Патофизиология Новицкого, Е.Д. Гольдберга Тома 1 и 2 - 2009 г.
|
|
|
|
ГЛАВА 5 РОЛЬ НАСЛЕДСТВЕННОСТИ, КОНСТИТУЦИИ И ВОЗРАСТА В ПАТОЛОГИИ
5.1. НАСЛЕДСТВЕННОСТЬ И ПАТОЛОГИЯ. ЭТИОЛОГИЯ И ПАТОГЕНЕЗ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
5.1.1. Изменчивость наследственных признаков как основа патологии
Наследственность как способность организмов передавать разнообразные (морфологические, функциональные, биохимические) признаки своим потомкам определяется стабильностью функционирования или консерватизмом генетического аппарата.
Основу надежности генотипа составляют следующие характеристики строения и функционирования генома человека: 1) дублированность его структурных элементов; 2) матричный принцип биосинтеза; 3) способность к репарации; 4) регуляция генной активности.
Дублированностью генетических элементов не исчерпываются компоненты стабильности генотипа. Гомеостаз самого генотипа заложен в матричном принципе биосинтеза ДНК (репликация) и РНК (транскрипция). Этот принцип обеспечивается двумя замечательными особенностями молекулы ДНК: двухспиральностью молекулярной структуры и способностью каждой из полинуклеотидных нитей-спиралей служить матрицей для синтеза новой нуклеотидной нити, которая комплементарна исходной нити и поэтому полностью соответствует ей. В процессе репликации самой ДНК обеспечивается точное воспроизведение генетической информации в ряду последовательных актов синтеза ДНК и последующих клеточных делений. В процессе транскрипции матричный синтез гарантирует точную, неискаженную трансформацию закодированной в ДНК генетической информации через нуклео-
тидные последовательности РНК в первичную аминокислотную последовательность специфических белков.
Эволюция обеспечила клетки разносторонними механизмами восстановления (или репарации) повреждений генетических структур (ДНК и хромосом). Абсолютно стабильного в организме ничего не может быть, в том числе не может быть абсолютно устойчивым генетический аппарат клеток. Первичная структура ДНК, хотя и с малой частотой, может изменяться при репликации ДНК. Эти события известны как «ошибки репликации». В гораздо большей степени ДНК повреждается от воздействия мутагенов.
К настоящему времени открыто несколько механизмов, с помощью которых устраняются те или иные повреждения ДНК. В их основе лежат ферментативные процессы.
Гомеостаз внутренней среды организма должен обеспечиваться, помимо вышеизложенных фундаментальных механизмов, надежностью генетического контроля генной активности. Механизмы такого контроля на молекулярном и надмолекулярном уровнях пока не раскрыты.
Стабильность функционирования или консерватизм генетического аппарата являются лишь одной стороной явления наследственности. Другой ее стороной является изменчивость (наследственная изменчивость). Лишь в своей совокупности наследственность и изменчивость обеспечили и сохранение жизни на Земле, и непрерывную биологическую эволюцию. Наследственная изменчивость организма обеспечивает необходимую ему приспособляемость к условиям существования как в пределах жизни одного индивида, так и в рамках существования биологического вида в целом.
Наследственное разнообразие человека - это результат длительной эволюции живой материи. При этом надо иметь в виду особенности эволюции человека как биологического, так и социального существа. У человека как социального существа естественный отбор протекал со временем все в более и более специфических формах, что, безусловно, расширяло наследственное разнообразие популяций. Сохранялось то, что могло отметаться у животных или, напротив, терялось то, что нужно животным. Например, более полноценное обеспечение себя пищей и возможность восполнять потребность в витамине С «позволили» человеку в процессе эволюции «утерять» ген фермента L-гулонолактоноксидазы, катализирующей у животных синтез
аскорбиновой кислоты. Наличие этого гена у животных ограждает их от развития цинги, а человек из-за такой «всеобщей врожденной ошибки метаболизма» подвержен авитаминозу С. В процессе эволюции человек и «приобретал» нежелательные признаки, имеющие прямое отношение к патологии. Большинство видов животных невосприимчиво к дифтерийному токсину и вирусу полиомиелита, потому что у них отсутствуют компоненты мембраны клеток, обеспечивающие восприятие того или другого патогенного фактора. У человека эти компоненты есть. Гены, их детерминирующие, уже идентифицированы. Например, ген, детерминирующий компоненты мембраны клеток для восприятия дифтерийного токсина, локализован в 5-й хромосоме, а для восприятия вируса полиомиелита - в 19-й хромосоме.
Ни у одного биологического вида, в том числе и у человека, нет резкой границы между наследственной изменчивостью, ведущей к нормальным вариациям признаков, и наследственной изменчивостью, определяющей патологические вариации (наследственные болезни). Большинство мутаций увеличивает полиморфизм человеческих популяций (группы крови, цвет волос, рост, разрез глаз и т.д.), но в ряде случаев мутации затрагивают жизненно важные функции, а это уже приводит к болезни. Таким образом, наследственная патология - это часть наследственной изменчивости, накопившейся за время эволюции человека. Человек, став биологическим видом homo sapiens (человек разумный), как бы заплатил за «сапиентацию» своего вида накоплением патологических мутаций. На основе этих положений формулируется одна из главных концепций медицинской генетики об эволюционном накоплении патологических мутаций в человеческих популяциях. Подтверждением этой концепции являются патологические мутации у животных, по своим проявлениям сходные с наследственными болезнями у человека (ахондроплазии, гемофилии, мышечные дистрофии и многие другие у коров, свиней, овец, кошек, собак, грызунов), а также наличие наследственных болезней у людей, живших несколько тысячелетий назад, о чем можно судить по раскопкам и произведениям искусства.
Основным источником многообразия наследственных признаков и их непрекращающейся эволюции служит мутационная изменчивость. Способность ДНК мутировать сформировалась в ходе эволюции и закрепилась в процессе естественного отбора, повидимому, так же, как и способность противостоять мутационным
изменениям, т.е. репарировать их. Следовательно, в организации ДНК заложена как вероятность ошибок ее репликации, так и возможность изменения ее первичной структуры. Вероятность «сбоя» в точности репликации молекулы ДНК невелика. Она составляет одно событие на 105-107 нуклеотидов. Однако, принимая во внимание исключительно большое число нуклеотидов в геноме (3,3 млрд нуклеотидов на гаплоидный набор), следует признать, что в сумме на геном клетки на одно ее поколение приходится несколько мутаций в структурных генах. Считается, что каждый индивид наследует 2-3 новые вредные мутации, которые могут вызывать летальный эффект.
Изменение нуклеотидной последовательности молекулы ДНК может отразиться на первичной (аминокислотной) структуре белка или на регуляции его синтеза. Так, большой опыт изучения молекулярной природы мутаций гемоглобина показывает, что значительная часть таких мутаций не изменяет функции гемоглобина. Эти мутации нейтральны и не подвергаются отбору. Другие мутации приводят к функциональным отклонениям в молекуле белка. Эти отклонения в определенных условиях могут оказаться полезными для организма, т.е. иметь адаптивное значение, поэтому сохраняются и преумножаются в последующих поколениях. Именно таким путем возникали и сохранялись в популяциях разнообразные варианты структурных, транспортных и ферментных белков организма. Свойственный организму широкий белковый полиморфизм, благодаря которому каждый индивид биохимически неповторим, обусловлен мутационной изменчивостью и отбором адаптивных белковых вариантов. Если структурные отклонения несовместимы с выполнением белком его функций, которые жизненно важны для клетки, для организма, мутация становится патологической и в дальнейшем либо исключается из популяции вместе с нежизнеспособной клеткой (организмом), либо сохраняется, обусловливая наследственную болезнь. В отдельных случаях гетерозиготные носители патологической мутации подвергаются положительному отбору. Примером этого служит ген серповидноклеточной анемии, который широко распространился в популяциях, проживающих в эндемичных по малярии районах, вследствие устойчивости гетерозиготных носителей «аномального» гена (мутантного аллеля) к малярийному плазмодию.
Различные признаки организма по-разному устойчивы к мутационным изменениям, что связано, по-видимому, со значением
признака и его эволюционным «возрастом». Такие признаки, как гистоновые белки, входящие в состав хромосом, или сократительные белки актин и тубулин, или ферментные белки репликации и транскрипции, весьма консервативны и одинаковы не только у разных представителей человечества, но и у биологических видов значительной филогенетической отдаленности. По-видимому, мутации в соответствующих генах летальны. Большинство же белков организма, особенно ферментных, существуют в нескольких изоформах и подвержены таким мутационным изменениям, которые ведут к патологии.
Патологические мутации различны по способности сохраняться и распространяться в популяциях. Одни из них, позволяющие их носителю сохранять плодовитость и не вызывающие серьезных неблагоприятных сдвигов в фенотипе, могут передаваться из поколения в поколение длительное время. Такие признаки сегрегируют (распределяются) в поколениях согласно законам Менделя, и обусловленный ими генетический груз в популяциях может долго сохраняться. Некоторые комбинации условно патологических рецессивных аллелей могут давать селективное преимущество индивидам (выживаемость, плодовитость). Частота таких аллелей в популяции будет повышаться до определенного уровня в ряду поколений, пока не наступит равновесие между мутационным процессом и отбором. Частота разных мутантных аллелей этого рода может быть неодинаковой в различных популяциях, что определяется популяционными закономерностями (эффект родоначальника, частота кровнородственных браков, миграция и экологические условия). Если вновь возникшая мутация имеет доминантное патологическое проявление и ведет к летальному генетическому исходу (индивид не оставляет потомства), то такой мутационный груз не передается следующему поколению. Это обычно доминантные формы тяжелых болезней, а также большая часть хромосомных болезней.
В целом эффекты генетического «груза» у человека выражены в эволюционно-генетических явлениях балансированного полиморфизма, летальности и сниженной фертильности.
На основе постоянно протекающих процессов изменения наследственности (мутаций) и отбора генотипов в процессе длительной эволюции человека в популяциях сформировался балансированный полиморфизм. Под этим названием понимают такое явление, когда в популяции представлены две или более формы
аллелей одного гена, причем частота редкого аллеля составляет не менее 1%. Поскольку возникновение мутаций - это редкое событие (1?10-7), то, следовательно, частоту мутантного аллеля в популяции более 1% можно объяснить селективным преимуществом этого аллеля для организма и постепенным накоплением в ряду поколений после его появления. Примерами балансированного полиморфизма являются группы крови АВ0, Rh, гены муковисцидоза, фенилкетонурии, первичного гемохроматоза. Генетическое многообразие человека основано на балансированном полиморфизме, формировавшемся в течение десятков и сотен тысячелетий. Такое многообразие является основой развития человека как биологического вида. Вероятность возникновения и фиксации в популяциях какой-либо мутации с положительным эффектом в эволюционно «отлаженном» человеческом организме существует и в настоящее время, но она крайне мала. Практически всегда новые мутации обладают отрицательным эффектом.
К эффектам мутационного груза относится летальность. Она проявляется на уровне гибели гамет, зигот, эмбрионов, плодов, детей. Наиболее интенсивно летальные эффекты выражены в человеческих популяциях на уровне зигот. Примерно 60% зигот погибает до имплантации, т.е. до клинически регистрируемой беременности. Исходы всех клинически зарегистрированных беременностей распределяются следующим образом: спонтанные аборты - 15%, мертворождения - 1% и живорождения - 84%. Из 1000 новорожденных не менее 5 умирают в возрасте до года по причине наследственной патологии, не совместимой с жизнью. Таков объем летального груза мутационной изменчивости в популяциях человека с медицинской точки зрения.
Для большинства наследственных болезней характерна сниженная фертильность, обусловленная нарушением репродуктивной функции. Это ведет к уменьшенному воспроизводству потомства (и больного, и здорового) в семьях с наследственной патологией.
Медицинские и социальные последствия мутационного процесса - это социальная дезадаптация больных, повышенная потребность в медицинской помощи и сниженная продолжительность жизни.
Социальная дезадаптация больных связана с их инвалидностью, чаще всего - с детского возраста. В течение многих лет наследственные больные относятся к категории инвалидов, которые не могут себя обслуживать. В среднем такие дети в интернатах живут
до 10 лет. Из 1 млн новорожденных примерно 5000 имеют высокую вероятность инвалидизации. Медицинскую помощь лицам с наследственными болезнями в поликлинических условиях оказывают в 5-6 раз чаще. Среди контингента детских больниц общего профиля от 10 до 20% - это дети с наследственной патологией, что в 5-10 раз выше, чем частота таких больных в популяции. Продолжительность жизни у больных с наследственной патологией зависит от формы болезни и уровня медицинской помощи. Хотя точные расчеты еще не сделаны, но для стран с хорошо развитой системой здравоохранения можно с большой уверенностью полагать, что не менее 50% всех пациентов с наследственными болезнями умирают в детском возрасте. В Канаде проведена комплексная оценка ожидаемой продолжительности жизни для всех больных с наследственной патологией (с разным возрастом начала болезней и тяжести течения). Она оказалась на 20 лет меньше средней по стране (50 лет вместо 70).
5.1.2. Мутации как этиологический фактор наследственной
патологии
В основе возникновения наследственных заболеваний, как правило, лежат мутации.
Мутации - это изменения генома, которые приводят к увеличению или уменьшению количества генетического материала, к изменению нуклеотидов и их последовательности. Организмы с такими изменениями называют мутантами. Факторы, вызывающие мутации, называют мутагенами.
По этиологии различают спонтанные и индуцированные мутации. Спонтанными (или естественными) называют мутации, возникшие самопроизвольно под влиянием естественных условий внешней и внутренней среды. Причиной таких мутаций может являться естественный фон радиации - космическое излучение, излучение земного шара, зданий, радиоактивных изотопов (К-40, который поступает с растительными продуктами питания; углерод-14, радон и продукты его распада). В этот перечень входят эндогенные химические мутагены, которые образуются в организме в процессе обмена веществ - перекиси и свободные радикалы (аутомутагены). Значительную роль в возникновении спонтанных мутаций играет возраст. У мужчин с возрастом в половых клетках накапливаются генные мутации. У женщин зависимость частоты
генных мутаций от возраста не отмечена, но выявлена четкая связь возраста матери с частотой хромосомных заболеваний у потомства. Индуцированные мутации - это мутации, вызванные специальными направленными воздействиями - физическими, химическими и биологическими мутагенами.
• Физические мутагены. На первом месте среди физических мутагенов находятся ионизирующая радиация и УФ-излучение. Особенность ионизирующего излучения состоит в том, что оно может индуцировать мутации в низких дозах, не вызывающих лучевого поражения.
• Химические мутагены. К этой группе относят кислоты, спирты, соли, тяжелые металлы и др. Химические мутагены содержатся в воздухе (сероводород, мышьяк, меркаптан, хром, фтор, свинец и др.), почве (пестициды и др. химикаты), воде и пищевых продуктах, в лекарствах. Сильнейшим мутагеном является конденсат сигаретного дыма, который содержит бензпирен. Конденсат дыма и поверхностная корочка, образующиеся при обжаривании рыбы и говядины, содержат пиролизаты триптофана, которые являются химическими мутагенами. Особенность химических мутагенов состоит в том, что их действие зависит от дозы и стадии клеточного цикла. Чем выше доза мутагена, тем сильнее мутагенный эффект. При этом наиболее чувствительна к действию мутагенов стадия синтеза ДНК (S-фаза).
• Биологические мутагены. Бактериальные токсины, вирусы (вирусы герпеса, гепатита, эпидемического паротита и др.). У беременных вирусные инфекции могут провоцировать возникновение мутаций у плода, что приводит к спонтанным абортам.
Мутации по типу клеток, в которых возникла мутация, делят на соматические и половые. Соматические мутации возникают в соматической клетке, носят случайный характер, могут возникать на любой стадии развития, начиная с зиготы. По наследству не передаются. Мутантными будет часть клеток организма. В этом случае говорят о мозаицизме (неполной форме болезни). Половые мутации (генеративные мутации) возникают в половой клетке, их последствия сказываются на судьбе потомства и служат причиной наследственных заболеваний. В этом случае мутантными будут все клетки потомства. Возникает полная форма болезни.
В зависимости от размеров повреждения генетического материала мутации делятся на геномные (численные хромосомные аберрации), хромосомные (структурные хромосомные аберрации), генные (изменения в отдельных генах).
Геномные мутации возникают при нарушении расхождения хромосом в мейозе и при нарушении оплодотворения. Выделяют два вида геномных мутаций - полиплоидия и анэуплоидия. Полиплоидия - кратное гаплоидному набору увеличение общего числа хромосом. В норме соматические клетки организма являются диплоидными, т.е. содержат 2n хромосом, половые клетки - гаплоидные. Полиплоидные клетки могут иметь количество хромосом 3n - триплоид, 4n - тетраплоид и т.д. Триплоид может возникнуть при нарушении оплодотворения в случаях: дигении - оплодотворение диплоидной яйцеклетки гаплоидным сперматозоидом, диандрии - оплодотворение гаплоидной яйцеклетки диплоидным сперматозоидом, диспермии - оплодотворение гаплоидной яйцеклетки двумя сперматозоидами. У человека полиплоидия диагностируется только у абортусов, плод с такой патологией нежизнеспособен. Анеуплоидия - уменьшение или увеличение числа отдельных хромосом (моносомия - присутствует одна хромосома в паре хромосом, трисомия - три хромосомы одной пары хромосом, тетрасомия - четыре хромосомы одной пары хромосом, пентасомия - пять хромосом одной пары хромосом и т.д.). Анэуплоидия - наиболее распространенная патология среди хромосомных болезней.
Хромосомные мутации проявляются изменениями структуры хромосом. К видам структурных хромосомных перестроек относятся: делеция - выпадение участка хромосомы, транслокация - обмен сегментами между негомологичными хромосомами, инверсия - поворот участка хромосомы на 180°, дупликация - удвоение отдельных участков хромосом и т.д. Эти хромосомные мутации не наследуются, передача потомству возможна только в случае сбалансированной транслокации.
Сбалансированная транслокация - это реципрокная (взаимная) транслокация без потери участков вовлеченных в нее хромосом, следовательно, нет патологических эффектов у носителя, индивид фенотипически здоров. Наиболее частый пример сбалансированной транслокации - робертсоновское слияние двух акроцентрических хромосом. Транслокация между двумя акроцентрическими хромосомами с потерей их коротких плеч приводит
к образованию одной метацентрической хромосомы вместо двух акроцентрических. Формально их носители имеют моносомию по коротким плечам двух акроцентрических хромосом. Однако они являются здоровыми, потому что потеря коротких плеч двух акроцентрических хромосом компенсируется работой таких же генов в остальных 8 акроцентрических хромосомах. Клиническая картина простых и транслокационных форм трисомий по акроцентрическим хромосомам одинакова.
Несбалансированные гаметы могут быть, например, с частичной дисомией, нуллисомией и т.д. Хромосомные мутации приводят к возникновению различных аномалий развития и хромосомных болезней.
Генные (или точечные) мутации определяются изменениями в структуре гена. Мутации транскрибируемых участков приводят к синтезу аномального белка, мутации нетранскрибируемых областей приводят к снижению скорости синтеза белка. Возникновение генных мутаций возможно в результате ошибок репликации (удвоения) и репарации (восстановления поврежденных участков) ДНК. По характеру влияния мутантного гена на формирование признака различают доминантные мутации - мутантный ген проявляет себя и в гетерозиготном состоянии и рецессивные мутации - мутантный ген проявляет себя только в гомозиготном состоянии.
По исходу различают летальные и нелетальные мутации. Летальность может проявляться на уровне гамет, зигот, эмбрионов, плодов, после рождения. Результатом действия патологической мутации в онтогенезе (фенотипический эффект) может быть прежде всего летальность на ранних стадиях развития зародыша - до имплантации. Механизмы такой летальности еще не выяснены, но наличие ее у человека не вызывает сомнений. Проявляется это в виде несостоявшегося зачатия (имплантации) у фертильных женщин при нормальной половой жизни. У молодых женщин зачатие наступает в среднем через 3 месяца регулярной половой жизни без контрацепции. Из несостоявшихся зачатий примерно половина обусловлена гибелью зиготы по генетическим причинам (генные, хромосомные и геномные мутации).
Динамические мутации (или мутации экспансии) у человека были открыты в начале 1990-х гг. Этот тип мутаций не встречается у животных, заключается в нарастании (экспансии) числа триплетных повторов, расположенных в регуляторной или кодирующей части
гена. Наиболее известными повторами, приводящими к развитию наследственной патологии, являются повторы цитозин-гуанингуанин и цитозин-аденин-гуанин. Увеличение числа данных повторов (от 5-40 в норме до 90-200) приводит к развитию тяжелых неврологических болезней (миотонические дистрофии, синдром ломкой Х-хромосомы и др.).
5.1.3. Феноменология проявления генов
Знание феноменологии проявления генов позволяет сформулировать представления о способах взаимосвязи между генами и признаками, которые составляют содержание «генетической физиологии развития». Об этом впервые было заявлено в 1930-е гг. отечественным генетиком Н.В. Тимофеевым-Ресовским, который к общим феноменам проявления генов относил: доминантность и рецессивность, гетерогенные гены (генокопии), полифенные (плейотропные) гены и феномены вариабельности проявления генов (пенетрантность, экспрессивность, поле действия гена). Позднее, спустя сорокалетие, известный американский генетик В.А. Маккьюсик сформулировал так называемые принципы клинической генетики, к которым наряду с явлениями доминантности и рецессивности отнес три главных - плейотропизм, клинический полиморфизм, генетическую гетерогенность.
В отношении феноменов доминантности и рецессивности следует иметь в виду три основных момента. Во-первых, они определяют свойства фенотипов, а не гена или аллеля. Во-вторых, представляют собой условные (эмпирические) термины, не предполагающие фундаментальных различий в генетических механизмах. Наконец, доминантность и рецессивность определяются чувствительностью методов, используемых для исследования фенотипа, а по мере приближения к первичному эффекту гена вообще теряют свою значимость. Примером последнего могут служить генетические компаунды, известные для некоторых наследственных болезней (гемоглобинопатии, муковисцидоз, мукополисахаридоз Гурлера- Шейе). Суть этого явления заключается в следующем: при использовании тонких методов биохимического анализа в таких случаях обнаруживается, что аллели одного гена нередко различаются, т.е. больные рецессивными заболеваниями являются не гомозиготами, а гетерозиготами, «состоящими» из двух мутаций одного и того же гена (составные гетерозиготы, или компаунды).
Под плейотропностью («плейотропизм») понимают влияние одного гена на развитие двух и более фенотипических признаков, т.е. одна мутация, приводящая к изменению активности фермента или структуры белка, важных для функционирования многих тканей разных органов индивида, определяет множественные эффекты. Иллюстрацией плейотропного действия мутаций может служить мутация гена фосфофруктокиназы (PFK). Данная мутация определяет недостаточность фермента PFK, что приводит к умеренной несфероцитарной гемолитической анемии средней тяжести, желтухе, увеличению селезенки. Эти фенотипические изменения объясняются сокращением времени жизни эритроцитов. Проявление этого дефекта ограничено только эритроцитами, так как у таких больных сохраняется нормальная активность фермента PFK в лейкоцитах, тромбоцитах и скелетных мышцах. Другой пример - мутация в гене фибриллина, локализованном в 15q21.1 (синдром Марфана), которая определяет чрезвычайно яркий многосимптомный фенотип: высокий рост, арахнодактилия, гиперподвижность суставов, подвывих хрусталика, аневризма аорты (рис. 5-1, см. цв. вклейку).
Клинический полиморфизм наследственных болезней проявляется в различии у индивидов времени начала болезни, в динамике появления симптомов, в их спектре, сочетании и степени выраженности, в течении болезни и ее исходе. Клиническая картина конкретной моногенной наследственной болезни может варьировать не только у индивидов из разных семей, но и у членов одной семьи, в которой разные больные имеют идентичный по происхождению патологический ген. Генетической причиной полиморфизма может быть явление взаимодействия главного гена и генов модификаторов (эпистаз - межаллельное взаимодействие генов, особенности инактивации и дозовая компенсация Х-хромосомы, цитоплазматический геном), с другой стороны - это могут быть и факторы внешней среды, в которых осуществляется развитие индивида. Рисунок 5-2 иллюстрирует влияние названных двух групп факторов (генетических и внешнесредовых) на фенотипы моногенных болезней. Так, фенилкетонурия в пространстве двух обозначенных координат занимает срединное положение, отражая заметное влияние средовых и случайных факторов, а также эффектов других генов на клинические проявления болезней. В то же время для болезни Тея-Сакса (относится к группе лизосомальных болезней накопления) влияние этих факторов менее выражено, а в
 Рис. 5-2. Потенциальное
влияние генотипических (G) и негенетических (E) факторов на фенотип
некоторых моногенных болезней (по Ч. Скрайверу и Уотерсу)
Рис. 5-2. Потенциальное
влияние генотипических (G) и негенетических (E) факторов на фенотип
некоторых моногенных болезней (по Ч. Скрайверу и Уотерсу)
клинических проявлениях недостаточности Г-6-ФДГ преобладающим модифицирующим фактором является внешняя среда.
С.Н. Давиденков (1925) был одним из первых, обративших внимание на важность явления генетической гетерогенности наследственных болезней: «...один и тот же фенотип может быть реализован различными генными комбинациями». К настоящему времени доказано, что сходное или идентичное фенотипическое проявление болезни часто обусловлено несколькими разными мутациями. Генетическая гетерогенность (генокопии) может определяться мутациями разных генов (межлокусная гетерогенность) или множественным аллелизмом отдельного конкретного гена (внутрилокусная гетерогенность). Так, межлокусная гетерогенность известна для наследственных форм эпилепсии - около 20 генов, в том числе митохондриальные (табл. 5-1), для врожденных хондродисплазий - более 10 разных генов.
Таблица 5-1. Гены эпилепсии человека (по Г. Эллену и соавт.)
Наименование болезни | Локализация в хромосоме | Ген |
Идиопатические генерализованные эпилепсии | ||
Ювенильная миоклональная эпилепсия | 6p | ? |
Семейные неопасные конвульсии (BFNC1) | 20q | ? |
Идиопатическая генерализованная эпилепсия (IGE) | 8q | ? |
Идиопатические парциональные эпилепсии | ||
Парциальная эпилепсия со слуховыми симптомами (EPT) | 10q | ? |
Ночная эпилепсия фронтальной доли | 20q | ? |
Миогенные нарушения с симптоматикой эпилепсии в качестве основного признака | ||
Болезнь Унферрихта-Лундборга (EPMT) | 21q22.3 | Цистанин В |
Миоклонус-эпилепсия, рваные красные мышечные волокна (MERRF) | Митохондриальный геном | тРНК(Lys) |
Синдром северной эпилепсии (EPMR) | 8q | ? |
Цероидный липофусциноз, ювенильный тип (CLN3) | 16q | CLN3 |
Миоклональная эпилепсия Лафора (MELF) | 6q23 | ? |
Околожелудочковая гетеротипия (РН) | Xq28 | ? |
Примером внутрилокусной генетической гетерогенности могут быть разные мутации в гене дистрофина: одни из них ведут к миопатии Дюшенна, другие - к миопатии Беккера, являющимся, как известно, разными клиническими формами в ряду нервномышечных заболеваний.
Наряду с генокопиями, хотя и редко, могут встречаться фенокопии генных болезней. Это те случаи, при которых повреждающие внешние факторы, действующие, как правило, внутриутробно, вызывают болезнь, сходную по клинической картине с наследственной. Противоположное состояние, когда при мутантном генотипе индивида в результате средовых воздействий (лекарства, диета и т.п.) болезнь не развивается, называют нормокопированием. Понятия о гено- и фенокопиях помогают врачу поставить правильный диагноз, а также более точно определить возможность возникновения заболевания и вероятность рождения больного ребенка. Понимание принципов нормокопирования дает врачу возможность вторичной профилактики наследственных болезней, а в каждом конкретном случае - предупредить развитие болезни у ребенка, унаследовавшего патологический ген.
К феноменам вариабельности проявления генов относятся пенетрантность и экспрессивность. Пенетрантность - это вероятность фенотипических проявлений патологического гена, способность гена реализоваться в признак. Она показывает, какой процент носителей патологического гена обнаруживает патологический фенотип. При 100% пенетрантности у всех людей, получивших патологический ген, разовьется заболевание, т.е. число носителей этого гена будет равным количеству больных. При слабой пенетрантности число носителей патологического гена будет превышать количество больных. Однако клинически здоровый носитель патологического гена может передать его своим потомкам. Так возникают «перескоки» заболеваний через поколение, например, при синдроме Марфана родители могут быть больны, дети фенотипически здоровы, внуки больны. Неполная пенетрантность определяется генотипическим окружением гена, т.е. человек может быть носителем патологического гена, но ген может не проявляться за счет модифицирующего влияния на него других генов генотипа.
Экспрессивность - это степень выраженности патологического гена. Например, при шестипалости шестой палец может быть коротким - наблюдается слабое проявление унаследованного признака.
5.1.4. Классификация наследственной патологии
Существуют наследственные, врожденные, семейные и спорадические заболевания.
Термин «наследственные болезни» не тождествен термину «врожденные болезни». Под врожденными болезнями понимают такие состояния, которые существуют уже при рождении ребенка. Врожденные болезни могут быть обусловлены наследственными и ненаследственными факторами. К последним относятся все врожденные пороки, возникающие за счет тератогенного действия внешних факторов, врожденные инфекции (сифилис, краснуха и др.). В то же время не все наследственные болезни являются врожденными. Некоторые заболевания проявляются в детском (миопатия Дюшенна, муковисцидоз), другие в зрелом (миотоническая дистрофия, хорея Гентингтона) и даже в пожилом (болезнь Альцгеймера) возрасте.
Термин «семейные болезни» также не является синонимом термина «наследственные болезни». Семейные болезни могут быть наследственными и ненаследственными. Этот термин не говорит ни о чем, кроме того, что заболевание встречается среди членов одной семьи, да и само понятие семьи включает родственников от двух до нескольких поколений. Болезнь может быть обусловлена влиянием одинакового вредного фактора, который действует в семье: неправильное питание, плохая освещенность, сырая квартира, одна и та же вредная профессия в семье (шахтеры, ткачи и др.).
Иногда подразделяют заболевания на семейные и спорадические. При этом подразумевается: для «семейного» - наличие заболевания в семье, а для «спорадического» - его отсутствие. Таким образом, при подобном определении большинство рецессивных заболеваний будет относиться к группе спорадических, поскольку в родословных (особенно если родословная не очень большая) часто не наблюдается других случаев этого заболевания. Термин «спорадический» можно с известной долей условности применять в случаях доминантных и хромосомных болезней. Спорадические случаи противопоставляются «унаследованным» от больного родителя, т.е. термин «спорадичность» подчеркивает первичное возникновение мутации.
Как известно, в зависимости от уровня организации наследственных структур различают генные, хромосомные и геномные
мутации, а в зависимости от типа клеток - гаметические и соматические.
Классификация наследственных болезней, в основу которой положены два варианта мутаций, - в половых и соматических клетках, была предложена В.А. Маккьюсиком (1988) и включает три формы наследственной патологии:
1. Болезни вследствие мутаций в половых клетках (собственно наследственные болезни). С учетом уровня организации наследственных структур среди них различают хромосомные (например, синдром Дауна), генные (мутации в отдельном гене, которые обобщаются в периодически издающемся каталоге В.А. Маккьюсика «Менделевская наследственность у человека») и мультилокусные (полигенные, мультифакториальные).
2. Болезни вследствие мутаций соматических клеток: опухоли, некоторые аутоиммунные болезни, некоторые пороки развития. Как и в первой группе болезней, среди них выделяют хромосомные, генные, мультифакториальные.
3. Болезни, представляющие комбинацию мутаций в половых и соматических клетках (например, семейная ретинобластома).
Наследственные болезни настолько разнообразны, что они встречаются в практике врача любой специальности. Следовательно, наследственная патология может быть классифицирована в соответствии с потребностями конкретных медицинских специальностей, она ничем не отличается от классификаций болезней по органному, системному принципу или по типу обмена веществ. Поскольку наследственные болезни едины по этиологическому принципу (мутации), то основу их классификации составляет системно-органный принцип: нервные, нервно-мышечные, психические, болезни опорно-двигательного аппарата, кожи, зубочелюстной системы, крови и др.
Естественно, что такой подход не однозначен. Например, больные нейрофиброматозом (доминантная мутация) обращаются к дерматологу (первоначально заболевание проявляется светлокоричневыми обширными пятнами и нейрофиброматозными узелками на коже), могут попасть к врачу-нейрохирургу (при развитии у больных опухолей мозга) и невропатологу (в связи с возникновением при данной патологии глубоких нейрофибром) (рис. 5-3). Больные с хореей Гентингтона являются пациентами и невропатолога, и психиатра; больные с гепатолентикулярной дегенерацией - и терапевта, и невропатолога. Можно найти очень
немного наследственных болезней, при которых избирательно поражается одна система. Даже моногенно детерминируемые болезни вследствие плейотропного действия гена и вторичных патогенетических факторов затрагивают разные органы и системы. Большинство генных мутаций, а тем более хромосомные и геномные, вызывают генерализованное повреждение какойлибо ткани (например, болезни соединительной ткани) или захватывают несколько органов. Вот почему многие наследственные болезни проявляются в виде синдромов или комплекса патологических признаков, на первый взгляд не связанных между собой.
В основе классификации наследственных болезней, проявляющихся в нарушении обмена веществ, лежат типы повреждения первичного звена обмена. Такая биохимическая классификация как бы объединяет генетический и патофизиологический (клинический) подход. По такому принципу различают наследственные болезни обмена углеводов, липидов, аминокислот, витаминов, пуринов и пиримидинов, биосинтеза гормонов и т.д.
Многочисленные клинические классификации наследственных заболеваний различных органов, систем организма и видов нарушения обмена веществ объединены главным общим свойством - они являются описательными, а потому часто изменяющимися как по структуре построения, так и по содержанию (включения или исключения тех или иных форм патологии). Накопленные наукой и медицинской практикой сведения о генетических основах болезней человека позволяют предложить рабочую классификацию - болезни, вызванные мутацией отдельного гена (генные болезни).
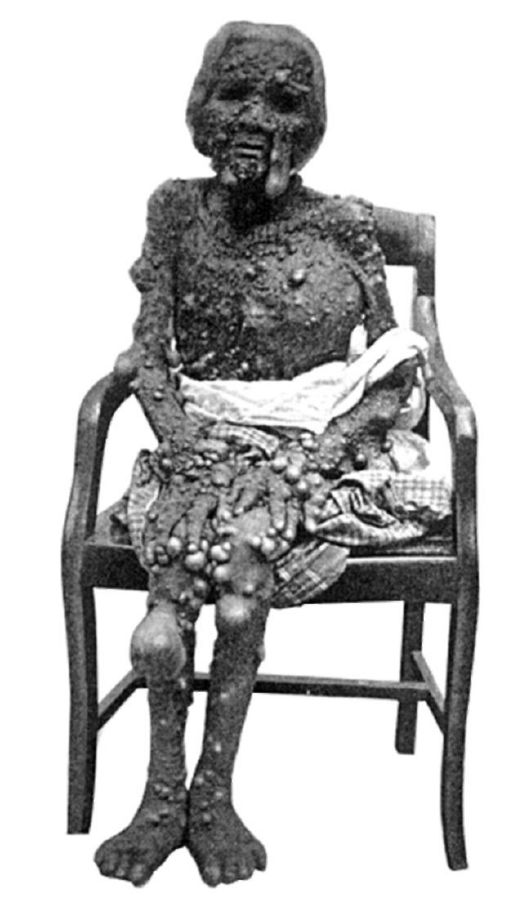 Рис. 5-3. Множественный нейрофиброз (по K. Kawashima, 1911)
Рис. 5-3. Множественный нейрофиброз (по K. Kawashima, 1911)
1. Синдромы, обусловленные хромосомными нарушениями (хромосомные болезни):
2. Мультифакториальные заболевания (болезни с наследственным предрасположением).
3. Генетические болезни соматических клеток.
4. В последнее время выделяют еще одну группу болезней - болезни с нетрадиционным типом наследования, обусловленные такими феноменами, как митохондриальная наследственность, геномный импринтинг, однородительская дисомия, экспансия тринуклеотидных повторов. О некоторых из них сведения будут представлены ниже.
5.1.5. Этиология и патогенез генных болезней
Генные болезни - это разнородная по клиническому проявлению группа заболеваний, обусловленных мутациями на генном уровне.
Основой для объединения их в одну группу являются этиологическая генетическая характеристика и соответственно закономерности наследования в семьях и популяциях. Поскольку мутации в индивидуальных генах - этиологический фактор генных болезней, то закономерности их наследования соответствуют менделевским правилам расщепления в потомстве, т.е. формальная генетика генных наследственных болезней ничем не отличается от «поведения» в семьях любых менделирующих признаков. Необходимо, однако, сразу сделать пояснения в отношении содержания понятий генных мутаций и менделирующей наследственности у человека.
Во-первых, согласно многочисленным исследованиям разных наследственных болезней и генома человека, в целом можно говорить о многообразии видов мутаций в одном и том же гене, которые являются причиной наследственных болезней. У человека описаны следующие виды генных мутаций, обусловливающие наследственные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеции, вставки (инсерции), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Любой из этих видов мутаций может вести к наследственным болезням. Даже одна и та же генная болезнь может быть обусловлена разными мутациями.
Во-вторых, современная генетика, принимая в полной мере менделизм, делает некоторые «поправки»: условность понятий о доминантности и рецессивности, неоднородность проявления ал-
леля, унаследованного от отца или матери, - импринтинг, сложный характер взаимодействия генов, гонадный мозаицизм и т.д. Более того, установлено, что мутации в разных частях одного гена ведут к разным болезням. Например, мутации в разных частях гена «RET-онкоген» ведут к четырем клинически разным наследственным болезням: двум формам множественной эндокринной неоплазии (ZA) и (ZB), семейной медуллярной тиреоидной карциноме, семейной болезни Гиршпрунга.
Мутации, вызывающие наследственные болезни, могут затрагивать структурные, транспортные, эмбриональные белки, ферменты.
Существует несколько уровней регуляции синтеза белков: претранскрипционный, транскрипционный, трансляционный. Можно предположить, что на всех этих этапах, осуществляемых соответствующими ферментативными реакциями, могут возникать наследственные аномалии. Если принять, что у человека примерно 80 тыс. генов, а каждый ген может мутировать и обусловливать другое строение белка, то, казалось бы, должно быть не меньшее число наследственных болезней. Более того, по современным данным, в каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в разных участках гена). На самом деле более чем для 50% белков изменения генетической природы: (первичная структура) приводят к гибели клетки, и мутация не реализуется в наследственную болезнь. Такие белки называются мономорфными. Они обеспечивают основные функции клетки, консервативно сохраняя стабильность видовой организации клетки. Так или иначе, но число генных болезней действительно большое. В настоящее время оно исчисляется тысячами (около 4,5 тыс.).
В группу генных болезней входят аутосомные болезни (доминантные и рецессивные) и сцепленные с полом (доминантные и рецессивные).
При доминантных аутосомных заболеваниях патологический ген находится в аутосоме и проявляет себя даже в гетерозиготном состоянии.
Особенности передачи доминантных аутосомных заболеваний следующие:
1. Лица мужского и женского пола поражаются в равной степени.
2. Передача патологического признака возможна от любого из родителей.
3. Частота индивидуального поражения среди потомков больного, как правило, составляет 50%.
4. Встречаются в каждом поколении (при условии 100% пенетрантности).
Аутосомно-доминантно наследуются: короткопалость, брахидактилия, многопалость, множественного полипоз кишечника, врожденный птоз век, ахондроплазия (рис. 5-4), врожденная куриная слепота (не поддающаяся лечению витамином А), болезнь Марфана (портрет Линкольна, арахнодактилия - паучьи пальцы, подвывих хрусталика и др.), хорея Гентингтона (проявляется в 35-40 лет, имеет 2 основных синдрома: хорея - гиперкинетические подергивания туловища, лица, шаркающая походка, симптом нарушения речи из-за подергивания языка и нёба; деменция - слабоумие) и др. Экспрессивность при хорее Гентингтона может варьировать от нистагма (толчкообразные движения глаз вертикально, в стороны, вращательно) до полной деменции (слабоумия), что свидетельствует о клиническом полиморфизме наследственных заболеваний.
Аутосомные рецессивные генные болезни проявляются только в гомозиготном состоянии, патологический ген находится в аутосоме.
Особенности передачи рецессивных аутосомных заболеваний таковы:
1. Лица мужского и женского пола поражаются в равной степени.
2. Как правило, родители больного фенотипически здоровы, являются гетерозиготами, носителями патологического гена.
3. При этом риск рождения больного ребенка - 25%.
4. Если болен один из родителей, дети обычно здоровы.
5. Нередко родители больного ребенка являются родственниками (выше вероятность быть носителями одного и того же рецессивного гена).
Аутосомно-рецессивно наследуются энзимопатии - наследственные дефекты обмена углеводов (пример - галактоземия), липидов (пример - сфинголипидозы), аминокислот (примеры - фенилкетонурия, альбинизм); витаминов, эритроцитарных ферментов, дефекты биосинтеза гормонов, коллагеновые болезни. Аутосомно-рецессивно наследуется муковисцидоз - заболевание, характеризующееся образованием в железах густого секрета, который закупоривает железистые протоки, в результате формируются кисты. Выделяют легочную и кишечную форму муковисцидоза.
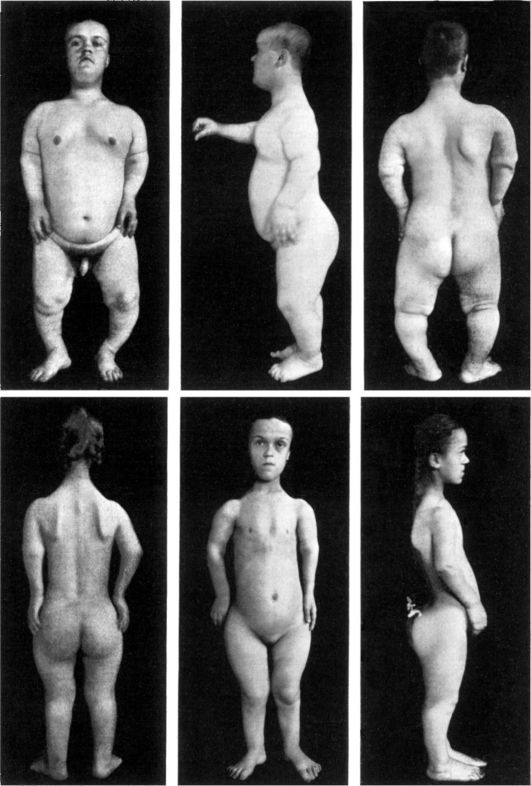 Рис. 5-4. Брат и сестра с ахондроплазией (по W. Falta, 1913)
Рис. 5-4. Брат и сестра с ахондроплазией (по W. Falta, 1913)
Например, в гене муковисцидоза описано свыше 900 вызывающих болезнь мутаций следующих типов: делеции, миссенс, нонсенс, сдвиг рамки считывания, нарушения сплайсинга. Более 400 патологических мутаций известно для гена фенилкетонурии (миссенс, нонсенс, делеции, нарушения сплайсинга). Аутосомно-рецессивно наследуется болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия). При этом заболевании увеличивается накопление меди в печени, головном мозгу, роговице глаз, что приводит к развитию цирроза печени, артритов, катаракт, неврологических нарушений, гемолитической анемии. В крови при этом наблюдается гипокупремия.
Особенности передачи Х-сцепленных доминантных болезней:
1. Поражаются и мужчины, и женщины. Больных женщин в 2 раза больше, чем больных мужчин.
2. Все дочери больного отца будут больными, сыновья здоровы.
3. Если мать гомозиготна по данному признаку, то все потомство будет больным, если гетерозиготна - больными будут 50% детей (сыновей и дочерей).
4. В среднем гетерозиготные женщины болеют менее тяжело, чем гемизиготные мужчины.
Примеры заболеваний, которые наследуются Х-сцепленно доминантно: дефект зубной эмали, аномалия волосяных фолликулов (фолликулярный гиперкератоз, приводит к полной или частичной утрате ресниц, бровей, волос головы, тяжелые формы развиваются только у мужчин) и др. Х-сцепленно доминантно наследуется гипофосфатемический рахит - нарушается реабсорбция фосфатов канальцами почек; у мальчиков наблюдаются гипофосфатемия, карликовость, рахит, у девочек - только гипофосфатемия.
Особенности передачи Х-сцепленных рецессивных болезней:
1. Передача патологического гена происходит от отца дочери, все дочери больного отца - фенотипически здоровые носители.
2. Женщина-носитель передаст патологический ген детям с вероятностью 50%.
3. Больной мужчина может получить патологический ген только от матери.
4. Женщина-носитель может получить патологический ген как от матери, так и от отца.
5. Женщины болеют редко. Рождение больной дочери возможно только в случае брака гемизиготного отца и гетерозиготной матери, происходит гомозиготирование - заболевание протекает
тяжело, часть плодов абортируется, часть новорожденных погибает на первом году жизни.
6. У гомозиготной же больной матери будут больными только сыновья, дочери будут носителями (в случае отсутствия патологического гена у отца).
Примеры Х-сцепленных рецессивных болезней: гемофилия А, гемофилия В; дальтонизм, сцепленный с полом ихтиоз, агаммаглобулинемия - болезнь Бруттона, недостаток глюкозо- 6-фосфатдегидрогеназы (Г-6-ФДГ), синдром Леша-Нихана - редкая аномалия метаболизма пуринов, связанная с недостаточностью фермента гипоксантин-гуанин-фосфорибозилтрансферазы, характеризуется тяжелой гиперурикемией, неврологическими расстройствами, идиотией, неукротимым стремлением к самоповреждениям - откусыванию пальцевых фаланг, кончика языка. Х-сцепленно рецессивно наследуется болезнь Менкеса (болезнь «курчавых волос») - нарушение метаболизма меди, проявляется тяжелыми поражениями ЦНС.
Особенностями передачи Y-сцепленных признаков является то, что признак передается от отца всем мальчикам. На Y-хромосоме локализован ряд генов, детерминирующий развитие семенников, отвечающих за сперматогенез, контролирующий интенсивность роста тела, конечностей и зубов, определяющий оволосение ушной раковины. Естественно, патологические мутации, затрагивающие формирование семенников и сперматогенез, наследоваться не могут, потому что такие индивиды стерильны. В последнее время установлено, что на Y-хромосоме расположен ген SRY, ответственный за дифференцировку пола. При генотипе XY в случае нарушения гена SRY может развиться женский фенотип. Транслокация гена SRY на Х-хромосому может привести к рождению мужчины с кариотипом ХХ. Для полного развития мужского фенотипа достаточно наличия в геноме только гена SRY, а не целой Y-хромосомы.
Патогенез генных болезней связан с первичным эффектом мутантного аллеля. Поэтому принципиальные позиции патогенеза генных болезней можно представить следующим образом: мутантный аллель - патологический первичный продукт (качественно или количественно) - цепь последующих биохимических процессов - клетки - органы - организм.
Это и есть главная общая закономерность развития генных болезней при всем их многообразии. В зависимости от того, какой
продукт контролируется конкретным геном и каков характер нарушения его при мутации, соответствующим образом развертывается патогенез болезни на молекулярном уровне.
Первичные эффекты любых (ядерных и митохондриальных) мутантных аллелей могут проявляться в четырех вариантах: 1) количественно избыточном синтезе полипептидной цепи (белка); 2) синтезе аномальной по первичной структуре полипептидной цепи (белка); 3) отсутствии синтеза полипептидной цепи (белка); 4) количественно недостаточном синтезе полипептидной цепи (белка). На основе первичного эффекта мутантного аллеля (при каждой болезни он всегда один и тот же) уже развертывается весь сложнейший патогенез генной болезни, проявляющийся в разнообразных фенотипических эффектах или клинической картине заболевания.
Если в результате мутации вырабатывается избыточное количество белка, то весь патогенез болезни в целом будет обусловлен именно гиперпродукцией генной активности. Так, при первичном гемохроматозе синтезируется избыточное количество глобина, что приводит к перенагруженности эритроцитов гемоглобином и соответственно железом. Увеличивается свертываемость крови, развивается гемосидероз паренхиматозных органов.
При другом варианте патологического эффекта мутантного гена вырабатывается аномальный белок. За этим следуют функциональные нарушения в системе, работу которой в физиологических условиях обеспечивает белок с нормальной структурой. Нарушения эти первоначально развертываются на молекулярном уровне. Примером такого варианта патогенеза болезни может быть серповидно-клеточная анемия. Замена гидрофильной глутаминовой кислоты на гидрофобный валин в структуре глобина изменяет функциональные свойства гемоглобина (пониженная растворимость, повышенная полимеризация). Он не может выполнять кислородакцепторную функцию и кристаллизуется при недостатке кислорода. В результате эритроциты приобретают серповидную форму (отсюда и название болезни), склеиваются и тромбируют капилляры и т.п.
Третий вариант патологического эффекта мутантного аллеля - отсутствие выработки первичного продукта. Это выражается в накоплении токсических продуктов-предшественников. Например, при фенилкетонурии в крови накапливается фенилаланин, поскольку из-за отсутствия фенилаланингидроксилазы печени он не
превращается в тирозин. Могут использоваться другие (обходные) пути обмена, часто также с патологическим исходом. В результате отсутствия первичного продукта гена может задерживаться какойлибо важный процесс, постоянно осуществляющийся в организме. Так, мутации генов, детерминирующих ферменты репарации ДНК, делают невозможным восстановление постоянно возникающих нарушений в структуре ДНК, что приводит к развитию злокачественных новообразований (пигментная ксеродерма, атаксиятелеангиэктазия).
Четвертый вариант первичного патологического эффекта мутантного аллеля - это недостаточность выработки нормального первичного продукта. Примерами являются гипокаталаземия (низкий уровень каталазы в крови, сопровождающийся рецидивирующими инфекциями, изъязвлением десен и слизистой оболочки полости рта) и β-талассемия (дефицит синтеза β-цепей глобина, обусловливающий нестабильность молекулы гемоглобина, укорочение жизни эритроцитов и развитие гемолитической анемии).
Выше были описаны общие закономерности патогенеза генных болезней на молекулярном уровне на примерах нарушения обмена веществ. Тот же самый принцип патогенеза (мутантный аллель - патологический первичный продукт) действует и для генов морфогенетического контроля, мутации в которых приводят к врожденным порокам развития (полидактилия, синдромы Холт- Орама, Нунена, Лоуренса-Муна, Меккеля, Робертса). Начальное звено врожденного порока развития связано с нарушением дифференцировки клеток. Запрограммированная в геноме дифференцировка клеток, а затем и органогенез осуществляются путем смены активации и выключения определенных генов в строго ограниченных временных (по отношению к онтогенезу) промежутках. Если первичный продукт морфогенетического гена аномальный, то необходимая для дальнейшего правильного развития органа дифференцировка клеток не последует. Естественно, что морфогенетических генов много, действуют они в разные периоды онтогенеза. Соответственно, мутации в них будут приводить к специфическим врожденным порокам развития.
Патогенез генных болезней не заканчивается на молекулярном уровне даже в его первичных звеньях. Для многих болезней основным звеном патогенеза является клеточный уровень. При этом речь идет не только о биологической аксиоме: во всех генетических процессах клетка является дискретной, самостоятельно регулируе-
мой единицей, и в ней осуществляются все процессы реализации генетической информации (транскрипция, трансляция, синтез белка). Клеточный уровень патогенеза генных болезней означает, что в определенных типах клеток разыгрываются основные патологические процессы, характерные для конкретной нозологической формы. Клетка как бы «не выпускает из себя патологические явления, а принимает удар от первичного патологического эффекта гена на себя». Точкой приложения являются отдельные структуры клетки, различные при разных болезнях (лизосомы, пероксисомы, мембраны).
Патогенез на клеточном уровне развертывается при болезнях накопления в связи с нарушением ферментативной активности в лизосомах. Так, накопление в клетках гликозоаминогликанов (мукополисахаридов), а в последующем и в основном межклеточном веществе приводит к развитию тяжелой группы заболеваний - мукополисахаридозов. Причиной избыточного содержания полимеров - гликозоамингликанов - является отсутствие их деградации в лизосомах (рис. 5-5, см. цв. вклейку). Нарушение распада гликозоамингликанов связано с дефектами в группе специфических ферментов, осуществляющих весь цикл деградации.
Другим примером болезней накопления могут быть гликогенозы. В клетках печени, почек, мышц и др. накапливаются полимеры гликогена, которые не подвергаются деградации даже тогда, когда организму необходима глюкоза в крови. Например, при гликогенозе I типа наследственный дефект фермента глюкозо-6- фосфатазы приводит к накоплению гликогена в печени и почках, глюкоза в кровь не поступает.
Другие внутриклеточные структуры - пероксисомы - также могут являться точкой приложения первичного действия мутантного гена. В этих случаях развиваются так называемые пероксисомные болезни. К этой группе относятся синдром Цельвегера, ризомелическая точечная остеохондродисплазия, болезнь Рефсума, акаталазия и др.
Мембраны клеток также могут быть ключевыми элементами патогенеза генных болезней. Так, нарушение синтеза рецепторов андрогенов приводит при хромосомном наборе ХУ к развитию женского фенотипа, наличию семенников в брюшной полости и повышенному уровню андрогенов (синдром тестикулярной феминизации). Клиника витамин D-резистентного рахита (аутосомнодоминантное заболевание) обусловлена дефектом рецепторов 1,25-
дигидроксихолекальциферолла. Патологические мутации (табл. 5-2) в гене рецептора липопротеинов низкой плотности (ЛПНП) вызывают дефекты клеточного рецептора. Эффект любой мутации - нарушение гомеостаза липидного обмена.
Таблица 5-2. Эффекты мутаций в гене рецептора ЛПНП на генный продукт
Класс мутаций | Последствия |
Нулевой | Нет белка-рецептора |
Дефект транспорта | Отсутствует или уменьшено число рецепторов на клеточной поверхности |
Дефект связывания | Нормальное число рецепторов, но отсутствует или уменьшено связывание ЛПНП |
Дефект интернализации | Нормальное число рецепторов и связывание ЛПНП; отсутствие или снижение эндоцитоза ЛПНП |
Дефект возвращения | Нормальное число рецепторов, связывание ЛПНП и эндоцитоз; отсутствует или уменьшено освобождение ЛПНП и возвращение рецептора на клеточную поверхность |
При муковисцидозе (синоним - кистозный фиброз) нарушается регуляция транспорта хлоридов через мембраны эпителиальных клеток, которая в норме осуществляется белком - продуктом гена, названным «кистофиброзным трансмембранным регулятором» (сокращенно CFTR). Одни мутации в гене CFTR ведут к снижению синтеза белка CFTR из-за незавершенности процессинга РНК, другие - к качественным изменениям мембранных хлоридных каналов. Одна первичная биохимическая аномалия (нарушение транспорта хлоридов) ведет к мультиорганному патологическому процессу (прогрессирующее поражение дыхательных путей, хронические синуситы, недостаточность экзогенной секреторной функции поджелудочной железы, стерильность у мужчин).
Клеточный уровень патогенеза генных болезней может проявляться не только в конкретных органеллах, но и в виде нарушения скоординированности функций интерфазного периода клетки и размножения. Так, мутации, затрагивающие области онкогенов,
ведут к снятию контроля размножения клеток (репрессия антионкогенов) и соответственно к злокачественному росту (наследственные раки толстого кишечника, ретинобластома).
Клетка может быть главным звеном при реализации молекулярного уровня патогенеза. Так, прекращение синтеза мышечного белка дистрофина при мутациях в этом гене приводит к постепенной деградации мышечных клеток. Это и является основным звеном патогенеза тяжелой наследственной болезни - миопатии Дюшенна.
Органный уровень патогенеза наследственных болезней, безусловно, является производным от молекулярного и клеточного. При различных болезнях мишенью патологического процесса служат разные органы - иногда в результате первичных процессов, иногда - вторичных. Так, отложение меди в печени и в экстрапирамидной системе мозга при гепато-лентикулярной дегенерации (болезнь Вильсона) является первичным процессом, а гемосидероз паренхиматозных органов при первичном гемохроматозе или талассемии развивается вторично вследствие усиленного распада эритроцитов. При алкаптонурии отложения гомогентизиновой кислоты в хрящах суставных поверхностей и клапанах сердца являются вторичным процессом, обусловленным высокой концентрацией гомогентизиновой кислоты в крови (она не превращается в малеилацетоуксусную кислоту в результате мутационно обусловленного отсутствия оксидазы гомогентизиновой кислоты). Это медленно ведет (примерно к 40 годам) к порокам сердца и тугоподвижности суставов.
В организме в целом взаимосвязь патогенетических процессов проявляется сочетанно на молекулярном, клеточном и органном уровнях. Патологический процесс, индуцированный первичным эффектом мутантного аллеля, приобретает целостность с закономерными межиндивидуальными вариациями. Тяжесть и скорость развития болезни при прочих равных условиях (пол ребенка, одинаковый характер мутации) зависят от генотипа организма (генымодификаторы) и условий среды. Патогенез любой наследственной болезни у разных индивидов, хотя и сходен по первичным механизмам и этапам, формируется строго индивидуально.
5.1.6. Этиология и патогенез хромосомных болезней
Этиологическими факторами хромосомной патологии являются все виды хромосомных мутаций и некоторые геномные мутации. Хотя последние в животном и растительном мире многообразны, у человека же обнаружены только три типа геномных мутаций: тетраплоидия, триплоидия, анеуплоидия. При этом из всех вариантов анеуплоидий встречаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий встречается только моносомия Х.
Что касается хромосомных мутаций, то все их типы (делеции, дупликации, инверсии, транслокации) обнаружены у человека. С клинико-цитогенетической точки зрения делеция в одной из гомологичных хромосом означает нехватку участка или частичную моносомию по этому участку, а дупликация - избыток или частичную трисомию. Современные методы молекулярной цитогенетики позволяют выявлять мелкие делеции на уровне гена, таким образом стирается грань между разделением генной и хромосомной патологии.
В основе классификации хромосомной патологии лежат три принципа. Соблюдение их позволяет точно охарактеризовать форму хромосомной патологии у обследуемого индивида и ее варианты.
Первый принцип - это характеристика хромосомной или геномной мутации (триплоидия, простая трисомия по хромосоме 21, частичная моносомия и т.д.) с учетом конкретной хромосомы. Его можно назвать этиологическим принципом.
Формы хромосомной патологии определяются типом геномной или хромосомной мутации, с одной стороны, и индивидуальным полиморфизмом хромосом - с другой. Нозологическое подразделение хромосомной патологии основывается, таким образом, на этиологическом и патогенетическом принципах: для каждой формы хромосомной патологии устанавливается, какая структура вовлечена (хромосома и ее сегмент) и в чем состоит генетическое нарушение (недостаток или избыток хромосомного материала). Дифференциация хромосомной патологии на основании клинической картины не имеет существенного значения, поскольку при разных хромосомных аномалиях имеется большая общность нарушений развития.
Второй принцип - это определение типа клеток, в которых возникла мутация (в гаметах или зиготе). Гаметические мутации ведут
к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию.
Если хромосомная аномалия возникает в зиготе или на ранних стадиях дробления (такие мутации называют соматическими в отличие от гаметических), то развивается организм с клетками разной хромосомной конституции (два типа и более). Такие формы хромосомных болезней называют мозаичными.
Третий принцип - это выявление поколения, в котором возникла мутация: возникла ли мутация заново в гаметах здоровых родителей (спорадические случаи - в 95% случаев) или родители уже имели такую аномалию (наследуемые или семейные формы - в 5% случаев). О наследуемых хромосомных болезнях говорят в тех случаях, когда мутация имеется в клетках родителя, в том числе и в гонадах. Это могут быть и случаи трисомии. Например, у индивидов с синдромами Дауна, трипло-Х происходит образование двух типов гамет - нормальных и дисомных. Такое происхождение дисомных гамет является следствием вторичного нерасхождения, т.е. нерасхождения хромосом у трисомного организма. Большая же часть наследуемых случаев хромосомных болезней связана с наличием у здоровых родителей робертсоновских транслокаций, сбалансированных реципрокных транслокаций между двумя (реже более) хромосомами и инверсиями. Клинически значимые хромосомные аномалии в этих случаях образованы в связи со сложными перестройками хромосом в процессе мейоза (конъюгация, кроссинговер).
Таким образом, для точной диагностики хромосомной болезни необходимо определить: 1) тип мутации; 2) вовлеченную хромосому; 3) полная это или мозаичная форма; 4) спорадический это случай или наследуемая форма. Такая диагностика возможна только при цитогенетическом исследовании пациента, а иногда его родителей, и сибсов.
Основным звеном патогенеза хромосомных заболеваний является несбалансированность генотипа в результате геномных и хромосомных мутаций, что проявляется: внутриутробной гибелью эмбрионов и плодов, развитием специфических синдромов, проявляющихся нарушениями физического и психического здоровья.
Патогенез хромосомных болезней, несмотря на хорошую изученность клиники и цитогенетики, даже в общих чертах остается открытым. Не разработана общая схема развития сложных патологических процессов, обусловленных хромосомными аномалиями
и приводящих к сложнейшим фенотипам хромосомных болезней. Ключевое звено в развитии хромосомной болезни ни при одной форме не выявлено. Некоторые авторы предполагают, что им может являться «несбалансированность генотипа» или «нарушение общего генного баланса». Однако такое определение ничего конструктивного не дает. Несбалансированность генотипа - это условие, а не звено патогенеза. Она должна реализовываться через какие-то специфические биохимические или клеточные механизмы в фенотип болезни.
Систематизация данных о механизмах нарушения при хромосомных болезнях показывает, что при любых трисомиях и частичных моносомиях можно выделить три типа генетических эффектов: специфические, полуспецифические и неспецифические.
Специфические эффекты должны быть связаны с изменением числа структурных генов, кодирующих синтез белка (при трисомии число их увеличивается, при моносомии - уменьшается). Многочисленные попытки найти специфические биохимические эффекты подтвердили это положение лишь для немногих генов или продуктов. При трисомии 21 обнаружено 50% повышение активности фермента супероксиддисмутазы (ген локализован в хромосоме 21). Подобный «эффект дозы гена» обнаружен для нескольких десятков генов при трисомиях по разным хромосомам. Однако биохимическое изучение фенотипа хромосомных болезней пока не привело к пониманию путей патогенеза, возникающих вследствие хромосомных аномалий врожденных нарушений морфогенеза в широком смысле слова. Обнаруженные биохимические отклонения пока трудно увязать с фенотипическими характеристиками болезней на органном и системном уровнях. Однако изменение числа аллелей гена не всегда вызывает пропорциональное изменение продукции соответствующего белка. При хромосомной болезни существенно меняется активность других ферментов или количество белков, гены которых локализованы не на вовлеченной в дисбаланс хромосоме. Ни в одном случае не обнаружено белка-маркера при хромосомных болезнях.
Полуспецифические эффекты при хромосомных болезнях могут быть обусловлены изменением числа генов, представленных и в норме в виде многочисленных копий. К таким генам относятся гены рибосомных и транспортных РНК, гистоновых и рибосомных белков, сократительных белков актина и тубулина. Эти белки в норме контролируют ключевые этапы метаболизма клетки, про-
цессов ее деления, межклеточных взаимодействий. Каковы фенотипические эффекты дисбаланса этой группы генов, как компенсируется их недостаток или избыток, пока неизвестно.
Неспецифические эффекты хромосомных аномалий связывают с измененным содержанием гетерохроматина в клетке. Важная роль гетерохроматина в клеточных делениях, клеточном росте и других биологических функциях не вызывает сомнений.
Таким образом, изучение неспецифических и частично полуспецифических эффектов способствует пониманию механизмов их реализации на клеточном уровне, безусловно, играющих важнейшую роль в патогенезе врожденных пороков развития.
Фенотипическое проявление хромосомных аномалий, т.е. формирование клинической картины синдрома, зависит от следующих главных факторов: 1) индивидуальности вовлеченной в аномалию хромосомы или ее участка (специфический набор генов); 2) типа аномалии (трисомия, моносомия; полная, частичная); 3) размера недостающего (при делеции) или избыточного (при частичной трисомии) материала; 4) степени мозаичности организма по аберрантным клеткам; 5) генотипа организма; 6) зависимости от условий среды (внутриутробной или постнатальной).
Для хромосомных болезней характерна множественность поражения - множественные врожденные пороки развития (МВПР):
1) черепно-лицевые дисморфии;
2) пороки развития внутренних и наружных органов;
3) умственная и физическая отсталость;
4) нарушение полового развития, бесплодие;
5) нарушение функций нервной и эндокринной систем.
Благодаря интенсивному изучению хромосом человека и хромосомных болезней на протяжении 35-40 лет сложилось учение о хромосомной патологии, которая имеет теперь большое значение в современной медицине. Данное направление медицины включает в себя не только хромосомные болезни, но и патологию внутриутробного периода (спонтанные аборты, выкидыши), а также соматическую патологию (лейкозы, лучевая болезнь). Число описанных типов хромосомных аномалий приближается к 1000, из них более 100 форм имеют клинически очерченную картину и называются синдромами.
К синдромам, связанным с числовыми аномалиями половых хромосом, относят:
1. Синдром Клайнфельтера (47,XXY; 48,XXYY; 48,XXXY; 49,XXXXY). Частота встречаемости - 1:1000 мальчиков. Число X-хромосом коррелирует со степенью умственной отсталости. Синдром описан в 1942 г. Проявления синдрома: высокий рост с непропорционально длинными конечностями, в детстве - хрупкое телосложение, у взрослых - ожирение, гипогенитализм (гипоплазия яичек и полового члена), недоразвитие вторичных половых признаков, иногда оволосение по женскому типу, в 50% случаев - гинекомастия. При гистологическом исследовании - гиалиноз и фиброз семенных канальцев, аспермия. Xарактерны снижение полового влечения, импотенция, бесплодие, отмечается склонность к алкоголизму, гомосексуализму, асоциальному поведению.
2. Синдром Шерешевского-Тернера (45,XО). Частота встречаемости - 1:3000 новорожденных. Проявления синдрома: отек кистей и стоп при рождении, кожная складка на шее, низкий рост (до 140 см), врожденные пороки сердца, аменорея, бесплодие, иногда снижение умственного развития. В основном социально адаптированы, могут получить специальность и работать.
3. Трисомия Х и полисомия Х. Частота встречаемости - 1:1000 девочек. Проявляется гипоплазией яичников и матки, бесплодием, иногда умственной отсталостью. С увеличением числа X-хромосом увеличиваются отклонения от нормы.
4. Полисомия Y. Популяционная частота - 1:1000 мальчиков. Xарактеризуется склонностью к асоциальному поведению, гомосексуализму.
Примеры синдромов, связанных с числовыми аномалиями аутосом:
1. Синдром Патау (трисомия по 13 хромосоме, 47,XX,+13 или 47,XY,+13). Популяционная частота - 1:7800 новорожденных. Впервые описан в 1960 г. Xарактеризуется микроцефалией, полидактилией, расщелиной губы и нёба, низко посаженными ушными раковинами, микрофтальмией, врожденными пороками сердца, дефектом межжелудочковой перегородки, аномалией почек, пороками развития органов пищеварения. Наблюдаются крипторхизм, гипоплазия наружных половых органов, удвоение матки и влагалища, двурогость матки, гипоспадия.
2. Синдром Дауна (трисомия по 21 хромосоме). Популяционная частота - 1:600-700. Проявления синдрома - плоское лицо, монголоидный разрез глаз, эпикант (кожная складка у внутреннего угла глаза), открытый рот, короткий нос, плоская переносица,
страбизм (косоглазие), пигментные пятна по краю радужки (пятна Брушфильда), плоский затылок, диспластические уши, аркообразное твердое нёбо, зубные аномалии, бороздчатый язык, гиперподвижность суставов, мышечная гипотония, врожденные пороки сердца, поперечная ладонная складка, умственная отсталость, иногда сочетается с эпилепсией (40%), лейкозом (8%). Развитие синдрома связывают с возрастом матери.
3. Синдром Эдвардса (трисомия по 18 хромосоме) - проявления похожи на синдром Патау. Популяционная частота - 1:6500.
Выделяют хромосомные синдромы, связанные со структурными перестройками хромосом. К этой группе относят синдром «кошачего крика» (моносомия 5р). Описан в 1963 г. Популяционная частота - 1:50 000 новорожденных. Встречаются цитогенетические варианты синдрома - от частичной до полной делеции короткого плеча хромосомы 5. Клиника синдрома: микроцефалия, необычный крик или плач, напоминающий мяуканье кошки, косоглазие, лунообразное лицо, широкая переносица, гипертелоризм, низко посаженные ушные раковины, умственная отсталость в стадии имбецильности.
При синдромах Патау, Эдвардса, «кошачьего крика» продолжительность жизни зависит от тяжести врожденных пороков развития внутренних органов. Большинство больных погибают в первые годы жизни.
5.1.7. Генетические факторы патогенеза мультифакториальных
заболеваний
Все существующие болезни человека (более 30 тыс.) в зависимости от роли наследственных (G) и средовых (Е) факторов в их развитии могут быть расположены на линии в данных (G-genome, геном; Е-environment, среда) координатах (рис. 5-6).
Первая группа - это наследственные болезни, при которых проявление патологического действия мутации как этиологического фактора практически не зависит от среды. Последняя может только менять выраженность симптомов болезни и тяжесть ее течения. К заболеваниям этой группы относятся хромосомные и генные наследственные болезни с полным проявлением (болезнь Дауна, нейрофиброматоз, гемофилия, фенилкетонурия, муковисцидоз, ахондроплазия и т.д.), а также врожденные аномалии развития полигенной природы. Болезнь может проявляться не обязательно в
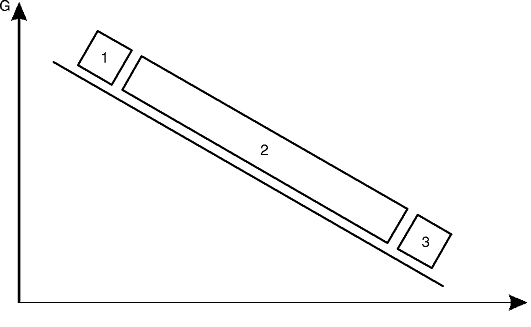
Рис. 5-6. Соотносительная роль генетических (G) и средовых (E) факторов в развитии болезней человека: 1 - «строго» наследственные болезни; 2 - болезни с наследственной предрасположенностью; 3 - ненаследственные болезни
детском, но и в любом возрасте - в соответствии с временными закономерностями генной экспрессии (например, средний возраст начала хореи Гентингтона равен 38-40 годам).
Во второй группе болезней наследственность может являться этиологическим или патогенетическим фактором, но для пенетрантности мутантных генов необходимы соответствующие факторы окружающей среды. Подобные заболевания и патологические реакции обычно проявляются неожиданно в разном возрасте при первом контакте со способствующими их развитию внешними факторами. К заболеваниям этой группы относятся сахарный диабет, атеросклероз, гипертоническая болезнь, туберкулез, экзема, псориаз, язвенная болезнь и др. Эту группу болезней называют болезнями с наследственным предрасположением или мультифакториальными заболеваниями (МФЗ).
В происхождении болезней третьей группы наследственность не играет этиологической роли (ненаследственные болезни). Сюда относятся большинство травм, инфекционных заболеваний, ожо-
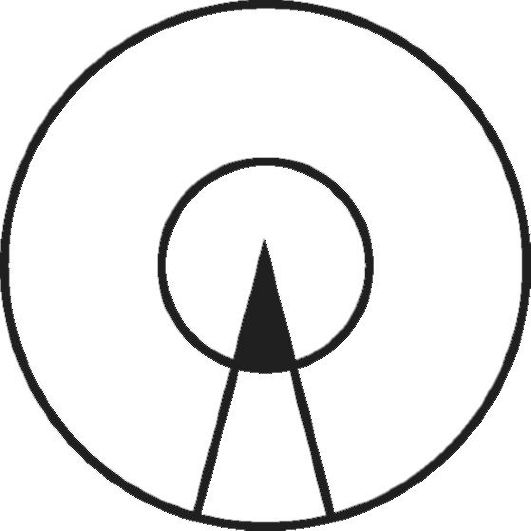 Рис. 5-7. Схема,
иллюстрирующая роль генетического предрасположения и неблагоприятных
факторов среды в возникновении заболевания. Площадь большого круга -
популяция, площадь внутреннего круга - индивидуумы данной популяции,
генетически предрасположенные к определенному типу заболевания. Сектор,
ограниченный двумя радиусами, - часть популяции, находящаяся в некоторых
определенных условиях среды, провоцирующих развитие данного
заболевания. Закрашенная часть сектора - индивидуумы, которые
действительно заболевают
Рис. 5-7. Схема,
иллюстрирующая роль генетического предрасположения и неблагоприятных
факторов среды в возникновении заболевания. Площадь большого круга -
популяция, площадь внутреннего круга - индивидуумы данной популяции,
генетически предрасположенные к определенному типу заболевания. Сектор,
ограниченный двумя радиусами, - часть популяции, находящаяся в некоторых
определенных условиях среды, провоцирующих развитие данного
заболевания. Закрашенная часть сектора - индивидуумы, которые
действительно заболевают
гов и т.д. Однако генетические факторы могут влиять на течение патологических и восстановительных процессов.
Схематически представление о роли генетических факторов и факторов среды, участвующих в возникновении и развитии МФЗ, показано на рис. 5-7. В этой схеме Г. Xарриса описана следующая ситуация: область, ограниченная внешней окружностью, - это популяция в целом; площадь внутреннего круга - те индивиды данной популяции, которые наследственно предрасположены к МФЗ определенного рода. Область, заключенная между двумя радиусами большей окружности, соответствует той части популяции, которая подвергается воздействию факторов среды, провоцирующих конкретное заболевание (в данном случае это меньшая группа); все остальные - та часть популяции (в этом случае большая), которая не подвергается воздействию этих факторов. Заболевание в действительности развивается у небольшой части популяции, т.е. у тех индивидов, у которых наследственное предрасположение сочетается с воздействием неблагоприятных условий среды.
В основе наследственной предрасположенности к болезням лежит широкий генетический балансированный полиморфизм популяций человека по ферментам, структурным и транспортным белкам, антигенам. Не менее 25-30% генетических локусов (из 80 тыс. генов) представлено в популяциях человека двумя аллелями и более. Эти гены не относятся к редким, их мутации широко распространены в популяциях человека и рассматриваются как обычные варианты аллельного полиморфизма. Индивидуальные комбинации аллелей («генетические ансамбли») невероятно многообразны.
Они обеспечивают генетическую уникальность каждого человека. Уникальность эта выражается не только в психофизиологических особенностях, но и в реакциях организма на патогенные факторы среды. Генетическая компонента наследственной предрасположенности к болезни может иметь моногенную или полигенную основу. В зависимости от этого выделяют два класса болезней с наследственным предрасположением: моно- и полигенные.
Моногенные болезни с наследственным предрасположением обусловлены мутациями отдельных генов. Эти болезни, как правило, наследуются по рецессивному типу. Причины сохранения этих форм наследственной патологии в популяциях человека, несмотря на пониженную адаптивность их носителя к тем или иным специфическим факторам среды, до конца не выяснены. Популяционногенетическое объяснение высоких концентраций таких мутаций заключается в признании сохранения полной приспособленности (в генетическом смысле) у гетерозиготных носителей. Наряду с этим у носителей таких генов должно быть селективное преимущество по сравнению с нормальными гомозиготами.
Патологическое действие «молчащих» генов проявляется под влиянием факторов окружающей среды. К настоящему времени известно более 40 локусов, мутации в которых могут вызывать болезни при дополнительном условии - действии «проявляющего» фактора, конкретного для этого гена. Некоторые примеры экопатологических реакций на факторы окружающей среды приведены в табл. 5-3.
Генные мутации, которые обусловливают возникновение таких молекулярных форм белков, патологическое действие которых выявляется не в обычных условиях, а только при взаимодействии со специфическими факторами внешней среды, называются экогенетическими вариациями. Например, у лиц с мутациями в локусе глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ) при лечении сульфаниламидными препаратами наблюдается гемолиз эритроцитов, у лиц с аномальной холинэстеразой введение дитилина приводит к длительной остановке дыхания.
Мультифакториальные болезни - это полигенные болезни с наследственным предрасположением. Они являются результатом взаимодействия генетических и средовых факторов, причем и те, и другие - многочисленны. Последнее отличает этот класс болезней от моногенных болезней с наследственным предрасположением, для которых как генетическая, так и средовая компонента пред-
Таблица 5-3. Примеры экогенетических патологических реакций на факторы окружающей среды
Фактор окружающей среды | Провоцирующий фактор | Генетически детерминированные системы | Патологическая реакция | |
Природноклиматические условия | Холодовое воздействие | ocj-ингибитор протеаз | Повышенный риск простудных заболеваний | |
Солнечная радиация | Ферменты репарации ДНК | Изъязвления кожи, рак | ||
Производственная среда | Запыленность | а,-ингибитор протеаз | Обструктивная болезнь легких | |
Гипоксия, нитрофураны | Глюкозо-6- фосфатдегидрогеназа | Гемолиз эритроцитов | ||
Красители бензидинового ряда | Ацетил трансфераза | Рак мочевого пузыря | ||
Фотоактивные вещества | Трансферрин | Фотодерматоз | ||
Бытовые вредности | Курение | а,-ингибитор протеаз | Обструктивная болезнь легких | |
Алкоголь | Альдегиддегидрогеназа | Патологическая чувствительность к алкоголю | ||
Пищевые продукты | Молоко | Лактоза | Лактаза | Непереносимость молока, диспепсия |
Галактоза | Галактозо-1-фосфатури- дил трансфераза | Галактоземия | ||
Недостаточность витамина С | L-гулонолактон- оксидаза | Цинга | ||
Конские бобы | Глюкозо-6- фосфатдегидрогеназа | Фавизм | ||
Фруктоза, сахароза | Альдолаза печени | Наследственная непереносимость фруктозы | ||
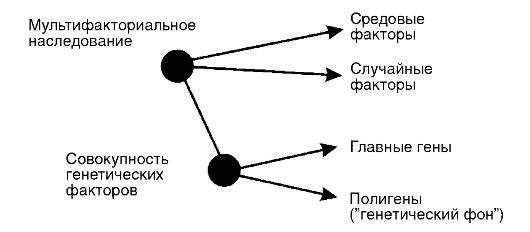 Рис. 5-8. Основные компоненты подверженности при мультифакториальных заболеваниях (по Ф. Фогелю и А. Матульски)
Рис. 5-8. Основные компоненты подверженности при мультифакториальных заболеваниях (по Ф. Фогелю и А. Матульски)
ставлены однофакторно - индивидуальный ген и специфический средовый фактор.
Взаимодействие наследственных и внешнесредовых факторов при полигенных болезнях с наследственным предрасположением представлено на рис. 5-8, где подчеркнута потенциальная роль одного или нескольких «главных» генов в структуре наследственной предрасположенности к МФЗ. Предполагается, что достаточно малое число главных генов может определять основной вклад в генетическую этиологию МФЗ. Совокупность всех других генов образует «генетический фон», который может изменять экспрессию главных генов. Важная роль в модификации всех генов полигенной системы принадлежит случайным (стохастическим) и средовым (систематическим) факторам.
Для объяснения природы болезней с наследственным предрасположением используется концепция подверженности, согласно которой подверженность имеет нормальное распределение в популяции и среди родственников первой степени родства. Кроме того, постулируется наличие «физиологического порога признака (болезни), вызываемого многими факторами», разделяющего индивидов на больных и здоровых (рис. 5-9). В рамках этой концепции разработан ряд моделей, которые позволяют оценить наследуемость как самой болезни, так и многообразных количественных признаков (биохимических, иммунологических, психофизиологических, соматических), имеющих патогенетическое значение для конкретной болезни.
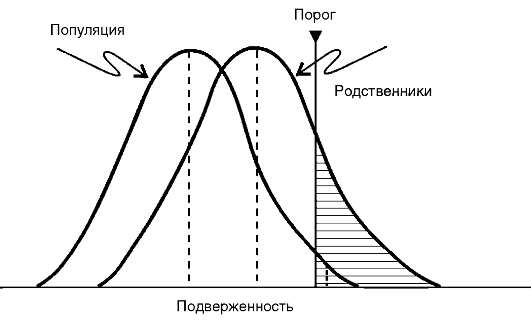 Рис. 5-9. Гипотетическое распределение порогового признака в популяции и среди родственников первой степени родства
Рис. 5-9. Гипотетическое распределение порогового признака в популяции и среди родственников первой степени родства
Под наследуемостью признака нормального или патологического понимают долю общей изменчивости его в популяции, которая может быть отнесена за счет генетических различий. Рассчитываемый коэффициент наследуемости (Н) позволяет оценить вклад генотипических факторов в детерминацию различий между больными и здоровыми. Основным материалом, анализ которого позволяет провести данные расчеты, служат сведения о распределении болезни (признака) в популяции (эпидемиологическое исследование) в семьях и среди близнецовых пар, принадлежащих данной популяции. Следовательно, генетико-эпидемиологический подход является ведущим в оценке соотносительной роли генетических и средовых факторов в развитии МФЗ. В таблице 5-4 приведены коэффициенты наследуемости для некоторых болезней и признаков.
Генетико-эпидемиологический подход в изучении МФЗ в настоящее время успешно дополнен молекулярно-генетическими исследованиями. Это стало возможным благодаря развитию технологий картирования генов, построению подробной генетической карты хромосом человека, разработке экспериментальных моделей болезней человека у животных. Результаты геномных исследова-
ний позволили обнаружить конкретные гены предрасположенности для многих МФЗ. Таким образом, появилась возможность подойти к детальному описанию структуры генетической компоненты подверженности для этой широко распространенной группы болезней. В таблицах 5-5, 5-6 приведен перечень генов предрасположенности к двум болезням мультифакториальной природы - артериальной гипертензии и бронхиальной астме.
Таблица 5-4. Коэффициент наследуемости (Н) болезней и признаков
 Таблица 5-5. Гены подверженности к эссенциальной гипертензии
Таблица 5-5. Гены подверженности к эссенциальной гипертензии
Название признака | Символ гена | Хромосомная локализация |
Субъединица эпителиального Na- канала (1β) | SCNN1B | 16p13-p12 |
Субъединица эпителиального Na- канала (1γ) | SCNN1G | 16p13-p12 |
Na+/H+- антипортер | APNH | 1p36/1-p35 |
Ренин | REN | 1q25-q32 |
Ангиотензин-I | AGT | 1q42-q43 |
Ангиотензин-1-конвертирующий фермент | ACE | 17q22-q24 |
Панкреатическая фосфолипаза А2 | PLA2 | 12q23-q24/1 |
Гипертензия, обусловленная геном, экспрессирующимся в почке | SAN | 16p13/11 |
Эндотелиальная синтаза окиси азота | NOS3 | 7q35-q36 |
Таблица 5-6. Гены подверженности к бронхиальной астме и атопии
Название признака | Символ гена | Хромосомная локализация |
Гиперреактивность бронхиального дерева | BHR1 | 5q |
β2-адренергический рецептор | ADRB2R | 5q32-q34 |
Интерлейкин-4 | JL4(IL36-4,-5,-9,-13,GM-CSF) | 5q31-5q33 |
Фактор некроза опухоли α | TNFA | 6p21.3-p21.1 |
HLA-комплекс | HLA-DR | 6p |
Уровень общего иммуноглобулина Е | IGEL | 11q12-q13 |
β-иммуноглобулиновый рецептор тучных клеток | FCER1b | 11q12-q12 |
Интерферон γ | igif | 12q15-q24.1 |
Невральная синтаза окиси азота | NOS1 | 12q24.2-q24.3 |
Эстераза D | ESD | 13q14.2-q14.3 |
α-Цепь антигенного рецептора Т-клеток | TCRA | 14q11-14q12 |
Однако необходимо иметь в виду, что в патогенезе мультифакториальных болезней могут участвовать аллели генов, вызывающих рецессивно передающиеся моногенные дефекты, никакого отношения к соответствующей болезни не имеющие. Такие сочетания отмечены у гетерозиготных носителей генов, вызывающих «болезни репарации» ДНК: анемию Фанкони, синдром Блюма, атаксию-телеангиэктазию и некоторые другие. У гетерозигот по этим генам существенно повышена частота возникновения рака. В этом случае такие гены играют роль генетически предрасполагающих факторов, по-видимому, через ослабление иммунологической системы элиминации соматических мутаций. Широкое распространение гетерозиготного носительства разных мутантных генов, возможно, еще недостаточно оценено как потенциальный фактор предрасположения.
Хотя в полной мере трудно количественно оценить значение наследственного предрасположения в патологии человека (заболеваемость, смертность, социальная дезадаптация), все же можно с уверенностью утверждать, что оно достаточно большое. Об этом говорят факты высокой частоты, длительности и тяжести течения широко распространенных болезней (гипертония, атеросклероз, аллергия, шизофрения, сахарный диабет, язвенная болезнь, псориаз, врожденные пороки развития). Безусловно, с возрастом значение наследственного предрасположения к развитию патологии возрастает, но и в детском возрасте оно является не менее важным. Речь идет не только о врожденных пороках развития, но и об атопических иммунных состояниях, целиакии, повышенной чувствительности к некоторым пищевым продуктам. В детском возрасте вклад наследственного предрасположения в развитие патологии составляет, по-видимому, не менее 10%, в среднем - существенно повышается, а в пожилом определяет до 25-50% болезненных состояний.
При разработке мер профилактики болезней с наследственным предрасположением и оценке их роли в патологии человека необходимо принимать во внимание, что закономерности их распространения достаточно сложны. Распространенность этой группы болезней варьирует в разных популяциях значительно. Причины вариаций можно объяснить различиями генетических и внешних факторов. В результате генетических процессов в популяциях человека (отбор, дрейф генов, миграция) гены «предрасположения» могут накапливаться или элиминироваться. Даже при равных условиях среды это может привести к разному уровню заболеваемости. В то же время при одинаковой частоте предрасполагающих аллелей или их сочетаний в популяциях частота болезней с наследственным предрасположением может быть разной, если условия среды отличаются.
Прогресс в изучении болезней с наследственным предрасположением достигнут в значительной степени благодаря осуществлению проекта «Геном человека». На основе «инвентаризации» генов предрасположенности и знания условий их патологического проявления могут разрабатываться профилактические мероприятия, включая своевременную диспансеризацию предрасположенных лиц.
5.1.8. Генетические болезни соматических клеток
К этой группе болезней относятся злокачественные новообразования, некоторые аутоаллергические заболевания и врожденные пороки развития. Показано значение соматических мутаций в механизмах старения.
Злокачественные новообразования были первой группой болезней, при которых постулировались многостадийность и необходимость ряда мутационных событий для возникновения злокачественной клетки. Классическим примером может служить ретинобластома - злокачественное офтальмологическое заболевание, развивающееся у детей. Двусторонние случаи ретинобластомы часто прослеживаются в родословных как доминантный признак. Большинство же односторонних опухолей - это спорадические случаи.
Длительное время такую закономерность трудно было объяснить с точки зрения классических менделевских правил наследования. Поэтому была предложена двухударная гипотеза возникновения ретинобластомы, которая и подтвердилась. Процесс развития опухоли представляется сейчас следующим образом. Заболевание обусловлено мутацией в гене ретинобластомы (13-я хромосома), который относится к группе генов - супрессоров опухолевого роста. Эти гены обладают рецессивным свойством, следовательно, для развития опухоли необходимо иметь мутации этого локуса в обеих гомологичных хромосомах.
В случае двусторонней ретинобластомы мутация в одном локусе наследуется от родителя, т.е. она имеется во всех клетках. Эта мутация сама по себе не является причиной ретинобластомы, хотя гетерозиготными по данной мутации оказываются все ретинальные клетки. Пока идентичный участок (локус) 13-й хромосомы, унаследованный от другого родителя, функционирует нормально, опухоль не образуется. Как только в гомологичном участке хромосомы одной из ретинальных клеток происходит мутация, пораженными оказываются оба аллельных локуса. Это приводит к развитию ретинобластомы. Таким образом, в данном случае, когда наследуется одна мутация, фенотипически она никак не проявляется в ретинальных клетках до появления мутации в гомологичной хромосоме. Вероятность возникновения соматической мутации того же локуса в одной из множества ретинальных клеток весьма высока - до 90%, поэтому ретинобластома развивается в обоих глазах.
Следовательно, становление наследственной формы болезни происходит в два этапа. На первом этапе мутация передается через половую клетку. На втором этапе клетка пополняется таким же изменением в гомологичной хромосоме в клетках сетчатки. Для возникновения спорадических случаев ретинобластомы необходимы также две мутации. Обе они должны возникнуть в соматических (ретинальных) клетках. Поскольку совпадение двух мутационных событий для одного и того же локуса - явление редкое, то маловероятно, что это случится в сетчатках обоих глаз. Вот почему спорадические случаи - это опухоли одного глаза.
Другая форма злокачественного новообразования, в развитии которой доказана роль последовательных генетических событий (мутаций, потери гетерозиготности) в соматических клетках, - колоректальный рак. Стадии превращения нормальной клетки в злокачественную и мутационные события в клетках, сопровождающие процесс развития колоректального рака, представлены на рис. 5-10.
Известна роль приобретенных в ходе индивидуального развития соматических мутаций в проявлении и клиническом полиморфизме унаследованных мутаций при ряде наследственных болезней и врожденных пороков развития. Подобная ситуация предполагает развитие патологии в два этапа: первично возникшая мутация в гаметических клетках предрасполагает к развитию болезни, но реализуется в болезнь при возникновении мутации в другом локусе. Недавно подобное явление было показано для аутосомнодоминантной поликистозной болезни почек (ген ее локализован в
 Рис. 5-10. Предполагаемая последовательность генетических событий в генезе колоректального рака
Рис. 5-10. Предполагаемая последовательность генетических событий в генезе колоректального рака
16-й хромосоме). Эта болезнь характеризуется образованием кист в почках, увеличением их размера, артериальной гипертензией и развитием почечной недостаточности. У некоторых больных развиваются кисты в других органах. У носителей унаследованной аутосомно-доминантной мутации гена PKD1 образуются изолированные очаговые кисты в почках. Но если дополнительно приобретается мутация в нормальном аллеле PKD1 (она тестировалась в кистозной ткани) или в рядом расположенном (сцепленном) гене TSC2 (ген туберозного склероза), то клиника кистозной болезни утяжеляется - множественные кисты формируются в печени, развиваются церебральные аневризмы. Такие же закономерности описаны при других наследственных болезнях (костная фиброзная дисплазия Олбрайта, синдром Клипе-ля-Тренове-Вебера, синдром Штурге-Вебера).
Старение, как об этом свидетельствуют многочисленные исследования, сопряжено с накоплением мутаций в соматических клетках. В последнее время в связи с успешным изучением митохондриального генома выдвинута гипотеза о том, что спонтанное возникновение и накопление мутаций в митохондриях с последующим цитоплазматическим распределением мутантных генов приводит к появлению клеток с различными биоэнергетическими свойствами и является звеном механизмов старения и развития дегенеративных процессов у человека.
Высказывается суждение и о том, что в качестве одной из причин возникновения аутоаллергических болезней могут быть соматические мутации. В целом следует признать, что, по-видимому, роль соматических мутаций в возникновении, а особенно в клиническом полиморфизме многих болезней более значима, чем ожидалось.
5.1.9. Болезни с нетрадиционным типом наследования
Как отмечалось выше, в последние годы обнаружен ряд новых закономерностей наследования признаков (нормальных и патологических), не соответствующих менделевским. К ним относятся: геномный импринтинг, экспансия триплетных повторов, митохондриальные болезни.
Совсем недавно у человека обнаружены однородительские дисомии. В норме ребенок наследует по каждой паре хромосом: одну хромосому от отца, другую - от матери. У индивидов с этим типом
наследования число хромосом по всем парам нормальное, однако одна пара представлена хромосомами от одного и того же родителя. Наиболее частый механизм возникновения однородительской дисомии у человека - редукция трисомии: в процессе гаметогенеза за счет нерасхождения хромосом возникает дисомия в гамете по определенной хромосоме, что при оплодотворении приводит к трисомии. По неясным пока причинам третья хромосома может элиминироваться на ранних стадиях дробления, а у зародыша останутся две хромосомы одного родителя. Следует отметить, что в целом частота возникновения однородительской дисомии материнского происхождения в 3 раза чаще отцовской. Если бы две копии генов, получаемых потомками от матери или от отца, функционировали в клетках одинаково, то никаких серьезных последствий в организме эти нарушения не вызывали бы. Однако оказалось, что некоторые хромосомы несут отдельные гены, экспрессия которых определяется полом передавшего их родителя. Это явление получило название феномена геномного импринтинга (от imprint - отпечаток).
Под геномным импринтингом понимают эпигенетический процесс, который дифференциально маркирует материнские и отцовские гомологичные хромосомы. В участках генома, подверженных импринтингу, экспрессируется (проявляется) только один из двух аллелей - отцовский или материнский, т.е. наблюдается моноаллельная экспрессия генов. Второй аллель, вследствие наличия на нем некоего отпечатка, импринтирован (выключен или подавлен) и не экспрессируется.
Такой способ регуляции генов свидетельствует о неэквивалентном вкладе родителей в геном потомков и приводит к разному фенотипическому проявлению мутаций у потомства, унаследованных от матери или отца.
У человека известно уже около 40 таких генов, и предполагается, что их число может достигать 200-500. Основную роль в процессе геномного импринтинга играет метилирование цитозиновых оснований ДНК с образованием 5-метилцитозина, способствующее выключению экспрессии генов с модифицированными нуклеотидами.
Так, известно, что в проксимальном участке хромосомы 15 имеются близко сцепленные, но противоположно импринтированные локусы, отвечающие за возникновение двух фенотипически разных синдромов - Прадера-Вилли и Энгельмана. Для синдрома
Прадера-Вилли, фенотипически проявляющегося умственной отсталостью, мышечной гипотонией, выраженным ожирением, гипогонадизмом, низким ростом и акромикрией, установлен кандидатный ген, ответственный за синтез полипептида N малого ядерного рибонуклеопротеина (SNRPN). Этот ген активно экспрессируется исключительно на отцовской хромосоме 15. Кандидатным геномом синдрома Энгельмана (синоним - синдром «счастливой куклы»), характеризующегося неадекватной счастливой улыбкой и глубокой умственной отсталостью с резкими кукольными судорожными движениями, является убиквитин - белковый лигазный ген (ИВЕЗА) E6-AP, который экспрессируется главным образом на материнской хромосоме 15.
Синдром Прадера-Вилли развивается при делеции участка 15 хромосомы отца (нет участка, который экспрессируется на отцовской хромосоме) или однородительской дисомии 15 хромосомы материнского происхождения (в этом случае гены импринтированы - выключены или подавлены).
Синдром Энгельмана («счастливой куклы») развивается при делеции участка 15 хромосомы матери или при однородительской дисомии 15 хромосомы отцовского происхождения.
В случае однородительской дисомии отсутствует потеря хромосомного материала, но нарушается нормальное метилирование ДНК, а следовательно, и транскрипция генов критического района, поэтому такое функциональное состояние равнозначно делеции.
В начале 1990-х гг. у человека был обнаружен новый тип мутаций, который до сих пор не зарегистрирован ни у одного вида млекопитающих. Он получил название динамических мутаций или мутаций экспансии (см. выше).
Общие характеристики этого класса болезней следующие:
1. Болезни с экспансией тринуклеотидных повторов представляют собой нейродегенеративные заболевания с поздним проявлением.
2. Отмечается прямая корреляция между числом тринуклеотидных повторов и тяжестью клинической картины.
3. Для болезней экспансии характерна генетическая антиципация - возрастание тяжести заболевания в последующих поколениях, что связано с тенденцией к возрастанию числа повторов у потомков.
Первое заболевание, при исследовании которого в 1991 г. был открыт феномен экспансии - синдром фрагильной (ломкой)
Х-хромосомы, или синдром Мартин-Белл. Проявления синдрома Мартин-Белл: умственная отсталость, аутизм, макроорхидизм (у взрослых), удлиненное лицо, прогнатия - выступающий подбородок, оттопыренные уши, пронзительная смешная речь, аномалии соединительной ткани, нарушение поведения.
Сейчас открыта целая группа болезней экспансии тринуклеотидных повторов.
Утверждение, что весь генетический материал человека находится в составе хромосом, не совсем верно, поскольку есть одно исключение - митохондриальный геном. Митохондриальная ДНК (мтДНК) представляет собой небольшую кольцевидную молекулу длиной 16 569 пар оснований. В отличие от ДНК ядерного генома она не связана с белками, имеет очень высокую «плотность генов» ввиду отсутствия интронов, содержит 13 генов, кодирующих белки (3 субъединицы цитохром-С-оксидазы, 6 компонентов АТФазы и др.), 22 гена транспортных РНК (тРНК) и 2 гена рибосомальных РНК. На рисунке 5-11 представлена схема структуры мтДНК и приведены примеры митохондриальных болезней, которые являются следствием мутации мт-генов. Эти болезни передаются только по материнской линии.
Наследование признаков, передаваемых через ДНК митохондрий, и связь мутаций мтДНК с болезнями человека были впервые показаны в 1988 г. С тех пор обнаружено большое число мутаций мтДНК, лежащих в основе целого ряда нейродегенеративных заболеваний, некоторых МФЗ, митохондриальных миопатий.
Примеры таких заболеваний:
• синдром MELAS, развивающийся в связи с мутацией в гене лейциновой тРНК митохондрий и проявляющийся митохондриальной энцефаломиопатией, лактатацидозом и инсультоподобными эпизодами. Вначале развивается сахарный диабет 2 типа с инсулинорезистентностью, который переходит в сахарный диабет 1 типа с инсулинонедостаточностью и наличием аутоантител к антигенам островков поджелудочной железы;
• наследственная нейроофтальмия Лебера (характеризуется билатеральной потерей зрения);
• синдром MERRF (характеризуется прогрессирующей дегенерацией нервной и мышечной ткани, что проявляется судорогами, атаксией, миопатией, потерей слуха);
• летальная инфантильная дыхательная недостаточность и др.
 Рис.
5-11. Структура митохондриального генома и примеры митохондриальных
болезней: APDP - болезнь Альцгеймера/болезнь Паркинсона; DEAF -
нейросенсорная потеря слуха; LHON - наследственная нейроофтальмия
Лебера; LDYT - LHON и дистония; MELAS - митохондриальная миотония,
энцефалопатия, молочнокислый ацидоз и приступы судорог; MERRF -
миоклональная эпилепсия в сочетании с необычно красными мышечными
волокнами; NARP - нейропатия, атаксия и пигментный ретинит; PEM -
летальная прогрессирующая энцефаломиопатия
Рис.
5-11. Структура митохондриального генома и примеры митохондриальных
болезней: APDP - болезнь Альцгеймера/болезнь Паркинсона; DEAF -
нейросенсорная потеря слуха; LHON - наследственная нейроофтальмия
Лебера; LDYT - LHON и дистония; MELAS - митохондриальная миотония,
энцефалопатия, молочнокислый ацидоз и приступы судорог; MERRF -
миоклональная эпилепсия в сочетании с необычно красными мышечными
волокнами; NARP - нейропатия, атаксия и пигментный ретинит; PEM -
летальная прогрессирующая энцефаломиопатия
5.1.10. Методы изучения и диагностики наследственных патологий
В настоящем разделе представлена краткая характеристика генетико-эпидемиологического подхода в изучении наследственных болезней (генетический, близнецовый и популяционностатистические методы), методы клинической диагностики (клиническое обследование и параклинические исследования), лабораторные методы диагностики (цитогенетические, биохимические, молекулярно-генетические) и методы моделирования наследственных болезней в эксперименте на животных.
Генетико-эпидемиологический подход в изучении роли наследственности в этиологии и патогенезе болезней человека предполагает совместное применение генеалогического, близнецового и популяционно-статистических методов.
Генеалогический метод - метод родословных, т.е. прослеживание болезни (или признака) в семье или роду с указанием типа родословных связей между членами родословной. Технически он складывается из двух этапов: составления родословной и генеалогического анализа. На первом этапе, используя специальные символы и правила, которые достаточно хорошо известны, составляется родословная.
Второй этап (генеалогический анализ) позволяет решать несколько задач. Во-первых, установить наследственный характер болезни или признака. Наследственную природу анализируемых болезней (признаков) предполагают, если они повторяются в родословной несколько раз (рис. 5-12). Во-вторых, в случае доказательства наследственного характера болезни возможно установление типа наследования (аутосомно-доминантный, аутосомнорецессивный, Х-сцепленный, Y-сцепленный, митохондриальный). Наконец, анализ родословных является обязательным условием успешного решения задач картирования генов на хромосомах, расшифровки механизмов взаимодействия генов, изучения интенсивности мутационного процесса. В клинической практике генеалогический анализ составляет основу осуществления медикогенетического консультирования.
Близнецовый метод. Возможности его использования при изучении наследственной патологии определяются представлениями о происхождении близнецов: партнеры однозиготной (монозиготной) пары близнецов являются генетически тождественными, поскольку образуются из одной зиготы; партнеры двузиготной
 Рис. 5-12. Габсбургская губа, прослеженная на протяжении столетий: а - император Максимилиан IV (1459-1519); б - император Карл V - внук (а) (1500-1558); в - эрцгерцог Карл Тешенский (1771-1847); г -
эрцгерцог Альбрехт - сын (в) (1817- 1895). Strohmayer, Nova Acta
Leopoldina, 5, 1937 (из книги К. Штерна «Основы генетики человека. М.:
Медгиз, 1965. CV.95)
Рис. 5-12. Габсбургская губа, прослеженная на протяжении столетий: а - император Максимилиан IV (1459-1519); б - император Карл V - внук (а) (1500-1558); в - эрцгерцог Карл Тешенский (1771-1847); г -
эрцгерцог Альбрехт - сын (в) (1817- 1895). Strohmayer, Nova Acta
Leopoldina, 5, 1937 (из книги К. Штерна «Основы генетики человека. М.:
Медгиз, 1965. CV.95)
(дизиготной) пары близнецов образуются из двух разных зигот, а потому их фенотипическое сходство не больше, чем у братьев и сестер, родившихся в разное время; пары разнополых близнецов всегда являются двузиготными; внутрипарное различие, обнаруживаемое по какой-либо нормальной или патологической особенности однозиготных близнецов, должно быть отнесено за счет различий, обусловленных факторами среды.
Близнецовый метод считается особенно эффективным для доказательства наследственной предрасположенности к конкретным заболеваниям (мультифакториальные болезни и комплексные признаки). В этом случае применяют два варианта сравнения: сравнение конкордантности (совпадения фенотипов) моно- и дизиготных близнецов; сравнение конкордантности вместе и порознь выросших монозиготных близнецов. Обобщенные результаты использования близнецового метода для понимания природы разных заболеваний представлены в табл. 5-7.
Популяционно-статистические методы. В отношении моногенных (менделевских) болезней эти методы в сравнительных ис-
следованиях различных групп населения (популяций, этносов) позволяют выявить и оценить наиболее важные факторы популяционной динамики (отбор, дрейф генов, инбридинг, давление мутаций), определяющие пространственную изменчивость (территориальную гетерогенность в распространенности) наследственных болезней. Например, показано, что накопление некоторых рецессивных болезней среди финноязычного населения Финляндии обусловлено длительной изоляцией субпопуляций и дрейфом генов. Накопление редких патологических мутаций среди евреевашкенази объясняется эффектом «родоначальника» и дрейфом генов. Для популяций Средней Азии, а также некоторых арабских популяций показано, что главным фактором, детерминирующим груз и структуру наследственных болезней, накопление аутосомнорецессивной патологии, являются неслучайные родственные браки (ассортативность браков).
Таблица 5-7. Конкордантность (%) близнецовых пар для разных групп болезней
Тип заболевания | Конкордантность близнецов | |
монозиготные | гетерозиготные | |
Болезни с наследственной предрасположенностью | 40-60 | 4-18 |
Аутосомно-доминантные болезни | 100 | 50 |
Аутосомно-рецессивные болезни | 100 | 25 |
Для других форм патологии - мультифакториальных заболеваний (болезней с наследственным предрасположением) популяционно-статистические методы, наряду с использованием современных молекулярно-генетических методов, позволяют на основе анализа ассоциаций и сцепления генетических маркеров с конкретными признаками и болезнями в сравниваемых группах «больные - здоровые» («случай - контроль») в популяции, «больные - здоровые» в ядерных семьях, принадлежащих данной популяции, осуществлять поиск «генов-кандидатов» исследуемой болезни.
Методы клинической диагностики. Наследственная патология имеет некоторые специфические характеристики. Для их выявления используется весь арсенал методов клинической диагностики (анамнез, осмотр, физические методы и пр.), параклинические ис-
следования (общеклиническая диагностика с использованием современной аппаратуры исследования отдельных органов и систем - R-логические, ультразвуковые, томографические и т.д.). Главный смысл их применения (и выбор оптимальных клинических приемов) - обнаружить специфические черты болезни, указывающие на наследственный характер поражения. К ним относятся: семейный характер заболевания; хроническое, прогредиентное, рецидивирующее течение; врожденный характер заболевания; «резистентность» к наиболее распространенным методам терапии; наличие патогномоничных признаков (или их сочетаний), свойственных только данной форме наследственной патологии. Именно последнее, обусловленное тем, что большинство мутантных генов, вызывающих наследственные болезни, обладает плейотропным эффектом и вовлечением в процесс многих органов и систем (неспецифичных ассоциаций), определяет необходимость применения параклинических методов исследования. Так, при таких наследственных болезнях соединительной ткани, как синдром Элерса-Данлоса, Марфана, для которых характерно нарушение сосудистой стенки (особенно аорты), подвывих хрусталика, пролапс митрального клапана, патология суставов, понадобятся R-логические, ультразвуковые, офтальмоскопические и некоторые другие параклинические методы исследования. Однако окончательная диагностика и выявление более тонких генетических вариантов исследованной патологии осуществляются с применением специальных лабораторных методов диагностики.
Лабораторные методы диагностики. Лабораторная диагностика наследственных болезней (феноили генотипирование индивидов) в основе своей может быть направлена на идентификацию одной из трех «ступеней» болезни. Во-первых, это выявление этиологической причины наследственной патологии, или характеристика генотипа, т.е. определение конкретной мутации у индивида (генной, хромосомной, геномной). Эти цели достигаются с помощью цитогенетических или молекулярно-генетических методов. Во-вторых, лабораторные методы позволяют регистрировать первичный продукт гена (биохимические, иммунологические методы). В-третьих, возможна регистрация специфических метаболитов измененного обмена, возникших в процессе реализации патологического действия мутации (биохимические, иммунологические, цитологические методы регистрации на уровне жидкостей - кровь, моча, секрет - или клеток).
Цитогенетические методы. Они предназначены для изучения структуры хромосомного набора или отдельных хромосом. Объектом цитогенетических наблюдений могут быть делящиеся соматические, мейотические и интерфазные клетки. Чаще исследования выполняются на соматических клетках: наиболее удобный объект - культура лимфоцитов периферической крови, но также и культура клеток из кусочков кожи (фибробласты), костного мозга, эмбриональных тканей, хориона, клеток амниотической жидкости.
Специальным образом полученные препараты из культуры клеток затем окрашиваются. Все методы окраски препаратов можно разделить на три группы: простые, дифференциальные, флюоресцентные.
Простая окраска (метод окраски по Гимзе или в русскоязычной литературе - «рутинная окраска») используется для ориентировочного определения числовых аномалий кариотипа. Структурные хромосомные аномалии (делеции, транслокации, инверсии), выявленные при простой окраске, должны быть идентифицированы с помощью дифференциальной окраски. Под этим методом понимают способность хромосом к избирательному окрашиванию по длине с фиксацией в виде чередования эу- и гетерохроматических районов (темные и светлые полосы).
Благодаря успехам в молекулярной генетике человека разработан принципиально новый метод изучения хромосом - метод флюоресцентной гибридизации in situ (FISH). Он основан на использовании однонитевого специфического участка ДНК («зонда»), специальным образом «помеченного» и способного, после присоединения к участку анализируемой хромосомы, присоединить флюоресцентные красители (красный, зеленый и другие цвета). С помощью люминесцентного микроскопа окрашенные хромосомы визуализируются на фоне неокрашенных. Метод позволяет расшифровать сложные хромосомные перестройки, а также локализовать ген.
Биохимические методы. Эти методы: направлены на вышвление биохимического фенотипа организма - от первичного продукта гена (полипептидной цепи) до конечных метаболитов в моче или поте. Поэтому существует огромное разнообразие методов. Но для целей диагностики наследственных болезней оправданными являются две биохимические стратегии, которые позволяют определить дальнейший ход обследования и выбор соответствующих биохими-
ческих методов и тестов: массовые и селективные программы первичной биохимической диагностики наследственных болезней.
Массовые просеивающие программы применяются в диагностике фенилкетонурии, врожденного гипотиреоза, адреногенитального синдрома, врожденных аномалий нервной трубки и болезни Дауна. Селективные диагностические программы предусматривают проверку, уточнение биохимических аномалий обмена для пациентов, у которых подозреваются генные болезни, с использованием простых качественных реакций или более точных методов (тонкослойная хроматография мочи и крови, газовая хроматография, флюорометрические методики и др.).
Молекулярно-генетические методы. Это большая и разнообразная группа методов, предназначенных для выявления вариаций в структуре исследуемого участка ДНК (аллеля, гена, региона хромосомы) вплоть до расшифровки первичной последовательности нуклеотидных оснований. Все разнообразие подходов для идентификации генов или определенных фрагментов ДНК и их вариаций основывается на двух основных методических разработках - технологии блот-гибридизации (от англ. blot - промокать) и амплификации отдельных участков ДНК.
В основу методики амплификации положена полимеразная цепная реакция (ПЦР), которая позволяет в течение нескольких часов выделить и размножить определенный фрагмент ДНК в количестве, превышающем исходное в 109 раз. Такая высокая степень направленного обогащения значительно упрощает работу с минимальными количествами ДНК-образцов. Реакция высокоспецифична и чувствительна, позволяет исследовать единичную копию гена в исходном материале.
В табл. 5-8 приводятся другие методы генотипирования, основанные главным образом на этих же двух технологиях, а также обозначены подходы к ДНК-диагностике болезней (прямая и косвенная диагностика).
Методы моделирования. Как и в других разделах биологии и медицины, в генетике человека и медицинской генетике широко применяются методы моделирования. Они разделяются на две группы: биологические и математические.
Таблица 5-8. Подходы к ДНК-диагностике наследственных болезней
Прямая диагностика мутаций | - Детекция крупных перестроек (мутаций) генов методами блот-гибридизации с использованием ДНК-зондов - Выявление крупных и мелких делеций генов ПЦР-амплификацией отдельных фрагментов, в том числе мультиплексной ПЦР - Детекция внутригенных мутаций, изменяющих сайты узнавания определенных рестриктаз (и вследствие этого - размер фрагментов, выявляемых блот-гибридизацией или ПЦР-амплификацией) - Аллельспецифическая гибридизация (амплификация) с использованием олигонуклеотидов, комлементарных нормальной или мутантной последовательности ДНК - Детекция конформационного полиморфизма одноцепочечной ДНК (single strand conformation polymorphism) - Метод детекции ошибок спаривания (cleavage mismatch detection) - Секвенирование гена или его фрагмента |
Косвенная (непрямая) молекулярная диагностика | Анализ косегрегации генетических маркеров (микро- и мини-сателлиты, ПДРФ), сцепленных с патологическим геном, и болезни в семьях |
Для изучения многих моногенных болезней человека используются животные, несущие мутации в гомологичных генах. Они являются удобными моделями для исследования молекулярных основ патогенеза и отработки оптимальных вариантов лечения. С этой целью во многих питомниках мира созданы и поддерживаются коллекции генетических линий животных (мышей, крыс, кроликов, собак и др.). Мировая коллекция мышей насчитывает несколько сотен линий с моногенно наследуемыми дефектами.
Для анализа экспрессии мутантных генов in vivo и оценки биологических свойств первичного генного продукта удобными оказались трансгенные животные. Трансгенных животных получают искусственным введением (трансгеноз) генетического материала (фрагмент гена или иная последовательность ДНК) в оплодотворенную яйцеклетку или в ранние зародыши млекопитающих. Дальнейшее развитие технологий трансгеноза позволило подойти к конструированию модельных генетических линий животных - на-
правленному получению генетических моделей наследственных болезней путем введения сайтспецифических модификаций в геном млекопитающих, или «выбиванию» (вырезанию) определенного гена, идентичного гену человека. Такие мыши, у которых экспериментально «вырезан» определенный ген из генома, называются нокаутными (от англ. knock out). На этих животных можно приближенно понять патогенез наследственной болезни и апробировать методы лечения.
Математическое моделирование применяют в тех случаях, когда сформулированные задачи не могут быть решены только путем анализа экспериментального материала или их решение математически оказывается более быстрым и точным, чем экспериментально. В области генетики популяций математическое моделирование позволяет, например, оценить удельный вес многочисленных факторов популяционной динамики (отбор, мутации, дрейф генов, инбридинг, миграции) в формировании «груза» наследственной патологии. Изучение таких сложных процессов, как взаимодействие наследственных факторов и факторов среды в развитии признака, закономерностей функционирования генома человека как интегративной системы, взаимодействие генов («генные сети») при формировании физиологических свойств организма, становится предметом исследования биоинформатики (компьютерной геномики).
5.2. РОЛЬ КОНСТИТУЦИИ В ПАТОЛОГИИ
Среди факторов, играющих роль в этиологии болезней, определенное значение имеет конституция человека (от лат. constitutio - строение), под которой подразумевается совокупность относительно устойчивых структурных и функциональных особенностей, оказывающих влияние на реактивность организма и его сопротивляемость к действию болезнетворных факторов.
Учение о конституции имеет многовековую историю. Поводом для его возникновения явилось стремление врачей выделить среди огромного количества людей, наделенных чрезвычайно разнообразными индивидуальными свойствами, какие-либо типовые структурные и функциональные особенности организма и установить их связь с развитием тех или иных заболеваний.
Основоположник учения о конституции и ее связи с развитием болезней - Гиппократ, который различал людей с сухим и влажным; сильным и слабым; вялым и упругим типами конституции. Кроме того, он подразделял людей по темпераменту на сангвиников, холериков, флегматиков и меланхоликов. В I-II столетиях нашей эры учение о конституции организма получило дальнейшее развитие в трудах Галена, который ввел понятие о habitus, подразумевая под этим особенности телосложения, влияющие тем или иным образом на развитие болезней.
Основное развитие учение о конституции получило в ХХ столетии.
5.2.1. Классификация типов конституции
Сложным явилось создание классификации типов конституции. Единой классификации не существует. Предложено более 40 ее разновидностей, причем за основу классификации в большинстве случаев принимались особенности телосложения, такие, как соотношение между ростом и весом, длиной туловища и конечностей, а также размеры и форма грудной клетки, степень развития мускулатуры и др. Значительно реже классификация типов конституции основывалась на функциональных особенностях нервной системы.
C. Sigaud (1914) предложил выделять 4 типа конституции - дыхательный, пищеварительный, церебральный и мышечный, в зависимости от преимущественного развития той или иной системы организма (рис. 5-13). Дыхательный (респираторный) тип характеризуется длинной грудной клеткой с острым эпигастральным углом и умеренным развитием брюшных внутренностей. У пищеварительного (дигестивного) типа грудная клетка короткая, эпигастральный угол тупой, вместе с тем увеличены размеры живота, усиленно развит жевательный аппарат, имеется склонность к ожирению. Для людей церебрального типа характерен большой череп с хорошо развитой лобной частью в сочетании с нежным тонким телосложением и короткими конечностями. Мышечному (мускульному) типу конституции присуще усиленное развитие мускулатуры, пропорциональное телосложение, широкая грудная клетка. Согласно представлениям К. Сиго, формирование типа конституции происходит главным образом в детском возрасте и зависит от тренировки органов и систем организма.
 Рис. 5-13. Типы конституции по C. Sigaud: а - дыхательный; б - пищеварительный; в - мышечный; г - церебральный
Рис. 5-13. Типы конституции по C. Sigaud: а - дыхательный; б - пищеварительный; в - мышечный; г - церебральный
Немецкий психиатр E. Kretchmer (1921) выделил 3 типа конституции - пикнический (пикник), лептосомный (астеник) и атлетический (атлет). Пикник характеризуется большой массой тела за счет избыточного отложения жира, короткой грудной клеткой, большим выступающим животом, длинным туловищем и сравнительно короткими конечностями. Астеник - высокий и тонкий, с длинными конечностями и относительно коротким туловищем, узкой грудной клеткой. Атлет - человек с хорошо развитой мускулатурой, с широкими грудной клеткой и плечами, узким тазом.
W. Sheldon (1940) взял за основу классификации степень развития дериватов зародышевых листков - эктодермы, эндодермы и мезодермы, и выделил, соответственно, 3 конституциональных типа: эндоморфный, эктоморфный и мезоморфный. Характеристика этих типов имеет сходство с конституциональными типами, выделенными E. Kretchmer: эндоморфный тип сходен с пикником, эктоморфный - с астеником, а мезоморфный - с атлетом. Эта классификация пользуется признанием на Западе.
В нашей стране наиболее популярна классификация М.В. Черноруцкого (1928), который выделил на основании особенностей телосложения два крайних типа - астеник и гиперстеник, и один промежуточный - нормостеник.
Астенический тип конституции характеризуется относительно коротким туловищем и длинными конечностями, узкой, плоской и сравнительно длинной грудной клеткой с острым эпигастральным углом, узкими плечами, тонкой длинной шеей, небольшим объемом живота; в целом продольные размеры значительно превалируют над поперечными. У людей с гиперстеническим типом конституции имеется обратное соотношение размеров тела по сравнению с астениками: относительно длинное туловище и короткие конечности, короткая шея, широкая короткая грудная клетка с увеличенным переднезадним размером, большой, часто выступающий вперед живот; в целом имеется нарастание поперечных размеров тела. Нормостеники характеризуются пропорциональным телосложением, широким плечевым поясом и выпуклой грудной клеткой, хорошим развитием мускулатуры. Заслугой М.В. Черноруцкого является то, что совместно со своими сотрудниками он изучил особенности обмена веществ и состояние некоторых внутренних органов у выделенных им конституциональных типов.
При сопоставлении различных классификаций можно отметить присутствие в большинстве их двух крайних вариантов: с одной стороны, тип с увеличенными продольными размерами тела, а с другой - люди с увеличенными поперечными размерами. Недостатком большинства классификаций является то, что в них не включены промежуточные (смешанные) типы конституции, к которым относится большинство людей.
Все перечисленные классификации типов конституции строились с учетом особенностей телосложения. Значительно реже в качестве основополагающих признаков использовались функциональные особенности организма. H. Eppinger, L. Hess (1910) предложили классифицировать конституциональные типы в зависимости от особенностей функционирования вегетативной нервной системы. Исходя из этого, они предложили подразделять всех людей на симпатикотоников и ваготоников. Эта классификация подверглась критике на том основании, что антагонизм между симпатической и парасимпатической системами проявляется не во всех отношениях.
И.П. Павлов (1925) выделил людей с различными типами высшей нервной деятельности на основе учета силы, подвижности и уравновешенности основных нервных процессов - возбуждения и торможения. Для их обозначения И.П. Павловым была использована предложенная Гиппократом классификация видов темпе-
 Рис. 5-14. Типы высшей нервной деятельности. Неизвестный художник XIX в., из архива кафедры патофизиологии СибГМУ
Рис. 5-14. Типы высшей нервной деятельности. Неизвестный художник XIX в., из архива кафедры патофизиологии СибГМУ
рамента - сангвиник, холерик, флегматик и меланхолик. Сангвиник характеризуется сильным уравновешенным подвижным типом высшей нервной деятельности; холерик - сильным неуравновешенным подвижным; флегматик - сильным уравновешенным инертным и меланхолик - слабым типом высшей нервной деятельности (рис. 5-14).
5.2.2. Типы конституции и болезни
В настоящее время не вызывает сомнений, что люди определенного телосложения более склонны к некоторым заболеваниям. Это заключение основывается на многочисленных наблюдениях, сделанных М.В. Черноруцким и рядом других авторов. Связь между типами конституции и соматическими заболеваниями отражена в табл. 5-9.
Таблица 5-9. Особенности обмена веществ и предрасположенность к заболеваниям у лиц с различными типами конституции (по М.В. Черноруцкому)
Тип конституции | Особенности обмена веществ | Предрасположенность к заболеваниям |
Астеники | Преобладание процессов диссимиляции над ассимиляцией; склонность к повышению основного обмена и алкалозу; ускоренная утилизация глюкозы при сахарной нагрузке; содержание холестерина и липидов в крови в пределах нормы или снижено | Склонность к птозу органов брюшной полости, язвенной болезни, тяжелому течению туберкулеза легких, гипотонии, патологической аменорее |
Гиперстеники | Преобладание процессов ассимиляции, склонность к понижению основного обмена и ацидозу; нарушение толерантности к глюкозе при сахарной нагрузке; повышенное содержание в крови липидов и холестерина | Предрасположенность к заболеваниям сердечно-сосудистой системы (атеросклерозу, инфаркту миокарда, гипертонии), сахарному диабету пожилых, ожирению, желчекаменной болезни |
Нормостеники | Равновесие процессов ассимиляции и диссимиляции; показатели обмена веществ и физиологических процессов близки к средней норме | Предрасположенность к заболеваниям верхних дыхательных путей и опорнодвигательного аппарата |
Имеются данные о связи между типом конституции и психическими заболеваниями. Впервые на существование такой связи указал психиатр E. Kretchmer. Согласно его наблюдениям, среди больных шизофренией превалируют астеники, а среди больных
маниакально-депрессивным психозом - пикники; кроме того, он и его соавторы утверждают, что развитие эпилепсии чаще имеет место у атлетов, чем у людей с другим типом конституции. В дальнейшем эти наблюдения получили некоторое подтверждение в исследованиях других авторов. W. Sheldon отметил, что истерия и депрессия чаще возникают у людей с мезоморфным или эндоморфным типами конституции, а тревожные состояния более характерны для лиц с эктоморфным типом телосложения.
Имеются также указания на более частое развитие неврозов у людей с сильным неуравновешенным и слабым типами высшей нервной деятельности.
Приведенные наблюдения указывают на важность учета типа конституции для профилактики многих заболеваний. Своевременное проведение профилактических мероприятий может предупредить развитие болезни.
5.2.3. Факторы, влияющие на формирование типа конституции
В прошлом по этому вопросу высказывались противоположные суждения. Сторонники одного из них (Ю. Тандлер, Р. Миллер, О. Негели, П. Матес и др.) считали, что тип конституции всецело зависит от генотипических особенностей данного человека, т.е. является наследственно обусловленным и в течение всей жизни остается неизменным. Согласно мнению C. Sigaud, тип конституции формируется в процессе жизни и зависит от вида деятельности, тренировки той или иной системы организма. А.А. Богомолец также рассматривал конституцию как совокупность фенотипических особенностей организма и считал, что в формировании типа конституции основную роль играют факторы внешней среды, условия жизни.
В настоящее время большинство ученых (П.Д. Горизонтов, А.Д. Адо, Н.Н. Зайко и др.) считают, что в формировании типа конституции главную роль играют наследственные особенности, но могут оказать влияние и факторы внешней среды (инфекции и интоксикации, избыточное питание или голодание, гиповитаминозы, физическая нагрузка, занятия спортом и др.).
5.3. ЗНАЧЕНИЕ ВОЗРАСТА В ВОЗНИКНОВЕНИИ И РАЗВИТИИ БОЛЕЗНЕЙ
В разные возрастные периоды люди по-разному реагируют на одни и те же воздействия. Каждому возрасту свойственны свои особенности и склонность к развитию определенных заболеваний. В онтогенезе человека (как и других млекопитающих) выделяют два этапа: пренатальный и постнатальный. Постнатальный этап развития, в свою очередь, делится на три периода: 1) период роста, когда формируются характерные для представителей данного вида морфологические и функциональные системы; 2) период зрелости, который характеризуется завершившимся формированием морфологических и функциональных систем; 3) период старости, при котором происходит постепенное ослабление всех физиологических функций и затухание жизненного процесса.
5.3.1. Возраст и болезни
Ранний детский возраст характеризуется пониженной реактивностью и резистентностью (см. главу 6), что обусловлено незаконченным развитием нервной, эндокринной и иммунной систем, незрелостью ферментных систем, несовершенством внешних и внутренних барьеров.
У новорожденного развитие корковых центров еще не закончено, возбудимость клеток коры низкая; регуляция метаболизма и функций организма осуществляется в основном подкорковыми центрами. Имеются слабость процессов внутреннего торможения и недостаточная способность к дифференцировке поступающих в ЦНС сигналов. Поэтому на воздействие различных безусловных раздражителей ребенок отвечает генерализованной реакцией. Болевая чувствительность у детей первого года жизни выражена слабо, что может затруднить своевременную постановку правильного диагноза при заболеваниях внутренних органов.
Дети первого года жизни, особенно новорожденные, характеризуются недостаточным проявлением защитно-приспособительных и компенсаторных реакций. Из-за незаконченного формирования механизмов теплорегуляции у детей раннего возраста легко возникают состояния перегревания и переохлаждения. Недостаточность аппарата регуляции водно-солевого обмена является причиной
частого развития состояний гипергидратации или обезвоживания (эксикоза) при нарушениях пищеварения, при гипервентиляционном синдроме и других заболеваниях.
Новорожденные тяжелее, чем взрослые, переносят кровопотерю. Вместе с тем дети первого года жизни проявляют большую устойчивость к гипоксии и не реагируют на нее учащением дыхания и пульса. Полностью эти реакции формируются к 6-7-му годам жизни.
Особенностью течения инфекционных заболеваний у детей является их недостаточная способность реагировать специфическими изменениями на действие возбудителя и как следствие этого имеют место стертость и искажение некоторых характерных симптомов, что может создать трудности для диагностики. Характерна также склонность к генерализованным реакциям и развитию осложнений, что связано с недостаточным проявлением защитных механизмов (незавершенность фагоцитоза, пониженное обезвреживание микробных токсинов, слабое развитие тканевых барьеров). Регионарные лимфатические узлы у ребенка начинают функционировать как тканевой барьер только после третьего месяца жизни. До этого срока микробы свободно проникают во внутреннюю среду организма. Вместе с тем в этом периоде жизни понижена чувствительность к возбудителям ряда инфекций (скарлатина, дифтерия, корь, краснуха, брюшной тиф) в связи с наличием пассивного иммунитета, обусловленного поступлением антител из организма матери через плаценту и с молоком при грудном вскармливании. Большую опасность для детей раннего возраста представляет гнойная инфекция, в особенности стафилококковая, а также вирусные инфекции, которые являются одной из частых причин заболевания пневмонией у детей.
Уровень иммуноглобулинов в крови новорожденного соответствует таковому у матери. Спустя 0,5-1 год после рождения восприимчивость к инфекционным заболеваниям повышается вследствие исчезновения из крови ребенка материнских антител. В то же время появляется способность к образованию собственных антител и к развитию аллергических реакций, которые в период новорожденности не выражены. Но в целом объем антителообразования в течение первого года жизни недостаточный.
Одним из частых заболеваний у детей раннего возраста является пневмония. Ее развитию способствует недостаточное расправление легких при вдохе вследствие слабой экскурсии грудной
клетки, ригидности стромы легких, частого образования ателектазов (т.е. спадения альвеол). Пневмония в большинстве случаев является интерстициальной и имеет генерализованный характер. Развитие пневмонии у новорожденных не сопровождается появлением выраженной одышки, что объясняется слабой возбудимостью дыхательного центра. Слабо выражен кашлевой рефлекс. К концу первого года жизни основные функции ребенка еще не устойчивы. Отсутствуют способность к ограничению патологического процесса, например воспаления, в пределах первично пораженного участка ткани, сохраняется склонность к диффузным реакциям и развитию сепсиса. Центральная нервная система ребенка легко истощаема.
В возрасте от одного года до 6-7 лет у детей постепенно совершенствуются все функции организма, ослабляется склонность к генерализованным реакциям при действии патогенных факторов, учащается заболеваемость детскими инфекциями, усиливаются проявления аллергии.
В период полового созревания (пубертатный период) может наблюдаться чрезмерная неустойчивость вегетативной нервной системы, психики и поведения. Возможны нарушения функции сердечно-сосудистой (ювенильная гипертония, акроцианоз) системы. В этом возрасте отмечается также повышенная частота развития туберкулеза и сахарного диабета. Может отклоняться от нормы процесс полового развития - он может быть ускорен или, напротив, замедлен.
По мере роста организма происходит развитие и совершенствование тканевых барьеров и иммунной системы, совершенствуется нервно-эндокринная регуляция. Поэтому у людей зрелого возраста реактивность и резистентность наиболее выражены. Отчетливо проявляются характерные особенности того или другого заболевания и различных патологических процессов. То же можно сказать в отношении развития защитно-приспособительных и компенсаторных реакций, что способствует благоприятному исходу болезни.
При продвижении от зрелого к пожилому и старческому возрасту снова происходит снижение реактивности и резистентности организма. У стариков уменьшается способность адаптироваться к воздействию изменяющихся физиологических и патогенных факторов внешней среды вследствие снижения метаболизма и функциональных резервов различных органов. Происходит угнетение
иммунологической реактивности. Снижается противоинфекционный иммунитет. Особенно часто наблюдается развитие заболеваний, вызываемых вирусами, гноеродными микроорганизмами, что связано не только с недостаточным образованием антител, но и с ослаблением активности фагоцитов и повышением проницаемости тканевых барьеров. Снижается противоопухолевая резистентность. Частому развитию злокачественных опухолей в пожилом и старческом возрасте способствуют не только ослабление иммунного надзора, но и накопление с возрастом соматических мутаций, с которыми связывают опухолевую трансформацию клеток. Возрастает частота сердечно-сосудистой патологии вследствие развития атеросклероза и разбалансированности в системе регуляции сосудистого тонуса. Повышается заболеваемость сахарным диабетом (диабет пожилых), деменцией; учащаются переломы костей в связи с развитием остеопороза.
Многие заболевания в этом возрасте имеют хроническое течение и нередко характеризуются недостаточно четкими проявлениями вследствие пониженной реактивности.
Таким образом, возрастная реактивность имеет существенное значение и должна обязательно учитываться при проведении профилактики, постановке диагноза и лечении пациентов любого возраста.
5.3.2. Старение
Старение является неотвратимой стадией развития всех живых существ, которым удалось прожить достаточно долго.
Процесс старения характеризуется развитием структурных и функциональных изменений, которые ограничивают способность организма поддерживать состояние гомеостаза при действии разнообразных стрессоров, вызывают снижение резистентности и обусловливают повышенную заболеваемость людей пожилого и старческого возраста, что приводит в итоге к полной утрате жизнеспособности.
Изменения в органах и тканях имеют генерализованный характер (табл. 5-10), хотя и могут развиваться неодновременно.
Внешними проявлениями старения являются дистрофические изменения кожи и ее придатков: она истончается, становится морщинистой, уменьшается количество потовых и сальных желез, седеют и выпадают волосы. Разрушаются зубы. Происходит умень-
Таблица 5-10. Изменения в различных системах организма в процессе старения
Вид системы | Характеристика возрастных изменений |
Нервная система | Снижается в той или иной степени масса мозга, уменьшается количество нейронов в коре головного мозга, мозжечка и ядрах подкорки; увеличивается количество клеток глии. В нейронах изменяется активность ряда ферментов, что ведет к нарушению синтеза и обмена нейромедиаторов, например дофамина. Снижаются память, скорость образования условных рефлексов, познавательные способности |
Органы чувств | Снижается острота зрения и слуха, нарушаются обоняние и вкусовая чувствительность |
Эндокринная система | Уменьшается секреция гормонов щитовидной, поджелудочной и половыми железами, корой надпочечников, аденогипофизом и эпифизом. Снижается реакция β-клеток поджелудочной железы на гипергликемию и чувствительность тканей к действию инсулина |
Сердечнососудистая система | Уменьшаются минутный объем и сердечный индекс, максимальная частота сердечных сокращений, скорость кровотока. Повышаются периферическое сопротивление сосудов и системное артериальное давление. Нарастает риск развития ишемической болезни сердца в связи с повышенным содержанием в крови холестерина, липопротеинов низкой и очень низкой плотности |
Органы дыхания | Снижаются максимальная вентиляция и жизненная емкость легких, увеличивается остаточный объем воздуха в легких |
Система органов пищеварения | Снижаются секреция пищеварительных ферментов и HCl, объем пищеварительных соков, двигательная функция желудка и кишечника; иногда возникают нарушения глотания |
Мочевыделительная система | Уменьшаются количество функционирующих нефронов, уровень клубочковой фильтрации, концентрационная способность почек; замедляется выведение с мочой лекарственных препаратов; ночной диурез превалирует над дневным (никтурия). У женщин нередко возникает недержание мочи, у мужчин - затруднение мочеиспускания в связи с аденомой предстательной железы |
Окончание табл. 5-10
Система гемостаза | Повышаются как прокоагулянтная, так и антикоагулянтная активность крови, однако последняя возрастает в меньшей степени, чем первая; снижается антиагрегационная способность стенок сосудов, что способствует образованию тромбов |
Иммунная система | Ослабевает клеточный и гуморальный иммунитет. Возможно развитие иммунодефицита. Усиливаются аутоиммунные реакции |
шение массы тела за счет снижения объема мышечной и костной ткани и уменьшения количества внутриклеточной жидкости. Снижается плотность костной ткани, она разрыхляется (остеопороз), в связи с этим возрастает частота переломов. Нередко возникает кифоз вследствие сплющивания тел позвонков. Снижается мышечная сила. Проблемы, с которыми сталкиваются старые люди, - это ходьба и подъем по лестнице, самообслуживание, понижение ясности зрения и остроты слуха, недержание мочи, нарушение интеллектуальных функций. В основе указанных изменений лежат атрофические процессы в различных тканях, замещение клеток, выполняющих специфические функции, соединительной и жировой тканями, в мозгу - глией. Этому способствуют генетические изменения в клетках и ухудшение кровоснабжения из-за прогрессирующего склероза сосудов и снижения функции сердца.
Проявления старения на уровне клеток. Как в стареющих фибробластах в культуре (in vitro), так и в клетках многих тканей при старении снижается скорость процессов транскрипции и трансляции на уровне генома, нарастает количество хромосомных мутаций; происходит увеличение количества лизосом, тогда как число митохондрий уменьшается. Понижается чувствительность клеток к различным ростовым факторам и гормонам, что связано с уменьшением числа рецепторов и нарушениями в пострецепторной передаче сигналов. Замедляется деградация белков, что связано со снижением активности внутриклеточных протеаз. Увеличивается количество структурно измененных белков. В клетках накапливается липофусцин.
В связи с пониженной доставкой кислорода, уменьшением в клетках количества митохондрий и нарушением синтеза дыхательных ферментов угнетаются процессы тканевого дыхания и окислительного фосфорилирования. В результате снижается синтез
макроэргических соединений, что ограничивает функциональные резервы клеток и возможность синтеза различных веществ. Ослабляется работа энергозависимых мембранных насосов. В связи с этим в клетках повышается содержание Na+, Ca2+ и Cl-, а содержание калия, магния и фосфора снижается.
Обнаруживается дисбаланс ферментативной активности - у одних ферментов она понижена, у других повышена; к числу последних относятся моноаминооксидаза, сукцинатдегидрогеназа, альдолаза и др.
Следует иметь в виду, что при старении изменяются некоторые показатели, характеризующие метаболические процессы. В частности, возрастает средняя концентрация глюкозы в крови (до 6,7 ммоль/л) и мочевины (до 8,9 ммоль/л), существенно снижается содержание альбуминов в плазме крови.
Биологическая наука, занимающаяся изучением изменений в организме при старении, называется геронтологией. Одной из задач, которую она пытается разрешить, является выяснение причин и механизмов старения.
Механизмы старения
По поводу механизмов старения существовали многочисленные гипотезы, но большинство из них имеет в настоящее время только исторический интерес. Так, И.И. Мечников высказал предположение, что старение является следствием эндогенной интоксикации организма веществами, образующимися в кишечнике при гнилостном брожении. Чтобы затормозить процесс старения, он предложил употреблять в пищу как можно больше молочнокислых продуктов и вести правильный образ жизни. Некоторым подтверждением этой гипотезы является высокое содержание молочнокислых продуктов в диете кавказских долгожителей.
Существующие в настоящее время теории и гипотезы относительно механизмов старения можно подразделить на две группы. Теории, относящиеся к одной из этих групп, рассматривают старение как генетически запрограммированный процесс, который развивается в «плановом» порядке, подобно другим стадиям онтогенеза. Другие считают, что старение является результатом накапливающихся в течение жизни повреждений в геноме и других клеточных структурах. Теории о связи старения с генетически запрограммированными процессами возникли на основе данных о
запрограммированной смерти клеток на ранних стадиях онтогенеза. С нею связаны, например, рассасывание хвоста головастиков, исчезновение клеток между пальцами у развивающихся млекопитающих и птиц.
В пользу теории запрограммированного старения свидетельствует ряд наблюдений. Л. Хайфлик около 40 лет тому назад обнаружил, что фибробласты человеческого эмбриона, находясь в условиях клеточной культуры, могут совершить около 60 клеточных циклов, после чего прекращают деление. В дальнейшем они еще сохраняют жизнеспособность в течение ряда месяцев, но затем погибают. Пролиферативный потенциал клеток мог быть изменен только в слабой степени путем добавления в питательную среду глюкокортикоидов и факторов роста. Фибробласты пациентов с ускоренным старением имели существенно укороченный срок жизни в культуре. Ограниченный срок жизни клеток в культуре был установлен не только для фибробластов, но и для других клеток от многих видов животных.
Одна из теорий программированного старения объясняет ограниченную способность клеток к делению существованием специфического гена, который экспрессируется после того, как клетка совершит соответствующее количество делений. Предполагается, что этот ген кодирует белок, который тормозит вступление клетки в фазу S митотического цикла, вследствие чего не происходит репликации ДНК. Так, установлено, что ингибитором пролиферации эндотелиальньгх клеток является интерлейкин (IL) 1α, при его инактивации способность этих клеток к делению резко возрастает. Но не доказано, что IL-1a может тормозить пролиферацию фибробластов или каких-либо других клеток.
Установлено, что при искусственном слиянии молодых и стареющих клеток образованные таким образом гибриды не размножаются, т.е. фенотип стареющих клеток является доминирующим. Это объясняют присутствием в старых клетках ингибитора, блокирующего митотический цикл. Природа этого ингибитора не установлена. Предполагается, что это продукт антионкогена, т.е. не исключено, что механизмы, участвующие в ограничении опухолевого роста, могут быть ответственными и за старение.
Высказывается также предположение, что старение является результатом уменьшения передачи информации некоторых необходимых для жизни генов.
Среди теорий, рассматривающих старение как результат накопления в клетках различных повреждений в процессе их жизни, заслуживают особого внимания теория ошибок (L. Orgel, 1963) и свободнорадикальная теория старения, предложенная Д. Харманом (1956) и Н.М. Эмануэлем (1958).
Согласно теории ошибок, в результате аномалий в структуре генов или погрешностей в процессе транскрипции и трансляции генной информации может произойти синтез аномальных белков, которые утрачивают способность выполнять соответствующие функции. Это может иметь катастрофические последствия, если такие белки играют роль ферментов, участвующих в белковом синтезе, например РНК-полимераза, аминоацил-1-РНК-синтетаза, рибосомальные белки. В результате погрешности в синтезе белков будут продолжать накапливаться. В пользу этой теории свидетельствует нарастание количества аномальных белков в стареющих клетках, но полного подтверждения она не получила, так как попытки вызвать преждевременное старение клеток посредством искусственного усиления продукции аномальных белков пока не увенчались успехом.
Согласно свободнорадикальной теории, повреждение генома и других клеточных структур в процессе старения происходит в основном под действием свободных радикалов кислорода, которые образуются при тканевом дыхании в качестве его побочного продукта. Большая их часть подвергается в клетках обезвреживанию с помощью системы антиоксидантов, но некоторые сохраняют активность и взаимодействуют с различными внутриклеточными структурами, вызывают мутации, стимулируют перекисное окисление липидов, нарушают структуру и функцию клеточных мембран. Полагают, что под действием кислородных радикалов в первую очередь повреждается ДНК митохондрий, так как она является менее защищенной, чем ядерная, из-за отсутствия в митохондриях гистонов и ферментов, осуществляющих репарацию поврежденных участков ДНК. Кроме того, именно в митохондриях образуется наибольшее количество радикалов кислорода в связи с утечкой электронов из дыхательной цепи. Во многих тканях старых индивидуумов были обнаружены различные виды мутаций митохондриальной ДНК (точковые мутации, делеции, дупликации).
Повреждения митохондриальной ДНК сопровождаются нарушением синтеза дыхательных ферментов. В результате снижается
образование макроэргов, необходимых для работы мембранных насосов, выполнения специфических функций и поддержания внутриклеточного гомеостаза, что завершается гибелью клетки. Справедливость этой гипотезы подтверждается наблюдениями, свидетельствующими о снижении функции митохондрий в процессе старения. Установлено, что с возрастом в клетках снижается мощность системы антиоксидантов и нарушается равновесие между про- и антиоксидантами. На основе всего этого сложилось мнение, что оксидантный стресс играет важную роль в процессах старения.
Предполагается также, что возрастные нарушения функции клеток в какой-то степени связаны с прогрессирующим накоплением в их цитоплазме и ядре пигмента липофусцина, который образуется путем неферментного гликозилирования долгоживущих белков и нуклеиновых кислот. Присоединившиеся к макромолекулам сахара могут окисляться и образовывать массивные перекрестные связи между белками, липидами и нуклеиновыми кислотами, что сопровождается нарушением их функции.
Можно предположить, что максимальный срок жизни, присущий тому или другому виду живых существ, запрограммирован в геноме клеток, но под действием оксидативного стресса и других повреждений он может в той или другой степени укорачиваться.
На уровне целого организма возрастные изменения в различных тканях и органах не являются равнозначными в смысле их влияния на продолжительность жизни. Главную роль играют возрастные изменения функции регуляторных систем (в особенности гипоталамуса), сердечно-сосудистой системы и других жизненно важных органов. Наиболее частыми причинами, укорачивающими срок жизни, являются развитие злокачественных опухолей и заболевания сердца и сосудов.
На продолжительность жизни оказывают влияние генетические факторы, степень сохранения интеллектуальной деятельности, выраженность инстинкта самосохранения, способность организма перестраиваться в зависимости от условий жизни, склонность к развитию атеросклероза в жизненно важных органах.
Наблюдения, проведенные на животных (главным образом, на грызунах), свидетельствуют о том, что положительное влияние на продолжительность жизни оказывает качественно полноценное, но количественно ограниченное питание, в особенности в первую половину жизни.
На продолжительность жизни, по-видимому, оказывает влияние функция эпифиза. Удаление его у крыс приводит к уменьшению продолжительности жизни, а введение животным пептидного экстракта эпифиза сопровождается противоположным эффектом.
Существует особый раздел медицины - гериатрия, которая ставит своей целью оказание медицинской помощи пожилым и старым людям, основываясь на знании особенностей физиологических и патологических процессов в позднем периоде жизни.