Патофизиология Новицкого, Е.Д. Гольдберга Тома 1 и 2 - 2009 г.
|
|
|
|
ГЛАВА 15 ПАТОФИЗИОЛОГИЯ СЕРДЕЧНО- СОСУДИСТОЙ СИСТЕМЫ
15.1. ОСНОВНЫЕ ФАКТОРЫ, ПРИВОДЯЩИЕ К НАРУШЕНИЮ ФУНКЦИИ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
В норме сердечно-сосудистая система функционирует как единое целое. Среди большого многообразия причин, вызывающих нарушение нормального функционирования сердечно-сосудистой системы можно выделить следующие группы факторов:
1) этиологические факторы, влияющие преимущественно на сосудистую стенку:
а) изменяющие структуру сосудистой стенки;
б) вызывающие нарушение тонуса сосудов;
2) факторы, индуцирующие преимущественно патологию сердца:
а) причины, приводящие к воспалительным и дистрофическим процессам;
б) наследственные факторы и нарушения эмбрионального развития системы кровообращения.
Итогом патогенного влияния этиологических факторов является развитие определенной патологии сердца или сосудов (стенокардии, аритмий, атеросклероза и т.д.). Прогрессирование имеющейся патологии сердечно-сосудистой системы может приводить к недостаточности кровообращения. Недостаточность кровообращения - это неспособность кровеносной системы обеспечивать адекватную перфузию органов и тканей.
Однако при некоторых патологических состояниях, особенно острых, следует дифференцировать следующие виды недостаточности кровообращения: сердечную, сосудистую и сердечно-сосудистую (смешанную). При этом подходы! к лечению сердечной или сосуди-
стой недостаточности могут не только отличаться, но и быть взаимоисключающими. Различают компенсированную и декомпенсированную стадии недостаточности кровообращения. Первая из них характеризуется тем, что количество крови, доставляемой тканям, оказывается достаточным в покое и недостаточным для выполнения какой-либо нагрузки. Декомпенсированная недостаточность кровообращения клинически проявляется даже в покое. Кроме того, выделяют острую и хроническую недостаточность кровообращения. Сосудистая недостаточность описана в разделе, посвященном патологии сосудов (см. раздел 15.2.3).
Изменения структуры сосудистой стенки. В той или иной степени выраженности эти изменения выявляются при всех заболеваниях сердечно-сосудистой системы. Они включают поражение сосудов воспалительной этиологии (эндартерииты, флебиты и другие васкулиты), патологические изменения, связанные с хроническими метаболическими нарушениями (амилоидоз, гиалиноз, кальциноз), склеротические поражения (атеросклероз) и др. Все эти нарушения усугубляют течение основного заболевания (например, васкулит при ревмокардите) или сами являются причиной поражения сердца (атеросклероз, вызывающий ишемическую болезнь сердца). Среди всех поражений сосудистого русла наиболее часто встречается атеросклероз.
Тонус кровеносных сосудов. Чрезвычайно важное значение для приспособления организма к меняющимся условиям внутренней и внешней среды играет тонус кровеносных сосудов. Регуляция сосудистого тонуса осуществляется нервными, гуморальными, а также местными механизмами и является предметом изучения нормальной физиологии. Однако целесообразно осветить некоторые аспекты местной ауторегуляции тонуса сосудов в связи с открытием ряда нейрогуморальных факторов, которые синтезируются сосудистой стенкой in situ (на месте). К таким биологически активным веществам относится мощный вазоконстриктор - эндотелин-I (пептид из 21 аминокислоты). Он секретируется эндотелием артерий из высокомолекулярного биологически неактивного предшественника биг-эндотелина-I (38 аминокислотных остатков) при участии специфического фермента эндотелинконвертазы. Синтез эндотелина может ингибироваться эндотелийрелаксирующим фактором (NO), простациклином, простагландином Е и предсердным натрийуретическим фактором (ПНУФ). Другие соединения (ангиотензин-II, катехоламины, тромбин), на-
оборот, усиливают биосинтез этого пептида. Молекулярный механизм действия эндотелина-I на гладкомышечные клетки артерий практически полностью идентичен действию ангиотензина-II.
Важную роль в ауторегуляции сосудистого тонуса, особенно при его повышении, играют простаноиды (производные арахидоновой кислоты) и прежде всего - тромбоксан А (ТхА2). Он секретируется тромбоцитами при контакте их с сосудистой стенкой и оказывает выраженное вазоконстрикторное действие. Выброс этого простаноида из тромбоцитов происходит при их соприкосновении с поврежденной артериальной стенкой. В этом случае функциональное предназначение ТхА2 сводится к возникновению сосудистого спазма, усилению тромбообразования и остановке кровотечения. Относительное увеличение его уровня по отношению к другому простаноиду - простациклину, являющемуся функциональным антагонистом ТхА2, приводит к формированию стойкого повышения артериального давления. Простациклин по своей способности расширять артерии превосходит все известные эндогенные вазодилататоры. Синтез простациклина происходит в клетках эндотелия и гладких мышцах сосудов. В основе вазодилататорного эффекта простациклина лежит его способность рецептор-опосредованным путем активировать фермент NO-синтазу в клетках эндотелия. Энзим NO-синтаза катализирует образование из L-аргинина свободного радикала окиси азота (NO'), названного эндотелийрелаксирующим фактором. В 1998 г. за открытие этого фактора американские ученые Роберт Фурчготт, Луис Игнарро и Ферид Мурад были удостоены Нобелевской премии. Эндотелийрелаксирующий фактор легко диффундирует через мембраны клеток эндотелия и достигает гладкомышечных клеток артерий, где активирует цитоплазматический фермент гуанилатциклазу. В результате уменьшается транспорт ионов кальция из внеклеточной среды в цитоплазму, а следовательно, уменьшается способность гладких мышц сосудов к тоническому сокращению. Вслед за снижением тонуса артерий происходит снижение системного артериального давления. Подобный гипотензивный эффект простациклина можно имитировать введением в кровоток донаторов NO' (нитроглицерин и нитропруссид натрия), которые широко применяются в клинической практике.
Важнейшим показателем сосудистого тонуса является систолическое артериальное давление, уровень которого зависит от величины ударного объема крови левого желудочка сердца, макси-
мальной скорости ее изгнания и растяжимости аорты. В норме систолическое артериальное давление составляет 100-140 мм рт.ст. Диастолическое артериальное давление определяется в первую очередь тонусом артерий мышечного типа, объемом циркулирующей крови и в меньшей степени фракцией выброса левого желудочка. У здоровых людей диастолическое артериальное давление колеблется в пределах 60-90 мм рт.ст. Нормальные суточные колебания систолического артериального давления не превышают 33 мм рт.ст., а диастолического - 10 мм рт.ст., тогда как при нарушениях сосудистого тонуса эти амплитудные характеристики могут существенно изменяться. Разность между систолическим и диастолическим артериальным давлением называется пульсовым давлением.
Среди поражений сердечно-сосудистой системы воспалительной и дистрофической природы следует особо выделить некоронарогенную патологию сердечной мышцы неревматической этиологии (миокардиодистрофии, миокардиты, кардиомиопатии, инфекционный эндокардит, перикардиты) и заболевания ревматической природы.
Роль наследственности и врожденных пороков развития в патогенезе сердечно-сосудистых заболеваний в наше время заметно возросла в связи с резким увеличением числа мутагенных факторов в быту и на производстве. Можно с сожалением констатировать, что со временем ситуация будет еще более усугубляться. Во-первых, неизбежно будут усиливаться техногенные воздействия на организм. Во-вторых, мутантные гены будут сохраняться в популяции в связи с успехами современной медицины, позволяющей спасти жизнь пациентам с генетической патологией. Среди причин врожденных пороков сердца можно выделить следующие: 1) хромосомные нарушения (аберрации); 2) мутация одного гена (частота - 2-3%); 3) факторы, вызывающие нарушения эмбрионального развития (алкоголизм родителей, краснуха, лекарственные препараты и др.) - 1-2%; 4) полигенно-мультифакториальное наследование - 90% случаев.
Существует более 38 различных врожденных пороков сердца, например дефект межжелудочковой перегородки, дефект межпредсердной перегородки, открытый атриовентрикулярный канал, открытый артериальный проток, тетрада Фалло, коарктация аорты, аномальный дренаж легочных вен, трехпредсердное сердце, синдром гипоплазии левого желудочка и др.
15.2. СОСУДИСТЫЕ НАРУШЕНИЯ
Можно выделить две основные группы сосудистых нарушений. Одна группа нарушений реализуется в изменении нормальной структуры сосудистой стенки различного генеза. Это могут быть дефекты развития сосудов, воспалительные, склеротические процессы в стенке сосудов. Вторая большая группа нарушений связана с изменением тонуса сосудов.
Классификация нарушений тонуса сосудов. В настоящее время различают два состояния, касающиеся изменения сосудистого тонуса:
1. Повышение тонуса сосудов - гипертензия, или гипертония.
2. Снижение тонуса сосудов - гипотензия (гипотония, или сосудистая недостаточность).
15.2.1. Артериальные гипертензии
Артериальная гипертензия - повышение внутрисосудистого давления в артериях большого круга кровообращения. Возникает в результате усиления работы сердца, увеличения периферического сопротивления или сочетания этих факторов. По данным комитета экспертов ВОЗ, артериальная гипертензия встречается у 15-25% взрослого населения, частота ее увеличивается с возрастом и регистрируется более чем у 50% людей старше 65 лет.
Артериальная гипертензия длительное время протекает без явных клинических симптомов. Однако достаточно скоро она может привести к возникновению острых нарушений мозгового кровообращения (транзиторная ишемическая атака, ишемический или геморрагический инсульт) и развитию гипертрофии миокарда. Кроме того, артериальная гипертензия является фактором риска атеросклероза и возникновения инфаркта миокарда.
По своему происхождению артериальная гипертензия бывает первичной и вторичной.
Первичная артериальная гипертензия (гипертоническая болезнь) - это стойкое повышение артериального давления, не связанное с органическим поражением органов и систем, регулирующих сосудистый тонус. Распространенным названием первичной артериальной гипертензии является термин «эссенциальная гипертония», что означает неясность ее этиологии. На долю гипертонической болезни приходится 90-95% общего числа артериальных гипертензий.
Вторичная артериальная гипертензия - это повышение артериального давления, представляющее собой лишь симптом другого диагностически подтвержденного заболевания (гломерулонефрит, стеноз почечных артерий, опухоль гипофиза или надпочечников и т.д.). В связи с этим вторичная гипертензия называется еще симптоматической. На долю подобного рода нарушений сосудистого тонуса приходится в среднем 5-10%. Различают эндокринную, нефрогенную, гемодинамическую, нейрогенную и лекарственную симптоматические гипертонии.
Симптоматические артериальные гипертензии
Нефрогенные артериальные гипертензии. Возникают при врожденных или приобретенных заболеваниях почек (аномалии развития, гломерулонефрит, пиелонефрит и др.), сопровождающихся расстройствами регионарного кровообращения (вазоренальная или реноваскулярная артериальная гипертензия) и поражением почечной паренхимы (ренопаренхиматозная или ренопривная артериальная гипертензия). Нарушения внутрипочечного кровотока вызывают ишемию почек, которая выступает в роли «пускового механизма», активирующего секрецию ренина в юкстагломерулярном аппарате (ЮГА). Следует отметить, что нарушение функции этой системы в большей степени характерно для вазоренальной (симптоматической) артериальной гипертензии, хотя и при эссенциальной гипертонии ренин-ангиотензин-альдостероновая система (РААС) активно вовлекается в патогенетическую цепь стойкого повышения артериального тонуса.
Ренин поступает в кровь и вызывает энзиматическое расщепление плазменного белка ангиотензиногена, относящегося к а2-глобулинам. В результате этого образуется декапептид ангиотензин-I, который под влиянием ангиотензинпревращающего фермента (ангиотензинконвертаза) переходит в октапептид ангиотензин-II, представляющий собой один из самых сильных вазоконстрикторов. Надо особо отметить, что ангиотензин-II вызывает стойкое и длительное повышение артериального давления, что связано с его достаточно медленным ферментативным расщеплением.
В настоящее время молекулярный механизм действия ангиотензина-II на сосудистую стенку хорошо изучен. Установлено, что он взаимодействует со специфическими рецепторами, рас-
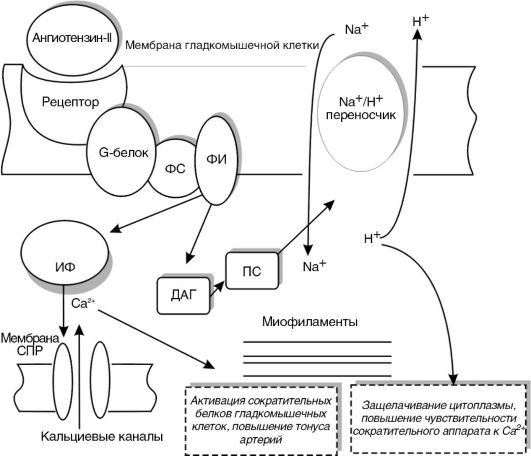 Рис. 15-1. Вазоконстрикторный
эффект ангиотензина-II. ФС - фосфолипаза С; ФИ - фосфоинозитол; ИФ -
инозитолтрифосфат; ДАГ - диацилглицерол; ПС - протеинкиназа С; СПР -
саркоплазматический ретикулум
Рис. 15-1. Вазоконстрикторный
эффект ангиотензина-II. ФС - фосфолипаза С; ФИ - фосфоинозитол; ИФ -
инозитолтрифосфат; ДАГ - диацилглицерол; ПС - протеинкиназа С; СПР -
саркоплазматический ретикулум
положенными на сарколемме гладкомышечных клеток. Активация этих рецепторов вызывает цепочку событий, очень сходную с теми, которые наблюдаются в условиях активации а-адренорецепторов (рис. 15-1).
Однако ангиотензин-II не только повышает тонус артерий, но и оказывает митогенное действие, вызывая усиленную пролиферацию гладкомышечных клеток и утолщение сосудистой стенки. По этой причине ангиотензин-II называют еще ростовым фактором. Указанный эффект опосредуется через активацию протеинкиназы С, тирозинкиназы и вызываемое ими фосфорилирование регуляторных белков. Функциональные и морфологические изменения артерий, индуцированные ангиотензином-II и другими эндогенными биологически активными веществами, получили название ремоделирования сосудистой стенки.
В последние годы рецепторы ангиотензина-II были обнаружены в надпочечниках. Стимуляция этих рецепторов вызывает усиление секреции альдостерона, который индуцирует задержку Na+ и воды в организме. Такие изменения водного и солевого обмена ведут к увеличению объема циркулирующей крови и повышению артериального давления.
Таким образом, ангиотензиновое звено патогенеза артериальной гипертензии включает в себя три основных компонента: 1) повышение тонуса артерий; 2) ремоделирование сосудистой стенки; 3) усиление секреции альдостерона.
Следует отметить, что одностороннее нарушение почечного кровообращения, как правило, приводит лишь к преходящей гипертензии. Однако если при этом у экспериментального животного удалить вторую («нормальную») почку, развивается стойкое повышение артериального давления. Дело в том, что паренхиматозная ткань почек секретирует не только ренин, но и ряд веществ, которые препятствуют повышению артериального давления, в частности простагландины, способные оказывать прямое сосудорасширяющее действие, ангиотензиназу, расщепляющую ангиотензин-II, и др. В результате первоначальное повышение артериального давления, вызванное гипоксией одной почки и выбросом ренина, сменяется его нормализацией. Именно этим можно объяснить тот факт, что удаление обеих почек в эксперименте вызывает возникновение стойкой гипертензии, получившей название ренопривной.
Артериальные гипертензии эндокринного происхождения выявляются главным образом при следующих заболеваниях: феохромоцитоме, первичном альдостеронизме (синдром Конна), болезни и синдроме Иценко-Кушинга, тиреотоксикозе.
Феохромоцитома. Так называется опухоль мозгового вещества надпочечников, продуцирующая значительные количества катехоламинов (адреналин, норадреналин, дофамин), концентрация которых у больных с феохромоцитомой в крови и моче увеличивается в 10-100 раз. Избыточной секрецией катехоламинов определяется гипертензионный синдром при данном заболевании. При феохромоцитоме выделяют три варианта артериальной гипертензии: стабильную, пароксизмальную (кризовую) и смешанный тип с пароксизмами на фоне стабильного повышения артериального давления.
Первичный альдостеронизм (синдром Конна). Морфологическим субстратом данного заболевания чаще всего являются солитарные или (реже) множественные аденомы и идиопатическая гиперплазия клубочковой зоны коры надпочечников, секретирующие альдостерон. В настоящее время идентифицированы уже три гена, регулирующих секрецию альдостерона. Мутация любого из них ведет к гиперальдостеронизму (так называемый семейный, или первичный, гиперальдостеронизм) и артериальной гипертензии. Основные симптомы болезни определяются гиперпродукцией альдостерона - минералокортикоиды способствуют реабсорбции в почках гидратированных ионов натрия, что ведет к задержке в организме воды и увеличению объема циркулирующей крови, а следовательно, к подъему артериального давления и формированию артериальной гипертензии.
Болезнь и синдром Иценко-Кушинга. Приводят к повышению в крови уровня глюкокортикоидов, которые при данной патологии играют решающую роль в формировании артериальной гипертензии. Первичное или вторичное (под влиянием АКТГ) повышение секреции глюкокортикоидов вызывает увеличение плотности адренорецепторов, локализованных в сердце и сосудах, а также повышение их чувствительности к катехоламинам; стимуляцию продукции ангиотензиногена в печени. Вслед за повышением адренореактивности сердца и сосудов отмечается увеличение тонуса сосудов и сердечного выброса. Результатом этих гемодинамических эффектов является повышение артериального давления.
Гипертиреоз. Возникает при гиперфункции щитовидной железы, в результате чего повышается уровень тироксина и трийодтиронина в крови. При этом подъем артериального давления происходит вследствие сочетанного возрастания периферического сопротивления артерий и увеличения минутного объема сердца. Последний эффект обусловлен тироксинзависимой тахикардией.
Гемодинамические гипертензии. Возникают в результате изменений в сердце или крупных сосудах. Чаще развивается систолическая гипертензия с увеличением пульсового давления. В одних случаях (коарктация аорты, неспецифический аортоартериит) гипертензия является региональной, в других (при вовлечении в патологический процесс барорецепторных зон эндотелия сосудов) - носит системный характер.
Коарктация аорты - врожденное сегментарное сужение нисходящей части грудной аорты, создающее два режима кровообра-
щения в большом круге: гипертензию верхней половины туловища и гипотензию нижней. У мужчин встречается в 4 раза чаще, чем у женщин. Неспецифический аортоартериит представляет собой системное сосудистое заболевание аутоиммунного генеза, приводящее к повышению ригидности аорты и магистральных артерий, а также к их стенозированию. Одним из клинических проявлений этого заболевания служит артериальная гипертензия, вызванная повышением периферического сосудистого сопротивления. Это патогенетическое звено играет важную роль также при потере эластичности сосудистой стенки у пациентов с распространенным атеросклерозом аорты и магистральных артерий.
Нейрогенные гипертензии. Развиваются при опухолях, ушибах и сотрясениях головного мозга, менингитах, менингоэнцефалитах, ишемии головного мозга, вызванной атеросклерозом ветвей брахиоцефальных артерий или сдавлением позвоночных артерий, которые связаны с остеохондрозом шейно-грудного отдела позвоночника. Патогенез подобных гипертоний обусловлен изменением тонуса высших вегетативных центров, регулирующих уровень артериального давления.
Лекарственные гипертензии. Многие лекарственные препараты, воздействуя на разные звенья регуляции артериального давления, могут вызывать его повышение. Следует прежде всего обратить внимание на глюкокортикоидные гормоны, широко применяемые в терапии различных системных заболеваний.
Гипертоническая болезнь
Этиология гипертонической болезни. Первостепенное значение в возникновении гипертензии имеет длительное психоэмоциональное перенапряжение. Об этом свидетельствуют частые случаи развития первичной гипертензии у лиц, переживших ленинградскую блокаду, а также у людей «стрессовых» профессий. Особую роль играют отрицательные эмоции. В отличие от представителей животного мира современный цивилизованный человек часто не имеет возможности «погасить» свое эмоциональное возбуждение двигательной активностью. Это способствует длительному сохранению в коре головного мозга очага застойного возбуждения и развитию артериальной гипертензии. На этом основании Г.Ф. Ланг и А.Л. Мясников назвали гипертоническую болезнь болезнью неотреагированных эмоций.
Гипертоническая болезнь - это «болезнь осени жизни человека, которая лишает его возможности дожить до зимы». Так писал академик А.А. Богомолец, подчеркивая тем самым предрасполагающую роль возраста в ее происхождении. Однако нередко первичная гипертензия развивается и в молодом возрасте (ювенильная форма гипертензии). Важно при этом отметить, что до 40 лет мужчины болеют чаще, чем женщины, а после 40 лет соотношение приобретает противоположный характер.
Важная роль в этиологии первичной гипертензии отводится наследственности. На роль генетических факторов указывают и факты высокой конкордантности по гипертонической болезни у однояйцевых близнецов, а также существование линий крыс, предрасположенных к развитию гипертензии (SHR - spontaneously hypertensive rats). Из 9 уже известных кандидатных генов артериальной гипертензии в последнее время особое внимание привлекает ген эндотелиальной NO-синтазы.
Успешное экспериментальное моделирование «солевой гипертензии» говорит в пользу этиологической роли избыточного потребления соли. Однако правильнее, видимо, считать этот фактор не столько главной причиной, сколько фактором риска. Считают даже, что длительное потребление более 5 г соли в день способствует развитию гипертензии только у лиц, имеющих наследственное предрасположение к ней.
Патогенез гипертонической болезни. Несмотря на то что эссенциальная и вторичная артериальные гипертензии существенно различаются по этиологическим факторам, механизмы их развития имеют много общего. Согласно концепции, разработанной Г.Ф. Лангом и А.Л. Мясниковым, «нервное перенапряжение при гипертонической болезни реализуется в расстройстве трофики определенных мозговых структур, управляющих артериальным давлением», прежде всего тех областей коры больших полушарий и подкорковых центров (гиппокамп, миндалевидное тело), экспериментальное раздражение которых вызывает повышение артериального давления. Так, было установлено, что ишемия головного мозга, вызванная у кролика перевязкой сонных артерий, способствует возникновению артериальной гипертензии. У высокоорганизованных животных (собаки, обезьяны) удалось получить рефлексогенную гипертензию путем «сшибки» пищевого и оборонительного рефлексов. В этом случае гипертензия явилась следствием невроза. Непосредственный механизм повышения артериального давления
связан с возникновением очага застойного возбуждения (патологической доминанты, по терминологии А.А. Ухтомского) вегетативных центров головного мозга, в первую очередь сосудодвигательного центра. Вазомоторные импульсы, возникающие в ядрах гипоталамуса, поступают в ядра продолговатого мозга, откуда по симпатическим путям передаются на сосуды резистивного типа, вызывая повышение их тонуса.
Важную роль в патогенезе гипертонической болезни играет «растормаживание» сосудодвигательного центра, расположенного в продолговатом мозге. В норме его активность рефлекторно подавляется импульсами, поступающими от синокаротидной зоны и рецепторов дуги аорты. При гипертонической болезни активность этого центра очень часто повышена.
Другим слагаемым гипертензивного эффекта, наблюдаемого при возбуждении высших вегетативных центров головного мозга, является выброс катехоламинов (адреналина и норадреналина) из мозгового вещества надпочечников в кровоток. Развитие гипертензии при этом наблюдается тогда, когда а-адренергические эффекты этих гормонов суммарно превышают присущее им влияние на β-адренергические рецепторы (рис. 15-2, 15-3).
Рассмотренные процессы формируются на первой стадии гипертонической болезни, которая называется транзиторной (преходящей) и клинически характеризуется непродолжительными эпизодами повышения артериального давления.
В дальнейшем снижается лабильность и появляется инертность нервных структур, составляющих сосудодвигательный центр, и развивается вторая стадия болезни - стабильная. В этой стадии поддержание высокого уровня артериального давления является длительным. Стимулирующим влиянием на сосудодвигательный центр во второй стадии гипертонической болезни обладают не только специфические раздражители, но и «посторонние», исходящие из соседних структур импульсы, даже подпорогового уровня. В происхождении стабильного повышения тонуса сосудов имеют значение формирующиеся в этой стадии «порочные круги»: почечный (с участием ЮГА), барорецепторный, гипофизарнонадпочечниковый и сосудистый (повышение чувствительности стенки сосудов к катехоламинам). В результате повышения артериального давления развивается парабиоз барорецепторов сосудов и выпадает их тормозной контроль над нейронами сосудодвигательного центра (рис. 15-4). В итоге тонус сосудов повышается
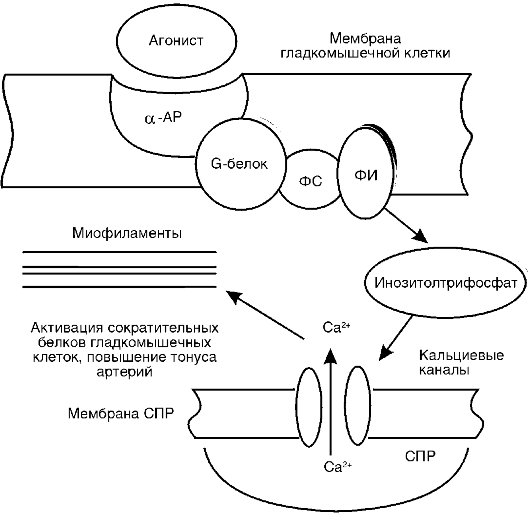 Рис. 15-2. Роль
альфа-адренорецепторов в регуляции тонуса артерий: α-AP -
α-адренорецептор; ФС - фосфолипаза С; ФИ - фосфоинозитол; СПР -
саркоплазматический ретикулум
Рис. 15-2. Роль
альфа-адренорецепторов в регуляции тонуса артерий: α-AP -
α-адренорецептор; ФС - фосфолипаза С; ФИ - фосфоинозитол; СПР -
саркоплазматический ретикулум
еще сильнее. Спазм сосудов приводит к гипоксии юкстагломерулярного аппарата почек и активации РААС. В свою очередь, ишемическая стимуляция аденогипофиза реализуется в секреции АКТГ и, следовательно, повышении содержания в крови гормонов коры надпочеников (минерало- и кортикостероидов). Поэтому высокий тонус сосудов поддерживается продолжительное время. В механизме гипертонии также играет роль повышение чувствительности стенки сосудов к катехоламинам, и даже небольшие дозы адреналина оказывают выраженный вазоконстрикторный эффект.
Таким образом, определяющую роль в патогенезе артериальной гипертензии играют изменения нейрогуморальной регуляции сосудистого тонуса. Конечным звеном этого патологического процесса является изменение функциональной активности ионотранспортирующих систем плазматической мембраны, что ведет к перегруз-
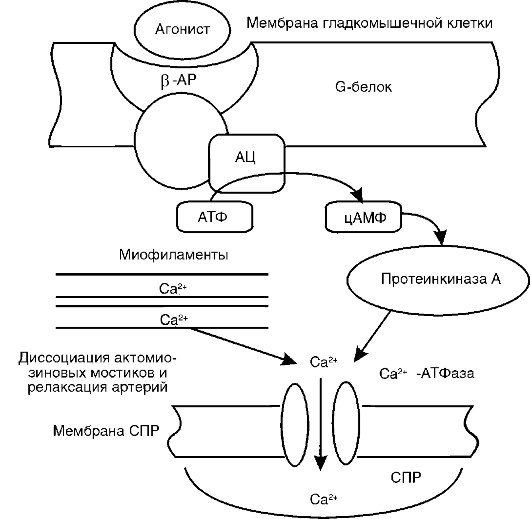 Рис. 15-3. Роль
бета-адренорецепторов в регуляции тонуса артерий: β-АР -
β-адренорецептор; СПР - саркоплазматический ретикулум; АЦ -
аденилатциклаза
Рис. 15-3. Роль
бета-адренорецепторов в регуляции тонуса артерий: β-АР -
β-адренорецептор; СПР - саркоплазматический ретикулум; АЦ -
аденилатциклаза
ке клеток ионами кальция и патологическому повышению тонуса кровеносных сосудов. Такая концепция патогенеза артериальной гипертензии была выдвинута Ю.В. Постновым и С.Н. Орловым, которые назвали ее мембранной концепцией.
Третья стадия гипертонической болезни - стадия органных изменений. В начале этой стадии можно обнаружить гипертрофию левого желудочка. В дальнейшем развивается кардиосклероз и присоединяется сердечная недостаточность. Со стороны внутренних органов отмечаются ишемические повреждения, которые индуцированы морфологическими изменениями стенки сосудов (гиалиноз, склероз, атеросклероз). Наиболее характерным является повреждение паренхимы почек с развитием хронической почечной недостаточности, задержкой жидкости в организме и прогрессированием артериальной гипертензии.
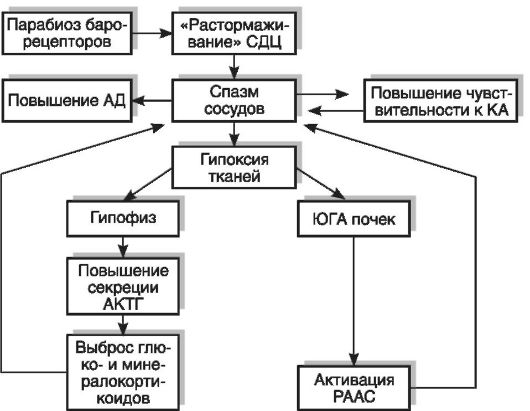 Рис. 15-4. «Порочные
круги» в патогенезе гипертонической болезни: АД - артериальное
давление; АКТГ - адренокортикотропный гормон; КА - катехоламины; СДЦ -
сосудодвигательный центр; РААС - ренинангиотензин-альдостероновая
система; ЮГА - юкстагломерулярный аппарат
Рис. 15-4. «Порочные
круги» в патогенезе гипертонической болезни: АД - артериальное
давление; АКТГ - адренокортикотропный гормон; КА - катехоламины; СДЦ -
сосудодвигательный центр; РААС - ренинангиотензин-альдостероновая
система; ЮГА - юкстагломерулярный аппарат
Артериальная гипертензия независимо от причин, ее обусловивших, протекает длительно и имеет тенденцию к неуклонному прогрессированию. Однако относительно «спокойное» развитие заболевания может осложняться возникновением так называемых гипертензивных (гипертонических) кризов.
Гипертензивный криз представляет собой внезапное повышение артериального давления, сопровождаемое появлением или усугублением уже имеющейся церебральной и/или кардиальной симптоматики. Систолическое артериальное давление при этом очень быстро может повыситься до 190-270 мм рт.ст., а диастолическое - до 120-160 мм рт.ст. Прямой зависимости между уровнем артериального давления и выраженностью симптоматики криза не наблюдается. Тяжесть криза определяется не столько величиной подъема артериального давления, сколько степенью развившейся дисфункции жизненно важных органов - головного мозга и сердца. Гипертензивный криз относится к разряду ургентных (неотложных) ситуаций, поскольку механизмы адаптации сердечнососудистой системы могут при этом оказаться несостоятельными
по причине быстрого развития гипертензии. Криз, как правило, проявляется сильнейшими головными болями, тошнотой, рвотой и может закончиться гибелью пациента от инфаркта миокарда или мозгового инсульта, поэтому требует экстренного медикаментозного вмешательства, направленного на нормализацию артериального давления.
15.2.2. Легочная гипертензия
Легочная гипертензия, или артериальная гипертензия малого круга кровообращения, - это патологическое состояние, характеризующееся повышением систолического, диастолического и среднего давления в легочной артерии. В малом круге кровообращения, как и в большом, артериальное давление может повышаться при усилении работы сердца (увеличение минутного объема правого желудочка) и/или росте сопротивления легочных сосудов. Однако артерии малого круга кровообращения обладают большой растяжимостью, и для заметного повышения артериального давления необходимо увеличение минутного объема кровообращения как минимум в 3 раза или уменьшение емкости сосудов малого круга кровообращения примерно на 2/3.
Различают первичную легочную гипертензию, которая развивается при заболеваниях кардиоваскулярной системы (артериит, митральный стеноз), вызывающих рост сосудистого сопротивления в малом круге, и вторичную легочную гипертензию, которая связана с уменьшением емкости сосудистого русла малого круга кровообращения и наблюдается при тромбоэмболии легочной артерии, заболеваниях легких, резекции легочной ткани, пневмотораксе и т.п.
Примером первичной легочной гипертензии может служить повышение артериального давления в сосудах малого круга при митральном стенозе и дефектах перегородок сердца. Основным звеном патогенеза легочной гипертензии при митральном стенозе является нарушение тока крови из левого предсердия в левый желудочек, что приводит к застою крови в системе легочных вен с последующим повышением артериального давления в артериях малого круга кровообращения согласно рефлексу Китаева (см. ниже). Артериальная легочная гипертензия при дефекте межпредсердной или межжелудочковой перегородки связана с шунтированием крови слева направо и гиперволемией малого круга кровообращения.
В случае вторичной легочной гипертензии, которая наиболее часто развивается при бронхолегочных заболеваниях, повышение давления в артериях малого круга обусловлено уменьшением объема кровяного русла у пациентов с пневмосклерозом и эмфиземой легких. Иногда артериальная легочная гипертензия возникает при острых легочных заболеваниях, например при тяжелом, затянувшемся приступе бронхиальной астмы.
При хронических заболеваниях легких, митральном стенозе и некоторых других заболеваниях давление в легочной артерии нередко оказывается повышенным в 4-6 и даже в 9 раз по сравнению с нормой (рис. 15-5).
Выраженность гипертензии малого круга зависит от действия двух факторов, первым из которых является механическое препятствие кровотоку, например стеноз митрального клапана, сдавление или окклюзия артериол, а вторым - рефлекторное сужение резистивных сосудов малого круга. Устранение митрального порока с помощью методов реконструктивной кардиохирургии во многих случаях сравнительно быстро приводит к обратному развитию легочной гипертензии. При этом у ряда больных давление в truncus pulmonalis снижается в течение нескольких месяцев после операции с 120-125 до 20-25 мм рт.ст.
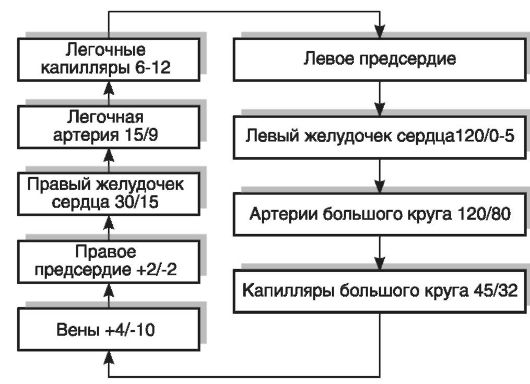 Рис. 15-5. Уровень давления крови в отделах большого и малого круга кровообращения (мм рт.ст.; систолическое/диастолическое)
Рис. 15-5. Уровень давления крови в отделах большого и малого круга кровообращения (мм рт.ст.; систолическое/диастолическое)
Легочная гипертензия приводит к перегрузке повышенным давлением правых отделов сердца с развитием «легочного сердца» (см. раздел 15.3.2.).
15.2.3. Сосудистая недостаточность
Классификация гипотоний. Выделяют две группы гипотоний: острую сосудистую недостаточность и хроническую недостаточность тонуса сосудов.
Острая сосудистая недостаточность - это патологическое состояние, основным звеном которого является уменьшение объема циркулирующей крови, приводящее к снижению артериального и венозного давления. Проявлениями этого процесса служат шок (см. раздел 4.3), коллапс и обморок.
Коллапс - вид острой сосудистой недостаточности, характеризующийся резким понижением артериального и венозного давления и снижением массы циркулирующей крови в сосудистой системе. По этиологии различают следующие виды коллапса:
1. Токсико-инфекционный. Развивается при кишечных инфекциях (дизентерия, сальмонеллез и др.), возбудители которых выделяют эндотоксин, высвобождающийся при гибели микробных тел. При интенсивной антибактериальной терапии может возникнуть одномоментная гибель огромного количества микробов. Высвобождающиеся при этом большие дозы эндотоксина вызывают поражение нервно-мышечного аппарата стенки сосудов с последующей их атонией, что и приводит в конечном итоге к состоянию коллапса.
2. Постгеморрагический. Возникает при острой массивной кровопотере и связан с быстрым уменьшением объема циркулирующей крови и снижением тонуса сосудов.
3. Панкреатический. Может наступить при тяжелой травме живота, сопровождающейся размозжением ткани поджелудочной железы и поступлением в кровь панкреатического сока. Установлено, что трипсин, действуя на гладкую мускулатуру сосудистой стенки, вызывает резкое снижение ее тонуса, что приводит к развитию коллапса.
4. Аноксический. Возникает при быстром снижении парциального давления кислорода во вдыхаемом воздухе. Развивающаяся в этом случае гипоксия вызывает снижение тонуса гладкой мускулатуры сосудистой стенки, что приводит, в свою очередь, к резкому расширению сосудов.
5. Ортостатический. Возникает у больных после длительного (многодневного) постельного режима при резком переходе из горизонтального положения в вертикальное.
Патогенез различных видов коллапса во многом идентичен (рис. 15-6). В развитии этого вида острой сосудистой недостаточности важную роль играет снижение тонуса емкостных сосудов (преимущественно мелких вен), в которых обычно сосредоточена основная масса (75-80%) крови. Такая ситуация может возникнуть вследствие прямого действия различных токсинов на гладкую мускулатуру сосудов или явиться результатом снижения тонической активности симпатоадреналовой системы в сочетании с ваготонией. Депонированная кровь не участвует в циркуляции, что приводит к снижению объема циркулирующей крови. Итогом этих
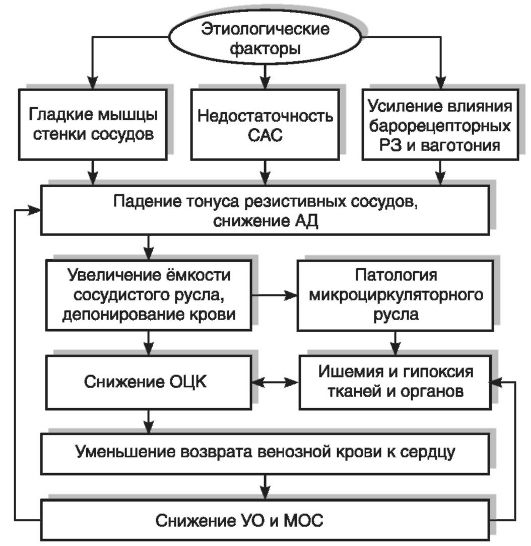 Рис. 15-6. Схема
патогенеза коллапса: САС - симпатоадреналовая система; РЗ -
рефлексогенные зоны; АД - артериальное давление; ОЦК - объем
циркулирующей крови, УО - ударный объем; МОС - минутный объем сердца
Рис. 15-6. Схема
патогенеза коллапса: САС - симпатоадреналовая система; РЗ -
рефлексогенные зоны; АД - артериальное давление; ОЦК - объем
циркулирующей крови, УО - ударный объем; МОС - минутный объем сердца
изменений является уменьшение венозного притока крови к сердцу и вторичное снижение сердечного выброса, в результате чего патологический процесс усугубляется. В дальнейшем при прогрессировании сосудистой недостаточности развивается гипоксия тканей, нарушаются окислительно-восстановительные процессы и возникает ацидоз. Сознание может быть сохранено.
Обморок - острая сосудистая недостаточность, проявляющаяся кратковременной потерей сознания. Возникает даже у абсолютно здоровых людей при эмоциональном возбуждении (страх, боль и пр.), когда имеет место остро возникающее повышение тонуса блуждающих нервов, а также при недостаточности барорецептивного рефлекса, обеспечивающего адаптивные реакции системы кровообращения.
Выделяют следующие виды обмороков:
1. Вазовагальный - развивается при отсутствии свежего воздуха (в душном помещении), эмоциональном возбуждении в результате резкого снижения тонуса сосудов и падения артериального давления. Проявления характеризуются потерей сознания из-за снижения мозгового кровотока.
2. Ортостатический - возникает при резкой перемене положения тела из горизонтального в вертикальное или при продолжительном неподвижном пребывании в вертикальном положении (на посту).
3. Синокаротидньгй - вызывается поворотом головы, застегиванием пуговицы тугого воротничка или при пальпации шеи (особенно у пожилых людей). Патогенез объясняется гиперчувствительностью каротидного синуса, стенозом сонной и позвоночной артерий.
4. Перераспределительный - развивается при внезапном расширении вен брюшной полости в результате резкого падения внутрибрюшного давления (например, после быстрой эвакуации асцитической жидкости). При данном виде обморока скорость перераспределения большой массы крови в венозное русло брюшной полости превышает скорость развития адаптационной тонической реакции вен.
5. Кашлевой - индуцируется приступом кашля, что сопровождается повышением внутригрудного давления и снижением возврата венозной крови к сердцу.
6. Кардиогенный - связан с нарушениями сердечного ритма и эффективности сердечного выброса на фоне пороков и др. патологии сердца.
7. Метаболический - вызывается гипервентиляцией. В патогенезе данного вида обморока играют роль обусловленное гипокапнией сужение просвета сосудов мозга и ишемия ЦНС.
Хроническая сосудистая недостаточность классифицируется следующим образом:
1) физиологическая гипотония, которая рассматривается как вариант конституциональной нормы, может развиваться у спортсменов и в процессе акклиматизации к условиям высокогорья;
2) патологическая гипотония. К этой группе относятся: а) первичная гипотония, являющаяся самостоятельным заболеванием (нейроциркуляторная дистония гипотонического типа, гипотоническая болезнь и другие синонимы); б) вторичная, или симптоматическая, гипотония, которая развивается при заболеваниях пищеварительной системы, длительном голодании, анемиях, гипофункции коры надпочечников и др.
В этиологии первичной хронической гипотонии играют роль психоэмоциональное напряжение и черепно-мозговые травмы. Способствуют и предрасполагают к ее развитию нарушение режима дня, питания, конфликтные ситуации, перенесенные инфекционные заболевания, пол, возраст (чаще регистрируется у женщин 30-40 лет). Основное звено патогенеза нейроциркуляторной дистонии гипотонического типа - нарушение корковой нейродинамики с ослаблением процессов возбуждения и преобладанием тормозных механизмов в ЦНС. В итоге развивается дисфункция коры и высших подкорковых нервных центров, регулирующих сосудистый тонус со снижением тонуса резистивных сосудов и стойким падением общего периферического сопротивления.
15.2.4. Атеросклероз
Атеросклероз - это хроническое заболевание, основные проявления которого связаны с образованием в стенке артерий специфических для данной патологии атероматозных бляшек, вызывающих нарушение кровотока в органах и тканях. Термин «атеросклероз» (от греч. athere - кашица, sclerosis - уплотнение) был предложен в 1904 г. Ф. Маршаном, который выделил это заболевание в качестве самостоятельной нозологической единицы из собирательного понятия «артериосклероз», объединявшего практически все вилы структурных изменений сосудистой стенки (медиокальциноз, гиалиноз, атеросклероз и др.).
Коварность атеросклероза состоит в том, что он длительное время протекает бессимптомно и клинически не проявляется до тех пор, пока не нарушается кровообращение соответствующего органа. Как правило, симптомы ишемии появляются при стенозировании просвета сосуда более чем на 50% (так называемый гемодинамически значимый стеноз). Чаще всего атеросклеротическим изменениям подвергаются аорта, артерии сердца, головного мозга, нижних конечностей и почек (рис. 15-7). Поэтому среди причин смерти на первом месте стоят ишемическая болезнь сердца, инфаркт миокарда, разрыв аневризмы аорты, ишемический или геморрагический инсульт.
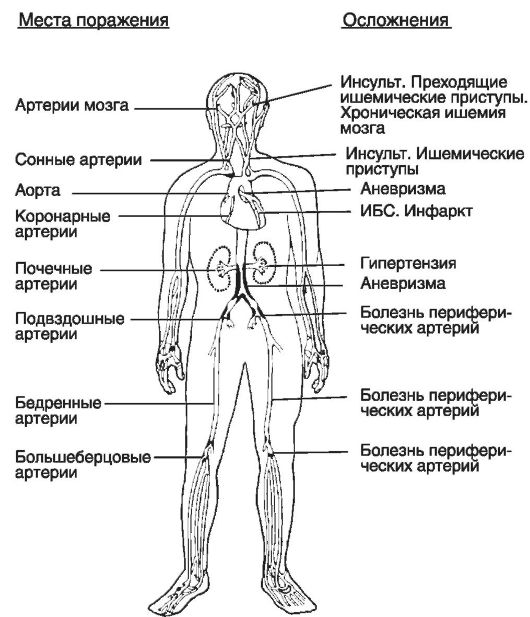 Рис. 15-7. Заболевания сердечно-сосудистой системы, вызванные атеросклерозом (по А.Н. Климову, 1981)
Рис. 15-7. Заболевания сердечно-сосудистой системы, вызванные атеросклерозом (по А.Н. Климову, 1981)
Этиология атеросклероза
В настоящее время господствует точка зрения, что атеросклероз - многофакторное заболевание. Факторы, которые способствуют развитию атеросклероза, принято называть факторами риска. Они могут проявиться, но могут и не оказывать своего действия. Чем больше факторов риска у человека, тем вероятнее развитие атеросклероза.
Возраст. Известно образное выражение немецкого ученого М. Burger о том, что «физиологический склероз стариков - судьба, а атеросклероз - болезнь». Эти два процесса имеют и достаточно четкие морфологические отличия. Однако важно отметить, что хотя атеросклероз не является результатом физиологического процесса старения организма, но имеются определенные соотношения между атеросклерозом и возрастом. Начальные признаки атеросклероза выявляются на секции уже в 20-летнем возрасте. Однако клинические проявления заболевания чаще всего обнаруживаются у людей старше 30-40 лет, когда уже имеется гемодинамически значимое стенозирование.
Пол. Фактором риска в этом смысле является принадлежность к мужскому полу. У мужчин риск развития атеросклероза в 3 раза выше, чем у женщин того же возраста при одинаковом уровне холестерина, а клинически выраженные формы атеросклероза возникают у женщин примерно на 10 лет позже, чем у мужчин. С этим в основном связана большая продолжительность жизни женского населения. Точный механизм большей защищенности женщин от атеросклероза пока не полностью ясен. Возможно, это связано с протекторной ролью эстрогенов. Например, развитие атеросклеротического процесса у женщин, перенесших в возрасте до 30 лет овариэктомию, как правило, ускоряется. Аналогичная тенденция обнаруживается у женщин в связи с наступлением менопаузы.
Наследственность. Атеросклероз нельзя отнести к «чисто наследственной» патологии. Вместе с тем генетические детерминанты оказывают свое влияние на результат воздействия факторов окружающей среды (образ жизни, характер питания, вредные привычки и др.). В настоящее время идентифицировано по меньшей мере 6 мутантных генов - кандидатов на роль эндогенных факторов, предопределяющих развитие атеросклероза. Подобные гены получили название кандидатных генов. Одним из них является ген, кодирующий рецептор липопротеинов низкой плотности
(ЛПНП). Американские ученые J. Goldstein и M. Brown разработали концепцию о том, что недостаток или дисфункция названных рецепторов приводят к развитию наследственной гиперхолестеринемии. Генетические нарушения в данном случае проявляются в гомо- и гетерозиготной форме. Наиболее опасны гомозиготные формы (один случай на миллион населения), при которых клеточные рецепторы к ЛПНП отсутствуют. У больных с такой патологией концентрация ЛПНП в крови может в 8-10 раз превышать их нормальный уровень. Механизм этого явления состоит в том, что аномалия в структуре рецептора ЛПНП препятствует утилизации последних клетками печени, а следовательно, приводит к увеличению концентрации циркулирующих атерогенных липидов. Это повышает вероятность развития раннего, резко выраженного и нередко фатального атеросклероза. В 1985 г. работа J. Goldstein и M. Brown была удостоена Нобелевской премии.
На роль кандидатных генов атеросклероза претендует также ген, кодирующий структуру транспортного белка эфира холестерина. Мутация этого гена сопровождается нарушением транспорта холестерина из крови в печень и отложением его избытка в интиме артерий.
Аномалия гена, детерминирующего аминокислотную последовательность в пептидной цепочке фермента супероксиддисмутазы, приводит к уменьшению антиоксидантной активности крови, накоплению продуктов перекисного окисления липидов, роль которых в атерогенезе несомненна.
Установлено, что важную роль в генезе атеросклероза играет ген, кодирующий структуру ангиотензинконвертазы - фермента, который катализирует биосинтез ангиотензина-II. Этот мутантный ген обозначили латинской буквой D. Установлено, что у людей, гомозиготных по данному гену, повышается активность ангиотензинконвертазы, в результате чего возрастает уровень ангиотензина- II, который вызывает вазоконстрикцию и способствует избыточной пролиферации гладкомышечных клеток, участвующих в формировании атеросклеротической бляшки. Более того, оказалось, что у людей, страдающих сахарным диабетом и имеющих генотип DD, резко увеличивается риск развития атеросклероза даже при нормальном уровне холестерина и липопротеинов крови.
Дислипопротеинемия. Оптимальный уровень общего холестерина в крови - 200-230 мг% (5,2-6,0 ммоль/л). При обычном питании в организм человека ежедневно поступает с пищей около
500 мг холестерина, кроме того, столько же образуется в самом организме, главным образом в печени.
На процесс атерогенеза влияет не столько уровень общего холестерина крови, сколько нарушение оптимального соотношения между разными фракциями липопротеинов. В связи с этим для клиники важным является определение трех основных показателей плазмы крови, взятой натощак: 1) общего холестерина;
2) холестерина липопротеинов высокой плотности (ЛПВП);
3) триацилглицеролов. Общий холестерин представляет собой сумму холестерина, входящего в состав трех липопротеинов: общий холестерин = холестерин ЛПВП + холестерин липопротеинов очень низкой плотности (ЛПОНП) + холестерин ЛПНП.
Во всех европейских странах уровень общего холестерина до 250 мг% (6,5 ммоль/л) принято считать умеренной гиперхолестеринемией, от 250 до 300 мг% (7,8 ммоль/л) - выраженной гиперхолестеринемией и свыше 300 мг% - высокой гиперхолестеринемией. Многочисленные популяционные исследования показали достоверное возрастание смертности населения от сердечно-сосудистых заболеваний по мере повышения уровня холестерина.
Кроме того, на практике чаще пользуются определением так называемого коэффициента атерогенности (К), который рассчитывают по формуле:
К = (холестерин общий - холестерин ЛПВП) / холестерин ЛПВП.
У здоровых людей значение этого коэффициента не превышает 2-3 (максимум 3,5), при прогрессировании атеросклероза может увеличиваться до 4, а иногда даже более 6-7 единиц.
Гипертензия. Сочетание атеросклероза и гипертонии встречается настолько часто, что неоднократно возникали суждения об этиологической и патогенетической общности этих заболеваний. Стойкий подъем кровяного давления любой этиологии намного ускоряет развитие атеросклероза. Это обусловлено тем, что спазм артерий сопровождается сдавлением проходящих в их стенке собственных кровеносных и лимфатических сосудов (vasa vasorum) и как следствие нарушением оттока липидов из интимы и медии. Кроме того, повышенный тонус сосудов стимулирует пролиферацию гладкомышечных клеток, что способствует формированию атеросклеротической бляшки. Вместе с тем атеросклероз может протекать без гипертонии, равно как известны случаи гипертони-
ческой болезни, когда в артериальной стенке атеросклеротические изменения вовсе не выявляются.
Курение. Установлено, что выкуривание 1-2 сигарет после еды сопровождается достаточно выраженным и продолжительным подъемом уровня холестерина и атерогенных липопротеинов в крови. Наряду с этим никотин вызывает спазм артерий, в том числе и v. vasorum, нарушая микроциркуляцию в сосудистой стенке, и стимулирует пролиферацию гладкомышечных клеток. Никотин повышает свертываемость крови и способствует тромбообразованию в области атеросклеротической бляшки.
Нерациональное питание. Употребление в пищу избыточного количества животных жиров и продуктов, богатых холестерином, в сочетании с недостатком в диете растительных жиров, витаминов и микроэлементов приводит к развитию гиперхолестеринемии, увеличению содержания атерогенных липопротеинов одновременно со снижением уровня ЛПВП. Наиболее богаты холестерином такие продукты, как зернистая икра, шоколад, мозги, печень, сливочное масло, сметана, сливки, яичные желтки. Кроме того, атерогенность жиров в большой степени зависит от трансили цисконфигурации жирных кислот. В частности, такие жиры, как маргарин, содержащие искусственные трансизомеры, могут увеличивать риск развития атеросклероза. Атерогенным действием обладают и легкоусвояемые углеводы (сахар, крахмал), что связано с их способностью быстро расщепляться до глюкозы, которая метаболизируется в ацетил-КоА, являющийся субстратом для синтеза холестерина и липидов, поэтому любое избыточное усиление его продукции неизбежно ведет к дислипопротеинемии и атерогенезу.
Ожирение. Известно, что для мужчин в возрасте 40-69 лет, у которых избыточная масса тела составляет 30% и более, показатель смертности от заболеваний, непосредственно связанных с атеросклерозом, увеличивается на 40%, а у женщин того же возраста - на 35%. Ожирение, как правило, сопровождается гиперхолестеринемией и увеличением концентрации атерогенных липопротеинов. При ожирении возникает «порочный круг»: избыточная масса тела способствует развитию гипертензии, сахарного диабета, дислипопротеинемии, прогрессированию осложнений атеросклероза. Перечисленные состояния приводят к ограничению физической активности, которое способствует увеличению избыточной массы тела.
Стресс и хроническое психоэмоциональное напряжение. Считается признанным, что чрезмерные, особенно длительные стрессовые
ситуации способствуют развитию атеросклероза. Достаточно красноречиво свидетельствуют об этом результаты секционных исследований более 10 тыс. людей, погибших в фашистском концлагере Дахау в 1940-1945 гг. Энергетическая ценность лагерного рациона в первые годы указанного периода составляла около 1000 ккал в сутки, а к концу войны - 600 ккал. Кроме того, бесчеловечный режим сопровождался постоянными стрессами, истязаниями, тяжелым физическим трудом, чувством абсолютной безысходности и комплексом тяжелых нервно-психических травм. При патологоанатомическом вскрытии погибших узников в их аорте, коронарных и церебральных артериях выявлялся значительный атероматоз, несмотря на полное отсутствие в рационе заключенных животных жиров, в том числе холестерина. Выраженность этих изменений не была связана с возрастом погибших, а находилась в прямой зависимости от длительности их пребывания в концлагере. Этот пример показывает, что значение чрезвычайно сильных и длительных отрицательных эмоций у человека может быть главенствующим фактором риска развития атеросклероза.
Полагают, что одним из механизмов атерогенного действия отрицательных эмоций является активация симпатоадреналовой системы с повышением концентрации катехоламинов в крови. В результате в жировых депо усиливаются процессы липолиза и происходит мобилизация жирных кислот, которые в нормальных условиях обеспечивают организм достаточным количеством энергии. Выделяясь же в избыточном количестве, они не утилизируются, а используются на синтез липидов, в том числе холестерина, триацилглицеролов и атерогенных липопротеинов.
Гиподинамия. Малоподвижный образ жизни постепенно снижает интенсивность метаболических процессов в организме, способствуя развитию ожирения и других нарушений обмена.
Гормональные нарушения. Среди эндокринных факторов, способствующих возникновению атеросклероза, ведущую роль играют сахарный диабет и гипотиреоз. Чем тяжелее протекает сахарный диабет, тем выше гиперхолестеринемия и дислипопротеинемия. При этой патологии в организме нарушается не только утилизация глюкозы, но и метаболизм ацетил-КоА, который в этом случае не окисляется в цикле Кребса, а используется для синтеза холестерина и липидов, что способствует формированию диабетической дислипопротеинемии с последующим развитием атеросклероза.
Известно, что гипофункция щитовидной железы является одной из причин прогрессирования атеросклероза. Это связано с тем, что тироксин и трийодотиронин стимулируют катаболизм липидов и их окисление в цикле Кребса, а недостаток названных гормонов способствует преобладанию процессов синтеза липидов над их распадом и как следствие увеличению содержания холестерина и атерогенных липопротеинов в плазме крови.
Подагра. При подагре поражаются не только суставы, но и сосуды (преимущественно почек, сердца, легких), что может создавать дополнительные условия для ускоренного развития атеросклеротических бляшек.
Жесткость воды. Приводятся данные о том, что в регионах, где население систематически употребляет слишком мягкую воду, среднепопуляционный уровень холестерина выше, чем в соседних регионах, где вода имеет обычную жесткость. По-видимому, отсутствие в воде определенных микроэлементов оказывает отрицательное влияние на липидный обмен.
Патогенез атеросклероза
Многочисленные теории патогенеза атеросклероза укладываются в рамки двух основных положений: 1) первичным при атеросклерозе является нарушение липидного обмена, а повреждение артериальной стенки - вторичным; 2) основным звеном патогенеза атеросклероза является повреждение клеточных, соединительнотканных и других структур артериальной стенки различной этиологии.
Первая теория патогенеза атеросклероза, получившая в литературе название холестериновой, была сформулирована Н.Н. Аничковым в 1915 г. Суть ее сам автор выразил словами: «без холестерина не может быть атеросклероза». Продолжая свои исследования, Н.Н. Аничков выдвинул комбинационную теорию происхождения атеросклероза, согласно которой холестерин рассматривается как фактор, непосредственно реализующий атеросклеротические изменения артерий. Способствуют отложению холестерина следующие явления: 1) нарушения липидного обмена и его регуляции (конституциональные, эндокринные и др.); 2) алиментарный фактор (избыток холестерина в пище); 3) «механические» влияния (главным образом гемодинамические) на стенки сосудов; 4) первичные изменения артерий (дистрофические, возрастные и др.).
Представления Н.Н. Аничкова о патогенезе атеросклероза легли в основу так называемой инфильтрационно-комбинационной теории, которая исходит из положения о том, что основная часть энергетических потребностей артериальной стенки восполняется за счет липидов плазмы крови, которые поступают в интиму и медию путем инфильтрации. В норме эти липиды проходят в адвентицию без задержки и удаляются через систему лимфатических сосудов. В тех же случаях, когда концентрация липидов в крови возрастает, они накапливаются в сосудистой стенке, вызывая развитие липидоза. Однако инфильтрационная теория не давала ответа на два важных вопроса: почему атеросклерозом поражаются только артерии и не поражаются вены и почему нередко развитие атеросклероза происходит при нормальном уровне холестерина и липопротеинов в крови? В связи с этим возникли другие теории атерогенеза.
Эндотелиальная теория утверждает, что «пусковым фактором» для возникновения атерогенной бляшки служит повреждение клеток эндотелия, а роль нарушений липидного обмена сводится к способствующим атерогенезу условиям. Например, известно, что ряд вирусов (Коксаки, Эпштейна-Барр, простого герпеса, цитомегаловирусной инфекции и др.) может поражать клетки сосудистого эндотелия, нарушая в них липидный обмен и создавая условия для формирования атеросклеротической бляшки. Повреждения внутренней оболочки сосудов могут индуцировать перекиси липидов (по A. Szczeklik, 1980), которые ингибируют в эндотелиальных клетках артерий фермент простациклин-синтетазу. В результате развиваются локальная недостаточность простациклина (вазодилататор) и относительное преобладание его функционального антагониста тромбоксана, проявляющего вазоконстрикторные свойства. Это приводит к адгезии тромбоцитов на поверхности эндотелия, способствующей повреждению клеток последнего и развитию бляшки.
В 1974 г. появилась моноклинальная теория, рассматривающая атероматозную бляшку как своеобразную «доброкачественную опухоль», образование которой вызвано вирусами и мутагенами. Автором этой теории явился американский ученый E. Bendit, обративший внимание на то, что для атеросклеротических поражений характерна пролиферация гладкомышечных клеток. Близка по своей сути к предыдущей теории мембранная гипотеза D. Jackson и A. Gotto (1976), объясняющая причину пролиферации гладкомы-
шечных клеток избыточным поступлением в них неэстерифицированного холестерина, который, встраиваясь в мембранные структуры клеток, способствует их гиперплазии.
В нашей стране под руководством академика А.Н. Климова активно развивается аутоиммунная теория патогенеза атеросклероза, согласно которой этот патологический процесс инициируют аутоиммунные комплексы, содержащие липопротеины в качестве антигена. Аутоиммунные комплексы характеризуются следующими особенностями: 1) вызывают повреждение эндотелия и тем самым ускоряют проникновение липопротеинов крови в сосудистую стенку; 2) продлевают циркуляцию липопротеинов в крови; 3) задерживают окисление и экскрецию холестерина с желчью, т.е. способствуют развитию гиперлипопротеинемии; 4) проявляют цитотоксическое действие, откладываясь и фиксируясь в стенке артерий.
Несмотря на существование различных теорий, наиболее популярной точкой зрения относительно патогенеза атеросклероза остается признание ключевой роли холестерина и атерогенных липопротеинов.
Современные представления о роли холестерина и липопротеинов в атерогенезе. Как известно, в крови липиды содержатся в таких двух основных формах, как хиломикроны и липопротеины.
ЛПНП и ЛПОНП осуществляют транспорт холестерина в клетки, участвуя в формировании атеросклеротических бляшек, поэтому их называют атерогенными. ЛПВП способны транспортировать холестерин из клеток эндотелия сосудов в печень, содействуя регрессии бляшек, в связи с чем их называют антиатерогенными. Эти различия в свойствах липопротеинов определяются их химическим составом. Так, в структуре ЛПНП находится основное (около 2/3) количество холестерина плазмы, в ЛПОНП - только 1/3 циркулирующего холестерина, а в ЛПВП - лишь следовые его количества. Кроме того, атерогенность липопротеинов во многом зависит от содержания в их структуре триацилглицеролов (они в основном содержатся в ЛПОНП), а также апопротеинов и фосфолипидов (последних очень много в составе ЛПВП).
Транспорт холестерина из плазмы в сосудистую стенку осуществляется, главным образом, в виде ЛПНП, более половины которых попадает внутрь клеток эндотелия при помощи мембранных рецепторов, остальные - нерецепторным путем. Установлено, что указанные рецепторы взаимодействуют с апопротеинами, распо-
ложенными на поверхности ЛПНП. В лизосомах эндотелиальных клеток ЛПНП распадаются, эфиры холестерина гидролизуются на холестерол и жирные кислоты, после чего жирные кислоты окисляются, а холестерол используется для «строительства» клеточных мембран. В ситуациях, когда ткани организма нуждаются в большом количестве холестерина (например, для синтеза клеточных мембран, стероидных гормонов, желчных кислот), активность клеточных ЛПНП-рецепторов возрастает, вследствие чего утилизация холестерина усиливается. В результате этого уменьшается содержание ЛПНП в крови и снижается вероятность транспорта холестерина в артериальную стенку. Если же ткани не нуждаются в дополнительных количествах холестерина, активность рецепторов ЛПНП снижается, концентрация ЛПНП в плазме возрастает, а вероятность формирования атеросклеротических бляшек увеличивается.
В отличие от липопротеинов низкой и очень низкой плотности ЛПВП выполняют в организме антиатерогенную функцию. Согласно транспортной гипотезе В.Н. Титова, они осуществляют обратньгй транспорт холестерина из сосудов, органов и тканей, переводя его в другие липопротеины или транспортируя прямо в печень, с последующим выведением с желчью в кишечник. В организме холестерин окисляется только в клетках печени. Следовательно, если содержание ЛПВП увеличивается, то одновременно усиливается окисление холестерина. Таким образом, чем больше в крови ЛПВП и холестерина в их составе, тем меньше вероятность развития атеросклероза и выше вероятность регресса атеросклеротических бляшек.
Вместе с тем сложную проблему атеросклероза нельзя свести к уровню холестерина и липопротеинов в крови. Неверно строить рациональную терапию исключительно на диете по принципу малого содержания холестерина. Такой подход к патогенетически обоснованной профилактике упрощен, поскольку, по данным ряда авторов, гиперхолестеринемия отсутствует у 50% больных атеросклерозом.
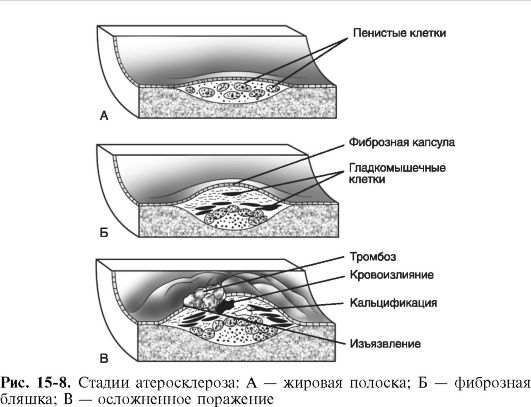
Морфогенез атеросклероза. В формировании атеросклеротической бляшки - морфологической основы атеросклероза - важную роль играют как нарушения липидного обмена (дислипопротеинемии), так и состояние сосудистой стенки. Бляшки могут расти вдоль сосуда, тогда они развиваются медленно, длительно и менее опасны, но могут располагаться и поперек сосуда - такие бляшки часто называют «летальными», поскольку даже единичные образования подобного типа могут привести к сосудистой катастрофе.
Предшественниками бляшки часто являются зоны липидной инфильтрации интимы, так называемые липидные полоски, через которые в сосудистую стенку проникают моноциты. В сосудистой стенке моноциты трансформируются в макрофаги, имеющие рецепторы к ЛПНП. В процессе переполнения этих клеток фагоцитированными липопротеинами они превращаются в пенистые клетки. Имеются данные, что пенистыми клетками могут становиться и переполненные липидами гладкомышечные клетки. Скопления пенистых клеток и составляют основу липидных полосок. Пенистые клетки могут разрушаться, высвобождая биологически активные вещества, стимулирующие пролиферацию гладкомышечных клеток и привлекающие их в субэндотелиальный слой из глубжележащих участков сосудистой стенки. В результате скопления гладкомышечных клеток наблюдается образование небольших выпячиваний эндотелия в просвет сосуда.
В процессе дальнейшего развития бляшек в них появляются соединительнотканные элементы: коллагеновые и эластические волокна, приводящие к уплотнению - склерозу. Этот процесс поддерживается за счет выделения из макрофагов медиаторов клеточного иммунитета и ростовых факторов, стимулирующих аутоиммунные реакции в интиме и пролиферацию фибробластов. В результате образуется плотная фиброзная бляшка.
Конечным этапом формирования бляшек является образование их «осложненных» форм (рис. 15-8). Выступающая в просвет сосуда атеросклеротическая бляшка насыщается солями кальция и нарушает ламинарный поток крови, который в этом месте становится турбулентным. Такая бляшка пропитывается липидами и становится рыхлой. Очевидно, что плотная фиброзная бляшка является потенциально менее опасной, чем ее рыхлая «осложненная» форма, которая вследствие слущивания покрывающего ее эндотелия, кальцификации и происходящего в ней клеточного распада таит угрозу образования пристеночного тромба или разрыва сосуда с кровоизлиянием.
15.3. ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОЙ ДЕЯТЕЛЬНОСТИ
Всю патологию сердца можно систематизировать в нескольких направлениях:
1. Коронарогенная патология сердца - ишемическая болезнь сердца.
 2. Некоронарогенная
патология: а) неревматической этиологии; б) заболевания перикарда; в)
заболевания ревматической этиологии.
2. Некоронарогенная
патология: а) неревматической этиологии; б) заболевания перикарда; в)
заболевания ревматической этиологии.
3. Аритмии.
Итогом неблагоприятного развития любой из перечисленных форм патологии сердца является нарушение его насосной функции и формирование сердечной недостаточности (см. раздел 15.3.2).
15.3.1. Патология коронарной перфузии
Ишемическое повреждение сердца
Заболевания, патогенетическую основу которых составляет ишемическое повреждение сердечной мышцы (коронарная болезнь сердца, инфаркт миокарда, атеросклеротический кардиосклероз), являются основной причиной смертности населения в современном обществе - по данным ВОЗ, 400-500 человек на 100 000 населения в возрасте 50-54 лет.
Термин «ишемическая болезнь сердца» (ИБС) или его синоним «коронарная болезнь сердца» был предложен комитетом экспертов ВОЗ в 1962 г. ИБС - термин собирательный, включающий многообразные клинические формы и проявления - как острые, так и хронические, как обратимые (преходящие), так и необратимые, заканчивающиеся некрозом сердечной мышцы. Ишемия миокарда (от греч. ischo - задерживать, останавливать и haemia - кровь) представляет собой состояние, при котором нарушается кровообращение мышцы сердца, появляется местное «малокровие», вследствие чего развивается коронарная недостаточность. Возникает несоответствие между потребностями миокарда в кислороде, с одной стороны, и уровнем оксигенации кардиомиоцитов - с другой. В результате возникшего дефицита кислорода в клетках сердечной мышцы нарушаются энергообразование, а также другие метаболические процессы, что приводит к изменению сократительной функции миокарда в зоне ишемии, появлению болевого синдрома (стенокардии).
Патогенез и клинические эквиваленты коронарной недостаточности
Механизм развития недостаточности венечных сосудов определяется взаимодействием трех основных факторов: наличием атеросклеротической бляшки, спазмом венечных сосудов и интракоронарным тромбозом.
1. Известно, что возникновение атеросклеротической бляшки в коронарных артериях является морфологической основой ишемического повреждения сердца в подавляющем большинстве случаев (90% и более). При уменьшении просвета венечной артерии на 70-80% возникают выраженные приступы стенокардии напряжения.
2. Спазм коронарных артерий - это сокращение их сосудистой стенки, которое препятствует нормальному кровотоку в сердечной мышце. Следует сказать, что спастическая реакция сосудов сердца на те или иные воздействия может вызвать болевой приступ даже при незначительном стенозе коронарных артерий и, более того, при полном отсутствии такового. В этом случае говорят о так называемой вариантной стенокардии Принцметала, впервые выделенной в отдельный синдром M. Prinzmetal с коллегами в 1959 г. и составляющей 3% всех случаев стенокардии. Причиной вариантной стенокардии является нарушение функционального состояния эндотелия сосудов сердца.
В дальнейшем развитие спазма коронарных артерий было документировано и при других формах стенокардии. Так, в 1976 г. Mudge и его коллеги зарегистрировали рефлекторное снижение кровотока в миокарде во время проведения холодового теста, который заключался в погружении одной руки пациента на 1 мин в воду с температурой 0 °С. Коронароспазм удалось устранить с помощью блокатора альфа-адренорецепторов - фентоламина. На основании этого был сделан вывод о том, что в патогенезе спазма венечных сосудов важную роль играет активация симпатической нервной системы. Участие парасимпатического звена вегетативной нервной системы в патогенезе стенокардии не расценивается столь однозначно. Так, было установлено, что медиатор парасимпатической системы ацетилхолин вызывает вазоконстрикцию венечных артерий с поврежденным эндотелием, тогда как «здоровые» коронарные сосуды в ответ на действие этого медиатора расширяются (ацетилхолин стимулирует образование NO).
Вместе с тем коронароспазм может развиваться и без участия вегетативной нервной системы. Факторами, вызывающими сужение сосудов, в этом случае могут быть вазоконстрикторные биологически активные вещества.
3. Важную роль в развитии коронарной недостаточности играет тромбоз венечных артерий. Чаще тромбы формируются на поверхности атеросклеротической бляшки, где нарушились структура и целостность эндотелиального слоя, в виде изъязвления и деструкции бляшки. Поскольку в таких ситуациях просвет коронарных артерий быстро перекрывается стремительно образующимся тромбом, а коллатеральное кровообращение не успевает компенсировать сниженный кровоток, то возникает нестабильная форма стенокардии (по прежней терминологии - предынфарктное состояние) и может развиться инфаркт миокарда или наступить внезапная сердечная смерть.
Следует отметить, что даже в центре зоны ишемии при коронарной недостаточности кровоток никогда не снижается до нулевых значений, а остается на уровне 10-30% нормы. На периферии же участка гипоперфузии кровоснабжение нередко составляет 50% от исходного уровня. Таким образом, коронароокклюзия никогда не приводит к аноксии (полному отсутствию кислорода), вызывая лишь гипоксию (недостаток кислорода).
Клиническими эквивалентами кратковременной (преходящей, транзиторной) ишемии миокарда являются стенокардия, бессимптомная ИБС, внезапная сердечная смерть, гибернация миокарда.
Стенокардия (anginapectoris). Клиническая картина стенокардии подробно описана B. Геберденом еще в 1768 г. Он же и предложил термин «angina pectoris», что переводится на русский язык как «грудное сжатие», «грудное стеснение» (от лат. ango - сжимать). Достаточно распространенным является также термин «грудная жаба».
Стенокардия, возникающая во время физических нагрузок, получила название стенокардии напряжения. Она развивается в тех случаях, когда коронарный кровоток, нарушенный по причине стенозирующего атеросклероза, остается тем не менее достаточным для обеспечения работы сердца в условиях функционального покоя. Однако при физической нагрузке, когда работа сердца усиливается и его потребность в кислороде возрастает, суженные коронарные артерии не могут обеспечить адекватный прирост кровотока и нормальную оксигенацию миокарда. Возникает относительная коронарная недостаточность, и развивается болевой синдром. Наиболее часто боли возникают в загрудинной области, имеют сжимающий характер. Нередко они локализуются слева от грудины, реже - в левой руке, начиная с плечевого сустава. Часто ангинозные боли иррадиируют в левое плечо, предплечье, кисть. Типичный приступ стенокардии длится от 1 до 5-10 мин. Обычно ангинозные боли быстро исчезают после прекращения физической нагрузки, но появляются вновь после ее возобновления.
Однако болевые приступы могут возникать и в состоянии покоя. Такая стенокардия получила название стенокардии покоя. Она свидетельствует о выраженном стенозирующем атеросклерозе коронарных артерий, при котором капиллярный резерв миокарда полностью исчерпан.
Нестабильная стенокардия - это стенокардия с нестабильным, прогрессирующим течением, часто заканчивающаяся внезапной сердечной смертью или инфарктом миокарда. К нестабильной стенокардии обычно относят впервые возникшую стенокардию или постинфарктную стенокардию, появившуюся через 48 ч после инфаркта, а также учащение приступов ангинозных болей. Различные формы нестабильной стенокардии длятся до месяца и в большинстве случаев переходят в стабильную форму, но в целом прогноз при нестабильной стенокардии намного хуже, чем при стабильном ее течении.
Бессимптомная (безболевая, «немая») ИБС. В последнее время большое практическое значение придается бессимптомной (без-
болевой) ишемии миокарда. К бессимптомной ИБС относят эпизоды транзиторной (кратковременной), обратимой ишемии миокарда, которые клинически ничем не проявляются. Поскольку в большинстве случаев бессимптомная ИБС прижизненно не выявляется, а следовательно, и не лечится, она часто может осложняться инфарктом миокарда и быть причиной внезапной коронарной смерти.
Наиболее распространенным методом диагностики стенокардии является электрокардиография. Однако электрокардиографические признаки ишемии миокарда (подъем или депрессия сегмента S-T) часто удается выявить только во время ангинозного приступа. В межприступном периоде электрокардиограмма (ЭКГ) пациента, страдающего ишемической болезнью сердца, может ничем не отличаться от ЭКГ здорового человека. Поэтому диагностика ИБС основана на данных суточного электрокардиографического мониторирования, когда удается записать ЭКГ во время эпизодов ишемии. Кроме того, ценность электрокардиографического исследования намного возрастает при проведении нагрузочных проб. Чаще используется велоэргометрическая проба, которая представляет собой электрокардиографическое исследование, проводимое в динамике ступенчато возрастающей физической нагрузки, выполняемой на велоэргометре. Под ее влиянием постепенно увеличивается работа сердца и повышается потребность миокарда в кислороде. Наряду с велоэргометрией в клинике используются и другие нагрузочные пробы (тредмил, фармакологические тесты, чреспищеводная электрокардиостимуляция и др.).
В последние годы все большее значение для диагностики транзиторной ишемии миокарда приобретает перфузионная гаммасцинтиграфия сердца, выполняемая в сочетании с нагрузочными тестами. Принципом этого метода является получение диагностического изображения с помощью гамма-камеры, позволяющей регистрировать излучение введенного в организм пациента радиоактивного фармакологического препарата (рис. 15-9).
«Золотым стандартом» выявления стенозирующего атеросклероза коронарных артерий является рентгеноконтрастная ангиография, суть которой состоит в получении серии рентгеновских изображений после интракоронарного введения больному рентгеноконтрастного вещества. Проведение такого исследования является обязательным для решения вопроса о кардиохирургической реваскуляризации миокарда.
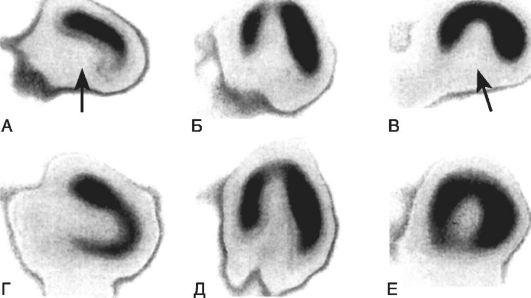 Рис. 15-9. Томограммы сердца после инъекции 199Tl
на пике физической нагрузки (А-В) и в покое (Г-Е) у пациента со
стенозирующим атеросклерозом правой коронарной артерии. Стрелками
обозначен дефект перфузии в области задней стенки левого желудочка,
который определяется на пике физической нагрузки и исчезает в покое
Рис. 15-9. Томограммы сердца после инъекции 199Tl
на пике физической нагрузки (А-В) и в покое (Г-Е) у пациента со
стенозирующим атеросклерозом правой коронарной артерии. Стрелками
обозначен дефект перфузии в области задней стенки левого желудочка,
который определяется на пике физической нагрузки и исчезает в покое
Внезапной коронарной смертью (внезапной сердечной смертью)
называют неожиданную смерть, возникшую мгновенно или в пределах 1 ч после появления первых симптомов коронарной катастрофы (ангинозные боли, аритмия). Чаще всего (более 90% случаев) внезапная сердечная смерть наступает у пациентов, имевших до этого заболевания сердца, но находившихся, с точки зрения врача, в относительно стабильном, не опасном для жизни состоянии. Непосредственными причинами внезапной сердечной смерти являются фибрилляция желудочков и желудочковая тахикардия (80% случаев), а также асистолия или резкая брадикардия (20%).
К причинам внезапной сердечной смерти у взрослых относят: постинфарктный кардиосклероз, кардиомиопатии (особенно гипертрофическую), миокардиты, аномалии проводящей системы (например, синдром Вольфа-Паркинсона-Уайта - WPW), синдром удлиненного интервала Q-T У лиц молодого возраста внезапная сердечная смерть может быть следствием спазма коронарных артерий даже при отсутствии коронарного атеросклероза. При патолого-анатомическом исследовании у них не удается выявить никаких морфологических изменений в сердечной мышце. В этом случае причиной внезапной сердечной смерти считается стрессорное повреждение сердца.
Гибернирующий миокард, иначе говоря, миокард, находящийся в состоянии спячки. Гибернация - это очаговая обратимая дисфункция миокарда. Этим термином в 1986 г. американский физиолог E. Braunwald обозначил ишемическое повреждение сердца, которое напоминает инфаркт миокарда, но в отличие от последнего характеризуется обратимостью электрофизиологических изменений. Гибернирующий участок миокарда сохраняет жизнеспособность, но перестает сокращаться. Он как бы балансирует между жизнью и смертью. Ишемические изменения в этом случае носят обратимый характер, и восстановление коронарного кровотока, как правило, сопровождается восстановлением сократимости кардиомиоцитов. Однако если состояние ишемии продлится слишком долго, в зоне гибернации могут произойти необратимые изменения, заканчивающиеся гибелью клеток миокарда. Клинически гибернирующий миокард сходен с инфарктом и характеризуется ангинозными болями, резистентными к нитроглицерину и длящимися более 30 мин. На ЭКГ при этом регистрируются признаки ишемии миокарда. Но в отличие от инфаркта миокарда в крови пациентов не удается обнаружить существенного повышения активности ферментов.
Гибель отдельных клеток в зоне ишемии наступает уже через 15 мин после прекращения коронарного кровотока, однако окончательное формирование зоны некроза завершается только через 6 ч от момента коронароокклюзии. Поэтому восстановление кровотока в течение 6 ч от момента начала ангинозного приступа сопровождается нормализацией сердечной деятельности или ведет к значительному уменьшению зоны некроза. Необратимые изменения в кардиомиоцитах, заканчивающиеся некрозом сердечной мышцы, клинически проявляются в виде инфаркта миокарда.
Инфаркт миокарда - это некроз определенного участка сердечной мышцы, который развивается в связи с резким и продолжительным уменьшением коронарного кровотока.
Чаще инфаркт миокарда поражает мышцу левого желудочка. Среди значительного числа причин, непосредственно вызывающих инфаркт миокарда («реализующие факторы»), первое место занимают стрессовые ситуации и длительная психоэмоциональная перегрузка. На втором месте находится физическое перенапряжение.
В первые сутки очаг некроза практически не отличается от неповрежденной ткани миокарда и имеет не сплошной, а мозаичный характер, поскольку среди погибших миокардиоцитов и некро-
тизированных участков встречаются частично и даже полностью нормально функционирующие клетки и группы клеток. На вторые сутки инфаркта миокарда зона некроза постепенно отграничивается от здоровой ткани и между ними формируется периинфарктная зона, т.е. область, расположенная на границе зоны некроза и здорового миокарда.
Состояние периинфарктной зоны имеет очень большое значение для дальнейшего течения, прогноза и исхода инфаркта миокарда. Если острая ишемия не прогрессирует или устраняется, то в периинфарктной зоне быстро восстанавливаются функция и структура всех клеточных элементов. При этом зона некроза ограничивается первоначальными размерами. При недостаточном кровоснабжении дистрофические процессы в периинфарктной зоне усиливаются, кардиомиоциты гибнут, очаг некроза расширяется. Полная или частичная обратимость повреждений может сохраняться в течение 3-5 ч с момента возникновения ишемии.
Установлено, что зона некроза, превышающая 50% и более массы миокарда левого желудочка, приводит к развитию тяжелой недостаточности кровообращения, часто не совместимой с жизнью.
Эволюция зоны некроза постепенно приводит, с одной стороны, к ограничению области инфаркта миокарда, а с другой - к развитию процессов миомаляции (размягчение мышечной ткани). При этом на периферии очага некроза формируется молодая грануляционная ткань, а внутри идет рассасывание погибших кардиомиоцитов. Этот период (примерно 7-10 суток от начала заболевания) является наиболее опасным в отношении разрыва сердца, когда на фоне клинического улучшения может наступить быстрая смерть больного. При благоприятном развитии инфаркта миокарда в течение 3 нед заболевания в зоне некроза начинает интенсивно формироваться соединительная ткань. Плотный рубец при трансмуральном инфаркте миокарда окончательно образуется только через 3-4 месяца. При мелкоочаговом инфаркте миокарда фиброзный рубец может образоваться через 2-3 недели.
При типичном начале инфаркта миокарда в основе его клинической картины лежат сильные боли, симптомы сердечной недостаточности, аритмии, повышение температуры, лейкоцитоз, увеличение СОЭ, гиперферментемия.
Боль. Этот важнейший симптом инфаркта чаще всего заставляет обратиться пациента к врачу, но, как ни странно, патогенез
болевого симптома при инфаркте миокарда до сих пор остается недостаточно изученным. Полагают, что основной причиной болей при этом заболевании является накопление в зоне ишемии лактата, который раздражает нервные окончания афферентных симпатических волокон. Кроме того, важная роль в генезе болей отводится простагландинам и брадикинину, которые в больших количествах накапливаются в ишемизированной ткани. Вместе с тем в организме существуют вещества - опиоидные пептиды, которые обладают обезболивающей активностью. Следовательно, выраженность болевой симптоматики во многом может определяться «балансом» между медиаторами болевого рефлекса (лактат, простагландины, брадикинин) и опиоидными пептидами.
В связи с этим нельзя не упомянуть о том, что существуют и так называемые безболевые инфаркты миокарда, при которых боль вообще отсутствует. Безболевой инфаркт миокарда протекает тяжелее по сравнению с типичным проявлением данного заболевания. В какой-то мере это объясняется диагностическими ошибками при его выявлении, а следовательно, поздно начатым лечением. На передний план клинической картины инфаркта в этом случае выходят симптомы сердечной недостаточности и аритмии.
Примерно в 50% наблюдений боли при инфаркте миокарда появляются внезапно. Суммарная продолжительность ангинозных болей, превышающая 1 ч, свидетельствует о возможном развитии инфаркта миокарда. Локализация болей при инфаркте миокарда в некоторой степени определяется расположением очага некроза в мышце сердца и может наблюдаться за грудиной, в левой руке, начиная с плечевого сустава и ниже, в челюсти, поддиафрагмальной области и т.д. В последнем случае говорят об абдоминальной (гастралгической) форме инфаркта, которая встречается довольно часто. В результате больные считают, что боли у них связаны с заболеванием органов брюшной полости. Нередко такой инфаркт миокарда может симулировать обострение язвенной болезни, острый холецистит, острый панкреатит и др.
Нитроглицерин при однократном приеме (в отличие от обычного приступа стенокардии) не дает облегчения. Больные вынуждены принимать его повторно, многократно, до 20-40 таблеток в сутки. Появление резистентных к нитроглицерину ангинозных болей служит важным признаком, позволяющим дифференцировать обычную стенокардию от инфаркта миокарда.
Симптомы сердечной недостаточности (одышка, тахикардия, отеки и гипотензия) относятся к типичным проявлениям инфаркта миокарда. Появление этих симптомов связано с нарушением насосной функции сердца, которая снижается прямо пропорционально размеру очага некроза. Если размер зоны инфаркта составляет 50% от массы левого желудочка, то такое поражение миокарда, как правило, несовместимо с жизнью, поскольку центральная гемодинамика в этом случае страдает настолько, что нарушается кровоснабжение жизненно важных органов.
Нарушения сердечного ритма практически всегда сопровождают развитие инфаркта миокарда, а при его безболевой форме могут стать ведущим симптомом заболевания (аритмический инфаркт миокарда).
Основной причиной аритмий в первые 6 ч инфаркта миокарда является изменение электрофизиологических свойств кардиомиоцитов в зоне ишемии. Из-за дефицита энергетических субстратов, вызванного гипоксией, они перестают сокращаться, но сохраняют способность к проведению электрического импульса по своим мембранам. Однако вследствие недостатка АТФ эффективность работы энергозависимых ионных насосов в клетках существенно страдает, а сами ионные каналы повреждаются. Это ведет к замедлению процессов деполяризации и реполяризации, что создает благоприятные условия для возникновения аритмий. На более поздних сроках инфаркта клетки рабочего миокарда в зоне ишемии гибнут, а более устойчивые к гипоксии клетки проводящей системы сохраняют свою жизнеспособность, но их электрофизиологические характеристики существенно меняются. Проведение импульса по волокнам Пуркинье, расположенным в некротизированном миокарде, замедляется, а у клеток проводящей системы появляется способность к спонтанной деполяризации. В итоге формируется источник аномального эктопического автоматизма сердца.
Следует подчеркнуть, что все вышеперечисленные симптомы инфаркта миокарда (боль, признаки сердечной недостаточности, аритмии) вообще могут отсутствовать. В этом случае говорят о бессимптомной («немая», silent) форме инфаркта миокарда, при которой больной не обращается за медицинской помощью, а заболевание часто остается незамеченным.
Повышение температуры, лейкоцитоз, увеличение СОЭ, гиперферментемия также являются характерными признаками, составляющими клиническую картину инфаркта миокарда. Коронарный
кровоток в зоне ишемии никогда не падает ниже 10% от нормального уровня, поэтому в кровь из пораженного миокарда поступают продукты распада кардиомиоцитов. При этом содержание данных веществ в плазме крови возрастает прямо пропорционально размерам очага некроза. В результате формируется симптомокомплекс, получивший название резорбционного синдрома. В частности, уже в конце первых - начале вторых суток начинает повышаться температура тела, что связано с резорбцией некротических масс. Для картины периферической крови в это время характерен нейтрофильный лейкоцитоз (до 15-109/л - 20-109/л и более) со сдвигом влево. СОЭ начинает возрастать спустя 1-3 суток после возникновения заболевания и сохраняется на повышенном уровне 3-4 недели, иногда дольше.
Для инфаркта миокарда характерна также гиперферментемия, т.е. повышение активности ферментов в плазме крови. При возникновении некроза они поступают из некротизированных миокардиальных клеток в кровь. При появлении инфаркта миокарда или подозрении на него активность ферментов крови: креатинфосфокиназы (КФК), аспартатаминотрансферазы (АсАТ, или АСТ), лактатдегидрогеназы (ЛДГ) должна определяться серийно. Чем больше очаг некроза, тем выше активность КФК в плазме крови. На этом принципе основан даже метод косвенного определения размеров инфаркта миокарда по математическим формулам.
Несмотря на то что КФК в диагностике инфаркта миокарда рассматривается как один из наиболее специфичных ферментов, его активность может повышаться и при других состояниях. Это обусловлено тем, что КФК в большом количестве содержится не только в миокарде, но и в скелетных мышцах. Повышенная активность КФК может отмечаться при внутримышечных инъекциях, тромбоэмболии легочной артерии, миокардитах, тахиаритмиях, повреждении мышц различного характера, даже после ушибов и тяжелой физической работы.
Информативным показателем развития инфаркта миокарда может быть также повышение в крови уровня миоглобина. Уровень миоглобина в крови больных крупноочаговым инфарктом миокарда может повышаться в 4-10 раз и более по сравнению с нормой (она колеблется от 5 до 80 нг/мл). Содержание миоглобина нормализуется через 20-40 ч после начала заболевания. По степени и длительности повышения уровня миоглобина в крови больных можно судить о величине зоны некроза и о прогнозе заболевания.
Однако если пациент попадает в стационар через трое суток после возникновения инфаркта миокарда, что часто бывает при гастралгической или безболевой форме данного заболевания, обнаружить гиперферментемию не удается. Если же у этих пациентов имеются рубцовые изменения в миокарде или блокада ножки пучка Гиса, регистрация ЭКГ не позволяет выявить признаки некроза миокарда. В этом случае для установления правильного диагноза инфаркта миокарда прибегают к проведению гамма-сцинтиграфии сердца (рис. 15-10).
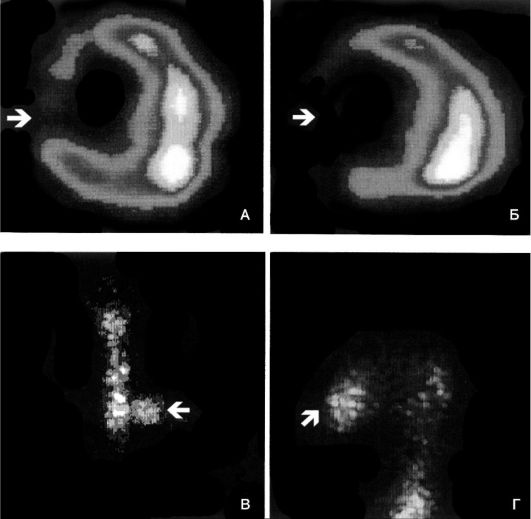 Рис. 15-10. Сцинтиграммы миокарда больного острым инфарктом миокарда, выполненные после инъекции 199ΊΊ, в покое (А) и через 4 ч после введения нуклида в левой косой проекции (Б), а также после инъекции 99тТс-пирофосфата
в передней (В) и левой боковых проекциях (Г). Стрелками обозначен
стабильный дефект перфузии в передней стенке левого желудочка и
включение 99тТс-пирофосфата в область инфаркта
Рис. 15-10. Сцинтиграммы миокарда больного острым инфарктом миокарда, выполненные после инъекции 199ΊΊ, в покое (А) и через 4 ч после введения нуклида в левой косой проекции (Б), а также после инъекции 99тТс-пирофосфата
в передней (В) и левой боковых проекциях (Г). Стрелками обозначен
стабильный дефект перфузии в передней стенке левого желудочка и
включение 99тТс-пирофосфата в область инфаркта
Осложнения инфаркта миокарда. Осложнения инфаркта весьма существенно отягощают его течение и часто являются непосредственной причиной летальности и инвалидизации пациентов при данном заболевании. Различают ранние и поздние осложнения острой коронарной патологии.
Ранние осложнения могут возникать в первые дни, часы и даже минуты инфаркта миокарда. К ним относятся кардиогенный шок, острая сердечная недостаточность, острая аневризма и разрывы сердца, тромбоэмболические осложнения, нарушения ритма и проводимости, перикардиты, острые поражения желудочнокишечного тракта.
Поздние осложнения возникают в подостром периоде рубцевания инфаркта миокарда. Это постинфарктный перикардит (синдром Дресслера), хроническая аневризма сердца, хроническая сердечная недостаточность и др.
Патогенез реперфузионного повреждения сердца
Первоначально предполагалось, что на определенном этапе полного восстановления функции ишемизированного миокарда можно легко добиться, возобновив коронарный кровоток. Исходя из этих соображений, отечественные кардиологи во главе с академиком Е.И. Чазовым разработали принципы тромболитической терапии инфаркта миокарда, эффективность которой оказалась наиболее высокой, если с момента коронароокклюзии проходило не более 6 ч. Для восстановления миокардиального кровообращения при хронической ИБС были разработаны различные методы хирургической реваскуляризации, среди которых наибольшее распространение получила операция аортокоронарного шунтирования, суть которой сводится к формированию сосудистого шунта, обеспечивающего кровоток в обход склерозированного участка венечной артерии.
Следует указать, что восстановление коронарной перфузии часто бывает недостаточно для полной нормализации сократимости сердца. Более того, в некоторых случаях реперфузия сердца может провоцировать гибель пациентов от желудочковой фибрилляции. Оказалось, что восстановление коронарного кровотока даже после непродолжительной ишемии может вызвать реперфузионное повреждение сердца, для которого характерны следующие проявления: а) сократительная дисфункция сердца; б) нарушения сердечного ритма; в) феномен невосстановленного кровотока.
Реперфузионная сократительная дисфункция сердца слагается из уменьшения силы сокращений миокарда и его неполного диастолического расслабления, в результате чего уменьшается сердечный выброс.
Основными механизмами реперфузионного повреждения миокарда являются так называемые кальциевый парадокс и кислородный парадокс.
Кальциевый парадокс - это перегрузка кардиомиоцитов ионами кальция. Ионы кальция в избытке проникают через сарколемму кардиомиоцитов, накапливаясь в саркоплазматическом ретикулуме и митохондриях. Механизм усиленного проникновения Са2+ через клеточную мембрану тесно связан с нарушением Na+/ Са2+ обмена. Если в норме основное поступление Са2+ в клетку происходит через медленные Са2+-каналы, то в условиях реперфузии резко активируется Na+/Са2+-транспорт (обмен внутриклеточного Na+ на внеклеточный Са2+), который осуществляется белком-переносчиком, расположенным на сарколемме. Кальциевая перегрузка кардиомиоцитов ведет к замедлению процесса расслабления сердца (реперфузионная контрактура), что неизбежно сопровождается уменьшением диастолического объема сердца и снижением сердечного выброса. Патогенез подобной сократительной дисфункции связан не только с замедлением релаксации кардиомиоцитов, но и с энергодефицитом, который вызван тем, что большая часть энергии, образующейся в митохондриях, расходуется на аккумуляцию Са2+ во внутриклеточных органеллах.
Кислородный парадокс - это токсическое действие кислорода, которое испытывает миокард в момент реоксигенации после ишемии. Дефицит кислорода приводит к восстановлению переносчиков электронов (НАДН-дегидрогеназа, убихинон, цитохромы) в дыхательной цепи митохондрий. В момент реоксигенации эти переносчики становятся донорами электронов для молекул кислорода. Последние при этом превращаются в свободные радикалы (активные формы кислорода). Активные формы кислорода повреждают молекулы ферментов, осуществляющих энергозависимый транспорт ионов в кардиомиоцитах. В результате происходит нарушение внутриклеточного ионного гомеостаза, развивается перегрузка кардиомиоцитов Са2+ и, как следствие, страдает сократительная функция сердца.
Таким образом, и кальциевый, и кислородный парадоксы приводят к перегрузке кардиомиоцитов ионами кальция. Более того,
в условиях реперфузии оба эти патологических процесса взаимно усиливают друг друга.
Реперфузионные нарушения сердечного ритма возникают в момент реоксигенации сердца и представлены главным образом желудочковыми аритмиями, патогенез которых также обусловлен кальциевым и кислородным парадоксами. Существует предположение, что в основе реперфузионных аритмий лежат не только кальциевый и кислородный парадоксы, но и изменения нейрогуморальных воздействий на сердце. Такие аритмии связаны с повышением тонической активности симпатоадреналовой системы и стимуляцией α-адренорецепторов миокарда эндогенным норадреналином. Все это приводит к еще большему повышению уровня внутриклеточного кальция.
Феномен невосстановленного кровотока (no reflow phenomenon) - это сохранение дефицита коронарной перфузии после возобновления магистрального кровотока в ветвях венечных артерий, питающих ишемизированные участки миокарда. В 1974 г. американский физиолог Kloner установил, что феномен невосстановленного кровотока развивается при этом не ранее чем через 1-2 ч после коронароокклюзии.
Главными факторами, препятствующими восстановлению коронарной микроциркуляции после реперфузии миокарда, являются: 1) набухание клеток эндотелия; 2) агрегация форменных элементов и повышение вязкости крови; 3) образование тромбов; 4) «краевое стояние» лейкоцитов у стенки микрососудов и инфильтрация ими сосудистой стенки. Удаление лейкоцитов из периферической крови в период, предшествующий реперфузии, препятствует формированию феномена невосстановленного кровотока.
Эндогенные механизмы защиты сердца при ишемии и реперфузии
Долгое время господствовало мнение, что клетки сердца абсолютно беззащитны в отношении ишемического повреждения. Ситуация изменилась в 1986 г., когда американские физиологи Murray и Jennings в экспериментах на собаках обнаружили так называемый феномен адаптации к ишемии (ischemic preconditioning). Суть этого явления сводится к повышению устойчивости миокарда к длительной ишемии в тех случаях, когда ей предшествовали несколько эпизодов 5-минутной ишемии. Результатом такого эксперимента явилось существенное повышение эффективности
коронарной реперфузии, которая привела к уменьшению размера очага инфаркта миокарда и повышению устойчивости сердца к аритмогенному действию ишемии и реперфузии. Клинические наблюдения подтвердили справедливость экспериментальных данных. Оказалось, что если инфаркту миокарда предшествовали приступы стенокардии, то эффективность тромболитической терапии значительно повышается. Размеры инфаркта у таких пациентов были меньше, чем у пациентов с инфарктом миокарда, возникшим внезапно, без предшествующих ангинозных приступов.
Многочисленные исследования показали, что механизм феномена ишемической адаптации тесно связан с активацией АТФзависимого К+-канала (К+АТф-канал). Во время ишемии из нервных окончаний и кардиомиоцитов, находящихся в зоне гипоперфузии, высвобождаются биологически активные вещества (аденозин, брадикинин, норадреналин, ангиотензин-II, опиоидные пептиды). Каждое из этих соединений стимулирует протеинкиназу С. Последняя и активирует К+АТф-канал. В результате отмечается тенденция к нормализации внутри- и внеклеточного баланса ионов. Повышение активности этого канала объясняется также снижением уровня АТФ (АТФ в норме подавляет К+АТф-канал).
Существует еще адаптация сердца к ишемии на уровне целого организма. Повышенная устойчивость миокарда к ишемии формируется при физических тренировках или периодическом действии на организм гипоксии, холода, кратковременного стресса и любых других экстремальных воздействий (Ф.З. Меерсон). Иными словами, особенностью подобной адаптации является развитие ее перекрестных эффектов. Например, при адаптации к холоду одновременно повышается устойчивость миокарда к ишемии. Однако между ischemic preconditioning и адаптацией сердца к ишемии на уровне целого организма существуют значительные различия. Так, кардиопротекторный эффект первого исчезает уже через 1 ч после прекращения последнего ишемического воздействия, в то время как защитный эффект адаптации к периодическим стрессорным воздействиям сохраняется в течение нескольких дней. Феномен адаптации к ишемии формируется в течение 30 мин, тогда как для формирования защитного эффекта адаптации к стрессу требуется по меньшей мере две недели. Важную роль в формировании долговременной адаптации играет повышение активности в кардиомиоцитах и эндотелиоцитах NO-синтазы.
Наряду с описанными механизмами, в процессе эволюции в клетках всех аэробных организмов сформировалась система противодействия токсическим эффектам свободных радикалов, образующихся в органах и тканях не только под влиянием реоксигенации, но и в нормальных условиях. Эта система получила название антиоксидантной (см. раздел 3.1.4).
15.3.2. Нарушения сократимости и насосной функции сердца
Сердечная недостаточность - неспособность сердца выполнять насосную функцию вследствие существенного снижения сократительной способности миокарда, а также поражения клапанов сердца или пороков развития системы кровообращения.
К основным причинам развития сердечной недостаточности относятся: 1) первичное поражение миокарда, приводящее к нарушению его сократимости. Возникает при ИБС (постинфарктный и атеросклеротический кардиосклероз), дилатационной кардиомиопатии, миокардитах, миокардиодистрофиях; 2) перегрузка давлением в фазу систолы. Это нарушение характеризуется увеличением работы сердца (например, при артериальной гипертензии или аортальном стенозе; 3) перегрузка объемом в фазу диастолы, сопровождающаяся увеличением работы сердца при аортальной или митральной недостаточности, дефекте межжелудочковой перегородки; 4) снижение наполнения желудочков (преимущественно диастолическая недостаточность). Развивается при гипертрофической кардиомиопатии, гипертоническом сердце (при отсутствии дилатации левого желудочка), изолированном митральном стенозе, констриктивном и экссудативном перикардите; 5) высокий сердечный выброс (при тиреотоксикозе, выраженной анемии и т.д.).
Классификация сердечной недостаточности. В зависимости от происхождения выделяют следующие формы сердечной недостаточности:
1) миокардиальная, которая обусловлена первичным поражением мышцы сердца физическими, химическими, биологическими факторами или дефицитом субстратов метаболизма;
2) перегрузочная, которая развивается на фоне повышенной работы миокарда по преодолению избыточного давления на путях изгнания крови из камер сердца (например, при гипертонической болезни); в связи с перегрузкой сердца увеличенным объемом кро-
ви (например, при наличии внутрисердечных шунтов) или при сочетании этих двух факторов (перегрузка объемом и давлением).
Очень часто течение миокардиальной сердечной недостаточности усугубляется присоединением ее перегрузочной формы.
По преимущественному поражению камер сердца сердечная недостаточность подразделяется на левожелудочковую, правожелудочковую и тотальную, каждая из которых, в свою очередь, может быть по характеру течения острой и хронической.
Левожелудочковая недостаточность встречается значительно чаще правожелудочковой, несмотря на то что левый желудочек более приспособлен к повышенным нагрузкам. Это связано с тем, что различные патологические состояния чаще приводят к перегрузке левого желудочка. При острой левожелудочковой недостаточности такими состояниями могут быть артериальная гипертензия (прежде всего гипертонический криз), инфаркт миокарда и обратимая ишемия левого желудочка, отрыв папиллярной мышцы с пролапсом митрального клапана и др. Острая левожелудочковая недостаточность клинически проявляется в виде сердечной астмы или отека легких. С морфологической точки зрения сердечная астма соответствует интерстициальному отеку, а отек легких - альвеолярному, или истинному, отеку легких.
Хроническая левожелудочковая недостаточность - это медленно формирующееся патологическое состояние, при котором нагрузка на левый желудочек превышает его способность совершать работу. Следует отметить, что этиологические факторы острой и хронической недостаточности сердца существенно различаются. Хроническая левожелудочковая недостаточность осложняет течение только хронических заболеваний сердца и сосудов.
Правожелудочковая недостаточность характеризуется развитием застойных явлений в большом круге кровообращения. При этом увеличивается кровенаполнение печени и соответственно ее размеры, нарушается экскреторная функция почек, происходит задержка воды в организме и появляются периферические отеки. Различают острую и хроническую правожелудочковую недостаточность. Наиболее частой причиной острой правожелудочковой недостаточности является распространение крупноочагового инфаркта левого желудочка на правые отделы сердца, реже - изолированный некроз миокарда правого желудочка. Очень часто причиной правожелудочковой недостаточности является легочная гипертензия, которая также может развиваться остро и хронически (см. раздел 15.2.2).
К весьма распространенным проявлениям перегрузки правого желудочка относятся легочное сердце и эмболия легочной артерии.
Понятие «легочное сердце» включает в себя легочную гипертензию, гипертрофию правого желудочка, его дилатацию и сердечную недостаточность. Однако в ряде случаев легочное сердце может проявляться только некоторыми из вышеназванных признаков. Так, длительное повышение артериального давления в легочных сосудах обычно приводит к гипертрофии правого желудочка с последующим развитием сердечной недостаточности по большому кругу. Если легочная гипертензия быстро прогрессирует, то гипертрофия правого желудочка не успевает развиться и дилатация непосредственно переходит в правожелудочковую недостаточность. Такая ситуация может иметь место и при тромбоэмболии легочной артерии, пневмотораксе, астматическом статусе и распространенной пневмонии, когда артериальное давление в малом круге повышается в течение нескольких суток или даже часов.
Синдром эмболии легочной артерии - это патологическое состояние, которое развивается при попадании эмболов в русло легочной артерии и характеризуется болями в грудной клетке, одышкой, цианозом с одновременным появлением тахикардии и признаков коллапса. Эмболия легочной артерии особенно опасна тем, что может вызвать рефлекторную остановку сердца и внезапную смерть больного. Если же пациент не погиб в первые минуты и часы, то спустя сутки-двое после эмболии формируется инфаркт легкого и так называемая инфаркт-пневмония, которая может послужить причиной развития легочного сердца.
Наиболее часто эмболия легочной артерии вызывается тромбами, мигрирующими из венозной системы и правых отделов сердца или из «левого сердца» при наличии дефектов межжелудочковой и межпредсердной перегородок. Наряду с этим причиной эмболии артерий малого круга кровообращения могут стать пузырьки воздуха или капли жира, попавшие в венозную систему при нарушении процедуры внутривенной инфузии или при травмах трубчатых костей.
При закупорке мелких ветвей легочной артерии выраженные клинические симптомы могут отсутствовать. Именно поэтому тромбоэмболия легочной артерии диагностируется только у 30% больных. Однако на вскрытии погибших пациентов, вместо ожидавшейся полной закупорки легочной артерии, иногда обнаруживали тромб
диаметром всего 3-4 мм в одной из ветвей легочной артерии (крупные сосуды), не закрывавший ни одну из них даже на треть. Этот факт подчеркивает важную роль нарушений нейрогуморальной регуляции гемодинамики в патогенезе легочной гипертензии.
По течению хроническая сердечная недостаточность делится на три стадии. Первая стадия - начальная. На этом этапе общие симптомы хронической сердечной недостаточности (одышка, тахикардия) появляются только во время физической нагрузки. Местные симптомы (цианоз) обычно отсутствуют. Во второй стадии появляются не только общие, но и местные симптомы. Помимо одышки и тахикардии при физической нагрузке появляются цианоз, признаки декомпенсации функционального состояния внутренних органов, асцит и анасарка. Третья стадия - конечная, дистрофическая. В этот период развивается тотальная застойная недостаточность, затрагивающая большой и малый круг кровообращения. Формируется почечная, печеночная и легочная недостаточность. Третья стадия является терминальной стадией хронической сердечной недостаточности.
Механизмы компенсации гемодинамики при сердечной недостаточности
Здоровый организм обладает многообразными механизмами, обеспечивающими своевременную разгрузку сосудистого русла от избытка жидкости. При сердечной недостаточности «включаются» компенсаторные механизмы, направленные на сохранение нормальной гемодинамики. Эти механизмы в условиях острой и хронической недостаточности кровообращения имеют много общего, вместе с тем между ними отмечаются существенные различия.
Как и при острой, так и при хронической сердечной недостаточности все эндогенные механизмы компенсации гемодинамических нарушений можно подразделить на интракардиальные: компенсаторная гиперфункция сердца (механизм Франка-Старлинга, гомеометрическая гиперфункция), гипертрофия миокарда и экстракардиальные: разгрузочные рефлексы Бейнбриджа, Парина, Китаева, активация выделительной функции почек, депонирование крови в печени и селезенке, потоотделение, испарение воды со стенок легочных альвеол, активация эритропоэза и др. Такое деление в некоторой степени условно, поскольку реализация как интра-, так и экстракардиальных механизмов находится под контролем нейрогуморальных регуляторных систем.
Механизмы компенсации гемодинамических нарушений при острой сердечной недостаточности. На начальной стадии систолической дисфункции желудочков сердца включаются интракардиальные факторы компенсации сердечной недостаточности, важнейшим из которых является механизм Франка-Старлинга (гетерометрический механизм компенсации, гетерометрическая гиперфункция сердца). Реализацию его можно представить следующим образом. Нарушение сократительной функции сердца влечет за собой уменьшение ударного объема крови и гипоперфузию почек. Это способствует активации РААС, вызывающей задержку воды в организме и увеличение объема циркулирующей крови. В условиях возникшей гиперволемии происходит усиленный приток венозной крови к сердцу, увеличение диастолического кровенаполнения желудочков, растяжение миофибрилл миокарда и компенсаторное повышение силы сокращения сердечной мышцы, которое обеспечивает прирост ударного объема. Однако если конечное диастолическое давление повышается более чем на 18-22 мм рт.ст., возникает чрезмерное перерастяжение миофибрилл. В этом случае компенсаторный механизм Франка-Старлинга перестает действовать, а дальнейшее увеличение конечного диастолического объема или давления вызывает уже не подъем, а снижение ударного объема.
Наряду с внутрисердечными механизмами компенсации при острой левожелудочковой недостаточности запускаются разгрузочные экстракардиальные рефлексы, способствующие возникновению тахикардии и увеличению минутного объема крови (МОК). Одним из наиболее важных сердечно-сосудистых рефлексов, обеспечивающих увеличение МОК, является рефлекс Бейнбриджа увеличение частоты сердечных сокращений в ответ на увеличение объема циркулирующей крови. Этот рефлекс реализуется при раздражении механорецепторов, локализованных в устье полых и легочных вен. Их раздражение передается на центральные симпатические ядра продолговатого мозга, в результате чего происходит повышение тонической активности симпатического звена вегетативной нервной системы, и развивается рефлекторная тахикардия. Рефлекс Бейнбриджа направлен на увеличение минутного объема крови.
Рефлекс Бецольда-Яриша - это рефлекторное расширение артериол большого круга кровообращения в ответ на разражение механо- и хеморецепторов, локализованных в желудочках и предсердиях.
В результате возникает гипотония, которая сопровождается бра-
дикардией и временной остановкой дыхания. В реализации этого рефлекса принимают участие афферентные и эфферентные волокна n. vagus. Этот рефлекс направлен на разгрузку левого желудочка.
К числу компенсаторных механизмов при острой сердечной недостаточности относится и повышение активности симпатоадреналовой системы, одним из звеньев которого является высвобождение норадреналина из окончаний симпатических нервов, иннервирующих сердце и почки. Наблюдаемое при этом возбуждение β-адренорецепторов миокарда ведет к развитию тахикардии, а стимуляция подобных рецепторов в клетках ЮГА вызывает усиленную секрецию ренина. Другим стимулом секреции ренина является снижение почечного кровотока в результате вызванной катехоламинами констрикции артериол почечных клубочков. Компенсаторное по своей природе усиление адренергического влияния на миокард в условиях острой сердечной недостаточности направлено на увеличение ударного и минутного объемов крови. Положительный инотропный эффект оказывает также ангиотензин-II. Однако эти компенсаторные механизмы могут усугубить сердечную недостаточность, если повышенная активность адренергической системы и РААС сохраняется достаточно продолжительное время (более 24 ч).
Все сказанное о механизмах компенсации сердечной деятельности в одинаковой степени относится как к лево-, так и к правожелудочковой недостаточности. Исключением является рефлекс Парина, действие которого реализуется только при перегрузке правого желудочка, наблюдаемой при эмболии легочной артерии.
Рефлекс Ларина - это падение артериального давления, вызванное расширением артерий большого круга кровообращения, снижением минутного объема крови в результате возникающей брадикардии и уменьшением объема циркулирующей крови из-за депонирования крови в печени и селезенке. Кроме того, для рефлекса Парина характерно появление одышки, связанной с наступающей гипоксией мозга. Полагают, что рефлекс Парина реализуется за счет усиления тонического влияния n.vagus на сердечно-сосудистую систему при эмболии легочных артерий.
Механизмы компенсации гемодинамических нарушений при хронической сердечной недостаточности. Основным звеном патогенеза хронической сердечной недостаточности является, как известно, постепенно нарастающее снижение сократительной функции ми-
окарда и падение сердечного выброса. Происходящее при этом уменьшение притока крови к органам и тканям вызывает гипоксию последних, которая первоначально может компенсироваться усиленной тканевой утилизацией кислорода, стимуляцией эритропоэза и т.д. Однако этого оказывается недостаточно для нормального кислородного обеспечения органов и тканей, и нарастающая гипоксия становится пусковым механизмом компенсаторных изменений гемодинамики.
Интракардиальные механизмы компенсации функции сердца. К ним относятся компенсаторная гиперфункция и гипертрофия сердца. Эти механизмы являются неотъемлемыми компонентами большинства приспособительных реакций сердечно-сосудистой системы здорового организма, но в условиях патологии могут превратиться в звено патогенеза хронической сердечной недостаточности.
Компенсаторная гиперфункция сердца выступает как важный фактор компенсации при пороках сердца, артериальной гипертензии, анемии, гипертонии малого круга и других заболеваниях. В отличие от физиологической гиперфункции она является длительной и, что существенно, непрерывной. Несмотря на непрерывность, компенсаторная гиперфункция сердца может сохраняться в течение многих лет без явных признаков декомпенсации насосной функции сердца.
Увеличение внешней работы сердца, связанное с подъемом давления в аорте (гомеометрическая гиперфункция), приводит к более выраженному возрастанию потребности миокарда в кислороде, чем перегрузка миокарда, вызванная повышением объема циркулирующей крови (гетерометрическая гиперфункция). Иными словами, для осуществления работы в условиях нагрузки давлением мышца сердца использует гораздо больше энергии, чем для выполнения той же работы, связанной с нагрузкой объемом, а следовательно, при стойкой артериальной гипертензии гипертрофия сердца развивается быстрее, чем при увеличении объема циркулирующей крови. Например, при физической работе, высотной гипоксии, всех видах клапанной недостаточности, артериовенозных фистулах, анемии гиперфункция миокарда обеспечивается за счет увеличения минутного объема сердца. При этом систолическое напряжение миокарда и давление в желудочках возрастают незначительно, и гипертрофия развивается медленно. В то же время при гипертонической болезни, гипертензии малого круга, стено-
зах клапанных отверстий развитие гиперфункции связано с повышением напряжения миокарда при незначительно измененной амплитуде сокращений. В этом случае гипертрофия прогрессирует достаточно быстро.
Гипертрофия миокарда - это увеличение массы сердца за счет увеличения размеров кардиомиоцитов. Существуют три стадии компенсаторной гипертрофии сердца.
Первая, аварийная, стадия характеризуется, прежде всего, увеличением интенсивности функционирования структур миокарда и, по сути, представляет собой компенсаторную гиперфункцию еще не гипертрофированного сердца. Интенсивность функционирования структур - это механическая работа, приходящаяся на единицу массы миокарда. Увеличение интенсивности функционирования структур закономерно влечет за собой одновременную активацию энергообразования, синтеза нуклеиновых кислот и белка. Указанная активация синтеза белка происходит таким образом, что вначале увеличивается масса энергообразующих структур (митохондрий), а затем - масса функционирующих структур (миофибрилл). В целом увеличение массы миокарда приводит к тому, что интенсивность функционирования структур постепенно возвращается к нормальному уровню.
Вторая стадия - стадия завершившейся гипертрофии - характеризуется нормальной интенсивностью функционирования структур миокарда и соответственно нормальным уровнем энергообразования и синтеза нуклеиновых кислот и белков в ткани сердечной мышцы. При этом потребление кислорода на единицу массы миокарда остается в границах нормы, а потребление кислорода сердечной мышцей в целом увеличено пропорционально возрастанию массы сердца. Увеличение массы миокарда в условиях хронической сердечной недостаточности происходит за счет активации синтеза нуклеиновых кислот и белков. Пусковой механизм этой активации изучен недостаточно. Считается, что определяющую роль здесь играет усиление трофического влияния симпатоадреналовой системы. Эта стадия процесса совпадает с длительным периодом клинической компенсации. Содержание АТФ и гликогена в кардиомиоцитах также находится при этом в пределах нормы. Подобные обстоятельства придают относительную устойчивость гиперфункции, но вместе с тем не предотвращают исподволь развивающихся в данной стадии нарушений обмена и структуры миокарда. Наиболее ранними признаками таких нарушений являются
значительное увеличение концентрации лактата в миокарде, а также умеренно выраженный кардиосклероз.
Третья стадия прогрессирующего кардиосклероза и декомпенсации характеризуется нарушением синтеза белков и нуклеиновых кислот в миокарде. В результате нарушения синтеза РНК, ДНК и белка в кардиомиоцитах наблюдается относительное уменьшение массы митохондрий, что ведет к торможению синтеза АТФ на единицу массы ткани, снижению насосной функции сердца и прогрессированию хронической сердечной недостаточности. Ситуация усугубляется развитием дистрофических и склеротических процессов, что способствует появлению признаков декомпенсации и тотальной сердечной недостаточности, завершающейся гибелью пациента. Компенсаторная гиперфункция, гипертрофия и последующая декомпенсация сердца - это звенья единого процесса.
Механизм декомпенсации гипертрофированного миокарда включает следующие звенья:
1. Процесс гипертрофии не распространяется на коронарные сосуды, поэтому число капилляров на единицу объема миокарда в гипертрофированном сердце уменьшается (рис. 15-11). Следовательно, кровоснабжение гипертрофированной сердечной мышцы оказывается недостаточным для выполнения механической работы.
2. Вследствие увеличения объема гипертрофированных мышечных волокон уменьшается удельная поверхность клеток, в связи с
 Рис. 5-11. Гипертрофия
миокарда: 1 - миокард здорового взрослого; 2 - гипертрофированный
миокард взрослого (масса 540 г); 3 - гипертрофированный миокард
взрослого (масса 960 г)
Рис. 5-11. Гипертрофия
миокарда: 1 - миокард здорового взрослого; 2 - гипертрофированный
миокард взрослого (масса 540 г); 3 - гипертрофированный миокард
взрослого (масса 960 г)
этим ухудшаются условия для поступления в клетки питательных веществ и выделения из кардиомиоцитов продуктов метаболизма.
3. В гипертрофированном сердце нарушается соотношение между объемами внутриклеточных структур. Так, увеличение массы митохондрий и саркоплазматического ретикулума (СПР) отстает от увеличения размеров миофибрилл, что способствует ухудшению энергоснабжения кардиомиоцитов и сопровождается нарушением аккумуляции Са2+ в СПР. Возникает Са2+-перегрузка кардиомиоцитов, что обеспечивает формирование контрактуры сердца и способствует уменьшению ударного объема. Кроме того, Са2+-перегрузка клеток миокарда повышает вероятность возникновения аритмий.
4. Проводящая система сердца и вегетативные нервные волокна, иннервирующие миокард, не подвергаются гипертрофии, что также способствует возникновению дисфункции гипертрофированного сердца.
5. Активируется апоптоз отдельных кардиомиоцитов, что способствует постепенному замещению мышечных волокон соединительной тканью (кардиосклероз).
В конечном итоге гипертрофия утрачивает приспособительное значение и перестает быть полезной для организма. Ослабление сократительной способности гипертрофированного сердца происходит тем скорее, чем сильнее выражены гипертрофия и морфологические изменения в миокарде.
Экстракардиальные механизмы компенсации функции сердца. В отличие от острой сердечной недостаточности роль рефлекторных механизмов экстренной регуляции насосной функции сердца при хронической сердечной недостаточности сравнительно невелика, поскольку нарушения гемодинамики развиваются постепенно на протяжении нескольких лет. Более или менее определенно можно говорить о рефлексе Бейнбриджа, который «включается» уже на стадии достаточно выраженной гиперволемии.
Особое место среди «разгрузочных» экстракардиальных рефлексов занимает рефлекс Китаева, который «запускается» при митральном стенозе. Дело в том, что в большинстве случаев проявления правожелудочковой недостаточности связаны с застойными явлениями в большом круге кровообращения, а левожелудочковой - в малом. Исключение составляет стеноз митрального клапана, при котором застойные явления в легочных сосудах вызваны не декомпенсацией левого желудочка, а препятствием току крови через
левое атриовентрикулярное отверстие - так называемым «первым (анатомическим) барьером». При этом застой крови в легких способствует развитию правожелудочковой недостаточности, в генезе которой рефлекс Китаева играет важную роль.
Рефлекс Китаева - это рефлекторный спазм легочных артериол в ответ на повышение давления в левом предсердии. В результате возникает «второй (функциональный) барьер», который первоначально играет защитную роль, предохраняя легочные капилляры от чрезмерного переполнения кровью. Однако затем этот рефлекс приводит к выраженному повышению давления в легочной артерии - развивается острая легочная гипертензия. Афферентное звено этого рефлекса представлено n. vagus, a эфферентное - симпатическим звеном вегетативной нервной системы. Негативной стороной данной приспособительной реакции является подъем давления в легочной артерии, приводящий к увеличению нагрузки на правое сердце.
Однако ведущую роль в генезе долговременной компенсации и декомпенсации нарушенной сердечной функции играют не рефлекторные, а нейрогуморальные механизмы, важнейшим из которых является активация симпатоадреналовой системы и РААС. Говоря об активации симпатоадреналовой системы у пациентов с хронической сердечной недостаточностью, нельзя не указать, что у большинства из них уровень катехоламинов в крови и моче находится в пределах нормы. Этим хроническая сердечная недостаточность отличается от острой сердечной недостаточности.
Механизмы декомпенсации сердечной недостаточности
Параллельно с интра- и экстракардиальными компенсаторными изменениями, которые развиваются при сердечной недостаточности, появляются и постепенно прогрессируют повреждения сердечной мышцы, приводящие к снижению ее сократительной способности. На определенной стадии процесса такие явления могут быть обратимыми. При продолжении или усилении действия причинного фактора, вызвавшего сердечную недостаточность, а также при срыве механизмов компенсации развиваются необратимые диффузные изменения миокарда с характерной клинической картиной декомпенсированной сердечной недостаточности.
Патогенез сердечной недостаточности представляется следующим образом. Многочисленный ряд примеров патологии сер-
дечной деятельности (кардиомиопатии, нарушения коронарной перфузии и др.) индуцирует кислородное голодание миокарда. Известно, что в условиях нормального кровоснабжения важным энергетическим субстратом для сердечной мышцы являются свободные жирные кислоты, глюкоза и молочная кислота. Гипоксия приводит к нарушению процессов аэробного окисления субстратов в цикле Кребса, к угнетению окисления НАДН в дыхательной цепи митохондрий. Все это способствует накоплению недоокисленных продуктов метаболизма свободных жирных кислот и глюкозы (ацил-КоА, лактат). Усиленное образование ацил-КоА в кардиомиоцитах негативно сказывается на энергетическом метаболизме клетки. Дело в том, что ацил-КоА является ингибитором аденилаттранслоказы - фермента, который осуществляет транспорт АТФ из митохондрий в саркоплазму. Аккумуляция ацил-КоА приводит к нарушению этого транспорта, усугубляя энергетический дефицит в клетке.
Единственным источником энергии для кардиомиоцитов становится анаэробный гликолиз, интенсивность которого в условиях гипоксии резко возрастает. Однако «коэффициент полезного действия» анаэробного гликолиза по сравнению с эффективностью энергопродукции в цикле Кребса намного ниже. В силу этого анаэробный гликолиз не в состоянии полностью возместить энергетические потребности клетки. Так, при анаэробном расщеплении одной молекулы глюкозы образуются всего две молекулы АТФ, в то время как при окислении глюкозы до углекислого газа и воды - 32 молекулы АТФ. Нехватка высокоэнергетических фосфатов (АТФ и креатинфосфата) приводит к нарушению энергозависимого процесса удаления ионов кальция из саркоплазмы кардиомиоцитов и возникновению кальциевой перегрузки миокарда.
В норме увеличение концентрации Ca2+ в кардиомиоцитах вызывает образование мостиков между цепочками актина и миозина, что является основой сокращения клеток. Вслед за этим происходит удаление избытка ионов кальция из саркоплазмы и развитие диастолы. Кальциевая перегрузка клеток миокарда при его ишемии ведет к остановке процесса сокращения - расслабления в стадии систолы, формируется контрактура миокарда - состояние, при котором кардиомиоциты перестают расслабляться. Возникшая зона асистолии характеризуется повышенным тканевым напряжением, что ведет к сдавлению коронарных сосудов и связанному с этим усугублению дефицита коронарного кровотока.
Ионы Са активируют фосфолипазу А2, которая катализирует расщепление фосфолипидов. В результате этого образуются одна молекула свободной жирной кислоты и одна молекула лизофосфатида. Свободные жирные кислоты обладают детергентоподобным действием и в случае избыточного их накопления в миокарде могут повреждать мембраны кардиомиоцитов. Еще более выраженный кардиотоксический эффект оказывают лизофосфатиды. Особенно токсичен лизофосфатидилхолин, который может провоцировать аритмии. В настоящее время роль свободных жирных кислот и лизофосфатидов в патогенезе ишемического повреждения сердца никем не оспаривается, однако молекулярная природа необратимого повреждения кардиомиоцитов не сводится только к накоплению этих веществ в клетках сердечной мышцы. Кардиотоксическими свойствами могут обладать и другие продукты метаболизма, например активные формы кислорода (АФК).
К АФК относятся супероксидный радикал (O2*-) и гидроксильный радикал O2*-, которые обладают высокой окислительной активностью. Источником АФК в кардиомиоцитах является дыхательная цепь митохондрий и прежде всего цитохромы, которые в условиях гипоксии переходят в восстановленное состояние и могут быть донорами электронов, «передавая» их молекулам кислорода с образованием не молекулы воды, как это происходит в норме, а супероксидного радикала (O2*-). Кроме того, образование свободных радикалов катализируется ионами металлов с переменной валентностью (прежде всего ионами железа), которые всегда присутствуют в клетке. АФК взаимодействуют с молекулами белков и полиненасыщенных жирных кислот, превращая их в свободные радикалы. Вновь образованные радикалы могут, в свою очередь, взаимодействовать с другими молекулами белков и жирных кислот, индуцируя дальнейшее образование свободных радикалов. Таким образом, реакция может принимать цепной и разветвленный характер. Если пероксидации подвергаются белки ионных каналов, то происходит нарушение процессов ионного транспорта. Если гидроперекиси образуются из молекул ферментов, последние теряют свою каталитическую активность.
Образование гидроперекисей полиненасыщенных жирных кислот, входящих в молекулярную структуру мембранных фосфолипидов, способствует изменению биологических свойств мембран. В отличие от жирных кислот гидроперекиси являются водорастворимыми веществами, и появление их в структуре гидрофобного
фосфолипидного матрикса клеточных мембран приводит к формированию пор, пропускающих ионы и молекулы воды. Кроме того, изменяется активность мембраносвязанных ферментов.
Процесс возникновения гидроперекисей жирных кислот является одним из звеньев перекисного окисления липидов (ПОЛ), которое включает в себя свободнорадикальное образование альдегидов и кетонов - продуктов ПОЛ. Согласно концепции Ф.З. Меерсона, продукты ПОЛ обладают кардиотоксическими свойствами, их накопление в клетке приводит к повреждению сарколеммы, а также лизосомальных и митохондриальных мембран. На заключительном этапе повреждения, предшествующем гибели клеток, особая роль отводится активации протеолитических ферментов. Обычно эти энзимы находятся в цитоплазме кардиомиоцитов в неактивном состоянии или локализованы внутри лизосом, мембраны которых изолируют их от структурных элементов клетки. В связи с этим в норме протеазы не оказывают цитотоксического действия. В условиях ишемии перегрузка кардиомиоцитов ионами кальция и закисление цитоплазмы за счет накопления лактата приводят к активации внутриклеточных протеаз. Кроме того, повышение проницаемости лизосомальных мембран под действием фосфолипаз и продуктов ПОЛ способствует выходу активных протеолитических ферментов в саркоплазму. Конечным звеном этой патогенетической цепочки является некроз кардиомиоцитов в зоне ишемии и их аутолиз.
Важно отметить, что первыми погибают только те кардиомиоциты, которые отличаются высокой интенсивностью энергетического метаболизма и соответственно повышенной потребностью в кислороде. В то же время фибробласты и клетки проводящей системы менее зависимы от доставки кислорода и сохраняют свою жизнеспособность. Функциональная активность фибробластов обеспечивает процессы рубцевания.
Клетки проводящей системы, сохраняя жизнеспособность в условиях кислородного голодания, существенно изменяют свои электрофизиологические характеристики, что может способствовать возникновению аритмий. В результате повреждения мембран и снижения образования АТФ изменяется активность К+/ Na+-АТФазы, что сопровождается усиленным поступлением натрия в кардиомиоциты и выходом из них калия. Это увеличивает электрическую нестабильность миокарда и способствует развитию аритмий.
Гипоксическая сократительная дисфункция сердца усугубляется нарушением процессов нейрогуморальной регуляции функционального состояния миокарда. Сердечные боли, приступы аритмии и другие нарушения являются для организма стрессором, т.е. воздействием чрезмерной силы, на которое организм, как и на любое стрессорное воздействие, реагирует активацией симпатоадреналовой системы. При этом происходит выброс катехоламинов из надпочечников и симпатических нервных терминалей. Однако, как и любой другой компенсаторный процесс, активация симпатоадреналовой системы в конце концов приобретает негативную окраску. Наступает период декомпенсации. Схематично последовательность событий представлена на рисунке 15-12.
В настоящее время установлено, что при хронической активации симпатоадреналовой системы происходят постепенная Са2+- перегрузка кардиомиоцитов и их контрактура, нарушается целостность сарколеммы. При гиперактивации адренергической системы формируется электрическая нестабильность миокарда. Последняя способствует возникновению фибрилляции желудочков сердца,
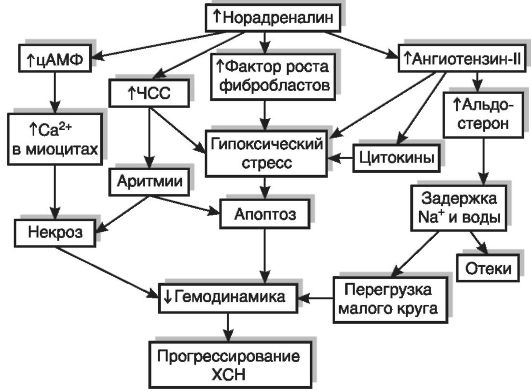 Рис. 15-12. Роль
симпатоадреналовой и ренин-ангиотензин-альдостероновой систем в
патогенезе хронической сердечной недостаточности: ХСН - хроническая
сердечная недостаточность; ЧСС - частота сердечных сокращений
Рис. 15-12. Роль
симпатоадреналовой и ренин-ангиотензин-альдостероновой систем в
патогенезе хронической сердечной недостаточности: ХСН - хроническая
сердечная недостаточность; ЧСС - частота сердечных сокращений
поэтому каждый третий пациент при хронической сердечной недостаточности погибает внезапно, иногда сердечная смерть наступает на фоне внешнего благополучия и положительной клинической динамики.
Адренергическая тахикардия сопровождается повышением потребности миокарда в кислороде, что наряду с Са2+-перегрузкой еще больше усугубляет энергетический дефицит в клетках миокарда. Включается защитно-приспособительный механизм, получивший название гибернации (спячки) кардиомиоцитов. Часть клеток перестает сокращаться и отвечать на внешние стимулы, потребляя при этом минимум энергии и экономя кислород для активно сокращающихся кардиомиоцитов. Таким образом, количество обеспечивающих насосную функцию сердца клеток миокарда может существенно уменьшиться, способствуя усугублению сердечной недостаточности.
Кроме того, гиперактивация симпатоадреналовой системы усиливает секрецию ренина почками, выступая в роли стимулятора РААС. Образующийся ангиотензин-II оказывает ряд негативных эффектов на сердечно-сосудистую систему. Он способствует увеличению адренореактивности сердца и сосудов, усиливая тем самым кардиотоксическое действие катехоламинов. Одновременно этот пептид увеличивает периферическое сопротивление кровеносных сосудов, что, безусловно, способствует увеличению постнагрузки на сердце и весьма негативно сказывается на гемодинамике. Кроме того, ангиотензин-II может самостоятельно или через активацию образования цитокинов (биологически активные вещества белковой природы, образующиеся в миокарде и других тканях) стимулировать программируемую гибель кардиомиоцитов («апоптоз»).
Наряду с отмеченным, повышение уровня ангиотензина-II негативно сказывается на состоянии водно-солевого гомеостаза, поскольку этот пептид активирует секрецию альдостерона. В результате в организме задерживается избыточное количество воды и натрия. Задержка натрия повышает осмолярность крови, в ответ на которую происходит активация секреции антидиуретического гормона, что приводит к уменьшению диуреза и еще большей гидратации организма. В итоге повышается объем циркулирующей крови и увеличивается преднагрузка на сердце. Гиперволемия ведет к раздражению механорецепторов, локализованных в устье полых и легочных вен, «включается» рефлекс Бейнбриджа, возникает
рефлекторная тахикардия, что еще больше увеличивает нагрузку на миокард и потребность сердечной мышцы в кислороде.
Создается «порочный круг», разорвать который можно только с помощью определенных фармакологических воздействий. Ко всему этому присоединяется повышение гидростатического давления в микрососудистом русле, что способствует выходу жидкой части крови в ткани и формированию отеков. Последние сдавливают ткани, что усугубляет нарушение микроциркуляции и еще больше усиливает тканевую гипоксию. При дальнейшем прогрессировании недостаточности кровообращения нарушаются и другие виды обмена, в том числе и белковый, что приводит к дистрофическим изменениям в органах и тканях, нарушению их функции. В конечной стадии хронической сердечной недостаточности развиваются кахексия, маскируемая отеками, гипопротеинемия, появляются признаки почечной и печеночной декомпенсации.
15.3.3. Некоронарогенная патология сердца
Некоронарогенная патология сердечной мышцы неревматической этиологии
Миокардиодистрофии - это группа некоронарогенных заболеваний миокарда, возникающих под влиянием экстракардиальных факторов, основными проявлениями которых служат нарушения метаболизма и сократительной функции сердечной мышцы. Понятие миокардиодистрофии было введено в клиническую практику в 1936 г. академиком Г.Ф. Лангом. В качестве причин миокардиодистрофии рассматриваются анемия, недостаточное питание, авитаминоз, поражения печени и почек, нарушения отдельных видов обмена веществ, заболевания эндокринной системы, системные заболевания, интоксикации, физическое перенапряжение, инфекции.
В развитии миокардиодистрофии выделяют три стадии. I стадия - стадия адаптивной гиперфункции миокарда. Для нее характерен гиперкинетический вариант кровообращения, возникающий вследствие повышения тонуса симпатического и подавления парасимпатического звеньев вегетативной нервной системы. Во II стадии формируются обменно-структурные изменения, приводящие к нарушению функции сердца и появлению клинических признаков недостаточности кровообращения. В III стадии развиваются тяжелые нарушения обмена веществ, структуры и функции сердечной мышцы, проявляющиеся стойкой недостаточностью кровообращения.
Миокардиты (неревматической этиологии) - это воспалительные поражения сердечной мышцы, возникающие вследствие прямого или опосредованного аллергическими реакциями повреждающего действия инфекционных или неинфекционных агентов. Миокардиты развиваются при бактериальных, риккетсиозных, спирохетозных, грибковых, вирусных и других инфекциях. К неинфекционным факторам, вызывающим миокардиты, относят некоторые лекарственные препараты - антибиотики и сульфаниламиды, лечебные сыворотки и вакцины.
Особое место среди различных видов воспалительных поражений миокарда занимает идиопатический миокардит Абрамова-Финдлена. Данная форма заболевания характеризуется тяжелым течением с развитием кардиомегалии и выраженной сердечной недостаточности. Причина возникновения этого заболевания невыяснена. Обсуждается возможная роль вирусной инфекции и аллергических реакций, возникающих как после перенесенной инфекции, так и после приема лекарственных препаратов. Прогноз при идиопатическом миокардите неблагоприятен. Больные погибают быстро, в сроки от 2-3 месяцев до года. Причиной смерти обычно бывают нарушения сердечного ритма или сердечная недостаточность.
Основные проявления миокардиодистрофии и миокардитов, несмотря на их различную этиологию, имеют много общего и определяются выраженностью структурно-функциональных изменений сердца. Обе группы заболеваний характеризуются кардиалгией, симптомами сердечной недостаточности (тахикардия, одышка, акроцианоз, отеки), а также нарушениями сердечного ритма и проводимости. При миокардитах, поскольку это воспалительный процесс, выявляются лейкоцитоз, эозинофилия, увеличение СОЭ, а при миокардиодистрофии подобные изменения не обнаруживаются.
Кардиомиопатии. Термин «кардиомиопатия» введен W. Brigden в 1957 г. для обозначения некоронарогенных заболеваний миокарда неизвестной этиологии. В 1968 г. рабочая группа ВОЗ определила кардиомиопатии как заболевания, характеризующиеся кардиомегалией и недостаточностью кровообращения. Кардиомиопатии подразделяются на дилатационные, гипертрофические и рестриктивные.
Дилатационная кардиомиопатия характеризуется значительным увеличением всех камер сердца и нарушением его систолической функции. Возможно, дилатационная кардиомиопатия является наследственно-детерминированным заболеванием. Так, ретро-
спективный анализ историй болезней 169 пациентов с дилатационной кардиомиопатией, проведенный в США, позволил установить положительный семейный анамнез в 7% случаев. Кроме того, были описаны случаи аутосомно-доминантного и аутосомнорецессивного наследования.
При патолого-анатомическом исследовании сердца выявляется значительная дилатация полостей. Масса сердца намного увеличена по сравнению с нормальной и может достигать 800-1000 г. Единственно возможное радикальное лечение дилатационной кардиомиопатии заключается в проведении трансплантации сердца. Симптоматическая терапия направлена на лечение сердечной недостаточности.
Гипертрофическая кардиомиопатия характеризуется выраженной гипертрофией миокарда с преимущественным нарушением его диастолической функции. Гипертрофическая кардиомиопатия относится к генетически обусловленным заболеваниям с аутосомнодоминантным характером наследования и высокой степенью пенетрантности. Течение заболевания может напоминать клапанные пороки сердца, гипертрофию миокарда при артериальной гипертензии или ишемической болезни сердца. Часто истинный диагноз устанавливается только во время патологоанатомического исследования, когда выявляются асимметричная гипертрофия межжелудочковой перегородки и уменьшение полости левого желудочка.
Патогенез гемодинамических изменений при гипертрофической кардиомиопатии обусловлен нарушениями диастолической функции левого желудочка, движения стенок которого становятся некоординированными и неравномерными. Гипертрофия миокарда в сочетании с гипоксией сердечной мышцы становится причиной электрофизиологической гетерогенности сердца и создает условия для возникновения аритмий. Именно поэтому у пациентов с гипертрофической кардиомиопатией чаще, чем при других видах кардиомиопатии, наступает фибрилляция и внезапная смерть.
Рестриктивная кардиомиопатия объединяет два заболевания, которые ранее описывались самостоятельно: эндомиокардиальный фиброз и фибропластический париетальный эндокардит Леффлера. Основным звеном патогенеза нарушений гемодинамики при рестриктивной кардиомиопатии, как и при гипертрофической кардиомиопатии, является нарушение диастолической функции миокарда. Однако при гипертрофической кардиомиопатии это проис-
ходит в результате перегрузки кардиомиоцитов ионами кальция, а при рестриктивной кардиомиопатии связано с утолщением эндокарда и фиброзным перерождением миокарда. Для рестриктивной кардиомиопатии характерны образование тромбов в полостях желудочков и поражение митрального клапана в виде прорастания створок фиброзной тканью с последующей кальцификацией.
Патогенетически обоснованное лечение рестриктивной кардиомиопатии должно быть направлено на борьбу с сердечной недостаточностью. Хирургическое лечение заключается в иссечении плотной фиброзной ткани и протезировании клапанов по показаниям.
Стрессорная кардиомиопатия - особая форма поражения миокарда. Характеризуется диффузными изменениями, которые возникают после длительного, многочасового экстремального воздействия на организм. В 1974 г. шведский физиолог Johansson для обозначения стрессорного повреждения сердца предложил использовать термин «стрессорная кардиомиопатия». Это заболевание характеризуется появлением дистрофических изменений в клетках миокарда вплоть до некроза отдельных кардиомиоцитов. В начале 1970-х гг. американским физиологом Бернардом Лауном было установлено, что стрессорная кардиомиопатия сопровождается снижением электрической стабильности сердца. Возникающая в результате стресса электрическая нестабильность сердца способствует возникновению тяжелых желудочковых аритмий, которые могут закончиться внезапной сердечной смертью (Б. Лаун). На вскрытии у таких пациентов при макроскопическом исследовании сердца очень часто не удается идентифицировать никаких патоморфологических изменений. Причиной стрессорной электрической нестабильности сердца является гиперактивация симпатоадреналовой системы. Патогенез стрессорного повреждения сердца очень сходен с патогенезом его ишемического повреждения.
Инфекционный эндокардит - заболевание, возникающее в результате инфекционного поражения эндокарда. Термин «инфекционный эндокардит» применяется с 1966 г. вместо ранее употреблявшихся терминов «бактериальный» и «затяжной септический эндокардит».
Основными возбудителями заболевания считаются зеленящий стрептококк и золотистый стафилококк. На долю этих микроорганизмов приходится около 80% случаев инфекционного эндокардита. Всего выявлено 119 микроорганизмов, способных привести
к развитию этого заболевания, которое начинается с сепсиса. При этом происходит бактериальное поражение клапанов сердца, чаще аортального и реже - митрального, трикуспидального и клапана легочной артерии. После внедрения микроорганизмов в ткань эндокарда происходит дополнительное отложение тромбоцитов и фибрина в этой зоне, что в определенной мере ограничивает контакт возбудителя с внутренней средой организма.
Формирование локальных очагов инфекции считается пусковым механизмом ряда патогенетически значимых процессов в организме, для которых характерны: 1) постоянное поступление инфекционного агента в кровеносное русло с развитием эпизодов бактериемии, вирусемии, проявляющееся усталостью, снижением массы тела, потерей аппетита, лихорадкой, развитием анемии, спленомегалией; 2) местное развитие микробных вегетаций, вызывающее нарушение функции сердца, абсцессы фиброзного клапанного кольца, перикардиты, аневризмы синуса Вальсальвы, перфорацию клапана; 3) отрыв фрагментов микробных вегетаций, попадание их в системный кровоток с развитием бактериальных эмболий.
Заболевания перикарда
Перикардиты - воспалительное поражение серозных оболочек, ограничивающих перикардиальную полость. По этиологии перикардиты подразделяют на инфекционные (туберкулезный, бактериальный, вирусный) и асептические (постинфарктный перикардит Дресслера, уремический и др.). Все перикардиты принято подразделять на экссудативные и сухие (слипчивые), патогенез которых имеет существенные различия.
Экссудативный перикардит обычно протекает остро и начинается с повышения температуры, развития лейкоцитоза и увеличения СОЭ. К этим симптомам воспаления присоединяются патологические проявления, связанные с накоплением экссудата в плевральной полости. В нормальных условиях в полости перикарда находится 2-5 мл жидкости. При выраженной экссудации и быстром увеличении количества жидкости в полости перикарда ее объем может составить 250-400 мл. Известны случаи, когда у хронических больных во время однократной пункции удаляли до 10 л экссудата. Если экссудат накапливается очень быстро, возникает опасность резкого нарушения гемодинамики - тампонады серд-
ца, которая развивается в результате сдавления сердца выпотом, с последующим падением сердечного выброса и формированием острой сердечной недостаточности. Она проявляется выраженной нарастающей одышкой до 40-60 дыханий в минуту, частым нитевидным пульсом, снижением систолического артериального давления.
Слипчивый перикардит часто называют констриктивным перикардитом, поскольку он характеризуется сдавлением миокарда патологически измененной околосердечной сумкой. Сухой перикардит может развиться после экссудативного (часто недиагностированного) перикардита, однако бывает и первичным. По мере развития заболевания в полости перикарда образуются вначале нежные спайки, которые не влияют на работу сердца и общую гемодинамику, но могут провоцировать болевой синдром. Изменение гемодинамики связано в первую очередь с нарушением заполнения сердца кровью в период диастолы. Это происходит вследствие сдавления фиброзной тканью верхней и нижней полых вен. Мощные спайки могут сдавливать и миокард, затрудняя его полное расслабление в фазу диастолы. Позже спайки, достигающие толщины 1 см и более, могут полностью облитерировать полость перикарда. На заключительных этапах заболевания в рубцовой ткани откладываются соли извести, возникает кальциноз, формируется «панцирное сердце».
Заболевания ревматической природы
Ревматизм - это системное заболевание соединительной ткани.
Происхождение этого заболевания продолжает вызывать споры и дискуссии, поскольку оно поражает всю соединительнотканную систему, органные проявления его могут быть самыми различными (артриты, васкулиты, ревмокардит и др.). Тем не менее наиболее часто болезнь поражает сердце и суставы. По образному выражению французского врача XIX столетия Лассега, «ревматизм лижет суставы и кусает сердце».
В этиологии ревматизма решающее значение придается β-гемолитическому стрептококку группы А. Это заболевание развивается в организме, особо реагирующем на стрептококковую инфекцию. Оно возникает у лиц с генетической недостаточностью иммунитета к стрептококку (наследственная предрасположенность), что привело к возникновению понятия «семейный ревматизм». Хотя стрептококк и рассматривается в качестве основного
этиологического фактора ревматизма, тем не менее с точки зрения классической инфекционной патологии его нельзя считать возбудителем данного заболевания. Более распространенными являются представления об инфекционно-аллергической природе ревматизма. У лиц с генетически детерминированной недостаточностью иммунитета к стрептококку обострение хронической инфекции приводит к накоплению высокого титра иммунных комплексов (стрептококковый антиген + антитело + комплемент). Циркулируя в кровеносной системе, они фиксируются в стенке сосудов микроциркуляторного русла и повреждают их. В результате облегчается поступление антигенов возбудителя и белков в соединительную ткань, что способствует ее деструкции (аллергические реакции немедленного типа). Из-за общности антигенного строения стрептококка и соединительной ткани сердца иммунные реакции в оболочках последнего повреждают их с образованием аутоантигенов и антикардиальных аутоантител. Ткани сердца связывают как противокардиальные, так и противострептококковые антитела. Одни аутоантитела при ревматизме реагируют с сердечным антигеном, другие перекрестно - с мембраной стрептококка. Образование иммунных комплексов при этом приводит к развитию хронического воспаления в сердце (ревмокардиту).
Кроме гуморального иммунитета, при ревматизме страдает и клеточный иммунитет. В результате образуется клон сенсибилизированных лимфоцитов-киллеров, несущих на себе фиксированные антитела к сердечной мышце и эндокарду. Эти лимфоциты способны повреждать ткани сердца по типу аллергической реакции IV или клеточно-опосредованного типа, т.е. гиперчувствительности замедленного типа.
Течение ревматизма имеет хронический характер, периоды ремиссии чередуются с периодами обострения. С каждой новой атакой ревматизма экстракардиальные проявления становятся менее яркими, а ведущее значение приобретают изменения, приводящие к формированию пороков сердца. Если после первой атаки порок обнаруживается только у 14-18% больных, то после второй и третьей - практически у всех пациентов.
Приобретенные пороки сердца
Приобретенные пороки сердца, которые формируются после повторных ревматических атак или возникают как осложнение
септического эндокардита, очень часто определяют исход заболевания.
Недостаточность митрального клапана. Этот порок характеризуется неполным смыканием створок митрального клапана. Гемодинамически при недостаточности митрального клапана имеется постоянный обратный ток крови в систолу из левого желудочка в левое предсердие, полости которых постепенно расширяются. В течение длительного времени основная нагрузка приходится на левые отделы сердца, затем возникает застой в малом круге кровообращения и к признакам левожелудочковой недостаточности присоединяется правожелудочковая (увеличение печени, отеки). Единственным симптомом начальной стадии заболевания является систолический шум при нормальных размерах сердца. На ЭКГ изменений может не быть, затем нарастают признаки гипертрофии левых отделов сердца (увеличение размеров и массы левого желудочка). При эхокардиологическом исследовании подтверждается деформация створок митрального клапана. Недостаточность митрального клапана наиболее часто бывает ревматической этиологии. Она может возникнуть при инфекционном эндокардите, коллагенозах (системная красная волчанка), бывает врожденной.
Митральный стеноз. Стеноз атриовентрикулярного отверстия, как правило, имеет ревматическое происхождение (реже бывает врожденным). Гемодинамически митральный стеноз наименее благоприятен, так как вся нагрузка в этом случае продолжительное время падает на левое предсердие. Сужение атриовентрикулярного отверстия («первый барьер») препятствует поступлению крови в левый желудочек, давление в левом предсердии повышается, возникает его гипертрофия. Эти компенсаторные механизмы облегчают прохождение крови через суженное митральное отверстие. Нарастание давления в левом предсердии приводит к ретроградному повышению его в легочных венах и капиллярах, формируется так называемая легочная гипертензия, развивается декомпенсация сердечной деятельности. Постепенно появляются морфологические изменения в сосудах легких, их склероз (органический «второй барьер»), стойкая легочная гипертензия и дилатация правых отделов, правожелудочковая недостаточность.
Ранним симптомом стеноза является одышка, при развитии легочной гипертензии нарастает цианоз. В дальнейшем возникают жалобы на сердцебиение, боли в области сердца. Аускультативно выслушивается диастолический шум, обусловленный прохождени-
ем струи крови через суженное атриовентрикулярное отверстие. Митральный стеноз относится к числу наиболее неблагоприятных пороков сердца, в силу чего часто требуется проведение комиссуротомии (от лат. commissura - соединение и греч. tome - разрез, рассечение) уже в детском возрасте, хотя возможна и длительная компенсация.
К осложнениям стеноза левого атриовентрикулярного отверстия относятся: сердечная недостаточность, острый отек легких, легочная гипертензия, нарушения ритма и проводимости, тромбоэмболии артерии большого круга кровообращения (сосуды головного мозга, селезенки, почек). Источником артериальных эмболий являются тромбы, образующиеся в левом предсердии.
Недостаточность аортального клапана. Недостаточность аортального клапана может быть ревматического генеза, часто возникает при септическом эндокардите, сифилисе. В результате нарушения замыкательной функции аортальных клапанов кровь из аорты в диастолу поступает в левый желудочек, который постепенно дилатируется и гипертрофируется, возникают признаки левожелудочковой недостаточности. Развитие дальнейших событий в организме аналогично явлениям, происходящим в стадии декомпенсации сердечной недостаточности по левым отделам сердца.
Стеноз устья аорты. Ревматический стеноз устья аорты чаще присоединяется к имеющейся недостаточности аортального клапана. В результате сращения створок по комиссурам возникает препятствие выбросу крови в аорту, в ответ на это миокард левого желудочка гипертрофируется. Жалобы долго отсутствуют, и единственным указанием на наличие порока является грубый систолический шум. Появление первых жалоб свидетельствует об уже имеющейся митральной недостаточности, после этого болезнь быстро прогрессирует.
Пороки трехстворчатого клапана. Пороки трехстворчатого клапана развиваются редко, как правило, при непрерывно рецидивирующем течении ревматизма. У таких больных трикуспидальная недостаточность или стеноз присоединяются к уже сформировавшимся порокам митрального и/или аортального клапанов. Клинически при недостаточности трехстворчатого клапана наблюдается цианоз лица, выслушивается систолический шум. Приобретенный стеноз правого атриовентрикулярного отверстия развивается редко, чаще он является врожденным.
15.3.4. Нарушения ритма сердца
Нарушения сердечного ритма (аритмии) - изменения нормальной частоты, регулярности и источника возбуждения сердца, а также расстройства проведения импульса, нарушения связи и/или последовательности между активацией предсердий и желудочков.
В соответствии с механизмом возникновения аритмий все нарушения сердечного ритма можно условно подразделить на три типа: 1) нарушения автоматизма; 2) нарушения возбудимости; 3) нарушения проводимости. Подобное деление в известном смысле условно, потому что в реальности часто приходится сталкиваться с аритмиями сочетанного характера. Например, при фибрилляции желудочков и предсердий могут иметь место как нарушение возбудимости, так и патология проведения сердечного импульса.
Нарушения сердечного автоматизма
Нарушения сердечного автоматизма - это аритмии, обусловленные нарушением электрофизиологической активности водителей сердечного ритма (синусового и атриовентрикулярного узлов). К этим аритмиям относятся: синусовая брадикардия, синусовая тахикардия, синусовая аритмия, атриовентрикулярная тахикардия, узловой ритм, идиовентрикулярный ритм.
Синусовая брадикардия - это уменьшение частоты сердечных сокращений до 50 уд./мин и менее при сохранении нормального ритма. Этиологическими факторами синусовой брадикардии являются: повышение тонуса блуждающего нерва, которое может наблюдаться у здоровых людей, чаще - у спортсменов (не требует лечения); первичное поражение синусового узла; повышение внутричерепного давления; гипотиреоз; гипотермия; инфаркт миокарда нижней локализации; передозировка β-адреноблокаторов или антагонистов кальция.
Синусовая тахикардия - это повышение частоты сердечных сокращений более 100 уд./мин при сохранении нормального ритма. Этиологические факторы: нормальная реакция на различные стрессорные факторы (волнение, беспокойство, страх, физическая нагрузка); патологические состояния, в частности - лихорадка, гипотония, тиреотоксикоз, анемия, гиповолемия, эмболия легочной артерии, ишемия миокарда, сердечная недостаточность, шок, митральный стеноз; прием некоторых лекарств (атропин, катехоламины, тиреоидные препараты) или некоторых биологически активных веществ (алкоголь, никотин, кофеин).
Синусовая аритмия - это периодически сменяющие друг друга эпизоды синусовой тахикардии или брадикардии при сохранении синусовой импульсации. По данным ЭКГ, комплекс QRS обычно не деформирован, интервалы R-R укорочены или удлинены, но равны. В норме она может быть следствием периодического изменения тонуса блуждающего нерва, так называемой дыхательной аритмией (повышение частоты сердечных сокращений при вдохе и снижение на выдохе). Этиологические факторы: эмоциональный стресс, климакс, тиреотоксикоз, миокардит.
Узловой ритм - это нарушение, при котором роль водителя ритма берет на себя атриовентрикулярный узел. При этой патологии частота сердечных сокращений снижается до 40-60 уд./мин. Причинами подобного нарушения автоматизма наиболее часто являются интоксикация, которая приводит к слабости синусового узла, или блокада внутрипредсердного проведения импульса.
Атриовентрикулярные - реципрокные пароксизмальные тахикардии - нарушения ритма, связанные с повышенной возбудимостью атриовентрикулярного узла. Эта группа нарушений ритма составляет 85% всех наджелудочковых аритмий. Электрофизиологический механизм данных аритмий представляет собой сочетание нарушения автоматизма и патологии проведения импульса (re-entry). Этиология реципрокных атриовентрикулярных пароксизмальных тахикардий остается до сих пор неизвестной, но почти у 1/3 всех пациентов, страдающих этим типом нарушений ритма, приступы сердцебиения связаны с психоэмоциональной нагрузкой.
Идиовентрикулярный ритм - это нарушение, при котором роль водителя ритма берут на себя ножки пучка Гиса или волокна Пуркинье. Ритм при этом урежается до 10-30 уд./мин. Такое нарушение автоматизма развивается при повреждении синусового и атриовентрикулярного узлов и ведет к нарушению центральной гемодинамики, что может закончиться гибелью пациента.
Нарушения возбудимости сердца
Нарушения возбудимости сердца лежат в основе таких видов аритмий, как экстрасистолии, желудочковые тахикардии, полиморфная желудочковая тахикардия, трепетание желудочков и предсердий, фибрилляция желудочков и предсердий, внезапная остановка сердца. Основные из них представлены на рис. 15-13.
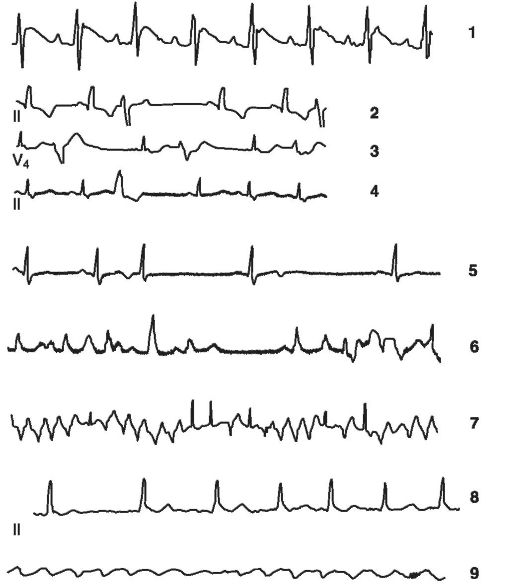 Рис. 15-13. Основные
виды нарушений сердечного ритма: 1 - нормальная ЭКГ; 2-4 - желудочковые
экстрасистолы; 5 - нижнепредсердная экстрасистолия; 6 - предсердная
пароксизмальная тахикардия; 7 - желудочковая пароксизмальная тахикардия;
8 - фибрилляция предсердий; 9 - фибрилляция желудочков
Рис. 15-13. Основные
виды нарушений сердечного ритма: 1 - нормальная ЭКГ; 2-4 - желудочковые
экстрасистолы; 5 - нижнепредсердная экстрасистолия; 6 - предсердная
пароксизмальная тахикардия; 7 - желудочковая пароксизмальная тахикардия;
8 - фибрилляция предсердий; 9 - фибрилляция желудочков
Экстрасистолия - внеочередное сокращение сердца. Зкстрасистолы, исходящие из синусового узла, называются номотопными. Однако и в этом случае источником экстрасистолы являются не клетки водителя ритма (пейсмекера), а расположенные в его окружении клетки, обладающие латентным автоматизмом, но не проявляющие пейсмекерной активности в нормальных условиях. Пейсмекерной активностью называют способность клеток к спонтанной деполяризации. Обычно пейсмекерная активность атриовентрикулярного узла, ножек пучка Гиса и волокон Пуркинье подавляется импульсами, поступающими из синусового узла,
но в условиях блокады проведения импульсов от предсердий к желудочкам сердца эти латентные пейсмекеры могут активироваться и вызывать появление экстрасистол. Гетеротопные экстрасистолы исходят из любого участка проводящей системы, исключая синусовый узел. Эктопические экстрасистолы имеют источник внеочередного возбуждения, локализующийся в миокарде за пределами проводящей системы сердца. Аналогичная ситуация часто складывается в очаге ишемии при инфаркте миокарда. В зависимости от локализации эктопического очага различают предсердные, атриовентрикулярные, левожелудочковые, правожелудочковые и перегородковые экстрасистолы.
Одиночные экстрасистолы не вызывают серьезных расстройств гемодинамики и клинически проявляются ощущением «перебоев» в работе сердца. Однако множественные и особенно политопные экстрасистолы, т.е. исходящие из нескольких эктопических центров, могут вызвать серьезные нарушения гемодинамики по двум причинам. Во-первых, многие экстрасистолы гемодинамически малоэффективны, поскольку процесс внеочередного сокращения может возникнуть в период, когда сердце еще не успело полностью расслабиться и, следовательно, конечный диастолический объем желудочков в этот момент остается сниженным, так же как и ударный объем. Во-вторых, после экстрасистолы следует компенсаторная пауза, т.е. удлиненная диастола, в период которой миокард находится в состоянии рефрактерности и не чувствителен к импульсу, поступающему из синусового узла. Наиболее выраженные нарушения гемодинамики наблюдаются при желудочковых экстрасистолах.
Желудочковые экстрасистолы - преждевременные желудочковые сокращения, обусловленные наличием очага автоматизма в желудочках. Этиологические факторы желудочковых экстрасистолий: ИБС и ее осложнения (в частности, острый инфаркт миокарда), кардиомиопатии, нарушения электролитного и кислотно-щелочного баланса, гипоксия, эндокринные заболевания (тиреотоксикоз), инфекции, прием некоторых лекарств (сердечные гликозиды и антиаритмические средства). Желудочковые экстрасистолы могут регистрироваться и у практически здоровых людей. Так, например, по данным суточного мониторирования ЭКГ, желудочковые экстрасистолы отмечаются в 70-80% случаев у лиц в возрасте старше 60 лет, причем наиболее часто обнаруживаются бессимптомные желудочковые экстрасистолы.
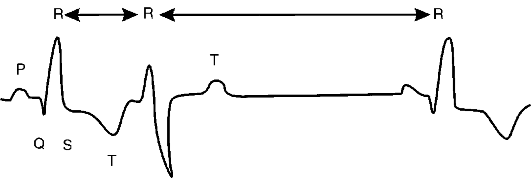 Рис. 15-14. Желудочковая экстрасистола. QRS деформирован, продолжительность более 0,12 с. Инверсия зубца Т
Рис. 15-14. Желудочковая экстрасистола. QRS деформирован, продолжительность более 0,12 с. Инверсия зубца Т
Электрокардиографически желудочковые экстрасистолы характеризуются (см. рис. 15-13) появлением преждевременных комплексов QRS, отличающихся от нормальных комплексов шириной более 0,12 с, деформацией, наличием предшествующего укороченного интервала R-R (рис. 15-14). Зубец Т, как правило, увеличен и так же, как сегмент S-T, расположен дискордантно, т.е. направлен в другую сторону по отношению к самому высокоамплитудному зубцу комплекса QRS. Экстрасистолическому комплексу QRS не предшествует зубец Р. Клинически желудочковые экстрасистолы проявляются как ощущение сердцебиения или дискомфорт в груди, ощущение перебоев в работе сердца (в связи с наличием компенсаторных пауз после желудочковых экстрасистол).
Среди желудочковых экстрасистол наиболее часто встречаются единичные, реже множественные, парные, политопные (возникающие из разных отделов миокарда) экстрасистолы, бигеминия (состояние, когда каждой нормальной систоле сопутствуют желудочковые экстрасистолы). Появление парных желудочковых экстрасистол увеличивает риск смерти. Особую опасность представляют ранние желудочковые экстрасистолы, при которых эктопический импульс приходится на так называемую ранимую фазу сердечного цикла. Ранимая фаза сердечного цикла - это интервал времени, когда процесс реполяризации еще полностью не завершился, сердце находится в состоянии относительной рефрактерности и любой экстрастимул, в том числе эктопический импульс, имеющий желудочковую локализацию, может вызвать появление не только желудочковой экстрасистолы, но и желудочковой фибрилляции, которая может закончиться гибелью пациента. Электрокардиографически ранимая фаза почти полностью соответствует зубцу Т, поэтому подобные экстрасистолы называют «экстрасистола R на
T» (R на Т). Появление ранних желудочковых экстрасистол является негативным прогностическим признаком, поскольку часто предшествует внезапной сердечной смерти.
Мерцательная аритмия (фибрилляция предсердий) - это отсутствие скоординированных сокращений предсердий, которое электрокардиографически характеризуется исчезновением зубца Р. Фибрилляция предсердий приводит к прекращению гемодинамически эффективных сокращений предсердий. Она проявляется нерегулярными мелкими колебаниями предсердий различной амплитуды и формы с частотой 350-600 в мин, которые не удается зарегистрировать на обычном электрокардиографе. Желудочковые сокращения также нерегулярны. Различают тахисистолическую (частота сердечных сокращений более 100 уд./мин), нормосистолическую (частота сердечных сокращений 60-90 уд./мин) и брадисистолическую (частота сердечных сокращений ниже 60 уд./мин) формы фибрилляции предсердий. Этиологические факторы: атеросклероз, гипотония, кардиомиопатия и ревматические заболевания сердца, тиреотоксикоз, иногда видимая причина отсутствует. При тахиаритмической форме мерцательной аритмии частота желудочковых сокращений может достигать 150-240 в мин, причем во время пароксизма может развиться резкая гипотония или отек легких вследствие перегрузки сердца и острой левожелудочковой недостаточности.
Трепетание предсердий - нарушение процессов возбуждения и проведения в предсердиях, которое электрокардиографически характеризуется исчезновением зубца Р и появлением вместо него частых низкоамплитудных колебаний, так называемых зубцов F, которые получили свое название от английского слова flatter - колебание. При этой патологии частота сокращений предсердий составляет более 220 в мин, а желудочков - 120-180 в мин. Одновременно возникают блокады атриовентрикулярного проведения 1:1, 2:1, 3:1, 4:1 и даже 5:1. Комплексы QRS нормальные, реже напоминают желудочковые экстрасистолы. Различают тахи-, нормо- и брадисистолическую формы трепетания предсердий. Этиологические факторы те же, что и при мерцательной аритмии.
Желудочковая тахикардия - частый и в основном регулярный ритм, берущий свое начало: а) в сократительном миокарде желудочков; б) в сети Пуркинье; в) в ножках пучка Гиса. Электрокардиографически желудочковая тахикардия характеризуется появлением серии из трех желудочковых экстрасистолий и более с дискордант-
но расположенным сегментом S-T по отношению к основному отклонению комплекса QRS. Интервалы: R-R могут быть регулярными или различаться по продолжительности. Часто желудочковая тахикардия представлена комплексом полиморфных желудочковых экстрасистолий. Большинство желудочковых тахикардий в своей основе обусловлено механизмом re-entry с локализацией критического участка циркуляции электрического возбуждения в субэндокардиальной области. В более редких случаях желудочковая тахикардия возникает вследствие нарушения автоматизма. Чаще всего желудочковая тахикардия носит пароксизмальный характер, но иногда имеет место стабильная многочасовая желудочковая тахикардия.
Причинами желудочковых тахикардий, как правило, являются тяжелые заболевания миокарда: хроническая ишемия и острый инфаркт миокарда, постинфарктный кардиосклероз, миокардиты, миокардиопатии, ревматические клапанные пороки сердца, синдром WPW, тяжелая сердечная недостаточность различной этиологии, синдром удлиненного интервала Q-T. Реже причинами желудочковых тахикардий могут быть: тиреотоксикоз, гипоксемия, нарушение кислотно-щелочного баланса, гипокалиемия, интоксикация препаратами наперстянки, хинидином, новокаинамидом, катехоламинами, циклопропаном.
Среди различных нарушений сердечного ритма желудочковая тахикардия занимает особое место, поскольку может привести к перегрузке сердца или перейти в фибрилляцию желудочков. Первое из этих осложнений чревато развитием острой левожелудочковой недостаточности, а второе - прекращением кровоснабжения жизненно важных органов и гибелью пациента. Именно поэтому появление у больных стойкой желудочковой тахикардии повышает риск внезапной сердечной смерти в 5-6 раз по сравнению с пациентами, не имеющими желудочковых аритмий.
Полиморфная (пароксизмальная) желудочковая тахикардия в большинстве случаев возникает в виде пароксизмов с частотой более 200 уд./мин. Обычно развивается вследствие неконтролируемой терапии антиаритмическими средствами, а также как проявление врожденного синдрома удлиненного интервала Q-T. Электрокардиографическая картина полиморфной желудочковой тахикардии представлена на рисунке 15-15, на котором видно, что желудочковые комплексы как бы «вьются» вокруг изоэлектрической оси. Появлению этой аритмии предшествуют брадикардия и удлинение
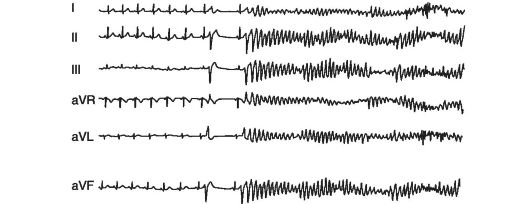 Рис. 15-15. Полиморфная желудочковая тахикардия (torsades de pointes)
Рис. 15-15. Полиморфная желудочковая тахикардия (torsades de pointes)
интервала Q-T. Полиморфная желудочковая тахикардия развивается по механизму триггерного автоматизма (см. ниже) и обычно носит обратимый характер, но может трансформироваться в фибрилляцию желудочков.
Причинами развития этой опасной для жизни аритмии могут быть: гипокалиемия, интоксикации, миокардит, ишемия, некоторые лекарственные средства и комбинация факторов. В частности, она может развиться даже при приеме антиаритмических препаратов (хинидин, новокаинамид, амиодарон, соталол и др.).
Синдром удлиненного интервала Q-T (long Q-T) может быть приобретенным и наследственным. Электрокардиографически он характеризуется удлинением интервала Q-T, брадикардией, возникновением полиморфной желудочковой тахикардии (рис. 15-16) и появлением волны U, следующей после зубца Т. Волну U из-за маленькой амплитуды не всегда удается зарегистрировать. Клинически синдром long Q-T проявляется внезапной потерей сознания и возникновением желудочковой тахикардии, которая может закончиться спонтанным восстановлением нормального сердечного ритма или, напротив, перейти в желудочковую фибрилляцию с нарушением центральной гемодинамики и гибелью пациента.
 Рис. 15-16. Синдром удлиненного интервала Q-T (long Q-T)
Рис. 15-16. Синдром удлиненного интервала Q-T (long Q-T)
Приобретенный синдром связан с употреблением некоторых лекарственных препаратов, врожденный - с мутациями генов, кодирующих структуру полипептидной цепочки быстрого Na+- канала или двух типов К+-каналов. Известно, что деполяризация кардиомиоцитов начинается с быстрой активации Na+-каналов, которая сменяется такой же быстрой их инактивацией. Весь цикл занимает несколько миллисекунд. Мутация гена, кодирующего белок Na+-канала, приводит к замедлению процесса инактивации этого канала. В результате возникает перегрузка кардиомиоцитов ионами Na, тормозится процесс восстановления нормального градиента ионов и замедляется реполяризация кардиомиоцитов. Эти события могут индуцировать появление желудочковых аритмий по механизму ранней постдеполяризации и проявляются на ЭКГ удлинением интервала Q-T.
Как известно, процесс реполяризации обеспечивается К+-каналами, которые при этом открываются. В настоящее время идентифицированы два гена, мутация которых приводит к инактивации этих каналов, что ведет к замедлению реполяризации. Наследственная форма синдрома long Q-T встречается достаточно редко.
Фибрилляция (и трепетание) желудочков - это хаотическое асинхронное возбуждение отдельных мышечных волокон или их небольших групп с остановкой сердца и прекращением кровообращения. Эти аритмии представляют наибольшую опасность, так как они при отсутствии экстренных мероприятий в течение 3-5 мин могут привести к летальному исходу. Электрокардиологически фибрилляция желудочков характеризуется появлением волн низкой амплитуды (менее 0,2 мВ) и различной формы с частотой от 300 до 600 в мин (рис. 15-17). Трепетание желудочков характеризуется на ЭКГ появлением волн с нерегулярными большими осцилляциями при частоте 150-300 в мин. При данных аритмиях невозможно выделить
 Рис. 15-17. Желудочковая фибрилляция: А - мелковолновая; Б - крупноволновая
Рис. 15-17. Желудочковая фибрилляция: А - мелковолновая; Б - крупноволновая
комплекс QRS, сегмент S-Т и зубец Т. Фибрилляция желудочков возникает при различных сердечно-сосудистых заболеваниях, особенно часто при острой коронарной недостаточности, ишемии миокарда, а также при тяжелом течении кардиомиопатии.
Следует особо отметить, что желудочковые аритмии имеют тенденцию трансформироваться в более тяжелые формы, например множественные желудочковые экстрасистолы - в пароксизмальную тахикардию, а последняя - в фибрилляцию сердца, которая может закончиться асистолией и внезапной сердечной смертью.
Внезапная остановка сердца может быть двух типов: а) асистолия желудочков, когда отсутствуют и сокращения желудочков, и их биоэлектрическая активность; б) электромеханическая диссоциация - крайне опасное состояние сердца, когда на ЭКГ регистрируется электрическая активность при отсутствии эффективного сокращения миокарда.
Причиной внезапной остановки сердца могут быть ИБС, тромбоэмболия легочных артерий, гипертрофия миокарда и кардиомиопатия, первичная или вторичная легочная гипертония, сердечная недостаточность, миокардиты, пороки сердца, синдром удлиненного интервала Q-T и ряд других заболеваний. Феномен электромеханической диссоциации развивается при ишемии миокарда, если она сопровождается выраженным нарушением внутриклеточного транспорта Са2+ на уровне СПР при сохраненной активности Na+/К+-АТФазы сарколеммы. В результате возникающий потенциал действия не влечет за собой сокращения миокарда, что обычно заканчивается гибелью пациента.
Внезапная сердечная смерть может наступить в любом возрасте, в том числе в молодом и даже в детском. По данным ВОЗ, частота внезапной сердечной смерти составляет 30 случаев в неделю на 1 млн населения, или около 12% всех случаев естественной смерти. В старших возрастных группах внезапная коронарная смерть наступает на фоне выраженных атеросклеротических изменений коронарных артерий, часто до этого клинически не проявлявшихся, а также на фоне бессимптомной ИБС. Непосредственными причинами внезапной сердечной смерти являются в основном фибрилляция желудочков и желудочковая тахикардия, а также асистолия или резкая брадикардия (на их долю приходится около 20% случаев).
Таким образом, внезапная остановка сердца является только одной из причин внезапной сердечной смерти. Последняя насту-
пает или мгновенно, или в течение 2 ч после появления первых симптомов коронарной катастрофы у негоспитализированных больных, имевших до этого заболевания сердца, но находившихся, с точки зрения врача, в относительно стабильном, не опасном для жизни состоянии. На вскрытии у таких пациентов не удается выявить признаки острого инфаркта миокарда. Фатальные нарушения ритма чаще развиваются на фоне электрической нестабильности миокарда, возникающей у больных с морфологическими изменениями в сердце. Однако внезапная сердечная смерть возможна и при отсутствии изменений структуры сердца. Причиной внезапной сердечной смерти в этом случае являются так называемые идиопатические аритмии, т.е. нарушения ритма неясной этиологии. Например, идиопатические желудочковые фибрилляции составляют приблизительно 1% всех случаев остановки сердца во внегоспитальных условиях. Причиной таких аритмий может явиться стресс-индуцированная электрическая нестабильность сердца (по Б. Лауну).
Нарушения проводимости
Нарушения проводимости включают в себя поперечную блокаду сердца, блокаду правой и/или левой ножек пучка Гиса, синдром Вольфа-Паркинсона-Уайта.
Поперечная блокада - это нарушение проведения возбуждения в области атриовентрикулярного узла. Поперечная блокада сердца, в свою очередь, подразделяется на блокаду I, II, III и IV степени. Первые три степени называют еще неполной, а последнюю - полной поперечной блокадой сердца.
Поперечная блокада I степени проявляется задержкой проведения импульса в атриовентрикулярном узле. Электрокардиографически она характеризуется удлинением интервала P-Q. Это расстройство сердечного ритма не сказывается на гемодинамике и чаще всего является следствием усиления вагусных влияний на миокард или результатом интоксикации сердечными гликозидами.
Поперечная блокада II степени характеризуется тем, что в структуре каждого последующего ЭКГ-цикла интервал P-Q удлиняется все больше и больше до тех пор, пока не происходит выпадения одного желудочкового комплекса (период Самойлова-Венкенбаха), после чего продолжительность интервала P-Q возвращается к норме, но тут же вновь начинает удлиняться. Таким образом, процесс но-
сит циклический характер. Возникновение периодов Самойлова- Венкенбаха связано с формированием сначала относительной, а затем абсолютной рефрактерности атриовентрикулярного узла. В последнем случае атриовентрикулярный узел оказывается неспособным к проведению возбуждения от предсердий к желудочкам. Очередное сокращение желудочков выпадает. В течение этой паузы возбудимость атриовентрикулярного узла восстанавливается до нормы, и весь цикл повторяется вновь. Клинически этот вид блокады проявляется ощущением «перебоев в работе сердца». Это расстройство проводимости не влияет на гемодинамику и также является следствием усиления тонической активности n. vagus или результатом интоксикации сердечными гликозидами.
Поперечная блокада III степени выражается в том, что через атриовентрикулярный узел проходит от предсердий к желудочкам только каждый второй или третий импульс. Частота сердечных сокращений значительно урежается, поэтому могут возникать серьезные нарушения гемодинамики.
Полная поперечная блокада - это состояние проводимости, при котором ни один импульс не проходит от предсердий к желудочкам. Предсердия при этом сокращаются в синусовом ритме, а желудочки - в идиовентрикулярном. Возникает выраженная брадикардия, которая вызывает тяжелые нарушения центральной гемодинамики, сопровождающиеся нарушением кровоснабжения головного мозга и эпизодами потери сознания продолжительностью от нескольких секунд до нескольких минут (синдром Морганьи-Эдемса-Стокса). Этот синдром опасен тем, что может закончиться гибелью пациента в результате асистолии. Единственным эффективным способом лечения этой патологии является имплантация искусственного водителя ритма.
Блокада правой и/или левой ножки пучка Гиса - опасное нарушение проведения импульсов по одной из ножек пучка Гиса. Опасность заключается в том, что при этой блокаде происходит асинхронное сокращение желудочков, что ведет к уменьшению ударного объема и развитию сердечной недостаточности. Это расстройство наиболее часто является результатом инфаркта миокарда в области межжелудочковой перегородки, реже - следствием ревматической гранулемы и других заболеваний сердца.
Синдром Вольфа-Паркинсона-Уайта (синдром WPW, синдром преждевременного возбуждения). Отличительной чертой этого синдрома является то, что возбуждение к желудочкам приходит двумя
путями: а) через атриовентрикулярный узел и б) по так называемому пучку Кента (аномальный дополнительный путь проведения импульса между предсердиями и желудочками). При этом происходит взаимное наложение проводимых импульсов и в 50% случаев возникает желудочковая тахиаритмия. Как известно, в норме волна возбуждения из синусного узла распространяется по предсердиям и достигает атривентрикулярного узла, где происходит задержка проведения импульса (атриовентрикулярная задержка), поэтому желудочки сокращаются после предсердий с небольшим опозданием. Однако у пациентов с синдромом WPW между предсердиями и желудочками имеется дополнительный путь проведения - пучок Кента, по которому импульс проходит без всякой задержки. По этой причине желудочки и предсердия могут сокращаться одновременно, что ведет к нарушению внутрисердечной гемодинамики и снижает эффективность насосной функции сердца.
Кроме того, опасность представляет и столкновение импульса из атриовентрикулярного узла с волной возбуждения, поступившей в желудочек по пучку Кента. Это может вызвать появление желудочковой экстрасистолы (внеочередного сокращения желудочка сердца). Если импульс поступит из атриовентрикулярного узла в тот момент, когда желудочки находятся в фазе относительной рефрактерности, т.е. тогда, когда процесс реполяризации еще полностью не завершен, то желудочковая экстрасистола может индуцировать появление желудочковой тахикардии или даже фибрилляции. В силу этого период относительной рефрактерности получил название ранимой фазы сердечного цикла. На ЭКГ этот период соответствует зубцу Т.
Выделяют три основных электрокардиографических признака синдрома WPW: а) укороченньгй интервал P-R на фоне синусового ритма; б) «растянутыгй» сверх нормы комплекс QRS с пологой начальной частью; в) вторичные изменения сегмента S-T, при которых зубец Т направлен дискордантно (в обратном направлении) по отношению к комплексу QRS.
Факторы, приводящие к нарушениям сердечного ритма
Все причины многочисленных тахи- и брадиаритмий можно условно подразделить на четыре группы: 1) нарушения нейрогенной и эндокринной (гуморальной) регуляции электрофизиологических процессов в специализированных или сократительных клет-
ках сердца; 2) органические поражения миокарда, его аномалии, врожденные или наследственные дефекты с повреждением электрогенных мембран и клеточных структур; 3) сочетание нарушений нейрогуморальной регуляции ритма и органических заболеваний сердца; 4) аритмии, вызванные лекарственными препаратами. Таким образом, практически любое заболевание кровеносной системы может осложниться нарушениями сердечного ритма. Однако в данном разделе рассматриваются только аритмии, связанные с нарушениями нейрогуморальной регуляции сердечного ритма или с употреблением некоторых лекарственных препаратов.
Нарушения нейрогенной и эндокринной регуляции электрофизиологических процессов в кардиомиоцитах и клетках проводящей системы сердца. Одной из основных причин нарушений сердечного ритма и проводимости является изменение физиологического соотношения между тонической активностью симпатических и парасимпатических элементов, иннервирующих сердце. Важно отметить, что повышение тонической активности симпатического звена вегетативной нервной системы способствует возникновению аритмий, в то время как стимуляция n. vagus, как правило, повышает электрическую стабильность сердца.
Описаны расстройства сердечного ритма, связанные с заболеваниями головного мозга, особенно часто с нарушениями мозгового кровообращения. Большой интерес вызывают спонтанные, психогенные по своей природе аритмии у больных неврозами, психопатиями, вегетативной дистонией. Число аритмий психосоматического генеза в наше время увеличивается.
В эксперименте на животных практически любую из известных форм аритмий - от простой синусовой тахикардии до фибрилляции желудочков - можно вызвать, воздействуя на некоторые отделы головного мозга: кору, лимбические структуры и в особенности гипоталамо-гипофизарную систему, с которой тесно связаны находящиеся в ретикулярной формации продолговатого мозга центры симпатической и парасимпатической регуляции сердечной деятельности. Одним из наиболее ярких примеров нарушений ритма, обусловленных дисбалансом симпатического и парасимпатического звеньев вегетативной нервной системы, является снижение электрической стабильности сердца при психоэмоциональном стрессе. По данным P. Reich et al. (1981), психологический стресс в 20-30% случаев предшествует появлению угрожающих жизни сердечных аритмий. Патогенез стрессиндуцированных аритмий весь-
ма сложен и до конца неясен. Вполне возможно, что он связан с прямым воздействием катехоламинов на миокард. Вместе с тем известно, что высокие концентрации адреналина в крови, активируя β-адренорецепторы почечных канальцев, способствуют усилению экскреции К+ и развитию гипокалиемии. Последняя вызывает нарушения процессов реполяризации, создавая условия для развития самих опасных желудочковых тахиаритмий, в том числе желудочковой фибрилляции и внезапной сердечной смерти. Фармакологическая или хирургическая симпатэктомия устраняет влияние различных типов стресса на ритм сердца и повышает электрическую стабильность миокарда. Такой же эффект оказывает и стимуляция блуждающего нерва, которая способствует угнетению высвобождения норадреналина из окончаний симпатических нервов и ослаблению адренореактивности сердца.
Говоря о роли эндокринных нарушений в патогенезе аритмий, следует указать, что избыточная продукция тиреоидных гормонов способствует увеличению количества адренорецепторов в миокарде и повышению их чувствительности к эндогенным катехоламинам. По этой причине у больных тиреотоксикозом, как правило, наблюдаются тахикардия и нарушения сердечного ритма, обусловленные повышением адренореактивности сердца. Одной из частых «эндокринных» причин нарушений электрической стабильности сердца является избыточное образование минералокортикоидов в коре надпочечников (первичный и вторичный альдостеронизм). Реже аритмии возникают при гиперсекреции глюкокортикоидных гормонов (болезнь и синдром Иценко-Кушинга) или длительном приеме их фармакологических аналогов.
Механизм аритмогенного эффекта минералокортикоидов и, прежде всего, наиболее активного из них - альдостерона - связан с дисбалансом Na+/K+ в организме. Альдостерон, действуя на почечные канальцы, вызывает задержку в организме Na+ и усиление экскреции К+, в результате чего возникает гипокалиемия, которая способствует нарушению процессов реполяризации и возникновению аритмий по триггерному механизму (см. ниже). Умеренное аритмогенное влияние глюкокортикоидов обусловлено тем, что природные (гидрокортизол, кортизол, кортикостерон) и синтетические (преднизолон, дексаметазон) гормоны этой группы не являются «чистыми» глюкокортикоидами, они обладают слабым сродством к рецепторам альдостерона в почечных канальцах. Именно этим свойством объясняется способность данных биоло-
гически активных веществ провоцировать аритмии у пациентов, получающих их длительное время.
Аритмии, вызванные лекарственными препаратами. Часто причиной аритмий являются лекарственные препараты, обладающие собственной аритмогенной активностью. В первую очередь это относится к сердечным гликозидам и диуретикам. Мочегонные препараты, усиливая экскрецию калия, способствуют возникновению гипокалиемии. Сердечные гликозиды (дигиталис и др.) имеют свойство накапливаться в организме, ингибируя при этом Na+/K+- АТФазу, локализованную на сарколемме кардиомиоцитов. Снижение активности этого фермента сопровождается снижением уровня К+ и увеличением концентрации Na+ в саркоплазме. Накопление натрия в цитоплазме кардиомиоцитов приводит к усилению Na+/ Ca2+-обмена, что сопровождается активным поступлением Са2+ в клетки миокарда и способствует усилению насосной функции сердца. Однако при этом формируется Са2+-перегрузка кардиомиоцитов. Кроме того, снижение внутриклеточной концентрации К+ вызывает замедление процессов реполяризации и тем самым способствует возникновению ранних деполяризаций и аритмий по механизму триггерного автоматизма.
Лекарственные аритмии могут быть вызваны и антиаритмическими препаратами. У больных с хронической сердечной недостаточностью, длительное время получавших блокаторы Na+-каналов (флекаинид, этацизин и др.) или блокатор К+-каналов D-соталол, повышается частота случаев внезапной сердечной смерти и сокращается общая продолжительность жизни. Было установлено, что D-соталол ингибирует К+-каналы, а это ведет к замедлению процесса реполяризации, возникновению ранних реполяризаций и опасных желудочковых аритмий по механизму триггерного автоматизма. Механизм аритмогенного действия блокаторов Na+-каналов у пациентов с хронической сердечной недостаточностью неизвестен.
Патогенез нарушений сердечного ритма
Следует выделить два основных механизма нарушений ритма сердечных сокращений: 1) патологию образования импульса и 2) дефекты проведения импульса. Однако чаще всего аритмии возникают при участии обоих механизмов.
Патология образования импульса может быть обусловлена нарушениями автоматизма и повышением возбудимости кардиомиоцитов.
Нарушения автоматизма синусового узла и латентных водителей ритма. Различают нарушения нормального автоматизма, т.е. автоматизма синусового узла, и появление аномального автоматизма, который обусловлен активацией пейсмекерной функции в клетках проводящей системы, не являющихся в норме водителями ритма (атриовентрикулярный узел, ножки пучка Гиса, волокна Пуркинье).
Как известно, в основе процесса любого автоматизма лежит медленная спонтанная диастолическая деполяризация, постепенно понижающая мембранный потенциал до порогового уровня, с которого начинается быстрая деполяризация мембраны, или фаза 0 потенциала действия (рис. 15-18). В кардиомиоцитах рабочего миокарда и в специализированных клетках потенциал покоя обеспечивается за счет высокой активности электрогенной Na+/K+- АТФазы, которая, в свою очередь, обеспечивает градиент ионов калия и натрия между цитоплазмой клетки и экстрацеллюлярным пространством. Кроме того, потенциал покоя поддерживается так называемым током утечки К+ из саркоплазмы во внеклеточное пространство. Оба эти процесса в совокупности поддерживают отрицательный заряд на внутренней поверхности сарколеммы. В сократительных кардиомиоцитах ток К+ направлен из клетки наружу и в состоянии покоя остается неизменным. В клетках проводящей системы сердца этот ток постепенно уменьшается, что и ведет к развитию медленной спонтанной диастолической деполяризации сарколеммы до пороговой. Особенно сильно выражена способность к подобной деполяризации в клетках синоатриального узла, именно поэтому данный узел является водителем ритма сердца.
Изменения нормального автоматизма сердца (времени медленной спонтанной деполяризации клеток синоатриального узла) приводят к возникновению синусовых аритмий. На продолжительность спонтанной деполяризации и, следовательно, на частоту сердечной деятельности оказывают влияние три механизма.
Первый из них (наиболее важный) - скорость спонтанной диастолической деполяризации. При ее возрастании пороговый потенциал возбуждения достигается быстрее и происходит учащение синусового ритма. Противоположнъгй эффект, т.е. замедление спонтанной диастолической деполяризации, ведет к замедлению синусового ритма.
Второй механизм, оказывающий влияние на уровень автоматизма синоатриального узла, - изменение величины мембранного
 Рис. 15-18. Потенциал
действия: А - кардиомиоцит; Б - клетка синоатриального узла; В -
волокно Пуркинье: 0 - стадия деполяризации; 1 - овершут; 2 - плато
потенциала действия; 3 - стадия реполяризации; 4 - потенциал покоя
Рис. 15-18. Потенциал
действия: А - кардиомиоцит; Б - клетка синоатриального узла; В -
волокно Пуркинье: 0 - стадия деполяризации; 1 - овершут; 2 - плато
потенциала действия; 3 - стадия реполяризации; 4 - потенциал покоя
потенциала покоя его клеток. Когда мембранный потенциал становится более отрицательным (при гиперполяризации клеточной мембраны, например при действии ацетилхолина), требуется больше времени для достижения порогового потенциала возбуждения, если, разумеется, скорость спонтанной диастолической деполяризации остается неизменной. Следствием такого сдвига будет уменьшение числа сердечных сокращений. При увеличении мембранного потенциала покоя, когда он становится менее отрицательным, частота сердечных сокращений, напротив, возрастает.
Третий механизм - изменение порогового потенциала возбуждения (фактически - чувствительности кардиомиоцитов к электрическому стимулу). Его уменьшение (более отрицательный) способствует учащению синусового ритма, а увеличение (менее отрицательный) - брадикардии. Величина порогового потенциала возбуждения кардиомиоцитов определяется свойствами Na+- каналов, а клеток проводящей системы - Ca2+- каналов. В связи с этим следует напомнить, что в основе фазы быстрой деполяризации в клетках рабочего миокарда лежит активация быстрых Na+- каналов, а в клетках специализированной ткани сердца - Ca2+- каналов.
Возможны и различные комбинации трех основных электрофизиологических механизмов, регулирующих автоматизм синоатриального узла.
Аномальный автоматизм (эктопический автоматизм) - это появление пейсмекерной активности в клетках сердца, не являющихся водителями сердечного ритма. В норме эктопическая активность подавляется импульсами, поступающими из синоатриального узла, но при блокаде проведения импульса по предсердиям главным водителем ритма сердца может стать атриовентрикулярный узел. Способность к спонтанной деполяризации в элементах этого узла менее выражена, чем в клетках синусового узла, поэтому в условиях поперечной блокады обычно развивается брадикардия.
Еще менее выражена способность к автоматизму у волокон Пуркинье. Однако эти волокна, как и другие клетки проводящей системы, более устойчивы к гипоксии, чем сократительные кардиомиоциты, в связи с чем не всегда погибают в зоне ишемии. Вместе с тем электрофизиологические свойства таких ишемизированных волокон Пуркинье существенно отличаются от параметров интактных волокон тем, что у них появляется пейсмекерная активность, а способность к проведению импульса существенно
снижается. Кроме того, спонтанная биоэлектрическая активность, возникающая в этих волокнах, в условиях патологии (например, при глубокой ишемии) перестает подавляться импульсами, поступающими из синусового узла, и может быть причиной возникновения желудочковых экстрасистол.
Повышение возбудимости кардиомиоцитов наиболее часто обусловливает возникновение аритмий по механизму триггерной (наведенной, пусковой) активности. Электрофизиологической основой триггерной активности (триггерного автоматизма) являются ранние и поздние постдеполяризации.
Ранняя постдеполяризация - это преждевременная деполяризация клеток миокарда и проводящей системы, которая появляется тогда, когда фаза реполяризации потенциала действия еще не завершена, потенциал мембраны еще не достиг диастолической величины, соответствующей потенциалу покоя (рис.15-19). Можно указать таких два важнейших условия возникновения ранних постдеполяризаций, как: удлинение фазы реполяризации потенциала действия и брадикардия. При замедлении реполяризации и соответственно увеличении общей продолжительности потенциала действия может возникнуть преждевременная спонтанная деполяризация в тот момент, когда процесс реполяризации еще не завершился. При уменьшении частоты основного ритма сердца (брадикардия) происходит постепенное возрастание амплитуды ранних надпороговых колебаний мембранного потенциала. Достигнув порога возбуждения, одна из них вызывает образование нового потенциала действия еще до завершения исходного (рис. 15-20). Этот преждевременный потенциал действия рассматривается как триггер-
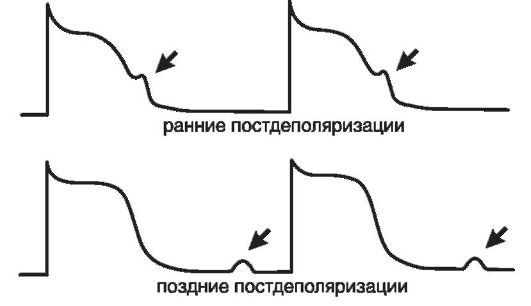 Рис. 15-19. Потенциал действия: триггерная активность
Рис. 15-19. Потенциал действия: триггерная активность
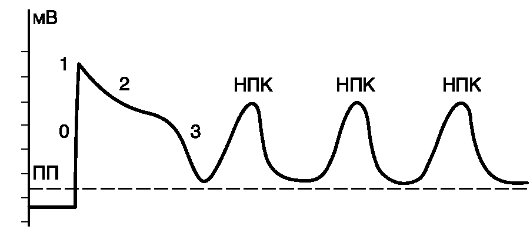 Рис. 15-20. Потенциал
действия и его надпороговые колебания: ПП - пороговый потенциал; 0, 1,
2, 3 - фазы трансмембранного потенциала; НПК - надпороговые колебания
трансмембранного потенциала
Рис. 15-20. Потенциал
действия и его надпороговые колебания: ПП - пороговый потенциал; 0, 1,
2, 3 - фазы трансмембранного потенциала; НПК - надпороговые колебания
трансмембранного потенциала
ный (наведенный), поскольку он обязан своим возникновением ранней постдеполяризации, исходящей от основного потенциала действия. В свою очередь, второй (наведенный) потенциал действия за счет своей ранней постдеполяризации может вызвать третий, тоже триггерный потенциал действия, а третий - четвертый триггерный потенциал действия и т.д. Если источник триггерной активности находится в желудочках, то на ЭКГ подобный тип нарушений образования импульсов проявляется, как желудочковая экстрасистолия или полиморфная желудочковая тахикардия.
Поскольку ранние постдеполяризации реализуются за счет активации Na+- и Са2+-каналов, супрессировать связанные с ними нарушения сердечного ритма можно с помощью блокаторов названных каналов. Кроме того, триггерный ритм, вызванный ранними постдеполяризациями, может быть подавлен с помощью электрокардиостимуляции с частотой, превышающей исходный ритм сердца. Возникновению ранних постдеполяризаций способствуют: гиперкатехоламинемия, гипокалиемия, ацидоз, ишемия, синдром удлиненного интервала Q-T. Часто подобный автоматизм является результатом применения антиаритмических препаратов, блокирующих К+-каналы (соталол, хинидин и др.).
Поздние (задержанные) постдеполяризации - это преждевременная деполяризация клеток миокарда и проводящей ткани, которая появляется сразу же после завершения фазы реполяризации, т.е. когда электрический заряд сарколеммы соответствует диастолическому потенциалу. Подпороговые колебания мембранного потенциала, которые в норме могут присутствовать, но никогда себя не проявляют, при патологических состояниях, вызывающих Са2+-перегрузку
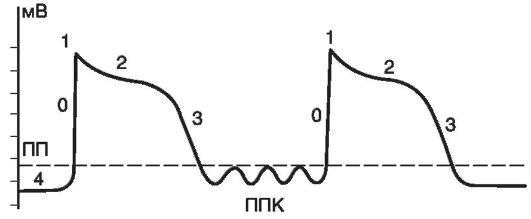 Рис. 15-21. Потенциал
действия и его подпороговые колебания: ПП - пороговый потенциал; 0, 1,
2, 3, 4 - фазы трансмембранного потенциала; ППК - подпороговые колебания
трансмембранного потенциала
Рис. 15-21. Потенциал
действия и его подпороговые колебания: ПП - пороговый потенциал; 0, 1,
2, 3, 4 - фазы трансмембранного потенциала; ППК - подпороговые колебания
трансмембранного потенциала
кардиомиоцитов, могут возрастать по амплитуде, достигая порога возбуждения (рис. 15-21). Повышение внутриклеточной концентрации ионов кальция вызывает активацию неселективных ионных каналов, обеспечивающих усиленное поступление катионов из внеклеточной среды в кардиомиоцит. При этом в клетку поступают главным образом ионы Na+, концентрация которых в экстрацеллюлярной жидкости намного превышает уровень К+ и Са2+. В результате отрицательный заряд внутренней поверхности клеточной мембраны уменьшается, достигая пороговой величины, вслед за чем возникает серия преждевременных потенциалов действия. В конечном итоге формируется цепь триггерных возбуждений.
Триггерная активность клеток сердца, связанная с задержанными постдеполяризациями, может возникнуть под действием сердечных гликозидов или катехоламинов. Очень часто она появляется при инфаркте миокарда. В отличие от ранних постдеполяризаций, возникновению (усилению) которых способствует брадикардия, задержанные постдеполяризации, наоборот, стимулируются учащением сердечного ритма. Это, по-видимому, связано с тем, что чем выше частота сердечных сокращений, тем большее количество ионов кальция поступает в клетку. Следует напомнить, что наиболее частой причиной увеличения концентрации Ca2+ в цитоплазме может быть активация Na+/Ca2+-обмена в условиях реперфузии миокарда.
Дефекты проведения импульса. Существует три основных типа нарушений проводимости: 1) замедление и/или блокада проведения; 2) повторный вход импульса (re-entry); 3) сверхнормальное (супернормальное) проведение.
Замедление проведения, блокада. Причиной замедленного проведения импульса или его блокады нередко бывает снижение количества потенциалзависимых Na+-каналов тех клеток, которым в нормальных условиях присуще свойство быстрой деполяризации (волокна Пуркинье и сократительные кардиомиоциты). Скорость проведения импульсов в этих клетках непосредственно связана с крутизной и амплитудой фазы деполяризации (фаза 0) потенциала действия, т.е. с такими характеристиками, которые как раз и определяются числом активных потенциалзависимых Na+-каналов мембраны. В свою очередь, существует тесная прямая зависимость между числом Na+-каналов, способных к открытию, и величиной мембранного потенциала покоя. Если под влиянием патологических воздействий этот потенциал понижается (приближается к нулевому значению), то уменьшается и скорость деполяризации, а соответственно замедляется проведение импульса. Так, при уменьшении потенциала покоя до уровня 50 мВ (в норме - 80-90 мВ) инактивируется около половины всех Na+-каналов. В этом случае возбуждение и проведение импульса становятся невозможными. Такая ситуация может иметь место в зоне ишемии инфаркта миокарда.
Однако в определенных случаях даже при значительном уменьшении потенциала покоя проведение импульса, правда, существенно замедленное, сохраняется (рис. 15-22). Такое проведение осуществляется медленными Са2+-каналами и «медленными» Na+-каналами, которые устойчивы к снижению потенциала покоя. В интактном кардиомиоците существуют только быстрые Na+-каналы, но в условиях ишемии одна половина этих каналов инактивируется, а другая половина может превратиться в аномальные «медленные» Na+-каналы. Таким образом, «быстрые» клетки превращаются в «медленные» кардиомиоциты, при прохождении через которые импульс может замедлить свое распространение или блокироваться. Причинами блокады могут быть: гипоксия и связанный с ней энергодефицит, вызывающий снижение активности Na+/К+-АТФазы и уменьшение потенциала покоя, а также гибель кардиомиоцитов и волокон Пуркинье в результате ишемии, апоптоза или дистрофии.
Повторный вход импульса (re-entry). Как возможный механизм сердечных аритмий существование re-entry было доказано еще в 1928 г. Этим термином обозначают явление, при котором импульс,
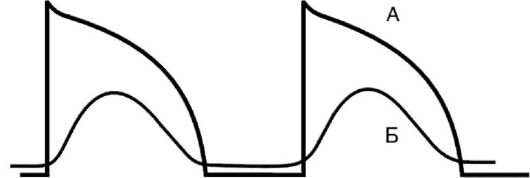 Рис. 15-22. Влияние
острой ишемии миокарда на потенциал действия кардиомиоцитов: А -
нормальный потенциал покоя; Б - «медленный» потенциал действия
Рис. 15-22. Влияние
острой ишемии миокарда на потенциал действия кардиомиоцитов: А -
нормальный потенциал покоя; Б - «медленный» потенциал действия
совершая движение по замкнутому кругу (петле, кольцу), возвращается к месту своего возникновения (circus movement).
Различают macro re-entry (макрориентри) и micro re-entry (микрориентри). При таком делении учитывают размеры петли (круга), в которой осуществляется повторный вход.
Для формирования macro re-entry с характерными для него свойствами требуются определенные условия:
а) существование двух каналов проведения, разделенных между собой функционально или анатомически (односторонняя блокада одного из них);
б) наличие потенциально замкнутой петли движения импульса;
в) замедление скорости распространения импульса, так что ни в одной точке петли волна возбуждения не встречается с зоной рефрактерности.
Пришедшая волна возбуждения медленно продвигается по ветви 1, но не попадает в веточку 2 (рис. 15-23), где имеется участок односторонней блокады. Медленно движущийся импульс вызывает деполяризацию всего мышечного сегмента с образованием потенциала действия. Затем он проникает ретроградно в ветвь 2, возбуждая ее на всем протяжении. К этому моменту исчезает рефрактерность ветви 1, в которую импульс входит повторно. Начи-
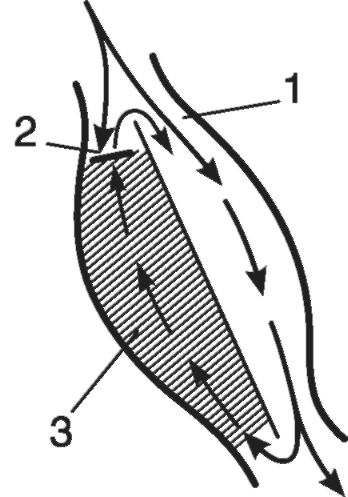 Рис. 15-23. Схема механизма re-entry. Участок
миокарда - задняя стенка левого желудочка: 1 - ортоградное
распространение импульса; 2 - односторонняя блокада проведения; 3 - зона
поврежденного миокарда с замедленным ретроградным распространением
возбуждения
Рис. 15-23. Схема механизма re-entry. Участок
миокарда - задняя стенка левого желудочка: 1 - ортоградное
распространение импульса; 2 - односторонняя блокада проведения; 3 - зона
поврежденного миокарда с замедленным ретроградным распространением
возбуждения
нается повторный круг с преждевременным возбуждением мышечного сегмента. Если такой процесс ограничивается одним re-entry, то на ЭКГ регистрируется экстрасистола. Если круговое движение импульса существует длительное время, возникает серия преждевременных ЭКГ-комплексов, т.е. приступ тахикардии.
При электрической кардиостимуляции отдела сердца, где существует петля re-entry, весь миокард одновременно переводится в состояние абсолютной рефрактерности, и циркуляция импульса прекращается. Наиболее наглядно это проявляется при дефибрилляции сердца.
Описанный механизм macro re-entry лежит, как полагают, в основе трепетания предсердий.
При другой разновидности повторного входа - micro re-entry - движение импульса происходит по малому замкнутому кольцу, не связанному с каким-либо анатомическим препятствием. Повидимому, многие сложные тахиаритмии, в частности фибрилляции, связаны с механизмом micro re-entry. Сочетания петель, лежащих в разных плоскостях, возникают у больных с желудочковыми тахикардиями в остром периоде инфаркта миокарда.
Очень часто морфологическим субстратом для возникновения re-entry являются волокна Пуркинье, находящиеся в зоне ишемии (рис. 15-24). Эти клетки устойчивы к гипоксии и могут не погибать в очаге инфаркта. Однако при этом они меняют свои электрофизиологические характеристики таким образом, что быстрые
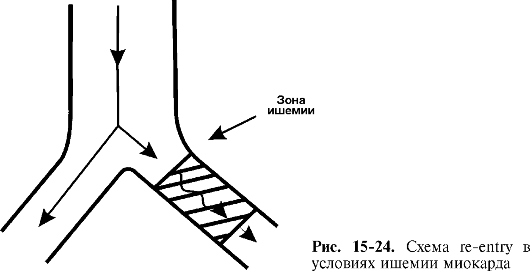 Na+-каналы
превращаются в «медленные». В этом случае проведение импульса
замедляется, и из зоны ишемии он выходит в тот момент, когда остальной
миокард уже находится в состоянии относительной рефрактерности и готов к
повторному возбуждению, но импульс из синусового узла еще не поступил.
Возникает феномен повторного входа (re-entry), когда миокард
дважды стимулируется одним и тем же импульсом: первый раз, когда он
поступает из синусового узла, и второй раз, когда он повторно выходит из
зоны ишемии. В этом случае разорвать петлю re-entry можно с помощью препаратов, блокирующих «медленные» Na+-каналы
в зоне ишемии (лидокаин, новокаинамид). Несомненным достоинством этих
антиаритмиков является то, что они проявляют высокое сродство именно к
аномальным Na+-каналам в зоне ишемии и практически не ингибируют быстрые Na+-каналы в клетках здорового миокарда, а значит, не влияют на электрофизиологические процессы в интактных кардиомиоцитах.
Na+-каналы
превращаются в «медленные». В этом случае проведение импульса
замедляется, и из зоны ишемии он выходит в тот момент, когда остальной
миокард уже находится в состоянии относительной рефрактерности и готов к
повторному возбуждению, но импульс из синусового узла еще не поступил.
Возникает феномен повторного входа (re-entry), когда миокард
дважды стимулируется одним и тем же импульсом: первый раз, когда он
поступает из синусового узла, и второй раз, когда он повторно выходит из
зоны ишемии. В этом случае разорвать петлю re-entry можно с помощью препаратов, блокирующих «медленные» Na+-каналы
в зоне ишемии (лидокаин, новокаинамид). Несомненным достоинством этих
антиаритмиков является то, что они проявляют высокое сродство именно к
аномальным Na+-каналам в зоне ишемии и практически не ингибируют быстрые Na+-каналы в клетках здорового миокарда, а значит, не влияют на электрофизиологические процессы в интактных кардиомиоцитах.