Патофизиология Новицкого, Е.Д. Гольдберга Тома 1 и 2 - 2009 г.
|
|
|
|
ГЛАВА 3 ПАТОФИЗИОЛОГИЯ КЛЕТКИ
Нарушение жизнедеятельности организма человека всегда так или иначе связано с изменением функционирования клеток. Клетки организма выполняют определенные функции. В совокупности они способны удовлетворять физиологические потребности организма в поддержании нормального гомеостаза. При воздействии избыточных физиологических или патологических стимулов в клетках может развиться процесс адаптации, следствием которого является достижение нового стационарного состояния, позволяющего им нормально функционировать в изменившихся условиях. Если резерв адаптационного ответа исчерпан, а адаптация не достигнута, наступает повреждение клетки. До определенного предела повреждение клетки обратимо, но даже если это нарушение имеет временный и обратимый характер, оно ухудшает состояние организма в целом. Если неблагоприятный фактор действует длительно или интенсивность его действия очень велика, наступает необратимое повреждение клетки и ее гибель.
При этом нарушение функций одних клеток может быть первопричиной развития болезни в целом, тогда как состояние других клеток может быть нарушенным вследствие неблагоприятных изменений в организме, связанных с развитием патологического процесса. Например, при инфаркте миокарда происходит нарушение функционирования, а затем гибель клеток сердечной мышцы вследствие острого недостатка кислорода. В результате того, что часть сердечной мышцы не участвует в сокращении, нарушается кровоснабжение организма, которое может привести к серьезной гипоксии и нарушению функций клеток других органов: почек, мозга, печени. При многих интоксикациях первично повреждаются клетки печени или почек, а нарушение функционирования этих органов сопровождается отравлением других клеток организма токсиче-
скими продуктами метаболизма. Первично или вторично повреждение клеток - оно в любом случае неблагоприятно отражается на состоянии организма и должно быть ликвидировано на раннем этапе своего развития. Но для того чтобы найти средство предупреждения или защиты клеток от болезнетворного воздействия, надо знать, почему и как повреждаются и гибнут клетки в живом организме.
3.1. ВИДЫ ПОВРЕЖДЕНИЙ И ГИБЕЛИ КЛЕТОК. УНИВЕРСАЛЬНЫЙ ОТВЕТ КЛЕТКИ НА ПОВРЕЖДЕНИЕ
Повреждение клетки - типический патологический процесс, основу которого составляют нарушения внутриклеточного гомеостаза, приводящие к нарушению структурной целостности клетки и ее функциональных способностей после удаления повреждающего агента. Так, например, на первом этапе нарушение функционирования клетки, вызванное действием неблагоприятных факторов, например недостатком кислорода или действием токсических соединений, может и не привести к повреждению клетки: как только восстановятся нормальные окружающие условия, клетка вновь вернется в состояние, близкое к исходному. Например, если в каком-нибудь участке миокарда кровоснабжение прекращается на короткий промежуток времени (не более 10-15 мин), а затем восстанавливается, то кардиомиоциты сохраняют способность к регенерации и нормальному функционированию. Если кровоснабжение не восстанавливается, то повреждение миокарда становится необратимым и кардиомиоциты на этом участке погибают.
Различают непосредственное (первичное) и опосредованное (вторичное) повреждения. Последнее возникает как следствие первичных нарушений постоянства внутренней среды организма.
В зависимости от скорости развития и выраженности основных проявлений повреждение клетки может быть острым и хроническим.
Острое повреждение развивается быстро, как правило, в результате однократного, но интенсивного повреждающего воздействия, в то время как хроническое повреждение протекает медленно и является следствием многократных, но менее интенсивных патогенных влияний.
В зависимости от периода жизненного цикла, на который приходится действие повреждающего агента, повреждение клетки может быть митотическим и интерфазным.
В зависимости от степени нарушения внутриклеточного гомеостаза повреждение бывает обратимым и необратимым (см. выше).
Выделяют два патогенетических варианта повреждения клеток:
1. Насильственный. Развивается в случае действия на исходно здоровую клетку физических, химических и биологических факторов, интенсивность которых превышает обычные возмущающие воздействия, к которым клетка адаптирована. Наиболее чувствительны к данному варианту повреждения функционально малоактивные клетки, обладающие малой мощностью собственных гомеостатических механизмов.
2. Цитопатический. Возникает в результате первичного нарушения защитно-компенсаторных гомеостатических механизмов клетки. В этом случае фактором, запускающим патогенетические механизмы повреждения, являются естественные для данной клетки возмущающие стимулы, которые в этих условиях становятся повреждающими. К цитопатическому варианту относятся все виды повреждения клетки, возникающего вследствие отсутствия какихлибо необходимых ей компонентов (гипоксическое, нервнотрофическое, при голодании, гиповитаминозах, недостаточности антиоксидантной системы, генетических дефектах и др.). К цитопатическому повреждению наиболее чувствительны те клетки, реактивность, а следовательно, и функциональная активность которых в естественных условиях очень высоки (нейроны, кардиомиоциты).
• Причинами повреждения клеток могут быть следующие факторы: гипоксия. Чрезвычайно важная и распространенная причина повреждения клеток. Уменьшение кровообращения (ишемия), возникающее при атеросклерозе, тромбозе, сдавлении артерий, является основной причиной гипоксии. Другой причиной может быть недостаточная оксигенация крови при сердечно-сосудистой или легочной патологии. Третьей причиной может являться нарушение транспорта кислорода, например при анемии, отравлении окисью углерода или действии метгемоглобинобразователей (нитраты и нитриты, хлорноватые и хлорноватистые соли, феррицианиды, лекарственные вещества - фенацетин, амидопирин, сульфаниламиды и др.) (подробнее см. раздел 16.2);
• физические агенты - механическая травма, температурные воздействия, колебания барометрического давления, ионизирующая и ультрафиолетовая радиация, электрический ток;
• химические агенты и лекарства. Повреждение клеток может быть вызвано как жизненно необходимыми химическими соединениями, такими, как, например, глюкоза или поваренная соль в гипертонических концентрациях, кислород в высоких концентрациях. Вещества, известные как яды (в частности, мышьяк, цианиды, соли ртути), могут вызывать гибель клеток в считанные минуты или часы. Гибель клеток может наступать при действии факторов внешней среды, «социальных» факторов - алкоголя, курения, наркотиков и др.;
• иммунологические реакции. Хотя иммунные реакции защищают организм от воздействия биологических агентов, в ряде случаев (аллергия, аутоиммунные реакции) они могут обусловливать повреждение клеток;
• генетические повреждения (например, наследственные мембранопатии, энзимопатии и др.);
• дисбаланс питания.
Первое событие, которое в конце концов приводит к повреждению клетки, - это взаимодействие повреждающего агента с мишенями-молекулами (табл. 3-1). Так, мишенями для ультрафиолетовых лучей могут быть ароматические группы белков, ферментов и рецепторов или нуклеотиды в молекулах ДНК и РНК. Мишенью для окиси углерода служат различные гемсодержащие ферменты. Мишенью при действии гипоксии оказываются митохондрии, которые перестают запасать энергию в форме АТФ, и т.д.
Таблица 3-1. Примеры повреждающих агентов, действующих на клетку
Действующие агенты | Основные мишени | Первичные процессы |
Токсины | Активные центры ферментов и рецепторов, ионные каналы | Инактивация ферментов, блокада рецепторов и ионных каналов |
Ультрафиолетовое излучение | Нуклеиновые кислоты и белки | Фотохимические реакции нуклеотидов и определенных аминокислот |
Окончание табл. 3-1
СВЧ миллиметрового диапазона | Молекулы воды | Ускорение процессов, лимитируемых диффузией в водной среде |
Гипоксия Митохондрии Снижение синтеза АТФ | ||
Гиперкалиемия | Клеточные мембраны | Увеличение мембранного потенциала*, гиперполяризация |
* Увеличение разницы потенциалов между наружной и внутренней поверхностью клеточной мембраны.
Взаимодействие повреждающего фактора c мишенью может приводить к повреждению самой мишени, что наблюдается, например, при действии ультрафиолетовых лучей на клетки. В других случаях мишень не повреждается действующим агентом, но временно перестает функционировать. Именно это приводит в конечном счете к повреждению клетки в целом. Например, при действии цианистого калия выключается функция цитохромоксидазы, которая в данном случае служит мишенью для яда. Но фермент не повреждается: если удалить цианид из окружающей среды, функция цитохромоксидазы восстановится. Причиной гибели клетки является последующее повреждение клеточных структур, вызванное длительным прекращением энергообеспечения.
Таким образом, между моментом взаимодействия повреждающего агента с мишенью и процессом повреждения определенных клеточных структур может произойти целая цепь последовательных событий.
Гибель клетки - это конечный результат ее повреждения. Существует два основных типа клеточной гибели - некроз и апоптоз. На сегодняшний день выделяют также третий тип смерти клеток - конечное дифференцирование, который, по мнению большинства современных ученых, является одной из форм апоптоза.
Некроз (от греч. nekros - мертвый) - это патологическая форма гибели клетки вследствие ее необратимого химического или физического повреждения (высокая и низкая температура, органические растворители, гипоксия, отравление, гипотонический шок, ионизирующее излучение и др.). Некроз представляет собой спектр морфологических изменений, являющихся результатом разрушающего действия ферментов на поврежденную клетку. Развивается два конкурирующих процесса: ферментативное переваривание клетки
(колликвационный, разжижающий некроз) и денатурация белков (коагуляционный некроз). Для проявления обоих этих процессов требуется несколько часов, поэтому в случае внезапной смерти, например, при инфаркте миокарда соответствующие морфологические изменения просто не успевают развиться. Этот вид гибели клеток генетически не контролируется.
Некрозу могут предшествовать периоды паранекроза и некробиоза.
Паранекроз - заметные, но обратимые изменения в клетке: помутнение цитоплазмы, вакуолизация, появление грубодисперсных осадков, увеличение проникновения в клетку различных красителей.
Некробиоз - состояние «между жизнью и смертью» (от necros - мертвый и bios - живой); изменения в клетке, предшествующие ее смерти. При некробиозе в отличие от некроза возможно возвращение клетки в исходное состояние после устранения причины, вызвавшей некробиоз.
Если некроз считается патологической формой клеточной гибели, возникающей в результате чрезмерного (резкого, сильного) повреждающего воздействия на клетку, то апоптоз противопоставляется ему как контролируемый процесс самоуничтожения клетки.
Апоптоз (от греч. аро - отделение и ptosis - падение) - это генетически контролируемая физиологическая форма гибели клетки. Биологическое значение апоптоза заключается в поддержании внутреннего гомеостаза организма на клеточном, тканевом и системном уровнях. Апоптоз ответствен за многочисленные физиологические и патологические процессы в организме:
1. Программированное разрушение клеток на стадии эмбриогенеза (автономный апоптоз). Различают три категории автономного апоптоза: морфогенетический, гистогенетический и филогенетический.
Морфогенетический апоптоз участвует в разрушении различных тканевых зачатков, что обеспечивается:
• гибелью клеток в межпальцевых промежутках;
• гибелью клеток «избыточного» эпителия при слиянии нёбных отростков, когда формируется твердое нёбо;
• гибелью клеток в дорсальной части нервной трубки во время смыкания, что необходимо для достижения единства эпителия двух сторон нервной трубки и связанной с ними мезодермы.
Нарушение морфогенетического апоптоза в этих трех локализациях приводит, соответственно, к развитию синдактилии, расщеплению твердого нёба и spina bifida.
Гистогенетический апоптоз имеет место при дифференцировке тканей и органов, например, при гормонально-зависимой дифференцировке половых органов из тканевых зачатков. Так, клетками Сертоли в яичках плода мужского пола синтезируется гормон, который вызывает путем апоптоза регрессию протоков Мюллера, из которых у женщин формируются маточные трубы, матка и верхняя часть влагалища.
Филогенетический апоптоз участвует в удалении рудиментарных структур у эмбриона, например пронефроса.
2. Гормонозависимая инволюция органов у взрослых, например отторжение клеток эндометрия во время менструального цикла, атрезия фолликулов в яичниках в менопаузе, регрессия молочной железы после прекращения лактации.
3. Стабилизация численности клеток и их популяций в активно пролиферирующих тканях, например клеток эпителия кишечника, крови и иммунной системы; удаление стареющих клеток, прошедших свой жизненный цикл.
4. Элиминация части опухолевых клеток во время спонтанной регрессии опухолей.
5. Гибель клеток иммунной системы (В- и Т-лимфоцитов) при гипосекреции цитокинов, аутореактивных Т-клеток в тимусе - при их клональной делеции.
6. Патологическая атрофия гормонозависимых органов, например атрофия предстательной железы после кастрации; истощение лимфоцитов в тимусе на фоне терапии глюкокортикоидами.
7. Патологическая атрофия паренхиматозных органов после обтурации выводящих протоков, например, в поджелудочной и слюнных железах, почках.
8. Гибель клеток, вызванная действием цитотоксических Т-лимфоцитов, в частности при отторжении трансплантата и болезни «трансплантат против хозяина».
9. Элиминация клеток, инфицированных вирусами (например, при вирусном гепатите фрагменты апоптотических клеток обнаруживаются в печени в виде телец Каунсильмана).
10. Элиминация поврежденных клеток при действии химических и физических факторов (высокая и низкая температура,
ионизирующее излучение, противоопухолевые препараты и др.) в дозе, недостаточной для развития некроза.
Апоптоз является активным процессом саморазрушения клетки, по морфологическим и другим признакам он существенно отличается от некроза (см. табл. 3-2). Наиболее характерные проявления апоптоза определяются тем, что первые события, связанные с его осуществлением, начинаются в ядре. К ним относятся конденсация хроматина с формированием скоплений (в виде ленты, комочков), прилежащих к ядерной мембране (маргинация хроматина), и появление вдавлений ядерной мембраны, приводящих к фрагментации ядра (кариорексису) и образованию апоптотических телец - внеклеточных фрагментов ядра, окруженных мембраной. В цитоплазме происходит конденсация и сморщивание гранул. Клеточная мембрана утрачивает ворсинчатость, образует пузыревидные вздутия, на ней экспрессируются различные молекулы, распознаваемые фагоцитами (фосфатидилсерин, тромбоспондин, десиалированные мембранные гликоконъюгаты). От поверхности апоптотической клетки отщепляются небольшие везикулы, наполненные содержимым цитоплазмы (митохондрии, рибосомы и др.) и окруженные мембранным липидным бислоем. Клетка постепенно уменьшается в объеме, округляется и теряет межклеточные контакты. Апоптотические клетки и их фрагменты (апоптотические тельца, везикулы) поглощаются макрофагами, нейтрофилами и другими соседними клетками, не являющимися «профессиональными» фагоцитами. В результате эндоцитоза содержимое апоптотических клеток не выделяется в межклеточное пространство, как это происходит при некрозе, при котором вокруг гибнущих клеток скапливаются их активные внутриклеточные компоненты, включая энзимы, закисляется среда, что способствует повреждению соседних клеток и развитию воспалительной реакции, т.е. апоптоз одиночной клетки не отражается на ее окружении.
В развитии апоптоза выделяют 3 стадии: сигнальную (индукторную), эффекторную и деградации (деструкции).
Пусковыми факторами апоптоза могут быть как внешние (внеклеточные) факторы, так и внутриклеточные сигналы. Сигнал воспринимается клеткой, далее последовательно передается молекулам-посредникам (мессенджерам) различного порядка и достигает ядра, где происходит включение программы клеточного «самоубийства».
Таблица 3-2. Дифференциальные признаки некроза и апоптоза
Признаки Некроз Апоптоз | ||
Пусковой фактор | Разрушение мембраны под действием патологических стимулов | Деградация ДНК под действием физиологических и патологических стимулов |
Распространенность Группа клеток Одиночная клетка | ||
Биохимические изменения в клетке | Активация лизосомальных ферментов | Активация эндонуклеаз, фрагментирующих ДНК |
Энергозависимость Нет Есть | ||
Целостность цитоплазматической и внутриклеточных мембран | Нарушена | Сохранена |
Морфологические изменения клетки | Увеличение размеров клетки, разрыхление мембраны, набухание (окноз) цитоплазмы, митохондрий, лизис ядра и гранул | Уменьшение размеров клетки, уплотнение и вздутие мембраны, кариопикноз, кариорексис, маргинация хроматина, конденсация и уплотнение гранул |
Воспалительный ответ | Есть | Нет |
Элиминация гибнущей клетки | Лизис клетки, фагоцитоз | Фрагментация клетки, поглощение фрагментов клетки (мембранных везикул, апоптотических телец) соседними клетками и фагоцитами |
Классическими индукторами экзогенного апоптоза являются стероидные гормоны (половые, тиреоидные, кальцитриол, минералокортикоиды, ретиноиды), антигены, антитела, митогены, цитокины (фактор некроза опухолей (TNF) α, интерлейкин (IL) 1, IL-10, интерферон (INF) γ, β-токоферол и др.). Их проапоптогенное действие осуществляется через ядерные рецепторы (например, GR - глюкокортикоидный рецептор), специализированные мембранные «рецепторы смерти» (Fas, TNF-RI, TNF-RII, DR-3, DR-5 и др.) и рецепторы, выполняющие иные функции, например функцию активации клетки (T-клеточный рецептор (TCR),
цитокиновые рецепторы), что сопровождается развитием активационного апоптоза.
Ситуация эндогенного запуска программы гибели клетки возможна при лишении ее ростовых факторов (IL-2, IL-3, IL-4, INF-α, колониестимулирующих факторов - гранулоцитарно-макрофагального (ГМ-КСФ), гранулоцитарного (Г-КСФ), эритропоэтина и др.), нарушении контактов с внеклеточным матриксом и другими клетками, накоплении нерепарируемых разрывов ДНК (например, при повреждении клетки вирусами, ионизирующей радиацией, ультрафиолетовым излучением, токсинами и др.). В последнем случае важная роль отводится ядерному белку р53 (см. ниже).
В результате запуска апоптогенным (экзогенным или эндогенным) сигналом программы активации генов-индукторов апоптоза (Р53, BAX, PIG, FAS/APO-1, IGF-BP3 и др.) и/или угнетения апоптозингибирующих генов (генов семейства BCL-2) в клетке изменяется набор внутриклеточных РНК и белков, синтезируются и активируются ферменты, способные разрушать клеточные белки (протеазы - каспазы, катепсины, кальпаины, гранзимы) и нуклеиновые кислоты (нуклеазы - Са2+/Мg2+-зависимая эндонуклеаза и др.). Основным проявлением деструктивных изменений клетки при апоптозе является деградация хроматина, основой которого служит расщепление ДНК.
В настоящее время выделены несколько основных механизмов реализации апоптоза:
1) Рецепторный. Осуществляется с помощью «рецепторов смерти» (см. выше) при активирующем взаимодействии с соответствующими лигандами, большинство из которых относится к суперсемейству фактора некроза опухолей. Взаимодействие рецептора с лигандом приводит к активации адапторных белков, ассоциированных с «доменами смерти» (FADD - Fas-associated death domain, TRADD - TNF-R-associated death domain), и прокаспазы 8, продукт которой - каспаза 8 (инициаторная) активирует каспазу 3 (эффекторную), что, в свою очередь, обусловливает активацию эндонуклеаз, фрагментирующих ДНК.
2) Митохондриальный. Участие митохондрий в апоптозе обеспечивается присутствием в их матриксе и межмембранном пространстве большого количества биологически активных веществ (цитохрома С (Cyt С); прокаспаз 2, 3, 9; апоптозиндуцирующего фактора (AIF), обладающих выраженным апоптогенным действием. Фактором активации апоптоза является выход данных веществ
в цитоплазму при снижении трансмембранного потенциала митохондрий вследствие открытия гигантских митохондриальных пор (выполняют роль Ca2+-, рН-, потенциал-, НАДФ2Н/НАДФ+- и редоксзависимых каналов) и повышения проницаемости митохондриальных мембран. К раскрытию пор приводят истощение в клетках восстановленного глутатиона, НАДФН, АТФ и АДФ, образование активных форм кислорода, разобщение окислительного фосфорилирования, увеличение содержания Ca2+ в цитоплазме. Поступление межмембранных белков и активация апоптоза возможны также при разрыве наружной мембраны митохондрий вследствие гиперполяризации внутренней мембраны.
3) р53-опосредованный. p53 - многофункциональный белок, играющий важную роль в мониторинге сигналов о состоянии клетки, целостности ее генома, активности систем ДНК-репарации. Повреждение ДНК ведет к накоплению белка р53 в клетке. Это определяет остановку клеточного цикла в фазах G1 и G2, предотвращает репликацию, активирует синтез и репарацию ДНК, а следовательно, создает условия для восстановления нативной структуры ДНК, препятствует появлению мутантных и анеуплоидных клеток в организме. В случае если имеется недостаточность систем ДНК-репарации и повреждения ДНК сохраняются, клетка подвергается апоптозу. В частности, белок р53 способен индуцировать транскрипцию таких апоптогенных факторов, как Bax, Fas- рецептор, DR-5 и др.
4) Перфорин-гранзимовый. Цитотоксические Т-лимфоциты (Т-киллеры) вызывают апоптоз клеток-мишеней (например, инфицированных клеток) с помощью белка перфорина. Полимеризуясь, перфорин образует в цитоплазматической мембране клеткимишени трансмембранные каналы, по которым внутрь клетки поступают секретируемые Т-киллером гранзимы (фрагментины) - смесь сериновых протеаз. Основным компонентом этой смеси является гранзим В - протеолитический фермент, активирующий каспазу 3.
Важную роль в процессе передачи апоптогенного сигнала и регуляции апоптоза играют следующие внутриклеточные факторы (мессенджеры):
• концентрация ионов Ca (Ca2+ активирует сериновые и цистеиновые протеазы, Ca2+/Mg2+-зависимую эндонуклеазу);
• протеинкиназы А (медиатор апоптоза) и С (ингибитор апоптоза);
• церамид (стимулирует киназы, фосфатазы);
• активные формы кислорода (обусловливают снижение трансмембранного потенциала митохондрий, увеличение внутриклеточной концентрации Ca2+, образование цАМФ);
• монооксид азота (опосредует изменение экспрессии р53, открытие гигантских пор в митохондриях и снижение митохондриального потенциала).
При различных патологических процессах в организме (инфекция, воспаление, иммунодефициты, гипо- и апластическая анемии, опухоли и др.) могут наблюдаться как ускорение, так и замедление апоптоза.
Примеры некоторых заболеваний, в патогенез которых включается апоптоз, представлены в табл. 3-3.
Таблица 3-3. Примеры заболеваний, связанных с угнетением или усилением апоптоза
 Универсальный ответ клетки на повреждение. Особенностью
развития патологических изменений в клетках в ответ на самые различные
неблагоприятные воздействия является сходство этих изменений, которое
позволило Д.Н. Насонову и В.Я. Александрову выдвинуть в 1940 г. теорию о
неспецифической реакции клеток на повреждение. Ее суть сводится к
следующему - каким бы ни был повреждающий агент и на какие бы клетки он ни действовал, ответ клеток по ряду показателей является одинаковым. К числу таких показателей относятся:
Универсальный ответ клетки на повреждение. Особенностью
развития патологических изменений в клетках в ответ на самые различные
неблагоприятные воздействия является сходство этих изменений, которое
позволило Д.Н. Насонову и В.Я. Александрову выдвинуть в 1940 г. теорию о
неспецифической реакции клеток на повреждение. Ее суть сводится к
следующему - каким бы ни был повреждающий агент и на какие бы клетки он ни действовал, ответ клеток по ряду показателей является одинаковым. К числу таких показателей относятся:
1) уменьшение дисперсности коллоидов цитоплазмы и ядра;
2) увеличение вязкости цитоплазмы, которому иногда предшествует ее некоторое уменьшение;
3) увеличение сродства цитоплазмы и ядра к ряду красителей. Во многих случаях обнаруживаются также набухание клетки,
изменение ионной проницаемости плазматической и внутриклеточных мембран, выход метаболитов из клетки, изменение флуоресценции, повышение кислотности цитоплазмы и т.д. Существование такого стереотипа изменений физико-химических свойств клеток при их повреждении связано с тем, что молекулярноклеточные механизмы повреждения сходны, хотя причины, вызвавшие повреждение, могут быть самыми разными. Практически у всех клеток при действии повреждающих агентов наблюдается резкое увеличение проницаемости клеточных мембран для ионов кальция. Это сопровождается активацией различных внутриклеточных ферментов и процессов: протеинкиназ, фосфолипаз, фосфодиэстеразы циклических нуклеотидов, системы биосинтеза белков и т.д. Эти изменения могут быть обратимыми, но в конце концов при сильном и длительном воздействии повреждающего фактора происходит стойкое нарушение функций клеток, а следовательно, ткани и органа в целом.
3.2. МЕХАНИЗМЫ ПОВРЕЖДЕНИЯ МЕМБРАННЫХ СТРУКТУР КЛЕТКИ
Наиболее ранние изменения свойств и поведения клеток при действии повреждающих агентов связаны с изменениями функционирования мембранных структур клетки: цитоплазматической мембраны, внутренней мембраны митохондрий, мембран эндоплазматического ретикулума и других внутриклеточных структур (см. табл. 3-4).
Биологические мембраны выполняют множество функций, нарушение любой из которых может привести к изменению жизнедеятельности клетки в целом и даже к ее гибели. На рис. 3-1 дано схематическое изображение типичной мембраны с указанием тех ее элементов, повреждение которых может иметь место в патологии и лежать в основе развития различных заболеваний.
Таблица 3-4. Ранние изменения в функционировании внутриклеточных структур при повреждении
Изменения Проявления | |
Увеличение проницаемости цитоплазматической мембраны | Увеличение электропроводности тканей Увеличение связывания красителей Изменение мембранного потенциала Выход К+ из клетки Выход метаболитов Увеличение объема (набухание) клеток Увеличение внутриклеточной концентрации Са2+ |
Нарушение структуры и функций митохондрий | Снижение потребления кислорода Увеличение проницаемости внутренней митохондриальной мембраны Набухание митохондрий Снижение Са2+-аккумулирующей способности |
Ацидоз | Активация ?+/Н+-обмена Повышение внутриклеточной концентрации Na+ Набухание клеток |
Повреждение эндоплазматического ретикулума | Выход Ca2+ в цитоплазму, нарушение системы синтеза белка |
Изменение активности ферментов и рецепторов | Активация ферментов лизосом Активация эндонуклеаз, апоптоз |
Повреждение генетического аппарата клетки | Повреждение рибосом |
Наиболее тяжелые последствия вызывает повреждение липидного слоя (или бислоя) мембраны. Липидный слой цитоплазматической и внутриклеточных мембран выполняет две основные функции - барьерную и матричную (структурную). Повреждение барьера приводит к нарушению регуляции внутриклеточных процессов и тяжелым расстройствам клеточных функций.
Изучение воздействия разного рода повреждающих агентов на изолированные клетки (например, эритроциты), митохондрии, фосфолипидные везикулы (липосомы), плоские бислойные липидные мембраны и другие модельные объекты показало, что в конечном счете существует всего четыре основных процесса, кото-
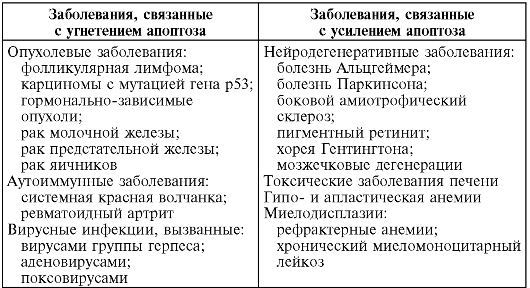
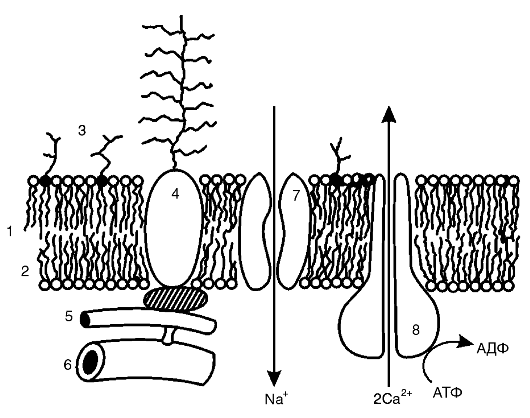 Рис. 3-1. Элементы
биологических мембран, подверженные повреждению: 1 - липидный бислой; 2
- монослой липидных молекул; 3 - гликолипиды; 4 - гликопротеины; 5 -
микрофиламенты; 6 - микротубулы; 7 - ионный канал; 8 - ионный насос
Рис. 3-1. Элементы
биологических мембран, подверженные повреждению: 1 - липидный бислой; 2
- монослой липидных молекул; 3 - гликолипиды; 4 - гликопротеины; 5 -
микрофиламенты; 6 - микротубулы; 7 - ионный канал; 8 - ионный насос
рые непосредственно обусловливают нарушение целостности липидного бислоя в патологии:
1) механическое (осмотическое) растяжение мембраны;
2) перекисное окисление липидов;
3) действие мембранных фосфолипаз;
4) адсорбция на липидном слое полиэлектролитов, включая некоторые белки и пептиды.
Понимание роли мембран в развитии того или иного патологического состояния предполагает знание химических и физических условий протекания перечисленных выше процессов и результатов их действия на мембранные структуры, включая знание молекулярных механизмов действия каждого из них и биологические по-
следствия повреждения мембран для жизнедеятельности клетки и организма в целом.
3.2.1. Нарушение барьерной функции биологических мембран
Важную роль в повреждении мембран играют процессы их механического растяжения в результате нарушения осмотического равновесия в клетках. Если поместить эритроциты в гипотонический раствор, то вода будет входить в клетки, они примут сферическую форму, а затем произойдет гемолиз. Митохондрии также набухают в гипотонических средах, но происходит разрыв только внешней мембраны; внутренняя остается целой, хотя теряет способность задерживать небольшие молекулы и ионы. В результате митохондрии утрачивают способность к окислительному фосфорилированию.
Сходные явления наблюдаются и в целых клетках и тканях в условиях патологии. Так, в результате активации фосфолипазы А2 мембран митохондрий при гипоксии они становятся проницаемыми для ионов калия. Если в этих условиях восстановить оксигенацию ткани, то на мембранах митохондрий восстановится мембранный потенциал (со знаком «минус» внутри) и митохондрии будут «насасывать» ионы калия, вслед за которыми в матрикс входит фосфат. Концентрация ионов внутри митохондрий возрастает, и органеллы набухают. Это приводит к растяжению мембран и их дальнейшему повреждению.
Молекулярные механизмы увеличения проницаемости липидного слоя мембран для ионов. При изучении молекулярных основ проницаемости липидного слоя широко используются модельные мембранные системы: изолированные мембранные структуры (эритроциты, митохондрии, везикулы саркоплазматического ретикулума), а также искусственные фосфолипидные мембраны (бислойные липидные мембраны и фосфолипидные везикулы - липосомы). Изучение такого рода систем показало, что сам по себе липидный слой практически непроницаем для ионов. При действии различных химических и физических факторов он становится проницаемым по одной из трех причин (или их комбинаций):
1. В липидном бислое, микровязкость которого близка к вязкости оливкового масла, появляется жирорастворимое вещество, способное связывать ионы. Механизм переноса ионов в этом случае напоминает «перевоз пассажиров в лодке с одного берега на
другой» и называется «челночным», или переносом с помощью подвижного переносчика. Примером подвижного переносчика может служить ионофорный антибиотик валиномицин, который образует комплекс с ионами калия, растворимый в липидной фазе мембраны. К числу подвижных переносчиков, возможно, относятся водорастворимые продукты перекисного окисления липидов, в присутствии которых, как оказалось, увеличивается проницаемость мембраны для ионов водорода.
2. В липидном слое появляются вещества, молекулы которых, собираясь вместе, образуют канал через мембрану. Сквозь такой канал ионы могут проходить с одной стороны мембраны на другую. Каналы образуются молекулами некоторых антибиотиков, например грамицидина А и полимиксина. Продукты перекисного окисления липидов также могут образовывать каналы в липидном слое, если в растворе есть ионы кальция. Продукты расщепления некоторых фосфолипидов (в частности, кардиолипина) фосфолипазой А2 образуют каналы для одновалентных катионов.
3. Электрическая прочность липидного слоя мембраны снижается, и ее участок разрушается электрическим током, который возникает под влиянием разности потенциалов, существующей на мембране. Такое явление носит название «электрического пробоя» (см. ниже). Формирование в мембране «пор» с индукцией пробоя мембраны лежит в основе нарушений барьерной функции мембраны при адсорбции на липидном бислое полиэлектролитов, чужеродных для клетки белков, антител.
Свободные радикалы. Свободнорадикальное (перекисное) окисление липидов (ПОЛ). Хорошо известно, что в органических молекулах (включая те, из которых состоит наш организм) электроны на внешней электронной оболочке располагаются парами: одна пара на каждой орбитали. Свободные радикалы отличаются от обычных молекул тем, что у них на внешней электронной оболочке имеется неспаренный (одиночный) электрон. Это делает их химически активными, поскольку они стремятся вернуть себе недостающий электрон, отняв его от окружающих молекул и тем самым повреждая их. Свободные радикалы вступают в реакции с неорганическими и органическими соединениями - белками, липидами, углеводами, нуклеиновыми кислотами, инициируют аутокаталитические реакции, в ходе которых молекулы, с которыми они реагируют, также превращаются в свободные радикалы. Таким образом, сво-
бодные радикалы - высокоактивные молекулы, способные разрушать структуры клетки.
Основным источником радикалов является молекулярный кислород. К кислородным радикалам относятся: NO* (оксид азота или нитроксид), RO* (алкоксильный радикал), RO*2 (перекисный или пероксидный радикал), O*2- (супероксидный анион-радикал или супероксид), HO*2 (гидроперекисный радикал, HO* (гидроксильный радикал).
В целом все радикалы, образующиеся в организме человека, можно разделить на природные и чужеродные. В свою очередь, природные радикалы можно разделить на первичные, вторичные и третичные (рис. 3-2).
Первичные радикалы - те радикалы, образование которых осуществляется при участии определенных ферментных систем (НАДФН-оксидазы, NO-синтазы, циклооксигеназы, липооксигеназы, монооксигеназы, ксантиноксидазы и др.). Прежде всего к первичным радикалам относятся семихиноны, образующиеся в реакциях таких переносчиков электронов, как коэнзим Q (обозначим радикал как Q*) и флавопротеины, O*2-, NO*.
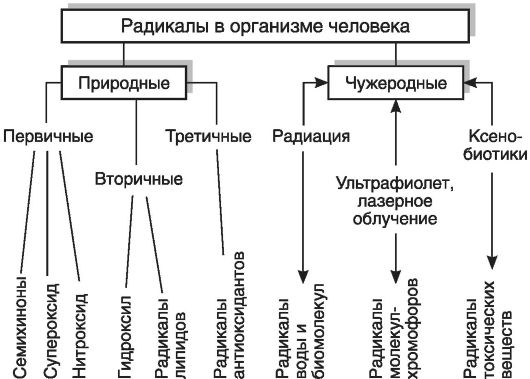 Рис. 3-2. Классификация радикалов в организме человека
Рис. 3-2. Классификация радикалов в организме человека
Из первичного радикала - O*2-, а также в результате других реакций в организме образуются весьма активные молекулярные соединения: перекись водорода (Н2О2), гипохлорит (HOCl), гидроперекиси липидов. Под действием ионов металлов переменной валентности, в первую очередь Fe2+, из этих веществ образуются вторичные радикалы (HO*, радикалы липидов), которые оказывают разрушительное действие на клеточные структуры.
Для защиты от повреждающего действия вторичных радикалов в организме используется большая группа веществ, называемых антиоксидантами (см. ниже), к числу которых принадлежат «ловушки» («перехватчики») свободных радикалов. Примером последних служат альфа-токоферол, тироксин, восстановленный убихинон (QH2) и женские стероидные гормоны. Реагируя с липидными радикалами, эти вещества сами превращаются в радикалы антиоксидантов, которые можно рассматривать как третичные радикалы.
Наряду с этими радикалами, постоянно образующимися в том или ином количестве в клетках и тканях организма человека, разрушительное действие могут оказывать радикалы, появляющиеся при таких воздействиях, как ионизирующее излучение, ультрафиолетовое облучение или даже освещение интенсивным видимым светом, например светом лазера. Такие радикалы можно назвать чужеродными. К ним принадлежат также радикалы, образующиеся из попавших в организм посторонних соединений, ксенобиотиков, многие из которых оказывают токсическое действие именно благодаря свободным радикалам, образующимся при метаболизме этих соединений.
Однако не следует считать, что свободные радикалы являются только повреждающим клетки фактором. Примером положительной роли этих соединений является система клеточного иммунитета. Например, фагоцитирующие лейкоциты (к которым относятся гранулоциты и моноциты крови и тканевые клетки - макрофаги), соприкасаясь с поверхностью бактерий в очаге воспаления, активируются и с помощью НАДФН-оксидазы - фермента, встроенного в мембрану клеток и внутриклеточных везикул-фагосом, генерируют из О2 супероксидный анион-радикал, обладающий бактерицидным действием (рис. 3-3). Нитроксид (NO*), выделяясь клетками-фагоцитами вместе с супероксид-радикалами, используется для борьбы с микробами грибковой природы. Для осуществления своих киллерных функций фагоциты используют также образующийся из перекиси водорода гипохлорит (OCl-). Реакция
 Рис. 3-3. Реакции супероксидного радикала
Рис. 3-3. Реакции супероксидного радикала
образования гипохлорита катализируется с помощью специального фермента - миелопероксидазы: Н2О2 + Cl- - Н2О + ОО-. Гипохлорит сам по себе не является свободным радикалом (относится к группе активных метаболитов кислорода нерадикальной природы), но взаимодействует с органическими молекулами через радикальные механизмы. При участии гипохлорита образуются такие высокоактивные молекулы, как гидроксильный радикал (Fe2+ + OCl- + H+ - Fe3+ + HO' + Cl-), синглетный кислород (Ю2). В активированных лейкоцитах гидроксильный радикал (HO') может образовываться также при разложении перекиси водорода в присутствии ионов двухвалентного железа (Н2О2 + Fe2+ - Fe3+ + HO' + HO'). Цитотоксическое действие OCl- и HO' заключается в их способности разрушать SH-гругты и другие аминокислотные остатки белков, индуцировать разрывы цепей ДНК и РНК, усиливать активность ПОЛ, протеиназ, белков системы комплемента, ингибировать белки деления и ферменты бактерий.
Свободные радикалы выполняют также и другие, в том числе регуляторные, функции. Так, для некоторых тканей, в частности мозга, характерен повышенный синтез простагландинов, тромбоксанов и лейкотриенов, образующихся из арахидоновой кислоты при индукции ПОЛ с участием супероксид-аниона. Радикал убихинона (коэнзима Q) - семихинон (HQ') участвует в цепи переноса электронов; при нарушении работы дыхательной цепи он может стать источником других радикалов, в первую очередь радикалов кислорода.
Кроме того, свободные радикалы активно участвуют в процессах передачи клеточного сигнала, могут выступать в качестве вто-
ричных мессенджеров в сигнальных каскадах, запускаемых ангиотензином II, эндотелином и др. Так, NO', образующийся клетками стенок кровеносных сосудов (эндотелия) при участии гемсодержащего фермента NO-синтазы, играет ключевую роль в регуляции тонуса сосудов и кровяного давления: его недостаток приводит к гипертензии, избыток - к гипотензии. Нарушение метаболизма NO вызывает заболевания, связанные с изменением кровяного давления. Радикалы, образующиеся в цитозоле клетки в ответ на стимуляцию факторами роста, участвуют в регуляции пролиферативного процесса.
В нормальных условиях радикалы кислорода не накапливаются в клетках. Состояние клеток, характеризующееся избыточным содержанием в них радикалов кислорода, называется окислительным стрессом. Окислительный стресс развивается тогда, когда окислительно-восстановительный гомеостаз (редокс-гомеостаз или баланс) в клетке нарушается. Этот дисбаланс может быть обусловлен гиперпродукцией активных форм кислорода или недостаточностью системы антиоксидантной защиты, в состав которой входят низкомолекулярные соединения растительного и животного происхождения (содержатся в плазме крови, в цитоплазме или мембранах клеток). Выделяют несколько основных групп антиоксидантов:
1) ферментативные - супероксиддисмутаза, каталаза, ферменты глутатионового цикла (глутатионпероксидаза, глутатионредуктаза, глутатион-S-трансфераза);
2) фенольные - витамин Е, коэнзим Q, флавоноиды (кверцетин, рутин, гесперетин и др.);
3) каротиноиды - жирорастворимые растительные пигменты, входящие в состав овощей и фруктов (морковь, шпинат, манго, абрикос и др.);
4) аскорбиновая кислота (витамин С) - содержится в свежих овощах, фруктах и ягодах (петрушка, молодая капуста, шиповник, черная смородина, лимон, апельсин, папайя, яблоко и др.), в организме в большом количестве обнаруживается в надпочечниках, гипофизе, вилочковой железе;
5) SH-содержащие соединения - глутатион, тио-, перокси- и глутаредоксины;
6) хелаторы ионов металлов переменной валентности - трансферрины, ферритин, церулоплазмин, металлотионеины, мочевая кислота и др.
По принципу антиокислительного действия выделяют антиоксиданты прямого (направленного) и непрямого (опосредованного) действия. Эффективность последних проявляется только в живых системах (in vivo), в то время как соединения направленного типа действия могут подавлять окислительные процессы с участием активных метаболитов кислорода как in vivo, так и in vitro.
В естественных условиях антиоксиданты (супероксиддисмутаза, каталаза, таурин и др.) защищают фагоциты от аутодеструкции собственными радикалами (супероксидом, гипохлоритом, гидроксильным радикалом), координируют генерацию воспалительных медиаторов нейтрофилами и макрофагами (простагландинов, IL-6, TNF-α и др.). Эффекты некоторых антиоксидантов представлены в табл. 3-5.
Таблица 3-5. Наиболее известные антиоксиданты
Антиоксидант | Характеристика |
Церулоплазмин | Окисляет Fe2+ до Fe3+ молекулярным кислородом |
Апо-белок трансферрина | Связывает Fe3+ |
Ферритин | Окисляет Fe2+ и депонирует Fe3+ |
Карнозин | Связывает Fe2+ |
Супероксиддисмутазы | Удаляют супероксид с образованием пероксида водорода |
Каталаза | Разлагает пероксид водорода с выделением кислорода |
Глутатионпероксидазы | 1. Удаляют пероксид водорода за счет окисления глутатиона 2. Удаляют гидроперекиси липидов |
Глутатионредуктаза | Восстанавливает окисленный глутатион |
Токоферол, тироксин, стероиды | Перехватывают радикалы липидов |
Аскорбиновая кислота | Регенерирует окисляющиеся токоферол и убихинон |
Глутатион | Используется для восстановления пероксидов |
Основные стадии цепного окисления. Реакция цепного окисления липидов играет исключительную роль в клеточной патологии. Она протекает в несколько стадий: инициирование, продолжение, разветвление и обрыв цепи (рис. 3-4).
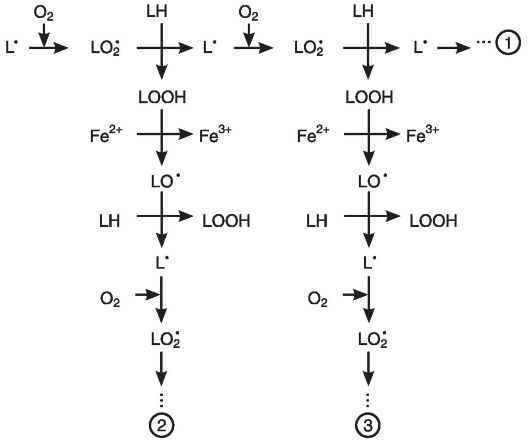 Рис. 3-4. Цепная реакция перекисного окисления липидов: 1-старая цепь окисления, 2, 3 - новые цепи окисления
Рис. 3-4. Цепная реакция перекисного окисления липидов: 1-старая цепь окисления, 2, 3 - новые цепи окисления
Инициирование цепной реакции начинается с того, что в липидный слой мембран или липопротеинов внедряется свободный радикал. Чаще всего это радикал гидроксила. Будучи небольшой по размеру незаряженной частицей, он способен проникать в толщу гидрофобного липидного слоя и вступать в химическое взаимодействие с полиненасыщенными жирными кислотами (их принято обозначать как LH), входящими в состав биологических мембран и липопротеинов плазмы крови. При этом образуются липидные радикалы:
HO' + LH - Н2О + L'.
Липидный радикал (L ) вступает в реакцию с растворенным в среде молекулярным кислородом, при этом образуется новый свободный радикал - радикал липоперекиси (LOO ):
L + О2 - LOO
Этот радикал атакует одну из соседних молекул фосфолипида с образованием гидроперекиси липида LOOH и нового радикала L :
LOO'+ LH - LOOН + L'
Чередование двух последних реакций как раз и представляет собой цепную реакцию ПОЛ (см. рис. 3-4).
Существенное ускорение пероксидации липидов наблюдается в присутствии небольших количеств ионов двухвалентного железа. В этом случае происходит разветвление цепей в результате взаимодействия Fe2+ с гидроперекисями липидов:
Fe2+ + LOOН - Fe3+ + НО- + LO'
Образующиеся радикалы LO' инициируют новые цепи окисления липидов (см. рис. 3-4):
LO' + LH - LOН + L'; L'+ О2 - LOO' - и т.д.
В биологических мембранах цепи могут состоять из десятка звеньев и более. Но, в конце концов, цепь обрывается в результате взаимодействия свободных радикалов с антиоксидантами (InH), ионами металлов переменной валентности (например, теми же Fe2+) или друг с другом:
LOO' + Fe2+ + H+ - LOOН + Fe3+
LOO' + InH - In'+ LOOH
LOO + LOO - молекулярные продукты
Повреждающее действие пероксидации липидов. На рис. 3-5 показаны основные мишени ПОЛ в мембранных структурах клеток. Повреждаются либо белковые структуры, либо липидный бислой в целом. В последнее время ученые уделяют все большее внимание взаимодействию мембран с нуклеиновыми кислотами в ядре и митохондриях. По-видимому, одним из результатов пероксидации липидов может стать повреждение этих молекул со всеми вытекающими последствиями.
Наиболее чувствительны к перекисному окислению липидов сульфгидрильные, или тиоловые, группы (SH) мембранных белков: ферментов, ионных каналов и насосов. В ходе окисления тиоловых групп образуются радикалы (S ), которые затем либо взаимодействуют друг с другом с образованием дисульфидов (SS), либо связываются с кислородом с образованием сульфитов и сульфатов (SO3 и SO4). Большую роль в патологии клетки играет также
 Рис. 3-5. Повреждающее действие перекисного окисления липидов на биологические мембраны
Рис. 3-5. Повреждающее действие перекисного окисления липидов на биологические мембраны
повреждение ионтранспортирующих ферментов (например, Ca2+, Мg2+-АТФазы), в активный центр которых входят тиоловые группы (рис. 3-5, 1). Инактивация Са2+-АТФазы приводит к замедлению откачивания из клетки ионов кальция и ускорению их «протечки» в клетку (где их концентрация меньше). Это вызывает рост уровня ионов кальция в цитоплазме и повреждение клеточных структур.
Окисление тиоловых групп мембранных белков приводит к появлению дефектов в мембранах клеток и митохондрий. Под действием электрического поля через такие дефекты в клетки входят ионы натрия, а в митохондрии - ионы калия. В результате происходит увеличение осмотического давления внутри клеток и митохондрий и их набухание. Это приводит к еще большему повреждению мембранных структур.
Наряду с белками и нуклеиновыми кислотами мишенью повреждающего действия ПОЛ служит сам липидный бислой. Было показано, что продукты ПОЛ делают липидную фазу мембран проницаемой для ионов водорода и кальция (рис. 3-5, 2-3). Это приводит к тому, что в митохондриях окисление и фосфорилирование разобщаются, и клетка оказывается в условиях энергетического голода. Одновременно из митохондрий в цитоплазму выходят ионы кальция, которые повреждают клеточные структуры.
Возможно, наиболее важный результат пероксидации - это уменьшение электрической стабильности липидного слоя, кото-
рое приводит к электрическому пробою мембраны собственным мембранным потенциалом (рис. 3-5, 4). Электрический пробой вызывает полную потерю мембраной ее барьерных функций.
Стабильность липидного слоя мембран и явление электрического пробоя. Как известно, мембраны обладают определенным сопротивлением R электрическому току I, которое при небольшой разности потенциалов φ между двумя сторонами мембраны является постоянной величиной. Иными словами, для мембраны соблюдается закон Ома: I = φ / R. Это означает, что зависимость между напряжением на мембране φ и током через мембрану I - линейная. Однако такая зависимость сохраняется при сравнительно небольших величинах |φ|: не выше 200-300 мВ. При определенной критической разности потенциалов ток резко увеличивается, что может стать причиной разрушения мембраны. Это явление называется электрическим пробоем.
В основе электрического пробоя мембраны лежит спонтанное (вследствие теплового движения молекул) зарождение в липидном бислое дефектов - пор, через которые могут проходить водорастворимые молекулы и ионы. При отсутствии разности потенциалов на мембране увеличения размеров спонтанно образовавшихся пор не происходит, так как данный процесс сопровождается ростом площади раздела фаз «липид - вода» и требует энергетических затрат на преодоление сил поверхностного натяжения на границе раздела фаз. Однако при увеличении разности потенциалов на мембране количество энергии, необходимое для образования и увеличения размеров поры, уменьшается, что способствует ее дальнейшему росту, который после преодоления некоторого энергетического барьера становится самопроизвольным и приводит к полному разрушению мембраны (рис. 3-6). При небольших мембранных потенциалах, существующих в живой клетке (-70 мВ на цитоплазматической мембране и -175 мВ на внутренней мембране митохондрий), этого не происходит, потому что энергетический барьер достаточно высок. Более того, в нормальных условиях, под действием сил поверхностного натяжения образовавшийся дефект «затягивается», и мембрана остается целой. Величина барьера снижается при увеличении поляризации мембраны. Потенциал, при котором начинается электрический пробой, называется потенциалом пробоя и обычно обозначается как U* или φ*. Величина потенциала пробоя различна для мембран с разным составом белков и липидов и может служить количественной мерой электрической
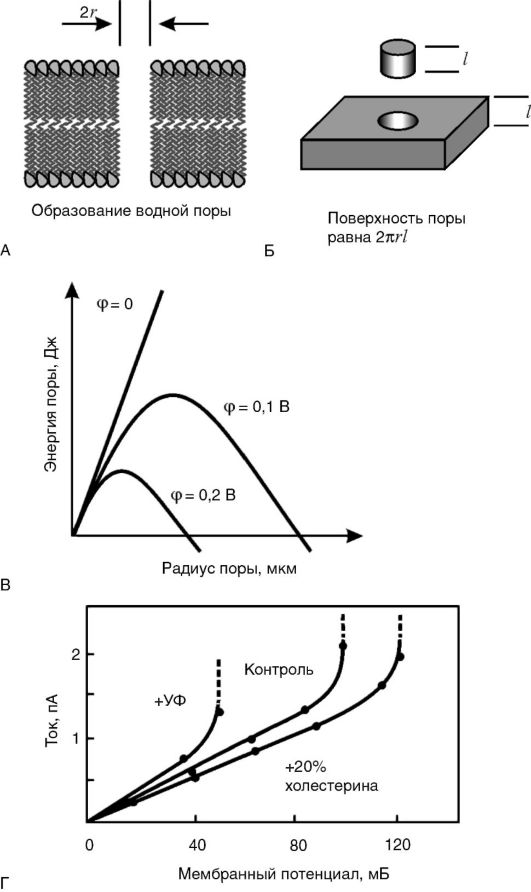 Рис. 3-6. Электрический
пробой мембран: А - появление в липидном бислое мембраны поры,
заполненной водой; Б - размер внутренней поверхности поры пропорционален
ее радиусу; В - энергия мембраны с порой в зависимости от ее радиуса
(величина потенциального барьера при росте поры уменьшается); Г -
возрастание тока в зависимости от потенциала пробоя
Рис. 3-6. Электрический
пробой мембран: А - появление в липидном бислое мембраны поры,
заполненной водой; Б - размер внутренней поверхности поры пропорционален
ее радиусу; В - энергия мембраны с порой в зависимости от ее радиуса
(величина потенциального барьера при росте поры уменьшается); Г -
возрастание тока в зависимости от потенциала пробоя
стабильности мембраны. Чем стабильнее мембрана, тем выше ее потенциал пробоя (т.е. |φ*|).
В живых клетках потенциал пробоя выше мембранного потенциала (|φ*|>|φ|), иначе мембраны пробивались бы своим собственным потенциалом и клетка не могла существовать. Однако запас электрической прочности невелик - 20-30 мВ. Это означает, что при |φ*|<|φ|, т.е. при снижении электрической прочности, может произойти «самопробой» мембраны.
Как уже указывалось выше, основными причинами нарушения барьерных свойств мембран при патологии являются их механическое (осмотическое) растяжение, активация ПОЛ, гидролиз фосфолипидов и адсорбция полиэлектролитов на поверхности. Изучение влияния этих факторов на электрическую прочность мембран показало, что все они снижают силы поверхностного натяжения на границе раздела фаз «липидный слой мембраны - окружающий водный раствор», а следовательно, величину потенциала пробоя (рис. 3-7). Таким образом, электрический пробой - это универсальный механизм нарушения барьерной функции мембран при патологии.
Мембранные системы защиты от электрического пробоя. Известны два фактора, с помощью которых живые клетки повышают электрическую стабильность своих мембранных структур:
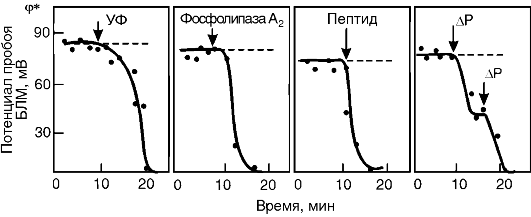 Рис. 3-7. Снижение
электрической прочности бислойной липидной мембраны (БЛМ) при действии
ультрафиолетового излучения (УФ), фосфолипазы А2, пептидов, при
растяжении мембраны, вызванном разностью гидростатического давления (ΔΡ)
Рис. 3-7. Снижение
электрической прочности бислойной липидной мембраны (БЛМ) при действии
ультрафиолетового излучения (УФ), фосфолипазы А2, пептидов, при
растяжении мембраны, вызванном разностью гидростатического давления (ΔΡ)
1. Асимметричный поверхностный потенциал. Поверхностный потенциал возникает на мембране в случае появления на поверхности липидного слоя заряженных химических группировок, например таких, как карбоксил или фосфат. Непосредственно на липидный бислой действует потенциал, равный разности величины мембранного потенциала (т.е. потенциала между водными средами, омывающими мембрану) и поверхностного потенциала (рис. 3-8). За счет неодинаковой плотности зарядов на поверхности мембраны реальная разность потенциалов, приложенная к липидному бислою, отличается от трансмембранной разности потенциалов. Это снижает вероятность пробоя мембраны собственным потенциалом.
2. Холестерин. Было показано, что включение молекул холестерина в фосфолипидный бислой весьма заметно увеличивает электрическую прочность мембран, т.е. повышает потенциал пробоя (см. рис. 3-6, Г). Особенно заметно действие холестерина на поврежденные мембраны. Защитные свойства холестерина против электрического пробоя мембраны можно объяснить его влиянием на вязкость липидного бислоя. Известно, что введение холестерина в фосфолипидный бислой повышает вязкость последнего в 2-3 раза. Это приводит к замедлению образования и роста дефектов (пор) в липидном бислое мембран, лежащих в основе явления электрического пробоя.
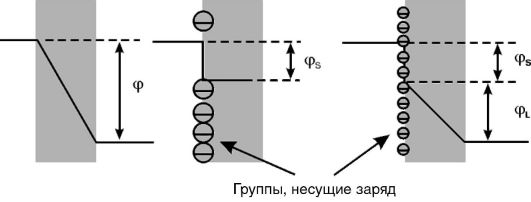 Рис. 3-8. Влияние поверхностного потенциала (cpS) на разность потенциалов на липидном слое мембран (cpL) при одном и том же мембранном потенциале (φ)
Рис. 3-8. Влияние поверхностного потенциала (cpS) на разность потенциалов на липидном слое мембран (cpL) при одном и том же мембранном потенциале (φ)
Критерии оценки нарушений барьерной функции цитоплазматической мембраны. Основными критериями, позволяющими судить о нарушении барьерных свойств цитоплазматической мембраны и увеличении ее проницаемости, являются: уменьшение электрического сопротивления ткани, проникновение водорастворимого красителя в цитоплазму, снижение мембранного потенциала покоя, нарушение ионного баланса, выход внутриклеточных метаболитов в окружающую среду, набухание клеток.
Уменьшение электрического сопротивления (импеданса) ткани. Методом оценки состояния как плазматической, так и внутриклеточных мембран может служить измерение электрического сопротивления - импеданса ткани, который включает в себя омическую и емкостную составляющие, поскольку каждая клетка представляет собой как бы систему конденсаторов (биологические мембраны) и резисторов (биологические мембраны, межклеточная жидкость и цитоплазма). При повреждении или старении клеток регистрируется уменьшение емкостного сопротивления тканей, связанное в основном с нарушением состояния мембран клеток. При набухании, или стрикции, клеток изменяется омическая (высокочастотная) составляющая импеданса. Для количественной оценки указанных нарушений Б.Н. Тарусовым предложено определение коэффициента жизнеспособности клеток (К) как отношения сопротивления ткани переменному току с частотой 104 Гц (R104) к сопротивлению ткани при действии тока с частотой 106 Гц (R106): К= R104 /R106.
Окраска цитоплазмы различными красителями. Водорастворимые красители плохо проникают через мембраны неповрежденных клеток, слабо связываются внутриклеточными структурами и потому слабо их прокрашивают. Увеличение проницаемости плазматической и внутриклеточных мембран приводит к возрастанию количества красителя, вошедшего в клетку и связавшегося с компонентами цитоплазмы. Следовательно, окрашивание клетки красителями усиливается при ее повреждении. На этом основаны многие гистохимические методы определения жизнеспособности клеток (с помощью нейтрального синего, эозина и др.).
Снижение мембранного потенциала покоя. Разность электрических потенциалов между содержимым клетки и окружающей средой (мембранный потенциал покоя) создается, как известно, в основном диффузией ионов калия из клетки в окружающую среду. Неравномерное распределение ионов между клеткой и окружающей
средой, лежащее в основе генерации электрических потенциалов на мембране, обеспечивается постоянной работой молекулярного ионного насоса (Na+/К+-АТФаза), встроенного в плазматическую мембрану клеток.
Так, внутри клеток содержание ионов калия в 20-40 раз выше, а ионов натрия - в 10-20 раз ниже, чем во внеклеточной жидкости. Благодаря различию в концентрации ионов в клетке и окружающей среде на плазматической мембране имеется разность потенциалов со знаком «минус» внутри клетки (около -70 мВ для нервных и мышечных клеток). Уменьшение поляризации мембраны при действии повреждающих факторов происходит как в результате неспецифического увеличения ионной проницаемости, так и при уменьшении градиентов концентрации ионов вследствие выключения ионных насосов.
Последнее происходит как при прямом повреждении Na+/K+- АТФазы, так и при снижении уровня АТФ вследствие нарушения биоэнергетических процессов в митохондриях. Например, установлено снижение мембранного потенциала покоя клеток печени у лабораторных животных при асфиксии. Снижение мембранного потенциала наблюдается также при холодовом, радиационном, аллергическом, токсическом и других повреждениях клеток и субклеточных структур.
Выход ионов калия из клеток. Благодаря разности потенциалов между внутренним содержимым клетки и окружающей жидкостью ионы калия входят в клетку. Этот постоянный поток К+ внутрь клетки компенсирует спонтанный выход калия наружу, который происходит в силу диффузии этих катионов из области с более высокой концентрацией калия в область с более низкой его концентрацией. Повреждение клетки сопровождается снижением содержания в ней АТФ, угнетением Na+/К+-АТФазы, падением электрического потенциала на плазматической мембране, повышением содержания внутриклеточного Ca2+ и выходом калия из клеток. Освобождение калия из клеток описано при механической травме, различных интоксикациях, аллергических состояниях, гипоксии, гипотермии и многих других повреждениях органов и тканей. Понижение содержания К+ в клетке может происходить также под влиянием больших доз минералокортикоидных гормонов, при действии некоторых лекарственных веществ, например сердечных гликозидов. В свою очередь, увеличение концентрации калия во внеклеточной среде приводит к снижению мембранного потенциа-
ла соседних неповрежденных клеток, что в случае электровозбудимых тканей может вызвать генерацию потенциалов действия. Так, увеличение концентрации калия в очаге инфаркта миокарда может стать одной из причин возникновения фибрилляции сердца.
Накопление ионов кальция в цитоплазме. В нормальных клетках концентрация ионов кальция в цитоплазме исключительно низка: 10-7 М или даже 10-8 М, тогда как в окружающей клетку среде содержится 10-3 М ионов кальция. При этом следует иметь в виду, что ионы кальция проходят в клетку не только самопроизвольно (процесс «утечки» через мембрану), но и в некоторых клетках через кальциевые каналы в мембране. Эти каналы могут открываться в ответ на деполяризацию мембраны (потенциалзависимые кальциевые каналы) или присоединение гормонов и медиаторов к мембранным рецепторам (рецепторуправляемые кальциевые каналы). Компенсирует вход Са2+ в клетку работа трех типов кальцийтранспортирующих систем: кальциевого насоса (Са2+/Мg2+-АТФаза) в мембране саркоплазматического ретикулума и плазмолемме, аккумуляции Са2+ в митохондриях и в некоторых клетках Na+/Ca2+- обменника, встроенного в плазмолемму.
При повреждении клетки нарушается работа митохондрий: снижается мембранный потенциал внутренней митохондриальной мембраны, прекращается окислительное фосфорилирование. Как следствие снижения мембранного потенциала уменьшается поглощение митохондриями ионов кальция. Снижение концентрации АТФ в клетке приводит к угнетению Са2+/Мg2+-АТФазы плазматической мембраны и мембраны саркоплазматического ретикулума. Увеличение концентрации Na+ в клетке вследствие угнетения натриевого насоса при недостатке АТФ приводит к выключению и даже обращению направления Na+/Са2+-обмена через плазматическую мембрану. В результате этого происходит увеличение концентрации кальция от 10-8 М - 10-7 M до 10-6 М - 10-5 М, что приводит к активации большого числа кальцийзависимых ферментов (протеинкиназ, фосфатаз, фосфолипаз, фосфодиэстеразы циклических нуклеотидов и др.), нарушениям цитоскелета (см. раздел 3.4), образованию нерастворимых включений кальция в матриксе митохондрий, повреждению внутриклеточных мембран и общей дезорганизации метаболизма. Морфологически это проявляется в замедлении броуновского движения различных включений внутри клетки (увеличение «вязкости протоплазмы») и возрастании светорассеяния; красители начинают легче проникать в
клетку и связываются в большом количестве с внутриклеточными структурами. Все эти признаки типичны для «неспецифической реакции клетки на повреждение» по Д.Н. Насонову и В.Я. Александрову (см. выше).
Выход метаболитов. Увеличение проницаемости мембраны клеток и ухудшение работы ионных насосов приводят к тому, что компоненты цитоплазмы выходят в окружающую среду. Вышедшие из клеток вещества отнюдь не безразличны для других клеток, тканей и органов. Так, среди веществ, выходящих из клеток, поврежденных в результате ишемии (нарушения кровотока) или ожога, имеются полипептиды, обладающие способностью вызвать остановку сердца (ишемический, ожоговый токсины). Обнаружение этих веществ осуществляется различными методами, включая измерение хемилюминесценции плазмы крови, интенсивность которой снижается в присутствии полипептидных токсинов.
Увеличение объема (набухание) клеток. Увеличение объема клеток - один из наиболее ранних признаков ее повреждения, который проявляется, например, при недостатке кислорода в ткани - тканевой гипоксии. Сохранение нормальной формы и объема клеток связано с состоянием цитоскелета и поддержанием определенного соотношения между осмотическим давлением белков и электролитов внутри и вне клетки. При этом форма клетки определяется в большей мере цитоскелетом, тогда как объем - поддержанием осмотического баланса. Поскольку все биологические мембраны хорошо проницаемы для воды, но плохо проницаемы для большинства растворенных в воде веществ, включая соли, клетки, так же как и внутриклеточные структуры, например митохондрии, обладают свойством осмометра: их объем изменяется при изменении концентрации ионов и молекул внутри и вне клетки или органеллы. В нормальных условиях соотношение концентраций всех ионов и молекул внутри и вне клетки строго поддерживается. Как только в цитоплазме начинает увеличиваться концентрация ионов или молекул, объем клетки возрастает, поскольку вода поступает внутрь. Выкачивание ионов мембранными насосами и обменниками сопровождается восстановлением ее объема за счет выхода вслед за ионами избытка воды.
Отек клетки связан с нарушением регуляции ее объема со стороны плазматической мембраны. В нормальных клетках концентрация белка выше, чем вне клеток, вследствие чего клетки млекопитающих обладают более высоким внутриклеточным
коллоидно-осмотическим (онкотическим) давлением, чем внеклеточная жидкость. Это неизбежно привело бы к увеличению объема клетки, если бы для уравновешивания этого «избыточного» давления не происходило удаление (выкачивание) ионов натрия из клетки за счет работы энергозависимой Na+/К+-АТФазы. Поскольку мембрана клеток хорошо проницаема для ионов хлора, то вместе с натрием выходит и хлор за счет разности потенциалов на мембране. Иначе говоря, натриевый насос удаляет из клетки NaCl и снижает концентрацию ионов в цитоплазме, что приводит к уменьшению клеточного объема. Этому процессу противостоит процесс самопроизвольного поступления натрия внутрь клетки через дефекты в липидном бислое, натриевые каналы, переносчики, сопрягающие вход натрия с транспортом сахаров и аминокислот в клетку, Na+/H+- и Na+/Ca2+-обменники, а также Na+/К+/2С1- котранспортер.
Таким образом, живая клетка находится в состоянии динамического равновесия, при котором «протечка» клеточной мембраны компенсируется постоянной работой ионной помпы (это так называемая гипотеза leak and pump).
При патологии может происходить либо увеличение ионной проницаемости клеточной мембраны (возрастание «протечки»), либо нарушение работы ионных насосов, например, при недостатке энергообеспечения вследствие гипоксии, действия цианидов или разобщителей окислительного фосфорилирования (динитрофенол). В опытах с изолированными клетками печени, почек и мозга было показано, что отравление солями ртути или других тяжелых металлов приводит к увеличению ионной проницаемости мембраны клеток (увеличению «протечки»), нарушению АТФ-зависимого транспорта и возрастанию объема клеток (т.е. набуханию ткани).
Второй механизм набухания клеток при гипоксии - увеличение внутриклеточной осмотической нагрузки, вызванное накоплением метаболитов (катаболитов), таких как неорганический фосфат, лактат и пуриновые нуклеозиды.
Набухание клеток - процесс, далеко не безразличный для функционирования клеток и ткани в целом. Первым результатом этого оказывается сдавливание кровеносных сосудов и затруднение кровообращения. Так, при ишемии происходит набухание клеток, и последующее общее возобновление кровообращения не сразу и не всегда приводит к восстановлению жизнедеятельности ткани, потому как кровь не проникает в мелкие кровеносные со-
суды, сдавленные набухшими клетками. То же происходит при трансплантации органов. Иногда применяется предварительное промывание пересаженного органа гипертоническим раствором, который восстанавливает прежний объем клеток и нормализует микроциркуляцию.
3.2.2. Нарушение структурных (матричных) свойств липидного бислоя
Наиболее изучены три характеристики липидного слоя мембран, от которых зависят его свойства как жидкой фазы (матрицы), обеспечивающей функционирование мембранных белков и рецепторов: поверхностный заряд, вязкость и площадь липидного слоя. Все эти характеристики исследуются с помощью флуоресцентных и спиновых зондов.
Перекисное окисление и действие мембранных фосфолипаз приводят к накоплению в липидной фазе мембран полиненасыщенных жирных кислот, которые придают мембране при нейтральных рН отрицательный заряд. Увеличение отрицательных зарядов на поверхности мембраны облегчает связывание с мембраной ионов и белковых молекул, несущих положительные заряды, и, наоборот, уменьшает взаимодействие мембраны с отрицательно заряженными молекулами или другими мембранами. Связывая больше Ca2+, мембраны с большим числом отрицательных зарядов на поверхности становятся более доступными для действия фосфолипаз, но при этом хуже связывают ионы двухвалентного железа, которые ускоряют пероксидацию липидов.
С другой стороны, при ПОЛ происходит повышение вязкости липидного слоя мембран. Возрастание вязкости приводит к нарушению функционирования мембранных рецепторов, ионных каналов, а также встроенных в мембраны ферментов, таких как Na+/К+-АТФаза и Ca2+/Mg2+-АТФаза. В свою очередь, это изменяет ионный баланс клетки и может привести к расстройствам метаболизма.
С помощью флуоресцентных зондов было показано, что при ПОЛ происходит уменьшение площади поверхности липидного слоя мембраны, а также площади, занимаемой фосфолипидами на поверхности липопротеинов плазмы крови. Это связано с окислением части жирнокислотных цепей фосфолипидов и выходом их в водную фазу. Одним из результатов такого явления оказывается
увеличение относительной концентрации холестерина в липидном монослое на поверхности липопротеинов, подвергнутых перекисному окислению. Липопротеины низкой плотности (ЛПНП) в результате этого переносят еще больше холестерина в клеточные мембраны сосудистой стенки, чем неокисленные ЛПНП, и их атерогенность возрастает. Липопротеины высокой плотности (ЛПВП), в норме обладающие антиатерогенным действием, в результате перекисного окисления полностью теряют способность акцептировать холестерин с мембран клеток, что способствует развитию атеросклероза.
3.3. ИЗМЕНЕНИЯ ВНУТРИКЛЕТОЧНОГО МЕТАБОЛИЗМА ПРИ ПОВРЕЖДЕНИИ
Ацидоз. Любое повреждение клетки сопровождается ацидозом ее цитоплазмы (рН падает до 6 и ниже). Ацидоз повреждения - это следствие накопления в клетке определенных продуктов метаболизма (например, молочной кислоты). Ацидоз в поврежденной ткани возникает при действии различных болезнетворных агентов: физических факторов, химических веществ (например, горчичного масла), бактериальных токсинов (дизентерийная палочка, гемолитический стафилококк). Ацидоз повреждения возникает в тканях также при гипоксии.
Активация протеаз. К числу протеаз с оптимумом активности в области нейтральных значений рН относятся: АТФ-зависимые, убиквитинзависимые, Са2+-зависимые (кальпаины) протеазы. Кальпаины присутствуют практически во всех клетках млекопитающих. Они локализуются вне лизосом в мембранных структурах в форме неактивного комплекса с ингибиторными протеинами (кальпастатины). Основные функции кальпаинов - репарация цитоскелета и клеточных мембран, разрушение рецепторных протеинов и их обновление, активация некоторых ферментов, участие в процессах митоза. Идентифицированы две изоформы ферментов - с высоким и низким сродством к кальцию; обе активируются данным ионом. В эксперименте показано, что стойкая, неуправляемая активация кальпаинов кальцием приводит к повреждению микрофиламентов цитоскелета тромбоцитов, мембраны эритроцитов, гибели клеток печени, кардиомиоцитов и т.д.
Активация эндонуклеаз. Как уже указывалось выше, при завершении клеткой жизненного цикла активируется процесс программированной физиологической клеточной гибели - апоптоз. На ранних этапах в апоптотической клетке проявляются морфологические изменения: «вскипание» клеточной и ядерной мембран, конденсация хроматина. Самым надежным маркером развивающегося процесса является активация эндонуклеаз (цитоплазматических и ядерных ДНКаз) - ферментов, расщепляющих хроматин на фрагменты (олигонуклеосомы). Активация эндонуклеаз отмечается, например, при гибели тимоцитов и лимфоцитов в облученном организме. Установлено, что в активации эндонуклеаз участвует кальций. Со стойким повышением содержания Са2+ в цитоплазме тимоцитов связана, например, фрагментация ДНК и гибель этих клеток при действии глюкокортикоидов. Кроме того, процесс Са2+-связанной фрагментации ДНК в тимоцитах лежит в основе атрофии тимуса экспериментальных животных при отравлении диоксином. Активация эндонуклеаз может быть также причиной гибели клеток печени, миокарда, почек при отравлениях многими химическими веществами.
3.4. НАРУШЕНИЕ СТРУКТУРЫ И ФУНКЦИЙ ВНУТРИКЛЕТОЧНЫХ ОРГАНЕЛЛ ПРИ ПОВРЕЖДЕНИИ
Высокую чувствительность к повреждающему воздействию болезнетворных факторов проявляют такие внутриклеточные структуры, как митохондрии, рибосомы и полисомы, лизосомы, элементы цитоскелета.
Митохондрии. Нарушение биоэнергетических функций митохондрий - одно из наиболее ранних проявлений повреждения клеток. Например, после прекращения кровообращения происходит нарушение окислительного фосфорилирования в митохондриях через 20-30 мин в печени и через 30-60 мин - в почках. Приблизительно в эти же сроки появляются и другие признаки повреждения клеток.
К признакам нарушений функций митохондрий относятся:
1. Снижение потребления кислорода. Уменьшение скорости потребления кислорода митохондриями, связанное с нарушением работы переносчиков электронов, наблюдается при действии
многих токсических соединений, например ионов тяжелых металлов, таких как ртуть или серебро, ряда гидрофобных соединений, различных производных углеводорода, при ПОЛ. Оно может быть также следствием набухания митохондрий и разрыва их наружной мембраны, в результате чего из митохондрий выходит цитохром С, который является одним из переносчиков электрона по дыхательной цепи.
2. Увеличение проницаемости внутренней митохондриальной мембраны. Происходит при повреждении митохондрий вследствие гипоксии или ПОЛ.
3. Снижение способности накапливать кальций. Параллельно разобщению окислительного фосфорилирования отмечается потеря способности митохондрий к накоплению ионов кальция. В присутствии избытка субстратов дыхания и при наличии кислорода и ортофосфата митохондрии печени способны накапливать в матриксе количество фосфорнокислого кальция, по массе превышающее массу митохондрий в сотни и даже в тысячу раз! Повреждение митохондрий приводит к падению разности потенциалов на митохондриальной мембране. Положительно заряженные ионы кальция, удерживаемые в матриксе электрическим полем, начинают выходить наружу из поврежденных митохондрий.
Разобщение окислительного фосфорилирования и выход кальция из митохондрий имеют самые драматические последствия для клетки (см. выше). Снижение уровня АТФ в клетке приводит к выключению ионных насосов плазмолеммы, вхождению в клетку из окружающей среды ионов натрия и кальция и выходу калия. Это обусловливает стимуляцию целого комплекса ферментных систем, активируемых ионами кальция, включая фосфолипазы, протеинкиназы, фосфатазы, протеазы, многие системы биосинтеза белков; метаболизм клетки вначале активируется, а затем дезорганизуется. Именно повреждение митохондрий является, согласно современным представлениям, тем переломным моментом, после которого изменения в клетке, вызванные повреждающим агентом, становятся необратимыми, и клетка погибает.
4. Набухание митохондрий. Весьма важным морфологическим признаком повреждения митохондрий является их набухание. Набухание митохондрий наблюдается, например, в клетках миокарда при недостаточности сердца, а также при многих инфекционных, гипоксических, токсических и других патологических процессах. Набухание митохондрий происходит при помещении клеток в ги-
потоническую среду, под влиянием ионизирующей радиации, бактериальных токсинов, при действии на клетку химических ядов и других патогенных агентов. Набухание приводит сначала к разрывам наружных мембран митохондрий, а затем - к их полному разрушению.
В опытах с изолированными митохондриями показано, что существует два типа набухания: пассивное и активное. В противоположность плазматической мембране, сравнительно хорошо проницаемой для ионов калия и хлора, внутренние мембраны митохондрий непроницаемы для ионов, за исключением Ca2+ и, возможно, ионов железа. В изотоническом растворе КС1 неповрежденные митохондрии сохраняют свой объем, несмотря на то что концентрация ионов калия и хлора внутри этих органелл существенно меньше, чем снаружи: осмотическое давление внутри создается и другими ионами, а также белками матрикса. Поэтому пассивное набухание митохондрий при отсутствии источников энергии (субстраты дыхания, кислород, АТФ) происходит только при воздействиях, которые увеличивают проницаемость мембран митохондрий одновременно для катионов и анионов (например, для К+ и С1-). К таким агентам относятся ионы тяжелых металлов (ртути, серебра, свинца). Таким же действием обладает усиление ПОЛ в мембранах митохондрий. При одновременном увеличении проницаемости для ионов калия и хлора они начинают диффундировать в митохондрии, что приводит к повышению осмотического давления, входу воды и набуханию органелл, которое называется пассивным, так как не зависит от дыхания и энергизации.
В условиях живой клетки чаще имеет место иной тип набухания - активное набухание, связанное с работой цепи переноса электронов. Повреждение митохондрий происходит под действием малых доз тяжелых металлов, активации собственной фосфолипазы в условиях гипоксии, при перекисном окислении липидов и обусловлено, прежде всего, повышением проницаемости внутренней мембраны для катионов. В присутствии источников энергии (субстраты дыхания и кислород, АТФ) на мембранах митохондрий генерируется разность потенциалов величиной около 170-180 мВ со знаком «минус» в матриксе, под действием которой ионы К+ поступают внутрь поврежденных митохондрий. Вместе с калием в матрикс поступает ортофосфат, который переносится в электронейтральной форме через внутреннюю мембрану с помощью специального белкового переносчика. Активное (т.е. связанное с
затратой энергии) накопление фосфата калия в матриксе сопровождается входом в него воды и набуханием митохондрий.
Рибосомы и полисомы. При токсических воздействиях на клетки происходит изменение конфигурации эндоплазматического ретикулума и связанных с ним рибосом. Например, при отравлении тринитротолуолом в клетках печени мембраны эндоплазматического ретикулума и расположенные на них рибосомы принимают форму различных завитков, не наблюдающихся в нормальных клетках. Синтез белков осуществляется на полисомах. Угнетение синтеза определенных белков, например синтеза гемоглобина при гипопластической анемии в клетках костного мозга, происходит на фоне уменьшения числа полисом и их распада на отдельные рибосомы.
Лизосомы. Лизосомная система, богатая ферментами, является специализированным инструментом клеток, используемым для осуществления таких важных метаболических и физиологических процессов, как катаболизм белков, глико- и липопротеидов, нуклеиновых кислот; накопление, трансформация и выведение из организма чужеродных веществ, в том числе лекарств, эндотоксинов; везикулярный транспорт и рециклизация рецепторов, ауто- и гетерофагоцитоз, апоптоз, адаптация и реконструкция клеточных структур и др. Особенно важной является роль лизосомного аппарата клетки при действии болезнетворных факторов. Лизосомы участвуют в защите клеток от бактерий, инородных тел, химических веществ, в воспалительных и иммунологических реакциях, в процессах дистрофии, некроза.
Различные повреждающие агенты, например эндотоксины бактерий брюшнотифозной группы, а также мелкие неорганические частицы (двуокись кремния, двуокись титана, алмазная пыль), попадая в лизосомы, разрушают их. При этом ферменты (гидролазы, оксидоредуктазы), заключенные в лизосомах, освобождаются в цитоплазму, активируются и вызывают повреждение субклеточных структур и макромолекул, что может привести клетку к гибели.
Однако активация лизосомальных ферментов может происходить не только под действием тех или иных специфических факторов, но и в результате ацидоза, характерного для неспецифической реакции клетки на повреждающее воздействие. Одним из процессов, вызывающих выход лизосомальных ферментов, является также активация пероксидации липидов в мембранах лизосом.
С повреждающим действием лизосомных факторов связывают развитие ряда заболеваний печени, почек, злокачественных новообразований, системной красной волчанки, ревматизма, ревматоидного артрита и др.
Увеличение численности и размеров лизосом является одной из форм структурно-функциональной адаптации клеток к повреждающему воздействию. Так, многие тяжелые металлы при поступлении в организм в течение длительного времени или в высоких концентрациях способны не только накапливаться в лизосомных везикулах различных клеток, но и индуцировать усиленное образование новых первичных лизосом и их последующее набухание. Это обеспечивает защиту клеток от повреждения путем аккумуляции и обособления токсичного металла в лизосомных везикулах, а в случае повреждения клеточных органелл - быструю ликвидацию дефектных структур с помощью лизосомного аутофагоцитоза и апоптоза.
Эффект индукции образования лизосом, морфологически выявляемый в виде увеличения их численности и размеров, продемонстрирован в гепатоцитах при циррозе печени у человека, в клетках почек и печени овец, отравленных медью, в эпителии проксимальных канальцев почек у крыс при многократном введении солей кадмия, в различных участках мозга при введении ацетата свинца крысам.
Цитоскелет. Цитоплазма клетки, помимо цитозоля и клеточных органелл, как правило, содержит еще и нитевидные белковые структуры, которые в массе формируют клеточный скелет. Цитоскелет - это система белковых нитей, заполняющих цитоплазму. Он отвечает за динамичную архитектуру клетки, ее подвижность, форму. Цитоскелет состоит из трех основных структур: микрофиламентов, микротубул и промежуточных филаментов. Каждая из них состоит из одного или двух основных белков: микрофиламенты - из актина, микротубулы - из тубулина, промежуточные филаменты - из специальных белков, различных в разных тканях (кератинов - в эпителии, десмина - в мышцах, виментина - в тканях внутренней среды: соединительная ткань, хрящ, кость и др., белков нейрофиламентов - в нейронах).
Цитоскелет - динамичное образование. Его строение может меняться за счет полимеризации и деполимеризации образующих его структур - нитей и микротубул. Благодаря такой полимеризациидеполимеризации цитоскелет непрерывно перестраивается, что
лежит в основе изменений формы, движений клеток, клеточного деления, внутриклеточного транспорта, секреции и др. К элементам цитоскелета могут присоединяться «моторные» молекулы: к актиновым микрофиламентам - молекулы миозина, к микротубулам - денеина и кинезина. При этом один конец такой молекулы прикрепляется к нити или микротубуле, а другой - к органелле или соседним элементам цитоскелета. В присутствии АТФ эти молекулярные «моторы» могут перемещать органеллы вдоль элементов цитоскелета и относительно друг друга. В нервных клетках микрофиламенты и микротубулы принимают участие в монтаже системы антероградного и ретроградного аксонального транспорта биологически активных веществ.
Как известно, белки синтезируются в перикарионе нейрона, а затем транспортируются с помощью этой системы на периферию клетки (в область синаптических контактов). Транспорт по аксону необходим и для обеспечения функций аксональной мембраны - для оборота глобулярных протеинов в мембране аксонов, выполняющих функции ионных каналов. Состояние шванновских клеток также контролируется трофическими веществами, переносимыми с аксональным током. С аксональным током перемещается весь набор ферментов, нейромедиаторов и их предшественников, необходимых для обеспечения синаптической передачи нервных импульсов.
Хорошо известно, что клетки способны переключаться с одной программы работы на другую под влиянием соответствующих сигналов: клетка может начать или прекратить деление, может дифференцироваться или включить программу самоубийства - апоптоза. В последние годы появились основания утверждать, что одним из факторов, регулирующих эти процессы, могут быть изменения натяжения цитоскелета.
При действии разнообразных альтерирующих агентов на изолированные клетки (культура ткани) выявляется отчетливое изменение формы их поверхности: появляются выпячивания цитоплазмы, называемые пузырьками (от blebs). Такое «пузырение» (или вскипание - blebbing) клеточной мембраны - один из ранних и надежных признаков разрушения сети цитоскелета. «Вскипание» мембраны инициируют вещества, нарушающие гомеостаз внутриклеточного кальция.
Механизм феномена объясняют следующим образом. Кальций вовлечен в процесс поддержания структуры цитоскелета как не-
посредственно, так и через ряд Са2+-связьгаающих белков и Са2+- зависимых ферментов. Особенно значим уровень Са2+ в цитоплазме для образования ассоциации белков цитоскелета с белками плазматической мембраны и взаимодействия различных элементов цитоскелета. Стойкое увеличение концентрации кальция в цитозоле, по-видимому, вызывает отщепление актина микрофиламентов от актинина - белка, служащего промежуточным звеном, который связывает микрофиламенты цитоскелета с белками плазматической мембраны.
Кроме того, Са2+ активирует протеазы, которые могут расщеплять актинсвязывающие белки, разрушая тем самым места прикрепления филаментов цитоскелета к клеточной мембране. Отщепление цитоскелета от мембраны приводит к ослаблению фиксации последней и ее «вскипанию», что и обнаруживается при действии на клетки различных токсинов (рис. 3-9). Вещества, связывающие внутриклеточныгй кальций и ингибиторы Са2+-зависимых протеаз, препятствуют «вскипанию» плазматической мембраны клеток, обработанных токсическими агентами.
 Рис. 3-9. Са2+-зависимые механизмы повреждения цитоскелета клеток, обработанных хинонами
Рис. 3-9. Са2+-зависимые механизмы повреждения цитоскелета клеток, обработанных хинонами
Другой механизм изменения конфигурации плазматической мембраны под влиянием избыточной концентрации ионов кальция может состоять в истощении внутриклеточных запасов макроэргов.
Белки цитоскелета повреждаются не только в результате повышения содержания Са2+ в цитоплазме, но и под влиянием факторов с иными механизмами действия. Так, непосредственно взаимодействуют с белками цитохалазины, фаллоидин (один из токсинов бледной поганки), а также колхицин, алкалоиды барвинка (цитостатики винбластин, винкристин).
Действие повреждающих веществ на тубулин митотического веретена делящихся клеток приводит к нарушению пролиферации последних, особенно клеток системы крови. Колхицин и алкалоиды барвинка в эксперименте, разрушая цитоскелет аксонов нервных клеток, существенно нарушают аксональный ток. Вероятно, аналогичные эффекты могут развиваться и при повреждении цитоскелета веществами, нарушающими внутриклеточный гомеостаз кальция.
3.5. ПОВРЕЖДЕНИЕ ГЕНЕТИЧЕСКОГО АППАРАТА КЛЕТКИ
Нуклеиновые кислоты весьма чувствительны к прямому действию повреждающих агентов физической (облучение ионизирующей радиацией, ультрафиолетом, видимым светом в присутствии некоторых окрашенных соединений - фотосенсибилизаторов), химической (сернистый и азотистый иприты, эпоксиды, этиленимин, метилсульфонат и т.д.), вирусной (аденовирусы, герпесвирусы, онкорнавирусы и др.) природы. Их повреждающее действие на ДНК называется генотоксическим.
В значительной мере повреждения нуклеиновых кислот исправляются в результате репарации. В противном случае возникают нарушения в геноме и работе системы биосинтеза белка. В последнее время многие необратимые изменения в клетках (например, при интоксикациях или в ходе процесса старения) связывают с повреждением генетического аппарата митохондрий.
Наиболее чувствительны к генотоксическому действию клетки, способные к делению (эмбриональные, герминативные, костного мозга, эпителия почек, кожи, слизистой желудочно-кишечного тракта и т.д.). Последствия нарушения нативной структуры ДНК
зависят от дозы повреждающего агента. Так, например, высокие дозы химических генотоксических веществ вызывают цитостатический эффект (гибель пула делящихся клеток), более низкие - канцерогенное, тератогенное, мутагенное действия, что зависит от ряда условий (табл. 3-6).
Таблица 3-6. Особенности действия генотоксических факторов
Токсический процесс | Чувствительная ткань | Время действия | Продолжительность действия и доза |
Канцерогенез и мутагенез | Любая пролиферирующая ткань, герминативные клетки | На всех стадиях митоза, гаметогенеза | Острое и хроническое, беспороговое |
Тератогенез | Зародышевые ткани | На ранних стадиях дифференцировки тканей | Только острое, в дозах выше пороговых |
3.6. ПОВРЕЖДЕНИЕ КЛЕТОК ПРИ ГИПОКСИИ
Наиболее распространенными причинами повреждения клетки являются недостаток кислорода (гипоксия) или же, напротив, избыточное образование его радикалов (окислительный стресс) (см. раздел 3.2.1).
Недостаток кислорода приводит к снижению синтеза митохондриями АТФ из АДФ и ортофосфата. Недостаток АТФ делает невозможным функционирование многих систем клетки, для которых необходима затрата энергии в форме макроэргических связей АТФ. Именно энергетический голод, а не само по себе отсутствие кислорода приводит к нарушению функционирования клеток, а затем и к их повреждению. Но и наличие кислорода еще не означает полного благополучия. Дело не только в том, есть ли кислород в клетках, но еще и в том, на что он расходуется. Наряду с окислением субстратов тканевого дыхания, конечным этапом которого является перенос электронов на кислород в цепи переноса электронов в митохондриях, в клетках существуют и альтернативные пути восстановления кислорода, приводящие к появлению радикалов кислорода и липидов.
В нормальных условиях под влиянием фермента цитохромоксидазы происходит четырехэлектронное восстановление молекулы кислорода с образованием двух молекул воды. Но возможно и одноэлектронное восстановление кислорода промежуточными компонентами дыхательной цепи - убихинонами. В ходе этого процесса образуются супероксид анион (O*2-) - свободный радикал, содержащий неспаренный электрон.
Нормоксия и аноксия на уровне отдельной клетки. Кислородный конус. В опытах с изолированными митохондриями показано, что скорость потребления кислорода этими частицами при наличии субстратов дыхания практически постоянна при всех концентрациях кислорода - вплоть до самых низких, соответствующих напряжению кислорода pO2 = 1-2 мм рт.ст. Причина этого явления заключается в высоком сродстве к кислороду конечного переносчика электронов по дыхательной цепи - цитохромоксидазы. Поэтому отдельная клетка «выбирает» весь кислород из окружающей среды до конца, не испытывая кислородного голода в весьма широком интервале pO2 - от 70 до 1-2 мм рт.ст. Это приводит к формированию так называемого кислородного конуса в тканях. Схематически кислородный конус представлен на рис. 3-10. Для простоты кровеносный сосуд изображен в виде трубки постоянно-
 Рис. 3-10. Кислородный конус в участке ткани
Рис. 3-10. Кислородный конус в участке ткани
го диаметра, а ткань - в виде однородной структуры, состоящей из одинаковых клеток, поглощающих кислород с постоянной скоростью. Кровь, протекающая по кровеносному сосуду, непрерывно отдает его окружающим тканям, в результате чего содержание кислорода снижается вдоль сосуда по ходу тока крови.
С другой стороны, кислород, диффундирующий от сосуда в толщу ткани, поглощается клетками, так что его напряжение (pO2) снижается по мере удаления от кровеносного сосуда. Там, где оно падает до 1-2 мм рт.ст. (т.е. практически до нуля), клетки как бы оказываются в состоянии полной аноксии. Во всем слое ткани ближе этой границы они не испытывают кислородного дискомфорта, т.е. находятся в состоянии нормоксии. Очевидно, что чем ниже было исходное содержание кислорода в данном участке сосуда, тем тоньше слой ткани, полностью «выедающей» весь кислород. Иначе говоря, по ходу тока крови толщина слоя клеток в состоянии нормоксии сужается, образуя тем самым конус из нормально обеспеченных кислородом клеток. Протяженность конуса увеличивается с ускорением тока крови, а ширина его уменьшается с увеличением потребления кислорода клетками.
Таким образом, в ткани часть клеток находится в состоянии нормоксии, а часть - аноксии. Доля клеток, которые лишены кислорода, от общего числа клеток в ткани может служить количественной характеристикой степени гипоксии в ткани.
Как кровоток, так и потребление кислорода клетками могут изменяться во времени, так что одна и та же клетка может в одни моменты быть в состоянии аноксии, а в другие - нормоксии. Тогда можно говорить и о степени гипоксии для данной клетки, имея в виду ту часть времени, которую данная клетка провела в условиях отсутствия кислорода.
Митохондрии - главная мишень при гипоксическом повреждении клеток. Пребывание клеток в состоянии аноксии в течение 30-90 мин (для разных тканей) приводит к их повреждению. Ученых давно волновал вопрос, какие структуры клеток при этом повреждаются первыми, предопределяя последующую гибель всей клетки. Накопленный к настоящему времени экспериментальный материал позволяет утверждать, что такими структурами являются биоэнергетические станции клетки - митохондрии. О повреждении митохондрий при длительной гипоксии в ткани свидетельствует снижение дыхательного контроля и их кальцийаккумулирующей способности (емкости) (рис. 3-11).
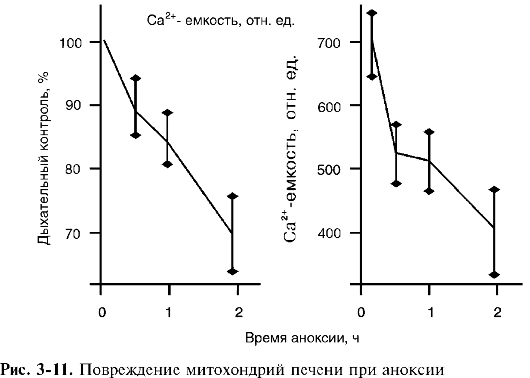 Ионы кальция и активация фосфолипазы при аноксии. Как
известно, фосфолипазы присутствуют практически во всех мембранных
структурах клетки, включая митохондрии, лизосомы, плазматическую
мембрану. Фосфолипазы катализируют гидролиз фосфолипидов, входящих в
состав клеточных мембран. Особое внимание исследователи уделяют
фосфолипазам А2 - группе липаз, основная функция которых
состоит в удалении из мембраны поврежденных фосфолипидов путем
высвобождения жирных кислот, подвергшихся пероксидации.
Ионы кальция и активация фосфолипазы при аноксии. Как
известно, фосфолипазы присутствуют практически во всех мембранных
структурах клетки, включая митохондрии, лизосомы, плазматическую
мембрану. Фосфолипазы катализируют гидролиз фосфолипидов, входящих в
состав клеточных мембран. Особое внимание исследователи уделяют
фосфолипазам А2 - группе липаз, основная функция которых
состоит в удалении из мембраны поврежденных фосфолипидов путем
высвобождения жирных кислот, подвергшихся пероксидации.
Фосфолипазы А2 являются Ca2+- и кальмодулинзависимыми ферментами и, следовательно, чувствительными к повышению концентрации кальция в цитоплазме. В мембранах фосфолипазы обычно находятся в малоактивном состоянии, поскольку активируются ионами кальция и ингибируются ионами магния, в то время как в цитоплазме поддерживается низкая концентрация кальция (10-7М и менее) и содержится относительно много ионов магния (около 10-3М). Увеличение проницаемости плазматической мембраны при повреждении клетки или, в некоторых клетках, при открывании кальциевых каналов, равно как и выключение ионных насосов за счет недостатка энергии в клетке, приводит
к увеличению концентрации кальция в цитоплазме. Повышение его концентрации до 10-6 М следует считать нормальным механизмом кальциевой регуляции внутриклеточных процессов, так как кальций является вторичным посредником при действии многих гормонов и медиаторов.
Умеренная активация фосфолипазы А2 - также нормальное физиологическое явление, поскольку служит первым звеном в цепи образования физиологически активных производных арахидоновой кислоты. Однако чрезмерное увеличение концентрации ионов кальция в цитоплазме и активация фосфолипазы приводят к усилению разрушения фосфолипидов мембран, потере мембранами их барьерных свойств и нарушению функционирования клеточных органелл и клетки в целом.
Повышение содержания кальция в цитоплазме, активацию фосфолипазы, ускорение оборота фосфолипидов с последующей гибелью клеток наблюдали в культуре кардиомиоцитов и гепатоцитов, инкубируемых в условиях аноксии. Аналогичные изменения гепатоцитов регистрировали при токсическом поражении печени четыреххлористым углеродом. Установлено, что хлорпромазин, угнетающий активацию фосфолипазы А2 комплексом Са2+-кальмодулин, защищает клетки от повреждающего действия гипоксии и цитотоксикантов, нарушающих внутриклеточный гомеостаз кальция.
В опытах с изолированными митохондриями было показано, что при инкубации этих органелл происходит их быстрое повреждение (за 15-20 мин при 37 °С), если в окружающей их среде нет кислорода и присутствуют ионы кальция в концентрациях, близких к 10-5 М, т.е. соизмеримых с концентрацией этих ионов в цитоплазме клеток в условии гипоксии.
Последовательность нарушений в клетке при гипоксии. В аэробных условиях ионов кальция вокруг митохондрий мало (10-6-10-7 М) и фосфолипаза А2 умеренно активна. При отсутствии кислорода исчезает электрический потенциал на мембране митохондрий, который удерживает ионы кальция в матриксе, и кальций выходит в цитоплазму. Связываясь с активным центром фосфолипазы А2 на наружной стороне внутренней мембраны митохондрий, ионы кальция активируют фермент. Гидролиз фосфолипидов приводит к потере мембраной ее барьерных свойств, и митохондрии теряют способность как к окислительному фосфорилированию, так и к закачиванию кальция в матрикс.
Последовательность изменений в клетке в результате прекращения доступа кислорода (аноксии) одинакова для самых различных тканей. Это показали опыты со срезами тканей, изолированными клетками и изолированными клеточными органеллами, в частности митохондриями. В клетках печени, находящихся в условиях аноксии при комнатной температуре, последовательность событий такова:
0-5 мин: снижение уровня АТФ в клетке в 2-4 раза, несмотря на активацию гликолиза;
5-15 мин: повышение концентрации Са2+ в цитоплазме клетки. Активация гидролитических ферментов, в том числе фосфолипазы А2 митохондрий. Содержание Са2+ в митохондриях повышается, так как они еще не повреждены;
15-30 мин: гидролиз митохондриальных фосфолипидов фосфолипазой А2 и нарушение барьерных свойств митохондриальной мембраны. Реоксигенация ткани на этой стадии приводит к активному набуханию митохондрий. Дыхательный контроль в митохондриях нарушен, окислительное фосфорилирование разобщено, способность митохондрий накапливать ионы кальция снижена;
30-60 мин: частичное восстановление функций митохондрий, временное повышение дыхательного контроля, способности накапливать кальций. Механизм компенсаторных процессов, приводящих к временному улучшению функций митохондрий, неизвестен, но связан с функцией клетки в целом, так как при анаэробной инкубации изолированных митохондрий это явление не наблюдается;
60-90 мин: необратимое повреждение митохондрий и полная гибель клеток.
При температуре тела человека все эти процессы протекают в 2-3 раза быстрее. Кроме того, в разных тканях они протекают с разной скоростью: быстрее всего в мозгу, медленнее - в печени, еще медленнее - в мышцах.
3.7. «ПОРОЧНЫЙ КРУГ» КЛЕТОЧНОЙ ПАТОЛОГИИ
Увеличение внутриклеточного содержания кальция и нарушение биоэнергетических функций митохондрий являются общими признаками для клеток, поврежденных в результате действия самых различных неблагоприятных факторов. Эти два события - не
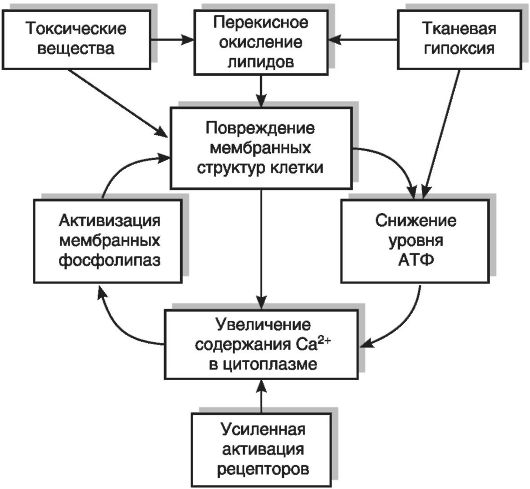 Рис. 3-12. «Порочный круг» клеточной патологии
Рис. 3-12. «Порочный круг» клеточной патологии
простое следствие других изменений в поврежденных клетках: они лежат в основе нарушения функций поврежденных клеток и могут рассматриваться как главные звенья в цепи событий, приводящих к развитию неспецифической реакции клеток на повреждение. Взаимоотношения между первичным повреждением клеточных структур, процессами биоэнергетики и содержанием кальция в цитоплазме представлены в виде схемы на рис. 3-12.
Согласно схеме, первичными мишенями действия повреждающих агентов служат мембранные структуры клетки, в которых могут подвергаться разрушению липидный бислой, рецепторы, белковые переносчики ионов и молекул, ионные каналы, а также встроенные в мембраны ферменты, включая ионные насосы. Увеличение проницаемости мембран и подавление работы ионных насосов, непосредственно вызванные действием повреждающих факторов (токсические соединения, свободные радикалы и про-
дукты липидной пероксидации, недостаток АТФ и т.д.), приводят к увеличению концентрации натрия и кальция в цитоплазме. Последнее сопровождается дисбалансом внутриклеточных сигнальных систем и активацией ряда ферментов, включая некоторые протеазы, фосфатазы, эндонуклеазы и фосфолипазу Гидролиз мембранных фосфолипидов фосфолипазой приводит к дальнейшему нарушению барьерных свойств липидного бислоя, что вызывает еще более выраженное увеличение уровня кальция в цитоплазме, набухание митохондрий и их повреждение. «Порочный круг» замыкается, и клетка может погибнуть.