Патофизиология: учебник / Литвицкий П.Ф. - 4-е изд., - 2009. - 496 с.
|
|
|
|
ГЛАВА 3. НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ
Отдельные гены, хромосомы и геном в целом постоянно претерпевают разнообразные изменения. Хотя существуют механизмы репарации (восстановления) ДНК, часть повреждений и ошибок сохраняется. Изменения в последовательности и числе нуклеотидов в ДНК обозначают как мутации.
IМутации - инициальное звено патогенеза наследственных заболеваний.
В широком смысле термином «мутация» обозначают любые изменения генетического материала (пара нуклеотидов, ген, аллели, хромосомы, ядерный и митохондриальный геном). В узком значении термин «мутация» соотносят с изменениями на уровне гена, то есть генные мутации. Мутагены - причины мутаций - факторы химической, физической или биологической природы. Мутагенез (мутационный процесс) - изменения, приводящие к возникновению мутаций. Различают генные, хромосомные и геномные мутации.
Мутации обнаруживают как в соматических клетках (фенотипически проявляются только в мутировавшей клетке или её потомстве), так и в половых клетках. Последние потенциально могут быть переданы по наследству и проявляться в фенотипе потомства, в том числе и в виде наследственных заболеваний.
Этиология и патогенез наследственных болезней
• Генные мутации
♦ По характеру изменений гена различают делеции, дупликации, инверсии, вставки, транзиции, миссенс- и нонсенс-мутации.
♦ По последствиям генных мутаций их классифицируют на нейтральные, регуляторные и динамические.
• Хромосомные мутации (аберрации) характеризуются изменением структуры отдельных хромосом. Последовательность нуклеотидов в генах обычно не меняется, но изменение числа или положения генов при аберрациях может привести к генетическому дисбалансу.
Различают внутрихромосомные, межхромосомные и изохромосомные аберрации.
• Изменения генома. Геномные мутации характеризуются изменением числа отдельных хромосом (моносомии и полисомии) или их гаплоидного набора (анеуплоидии и полиплоидии).
• Мутагены классифицируют по происхождению (источнику) на эндогенные и экзогенные, а по природе на физические, химические и биологические.
♦ Экзогенные мутагены. К ним относятся многочисленные факторы внешней среды (например, радиационное излучение, алкилирующие агенты, окислители, многие вирусы).
♦ Эндогенные мутагены образуются при жизнедеятельности организма (например, свободные радикалы).
♦ Физические мутагены - ионизирующее излучение и температурный фактор.
♦ Химические мутагены - сильные окислители или восстановители (например, нитраты, нитриты, активные формы кислорода), алкилирующие агенты, пестициды (например, гербициды, фунгициды); некоторые пищевые добавки (например, ароматические углеводороды, цикламаты), продукты переработки нефти, органические растворители, лекарственные средства (например, цитостатики, содержащие ртуть средства, иммунодепрессанты).
♦ Биологические мутагены - вирусы (например, кори, краснухи, гриппа и др.); Аг некоторых микроорганизмов, транспозоны, онкогены.
• Частота мутаций. Средняя частота возникновения мутаций в структурных локусах оценена в пределах от 10-5 до 10-6 на одну гамету за каждое поколение. Весь геном содержит 3х109 пар оснований, около 23 тыс. генов. Следовательно, каждое последующее поколение приобретает несколько десятков мутаций. В Каталоге наследственных заболеваний человека OMIM перечислено около 7000 моногенных болезней (вызываемых мутациями конкретного гена). Для значительного числа пора- жённых генов идентифицированы разные аллели, количество которых для некоторых болезней достигает десятков и сотен.
Наследственные формы патологии
Для наследственных форм патологии приняты определения, перечисленные ниже.
• Наследственные - болезни, причиной которых является генная, хромосомная или геномная мутация. Они, как правило (но не всегда) передаются от родителей потомкам.
• Генные - болезни, вызываемые генными мутациями.
• Хромосомные - болезни, возникающие вследствие хромосомных
и геномных мутаций.
• Болезни с наследственной предрасположенностью (мультифактори-
альные, многофакторные) - болезни, развивающиеся в результате взаимодействия определённых комбинаций аллелей разных локусов и воздействий факторов окружающей среды.
• Генетические болезни соматических клеток: злокачественные ново-
образования (изменения в генетическом материале являются этиологическими для злокачественного роста) и врождённые пороки, развившиеся вследствие мутаций.
• Семейные - болезни, наблюдающиеся у двух и более членов семьи в одном или нескольких поколениях. Термин применяют для нозологических единиц, когда с высокой степенью вероятности подозревают их наследуемую природу, но наличие генетического дефекта не установлено.
• Врождённые - болезни, проявившиеся при рождении (они могут
быть наследственными и ненаследственными).
• Врождённый порок развития - морфологический дефект органа, части его или большой области тела, возникший в результате нарушенного органогенеза. Врождённые пороки развития могут быть наследственными и приобретёнными (под действием тератогенов во внутриутробном периоде).
ГЕННЫЕ БОЛЕЗНИ
Типы наследования. Для любого моногенного заболевания существенной характеристикой является тип наследования: аутосомно-доминант- ный, аутосомно-рецессивный, сцепленный с хромосомой X (доминантный и рецессивный), голандрический (сцепленный с хромосомой Y) и митохондриальный.
♦ При заболеваниях с рецессивным типом наследования фенотип гетерозиготы может не отличаться от нормы (т.е. иметь слабые проявления заболевания или не иметь их).
♦ При заболеваниях с доминантным типом наследования пациенты в гетерозиготном состоянии имеют практически ту же клиническую картину, что и в гомозиготном состоянии, но проявления болезни у гомозигот тяжелее.
Аутосомно-доминантный тип наследования
Примеры: синдром Марфана, гемоглобиноз M, хорея Хантингтона, полипоз толстой кишки, семейная гиперхолестеринемия, нейрофиброматоз, полидактилия. Родословная с аутосомно-доминантным типом наследования (синдром Марфана в 5 поколениях) представлена на рис. 3-1А.
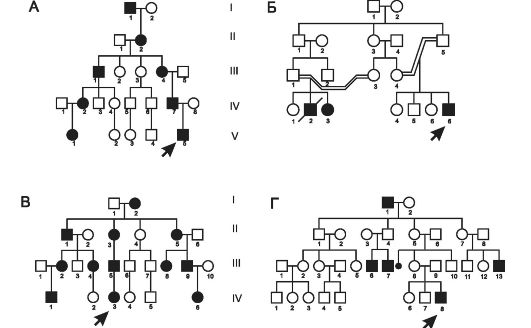 Рис. 3-1. Родословные с разными типами наследования заболеваний. А
- аутосомно-доминантный; Б - аутосомно-рецессивный; В - доминантный
Х-сцепленный; Г - рецессивный Х-сцепленный. Римские цифры - поколения.
Кружок - пол женский, квадрат - пол мужской, тёмный кружок или квадрат -
больной, наискось перечёркнутый тёмный кружок или квадрат - умерший
больной. Стрелкой указан пробанд - больной или носитель изучаемого
признака.
Рис. 3-1. Родословные с разными типами наследования заболеваний. А
- аутосомно-доминантный; Б - аутосомно-рецессивный; В - доминантный
Х-сцепленный; Г - рецессивный Х-сцепленный. Римские цифры - поколения.
Кружок - пол женский, квадрат - пол мужской, тёмный кружок или квадрат -
больной, наискось перечёркнутый тёмный кружок или квадрат - умерший
больной. Стрелкой указан пробанд - больной или носитель изучаемого
признака.
Особенности наследования: ❖ один из родителей пациента, как правило, болен; ❖ выраженность и количество проявлений зависят от действия факторов среды; ❖ частота патологии у лиц мужского и женского пола одинакова; ❖ в каждом поколении имеются больные (так называемый вертикальный характер распределения болезни); ❖ вероятность рождения больного ребёнка равна 50% (независимо от пола ребёнка и количества родов); ❖ непоражённые члены семьи, как правило, имеют здоровых потомков (поскольку не имеют мутантного гена).
Аутосомно-рецессивный тип наследования
Примеры: фенилкетонурия, адреногенитальный синдром, кожно-глазной альбинизм, галактоземия, гликогенозы, гиперлипопротеинемии, муковисцидоз. Родословная с аутосомно-рецессивным типом наследования (муковисцидоз в 4 поколениях) представлена на рис. 3-1Б. Особенности наследования: ❖ родители больного, как правило, здоровы; заболевание может обнаруживаться у других родственников (например, у двоюродных или троюродных братьев/сестёр больного);
❖ однообразные проявления болезни (в связи с высокой пенетрантностью); ❖ симптомы болезни обычно выявляются уже в детском возрасте; ❖ частота патологии у лиц мужского и женского пола равная; ❖ в родословной патология проявляется по горизонтали, часто у сибсов; ❖ заболевание отсутствует у единокровных (дети одного отца от разных матерей) и единоутробных (дети одной матери от разных отцов) братьев и сестёр; ❖ появление аутосомно-рецессивной патологии более вероятно при кровнородственных браках за счёт большей вероятности встречи двух супругов, гетерозиготных по одному и тому же патологическому аллелю, полученному от их общего предка.
Сцепленное с хромосомой X доминантное наследование
Примеры: одна из форм гипофосфатемии - витамин D-резистент- ный рахит, болезнь Шарко-Мари-Тута X-сцепленная доминантная, рото-лице-пальцевой синдром типа I. Родословная с доминантным X-сцепленным типом наследования витамин D-резистентного рахита в четырёх поколениях представлена на рис. 3-1В. Особенности наследования: ❖ поражение лиц мужского и женского пола;
❖ у мужчин более тяжёлое течение заболевания; ❖ передача больным мужчиной патологического аллеля только дочерям, но не сыновьям (сыновья получают от отца хромосому Y); ❖ передача больной женщиной заболевания и сыновьям, и дочерям с равной вероятностью.
Сцепленное с хромосомой X рецессивное наследование
Примеры заболеваний: гемофилия A, гемофилия B, дальтонизм, мышечная дистрофия Дюшенна-Беккера, болезнь Хантера (мукопо-
лисахаридоз типа II), гипогаммаглобулинемия брутоновского типа. Родословная с рецессивным X-сцепленным типом наследования (гемофилия A в 4 поколениях) представлена на рис. 3-1Г. Признаки заболевания: ❖ больные рождаются в браке фенотипически здоровых родителей; ❖ заболевание наблюдается исключительно у лиц мужского пола; ❖ матери больных - облигатные носительницы патологического гена; ❖ сын никогда не наследует заболевание от отца;
❖ у носительницы мутантного гена вероятность рождения больного ребёнка равна 25% (50% родившихся мальчиков - больные).
Голандрический, или сцепленный с хромосомой Y, тип наследования
Примеры: гипертрихоз ушных раковин, избыточный рост волос на средних фалангах пальцев кистей, азооспермия.
Особенности наследования: ❖ передача признака от отца всем сыновьям (только сыновьям, дочери никогда не наследуют признак от отца);
❖ «вертикальный» характер наследования признака; ❖ вероятность наследования для лиц мужского пола равна 100%;
Митохондриальное наследование
Примеры заболеваний («митохондриальные болезни»): атрофия зрительного нерва Лебера, синдромы Лея (митохондриальная миоэнцефалопатия), MERRF (миоклоническая эпилепсия), кардиомиопатия дилатационная семейная.
Особенности наследования: ❖ наличие патологии у всех детей больной матери; ❖ рождение здоровых детей у больного отца и здоровой матери (объясняется тем, что митохондриальные гены наследуются от матери).
ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные болезни выявляются у новорождённых с частотой около 6:1000. Инициальное звено патогенеза - геномная или хромосомная мутация. Тяжесть нарушений обычно прямо коррелирует со степенью хромосомного дисбаланса: чем больше хромосомного материала вовлечено в аберрацию, тем раньше проявляется хромосомный дисбаланс в онтогенезе и тем значительнее нарушения физического и психического развития индивида.
Особенности: ❖ большинство геномных мутаций (полиплоидии, трисомии по крупным хромосомам [рис. 3-2], моносомии по аутосомам) летальны; ❖ мутации в гаметах приводят к развитию так называемых полных форм хромосомных болезней, когда изменения кариотипа выявляются во всех клетках организма; ❖ мутации в соматических клетках на ранних этапах эмбриогенеза приводят к развитию мозаи-
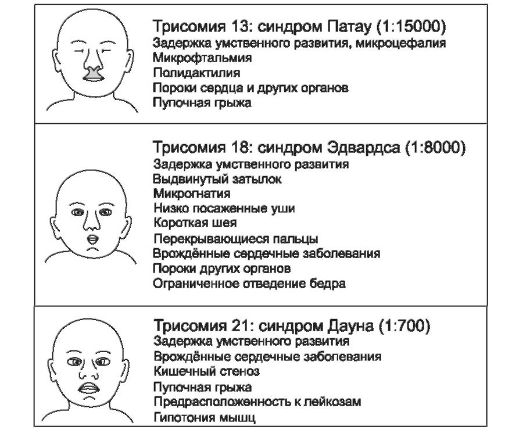 Рис. 3-2. Характеристика наиболее частых аутосомных трисомий [по 4].
Рис. 3-2. Характеристика наиболее частых аутосомных трисомий [по 4].
цизма: часть клеток организма имеет нормальный кариотип, а другая часть - аномальный.
Аномалии половых хромосом. Нарушение расхождения половых хромосом приводит к образованию аномальных гамет: у женщин - XX и 0 (в последнем случае гамета не содержит половых хромосом); у мужчин - XY и 0. При слиянии половых клеток в подобных случаях возникают количественные нарушения половых хромосом. При болезнях, вызванных дефицитом или избытком Х хромосом, нередко наблюдается мозаицизм.
• Синдром Кляйнфелтера: ❖ Частота: 2-2,5 на 1000 новорождённых мальчиков. ❖ Кариотип: разнообразные цитогенетические варианты (47,XXY; 48,XXXY; 49,XXXXY и др.), но чаще встречается вариант 47,XXY. ❖ Проявления: высокий рост, непропорционально длинные конечности, отложение жира по женскому типу, евнухоидное телосложение, скудное оволосение, гинекомастия, гипогенитализм, бесплодие (в результате нарушения сперматогенеза, снижения продукции тестостерона и увеличения продукции женских половых гормонов), снижение интеллекта (чем больше в кариотипе добавочных хромосом, тем более выражено). ❖ Лечение мужскими половыми гормонами направлено на коррекцию вторичных половых признаков, но и после терапии больные остаются бесплодными.
• Трисомия X - наиболее частый синдром из группы полисомий X; частота 1:1000 новорождённых девочек, кариотип 47,XXX; пол -
женский, фенотип женский; как правило, физическое и психическое развитие у женщин с этим синдромом не имеет отклонений от нормы.
• Синдром Шерешевского-Тёрнера. ❖ Частота синдрома: 1:3000 но- ворождённых девочек ❖ Кариотип: 45,Х0, но встречаются и другие варианты. ❖ Проявления: низкий рост, короткая шея с избытком кожи или крыловидной складкой, широкая, часто деформированная грудная клетка, деформация локтевых суставов, недоразвитие первичных и вторичных половых признаков, бесплодие. ❖ Раннее лечение женскими половыми гормонами может оказаться эффективным.
БОЛЕЗНИ С НАСЛЕДСТВЕННЫМ ПРЕДРАСПОЛОЖЕНИЕМ
Болезни с наследственным предрасположением называют также многофакторными (мультифакториальными), так как их возникновение определяется взаимодействием наследственных факторов и факторов внешней среды. К болезням с наследственным предрасположением относятся ишемическая болезнь сердца (ИБС), гипертоническая болезнь, бронхиальная астма, психические заболевания, СД, ревматические болезни, язвенная болезнь желудка, врождённые пороки развития (ВПР) и многие другие. Болезни с наследственным предрасположением классифицируют - в зависимости от числа генов, определяющих предрасположенность, - на моногенные и полигенные.
• Моногенные болезни с наследственным предрасположением детерминируются одним мутантным геном и возникают при действии конкретного и обязательного фактора внешней среды. Пример - непереносимость лактозы: при мутантной форме гена лактазы употребление молока приводит к развитию кишечного дискомфорта и поноса.
• Полигенные болезни. Предрасположенность к развитию полигенных болезней детерминируется взаимодействием нормальных и изменён- ных (мутировавших) генов, хотя каждый из них по отдельности не приводит к развитию заболевания. Индивид с такой комбинацией генов под действием определённого фактора окружающей среды достигает «порога возникновения» болезни и заболевает.
Характеристика многофакторных болезней: ❖ наследование не отвечает менделевским закономерностям; ❖ патогенез зависит от «удельного вклада» генетических и средовых факторов; эта зависимость различна как для разных заболеваний, так и для каждого человека; ❖ характерно наличие большого числа клинических вариантов; ❖ наблюдается более высокая конкордантность по заболеванию у монозиготных близнецов в сравнении с дизиготными.
Врождённые пороки развития
Аномалии развития (в том числе врождённые пороки - ВПР) и их причины изучает тератология. Распространённость ВПР составляет 2-3% от общего количества родившихся живыми детей.
• Типы ВПР. В зависимости от времени воздействия повреждающих факторов выделяют гаметопатии, бластопатии, эмбриопатии и фетопатии.
♦ Гаметопатии - результат воздействия на половые клетки (в основе лежат мутации в половых клетках).
♦ Бластопатии - следствие поражения бластоцисты - зародыша первых 15 сут после оплодотворения (до завершения формирования зародышевых листков). Результатом бластопатий являются двойниковые пороки (сросшиеся близнецы), циклопия (наличие одного или двух слившихся глазных яблок в единственной орбите по срединной линии лица).
♦ Эмбриопатии - результат воздействия тератогенного фактора на эмбрион в период с 16-го дня до 8 недели беременности. К этой группе относятся талидомидные, диабетические, алкогольные и некоторые медикаментозные эмбриопатии, а также ВПР, развившиеся под влиянием вируса краснухи.
♦ Фетопатии - следствие повреждения плода от 9-й недели до момента рождения. К фетопатиям относятся, например, крипторхизм, открытый боталлов проток или пренатальная гипоплазия какого-либо органа или плода в целом.
• Категории ВПР:
♦ агенезия - полное отсутствие органа (например, тимуса, почки, глаз);
♦ аплазия и гипоплазия - отсутствие или значительное недоразвитие органа при наличии его сосудистой ножки и нервов (например, одной почки, селезёнки, лёгкого, кишечника);
♦ атрезия - полное отсутствие канала или естественного отверстия (например, атрезия наружного слухового прохода, пищевода, ануса);
♦ гетеротопия - перемещение клеток, тканей или части органа в другую ткань (например, клеток поджелудочной железы в дивертикул Меккеля, хромаффинных клеток в ткань лёгких);
♦ персистирование - сохранение эмбриональных структур, исчезающих в норме к определённому этапу развития (например, открытый артериальный проток у годовалого ребёнка, крипторхизм);
♦ стеноз - сужение просвета отверстия или канала (например, клапанного отверстия сердца, привратника желудка, фрагмента кишечника);
♦ удвоение (утроение) - увеличение числа органов или его части (например, удвоение матки, мочеточников);
♦ эктопия - необычное расположение органа (например, почки в малом тазу, сердца - вне грудной клетки).
• Уродства (как правило, дефекты морфогенеза) - наиболее тяжёлые проявления ВПР.
• Дисплазии (мальформации, деформации, дизрупции) - морфологические врождённые изменения, выходящие за пределы общепринятой нормы.
• Малые аномалии развития (стигмы дизэмбриогенеза: синдактилия, ямочки на щеках, аномалии ушных раковин, искривление мизинца и др.) - врождённые дефекты, не требующие косметической или медицинской коррекции.
• Клинически значимые пороки развития - врождённые аномалии, требующие тех или иных форм медицинского вмешательства (квалифицированной диагностики, медицинской коррекции). Степень тяжести врождённого порока может быть различной: от малых аномалий (например, полидактилия) до очень тяжёлых системных поражений (гидроцефалия, болезнь Дауна).
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
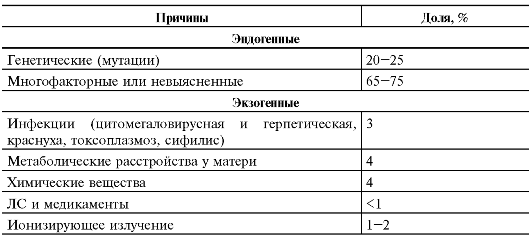
На развитие организма оказывают влияние как генетические факторы, так и факторы окружающей среды. Факторы, приводящие к развитию ВПР, обозначают как тератогены. Большинство врождённых пороков обусловлено воздействием факторов внешней среды, генетическими дефектами или их сочетанием (табл. 3-1). В ряде случаев не удаётся установить причину врождённого дефекта (спорадические болезни).
Таблица 3-1. Причины врождённых аномалий
 Тератогенные агенты
Тератогенные агенты
• Ионизирующее излучение. Доза облучения и срок гестации определяют степень и характер аномалий плода. Так у детей, рождён- ных после атомных взрывов в Хиросиме и Нагасаки (внутриутробное облучение), наблюдали различные аномалии ЦНС и лейкозы. Однако, эти поражения возникали в случаях, когда плод подвергался облучению до 16 недель гестации, в период органогенеза; при облучении на более поздних сроках происходит задержка роста на фоне нормального умственного развития.
♦ Стадия внутриутробного развития плода (рис. 3-3). Степень воздействия на эмбрион зависит от срока беременности на момент воздействия: ❖ 2-4 нед. после оплодотворения: плод либо развивается нормально, либо гибнет; ❖ 4-12 нед.: возникают микроцефалия, умственная отсталость, катаракта, задержка роста, микрофтальмия; ❖ 12-16 нед: развивается умственная отсталость или задержка роста; ❖ после 20 нед: повреждение волосяных фолликулов, поражение кожи и слизистых оболочек, угнетение красного костного мозга.
♦ Доза: ❖ дозу облучения 5-10 рад считают нетератогенной; ❖ 10- 25 рад - возможно повреждающее действие на плод; ❖ более 25 рад - часто возникают структурные пороки развития, задержка роста и гибель плода. После воздействия такой дозы рекомендуют прерывание беременности (медицинский аборт).
• Лекарственные препараты (ЛС). Американская Федеральная Комиссия по пищевым продуктам и ЛС (FDA) предложила все ЛС подразделять на 5 категорий:
♦ A. ЛС совершенно безвредны для плода (например, витамины).
♦ B. Опыты на животных не выявили тератогенности, но нет контрольных исследований на беременных. В эту категорию также входят ЛС, оказывающие повреждающее воздействие на животных, но не на человека (например, пенициллин, дигоксин, адреналин, тербуталин).
♦ C. Исследования на животных показали или тератогенное, или эмбриотоксическое воздействие ЛС на плод, но исследования на людях не проводились. Эти ЛС можно применять только в тех случаях, когда польза от их применения перевешивает потенциальный риск для плода (фуросемид, гуанидин, верапамил).
♦ D. Есть доказательства тератогенности ЛС. Однако, польза от его применения при определённых обстоятельствах превышает риск для плода (например, фенитоин).
♦ X. Исследования на животных и людях выявили очевидную опасность для плода. ЛС этой категории противопоказаны беремен-
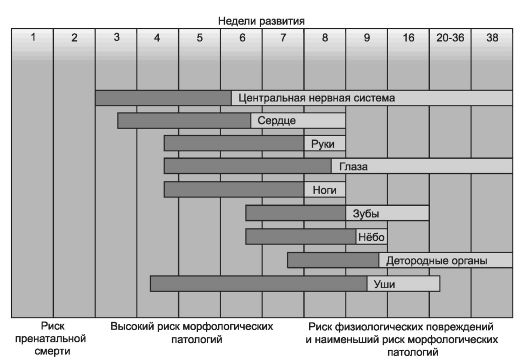 Рис. 3-3. Критические сроки развития возможных пороков развития по системам органов, [по 4].
Рис. 3-3. Критические сроки развития возможных пороков развития по системам органов, [по 4].
ным или женщинам, желающим забеременеть (например, изотретиноин).
• Алкоголь - один из наиболее распространённых тератогенов. Количество употребляемого алкоголя коррелирует со степенью вредного воздействия на плод. Выраженность поражения (сильная, слабая или её отсутствие) во многом зависит от генетической предрасположенности. В настоящее время нет данных о безопасной дозе потребления алкоголя во время беременности. В связи с этим рекомендован полный отказ от алкоголя во время беременности.
• Наркотики:
♦ Марихуана. У женщин, курящих марихуану во время беременности, повышена частота выкидышей и преждевременных родов.
♦ Героин. Побочные продукты синтеза, встречающиеся в недостаточно очищенном героине, часто обладают выраженным тератогенным эффектом. Основное неблагоприятное действие на плод при употреблении героина состоит в развитии выраженного «синдрома отмены» у новорождённого, что в 3-5% случаев приводит к гибели ребёнка. Метадон (аналог героина) обладает такими же свойствами.
♦ Фенилциклидин (ангельская пыль) иногда вызывает развитие дефектов лица у плода.
♦ Кокаин. При употреблении беременной кокаина увеличивается риск развития врождённых аномалий, гибели плода и рождения детей с малой массой тела.
• Гипертермия. Длительный подъём температуры (до 38,9 °С и выше) у женщины в период с 4 по 14 нед. беременности обладает большим тератогенным эффектом, чем кратковременные подъёмы до тех же цифр.
• Вещества, загрязняющие окружающую среду, можно рассматривать как тератогены, хотя изучение их влияния представляет большие трудности.
• Инфекционные заболевания матери. Воздействие вирусных, паразитарных и бактериальных инфекций на плод во время беременности - одна из частых причин развития врождённых дефектов. Большинство детей, матери которых были инфицированы в I триместре беременности, рождается с различными пороками развития и малой массой тела.
♦ Вирус краснухи. При заражении краснухой на первом месяце беременности вероятность развития аномалий плода составляет 50%. Риск снижается до 22% при инфицировании на втором месяце и до 6-10% на третьем-четвёртом месяце беременности.
♦ Цитомегаловирус поражает плод в 1-2% случаев всех беременностей. От 1 до 3 из 10 000 новорождённых страдает серьёзными пороками развития.
♦ Вирус простого герпеса 2-го типа. Хотя герпетическая инфекция встречается довольно часто, её передача от больной беременной плоду происходит менее чем в 0,02% случаев. Ещё реже возникают пороки развития, возможно из-за того, что инфицирование плода в I триместре беременности обычно приводит к его гибели.
♦ Токсоплазма. Количество детей с врождённым токсоплазмозом колеблется от 1 до 6 на 1 000 новорождённых. Внутриутробное заражение плода происходит у 30% инфицированных беременных.
♦ Treponema pallidum способна проходить через плацентарный барьер на любом сроке беременности, но заражение плода редко происходит до 16-18 нед. гестации. Последствия внутриутробного инфицирования: преждевременные роды или выкидыш, гибель плода, смерть 50% заражённых новорождённых, врождённый сифилис.
♦ Вирус ветряной оспы. Первичное инфицирование проявляется в виде ветряной оспы, рецидив заболевания называют опоясывающим лишаем. Во время беременности трансплацентарная передача вируса плоду в 5% случаев происходит в I триместре беременности и приблизительно в 24% случаев, если заражение женщины произошло на последнем месяце беременности. При опоясывающем лишае инфицирования плода не происходит.
♦ Энтеровирусы. Инфицирование матери вирусом Коксаки вызывает пороки развития или гибель плода в 40% случаев.
Методы диагностики
• Клинико-синдромологический метод позволяет выявлять морфологические, биохимические и функциональные признаки наследственных форм патологии (например, дефицит плазменного фактора VIII при подозрении на гемофилию A; кариотип 45,Х0 при подозрении на синдром Шерешевского-Тёрнера; поражения скелета, сердечнососудистой системы и глаз при подозрении на синдром Марфана).
• Клинико-генеалогический метод позволяет выявить патологические признаки и проследить особенности их передачи в поколениях при составлении родословной.
♦ Составление родословной начинают со сбора сведений о семье консультирующегося или пробанда. Терминология: пробанд - больной или носитель изучаемого признака, сибсы (братья и сёс- тры) - дети одной родительской пары, семья - в узком смысле родительская пара и их дети, но иногда и более широкий круг кровных родственников, хотя в последнем случае лучше применять термин род.
♦ Близнецовый метод базируется на сравнительном анализе частоты определённого признака в разных группах близнецов, а также в сопоставлении с партнёрами монозиготных пар между собой и общей популяцией. Идентичность близнецов по анализируемому признаку обозначают как конкордантность, а отличие - как дискордантность. Роль наследственности и факторов среды в возникновении патологии у близнецов оценивают по специальным формулам.
• Цитогенетическая диагностика основана на микроскопическом изучении хромосом с целью выявления структурных нарушений в хромосомном наборе (кариотипирование). В качестве материала используют тканевые культуры с большим числом делящихся клеток, чаще лимфоциты периферической крови. Хромосомы на стадии метафазы изучают при помощи специальных методов окрашивания и составляют идиограммы (систематизированные кариотипы с расположением хромосом от наибольшей к наименьшей), что позволяет выявлять геномные и хромосомные мутации.
• Биохимическая диагностика базируется на изучении биохимических показателей, отражающих сущность болезни (например, активность ферментов, наличие патологических метаболитов, концентрация компонентов ферментативной реакции).
• Молекулярная диагностика. При помощи методов ДНК-диагностики устанавливают последовательность расположения отдельных нуклеотидов, выделяют гены и их фрагменты, устанавливают их наличие в изучаемых клетках. К числу наиболее эффективных методов относятся гибридизация ДНК, клонирование ДНК, полимеразная цепная реакция.
♦ Гибридизация ДНК. Для определения порядка расположения нуклеотидов в исследуемом генетическом материале изучаемую ДНК помещают в специальную среду, где происходит контакт ДНК с нитями другой нуклеиновой кислоты. В случае комплементарности каких-либо двух нитей происходит их «сшивка». При специальных исследованиях используют генетические «зонды» - фрагменты меченной радиоактивным изотопом однонитевой ДНК с известной последовательностью нуклеотидов.
♦ Блот-гибридизация. Для выявления интересующих (в том числе мутантных) генов ДНК подвергают рестрикции, разделяют по молекулярной массе, денатурируют и переносят на носитель (нейлоновую или иную мембрану). Фиксированную на носителе в виде пятна ДНК гибридизируют с меченым радиоактивным изотопом ДНКили РНК-зондом. В результате определяют положение аномального фрагмента ДНК.
♦ Клонирование ДНК. С помощью специализированных ферментов (ДНК-рестриктаз) подразделяют нить ДНК на отдельные группы генов или на единичные гены. Для изучения признаков (в том числе патологических), кодируемых данными генами, особенностей транскрипции и трансляции создают нужное количество копий данного гена.
♦ Полимеразная цепная реакция (специфическая амплификация ДНК). Применяют для изучения локусов предполагаемых мутаций и других особенностей структуры ДНК. Для исследования можно использовать любой биологический материал, содержащий ДНК (например, кусочек ткани, капля или пятно крови, смыв полости рта, луковица корня волос). На первом этапе исследуемую ДНК подвергают отжигу: расщепляют на две нити при нагревании до 95-98 °C. Затем одну из нитей гибридизируют и стимулируют синтез последовательности, комплементарной исследуемой ДНК (с помощью ДНК-полимеразы). В первом цикле полимеразной цепной реакции гибридизацию выполняют с исследуемым фрагментом ДНК, а в последующих - с вновь синтезированными. При каждом цикле реакции число синтезированных копий участка ДНК увеличивается двукратно. Циклы повторяют до накопления нужного количества ДНК.
Принципы лечения
Лечение наследственных болезней базируется на трёх принципах: этиотропном, патогенетическом и симптоматическом.
• Этиотропная терапия направлена на устранение причины заболевания. С этой целью разрабатываются, апробируются и частично могут быть применены методы коррекции генетических дефектов, называемые генной терапией.
• Патогенетическая терапия имеет целью разрыв звеньев патогенеза. Для достижения этой цели применяют несколько методов.
♦ Заместительная терапия - введение в организм дефицитного вещества (не синтезирующегося в связи с аномалией гена, который контролирует продукцию данного вещества; например, инсулина при СД, антигемофильного глобулина человека при гемофилии).
♦ Коррекция метаболизма путём: ❖ ограничения попадания в организм веществ, метаболически не усваивающихся (например, фенилаланина или лактозы); ❖ выведения из организма метаболитов, накапливающихся в нём в избытке (например, фенилпировиноградной кислоты или холестерина); ❖ регуляции активности ферментов (например, подавление активности КФК при
отдельных видах миодистрофий, активация липопротеинлипазы крови при гиперхолестеринемии). ♦ Хирургическая коррекция дефектов (например, создание шунта между нижней полой и воротной венами у пациентов с «гепатотропными» гликогенозами).
• Симптоматическая терапия. Направлена на устранение симпто-
мов, усугубляющих состояние пациента (например, применение веществ, снижающих вязкость секретов экзокринных желёз при муковисцидозе; хирургическое удаление дополнительных пальцев и перемычек кожи между ними при поли- и синдактилии; выполнение пластических операций при дефектах лица, пороках сердца и крупных сосудов).
Профилактика
Всем семьям, имеющим случаи наследственных заболеваний, т.е. при повышенной вероятности рождения ребёнка с патологией необходимо проводить медико-генетическое консультирование, задачи которого - выявление генетических заболеваний и определение возвратного риска.
• Выявление генетических заболеваний. В первую очередь необходима
точная диагностика, позволяющая определить природу заболевания и отдифференцировать состояния, имеющие сходную клиническую картину.
• Определение возвратного риска. При установлении точного диагноза
становится возможным рассчитать вероятность повторного случая заболевания. В связи с этим необходима пренатальная диагностика.
• Анализ родословной (см. рис. 3-1) - первый этап медико-генетичес-
кого консультирования. Необходимо собрать полную информацию о состоянии здоровья всех членов семьи (не менее четырёх поколений).