Патофизиология: учебник / Литвицкий П.Ф. - 4-е изд., - 2009. - 496 с.
|
|
|
|
ГЛАВА 4. ПАТОЛОГИЯ КЛЕТКИ
Клетки - основные структурно-функциональные элементы тканей, органов и организма в целом - для выполнения своих функций поддерживают собственный гомеостаз, осуществляют обмен веществ и энергии, реализуют генетическую информацию, передают её потомству и прямо или опосредованно (через межклеточный матрикс и жидкости) обеспечивают функции организма. Любая клетка (рис. 4-1) либо функционирует в границах нормы (гомеостаз), либо приспосабливается к жизни в изменившихся условиях (адаптация), либо гибнет при превышении её адаптивных возможностей (некроз) или действии соответствующего сигнала (апоптоз).
• Гомеостаз (гомеокинез) - динамическое равновесие в данной клетке, с другими клетками, межклеточным матриксом и гуморальными
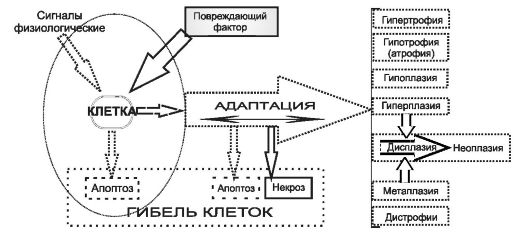 Рис. 4-1. Гомеостаз, адаптация и типовые формы патологии клеток. Слева
в овале - границы нормы. Существенное свойство типовых патологических
процессов - их обратимость. Если степень повреждения выходит за пределы
адаптивных возможностей, процесс становится необратимым (примеры -
некроз, апоптоз, дисплазия, опухолевый рост).
Рис. 4-1. Гомеостаз, адаптация и типовые формы патологии клеток. Слева
в овале - границы нормы. Существенное свойство типовых патологических
процессов - их обратимость. Если степень повреждения выходит за пределы
адаптивных возможностей, процесс становится необратимым (примеры -
некроз, апоптоз, дисплазия, опухолевый рост).
факторами, обеспечивающее оптимальную метаболическую и информационную поддержку. Жизнь клетки в условиях гомеостаза - постоянное взаимодействие с различными сигналами и факторами.
• Адаптация - приспособление в ответ на изменения условий существования клеток (в том числе на воздействие повреждающего фактора).
• Гибель клетки - необратимое прекращение жизнедеятельности. Происходит либо вследствие генетически программированного процесса (апоптоз), либо в результате летального повреждения (некроз).
• Типовые формы патологии клеток: дистрофии, дисплазии, метаплазия, гипотрофия (атрофия), гипертрофия, а также некроз и патологические формы апоптоза.
Повреждение Повреждающие факторы
• Эффект повреждающего фактора может быть обратимым или необратимым (рис. 4-2).
• Природа повреждающего фактора трояка: физическая, химическая или биологическая (включая социальную).
• Генез. По происхождению повреждающие факторы подразделяют на экзогенные и эндогенные.
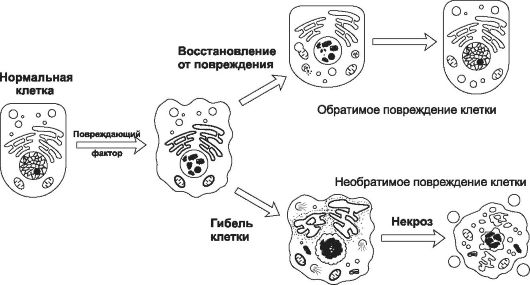 Рис. 4-2. Признаки обратимого и необратимого повреждения. [по 4].
Рис. 4-2. Признаки обратимого и необратимого повреждения. [по 4].
♦ Экзогенные факторы (действуют на клетку извне):
❖ физические воздействия (механические, термические, лучевые, электрический ток);
❖ химические агенты (кислоты, щёлочи, этанол, сильные окислители);
❖ инфекционные факторы (вирусы, риккетсии, бактерии, эндо- и экзотоксины микроорганизмов, гельминты и др.).
♦ Эндогенные агенты (образуются и действуют внутри клетки):
❖ физической природы (например, избыток свободных радикалов; колебания осмотического давления);
❖ химические факторы (например, накопление или дефицит ионов H+, K+, Ca2+, кислорода, углекислого газа, перекисных соединений, метаболитов и др.);
❖ биологические агенты (например, белки, лизосомальные ферменты, метаболиты, Ig, цитотоксические факторы; дефицит или избыток гормонов, ферментов, простагландинов - Пг).
• Эффекты повреждающих факторов достигаются прямо (первичные факторы повреждения) или опосредованно (при формировании цепи вторичных патологических реакций - вторичные факторы повреждения).
МЕХАНИЗМЫ ПОВРЕЖДЕНИЯ КЛЕТОК
К наиболее важным механизмам клеточной альтерации относятся:
♦ расстройства энергетического обеспечения клетки;
♦ повреждение мембран и ферментов;
♦ активация свободнорадикальных и перекисных процессов;
♦ дисбаланс ионов и воды;
♦ нарушения в геноме или экспрессии генов;
♦ расстройства регуляции функций клеток.
Расстройства энергетического обеспечения клетки
Энергоснабжение клетки может расстраиваться на этапах ресинтеза, транспорта и утилизации энергии АТФ. Главная причина расстройств - гипоксия (недостаточное снабжение клеток кислородом и нарушение биологического окисления).
• Ресинтез АТФ нарушается в результате дефицита кислорода и субстратов метаболизма, снижения активности ферментов тканевого дыхания и гликолиза, а также повреждения и разрушения митохондрий (в которых осуществляются реакции цикла Кребса и со- пряжённый с фосфорилированием АДФ перенос электронов к молекулярному кислороду).
• Транспорт энергии. Заключённая в макроэргических связях энергия АТФ поступает к эффекторным структурам (миофибриллы, ион-
ные насосы и др.) с помощью АДФ-АТФ-транслоказы и КФК. При повреждении этих ферментов или мембран клеток нарушается функция эффекторных структур.
• Утилизация энергии может быть нарушена преимущественно за счёт уменьшения активности АТФаз (АТФаза миозина, Na+K+-АТФаза плазмолеммы, протонная и калиевая АТФаза, Са2+-АТФаза и др.), КФК, адениннуклеотидтрансферазы.
Повреждение мембран
Повреждение клеточных мембран происходит за счёт следующих процессов:
• Активация гидролаз. Под влиянием патогенных факторов активность мембраносвязанных, свободных (солюбилизированных) и лизосомальных липаз, фосфолипаз и протеаз может значительно увеличиться (например, при гипоксии и ацидозе). В результате фосфолипиды и белки мембран подвергаются гидролизу, что сопровождается значительным повышением проницаемости мембран.
• Расстройства репарации мембран. При воздействии повреждающих факторов репаративный синтез альтерированных или утраченных мембранных макромолекул (а также их синтез de novo) подавляется, что приводит к недостаточному восстановлению мембран.
• Нарушения конформации макромолекул (их пространственной структуры) приводит к изменениям физико-химического состояния клеточных мембран и их рецепторов, что приводит к искажениям или потере их функций.
• Разрыв мембран. Перерастяжение и разрывы мембран набухших клеток и органоидов в результате их гипергидратации (следствие значительного увеличения осмотического и онкотического давления) - важный механизм повреждения мембран и гибели клетки.
• Свободнорадикальные и перекисные реакции - в норме это необходимое звено транспорта электронов, синтеза Пг и лейкотриенов, фагоцитоза, метаболизма катехоламинов и др. В свободнорадикальные реакции вовлекаются белки, нуклеиновые кислоты и, особенно, липиды, учитывая наличие большого их числа в мембранах клеток (свободнорадикальное перекисное окисление липидов - СПОЛ). При действии патогенных факторов генерация свободных радикалов и СПОЛ значительно возрастает, что усиливает повреждение клеток.
♦ Этапы СПОЛ: образование активных форм кислорода - генерация свободных радикалов органических и неорганических веществ - продукция перекисей и гидроперекисей липидов.
Активные формы кислорода - ❖ синглетный (Ό2) ❖ супероксидный радикал (O2-)
❖ пероксид водорода (H2O2) ❖ гидроксильный радикал (OH-).
♦ Прооксиданты и антиоксиданты. Интенсивность СПОЛ регулируется соотношением активирующих (прооксидантов) его и подавляющих (антиоксидантов) факторов.
❖ Прооксиданты - легко окисляющиеся соединения, нейтрализующие свободные радикалы (нафтохиноны, витамины A и D, восстановители - НАДФH2, НАДH2, липоевая кислота, продукты метаболизма Пг и катехоламинов).
❖ Антиоксиданты - вещества, ограничивающие или даже прекращающие свободнорадикальные и перекисные реакции (ретинол, каротиноиды, рибофлавин, токоферолы, маннитол, супероксиддисмутаза, каталаза).
♦ Детергентные эффекты амфифилов. В результате активации липопероксидных реакций и гидролаз накапливаются гидроперекиси липидов, свободные жирные кислоты и фосфолипиды - амфифилы (вещества, способные фиксироваться как в гидрофобной, так и в гидрофильной зоне мембран). Это ведёт к формированию обширных амфифильных кластеров (простейшие трансмембранные каналы), микроразрывам и разрушению мембран.
Дисбаланс ионов и воды
Внутриклеточная жидкость содержит примерно 65% всей воды организма и характеризуется низкими концентрациями Na+ (10 ммоль/л), Cl- (5 ммоль/л), HCO3- (10 ммоль/л), но высокой концентрацией K+ (150 ммоль/л) и PO43- (150 ммоль/л). Низкая концентрация Na+ и высокая концентрация K+ обусловлены работой Na+,K+-АТФазы, выкачивающей Na+ из клеток в обмен на K+. Клеточный дисбаланс ионов и воды развивается вслед за расстройствами энергетического обеспечения и повреждением мембран.
К проявлениям ионного и водного дисбаланса относятся: ❖ изменение соотношения отдельных ионов в цитозоле; ❖ нарушение трансмембранного соотношения ионов; ❖ гипергидратация клеток; ❖ гипогидратация клеток; ❖ нарушения электрогенеза.
• Изменения ионного состава обусловлены повреждениями мембранных АТФаз и дефектами мембран. Так, вследствие нарушения работы Na+,K+-АТФазы происходит накопление в цитозоле избытка Na+ и потеря клеткой K+.
• Осмотическое набухание и осмотическое сморщивание клеток. Состояние клеток при изменении осмотичности рассмотрено на рис. 4-3.
• Гипергидратация. Основная причина гипергидратации повреждён- ных клеток - повышение содержания Na+, а также органических веществ, что сопровождается увеличением в них осмотического давления и набуханием клеток. Это сочетается с растяжением и
• микроразрывами мембран. Такая картина наблюдается, например, при осмотическом гемолизе эритроцитов (рис. 4-3). Гипогидратация клеток наблюдается, например, при лихорадке, гипертермии, полиурии, инфекционных заболеваниях (холере, брюшном тифе, дизентерии). Эти состояния ведут к потере организмом воды, что сопровождается выходом из клеток жидкости, а также органических и неорганических водорастворимых соединений.
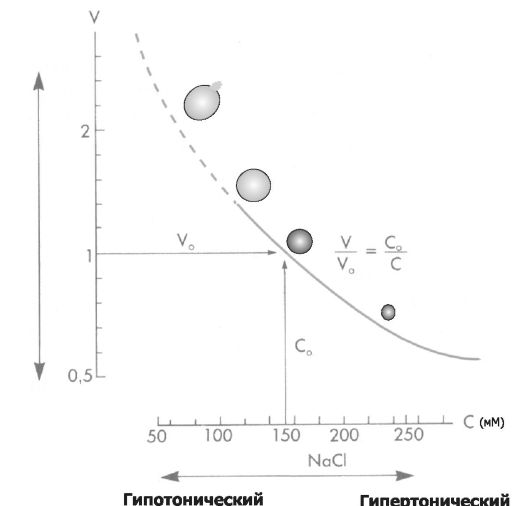 Рис.
4-3. Состояние взвешенных в растворе NaCl эритроцитов. По оси абсцисс:
концентрация (С) NaCl (ммоль/л); по оси ординат: объём клеток (V). При
концентрации NaCl 154 ммоль/л объём клеток такой же, как и в плазме
крови (изотонический раствор NaCl), При увеличении концентрации NaCl
(гипертонический раствор NaCl) вода выходит из эритроцитов, и они
сморщиваются. При уменьшении концентрации NaCl (гипотонический раствор
NaCl) вода входит в эритроциты, и они набухают. При гипотоничности
раствора, примерно в 1,4 раза превышающей значение изотонического
раствора, происходит разрушение мембраны. [5].
Рис.
4-3. Состояние взвешенных в растворе NaCl эритроцитов. По оси абсцисс:
концентрация (С) NaCl (ммоль/л); по оси ординат: объём клеток (V). При
концентрации NaCl 154 ммоль/л объём клеток такой же, как и в плазме
крови (изотонический раствор NaCl), При увеличении концентрации NaCl
(гипертонический раствор NaCl) вода выходит из эритроцитов, и они
сморщиваются. При уменьшении концентрации NaCl (гипотонический раствор
NaCl) вода входит в эритроциты, и они набухают. При гипотоничности
раствора, примерно в 1,4 раза превышающей значение изотонического
раствора, происходит разрушение мембраны. [5].
• Нарушения электрогенеза (изменения характеристик мембранного потенциала - МП и потенциалов действия - ПД) имеют существенное значение, поскольку они нередко являются одним из важных признаков наличия и характера повреждения клеток. Примером могут служить изменения ЭКГ при повреждении клеток миокарда, электроэнцефалограммы при патологии нейронов головного мозга, электромиограммы при изменениях в мышечных клетках.
Генетические нарушения
Изменения в геноме и экспрессии генов - существенный фактор повреждения клетки. К таким нарушениям относятся мутации, дерепрессии и репрессии генов, трансфекции, нарушения митоза.
• Мутации (так, мутация гена инсулина приводит к развитию сахарного диабета).
• Дерепрессия патогенного гена (дерепрессия онкогена сопровождается трансформацией нормальной клетки в опухолевую).
• Репрессия жизненно важного гена (подавление экспрессии гена фенилаланин 4-монооксигеназы обусловливает гиперфенилаланинемию и развитие олигофрении).
• Трансфекция (внедрение в геном чужеродной ДНК). Например, трансфекция ДНК вируса иммунодефицита приводит к возникновению СПИДа.
• Нарушения митоза (так, деление ядер эритрокариоцитов без деления цитоплазмы наблюдается при мегалобластных анемиях) и мейоза (нарушение расхождения половых хромосом ведёт к формированию хромосомных болезней).
ПРОЯВЛЕНИЯ ПОВРЕЖДЕНИЙ КЛЕТОК
Любое повреждение клетки вызывает в ней разной степени выраженности специфические и неспецифические изменения. Специфические изменения развиваются при действии определённого патогенного фактора на различные клетки или в определённых видах клеток при действии разных повреждающих агентов.
• Патогенные факторы, вызывающие специфические изменения в различных клетках: осмотическое давление, разобщители, гиперальдостеронемия и др.
♦ Осмотическое давление. Повышение осмотического давления в клетке всегда сопровождается её гипергидратацией, растяжением мембран и нарушением их целостности (феномен «осмотическая деструкция клеток»).
♦ Разобщители. Под влиянием разобщителей окисления и фосфорилирования (например, высших жирных кислот - ВЖК или Ca2+) снижается или блокируется сопряжение этих процессов и эффективность биологического окисления.
♦ Гиперальдостеронемия. Повышенное содержание в крови и интерстиции альдостерона ведёт к накоплению в клетках Na+.
• Группы клеток, реагирующие специфическими изменениями на действие различных повреждающих агентов:
♦ Мышечные элементы на влияние разнообразных патогенных факторов значительной силы реагируют развитием их контрактуры.
♦ Эритроциты при различных повреждениях подвергаются гемолизу с выходом Hb.
Неспецифические изменения (стереотипные, стандартные) развиваются при повреждении различных видов клеток и действии на них широкого спектра патогенных агентов. Примеры: ацидоз, чрезмерная активация свободнорадикальных и перекисных реакций, денатурация молекул белка, повышение проницаемости клеточных мембран, дисбаланс ионов и воды, снижение эффективности биологического окисления.
Типовые формы патологии
Основными типовыми формами патологии клеток являются их гипотрофия и атрофия, гипертрофия и дистрофии, дисплазии, метаплазия, а также некроз и апоптоз.
Гипотрофия и атрофия. Гипотрофия характеризуется уменьшением размеров и массы клетки, крайней степенью чего является атрофия. Гипотрофия и атрофия обычно сочетаются с уменьшением количества клеток - гипоплазией. Это приводит к уменьшению объёма органа, истончению кожи и слизистых оболочек. Пример: уменьшение массы и числа клеток в ишемизированной ткани или органе. Гипертрофия. Для гипертрофии характерно увеличение размеров и массы клетки. Нередко это сопровождается увеличением числа клеток (гиперплазией). Выделяют физиологическую и патологическую гипертрофию.
• Физиологическая гипертрофия носит адаптивный характер (например, гипертрофия скелетных мышц у спортсменов).
• Патологическая гипертрофия имеет (наряду с адаптивным) патологическое значение. Различают рабочую, викарную и нейрогуморальную патологическую гипертрофию, сочетающуюся с ремоделированием органа или ткани.
♦ Рабочая гипертрофия развивается при постоянно повышенной нагрузке (например, патологическая гипертрофия миокарда при гипертонической болезни).
♦ Викарная (заместительная) гипертрофия развивается в одном из парных органов при удалении второго.
♦ Нейрогуморальная гипертрофия развивается при нарушении нейрогуморальной регуляции (например, акромегалия, гинекомастия).
Дистрофии
Клеточные дистрофии - нарушения обмена веществ, сопровождающиеся расстройством функций клеток.
• Механизмы дистрофий разнообразны:
❖ синтез аномальных (в норме не встречающихся в клетке) веществ (например, белково-полисахаридного комплекса амилоида);
❖ избыточное превращение одних соединений в другие (например, углеводов в жиры при сахарном диабете);
❖ декомпозиция (фанероз): распад субклеточных структур и веществ (например, белково-липидных комплексов мембран при хронической гипоксии);
❖ инфильтрация клеток и межклеточного вещества органическими и неорганическими соединениями (например, липопротеинами низкой плотности - ЛПНП и Ca2+ интимы артерий при атеросклерозе).
• Классификация. Основным критерием классификации клеточных дистрофий является преимущественное нарушение метаболизма отдельных классов веществ. В связи с этим критерием различают диспротеинозы (белковые дистрофии), липидозы (жировые дистрофии), диспигментозы (пигментные дистрофии), углеводные и минеральные дистрофии. В отдельную группу выделяют тезаурисмозы (болезни накопления).
❖ Диспротеинозы. Для белковых дистрофий характерно изменение физико-химических свойств клеточных белков. Выделяют зернистую, гиалиново-капельную и гидропическую дистрофии.
❖ Липидозы. Для жировых дистрофий характерно увеличение содержания внутриклеточных липидов и их перераспределение в тканях и органах. Выделяют первичные и вторичные липидозы.
❖ Первичные липидозы наблюдаются, как правило, при генетически обусловленных ферментопатиях (например, ганглиозидозы, цереброзидозы, сфинголипидозы).
❖ Вторичные липидозы развиваются в результате воздействия различных патогенных факторов, таких как гипоксия, тяжёлые инфекции, системные заболевания, отравления (в том числе некоторыми ЛС - цитостатиками, антибиотиками, барбитуратами).
❖ Углеводные дистрофии. Характеризуются нарушениями обмена полисахаридов (гликогена, мукополисахаридов) и гликопротеинов (муцина, мукоидов).
❖ Полисахариды. При нарушениях метаболизма полисахаридов в клетках можно наблюдать уменьшение содержания углеводов (например, гликогена при СД), отсутствие углеводов (агликогенозы; например, при циррозе печени или хронических гепатитах) и накопление избытка углеводов (например, гликогеноз фон Гирке - нефромегалический синдром - гликогенная инфильтрация клеток почек).
❖ Гликопротеины. Углеводные дистрофии, связанные с нарушением метаболизма гликопротеинов, характеризуются, как правило, накоплением муцинов и мукоидов, имеющих слизистую консистенцию (в связи с этим их называют также слизистыми дистрофиями).
♦ Диспигментозы. Пигментные дистрофии классифицируют в зависимости от их происхождения (первичные и вторичные), механизма развития, структуры пигмента, проявлений и распро- странённости (местные и системные). Примеры:
❖ Частицы сажи, угля и т.п. накапливаются в макрофагах лёгких в результате пребывания в загрязнённой атмосфере. В связи с этим ткань лёгких приобретает тёмно-серый цвет.
❖ Гемосидерин. При гемолизе эритроцитов происходят освобождение Hb, его захват макрофагами печени, селезёнки, красного костного мозга и превращение в пигмент бурого цвета - гемосидерин.
♦ Минеральные дистрофии. Из минеральных дистрофий наибольшее клиническое значение имеют нарушения обмена кальция, калия, железа, цинка, меди в виде отложения солей этих химических элементов (например, кальцинозы, сидерозы, отложение меди при гепатоцеребральной дистрофии).
♦ Тезаурисмозы (от греч. thesauros - сокровищница) - болезни накопления промежуточных продуктов обмена углеводов, гликозаминогликанов, липидов и белков. Большинство тезаурисмозов - результат наследственных ферментопатий. В зависимости от типа накапливающихся веществ тезаурисмозы подразделяют на липидные (липидозы), гликогеновые (гликогенозы), аминокислотные, нуклеопротеиновые, мукополисахаридные (мукополисахаридозы), муколипидные (муколипидозы). В отдельные группы выделяют болезни накопления лизосомные и пероксисомные. Примеры:
❖ Тэя-Сакса болезнь - врождённая недостаточность лизосомальной гексозаминидазы А нейронов - характеризуется накоплением ганглиозидов в цитоплазме нервных клеток.
❖ Цереброгепаторенальный синдром (синдром Целлвегера) - типичная лизосомная болезнь накопления, развивающаяся вследствие дефектов генов, кодирующих белки пероксисом (в плазме крови и тканях увеличено содержание длинноцепочечных жирных кислот).
❖ Болезнь Гоше - накопление в фагоцитирующих клетках селезён- ки и красного костного мозга избытка глюкоцереброзидов.
❖ Гликогенозы - накопление в цитоплазме клеток внутренних органов разных форм аномального гликогена.
Метаплазия
Метаплазия - замещение клеток, свойственных данному органу, нормальными клетками другого типа. Примеры:
♦ Хронические воспалительные заболевания лёгких, дефицит витамина А, курение приводят к появлению среди клеток мерцательного эпителия бронхов островков многослойного плоского эпителия.
♦ При хроническом цервиците возможно замещение однослойного цилиндрического эпителия многослойным плоским.
♦ В результате забрасывания (рефлюкса) кислого содержимого желудка многослойный плоский эпителий слизистой оболочки пищевода замещается однослойным эпителием, характерным для тонкой кишки (пищевод Баррета).
Метаплазию рассматривают как пограничное состояние (на грани нормального). В ряде случаев участки метаплазии становятся диспластическими, что чревато их опухолевой трансформацией. Дисплазии - нарушения дифференцировки клеток, сопровождающиеся стойкими изменениями их структуры, метаболизма и функции (клеточный атипизм). В отличие от метаплазий, для дисплазий характерно появление признаков клеточного атипизма при сохранной структуре и архитектуре ткани. Дисплазии предшествуют опухолевому росту (предопухолевые состояния).
ГИБЕЛЬ КЛЕТКИ
Клетки погибают как в норме, так и в условиях патологии. Различают два принципиально разных варианта смерти клеток - некроз (гибель клетки вследствие её значительного - летального - повреждения) и апоптоз (гибель клетки в результате включения специальной программы смерти).
Некроз
Некроз (от греч. necros - мёртвый) - патологическая гибель клеток в результате действия на них повреждающих факторов.
Некроз является завершающим этапом клеточных дистрофий или следствием прямого действия на клетку повреждающих факторов значительной (разрушающей) силы. Основные звенья патогенеза некроза те же, что и повреждения клеток, но при развитии некроза они максимально интенсифицированы и развиваются на фоне недостаточности адаптивных механизмов (защиты и регенерации повреждённых структур, компенсации нарушенных процессов). О необратимости повреждения клетки свидетельствуют, как правило, разрывы плазмолеммы и выраженные изменения структуры ядра (кариорексис - разрывы
ядерной мембраны, фрагментация ядра; кариолизис - распыление хроматина; кариопикноз - сморщивание содержимого ядра).
♦ Паранекроз и некробиоз. Некрозу предшествуют паранекроз (сходные с некротическими, но ещё обратимые изменения метаболизма и структуры клеток) и некробиоз (совокупность необратимых дистрофических изменений, ведущих к некрозу).
♦ Лизис и аутолиз. Некротизированные клетки подвергаются деструкции (лизису). Если разложение осуществляется при помощи лизосомных ферментов и свободных радикалов погибших клеток, процесс называется аутолизом.
♦ Гетеролизис. Разрушение повреждённых и погибших клеток при участии других (неповреждённых) клеток (мигрирующих в зону альтерации фагоцитов, а также попавших в неё микробов) обозначают как гетеролизис.
• Этиология и патогенез некроза. Выделяют несколько основных этиологических факторов некроза - травматические, токсические, трофоневротические, циркуляторные и иммуногенные. Развивающиеся в связи с действием этих факторов ишемия, венозная гиперемия и лимфостаз сопровождаются гипоксией и активацией механизмов повреждения клеток, что приводит, в конце концов, к некрозу.
♦ Травматический некроз. Является результатом прямого действия на ткань физических (механических, температурных, вибрационных, радиационных) и др. факторов.
♦ Токсический некроз. Развивается при действии на ткани токсинов, чаще микробных.
♦ Трофоневротический некроз развивается при нарушении кровоснабжения или иннервации тканей при поражении периферической нервной системы. Примером трофоневротического некроза могут служить пролежни.
♦ Иммуногенный некроз - результат цитолиза в ходе аутоагрессивных иммунных и аллергических реакций. Примером может служить фибриноидный некроз при феномене Артюса. Цитолиз с участием T-лимфоцитов-киллеров, NK-клеток и фагоцитов приводит к некрозу участков печени при хроническом гепатите.
♦ Циркуляторный некроз. Вызван недостаточностью циркуляции крови в кровеносных и лимфатических сосудах в результате их тромбоза, эмболии, длительного спазма, сдавления извне. Недостаточная циркуляция в ткани вызывает её ишемию, гипоксию и некроз.
Апоптоз
Апоптоз (от греч. apoptosis - опадание листьев) - программируемая гибель клетки.
В этом принципиальное отличие апоптоза от некроза. Апоптоз является компонентом многих физиологических процессов, а также наблюдается при адаптации клетки к факторам среды. Биологическая роль апоптоза заключается в поддержании равновесия между процессами пролиферации и гибели клеток. Апоптоз - энергозависимый процесс. Нарушения или блокада апоптоза может стать причиной патологии (роста опухолей, реакций иммунной аутоагрессии, иммунодефицитов и др.).
Примеры апоптоза
♦ Запрограммированная гибель клеток в ходе эмбрионального развития, гистогенеза и морфогенеза органов. Пример: гибель нейробластов (от 25 до 75%) на определённых этапах развития мозга.
♦ Смерть клеток, выполнивших свою функцию (например, иммунокомпетентных клеток по завершении иммунного ответа или эозинофилов после дегрануляции).
♦ Ликвидация аутоагрессивных T-лимфоцитов на определённых этапах развития тимуса или после завершения иммунного ответа.
♦ Старение сопровождается гормонозависимой инволюцией и апоптозом клеток эндометрия, атрезией фолликулов яичников у женщин в менопаузе, а также - ткани простаты и яичек у пожилых мужчин.
♦ Трансфекция - внедрение в клетку фрагмента нуклеиновой кислоты вируса (например, при вирусном гепатите, миокардите, энцефалите, СПИДе) нередко вызывает её апоптоз.
♦ Опухолевый рост закономерно сопровождается апоптозом большого числа трансформированных клеток.
Механизм апоптоза
• В ходе апоптоза выделяют четыре стадии - инициация, программирование, реализации программы, удаление погибшей клетки. Стадия инициации. На этой стадии информационные сигналы воспринимаются клеточными рецепторами и передаются сигналы внутрь клетки.
♦ Трансмембранные сигналы подразделяют на «отрицательные», «положительные» и смешанные. ❖ «Отрицательный» сигнал означает прекращение действия на клетку либо отсутствие в ткани факторов роста или цитокинов, регулирующих деление и созревание клетки, а также гормонов, контролирующих развитие клеток. ❖ «Положительный» сигнал подразумевает воздействие на клетку агента, запускающего программу апоптоза. Например, связывание ФНО с его мембранным рецептором CD95 активирует программу смерти клетки. ❖ Смешанный сигнал - комбинация сигналов первой и второй групп. Так, апоптозу подвергаются лимфоциты, стимулированные митогеном, но не контактировавшие с чужеродным Аг; погибают и лимфоциты, на которые воз-
действовал Аг, но они не получили других сигналов (например, митогенного).
♦ Среди внутриклеточных стимулов апоптоза наибольшее значение имеют: ❖ избыток H+ и свободных радикалов; ❖ повышенная температура; ❖ внутриклеточные вирусы и ❖ гормоны, обеспечивающие свой эффект через ядерные рецепторы (например, глюкокортикоиды).
• Стадия программирования (контроля и интеграции процессов апоптоза). Выделяют два варианта реализации стадии программирования: прямая активация эффекторных каспаз и эндонуклеаз (минуя геном клетки) и опосредованная их активация через экспрессию определённых генов.
♦ Прямая передача сигнала. Осуществляется через адапторные белки, гранзимы и цитохром С. Прямая передача сигнала наблюдается в безъядерных клетках (например, эритроцитах).
♦ Опосредованная через геном передача сигнала. На этой стадии специализированные белки либо блокируют потенциально летальный сигнал, либо реализуют сигнал к апоптозу путём активации исполнительной программы.
❖ Белки-ингибиторы апоптоза (продукты экспрессии антиапоптозных генов Bcl-2, Bcl-XL) блокируют апоптоз (например, путём уменьшения проницаемости мембран митохондрий, в связи с чем уменьшается вероятность выхода в цитозоль одного из пусковых факторов апоптоза - цитохрома C).
❖ Белки-промоторы апоптоза (например, белки, синтез которых контролируется генами Bad, Bax, антионкогенами Rb или p53) активируют эффекторные цистеиновые протеазы (каспазы и эндонуклеазы).
• Стадия реализации программы (исполнительная, эффекторная) заключается в гибели клетки, осуществляемой посредством активации протеаз и эндонуклеаз. Непосредственными исполнителями «умертвления» клетки являются Ca2+,Mg2+-зависимые эндонуклеазы (катализируют распад нуклеиновых кислот) и эффекторные каспазы (расщепляют белки). При этом в клетке формируются и от неё отпочковываются фрагменты, содержащие остатки органелл, цитоплазмы, хроматина и цитолеммы - апоптозные тельца.
• Стадия удаления фрагментов погибших клеток. На поверхности апоптозных телец имеются лиганды, с которыми взаимодействуют рецепторы фагоцитирующих клеток. Фагоциты обнаруживают, поглощают и разрушают апоптозные тельца (гетеролизис). В результате содержимое разрушенной клетки не попадает в межклеточное пространство и при апоптозе отсутствует воспалительная реакция.
НЕКРОПТОЗ
В последние годы описан еще один вариант смерти клеток, отличающийся как от апоптоза, так и от некроза. Он обозначен как некроптоз. Программа некроптоза может быть стимулирована, подобно апоптозу, лигандами клеточных рецепторов из семейства фактора некроза опухолей (ФНОα). Однако гибель клетки происходит без активации протеаз, относящихся к каспазам (некроптоз развивается при полном подавлении активности каспаз).
Механизм разрушения клетки при некроптозе в большей мере подобен аутолизу. Считают, что некроптоз является одним из своеобразных механизмов гибели нервных клеток при инсультах.
Адаптация клеток
МЕХАНИЗМЫ АДАПТАЦИИ КЛЕТОК К ПОВРЕЖДЕНИЮ
Комплекс адаптивных реакций клеток подразделяют на внутриклеточные и межклеточные.
Внутриклеточные адаптивные механизмы
Внутриклеточные механизмы адаптации реализуются в самих повреж- дённых клетках. К этим механизмам относят: ❖ компенсацию нарушений энергетического обеспечения клетки; ❖ защиту мембран и ферментов клетки; ❖ уменьшение или устранение дисбаланса ионов и воды в клетке; ❖ устранение дефектов реализации генетической программы клетки;
• компенсацию расстройств регуляции внутриклеточных процессов;
• снижение функциональной активности клеток; ❖ действие белков теплового шока; ❖ регенерацию; ❖ гипертрофию; ❖ гиперплазию.
• Компенсация энергетических нарушений обеспечивается активацией процессов ресинтеза и транспорта АТФ, снижением интенсивности функционирования клеток и пластических процессов в них.
• Устранение дисбаланса ионов и воды в клетке осуществляется путём активации буферных и транспортных клеточных систем.
• Ликвидация генетических дефектов достигается путём репарации ДНК, устранения изменённых фрагментов ДНК, нормализации транскрипции и трансляции.
• Компенсация расстройств регуляции внутриклеточных процессов заключается в изменении числа рецепторов, их чувствительности к лигандам, нормализации систем посредников.
• Снижение функциональной активности клеток позволяет сэкономить и перераспределить ресурсы и, тем самым, увеличить возможности компенсации изменений, вызванных повреждающим фактором. В результате степень и масштаб повреждения клеток при действии
патогенного фактора снижаются, а после прекращения его действия отмечается более интенсивное и полное восстановление клеточных структур и их функций.
• Белки теплового шока (HSP, от Heat Shock Proteins; белки стресса) интенсивно синтезируются при воздействии на клетки повреждающих факторов. Эти белки способны защитить клетку от повреждений и предотвратить её гибель. Наиболее распространены HSP с молекулярной массой 70 000 (hsp70) и 90 000 (hsp90). Механизм действия этих белков многообразен и заключается в регуляции процессов сборки и конформации других белков.
Межклеточные адаптивные механизмы
Межклеточные (системные) механизмы адаптации реализуются непов- реждёнными клетками в процессе их взаимодействия с повреждёнными.
• Механизмы взаимодействия клеток:
♦ обмен метаболитами, местными цитокинами и ионами; ❖ реализация реакций системы ИБН;
♦ изменения лимфо- и кровообращения;
♦ эндокринные влияния;
♦ нервные воздействия.
• Примеры
♦ Гипоксия. Уменьшение содержания кислорода в крови и клетках стимулирует активность нейронов дыхательного центра, деятельность сердечно-сосудистой системы, выброс эритроцитов из костного мозга. В результате увеличивается объём альвеолярной вентиляции, перфузия тканей кровью, число эритроцитов в периферической крови, что уменьшает или ликвидирует недостаток кислорода и активирует обмен веществ в клетках.
♦ Гипогликемия. Повреждение клеток в условиях гипогликемии может быть уменьшено в результате инкреции глюкагона, адреналина, глюкокортикоидов, соматотропного гормона (СТГ), способствующих повышению уровня глюкозы в плазме крови и транспорта глюкозы в клетки.
♦ Ишемия. Снижение кровоснабжения артериальной кровью какого-либо участка ткани, как правило, сопровождается увеличением притока крови по коллатеральным (обходным) сосудам, что восстанавливает доставку к клеткам кислорода и субстратов метаболизма.
Повышение устойчивости клеток к повреждению
Мероприятия и средства, повышающие устойчивость интактных клеток к действию патогенных факторов и стимулирующие адаптивные механизмы при повреждении клеток, подразделяют:
♦ по целевому назначению на лечебные и профилактические;
♦ по природе на медикаментозные, немедикаментозные и комбинированные;
♦ по направленности на этиотропные, патогенетические и саногенетические.
Профилактические и лечебные мероприятия
• Немедикаментозные агенты. Немедикаментозные средства применяют с целью профилактики повреждения клетки. Эти средства повышают устойчивость клеток к ряду патогенных агентов.
Пример. Тренировка организма (по определённой схеме) умеренной гипоксией, стрессорными факторами, физическими нагрузками и охлаждением увеличивает резистентность к значительной гипоксии, ишемии, холоду, инфекционным и другим агентам. В основе увеличения резистентности клеток при тренировке лежит повышение надёжности и мощности регулирующих систем, механизмов энергетического и пластического обеспечения клеток, их компенсаторных, восстановительных и защитных реакций, механизмов синтеза белков и репарации ДНК, процессов формирования субклеточных структур и других изменений.
• Медикаментозные средства. Лекарственные средства (ЛС) применяют, в основном, для активации адаптивных механизмов уже после воздействия патогенного агента. Большинство ЛС применяют с целью этиотропной или патогенетической терапии.
К основным воздействиям, имеющим целью уменьшить силу патогенного действия на клетки или блокировать механизм развития патологического процесса, относят: снижение степени или устранение нарушений энергетического обеспечения клеток; коррекцию и защиту механизмов трансмембранного переноса, внутриклеточного распределения ионов и контроля объёма клеток; предотвращение повреждения генетического аппарата клетки; ? коррекцию механизмов регуляции и интеграции внутриклеточных процессов.
• Комбинированные воздействия дают наибольший эффект (как лечебный, так и профилактический).
Общие принципы терапии и профилактики
К общим принципам терапии и профилактики относят этиотропный, патогенетический и саногенетический принципы.
• Этиотропные воздействия направлены на предотвращение действия (профилактика) или на устранение, прекращение, уменьшение силы или длительности влияния патогенных факторов на клетки, а также устранение условий, способствующих реализации этого действия (лечение).
• Саногенетические мероприятия имеют целью активацию адаптивных механизмов (компенсации, защиты, восстановления и приспособления клеток) к изменившимся условиям, что предотвращает развитие заболевания (профилактика) или ускоряет выздоровление организма (лечение).
• Патогенетические воздействия направлены на разрыв звеньев патогенеза путём защиты механизмов энергоснабжения клеток, коррекции трансмембранного переноса, внутриклеточного распределения ионов и контроля объёма клеток; предотвращения действия факторов, вызывающих изменения в генетическом аппарате клеток.