Патофизиология Новицкого, Е.Д. Гольдберга Тома 1 и 2 - 2009 г.
|
|
|
|
ЧАСТЬ III ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ ГЛАВА 14 ПАТОФИЗИОЛОГИЯ СИСТЕМЫ КРОВИ
Патологические сдвиги в системе крови выявляются при морфологических и функциональных нарушениях в органах, принимающих участие в процессах гемопоэза и кроверазрушения, а также при расстройствах их регуляции в результате прямого действия различных повреждающих факторов, при ряде инфекционных заболеваний и собственно болезнях системы крови.
14.1. КРАТКИЕ СВЕДЕНИЯ О КРОВЕТВОРЕНИИ. ГЕМОПОЭЗИНДУЦИРУЮЩЕЕ МИКРООКРУЖЕНИЕ
Согласно современной схеме кроветворения (рис. 14-1, см. цв. вклейку), предложенной А.И. Воробьевым и И.Л. Чертковым (1973), все клетки крови подразделяются на 3 большие отдела: родоначальные (или стволовые) кроветворные (1-2% от общей массы клеток крови), промежуточные (25-40%) и зрелые (60- 75%).
В пределах этих 3 отделов все клетки крови дополнительно разделены на 6 классов:
I. Полипотентные стволовые кроветворные клетки.
II. Полиолигопотентные коммитированные клетки-предшественницы.
III. Моноолигопотентные коммитированные клетки-предшественницы.
IV. Бласты.
V. Созревающие клетки.
VI. Зрелые клетки.
Стволовые кроветворные клетки (СКК) - гетерогенная популяция родоначальных морфологически нераспознаваемых клеток
кроветворной системы. По степени дифференцированности и пролиферативному потенциалу выделяют полипотентные стволовые (I класс), полиолигопотентные (II класс) и моноолигопотентные (III класс) коммитированные клетки-предшественницы.
Полипотентные стволовые кроветворные клетки (ПСКК) обладают способностью к дифференцировке в различных направлениях.
Еще со времен А.А. Максимова (1923) признавалось, что стволовая клетка полипотентна и что ее дифференцировка в определенном направлении осуществляется под влиянием воздействий, обусловливающих ее трансформацию в сторону гранулоцитарных, эритроидных, макрофагальных и мегакариоцитарных форм клеток. Установлено также, что стволовая клетка иммунокомпетентна и способна образовывать клетки иммунного ответа (лимфоидные клетки).
Считается, что стволовые клетки находятся в костном мозгу (1 на 8000 кроветворных клеток, 30?103 на мышь), являющемся основным их поставщиком в постэмбриональный период. Из костного мозга стволовые клетки могут поступать в кровь и циркулировать в кровяном русле; не исключена возможность их поступления и из селезенки. В тимусе и лимфатических узлах стволовые клетки отсутствуют.
Проблема превращения ПСКК в коммитированные клеткипредшественницы окончательно не решена. Согласно стохастической модели кроветворения J.E. Till et al. (1964), процесс коммитирования происходит случайно и не зависит от внешних воздействий. В то же время, согласно теории J.J. Trentin (1976), созревание ПСКК и превращение их в зрелые элементы протекают под влиянием гемопоэзиндуцирующего микроокружения.
Согласно гипотезе R. Schofield (1978), в кроветворной ткани существуют специализированные образования - «ниши», в которых ПСКК находятся в заторможенном состоянии и не реагируют на действие внешних стимулов. Покинув «нишу», стволовые клетки попадают под влияние гемопоэтических факторов и необратимо дифференцируются. При этом гипотеза предполагает, что процесс выхода родоначальных элементов из данных образований происходит случайно.
Вышесказанному принципиально не противоречит и теория клональной сукцессии (Кау, 1965), согласно которой стволовые кроветворные клетки обладают высоким, но не безграничным пролиферативным потенциалом.
По теории И.Л. Черткова и Н.И. Дризе (1998), ПСКК закладываются только в эмбриогенезе и расходуются последовательно, образуя сменяющие друг друга клоны клеток, аналогично тому, как это происходит в яичниках. Считается, что на всех кроветворных территориях в течение жизни мыши функционирует около 6000 клонов.
У человека ПСКК выявляются во фракциях клеток, содержащих маркер примитивных клеток CD34. При этом преобладающим мембранным иммунофенотипом ПСКК является фенотип CD34+CD45+(low)CD38+HLA-DR+CD71+.
Большинство полипотентных клеток (около 90%) находится вне митотического цикла в стадии покоя - G0; многие из стволовых клеток существуют в конце GI-фазы клеточного цикла и способны к быстрому переходу в фазу синтеза ДНК - S-фазу.
Показано, что из одной начавшей дифференцировку стволовой клетки может образовываться около 1 млн эритроцитов и 100 тыс. гранулоцитов и макрофагов.
Согласно современным представлениям, полипотентная стволовая клетка является округлым мононуклеаром, близким по морфологии к костно-мозговому лимфоциту относительно небольшого размера (8-10 мкм), с высоким ядерно-цитоплазматическим соотношением, который не обладает прилипающей способностью и фагоцитарной активностью.
Колониеобразующие единицы селезенки (КОЕс). Первый метод, позволивший по существу доказать наличие в кроветворной ткани ПСКК, был предложен в 1961 г. J.E. Till и E.A. McCulloch, которые продемонстрировали способность кроветворных клеток при трансплантации смертельно облученным мышам образовывать в их селезенке колонии нескольких гистологических типов: эритроидные (42%), гранулоцитарные (21%), мегакариоцитарные (21%) и смешанные (16%); лимфоидные колонии в селезенке не образовывались. Было показано, что каждая такая колония представляет собой клон-потомство одной клетки - колониеобразующей единицы в селезенке - КОЕс. Для этого донорские кроветворные клетки метили облучением в низкой дозе (2 Гр). Метка (кольцевая хромосома) обнаруживалась в клетках всех колониальных линий, развивающихся в селезенке облученной мыши-реципиента. Позднее с помощью хромосомного маркера была обнаружена способность КОЕс дифференцироваться в лимфоциты, поскольку кольцевая хромосома выявлялась не только в клетках селезеночных колоний,
но и в лимфоцитах лимфатических узлов, тимуса и костного мозга облученных мышей.
Считается, что КОЕс относятся к категории более зрелых полипотентных клеток-предшественниц. При этом сама их популяция гетерогенна, т.е. отдельные КОЕс различаются по физическим константам (диаметру, плавучей плотности и др.), функциональным особенностям, радиорезистентности и др.
Полиолигопотентные коммитированные (или полустволовые) клетки-предшественницы. Этот класс составляют преимущественно клетки-предшественницы миелопоэза - КОЕ-ГЭММ - клетки, дающие смешанные колонии из гранулоцитов, эритроцитов, макрофагов и мегакариоцитов. Они определяются методами «клональных культур» in vitro либо в диффузионных камерах in vivo. Предположительно к этому классу клеток относится также клеткапредшественница лимфопоэза. Кроме того, в этот класс входят клетки-предшественницы, более ограниченные в дифференцировке, т.е. способные образовывать смешанные колонии из двух типов клеток, например из гранулоцитов и макрофагов (КОЕ-ГМ).
Моноолигопотентные коммитированные клетки-предшественницы дают начало отдельным росткам миелопоэза. К ним относятся КОЕ-Г - клетки-предшественницы гранулоцитов и более зрелые их потомки: КОЕ-Н, КОЕ-Эо и КОЕ-Ба - клетки-родоначальницы соответственно нейтрофильного, эозинофильного и базофильного (включая тучные клетки) рядов дифференцировки гранулоцитов; КОЕ-М - клетки-предшественницы моноцитопоэза (макрофагов); КОЕ-Мгкц - клетки-предшественницы мегакариоцитов. Клетками-предшественницами красного ряда являются бурстобразующие единицы: БОЕ-Э незрелая, нечувствительная к эритропоэтину, и БОЕ-Э зрелая, чувствительная к эритропоэтину. Зрелая БОЕ-Э дифференцируется в КОЕ-Э, дающую начало in vitro небольшим эритроидным колониям. К этому классу клеток относят также преТ- и преВ-клетки, дифференцирующиеся в направлении Т- и В-линий лимфоидных клеток.
Пролиферация коммитированных клеток всех типов (поли- и моноолигопотентных) регулируется ростовыми факторами, секреция которых зависит от существующего запроса организма, т.е. представляет собой уже не стохастический, а детерминированный процесс. По мере созревания клеток снижается их пролиферативный потенциал, но повышается пролиферативная активность. В целом, по мнению А.И. Воробьева и соавт. (1995), в этом отделе
взаимоотношения между отдельными типами клеток строятся не только в вертикальном срезе, но и в горизонтальном. Возможен пропуск некоторых стадий дифференцировки, что может определяться возросшей потребностью организма в клетках определенного типа.
К отделу промежуточных клеток относятся бласты (IV класс) и созревающие клетки (V класс).
Бласты представляют собой активно пролиферирующие клетки, распознаваемые не только по иммунофенотипическим, но и по морфологическим и цитохимическим признакам, что позволяет различать их с помощью методов дифференциальной окраски. К ним относятся миелобласты, монобласты, мегакариобласты, эритробласты, лимфобласты.
Созревающие клетки еще не полностью дифференцированы, но часть из них уже утрачивает способность к пролиферации. К пролиферирующим клеткам этого класса относятся клетки гранулоцитарного ряда - промиелоциты, нейтрофильные, эозинофильные и базофильные миелоциты; промоноцит; промегакариоцит; мегакариоцит; клетки эритроидного ряда - пронормобласты, базофильный и полихроматофильный нормобласты; пролимфоциты. Непролиферирующими клетками являются нейтрофильные, эозинофильные и базофильные метамиелоциты и палочкоядерные гранулоциты, оксифильный нормобласт и ретикулоцит.
Зрелые клетки (VI класс) являются непролиферирующими специализированными клетками крови, выполняющими строго определенные функции в организме (фагоцитарную, про- и антивоспалительную, трофическую, гемопоэтическую и др.). Они представлены сегментно-ядерными нейтрофилами, эозинофилами и базофилами, тучными клетками, моноцитами, тромбоцитами, эритроцитами, Т- и В-лимфоцитами, натуральными киллерами.
В тканях созревшие моноциты превращаются в макрофаги. В-лимфоциты способны дифференцироваться последовательно в плазмобласты, проплазмоциты и плазматические клетки.
К зрелым клеткам относятся также 3 популяции дендритных клеток, различающихся по происхождению: дендритные клетки макрофагального происхождения (миелоидные), дендритные клетки лимфоидного происхождения и клетки Лангерганса, происходящие напрямую из CD34-позитивных ПСКК.
Гемопоэзиндуцирующее микроокружение (ГИМ). Согласно современным представлениям, ГИМ имеет решающее значение в
регуляции кроветворения, выполняя роль локальной регуляторной системы. В формировании ГИМ принимают участие различные клеточные элементы и продукты их жизнедеятельности, входящие в состав как стромы, так и паренхимы кроветворных органов. К компонентам микроокружения следует в первую очередь отнести отдельные субпопуляции Т-лимфоцитов и макрофагов (мобильные элементы), фибробласты с продуцируемыми ими компонентами экстрацеллюлярного матрикса, резидентные макрофаги, адипоциты, эндотелиальные клетки, элементы микроциркуляторного русла и нервные волокна.
Элементы ГИМ осуществляют контроль за процессами кроветворения как через продуцируемые цитокины, так и благодаря непосредственным контактам с гемопоэтическими клетками (межклеточное взаимодействие). Межмембранное связывание служит при этом для сообщения регуляторной информации, передачи необходимых веществ, миграции и последующего хоминга клетокпредшественниц в специфических участках кроветворной ткани, а также представления гемопоэтических ростовых факторов в биологически доступной форме.
Необходимо отметить, что такой контроль может быть не только положительным, но и отрицательным (ингибиция пролиферации и дифференцировки) в зависимости от субпопуляции клеток микроокружения и от их функционального состояния.
К раннедействующим гемопоэтинам, которые самостоятельно либо в сочетании с другими факторами участвуют в стимуляции процессов пролиферации и дифференцировки ПСКК и полустволовых клеток, относятся интерлейкин (IL) 3, вырабатываемый активированными Т-лимфоцитами, фактор стволовых клеток (SCF), IL-1, IL-6, IL-11 и РИЗ-лиганд, которые продуцируются макрофагами, стромальными механоцитами, эндотелиальными и жировыми клетками, а также ГМ-КСФ (гранулоцитарно-макрофагальный колониестимулирующий фактор), способность к синтезу которого обнаружена практически у всех клеточных элементов ГИМ.
К позднедействующим гемопоэтинам, продуцируемым макрофагами, фибробластами и эндотелиальными клетками и контролирующим процессы пролиферации и дифференцировки коммитированных клеток-предшественниц гемопоэза и более поздних клеток, относят Г-КСФ (гранулоцитарный колониестимулирующий фактор), М-КСФ (макрофагальный колониестимулирующий фактор), Мег-КСФ (колониестимулирующий фактор мегакарио-
цитов), которые участвуют в регуляции соответственно грануло-, моноцито- и тромбоцитопоэза. Кроме того, клетки стромы и специализированные макрофаги вырабатывают коллаген I, III и IV типов, ретикулиновые волокна, фибронектин, ламинин, тенасцин и другие белковые компоненты нитчатой сети внеклеточного матрикса.
Т-лимфоциты вырабатывают линейно-рестриктированный цитокин IL-5, контролирующий продукцию эозинофилов. Как резидентные костно-мозговые макрофаги, так и моноциты секретируют эритропоэтин (ЭПО) и IL-6, которые стимулируют пролиферацию эритроидных прекурсоров, причем эта их способность возрастает при активации Т-лимфоцитами, продуктами деструкции эритроцитов и другими факторами. Тромбопоэтин, секретируемый эндотелиоцитами микроциркуляторного русла, стимулирует конечную фазу созревания мегакариоцитов, отшнуровку от цитоплазмы мегакариоцитов и активацию тромбоцитов.
Комплекс входящих в состав основного вещества соединительной ткани гликозаминогликанов и указанных выше экстрацеллюлярных белков рассматривается как структура, обеспечивающая концентрацию гемопоэтических ростовых факторов и модуляцию их функций. Таким образом, основное вещество соединительной ткани костного мозга представляет собой физиологически весьма активную среду, что дает основание рассматривать ее в качестве важнейшего регулятора кроветворения.
14.2. ИЗМЕНЕНИЯ КОЛИЧЕСТВЕННОГО И КАЧЕСТВЕННОГО СОСТАВА ЭРИТРОЦИТОВ
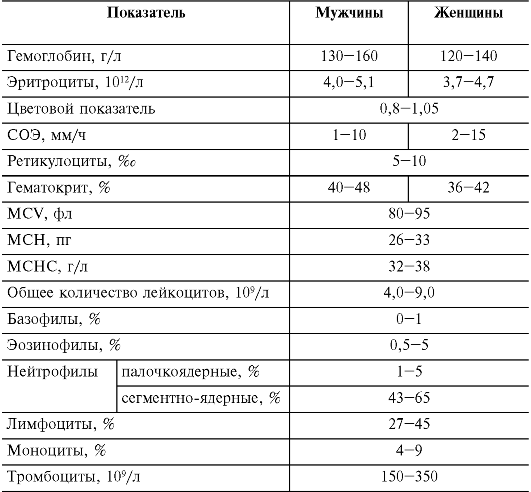
В норме содержание эритроцитов в периферической крови у мужчин составляет в среднем (4,0-5,1)-1012/л, у женщин - (3,7- 4,7)-1012/л; уровень гемоглобина соответственно 130-160 г/л и
120-140 г/л (см. табл. 14-1).
У здоровых людей количество образующихся в костном мозгу эритроцитов равно числу выходящих из циркуляции (гемолизирующихся) клеток, в связи с чем уровень их в крови практически постоянен. При различных заболеваниях эритроцитарный баланс может нарушаться, что приводит к увеличению числа эритроцитов в крови (эритроцитозу) или к его уменьшению (анемии).
Таблица 14-1. Количественные показатели периферической крови здорового человека
 Примечание.
MCV - средний объем эритроцитов; MCH - среднее (относительное)
содержание гемоглобина в эритроците; MCHC - средняя концентрация
(абсолютное содержание) гемоглобина в эритроците.
Примечание.
MCV - средний объем эритроцитов; MCH - среднее (относительное)
содержание гемоглобина в эритроците; MCHC - средняя концентрация
(абсолютное содержание) гемоглобина в эритроците.
14.2.1. Анемии. Патологические формы эритроцитов
Анемия, или малокровие - патологическое состояние, характеризующееся уменьшением концентрации гемоглобина и в подавляющем большинстве случаев числа эритроцитов в единице объема крови. При
тяжелых формах анемий в крови могут появляться патологические формы эритроцитов (см. ниже).
От истинной анемии следует отличать гидремию - увеличение объема плазмы вследствие «разжижения» крови (при беременно-
сти, микседеме, почечной недостаточности с олиго- и анурией, застойной спленомегалии и др.), сопровождающееся относительным снижением концентрации гемоглобина и количества эритроцитов в единице объема крови.
Анемия может не выявляться при состояниях, связанных со сгущением крови вследствие дегидратации организма (при длительной диарее, многократной рвоте, стенозе привратника, ожоговой болезни и др.). В этом случае за счет уменьшения жидкой части крови концентрация гемоглобина и масса эритроцитов в крови могут оставаться в пределах нормы («скрытая» анемия).
Этиология анемий включает острые и хронические кровотечения, инфекции, воспаления, интоксикации (солями тяжелых металлов), глистные инвазии, злокачественные новообразования, авитаминозы, заболевания эндокринной системы, почек, печени, желудка, поджелудочной железы. Анемии часто развиваются при лейкозах, особенно при острых их формах, при лучевой болезни. Кроме того, играют роль патологическая наследственность и нарушения иммунологической реактивности организма.
Общими симптомами для всех форм анемий, возникновение которых связано с основным звеном патогенеза малокровия - гипоксией, являются бледность кожных покровов и слизистых оболочек, одышка, сердцебиение, а также жалобы на головокружение, головные боли, шум в ушах, неприятные ощущения в области сердца, резкую общую слабость и быструю утомляемость. В легких случаях малокровия общие симптомы могут отсутствовать, так как компенсаторные механизмы (усиление эритропоэза, активация функций сердечно-сосудистой и дыхательной систем) обеспечивают физиологическую потребность тканей в кислороде.
Классификация анемий. В основу существующих классификаций анемий положены их патогенетические признаки с учетом особенностей этиологии, данные о содержании гемоглобина и эритроцитов в крови, морфологии эритроцитов, типе эритропоэза и способности костного мозга к регенерации.
По механизму развития выделяют три основных вида анемий:
1. Анемии вследствие кровопотерь (постгеморрагические):
• острые постгеморрагические;
• хронические постгеморрагические.
2. Анемии вследствие нарушения кровообразования. Анемии, связанные с нарушением образования гемоглобина:
• связанные с дефицитом железа (железодефицитные анемии);
• связанные с нарушением синтеза или утилизации порфиринов (сидеробластные анемии).
Анемии, связанные с нарушением синтеза ДНК (мегалобластные анемии):
• связанные с дефицитом витамина В12 (В12-дефицитные анемии);
• связанные с дефицитом фолиевой кислоты (фолиеводефицитные анемии).
Гипо- и апластические анемии:
• наследственные формы;
• приобретенные формы.
Анемии, ассоциированные с заболеваниями внутренних органов:
• при эндокринных заболеваниях;
• при заболеваниях печени;
• при заболеваниях почек. Анемии хронических заболеваний:
• при хронических инфекционных заболеваниях;
• при системных заболеваниях соединительной ткани;
• при опухолевых заболеваниях.
Анемии при опухолевых и метастатических поражениях костного мозга.
3. Анемии вследствие повышения кроверазрушения (гемолитические):
Наследственные гемолитические анемии:
• связанные с нарушением структуры мембраны эритроцитов (мембранопатии);
• связанные с нарушением активности ферментов эритроцитов (энзимопатии);
• связанные с нарушением синтеза или структуры гемоглобина (гемоглобинопатии):
- связанные с нарушением синтеза полипептидных цепей глобина;
- обусловленные носительством аномальных гемоглобинов. Приобретенные гемолитические анемии:
• связанные с воздействием антител (аутоиммунные, гетероиммунные, трансиммунные, изоиммунные);
• связанные с изменением структуры мембраны эритроцитов, обусловленным соматической мутацией;
• связанные с повреждением мембраны эритроцитов: механическими, физическими и химическими факторами;
• обусловленные недостатком витаминов (витамина Е и др.);
• обусловленные разрушением эритроцитов паразитами (малярийным плазмодием, бабезиями и др.).
Основой классификации анемий по степени тяжести является уровень снижения содержания гемоглобина и эритроцитов в единице объема крови (табл. 14-2).
Таблица 14-2. Классификация анемий по степени тяжести (Е.Д. Гольдберг, 1989)
Степень тяжести | Количество гемоглобина, г/л | Количество эритроцитов, 1012/л |
Легкая | >100 | >3,0 |
Средняя | 100-66 | 3,0-2,0 |
Тяжелая | <66 | <2,0 |
Морфологическими критериями, заложенными в основу классификаций анемий, являются величины цветового показателя (ЦП), среднего диаметра эритроцитов (СДЭ) и тип кроветворения (рис. 14-2).
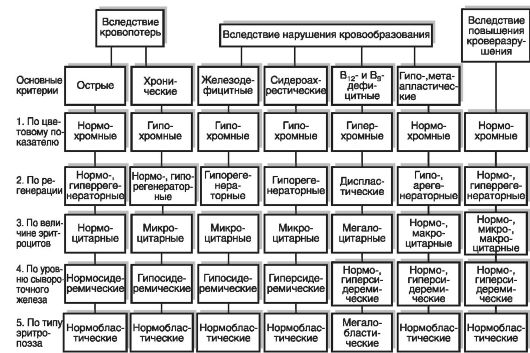 Рис. 14-2. Гематологическая характеристика анемий
Рис. 14-2. Гематологическая характеристика анемий
По цветовому показателю анемии делят на гипохромные (ЦП ниже 0,8), нормохромные (ЦП от 0,8 до 1,05) и гиперхромные (ЦП выше 1,05).
По величине СДЭ различают микроцитарные - СДЭ ниже 7,2 мкм (железодефицитные и хроническая постгеморрагическая анемии, болезнь Минковского-Шоффара; нормоцитарные - СДЭ в пределах 7,2-8,0 мкм (острая постгеморрагическая и большинство гемолитических анемий); макроцитарные - СДЭ выше 8,0 мкм (гемолитическая болезнь новорожденных, гипо- и апластические анемии). В группу макроцитарных анемий входят и мегалоцитарные (мегалобластические) анемии, при которых СДЭ превышает 9,5 мкм (В12-дефицитные, фолиеводефицитные анемии).
По типу кроветворения анемии можно подразделить на две группы: с нормобластическим типом кроветворения (нормальный эритропоэз: эритробласт -> пронормобласт -> нормобласт базофильный -> нормобласт полихроматофильный -> нормобласт оксифильный -> эритроцит) (постгеморрагические, гемолитические, гипо- и апластические анемии) и мегалобластическим типом кроветворения (промегалобласт -> мегалобласт базофильный -> мегалобласт полихроматофильный -> мегалобласт оксифильный -> мегалоцит) (В12-дефицитные, фолиеводефицитные анемии) (рис. 14-3, см. цв. вклейку).
По способности костного мозга к регенерации различают анемии регенераторные - нормо- и гиперрегенераторные (острая постгеморрагическая и большинство гемолитических анемий), гипорегенераторные (железодефицитные, В12-дефицитные анемии) и арегенераторные (гипо- и апластические). Показателем достаточной регенераторной способности костного мозга служит развивающийся ретикулоцитоз. В норме на суправитально окрашенных мазках периферической крови выявляется 5-10%о ретикулоцитов (расчет ведется на 1000 эритроцитов). При анемиях с достаточной функцией костного мозга их число составляет от 11% до 50%, с гиперрегенерацией - 50-100% и выше, при гипорегенераторных анемиях - 5-10%. При арегенераторных анемиях ретикулоциты выявляются в виде единичных экземпляров (до 5%) или отсутствуют.
Оценить функциональное состояние костного мозга при анемиях помогает также лейко-эритробластическое отношение (ЛЭО), которое можно определить на мазках костного мозга при подсчете миелограмм. В норме оно составляет 2:1-4:1. Увеличение ЛЭО при
низкой клеточности костного мозга свидетельствует о редукции красного ростка (гипо- и апластические анемии), снижение ЛЭО (до 1:1 или даже до 1:2-1:3) при нормальной или повышенной клеточности костного мозга указывает на гиперплазию эритроидного ростка, что обусловливается компенсаторной активацией эритропоэза (гемолитические анемии) или нарушением созревания эритроидных клеток и задержкой незрелых эритрокариоцитов в костном мозгу (железодефицитная анемия, мегалобластные анемии). При тяжелых формах малокровия (пернициозная анемия) ЛЭО может доходить до 1:8.
Патологические формы эритроцитов
При анемиях в периферической крови на фиксированных или суправитально окрашенных мазках могут встречаться эритроциты и эритроидные формы костного мозга, не выявляемые у здоровых людей (табл. 14-3).
Таблица 14-3. Особенности морфологии эритроцитов при анемиях
Вариант патологических изменений | Характеристика патологических изменений |
Изменение размеров эритроцитов (анизоцитоз) | Микроциты - эритроциты диаметром менее 6,5 мкм |
Макроциты - эритроциты диаметром от 8 до 10 мкм | |
Мегалоциты - эритроциты диаметром 10 мкм и более | |
Изменение формы эритроцитов | Акантоциты - эритроциты с неравномерно распределенными по поверхности роговидными выростами |
(пойкилоцитоз) | Каплевидные эритроциты - клетки в форме «капли» |
Мишеневидные эритроциты - клетки в форме «мишени» с центральным расположением гемоглобина | |
Дегмациты - «надкусанные» эритроциты | |
Овалоциты (эллиптоциты) - клетки овальной (эллипсовидной) формы | |
Серповидные эритроциты (дрепаноциты) - клетки в форме «серпа» («полумесяца») | |
Стоматоциты («улыбающиеся» эритроциты) - клетки с центральным просветлением в форме «рта» | |
Сфероциты - эритроциты шаровидной формы |
Окончание табл. 14-3
Шизоциты - осколки разрушенных эритроцитов диаметром 2-3 мкм неправильной формы | |
Шлемовидные эритроциты - фрагменты разрушенных эритроцитов в форме «шлема» | |
Эхиноциты - эритроциты с равномерно распределенными по поверхности шиповидными выростами | |
Изменение окраски эритроцитов (анизохромия) | Гипохромия - снижение плотности окраски эрироцитов |
Гиперхромия - интенсивная окраска эритроцитов | |
Полихроматофилы - эритроциты серо-фиолетового цвета | |
Включения в эритроцитах | Базофильная зернистость (пунктация) - рассеянные в цитоплазме эритроцитов гранулы темно-синего цвета (агрегаты рибосом, митохондрий) |
Ретикулоциты - молодые эритроциты с остатками цитоплазматических органелл, выявляемых при суправитальной окраске в виде нитей и зерен синеголубого цвета (зернисто-сетчатая субстанция) | |
Кольца Кабо - нитевидные остатки ядерной мембраны в форме «кольца» или «восьмерки» синефиолетового цвета | |
Тельца Жолли - остатки ядерного хроматина округлой формы сине-фиолетового цвета | |
Тельца Гейнца - преципитаты гемоглобина округлой формы синего цвета, выявляемые в эритроцитах при суправитальной окраске | |
Гемоглобиновая дегенерация Эрлиха - краснорозовые уплотнения (глыбки) гемоглобина вследствие его коагуляции |
Появление их свидетельствует о компенсаторных усилиях эритропоэза или о нарушении созревания клеток эритроидного ряда в костном мозгу (регенеративные формы эритроцитов) либо о дегенеративных изменениях эритроцитов, возникающих в результате нарушения кровообразования в костном мозгу (дегенеративные формы эритроцитов).
К группе регенеративных форм эритроцитов относят незрелые формы эритропоэза - ядросодержащие эритроциты (нормобласты, мегалобласты), эритроциты с остатками ядерной субстан-
ции (тельца Жолли, кольца Кабо). Цитоплазматическую природу (остатки базофильной субстанции) имеют полихроматофильные эритроциты, ретикулоциты (выявляются на суправитально окрашенных препаратах), базофильная зернистость эритроцитов (см.
рис. 14-3).
К группе дегенеративных форм эритроцитов относят клетки с измененной величиной (анизоцитоз), формой (пойкилоцитоз), различным содержанием гемоглобина в эритроцитах (анизохромия), гемоглобиновую дегенерацию Эрлиха, вакуолизацию эритроцитов. На суправитально окрашенных мазках в эритроцитах обнаруживаются тельца Гейнца, а также иссиня-темные эритроциты - дегенеративная полихромазия (см. рис. 14-3).
Анемии вследствие кровопотерь (постгеморрагические)
Различают острую и хроническую постгеморрагическую анемии. Первая является следствием быстрой потери значительного количества крови, вторая развивается в результате длительных постоянных кровопотерь даже в незначительном объеме.
Кровопотеря. Выход значительного количества крови из сосудистого русла (кровопотеря) возникает в результате нарушения целостности стенки кровеносных сосудов вследствие травмы, болезни или оперативного вмешательства и характеризуется сложным комплексом патологических и компенсаторно-приспособительных реакций организма (рис. 14-4).
В механизме расстройства функций организма при кровопотере основную роль играют следующие факторы: уменьшение объема циркулирующей крови (ОЦК), падение артериального давления, гипоксемия, гипоксия органов и тканей.
Кровопотеря является мощным стрессовым воздействием, активирующим симпатоадреналовую систему. Выраженность ответной реакции организма при этом зависит от скорости и объема кровопотери.
Одномоментная кровопотеря в объеме 10-15% всей массы крови у взрослого человека не вызывает опасных для жизни гемодинамических нарушений. Выброс надпочечниками катехоламинов приводит к уменьшению объема сосудистого русла вследствие спазма емкостных сосудов кожи, легких, органов желудочно-кишечного тракта и т.д., что наряду с мобилизацией в кровоток межтканевой жидкости компенсирует возникающий в условиях легкой крово-
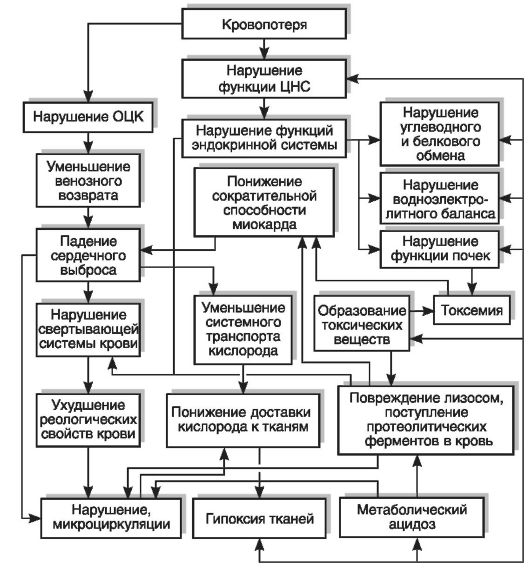 Рис. 14-4. Основные нарушения функции органов и систем при острой кровопотере (по В.Н. Шабалину, Н.И. Кочетыгову)
Рис. 14-4. Основные нарушения функции органов и систем при острой кровопотере (по В.Н. Шабалину, Н.И. Кочетыгову)
потери дефицит ОЦК. В результате наступающей «централизации кровообращения» кровоток в жизненно важных органах (головной и спинной мозг, сердце, надпочечники) остается, как правило, в пределах нормы.
В случае острой кровопотери (более 15% ОЦК) значительно сокращается венозный приток к правому сердцу, что, в свою очередь, приводит к уменьшению сердечного выброса, прогрессирующему падению артериального давления и замедлению кровотока. В ответ на снижение показателей центральной гемодинамики возникает системная вазоконстрикция, происходит выброс депонированной крови, развивается тахикардия, включается ряд других механиз-
мов компенсации гиповолемии, что до определенного времени (до тех пор пока кровопотеря не превысит 40-45% ОЦК) позволяет поддерживать артериальное давление на субкритическом уровне (90-85/45-40 мм рт.ст.).
Продолжающееся кровотечение приводит к истощению адаптационных систем организма, участвующих в борьбе с гиповолемией, - развивается геморрагический шок. Защитные рефлексы системы макроциркуляции при этом оказываются уже недостаточными для обеспечения адекватного сердечного выброса, вследствие чего систолическое давление быстро падает до критических цифр (50-40 мм рт.ст.). В конечном счете нарушается кровоснабжение органов и систем организма, развивается кислородное голодание и наступает смерть в связи с параличом дыхательного центра и остановкой сердца.
Основным звеном патогенеза необратимой стадии геморрагического шока является декомпенсация кровообращения в микроциркуляторном русле. Нарушение системы микроциркуляции имеет место уже на ранних стадиях развития гиповолемии. Длительный спазм емкостных и артериальных сосудов, усугубленный прогрессирующим снижением артериального давления при непрекращающемся кровотечении, рано или поздно приводит к полной остановке микроциркуляции. Наступает стаз, в спазмированных капиллярах образуются агрегаты эритроцитов. Возникающие в динамике кровопотери уменьшение и замедление кровотока сопровождаются повышением концентрации фибриногена и глобулинов плазмы крови, что увеличивает ее вязкость и способствует агрегации эритроцитов. В результате быстро возрастает уровень токсических продуктов метаболизма, который становится анаэробным. Метаболический ацидоз в известной степени компенсируется дыхательным алкалозом, развивающимся вследствие рефлекторно возникающей гипервентиляции. Грубые нарушения сосудистой микроциркуляции и поступление в кровь недоокисленных продуктов обмена могут привести к необратимым изменениям в печени и почках, а также пагубно сказаться на функционировании сердечной мышцы даже в период компенсируемой гиповолемии.
В случае своевременной остановки кровотечения и сохранения жизнеспособности организма непосредственно после кровопотери восстановление утраченного объема крови обеспечивается главным образом за счет активного поступления в сосудистое русло тканевой жидкости. Удержанию жидкости в кровеносном русле
и наступающему при этом увеличению ОЦК способствуют восполнение дефицита белков плазмы (за счет мобилизации лимфы), являющихся наряду с электролитами основными водосвязывающими структурами крови, а также повышение выделения антидиуретического гормона и альдостерона.
Компенсаторная аутогемодилюция неизбежно приводит к снижению дыхательной емкости крови вследствие уменьшения содержания эритроцитов. В то же время газотранспортная функция крови существенно не страдает даже при потере до 50% ОЦК, так как для поддержания жизни достаточно и трети имеющегося в норме гемоглобина. В связи с этим уменьшение кислородной емкости крови при кровопотере не имеет решающего значения для организма. Уже в первые часы после кровотечения печень начинает активно продуцировать белки, которые поступают в кровоток и повышают онкотическое давление крови.
Острая кровопотеря до 15-22% ОЦК условно считается легкой, до 25-35% ОЦК - средней степени тяжести, до 50% ОЦК - тяжелой. Исход кровотечений определяется также состоянием реактивности организма - совершенством систем адаптации, полом, возрастом, сопутствующими заболеваниями и т.д. Дети, особенно новорожденные и грудного возраста, переносят кровопотерю значительно тяжелее, чем взрослые.
Внезапная потеря 50% ОЦК является смертельной. Медленная (в течение нескольких дней) кровопотеря такого же объема крови менее опасна для жизни, поскольку компенсируется механизмами адаптации. Острые кровопотери до 25-50% ОЦК рассматриваются как угрожающие для жизни в связи с возможностью развития геморрагического шока. При этом особенно опасны кровотечения из артерий.
Острая постгеморрагическая анемия - состояние, развивающееся при скоротечной потере значительного объема крови в результате наружного или внутреннего кровотечения вследствие травм, желудочных, кишечных, маточных кровотечений, при разрыве фаллопиевой трубы при внематочной беременности и др.
В основе острой постгеморрагической анемии лежат гиповолемия, в результате которой могут развиться коллапс и шок, и уменьшение массы циркулирующих эритроцитов, приводящее к нарушению процессов оксигенации тканей организма.
Острая кровопотеря, совместимая с жизнью (до 30% ОЦК), сопровождается последовательным включением защитно-приспосо-
бительных механизмов, направленных на восстановление ОЦК. Выделяют следующие фазы компенсаторных реакций: сосудисторефлекторную, гидремическую, костно-мозговую.
Сосудисто-рефлекторная фаза длится 8-12 ч от начала кровопотери и характеризуется спазмом периферических сосудов вследствие выброса надпочечниками катехоламинов, что приводит к уменьшению объема сосудистого русла («централизации» кровообращения) и способствует сохранению кровотока в жизненно важных органах (см. выше). Вследствие активации ренинангиотензин-альдостероновой системы активируются процессы реабсорбции натрия и воды в проксимальных канальцах почек, что сопровождается снижением диуреза и задержкой воды в организме. В этот период в результате равнозначной потери плазмы крови и форменных элементов, компенсаторного поступления депонированной крови в сосудистое русло содержание эритроцитов и гемоглобина в единице объема крови и величина гематокрита остаются близкими к исходным («скрытая» анемия). Ранними признаками острой кровопотери являются лейкопения и тромбоцитопения. В ряде случаев возможно увеличение общего количества лейкоцитов.
Гидремическая фаза развивается на 1-2-й день после кровопотери. Проявляется мобилизацией тканевой жидкости и поступлением ее в кровяное русло, что приводит к восстановлению объема плазмы. «Разбавление» крови сопровождается прогрессирующим снижением количества эритроцитов и гемоглобина в единице объема крови. Анемия носит нормохромный, нормоцитарный характер.
Костно-мозговая фаза развивается на 4-5-й день после кровопотери. Определяется усилением процессов эритропоэза в костном мозгу в результате гиперпродукции клетками юкстагломерулярного аппарата почек в ответ на гипоксию эритропоэтина, стимулирующего активность коммитированной (унипотентной) клетки-предшественницы эритропоэза - КОЕ-Э. Критерием достаточной регенераторной способности костного мозга (регенераторная анемия) служит повышение содержания в крови молодых форм эритроцитов (ретикулоцитов, полихроматофилов), что сопровождается изменением размеров эритроцитов (макроцитозом) и формы клеток (пойкилоцитозом). Возможно появление эритроцитов с базофильной зернистостью, иногда - единичных нормобластов в крови. Вследствие усиления гемопоэтической функции костного мозга развивается умеренный лейкоцитоз (до 12-109/л) со
сдвигом влево до метамиелоцитов (реже до миелоцитов), увеличивается количество тромбоцитов (до 500-109/л и более). В костном мозгу ЛЭО может достигать 1:1.
Восстановление массы эритроцитов происходит в течение 1-2 месяцев в зависимости от объема кровопотери. При этом расходуется резервный фонд железа в организме, что может стать причиной железодефицита. Анемия в этом случае приобретает гипохромный, микроцитарный характер.
Хроническая постгеморрагическая анемия развивается в результате небольших повторных кровотечений (язвы, опухоли желудка и кишечника, геморрой, дисменоррея, геморрагические диатезы, легочные, почечные, носовые кровотечения и др.). Протекает по типу гипохромной, железодефицитной анемии (см. ниже). В мазках крови обнаруживаются анизоцитоз, пойкилоцитоз, анизохромия эритроцитов, микроциты. Выявляется лейкопения за счет нейтропении, иногда со сдвигом влево.
Анемии вследствие нарушения кровообразования
Группа анемий, объединенных одним общим механизмом развития, который заключается в нарушении или полном прекращении эритропоэза в результате дефицита веществ, необходимых для осуществления нормального кроветворения, носит название дефицитных анемий. Сюда относят дефицит микроэлементов (железо, медь, кобальт), витаминов (В12, B6, В2, фолиевая кислота) и белков.
При замещении костномозговой полости жировой, костной или опухолевой тканью (метастазы опухолей в костный мозг, лейкоз), а также при действии физических (ионизирующая радиация) и химических факторов, некоторых микробных токсинов и лекарственных препаратов развиваются анемии в результате сокращения площади кроветворения.
Железодефицитные анемии. Анемии, обусловленные дефицитом железа в организме, относятся к числу наиболее распространенных заболеваний в мире и составляют 80-95% всех форм малокровия. Наиболее часто они встречаются у детей младшего возраста, девушек-подростков и женщин детородного возраста.
Этиология. Железодефицитная анемия может быть обусловлена самыми разнообразными причинами: недостаточным поступлением железа с пищей, нарушением всасывания его в тонком кишеч-
нике, повышенной потребностью в период роста, беременностью, лактацией, кровотечениями из различных органов и др. Однако наиболее частой причиной железодефицитной анемии являются кровопотери и в первую очередь длительные постоянные кровотечения даже с небольшими потерями крови. В этих случаях количество теряемого железа превышает его поступление с пищей. Дефицит железа в организме развивается при суточной потере его в количестве, превышающем 2 мг. Схема метаболизма железа представлена на рис. 14-5.
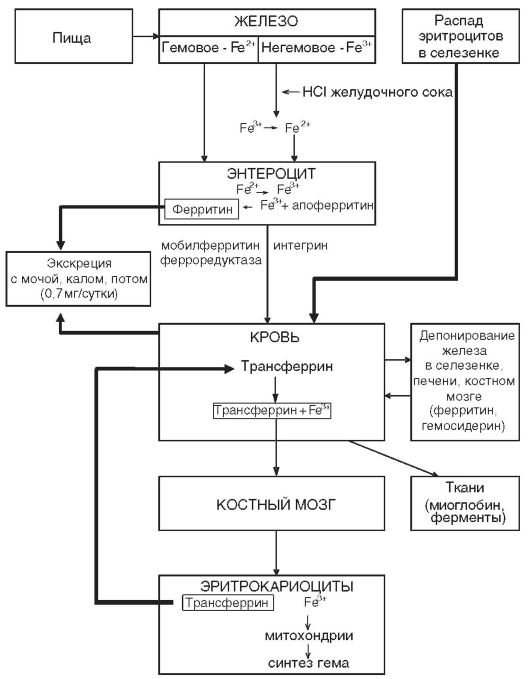 Рис. 14-5. Схема метаболизма железа в организме
Рис. 14-5. Схема метаболизма железа в организме
По патогенетическому принципу с учетом основных этиологических причин железодефицитные анемии делят на пять основных подгрупп (Л.И. Идельсон): 1) связанные с повышенной потерей железа; 2) связанные с недостаточным исходным уровнем железа; 3) связанные с повышенным расходованием железа; 4) связанные с нарушением всасывания железа и недостаточным поступлением его с пищей (алиментарные); 5) связанные с нарушением транспорта железа (табл. 14-4).
Таблица 14-4. Причины и механизмы развития железодефицитных состояний
Этиологические факторы | Патогенез | |
группы факторов | характеристика | |
Особенные периоды жизни | Дети недоношенные и новорожденные Дети первых лет жизни | Недостаточный исходный уровень железа |
Интенсивный рост (пубертатный период) Беременность Лактация | Повышенное расходование железа | |
Патологические состояния | Хроническая кровопотеря: при частых лечебных кровопусканиях, донорстве; при заболеваниях сердечнососудистой системы (гипертоническая болезнь, геморрагическая телеангиэктазия и др.); при патологии желудочнокишечного тракта (варикозное расширение вен пищевода, диафрагмальная грыжа, язва желудка и двенадцатиперстной кишки, язвенный колит, дивертикулез, геморрой и др.); из органов мочеполовой системы (алкогольная нефропатия, туберкулез почек, почечнокаменная болезнь, полипы и рак мочевого пузыря, обильные меноррагии, эндометриоз, миома матки и др.); из органов дыхательной системы (рак легкого, туберкулез, бронхоэктазия и др.); | Повышенная потеря железа |
Окончание табл. 14-4
при заболеваниях системы крови (лейкозы, апластическая анемия и др.); при патологии системы гемостаза (аутоиммунная тромбоцитопения, гемофилии, ДВС-синдром и др.) | ||
Патологические состояния и болезни | Патология желудочно-кишечного тракта: резекция желудка и кишечника; гипосекреция желудочного сока; хронический энтерит; дисбактериозы; глистные инвазии и др. | Нарушение всасывания железа |
Наследственная атрансферринемия Приобретенная гипотрансферринемия (при нарушении белоксинтезирующей функции печени) | Нарушение транспорта железа | |
Алкоголизм | Комбинация факторов: недостаточное поступление железа; нарушение транспорта железа; нарушение всасывания железа; потеря железа | |
Неблагоприятные воздействия | Нерациональное питание: голодание; вегетарианская диета; искусственное вскармливание грудных детей | Недостаточное поступление железа |
Избыточные физические нагрузки | Повышенное расходование железа |
Патогенез. Основным звеном патогенеза заболевания является снижение содержания железа в сыворотке крови, костном мозгу и депо. В результате нарушается синтез гемоглобина, возникают гипохромная анемия и трофические расстройства в тканях, признаками которых являются: сухость и вялость кожи, ломкость ногтей, выпадение волос, атрофия слизистой оболочки языка, повышенное разрушение зубов, дисфагия, извращение вкуса, мы-
шечная слабость и др. (сидеропенический синдром Вальденстрема). В патогенезе клинических проявлений болезни в еще большей степени, чем недостаточное снабжение тканей кислородом, имеет значение нарушение активности железосодержащих ферментов в тканях организма (цитохром С, цитохромоксидаза, сукцинатдегидрогеназа, пероксидаза, митохондриальная моноаминооксидаза, α-глицерофосфатоксидаза). Признаки гипоксии тканей появляются лишь при значительной выраженности малокровия, когда наступает истощение компенсаторных механизмов, обеспечивающих на ранних этапах развития дефицита железа нормализацию отдачи кислорода из гемоглобина тканям.
Картина крови. Основным признаком железодефицитной анемии является гипохромия со снижением цветового показателя ниже 0,8 и, соответственно, уменьшением содержания гемоглобина ниже 110 г/л. Количество эритроцитов, как правило, остается на исходном уровне, но в ряде случаев может оказаться сниженным до (2,0-1,5)-1012/л вследствие нарушения процессов пролиферации клеток эритроидного ряда в костном мозгу и усиления неэффективного эритропоэза (в норме разрушение эритронормобластов в костном мозгу не превышает 10-15%). Содержание ретикулоцитов колеблется в пределах нормы, но при значительной кровопотере бывает несколько повышенным. Важным морфологическим признаком железодефицитных анемий является анизоцитоз эритроцитов с преобладанием микроцитов.
В костном мозгу отмечаются нарушение процессов гемоглобинизации эритрокариоцитов, сопровождающееся увеличением количества базофильных и полихроматофильных нормобластов при параллельном снижении числа их оксифильных форм, а также резкое уменьшение количества сидеробластов - нормобластов, содержащих единичные гранулы железа в цитоплазме (в норме до 20-40%).
В диагностике железодефицитной анемии решающее значение имеют показатели обмена железа (сывороточное железо, железосвязывающая способность сыворотки, общий запас железа в организме и др.). Количество железа в сыворотке крови при выраженной железодефицитной анемии падает до 5,4-1,8 мкМ/л при норме 12,5-30,4 мкМ/л (мужчины; у женщин этот показатель на 10-15% ниже). Увеличивается железосвязывающая способность сыворотки. В норме одна треть трансферрина насыщена железом, а две трети - свободны. Под общей железосвязывающей способностью сыворотки понимается не абсолютное количество трансферрина, а коли-
чество железа, которое может связаться с трансферрином (в норме 54,0-72,0 мкМ/л). Содержание ферритина в сыворотке крови, по результатам радиоиммунологических методов исследования, при железодефицитных анемиях снижается до 9,0-1,5 мкг/л (в норме - 12-300 мкг/л). Об уровне депонированного железа можно судить по содержанию железа в суточной моче после однократного введения больному 500 мг десферала (продукт метаболизма актиномицетов, избирательно выводящий ион железа из организма). В норме этот показатель соответствует 0,6-1,3 мг железа, а при железодефицитной анемии снижается до 0,2 мг в сутки и менее.
В12-дефицитные и фолиеводефицитные анемии. Витамин В12 и фолиевая кислота - кофакторы синтеза ДНК. Их дефицит сопровождается нарушением процессов пролиферации клеток с высоким кругооборотом - клеток крови, клеток кишечного эпителия и как следствие развитием анемии, характеризующейся наличием в костном мозгу мегалобластов, расстройствами пищеварения. Сочетанный дефицит витамина В12 и фолиевой кислоты встречается редко, чаще наблюдается изолированный дефицит витаминов.
Этиология. Дефицит витамина В12 чаще развивается в результате нарушения его всасывания при снижении секреции внутреннего фактора Касла (рис. 14-6) вследствие атрофии слизистой желудка либо после резекции желудка (агастрические анемии). У большинства больных с дефицитом витамина В12 обнаруживаются антитела, направленные против обкладочных клеток желудка и внутреннего фактора Касла. В12- и фолиеводефицитные состояния могут развиваться также при инвазии широким лентецом, поглощающим большое количество витамина В12, при беременности, нарушении всасывания витамина В12 в кишечнике, реже - при недостатке поступления с пищей (табл. 14-5).
Таблица 14-5. Причины развития мегалобластных анемий


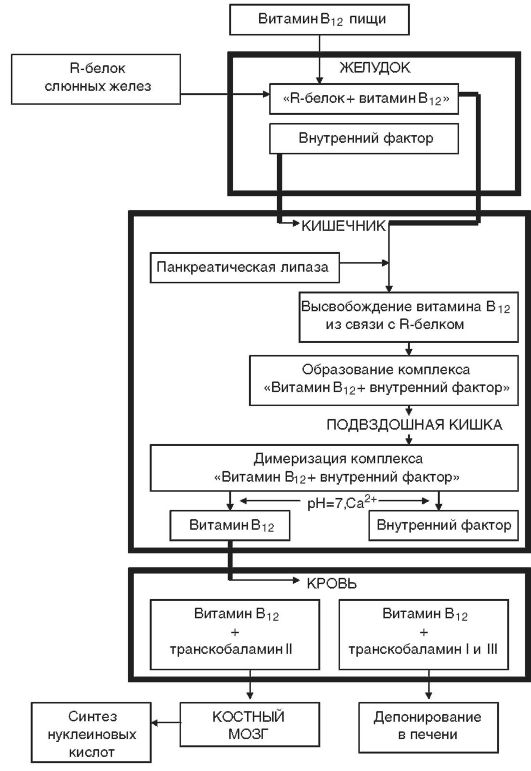 Рис. 14-6. Схема метаболизма витамина В12 в организме
Рис. 14-6. Схема метаболизма витамина В12 в организме
Пернициозная анемия (болезнь Аддисона-Бирмера). Представляет собой одну из форм заболевания, связанного с дефицитом фактора Касла и связанной с ним недостаточностью витамина В12. Различают наследственную и приобретенную формы болезни (см. табл. 14-5). Приобретенная форма анемии чаще развивается у лиц пожилого возраста (в среднем в возрасте 60 лет), редко встречается у детей в возрасте до 10 лет (ювенильная форма). Заболевание характеризуется поражением трех систем: пищеварительной (воспаление и атрофия сосочков языка, гистаминрезистентная ахилия, связанная с глубокой атрофией слизистой желудка, в peзультате чего железы дна и тела желудка прекращают выработку внутреннего фактора Касла - гастромукопротеина), нервной (фуникулярный миелоз - дегенерация задних и боковых столбов спинного мозга, нарушение кожной и вибрационной чувствительности, мышечно-суставного чувства, изменение ахилловых, коленных и др. рефлексов) и кроветворной (гиперхромная макроцитарная мегалобластическая анемия).
Патогенез. Расстройства пищеварения и переход на мегалобластический тип кроветворения обусловливаются резким снижением активности В12-зависимых энзимов, участвующих в метаболизме фолатов (соли фолиевой кислоты), необходимых для синтеза ДНК. При этом обнаруживается yменьшение активности метилтрансферазы, сопровождающееся кумуляцией в клетках неактивного метилтетрагидрофолата и нарушением синтеза ДНК (синтез РНК не страдает) (рис. 14-7), что приводит к удлинению S-фазы клеточного цикла и патологии деления и созревания эпителиальных клеток желудочно-кишечного тракта и миелокариоцитов. В костном мозгу развивается мегалобластоз.
Нарушение кроветворения связано с замедлением темпа мегалобластического эритропоэза в результате удлинения времени митотического цикла и сокращения числа митозов: вместо трех митозов, свойственных нормобластическому эритропоэзу, регистрируется только один. Срок жизни эритроцитов сокращается до 30-40 дней (в норме - 90 дней). Часть клеток погибает на ранних стадиях развития. Распад мегалобластов в костном мозгу, наряду с их замедленной дифференциацией, и мегалоцитов в крови приводит к тому, что процессы кроветворения не компенсируют процессы кроверазрушения. Развивается мегалобластическая анемия.
В основе неврологических расстройств (фуникулярный миелоз) лежит нарушение превращения метилмалонил-КоА в сукцинил-
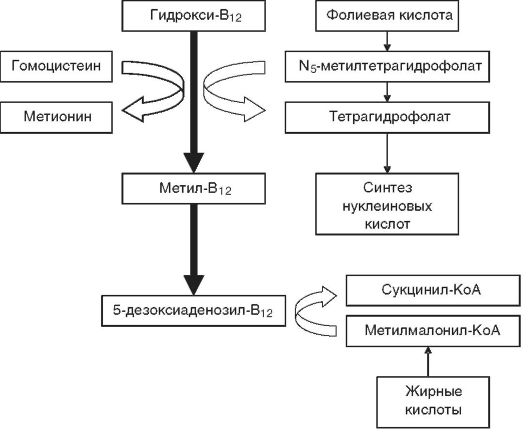 Рис. 14-7. Схема биохимических реакций, протекающих в организме с участием витамина В12 и фолиевой кислоты (по В.В. Долгову и соавт., 2001)
Рис. 14-7. Схема биохимических реакций, протекающих в организме с участием витамина В12 и фолиевой кислоты (по В.В. Долгову и соавт., 2001)
КоА при недостатке 5-дезоксиаденозил-В12 (см. рис. 14-7) и накопление в нервной ткани токсичных метилмалоновой и пропионовой кислот, вызывающих жировую дистрофию нервных клеток и демиелинизацию нервных волокон спинного мозга и периферических нервов.
При монодефиците фолиевой кислоты нарушения метаболизма жирных кислот в нервной ткани, а следовательно, признаки неврологических расстройств отсутствуют.
Картина крови. В периферической крови обнаруживаются гиперхромная анемия (цветовой показатель 1,2-1,5), явления пойкилоцитоза с тенденцией к овалоцитозу, анизоцитоза с выраженным макроцитозом и мегалоцитозом. Характерны явления анизохромии и гиперхромии эритроцитов; могут обнаруживаться полихроматофильные и оксифильные мегалобласты, эритроциты с тельцами Жолли, кольцами Кабо, азурофильной зернистостью. Средний диаметр эритроцитов увеличивается до 8,2-9,5 мкм, их средний объем превышает 100 фл (110-160 фл). Регистрируется умеренная лей-
копения с нейтропенией при полном отсутствии малочисленных форм гранулоцитов - эозинофилов и базофилов (анэозинофилия, абазофилия). Встречаются гиперсегментно-ядерные нейтрофилы (дегенеративный ядерный сдвиг вправо), редко - гигантские формы нейтрофилов. Количество тромбоцитов уменьшается, часть их имеет крупные размеры (6-10 мкм и более).
Костный мозг гиперклеточный за счет накопления незрелых (негемоглобинизированных) ядросодержащих форм клеток красного ряда. На препаратах костного мозга обнаруживаются мегалоциты и мегалобласты, гигантские формы метамиелоцитов.
Гипо- и апластические анемии. Гипопластические анемии относятся к числу анемий, обусловленных депрессией костно-мозгового кроветворения без признаков гемобластоза и метаплазии.
Апластические анемии могут быть наследственными и приобретенными. Последние развиваются при действии на организм некоторых химических и лекарственных веществ (бензол, бензин, пары ртути и различных кислот, красители, сульфаниламиды, антибиотики, цитостатические препараты, препараты золота, висмута, мышьяка и др.), ионизирующей радиации, при ряде инфекций (герпесвирусные инфекции, туберкулез), аутоиммунных заболеваниях (системная красная волчанка, ревматоидный артрит), эндокринопатиях (дисфункция щитовидной железы, яичников, тимуса), а также при стрессе, голодании, расстройствах пищеварения. Описаны случаи апластической анемии у жителей Хиросимы и Нагасаки, перенесших острое лучевое поражение после взрыва атомной бомбы.
Патогенез анемии до конца неизвестен. Считается, что при апластических анемиях имеет место дефицит частично детерминированных (плюрипотентных) стволовых клеток (КОЕ-ГЭММ) в результате их некроза или апоптоза при действии повреждающих факторов, потери способности к пролиферации, патологии гемопоэзиндуцирующего микроокружения (с нарушением процессов не только образования, но и созревания СКК), образования аутоантител.
Апластические анемии могут быть тотальными, протекающими с редукцией одновременно красного и белого ростков кроветворения (анемия Фанкони, анемия Эстрена-Дамешека), и парциальными, с избирательным угнетением красного ростка кроветворения (анемия Блекфена-Даймонда, парциальная красно-клеточная аплазия). Для тотальной апластической анемии характерна панцитопения (низкое содержание всех форм клеток в крови), сочетаю-
щаяся с панмиелопатией (низкое содержание всех форм клеток в костном мозгу), для парциальной - дефицит эритроидных клеток в крови и костном мозгу.
Заболевание чаще начинается постепенно, в крови отмечается снижение содержания гемоглобина (до 30-20 г/л), эритроцитов, ретикулоцитов. Анемия, как правило, нормохромная, макроцитарная. Характерно ускорение СОЭ до 30-50 мм/ч. При тотальной форме аплазии лейкопения сопровождается абсолютной нейтропенией, относительным лимфоцитозом. Содержание тромбоцитов уменьшается до (60-30)-109/л и ниже, удлиняется время кровотечения, развивается геморрагический синдром.
В костном мозгу выявляются резкое снижение количества ядросодержащих элементов, торможение созревания клеток, почти полное исчезновение мегакариоцитов. При парциальной апластической анемии гранулоцитарный и мегакариоцитарный ростки сохраняются без существенных изменений.
Среди анемий, связанных с нарушенным кровообразованием, выделяют также анемии хронических заболеваний (гипохромные) и анемии, ассоциированные с заболеваниями внутренних органов (нормохромные). При этом выраженность анемического синдрома прямо пропорциональна продолжительности и тяжести основного заболевания, признаки которого чаще превалируют над гипоксическими проявлениями анемии.
Анемия хронических заболеваний (АХЗ) развивается при инфекционно-воспалительных заболеваниях (менингит, пневмония, туберкулез, инфекционный эндокардит, остеомиелит, сифилис, ВИЧ-инфекция, грибковые инфекции и др.), системных заболеваниях соединительной ткани (ревматизм, ревматоидный артрит, системная красная волчанка, дерматомиозит и др.) и опухолях (множественная миелома, неходжкинские лимфомы, лимфогранулематоз, рак легкого, рак молочной железы, рак яичников и др.). К АХЗ не относятся анемии, возникающие при заболеваниях эндокринной системы, печени и почек (даже если они являются хроническими). В патогенезе АХЗ (рис. 14-8) основное значение имеют: нарушение метаболизма железа, недостаточная продукция эритропоэтина, угнетение эритропоэза, укорочение продолжительности жизни эритроцитов (в среднем до 80 дней). Развитие АХЗ у больных со злокачественными новообразованиями, наряду с цитокинопосредованным угнетением эритропоэза, связано также с метастатическим поражением костного мозга и миелофиброзом.
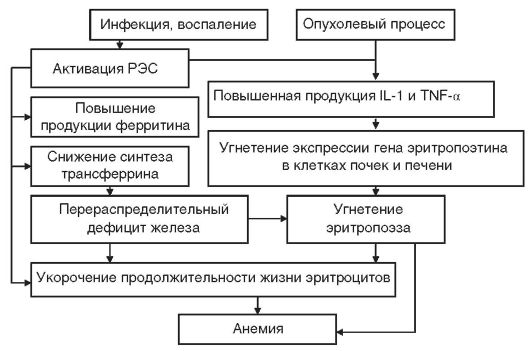 Рис. 14-8. Схема патогенеза анемии хронических заболеваний. РЭС - ретикулоэндотелиальная система
Рис. 14-8. Схема патогенеза анемии хронических заболеваний. РЭС - ретикулоэндотелиальная система
Анемии, ассоциированные с заболеваниями внутренних органов, включают анемии при эндокринных заболеваниях, заболеваниях печени и почек.
К числу анемий при эндокринных заболеваниях относятся анемии при заболеваниях щитовидной и паращитовидных желез, надпочечников, половых желез, гипопитуитаризме и др., в основе патогенеза которых лежит депрессия эритропоэза при дефиците или, напротив, гиперсекреции ряда гормонов. В частности, такие гормоны, как тироксин, кортизол, тестостерон в очень высоких концентрациях вызывают угнетение пролиферативной активности эритроидных прекурсоров. В случае сгущения крови в результате дегидратации (при надпочечниковой недостаточности, гипотиреозе) диагностика анемии может быть затруднена.
К анемиям при заболеваниях печени относятся анемии, возникающие при диффузных поражениях органа (циррозе, хроническом гепатите, гемохроматозе и др.). Патогенез анемии при заболеваниях печени отличается многообразием патогенетических факторов, что определяется особенностями патогенеза основного заболевания. Выделяют следующие механизмы развития анемии:
• угнетение процессов кроветворения в костном мозгу вследствие прямого токсического влияния на клетки-предшест-
венницы гемопоэза алкоголя (при алкогольном поражении печени) и эндогенных токсинов (при нарушениях обезвреживающей и клиренсной функции печени), при нарушениях метаболизма железа и депонирования витамина В12 и фолиевой кислоты в пораженной печени;
• укорочение продолжительности жизни эритроцитов в результате прямого повреждающего действия токсических продуктов экзогенного (алкоголь) и эндогенного (при эндотоксемии) происхождения, гиперспленизма, при нарушениях внутриклеточного метаболизма эритроцитов (например, в связи с дефицитом в клетках НАДФ+) и их способности к деформации (вследствие патологии клеточной мембраны при изменениях фракционного состава фосфолипидов, снижении содержания сиаловых кислот);
• кровотечения из расширенных вен желудочно-кишечного тракта (при циррозе печени), носовые, геморроидальные и иной локализации (при формирующейся недостаточности синтеза факторов свертывания крови вследствие нарушений белкового обмена).
В подавляющем большинстве случаев при заболеваниях печени регистрируется нормохромная нормоцитарная анемия, при присоединяющемся дефиците железа - микроцитарная нормоили гипохромная, при недостаточности витамина В12 и фолиевой кислоты, метастазах рака желудка в печень - макроцитарная анемия нормоили гиперхромного типа.
Анемия при заболеваниях почек может выявляться у больных острым гломерулонефритом, интерстициальным нефритом, хронической почечной недостаточностью. Патогенез анемии при заболеваниях почек определяется снижением продукции эритропоэтина клетками юкстагломерулярного аппарата, депрессией кроветворения в костном мозгу (в результате нарушения пролиферативной активности эритроидных клеток, торможения процессов синтеза гема) и сокращением срока жизни эритроцитов (до 40-50 дней) при действии токсических продуктов азотистого обмена.
Анемии вследствие повышенного кроверазрушения (гемолитические анемии)
В обширную группу гемолитических анемий входят разнообразные заболевания, объединенные лишь одним общим признаком - укорочением продолжительности жизни эритроцитов. Механизм развития
этого вида анемии связан с повышенным разрушением (гемолизом) эритроцитов периферической крови или (значительно реже) с гибелью созревающих клеток эритроидного ряда в костном мозгу.
В норме высвобождающийся при физиологическом гемолизе (до 10% эритроцитов в сутки) гемоглобин полностью связывается с белком плазмы - гаптоглобином, который не проникает через гломерулярный фильтр, что препятствует потере железа с мочой. Образовавшийся комплекс гемоглобина с гаптоглобином захватывается и разрушается макрофагами - клетками ретикулоэндотелиальной системы (РЭС). Высвобождающийся при этом гем метаболизируется в неконъюгированный (непрямой) билирубин, который выделяется из макрофагов, связывается в кровотоке с альбумином (с образованием альбумин-билирубинового комплекса) и доставляется в печень, где преобразуется в конъюгированный (прямой) билирубин, выделяемый из желчи в кишечник. В кишечнике под влиянием микрофлоры прямой билирубин восстанавливается до уробилиногена, далее разрушающегося в печени с образованием моно- и дипироллов, и стеркобилиногена (пигмент кала), основная часть которого выделяется с калом, незначительная (после всасывания из толстой кишки через геморроидальные вены в кровь) - с мочой.
Гемоглобинсвязывающая способность гаптоглобина составляет 100 мг% (100 мг в 100 мл крови). При выраженном гемолизе, превышающем резервную гемоглобинсвязывающую емкость гаптоглобина, свободный гемоглобин поступает в почки. В почечных канальцах осуществляется реабсорбция гемоглобина с дальнейшим его окислением в эпителии канальцев до гемосидерина. Нагруженные гемосидерином клетки слущиваются и выделяются с мочой (гемосидеринурия). При гемоглобинемии свыше 125-135 мг% реабсорбция гемоглобина оказывается недостаточной, что приводит к появлению в моче свободного гемоглобина (гемоглобинурии).
Гемолиз эритроцитов при гемолитических анемиях может происходить внутриклеточно (так же как и физиологический гемолиз), или непосредственно в сосудах. В связи с этим выделяют 2 типа патологического гемолиза:
1. Внутриклеточный гемолиз - разрушение «маркированных» иммуноглобулином (Ig) G эритроцитов в РЭС при наследственной патологии мембраны эритроцитов, нарушениях активности ферментов, синтеза гемоглобина, при несовместимости по эритроцитарным антигенам между матерью и плодом и при гемотрансфузиях.
2. Внутрисосудистый гемолиз - комплементозависимый лизис «маркированных» IgM (реже IgG) эритроцитов непосредственно в кровотоке (в сосудах) при действии каких-либо внешних факторов, которые вызывают прямое или опосредованное повреждение клеток. Причиной этого может быть разрушение мембраны эритроцитов вследствие механической травмы (при окклюзии сосудов, гемодиализе, протезах клапанов сердца и др.), под влиянием физических (ионизирующая радиация, высокая температура), токсических (при действии экзо- и эндотоксинов), инфекционных и иммунных (при образовании антаэритроцитарных аутоантител) патологических факторов.
В результате повышенного гемолиза эритроцитов в крови накапливается большое количество непрямого билирубина, что приводит к развитию желтухи. Помимо этого главным признаком повышенного внутриклеточного гемолиза является увеличение селезенки (спленомегалия), в случаях внутрисосудистого разрушения эритроцитов ведущим симптомом становится появление гемоглобина в моче (гемоглобинурия), что сопровождается изменением ее окраски вплоть до черного цвета (табл. 14-6).
Таблица 14-6. Дифференциальные признаки внутрисосудистого и внутриклеточного гемолиза
Признаки гемолиза | Виды гемолиза | |
внутрисосудистый | внутриклеточный | |
Локализация гемолиза | Сосуды | РЭС |
Локализация гемосидероза | Канальцы почек | Селезенка, печень, костный мозг |
Желтушность кожи и слизистых оболочек | Умеренная | Выраженная |
Увеличение размеров печени и селезенки | Незначительное | Значительное |
Ведущие лабораторные признаки | Нормохромная анемия, ретикулоцитоз, гиперсидеремия, гиперплазия эритроидного ростка в костном мозгу | |
Гемоглобинемия Гемоглобинурия Гемосидеринурия | Гипербилирубинемия Повышенное содержание стеркобилиногена в кале и уробилиногена в моче | |
Все формы малокровия, связанные с повышенной гибелью эритроцитов периферической крови, относятся к группе регенераторных анемий с нормобластическим типом эритропоэза.
Наследственные гемолитические анемии. Данные анемии делят на три большие группы:
1. Мембранопатии эритроцитов с характерной морфологией клеток (сфероцитоз, эллиптоцитоз, стоматоцитоз, акантоцитоз и др.).
2. Энзимопенические (ферментопенические) анемии, или эритроцитарные энзимопатии (связанные с дефицитом ферментов пентозофосфатного цикла - глюкозо-6-фосфатдегидрогеназы и др.; связанные с дефицитом ферментов гликолиза - пируваткиназы и др.; связанные с нарушением метаболизма нуклеотидов - дефицит пиримидин-5-нуклеотидазы и др.).
3. Гемоглобинопатии («качественные» гемоглобинопатии - HbS, С, Д, Е и др. и «количественные» гемоглобинопатии - талассемии).
Мембранопатии. Основным патогенетическим звеном гемолитических анемий этой группы является генетический дефект белковолипидной структуры мембраны эритроцитов, что приводит к изменению формы и эластичности клеток. В результате нарушается способность эритроцитов деформироваться в узких участках кровотока, в частности при переходе из межсинусных пространств селезенки в синусы. В процессе циркуляции эритроциты постепенно теряют оболочку и в конечном счете разрушаются макрофагами РЭС. Из группы мембранопатий наиболее часто встречаемым заболеванием является наследственный микросфероцитоз (болезнь Минковского-Шоффара), в основе которого лежит наследственный дефект белков мембраны (анкирина, спектрина, белка полосы 3, 4.2), способствующий повышенной ее проницаемости для ионов натрия. Избыток натрия, а вместе с ним и воды увеличивает объем эритроцитов и придает им характерную шаровидную форму (сфероцитоз). Утрата части клеточной оболочки приводит к уменьшению размеров эритроцитов и образованию микросфероцитов (микросфероцитоз). В результате прогрессирующей фрагментации мембраны после двух-трех последующих прохождений через селезеночные синусы микросфероциты подвергаются внутриклеточному гемолизу. Одной из причин укорочения продолжительности жизни микросфероцитов (до 7-14 дней) служит также истощение их ферментных ресурсов (расход АТФ, глюкозы) в процессе удаления из клеток избытка воды.
Аномалия передается с аутосомной хромосомой и наследуется по доминантному типу, т.е. болезнь проявляется не только у гомозигот, но и у гетерозигот. Гемолитические кризы возникают при воздействии холода, эмоциональном стрессе, беременности, инфекциях. Центральное место в клинической картине занимают три ведущих симптома (триада Шоффара): желтуха, бледность кожи и слизистых, спленомегалия (у 75-80% больных).
Энзимопатии обусловлены наследственным дефицитом ряда ферментов эритроцитов. В мире насчитывается несколько сотен миллионов человек (примерно 1/20 человечества) - носителей наследственного дефицита глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ). При недостатке Г-6-ФДГ блокируется реакция окисления глюкозо-6-фосфата в пентозофосфатном цикле, вследствие чего уменьшается образование восстановленной формы глутатиона, предохраняющего SH-группы глобина и мембраны эритроцитов от повреждающего действия различного рода окислителей. Это сопровождается снижением устойчивости эритроцитов к действию активных форм кислорода, окислительной денатурацией гемоглобина и белков мембраны эритроцитов с последующим внутрисосудистым гемолизом клеток.
Описано около 90 различных мутантных форм Г-6-ФДГ, из которых основными являются европейская форма дефицита (активность фермента в пределах 90% от нормы), африканская (10-15%) и средиземноморская (менее 1%). Дефицит Г-6-ФДГ наследуется как сцепленный с Х-хромосомой признак, в связи с чем заболевают в основном мужчины.
Клинически носительство дефицита Г-6-ФДГ проявляется острыми гемолитическими кризами при приеме некоторых лекарств, обладающих окислительными свойствами: хинин, ПАСК, сульфаниламиды, производные салициловой кислоты и др., при употреблении в пищу конских бобов и стручковых растений (фавизм), а также на фоне заболевания вирусным гепатитом или гриппом. В период гемолитического криза у больных выявляются признаки внутрисосудистого гемолиза - повышение температуры тела, бледность, умеренная желтушность кожи и склер, головная боль, рвота, диарея. Вследствие гемоглобинурии возможно развитие острой почечной недостаточности.
Гемоглобинопатии (гемоглобинозы) связаны с наследственным нарушением синтеза гемоглобина. «Качественные» гемоглобинопатии сопровождаются изменением первичной структуры молекулы
гемоглобина, «количественные» гемоглобинопатии характеризуются нарушением количественного соотношения HbA и HbF в крови из-за недостаточности образования отдельных полипептидных цепей глобина. Как и носительство дефицита Г-6-ФДГ, наследственные гемоглобинопатии относятся к числу наиболее распространенных в человеческой популяции генетических аномалий. Среди известных форм гемоглобинопатий наибольшее значение в практическом отношении представляют гемоглобиноз S (серповидноклеточная анемия) и талассемия.
Гемоглобиноз S. Заболевание возникает в связи с наследованием патологического гемоглобина S, в котором гидрофильная глутаминовая кислота в 6-м положении β-цепи глобина замещена на гидрофобный валин. Это приводит к смене электрического заряда и полимеризации гемоглобина в условиях гипоксии, снижению его растворимости с образованием тактоидов (веретенообразных остроконечных кристаллов), которые растягивают оболочку эритроцитов. В результате клетки приобретают форму «серпа», теряют пластичность, повышают вязкость крови, замедляют кровоток, вызывают стаз. Стаз, в свою очередь, приводит к развитию гипоксемии, еще более повышая уровень «серпления» эритроцитов. Острыми концами серповидные эритроциты могут повреждать другие измененные и неизмененные эритроциты:, что сопровождается внутрисосудистым гемолизом. Часть серповидных эритроцитов разрушается в селезенке. Средняя продолжительность жизни эритроцитов при серповидно-клеточной анемии не превышает 17 дней.
Тяжелая анемия проявляется лишь у гомозиготных по HbS носителей. Усиление образования серповидных эритроцитов с развитием гемолитического криза отмечается при действии низких температур, патологических состояниях, сопровождающихся ацидозом, инфекциях, дегидратации, лихорадке, голодании, заболеваниях легких, в условиях гипоксии. Вследствие компенсаторной спленомегалии у ряда пациентов в силу неизвестных причин вероятна массивная секвестрация эритроцитов в селезенке, что может стать причиной развития гипотензии и внезапного летального исхода. У гетерозигот заболевание протекает, как правило, бессимптомно. Поскольку серповидные эритроциты являются непригодными для жизнедеятельности малярийных плазмодиев, люди - носители аномального HbS - обладают резистентностью к малярии.
Талассемия (средиземноморская анемия) связана со снижением или отсутствием синтеза α-, β-, δ- или γ-цепей глобина. В за-
висимости от этого различают α-, β-, δ- и γ-талассемию. Чаще всего встречается нарушение синтеза β-глобиновых цепей - β-талассемия. В этом случае содержание HbA1 (α2β2) уменьшается, а уровень HbF (α2γ2) и HbA2 (α2δ2), напротив, возрастает. Недостаточный синтез β-цепей приводит к избыточному образованию α-цепей. Лишние α-цепи способствуют появлению нестабильного гемоглобина, который преципитирует и выпадает в эритроците в виде «телец включения», придавая им форму мишеней. Кроме того, образующиеся в избытке α-цепи вступают в соединение с SH-группами мембраны и повышают ее проницаемость, нарушаются процессы ассимиляции железа и синтеза гемоглобина. Это обусловливает раннюю гибель эритроцитов в результате внутриклеточного гемолиза с развитием гипохромной анемии.
Развернутая картина тяжелой гемолитической анемии возникает при гомозиготном наследовании нарушения синтеза β-цепей - болезни Кули, проявляющейся физическим и умственным недоразвитием, бледной желтушной окраской кожи с признаками гемосидероза, придающего коже зеленовато-коричневый оттенок, деформацией костей черепа (башенный череп, увеличение верхней челюсти, нарушение прикуса; на рентгенограмме - расширение костно-мозгового канала трубчатых костей, поперечная исчерченность плоских костей черепа - игольчатый периостоз), язвами нижних конечностей, выраженной гепато- и спленомегалией.
Приобретенные гемолитические анемии. Среди заболеваний этой группы выделяют иммунные гемолитические анемии и анемии, связанные с воздействием прямых гемолизинов и других повреждающих факторов.
Иммунные гемолитические анемии. Данные анемии характеризуются образованием антител, действие которых направлено против антигенов, находящихся на поверхности эритроцитов.
Аутоиммунные гемолитические анемии (АИГА) возникают в результате образования аутоантител к поверхностным антигенам эритроцитов, что сопровождается внутриклеточным или внутрисосудистым гемолизом. Выработка антиэритроцитарных аутоантител может быть связана с изменением антигенной структуры мембраны эритроцитов в результате воздействия различных повреждающих факторов либо обусловлена нарушениями в самой иммунокомпетентной системе больного. В основе патологического процесса большинства форм АИГА лежит срыв иммунологической толерантности. Считается, что это возникает лишь в случаях
воздействия малых доз толерогена и при дисфункции Т-клеток в условиях нормального функционирования В-звена иммунитета. Антиэритроцитарные аутоантитела могут уничтожающе действовать на эритроциты крови, эритронормобласты костного мозга и даже на самые ранние клетки-предшественницы эритроцитов периферической крови. По серологическому типу выделяют АИГА с неполными тепловыми агглютининами, с тепловыми гемолизинами, с полными холодовыми агглютининами и двухфазными гемолизинами (антителами типа Доната-Ландштейнера).
АИГА, вызываемые тепловыми аутоантителами, развиваются либо без видимых причин (идиопатическая форма), либо на фоне различных заболеваний - лимфогранулематоза, хронического лимфолейкоза, системной волчанки (симптоматическая форма), а также при приеме некоторых лекарств (пенициллин) в результате образования IgG (реже IgA и IgM) к эритроцитарному Rh-антигену с последующей деструкцией эритроцитов в селезенке (внутриклеточный гемолиз).
Действие холодовых аутоантител проявляется при температуре ниже 32 °С. АИГА, вызываемые холодовыми антителами, могут быть первичными и вторичными, развивающимися при инфекциях (микоплазменная, цитомегаловирусная), коллагенозах (ревматоидный артрит), гемобластозах (хронический лимфолейкоз, макроглобулинемия Вальденстрема). В основе патогенеза лежат активация комплемента IgM к I-антигену эритроцитов и внутрисосудистый лизис эритроцитов в мелких сосудах отдаленных от сердца участков тела (в пальцах стоп, кистей), где температура ниже, чем в других участках тела. Период гемолитического криза характеризуется синдромом Рейно - фазовыми изменениями кожных покровов пальцев рук и ног с последовательным их побледнением, цианозом и гиперемией, обусловленными приступообразным спазмом артерий и артериол в ответ на холодовое воздействие.
Примером АИГА с двухфазными антителами является пароксизмальная холодовая гемоглобинурия, идипатическая и вторичная (при вирусных инфекциях, третичном сифилисе). В основе развития анемии лежит двухфазная реакция с участием IgG к Р-антигену эритроцитов (антител Доната-Ландштейнера). В первой фазе IgG при охлаждении организма связываются с эритроцитами и фиксируют комплемент, во второй фазе (при 37 °С) происходит активация комплемента с индукцией внутрисосудистого гемолиза. Гемолитический криз развивается через несколько часов
после переохлаждения при согревании больного и проявляется лихорадкой, ознобом, болями в животе и поясничной области, тошнотой, рвотой, потемнением (почернением) мочи.
Гетероиммунные (гаптеновые) гемолитические анемии формируются в связи с появлением на поверхности эритроцитов больного нового антигена (гаптена). Гаптенами могут служить лекарственные препараты и вирусы.
Изоиммунные гемолитические анемии характеризуются тем, что антитела против антигенных детерминант эритроцитов попадают в организм больного извне. Примером являются посттрансфузионные гемолитические анемии и гемолитическая болезнь новорожденного.
Гемолитическая болезнь новорожденного (ГБН) или эритробластоз плода развивается в результате несовместимости матери и плода по антигенам эритроцитов системы Rh (D) (у Rh- положительных детей от Rh-отрицательных матерей) или по антигенам эритроцитов системы АВ0 (у детей с группой крови А, В или АВ, матери которых имеют группу крови 0). Первая беременность Rh-отрицательной матери Rh-положительным плодом обычно протекает нормально. Во время родов происходит иммунизация матери антигенами эритроцитов плода с выработкой антиэритроцитарных антител (анти Rh (D)-IgG), которые в период второй беременности Rh-положительным плодом фиксируются на эритроцитах плода и обусловливают гибель эритроцитарных клеток путем внутриклеточного гемолиза с развитием эритробластоза плода. Основными симптомами ГБН являются желтуха, гепато- и спленомегалия, в тяжелых случаях - отеки, асцит (из-за недостаточности кровообращения). Наиболее опасным симптомом анемии служит «ядерная желтуха» с признаками поражения нервной системы вследствие токсического действия непрямого билирубина, к которым относятся нистагм, судорожные подергивания, крик высокой тональности. Встречаются случаи мертворождения.
Трансиммунные гемолитические анемии развиваются при проникновении в организм новорожденного антиэритроцитарных антител матери, страдающей аутоиммунной гемолитической анемией.
Анемии при действии прямых гемолизинов и других повреждающих факторов. Эта группа анемий объединяет гемолитические состояния, при которых полноценные в морфофункциональном отношении эритроциты разрушаются под действием гемолитических
(фенилгидразин, свинец, бензол, мышьяковистый водород, анилиновые красители, змеиный и грибной яды и др.), бактериальных (токсины гемолитического стрептококка, стафилококка и др.), паразитарных (малярия, бабезиоз) и других факторов. Патогенез этих анемий различен - разрушение мембраны эритроцитов, истощение их ферментных систем и т.д.
14.2.2. Эритроцитозы
Под эритроцитозом понимают увеличение содержания эритроцитов в крови. Выделяют две группы эритроцитозов: относительные (увеличение содержания эритроцитов и гемоглобина в единице объема крови без повышения их абсолютного количества) и абсолютные (увеличение абсолютного количества эритроцитов в крови).
Относительные эритроцитозы имеют, как правило, преходящий характер. Они подразделяются на:
• гемоконцентрационные - возникают при уменьшении объема плазмы (сгущении крови) вследствие дегидратации организма (при неукротимой рвоте, диарее, обильном потоотделении, ожоговой болезни и др.);
• стресс-эритроцитозы - развиваются за счет «выброса» эритроцитов из органов-депо (при стресс-реакции, в сосудисторефлекторную фазу компенсаторных реакций на фоне острой кровопотери, при синдроме Гайсбека (или ложной полицитемии курильщиков), гипертензии и др.).
Абсолютные эритроцитозы обусловливаются увеличением эритропоэтической функции костного мозга.
1. На фоне повышенной продукции эритропоэтина в организме:
• гипоксические - развиваются в результате повышенной продукции эритропоэтина клетками юкстагломерулярного аппарата почек в ответ на долговременную гипоксию: при снижении парциального давления кислорода в воздухе (у людей, занимающихся кессонными работами, при высокогорной болезни и др.), при заболеваниях органов дыхания (бронхиальная астма, эмфизема, интерстициальная пневмония, диффузный пневмосклероз и др.), патологии сердечно-сосудистой системы (пороки сердца, гипертрофическая кардиомиопатия, геморрагическая телеангиэктазия и др.), локальной ишемии почек (киста почек, гидронефроз, повреждение почечных сосудов и др.);
• опухолевые - развиваются за счет продукции эритропоэтина опухолевыми клетками: при феохромоцитоме, гипернефроме, гепатоцеллюлярной карциноме, раке желудка и др.
2. При нормальной продукции эритропоэтина клетками юкстагломерулярного аппарата почек - миелопролиферативные, возникающие при эритремии (или истинной полицитемии) (см. раздел 14.3.6) за счет опухолевой гиперплазии эритроидного ростка вследствие дефекта клетки-предшественницы миелопоэза.
К группе абсолютных эритроцитозов относятся также эндокринные эритроцитозы, возникающие вследствие способности ряда гормонов оказывать прямое или опосредованное (через усиление продукции эритропоэтина клетками юкстагломерулярного аппарата почек) стимулирующее влияние на эритропоэз: при тиреотоксикозе, синдроме Иценго-Кушинга, гиперальдостеронизме, гиперандрогенемии и др.
Описаны наследственные (семейные) эритроцитозы.
14.3. ИЗМЕНЕНИЯ КОЛИЧЕСТВЕННОГО И КАЧЕСТВЕННОГО СОСТАВА ЛЕЙКОЦИТОВ
Общее количество лейкоцитов в крови здорового взрослого человека в условиях покоя и натощак колеблется от 4,0-109/л до 9Т09/л (4000-9000 в 1 мкл). Нарушения количественного состава лейкоцитов в периферической крови могут носить реактивный (временный) характер (лейкоцитозы, лейкемоидные реакции, лейкопении) и иметь опухолевую природу (лейкозы, лимфомы). В ряде случаев они сопровождаются изменением морфологических и функциональных свойств лейкоцитов. В свою очередь, качественные дефекты лейкоцитов могут формироваться не только на фоне изменений количества лейкоцитов, но и носить автономный характер. Их идентификация имеет решающее значение в дифференциальной диагностике отдельных видов патологии системы крови.
14.3.1. Патологические формы лейкоцитов
Патологические формы лейкоцитов подразделяют на регенеративные (обнаруживаемые в норме только в костном мозгу) и дегенеративные (деструктивно измененные) формы (табл. 14-7, рис.
14-9, 14-10, см. цв. вклейку). Дегенеративные изменения лейкоцитов могут быть следствием прямого или опосредованного повреждающего воздействия болезнетворных факторов на зрелые клетки крови, а также дизрегуляции кроветворной функции костного мозга в результате патологии гемопоэтического микроокружения и ранних клеток-предшественниц гемопоэза. Признаки дегенерации в лейкоцитах могут обнаруживаться при инфекциях, воспалении, в условиях экзогенной и эндогенной интоксикации, при ожогах, действии ионизирующего излучения, недостаточности витамина В12 и фолиевой кислоты, агранулоцитозе, лейкемоидных реакциях, лейкозах, миелодиспластическом синдроме, на фоне терапии цитостатическими препаратами, глюкокортикоидами и др.
Таблица 14-7. Виды дегенеративных изменений лейкоцитов
Вариант патологических изменений лейкоцитов | Морфологическая характеристика |
Анизоцитоз | Уменьшение размеров (микроформы) и увеличение размеров клеток (макрополициты - гигантские лейкоциты) |
Патология ядра: | |
гиперсегментация | Увеличение числа сегментов в ядрах нейтрофильных гранулоцитов (свыше 5 при норме 2-5), эозинофилов (свыше 3 при норме 2-3) |
гипосегментация | Нарушение сегментации ядра зрелых гранулоцитов - уменьшение количества сегментов или полное отсутствие сегментации (ядра округлые или в форме эллипса, боба, земляного ореха, гимнастической гири, пенсне) |
пикноз Уплотнение хроматина | |
рексис | Распад ядра на отдельные части, исчезновение межсегментарных «нитей» в зрелых гранулоцитах |
фрагментация | Образование фрагментов ядерного хроматина (микроядер) |
лизис | Растворение ядерной оболочки |
хроматинолиз | Разжижение хроматина |
вакуолизация | Бесцветные пятна («дырки») в хроматине |
голые ядра лимфоцитов | Лимфоциты без цитоплазмы |
Окончание табл. 14-7
формы Риддера | Двуядерные лимфоциты |
тени Боткина-Гумпрехта | Раздавленные ядра лимфоцитов |
Патология цитоплазмы: | |
«истощение» зернистости | Дефицит или отсутствие специфических гранул |
токсогенная зернистость | Крупные грубые базофильные гранулы |
азурофильная зернистость | Множественные, перекрывающие ядра клеток или единичные крупные азурофильные гранулы в цитоплазме зрелых лейкоцитов |
вакуолизация | Бесцветные пятна («дырки») в цитоплазме |
тельца Князькова-Деле | Округлые или овальные аморфные включения голубого цвета |
палочки Ауэра | Палочки вишневого цвета (слипшаяся азурофильная зернистость) |
Кроме того, дефекты: морфологии лейкоцитов могут иметь наследственную природу. Примером является аутосомно-доминантная аномалия Пельгера-Хюэта, характеризующаяся гипосегментацией ядер нейтрофилов вследствие дефекта генетического контроля постмитотической стадии созревания гранулоцитов с формированием псевдомиелоцитов (нейтрофилов с округлыми крупноглыбчатыми, пикнотичными ядрами), клеток с ядрами в форме «гири», «пенсне», «палочки», двусегментированных клеток. К наследственным сочетанным дефектам морфологии, функции и количества лейкоцитов относится аутосомно-рецессивный синдром Чедиака-Хигаси. При этом заболевании в крови на ранних стадиях отмечается нейтропения, в последующем - панцитопения. В нейтрофилах, эозинофилах, моноцитах, лимфоцитах обнаруживаются гигантские гранулы (диаметром более 5 мкм), проявляющие положительную реакцию на пероксидазу и обусловливающие нарушение хемотаксиса и микробицидной функции клеток. Имеет место также дефект плотных гранул в тромбоцитах. В костном мозгу - признаки гиперплазии гранулоцитарного ростка.
14.3.2. Функциональные дефекты лейкоцитов
Нарушения функциональных свойств лейкоцитов могут быть наследственными и приобретенными. Они связаны, главным об-
 Рис. 14-11. Патогенетические факторы дисфункции нейтрофилов
Рис. 14-11. Патогенетические факторы дисфункции нейтрофилов
разом, с дефектами нейтрофильных гранулоцитов вследствие нарушения их маргинации, адгезии, миграции и микробицидных свойств (рис. 14-11).
14.3.3. Лейкоцитозы
Лейкоцитоз - увеличение общего количества лейкоцитов (более 9,0-109/л) или числа их отдельных морфологических форм. Лейкоцитоз носит временный характер и исчезает вместе с причиной,
его обусловившей; это не самостоятельное заболевание, а реакция крови на соответствующие этиологические факторы. В зависимости от природы этих факторов различают физиологические и патологические лейкоцитозы.
К физиологическим лейкоцитозам относят алиментарный (пищеварительный), развивающийся через 2-3 ч после приема пищи; миогенный - при мышечном напряжении; эмоциональный - вследствие психического возбуждения, а также лейкоцитоз новорожденных (в течение первых двух дней жизни), беременных (развивающийся с 5-6-го мес беременности) и рожениц (отмечающийся ко второй неделе после родов). Кратковременный физиологический лейкоцитоз имеет перераспределительный характер и связан с мобилизацией в кровяное русло резерва зрелых лейкоцитов из органов-депо; длительный (новорожденных, беременных) - обусловлен активацией процессов образования лейкоцитов в костном мозгу.
Среди патологических лейкоцитозов различают: инфекционный - при пневмонии, менингите, скарлатине и ряде других инфекционных заболеваний; воспалительный (особенно при гнойных воспалительных процессах) - при различного рода травмах: повреждении электрическим током, действии высокой и низкой температуры и т.д.; токсогенный - при действии вредных веществ как экзогенного (бензол, мышьяковистый водород, анилин и др.), так и эндогенного происхождения (при уремии, диабетической коме); постгеморрагический - наступающий после острых кровопотерь; новообразовательный - при распаде опухолей; лейкемический - при острых и хронических лейкозах. Механизм их возникновения связан с повышением лейкопоэтической функции костного мозга, и лишь один вид патологического лейкоцитоза - центрогенный (при шоковых состояниях, эпилепсии, агонии; послеоперационный) имеет перераспределительный характер.
В зависимости от увеличения содержания тех или иных видов лейкоцитов в крови различают нейтрофильный лейкоцитоз, эозинофилию, базофилию, лимфоцитоз и моноцитоз.
Нейтрофильный лейкоцитоз (нейтрофилия) - увеличение содержания нейтрофилов свыше 70% в гемограмме. Отмечается при острых инфекционных заболеваниях, гнойных воспалительных процессах, инфаркте миокарда, укусах ядовитых насекомых, после острой кровопотери, а также при алиментарном и эмоциональном физиологических лейкоцитозах. Важное практическое значение имеет определение степени ядерного сдвига в лейкоцитарной фор-
муле. По этому признаку различают шесть видов нейтрофильного лейкоцитоза (табл. 14-8):
1) без ядерного сдвига - при острой кровопотере, стрессреакции;
2) с гипорегенеративным ядерным сдвигом влево - при легком течении ряда инфекций и воспалений;
3) с регенеративным ядерным сдвигом влево - при гнойносептических процессах;
4) с гиперрегенеративным ядерным сдвигом влево - признак неблагоприятного течения инфекционных и гнойно-септических заболеваний;
5) с дегенеративным ядерным сдвигом влево - показатель угнетения функциональной активности костного мозга, может иметь место при тяжелом течении инфекционных заболеваний, при эндогенной интоксикации и т.д.;
6) с дегенеративным ядерным сдвигом вправо - при лучевой болезни, злокачественной анемии Аддисона-Бирмера; в ряде случаев у практически здоровых людей.
Таблица 14-8. Классификация нейтрофилий в зависимости от характера и
степени ядерного сдвига Варианты нейтрофилии Характеристика | |
Без сдвига | Увеличение общего числа нейтрофилов (>70%) за счет сегментно-ядерных форм клеток (>65%) |
Со сдвигом влево: | |
гипорегенераторным | Увеличение общего числа нейтрофилов (>70%) за счет сегментно-ядерных (>65%) и палочкоядерных (>5%) форм клеток |
регенераторным | Увеличение общего числа нейтрофилов (>70%) за счет сегментно-ядерных (>65%), палочкоядерных (>5%) форм клеток и метамиелоцитов (>0,5%) |
гиперрегенераторным | Увеличение общего числа нейтрофилов (>70%) за счет сегментно-ядерных (>65%), палочкоядерных (>5%) форм клеток, метамиелоцитов (>0,5%) и более молодых клеток (миелоцитов, промиелоцитов, миелобластов). Анэозинофилия |
Окончание табл. 14-8
дегенеративным | Увеличение общего числа нейтрофилов (>70%) за счет сегментно-ядерных (>65%) и палочкоядерных (>5%) форм клеток, появление в крови нейтрофилов с признаками дегенерации (вакуолизация ядра и цитоплазмы, токсогенная зернистость, кариорексис и др.) |
Со сдвигом вправо | Увеличение общего числа нейтрофилов (>70%) за счет сегментно-ядерных форм клеток (>65%) на фоне повышенного (>5%) содержания в крови гиперсегментно-ядерных нейтрофилов (более 5 ядерных сегментов) |
Эозинофилия - увеличение содержания эозинофилов свыше 5% в гемограмме. По современным представлениям эозинофилия является своеобразной реакцией организма на чужеродные белки, гистамин, инвазию паразитами и связана с антитоксической, антигистаминной (посредством гистаминазы - фермента гранул эозинофилов), фагоцитарной (фагоцитоз иммунных комплексов) и противогельминтной (экзоцитоз личинок, разрушение миелина нервных волокон паразитов) функцией эозинофилов.
Развитие эозинофилии имеет место при различных аллергических заболеваниях и синдромах (бронхиальная астма, отек Квинке, крапивница и др.); при паразитарных заболеваниях (описторхоз, аскаридоз, лямблиоз и др.), некоторых кожных болезнях (псориаз, экзема), коллагенозах (ревматизм, дерматомиозит), гемобластозах (хронический миелолейкоз, лимфогранулематоз), некоторых эндокринопатиях (гипофизарная кахексия, микседема и др.), ряде инфекционных заболеваний (скарлатина, сифилис, туберкулез), при применении некоторых лекарственных препаратов (антибиотики, сульфаниламиды и др.); описаны также наследственные формы эозинофилии.
Базофилия (более 1% базофилов в гемограмме) - редкая форма лейкоцитоза, встречающаяся при анафилактических и реагиновых аллергических реакциях (крапивница, отек Квинке, пищевая и лекарственная аллергия и др.), что обусловливается способностью базофилов фиксировать IgE и IgG, высвобождать медиаторы гранул (факторы хемотаксиса нейтрофилов и эозинофилов, гепарин, гистамин, серотонин и др.). Базофилия обнаруживается также при
вакцинации, гемолитических анемиях, гемофилии, эндокринопатиях (сахарный диабет, микседема и др.), хроническом миелолейкозе.
Лимфоцитоз - увеличение содержания лимфоцитов свыше 45% в гемограмме. Физиологический лимфоцитоз характерен для детей первых 10 лет жизни, а также отмечается у вегетарианцев и после физических нагрузок (миогенный). В условиях патологии лимфоцитоз развивается при ряде инфекционных заболеваний (брюшной тиф, свинка, коклюш, малярия, бруцеллез, инфекционный мононуклеоз, туберкулез, сифилис и др.), что связано с формированием противоинфекционного иммунитета, а также при алиментарной дистрофии, бронхиальной астме и некоторых эндокринных расстройствах (евнухоидизм, микседема, акромегалия).
Моноцитоз - увеличение содержания моноцитов свыше 9% в гемограмме. Обнаруживается при персистентных бактериальных и вирусных инфекциях (туберкулез, инфекционный мононуклеоз, корь, краснуха и др.), воспалительных заболеваниях (неспецифический язвенный колит, спру, коллагенозах и др.), гемобластозах, раке молочной железы и яичников, после спленэктомии и др.
14.3.4. Лейкемоидные реакции
Лейкемоидные реакции - патологические реакции системы крови, характеризующиеся изменениями в периферической крови (увеличением общего количества лейкоцитов до 30-109/л и выше, появлением незрелых форм лейкоцитов), сходными с таковыми при лейкозах и исчезающими после купирования вызвавшего их первичного процесса. При этом клеточный состав костного мозга (в отличие от лейкозов) остается нормальным. Выделяют две большие группы лейкемоидных реакций: миелоидного и лимфатического (моноцитарно-лимфатического) типов (см. табл. 14-9). В свою очередь, лейкемоидные реакции миелоидного типа подразделяют на нейтрофильные лейкемоидные реакции (при инфекционновоспалительных заболеваниях, интоксикациях, опухолях) и так называемые большие эозинофилии крови (при паразитарных инвазиях, аллергических заболеваниях, коллагенозах и др.). Среди лейкемоидных реакций моноцитарно-лимфатического типа наиболее важной в практическом отношении является лейкемоидная реакция с картиной острого лимфобластного лейкоза при инфекционном мононуклеозе, при которой в периферической
крови обнаруживаются «атипичные мононуклеары» - трансформированные вирусом Эпштейна-Барр или другими инфекционными возбудителями (вирус простого герпеса, цитомегаловирус, Toxoplasma gondii и др.) мононуклеарные лейкоциты (лимфоциты, моноциты), сходные по морфологии с бластными клетками.
14.3.5. Лейкопении
Лейкопения - уменьшение общего количества лейкоцитов ниже 4,0-109/л. Наиболее часто развитие лейкопении связано с уменьшением абсолютного числа нейтрофилов (нейтропения). Лимфоцитопения может иметь место при лимфогранулематозе, пневмонии, сепсисе, коллагенозах и некоторых других заболеваниях, но редко является причиной лейкопении. Моноцитопения, эозинопения, хотя и имеют существенное диагностическое значение, но не отражаются на общем количестве лейкоцитов.
В основе патогенеза лейкопении (нейтропении) лежат три механизма: 1) угнетение лейкопоэтической функции костного мозга; 2) повышенное разрушение нейтрофилов; 3) перераспределение нейтрофилов.
Нейтропении, обусловленные угнетением лейкопоэтической функции костного мозга. Развитие их в основном связано:
1) с нарушением пролиферации и дифференцировки стволовых гемопоэтических клеток при «внутреннем» дефекте клетокпредшественниц грануломоноцитопоэза - потере способности их к дифференцировке в клетки нейтрофильного ряда при сохраняющейся способности к нормальной дифференцировке в эозинофильные, базофильные и моноцитарные клетки, при дефиците веществ, необходимых для деления и созревания кроветворных клеток (белки, аминокислоты, витамины В12, фолиевая кислота и др.), а также вследствие аутоиммунных механизмов, связанных с образованием антиКОЕ-ГМ антител и аутореактивных Т-лимфоцитов;
2) с разрушением клеток-предшественниц нейтрофилов в костном мозгу при действии токсических веществ и лекарственных препаратов;
3) с патологией гемопоэзиндуцирующего микроокружения, в том числе в случаях выпадения стимулирующей дифференцировку стволовых клеток функции Т-лимфоцитов (при аплазии тимуса), гипосекреции клетками ГИМ факторов роста (ГМ-КСФ, Г-КСФ, IL-3, М-КСФ и др.;
4) с уменьшением площади гранулоцитопоэза в результате замещения кроветворной ткани костного мозга опухолевой (при лейкозах и карцинозах - метастазах рака в костный мозг), фиброзной, костной, жировой тканью.
Кроме того, формирование данного рода нейтропений может быть обусловлено наследственным дефектом механизма обратной связи, контролирующего процесс образования, созревания нейтрофилов в костном мозгу и их элиминацию на периферию.
Нейтропении, обусловленные повышением разрушения нейтрофилов. Разрушение нейтрофилов в крови может происходить под влиянием антител типа лейкоагглютининов, которые образуются при переливании крови (особенно лейкоцитарной массы), при действии некоторых лекарственных препаратов, являющихся аллергенами-гаптенами (сульфаниламиды, амидопирин и др.), токсических факторов инфекционного происхождения (тяжелые инфекционные заболевания, обширные воспалительные процессы), при заболеваниях, сопровождающихся увеличением количества циркулирующих иммунных комплексов в крови (аутоиммунные заболевания, лимфомы, опухоли, лейкозы и др.). Причиной наследственных нейтропений этой группы может быть преждевременная гибель клеток вследствие цитогенетической аномалии (тетраплоидия). Наряду с этим нейтропения может развиваться вследствие повышенного разрушения циркулирующих нейтрофилов в селезенке при заболеваниях, сопровождающихся гиперспленизмом (коллагенозы, цирроз печени, гемолитическая анемия, болезнь Фелти и др.).
Нейтропения, связанная с перераспределением нейтрофилов, носит временный характер и, как правило, сменяется лейкоцитозом. Ее формирование отмечается при шоке, неврозах, острой малярии и некоторых других состояниях в результате скопления клеток в расширенных капиллярах органов-депо (легкие, печень, кишечник). Перераспределительная нейтропения может обусловливаться также избыточной адгезией нейтрофилов на эндотелиоцитах вследствие активации эндотелия с последующей миграцией гранулоцитов в ткани под влиянием IL-8. Данный механизм лежит в основе хронической идиопатической нейтропении, характеризующейся повышенным содержанием IL-8 и растворимых лейкоцитарных адгезивных молекул к эндотелию (sELAM, sICAM, sVCAM) в сыворотке крови.
Таблица 14-9. Варианты лейкемоидных реакций и их характеристика
Вариант ЛР | Причины развития | Картина крови |
1. Миелоидного типа | ||
Нейтрофильные: | ||
Псевдобластная | «Выход» из иммунного агранулоцитоза Первичный туберкулез Тяжелые токсикоинфекции (дифтерия, столбняк и др.) Сепсис | Появление большого числа бластоподобных клеток |
Промиелоцитарная | Появление большого числа типичных промиелоцитов | |
С картиной хронического миелолейкоза | Инфекции (бактериальные, вирусные, грибковые) Воспаление (хронические васкулиты, дерматиты, подагра, миозиты и др.) Интоксикация (при эндокринных расстройствах, нарушениях метаболизма, уремии, отравлениях) Злокачественные новообразования (рак молочной железы, почек, печени, легких) Лимфогранулематоз | Нейтрофилия с гиперрегенераторным ядерным сдвигом влево при нормальном содержании эозинофилов и базофилов, дегенеративные изменения нейтрофилов (токсогенная зернистость, кариопикноз) |
Большая эозинофилия крови | Паразитозы (филяриоз, лямблиоз, описторхоз и ДР-) Аллергические заболевания (бронхиальная астма, аллергический ринит, лекарственная аллергия при приеме антибиотиков, сульфаниламидов, ацетилсалициловой кислоты) | Увеличение числа эозинофилов (>15%) и изменение их морфологии (вакуолизация ядра, цитоплазмы) |
Продолжение табл. 14-9
Коллагенозы (ревматоидный артрит, узелковый периартериит, системная склеродермия, системная красная волчанка) Пристеночный фибропластический эндокардит (эндокардит Леффлера) Иммунодефицитные синдромы (синдром Вискотта-Олдрича, дефицит IgA) Злокачественные новообразования (рак щитовидной железы, желудка, гипернефроидный рак почки, лимфогранулематоз, лимфома Ходжкина, хронический миелолейкоз) Выделяют также идиопатические и наследственные формы | ||
2. Моноцитарно-лимфатического типа | ||
С картиной острого лимфобластного лейкоза | Инфекционный мононуклеоз (болезнь Филатова) | ОКЛ - 20-109/л и более, увеличение числа лимфоцитов и моноцитов, «атипичных мононуклеаров» (>10%), нейтропения |
Острый инфекционный лимфоцитоз | Энтеровирусная инфекция, вызванная вирусом Коксаки Болезнь кошачьих царапин Бактериальные инфекции (коклюш, иерсиниоз, туберкулез и др.) Протозойная инвазия (токсоплазмоз, малярия) | ОКЛ - 15-100? 109/л и более, лимфоцитоз (>60%) без изменений морфологии клеток, моноцитоз |
Окончание табл. 14-9
Стресс-лимфоцитоз | Сердечно-сосудистая патология (кардиоваскулярный коллапс, острая сердечная недостаточность, инфаркт миокарда, септический шок и др.) Реакции гиперчувствительности немедленного типа Хирургические вмешательства Травмы Эпилепсия | Кратковременный лимфоцитоз до 5-109/л и более |
Персистирующий лимфоцитоз | Ревматоидный артрит Злокачественные новообразования (тимома) Хронические воспалительные заболевания (саркоидоз, гранулематоз Вегнера и др.) Реакции гиперчувствительности замедленного типа Гипоспленизм Курение | Длительный лимфоцитоз до 3,8-109/л и более |
Реактивный моноцитоз | Инфекционно-воспалительные заболевания (туберкулез, хронический пиелонефрит, саркоидоз, спру) Злокачественные новообразования (рак молочной железы и яичников, лимфогранулематоз, миеломная болезнь) | Увеличение числа моноцитов (>0,8-109/л) |
Примечание. ОКЛ - общее количество лейкоцитов.
Агранулоцитоз - клинико-гематологический синдром, характеризующийся полным или почти полным отсутствием нейтрофильных гранулоцитов в крови. Условно за агранулоцитоз принимают состояние, при котором уровень гранулоцитов ниже 0,75-109/л и/или общее количество лейкоцитов менее 1-109/л. Иногда этим термином обозначают тяжелую нейтропению.
Наиболее часто развитие агранулоцитоза связано с приемом медикаментов (цитостатические препараты, амидопирин, аминазин, антитиреоидные средства, сульфаниламиды, некоторые антибиотики и др.). Во многих случаях этиологические факторы, приводящие к возникновению тяжелой гранулоцитопении, остаются неустановленными («генуинные», или идиопатические агранулоциозы).
По механизму развития агранулоцитозы подразделяют на миелотоксический и иммунный (см. табл. 14-10). В основе миелотоксического агранулоцитоза лежит угнетающее действие медикаментозных препаратов и других повреждающих факторов на пролиферативную активность гранулоцитарных элементов костного мозга, вследствие чего развивается гипоплазия гранулоцитопоэза; возможность возникновения тяжелой гранулоцитопении при этом определяется суммарной дозой принятого препарата. Миелотоксический агранулоцитоз обычно сочетается с анемией и тромбоцитопенией.
Ведущее значение в патогенезе иммунный (гаптеновых) агранулоцитозов имеет появление в организме антител (агглютинины, лизины и т.д.), действие которых направлено против собственных гранулоцитов периферической крови или их клеток-предшественниц в костном мозгу. Считается, что медикаментозные препараты выступают в роли гаптенов, образующих комплексные соединения с белками плазмы и мембран лейкоцитов. Вырабатываемые на образовавшийся «чужеродный» комплекс (антиген) антитела, фиксируясь на поверхности клеток, вызывают их разрушение. Как правило, при иммунном агранулоцитозе снижается содержание только лейкоцитов.
Классическим клиническим проявлением агранулоцитоза независимо от причин и механизмов его развития является язвеннонекротическая ангина (angina agranulocytotica), развивающаяся вследствие подавления защитных реакций организма (снижения резистентности к бактериальной флоре).
14.3.6. Лейкозы
Одной из наиболее опасных для жизни человека патологий системы крови являются лейкозы, относящиеся к группе гемобластозов - злокачественных новообразований кроветворной (миелоидной и лимфоидной) ткани. В отличие от лейкоцитозов, лейкемоидных реакций и лейкопений лейкоз является не реактивным состоянием, а болезнью системы крови.
Лейкоз - это опухоль, исходящая из кроветворных клеток костного мозга, в основе развития которой лежит неконтролируемый рост клеток с преобладанием процессов их пролиферации над дифференцировкой и образованием очагов патологического кроветворения в органах и тканях, в норме не участвующих в гемопоэзе. При этом утратившие способность к созреванию лейкозные клетки могут проходить значительно большее, чем нормальные клетки крови, число циклов деления, что и создает огромную клеточную продукцию, характеризующую лейкоз.
Этиология лейкозов до настоящего времени точно не установлена. Об опухолевой природе лейкозов свидетельствует наличие общих закономерностей, объединяющих лейкозы и опухоли: нарушение способности клеток к дифференцировке; морфологическая и метаболическая анаплазия клеток; способность к метастазированию; общие этиологические факторы, способствующие развитию лейкозов и опухолей, и др.
К возможным этиологическим факторам, вызывающим развитие лейкозов, относят ионизирующую радиацию, ряд химических веществ, вирусы. Определенное значение в развитии лейкозов придается генетическим факторам, наследственной и приобретенной иммунной недостаточности, действию бластомогенных метаболитов триптофана и тирозина.
Соответственно существует несколько теорий происхождения лейкозов.
Радиационная теория. Роль ионизирующих излучений в возникновении лейкозов доказана в эксперименте. Как однократное (в дозе 2 Гр и выше), так и хроническое (в течение 2-3 месяцев) облучение лучами Рентгена в малых дозах может индуцировать лейкоз у лабораторных животных (крысы, мыши). Прослежено повышение заболеваемости острым и хроническим миелолейкозом у жителей Хиросимы и Нагасаки, у рентгенологов и радиологов. Приводятся данные об увеличении частоты лейкозов у больных, леченных
большими дозами лучей Рентгена, иттрия, радия по поводу злокачественных новообразований и анкилозирующего спондилоартрита, а также у детей, получавших облучение вилочковой железы в раннем возрасте, и др. Описано учащение случаев острых лейкозов среди больных эритремией после лечения их радиоактивным фосфором.
Теория химического лейкозогенеза. Экспериментально доказана возможность индуцирования лейкозов у животных введением канцерогенных веществ (диметилбензантрацен, метилхолантрен и др.). Также в эксперименте показана возможность стимуляции лейкозогенеза метаболитами триптофана и тирозина (М.Л. Раушенбах). Однако роль этих веществ в лейкозогенезе человека не доказана. В то же время накоплены данные, указывающие на увеличение риска заболевания лейкозами (как правило, острыми) у людей, имеющих длительный профессиональный контакт с бензолом и летучими органическими растворителями (шоферы, работники кожевенной и обувной промышленности и т.д.). В последние годы отмечено заметное учащение случаев острого лейкоза у больных злокачественными новообразованиями, леченных такими цитостатическими препаратами, как циклосфосфан, хлорбутин, метотрексат, миелосан, адриамицин и др. К лекарственным препаратам, способным индуцировать лейкозы, относятся также бутадион, левомицетин и некоторые другие.
Вирусная теория связывает возникновение лейкозов с активацией (под действием радиации и химических факторов) латентных лейкозогенных вирусов. Несомненно доказанным является вирусное происхождение лейкозов у многих видов животных - птиц, мышей, крыс, хомячков, кошек, крупного рогатого скота. К настоящему времени выделено и детально охарактеризовано несколько типов вирусов, вызывающих различные виды лейкозов у животных. Как правило, это РНК-содержащие вирусы, а также ДНК-содержащие вирусы, которые относятся к герпесвирусам (подробнее см. раздел 13.3).
Вопрос о роли вирусов в происхождении лейкозов у человека остается во многом спорным. Против вирусной этиологии лейкозов у человека свидетельствует, прежде всего, факт невозможности прямой перевивки лейкозов при случайном переливании крови лиц, больных лейкозом, и отсутствие убедительных доказательств контагиозности лейкозов. Не описаны также случаи передачи лейкоза от больной матери плоду и новорожденному в период вскармливания грудью.
Таблица 14-10. Дифференциальные критерии лекарственных агранулоцитозов
Критерии | Вид агранулоцитоза | ||
миелотоксический | иммунный | ||
Патогенез | Вследствие прямого повреждения клетокпредшественниц гемопоэза | Вследствие иммунного разрушения циркулирующих нейтрофилов | Вследствие иммунного разрушения клетокпредшественниц гранулоцитопоэза |
Начало | Постепенное | Острое | Постепенное |
Клинические проявления | Лихорадка, язвеннонекротическое поражение слизистых оболочек ротоглотки и желудочнокишечного тракта, некротическая энтеропатия | Лихорадка, озноб, крапивница, отек Квинке, изъязвление слизистых оболочек ротоглотки, верхних дыхательных путей, желудочно-кишечного тракта, увеличение размеров лимфоузлов, печени и селезенки, грибковые инфекции | |
Связь с дозой медикамента | Присутствует | Отсутствует | |
Количество лейкоцитов | Снижение общего количества лейкоцитов, нейтропения до (0,2-0,1)-109/л, моноцитоз, лимфоцитоз, увеличение числа плазматических клеток (только при иммунном агранулоцитозе) | ||
Количество тромбоцитов | Снижено | В норме | |
Анемия | Определяется | Не определяется | |
Картина костного мозга | Тотальное угнетение гемопоэза | Гиперплазия гранулоцитарного ростка | Гипоплазия гранулоцитарного ростка |
Антилейкоцитарные антитела | Отсутствуют | Присутствуют | |
Генетическая теория располагает достаточно убедительными аргументами, указывающими на возможность наследственной предрасположенности к лейкозам. Известны случаи семейных лейкозов, доказана роль этнических особенностей в развитии лимфолейкоза. К возникновению лейкозов предрасполагают болезни, характеризующиеся спонтанными разрывами хромосом и нерасхождением соматических или половых хромосом (болезнь Дауна, анемия Фанкони, синдромы Кляйнфелтера, Тернера и др.). Получены линии мышей, у которых частота спонтанных лейкозов близка к 100%.
Патогенез лейкозов. Согласно мутационно-клоновой теории, в основе происхождения лейкозов лежат мутация и опухолевая трансформация ранних клеток-предшественниц гемопоэза (клеток II и III классов) под влиянием лейкозогенного фактора (ионизирующей радиации, химических веществ, вирусов и др.). В результате происходит выход кроветворных клеток из-под контроля регулирующих систем макроорганизма с активацией их деления на фоне подавления дифференцировки. Формируется клон опухолевых (лейкозных) клеток - потомков одной первоначально мутировавшей клетки (моноклоновая опухоль), которые заселяют (инфильтрируют) костный мозг. В моноклоновой стадии опухолевые клетки чувствительны к химиотерапии. В процессе развития лейкоза (опухолевая прогрессия) происходят качественные изменения составляющих субстрат опухоли клеток, обусловленные нестабильностью их генетического аппарата, что проявляется нарушениями структуры хромосом, анэуплоидией, переходом части ранее неактивных в клетке генов в активное состояние (феномен дерепрессирования генов). Эти изменения ведут к появлению новых клонов опухолевых клеток, среди которых в процессе жизнедеятельности организма, а также под воздействием лечебных средств, применяемых в химиотерапии заболевания, «отбираются» наиболее автономные клоны. В результате моноклоновая опухоль превращается в поликлоновую злокачественную опухоль. На этой стадии развития лейкоза лейкозные клетки становятся устойчивыми к цитостатической терапии, метастазируют в органы и ткани, в норме не участвующие в гемопоэзе, образуя очаги экстрамедуллярного кроветворения (табл. 14-11).
Таблица 14-11. Основные стадии патогенеза лейкозов
Стадия Характеристика | |
Инициация | Лейкозогенный фактор (радиация, вирусы и др.) действует на стволовые кроветворные клетки II-III классов, вызывая их опухолевую трансформацию в результате мутационного превращения протоонкогенов в онкогены и инактивации антионкогенов |
Промоция | Активация и гиперпролиферация лейкозных клеток при действии промотора с формированием клона лейкозных клеток, идентичных по фенотипу и генотипу (моноклоновая стадия) |
Инфильтрация | «Расселение» лейкозных клеток в костном мозгу с угнетением нормального гемопоэза |
Прогрессия | Формирование множества клонов лейкозных клеток, различающихся по фенотипу и генотипу (поликлоновая стадия), и естественный отбор наиболее автономных из них, что ведет к «озлокачествлению» заболевания |
Метастазирование | Образование очагов патологического кроветворения вне костного мозга за счет способности лейкозных клеток к инвазии, интра- и экстравазации, миграции по сосудистой системе, имплантации и пролиферации в различных тканях и органах |
О клоновой природе лейкозов свидетельствуют возможность перевивки лейкоза мышам путем введения одной лейкозной клетки; продукция гомогенного иммуноглобулина при парапротеинемических гемобластозах (миеломная болезнь, макроглобулинемия Вальденстрема и др.); однотипность лейкозных клеток (несущих на поверхности иммуноглобулины одного класса и подкласса) при хроническом лимфолейкозе; наличие специфических хромосомных изменений в опухолевых клетках (транслокации, делеции). Частным подтверждением клонового происхождения лейкозов является обнаружение в 80-90% случаев хронического миелолейкоза аномальной филадельфийской (Ph') хромосомы (рис. 14-12) в миелоидных клетках, включая гранулоцитарный, эритроидный и мегакариоцитарный ростки. Этот факт служит неоспоримым доказательством происхождения хронического миелолейкоза из одного патологического клона, родоначальницей которого является
 Рис. 14-12. Аномальная Ph'-хромосома в клетке больного хроническим миелолейкозом
Рис. 14-12. Аномальная Ph'-хромосома в клетке больного хроническим миелолейкозом
плюрипотентная стволовая клетка-предшественница миелопоэза (КОЕ-ГЭММ).
Общие нарушения в организме при лейкозах проявляются следующими клиническими синдромами: анемическим (головокружение, слабость, повышенная утомляемость, одышка и т.д.), геморрагическим (кровотечения из десен, носа, кишечника, кровоизлияния в жизненно важные органы), инфекционным (рецидивирующие инфекции вследствие угнетения фагоцитоза, микробицидной функции лейкоцитов, синтеза антител и т.д.), интоксикационным (тошнота, рвота, снижение аппетита, уменьшение массы тела и т.д.) и гиперпластическим (увеличение размеров и нарушение функции различных органов) (табл. 14-12).
Таблица 14-12. Патогенез основных клинических синдромов лейкозов

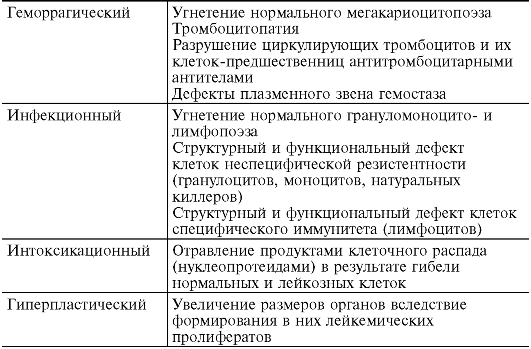 Причинами
смерти при лейкозах являются резкое малокровие и тяжелая общая
интоксикация, поражение жизненно важных органов (лейкозная инфильтрация,
обширные кровоизлияния). Непосредственной причиной смерти больных могут
стать инфекционные осложнения (пневмонии, сепсис, перитонит).
Причинами
смерти при лейкозах являются резкое малокровие и тяжелая общая
интоксикация, поражение жизненно важных органов (лейкозная инфильтрация,
обширные кровоизлияния). Непосредственной причиной смерти больных могут
стать инфекционные осложнения (пневмонии, сепсис, перитонит).
Классификация лейкозов, особенности кроветворения и клеточного состава периферической крови при различных видах лейкозов
По патогенетическому принципу лейкозы подразделяют на острые и хронические.
Острый лейкоз - это опухоль, исходящая из костного мозга, с полной утратой способности кроветворных клеток к дифференцировке на
уровне клеток IV класса - бластов, составляющих морфологический субстрат опухоли. Гематологическая картина острого лейкоза характеризуется появлением большого числа бластных клеток в крови (до нескольких десятков процентов) и так называемым лейкемическим зиянием (hiatus leukaemicus), проявляющимся наличием бластных и (в небольшом количестве) зрелых клеток в крови при практически полном отсутствии их промежуточных форм.
Хронический лейкоз - опухоль, исходящая из костного мозга, с частичной задержкой способности кроветворных клеток к дифференцировке. При хронических лейкозах клетки сохраняют способность к дифференцировке до стадии созревающих или зрелых клеток, т.е. морфологическим субстратом опухоли являются клетки V и VI классов.
Диагностика лейкозов требует комплексного подхода. Она основывается прежде всего на анализе мазков периферической крови и костного мозга. Для дифференциальной диагностики отдельных форм и цитологических вариантов острых и хронических лейкозов применяют цитохимические, цитологические, иммунологические, цитогенетические и молекулярно-генетические методы.
Острые лейкозы. Острые лейкозы характеризуются высокой скоростью развития опухолевого процесса, что при отсутствии необходимого лечения быстро приводит к гибели больного. Выделяют следующие стадии клинического течения острых лейкозов: первая атака, развернутая стадия, терминальная стадия или выздоровление.
Стадия первой атаки охватывает период времени от проявления первых клинико-гематологических симптомов заболевания, установления диагноза и начала лечения до получения эффекта от проводимой терапии. Критерием лабораторной диагностики острых лейкозов служит наличие в костном мозгу свыше 30% бластных клеток.
В зависимости от содержания бластных клеток в периферической крови острые лейкозы подразделяются на:
• алейкемические - бластные клетки в крови отсутствуют;
• сублейкемические - бластные клетки в крови обнаруживаются в небольшом количестве (3-5%);
• лейкемические - бласты составляют основную массу клеток крови.
Общее количество лейкоцитов (ОКЛ) в периферической крови при острых лейкозах может быть различным:
• при алейкемическом варианте - менее 6-109/л;
• при сублейкемическом варианте - (6-60)-109/л;
• при лейкемическом варианте - более 60-109/л.
Уже на ранних стадиях болезни отмечаются нормохромная анемия и тромбоцитопения, развитие которых обусловлено угнетением нормального гемопоэза вследствие лейкемической трансформации кроветворения.
Развернутая стадия острого лейкоза (стадия развернутых клинических проявлений) характеризуется чередованием ремиссий и рецидивов.
Ремиссия - исчезновение проявлений патологического процесса под влиянием цитостатической терапии. Различают полную и неполную ремиссию.
Полная ремиссия характеризуется нормализацией клинических показателей, картины периферической крови и костного мозга в течение не менее 1 месяца.
Клинико-лабораторные критерии полной ремиссии:
• костно-мозговой - содержание в костном мозгу не более 5% бластных клеток и не более 30% лимфоцитов при нормальной его общей клеточности;
• кровяной - отсутствие бластов в периферической крови, количество гранулоцитов более 1,5-109/л, количество тромбоцитов более 100-109/л, содержание гемоглобина более 100 г/л;
• клинический - исчезновение патологических симптомов;
• субъективный - отсутствие жалоб.
Неполная ремиссия - состояние, при котором нормализуются клинические показатели и гемограмма, но в пунктате костного мозга сохраняются бластные клетки в количестве не более 20%.
Рецидив проявляется возвратом активной стадии заболевания после ремиссии. Рецидив может быть:
костно-мозговым, который подразделяется на:
а) алейкемический - характеризуется обнаружением бластов в костном мозгу (свыше 20%) при отсутствии их в периферической крови;
б) лейкемический - характеризуется обнаружением бластов не только в костном мозгу (свыше 20%), но и в периферической крови;
внекостно-мозговым (местным) - присутствие лейкемических инфильтратов вне костного мозга (в лимфоузлах, селезенке, лейкемидах кожи и др.).
Терминальная стадия острого лейкоза представляет собой завершающий этап опухолевой прогрессии при полном истощении нормального кроветворения и резистентности к цитостатической терапии. Причиной гибели пациентов чаще всего являются инфекционно-воспалительные осложнения (перитонит, пневмония, сепсис и др.), кровотечения, кровоизлияния во внутренние органы.
Под выздоровлением подразумевают полную ремиссию, сохраняющуюся в течение 5 лет и более.
Составляющие субстрат опухоли бластные клетки при различных вариантах острого лейкоза морфологически трудноразличимы. Для определения линейной принадлежности и степени зрелости бластных клеток с целью дифференциальной диагностики отдельных форм и цитологических вариантов острых лейкозов применяются цитохимическое исследование и метод идентификации поверхностных, цитоплазматических и ядерных антигенов (т.е. иммунофенотипа) бластных клеток костного мозга и крови. В качестве базисных цитохимических методов диагностики острых лейкозов проводят определение в бластах содержания и характера распределения липидов, гликогена, кислых сульфатированных мукополисахаридов, активности миелопероксидазы, кислой фосфатазы, неспецифической эстеразы (α-нафтилацетатэстеразы), хлорацетатэстеразы (табл. 14-13).
В 1975 г. гематологами Франции, США и Великобритании была создана Франко-американо-британская классификация острых лейкозов, базирующаяся на цитоморфологических признаках бластных клеток - ФАБ-классификация, получившая наиболее широкое применение в практике. Согласно цитоморфологической ФАБклассификации, острые лейкозы подразделяют на две группы - миелоидные и лимфобластные лейкозы, в структуре которых выделены следующие цитологические варианты:
1. Острые миелоидные лейкозы (ОМЛ)
М0 - острый миелобластный лейкоз с минимальными признаками дифференцировки
М1 - острый миелобластный лейкоз без признаков вызревания М2 - острый миелобластный лейкоз с признаками вызревания М3 - гипергранулярный острый промиелоцитарный лейкоз М3v - микрогранулярный острый промиелоцитарный лейкоз М4 - острый миеломонобластный лейкоз М5а - острый монобластный лейкоз без признаков вызревания М5b - острый монобластный лейкоз с признаками вызревания М6 - острый эритробластный лейкоз (эритролейкоз) М7 - острый мегакариобластный лейкоз
2. Острые лимфобластные лейкозы (ОЛЛ) L1 - микролимфобластный ОЛЛ
L2 - ОЛЛ с типичными бластами L3 - макролимфобластный ОЛЛ
Таблица 14-13. Цитохимические особенности бластных клеток при острых лейкозах различного происхождения
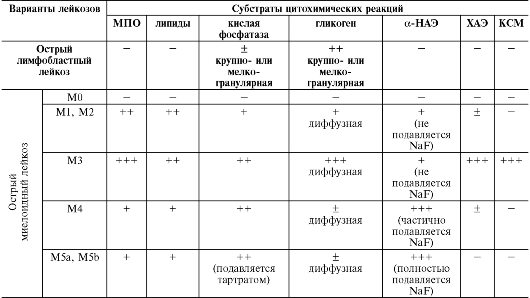 Окончание табл. 14-13
Окончание табл. 14-13
 Примечание. МПО - миелопероксидаза; -НАЭ--нафтилацетатэстераза; ХАЭ - хлорацетатэстераза; КСМ - кислые
Примечание. МПО - миелопероксидаза; -НАЭ--нафтилацетатэстераза; ХАЭ - хлорацетатэстераза; КСМ - кислые
сульфатированные мукополисахариды; «-» - отрицательная реакция; «+» - положительная реакция в единичных клетках; «+» - слабая реакция; «++» - умеренная реакция; «+++» - интенсивная реакция.
Выделенные нозологические формы острых лейкозов различаются по клиническим признакам и, что особенно важно, по ответу на цитостатическую медикаментозную терапию. У взрослых больных чаще встречаются миелобластный и лимфобластный, у детей - лимфобластный и (реже) недифференцированный варианты острого лейкоза.
Острый недифференцированный лейкоз, вариант М0. Морфологический субстрат опухоли представлен клетками II-III классов по современной схеме кроветворения, которые морфологически напоминают лимфобласты, но отличаются цитохимической интактностью (см. табл. 14-13). Из хромосомных аномалий для МО типичны моносомия 7, трисомия 4, 8, 13.
Острый миелобластный лейкоз. Представляет собой опухоль, исходящую из клетки-предшественницы миелопоэза и состоящую преимущественно из родоначальных клеток гранулоцитарного ряда - миелобластов. Выделяют 2 варианта ОМЛ - М1 и М2, для которых свойственны транслокации хромосом соответственно t(9;22) и t(8;21). В костном мозгу и крови у больных ОМЛ обнаруживаются многочисленные миелобласты средних размеров с узким ободком незернистой цитоплазмы и крупные миелобласты с обширной (иногда вакуолизированной) цитоплазмой с азурофильными гранулами и 1-2 палочками Ауэра (они могут определяться в более зрелых клетках гранулоцитарного ряда). Для зрелых нейтрофилов характерна гиперили гипосегментация ядер, «истощение» цитоплазматической зернистости.
Острый промиелоцитарный лейкоз. Для лейкозных клеток при этой форме острого лейкоза характерны транслокации хромосом: t(15;17), t(5;17), t(11;17). Атипичные промиелоциты содержат большое количество крупных (при гипергранулярном варианте - М3) или мелких (при микрогранулярном варианте - M3v) гранул и палочки Ауэра (нередко их 1О-2О и более). При слиянии палочек Ауэра образуются «faggot cells» - клетки с «пучком прутьев». Ядра атипичных промиелоцитов имеют рыхлую структуру хроматина, неправильную форму со смазанными контурами (иногда их форма неразличима из-за обилия гранул в цитоплазме).
Острый монобластный лейкоз. Существует два его варианта - без признаков вызревания клеток (М5а) и с признаками вызревания клеток (М5Ь), при которых обнаруживаются транслокационные перестройки хромосом t(9;11) и t(4;11). При М5а субстрат опухоли составляют монобласты крупных размеров с обширной
серо-голубой цитоплазмой, скудной азурофильной зернистостью и вакуолизацией. При М5b преобладающим типом клеток в костном мозгу и крови являются промоноциты (количество монобластов не превышает 10-15%).
Острый миеломонобластный лейкоз или вариант М4 острого лейкоза. В его основе лежат перестройки хромосомы 16: inv (16), t(16;16), t (8;16). Для М4 характерно присутствие в костном мозгу одновременно двух типов атипичных бластов - миелобластов и монобластов. В крови могут обнаруживаться также промоноциты.
Острый эритробластный лейкоз или М6. Наиболее частой цитогенетической аномалией при М6 является делеция длинного плеча хромосомы 5 или 7. Субстрат опухоли составляют клетки эритроидного ряда: эритронормобласты, гигантские, многоядерные эритрокариоциты с мегалобластическим оттенком, мегалобласты, аномальные эритроидные клетки с вакуолизированной и отростчатой цитоплазмой, с кариорексисом. В крови отмечаются выраженный анизо- и пойкилоцитоз, гиперхромия эритроцитов, появление в кровотоке эритроцитов с базофильной зернистостью, кольцами Кабо, эритробластов, полихроматофильных и оксифильных эритронормобластов.
Острый мегакариобластный лейкоз, вариант М7. Характерными цитогенетическими признаками М7 являются inv (3), t(3;3), t(1;22). В костном мозгу и крови выявляются мегакариобласты и их осколки. В крови обнаруживается тромбоцитоз (более 1000-109/л). Тромбоциты имеют гигантские размеры, могут склеиваться в «кучки», имеется качественный дефект клеток - «серые» тромбоциты (в результате дефекта α-гранул).
Острый лимфобластный лейкоз. Это опухоль, возникающая из клетки-предшественницы лимфопоэза. В костном мозгу при ОЛЛ выявляется тотальная лимфобластная метаплазия. Картина периферической крови при ОЛЛ характеризуется присутствием бластных клеток. Согласно ФАБ-классификации, в зависимости от цитоморфологических признаков лимфоидных клеток выделяют три цитологических варианта ОЛЛ: L1 (микролимфобластный), представленный однородной популяцией клеток малых размеров; L2 (с типичными бластами), характеризующийся неоднородностью клеток, среди которых преобладают крупные клетки, реже выявляются клетки средних и малых размеров; L3 (макролимфобластный), при котором обнаруживаются клетки только крупных размеров.
В 1995 г. Европейской группой по иммунологической характеристике лейкозов (European Group for the immunological characterization of leukemias, EGIL) была предложена классификация острых лимфобластных лейкозов по иммунологическому принципу. В соответствии с EGIL-классификацией их подразделяют на Т- и В-линейные лейкозы (табл. 14-14).
Таблица 14-14. EGIL-классификация острых лимфобластных лейкозов
(ОЛЛ)
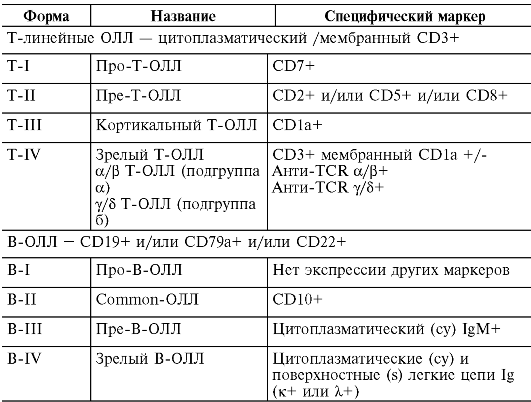 Хронические лейкозы. Несколько упрощенно классификацию хронических лейкозов можно представить в следующем виде. 1. Хронические миелопролиферативные лейкозы: Хронический миелолейкоз
Хронические лейкозы. Несколько упрощенно классификацию хронических лейкозов можно представить в следующем виде. 1. Хронические миелопролиферативные лейкозы: Хронический миелолейкоз
• Ph-позитивный (типичный, взрослый тип)
• Ph-негативный (атипичный, ювенильный тип) Хронический миеломоноцитарный лейкоз Хронический нейтрофильный лейкоз Хронический эозинофильный лейкоз Хронический базофильный лейкоз
Хронический тучноклеточный лейкоз Истинная полицитемия (эритремия) Тромбоцитемия
Хронический идиопатический миелофиброз (сублейкемический миелоз)
2. Хронические лимфопролиферативные лейкозы:
В-клеточные хронические лимфолейкозы Хронический лимфолейкоз Волосатоклеточный лейкоз Парапротеинемические гемобластозы
• Множественная миелома (миеломная болезнь)
• Макроглобулинемия Вальденстрема
• Болезни тяжелых цепей
Т- и ЕК-клеточные (из ЕК-клеток - естественных киллеров) хронические лимфолейкозы
Болезнь Сезари (лимфоматоз кожи)
Лейкоз из больших гранулосодержащих лимфоцитов (ЕКклеток)
В отличие от острых лейкозов моноклоновая («доброкачественная») стадия хронических лейкозов является более продолжительной (годы, десятилетия). В развитии хронических лейкозов выделяют хроническую фазу, которая характеризуется длительным компенсированным течением, и фазу бластной трансформации, проявляющуюся бластным кризом с резким увеличением количества бластных клеток в костном мозгу и периферической крови (более 30%), прогрессированием анемии, тромбоцитопении и формированием внекостно-мозговых лейкемических инфильтратов.
Клинико-лабораторная картина хронических лейкозов характеризуются разнообразием признаков, строго специфичных для определенной формы заболевания. Наиболее распространенными вариантами хронических лейкозов являются хронический миелолейкоз, хронический миеломоноцитарный лейкоз, эритремия, сублейкемический миелоз, хронический лимфолейкоз, парапротеинемические гемобластозы и лимфоматоз кожи.
Хронический миелолейкоз. Одно из самых частых заболеваний в группе лейкозов. Морфологическим субстратом хронического миелолейкоза (ХМЛ) являются зрелые и созревающие клетки гранулоцитарного ростка кроветворения (лейкоэритробластическое отношение в костном мозгу увеличивается до 20:1 при 2:1-4:1 в норме). По современным представлениям ХМЛ возникает в резуль-
тате соматической мутации в стволовой клетке-предшественнице миелопоэза, что в 85-95% случаев приводит к образованию хромосомного маркера - Ph'-хромосомы (см. рис. 14-12), являющейся продуктом делеции хромосомы 22 или транслокации дистальной части длинного плеча хромосомы 22 на хромосому 9. У детей ХМЛ встречается в двух вариантах - с филадельфийской хромосомой в кариотипе лейкозных клеток (взрослый тип заболевания) и без нее (ювенильный тип).
Течение хронического миелолейкоза как у взрослых, так и у детей разделяют на стадии или фазы заболевания в зависимости от степени зрелости клеточного состава крови. В хронической фазе типичного ХМЛ общее количество лейкоцитов в периферической крови колеблется от 50-109/л и более (у 25% больных - более 350-109/л). Отмечается нейтрофильный лейкоцитоз с выраженным ядерным сдвигом влево: обнаруживаются единичные миелобласты (2-3%), промиелоциты, миелоциты, метамиелоциты, палочко- и сегментно-ядерные формы гранулоцитов. Количество промиелоцитов и миелоцитов увеличивается по мере прогрессирования болезни при одновременном уменьшении числа палочкоядерных и сегментно-ядерных форм гранулоцитов. Отмечаются выраженная эозинофилия и базофилия (эозинофильно-базофильная ассоциация). В гранулоцитах обнаруживаются признаки дегенерации - псевдопельгеризация, низкая активность щелочной фосфатазы. У детей ювенильная (Ph-негативная) форма ХМЛ характеризуется высоким моноцитозом и тромбоцитопенией. В период бластной трансформации происходит резкое «омоложение» лейкоцитарной формулы за счет увеличения числа промиелоцитов и миелобластов (не менее 30%), прогрессирует цитопения (анемия, лейко-и тромбоцитопения), возникают лейкемические инфильтраты в коже, лимфоузлах, миокарде и других органах. При кариологическом исследовании выявляется поликлоновость патологических клеток (анэуплоидия), которая является основным признаком терминальной стадии - новым этапом опухолевой прогрессии.
Хронический миеломоноцитарный лейкоз характеризуется гиперклеточностью костного мозга за счет увеличения количества незрелых и зрелых клеток гранулоцитарного и моноцитарного ряда (промоноцитов, моноцитов). Миелобласты и монобласты составляют не более 5% от общего числа миелокариоцитов. В периферической крови обнаруживаются незрелые клетки нейтрофильного ряда (не более 10% от общего количества лейкоцитов), промоно-
циты, реже - бласты. В гранулоцитах - псевдопельгеризация, «истощение» зернистости в цитоплазме, отрицательная реакция на миелопероксидазу, низкая активность лейкоцитарной щелочной фосфатазы. В сыворотке крови и моче устанавливается высокое содержание лизоцима.
Эритремия (истинная полицитемия, болезнь Вакеза). Заболевание опухолевой природы, характеризующееся относительно доброкачественным течением. Источником роста опухоли является клетка-предшественница миелопоэза, основной субстрат опухоли - эритроциты. Наиболее характерны изменения со стороны периферической крови: количество эритроцитов достигает (6-12)-1012/л, уровень гемоглобина - 180-220 г/л, показатель гематокрита увеличивается до 60-80%. Уровень эритропоэтина в крови и моче в отличие от симптоматических эритроцитозов остается нормальным. Имеются лейко- и тромбоцитоз, уменьшается СОЭ, возрастает вязкость крови. Важным диагностическим признаком является увеличение массы циркулирующих эритроцитов, опосредующее гиперемию кожи и слизистых, окклюзию микрососудов и связанные с ней головные боли, боли в суставах, позвоночнике, эпигастрии и др.
Сублейкемический миелоз характеризуется гиперклеточностью костного мозга за счет гиперплазии гранулоцитарного, эритроидного и мегакариоцитарного ростков, а также миелофиброзом и миелосклерозом, при прогрессировании которых клеточность костного мозга постепенно снижается. В крови обнаруживаются незрелые клетки гранулоцитарного ряда, гипо- и гиперсегментированные нейтрофилы, миелобласты (1-5%). Количество тромбоцитов варьирует. Нередко выявляются крупные гипогранулярные тромбоциты:, аномальные мегакариоциты и их фрагменты. Гематологическими признаками сублейкемического миелоза, позволяющими дифференцировать его с ХМЛ, наряду с выраженным фиброзом и склерозом костного мозга, являются: увеличение общего количества лейкоцитов в крови не более 50-109/л (при ХМЛ более 50-109/л), умеренная базофилия при нормальном содержании эозинофилов (при ХМЛ сочетанное увеличение количества базофилов и эозинофилов - эозинофильно-базофильная ассоциация) и высокая активность щелочной фосфатазы в нейтрофилах (при ХМЛ активность лейкоцитарной щелочной фосфатазы низкая).
Хронический лимфолейкоз. Это опухоль иммунокомпетентной ткани, состоящая преимущественно из зрелых лимфоцитов, пред-
ставленных в большинстве случаев В-клетками. Характерен лейкоцитоз; в мазках крови преобладают зрелые узкоцитоплазменные лимфоциты, содержание которых может доходить до 80% и более. Важным признаком служит появление дегенеративных форм лимфоцитов - теней Гумпрехта (результат раздавливания качественно неполноценных лимфоцитов при приготовлении гематологических мазков), голых ядер лимфоцитов и форм Ридера. Количество лимфоцитов в костном мозгу составляет не менее 30% всех миелокариоцитов. Разрастание лимфоидной ткани имеет место в лимфатических узлах, селезенке и печени, что сопровождается увеличением указанных органов.
Функциональная неполноценность образующих опухоль лимфоцитов приводит к нарушению иммунологического гомеостаза у больных, что, в свою очередь, становится причиной аутоиммунных конфликтов (аутоиммунные гемолитические анемии и тромбоцитопении); инфекционных осложнений (вследствие нарушения антителообразования) и т.д.
В отличие от хронического миелолейкоза бластные кризы наблюдаются крайне редко, не развивается также вторичной резистентности к цитостатическим препаратам.
Парапротеинемичские гемобластозы. К парапротеинемическим гемобластозам относятся миеломная болезнь (множественная миелома, плазмоцитома), макроглобулинемия Вальденстрема и болезни тяжелых цепей - опухоли В-клеточного происхождения. Главной особенностью данных лейкозов является сохранение способности В-клеток к дифференцировке до стадии иммуноглобулинсекретирующих клеток. При этом секретируемые ими иммуноглобулины отличаются однообразием структуры (моноклональные парапротеины), что объясняется их происхождением из одного клона опухолевых клеток. Парапротеины соответствуют разным вариантам нормальных иммуноглобулинов (чаще IgG или IgM), отличаясь от них строгой однотипностью тяжелых и легких цепей, либо представляют собой структурно аномальные молекулы иммуноглобулинов (изолированные фрагменты тяжелых цепей, свободные легкие цепи).
При множественной миеломе имеет место диссеминированная злокачественная пролиферация клона В-лимфоцитов на уровне плазматических клеток. Множественная миелома составляет около 1% всех злокачественных новообразований, частота встречаемости миеломной болезни колеблется в разных этнических группах
от 1 до 10 на 100 000 населения. Плазматические клетки наиболее часто пролиферируют диффузно по костному мозгу, но иногда формируют солитарную опухоль, называемую плазмоцитомой. Из-за остеолитических повреждений, обусловленных продукцией опухолевыми клетками остеокластактивирующих факторов (IL-1, TNF-α, TNF-β), развивается гиперкальциемия и связанные с ней поражения нервной, сердечно-сосудистой системы, почек, желудочно-кишечного тракта, формируются тромбоцитопения, анемия, лейкопения. Одновременно подавляется формирование нормальных плазматических клеток, что приводит к нарушению образования других иммуноглобулинов, развивается синдром возвратных инфекций.
В случае выявления парапротеинов при электрофорезе сыворотки крови обязательным является электрофоретическое исследование мочи. В 20% случаев миеломной болезни опухоль продуцирует только легкие цепи иммуноглобулинов, которые из-за низкой молекулярной массы быстро фильтруются в почках и могут не обнаруживаться в сыворотке, но выявляются в моче (протеинурия Бенс-Джонса). В связи с этим для подтверждения диагноза методом электрофореза определяют белок Бенс-Джонса в моче, который располагается недалеко от старта соответственно М-градиенту в сыворотке, между γ- и β-глобулинами.
Макроглобулинемия Вальденстрема характеризуется тем, что опухолевые В-лимфоциты продуцируют макромолекулярные моноклональные парапротеины класса IgМ. Из-за накопления высокомолекулярных белков характерны повышение вязкости крови, нарушение микроциркуляции, сладж-синдром, предрасположенность к тромбозам, геморрагический синдром. Болеют преимущественно мужчины после 60 лет. Картина крови характеризуется анемией, в патогенезе которой играют роль опухолевое подавление эритропоэза, кровопотери, нередко развивается лейкопения с нейтропенией, моноцитоз, тромбоцитопения присоединяется по мере прогрессирования заболевания. СОЭ всегда резко увеличена, в сыворотке гиперпротеинемия, а на электрофореграмме - М-градиент за счет IgМ. В моче белок Бенс-Джонса встречается примерно в 80% случаев, но количество его значительно меньше, чем при множественной миеломе. В отличие от миеломной болезни при макроглобулинемии Вальденстрема обычно обнаруживаются увеличение лимфоузлов и гепатоспленомегалия, но лимфоидная инфильтрация прогрессирует, как правило, относительно медленно.
При этом поражение костей и гиперкальциемия отмечаются редко. Проявления болезни исчезают при замещающей гемотрансфузии.
Болезни тяжелых цепей представляют собой В-клеточные лимфатические опухоли с разнообразной клинической и морфологической картиной и секрецией фрагментов тяжелых цепей различных классов иммуноглобулинов. Это обычно α-цепи, но могут быть также γ- или μ-цепи. Выделяют две формы течения болезни: абдоминальную и легочную. Абдоминальная форма характеризуется диффузной инфильтрацией слизистой тонкой кишки и мезентериальных лимфатических узлов лимфоидными и плазматическими клетками, макрофагами, тучными клетками. Поражение желудочно-кишечного тракта приводит к атрофии ворсинчатого эпителия и развитию синдрома мальабсорбции. Легочная форма протекает с бронхопульмональными поражениями и медиастинальной лимфаденопатией. Протеинурия отсутствует. Диагностика основана на иммунохимическом анализе сывороточных белков, позволяющем выявить тяжелые цепи иммуноглобулинов.
Болезнь Сезари (лимфоматоз кожи) проявляется генерализованной эритродермией, алопецией, поражением век, дистрофией ногтей, интенсивным зудом кожи. У 30% больных отмечается спленомегалия, у 60% - лимфаденопатия смешанной природы (одни лимфатические узлы увеличиваются реактивно вследствие присоединения кожных инфекций, другие - в связи с их лейкемической инфильтрацией). При исследовании трепанобиоптатов костного мозга обнаруживаются очаги лимфоидной инфильтрации из малых лимфоцитов и клеток Сезари - атипичных Т-лимфоцитов со светлой цитоплазмой и складчатыми (конволютными) ядрами. Содержание гемоглобина, количество эритроцитов и тромбоцитов в крови обычно сохраняется в пределах нормы. Характерен лимфоцитоз (до 70-109/л), наличие клеток Сезари в мазках крови.
14.4. ИЗМЕНЕНИЯ ФИЗИКО-ХИМИЧЕСКИХ
СВОЙСТВ КРОВИ
14.4.1. Изменения количества крови
Общее количество крови взрослого человека составляет у мужчин 5,2 л, у женщин - 3,9 л (около 6-7,5% массы тела), большая часть которой находится в циркуляции (ОЦК 3,5-4,0 л), меньшая - в депонированном состоянии.
Периферическая кровь состоит из форменных элементов (эритроциты, лейкоциты, тромбоциты) и жидкой части - плазмы в соотношении 45:55.
Гиперволемия - увеличение общего объема крови. Различают три формы гиперволемии (табл. 14-15).
Гиперволемия простая, когда при увеличении общего объема крови сохраняется нормальное соотношение между объемами форменных элементов и плазмы. Отмечается в ранние сроки после переливания большого количества крови, при интенсивной физической нагрузке, когда в сосудистое русло поступают депонированная кровь и тканевая жидкость, при высокой температуре окружающей среды.
Гиперволемия олигоцитемическая, когда увеличение общего объема крови связано с увеличением объема плазмы крови (гидремия). Наблюдается при нарушениях выведения воды из организма (диффузный гломерулонефрит, острая и хроническая почечная недостаточность), в период схождения отеков при сердечной недостаточности, патологии почек, а также после введения кровезаменяющих жидкостей (кратковременная гиперволемия).
Гиперволемия полицитемическая, когда увеличение общего объема крови связано с преимущественным увеличением количества эритроцитов. Наблюдается при гипоксиях различного генеза - у жителей высокогорья (понижение атмосферного давления), у больных с эмфиземой легких и врожденными пороками сердца (как компенсаторная реакция костного мозга на гипоксию), при эритремии. При этом ОЦК может возрасти на 40-60% за счет увеличения массы эритроцитов.
Таблица 14-15. Типовые формы изменений объема циркулирующей крови и гематокрита
Вид нарушений | Показатель | |
ОЦК, % массы тела | гематокрит, % | |
Нормоволемия простая (нормоцитемическая) | 6,9-7,5 | 40-45 |
Нормоволемия олигоцитемическая | 6,9-7,5 | <40 |
Нормоволемия полицитемическая | 6,9-7,5 | >45 |
Гиперволемия простая (нормоцитемическая) | >7,5 | 40-45 |
Окончание табл. 14-15
Гиперволемия олигоцитемическая | >7,5 | <40 |
Гиперволемия полицитемическая | >7,5 | >45 |
Гиповолемия простая (нормоцитемическая) | <6,9 | 40-45 |
Гиповолемия олигоцитемическая | <6,9 | <40 |
Гиповолемия полицитемическая | <6,9 | >45 |
Гиповолемия (или олигемия) - уменьшение общего объема крови - может встречаться в трех вариантах.
Гиповолемия простая, когда при уменьшении общего объема крови пропорционально уменьшается количество плазмы и форменных элементов крови. Регистрируется в ранние сроки после кровопотери.
Гиповолемия полицитемическая, когда уменьшение общего объема крови связано с уменьшением объема плазмы. При этом имеют место относительное увеличение содержания эритроцитов в 1 мкл, сгущение и повышение вязкости крови. Развивается при обезвоживании организма (профузные поносы и рвота, перегревание организма, интенсивное потоотделение, отек легких, ожоговый шок).
Гиповолемия олигоцитемическая, при которой уменьшение объема крови связано главным образом с уменьшением содержания эритроцитов. Выявляется после острых кровопотерь, при анемиях, когда объем крови восстанавливается за счет поступления в сосудистое русло тканевой жидкости (олигоцитемическая нормоволемия).
14.4.2. Изменения вязкости и осмотического давления крови
Вязкость крови определяется по отношению к вязкости воды и зависит от содержания форменных элементов (главным образом эритроцитов) и белков плазмы. Если принять вязкость воды за 1, то средняя относительная вязкость крови у здорового взрослого человека составляет 4,5 (3,5-5,4), а вязкость плазмы - 2,2 (1,9- 2,6). При этом вязкость венозной крови выше, чем артериальной, что связано с поступлением в эритроциты углекислоты, обусловливающей увеличение размера клеток.
У новорожденных в первые сутки после рождения вязкость крови выше, чем у взрослого человека, и достигает 10-14 за счет
высокого содержания зрелых эритроцитов [(5,4-7,2)-1012/л], ретикулоцитов (от 8-13%с до 42%с), эритро- и нормобластов (до нескольких десятков процентов) в результате гипоксической стимуляции эритропоэза в период внутриутробного развития и в родах. К 5-7 дню после рождения вязкость крови снижается в связи с установлением внешнего дыхания (гипоксия сменяется гипероксией), разрушением HbF-содержащих эритроцитов. К концу первого месяца жизни она приближается к цифрам, характерным для взрослого человека.
У взрослых вязкость крови увеличивается с возрастом. Ее повышение отмечается также на фоне обильного белкового питания, при дегидратации, истинной полицитемии, опорожнении депо (селезенка, печень, легкие, костный мозг и др.), нарушении деформируемости и агрегации эритроцитов, активации факторов гемокоагуляции и др. Увеличение вязкости крови является неблагоприятным прогностическим признаком у больных атеросклерозом и людей, предрасположенных к таким заболеваниям, как ишемическая болезнь сердца (стенокардия, инфаркт миокарда), облитерирующий эндартериит, инсульт.
Осмотическое давление крови - сила, с которой растворитель (для крови это вода) переходит через полунепроницаемую мембрану из менее концентрированного в более концентрированный раствор. Осмотическое давление крови вычисляют криоскопическим методом путем определения точки замерзания крови (депрессии), которая равна 0,56-0,58 °С. Осмотическое давление крови при температуре 37 °С составляет 7,5-8,1 атм. Оно обусловлено растворенными в ней осмотически активными веществами, главным образом (на 99,5%) неорганическими веществами (около 60% осмотического давления создается солями натрия (NaСl)), безазотистыми органическими веществами (глюкоза) и мелкодисперсными белками (альбуминами). Создаваемое белками осмотическое давление называется онкотическим давлением, в норме оно не превышает 0,03-0,04 атм (или 25-30 мм рт.ст.).
Осмотическое давление крови играет важную роль в регуляции распределения воды между тканями и сосудами, межтканевой жидкостью и клетками. Функции клеток организма могут осуществляться лишь при его относительной стабильности, которая обеспечивается нейрогуморальными механизмами - антидиуритической и антинатрийуритической системами (см. разд. 12.8.1). На осмотическое давление крови могут оказывать влияние продукты
переваривания белков, жиров и углеводов, всасывающиеся в кровь и лимфу, а также низкомолекулярные продукты: метаболизма клеток. При повышении осмотического давления на фоне увеличения концентрации солей в крови (при гипертонической дегидратации, гиперосмолярной гипергидрии) эритроциты сморщиваются в результате обезвоживания. При снижении осмотического давления крови (на фоне гипоосмолярной гипергидрии, гипотонической дегидратации) эритроциты набухают посредством поглощения воды и подвергаются гемолизу, что приводит к развитию гемолитической анемии.
14.4.3. Изменение скорости оседания эритроцитов
Скорость оседания эритроцитов (СОЭ) - скорость разделения стабилизированной антикоагулянтами крови на два слоя: верхний - из прозрачной плазмы и нижний - из осевших эритроцитов. Основное влияние на скорость оседания эритроцитов, взвешенных в плазме, оказывает их агрегация, сила которой зависит от поверхностного заряда эритроцитов и концентрации в плазме асимметричных молекул (белков). Агрегация приводит к образованию скоплений и слипанию эритроцитов («монетные столбики»), смещающихся в нижние слои при отстаивании крови.
Величина СОЭ зависит от возраста и пола. В норме СОЭ составляет у женщин от 2 до 15, у мужчин - от 1 до 10 мм/ч. У новорожденных СОЭ не превышает 1-2 мм/ч, что в значительной степени связано с низкой концентрацией глобулинов и фибриногена, а также высоким содержанием эритроцитов в крови. Начиная со второго месяца после рождения, СОЭ возрастает и к концу первого года жизни приближается к 4-10 мм/ч. У девочек с появлением менструаций СОЭ может достигать 15 мм/ч.
При патологии величина СОЭ может изменяться, что зависит от следующих факторов:
1. От изменения соотношения различных фракций белков крови. При увеличении концентрации мелкодисперсных альбуминов в крови СОЭ уменьшается. Повышенное содержание крупнодисперсных белков (α-глобулины, γ-глобулины, фибриноген) при стрессе, интоксикации, воспалительных, инфекционных, онкологических заболеваниях ведет к увеличению СОЭ - слабозаряженные крупнодисперсные белки, адсорбируясь на отрицательно заряженных эритроцитах, уменьшают их поверхностный заряд и способствуют
тем самым сближению, агглютинации и более быстрому оседанию последних (табл. 14-16), увеличивают массу оседающих эритроцитов. Особенно выраженное ускорение СОЭ (60-80 мм/ч) характерно для заболеваний, сопровождающихся продукцией и накоплением в крови моноклональных иммуноглобулинов (парапротеинов), - при парапротеинемических гемобластозах (множественная миелома, макроглобулинемия Вальденстрема и др.) и симптоматических парапротеинемиях, сопутствующих злокачественным новообразованиям, хроническому гепатиту, циррозу печени, туберкулезу, амилоидозу, коллагенозам.
Таблица 14-16. Влияние белков плазмы на скорость оседания эритроцитов
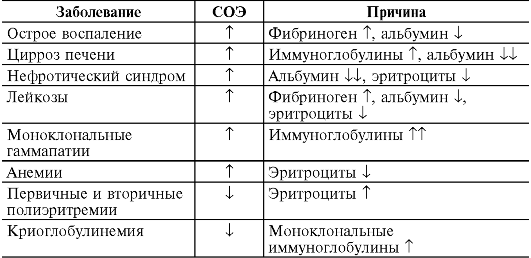 2. От объема, числа и диаметра эритроцитов. Их увеличение замедляет, а уменьшение ускоряет оседание эритроцитов.
2. От объема, числа и диаметра эритроцитов. Их увеличение замедляет, а уменьшение ускоряет оседание эритроцитов.
3. От содержания холестерина, лецитина, желчных кислот и желчных пигментов в крови. Холестерин, адсорбируясь на эритроцитах, ускоряет, а лецитин, желчные кислоты и пигменты, напротив, замедляют СОЭ.
4. От рН крови. При увеличении рН (алкалозе) отмечается ускорение, при уменьшении рН (ацидозе) - замедление СОЭ. При гиперкапнии (асфиксия, сердечная декомпенсация) СОЭ замедляется вследствие увеличения диаметра эритроцитов и уменьшения их относительной плотности.
5. От вязкости крови. Гидремия приводит к ускорению оседания эритроцитов, с увеличением вязкости крови (при обезвоживании) СОЭ замедляется.
Большое влияние на СОЭ оказывают прием некоторых лекарств и терапевтические мероприятия. Так, ускорение оседания эритроцитов отмечается при специфической и неспецифической раздражающей терапии, вакцинотерапии, переливании крови, длительных приемах соды, витамина А, контрацептивов и т.д. Замедление СОЭ наблюдается при приеме салициловых, ртутных и кальциевых препаратов, диуретиков, снотворных и противомалярийных средств.
Ускорение оседания эритроцитов отмечается также при сухоядении, голодании, что связано с увеличением в крови содержания фибриногена и глобулинов из-за распада белков тканей.
В физиологических условиях СОЭ ускоряется при интенсивной физической работе за счет распада миоглобина, во время беременности и в послеродовом периоде (в течение нескольких недель после родов) - в результате увеличения объема плазмы, повышения концентрации глобулинов, холестерина и падения уровня кальция в крови.
14.4.4. Изменение резистентности эритроцитов
Резистентность (стойкость) эритроцитов - способность их противостоять различным разрушительным воздействиям: осмотическим, механическим, химическим, физическим и пр. Наибольшее практическое значение имеет определение осмотической резистентности - устойчивости эритроцитов в гипотонических растворах. Осмотическая резистентность эритроцитов определяется по соотношению площади поверхности клетки к ее объему. Объемные эритроциты (сфероциты, стоматоциты) характеризуются пониженной, а плоские (гипохромные, мишеневидные) эритроциты - повышенной осмотической резистентностью.
Как уже указывалось выше (см. разд. 14.4.2), в гипертонических солевых растворах эритроциты теряют воду и сморщиваются, а в гипотонических - поглощают воду и набухают. При значительном набухании происходит гемолиз. Изотоническим раствором для эритроцитов является 0,85% раствор хлорида натрия. В 0,48-0,44% растворах NaCl разрушаются наименее резистентные эритроциты (минимальная осмотическая резистентность, верхняя граница резистентности). При концентрации 0,32-0,28% полностью гемолизируются все эритроциты (максимальная осмотическая резистентность, нижняя граница резистентности).
Уменьшение осмотической резистентности эритроцитов (повышение показателей минимальной и максимальной резистентности) отмечается при аутоиммунной гемолитической анемии, обусловленной тепловыми антителами, гемолитической болезни новорожденных и наследственных микросфероцитозе, стоматоцитозе, а также (в слабой степени выраженности) при токсикозах, бронхопневмониях, гемобластозах, циррозах печени и др. Увеличение осмотической резистентности эритроцитов имеет место при механической желтухе, в некоторых случаях полицитемии и железодефицитной анемии, а также при талассемии, гемоглобинозе S и после массивных кровопотерь.
14.4.5. Изменения количественного и качественного состава гемоглобина
Гемоглобин - основной компонент эритроцитов (составляет около 95% сухого остатка). По химической природе гемоглобин относится к хромопротеидам и имеет в своем составе белок (глобин) и комплексное соединение железа и протопорфирина IX (гем).
Различие аминокислотного состава полипептидных цепей глобина определяет гетерогенность молекулы гемоглобина. В эритроцитах человека на разных этапах развития в норме определяются 6 типов гемоглобина: эмбриональный (Gower 1, Gower 2, Portland), фетальный (HbF), взрослый (НЬА1, НЬА2). Время появления гемоглобинов в организме и их количественное соотношение представлено в табл. 14-17.
Таблица 14-17. Типы гемоглобина в онтогенезе человека (по А.В. Папаян, Л.Ю. Жуковой, 2001)
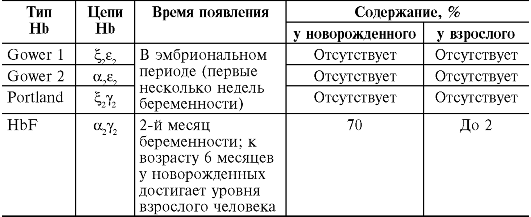
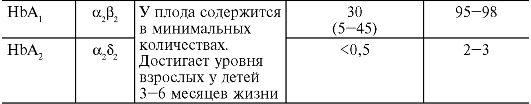 У взрослого человека основную массу гемоглобина в эритроцитах составляет гемоглобин А (гемоглобин взрослых): А1 и А2.
Около 1-2% приходится на гемоглобин F. Увеличение содержания HbF в
крови отмечается при гетерозиготном и гомозиготном вариантах
β-талассемии, у больных гемоглобинопатиями с дефектами β-цепей (HbSS, SС
и др.), при апластических анемиях, лейкозах. При α-талассемии могут
обнаруживаться тетрамеры γ- или β-цепей. Уровень HbА2
повышается (свыше 3,4%) также у носителей гена β-талассемии, при
мегалобластных анемиях, связанных с недостаточностью витамина В12 и фолиевой кислоты. Снижение уровня HbА2 характерно для α-талассемии, железодефицитной и сидеробластной анемий.
У взрослого человека основную массу гемоглобина в эритроцитах составляет гемоглобин А (гемоглобин взрослых): А1 и А2.
Около 1-2% приходится на гемоглобин F. Увеличение содержания HbF в
крови отмечается при гетерозиготном и гомозиготном вариантах
β-талассемии, у больных гемоглобинопатиями с дефектами β-цепей (HbSS, SС
и др.), при апластических анемиях, лейкозах. При α-талассемии могут
обнаруживаться тетрамеры γ- или β-цепей. Уровень HbА2
повышается (свыше 3,4%) также у носителей гена β-талассемии, при
мегалобластных анемиях, связанных с недостаточностью витамина В12 и фолиевой кислоты. Снижение уровня HbА2 характерно для α-талассемии, железодефицитной и сидеробластной анемий.
Мутации в генах, ответственных за синтез гемоглобина, сопровождаются образованием аномальных гемоглобинов, что характерно, в частности, для серповидно-клеточной анемии (HbS), гомозиготных гемоглобинопатий (HbСС, HbEE, HbDD и др.).
Гемоглобин определяет цвет крови. В крови гемоглобин существует в двух основных формах: оксигемоглобин (HbO2), придающий артериальной крови ярко-красный цвет, и дезоксигемоглобин (восстановленный гемоглобин, HbH), обусловливающий темнокрасную с синеватым оттенком окраску крови. Некоторые патологические формы гемоглобина, неспособные к переносу кислорода к тканям, могут изменять цвет крови. К ним относятся метгемоглобин (гемиглобин, HbMet) и сульфгемоглобин (SHb), образующиеся в результате токсического действия различных химических веществ (нитраты и нитриты, анилин, бензол, пиридин и др.). Их физиологический уровень в крови не превышает 1%. Присутствие в крови HbMet, SHb свыше 15% придает крови коричневый цвет («шоколадная кровь»). В противоположность HbMet и SHb, карбоксигемоглобин (HbCO), формирующийся при отравлении угарным газом (СО) и карбонилами металлов (Ni(CO)4; Fe(CO)5), имеет яркий вишнево-красный цвет, и его присутствие нельзя ви-
зуально определить по цвету крови. Для определения содержания СО в крови проводятся спектрофотометрический анализ крови, а также цветовые химические пробы с формалином, дистиллированной водой, меняющими ярко-красный цвет СО-содержащей крови на малиновый, или реакция с 50% раствором КОН, придающим крови в присутствии СО коричневато-красный оттенок.
14.4.6. Активация протеолитических систем плазмы крови
К протеолитическим системам плазмы крови относятся системы комплемента, калликреин-кининовая, а также фибринолитическая и свертывания крови (подробнее см. ниже раздел 14.5). Все они играют определенную роль в физиологических процессах, а также участвуют в развитии некоторых компенсаторных приспособительных реакций организма при действии на него различных повреждающих факторов. И только в случаях, когда активация этих систем становится неоптимальной, несоответствующей данным конкретным условиям, они превращаются в патогенный фактор, обусловливающий развитие патологического процесса.
Калликреин-кининовая система. Активация этой системы приводит к образованию кининов (рис. 14-13). Кинины - группа биологически активных нейровазоактивных полипептидов. Наиболее изученными являются калликреин-кининовая система плазмы крови и один из кининов - нонапептид брадикинин.
Физиологическое значение кининов основано на том, что они оказывают непосредственное влияние на тонус и проницаемость
 Рис. 14-13. Активация калликреин-кининовой системы
Рис. 14-13. Активация калликреин-кининовой системы
сосудистой стенки, вызывая расширение прекапиллярных сосудов и увеличивая проницаемость капилляров. В связи с этим кинины играют особую роль в органах, периодически экскретирующих значительные количества жидкости (слюнные железы, поджелудочная железа, потовые железы, желудок, кишечник).
Активация калликреин-кининовой системы происходит при действии на организм различных повреждающих факторов, нарушающих целостность клеток и тканей и приводящих, как правило, к активации фактора Хагемана. Это - травмы, токсины, облучение, накопление продуктов обмена веществ (например, кристаллов мочекислого натрия), ишемия и др. Обычно в результате местных повреждающих воздействий развивается воспаление. В его развитии определенную роль играет увеличение содержания кининов, которые через изменение сосудистой реакции оказывают влияние на интенсивность и характер воспаления, а также участвуют в формировании чувства боли. Участвуют кинины и в развитии общих реакций организма на повреждение, причем главным образом в формировании компенсаторно-приспособительных механизмов, и только в случаях неадекватного их образования кинины могут стать патогенетическим фактором различных расстройств.
Одно из таких компенсаторно-приспособительных влияний выявляется в генерализованном действии на гемодинамику. При определенной концентрации кинины уменьшают периферическое сопротивление сосудов малого и большого кругов кровообращения, что увеличивает возврат крови к сердцу, а это, в свою очередь, увеличивает ударный объем обоих желудочков сердца. Этот механизм может включаться при срочных или длительных адаптивных реакциях организма в условиях действия на него различных факторов в виде эмоциональных или физических нагрузок, тепла, гипоксии и др. При острой ишемии и инфаркте миокарда компенсаторная роль увеличенного образования кининов сводится к расширению сосудов миокарда и увеличению сердечного выброса, а также к развитию гипотензии, что облегчает работу сердца и вызывает перераспределение крови. Неадекватность активации калликреин-кининовой системы может стать патогенетическим фактором развития фатальной гипотензии, шока, болевого эффекта (кардиогенный шок).
Кинины принимают участие в развитии реакций при аллергической альтерации тканей. Аллергическое воспаление, как и обычное, также сопровождается увеличением концентрации кининов.
Их обнаруживают в довольно значительной концентрации в экссудате суставов при ревматоидном артрите. Кроме того, увеличение их содержания в крови и спинно-мозговой жидкости выявляется у собак с экспериментальным аллергическим энцефаломиелитом, в миокарде и плазме крови кроликов с экспериментальным аллергическим миокардитом.
Установлено 10-15-кратное увеличение содержания кининов в крови больных людей во время обострения бронхиальной астмы. Очевидно, кинины играют определенную роль в развитии бронхоспазма, так как обладают способностью вызывать при определенной концентрации спазм гладкой мускулатуры бронхиол. Сокращение гладкомышечных клеток при взаимодействии кининов со специфическими мембранными рецепторами приводит к активации кальциевых каналов и поступлению кальция в цитоплазму, где он и стимулирует процесс сокращения. Это действие усиливается на фоне снижения активности β-адренергических рецепторов, что, в частности, имеет место у больных бронхиальной астмой. В такой ситуации концентрация кининов, недостаточная для индукции бронхоспазма у здорового человека, способна вызвать его у больного, имеющего пониженную активность β-адренергических рецепторов.
Активация калликреин-кининовой системы обнаружена при шоках различной этиологии, ревматизме, нефритах, артритах, карциноидном и демпинг-синдромах, атеросклерозе, гипертонической болезни и ряде других заболеваний. Соотношение защитного и патогенного компонентов в каждом конкретном случае различно. Применяя ингибиторы протеолиза, можно ограничить активность калликреин-кининовой системы, а следовательно, выраженность соответствующих симптомов и интенсивность развития патологического процесса.
Комплемент - система функционально связанных сывороточных белков (C1, C2 и т.д.), активация которых приводит к образованию биологически активных веществ, участвующих в защитных реакциях организма. Различают два пути активации комплемента: классический и альтернативный.
Классическийпутьактивируетсякомплексом«антиген+антитело» (рис. 14-14). В процессе активации происходит расщепление ряда компонентов комплемента (С) с образованием активных продуктов. Некоторые из них удаляются из цепи активации, другие объединяются. Конечным этапом активации является образова-
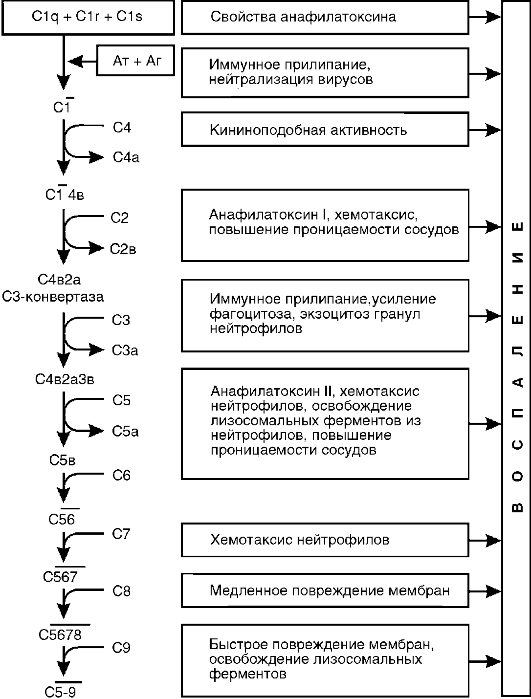 Рис. 14-14. Классический путь активации комплемента и биологические эффекты продуктов его активации
Рис. 14-14. Классический путь активации комплемента и биологические эффекты продуктов его активации
ние комплекса С5-9, оказывающего цитотоксическое действие на клетки-мишени (клетки тканей, микробы с фиксированными на них антителами). Если проанализировать эффекты различных компонентов С, то окажется, что все они участвуют в развитии того или иного компонента воспалительной реакции. Поэтому сложилось представление, что биологический смысл активации С
заключается в подключении к иммунной (специфической) реакции неспецифических механизмов зашиты - фагоцитоза, воспаления, при помощи которых образовавшийся комплекс фиксируется и фагоцитируется.
Альтернативный путь активации С является важнейшим механизмом противоинфекционной защиты и активируется бактериальными полисахаридами. Он включается быстро и без участия иммунных механизмов. В отличие от классического пути активация начинается с расщепления С с участием ряда дополнительных факторов. Процессы активации комплемента контролируются ингибиторами различных звеньев этой системы. Наиболее изучены С1- и С3-ингибиторы. Cl-ингибитор эстеразы блокирует спонтанную активацию С1. Кроме того, он ограничивает активность калликреин-кининовой и фибринолитической систем.
Неконтролируемая активация комплемента приводит к развитию патологических процессов. Возможны генетически детерминированные дефициты отдельных ингибиторов, передающиеся по аутосомно-рецессивному типу. Так, при дефиците Cl-ингибитора различные, даже не очень выраженные повреждения запускают начальную цепь классического пути активации комплемента до С3, которая обрывается С3-ингибитором. В результате формируется врожденный ангионевротический отек в связи с образованием С2в фрагмента, обладающего кининоподобной активностью (см. рис. 14-14). При дефиците С3-ингибитора усиливается действие С3 и нарушается функционирование альтернативного пути активации, что приводит к снижению противоинфекционной защиты с развитием тяжелых бактериальных инфекций (пневмония, отиты, гаймориты, менингиты).
Встречаются случаи дефицитов отдельных компонентов системы С. Они обычно передаются также по аутосомно-рецессивному типу и служат причиной бактериальных инфекций и волчаночноподобных синдромов.
Избыточная активация того или иного компонента комплемента является патогенетическим фактором ряда патологических процессов. Она лежит в основе многих случаев неиммунологических аллергических реакций (псевдоаллергических) на лекарственные препараты, принимает в той или иной степени участие в развитии некоторых видов шока, особенно септического. Последнее связано с выраженными активирующими свойствами эндотоксина. Эндотоксин является универсальным активатором ряда протеоли-
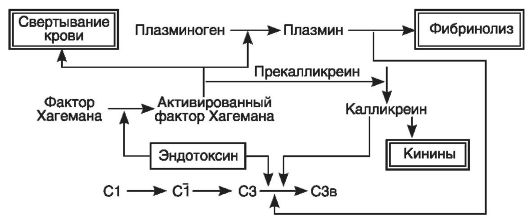 Рис. 14-15. Эндотоксины как активаторы протеолитических систем плазмы крови
Рис. 14-15. Эндотоксины как активаторы протеолитических систем плазмы крови
тических систем (рис. 14-15) плазмы крови. И если эта активация не носит лавинообразного характера, то только потому, что она ограничивается различными ингибиторами и механизмами обратной связи.
Активирующим действием обладают и некоторые эндогенно образующиеся ферменты. Так, трипсин, плазмин, калликреин могут запускать альтернативный путь активации комплемента. Все эти энзимы обычно активируются при различных повреждающих воздействиях.
14.5. ПАТОЛОГИЯ СИСТЕМЫ ГЕМОСТАЗА. ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ И СИНДРОМЫ. ТРОМБОФИЛИИ
Система гемостаза - совокупность биологических и биохимических механизмов, обеспечивающих сохранение жидкого состояния циркулирующей крови, поддержание целостности кровеносных сосудов и купирование кровотечения при их повреждении. От функционирования системы гемостаза в значительной степени зависят состояние микроциркуляции в органах и тканях и уровень их кровоснабжения. Патология этой системы проявляется кровоточивостью либо развитием тромбозов сосудов, ишемий и инфарктов органов.
Осуществляется гемостаз тремя взаимодействующими между собой морфофункциональными компонентами: стенками крове-
носных сосудов, клетками крови (в первую очередь тромбоцитами) и плазменными системами - свертывающей, антикоагулянтной, фибринолитической (плазминовой) и калликреин-кининовой.
Первыми на повреждение реагируют кровеносные сосуды и тромбоциты. Именно этой реакции отводится ведущая роль в предупреждении и остановке кровотечений из поврежденных микрососудов. Поэтому сосудисто-тромбоцитарная реакция на повреждение обозначается как первичный гемостаз, а последующее свертывание крови при участии плазменных факторов - как вторичный, хотя оба эти механизма взаимно потенцируют друг друга и функционируют сопряженно.
14.5.1. Сосудисто-тромбоцитарный гемостаз
14.5.1.1. Функции эндотелия
В нормальных условиях эндотелий кровеносных сосудов обладает высокой тромборезистентностью, поддерживает жидкое состояние крови и препятствует образованию тромбов, что обеспечивается следующими его свойствами:
• отрицательным зарядом и контактной инертностью внутренней (обращенной в просвет сосуда) поверхности эндотелиальных клеток;
• синтезом и высвобождением в кровь веществ, инактивирующих тромбоциты (простациклин, оксид азота - NO) и обладающих фибринолитической активностью (тканевой активатор плазминогена - t-PA (tissue plasminogen activator));
• наличием АДФазы - фермента, который расщепляет АДФ, являющийся одним из основных стимуляторов агрегации тромбоцитов;
• синтезом гепарина и гепариноподобных сульфатированных гликозаминогликанов (дерматансульфат, гепарансульфат), усиливающих действие ингибиторов протеаз свертывания крови - естественных антикоагулянтов, в частности антитромбина III (гепарин образует комплекс с антитромбином III на эндотелии);
• способностью связывать и инактивировать тромбин, что обеспечивается присутствием на мембране эндотелиоцитов особого гликопротеина - тромбомодулина. В результате тромбин утрачивает способность вызывать свертывание крови, но сохраняет свое активирующее действие на систему двух
важнейших антикоагулянтов - протеинов С и S (см. ниже). Благодаря связыванию тромбина с тромбомодулином на цитоплазматической мембране эндотелия тромбин трансформируется из главного фактора свертывания крови в противосвертывающий агент;
• элиминацией из крови активированных факторов свертывания крови и их метаболитов;
• метаболизмом биологически активных веществ, прямо или косвенно влияющих на систему гемостаза и стенку кровеносных сосудов (простагландины, тромбоксан А2 (ТхА^, фактор активации тромбоцитов (ФАТ), плазменные кинины и др.).
Вместе с тем эндотелий обладает уникальной способностью менять свой антитромботический потенциал на тромбогенный, что происходит при его повреждении экзо- и эндотоксинами, антителами и иммунными комплексами (при иммунных васкулитах и инфекционно-иммунных процессах), медиаторами воспаления (цитокины, фактор некроза опухоли), лейкоцитарными протеазами (эластазой и др.), при повреждающем действии Н2О2 и ряде метаболических поражений сосудов (диабет, гиперлипидемии, гипергомоцистеинемия и др.). В последнем случае эндотелий сохраняет морфологическую целостность, но утрачивает тромборезистентность.
При гибели эндотелиальных клеток обнажается субэндотелий, содержащий фибриллярный белок - коллаген, при контакте с которым происходят фиксация, распластывание и активация тромбоцитов на стенке сосуда (рис. 14-16) с последующим образованием в районе повреждения эндотелия тромбоцитарного (белого) тромба. С другой стороны, коллаген активирует плазменные факторы свертывания крови с образованием коагуляционных (красных) тромбов.
 Рис. 14-16. Тромбоциты: интактный (слева), при распластывании (в центре), с псевдоподиями (справа)
Рис. 14-16. Тромбоциты: интактный (слева), при распластывании (в центре), с псевдоподиями (справа)
Повреждение эндотелия является также важным фактором развития и прогрессирования атеросклероза, утолщения сосудистой стенки и возникновения облитерирующих заболеваний артерий. Дефекты субэндотелия и его обеднение коллагеном лежат в основе ряда наследственных мезенхимальных дисплазий, протекающих с кровоточивостью и развитием аневризм сосудов и их шунтов (телеангиэктазия, или болезнь Рандю-Ослера, аневризмы аорты при синдроме Марфана и др.).
14.5.1.2. Тромбоциты и их участие в гемостазе
Тромбоциты играют главную роль в поддержании нормальной жизнедеятельности и функции эндотелиальных клеток и в осуществлении первичного гемостаза при повреждении сосудов.
Образуясь путем отшнуровывания от цитоплазмы полиплоидных клеток костного мозга - мегакариоцитов, тромбоциты в виде округлых или овальных плоских дисков (кровяные пластинки) диаметром 2-4 мкм поступают в кровь, в которой у человека их количество колеблется в пределах (150-400)-109/л. Продолжительность жизни тромбоцитов составляет 8-11 дней. Тромбоциты частично депонируются в селезенке и печени (20-25% клеток), откуда осуществляется вторичный их выход в кровоток. Разрушаются тромбоциты в селезенке, печени и легких.
Тромбоциты выполняют различные функции в организме. Они способны адсорбировать и транспортировать биологически активные вещества, циркулирующие иммунные комплексы (сорбционнотранспортная функция); оказывать провоспалительное действие путем высвобождения кислых гидролаз, серотонина, бактерицидного, хемотаксического и других факторов, содержащихся в гранулах тромбоцитов (воспалительная функция); поглощать и разрушать чужеродные частицы, вирусы и антитела (защитная функция).
Участие тромбоцитов в гемостазе определяется следующими их свойствами:
• поддержание нормальной структуры, функции и адекватной проницаемости стенок микрососудов (ангиотрофическая функция);
• реэндотелизация сосудистой стенки при повреждении путем высвобождения ростового фактора, стимулирующего размножение эндотелиоцитов и гладкомышечных клеток, образование коллагена (репарационная функция);
• поддержание спазма поврежденных сосудов путем секреции вазоактивных веществ - серотонина, катехоламинов, β-тромбоглобулина и др. (ангиоспастическая функция);
• регуляция свертывания крови и фибринолиза, экспонирование тромбогенной фосфолипидной поверхности для взаимодействия плазменных факторов и антикоагулянтов (коагуломодулирующая функция);
• уплотнение (ретракция) тромба (ретрактильная функция);
• прилипание к субэндотелиальным структурам поврежденной сосудистой стенки (адгезия), образование скоплений (агрегация) (адгезивно-агрегационная функция).
Адгезия и агрегация тромбоцитов осуществляются посредством поверхностных рецепторов - гликопротеинов (ГП), встроенных в фосфолипидную мембрану тромбоцитарных клеток: ГП Ia/IIa- рецепторов к коллагену; ГП Iβ, взаимодействующего с фактором Виллебранда; ГП IIβ/IIIα, связывающихся с фибриногеном, АДФ, адреналином, арахидоновой кислотой, простагландинами и др. агонистами агрегации; ГП IIIβ (или ГП IV)-рецептора для тромбоспондина. Адгезивно-агрегационная функция тромбоцитов в значительной степени зависит от транспорта в клетки Са2+ и образования арахидоновой кислоты и циклических производных простагландинов из фосфолипидов мембраны тромбоцитов. Из внутренних органелл, локализующихся в цитоплазме (гиаломере) тромбоцитов, наиболее важны в функциональном отношении система микротрубочек и микрофиламентов, содержащих сходный с актомиозином сократительный белок (тромбастенин), и гранулярный аппарат (грануломер), состоящий из гранул высокой плотности (плотные гранулы, или плотные тельца), α-гранул и лизосом. При активации тромбоцитов содержимое гранул (табл. 14-18) высвобождается. Высвободившиеся компоненты гранул опосредуют агрегацию тромбоцитов и образование тромба.
Таблица 14-18. Компоненты гранул тромбоцитов
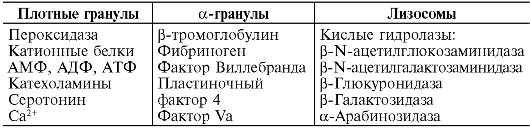

Примечание. ФСФ - фибринстабилизирующий фактор.
14.5.1.3. Механизм сосудисто-тромбоцитарного гемостаза
Активация сосудисто-тромбоцитарного (первичного) гемостаза обусловливает полную остановку кровотечения из капилляров и венул и временную остановку кровотечения из вен, артериол и артерий путем формирования первичной гемостатической пробки, на основе которой при активации вторичного (коагуляционного) гемостаза формируется тромб. Ключевыми механизмами тромбообразования являются: повреждение сосудистого эндотелия; локальный ангиоспазм; адгезия тромбоцитов к участку обнаженного субэндотелия; агрегация тромбоцитов; активация свертывающей способности крови при снижении ее литических свойств.
Стадии сосудисто-тромбоцитарного гемостаза (рис. 14-17):
1. Повреждение эндотелия и первичный спазм сосудов.
На повреждение микрососуды отвечают кратковременным спазмом, в результате чего кровотечение из них в первые 20-30 с не возникает. Эта вазоконстрикция определяется капилляроскопически при нанесении укола в ногтевое ложе и регистрируется по начальной задержке появления первой капли крови при проколе кожи скарификатором. Она обусловлена рефлекторным спазмом сосудов за счет сокращения гладкомышечных клеток сосудистой стенки и поддерживается вазоспастическими агентами, секретируемыми эндотелием и тромбоцитами - серотонином, ТхА2, норадреналином и др.
Повреждение эндотелия сопровождается снижением тромборезистентности сосудистой стенки и обнажением субэндотелия,
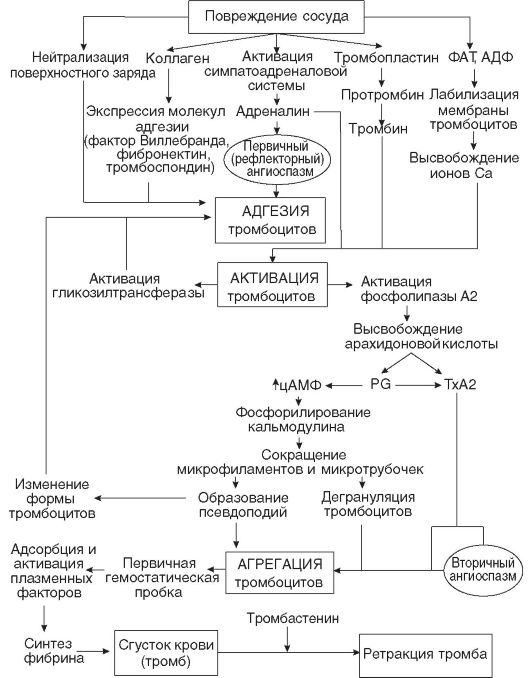 Рис. 14-17. Схема сосудисто-тромбоцитарного гемостаза. PG - простогландины, ТхА - тромбоксан ФАТ - фактор активации тромбоцитов
Рис. 14-17. Схема сосудисто-тромбоцитарного гемостаза. PG - простогландины, ТхА - тромбоксан ФАТ - фактор активации тромбоцитов
который содержит коллаген и экспрессирует адгезивные белки - фактор Виллебранда, фибронектин, тромбоспондин.
2. Адгезия тромбоцитов к участку деэндотелизации осуществляется в первые секунды после повреждения эндотелия посредством сил электростатического притяжения в результате снижения величины поверхностного отрицательного заряда сосудистой стенки при нарушении ее целостности, а также рецепторов тромбоцитов к коллагену (ГП Ia/IIa) с последующей стабилизацией образовавшегося соединения белками адгезии - фактором Виллебранда, фибронектином и тромбоспондином, образующих «мостики» между комплементарными им ГП тромбоцитов (см. выше - 14.5.1.2.) и коллагеном.
3. Активация тромбоцитов и вторичный спазм сосудов.
Активацию вызывают тромбин, образующийся из протромбина под влиянием тканевого тромбопластина, ФАТ, АДФ (высвобождаются одновременно с тромбопластином при повреждении сосудистой стенки), Са2+, адреналин. Активация тромбоцитов является сложным метаболическим процессом, связанным с химической модификацией тромбоцитарных мембран и индукцией в них фермента гликозилтрансферазы, который взаимодействует со специфическим рецептором на молекуле коллагена и обеспечивает тем самым «посадку» тромбоцита на субэндотелий. Наряду с гликозилтрансферазой активируются и другие мембраносвязанные ферменты, в частности фосфолипаза А2, обладающая наибольшей аффинностью по отношению к фосфатидилэтаноламину. Гидролиз последнего запускает каскад реакций, включающих высвобождение арахидоновой кислоты и последующее образование из нее под действием фермента циклооксигеназы короткоживущих простагландинов (PGG2, PGH2), трансформирующихся под влиянием фермента тромбоксансинтетазы в один из самых мощных индукторов агрегации тромбоцитов и вазоконстрикторов - ТхА2.
Простагландины способствуют накоплению в тромбоцитах цАМФ, регулируют фосфорилирование и активацию белка кальмодулина, транспортирующего ионы Са2+ из плотной тубулярной системы тромбоцитов (эквивалент саркоплазматического ретикулума мышц) в цитоплазму. В результате происходит активация сократительных белков актомиозинового комплекса, что сопровождается сокращением микрофиламентов тромбоцитов с образованием псевдоподий. Это еще более усиливает адгезию тромбоцитов к поврежденному эндотелию. Наряду с этим за счет Са2+-индуцированного сокращения микротрубочек гранулы тромбоцитов «подтягивают-
ся» к плазматической мембране, происходит слияние мембраны депонирующих гранул со стенкой мембраносвязанных канальцев, через которые происходит опорожнение гранул. Реакция высвобождения компонентов гранул осуществляется в две фазы: первая фаза характеризуется выбросом содержимого плотных гранул, вторая - α-гранул (см. табл. 14-18).
ТхА2 и освобождаемые из плотных гранул тромбоцитов вазоактивные вещества вызывают вторичный спазм сосудов.
4. Агрегация тромбоцитов.
ТхА2 и высвобождаемые при дегрануляции тромбоцитов АДФ, серотонин, β-тромбоглобулин, пластиночный фактор 4, фибриноген и др. компоненты плотных гранул и α-гранул обусловливают слипание тромбоцитов друг с другом и с коллагеном. Кроме того, появление в кровотоке ФАТ (при разрушении эндотелиоцитов) и компонентов тромбоцитарных гранул приводит к активации интактных тромбоцитов, их агрегации друг с другом и с поверхностью адгезированных на эндотелии тромбоцитов.
Агрегация тромбоцитов не развивается при отсутствии внеклеточного Са2+, фибриногена (обусловливает необратимую агрегацию тромбоцитов) и белка, природа которого пока не выяснена. Последний, в частности, отсутствует в плазме крови больных тромбастенией Гланцмана.
5. Образование гемостатической пробки.
В результате агрегации тромбоцитов образуется первичная (временная) гемостатическая пробка, закрывающая дефект сосуда. В отличие от сгустка крови тромбоцитарный агрегат не содержит нитей фибрина. Впоследствии на поверхности агрегата из тромбоцитов адсорбируются плазменные факторы свертывания и запускается «внутренний каскад» коагуляционного гемостаза, завершающийся выпадением нитей стабилизированного фибрина и формированием на основе тромбоцитарной пробки сгустка крови (тромба). При сокращении тромбастенина (от греч. stenoo - стягивать, сжимать) тромбоцитов тромб уплотняется (ретракция тромба). Этому также способствует снижение фибринолитической активности крови, ответственной за лизис фибриновых сгустков.
Наряду с «внутренним каскадом» в процесс тромбообразования включается и «внешний каскад» свертывания крови, связанный с высвобождением тканевого тромбопластина. Кроме того, тромбоциты могут самостоятельно (при отсутствии контактных факторов) запускать свертывание крови путем взаимодействия экспони-
рованного на их поверхности фактора Уа с фактором плазмы Ха, катализирующим превращение протромбина в тромбин.
Таким образом, тромбоциты выполняют роль поверхности, на которой формируется тромб. При отсутствии этой поверхности тромбообразование в артериальной циркуляции невозможно изза высокой скорости кровотока и связанного с ней разведения и удаления активированных белков свертывания крови из области повреждения сосуда.
Для оценки сосудисто-тромбоцитарного гемостаза определяют:
• резистентность (ломкость) сосудов с помощью манжеточной пробы (в норме не более 10 петехий, образующихся в круге диаметром 5 см на ладонной поверхности предплечья при дозированном повышении венозного давления);
• время кровотечения из прокола кожи на ладонной поверхности верхней трети предплечья по методу Айви (в норме 5- 8 мин) или из мочки уха - проба Дьюка (норма 2-4 мин);
• количество, размеры, спонтанную и индуцированную (АДФ, адреналином, коллагеном, арахидоновой кислотой и др.) агрегацию тромбоцитов;
• уровень фактора Виллебранда в плазме крови (при использовании метода фотоэлектроколориметрии - 80-120%, при использовании агрегометра - не менее 40%);
• ретракцию кровяного сгустка (в норме 48-60%).
При уменьшении количества тромбоцитов в крови, а также при ряде качественных дефектов тромбоцитов эндотелий становится неполноценным, вакуолизируется, слущивается, повышается ломкость микрососудов. Одновременно нарушается адгезивноагрегационная функция тромбоцитов. Это приводит к удлинению и усилению кровоточивости из поврежденных микрососудов. Исследование различных видов агрегации тромбоцитов (агрегатометрия), изучение их ультраструктуры (определение наличия плотных гранул и α-гранул), определение структуры и функции основных рецепторов этих клеток и фактора Виллебранда позволяют уточнить природу тромбоцитопатии.
С другой стороны, повышение количества тромбоцитов, их адгезивности и агрегации (так называемый синдром вязких, или липких, тромбоцитов), содержания и мультимерности фактора Виллебранда способствуют возникновению у больных тромбозов, ишемий и инфарктов органов, облитерирующих заболеваний артерий конечностей (см. раздел 14.5.6).
Помимо тромбоцитов, в образовании внутрисосудистых тромбов принимают участие и другие клетки крови, в частности эритроциты и лейкоциты. Способность указанных клеток к индукции тромботического процесса связана не только с пассивным захватом их фибриновой сетью, но и с активным воздействием на гемостатический процесс. Последнее особенно наглядно выявляется при гемолизе эритроцитов, сопровождающемся обильным «наводнением» плазмы АДФ и развитием необратимой агрегации тромбоцитов. Нередко причиной развития артериального тромбоза являются эритроцитоз, приводящий к увеличению вязкости крови и застою ее в системе микроциркуляции, сфероцитоз и серповидно-клеточная анемия, при которой закупорка мелких сосудов может произойти вследствие потери эритроцитами эластичности и деформируемости. Имеются доказательства того, что эритроциты в силу крупных размеров оттесняют циркулирующие рядом с ними в потоке крови тромбоциты к периферии и облегчают адгезию последних к субэндотелию.
Роль лейкоцитов в механизмах тромбообразования изучена менее подробно, однако известно, что в лейкоцитах активно синтезируются продукты липоксигеназного пути метаболизма арахидоновой кислоты, и в частности лейкотриены, которые способны оказывать существенное влияние на активность тромбоцитарной тромбоксансинтетазы с образованием ТхА2. К тому же в нейтрофилах и других клетках гранулоцитарного ряда синтезируется тромбоцитактивирующий фактор, который тоже может стимулировать повышенную агрегацию тромбоцитов и развитие тромбоза.
Из других внутриклеточных компонентов лейкоцитов, высвобождение которых при острых или хронических воспалительных процессах, а также сепсисе способно активировать циркулирующие в крови интактные тромбоциты и запускать внутрисосудистую агрегацию, наибольшее значение имеют супероксидные и гидроксильные анионрадикалы, лизосомальные гидролазы, ферменты, расщепляющие гепарин, протеиназы типа нейтрофилина и др.
К тромбогенным компонентам лимфоцитов относятся лимфокины, высвобождающиеся, к примеру, из Т-эффекторов при реакциях замедленного типа.
14.5.2. Коагуляционный гемостаз
В процессе коагуляционного (вторичного) гемостаза на субэндотелии на основе тромбоцитарного агрегата формируется сгусток
крови, который на завершающей стадии подвергается сжатию (ретракции). Таким образом, первичная или временная гемостатическая пробка, представляющая собой рыхлый тромбоцитарный агрегат, превращается во вторичную гемостатическую пробку (тромб), в которой тромбоцитарный агрегат консолидируется фибрином и подвергается дополнительному уплотнению. Вторичный гемостаз обеспечивает полную остановку кровотечения из артериол, артерий и вен. Активации плазменного звена гемостаза в венах при отсутствии предварительной активации сосудисто-тромбоцитарного гемостаза благоприятствует гемодинамическая ситуация, создающаяся вблизи венозных клапанов и в местах бифуркаций с замедленным турбулентным потоком крови.
Вторичный (коагуляционный) гемостаз - многоступенчатая реакция, в которой принимает участие ряд белков, обозначаемых как факторы свертывания крови (см. табл. 14-19). Одни из этих белков являются протеазами (факторы II, VII, IX, X,XI, XII, XIII), другие - акцелераторами (ускорителями) ферментных реакций (факторы У и VIII), третьи - конечным субстратом процесса (фактор I, или фибриноген). Взаимодействие факторов свертывания крови, их активация, а затем и инактивация практически на всем протяжении процесса происходят на фосфолипидных мембранах (ФЛМ) клеток (тромбоциты, эритроциты и др.). При этом способностью к фиксации и активации плазменных факторов свертывания, а также инактивирующих их факторов антисвертывающей системы обладают только обращенные к наружной стороне мембраны головки отрицательно заряженных фосфолипидов - фосфатидилсерина, фосфатидилэтаноламина и др.
Синтез большинства факторов свертывания (см. табл. 14-19), а также двух основных физиологических антикоагулянтов - протеинов С и S - осуществляется паренхиматозными клетками печени - гепатоцитами. При этом для того, чтобы факторы II, VII, IX, X и протеины С и S могли участвовать в гемостазе, они должны подвергнуться γ-карбоксилированию витамин К-зависимой карбоксилазой.
14.5.2.1. Механизм коагуляционного гемостаза
Различают три этапа процесса свертывания крови (рис. 14-18). Первый этап завершается образованием активного протромбиназного комплекса на ФЛМ, в состав которого входят факторы X, У
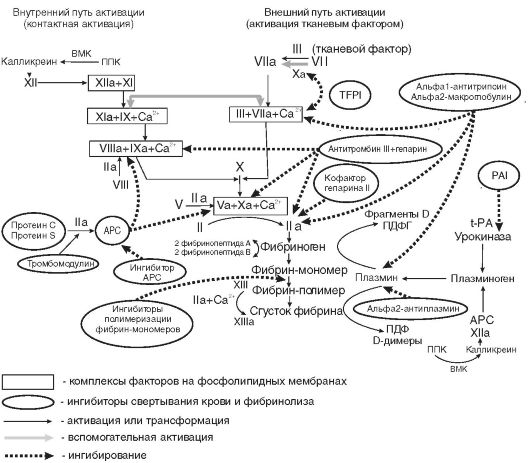 Рис. 14-18. Схема
коагуляционного гемостаза (по З.С. Бар кагану, А.П. Момоту, 1999-2001)
АРС - активированный протеин С; ВМК - высокомолекулярный кининоген; ППК -
плазменный прекалликреин; TFPI - ингибитор внешнего пути свертывания;
t-PA - тканевой активатор плазминогена; PAI - ингибитор активатора
плазминогена; ПДФГ - продукты деградации фибриногена; ПДФ - продукты
деградации фибрина; VII - фактор неактивный; Vila - фактор активный
Рис. 14-18. Схема
коагуляционного гемостаза (по З.С. Бар кагану, А.П. Момоту, 1999-2001)
АРС - активированный протеин С; ВМК - высокомолекулярный кининоген; ППК -
плазменный прекалликреин; TFPI - ингибитор внешнего пути свертывания;
t-PA - тканевой активатор плазминогена; PAI - ингибитор активатора
плазминогена; ПДФГ - продукты деградации фибриногена; ПДФ - продукты
деградации фибрина; VII - фактор неактивный; Vila - фактор активный
и Са2+. Второй этап характеризуется образованием тромбина - активной формы фактора II. На третьем этапе (конечная фаза свертывания крови) происходят формирование и стабилизация сгустка фибрина.
Первый этап каскадно-комплексной схемы свертывания крови включает активацию коагуляционного гемостаза по внутреннему и внешнему механизмам.
Внутренний (контактный) механизм характеризуется последовательной активацией факторов XII, XI, IX, VIII, X. В результате повреждения сосудистой стенки на поверхности тромбоцитарного агрегата образуется комплекс из трех белков - фактора XII (фактор Хагемана), плазменного прекалликреина (ППК) и высокомолекулярного кининогена (ВМК). После связывания с ВМК и калликреином (образуется из ППК под влиянием ВМК) фактор XII превращается в активную протеазу - XIIa, которая взаимодействует с неактивным фактором XI на ФЛМ и активирует его - образуется фактор XIa. Далее фактор XIa комплексируется с неактивным фактором IX и Ca2+ на ФЛМ, что в условиях вспомогательной активации фактором VIIa сопровождается формированием IXa, последующее взаимодействие которого с активной формой фактора VIII-VIIIa (ее образование происходит под действием тромбина - IIa) и Ca2+ на ФЛМ приводит к активации фактора Х.
Внутренний механизм первого этапа свертывания протекает намного медленнее, чем внешний. Он определяется:
• временем свертывания крови (5-11 мин в норме);
• каолиновым временем - временем свертывания рекальцифицированной цитратной плазмы в условиях контактной (каолин) активации факторов XII и XI (77-116 с при использовании нефракционированного каолина и 60-98 с при применении легкой фракции каолина);
• активированным парциальным тромбопластиновым временем (АПТВ) - временем свертывания рекальцифицированной цитратной плазмы в условиях контактной (каолин) и фосфолипидной (кефалин) активации факторов XII, XI, IX, VIII (в норме соответствует 30-42 с).
Внешний механизм активации гемостаза предполагает наличие в крови внешнего (в обычных условиях не присутствующего в крови) фактора III (тканевого фактора - ТФ, или тканевого тромбопластина), высвобождающегося из эндотелиоцитов и гладкомышечных клеток поврежденных сосудов. Под его влиянием происходит
активация фактора VII c образованием VIIa. Реакция стимулируется следовыми количествами плазменных протеиназ - IIa, VIIa, IXa, Xa, циркулирующих в крови. Взаимодействие факторов III и VIIa на ФЛМ в присутствии ионов Са2+ сопровождается активацией фактора Х с образованием Ха.
Свертывание крови по внешнему пути, который в пробирке имитируется добавлением к рекальцифицированной плазме тканевого тромбопластина, обозначается как протромбиновый (тромбопластиновый) тест. Нормальное время свертывания плазмы в присутствии тканевого тромбопластина (протромбиновое время - ПВ) колеблется в пределах 12-15 с. На основе ПВ рассчитываются протромбиновое отношение - ПО (отношение ПВ исследуемой плазмы к ПВ нормальной плазмы; в норме 0,7-1,1) и международное нормализованное отношение - МНО (ПОМИЧ, где МИЧ - международный индекс чувствительности тромбопластина; в норме от 1,0 до 1,4).
Таблица 14-19. Плазменные факторы свертывания крови
Номер, наименование и природа фактора | Место образования. Содержание в плазме | Факторы активации и механизм действия |
I Фибриноген (структурный белок) | Гепатоциты 1,8-4,0 г/л (80-120% активности) | Под действием тромбина превращается в фибрин (Ia - основное вещество тромба) Участвует в агрегации тромбоцитов Способствует репарации тканей |
II Протромбин (профермент сериновой протеазы тромбина) | Гепатоциты (в присутствии витамина К) Около 0,1 г/л | Под действием активной протромбиназы превращается в тромбин (IIa) Активирует фибриноген с образованием фибрина |
III Тканевой тромбопластин или апопротеин III (трансмембранный белок) | Эндотелиоциты, макрофаги Не содержится (высвобождается при повреждении стенки сосуда, тканей) | Кофактор фактора VII, запускает внешний путь свертывания крови |
Продолжение табл. 14-19
IV Ионы кальция - Са2+ | Гранулы тромбоцитов (плотные тельца), всасывается из кишечника 1,1-1,4 ммоль/л | Участвует в образовании комплексов плазменных факторов и липидов Входит в состав активной протромбиназы Способствует агрегации тромбоцитов Связывает гепарин Принимает участие в образовании первичной гемостатической пробки и ретракции тромба Тормозит фибринолиз |
V Проакцелерин или лабильный фактор (церулоплазмино- подобный связывающий белок) | Гепатоциты, мегакариоциты, тромбоциты Около 0,01 г/л (70-150% активности) | Активируется фактором IIа Входит в состав активной протромбиназы Создает оптимальные условия для взаимодействия факторов Ха и II |
VII Проконвертин или стабильный фактор (профермент сериновой протеазы) | Гепатоциты (в присутствии витамина К) Около 0,005 г/л (80-120% активности) | Активируется фактором III Активирует факторы IX, Х (участвует в образовании протромбиназы по внешнему пути) |
VIII:C Антигемофильный глобулин (церулоплазминоподобный связывающий белок) | Гепатоциты 0,01-0,02 г/л (60-250% активности) | Активируется тромбином Создает оптимальные условия для взаимодействия факторов Ка и X |
VIII:ΒΦ Фактор Виллебранда (структурный белок) | Эндотелиоциты, мегакариоциты 80-120% активности | Стабилизирует фактор VIII Способствует адгезии тромбоцитов |
Окончание табл. 14-19
IX Фактор Кристмаса или компонент плазменного тромбопластина, РТС-фактор (профермент сериновой протеазы) | Гепатоциты (в присутствии витамина К) Около 0,003 г/л (70-130% активности) | Активируется факторами XIa, VIIa Активирует фактор X |
X Фактор Стюарта-Прауэра (профермент сериновой протеазы) | Гепатоциты (в присутствии витамина К) Около 0,01 г/л (80-120% активности) | Активируется факторами VIIIа и VIIа Входит в состав активной протромбиназы Переводит протромбин в тромбин (IIa) |
XI Плазменный предшественник тромбопластина или PTA- фактор, фактор Розенталя (профермент сериновой протеазы) | Гепатоциты Около 0,005 г/л (70-130% активности) | Активируется фактором Активирует фактор IX |
XII Фактор Хагемана или контактный фактор (профермент сериновой протеазы) | Гепатоциты Около 0,03 г/л (70-150% активности) | Активируется калликреином и ВМК Запускает внутренний путь свертывания крови Активирует ППК, систему фибринолиза |
XIII Фибринстабилизирующий фактор (профермент трансглутаминазы) | Гепатоциты, мегакариоциты 0,01-0,02 г/л (70-130% активности) | Активируется тромбином и Ca2+ Стабилизирует фибрин Способствует репарации тканей |
Плазменный прекалликреин (ППК) или фактор Флетчера (профермент плазменного калликреина) | Гепатоциты Около 0,05 г/л (60-150% активности) | Активируется ВМК, фактором XIIa Активирует факторы VII, XII, ВМК, плазминоген |
Высокомолекулярный кининоген (ВМК) или фактор Фитцджеральда (гликопротеин) | Гепатоциты Около 0,06 г/л (80-130% активности) | Активирует факторы XI, XII, плазминоген, ППК |
Внутренний и внешний механизмы гемостаза тесно взаимосвязаны, их разделение является в некоторой степени условным. Так, фактор VIIa активирует факторы свертывания XII и (в присутствии тканевого тромбопластина и ионов кальция) IX (рис. 14-18). В свою очередь, фактор VII может быть активирован факторами XIIa и XIa. Предполагается, что внешний механизм обеспечивает фоновое свертывание крови. Инициация внешнего пути гемостаза протекает быстрее (12-15 с), чем внутреннего механизма (30-42 с). Это приводит к формированию базового количества тромбина, достаточного для последующей активации факторов внутреннего каскада гемокоагуляции.
После активации фактора X внутренний и внешний пути имеют одинаковое (общее) развитие, и поэтому дальнейшие превращения факторов свертывания крови обозначают как общий путь свертывания крови.
Второй этап характеризуется активацией фактора V и образованием активного протромбиназного комплекса (активной протромбиназы) на ФЛМ из факторов Vа, Ха и Са2+, способствующего превращению протромбина (фактор II) в тромбин (фактор IIa).
Третий этап - конечная фаза свертывания крови, характеризующаяся трансформацией растворенного в плазме фибриногена в фибрин, образующий каркас сгустка крови. На этом этапе происходит отщепление тромбином от молекулы фибриногена двух фибринопептидов А и двух фибринопептидов В с образованием фибрин-мономеров, полимеризующихся в димеры и далее - в тетрамеры и более крупные олигомеры, трансформирующиеся в волокна фибрина, образующие сгусток (см. рис. 14-19). Стабилизация сгустка фибрина осуществляется фактором XIII, активирующимся под действием тромбина (IIa) в присутствии ионов кальция, в результате чего водородные связи между мономерами фибрина трансформируются в ковалентные связи, и образующиеся сгустки фибрина становятся более прочными и устойчивыми к различным растворителям (мочевине, монохлоруксусной кислоте и др.).
Повышенное содержание в плазме крови промежуточных продуктов превращения фибриногена в фибрин служит показателем активации системы свертывания крови и наличия тромбинемии.
• Для оценки конечного этапа свертывания крови используются: тромбиновый тест, с помощью которого определяют время свертывания цитратной плазмы под влиянием стандартизи-
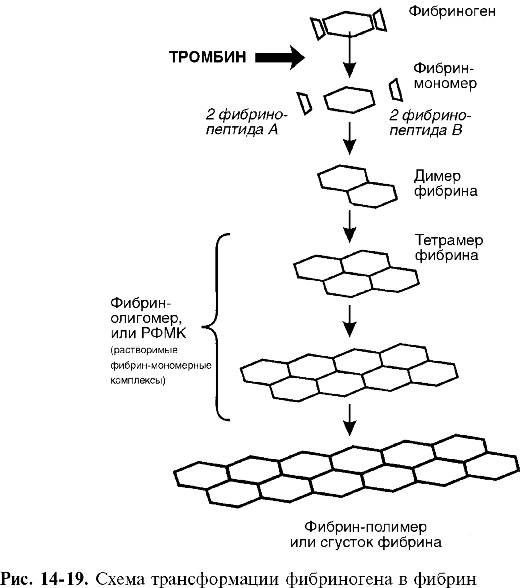 рованного на контрольной (нормальной) плазме тромбина (в норме 14-16 с);
рованного на контрольной (нормальной) плазме тромбина (в норме 14-16 с);
• определение содержания фибриногена в плазме крови хронометрическим методом (по Клаусу - с помощью коагулометра) или гравиметрическим методом (по Рутбергу - по весу сгустка крови). У здорового человека концентрация фибриногена в плазме составляет 2,0-4,0 г/л.
14.5.3. Противосвертывающие механизмы и система фибринолиза
В свертывающей системе крови действуют силы как аутокатализа, или самоускорения, так и самоторможения. Жидкое со-
стояние крови поддерживается за счет ее движения (снижающего концентрацию реагентов), адсорбции факторов свертывания эндотелием и, наконец, самостоятельно синтезируемых и постоянно находящихся в крови естественных (первичных) антикоагулянтов (табл. 14-20).
Таблица 14-20. Первичные (физиологические) антикоагулянты и механизмы их действия
 * Серпины, ингибирующие сериновые протеазы - тромбин и (в меньшей степени) другие факторы свертывания; ** - на долю а1-антитрипсина приходится 90-92% общей антипротеазной активности плазмы.
* Серпины, ингибирующие сериновые протеазы - тромбин и (в меньшей степени) другие факторы свертывания; ** - на долю а1-антитрипсина приходится 90-92% общей антипротеазной активности плазмы.
Многие факторы свертывания крови и их фрагменты, образующиеся в процессе гемокоагуляции, выступают в роли вторичных антикоагулянтов. В частности, противосвертывающим действием обладают фибрин и продукты расщепления фибриногена плазмином, тормозящие конечную фазу свертывания крови.
В патологических условиях в крови могут появляться в высоком титре иммунные ингибиторы факторов свертывания крови - антитела к факторам VIII, IX и другим, а также к ФЛМ, на которых взаимодействуют и активируются факторы свертывания крови (антифосфолипидный синдром - см. ниже).
Фибринолитическая (плазминовая) система, как и система свертывания крови, активируется как по внешнему, так и по внутреннему механизму. Основным внешним активатором этой системы является продуцируемый в эндотелии, а также в ряде тканей тканевой активатор плазминогена (t-PA), на долю которого приходится около 70% всего активаторного эффекта. Еще около 15% внешнего механизма активации приходится на фермент урокиназу, который вырабатывается в почках и в наибольшей своей части выделяется с мочой, а в кровь попадает в значительно меньшем количестве. При патологии в роли дополнительных активаторов фибринолиза могут выступать тканевые и лейкоцитарные протеазы, бактериальные ферменты (стрептокиназа, стафилокиназа и др.), экзогенные протеолитические ферменты (протеазы змеиных и грибных ядов, ядов насекомых и др.).
Внутренний механизм активации фибринолиза осуществляется в основном комплексом «фактор ХПа + калликреин + ВМК» (так называемый ХНа-калликреинзависимый фибринолиз) и протеинами C и S.
Внешний и внутренний механизмы активации фибринолиза замыкаются на плазминогене, который трансформируется в активный фермент - плазмин (ранее он обозначался как фибринолизин).
Плазмин фиксируется в основном на сгустках фибрина в тромбах, в связи с чем лизис фибрина преобладает над лизисом растворенного в плазме фибриногена. Кроме того, действию плазмина на фибриноген препятствует содержащийся в плазме мощный ингибитор этого фермента - a2-антиплазмин. Однако при чрезвычайно сильной активации плазминогена происходит истощение a2-антиплазмина, и в плазме крови обнаруживается большое количество продуктов как фибринолиза, так и фибриногенолиза. Как видно из схемы на рис. 14-20, эти продукты не идентичны друг другу. В результате расщепления фибриногена в плазме нарастает количество конечного продукта этого процесса - фрагмента D, тогда как при расщеплении фибрина увеличивается концентрация фрагментов D-D (димера) и D-E-D.
Путем раздельного определения концентрации в плазме фрагментов D и D-димеров можно получить представление, в какой степени у больного активированы фибринолиз и фибриногенолиз. Более того, при проведении таких анализов учитывается, что для нарастания в крови продуктов фибринолиза, т.е. D-димера, должно раньше произойти свертывание крови - образование фибрина,
 Рис. 14-20. Схема расщепления плазмином фибриногена (А) и фибрина (Б)
Рис. 14-20. Схема расщепления плазмином фибриногена (А) и фибрина (Б)
а затем его расщепление до фрагмента D-D. Поэтому увеличение концентрации в плазме D-димера служит ценным показателем как интенсивного внутрисосудистого свертывания крови, так и сопряженного с этим процессом фибринолиза. Нарастание содержания в крови D-димера является важным маркером массивного тромбоза кровеносных сосудов, тромбоэмболии, диссеминированного внутрисосудистого свертывания крови.
14.5.4. Геморрагические диатезы и синдромы
К геморрагическим диатезам и синдромам относят все те виды патологии, для которых характерна склонность к кровоточивости системного характера.
14.5.4.1. Типы кровоточивости
В настоящее время выделяют 5 типов кровоточивости, связанных с патологией гемостаза:
1. Петехильно-синячковый (микроциркуляторный) тип характеризуется появлением безболезненных точечных (петехии) или мелкопятнистых (экхимозы) кровоизлияний в кожу и «синяков» при незначительных ушибах, в местах давления и трения одежды, в местах инъекций, при измерении артериального давления (по нижнему краю манжеты и в локтевом сгибе) и т.д. Часто сочетается с повышенной кровоточивостью слизистых оболочек (носовые кровотечения, меноррагии). Петехиально-синячковая кровоточивость характерна для тромбоцитопений, тромбоцитопатий, болезни Виллебранда, гиповитаминоза С, дисфункций тромбоцитов эндокринного (дизовариального) генеза. В связи с вторичной патологией тромбоцитов и их дефицитом отмечается также при острых лейкозах, гипо- и апластических анемиях, уремии.
2. Гематомный тип характеризуется обширными, болезненными кровоизлияниями в подкожную клетчатку, мышцы, суставы, под надкостницу, отсроченными геморрагиями после травм и хирургических вмешательств, в том числе при порезах, после экстракции зубов, а также носовыми, почечными и желудочнокишечными кровотечениями. Особенно характерно поражение опорно-двигательного аппарата - деформация суставов, ограничение подвижности в суставах, атрофия мышц конечностей и т.д. Такой тип кровоточивости типичен для гемофилий.
3. Смешанный (петехиально-гематомный) тип характеризуется петехиально-синячковыми высыпаниями, сочетающимися с обширными кровоизлияниями и гематомами (чаще в подкожную и забрюшинную клетчатку, нередко с парезом кишечника) при отсутствии поражений суставов и костей (в отличие от гематомного типа) либо с единичными геморрагиями в суставы. Отмечается при дефиците факторов протромбинового комплекса (факторов II, V, VII, Х), передозировке антикоагулянтов непрямого действия, передозировке гепарина и препаратов фибринолитического действия (стрептокиназа и др.), болезни Виллебранда и синдроме диссеминированного внутрисосудистого свертывания (ДВС) крови.
4. Васкулитно-пурпурный тип проявляется симметричной папулезно-геморрагической сыпью диаметром до 0,5-1 см на нижних или (реже) верхних конечностях и в области нижней части туловища (ягодицы), формирующейся в результате диапедеза эритроцитов через стенку сосуда в связи с повышенной проницаемостью последней. Одновременно могут возникать крапивница, артралгии, острая боль в животе с кишечным кровотечением - меленой (абдоминальная форма), признаки гломерулонефрита. Такой тип кровоточивости специфичен для геморрагического васкулита (болезни Шенлейна-Геноха) и некоторых других системных васкулитов иммунного и инфекционного генеза. Характерной чертой является длительно сохраняющаяся на месте геморрагий синюшно-коричневая пигментация. При других типах кровоточивости остаточной гиперпигментации нет.
5. Ангиоматозный тип характеризуется рецидивирующей кровоточивостью постоянной несимметричной локализации из телеангиэктазов (ангиом мелких сосудов). Такой тип кровоточивости отмечается при телеангиэктазиях (болезнь Рандю-Ослера), при которых сосудистая стенка утрачивает способность к активации факторов гемокоагуляции и тромбообразованию in vivo. При этом сохраняется способность крови к образованию сгустков при контакте с чужеродной поверхностью in vitro.
Выделяют также типы кровоточивости, не связанные с патологией гемостаза:
1. Невритический - кровоточивость из строго определенных областей тела (кровавые слезы, кровавые «очки»), возникающая после тяжелого стресса либо вследствие самовнушения.
2. Имитационный - кровоточивость, связанная с преднамеренным приемом антикоагулянтов (чаще всего непрямого дей-
ствия - дикумарола, варфарина и др.) либо преднамеренной самотравматизацией.
3. Пурпура жестокого обращения - кровоточивость при избиении, щипках, укусах.
В целом геморрагические диатезы и синдромы подразделяют на наследственные (врожденные) и приобретенные формы.
14.5.4.2. Типы геморрагических диатезов и синдромов
В зависимости от того, нарушения в каком звене системы гемостаза стали причиной кровоточивости, все заболевания этой группы подразделяют на следующие типы:
1. Вазопатии, обусловленные поражением микрососудов наследственной или приобретенной (при гиповитаминозах (цинга), инфекционных, иммуновоспалительных и др. заболеваниях) природы.
К числу иммуновоспалительных заболеваний соединительной ткани приобретенной этиологии относится геморрагический васкулит, или болезнь Шенлейна-Геноха. Наиболее часто встречается у детей 2-8 лет и подростков (чаще болеют мальчики), реже - у взрослых. Болезнь развивается после перенесенной инфекции (стрептококковая ангина), травмы, переохлаждения, вакцинации, в связи с пищевой и лекарственной аллергией. Заболевание характеризуется поражением микрососудов кожи и внутренних органов циркулирующими иммунными комплексами и активированными компонентами комплемента, обусловливающими при фиксации на стенке сосудов лизис эндотелиальных клеток с развитием асептического воспаления. Повышенная проницаемость сосудов приводит к геморрагическому синдрому (васкулитно-пурпурный тип кровоточивости). Выделяют кожно-суставную и абдоминальную формы болезни. Кожно-суставная форма заболевания характеризуется общим недомоганием, повышением температуры тела, бледностью, появлением полиморфной симметрично расположенной папулезно-геморрагической сыпи различной локализации (в области головы, кистей, стоп, коленных и голеностопных суставов, ягодиц и др.), явлениями полиартрита (припухлость, болезненность суставов). В некоторых случаях кожные высыпания являются единственным проявлением болезни. Абдоминальный синдром встречается у 2/3 больных и выражается сильными болями в животе без четкой локализации, обусловленными кровоизлияниями в субсерозный слой, брюшину и стенку кишечника. Возмож-
ны желудочные и кишечные кровотечения (рвота кровью, мелена), поражение почек по типу острого гломерулонефрита с гематурией, протеинурией, дизурическими явлениями. Важное значение в диагностике данного заболевания имеет определение фактора Виллебранда в крови, уровень которого повышается в 1,5-3 раза вследствие его высвобождения из поврежденных эндотелиальных клеток. Количество тромбоцитов, время свертывания крови и длительность кровотечения при геморрагическом васкулите сохраняются в пределах нормы. Симптом жгута - положительный.
В особую подгруппу выделяют генетически обусловленные вазопатии и приобретенные вазопатии невоспалительной природы (при сердечной недостаточности, патологии печени), обусловленные локальным расширением сосудов кожи и слизистых оболочек - телеангиэктазией (от греч. telos - окончание, angeion - сосуд, ektasis - расширение). Возможны висцеральные ангиомы, артериовенозные аневризмы, шунты. Примером наследственной аутосомно-доминантной телеангиэктазии является болезнь Рандю- Ослера (геморрагический ангиоматоз), в основе которой лежат истончение субэндотелия, связанное с дефектом синтеза коллагена, и мутации генов, кодирующих белки TGF-b-рецепторного комплекса эндотелиальных клеток, такие, как TGF-b-связывающий гликопротеин эндоглин (endoglin - ENG) (9q33-34) и TGF-β- рецептор-ассоциированную серин-треониновую киназу (activin receptor-likekinase 1-ALK-1)(12q11-14), стабилизирующиекапилляры. У подавляющего большинства больных болезнь впервые проявляется в возрасте 6-10 лет повторяющимися, упорными носовыми кровотечениями из телеангиэктазов, локализующихся в носовой полости (ангиоматозный тип кровоточивости); возможна рвота кровью (рвота цвета «кофейной гущи») вследствие заглатывания крови и образования ангиом и артериовенозных фистул (т.е. прямых соединений между артериями и венами вследствие прогрессирующего исчезновения капиллярного ложа) в слизистой оболочке желудка. Ангиомы могут образовываться также в легких, почках, печени и др. внутренних органах. Узловатые, петлеобразные или «паукообразные» телеангиэктазы на слизистых оболочках, в области губ и на коже, возвышающиеся над поверхностью и бледнеющие при надавливании, являются ведущим диагностическим признаком заболевания. При этом гемостазиологические показатели сохраняются в пределах нормы. С возрастом имеется тенденция к увеличению количества телеангиэктазов и учащению геморрагий,
которые становятся более обильными. Обычная разовая кровопотеря из телеангиэктазов составляет от нескольких капель до 500 мл и более. Острые массивные кровопотери могут привести к летальному исходу.
2. Геморрагические диатезы и синдромы, обусловленные нарушениями тромбоцитарного гемостаза. К нарушениям тромбоцитарного гемостаза относятся тромбоцитопении - уменьшение количества тромбоцитов менее 150-109/л, и тромбоцитопатии - качественные дефекты тромбоцитов, связанные с нарушением их структуры и функций.
Клинические проявления тромбоцитопений различаются в зависимости от степени их выраженности:
• увеличение времени кровотечений при ранениях, травмах, после хирургических вмешательств - при снижении количества тромбоцитов до 120-109/л и более;
• петехии, экхимозы, синяки - при снижении количества тромбоцитов до (50-20)-109/л;
• спонтанные кровотечения (носовые, маточные, желудочнокишечные и др.) - при снижении количества тромбоцитов менее 20-109/л.
Кровоточивость при тромбоцитопатиях обусловливается нарушением адгезивно-агрегационных свойств тромбоцитов.
Тромбоцитопении по патогенезу классифицируются на 4 основные группы:
1. Тромбоцитопении вследствие патологии тромбоцитопоэза. К этой группе относятся наследственные тромбоцитопе-
нии (аутосомно-доминантная тромбоцитопения, аутосомнодоминантный синдром Мэя-Хегглина, Х-сцепленные синдромы Вискотта-Олдрича, Фанкони и др.) и приобретенные (симптоматические или вторичные) формы тромбоцитопений, которые обнаруживаются при гипо- и апластических состояниях костного мозга (при некоторых эндокринных заболеваниях, отравлениях, уремии, инфекциях, лучевой болезни), гиповитаминозах (дефицит витамина В12, фолиевой кислоты), опухолевых заболеваниях (лейкозы, метастазы внекостно-мозговых опухолей в костный мозг), миелофиброзе.
2. Тромбоцитопении вследствие повышения разрушения тромбоцитов.
В данную группу включены иммунные тромбоцитопении:
• аутоиммунные, связанные с образованием аутоантител к неизмененным тромбоцитам с последующим разрушением клеток
комплементом или фагоцитами (идиопатическая тромбоцитопеническая пурпура, или болезнь Верльгофа);
• гетероиммунные, связанные с образованием антител против измененных антигенов тромбоцитов или фиксированных на мембране тромбоцитов чужеродных антигенов - гаптенов (гаптеновые тромбоцитопении - лекарственные или ассоциированные с вирусной инфекцией);
• трансиммунные, отмечающиеся при трансплацентарной передаче плоду антитромбоцитарных антител от матери с болезнью Верльгофа;
• изоиммунные, формирующиеся при несовместимости по антигенам тромбоцитов матери и плода или донора и реципиента при гемотрансфузиях.
3. Тромбоцитопении вследствие повышения потребления тромбоцитов.
Частными примерами данного рода патологии являются тромботическая тромбоцитопеническая пурпура (ТТП) и гемолитикоуремический синдром (ГУС).
Основные звенья патогенеза ТТП и ГУС: агрегация тромбоцитов и окклюзия сосудов тромботическими массами, приводящая к ишемии. Оба заболевания характеризуются тяжелой тромбоцитопенией потребления и микроангиопатической гемолитической анемией с характерной картиной шизоцитоза в мазках периферической крови (шизоциты - «обрезанные» эритроциты). Развитие гемолитической анемии опосредуется механическим повреждением эритроцитов нитями фибрина при прохождении их по частично закупоренным микрососудам. Механизм агрегации тромбоцитов до конца неизвестен. Предполагается, что активация тромбоцитов и тромбообразование в микрососудах могут быть связаны с чрезмерным высвобождением, аномальной фрагментацией и образованием необычно крупных мультимеров фактора Виллебранда при повреждении сосудистого эндотелия.
ГУС развивается преимущественно у детей раннего возраста (в 70% случаев у детей до 3 лет) и, как правило, связан с перенесенной кишечной (шигеллез, коли-инфекция) или респираторной инфекцией. Внутрисосудистая агрегация тромбоцитов ограничивается почти исключительно бассейном почечных сосудов. Микротромботическое повреждение сосудов клубочков, интерстиция почек и субэндотелиальное отложение фибрина вследствие локального внутрисосудистого свертывания, приводящее к развитию острой
почечной недостаточности, обусловливаются микробными токсинами (веротоксин-1, -2, шигатоксин), нейраминидазой, вазоактивными аминами. Наряду с инфекционными формами выделяют неинфекционные формы ГУС (идиопатический, лекарственный, ассоциированный с системной красной волчанкой, гломерулонефритом и др.). Особую группу составляют редкие наследственные аутосомно-рецессивные и аутосомно-доминантные формы болезни. Причины наследственных форм ГУС неизвестны. Предполагается, что они могут быть обусловлены врожденным дефектом системы комплемента, дефицитом простациклина, антитромбина III, врожденной аномалией метаболизма витамина В12 с метилмалоновой ацидурией и гомоцистинурией.
ТТП наблюдается у лиц любого возраста, чаще у молодых женщин. Описаны семейные случаи, а также случаи ТТП при беременности, ВИЧ-инфекции, подостром бактериальном эндокардите, системной красной волчанке, опухолях (в том числе после цитостатической противоопухолевой терапии), приеме оральных контрацептивов и др. ТТП характеризуется сходными с ГУС клиническими проявлениями. Дифференциальным признаком ТТП является обязательная выраженная неврологическая симптоматика. Симптомы острой почечной недостаточности при ТТП обнаруживаются реже, чем при ГУС.
К тромбоцитопениям потребления относят также ДВС-синдром (см. раздел 14.5.5).
4. Перераспределительные тромбоцитопении.
В данном случае дефицит тромбоцитов в крови формируется за счет их депонирования в селезенке на фоне спленомегалии и гиперспленизма (при циррозе печени, портальной гипертензии, инфекционных и паразитарных заболеваниях и т.д.).
Тромбоцитопатии могут быть наследственными (первичными) и симптоматическими (вторичными). В основе первичной дисфункции тромбоцитов, обусловливающей развитие геморрагических диатезов, лежат следующие основные патогенетические факторы:
1) дефекты поверхностной мембраны, связанные с отсутствием или блокадой на мембране тромбоцитов рецепторов, взаимодействующих со стимуляторами (агонистами) их адгезии и агрегации (тромбастения Гланцмана - аутосомно-рецессивный дефицит ГП ΙΙβ/IIIa, тромбодистрофия Бернара-Сулье - аутосомнорецессивный дефицит ГП Iβ, сочетающийся с увеличением размеров тромбоцитов);
2) нарушение дегрануляции (реакции освобождения) тромбоцитов;
3) дефицит стимуляторов агрегации в гранулах тромбоцитов:
• болезни отсутствия плотных гранул (Х-сцепленный синдром Вискотта-Олдрича, аутосомно-рецессивные синдромы Хермански-Пудлака, Чедиака-Хигаси, связанные с дефицитом АДФ, АТФ, Са2+ и др.);
• болезни отсутствия α-гранул (синдром «серых» тромбоцитов, связанный с дефицитом фибриногена, пластиночного фактора 4, ростового фактора и др.);
• дефицит, снижение активности и структурная аномалия (нарушение мультимерности) фактора Виллебранда. Примером является наследуемая, как правило, по аутосомно-доминантному типу болезнь Виллебранда, характеризующаяся нарушением адгезивности и ристомицин-агрегации тромбоцитов.
Первичные нарушения агрегации тромбоцитов могут быть опосредованы также блокадой образования циклических простагландинов и ТхА2, мобилизации ионов кальция из тубулярной системы тромбоцитов.
К приобретенным относятся тромбоцитопатии при опухолевых процессах, в том числе лейкозах, ДВС-синдроме, заболеваниях печени и почек, недостатке витаминов В12 и С, действии ионизирующей радиации и др. В особую группу вторичных тромбоцитопатий выделяют ятрогенные (лекарственные) тромбоцитопатии, вызываемые рядом лекарственных воздействий, одни из которых (аспирин и др.) блокируют образование в тромбоцитах мощных циклических простагландиновых стимуляторов агрегации, в частности ТхА2, другие блокируют рецепторы IIβ/IIIa (тиенопиридины и др.), третьи нарушают транспорт в тромбоциты ионов кальция либо стимулируют образование цАМФ.
3. Нарушения свертываемости крови (коагулопатии). В эту группу включаются наследственные и приобретенные коагулопатии.
Среди наследственных коагулопатий доминируют (около 97%) гемофилии А и В.
В основе обоих видов гемофилии лежит мутация локусов синтеза фактора VIII (гемофилия А) или фактора IX (гемофилия В) в Х-хромосоме. Болезнь при обеих формах наследуется по рецессивному, сцепленному с полом типу; носителями болезни являются женщины, а больными - только лица мужского пола (исключения из этого правила крайне редки). Все дочери больного гемофилией,
получившие патологическую X-хромосому от отца, являются кондукторами болезни, а сыновья женщин-носительниц гемофилии в 50% случаев имеют шанс стать больными, дочери (в 50% случаев) - носителями патологического гена.
Для гемофилии характерны кровоточивость гематомного типа - болезненные кровоизлияния в крупные суставы (гемартрозы), мышцы, забрюшинную клетчатку, в область черепа, гематурия, тяжелые отсроченные посттравматические и послеоперационные кровотечения, в том числе при малых травмах и вмешательствах (при порезах, удалении зубов и т. п.). Поскольку факторы VIII и IX участвуют только во внутреннем механизме свертывания крови, при гемофилии удлинены общее время свертывания цельной крови, время свертывания рекальцифицированной цитратной плазмы и активированного парциального тромбопластинового времени (АПТВ), тогда как протромбиновый показатель и тромбиновое время свертывания остаются нормальными.
К группе наследственных коагулопатий относят также болезнь Виллебранда, при которой (см. выше) тромбоцитопатия сочетается с дефицитом фактора VIII. В этом случае заболевание проявляется петехиально-гематомной кровоточивостью в связи с сочетанным нарушением адгезивно-агрегационных свойств тромбоцитов и снижением коагуляционной активности крови. Это обусловлено тем, что фактор Виллебранда является переносчиком плазменного фактора VIII. При отсутствии фактора Виллебранда фактор VIII подвергается ускоренному разрушению в крови, что и обусловливает его дефицит и связанную с ним спонтанную гематомную кровоточивость.
Основными причинами приобретенных коагулопатий являются: 1) гипо- и авитаминозы К при недостаточном поступлении витамина К с пищей, нарушениях всасывания (обтурационная желтуха, энтерит) и синтеза (кишечный дисбактериоз) витамина К в кишечнике, приеме антагонистов витамина К, что сопровождается патологией синтеза и γ-карбоксилирования факторов протромбинового комплекса (II, VII, IX, X) в печени; 2) заболевания печени (истощение депо витамина К и нарушение синтеза факторов свертывания при токсических и вирусных гепатитах, циррозе печени); 3) заболевания почек (потеря прокоагулянтов с мочой при нефротическом синдроме); 4) образование и накопление в организме аутоантител (в основном класса IgG) - ингибиторов факторов II, V, VIII, IX, X, XIII (при аутоиммунных заболеваниях - системная
красная волчанка, ревматоидный артрит, неспецифический язвенный колит и др., иммунизации экзогенными факторами в процессе заместительной терапии гемофилий, на фоне лечения антибиотиками) и аномальных белков - парапротеинов, криоглобулинов (см. раздел 12.6.5); 5) коагулопатия потребления (ДВС-синдром).
14.5.5. ДВС-синдром
ДВС-синдром (синдром диссеминированного внутрисосудистого свертывания, тромбогеморрагический синдром) - одна из наиболее частых форм патологии гемостаза, характеризующаяся распространенным внутрисосудистым свертыванием крови с образованием в ней множества микросгустков фибрина и агрегатов клеток крови (тромбоцитов, эритроцитов), блокирующих микроциркуляцию в жизненно важных органах и вызывающих в них глубокие дистрофические изменения. ДВС-синдром сопровождается активацией и истощением свертывающей, фибринолитической, калликреин-кининовой плазменных систем и тромбоцитарного гемостаза (тромбоцитопения потребления), сочетанием микротромбирования кровеносных сосудов с выраженной кровоточивостью.
ДВС-синдром неспецифичен и универсален. Процесс имеет острое (молниеносное), подострое и хроническое (затяжное) течение. Острое и подострое течение наблюдается при всех видах шока, травмах (например, при синдроме длительного сдавления), термических и химических ожогах, остром внутрисосудистом гемолизе (в том числе при переливании несовместимой крови и массивных гемотрансфузиях), тяжелых инфекциях и сепсисе, массивных деструкциях и некрозах в органах, при ряде форм акушерскогинекологической патологии (эмболия сосудов матери околоплодными водами, криминальные аборты, инфекционно-септические осложнения в родах и при прерывании беременности и др.), при травматичных хирургических вмешательствах, большинстве терминальных состояний, а также при укусах ядовитых змей. Хроническое течение отмечается при инфекционных (персистирующие бактериальные и вирусные инфекции), иммунных (системная красная волчанка и др.), опухолевых заболеваниях (лейкозы, рак), при обезвоживании организма, массивном контакте крови с чужеродной поверхностью (экстракорпоральное кровообращение, хронический гемодиализ, протезы магистральных сосудов и клапанов сердца и др.), хроническом гемолизе.
Для ДВС-синдрома характерны симптомы полиорганной недостаточности в результате поражения и дисфункции органовмишеней: 1) развитие острой легочной недостаточности (вплоть до легочного дистресс-синдрома); 2) острая почечная или гепаторенальная недостаточность со снижением диуреза и уремией; 3) церебральная симптоматика, связанная с ишемией мозга; 4) поражение слизистой оболочки желудка и кишечника, сопровождающееся обильными кровотечениями, развитием острых гипоксических язв, вторичным инфицированием организма кишечной микрофлорой; 5) надпочечниковая, или плюригландулярная, эндокринная недостаточность с нестабильной гемодинамикой; 6) синдром системной воспалительной реакции с накоплением в крови цитокинов и других метаболитов. В целом клинические проявления ДВС-синдрома зависят от фазы патологического процесса.
Острый и подострый ДВС-синдромы проходят в своей эволюции три фазы: I - стадию гиперкоагуляции, которая тем короче (вплоть до нескольких минут), чем острее протекает процесс;
II - переходную стадию, при которой показания коагуляционных тестов разнонаправлены: одни из них продолжают выявлять гиперкоагуляцию, тогда как другие обнаруживают гипокоагуляцию;
III - стадию резко выраженной гипокоагуляции вплоть до несвертываемости крови. Острый ДВС-синдром имеет, как правило, кратковременные первую и отчасти вторую фазы, диагностируется в фазе гипокоагуляции и профузных геморрагий. При подостром течении ДВС-синдрома возможны повторные чередования фаз, а при хроническом - длительная стабилизация процесса в первых двух фазах. При остром и подостром течении активируются и прогрессивно истощаются не только факторы свертывания крови, но и важнейшие физиологические антикоагулянты (антитромбин III, протеины С и S), компоненты фибринолиза (плазминоген и его активаторы) и калликреин-кининовой системы (прекалликреин, высокомолекулярный кининоген), прогрессирует тромбоцитопения потребления, содержание фибриногена снижается, нарастает концентрация продуктов расщепления фибриногена и фибрина, D-димеров, количество фрагментированных эритроцитов (шизоциты) в крови. Ввиду этого специфическим признаком острого и подострого ДВС-синдрома в отличие от хронического ДВС-синдрома и тромбозов иного генеза является обязательное сочетание тромбоцитопении и дефицита антитромбина III, часто сопровождающееся снижением уровня плазминогена в крови. При хроническом ДВС-
синдроме (к примеру, на фоне антифосфолипидного синдрома, гипергомоцистеинемии и др.) количество тромбоцитов сохраняется в норме или повышается за счет компенсаторной активации тромбоцитопоэза вследствие длительного потребления тромбоцитов.
Лечение ДВС-синдрома направлено на устранение причины, восстановление микроциркуляции в органах и нормализацию их функции, предупреждение рецидивов. При успешной компенсации вышеуказанных нарушений путем трансфузии свежезамороженной плазмы и кровезаменителей и прекращении внутрисосудистого свертывания крови все перечисленные сдвиги подвергаются обратному развитию.
14.5.6. Тромбофилические состояния
Термином «тромбофилии» обозначаются все наследственные (генетически обусловленные) и приобретенные нарушения гемостаза, которым свойственна предрасположенность к раннему появлению и рецидивированию тромбозов и облитераций кровеносных сосудов, ишемиям и инфарктам органов.
Согласно принятой в настоящее время классификации 3.С. Баркагана (1996), тромбофилии подразделяют на ряд основных групп. Для каждого вида тромбофилий характерны определенные сдвиги в отдельных звеньях гемостаза и лабораторные признаки, позволяющие выявить патогенетические факторы этих нарушений.
В первую группу гематогенных тромбофилий включаются все гемореологические формы, при которых наклонность к тромбозам связана со сгущением крови, повышением ее вязкости, гематокритного показателя, содержания гемоглобина и эритроцитов. Наиболее характерны такие тромбофилические состояния для истинной полицитемии, полиглобулий - идиопатических и вторичных, развивающихся при гипоксии, обильной потере организмом воды (интенсивное потоотделение, профузные поносы и т.п.), нарушении физиологической гемодилюции (при токсикозах беременности), увеличении концентрации белка (гиперфибриногенемия) и появлении аномальных протеинов (парапротеинов) в крови. К этой группе относятся также тромбофилии, обусловленные изменениями формы и деформируемости эритроцитов (при мембранопатиях, гемоглобинопатиях, ферментопатиях).
Во вторую группу включаются тромбофилии, обусловленные нарушениями сосудисто-тромбоцитарного гемостаза вследствие
значительного (до 1200-109/л) повышения количества тромбоцитов в крови (первичные и симптоматические тромбоцитозы), а также тромбозы, связанные с повышенной адгезивностью и агрегацией тромбоцитов (синдром вязких тромбоцитов - первичный и при атеросклерозе, диабете, приеме противозачаточных гормональных препаратов и др.). В эту же группу входят тромбофилии, связанные с гиперпродукцией и повышением мультимерности фактора Виллебранда.
В третью группу тромбофилий включают все формы, обусловленные наследственным или приобретенным дефицитом или аномалиями важнейших физиологических антикоагулянтов - антитромбина III, протеина С, протеина S, TFPI, комбинированные формы антикоагулянтной недостаточности.
К четвертой группе тромбофилий относят формы, обусловленные дефицитом, гиперпродукцией или наследственными аномалиями плазменных факторов свертывания крови. В последнем случае эти факторы утрачивают чувствительность к физиологическим антикоагулянтам или компонентам системы фибринолиза. Наиболее частая из этих форм - аномалия Лейден, при которой фактор Va утрачивает способность инактивироваться протеином С. Эта форма обозначается как резистентность фактора Va к активированному протеину С. Она очень распространена у европейцев и нередко встречается в российской популяции. Другими достаточно частыми тромбофилиями этой группы являются аномалии фактора II и ряд аномалий фибриногена (дисфибриногенемии), гиперпродукция и повышение активности факторов VII (при гестозе, преэклампсии) и VIII. Намного реже тромбозы бывают связаны с дефицитом или аномалиями фактора XII (фактор Хагемана), при которых, как и при дисфибриногенемиях, наблюдается сочетание гипокоагуляции с нарушениями фибринолиза, формирующими тромбофилическое состояние.
Пятая группа тромбофилий представлена генетически обусловленными или приобретенными нарушениями фибринолиза, в основе которых лежат недостаточная продукция в эндотелии тканевого плазминогенового активатора или повышение содержания в плазме его ингибиторов, дефицит или аномалии плазминогена.
К шестой группе относят тромбофилии метаболического генеза, связанные со снижением антитромботического потенциала эндотелия и комплексными нарушениями во всех звеньях системы гемостаза. В эту группу входят тромбофилии при атеросклерозе,
гиперлипидемиях, гипергомоцистеинемии, диабетической ангиопатии и ряде других эндокринных нарушений.
К седьмой группе относятся аутоиммунные и инфекционноиммунные тромбофилии, среди которых доминирует антифосфолипидный синдром (АФС, синдром Хьюза), объединяющий группу аутоиммунных нарушений, характеризующихся наличием в высоком титре в крови аутоантител классов IgG и IgM к отрицательно заряженным мембранным фосфолипидам. Среди мембранных фосфолипидов основными мишенями антифосфолипидных антител являются несущие отрицательный заряд кардиолипин, фосфатидилсерин, фосфатидилэтаноламин, фосфатидиловая кислота и связанные с ними гликопротеины - β2-гликопротеин-1, аннексин V и протромбин (фактор II). Антифосфолипидные антитела блокируют фосфолипиды и фосфолипидно-белковые комплексы как свободных фосфолипидных микровезикул плазмы крови, так и лабилизированных клеточных ФЛМ эндотелия, тромбоцитов и др. клеток. Это сопровождается, с одной стороны, снижением тромборезистентности эндотелия и активацией сосудистотромбоцитарного гемостаза, с другой - дисбалансом в системе коагуляционного гемостаза, что в 75% случаев проявляется формированием тромбогенных осложнений. При этом активация факторов Vа, Ха и протромбина вследствие высвобождения тканевого тромбопластина в условиях повреждения эндотелия и аутоиммунной активации макрофагов сочетается с депрессией противосвертывающих механизмов. В 20-25% случаев АФС регистрируется, напротив, кровоточивость, связанная с дефицитом и экранированием ФЛМ, на поверхности которых происходит взаимодействие факторов свертывания крови.
Лабораторными признаками АФС являются:
• обнаружение антител к β2-гликопротеину-1, антикардиолипиновых антител, волчаночного антикоагулянта (группа антифосфолипидных антител, препятствующих связыванию витамин-К-зависимых факторов свертывания с отрицательно заряженными ФЛМ) в крови при удлинении показателя одного из скрининговых гемостазиологических тестов (АПТВ, каолиновое время, стандартное ПВ, ПВ со змеиными ядами) и его коррекции при добавлении экзогенных фосфолипидов;
• удлинение показателей двух скрининговых гемостазиологических тестов и более без иммунологического подтверждения наличия антифосфолипидных антител в крови.
АФС отмечается при системной красной волчанке, хронических вирусных инфекциях, лимфопролиферативных заболеваниях. Тромбирование микрососудов плаценты у женщин, страдающих АФС, приводит к «привынному» невышашиванию беременности (два случая и более спонтанного прерывания беременности или мертворождения).
В эту же седьмую группу тромбофилий включают множественные тромбозы и инфаркты органов при затяжном бактериальном эндокардите, миокардиопатиях, ряде вирусно-иммунных васкулитов (в том числе при тромботической тромбоцитопенической пурпуре).
К восьмой группе относятся паранеопластические тромбофилические синдромы при всех формах опухолевых заболеваний.
В особую девятую группу включены медикаментозные и другие ятрогенные тромбофилии, патогенез которых весьма неоднороден. В частности, сюда входят частые тромбозы при лечении L-аспарагиназой, приеме эстрогенных противозачаточных препаратов и полихимиотерапии, гепариновая тромбоцитопения с рикошетными тромбозами, тромбозы сосудов печени при трансплантациях костного мозга и ряд других форм.
К десятой группе относятся комбинированные формы тромбофилий, представленные сочетанием двух вышеуказанных нарушений и более.
Заканчивая рассмотрение тромбофилий, следует особо подчеркнуть, что нельзя ставить знак равенства между понятиями «гиперкоагуляционное» и «тромбофилическое» состояние, поскольку многие виды тромбофилий (при дефиците фактора XII, дисфибриногенемиях, антифосфолипидном синдроме и др.) протекают со снижением, а не с повышением свертываемости крови. При ряде форм этой патологии параметры обычной коагулограммы остаются в нормальных пределах. В силу этого распознавание различных тромбофилий требует применения специальных методов исследования, выявляющих типичные для каждой из этих форм нарушения гемостаза (увеличение концентрации гемоглобина, фибриногена, растворимого фибрина, активности фактора VIII, количества эритроцитов, тромбоцитов, изменение активированного тромбопластинового и тромбинового времени, высокий уровень гомоцистеина, наличие волчаночного антикоагулянта в плазме, мутаций Лейдена и др.). В целом четкое разграничение и дифференциальная диагностика различных тромбофилий имеют исключительно большое клиническое значение, поскольку позволяют проводить их эффективную профилактику и терапию с полным излечением больных.