Фармакология / Под ред. проф. Р.Н. Аляутдина. - 4-е изд., перераб. и доп. - 2008. - 832 с. : ил.
|
|
|
|
ГЛАВА 1 ФАРМАКОКИНЕТИКА
Фармакокинетические процессы - всасывание, распределение, депонирование, биотрансформация и выведение - связаны с проник- новением ЛВ через биологические мембраны (в основном через цитоплазматические мембраны клеток) и через межклеточные промежутки.
Существуют следующие способы проникновения веществ через биологические мембраны: пассивная диффузия, перенос веществ через мембраны c помощью транспортных систем (активный транс- порт, облегченная диффузия) (рис. 1-1), пиноцитоз.
Пассивная диффузия. Путем пассивной диффузии вещества про- никают через мембраны по градиенту концентрации (если концентрация вещества с одной стороны мембраны выше, чем с другой, вещество перемещается через мембрану от большей концентрации
к меньшей). Этот процесс не требует затраты энергии. Поскольку биологические мембраны в основном состоят из липидов, через них, как правило, легко проникают незаряженные соединения, хорошо растворимые в липидах, т.е. липофильные неполярные вещества. При этом пассивная диффузия веществ через липиды зависит от их относительной липофильности, т.е. коэффициента распределения веществ между органическим растворителем и водой. Как правило, вещества с высоким коэффициентом распределения проникают через липиды мембран лучше веществ с низкими значе- ниями этого коэффициента. Заряженные соединения, хорошо рас- творимые в водной среде и малорастворимые в липидах, т.е. гидрофильные полярные вещества непосредственно через липиды мембран практически не проникают.
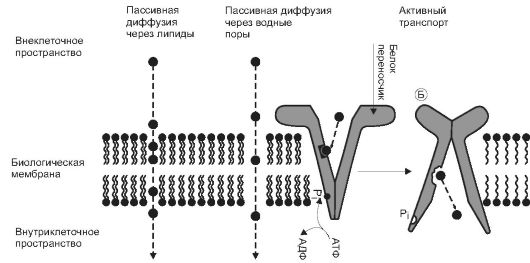
Рис. 1-1. Основные способы проникновения веществ через биологические мем- браны
Многие ЛВ являются слабыми кислотами или слабыми основа- ниями, т.е. слабыми электролитами. В водной среде такие вещества частично ионизированы. Поскольку путем пассивной диффузии через двойные липидные слои мембран легко проходят только неионизиро- ванные молекулы, проникновение слабых кислот и слабых оснований через мембраны зависит от степени их ионизации.
Степень ионизации слабых кислот и слабых оснований определя- ется значениями рН среды и константой ионизации (Ка) веществ.
Слабые кислоты в большей степени ионизированы в щелочной среде, а слабые основания - в кислой.

Константа ионизации (Ка) характеризует способность вещества к ионизации при определенном значении рН среды (численно равна концентрации водородных ионов в среде, при которой ионизирована половина молекул данного вещества).
На практике для характеристики способности веществ к иониза- ции используют показатель рКа, который является отрицательным логарифмом Ка (-lg Ka). Показатель рКа численно равен значению рН среды, при котором ионизирована половина молекул данного веще- ства. Значения рКа слабых кислот и слабых оснований варьируют в широких пределах. Чем меньше рКа слабой кислоты, тем легче она ионизируется даже при относительно низких значениях рН среды. Так, ацетилсалициловая кислота (рКа=3,5) при рН=4,5 ионизиро- вана более чем на 90%, а степень ионизации аскорбиновой кислоты (рКа=11,5) при том же значении рН составляет доли процента (рис. 1-2). Для слабых оснований существует обратная зависимость. Чем выше рКа слабого основания, тем в большей степени оно иони- зировано даже при относительно высоких значениях рН среды.
Степень ионизации слабой кислоты или слабого основания можно рассчитать по формуле Гендерсона-Гассельбальха:
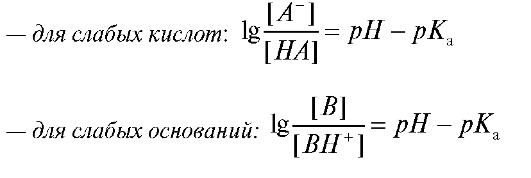
Эта формула позволяет определить степень проникновения ЛВ (слабых кислот или слабых оснований) через мембраны, разделяющие среды организма с различными значениями рН, например при всасы- вании ЛВ из желудка (рН = 1,0-2,0) в плазму крови (рН = 7,4) или при реабсорбции ЛВ из почечных канальцев (рН = 5,0-8,0).
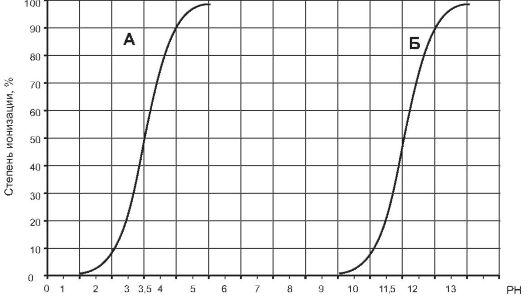
Рис. 1-2. Зависимость степени ионизации слабых кислот от рН среды и рКа соединений: А - ацетилсалициловая кислота (рКа = 3,5); Б - аскорбиновая кис- лота (рКа = 11,5)
Изменяя рН среды, можно изменить (увеличить или уменьшить) степень проникновения слабых кислот и слабых оснований через мембраны. Это может быть использовано в определенных клиничес- ких ситуациях, например, для ускорения выведения некоторых ЛВ почками.
Пассивная диффузия гидрофильных полярных веществ возможна через водные поры (аквапорины), белковые молекулы в мембра- не клеток, проницаемые для воды и растворенных в ней веществ (см. рис. 1-1). Однако такая пассивная диффузия (пассивная диффузия в водной среде) не имеет существенного значения для проникновения ЛВ через мембраны. Это объясняется тем, что диаметр водных пор невелик (приблизительно 0,3-0,4 нм), и через них проникают только вода и небольшие гидрофильные молекулы (например, мочевина или глицерин). Диаметр молекул большинства гидрофильных ЛВ превы- шает 1 нм, поэтому они не проходят через водные поры в мембране и, следовательно, не проникают в клетки.
Перенос веществ через мембраны с помощью специальных транспорт- ных систем. Специальными транспортными системами обычно явля- ются белковые молекулы (белки-переносчики), которые пронизывают клеточную мембрану и имеют специфические места связывания для определенных, как правило близких по структуре, веществ, что
обеспечивает их избирательный транспорт через мембраны. Белкипереносчики транспортируют вещества, которые не проникают через мембраны путем пассивной диффузии вследствие гидрофильности и больших размеров молекул.
Процесс переноса веществ через мембраны против градиента кон- центрации, требующий затраты энергии, обозначается как активный транспорт. Вещество связывается с белком-переносчиком с одной стороны мембраны. При участии энергии АТФ происходит изме- нение конформации белковой молекулы и перенос вещества через мембрану. Затем уменьшение силы связывания между переносчиком и транспортируемым веществом приводит к его высвобождению (см. рис. 1-1).
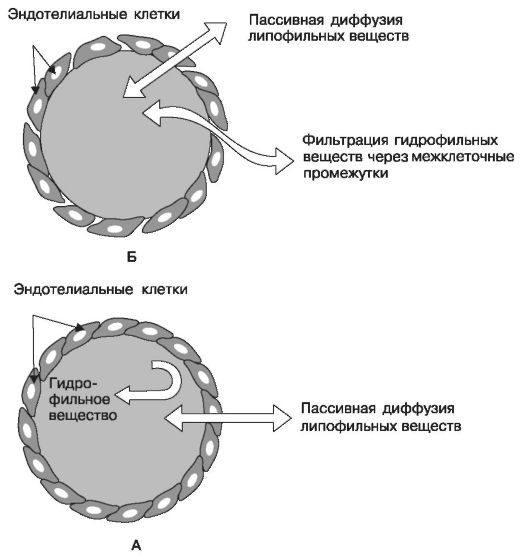
Рис. 1-3. Проникновение веществ через стенки капилляров мозга (А) и капилля- ров скелетных мышц (Б)
Активный транспорт веществ через мембраны характеризует- ся специфичностью (с белками-переносчиками связываются лишь определенные вещества) и насыщаемостью (при достижении определенной концентрации количество переносимого в единицу времени вещества, достигает предельной величины), происходит против градиента концентрации и требует затрат энергии (поэтому угнетается метаболическими ядами).
Перенос веществ через мембраны по градиенту концентрации (от большей концентрации к меньшей) называется облегченной диф- фузией. При этом изменение конформации белка-переносчика и, следовательно, перенос и высвобождение вещества с другой стороны мембраны происходит без потребления энергии. Подобно активному транспорту, облегченная диффузия - специфичный по отношению к определенным веществам и насыщаемый процесс.
Активный транспорт и облегченная диффузия обеспечивают транспорт через клеточные мембраны таких необходимых для жиз- недеятельности клеток веществ, как аминокислоты, сахара, пиримидиновые и пуриновые основания, железо, витамины. И только ЛВ, близкие к ним по химической структуре, способны проникать через клеточные мембраны с помощью тех же (специфичных) транспортных систем. Например, транспорт леводопы (диоксифенилаланина) через гематоэнцефалический барьер происходит при участии транспортно- го белка, переносящего через мембраны ароматические аминокислоты (см. главу «Противопаркинсонические средства»).
Кроме белков, которые переносят вещества внутрь клеток, сущест- вуют АТФ-зависимые транспортные белки P-гликопротеины, способствующие удалению из клеток чужеродных соединений. Эти белки нахо- дятся в мембранах энтероцитов, эндотелиальных клеток, гепатоцитов, эпителиальных клеток почечных канальцев. Они препятствуют всасы- ванию, проникновению через гистогематические барьеры и ускоряют выведение некоторых ЛВ из организма. Многие противоопухолевые вещества удаляются из клеток при участии Р-гликопротеинов, обна- руженных в мембранах клеток злокачественных опухолей, что нередко является причиной неэффективности противоопухолевой терапии. Некоторые ЛВ снижают (хинидин, лидокаин, верапамил) или повыша- ют (препараты зверобоя) активность Р-гликопротеинов. Так, хинидин, ингибируя Р-гликопротеин, осуществляющий перенос дигоксина из энтероцитов в просвет кишечника, повышает его концентрацию в крови, что увеличивает риск интоксикации этим препаратом.
Пиноцитоз (от греч. pino - пью). Крупные молекулы вещетва сопри- касаются с наружной поверхностью мембраны и окружаются ею с образованием пузырька (вакуоли), который отделяется от мембраны и погружается внутрь клетки. Далее содержимое пузырька может высвобождаться внутри клетки или наружу путем экзоцитоза.
Опосредованный рецепторами эндоцитоз. Вещество связывается с рецепторами, локализованными в клеточной мембране, в результате образуются комплексы вещество-рецептор, которые захватываются клетками при участии специальных цитоплазматических белков. Таким образом некоторые крупномолекулярные вещества, например инсулин, могут проникать внутрь клеток.
Парацеллюлярный транспорт. Большинство гидрофильных ЛВ вса- сывается, распределяется по органам и тканям и выводится из организма, не проникая через мембраны клеток. Гидрофильные вещества, рас- творяясь в интерстициальной жидкости, способны проникать в кровь, а из крови - в интерстициальную жидкость через межклеточные промежутки. Такой способ проникновения зависит от величины межкле- точных промежутков и обозначается как парацеллюлярный транспорт. Если перемещение гидрофильных веществ происходит под давлением (гидростатическим или осмотическим), используют термин «фильтра- ция». При этом исключается проникновение веществ, диаметр молекул которых превышает размер межклеточных промежутков.
Межклеточные промежутки в различных тканях не одинаковы по величине, поэтому гидрофильные ЛВ при различных путях введения всасываются в неодинаковой степени и неравномерно распределя- ются в организме. Промежутки между эпителиальными клетками слизистой оболочки кишечника невелики, что затрудняет всасывание гидрофильных ЛВ из кишечника в кровь. Аналогичными свойствами обладает цилиарный эпителий дыхательных путей, поэтому гидро- фильные соединения плохо всасываются с поверхности легких.
Промежутки между эндотелиальными клетками сосудов перифе- рических тканей (скелетных мышц, подкожной клетчатки, внутренних органов) имеют достаточно большие размеры (приблизительно 2 нм и более) и пропускают большинство гидрофильных ЛВ, что обеспечивает достаточно быстрое их проникновение из тканей в кровь и обратно. Эндотелиальные клетки сосудов головного мозга, наоборот, плотно прилегают к друг другу, образуя барьер (гематоэнцефалити- ческий барьер, ГЭБ), препятствующий проникновению гидрофильных полярных веществ (см. рис. 1-3).
1.1. ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
Всасывание (абсорбция, от лат. absorbeo - всасываю) - процесс, в результате которого вещество поступает с места введения в кровенос- ную и/или лимфатическую систему. Всасывание ЛВ начинается сразу после его введения. От пути введения ЛВ зависят скорость и степень его всасывания, и в конечном итоге скорость развития фармакологи- ческого эффекта, его величина и продолжительность.
Пути введения лекарственных средств
Различают энтеральные (через пищеварительный тракт) и паренте- ральные (минуя пищеварительный тракт) пути введения ЛС.
Энтеральные пути введения
К энтеральным (от греч. ento - внутри и enteron - кишка) путям введения относятся сублингвальный (под язык); трансбуккальный (за щеку); пероральный (внутрь, per os); ректальный (через прямую кишку, per rectum).
Сублингвальное и трансбуккальное введение. При сублингвальном и трансбуккальном путях введения через слизистую оболочку ротовой полости хорошо всасываются (путем пассивной диффузии) липо- фильные неполярные и незначительно - гидрофильные полярные вещества.
Сублингвальный и трансбуккальный пути введения ЛВ имеют следующие преимущества перед другими путями введения:
• простота и удобство для больного;
• отсутствие воздействия на ЛВ хлористоводородной кислоты;
• поступление ЛВ в общий кровоток, минуя печень, что предо- твращает их преждевременное разрушение и выделение с желчью (отсутствие эффекта первого прохождения через печень);
• быстрота всасывания ЛВ вследствие хорошего кровоснабжения слизистой оболочки полости рта, а следовательно, быстрота раз- вития терапевтического эффекта (возможность применения при неотложных состояниях).
Однако из-за небольшой всасывающей поверхности слизистой оболочки полости рта эти пути введения приемлемы лишь для высо- коактивных веществ, применяемых в малых дозах, таких как нитро-
глицерин, некоторые стероидные гормоны. Так, для устранения при- ступа стенокардии сублингвально применяют таблетки, содержащие 0,5 мг нитроглицерина - эффект развивается через 1-2 мин.
Пероральное введение. При введении внутрь лекарственные вещества всасываются в основном путем пассивной диффузии. Таким образом легко всасываются липофильные неполярные вещества. Всасывание гидрофильных полярных веществ ограничено из-за небольшой вели- чины межклеточных промежутков в эпителии ЖКТ. Некоторые гид- рофильные ЛВ (например, леводопа) всасываются при участии транспортных белков.
Всасывание слабокислых соединений (ацетилсалициловой кисло-
ты, барбитуратов и др.) начинается в желудке, в кислой среде которого
большая часть вещества не ионизирована. Однако основным местом
всасывания всех ЛВ, включая слабые кислоты, является кишечник. Этому
способствуют большая всасывающая поверхность слизистой оболочки
кишечника (
На всасывание ЛВ оказывает также влияние их способность рас- творяться в воде. Для достижения места всасывания вещество должно раствориться в содержимом кишечника (в водной среде). Поэтому из ЖКТ лучше всасываются вещества, обладающие не только липофиль- ными (что облегчает их проникновение через мембраны), но и, в определенной степени, гидрофильными свойствами. Так, прием внутрь слабых кислот и оснований в виде солей улучшает их всасывание, так как соли лучше растворяются в воде.
Определенное значение имеют также размер частиц ЛВ и лекарс- твенная форма. В большинстве случаев наилучшие условия для более полного и быстрого всасывания ЛВ создаются при их введении в виде жидких лекарственных форм (водных растворов, суспензий, микстур). Некоторые малорастворимые в воде вещества назначают в виде водно-спиртовых растворов. Всасывание ЛВ при их назначении в форме суспензий зависит от размера частиц вещества. Чем выше степень диспергирования вещества (и, следовательно, меньше размер частиц), тем выше скорость его всасывания. Степень диспергирова- ния вещества повышается при добавлении к суспензии поверхностноактивных веществ.
Большинство ЛС для приема внутрь применяют в виде твердых лекарственных форм (таблеток, капсул). В этом случае процесс всасы- вания ЛВ во многом зависит от скорости, с которой твердые лекарственные формы распадаются в кишечнике. Быстрая распадаемость (дезинтеграция) таблеток или капсул способствует более полному и быстрому всасыванию ЛВ. Для ускорения распадаемости таблеток в их состав включают специальные дезинтегрирующие вещества, спо- собствующие разрушению таблеток. После дезинтеграции таблеток (или капсул) процесс всасывания ЛВ зависит от скорости его раство- рения в содержимом кишечника и поступления к месту всасывания. Увеличение количества и уменьшение размеров микрочастиц, содер- жащих лекарственное вещество, ускоряют его всасывание.
Поскольку таблетки в ЖКТ распадаются достаточно медленно, различие в скорости и степени всасывания одного и того же вещества при введении его в виде таблеток и растворов может быть достаточно существенным. Так, через 30 мин после приема ацетилсалициловой кислоты в виде таблеток ее концентрация в плазме крови оказывает- ся в 2 раза ниже, чем при применении этого вещества в той же дозе в виде раствора. Для замедления всасывания и создания более посто- янной концентрации ЛВ в крови используют лекарственные формы с замедленным (контролируемым) высвобождением ЛВ. Таким образом можно получить препараты пролонгированного действия, которые действуют гораздо дольше (блокатор кальциевых каналов нифедипин в обычных лекарственных формах назначают 3 раза в сутки, а его про- лонгированные формы - 1-2 раза в сутки).
В ЖКТ лекарственные вещества подвергаются воздействию хло- ристоводородной кислоты и пищеварительных ферментов. Например, бензилпенициллин разрушается хлористоводородной кислотой желу- дочного сока, а инсулин и другие вещества полипептидной структуры - протеолитическими ферментами. Чтобы избежать разрушения некоторых веществ в кислой среде желудка, их назначают в специ- альных лекарственных формах (таблетках или капсулах с кислотоустойчивым покрытием). Такие лекарственные формы без изменения проходят через желудок и распадаются лишь в тонком кишечнике (так назывемые кишечнорастворимые лекарственные формы).
Количество и качественный состав содержимого ЖКТ также влияет на всасывание ЛВ. Жирная пища, задерживая опорожнение желудка и, следовательно, поступление ЛВ в кишечник, замедляет и уменьшает всасывание большинства ЛВ. Исключение составляют липофильные
плохо растворимые в воде вещества (например, жирорастворимые вита- мины), которые лучше всасываются в присутствии жиров. Компоненты пищи способны нарушать всасывание ЛВ. Так, например, ионы каль- ция, содержащиеся в молочных продуктах, образуют с антибиотиками тетрациклинового ряда плохо всасывающиеся комплексы, а компонент чая танин образует с препаратами железа нерастворимые танаты.
Некоторые лекарственные вещества существенно влияют на вса- сывание других ЛВ, принимаемых одновременно. Активированный уголь и другие энтеросорбенты подавляют всасывание практически всех ЛВ. Антациды препятствуют всасыванию противомикробных средств группы фторхинолонов, так как магний, кальций, алюми- ний, входящие в состав антацидов, образуют с фторхинолонами невсасывающиеся хелатные комплексы. Кроме того, антациды и антисекреторные средства (снижающие секрецию соляной кислоты), повышая рН содержимого желудка, нарушают всасывание слабых кислот (например, ацетилсалициловой кислоты, сульфаниламидов), но улучшают всасывание слабых оснований. Влияния одного ЛВ на всасывание другого можно избежать, если между их приемами сделать перерыв (2 ч и более).
Всасывание ЛВ при приеме внутрь зависит также от моторики ЖКТ. Поскольку кишечник является основным местом всасывания веществ, большинство ЛВ, в особенности слабые основания (про- пранолол, кодеин), которые находятся в щелочной среде кишечника преимущественно в неионизированной форме, всасываются более интенсивно при ускорении опорожнения желудка (например, при применении прокинетика метоклопрамида).
Усиление моторики кишечника может нарушить всасывание мед- ленно всасывающихся веществ, например дигоксина, поэтому его всасывание резко уменьшается при одновременном приеме с прокинетиком метоклопрамидом. При приеме ЛВ, угнетающих перис- тальтику кишечника, таких как М-холиноблокаторы (атропин), всасывание медленно всасывающихся веществ (дигоксина, препаратов железа) может резко увеличиться, что повышает риск возникновения их токсических эффектов.
Из тонкого кишечника вещества всасываются в воротную вену, с током крови поступают сначала в печень и лишь затем в системный кровоток (рис. 1-4). В печени многие ЛВ частично подвергаются био- трансформации (большинство веществ при этом инактивируется) и/или выделяются с желчью в просвет кишечника, поэтому в сис-
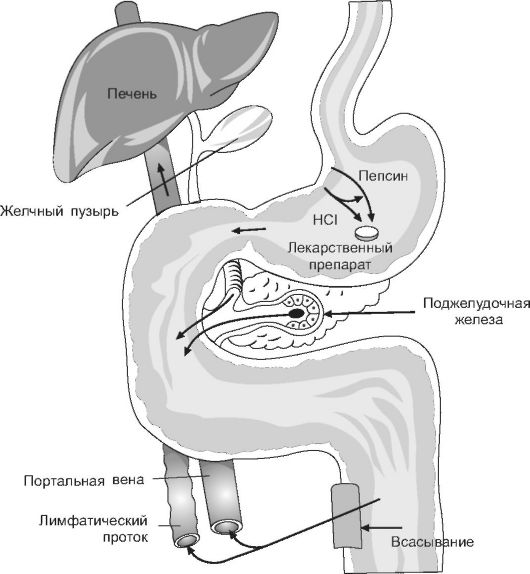
Рис. 1-4. Всасывание лекарственных веществ при введении внутрь
темный кровоток поступает лишь часть всосавшегося вещества. Этот процесс называется эффектом первого прохождения через печень или элиминацией при первом прохождении через печень (термин «элиминация» включает биотрансформацию и выведение ЛВ).
Большинство ЛВ оказывает действие только после достижения системного кровотока и последующего распределения по органам и тканям. О количестве ЛВ, которое достигло места своего действия, судят по общему количеству неизмененного вещества, поступившего в системный кровоток (считается, что существует прямая корреляция между количеством вещества в системном кровотоке и в органах и тканях, где оно оказывает свое действие). Для этого используют термин биодоступность.
Биодоступность определяют как часть введенной дозы ЛВ, кото- рая в неизмененном виде достигла системного кровотока, и обычно выражают в процентах. Биодоступность вещества при внутривен- ном введении принимают равной 100%. Биодоступность ЛВ при приеме внутрь, как правило, меньше 100%. В справочной литературе обычно приводят значения биодоступности ЛВ при введении внутрь.
Уменьшение биодоступности ЛВ при приеме внутрь может быть связано с различными причинами. Одни ЛВ частично разруша- ются под влиянием хлористоводородной кислоты и/или пищеварительных ферментов ЖКТ, другие неполностью всасываются в кишечнике (например, гидрофильные полярные соединения) или подвергаются метаболизму в стенке кишечника (например, леводопа, которая под действием ДОФА-декарбоксилазы превращается в дофамин).
Кроме того, многие вещества подвергаются интенсивной элимина- ции при первом прохождении через печень и поэтому имеют низкую биодоступность. Соответственно, дозы таких ЛВ при введении внутрь обычно превышают дозы, необходимые для достижения того же эффек- та при парентеральном или сублингвальном введении. Так, нитрогли- церин, который практически полностью всасывается из кишечника, но при первом прохождении через печень элиминирует более чем на 90%, назначают сублингвально в дозе 0,5 мг, а внутрь в дозе 6,4 мг.
Причиной низкой биодоступности препарата может быть неполное высвобождение действующего вещества из таблетированных лекар- ственных форм (или капсул). Поэтому даже препараты, содержащие одно и то же вещество в одинаковой дозе и в одной лекарственной форме при одном и том же пути введения (т.е. препараты, обладающие так называемой фармацевтической эквивалентностью), могут иметь разную биодоступность.
Фармацевтически эквивалентные препараты, производимые в различных условиях, могут различаться не только по биодоступ- ности, но также по скорости всасывания ЛВ. При этом препараты с одинаковой биодоступностью, имеют разную константу скорости абсорбции (время достижения максимальной концентрации веще- ства в крови при введении таких препаратов может существенно различаться). Это, как правило, связано с различиями физических свойств субстанции (размеров микрочастиц, степени гидратации и др.), технологии производства препарата (в том числе способов
таблетирования, характера и количества вспомогательных веществ и т.д.).
В связи с этим для характеристики идентичности фармацевти- чески эквивалентных препаратов из различных партий и, в особенности, препаратов, производимых различными фармацевтическими предприятиями, введено понятие биоэквивалентность. Два препарата считают биоэквивалентными, если они обеспечивают одинаковую биодоступность ЛВ и одинаковую скорость достижения его макси- мальной концентрации в крови (т.е. одинаковую скорость поступления ЛВ в системный кровоток из места введения). Сравнение по биоэквивалентности имеет большое значение при получении воспро- изведенных лекарственных препаратов, или «дженериков», при этом воспроизведенный препарат должен быть эквивалентным оригинальному, выпускаемому фирмой-разработчиком.
Пероральный путь введения, так же как сублингвальный, имеет ряд преимуществ перед парентеральными путями введения: наиболее прост и удобен для больного, не требует стерильности препаратов и спе- циально обученного персонала. Однако внутрь можно назначать вещес- тва, не разрушающиеся в ЖКТ в значительной степени. На степень вса- сывания оказывает также влияние относительная липофильность ЛВ. К недостаткам этого пути введения можно также отнести зависимость всасывания ЛВ от состояния слизистой оболочки и моторики кишеч- ника, рН среды и состава содержимого кишечника (возможность взаимодействия с компонентами пищи и другими ЛВ), а также частичная инактивация многих ЛВ при первом прохождении через печень.
Кроме того, сами ЛВ могут оказывать влияние на процесс пище- варения и всасывание компонентов пищи, в том числе на усвоение витаминов. Например, осмотические слабительные средства затруд- няют всасывание питательных веществ из кишечника, а антацидные средства, нейтрализуя хлористоводородную кислоту желудочного сока, нарушают процесс переваривания белков.
Назначение лекарственных препаратов внутрь невозможно у неко- торых больных (например, при отказе больного от лечения, нарушении акта глотания, отсутствии сознания, у детей раннего возраста). В этих случаях ЛС можно вводить с помощью тонкого желудочного зонда через носовые ходы или через рот в желудок и/или двенадцати- перстную кишку.
Ректальное введение. Введение ЛС в прямую кишку (ректально) используют при невозможности перорального приема (например,
при рвоте), при неприятном вкусе и запахе ЛВ или его разрушении в желудке и верхних отделах кишечника. Этот путь введения часто используется в педиатрической практике.
Ректально ЛВ назначают в форме суппозиториев или в лекарс- твенных клизмах (средний объем 50 мл). Если ЛВ обладает местнораздражающим действием на слизистую оболочку прямой кишки, его предварительно смешивают со слизями.
Из прямой кишки ЛВ быстро поступает в системный кровоток, при этом более 50% введенной дозы всасывается, минуя печень. Ректальный путь не используют для введения высокомолекулярных ЛВ (белков, жиров, полисахаридов). Некоторые вещества вводят ректально для местного воздействия на слизистую оболочку прямой кишки (например, ректальные суппозитории с бензокаином).
Парентеральные пути введения
К парентеральным путям введения относят: внутривенный; внут- риартериальный; интрастернальный (в грудину); внутримышечный; подкожный; внутрибрюшинный; под оболочки мозга; некоторые другие.
Внутривенное введение. При этом пути введения ЛВ сразу попадают в системный кровоток, что объясняет короткий латентный период их действия.
В вену вводят только водные растворы лекарственных веществ. Во избежание резкого повышения концентрации большинство ЛВ вводят медленно (в течение 1 мин), часто после предваритель- ного разведения, например раствором натрия хлорида. Большие объемы растворов вводят капельным (инфузионным) способом. В этих случаях используются специальные системы с капельницами, позволяющие регулировать скорость введения (обычно составляет 20-60 капель в минуту, что соответствует приблизительно 1-3 мл раствора в минуту). Внутривенно можно вводить гипертонические растворы. Из-за риска закупорки сосудов (эмболии) недопустимо внутривенное введение масляных растворов, суспензий, водных рас- творов с пузырьками газа.
Основной недостаток внутривенного введения - действие вещес- тва, попавшего в системный кровоток, не может быть быстро прекращено в случае необходимости (например, при передозировке или непереносимости препарата).
Внутривенный путь введения обычно используют при оказании неотложной медицинской помощи, но можно применять планово и для курсового лечения в условиях стационара и амбулаторно.
Внутриартериальное введение. Введение ЛВ в артерию, кровоснабжа- ющую определенный орган, дает возможность создать в нем высокую концентрацию действующего вещества. Внутриартериально вводят рентгеноконтрастные и противоопухолевые препараты. В некоторых случаях внутриартериально вводят антибиотики.
Интрастернальное введение (введение в грудину) применяют при невозможности внутривенного введения, например у детей, пациен- тов старческого возраста.
Внутримышечное введение. ЛВ обычно вводят в ягодичную мышцу (верхний наружный квадрант ягодицы). Внутримышечно вводят как липофильные, так и гидрофильные соединения. Всасывание гидрофильных ЛВ происходит в основном путем фильтрации через межклеточные промежутки в эндотелии сосудов скелетных мышц. Липофильные ЛВ всасываются в кровь путем пассивной диффузии через мембраны эндотелиальных клеток. Скорость всасывания зави- сит от интенсивности кровотока в месте введения. Мышечная ткань имеет хорошее кровоснабжение, поэтому всасывание ЛВ происходит довольно быстро, что позволяет в большинстве случаев создать доста- точно высокую концентрацию ЛВ в крови через 5-10 мин.
Внутримышечно вводят водные растворы (до 10 мл), а для обес- печения длительного эффекта - масляные растворы и суспензии (рис. 1-5). Внутримышечно нельзя вводить гипертонические растворы и раздражающие вещества.
Подкожное введение. При введении под кожу липофильные и гидро- фильные вещества всасываются такими же способами, что и при внутримышечном введении (т.е. путем пассивной диффузии и фильтрации), однако в связи с менее интенсивным кровоснабжением из подкожной клетчатки вещества всасываются медленнее, чем из мышечной ткани. Для ускорения всасывания ЛВ используют согревающие компрессы и местный массаж, что стимулирует кровоток в месте введения. Для уско- рения всасывания можно одновременно с ЛВ вводить гиалуронидазу, фермент разрушающий мукополисахариды соединительной ткани (при этом увеличивается площадь всасывания ЛВ). При подкожном введе- нии веществ, всасывание которых является нежелательным (например, местных анестетиков), одновременно вводят сосудосуживающие вещества (адреналин), что уменьшает кровоток в месте введения.
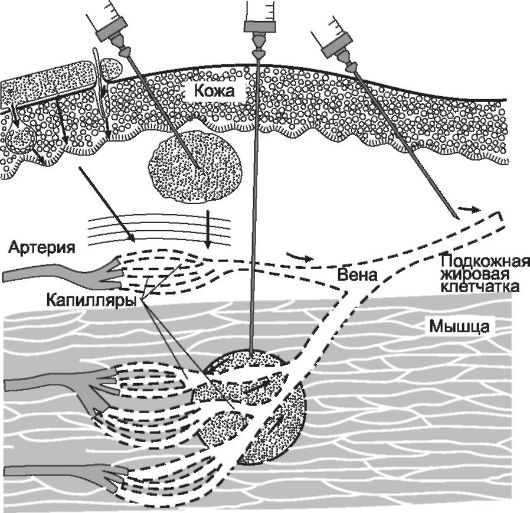
Рис. 1-5. Парентеральные пути введения лекарственных веществ
Подкожно вводят водные растворы (до 2 мл) и с осторожностью мас- ляные растворы и суспензии, которые обеспечивают более медленное всасывание ЛВ в кровь. В подкожную клетчатку можно имплантиро- вать силиконовые контейнеры, таблетированные стерильные лекарственные формы. Благодаря медленному высвобождению веществ из этих лекарственных форм достигается достаточно постоянная кон- центрация ЛВ в крови в течение недель и даже месяцев. Таким образом вводят некоторые контрацептивы, препараты тестостерона. Подкожно нельзя вводить вещества с раздражающим действием и гипертоничес- кие растворы.
Внутрибрюшинное введение. Вещества вводят в полость брюши- ны между ее париетальным и висцеральными листками. Этот путь используют, например, для введения антибиотиков во время хирурги- ческих вмешательств на органах брюшной полости.
Введение под оболочки мозга. ЛВ можно вводить субарахноидаль- но или субдурально. Так при инфекционных поражениях тканей и оболочек мозга вводят антибиотики, плохо проникающие через ГЭБ. Субарахноидальное введение местных анестетиков используют для спинномозговой анестезии.
Внутривенно, внутриартериально, интрастернально, внутримы- шечно, подкожно и под оболочки мозга вводят только стерильные лекарственные формы; введение осуществляет квалифицированный медицинский персонал.
Ингаляционное введение (от лат. inhalare - вдыхать). Ингаляционно вводят газообразные вещества, пары легко испаряющихся жидкостей, аэрозоли (воздушные взвеси мелкодисперсных частиц жидких или твердых веществ обычно диаметром от 1 до 10 мкм).
Всасывание ЛВ в кровь с большой поверхности легких происходит очень быстро, при этом лучше всасываются липофильные неполярные соединения. Этот способ используют для введения средств для инга- ляционного наркоза (газообразных веществ и легко испаряющихся жидкостей).
Ингаляционное введение в виде аэрозолей используют в основ- ном для местного воздействия на слизистую оболочку и гладкие мышцы дыхательных путей, при этом мелкие частицы (менее 2 мкм) достигают альвеол, а более крупные (6 мкм и более) задер- живаются эпителием бронхиол и верхних дыхательных путей. Ингаляционное введение - один из самых распространенных способов введения бронхорасширяющих средств и препаратов глюкокортикоидов при лечении бронхиальной астмы (в данном случае всасывание веществ в кровь нежелательно, так как приво- дит к появлению системных побочных эффектов). Из дыхательных путей частицы веществ удаляются с помощью мукоцилиарного транспорта, при этом значительное количество вещества дости- гает ротовой полости, проглатывается и может всасываться из кишечника. Поэтому для предупрежедения резорбтивного дейс- твия веществ при ингаляционном введении в виде аэрозолей используют или плохо всасывающиеся гидрофильные соединения (например, ипратропия бромид) , или вещества, подвергающиеся интенсивной пресистемной элиминации, такие как сальбутамол или глюкокортикоиды (беклометазон, будесонид и др.).
Интраназальное введение. ЛВ вводят в полость носа в виде капель или специальных интраназальных спреев. Всасывание происходит со слизистой оболочки полости носа. Таким путем вводят препа- раты некоторых пептидных гормонов, назначаемых в малых дозах. Например, десмопрессин, аналог антидиуретического гормона задней доли гипофиза, применяют интраназально при несахарном диабете в дозе 10-20 мкг.
Трансдермальное введение. Некоторые липофильные ЛВ в виде дози- рованных мазей или пластырей (трансдермальные терапевтические системы) наносят на кожу. Они всасываются с ее поверхности, попа- дают в системный кровоток, минуя печень, и оказывают резорбтивное действие. Таким путем вводят нитроглицерин для предупреждения приступов стенокардии, скополамин при морской и воздушной болез- ни, никотин для отвыкания от курения. С помощью трансдермальных лекарственных форм можно длительно поддерживать постоянную терапевтическую концентрацию ЛВ в крови и таким образом обеспе- чить продолжительный лечебный эффект. Так, пластыри, содержа- щие нитроглицерин, оказывают антиангинальное действие (лечебный эффект при стенокардии) в течение 12 ч.
Всасывание ЛВ, в том числе гидрофильных веществ с поверхности кожи значительно повышается под действием диметилсульфоксида (димексида*), который применяется иногда вместе с мазями и крема- ми, содержащими противовоспалительные средства.
Возможно введение ионизированных ЛВ с помощью ионофоре- за (ионофоретическое введение). Всасывание таких веществ после нанесения их на кожу или слизистые оболочки происходит под воз- действием слабого электрического поля. Ионофоретический способ введения нередко применяют в стоматологии.
Кроме того, ЛВ наносят на кожу или слизистые оболочки для получения местного действия. В таких случаях используют специаль- ные лекарственные формы для наружного применения (мази, кремы, растворы для наружного применения и т.д.). Всасывание ЛВ в кровь в этом случае нежелательно.
ЛВ можно вводить также в полость плевры (противотуберкулезные средства), в полость суставной сумки (гидрокортизон при ревматоид- ном артрите), в тело и в просвет органа (введение окситоцина в шейку и тело матки для остановки послеродовых кровотечений).
1.2. РАСПРЕДЕЛЕНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ В ОРГАНИЗМЕ
После поступления в системный кровоток ЛВ распределяются в различные органы и ткани. Характер распределения во многом определяется способностью ЛВ растворяться в воде или липидах (т.е. относительной гидрофильностью или липофильностью), а также кровоснабжением органов и тканей.
Гидрофильные полярные вещества распределяются в организме неравномерно. Большинство гидрофильных ЛВ не проникают внутрь клеток и распределяются в основном в плазме крови и интерстици- альной жидкости, куда они попадают из плазмы крови через межклеточные промежутки в эндотелии капилляров (низкая скорость кровотока и большая суммарная площадь эндотелия капиллярной сети способствуют проникновению веществ). Проникают гидрофильные вещества через межклеточные промежутки путем диффузии в водной фазе и/или фильтрации.
В эндотелии капилляров мозга межклеточные промежутки отсутс- твуют (между клетками имеются так называемые плотные контакты). Такой непрерывный слой эндотелиальных клеток образует гемато- энцефалитический барьер, препятствующий распределению гидрофильных полярных веществ (в том числе ионизированных молекул) в ткани мозга (см. рис. 1-3). Определенную барьерную функцию выпол- няют, по-видимому, и клетки глии, а также транспортные белки, такие как Р-гликопротеины, ограничивающие поступление некоторых веществ (дигоксина , циклоспорина , лоперамида) в ЦНС. Лишь немногие гидрофильные ЛВ (например, леводопа) проникают через ГЭБ с помощью активного транспорта.
Однако есть участки головного мозга, не защищенные ГЭБ. Пусковая зона рвотного центра доступна для действия веществ, не проникающих через ГЭБ, таких как антагонист дофаминовых рецепторов домпери- дон. Это позволяет применять домперидон в качестве противорвотного средства, не оказывающего влияния на другие отделы головного мозга. При воспалении мозговых оболочек ГЭБ становится более проницае- мым для гидрофильных ЛВ (это позволяет вводить внутривенно бензилпенициллин для лечения бактериального менингита).
Кроме ГЭБ, в организме есть другие г и с т о г е м а т и ч е с к и е б а р ь е р ы , которые являются препятствием для распределения гидрофильных ЛВ. К ним относятся гематоофтальмический барьер, не пропускающий гидрофильные полярные ЛВ в ткани глаза, гема- тотестикулярный и плацентарный барьеры. Плацентарный барьер во время беременности препятствует проникновению некоторых гидро- фильных полярных веществ из организма матери в организм плода.
Относительно равномерно распределяются в организме липо- фильные неполярные вещества. Они проникают путем пассив- ной диффузии через мембраны клеток и распределяются как во внеклеточной, так и во внутриклеточной жидкостях организма.
Липофильные ЛВ проходят через все гистогематические барьеры, в том числе диффундируют непосредственно через мембраны эндоте- лиальных клеток капилляров в ткани головного мозга. Липофильные ЛВ легко проходят через плацентарный барьер. Многие ЛС могут оказывать нежелательное действие на плод, и поэтому прием пре- паратов беременными женщинами должен находиться под строгим врачебным контролем.
Распределение слабых кислот и слабых оснований между внекле- точной (экстрацеллюлярной) и внутриклеточной жидкостями организма зависит от их рКа и рН среды. рН внутриклеточной жидкости равен приблизительно 7,0 и незначительно отличается от рН внеклеточной жидкости (7,4), поэтому проникновение слабых электролитов через мембраны клеток, а следовательно, и характер их распределения в значительной степени определяется значениями рКа. Слабые кисло- ты со значениями рКа менее 8,0 в основном остаются во внеклеточной жидкости, а слабые основания со значениями рКа более 6,0 могут проникать через мембраны и накапливаться внутри клеток. Подобное распределение характерно для местного анестетика лидокаина, кото- рый, являясь слабым основанием (рКа=7,8), в неионизированной форме проникает через клеточную мембрану внутрь аксона, частично ионизируется в более кислой внутриклеточной среде и оказывает там свое действие [см. раздел «Местноанестезирующие средства (местные анестетики)»].
На распределение ЛВ влияет также интенсивность кровоснабже- ния органов и тканей. ЛВ распределяются быстрее в хорошо кровос- набжаемые органы, такие как сердце, печень, почки, головной мозг и достаточно медленно - в ткани с относительно плохим кровоснабжением (подкожную клетчатку, жировую и костную ткани).
1.3. ДЕПОНИРОВАНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
В ОРГАНИЗМЕ
При распределении в организме некоторые ЛВ могут задерживать- ся и накапливаться в различных тканях. Происходит это в основном вследствие обратимого связывания ЛВ с белками, фосфолипидами и нуклеопротеинами клеток. Этот процесс носит название депони- р о в а н и е . Вещества могут депонироваться в различных тканях, что отчасти зависит от физико-химических свойств ЛВ. В соедини- тельной ткани могут накапливаться полярные соединения, жиро-
вая ткань - основное место депонирования липофильных веществ. Концентрация вещества в месте его депонирования (в депо) может быть очень высокой. Так, концентрации противомалярийного средс- тва хлорохина в печени, где он избирательно накапливается, в 1000 раз превышают его концентрации в плазме крови.
Некоторые вещества, избирательно накапливаясь в определенных органах и тканях, оказывают там специфическое действие. Например, йод, необходимый для синтеза тиреоидных горомонов, концентриру- ется в щитовидной железе, а фтор, принимающий участие в формировании костной ткани, накапливается в костях и зубах.
Депонирование некоторых ЛВ может привести к развитию побочных эффектов. Тетрациклины, связываясь с кальцием, накапливаются в кос- тной ткани, в том числе в зубах, что может привести к нарушению формирования скелета при внутриутробном развитии плода и пигментации и повреждению зубов у маленьких детей. Поэтому назначение тетрацик- линов противопоказано беременным женщинам и детям до 8 лет.
Однако действие большинства ЛВ развивается не в местах их депо- нирования. Из депо вещества постепенно высвобождаются в кровь и распределяются в другие органы и ткани, достигая места своего действия. При этом депонирование может привести или к удлине- нию (пролонгированию) действия препарата, или к возникновению эффекта последействия. Эффект последействия возникает, например, при введении средства для внутривенного наркоза тиопентала натрия, высоколипофильного соединения, в большом количестве накаплива- ющегося в жировой ткани. Сразу после введения тиопентал распреде- ляется в головной мозг и вызывает непродолжительный наркоз (около 15 мин), после прекращения которого развивается посленаркозный сон (в течение 2-3 ч), связанный с центральным действием препарата, высвобождаемого из жирового депо.
Самый распространенный вид депонирования ЛВ - связывание с белками плазмы крови. Слабокислые соединения (нестероидные противовоспалительные средства, сульфаниламиды) связываются в основном с альбуминами, а слабые основания - с α1-кислым гли- копротеином и другими белками плазмы крови. Некоторые вещества (глюкокортикоиды, препараты железа) избирательно связываются с определенными плазменными белками (транскортином, трансфер- рином).
Связывание ЛВ с белками плазмы крови - обратимый процесс, который может быть представлен следующим образом:
ЛВ + белок ↔ комплекс ЛВ-белок
Комплексы ЛВ-белок не проникают через мембраны клеток и через межклеточные промежутки в эндотелии сосудов (не фильтруются они и в капиллярах почечных клубочков) и поэтому служат своеобразным резервуаром (депо) данного вещества в крови.
Связанные с белками ЛВ не достигают места своего действия и поэтому не проявляют фармакологической активности. Но поскольку это связывание обратимо, часть ЛВ постепенно, по мере снижения концентрации свободного вещества в плазме крови высвобождается из комплекса с белком и оказывает фармакологическое действие. Иногда оно развивается медленнее, чем при применении ЛВ, не связы- вающихся с белками плазмы крови. При связывании с плазменными белками замедляется также проникновение ЛВ в печень и фильтра- ция в почках, что приводит к снижению скорости биотрансформации и выведения ЛВ и, следовательно, к пролонгированию их действия.
Для большинства ЛВ связывание с белками плазмы крови неспе- цифично. Разные ЛВ могут связываться с одними и теми же белками с достаточно высоким аффинитетом, при этом они конкурируют за места связывания на белковых молекулах и могут вытеснять друг друга. В таких случаях большое значение имеет степень связывания веществ с белками при их терапевтических концентрациях в крови. Например, толбутамид (гипогликемическое средство, применяемое при сахарном диабете) приблизительно на 96% связывается с белками плазмы крови, т.е. в свободном (активном) состоянии в крови находится только около 5% вещества. При одновременном назначении сульфаниламидов, также интенсивно связывающихся с белками плазмы крови, происходит быс- трое вытеснение толбутамида из мест связывания, что приводит к значительному повышению концентрации свободного вещества в крови. В результате, как правило, развивается чрезмерное гипогликемическое действие, но менее продолжительное, так как одновременно ускоряется биотрансформация толбутамида и его выведение из организма. Особую опасность представляет одновременное назначение сульфанилами- дов и антикоагулянта варфарина, связывающегося с белками плазмы крови на 99%. Быстрое повышение концентрации свободного варфарина (препарата с малой широтой терапевтического действия) может привести к резкому снижению свертываемости крови и кровотечениям.
Вытеснение из связи с белками не приводит к клинически значи- мому изменению концентрации свободного вещества в крови, если
ЛВ связывается с белками менее чем на 90%. Значение имеют также другие факторы, такие как медленное вытеснение вещества, депони- рование вещества в тканях, что уменьшает концентрацию свободного ЛВ в крови и, следовательно, устраняет причину его токсического действия. Поэтому лишь вытеснение немногих ЛВ из связи с белками плазмы крови приводит к клинически значимым последствиям.
1.4. БИОТРАНСФОРМАЦИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
Биотрансформация (метаболизм) - изменение химической струк- туры и физико-химических свойств ЛВ под действием ферментов организма. Основная направленность этого процесса - удаление чужеродных соединений, в том числе ЛВ, из организма путем пре- вращения неполярных липофильных веществ в полярные гидрофильные соединения. Так как полярные гидрофильные вещества в отличие от липофильных не реабсорбируются в почечных канальцах, они быстро выводятся почками, а некоторые из них выводятся с желчью в просвет кишечника.
Биотрансформация липофильных ЛВ в основном происходит под действием ферментов печени, локализованных в мембране эндо- плазматического ретикулума гепатоцитов. Эти ферменты называются микросомальными, так как они оказываются связанными с мелкими субклеточными фрагментами гладкого эндоплазматическо- го ретикулума (микросомами), которые образуются при гомогенизации печеночной ткани или тканей других органов и могут быть выделены центрифугированием (осаждаются в так называемой «микросомальной» фракции). Основное место локализации микросомальных фер- ментов - гепатоциты, но обнаружены они также и в других органах (кишечнике, почках, легких, головном мозге).
В плазме крови, а также в печени, стенке кишечника, почках, лег- ких, коже, слизистых оболочках и других тканях имеются н е м и к - росомальные ферменты, локализованные в цитозоле или митохондриях.
Различают два основных вида метаболизма ЛВ:
• несинтетические реакции (метаболическая трансформация);
• биосинтетические реакции (конъюгация).
Большинство ЛВ сначала метаболизируется при участии реакций метаболической трансформации с образованием реакционноспо- собных метаболитов, затем вступающих в реакции конъюгации.
При конъюгации к ЛВ или их метаболитам присоединяются остатки эндогенных соединений (глюкуроновой кислоты и др.) или химичес- кие группировки (ацетильные, метильные), поэтому реакции конъюгации обозначают термином «биосинтетическая трансформация».
Метаболическая трансформация
Реакции метаболической трансформации включают окисление, восстановление, гидролиз.
Окисление. Многие липофильные соединения подвергаются окислению в печени под действием микросомальной системы ферментов, известных как оксидазы смешанных функций (или монооксигеназы), основным компонентом которой является цитохром Р-450 (гемопротеин, связывающий ЛВ и кислород в своем активном центре). Реакция протекает при участии цитохром Р-450 редуктазы и НАДФН, который является донором электронов. В результате после восстановления молекулярного кислорода происходит присоединение одного атома кислорода к субстрату (ЛВ) с образованием окисленного метаболита и включение другого атома кислорода в молекулу воды.
RH + O2 + НАДФН + H+ → ROH + H2O + НАДФ+,
где RH - лекарственное вещество, а ROH - метаболит.
Кислород может быть включен в молекулу субстрата в составе гид- роксильной группы (реакция гидроксилирования), эпоксидной группы (реакция эпоксидации), может замещать аминогруппу (реакция дезаминирования) или атом серы. В реакциях дезалкилирования метаболиты образуются при включении кислорода в алкильную группу, которая отделяется от молекулы субстрата. Примеры реакций микросомаль- ного окисления приведены в табл. 1-1.
Оксидазы смешанных функций в целом обладают низкой суб- стратной специфичностью и могут метаболизировать многие химические соединения. В то же время отдельные изоформы (изоферменты) цитохрома Р-450 (Cytochrome P-450, CYP) отличаются определенной специфичностью (каждая из них участвует в метаболизме относительно небольшого количества веществ). В настоящее время известно более тысячи изоферментов цитохрома Р-450, подразделяемых на семейства и подсемейства. Изоформы, имеющие более 40% общего аминокислотно-
Таблица 1-1
Основные реакции метаболизма (биотрансформации) лекарственных веществ
Реакции биотрансформации Лекарственные вещества | |
Микросомальное окисление | |
Ароматическое гидроксилирование | Фенобарбитал, фенитоин, пропранолол, варфарин |
Алифатическое гидроксилирование | Толбутамид, ибупрофен, дигитоксин, барбитураты |
N-окисление | Морфин, хинидин, парацетамол |
S-окисление | Хлорпромазин, циметидин |
Дезаминирование | Диазепам, амфетамин, эфедрин |
Дезалкилирование | Морфин, кодеин, кофеин, теофиллин |
Немикросомальное окисление | |
Окислительное дезаминирование | Норэпинефрин, серотонин |
Ароматическое гидроксилирование | Аллопуринол |
Декарбоксилирование | Леводопа |
Восстановление | |
Нитрогруппы | Хлорамфеникол, нитразепам |
Карбонильной группы | Налоксон |
Дегалогенирование | Галотан |
Гидролиз | |
Сложных эфиров | Прокаин, ацетилсалициловая кислота, эналаприл, суксаметония бромид |
Амидов Прокаинамид, индометацин | |
Биосинтетические реакции | |
Конъюгация с остатком глюкуроновой кислоты (образование эфиров, тиоэфиров или амидов глюкуроновой кислоты) | Парацетамол, хлорамфеникол, диазепам, морфин, дигоксин, салициловая кислота |
Конъюгация с остатком серной кислоты (образование сульфатов) | Парацетамол, стероиды |
Конъюгация с глицином | Салициловая кислота |
Конъюгация с глутатионом | Этакриновая кислота, доксорубицин |
Ацетилирование | Сульфаниламиды, изониазид |
Метилирование | Катехоламины, каптоприл |
го состава, объединены в семейства и обозначаются арабскими цифрами (CYP1, CYP2, CYP3 и т. д.). Подсемейства, обозначаемые латинскими буквами, объединяют изоформы с идентичностью аминокислотного
состава более 55% (CYP2D, CYP3A и т.д.) Отдельные изоферменты обозначают арабскими цифрами, следующими за латинскими буквами (CYP1A2, CYP2D6, CYP3A4). ЛВ могут быть субстратами двух и более изоферментов, при этом различные изоферменты способны метаболизировать одно вещество в различных участках его молекулы. В табл. 1-2 приведены основные изоферменты цитохрома Р-450 печени человека, принимающие участие в метаболизме ЛС, и примеры ЛВ, являющихся субстратами этих изоферментов. Наибольшее количество ЛВ метаболизируется при участии CYP3A4.
Окисление некоторых ЛВ происходит при участии немикросомальных ферментов, локализованных в цитозоле, митохондриях, лизосомах и цитоплазматических мембранах клеток. Для этих ферментов характерна субстратная специфичность. Так, моноаминоксидаза типа А (МАО-А) осуществляет окислительное дезаминирование катехоламинов (норадреналина, адреналина, серотонина и др.), под действием алкогольдегидрогеназы этанол окисляется до ацетальдегида, под действием ксантиноксидазы происходит гидроксилирование пуриновых соединений (аллопуринола, теофиллина).
Восстановление лекарственных веществ заключается в присоединении к его молекуле атома водорода или удалении атома кислорода. Эти реакции могут протекать при участии микросомальных (восстановление хлорамфеникола) и немикросомальных (восстановление хлоралгидрата) ферментов. Некоторые ЛВ (например, месалазин) восстановливаются в кишечнике под действием редуктаз, продуцируемых кишечными бактериями.
Гидролиз большинства ЛВ осуществляют немикросомальные фер- менты (эстеразы, амидазы, фосфатазы) в плазме крови и тканях (в основном в печени). Вследствие присоединения воды происходит разрыв эфирных, амидных и фосфатных связей в молекулах ЛВ. Гидролизу подвергаются сложные эфиры (суксаметоний, прокаин, бензокаин, ацетилсалициловая кислота) и амиды (прокаинамид, индометацин). Некоторые ЛВ гидролизуются под действием микросомальных ферментов, например амидаз (местные анестетики из группы амидов). Микросомальный фермент эпоксидгидролаза гидролизует высокореактивные метаболиты, образующиеся при микросомальном окислении некоторых ЛВ (например, карбамазепина) с образованием неактивных соединений.
В процессе метаболической биотрансформации обычно происходит снижение активности и токсичности исходных веществ. Однако метабо-
Таблица 1-2
Основные изоферменты цитохрома Р-450, участвующие в метаболизме лекарственных веществ, их индукторы и ингибиторы
Изоферменты | Субстраты | Индукторы | Ингибиторы |
CYP1A2 | Кофеин, теофиллин, парацетамол, варфарин, тамоксифен, кломипрамин | Фенобарбитал, омепразол, рифампицин, вещества, содержащиеся в сигаретном дыме и жареной пище (бензопирены, метилхолантрены), броколли, брюссельская капуста | Ципрофлоксацин, циметидин, кларитромицин, эритромицин |
CYP2C9 | Ибупрофен, фенитоин, толбутамид, варфарин | Рифампицин, фенобарбитал | Диклофенак, сульфаниламиды циметидин, этанол (однократно) |
CYP2C19 | Диазепам, напроксен, пропранолол, омепразол | Рифампицин, фенобарбитал | Омепразол, флуоксетин |
CYP2D6 | Кодеин, клозапин, омепразол, метопролол, тимолол, галоперидол, трициклические антидепрессанты | Не известны | Амиодарон, галоперидол, флуоксетин, хини- дин, циметидин |
CYP2E1 | Этанол, парацетамол, галотан, энфлуран | Этанол (хронический прием), изониазид | Дисульфирам, ритонавир |
CYP3A4/3A5 | Амиодарон, варфарин, верапамил, диазепам, дилтиазем, кетоконазол, кортикостероиды, кокаин ловастатин, лидокаин, лозартан, макролиды, мидазолам, нифедипин, прогестерон, ритонавир, спиронолактон, сульфаметоксазол, тестостерон, циклоспорин, хинидин, этинилэстрадиол | Барбитураты, рифампицин, фенитоин, карбамазепин, глюкокортикоиды, фенилбутазон, трава зверобоя | Кетоконазол, метронидазол, омепразол, циметидин, хинидин, ципрофлоксацин, эритромицин, кларитромицин, хинидин, фуранокумарины сока грейпфрута |
литы, образуемые в результате несинтетических реакций, могут обладать такой же, а иногда и более высокой активностью, чем исходные соединения. Образование активных метаболитов обеспечивает длительное действие некоторых ЛВ (например, диазепама). Примером ЛВ, неактивных в исходном состоянии и активируемых в процессе метаболизма, являются предшественники лекарств (пролекарства). Например, антигипертензивные средства из группы ингибиторов ангиотен-превращающего фермента (эналаприл, фозиноприл) гидролизуются в организме с образованием активных соединений. С помощью пролекарств могут решаться проблемы с доставкой ЛВ к месту его действия. Так, предшественник дофамина леводопа в отличие от дофамина проникает в ЦНС, где под влиянием ДОФА-декарбоксилазы превращается в дофамин.
В некоторых случаях в процессе метаболической трансформации образуются токсичные соединения. Примером является образование промежуточного токсичного метаболита (N-ацетил-пара-бензохино- нимина) при микросомальном окислении анальгетика парацетамола. Инактивация этого метаболита происходит в результате связывания с глутатионом, однако при истощении запасов глутатиона (в основном вследствие передозировки препарата) он оказывает токсическое действие на печень.
Биосинтетическая трансформация
В процессе б и о с и н т е т и ч е с к и х р е а к ц и й к функциональным группировкам (аминогруппам, гидроксильным, карбоксильным группам) молекул ЛВ или их метаболитов присоединяются остатки эндогенных соединений (глюкуроновой или серной кислоты, глутатиона, глицина др.) или высокополярные химические группы (ацетильные, метильные). Эти реакции протекают при участии ферментов (в основном, трансфераз) печени, а также ферментов других тканей (легких, почек). Содержатся ферменты в эндоплазматическом ретикулуме гепатоцитов (микросомальные ферменты) или в цитозольной фракции.
Наиболее общей реакцией является конъюгация с глюкуроновой кислотой. Присоединение остатков глюкуроновой кислоты (образование глюкуронидов) происходит при участии микросомального фермента уридилдифосфат-глюкуронилтрансферазы (цитохром Р-450-содержащий фермент), обладающего низкой субстратной специфичностью, вследствие чего этот фермент метаболизирует многие ЛВ
и их метаболиты (а также некоторые эндогенные вещества, например билирубин).
В реакцию конъюгации с глутатионом вступают некоторые реакционноспособные вещества (эпоксиды, хиноны), в том числе проме- жуточные метаболиты, образующиеся в результате микросомального окисления (например, парацетамола), в результате чего резко снижается их токсичность.
В процессе конъюгации образуются высокополярные гидрофильные соединения, которые быстро выводятся почками или с желчью в просвет кишечника. Конъюгаты, как правило, менее активны и ток- сичны, чем исходные ЛВ или их метаболиты.
Факторы, влияющие на биотрансформацию лекарственных веществ
Активность ферментов, метаболизирующих ЛВ, а следовательно, скорость их биотрансформации зависит от пола, возраста, состояния организма, одновременного назначения других ЛС, а также от некоторых веществ, содержащихся в продуктах питания.
У мужчин активность микросомальных ферментов выше, чем у женщин, так как синтез этих ферментов стимулируется мужскими половыми гормонами. Такие вещества, как этанол, эстрогены, бензодиазепины, салицилаты метаболизируются быстрее у мужчин, чем у женщин.
В эмбриональном периоде отсутствует большинство ферментов метаболизма ЛВ. У новорожденных в первые 2-4 нед жизни активность многих ферментов (в частности, ферментов, участвующих в реакциях конъюгации) снижена и достигает достаточного уровня лишь через 1-6 мес. Поэтому детям в первые недели жизни не рекомендуется назначать такие ЛВ, как хлорамфеникол, так как вследствие недостаточной активности микросомальных ферментов, замедлены процессы конъюгации его токсичного метаболита.
В старческом возрасте снижается активность некоторых микросомальных ферментов, печеночный кровоток и масса печени, вследствие чего уменьшается скорость метаболизма многих ЛВ (лицам старше 60 лет, такие препараты назначают в меньших дозах).
При заболеваниях печени снижается активность микросомальных ферментов и замедляется биотрансформация многих ЛВ, что приводит к усилению и удлинению их действия. Уменьшение скорости кровото-
ка также существенно замедляет метаболизм некоторых ЛВ (морфина, лидокаина), поэтому при сердечной недостаточности обычные дозы этих препаратов могут вызвать токсические эффекты. Нарушения функций щитовидной железы повышают (при гипертиреозе) или снижают (при гипотиреозе) биотрансформацию ЛВ. Сахарный диабет и другие нарушения функции эндокринной системы также влияют на лекарственный метаболизм.
Синтез микросомальных ферментов может повышаться (индукция ферментов) под действием различных ЛВ, некоторых веществ, содержащихся в продуктах питания, сигаретном дыме, окружающей среде и т.д. Индукции могут подвергаться ферменты, участвующие как в несинтетических реакциях, так и в реакциях конъюгации (в основном, с глюкуроновой кислотой).
При воздействии индукторов микросомальных ферментов повышается скорость биотрансформации ЛВ, которые метаболизируются этими ферментами (см. табл. 1-2), что приводит к ослаблению их тера- певтического действия. Поскольку CYP3A4 участвует в метаболизме многих ЛВ (более 60% препаратов, применяемых в клинической практике), индукция этого изофермента достаточно часто может иметь нежелательные последствия. Некоторые ЛВ (например, фенобарбитал, рифампицин) являются универсальными индукторами, повышая активность нескольких изоферментов цитохрома Р-450, в том числе CYP3A4 и, как следствие, ослабляют терапевтический эффект многих ЛВ. Например, эффективность пероральных контрацептивов может снизиться на фоне лечения рифампицином или фенобарбиталом из-за ускорения метаболизма входящих в их состав эстрогенов и прогестинов.
Если в процессе биотрансформации ЛВ образуются токсичные метаболиты, индукция метаболизирующих это вещество ферментов приводит к повышению риска его токсического действия. Так, индукция CYP2E1 при хроническом употреблении алкоголя (см. табл. 1-2) увеличивает токсичность парацетамола.
Как правило, действие индукторов синтеза микросомальных ферментов (в том числе фенобарбитала) развивается медленно (в течение нескольких недель). Рифампицин оказывает более быстрый эффект, существенно повышая активность ферментов уже через несколько дней от начала применения.
Препараты некоторых лекарственных растений могут ускорять метаболизм ЛВ. Например, препараты зверобоя, применяемые в
качестве легких антидепрессантов, вызывают индукцию изофермента CYP3A4 и поэтому ослабляют или предупреждают действие ЛВ, которые метаболизируются при участии этого изофермента.
Полициклические ароматические углеводороды (бензопирены, метилхолантрены), содержащиеся в табачном дыме, некоторые вещества, применяемые в промышленности (например, полихлорированные бифенилы) или побочные продукты химического синтеза (диоксин) вызывают индукцию изофермента CYP1A2. Индукция изофермента CYP2E1 развивается при хроническом употреблении алкоголя (см. табл. 1-2).
В некоторых случаях может увеличиваться скорость метаболизма самого индуктора (аутоиндукция), вследствие чего ослабляются его фармакологические эффекты. Аутоиндукция весьма характерна для барбитуратов (в частности, фенобарбитала) и является причиной развития толерантности при их длительном приеме.
Некоторые ЛВ снижают активность микросомальных ферментов, в результате повышается концентрация в крови веществ, метаболизируемых этими ферментами. Это может привести к развитию токсических эффектов. Например, циметидин, некоторые макролидные антибиотики, кетоконазол, ципрофлоксацин, ингибируя цитохром CYP3A4/3А5, замедляют микросомальное окисление варфарина, что может усилить его антикоагулянтный эффект и спровоцировать кровотечение. Токсическое действие теофиллина, который метаболизируется при участии цитохрома CYP1A2, резко усиливается при его одновременном назначении с антибактериальным препаратом ципрофлоксацином, ингибирующим этот фермент. Фуранокумарины, содержащиеся в грейпфрутовом соке, ингибируют CYP3A4/3А5, в результате может усилиться действие многих
ЛВ (см. табл. 1-2).
В отличие от индукторов метаболизма ингибиторы ферментов действуют быстрее (эффект ингибирования отмечен через 24 ч после приема соответствующего препарата).
При одновременном применении ЛВ с индукторами или ингибиторами их метаболизма необходимо корректировать назначаемые дозы этих веществ.
Изменение активности ферментов метаболизма ЛВ могут определяться генетическими факторами. Такие изменения имеют в своей основе передающиеся из поколения в поколение мутации генов, кодирующих синтез данных ферментов. Данный феномен носит название
генетического полиморфизма и имеет следствием значительные межиндивидуальные различия в метаболизме ЛВ. При этом у определенного процента больных, принимающих данное ЛВ, активность метаболизирующих ферментов может быть повышена, процесс биотрансформации ЛВ ускоряется, и его действие снижается. И наоборот, активность ферментов может быть снижена (недостаточность ферментов), вследствие чего биотрансформация ЛВ будет происходить медленнее, и действие его будет усиливаться вплоть до появления токсических эффектов. Часто отмечают генетический полиморфизм изоферментов цитохрома Р-450.
Известны случаи генетического полиморфизма ферментов, не имеющих отношения к системе цитохрома Р-450. Например, при аце- тилировании противотуберкулезного препарата изониазида у определенного процента больных в популяции выявляют недостаточность фермента N-ацетилтрансферазы («медленные ацетиляторы»), а у других больных активность этого фермента повышена («быстрые ацетиляторы»). У «медленных ацетиляторов» концентрация изониазида в плазме крови в 4-6 раз выше, чем у «быстрых ацетиляторов», что может быть причиной токсического действия препарата. Примеры влияния генетической недостаточности некоторых ферментов на действие ЛВ приведены в табл. 1-3.
Таблица 1-3
Реакции организма на лекарственные вещества при генетической недостаточности некоторых ферментов
Фермент | Особые реакции | Лекарственные вещества | Распространенность |
Глюкозо-6- фосфатдегидрогеназа эритроцитов | Гемолиз эритроцитов вследствие образования хинона. Гемолитическая анемия | Хинин, хинидин, сульфаниламиды, хлорамфеникол | Тропические и субтропические страны (до 100 млн человек) |
N-ацетил- трансфераза печени | Повышение частоты побочных эффектов. | Изониазид, сульфаниламиды, прокаинамид | Европеоиды (до 50% населения) |
Псевдохолинэстераза плазмы крови | Удлинение расслабляющего действия на скелетные мышцы (6-8 ч вместо 5-7 мин) | Суксаметоний | Европеоиды (0,04% населения), эскимосы (1% населения) |
1.5. ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ ИЗ ОРГАНИЗМА
Лекарственные вещества и продукты их биотрансформации в основном выводятся из организма через почки (почечная экскреция), а также через ЖКТ (с желчью в просвет кишечника).
Почечная экскреция. Выведение ЛВ и их метаболитов почками происходит при участии трех основных процессов: клубочковой фильтрации, активной секреции в проксимальных канальцах и канальцевой реабсорбции.
Клубочковая фильтрация. При фильтрации плазмы крови лекарственные вещества (за исключением высокомолекулярных соединений), под гидростатическим давлением поступают в просвет почечных канальцев через межклеточные промежутки эндотелия капилляров почечных клубочков. Если эти вещества не реабсорбируются в почечных канальцах, они выводятся с мочой. Вещества, связанные с плазменными белками, не фильтруются в почечных клубоч- ках, что приводит к замедлению клубочковой фильтрации веществ, которые связываются с белками плазмы крови.
Активная секреция. Путем активной секреции в просвет канальцев выделяется большая часть веществ, экскретируемых почками. Вещества секретируются в проксимальных канальцах с помощью специальных транспортных систем против градиента концентрации (этот проццесс требут затраты энергии), в результате ЛВ удаляется из плазмы крови практически полностью. Существуют отдельные транспортные системы для органических кислот (пенициллинов, салицилатов, сульфаниламидов, тиазидных диуретиков, фуросемида и др.), участвующие также в секреции глюкуронидов и сульфатов, и транспортные системы для органических оснований (морфина, хинина, допамина, серотонина и др.). В процессе секреции органические кислоты (и органические основания) могут конкурентно вытеснять друг друга из связи с транспортными белками, вследствие чего экскреция вытесняемого вещества снижается.
В отличие от клубочковой фильтрации канальцевая секреция обеспечивает эффективное выведение из организма веществ, связанных с белками плазмы крови. Это объясняется тем, что канальцевая секреция ЛВ не сопровождается одновременным выведением воды, поэтому концентрация свободного вещества в плазме крови снижается, и происходит диссоциация связанных с белками молекул вещества.
Пенициллин, который на 80% связывается с белками плазмы крови, очень медленно фильтруется в клубочках, но практически полностью выводится путем активной секреции в проксимальных канальцах.
Канальцевая реабсорбция (обратное всасывание). Через мембраны почечных канальцев ЛВ реабсорбируются путем пас- сивной диффузии по градиенту концентрации. Таким образом, реабсорбируются липофильные неполярные соединения, так как они легко проникают через мембраны эпителиальных клеток почечных канальцев. Гидрофильные полярные вещества (в том числе ионизированные соединения) в почках практически не реабсорбируются и выводятся из организма.
Выведение почками слабых кислот и слабых оснований находится в прямой зависимости от степени их ионизации и, следовательно, от рН мочи (варьирует от 4,5 до 8,0). Кислая реакция мочи способствует экскреции слабых оснований (алкалоидов никотина, атропина, хинина) и затрудняет выведение слабых кислот (барбитуратов, ацетилсалициловой кислоты). Чтобы ускорить выведение почками слабых оснований, следует изменить реакцию мочи в кислую сторону (снижение рН мочи). Обычно в таких случаях назначают аммония хлорид. И наоборот, при необходимости увеличения экскреции слабых кислот назначают натрия гидрокарбонат и другие соединения, сдвигающие реакцию мочи в щелочную сторону (повышение рН мочи). Внутривенное введение натрия гидрокарбоната, в частности, используют для ускорения выведения барбитуратов или ацетилсалициловой кислоты в случае их передозировки.
Реабсорбция некоторых эндогенных веществ (аминокислот, глюкозы, мочевой кислоты) осуществляется путем активного транспорта в дистальных канальцах.
Выведение через желудочно-кишечный тракт. Многие ЛВ (дигоксин, тетрациклины, пенициллины, рифампицин и др.) выделяются с желчью в просвет кишечника в неизмененном виде или в виде метаболитов и конъюгатов. Выделение веществ из гепатоцитов в желчь происходит с помощью активного транспорта при участии специальных транспортных систем для органических анионов и катионов (Р-гликопротеина и других транспортных белков). При этом часть ЛВ может неоднократно повторно всасываться и вновь выделяться с желчью в просвет кишечника. Этот циклический процесс называется энтерогепатической (кишечно-печеночной) циркуляцией. Некоторые ЛВ (морфин, этинилэстрадиол) выделяются с желчью в виде конъюга-
тов с глюкуроновой кислотой (глюкуронидов), гидролизующихся в кишечнике с образованием активных веществ, которые снова реаб- сорбируются и поступают в печень. Таким образом, энтерогепатическая циркуляция способствует пролонгированию действия ЛВ.
Некоторые ЛВ плохо всасываются в ЖКТ и полностью выводятся из организма через кишечник. Их, в основном, применяют для лечения или профилактики кишечных инфекций и дисбактериоза (например, неомицин, нистатин).
Другие пути выведения. Газообразные и летучие вещества выделяются легкими. Таким путем из организма выводятся средства для ингаляционного наркоза. Некоторые вещества могут выделяться потовыми, бронхиальными и слюнными железами (пенициллины, йодиды), железами желудка (хинин) и кишечника (слабые органические кислоты), слезными железами (рифампицин). При выделении слюнными железами и железами желудка и кишечника ЛВ могут всасываться вновь. Известно, что концентрация некоторых ЛВ в слюне коррелирует с их концентрацией в плазме крови, что может быть использовано для фармакокинетических исследований.
Многие вещества (ацетилсалициловая кислота, барбитураты, антитиреоидные средства, кофеин, этанол, никотин) выделяются молочными железами в период лактации, при этом интенсивность выделения зависит от липофильности веществ и степени их ионизации. Поскольку грудное молоко имеет более кислую реакцию (рН=6,5), чем плазма крови (рН=7,4), концентрация слабых оснований в нем достигает более высоких, а концентрация слабых кислот более низких значений, чем в плазме. В связи с этим такие вещества, как никотин, морфин, снотворные средства из группы бензодиазепинов и другие слабоосновные соединения, могут накапливаться в грудном молоке и во время кормления, выделяясь молочными железами, попасть в организм ребенка. Поэтому кормящим матерям противопоказано назначение ЛВ, проникающих в грудное молоко, в особенности тех, которые могут оказывать неблагоприятное воздействие на ребенка.
Для характеристики совокупности процессов, в результате которых активное ЛВ удаляется из организма, введено понятие «элимина- ция», объединяющее два процесса: биотрансформацию и выведение. Количественно процесс элиминации характеризуется рядом фармакокинетических параметров (см. раздел «Математическое моделирование фармакокинетических процессов»).
1.6. МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ФАРМАКОКИНЕТИЧЕСКИХ ПРОЦЕССОВ
Величина и продолжительность фармакологического эффекта во многом определяются концентрацией ЛВ в органах или тканях, где оно оказывает свое действие. Поэтому очень важно поддерживать определенную (терапевтическую) концентрацию ЛВ в месте его действия. Однако в большинстве случаев концентрацию вещества в тканях определить практически невозможно, поэтому при фармакокинетических исследованиях определяют концентрации ЛВ в плазме крови, которые для большинства веществ коррелируют с их концентрациями в органах-мишенях.
В результате всасывания, распределения, депонирования и элиминации (биотрансформации и выведения) ЛВ его концентрация в плазме крови изменяется. Эти изменения могут быть отражены графически. Концентрацию ЛВ измеряют в плазме крови сразу и через определенные промежутки времени после его введения и на основании полученных данных строят кривую изменения концентрации ЛВ во времени - фармакокинетическую кривую (рис. 1-6).
Для количественной оценки влияния процессов всасывания, распределения, депонирования и элиминации на концентрацию ЛВ в крови используют математические фармакокинетические модели. Различают однокамерные, двухкамерные и многокамерные фармакокинетические модели.
В однокамерной модели организм условно представляют в виде камеры, заполненной жидкостью. Вещество может поступать в камеру постепенно как при приеме внутрь или других внесосудистых путях введения или мгновенно как при быстром внутривенном введении (рис. 1-7).
После поступления вещества в камеру в количестве D оно, распределяясь мгновенно и равномерно, занимает объем камеры, при этом концентрация вещества в камере обозначается как начальная кон- центрация - С0. Объем распределения вещества в камере Vd (volume of distribution) равен D/C0.
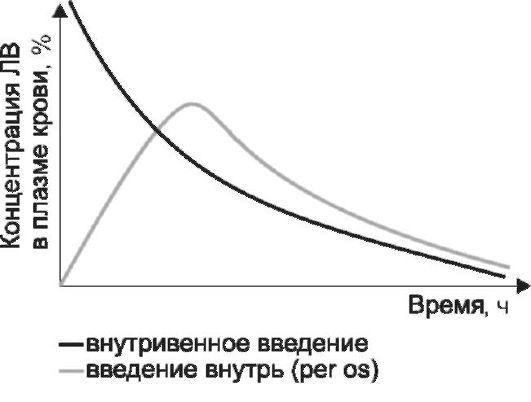
Рис. 1-6. Изменение концентрации лекарственного вещества во времени при внутривенном и внесосудистом введении
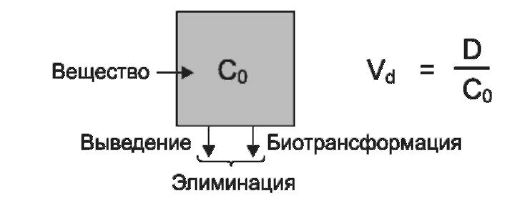
Рис. 1-7. Однокамерная фармакокинетическая модель: Vd (volume of distribution) - объем распределения вещества в камере; D - количество вещества, введенное в камеру; С0 - начальная концентрация вещества в камере
В клинической практике применяют параметр, получивший название кажущийся объем распределения (apparent volume of distribution, Vd).
Кажущийся объем распределения - гипотети- ческий объем жидкости организма, в котором ЛВ распределено равномерно и находится в концентрации, равной концентрации данного вещества в плазме крови (Cp). Соответственно, кажущийся объем распределения можно выразить уравнением:
Vd = Q/Cp,
где Q - количество вещества в организме при его концентрации в плазме крови Cp .
Если допустить, что вещество после внутривенного введения в дозе D мгновенно и равномерно распределилось в организме, кажущийся объем распределения можно определить как:
Vd = D/C0,
где C0 - начальная концентрация вещества в плазме крови, D - доза.
Кажущийся объем распределения позволяет судить о том, в каком соотношении распределяется вещество между жидкостями организма (плазмой крови, интерстициальной, внутриклеточной жидкостями). Так, если величина Vd какого-либо вещества приблизительно равна 3 л (средний объем плазмы крови), это означает, что данное вещество преимущественно находится в плазме крови. Такой объем распределе-
ния характерен для крупномолекулярных соединений, практически не проникающих в клетки крови и через эндотелий сосудов (веществ, которые не выходят за пределы сосудистого русла), например, Vd гепарина приблизительно равен 4 л.
Если Vd равен 15 л (сумма средних объемов плазмы крови и интерстициальной жидкости), вещество преимущественно содержится в плазме крови и интерстициальной жидкости, т.е. не проникает внутрь клеток. Предположительно это гидрофильное соединение, которое не проникает через клеточные мембраны. К таким ЛВ относятся аминогликозидные антибиотики (гентамицин, тобрамицин). Поэтому эти антибиотики практически не оказывают действие на микроорганизмы, находящиеся внутри клеток, т.е. неэффективны в отношении внутриклеточных инфекций.
Некоторые ЛВ имеют объем распределения порядка 40 л (средний объем всех жидкостей организма). Это означает, что эти ЛВ присутствуют как во внеклеточной, так и во внутриклеточной жидкостях организ- ма, т.е. проникают через мембраны клеток. В основном так распределяются в организме липофильные неполярные соединения.
Если величина Vd вещества значительно превышает объем жидкостей организма, наиболее вероятно, что это вещество депонировалось в периферических тканях, и его концентрация в плазме крови чрезвычайно мала. Большие значения объема распределения (приблизитель- но 1600 л) характерны для трициклических антидепрессантов имипрамина и амитриптилина. Подобные ЛВ не могут быть эффективно удалены из организма с помощью гемодиализа.
После мгновенного и равномерного распределения вещества в объеме камеры и достижения концентрации C0, его концентрация в камере постепенно снижается при участии двух процессов биотрансформации и экскреции, которые объединяются термином элиминация (см. рис. 1-7).
Скорость элиминации большинства ЛВ зависит от их концентрации в крови (чем ниже концентрация вещества, тем меньше скорость элиминации). При этом кривая изменения концентрации вещества во времени имеет экспоненциальный характер (рис. 1-8). Такая элиминация соответствует кинетике 1-го порядка, когда в единицу времени элиминируется определенная часть вещества.
Основными параметрами, характеризующими процесс элиминации, являются константа скорости элиминации (ke1, ke) и период полуэлиминации (t1/2).
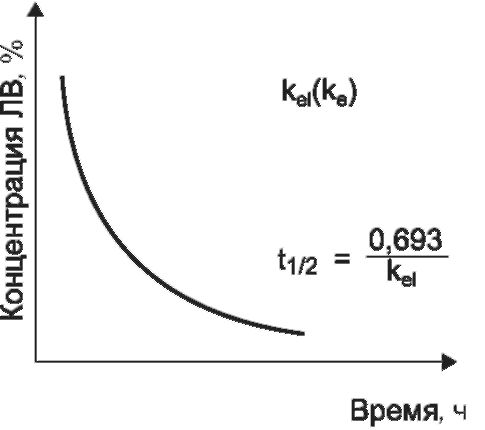
Рис. 1-8. Элиминация вещества, соответствующая кинетике первого порядка. kel(ke) - констнта скорости элиминации 1-го порядка; t1/2- период полуэлиминации
Константа скорости элиминации 1-го порядка показывает, какая часть вещества элиминируется из организма в единицу времени (размерность мин-1, ч-1). Например, если kel вещества, введенного внутривенно в дозе 100 мг, составляет 0,1 ч-1, через 1 ч его количество в крови будет составлять 90 мг, через 2 ч - 81 мг и т. д.
Элиминация отдельных ЛВ (например, этанола, фенитоина) соответствует кинетике нулевого порядка. Скорость такой элиминации не зависит от концентрации вещества в плазме крови и является постоянной величиной, т.е. в единицу времени элиминируется определенное количество вещества (например, за 1 ч элиминируется 10 г чистого этанола). Это связано с тем, что при терапевтических концентрациях названных веществ в крови происходит насыщение метаболизирующих их ферментов. Поэтому увеличение концентрации веществ в крови не приводит к повышению скорости их элиминации.
Период полуэлиминации (t1/2) - время, за которое концентрация вещества в плазме крови снижается на 50% (рис. 1-9). Для большинства ЛВ (если их элиминация подчиняется кинетике 1 -го поряд- ка) - величина постоянная в определенных пределах и не зависящая от дозы. Поэтому, если за один период полуэлиминации из плазмы крови удаляется 50% внутривенно введенного ЛВ, то за 2 периода - 75%, а за 3,3 периода - 90%. Этот параметр используют для подбора интервалов
между введениями ЛВ, необходимых для поддержания его постоянной концентрации в крови.
Период полуэлиминации связан с константой скорости элиминации следующим соотношением:
t1/2 = ln 2 / kel = 0,693 / kel
Если сразу после внутривенного введения ЛВ производить измерения его концентрации в
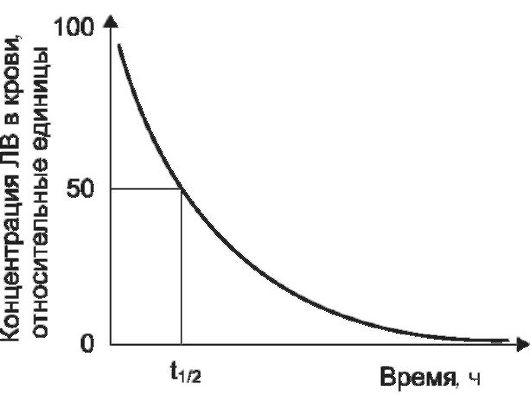
Рис. 1-9. Определение периода полуэлиминации. t,,2 - период полуэлиминации
плазме крови через короткие интервалы времени, можно получить двухфазный характер изменения концентрации вещества в крови (см. рис. 1-11).
Такой же характер кривой можно получить с помощью двухкамерной фармакокинетической модели (рис. 1-10). В этой модели организм представляют в виде двух сообщающихся между собой камер. Одна из камер этой модели называется центральной и представляет плазму крови и хорошо кровоснабжаемые органы (сердце, печень, почки, легкие), а другая, называемая периферической, представляет плохо кровоснабжаемые ткани (кожу, жировую и мышечную ткани). Вещество вводят в центральную камеру, где оно мгновенно и равномерно распределяется и затем проникает в периферическую камеру. Этот период обозначается как фаза распределения, или α-фаза. Затем вещество перераспределяется из периферической камеры в центральную и удаляется из нее вследствие элиминации. Эта фаза (фаза элиминации) обозначается как β-фаза. α-Фаза характеризуется параметром, который называется периодом полураспределения - t1/2α, а характеристикой β-фазы служит собственно период полуэлиминации, обозначаемый как (см. рис. 1-11). Период полураспределения, как правило, меньше периода полуэлиминации, так как вещество распределяется из центральной камеры в периферическую быстрее, чем элиминируется.
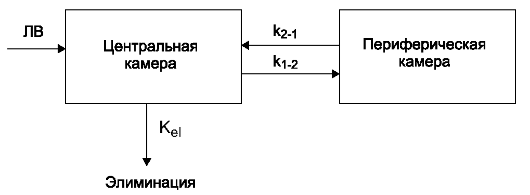
Рис. 1-10. Двухкамерная фармакокинетическая модель. ЛВ - лекарственное вещество
Клиренс - фармакокинетический параметр, характеризующий скорость освобождения организма от лекарственного вещества.
Поскольку освобождение организма от ЛВ происходит за счет процессов биотрансформации (метаболизма) и экскреции, различают метаболический и экскреторный клиренсы. Метаболический клиренс (Clmet) и экскреторный клиренс (Cexcr) в сумме составляют системный (общий) клиренс (Clt, total clearance):
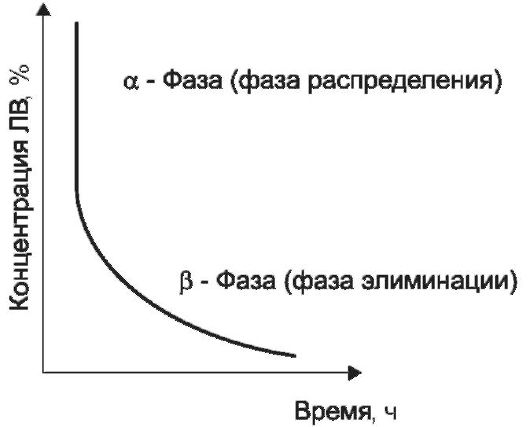
Рис. 1-11. Характер элиминации вещества в двухкамерной модели
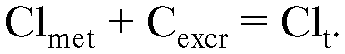
Системный клиренс численно равен объему распределения, освобождаемому от вещества в единицу времени (размерность - объем в единицу времени, например мл/мин, л/ч, иногда с учетом массы тела, например мл/кг/мин):
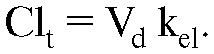
Значения клиренса прямо пропорциональны скорости элиминации вещества и обратно пропорциональны его концентрации в биологической жидкости (в крови плазме крови и др.):
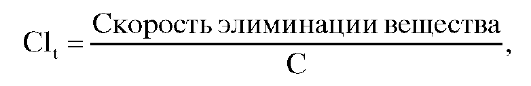
где С - концентрация вещества.
В зависимости от путей элиминации ЛВ различают почечный клиренс (Clren), печеночный клиренс (Clhep), а также клиренс, осуществляемый другими органами (легкими, слюнными, потовыми и молочными железами, внепеченочный метаболизм). Наиболее важными состав- ляющими системного клиренса являются почечный и печеночный клиренсы.
Почечный клиренс численно равен объему плазмы крови, освобождаемому от ЛВ в единицу времени, и зависит от интенсивности процессов клубочковой фильтрации, канальцевой секреции и реабсорбции. Почечный клиренс можно определить при постоянной концентрации вещества в плазме крови:
где Cu - концентрация вещества в моче, Cp - концентрация вещества в плазме крови и Vu - скорость мочеотделения.
Печеночный клиренс зависит от процессов биотрансформации ЛВ и экскреции неизмененного ЛВ с желчью. Значения почечного и печеночного клиренса следует учитывать при назначении ЛВ больным с недостаточностью почек или печени соответственно.
Оптимизация дозирования лекарственных веществ
Для достижения оптимального терапевтического эффекта ЛВ необходимо постоянно поддерживать его терапевтическую концентрацию в крови. Постоянно поддерживаемый уровень вещества в плазме крови обозначается как стационарная концентрация (Css steady-state concentration). Стационарная концентрация устанавливается при достижении равновесия между процессом поступления вещества в системный кровоток и процессом его элиминации (когда скорость поступления равна скорости элиминации). Наиболее простой способ достижения стационарной концентрации ЛВ - внутривенное капельное введение (рис. 1-12). При внутривенном капельном введении ЛВ величина Css зависит от скорости введения, которую можно определить по формуле:
D/T = CltCss,
где D - доза, Clt - общий клиренс, T - время введения, Css - стационарная концентрация
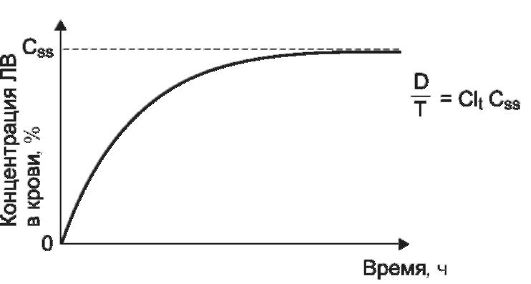
Рис. 1-12. Достижение стационарной концентрации вещества при внутривенном капельном введении: Css - стационарная концентрация вещества в крови; Clt - системный клиренс; D/T - скорость введения вещества
ЛВ необходимо вводить с такой скоростью, чтобы поддерживать его терапевтическую концентрацию в крови. Существует диапа- зон терапевтических концентраций (рис. 1-13). Нижняя граница этого диапазона - минимальная эффективная концентрация
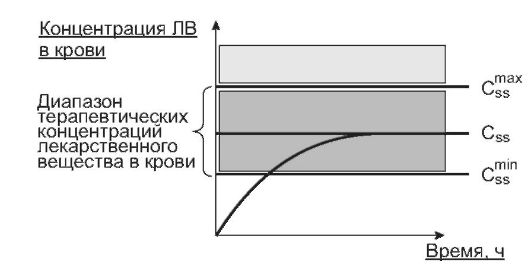
Рис. 1-13. Диапазон стационарных терапевтических концентраций вещества в крови: Cssmin - минимальная эффективная концентрация; Cssmax - максимальная безопасная концентрация; Css - средняя терапевтическая концентрация
(Cssmm), ниже которой вещество не оказывает необходимого действия, верхняя граница - максимальная безопасная концентрация (Cssmax), выше которой находится область токсических концентраций. Обычно поддерживают среднюю концентрацию этого диапазона, т.е. среднюю терапевтическую концентрацию вещества в крови. Значения средних терапевтических концентраций ЛВ приведены в справочной литературе.
Время достижения стационарной терапевтической концентрации вещества в крови зависит от его периода полуэлиминации. Через один t1/2 достигается 50%, через 2 t1/2 - 75% и через 3,3 t1/2 - 90% от стационарного уровня вещества в крови. Поэтому при необходимости получения быстрого терапевтического эффекта, особенно если вещество имеет достаточно большой t1/2, сначала вводят большую нагрузочную дозу пре- парата (для достижения стационарной терапевтической концентрации), а затем вещество вводят инфузионно с определенной скоростью для поддержания стационарной концентрации. Однако чаще вещества назначают отдельными дозами через определенные интервалы времени (наиболее часто внутрь). В таких случаях концентрация вещества в крови не остается постоянной, а меняется относительно стационарного уровня, причем эти колебания не должны выходить за пределы диапазона терапевтических концентраций. Поэтому после назначения нагрузочной дозы, которая обеспечивает быстрое достижение стационарной терапевтической концентрации, вводят меньшие по величине поддерживающие дозы; при этом допускаются лишь небольшие колебания концентрации вещества в крови относительно его стационарного терапевтического уровня (рис. 1-14). Нагрузочную и поддерживающую дозы ЛВ для каждого конкретного больного можно
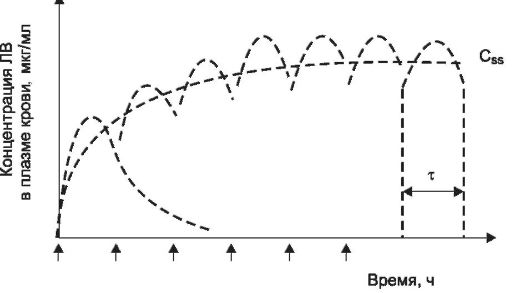
Рис. 1-14. Изменение концентрации лекарственного вещества в плазме крови при многократном введении (через равные промежутки времени вводятся поддерживающие дозы вещества): τ - интервал времени между введением поддерживающих доз
рассчитать по формулам, в которых использованы фармакокинетические параметры, представленные в этом разделе: объем распределения, t1/2 и др.
Кроме того, при введении веществ внутрь учитывают их биодоступность (часть введенной дозы вещества, которая в неизмененном виде достигла системного кровотока). Биодоступность веществ при введении внутрь зависит от многих факторов (см. стр. 49) и определяется следующим образом. Вещество вводят больному внутривенно и измеряют его концентрации в крови через определенные промежутки времени. На основании полученных данных вычерчивают кривую изменения концентрации вещества во времени при внутривенном введении. Затем тому же больному это вещество вводят внутрь в той же дозе и определяют его концентрации в крови через определенные интервалы времени. По результатам измерения строят кривую изменения концентрации вещества во времени при введении внутрь (рис. 1-15). Затем измеряют площади под кривыми «концент- рация-время» (AUC, Area Under the Curve). Биодоступность вещества определяют по формуле:
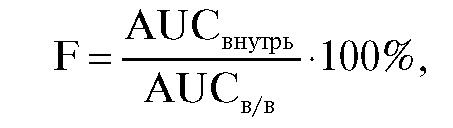
где F - биодоступность (Fraction); AUC - площадь под кривой концентрация - время; в/в - внутривенное введение.
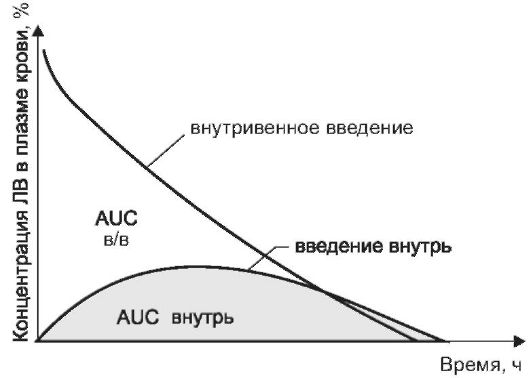
Рис. 1-15. Определение биодоступности лекарственного вещества при введении внутрь. AUC (Area Under the Curve) - площадь под кривой концентрация-время