Детская неврология : учебник : в двух томах / А. С. Петрухин. - Т. 2. - 560 с. : ил.
|
|
|
|
ГЛАВА 3. ФАКОМАТОЗЫ
Факоматозы (от греч. phakos - пятно) - это гетерогенная группа наследственных нейрокожных заболеваний, отличительной чертой которых является поражение производных эктодермы - кожи и ее дериватов, нервной системы, сетчатки, висцеральных органов. К нейрокожным синдромам относят более 30 заболеваний. Из них наиболее известны и хорошо изучены нейрофиброматоз Реклингхаузена, туберозный склероз, энцефалотригеминальный ангиоматоз Штурге- Вебера, ретиноцеребеллярный ангиоматоз Гиппеля-Линдау. К более редким относятся гипомеланоз Ито, атаксия-телеангиэктазия, болезнь базально-клеточного невуса, синдром недержания пигмента, прогрессирующая лицевая гемиатрофия (синдром Пари-Ромберга) и др.
Трудности диагностики факоматозов связаны с выраженным клиническим полиморфизмом и возраст-зависимым дебютом симптомов. Пациенты с факоматозами в течение жизни наблюдаются врачами практически всех специальностей, поэтому только информированность специалистов о характере и особенностях течения данных заболеваний, а также согласованность их диагностических и лечебных мероприятий могут обеспечить выбор правильной тактики ведения больных.
3.1. Туберозный склероз
Туберозный склероз (болезнь Бурневилля-Прингла) - наслед- ственное заболевание из группы факоматозов, характеризующееся системным поражением нервной системы, кожи, внутренних органов, органов зрения, костной и эндокринной системы и связанное с нарушением пролиферации, миграции и дифференциации клеток нейроглии.
Изменения в головном мозге наиболее наглядно демонстрируют дизонтогенетическую теорию развития этого заболевания. Считается, что уже на 13-17-й нед внутриутробного развития образуются аномальные гигантские нейроглиальные клетки, которые в процессе своей миграции могут останавливаться в нетипичных местах. Расположенные субэпендимально, они образуют субэпендимальные узлы; в белом веществе полушарий - нейрональные гетеротопии; субкортикальнокортикальные и субкортикальные туберсы (рис. 3.1). Мутантные гены в первые недели гестации вызывают нарушение функций в клетках герминативного матрикса, в результате этого образуются гигантские нейроглиальные клетки.
Туберозный склероз - генетически гетерогенное состояние, с локусом на 2 хромосомах. TSCl-ген 9q34.3 (участок 34 длинного плеча 9 хромосомы) кодирует белок гамартин. TSC2 - ген 16pl3.3 (участок 13 короткого плеча 16 хромосомы) кодирует белок туберин. На каждый локус приходится примерно 50% семейных случаев. Белки гамартин и туберин являются супрессорами генов опухоли. Тип наследования - аутосомно-доминантный. В 60-70% заболевание возникает спорадически, в результате спонтанных мутаций. Его отличают вариабельная экспрессивность, 100% пенетрантность. Частота встречаемости туберозного склероза в среднем составляет 1:4700 новорожденных.
В мозге находят три разновидности повреждений: кортикальные туберсы, субэпендимальные узлы и нарушение миелинизации. Туберсы могут располагаться в извилинах любой доли мозговых полушарий, реже в мозжечке, стволе или спинном мозге. При гистологическом исследовании они представляют собой зоны склероза, которые состоят из атипичных гигантских астроцитов и крупных вакуолизированных «клеток-монстров». В половине случаев внутри глии депонируется кальций. Субэпендимальные узлы располагаются в стенках желудочков. Фактически они являются доброкачественными новообразованиями, производными стенок боковых желудочков или передней части III желудочка; гистологически - гигантоклеточными астроцитомами. Часто с возрастом происходит кальцификация этих узелков, что и вызывает окклюзионную гидроцефалию. В белом веществе встречаются линейные гетеротопии, повторяющие след нормальной миграционной дорожки примитивных нейронов от перивентрикулярного герминативного слоя к поверхности коры. Субэпендимальные гигантоклеточные опухоли появляются у детей 5-10 лет. Характерными особенностями данных образований являются их тенденция к медленному росту, расположение в области отверстия Монро и большие размеры. Размеры опухоли и прогрессирующий рост отличают ее от субэпендимальных узлов. Опухоль часто содержит кровеносные сосуды, чем объясняется возможность кровоизлияний, частично
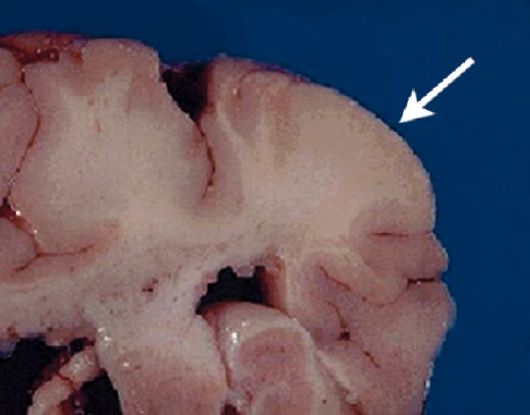
Рис. 3.1. Туберс головного мозга (стрелка)
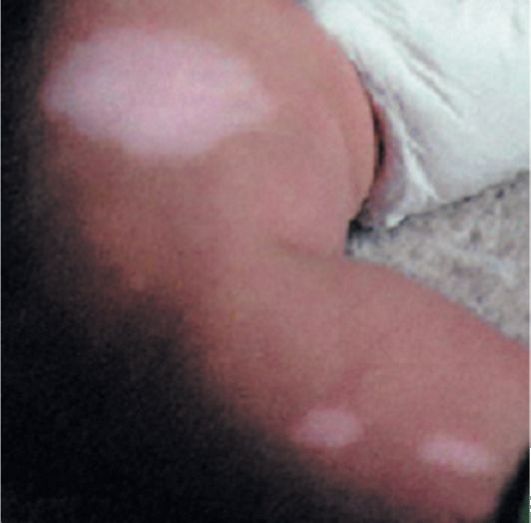
Рис. 3.2. Депигментированные пятна при туберозном склерозе
накапливает соли кальция. Располагаясь в области отверстия Монро, опухоль может вызывать окклюзионную гидроцефалию.
Манифестные симптомы при ТС отличаются выраженным полиморфизмом и возраст-зависимым дебютом.
Изменения кожи и ее производных встречаются в 100% случаев.
• Гипопигментированные пятна (рис. 3.2). Число пятен колеблется от 3-4 до 100 и более. Они располагаются диффузно, асимметрично, появляются с рождения или в более позднем возрасте, большей частью в первые 3 года жизни. Содержат клетки со сниженным содержанием меланина. Для диагностики гипопигментированных пятен показано исследование с применением лампы Вуда - участки кожи со сниженным содержанием меланина при освещении лампой ярко светятся по сравнению с тусклой здоровой кожей. Еще одним ярким клиническим симптомом заболевания являются депигментированные волосы, брови и ресницы (рис. 3.3).
• Гиперпигментированные пятна цвета «кофе с молоком» встречаются реже. Они имеют овальную или округлую форму, размеры в пределах 1-5 см, число пятен не превышает 5.
• Аденома сальных желез (ангиофиброма лица) чаще появляется в возрасте 3-11 лет, в среднем в 4-7 лет. Внешний вид ангиофибром напоминает пятна или узелки с гладкой блестящей поверхностью (рис. 3.4). Излюбленные места локализации ангиофибром - центр носогубных складок, крылья носа, щеки и подбородок.
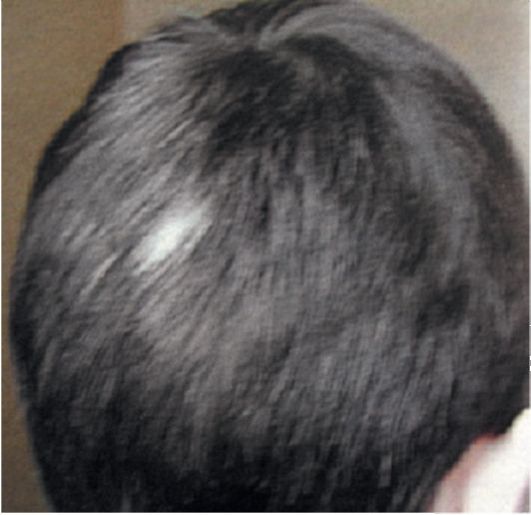
Рис. 3.3. Белые пряди волос, (фото из архива М.Ю. Дорофеевой, ФГУ «Московский НИИ педиатрии и детской хирургии»)
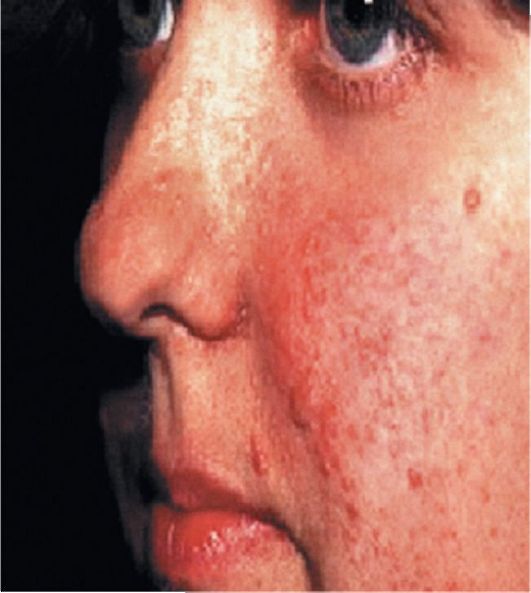
Рис. 3.4. Ангиофиброма лица (аденома Прингла)
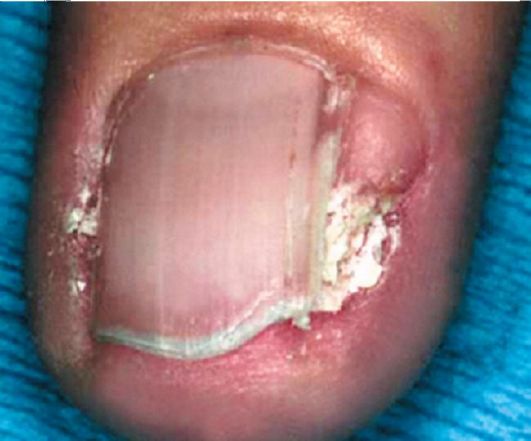
Рис. 3.5. Околоногтевые фибромы (опухоли Коэнена)
Патогномоничным является их билатерально-симметричное расположение - «крылья бабочки».
• Фиброзные бляшки - патогномоничный симптом туберозного склероза. Как правило, они располагаются на лбу и волосистой части головы унилатерально, появляются позже ангиофибром. Бляшки имеют разную консистенцию, выступают над поверхностью кожи, шероховатые на ощупь.
• Участки «шагреневой кожи» обычно расположены асимметрично на спине в поясничнокрестцовом отделе. Их размер - от нескольких миллиметров до 10 см и более. Пятна слегка выступают над поверхностью кожи, имеют желто-коричневую или розовую окраску и внешне напоминают свиную кожу или апельсиновую кожуру.
• Околоногтевые фибромы представляют собой тусклые, красные или цвета кожи узелки, расположенные на пальцах
или латеральной поверхности ногтевого ложа под ногтевой пластинкой. Чаще встречаются на пальцах стопы, преимущественно у женщин после пубертата. Размер фибром варьирует от 1 до 10 мм. Эти образования склонны к росту даже после удаления (рис. 3.5). Глазные симптомы выявляются у 50% больных. Часто встречаются гамартомы сетчатки (факомы). По внешнему виду эти опухоли разделяются на кальцинированные (симптом «тутовой ягоды») и некальцинированные. Факомы располагаются поверх сосудов сетчатки, по
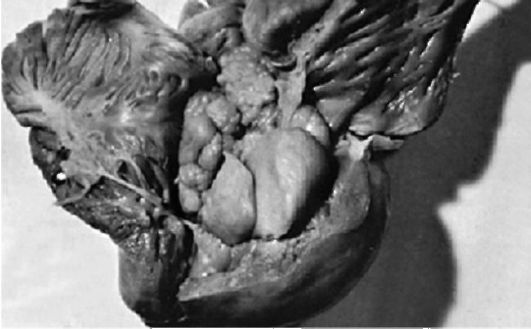
Рис. 3.6. Рабдомиома (макропрепарат)
краю диска зрительного нерва и на периферии. Другие изменения включают ангиофибромы век, депигментацию радужной оболочки или сетчатки глаза, атипичную колобому, катаракту, отек диска зрительного нерва, атрофию зрительного нерва, выпадение полей зрения (гомонимная гемианопсия, скотомы), непаралитическое косоглазие,
парез VI нерва, кровоизлияния в стекловидное тело. Глазные симптомы манифестируют в первые 2 года жизни.
Кардиальные симптомы. Рабдомиома (рис. 3.6) часто появляется внутриутробно или в первые месяцы жизни. Она располагается интрамурально или пролабирует в полость, становится причиной смерти от сердечной недостаточности. Кардиологические симптомы включают нарушения сердечного ритма (тахикардия, миграция водителя ритма, АВ-блокада, развитие синдрома Вольфа-Паркинсона-Уайта, фибрилляция желудочков) и сократительной функции миокарда при интрамуральном расположении опухоли, сердечную недостаточность вследствие обструкции камер сердца опухолью. Рабдомиома является доброкачественной опухолью (фетальной гамартомой), четко отграниченной от окружающих тканей. Случаи малигнизации не описаны. Наиболее эффективными методами диагностики рабдомиом являются двумерная эхокардиография (Эхо-КГ) и быстрые режимы МР-исследования.
Изменения внутренних органов включают в себя ангиомиолипомы и кисты почек, почечно-клеточную карциному, лимфангиомиоматоз легких, ангиомиолипомы надпочечников, печени, ректальные полипы. Характерной особенностью этих изменений является их множественный характер, двустороннее поражение парных органов, длительное бессимптомное течение.
Костно-суставные изменения представляют собой участки склероза и кисты в различных костях, деструкции плоских костей; редко возникают остеолиз и остеопороз поясничного отдела позвоночника и головок бедренных костей.
Симптомы поражения нервной системы включают эпилептические приступы, нарушение поведения, задержку психического развития, нарушения сна.
Психические нарушения представлены гиперактивностью, аутизмом, агрессивностью. Аутизм характеризуется невозможностью вербальных и невербальных коммуникаций, стереотипными движениями и отсутствием целенаправленной деятельности. Большая часть детей с аутизмом страдает инфантильными спазмами и тяжелой степенью умственной отсталости. Характер и выраженность психических нарушений зависит от расположения и количества кортикальных туберсов. Чем больше кортикальных туберсов, тем тяжелее степень умственной отсталости и более резистентны к противоэпилептической терапии приступы.
Судорожные приступы возникают у 92% пациентов с туберозным склерозом. Дебют приступов приходится чаще на первый год жизни, особенно на первые месяцы. Основным типом припадков являются инфантильные спазмы и фокальные моторные приступы. Возможна вторичная генерализация приступов, сопровождающаяся потерей сознания и генерализованными тонико-клоническими судорогами. Возможно развитие приступов по пути «простой парциальный - сложный парциальный» или «простой парциальный - сложный парциальный - вторично-генерализованный приступ». Инфантильные спазмы при туберозном склерозе входят в структуру симптоматической формы синдрома Веста и сопровождаются всеми характерными для симптоматики эпилепсии признаками: задержка психомоторного развития до дебюта припадков, наличие нескольких типов приступов (инфантильные спазмы, фокальные приступы, вторично-генерализованные приступы), структурные изменения мозга, выявляемые при КТ и/или МРТ головного мозга, резистентность приступов к противоэпилептической терапии и неблагоприятный прогноз. Инфантильные спазмы носят, как правило, асимметричный характер с вовлечением в приступ одной половины тела. Парциальный компонент асимметричных инфантильных спазмов представлен девиацией глаз, головы, ритмическими подергиваниями глазных яблок, насильственной улыбкой на лице, которые предшествуют спазму или сопровождают его.
Наличие задержки развития в сочетании с кожными изменениями является показанием для проведения нейровизуализации для уточнения характера повреждения головного мозга и постановки диагноза.
ЭЭГ-паттерны туберозного склероза: гипсаритмия, паттерн «вспышка-угнетение», мультифокальная эпилептиформная активность, генерализованная пик-волновая активность с частотой 2-
2,5 Гц (рис. 3.7).
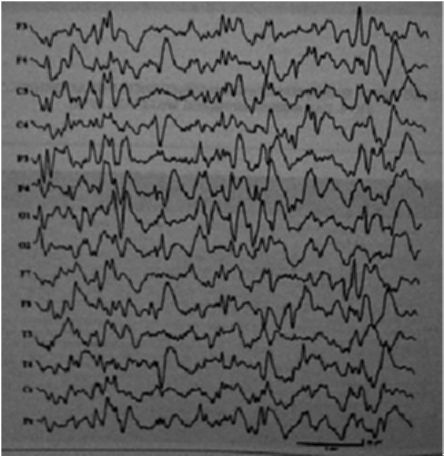
Рис. 3.7. ЭЭГ: гипсаритмия
Особенности психической сферы. Умственная отсталость от умеренной до глубокой степени отмечается в 48% случаев, нередко связана с некупируемыми судорогами на первом году жизни. Также выявляются изолированная задержка речевого развития, алалия и мутизм. Аутизм отмечается в 50% случаев По мнению А. Болтонна и Дж. Грифитса, аутизм сочетается с туберсами височных долей. Диагностика
Таблица 8. Современные диагностические критерии туберозного склероза
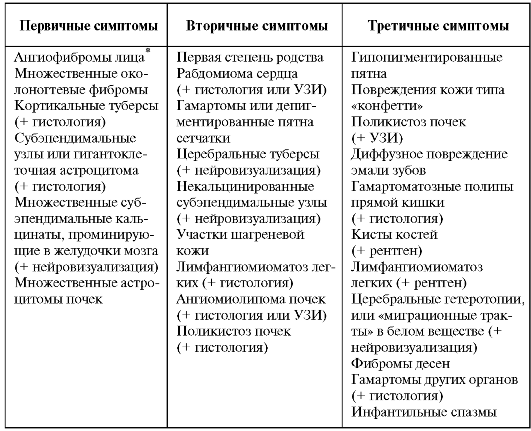
Гистологического подтверждения не требуется в том случае, если признак клинически очевиден.
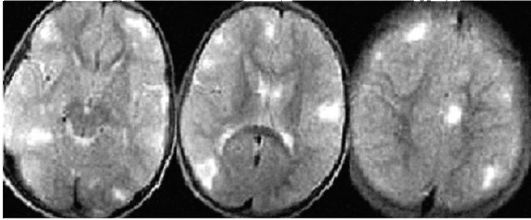
Рис. 3.8. МРТ головного мозга: кортикальные туберсы
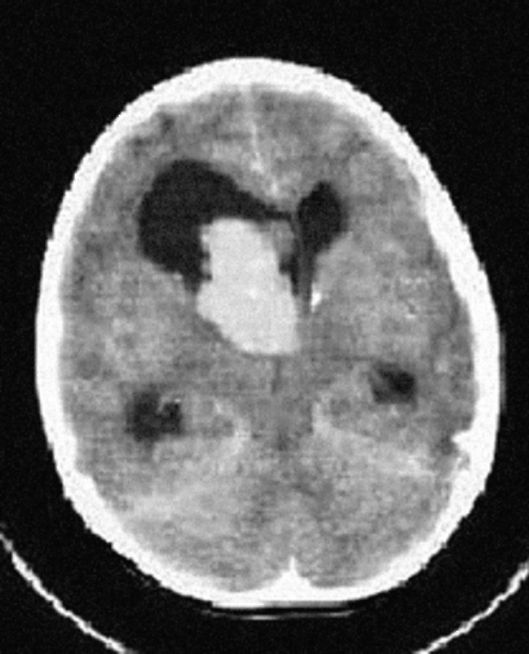
Рис. 3.9. КТ головного мозга: гигантоклеточная астроцитома, окклюзионная гидроцефалия
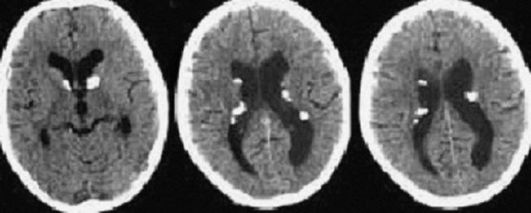
Рис. 3.10. КТ головного мозга: субэпендимальные узлы, вентрикуломегалия
На основании клинических и радиологических симптомов разработана схема диагностики.
• Несомненный диагноз: 1 первичный признак, 2 вторичных или 1 первичный в сочетании с 2 третичными признаками.
• Вероятный диагноз: сочетание 1 вторичного и 1 третичного признаков, или 3 третичных признаков.
• Предположительный диагноз: 1 вторичный признак или 2 третичных. Нейровизуализация (рис. 3.8-3.10).
КТ выявляет кальцификаты, расположенные вдоль стенки боковых и III желудочков, кальцификаты в коре и белом веществе. КТ не выявляет кортикальные туберсы, если они не кальцифицированы. МРТ визуализирует кортикальные туберсы, лейкопатию, участки гетеротопий и гамартомы. Кортикальные туберсы обнаруживаются у 95-100% пациентов с туберозным склерозом. Они представлены на МРТ в виде утолщенного участка коры, гиперинтенсивного на Т2-взвешенных изображениях. Субэпендимальные узлы гипоинтенсивны на Т2-взвешенных томограммах, степень их кальцификации увеличивается с возрастом. Поэтому для полноценного обследования ребенка с туберозным склерозом необходимы и КТ, и МРТ. Роль методов нейровизуализации состоит в подтверждении клинически заподозренного туберозного склероза, оценке степени поражения, выявлении аномалий и наблюдении за динамикой болезни.
Лечение. Специфического лечения не существует.
Антиэпилептические препараты: базовые препараты вигабатрин (сабрил) 50-100 мг/кг/сут, вальпроаты (депакин) 50-100 мг/кг/сут, топирамат (топамакс) 5-10 мг/кг/сут. Комбинированная терапия вклю- чает комбинацию базовых препаратов с бензодиазепинами (клоназепамом) 0,25-2 мг/сут, фенобарбиталом 5-15 мг/кг/сут, ламикталом 0,2- 5 мг/кг/сут, суксилепом 15-30 мг/кг/сут, карбамазепином (финлепсин) при асимметрии приступов 10-15 мг/кг/сут.
Гормональная терапия: кортикостероидные гормоны (АКТГ, синактендепо в/м; преднизолон, дексаметазон перорально) 1-2,5 мг/кг/сут.
Хирургическое лечение. У некоторых больных резекция единичного кортикального эпилептогенного туберса стереотаксическими методами или открытой краниотомией способствует значительному урежению приступов. Резекция интравентрикулярных опухолей показана при окклюзионной гидроцефалии.
Пренатальная диагностика и профилактика. УЗИ, МРТ для выявления рабдомиомы сердца и туберсов имеет диагностическую ценность со II триместра беременности. Генетический диагноз туберозного склероза нецелесообразен, поскольку 2/3 случаев возникают спорадически или вызваны мозаицизмом и не определяются скринингом лейкоцитов. Если оба родителя клинически здоровы, риск повторения туберозного склероза у второго ребенка равен 1:22 после одного больного и 1:3 после двух больных детей.
Рекомендуемая литература
1. Мухин К.Ю., Петрухин А.С. Эпилептические синдромы: Справочное руководство. - М., 2005.
2. Мухин К.Ю., Петрухин А.С., Глухова Л.Ю. Эпилепсия // Атлас электроклинической диагностики. - М.: Альварес Паблишинг, 2004.
3. Петрухин А.С. Неврология детского возраста: Учебник. - М.: Медицина, 2004.
4. Темин П.А., Дорофеева М.Ю. Туберозный склероз: Методическое руководство. - М.: Московский НИИ педиатрии и детской хирургии МЗ РФ.
5. Menkes J.H., Sarnat H.B., Child Neurology. - Lippincott Williams & Wilkins, 2000. - P. 859-884.
6. Tuberous Sclerosis - The Knowledge Database of the Swedish National Board of Health and Welfare on rare diseases.
7. TheFetus.net - Cardiac rhabdomyoma http://scielo.isciii.es/pdf/aue/ v28n2/nota2.pdf
8. Medicine - Tuberous Sclerosis : Article by David Neal Franz, MD http://wvww.njmonline.nl/njm/getpdf.php?t=a&id=328
3.2. Нейрофиброматоз
Термин «нейрофиброматоз» (НФ) объединяет несколько форм болезни с разными типами наследования, прогнозом и клинической картиной. Выделяют центральный, периферический и сегментарный нейрофиброматоз.
Периферический нейрофиброматоз (болезнь Реклингхаузена), I тип (НФ-I) - аутосомно-доминантное заболевание с частотой встречаемости - 1 на 3000-5000 в общей популяции. Ген НФ-I кодирует белок - опухолевый супрессор, который инактивируется у пациентов с этим заболеванием. В результате в течение болезни появляются опухоли разного происхождения и локализации: менингиомы, глиомы, астро-
цитомы, гамартомы зрительных нервов, гипоталамо-селлярной области, ствола мозга, черепных нервов, мозжечка и др., а также различные нейрофибромы.
Первые описания пациентов с дермальными опухолями при НФ появились в трактате «История монстров» в 1642 г. (рис. 3.11), но полное классическое описание НФ дал в 1882 г. Фридрих фон Реклингхаузен, и клинические критерии этого заболевания остались с тех пор неизменными.
Клинические и диагностические критерии НФ-I
• Кожные пятна цвета «кофе с молоком», в количестве не менее шести, диаметром более 5 мм у детей раннего возраста и более 15 мм у взрослых пациентов. Пятна появляются уже на первом году жизни и в дальнейшем их количество увеличивается.

Рис. 3.11. Изображение больного, «Monstrorum Historia», 1642 г., «Homuncio»
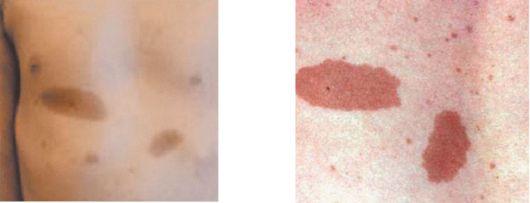
Рис. 3.12. Кожные пятная цвета «кофе с молоком»
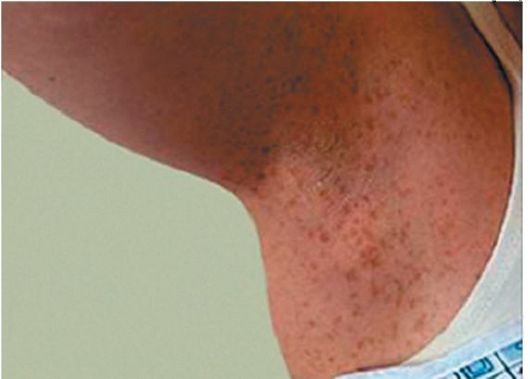
Рис. 3.13. Мелкие гиперпигментированные пятна в виде веснушек
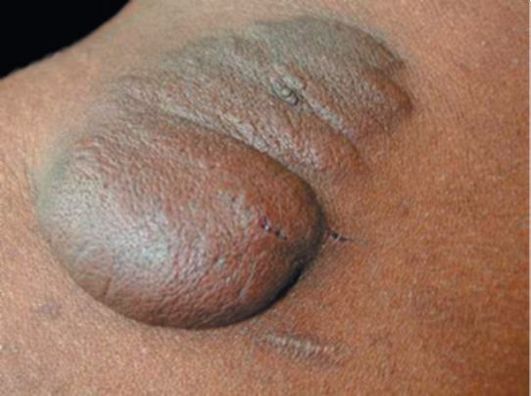
Рис. 3.14. Нейрофибромы
• Нейрофибромы более двух любого типа или одна плексиформная нейрофиброма. Они могут поражать различные ткани: кожу, спинномозговые ганглии, корешки и периферические нервы, нервные сплетения, спинной мозг. Выделяют 3 типа нейрофибром: 1-й тип - кожные нейрофибромы, то есть мягкие по консистенции и подвижные кожные опухоли фиолетового или красного цвета, на широком основании или на ножке, размером от нескольких миллиметров до метра в диаметре; 2-й тип - это подкожные нейрофибромы плотной «резиновой» консистенции, неподвижные, овальные, размером от нескольких миллиметров до 3-4 см в поперечнике; 3-й тип - плексиформные нейрофибромы, сочетающие в себе признаки кожных и подкожных нейрофибром. Они прорастают здоровые ткани, покрыты гиперпигментированной кожей или кожей с участками гипертрихоза (рис. 3.12, 3.13). Нейрофибромы (рис. 3.14) могут появляться с рождения или в течение всей жизни пациентов; с возрастом их количество
и размеры увеличиваются. Клиническими спутниками нейрофибром при их локализации в области сплетений, корешков и периферических нервов являются периферические невропатии; при бульбарной локализации - нарушение дыхания, глотания; при орбитальной (периорбиталь- ной) локализации - стойкий птоз и зрительные нарушения. Параспинальные нейрофи- бромы прорастают в грудную клетку, средостение, гортань, малый таз, брюшную полость, нарушая функцию внутренних органов. Нейрофибромы конечностей иногда достигают огромных размеров, приводят к «парциальному» гигантизму и требуют ампутации конечности как единственного эффективного метода коррекции.
• Веснушки в подмышечных и подколенных областях, небольших размеров - 1-3 мм, неотличимы по внешнему виду от гиперпигментированных пятен (рис. 3.13).
• Оптическая глиома возникает у 15% пациентов с НФ-I, при этом только в половине случаев глиомы проявляются клинически. Глиомы располагаются униили билатерально, локализуются в области хиазмы, зрительных нервов и зрительных трактов. Дебютирует опухоль, как правило, снижением остроты зрения, которое объясняется двумя механизмами: 1 - сдавлением зрительного нерва; 2 - сдавлением зрительного перекреста расширенным III желудочком вследствие окклюзионной гидроцефалии при опухолях различной внутричерепной локализации. В последнее время все чаще встречаются сообщения о спонтанном регрессе новообразований при НФ, с параллельным улучшением зрения. При локализации в области хиазмы и гипоталамуса глиома может проявлять себя преждевременным половым развитием. Прогноз менее благоприятный при локализации опухоли в области хиазмы (рис. 3.15).
• Узелки Лиша - пигментные гамартомы сетчатки (рис. 3.16), в количестве двух и более. Они появляются в раннем детстве и присутствуют практически у всех взрослых пациентов.
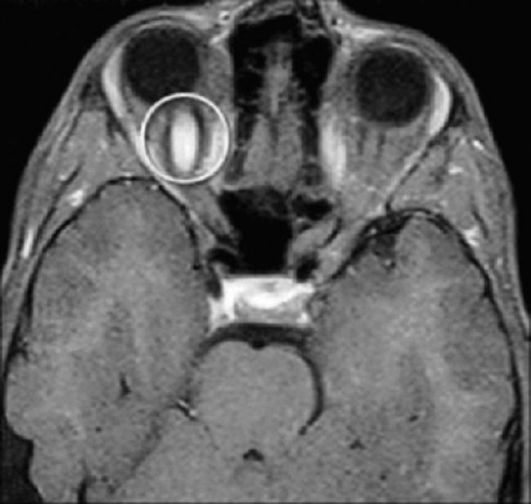
Рис. 3.15. Глиома зрительного нерва (МРТ головного мозга)
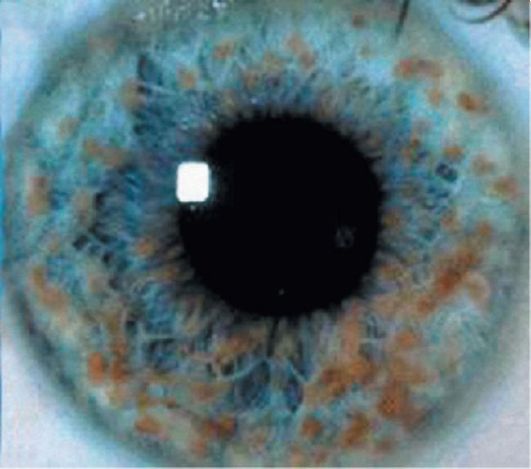
Рис. 3.16. Узелки Лиша при НФ-I
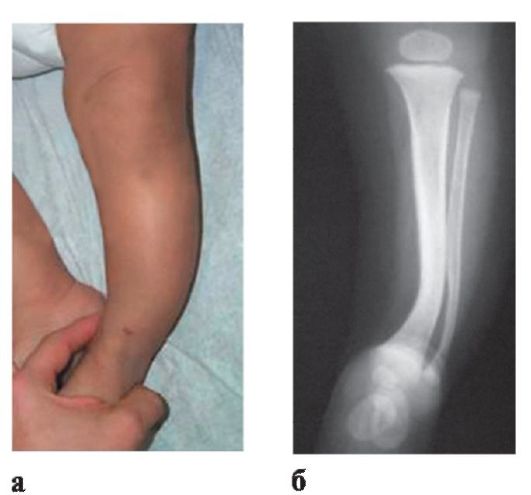
Рис. 3.17. Костные нарушения НФ-I: а - внеший вид; б -рентгенограмма
• Специфические костные нарушения: сфеноидальная дисплазия, встречающаяся изолированно или в сочетании с орбитальной (периорбитальной) плексиформной нейрофибромой; вертебральная дисплазия с латеральным менингоцеле, истончение коркового слоя длинных трубчатых костей; кифосколиоз; низкорослость; макроцефалия; псевдоартроз, варусная и вальгусная деформация голеней; кисты костей (рис. 3.17).
• Первая степень родства (родители, сибсы с НФ-I).
Неврологические симптомы при НФ-I неспецифны: задержка психоречевого развития, школьная дезадаптация, очаговая неврологическая симптоматика, судороги. Эпилепсия возникает в связи с опухолями или дисгенезиями головного мозга. Судороги могут быть фокальными или генерализованными. Эпилептические приступы у детей представляют собой симметричные инфантильные спазмы с типичной гипсаритмией на ЭЭГ в межприступный период. После 1-го года жизни преобладают фокальные приступы, сопровождающиеся легкой задержкой развития и хорошо поддающиеся противоэпилептической терапии.
Опухоли ЦНС. Астроцитомы (в основном пилоцитарные) при НФ-I встречаются чаще, чем в общей популяции. Вторым по частоте поражения после зрительного анализатора является ствол мозга. Стволовые опухоли при НФ-I также имеют некоторые особенности. Если в общей популяции преобладает локализация в мосте мозга, то при НФ-I астроцитомы оккупируют продолговатый мозг, затем средний мозг и лишь в последнюю очередь - мост мозга. У некото-
рых пациентов в результате ликворного блока на уровне водопровода мозга возникает окклюзионная гидроцефалия. Злокачественные опухоли (нейрофибросаркомы) являются основной причиной смерти пациентов с НФ-I. Частота возникновения злокачественных новообразований при НФ-1 колеблется от 3 до 13%, риск малигнизации повышается с возрастом.
Дисплазия сосудов при НФ-I выражается излишней пролиферацией клеток интимы, приводящей к стенозу или обтурации просвета сосуда. Подобные изменения типично располагаются в каротидных артериях, проксимальных ветвях средней мозговой и передней мозговой артерий.
Диагностика. Диагностика основана на клиническом осмотре, данных офтальмологического обследования, нейровизуализации (КТ, МРТ). Следует отметить преимущество МРТ перед КТ, которое объясняется возможностью выбора нескольких проекций срезов - фронтальной, сагиттальной и аксиальной, а также лучшей контрастностью изображения. Неотъемлемым компонентом нейрорадиологической диагностики НФ-I являются динамические исследования с применением контрастирования.
Лечение при НФ-I симптоматическое: оперативное лечение опухолей, противоэпилептическая терапия в случае появления эпилептических приступов, корректоры поведения, ноотропные препараты в случаях задержки развития, лечение костных деформаций, улучшение минерализации костной ткани и др. Следует заместить, что удаление нейрофибром нередко способствует усилению роста новообразований, поэтому в большинстве случаев рекомендуется выжидательная тактика. Показаниями к оперативному лечению служат: болевой синдром, значительный неврологический или косметический дефект, подозрение на малигнизацию опухоли.
Центральный нейрофиброматоз - тип II (НФ-II) представляет собой отличное по клиническим проявлениям, прогнозу и типу наследования заболевание. Ген НФ-II локализован на хромосоме 22, тип наследования - аутосомно-доминантный. Частота встречаемости 1 на 50 000 в общей популяции. Отличительной особенностью НФ-II являются шванномы черепных нервов, чаще - VIII нерва. Глиомы этого нерва являются облигатным и патогномоничным признаком НФ-II. Могут встречаться и другие виды опухолей другой локализации: менингеомы, гамартомы, глиомы, нейрофибромы, шванномы (рис. 3.18). Кожные проявления при НФ-II встречаются значительно реже по сравнению с НФ-I. Гиперпигментированные пятна на
коже обнаруживаются менее чем у половины пациентов. Количество пятен не превышает 5. Кожные нейрофибромы при НФ-II малых размеров и наблюдаются у 65% пациентов. Они важны для ранней диагностики НФ-II, поскольку шванномы слухового нерва манифестируют поздно - после 10-15 лет. Катаракта развивается более чем у половины пациентов с НФ-II и может являться первым симптомом заболевания. Узелки Лиша, костные дисплазии и опти- ческие глиомы для НФ-II нехарактерны. Поэтому до II-IV декады жизни НФ-II течет бессимптомно. Иногда только нейрорадиологическое исследование может выявить данное заболевание. Основные клинические и диагностические критерии НФ-II
A. Билатеральные новообразования VIII черепного нерва.
Б. Родственники I степени родства (родители, дети, сибсы) и следующие симптомы:
- унилатеральное новообразование VIII черепного нерва;
- два признака из перечисленных ниже:
■ менингеома;
■ нейрофиброма;
■ глиома;
■ шваннома;
■ церебеллярные кальцинаты.
B. Два признака из перечисленных ниже:
- унилатеральная вестибулярная шваннома;
- множественные менингеомы;
- признаки, перечисленные в пункте «Б».
Шванномы и менингиомы нетипичны для детей и подростков, поэтому при их обнаружении необходимо провести МРТ с контра- стированием для поиска других бессимптомных новообразований и исключить НФ-II. Проявлением НФ-II у детей могут быть мно-
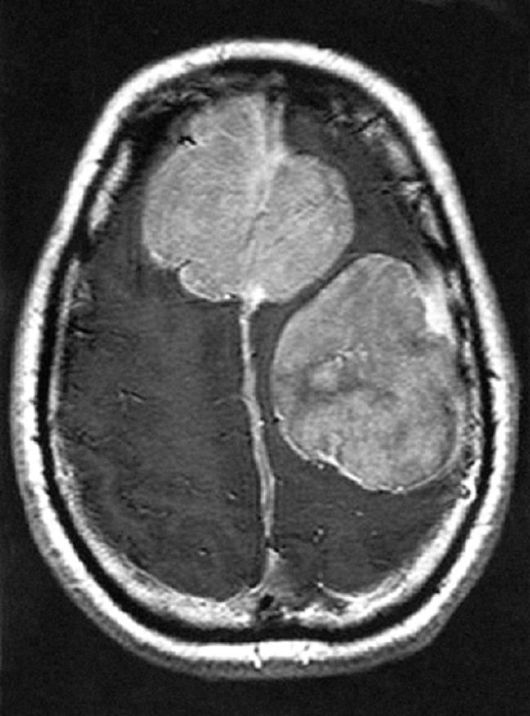
Рис. 3.18. Опухоли полушарий при НФ-II
жественные параспинальные шванномы и спинальные эпендимомы. Интраспинальные и параспинальные шванномы выявляются у пациентов до 15 лет и, являясь экстрамедуллярными опухолями, проявляют себя симптомами сдавления спинного мозга. Эпендимомы в отличие от шванном расположены интрамедуллярно.
Интраспинальные менингеомы, как правило, расположены интрадурально или экстрамедуллярно. Излюбленная локализация - грудной отдел позвоночника. Обладая прогрессивным ростом, менингеомы вызывают компрессию спинного мозга на соответствующем уровне, а также могут подвергать деструкции прилежащие костные структуры.
Лечение. Объем лечебных манипуляций при НФ-II определяется количеством, распространенностью, локализацией новообразований и включает в себя хирургическую коррекцию при нарушении функции какого-либо органа или системы.
Пациенты с НФ любого типа, а также их родственники, особенно 1-й степени родства, должны проходить динамическое обследование в течение всей жизни. Только такой подход может обеспечить адекватную терапию и позволит проводить медико-генетическое консультирование.
3.3. Синдром Штурге-Вебера
Энцефалотригеминальный ангиоматоз, или синдром Штурге-Вебера, или менингофациальный ангиоматоз характеризуется ангиоматозом лица, сосудистой оболочки глаз и твердой мозговой оболочки (рис. 3.19). Большинство случаев спорадические, но описаны и семейные варианты.
Штурге и Вебер в 1879 г. независимо описали это заболевание. Авторы выделили основной клинический симптом - темно-розовое сосудистое пятно на одной стороне лица и головы, иногда распространяющееся на язык, губы, нёбо, глотку, дно ротовой полости, заднюю поверхность шеи. Заболевание может сопровождаться экзофтальмом и эпилепсией.
Этиология. Причиной заболевания принято считать нарушение венозной канализации, происходящей между листками эктодермы на 4-8-й нед внутриутробного развития.
Клиническая картина. Кожные изменения в виде ангиоматозного невуса локализуются на лице, обычно с одной стороны, но могут быть и двусторонними. Невус имеет цвет портвейна и выражен уже при рож-
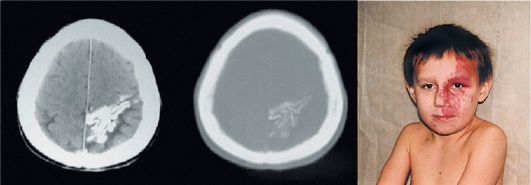
Рис. 3.19. Энцефалотригеминальный ангиоматоз, внешний вид больного и КТ головного мозга
дении. Он локализован в верхней и средней части лица, иногда распространяется на нижнюю часть лица, шею, грудную клетку, конечности (также на ипсилатеральной стороне), не изменяется в течение жизни.
Неврологические нарушения. Эпилептические приступы возникают у 75 -90% пациентов. Приступы обычно фокальные, часто по типу «джексоновского марша», могут быть вторично генерализованными. У 90% пациентов на 1-м году жизни регистрируются инфантильные спазмы, которые затем могут трансформироваться в тонические, атонические и миоклонические приступы. Эпилептические приступы при синдроме Штурге-Вебера резистентны к противоэпилептической терапии и в ряде случаев приводят к эпилептическому статусу. Тяжесть эпилепсии при синдроме Штурге-Вебера определяется степенью поражения головного мозга. Характерны постприступные парезы и параличи, которые регрессируют спустя несколько часов после приступа. При неврологическом осмотре выявляются контра- латеральный гемипарез, гемианопсия, гемиатрофия.
Глазные изменения характеризуются униили билатеральной глаукомой, ангиомой сосудистой оболочки глаза, гетерохромией радужной оболочки.
Очень редко ангиомы расположены во внутренних органах - в кишечнике, почках, селезенке, яичниках, щитовидной и поджелу- дочной железах, легких. Комбинация синдрома Штурге-Вебера и ангиоматоза внутренних органов выделяется некоторыми авторами в отдельный синдром Клиппеля-Треноне-Вебера.
Нейрорадиологические симптомы энцефалотригеминального ангиоматоза включают венозные ангиомы мягкой мозговой оболочки, расположенные над затылочной или теменно-затылочной обла-
стью, а также поражение церебральной гемисферы (атрофические изменения, ангиомы, участки кровоизлияний), возникающие в подлежащих отделах головного мозга, расширение сосудистых сплетений боковых желудочков, расширение субэпендимальных вен и сосудов перивентрикулярной зоны, гемиатрофия полушарий, атрофическая редукция белого вещества, экстра-, интрацеребральные ангиомы.
Основными методами диагностики энцефалотригеминального ангиоматоза являются нейрорадиологические исследования - КТ и МРТ головного мозга. КТ более информативна для обнаружения интракраниальных кальцинатов; МР-исследование лучше визуализирует ангиомы, изменения белого вещества и сосудистых сплетений. Всем пациентам рекомендуется проведение МРТ с контрастированием.
Ангиография, являясь достаточно инвазивным методом исследования, используется только в качестве прехирургической диагностики. Показания для исследования включают коррекцию косметических дефектов кожи и повторные интракраниальные кровоизлияния.
При ЭЭГ выявляют межполушарную асимметрию и эпилептические паттерны - фокальные и генерализованные.
Лечение синдрома Штурге-Вебера симптоматическое, оперативное и консервативное. Консервативная терапия направлена на коррекцию нервно-психического статуса пациентов и купирование эпилептических приступов. Целью оперативного лечения является устранение косметических дефектов и предотвращение повторных кровоизлияний.
3.4. Синдром Гиппеля-Линдау, или ретиноцеребеллярный ангиоматоз
Синдром Гиппеля-Линдау, или ретиноцеребеллярный ангиоматоз - это аутосомно-доминантное заболевание с неполной пенетрантностью, характеризующееся наличием ангиом сетчатки, мозжечковых и спинальных гемангиобластом, почечно-клеточной карциномы, феохромоцитомы, ангиом печени и почек, папиллярной цистаденомы придатков яичек, опухолей эндолимфатического мешочка внутреннего уха, кист поджелудочной железы, почек, печени и придатков яичек.
Причиной синдрома Гиппеля-Линдау является аномальный ген, локализованный на хромосоме 3р25-р26, отвечающий за синтез бел- ка - опухолевого супрессора. Симптомы обычно манифестируют в возрасте 30-40 лет. Чаще сначала появляется ангиома сетчатки, а затем развивается мозжечковая симптоматика. Заболевание протекает
с реактивными воспалительными изменениями сетчатки с экссудацией и геморрагиями, сопровождающимися в дальнейшем отслойкой сетчатки, глаукомой, катарактой и увеитом. В течение некоторого времени пациенты наблюдаются у офтальмолога по поводу болей в глазу и снижения зрения, позже возникает общемозговая симптоматика (головная боль, рвота), а затем признаки очагового поражения мозжечка - адиадохокинез, дисметрия, атаксия в позе Ромберга.
Причиной появления общемозговых и очаговых симптомов является опухоль мозжечка и нарастающая внутричерепная гипертензия. Иногда заболевание дебютирует спинальными симптомами - потерей проприоцептивной и поверхностной чувствительности по сегментарному и проводниковому типу, которые связаны с компрессией спинальной гемангиобластомы на спинной мозг. При локализации опухоли во внутреннем ухе заболевание сопровождается потерей слуха.
Частота встречаемости висцеральной патологии при синдроме Гиппеля-Линдау колеблется от 10-15% (феохромоцитома) до 40% (почечно-клеточная карцинома).
Диагностические критерии:
• 2 и более гемангиобластом ЦНС;
• 1 гемангиобластома в сочетании с висцеральной патологией;
• один из перечисленных выше клинических симптомов в сочетании с семейным анамнезом.
Для подтверждения диагноза при синдроме Гиппеля-Линдау проводят нейрорадиологические исследования (КТ, МРТ) с контрастированием. Гемангиобластомы мозжечка обнаруживают в каждом втором случае. У 20-40% пациентов это солидная опухоль, в остальных случаях - васкулярный опухолевый узел, который располагается в стенке крупной ликворной кисты, чаще локализованной в гемисфере мозжечка. Гемангиобластома практически неотличима от кисты, и только введение контраста и накопление его клетками опухоли облегчает постановку диагноза. Редко опухоли располагаются в стволе или полушариях большого мозга, для их диагностики более информативным методом служит МРТ. Спинальные гемангиобластомы представлены солидными опухолями или псевдосирингомиелическими полостями. Всем пациентам со спинальными образованиями показаны контрастные МР-исследования.
Лечение синдрома Гиппеля-Линдау симптоматическое. При гемангиобластомах показана хирургическая резекция, но после удаления опухоли склонны к рецидивированию.
3.5. Атаксия-телеангиэктазия (синдром Луи-Бар)
Атаксия-телеангиэктазия - заболевание с аутосомно-рецессивным типом наследования, характеризующееся нарушением репарации ДНК, тяжелым иммунодефицитом, мозжечковой дегенерацией, телеангиэктазиями различной локализации, предрасположенностью к онкологическим заболеваниям, чувствительностью к радиационным воздействиям. Частота возникновения 1 на 40 000. Ген локализован на хромосоме 11.
Клиническая картина характеризуется мозжечковой атаксией (которая обычно становится заметной для окружающих в период приобретения ребенком навыков ходьбы), дизартрией, глазодвигательными нарушениями, хореоатетозом, миоклониями, а также эндокринными нарушениями. Характерные кожные изменения (телеангиэктазии) появляются в 3-6 лет на конъюнктиве глаз, а затем распространяются на лицо, шею, нёбо, ушные раковины, дорсальную поверхность
рук.
Дефекты иммунитета проявляются рецидивирующими бактериальными и вирусными инфекциями, лимфомами, лейкозом и злокачественными опухолями, которые в сумме поражают 10-15% больных с атаксией-телеангиэктазией.
Нейрорадиологические исследования выявляют прогрессирующую атрофию мозжечка, преимущественно червя, вторичную атро- фическую дилатацию IV желудочка, постгеморрагические очаги в паренхиме мозга, появление которых связано с кровоизлияниями из эктазированных сосудов.
Наличие у ребенка атрофических изменений мозжечка при проведении КТ или МРТ головного мозга, атаксия и прогрессирующий неврологический дефицит требуют исключения синдрома атаксиителеангиэктазии.
3.6. Гипомеланоз Ито
Гипомеланоз Ито по частоте занимает 3-е место среди факоматозов после туберозного склероза и нейрофиброматоза. В некоторых случаях встречается аутосомно-доминантное наследование. Патогенез не выяснен. Гипомеланоз Ито характеризуется наличием на коже гипо- и депигментированных зон с нечеткими границами, полосок и пятен. У 40% пациентов встречаются пятна цвета «кофе с молоком», ангиомы, «монгольское» голубое пятно, диффузная алопеция, гетерохромия радужной оболочки и волос (рис. 3.20). Скелетно-мышечные аномалии включают гемигипертрофию, кифоз, сколиоз, гиперлордоз,
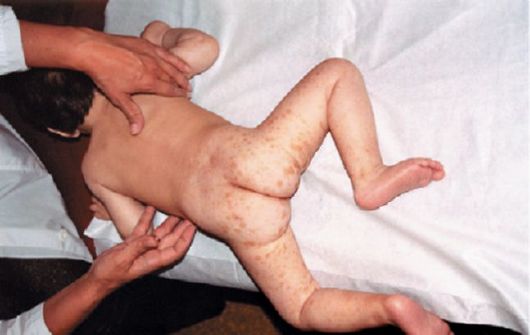
Рис. 3.20. Недержание пигмента (гипомеланоз Ито)
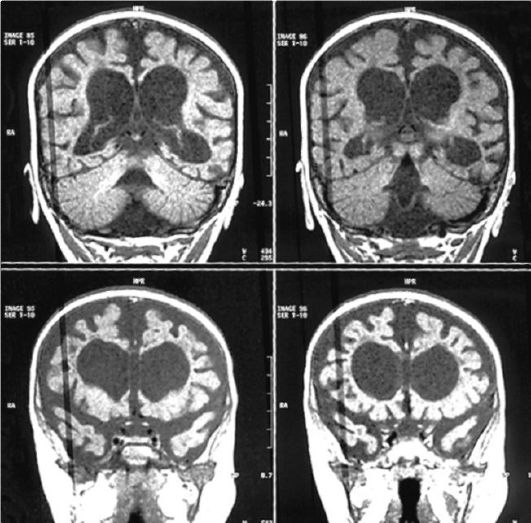
Рис. 3.21. МРТ головного мозга при гипомеланозе Ито. Атрофия мозга
рекурвацию коленных суставов, рудиментарные ребра, варусные и вальгусные деформации стоп.
Неврологические симптомы встречаются у 50% пациентов и проявляются эпилептическими приступами и задержкой психического развития. Дебют эпилептических приступов приходится на 1-й год жизни ребен- ка. Основным типом эпилептических пароксизмов являются инфантильные спазмы, фокальные моторные приступы и миоклонии. Приступы резистентны к противоэпилептической терапии. Наиболее информативным методом диагностики церебраль- ной патологии является МРТ головного мозга. При исследовании обнаруживают широкий спектр аномалий: полимикрогирию, нейрональные гетеротопии, гемимегалэнцефалию, лиссэнцефалию, фокальные корковые дисплазии, гипоплазию мозолистого тела, а также атрофию вещества головного мозга (рис. 3.21).
Рекомендуемая литература к главе 3
1. Comez MR. Neurocutaneous Diseases: a Practical. Approach. Butterworths. - Boston, 1987. - C. 329-334
2. Балязин В.А. и др. Нейрокожные синдромы. Клиника, диагностика. - Джангар, 2001. - 96 с.