствительности, изменением ЦСЖ;
• боковый амиотрофический склероз: слабость постоянна, отмечаются атрофия, фасцикуляции, повышение сухожильных рефлексов, симптом Бабинского;
• глазная форма миопатии: характерны птоз и симметричное ограничение движений глазных яблок; легкая слабость мышц глотки, шеи, конечностей и лица;
• митохондриальные миопатии;
• нейроэндокринные синдромы;
• другие заболевания ЦНС (опухоли, сосудистые заболевания головного и спинного мозга): характерны нарушения рефлексов, проводниковые расстройства;
• астеноневротические реакции, синдром хронической усталости и др.
Лечение. Общие принципы:
1. При генерализованной форме больного госпитализируют и ограничивают физическую нагрузку до подбора антихолинес- теразной терапии.
2. Противопоказаны средства, блокирующие нервно-мышечную передачу, а также оказывающие угнетающее действие на ЦНС,
и особенно - на дыхательный центр (хинин, хинидин, пропранолол, лидокаин, аминогликозиды, полимиксин, морфин, барбитураты, транквилизаторы). 3. Задачи лечения зависят от тяжести заболевания. Антихолинестеразные препараты (АХЭП) - препараты выбора при миастении, тормозят разрушение ацетилхолина и способствуют его накоплению в синаптической щели, воздействуя на холинергические синапсы, не проникают через ГЭБ (табл. 11). Побочные эффекты обусловлены одновременным влиянием на вегетативные холинергические синапсы и зависят от дозы и тонуса ВНС. Их можно уменьшить, если принимать ингибиторы АХЭ чаще, но в меньших дозах и во время еды, что замедляет всасывание. В некоторых ситуациях (менструация, инфекции, ремиссия) чувствительность к АХЭП повышается, и их дозу уменьшают. Больных обучают регулировать дозу самостоятельно. Относительными противопоказаниями к применению АХЭП являются бронхиальная астма, тяжелый атеросклероз, ИБС, эпилепсия.
Таблица 11. Антихолинергические препараты
Препараты | Время действия | Области применения |
Прозерин (неостигмин) | Начало действия через 20-40 мин, дли- тельность 2-4 ч | Применяется преимущественно для проведения медикаментозных проб и в острых состояниях |
Калимин 60 Н, калимин-форте (пиридостигми- на бромид) | Начало через 45 мин, действует 4-8 ч Интервал между приемами 5-5,5 ч. | Наиболее широко применяется, хорошо переносится, эффективен при всех формах, в том числе бульбарных. Калимин форте (парентеральный) - при нарушении витальных функций и при стойком бульбарном параличе. При переводе больных на парентеральное введение препаратов учитывается, что 1 таблетка калимина (60 мг) равноценна 1 мл 0,05% раствора прозерина |
Вспомогательная терапия: препараты калия (пролонгируют действие АХЭП); диета, богатая калием (печеный картофель, курага, бананы и др.); калийсберегающие препараты (верошпирон); хлорид калия 3,0 г/сут в растворах, порошках, таблетках с целью предупреждения передозировки АХЭП; препараты кальция; тонизирующие средства (экстракты элеутерококка, родиолы, левзеи, пантокрин); поливитамины, эуфиллин (блокатор фосфодиэстеразы, увеличивающий содержание цАМФ в пресинаптической мембране), анаболики (рибоксин, ретаболил).
Патогенетическая терапия - тимэктомия. Эффективность - 70-90%, возможны ремиссии. Показаниями к оперативному лечению являются:
а) злокачественные формы миастении;
б) прогрессирующая форма миастении;
в) миастеническое состояние в зависимости от степени выраженности дефекта.
Противопоказания к тимэктомии:
а) тяжелые декомпенсированные соматические заболевания;
б) пожилой возраст.
Предоперационная подготовка включает общеукрепляющую терапию, плазмаферез, по показаниям - глюкокортикоиды, лучевую терапию (противопоказана детям и подросткам).
Глюкокортикоиды (преднизолон, дексаметазон) показаны при неэф- фективности других методов. Их назначают ежедневно или через день, по 60-150 мг/сут (1-1,5 мг/кг/сут) утром, сразу после завтрака, через день; при выраженном обострении ежедневно (до компенсации витальных нарушений), через 5-7 дней (до терапевтического эффекта) переходят на схему через день. Поддерживающая доза - через день 20-30 мг в сутки, принимается в течение нескольких месяцев. Примерно у 75% больных кортикостероидная терапия приводит к существенному улучшению. После стабильного улучшения дозу кортикостероидов медленно (в течение нескольких месяцев) снижают до поддерживающей (5-15 мг ежедневно или 10-30 мг через день). Иногда удается полностью отменить кортикостероиды. Чтобы избежать первоначального ухудшения, лечение можно начинать с низких доз (25 мг преднизолона через день) с постепенным увеличением дозы на 12,5 мг в каждый третий прием, пока суточная доза не достигнет 100 мг или не будет получен хороший эффект. Улучшение отмечается спустя 6-7 нед лечения. Дозу в этих случаях начинают снижать не раньше чем через 3 мес после первого приема.
Плазмаферез назначают при обострениях, миастенических кризах, предоперационной подготовке, неэффективности кортикостероидной терапии. Проводится 3-5 сеансов через день, затем 2-3 раза в неделю. Плазмаферез проводят с заменой плазмы или использованием белков-заменителей. Гемосорбция и энтеросорбция проводятся у больных с генерализованной формой миастении с целью выведения антител, а при смешанных кризах и неэффективности массивной медикаментозной терапии - с целью детоксикации.
Цитостатики (азатиоприн, циклофосфан и циклоспорин) назначают под контролем анализов крови. Препараты иммуноглобулина G (в/в 0,4 г/кг/сут ежедневно в течение 5 дней; или 3-5 г на курс) эффективны при интеркуррентных инфекциях, во время миастенического или смешанного криза.
Лечение кризов направлено на компенсацию витальных нарушений, купирование обострения и устранение метаболических нарушений. При лечении миастенического криза вводят парентерально АХЭП (калимин-форте по 1-1,5 мл в/в или в/м каждые 4-5 ч или прозерин по 1,5-2 мл каждые 3 ч). ИВЛ с полной отменой АХЭП, назначение иммуносупрессивной терапии на фоне антибактериальных пре- паратов проводят с целью профилактики интеркуррентных инфекций. Отключение от аппарата проводят только после 30 мин самостоятельного дыхания, при компенсации дыхательных расстройств и на фоне калимина-форте на 5-6 ч. Большие дозы глюкокортикоидов назначают по альтернирующей схеме (пульс-терапия - 1000-2000 мг в/в капельно через день) с последующим переводом на пероральный прим. Также стабилизируют сердечно-легочную деятельность. Проводят плазмафе- рез, внутривенные инфузии нормального человеческого иммуноглобулина. Холинергический криз купируют атропином, реактиваторами холинэстеразы (дипироксим); применяют дезинтоксикацию.