Патофизиология Новицкого, Е.Д. Гольдберга Тома 1 и 2 - 2009 г.
|
|
|
|
ГЛАВА 12 ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
Обмен веществ - это основа жизнедеятельности организма, существенный и непременный признак жизни. Сущность обмена веществ заключается в совокупности физиологических и биохимических реакций, идущих в живых организмах, включая усвоение из внешней среды органических и неорганических соединений (ассимиляция) и их расщепление (диссимиляция) вплоть до образования и выделения конечных продуктов обмена.
В результате химических превращений белков, жиров и углеводов, поступающих в организм, происходит постепенное упрощение органических соединений, что дает возможность синтезировать новые, необходимые для организма вещества, кроме того, освобождается энергия, заключенная в молекулах сложных органических соединений.
Для удобства изучения обмен веществ можно условно разделить на отдельные виды: обмен энергии и основной обмен, углеводный обмен, жировой и холестериновый, белковый и нуклеиновый и др.
12.1. ПАТОФИЗИОЛОГИЯ ЭНЕРГЕТИЧЕСКОГО И ОСНОВНОГО ОБМЕНОВ
12.1.1. Нарушения обмена энергии
Сущность обмена энергии в живом организме состоит в том, что энергия, заключенная в молекулах углеводов, жиров и белков, освобождается при их расщеплении и используется для функционирования клеток. При этом энергия расщепления может накапливаться в химических соединениях, называемых макроэргами. Носителями энергии служат различные фосфорные соединения, в которых связь остатка фосфорной кислоты является макроэр-
гической. Главное место среди них принадлежит АТФ. В форме этого соединения в организме аккумулируется около 55% энергии, освобождающейся при распаде органических молекул, остальная часть превращается в тепло. Синтез АТФ в основном идет в митохондриях клеток в реакциях биологического окисления.
Процесс одновременного освобождения энергии окисления и использования этой энергии в синтезе фосфатных макроэргов называется процессом сопряженного фосфорилирования. При действии факторов, разобщающих реакции окисления субстратов и образования макроэргов, снижается синтез АТФ и повышается теплопродукция. Это явление называется состоянием разобщения. К разобщающим факторам относятся:
1) гормоны щитовидной железы в больших концентрациях;
2) снижение температуры окружающей среды;
3) бактерии и их токсины, вирусы;
4) гипервитаминоз С (усиливается свободное окисление);
5) 2,4-а-динитрофенол и др.
Факторы, способствующие повышению образования АТФ при высвобождении энергии в реакциях биологического окисления, называются сопрягающими. К сопрягающим факторам следует отнести гормоны щитовидной железы в физиологических дозах, паратгормон, витамины Е, К, Н, витамины группы В - В1, В2, В12; ионы К+, Mg2+, Са2+.
12.1.2. Нарушения основного обмена
Основной обмен - то количество энергии, которое необходимо для поддержания нормальных функций организма при полном мышечном и психическом покое, натощак (через 12-18 ч после приема пищи), при температуре окружающей среды 20-22 °С. Основной обмен у взрослого человека составляет около 1600-1700 ккал/сутки (соответствует 6700-7120 кДж/сутки).
В течение жизни основной обмен изменяется и зависит от возраста, пола, массы тела и роста. У детей в возрасте 3-5 лет и в период полового созревания он выше, чем у взрослых, у пожилых людей он снижается. У женщин основной обмен ниже, чем у мужчин, на 5-10%. Основной обмен также зависит от времени суток, сезона (зимой выше, чем летом), климата (у коренного населения Севера выше, чем у жителей южных районов). На основной обмен влияют центральная нервная и эндокринная системы и другие
факторы. Оценивается основной обмен методами прямой и непрямой калориметрии.
Метод прямой калориметрии основан на прямом определении количества калорий, выделяемых человеком в специальной камере (калориметре). Количество калорий оценивается путем определения нагревания содержащейся в стенках калориметра воды.
Метод непрямой калориметрии основан на определении количества поглощаемого кислорода и выделяемой углекислоты в единицу времени (минута, час, сутки). Калорическая ценность одного литра поглощенного кислорода находится в зависимости от величины дыхательного коэффициента. Дыхательный коэффициент - это отношение объема углекислого газа в выдыхаемом воздухе к потребляемому кислороду. Дыхательный коэффициент изменяется в зависимости от состава пищи, употребляемой пациентом. Наибольшая калорическая ценность 1 л кислорода отмечается у пациентов, использующих углеводную пищу. При такой диете дыхательный коэффициент может приближаться к 1,0. При употреблении белковой и жирной пищи понижается калорическая ценность поглощаемого кислорода и снижается дыхательный коэффициент. При окислении глюкозы дыхательный коэффициент равен 1,0, при окислении жиров снижается до 0,7.
Среди причин снижения основного обмена выделяются следующие:
1) голодание, кахексия;
2) анемии;
3) гипоксии различного генеза (тяжелой степени);
4) гипофункция эндокринных желез: щитовидной, половых, надпочечников, гипофиза;
5) гиперинсулинизм;
6) поражение ЦНС - некоторые психические заболевания, прогрессивный паралич, старческая деменция, олигофрения и др.;
7) состояние сна.
Причинами повышения основного обмена являются:
1) эмоциональное возбуждение, стресс;
2) расстройства функций ЦНС - некоторые формы неврозов, поражения стволового отдела мозга, повышение тонуса симпатической нервной системы;
3) лихорадочные и лихорадоподобные состояния;
4) патология эндокринной системы: сахарный диабет, повышение продукции гормонов: тиреотропного, адренокортико-
тропного (при базофильной аденоме гипофиза), соматотропного (при эозинофильной аденоме гипофиза), половых (при опухолях тестикул, сетчатой зоны коры надпочечников); тиреоидных (при диффузном токсическом зобе), катехоламинов (при феохромоцитоме);
5) сенсибилизация организма при введении чужеродного белка, различных антигенов;
6) умеренная активация сердечной деятельности и дыхания (физическая нагрузка, гипоксия легкой степени).
Значительные изменения основного обмена наблюдаются также при травмах, кровоизлияниях, опухолях мозга, поражении вегетативных отделов гипоталамуса.
12.2. ГОЛОДАНИЕ
Голодание - состояние организма при недостаточном или полном прекращении поступления пищи, а также при нарушении переваривания и всасывания пищевых веществ. Выраженная пищевая недостаточность в большинстве развитых стран встречается редко, хотя в определенной степени она может наблюдаться у неимущих или пожилых людей, в группах с особыми потребностями в питании (дети, беременные или кормящие женщины, больные и выздоравливающие, алкоголики), а также у лиц, потребляющих ограниченное количество пищи по желанию, болезни или в силу необходимости.
В природе голодание как физиологическое явление имеет широкое распространение: во время зимней (и летней) спячки у ряда млекопитающих, при холодовом оцепенении у рептилий, рыб и др. При этом голодание сочетается с глубоким торможением жизнедеятельности и с резким снижением интенсивности обмена веществ, что позволяет животному длительно поддерживать жизнь при ничтожных тратах энергии в периоды года, не благоприятные для активного образа жизни. Временное голодание связано с биологически выработанными специальными видовыми реакциями приспособления организмов. В процессе эволюции у человека сформировалась и генетически закрепилась способность переносить относительно длительные периоды голодания.
Научное изучение проблемы голодания стало проводиться в XIX в. после появления основных представлений об обмене ве-
ществ и энергии в организме. Первые сведения по этой проблеме связаны с экспериментальными работами В.В. Реньо и Ж. Рейзе (Франция), В. Шмидта, К. Фойта, М. Рубнера (Германия), в России - В.А. Манассеина. Большое значение имели выполненные в 80-90-х гг. XIX в. работы В.В. Пашутина и его учеников (П.М. Альбицкий, А.В. Репрев, П.П. Авроров, А.А. Лихачев и др.), а также более поздние исследования ученых школы Ф. Бенедикта (США). Большой материал по голоданию человека был собран в различных странах мира во время Первой и Второй мировых войн.
Различают следующие формы голодания: полное - при полном отсутствии пищи, но с приемом воды, и абсолютное, если отсутствует и прием воды; неполное голодание (недоедание) - недостаточное по отношению к общему расходу энергии (в данных условиях) питание; частичное, или качественное, голодание (неполноценное, или одностороннее, питание) - недостаточное поступление с пищей одного или нескольких пищевых веществ при достаточной энергетической ценности. Выделяют белковое, липидное, углеводное, минеральное, водное, витаминное частичное голодание, а также ограничение поступления пищевых волокон (клетчатки) - компонентов мембран растительных клеток.
В естественных условиях разграничение неполного и частичного голодания затруднительно, поскольку недоедание обычно сочетается с нарушением состава пищи. Чистые формы частичного голодания чаще наблюдаются только в экспериментальных условиях.
Самым тяжелым видом голодания у людей является полное голодание без приема воды (абсолютное), приводящее к гибели организма в течение 4-7 суток при явлениях обезвоживания и интоксикации. При полном голодании, но с поступлением воды (количественное, или общее, голодание) отмечены случаи продолжения жизни человека дольше 70 суток. При полном голодании жизнь поддерживается за счет утилизации в процессах обмена и выработки энергии имеющихся запасов питательного материала (липиды, углеводы) и продуктов, освобождающихся при постепенной атрофии части собственных тканей организма. Общее образование энергии в течение периода голодания постепенно понижается в соответствии с падением массы тела, но при расчете на единицу массы образование энергии, снижаясь на 20% вначале, затем мало изменяется в течение всего оставшегося периода голодания, в связи с чем регуляторные механизмы продолжают
функционировать почти до периода агонии. Поэтому все условия, повышающие метаболизм (мышечная деятельность, понижение окружающей температуры, ведущее к увеличению теплопродукции, перегревание организма, обезвоживание, гормональная активация обмена и др.), ускоряют гибель при голодании.
В.В. Пашутиным были проанализированы изменения массы тела собаки при полном голодании (рис. 12-1) с кривыми теплообразования, количеством выделения СО2, азота мочи и др. Эти
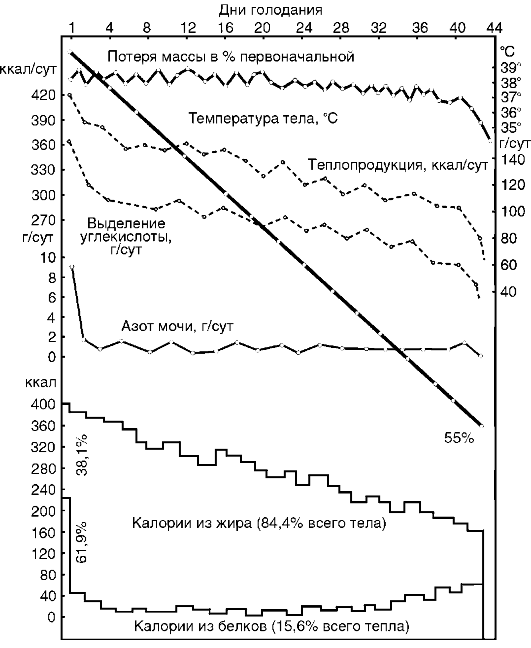 Рис. 12-1. Кривые потери массы, теплопроизводства, выделения СО2
и азота мочи при полном голодании собаки. Внизу - количество калорий,
образованных из жира и белка при полном голодании собаки (по В.В.
Пашутину)
Рис. 12-1. Кривые потери массы, теплопроизводства, выделения СО2
и азота мочи при полном голодании собаки. Внизу - количество калорий,
образованных из жира и белка при полном голодании собаки (по В.В.
Пашутину)
данные свидетельствуют о незначительном понижении основного обмена у животного, несмотря на прогрессирующее падение массы тела. При этом разрушается 40-45% начального количества белковых веществ, отмечается некоторое уменьшение температуры тела.
Интенсивность обмена у разных видов гомойотермных животных находится в зависимости от удельной поверхности, т.е. от соотношения между поверхностью тела и его массой, поэтому мелкие животные погибают при голодании обычно быстрее (мыши - в течение 2-4 дней, крысы - 9 дней, лошади могут выдерживать до 80 дней). Молодые растущие животные и дети погибают при голодании быстрее, чем взрослые. Суточные потери массы новорожденных при голодании в 2-3 раза больше, чем у более старших детей, что объясняется как меньшими размерами тела, так и менее совершенной регуляцией метаболизма. Обычно чем больше запасы жировой ткани, тем дольше организм может переносить голодание, при этом, однако, большое значение имеют индивидуальные особенности. Мужчины переносят голодание тяжелее женщин.
В развитии полного голодания различают три периода: 1) период начального приспособления, который длится 1-2 дня; 2) период относительно равномерного расходования собственных белков, энергетических субстратов и приспособления организма к жизни в условиях голодания («стационарный» период, наиболее длительный) и 3) «терминальный» период, длящийся последние 3-5 дней, заканчивающийся комой и смертью, - предагональные нарушения обмена и функций организма. Иногда второй период полного голодания разделяют на две фазы: первая длится неделю, вторая - несколько недель. В первую фазу полного голодания биоэнергетика организма поддерживается за счет распада углеводов, липидов и образующихся ацетоновых тел, во вторую - в основном за счет распада белков.
Жизненные функции организма в течение первого и второго периодов голодания сохраняются в пределах, близких к норме. Чувство голода, особенно выраженное в первые дни, в дальнейшем ослабевает. Температура тела колеблется в пределах нижней границы нормы, артериальное кровяное давление вначале может повышаться, позже снижается, развивающаяся первоначально тахикардия затем сменяется брадикардией, моторная деятельность желудка и кишечника вначале резко повышается (голодные сокращения, иногда спастического характера), в дальнейшем угнетается. Однако И.П.
Разенков показал, что у собак при голодании секреция желудочного сока, богатого белками, продолжается длительное время. Мочеотделение (при голодании с водой) происходит регулярно, количество мочи снижено, водный баланс положительный: происходит накопление воды в организме, появляются голодные отеки. У голодающих людей наблюдается вначале повышенная раздражительность, часто головные боли, беспокойный сон, позднее - понижение возбудимости, вялость, апатия, сонливость. Однако у человека отмечено сохранение умственной деятельности даже после 30-40 дней голодания. Мышечная активность значительно падает, но выполнение физической работы возможно в течение обоих периодов.
В первом периоде голодания основной обмен несколько снижается, дыхательный коэффициент близок к единице, что свидетельствует о включении в метаболизм углеводов. За первые сутки исчерпываются запасы гликогена, концентрация инсулина в крови снижается в 10-15 раз по сравнению с периодом пищеварения, концентрации глюкагона и кортизола увеличиваются. В результате изменения гормонального статуса и действия внутриклеточных механизмов регуляции нарастает скорость мобилизации жиров и глюконеогенеза из аминокислот и глицерина. Содержание глюкозы в крови уменьшается до нижних пределов нормы (3,5 ммоль/л) и на этом уровне поддерживается и в последующие периоды голодания (за счет глюконеогенеза).
Второй период голодания связан с продолжением мобилизации жиров. Концентрация жирных кислот в крови увеличивается в 3-4 раза по сравнению с постабсорбтивным состоянием, уровень кетоновых тел в крови через неделю голодания повышается в 10-15 раз. При такой концентрации ацетоуксусная кислота активно декарбоксилируется с образованием ацетона, который выводится с выдыхаемым воздухом и через кожу: уже на 3-4-й дни изо рта и от кожи голодающего исходит запах ацетона. Энергетические потребности мышц и большинства других органов удовлетворяются за счет жирных кислот и кетоновых тел. При низком уровне инсулина глюкоза в мышечные клетки не проникает, потребителями глюкозы являются инсулинонезависимые клетки, и прежде всего клетки мозга, но и в этой ткани биоэнергетика частично обеспечивается кетоновыми телами. Глюконеогенез продолжается за счет распада тканевых белков.
Интенсивность обмена веществ в целом снижена: через неделю голодания потребление кислорода уменьшается примерно
на 40%, происходят торможение окислительных процессов в митохондриях и угнетение окислительного фосфорилирования с образованием АТФ, т.е. развивается гипоэнергетическое состояние. Снижается превращение ацетил-КоА в цикле трикарбоновых кислот, в связи с этим он включается в синтез холестерола через образование гидроксиметилглутарил-КоА и ацетоновых тел, поэтому у голодающих отмечено накопление холестерола. Дыхательный коэффициент снижается за счет участия в метаболизме липидов, потери которых определяют характерный внешний вид голодающих (обилие кожных складок на животе, конечностях, шее, грудной клетке). Выделение азота с мочой, сниженное на 2-3-й дни, к 5-6-му дням возрастает, но держится на уровне 5,0 г мочевины в сутки (норма - 25-30 г) в течение нескольких недель, что соответствует отрицательному азотистому балансу с расщеплением примерно 20-25 г собственных тканевых белков в сутки. При снижении скорости распада белков уменьшается и активность глюконеогенеза. В этой фазе голодания основным источником энергии для мозга становятся ацетоновые тела. Если в это время голодающему вводить аланин или другие гликогенные аминокислоты, уровень глюкозы в крови повышается, а концентрация кетоновых тел снижается.
При продолжении голодания нарастает атрофия органов (рис. 12-2): масса жировой ткани уменьшается на 97%, селезенки - на 60%, печени - на 50%, тестикул - на 40%, мышц - на 31%. В наименьшей степени снижается масса сердечной мышцы и мозга - на 3-4%. Сохранение массы этих жизненно важных органов обусловлено адаптационными биологическими механизмами. Распад тканевых нуклеопротеидов вызывает повышенное выделение с мочой мочевой кислоты, а также усиленную экскрецию солей калия, кальция, фосфора. Указанные особенности метаболизма у голодающих сочетаются с изменениями функций многих органов и систем. Прогрессирующие процессы торможения в нервной системе проявляются нарастанием апатии и сонливостью; со стороны сердечно-сосудистой системы выявляются брадикардия и повышение кровяного давления; число дыхательных движений за единицу времени уменьшается; изменяется клеточный состав крови: снижается количество эритроцитов и лейкоцитов; отмечается гипопротеинемия; в результате мобилизации жира из жировых депо развивается выраженная триацилглицеролемия вплоть до появления липемии (мутная плазма). Гипопротеинемия способствует
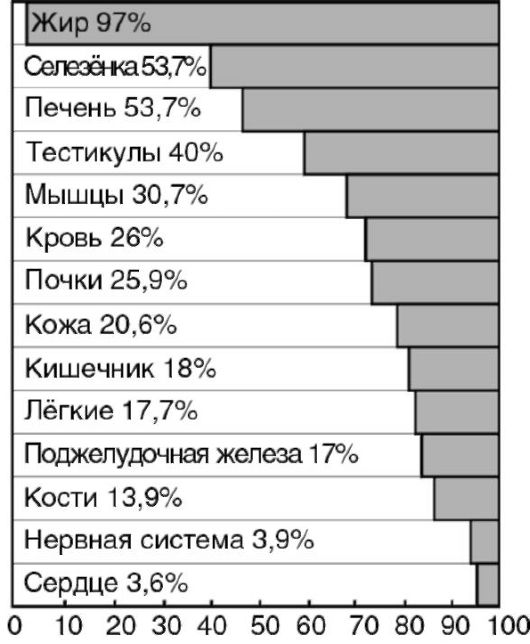 Рис. 12-2. Степень потери в массе органов при полном голодании собаки
Рис. 12-2. Степень потери в массе органов при полном голодании собаки
развитию отеков («голодные отеки») из-за падения осмотической и онкотической активности белков, особенно альбуминов.
Иммунологическая и аллергическая реактивность голодающего организма резко снижается, меняется его восприимчивость к действию различного ряда инфекционных возбудителей. Этим объясняется тот факт, что у голодающих жителей блокадного Ленинграда практически не регистрировались такие болезни, как бронхиальная астма, ангина; значительно изменилась клиническая картина пневмонии, кишечных и ряда других заболеваний.
Терминальный период голодания
наступает при расходовании 1/3-1/2 части всех белков (в норме 15 кг) и характеризуется усиленным распадом тканей. Развивается глубокое угнетение центральной нервной системы, нарастают слабость, апатия, переходящие в глубокую кому. Температура тела постепенно падает, опускаясь к моменту смерти до 30-28 °С. Выделение азотсодержащих продуктов с мочой (мочевина, креатинин, мочевая кислота, аминокислоты, пептиды), содержание калия, фосфора в моче, явления ацидоза и ацетонемии возрастают. Смерть наступает от интоксикации и истощения запасных веществ организма, в крови прогрессирующе нарастает содержание биогенных аминов, ряда гормонов (катехоламины) и других биологически активных веществ.
Откармливание при голодании возможно даже в терминальном периоде. Сначала применяется искусственное стимулирование сокоотделения в желудке и введение жидкой пищи в ограниченном количестве, затем постепенно переходят к более энергичному питанию после восстановления возбудимости пищевого центра. Перегрузка пищей в первые дни откармливания может вызвать тяжелые расстройства желудочно-кишечного тракта (рвота, поносы) и интоксикацию.
12.2.1. Лечение голоданием
Никаких отрицательных последствий после перенесенного кратковременного голодания не отмечается. При часто повторяющихся голоданиях (4-5 раз) масса тела восстанавливается с каждым разом труднее, развивается жировая дистрофия печени.
Голодание с лечебной целью применялось в древней Индии, Греции, Египте, в начале 80-х гг. XIX в. стало использоваться и в Европе для лечения сахарного диабета, ревматизма, подагры, при заболеваниях почек, гипертонии, бронхиальной астме, некоторых кожных, нервно-психических болезнях и др. По окончании лечебного голодания применяется ахлоридная диета (фруктовые соки, кефир, позже растительное масло) с максимальным содержанием витаминов и минеральных солей. При постоянном увеличении объема пищи и ее энергетической ценности осторожность связана с тем, что желудочнокишечный тракт в период лечебного голодания находится в состоянии гипофункции и атрофии. Период откармливания продолжается около 2 недель. Курс повторного лечебного голодания переносится значительно легче, но обязательным требованием является полное восстановление после предыдущего голодания.
12.2.2. Белково-калорийная недостаточность
При длительном неполном голодании в форме белковокалорийной недостаточности развивается выраженная гипопротеинемия, нарушается соотношение между различными белковыми фракциями плазмы крови (в первую очередь резко сокращается фракция альбуминов). Из-за нарушения секреторной деятельности желудочно-кишечного тракта пищеварительная система не в состоянии усваивать даже те питательные вещества, которые всетаки поступают в организм. Развивается прогрессирующая дистрофия органов и тканей. Резкое снижение содержания белка в сыворотке крови становится причиной так называемых голодных отеков. При продолжающейся белково-калорийной недостаточности развиваются тяжелейшие расстройства со стороны эндокринной, иммунной и нервной систем, неизбежно приводящие к распаду личности (алиментарный маразм) и гибели организма.
У детей белково-калорийная недостаточность протекает значительно тяжелее, чем у взрослых, что связано с большей и постоянно растущей потребностью в белках.
Одним из видов нарушения питания, связанного с недостаточным содержанием в пище белков и потреблением низкокалорийных продуктов, является квашиоркор (kwashiorkor) (рис. 12-3).
B настоящее время это заболевание довольно часто встречается среди некоторых африканских племен, где оно является одной из основных причин детской смертности. Само название болезни на языке племени ашанти (Западная Африка) означает «красный (золотой) мальчик, родившийся в воскресенье», на языке га (Гана) - «отверженный ребенок». Квашиоркор развивается в тех случаях, когда после длительного кормления грудным молоком ребенка отнимают от груди и переводят на традиционную для местного населения пищу, которая бедна белками, особенно незаменимыми аминокислотами, и плохо усваивается организмом. В результате ребенок не получает с пищей как необходимого количества белка, так и калорий. Основными симптомами заболевания являются: истощение (кахексия), отставание в росте, атрофия мышц, голодные отеки, диарея, поражения кожи (депигментация, красноватый оттенок, дерматиты и др.), потеря аппетита, апатия, малоподвижное лицо, а также нарушение иммунитета и сдвиги в системе крови (цитопеническое состояние) - все это факторы, способствующие присоединению инфекций. При переводе больных детей на полноценное питание возможно выздоровление.
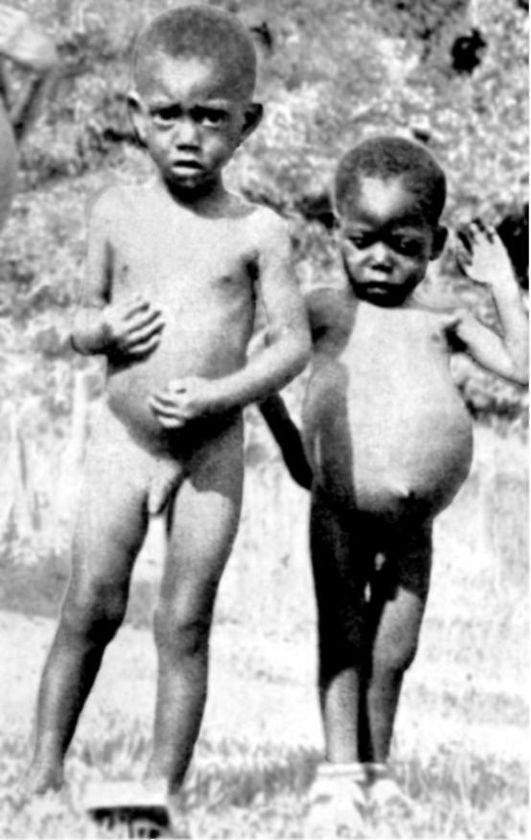 Рис. 12-3. Ребенок (справа), страдающий тяжелой формой неполноценного питания
Рис. 12-3. Ребенок (справа), страдающий тяжелой формой неполноценного питания
12.3. ПАТОФИЗИОЛОГИЯ ОБМЕНА ВИТАМИНОВ
Витамины - это группа органических веществ (незаменимые пищевые факторы, не синтезируемые в организме в необходимом количестве, не включающиеся в структуру тканей и не используемые организмом в качестве источника энергии), абсолютно необ-
ходимых для нормального развития и жизнедеятельности организма животных и человека. Они обеспечивают оптимальную скорость протекания биохимических реакций и физиологических процессов за счет выполнения непосредственно или в составе более сложных соединений каталитических и регуляторных функций.
Выделяют две группы витаминов: жирорастворимые (А, D, Е и К) и водорастворимые (В1, В2, пантотеновая кислота, РР, В6, В9, В12, Н, С и Р). Кроме того, существуют так называемые витаминоподобные вещества (холин, липоевая, пангамовая, оротовая кислоты, инозит, убихинон, карнитин, витамин U и др.), обладающие некоторыми свойствами витаминов, но не являющиеся строго обязательными пищевыми факторами (нутриентами). Дефицит витаминоподобных веществ не проявляется определенным специфическим симптомокомплексом.
Заболевания, развивающиеся при глубоком дефиците витаминов в организме (отсутствии поступления с пищей некумулируемых и несинтезируемых эндогенно витаминов либо нарушении их обмена) и характеризующиеся клиническими симптомами, принято называть авитаминозами (цинга, бери-бери, пеллагра, рахит и др.). В случае если витамины поступают с пищей и/или усваиваются в недостаточном количестве либо они запасаются или частично синтезируются в организме, клиническая картина заболевания (гиповитаминоз) менее специфична и характеризуется преобладанием общих, неспецифических проявлений умеренного дефицита витаминов (головокружение, раздражительность, головная боль, ослабление памяти, физическая и умственная утомляемость и т.д.). Недостаток витамина сначала ведет к мобилизации его запасов в организме и истощению тканевых депо (печень и жировая ткань для жирорастворимых витаминов), затем к биохимическим нарушениям (субклинический дефицит) и, наконец, к истинной недостаточности с выраженной клинической картиной заболевания. При недостаточности питания может иметь место дефицит не одного витамина, а нескольких (полигипоили полиавитаминоз), приводящий к развитию сложной клинической картины. Гипервитаминозы - патологические состояния, вызванные поступлением в организм чрезмерно большого количества витаминов и характеризующиеся нарушениями физиологических процессов, связанными со специфической ролью витаминов в обмене веществ, а отчасти имеющие характер неспецифического отравления. Более других токсичны жирорастворимые витамины А и D.
Гипо- и авитаминозы у человека и животных называют экзогенными (первичными), если их причиной является недостаточное поступление или полное отсутствие витаминов (или их предшественников) в пище при нормальных потребностях. Важнейшими причинами эндогенных (вторичных) гипо- и авитаминозов являются:
1) недостаточный синтез некоторых витаминов кишечной микрофлорой;
2) нарушения процессов всасывания витаминов при заболеваниях желудочно-кишечного тракта, печени, поджелудочной железы;
3) усиленный распад или конкурентная утилизация витаминов в кишечнике при наличии кишечных паразитов и патогенной микрофлоры;
4) поступление в организм антивитаминов (пищевых, лекарственных и др.);
5) повышенные потери витаминов;
6) повышенная потребность в витаминах при некоторых физиологических и патологических состояниях (беременность, лактация, физическая и нервно-психическая нагрузка, тиреотоксикоз и др.);
7) нарушения обмена витаминов, связанные с заболеваниями органов, которые принимают участие в метаболизме витаминов, или с недостатком необходимых для метаболизма витаминов веществ;
8) врожденные нарушения обмена витаминов.
К врожденным нарушениям обмена и функций витаминов относятся витаминзависимые состояния (корригируются мегадозами витаминов) и витаминрезистентные состояния (введение даже высоких доз соответствующих витаминов не устраняет явлений их недостаточности). Клиническая картина врожденных нарушений обмена и функций витаминов (пиридоксинзависимый судорожный синдром, пиридоксинзависимая анемия, витамин D-резистентный рахит, витамин D-зависимый рахит и др.) мало отличается от клинической картины алиментарного авитаминоза. Причинами развития подобных заболеваний являются генетические дефекты, вызывающие нарушения как собственно обмена витаминов, который осуществляется при участии специфических белков и ферментов (нарушения процессов всасывания, транспорта, превращений витаминов в коферменты или в активные формы, взаимодействия кофермента с апоферментом, т.е. дефект образования холофер-
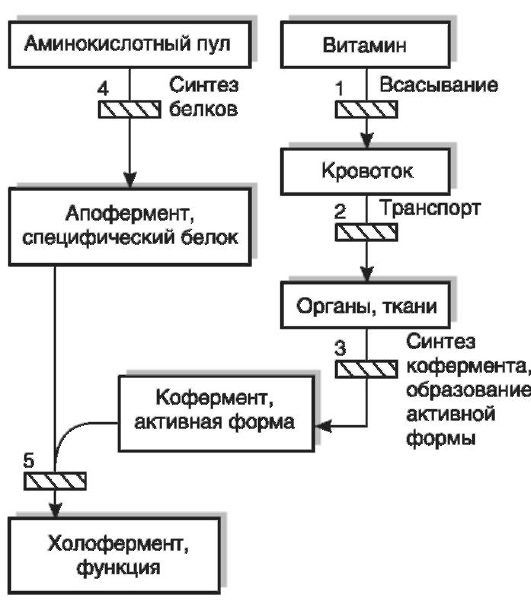 Рис. 12-4. Возможные
нарушения обмена и функций витаминов: 1 - нарушение всасывания в
кишечнике; 2 - нарушение транспорта к органам и тканям; 3 - нарушение
превращений в коферменты или активные формы; 4 - нарушение синтеза
апоферментов или специфических белков, взаимодействующих с витамином при
реализации его функций; 5 - нарушение взаимодействия кофермента или
иной активной формы витамина с апоферментом или специфическим белком (по
В.Б. Спиричеву, Ю.И. Барашневу, 1977)
Рис. 12-4. Возможные
нарушения обмена и функций витаминов: 1 - нарушение всасывания в
кишечнике; 2 - нарушение транспорта к органам и тканям; 3 - нарушение
превращений в коферменты или активные формы; 4 - нарушение синтеза
апоферментов или специфических белков, взаимодействующих с витамином при
реализации его функций; 5 - нарушение взаимодействия кофермента или
иной активной формы витамина с апоферментом или специфическим белком (по
В.Б. Спиричеву, Ю.И. Барашневу, 1977)
мента), так и нарушения синтеза тех белков, при взаимодействии с которыми витамины или их активные формы осуществляют свои специфические функции в обмене веществ (наследственный дефект синтеза апофермента) (рис. 12-4).
Согласно данным эпидемиологических исследований, рацион питания значительной части населения России характеризуется недостаточным содержанием эссенциальных факторов питания: витаминов, микроэлементов и др. Недостаток витамина С, по обобщенным данным, выявляется у 80-90% обследуемых; недостаточная обеспеченность витаминами В1, В2, В6, фолиевой кислотой - у 40-80%; 40-55% населения испытывают дефицит каротина. Дефицит витаминов является постоянно действующим неблагоприятным фактором у всех групп населения во всех регионах страны и зачастую носит характер полигиповитаминоза (витамин С, витамины группы В, каротин), сочетающегося у значительной части детей, беременных и кормящих женщин с недостатком железа и развитием железодефицитной анемии. В ряде регионов полигиповитаминоз сочетается с недостаточным поступлением йода, селена, кальция, фтора и ряда других макро- и микроэлементов, что требует применения в терапии витамин-минеральных комплексов.
Знание этиологии и патогенеза нарушений обмена витаминов необходимо для проведения дифференциальной диагностики и патогенетической терапии данных состояний.
12.3.1. Жирорастворимые витамины Витамины группы А
Термин «витамин А» (антиксерофтальмический фактор) объединяет группу близких по химической структуре соединений: ретинола, ретиналя, ретиноевой кислоты, их эфиров.
Характерными симптомами недостаточности витамина А у человека и животных являются специфические поражения глаз, кожи, слизистых оболочек, торможение роста, снижение массы тела, общее истощение организма. Поражение эпителия кожи проявляется его ускоренной пролиферацией и патологическим ороговением, что сопровождается развитием фолликулярного папулезного гиперкератоза («жабья кожа»). Нарушение эпителизации вызывает сухость кожи, шелушение, развитие вторичных инфекций. При авитаминозе А поражается эпителий слизистой оболочки мочеполовой и дыхательной систем, а также пищеварительного тракта, что вызывает (усугубляет) нарушение всасывания жирорастворимых витаминов. Снижение барьерной функции кожи и слизистых оболочек способствует проникновению в организм болезнетворных микробов и возникновению дерматитов, ларинготрахеобронхитов, пневмоний, циститов, пиелитов и т.д. Ороговение эпителия слезного канала приводит к его закупорке, развитию сухости конъюнктивы и роговой оболочки глаза (ксерофтальмия) с последующим конъюнктивитом, отеком, изъязвлением и размягчением роговицы (кератомаляция), так как глазное яблоко не омывается слезной жидкостью, обладающей бактерицидными свойствами. Может развиться атрофия слезных желез. На склерах образуются очаги гиперкератоза (бляшки Бито). Исходом кератомаляции может быть помутнение роговицы (бельмо), приводящее к слепоте (амблиопия). Специфическим симптомом авитаминоза (гиповитаминоза) А является куриная, или сумеречная, слепота (гемералопия). Недостаточность витамина А проявляется прежде всего нарушением темновой адаптации - увеличением промежутка времени, необходимого для адаптации после перехода из освещенного помещения в темное. Наряду с этим увеличивается зрительный порог, т.е. минимальная интенсивность света, которая вызывает зрительные ощущения.
Витамин А обладает широким спектром биологического действия. Он участвует в процессах фоторецепции, необходим для роста тела, воспроизведения потомства, поддержания иммуно-
логического и гематологического статуса, пролиферации и дифференцировки тканей (эпителии кожных покровов, слизистых, сперматогенный эпителий, костная ткань); влияет на многие стороны обмена веществ: тканевое дыхание и энергетический обмен в тканях (стабилизирует митохондриальные мембраны и активирует синтез убихинона), обмен белков (воздействует на инициацию репликации), углеводов (участвует в синтезе мембранных гликопротеинов и гликолипидов), липидов (влияет на превращение мевалоновой кислоты в холестерин), нуклеиновых кислот. Большинство метаболических эффектов витамина А связано с его влиянием на стабильность и проницаемость клеточных мембран.
Развитие гемералопии при гиповитаминозе А связано с участием витамина в фотохимическом акте зрения. Светочувствительным пигментом сетчатки (палочек) является сложный белок родопсин, состоящий из липопротеина опсина и простетической группы, представленной 11-цис-ретиналем (рис. 12-5). На свету родопсин расщепляется на опсин и ретиналь, последний подвергается превращению в трансформу. Фотоизомеризация ретиналя вызывает местную деполяризацию мембраны, что приводит к возникновению электрического импульса, который распространяется по нервному волокну. Цикл превращений светочувствительного пигмента завершается в темноте регенерацией родопсина (соединением опсина и 11-цис-ретиналя, который может синтезироваться из цис-ретинола или транс-ретиналя) и восстановлением чувствительности к свету слабой интенсивности. Потери ретиналя в цикле должны восполняться за счет поступления в организм ретинола с пищей. При дефиците витамина А нарушается темновая фаза цикла - восстановление родопсина. Развивается дегенерация наружных сегментов палочек.
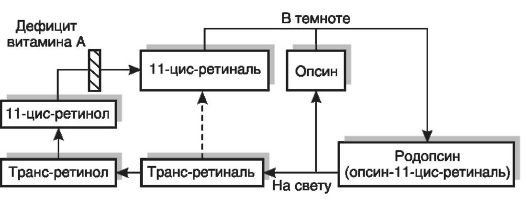 Рис. 12-5. Механизм нарушения сумеречного зрения при гиповитаминозе А
Рис. 12-5. Механизм нарушения сумеречного зрения при гиповитаминозе А
Глубокий дефицит витамина А может оказывать влияние на цветоощущение, так как он входит и в состав йодопсинов (светочувствительных пигментов колбочек, отвечающих за цветовое зрение). Дефицит витамина А обусловливает нарушение синтеза хондроитинсульфатов, влияющее на формирование соединительной ткани, в том числе костной, из-за увеличения распада фосфоаденозинфосфосульфата под действием лизосомальных сульфатаз, подавляемых ретиноидами.
В условиях гиповитаминоза А замедляется синтез гликопротеинов, нарушаются рецепторный состав клеточных поверхностей, выработка гормонов, секретов, разрыхляется гликокаликс, нарушается структура межклеточного вещества, снижаются адгезивные свойства клеток. В клетках тормозится синтез РНК, падает активность ферментов, обеспечивающих защиту липидов от окисления. При недостатке витамина А вследствие нехватки ростовых факторов и медиаторов задерживается пролиферация эпителиальных и мезенхимных клеточных популяций, возможно, активируется апоптоз. Развивается функциональная неполноценность железистых эпителиев с их метаплазией. Страдает иммунитет, сперматогенез прекращается на стадии мейоза.
Дефицит витамина А ведет к нарушению противосвертывающих механизмов крови (рост толерантности плазмы к гепарину, гиперфибриногенемия).
У новорожденных практически нет запасов витамина А, поэтому при отсутствии его в рационе питания быстро развивается авитаминоз с соответствующим симптомокомплексом, вплоть до летального исхода.
Вторичный гиповитаминоз А возможен при печеночной недостаточности, так как в клетках печени ретинолэстераза освобождает ретинол, который транспортируется в крови в связанном с белком состоянии; при протеинурии - вследствие потери ретинолсвязывающего белка; при алкоголизме - вследствие недостатка цинка и ниацина, необходимых для метаболизма витамина А, а также нарушения всасывания ретинола из-за алкогольной мальабсорбции. Нарушение превращения провитаминов в витамин А с развитием каротинемической псевдожелтухи может возникать при гипотиреозе, так как активность β-каротиндиоксигеназы (фермента, превращающего β-каротин в ретиналь) стимулируется гормонами щитовидной железы.
Недостаточность витамина А может развиваться при врожденном нарушении превращения β-каротина в ретиналь, связанном с генетическим дефектом β-каротиндиоксигеназы.
Генетический дефект, при котором увеличивается потребность в витамине А для поддержания нормального состояния эпителиальной ткани, лежит в основе фолликулярного кератоза Дарье. Наряду с изменениями кожи и слизистой оболочки полости рта у больных с данной патологией отмечаются отставание умственного развития и психозы.
Поступление в организм больших доз витамина А, во много раз превышающих физиологическую потребность, вызывает характерную картину интоксикации, известную под названием гипервитаминоз А. В клинической картине острой формы отравления у животных, часто заканчивающейся смертью, преобладают судороги и параличи. Хроническая форма характеризуется остановкой роста, потерей массы тела, спонтанными переломами длинных трубчатых костей (вследствие торможения остеогенеза и активации хондролитических процессов), кровоизлияниями во внутренние органы, дегенеративными изменениями эндокринных желез, печени, селезенки.
Гипервитаминоз А у человека может возникнуть в результате употребления продуктов, содержащих большое количество витамина А, либо токсических доз препаратов витамина А (50 тыс. МЕ в сутки при продолжительном применении или 1-6 млн МЕ однократно). Острый гипервитаминоз А чаще всего выражается в головокружении, тяжелой головной боли, сонливости, ступоре, диспепсических явлениях (понос, рвота), шелушении кожи. Хроническое отравление витамином А влечет за собой головную боль, сухой дерматит, выпадение волос, боль в суставах и костях при ходьбе и надавливании, отек вдоль трубчатых костей, повышенную ломкость костей, кальциноз связок, анорексию, потерю массы тела, гепатоспленомегалию, геморрагический синдром. Нередко наблюдаются симптомы экзофтальмии и повышения давления спинно-мозговой жидкости, отек соска зрительного нерва, связанный с развитием ликворной гипертензии и сдавлением нервов в отверстиях костей черепа. Высокие дозы витамина А эмбриотоксичны. На фоне гипервитаминоза А наряду с активацией фибринолиза (ретинол стимулирует продукцию активатора плазминогена клетками эндотелия), сопровождаемой гипофибриногенемией, проявляется рост антитромбиновой активности.
При гипервитаминозе А в клетках нарушаются окислительно-восстановительные реакции, ускоряются процессы гликозилирования; в липидном слое мембран, изменяющих свои физико-химические свойства, появляются участки, обогащенные витамином А, гипертрофируется комплекс Гольджи, разрушаются мембраны митохондрий, цитоплазматического ретикулума, нарушается структура гликокаликса и межклеточных контактов. Клетки разобщаются, усиливается фагоцитоз. Развиваются слизистая метаплазия ороговевающих эпителиев, нарушение зрения, генерализованное аутоиммунное воспаление, дегенеративные изменения многих органов и систем (некроз гепатоцитов и клеток почечного канальцевого эпителия, фиброз печени).
Как гипо-, так и гипервитаминоз А сопровождаются активацией свободнорадикальных процессов, поскольку ретинол в гидрофобной области мембран клеток выполняет роль стерического регулятора, определяющего доступность ненасыщенных жирных кислот, которые входят в состав фосфолипидов, для окисления.
Витамин А содержится в основном в продуктах животного происхождения (печень, цельное молоко, сливки, сметана, сливочное масло, сыр). Особенно богаты им печень и внутренний жир некоторых видов рыб (палтус, треска) и морского зверя (киты, моржи, тюлени, дельфины), белого медведя. Основным пищевым источником каротинов (провитаминов А) являются продукты растительного происхождения: морковь, красный перец, томаты, зелень петрушки, салат, шпинат, щавель, облепиха, шиповник и др. Суточная потребность в витамине А для взрослого человека - 1,5-3,0 мг, или 2-5 мг β-каротина; для детей выше, чем для взрослых. Повышается при беременности, лактации, утомлении, продолжительном напряжении зрения.
Витамины группы D
Витамин D (кальциферол) - антирахитический фактор, его открытие связано с поисками лечебного препарата для профилактики и лечения рахита. Основным источником витамина D является эндогенный синтез: до 80% необходимого количества витамина D3 (холекальциферола) организм может синтезировать в коже (в макрофагальных клетках дермы) под воздействием ультрафиолетовых лучей из 7-дегидрохолестерина (провитамина D3). В растениях и дрожжах содержится эргостерин (провитамин D2) - предшественник эргокальциферола (витамина D2).
Дефицит кальциферола, часто проявляющийся у людей в современных условиях, неразрывно связан с образом жизни значительной части населения - скоплением людей в больших городах, сокращением времени пребывания на свежем воздухе. Уменьшение содержания витамина D в организме наблюдается при недостаточном поступлении его с продуктами питания при одновременном снижении эндогенного синтеза. Велика вероятность развития гиповитаминоза D у недоношенных детей, так как женское молоко содержит недостаточное его количество, а через плаценту витамин поступает в основном в последнем триместре беременности. Эндогенный гипо- и авитаминоз D может возникать при нарушении переваривания и (или) всасывания липидов, в том числе жирорастворимых витаминов (механическая желтуха, холестаз, панкреатическая недостаточность); нарушении образования активных форм витамина: гидроксилирования в печени (при циррозе) и в почках (при хронической почечной недостаточности); усилении распада (например, под влиянием ряда лекарственных препаратов, индуцирующих активность ферментов системы микросомального окисления).
Уменьшение синтеза и/или поступления витамина D с пищей, нарушения метаболизма холекальциферола вызывают «кальципенический» рахит у детей и остеомаляцию у взрослых. Нарушения кальций-фосфорного обмена, играющие главную роль в патогенезе рахита, приводят к нарушению минерализации костной ткани, особенно в зонах роста трубчатых и черепных костей. Помимо нарушения минерализации остеоида, при рахите наблюдаются усиленное рассасывание костной ткани, вымывание кальция и фосфата из кости, что обусловлено гиперсекрецией паратгормона (развитие вторичного гиперпаратиреоза), стимулируемой низким уровнем кальция в крови. Ухудшение энергообеспечения процессов костеобразования и минерализации костной ткани связано с нарушением всасывания и метаболизма цитрата при недостатке витамина D. Рахитические кости медленнее растут, не способны выдерживать нормальную статическую и динамическую нагрузки, легко подвергаются механическим деформациям, что обусловливает внешние проявления рахита со стороны скелета: искривление нижних конечностей, рук и позвоночника, утолщение эпифизов длинных трубчатых костей («рахитические браслеты»), «рахитические четки» на грудных концах ребер, «куриная грудь» с выступающей грудиной и сдавленными с боков ребрами, задержка
закрытия родничков и прорезывания зубов, «лоб Сократа» (периостальные наслоения остеоида в области лобных и теменных бугров), краниотабес (уплощение затылочных костей) (рис. 12-6, 12-7). К ранним клиническим проявлениям рахита относятся раздражительность, мышечная гипотония (связанная не только с изменением нервной регуляции, но и с нарушением структуры и метаболизма мышечных волокон), тетания (в связи с системной гипокальциемией), облысение затылка, развитие бронхолегочных заболеваний, связанное с ухудшением вентиляции легких и снижением иммунитета.
Недостаток витамина D у взрослых вызывает нарушение периостального окостенения (остеоид вырабатывается, но не минерализуется) - развивается остеомаляция. У больных отмечаются гипоцитратемия, гипофосфатемия, в тяжелых случаях - гипо-
 кальциемия,
развивается гиперпаратиреоз. При развитии злокачественных
новообразований остеомаляция может быть связана с торможением
образования кальцитриола в почках пептидными факторами, синтезированными
в опухолевых тканях.
кальциемия,
развивается гиперпаратиреоз. При развитии злокачественных
новообразований остеомаляция может быть связана с торможением
образования кальцитриола в почках пептидными факторами, синтезированными
в опухолевых тканях.
Физиологически активной формой является не сам витамин D, а его метаболиты (в настоящее время их известно более 40: 25-ОНD3, 1а,25-(ОН)2D3, 24,25-(ОН)2D3, 25,26-(ОН)2D3, 1,24,25-(ОН.уЭ3 и др.), образующиеся в результате обменных превращений в тканях. Образование кальцитриола (1,25-дигидроксихолекальциферол [1,25-(ОН)2D3]), поддерживающего уровень кальция и фосфора в сыворотке крови в узких физиологических границах, происходит в организме в несколько этапов. Фотохимически синтезированный в коже витамин D3 под воздействием гидроксилаз подвергается двум реакциям гидроксилирования: сначала в печени в 25-м положении (25-гидроксилаза), затем в почках в 1-м положении (1а-гидроксилаза) (рис. 12-8). Образование активной формы витамина D3 в почках контролируется через систему обратных связей с обменом Са, Р, секрецией паратиреоидного гормона, кальцитонина, соматостатина, пролактина, половых гормонов и инсулина. Избыток 1,25-(ОН)2D3 в организме ингибирует его образование.
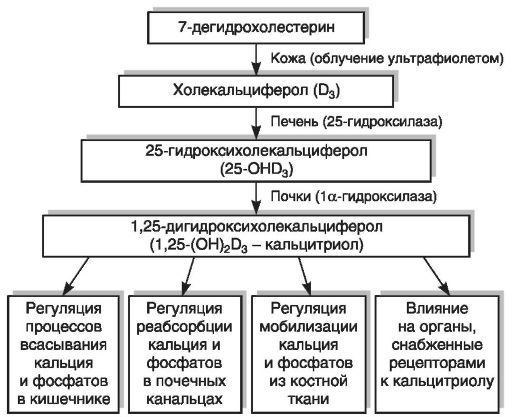 Рис. 12-8. Метаболизм и функции витамина D3
Рис. 12-8. Метаболизм и функции витамина D3
Дефицит витамина D ведет к понижению концентрации кальция и фосфора в сыворотке крови и нарушениям деятельности многих систем организма: нервной, мышечной, костной, репродуктивной, иммунной. Регулирующий эффект витамина D в отношении кальций-фосфорного обмена достигается за счет воздействия кальцитриола на следующие процессы: реабсорбцию кальция и фосфатов в почечных канальцах, мобилизацию элементов из костной ткани, всасывание Са2+ и фосфатов в кишечнике (наиболее весомый вклад в антирахитическое действие). В клетках кишечника под влиянием кальцитриола происходит синтез Сасвязывающего белка (кальбиндин, или холекальцин) и стимуляция транспорта Са2+. Остеогенез и ремоделирование костной ткани регулируются 24,25-(ОН)2D3. Рецепторы к 1,25-(ОН)2D3 обнаружены, помимо органов-мишеней (кишечник, костная ткань, почки), и в ряде других органов и тканей: в коже, мозгу, гипофизе, половых, молочных, паращитовидных железах и др. Биологические эффекты кальцитриола реализуются через взаимодействие его с рецепторами в ядре клеток, следствием чего является избирательная стимуляция процесса транскрипции ДНК, приводящая к синтезу молекул матричной РНК и трансляции кальцийсвязывающих белков и гормонов, регулирующих обмен Са2+. Витамин D не только влияет на процессы, непосредственно связанные с минеральным обменом, но также регулирует пролиферацию и дифференцировку различных типов клеток, в том числе клеток иммунной системы (макрофаги, Т- и В-лимфоциты). Активированные цитокинами макрофаги могут синтезировать кальцитриол, который усиливает их способность к завершению фагоцитоза.
Избыток витамина D, к которому особенно чувствительны дети, оказывает токсическое действие на организм. К наиболее вероятным причинам гипервитаминоза D относятся: продолжительная передозировка или однократный прием токсической дозы витамина, активация почечного (при первичном гиперпаратиреозе) и внепочечного синтеза 1,25-(ОН)2D3 (в цитокинактивированных макрофагах при саркоидозе). Гипервитаминоз D проявляется глубокими нарушениями гомеостаза Са2+ и фосфатов, которые не способны корректироваться нейрогуморальными системами организма. При избытке витамина D развивается усиленная резорбция костной ткани, которая не сопровождается соответствующей реутилизацией минеральных веществ, так как подавлены процесс образования кости и ее минерализация. Это не связано с влия-
нием паратиреоидного гормона (при гипервитаминозе активность паращитовидных желез снижена). Остеопения, которая развивается в этих условиях, является результатом торможения дифференцировки хондроцитов, дефицита остеобластов и остеонекроза, приводящего к дезинтеграции комплекса органического матрикса и минерального компонента. Развиваются гиперкальциемия, гиперкальциурия, отмечаются отложения труднорастворимых солей (почечные конкременты, кальцификация почек, а также кровеносных сосудов, печени, легких, сердечной мышцы, стенок кишечника, приводящая к тяжелому и стойкому нарушению функций этих органов), снижение активности щитовидной железы и гонад, мышечная ригидность, гипертензия. Наблюдаются также неспецифические симптомы отравления: тошнота, диарея, потеря аппетита, головная боль, слабость. У детей гипервитаминоз D вызывает раннее зарастание родничков (с возможным формированием микроцефалии), прекращение роста скелета в длину, утолщение диафизов и эпифизов. Летальный исход при тяжелом гипервитаминозе D может быть вызван почечной недостаточностью, сдавлением мозга, ацидозом и гиперкальциемическими аритмиями.
В эксперименте показано, что избыток витамина D и его метаболитов может оказывать прямое повреждающее действие на мембраны клеток, усиливая в них процессы перекисного окисления липидов.
К наследственным нарушениям обмена и функций витамина D относятся витамин D-зависимый наследственный рахит I типа (дефект почечной 1а-гидроксилазы) и витамин D-зависимый наследственный рахит II типа (дефект тканевых рецепторов кальцитриола).
Наибольшее количество витамина D3 содержится в продуктах животного происхождения - сливочном масле, желтке яиц, печени, рыбьем жире. Из растительных продуктов наиболее богаты витамином D2 растительные масла (подсолнечное, оливковое и др.). Суточная потребность в витамине D для взрослых - 10-25 мкг.
Витамины группы Е
Важнейшие проявления недостаточности витаминов группы Е
(токоферолы, антистерильный фактор, витамин размножения) у экспериментальных животных (крысы, кролики, собаки, кошки и др.) - нарушения эмбриогенеза и дегенеративные изменения
репродуктивных органов, приводящие к стерильности. У самок процесс оплодотворения яйца не нарушен, но развивающиеся патологические изменения в матке, дегенерация эмбриональной сосудистой системы приводят к внутриутробной гибели плода. У самцов происходит атрофия семенников с дегенерацией сперматогенных клеток, приводящая к полной или частичной стерильности. Наряду с этим отмечаются дистрофия скелетных и гладких мышц, миокарда (фрагментация мышечных волокон, микронекрозы, деструкция митохондрий, нарушение образования креатинфосфата); гипотонус, резкое органичение подвижности животных; жировое перерождение печени; энцефаломаляция, демиелинизация и глиоз в спинном мозгу, атаксия, гипорефлексия, дизартрия, параличи конечностей.
Витамин Е депонируется в организме, поэтому признаки его недостаточности у человека обнаруживаются редко, за исключением населения тех стран, где растительные жиры (основные источники витамина в пище человека) почти не используются для питания. Уменьшение содержания витамина Е в сыворотке крови отмечается при недостаточности питания, нарушении всасывания (муковисцидоз, атрезия желчных протоков, стеаторея и др.), анемии при квашиоркоре, гемолитической анемии у недоношенных, абеталипопротеинемии, отравлении некоторыми химическими веществами (например, акронитрилом), лучевом поражении, ишемической болезни сердца, злокачественных новообразованиях, туберкулезе легких, неспецифических воспалительных процессах в легких, ожоговой болезни, язвенной болезни желудка и двенадцатиперстной кишки, проведении химиотерапии (стрептомицином, тубазидом).
Основная функция токоферолов, наиболее активных природных жирорастворимых антиоксидантов, - регуляция интенсивности свободнорадикальных реакций в клетках, выражающаяся в ограничении скорости процессов перекисного окисления ненасыщенных жирных кислот в липидах биологических мембран. Токоферолы являются синергистами селена как кофактора глутатионпероксидазы, участвующей в нейтрализации гидроперекисей липидов. Витамин Е защищает от перекисного окисления ненасыщенную боковую цепь витамина А, повышая его биологическую активность, восстанавливает коэнзим Q, принимающий участие в окислительном фосфорилировании, регулирует активность фосфолипазы А2, участвующей в метаболизме арахидоновой кислоты, - предшественницы простагландинов и лейкотриенов. Регуляцию
биосинтеза ферментов витамин Е осуществляет, возможно, на уровне транскрипции матричных РНК.
Витамин Е, являясь антиоксидантом, стабилизирует клеточные мембраны и обеспечивает нормальное течение биохимических процессов, поэтому недостаточное содержание токоферолов в организме приводит к формированию различных проявлений так называемой мембранной патологии. Токоферолы реагируют с активными формами кислорода (гася центры инициации свободнорадикальных процессов), свободными радикалами ненасыщенных жирных кислот и их перекисями, обезвреживая их (обрывая уже инициированную цепную реакцию перекисного окисления липидов - ПОЛ или замедляя ее), свободными жирными кислотами, повышение содержания которых рассматривается как патогенетический механизм повреждения клеточных структур, в частности скелетных и сердечной мышц. Мембраностабилизирующее действие токоферолов обусловлено также их способностью предохранять от окисления SH-группы мембранных белков, в том числе ферментов, и образовывать в качестве структурного компонента мембран гидрофобные комплексы с ненасыщенными жирными кислотами, защищая мембраны от окисления.
Недостаточность токоферолов проявляется разнообразными симптомами, связанными со структурно-функциональными нарушениями мембран: гемолитическая анемия у недоношенных детей, атрофия семенников и бесплодие, рассасывание плода на ранних сроках беременности, мышечная дистрофия, развитие морфологических изменений в клетках паренхиматозных и эндокринных органов, сопровождающихся нарушением их функций (гибель клеток слизистой оболочки кишечника, появление участков некроза в печени, ткани мозга, особенно мозжечка, щитовидной железе и др.). При недостатке витамина Е наиболее выраженно повреждаются клетки быстро пролиферирующих тканей (зародышевые ткани, гемопоэтические клетки, сперматогенный эпителий, гепатоциты, слизистая оболочка кишечника, эндокринные органы), в которых осуществляется интенсивный мембраногенез, сопровождающийся снижением антиоксидантных резервов, а также клетки, в которых интенсивно образуются активные формы кислорода (нейроны и миоциты). Непосредственная причина мышечной дистрофии - высвобождение лизосомальных гидролаз, связанное, возможно, с нарушением обмена полиненасыщенных жирных кислот в лизосомальных мембранах.
Недостаток витамина Е сопровождается снижением содержания иммуноглобулина Е и количества Т- и В-лимфоцитов в периферической крови. Токоферолы участвуют в синтезе нуклеиновых кислот, гема микросомальных цитохромов и других гемсодержащих белков. При недостаточности витамина Е происходит снижение интенсивности тканевого дыхания, что объясняется влиянием а-токоферола на синтез убихинона и его участием в предохранении сульфгидрильных групп белков от окисления. При дефиците витамина Е снижается активность ферментных систем, функционально связанных с мембранами клеток, содержание в тканях креатинфосфата и АТФ. Дефицит токоферолов сопровождается нарушением образования тромбоксанов и простациклинов (за счет торможения накапливающимися перекисями активности простагландинсинтазы).
Гиповитаминоз Е возможен при наследственной абеталипопротеинемии в связи с нарушением транспорта витамина.
Симптомы гипервитаминоза Е у животных сходны с симптомами гиповитаминоза Е (поражение скелетных мышц, семенников, размягчение мозга). Ранние признаки интоксикации витамином Е у человека - резкое повышение содержания его в сыворотке крови и преходящая креатинурия. При значительной передозировке витамина наблюдаются замедление активации протромбиназы, тромбоцитопатии, гипокоагуляция, геморрагии, гипогликемия, диспепсия, головная боль, слабость, повышенная мышечная утомляемость, мышечные судороги. Избыток токоферолов может активировать ПОЛ, эти соединения способны образовывать свободные радикалы.
Источниками витамина Е для человека являются растительные масла (подсолнечное, хлопковое, кукурузное и др.), а также салат, капуста и семена злаков; из продуктов животного происхождения витамин Е содержится в мясе, сливочном масле, яичном желтке и др. Суточная потребность в витамине Е составляет около 5 мг.
Витамины группы К
К витаминам группы К (нафтохиноны, антигеморрагический фактор) относятся витамины К1 (филлохинон) и К2 (менахинон). Активные метаболиты нафтохинонов, возможно, представляют собой их гидрохиноновые производные.
Авитаминоз К у человека встречается нечасто, смешанная пища довольно богата витамином, который, кроме того, синтезируется микрофлорой кишечника. Витамин К-дефицитные состояния могут быть вызваны подавлением кишечной микрофлоры антибиотиками и сульфаниламидами, недостаточным поступлением витамина с пищей, нарушением всасывания жиров, в том числе жирорастворимых витаминов (энтерит, недостаток желчи при циррозе, механической желтухе), а также нерациональным лечением тромбозов и эмболий антикоагулянтами кумаринового ряда, являющимися антивитаминами К.
К-витаминная недостаточность проявляется снижением содержания в крови активных факторов свертывания крови II (протромбин), VII (проконвертин), IX (фактор Кристмаса) и Х (фактор Стюарта-Прауэра), вследствие чего удлиняется время свертывания крови, тромбопластиновое и протромбиновое время, снижается протромбиновый индекс, в плазме накапливаются некарбоксилированные предшественники факторов свертывания. При снижении протромбинового индекса ниже 35% развиваются геморрагические явления вплоть до обильных кровотечений, в первую очередь в областях тела, подвергшихся травмам (операционные раны, ушибы, гематомы в области пункции вен и т.п.), а также самопроизвольные паренхиматозные и капиллярные кровотечения. При более глубокой гипопротромбинемии развивается тяжелый геморрагический диатез с гематурией, наличием крови в рвотных массах, гематомами в различных областях тела, гемартрозом и т.д. Недостаточность витамина К у недоношенных детей, особенно подвергшихся асфиксии в родах, проявляется кровоизлияниями в желудочно-кишечном тракте, меленой, носовыми и нёбными кровотечениями, кровотечениями из культи пуповины. Без лечения погибает около 30% заболевших. При летальных исходах обнаруживают кровоизлияния в надпочечниках, печени, мозгу, легких и других органах. Низкое содержание витамин К-зависимых факторов свертывания у новорожденных связано с отсутствием в первую неделю жизни в кишечнике микрофлоры, небольшим запасом витамина в печени и недостаточным содержанием витамина К в женском молоке.
Витамин К принимает участие в посттрансляционной модификации в печени белков, участвующих в сложном процессе свертывания крови: факторов II, VII, IX и X, что обеспечивает их физиологическую активность, а также влияет на состояние эн-
дотелия кровеносных сосудов. Посттрансляционная модификация белков свертывания крови, требующая наличия витамина К, заключается в γ-карбоксилировании остатков глутаминовой кислоты в их молекулах при участии γ-глутамилкарбоксилазы. Витамин К функционирует в качестве кофактора карбоксилирования. Предположительно его роль сводится либо к транспорту НСО3-, включающихся в γ-положение остатков глутаминовой кислоты, либо к активации водорода γ-углеродного атома глутаминовой кислоты, либо к активации γ-глутамилкарбоксилазы. Постсинтетическое карбоксилирование остатков глутаминовой кислоты необходимо для оптимального связывания факторами свертывания крови ионов Са2+, посредством которого осуществляется прикрепление белков к полианионным поверхностям, что дает им возможность выполнять прокоагулянтные функции. Витамин К участвует также в карбоксилировании неколлагеновых белков костей, почечных белков, антитромботических белков С и S. Витамин К необходим для кальцификации остеоидной матрицы. Витамин К-зависимые Са2+-связьгаающие белки участвуют в почечной реабсорбции Са2+. Дефицит витамина К может вызывать изменение агрегационной активности эритроцитов, связанное с дестабилизацией эритроцитарных мембран.
Повышенная дозировка витаминов К и К2 вызывает у животных снижение концентрации гемоглобина и количества эритроцитов и в 1,5-2 раза повышает содержание протромбина в крови; у людей обнаружено преходящее повышение свертывания крови. Гемолиз эритроцитов связан с повышением содержания метгемоглобина. Передозировка витамина К у новорожденных (и особенно недоношенных) может вызвать гемолитическую анемию, гепатоцеллюлярное поражение и ядерную желтуху вследствие повышенного содержания билирубина в крови. Водорастворимые аналоги витамина К при передозировке вызывают гемолитическую анемию и поражение печени.
К растительным продуктам, богатым витамином К, относятся капуста, шпинат, салат, тыква, зеленые томаты, арахисовое масло, ягоды рябины и т.д. В животных продуктах, кроме печени свиньи, он почти нигде не содержится. Суточная потребность в витамине К точно не установлена, поскольку он синтезируется микроорганизмами кишечника (до 1,5 мг в сутки); считается достаточным количество 1 мг.
12.3.2. Водорастворимые витамины
Витамин С
Необходимым пищевым фактором для человека, обезьян, морских свинок, некоторых птиц и рыб является витамин С (аскорбиновая кислота, антискорбутный, антицинготный фактор), отсутствие которого в рационе в течение даже короткого периода времени приводит к тяжелым последствиям для организма. Все другие животные не нуждаются в пищевом витамине С, поскольку у них он синтезируется в печени из глюкозы.
Аскорбиновая кислота активно всасывается в тощей кишке, формы гиповитаминоза С, вызванные нарушением ее абсорбции, неизвестны. Недостаточное поступление в организм витамина С вызывает развитие скорбута (цинги) - болезни, сопутствующей человечеству на протяжении всей его истории.
К основным симптомам С-витаминной недостаточности относятся: ломкость кровеносных сосудов, общая слабость, апатия, повышенная утомляемость; снижение аппетита, задержка роста; восприимчивость к инфекциям, анемия; ослабление фиксации зубов в лунках, геморрагический парадонтит с гингивитом. При тяжелой цинге развивается некротический процесс в области зева, мягкого нёба, пищевода, челюстных костей. Поражения кровеносных сосудов служат причиной мелких точечных кровоизлияний под кожу (петехий), сопровождающихся перифолликулярным гиперкератозом с характерными папулами, имеющими кровяной венчик. Крупные подкожные экхимозы при тяжелой цинге могут изъязвляться. Отмечаются кровотечения и кровоизлияния в слизистых и серозных оболочках и внутренних органах, кровоизлияния в толщу мышц (преимущественно икроножных и бедра), в оболочки периферических нервов, между фасциями и вокруг сухожильных влагалищ. Ослабевает прикрепление надкостницы к костям, наблюдаются поднадкостничные кровоизлияния, субпериостальные переломы, серозный и серозно-геморрагический выпот в суставы, преимущественно коленные. Развиваются одышка, сердцебиение, ослабление сердечного толчка, увеличение поперечника сердца. Возможны увеличение селезенки, жировая инфильтрация печени. Угнетена моторная и секреторная деятельность желудка и кишечника. Терминальная стадия скорбута сопровождается кахексией, контрактурами, внутричерепными кровоизлияниями, возможен летальный исход.
У детей раннего возраста скорбут проявляется в виде болезни Меллера-Барлоу. Нарушение остеогенеза приводит к деформации грудной клетки с образованием болезненных цинготных «четок» в местах перехода хрящевой части ребра в костную, искривлению трубчатых костей нижних конечностей. В тяжелых случаях отмечаются геморрагический выпот в полости голеностопных и коленных суставов, отек голеней. Наблюдаются длительный субфебрилитет, болезненность конечностей при движении, припухание диафизов, отслойка надкостницы с поднадкостничными гематомами, позже обызвествляющимися. Изменения в костях и кровоизлияния в костный мозг приводят к нарушению гемопоэза.
Витамин С участвует в реакциях гидроксилирования триптофана (синтез серотонина), диоксифенилаланина (образование норадреналина), оксифенилпирувата (синтез гомогентизиновой кислоты), стероидов (синтез кортикостероидов), β-бутиробетаина (синтез карнитина). Активируя пролилгидроксилазу и лизилгидроксилазу, он обеспечивает гидроксилирование остатков пролина и лизина в проколлагене (образование коллагена). Кроме того, аскорбиновая кислота участвует в обмене железа, обеспечивая переход трехвалентного железа в двухвалентное в кишечнике и тем самым облегчая его всасывание, а также высвобождая железо из трансферрина в крови, что ускоряет его поступление в ткани. Аскорбиновая и дегидроаскорбиновая кислоты, составляя окислительно-восстановительную систему, участвуют в окислительно-восстановительных реакциях. Витамин С в зависимости от дозы может выступать в роли антиоксиданта или прооксиданта. Антиокислительные эффекты витамина проявляются нейтрализацией активных форм кислорода, реактивацией витамина Е, предохранением от окисления SH-групп белков, в том числе ферментов. Аскорбиновая кислота принимает участие в окислительном распаде тирозина и гемоглобина, активации гексокиназы.
Геморрагические явления и специфические изменения в костной и хрящевой тканях при недостаточности витамина С вызваны нарушениями постсинтетической модификации коллагена и изменениями в синтезе гликозаминогликанов (хондроитинсульфатов и гиалуроновой кислоты). Наиболее сильно поражается коллаген базальных мембран кровеносных сосудов, насыщенный гидроксипролиновыми остатками. Молекулы тропоколлагена, в которых не подверглись гидроксилированию пролиновые и лизиновые остатки, не имеют достаточной механической прочности и легко
расщепляются коллагеназами. Клиническая картина цинги определяется также нарушениями со стороны эндокринной системы (гипофункция надпочечников), иммунной системы (снижение выработки антител лимфоцитами и продукции защитных белков нейтрофилами), усилением распада и торможением синтеза белков, снижением активности гексокиназы. Гипохромная анемия при скорбуте может быть связана со снижением всасывания железа и повышенной кровоточивостью. Гиповитаминоз С сопровождается, как правило, дефицитом витамина Р, что усугубляет нарушения сосудистой проницаемости.
В больших количествах витамин С содержится в продуктах растительного происхождения: перце, салате, капусте, хрене, укропе, ягодах рябины, черной смородины, цитрусовых и т.д. Картофель относится к основным повседневным источникам витамина С. Из непищевых источников витамином С богаты хвоя, листья черной смородины, экстракты из которых могут полностью удовлетворить потребность организма. Суточная потребность в витамине С для человека составляет 50-100 мг, она возрастает при физической и нервно-психической нагрузке, пребывании в условиях низкой и высокой температуры, при облучении, курении, беременности и лактации, инфекционных заболеваниях, хронических заболеваниях желудочно-кишечного тракта, при приеме антибиотиков и сульфаниламидов.
Витамин В1
Авитаминоз Вр проявляющийся заболеванием бери-бери, ранее широко распространенный в странах Дальнего Востока, сейчас встречается значительно реже. Различают несколько клинических типов недостаточности витамина В1 (тиамина, антиневритного фактора): сухую (полиневритную, паралитическую) форму, протекающую с преобладанием неврологических поражений (парезы, параличи) (рис. 12-9, 12-10); отечную - с преимущественным поражением сердечно-сосудистой системы, хотя явления полиневрита при ней также отмечаются, и остро протекающую кардиальную форму болезни, нередко приводящую к летальному исходу в результате развития острой сердечной недостаточности. Зачастую в клинике наблюдаются частичные взаимопереходы форм заболевания. Бери-бери у человека нередко представляет собой полиавитаминоз, при котором организм испытывает недостаток также в витаминах В2, В6, РР, С и др.
 Рис. 12-9. Паралич левой ноги при бери-бери (по R. Berg, 1927)
Рис. 12-9. Паралич левой ноги при бери-бери (по R. Berg, 1927)
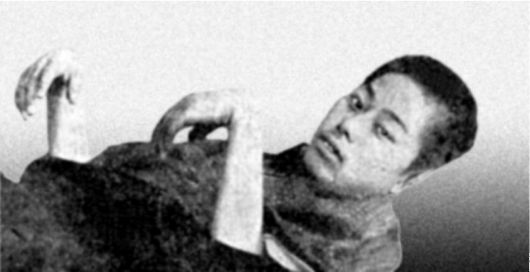 Рис. 12-10. Паралич кисти при бери-бери (по
Рис. 12-10. Паралич кисти при бери-бери (по
R. Berg, 1927)
При отсутствии или недостаточности тиамина клинические симптомы преимущественно связаны с нарушениями деятельности нервной и сердечно-сосудистой систем, а также секреторной и моторной функций пищеварительного тракта. Со стороны периферической нервной системы наблюдается симптоматика распространенного дегенеративного демиелинизирующего полиневрита с преимущественными поражениями нервов нижних конечностей, выражающаяся в расстройстве чувствительности и болях по ходу нервов. Возможно развитие контрактур, параличей нижних, а затем и верхних конечностей. Отмечаются изменения в психике: потеря памяти на недавние события, галлюцинации, фобические неврозы, снижение интеллекта. Нарушения деятельности сердечно-сосудистой системы проявляются одышкой, сердцебиением, болями в области сердца, отеками.
Вторичные гиповитаминозы В1 могут возникать в случае повы-
шенного расхода витамина при обычном его поступлении с пищей (тиреотоксикоз, избыток углеводов в диете), а также при нарушении процессов всасывания из желудочно-кишечного тракта, усиленном выведении витамина после длительного применения диуретических средств, ослаблении процессов тканевого фосфорилирования тиамина. Синдром Вернике-Корсакова (форма тиами-
новой недостаточности, развивающаяся у алкоголиков) характеризуется энцефалопатией Вернике (мозжечковая атаксия, судороги, арефлексия, нистагм, дезориентация, спутанность сознания, паралич глазных мышц) и корсаковским психозом (неспособность усваивать информацию, ретроградная амнезия, дезориентировка во времени и пространстве, слабая способность сосредоточиваться, болтливость, иногда эйфория).
Модель бери-бери была получена на курицах (модель Х. Эйкмана), голубях и лисицах (паралич Частека).
Молекулярной основой обменных нарушений, наблюдаемых при развитии В1-гиповитаминоза, является снижение функциональной активности тиаминзависимых ферментов, занимающих ключевое положение в клеточном метаболизме (рис. 12-11). Тиаминдифосфат (тиаминпирофосфат) - коферментная форма витамина В1 - входит в состав пируват- и α-кетоглутаратдегидрогеназных ферментативных комплексов, катализирующих окислительное декарбоксилирование пировиноградной и α-кетоглутаровой кислот. Ацетил-КоА, образующийся в результате окислительного декарбоксилирования пировиноградной кислоты, либо попадает в цикл трикарбоновых кислот (ЦТК), где происходит его полное окисление с образованием СО2, Н2О и АТФ, в том числе в цепи переноса электронов, либо принимает участие в синтезе холестерина, фосфолипидов, жирных и желчных кислот, ацетилхолина и стероидных гормонов. α-Кетоглутаратдегидрогеназный комплекс, катализирующий окислительное декарбоксилирование α-кетоглутаровой кислоты, является звеном цикла трикарбоновых кислот, обеспечивающего окисление продуктов расщепления белков, липидов и углеводов. Транскетолаза (ключевой фермент пентозофосфатного пути окисления углеводов), в состав которой входит тиаминдифосфат, участвует в переносе гликольальдегидного радикала от кетосахаров на альдосахара. Нормальное функционирование пентозного цикла, являющегося основным источником НАДФ2Н и рибозо-5-фосфата, представляет собой необходимое условие для осуществления биосинтеза нуклеиновых кислот, белков и липидов. Тиаминдифосфат также участвует в окислительном декарбоксилировании α-кетокислот, образующихся из аминокислот с разветвленной боковой цепью, что играет важную роль в процессах катаболизма белка.
Наряду с алиментарной недостаточностью витамина В1 известны заболевания, обусловленные врожденными нарушениями обмена
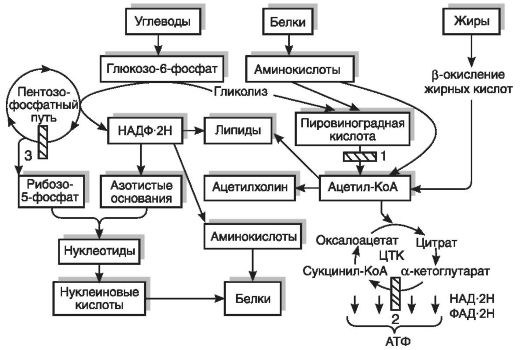 Рис. 12-11. Нарушения
синтеза нуклеиновых кислот, белков, липидов, ацетилхолина, образования
АТФ при гиповитаминозе 1 - дефицит пируватдегидрогеназы; 2 - дефицит
α-кетоглутаратдегидрогеназы; 3 - дефицит транскетолазы (по В.Б.
Спиричеву, Ю.И. Барашневу, 1977). ЦТК - цикл трикарбоновых кислот
Рис. 12-11. Нарушения
синтеза нуклеиновых кислот, белков, липидов, ацетилхолина, образования
АТФ при гиповитаминозе 1 - дефицит пируватдегидрогеназы; 2 - дефицит
α-кетоглутаратдегидрогеназы; 3 - дефицит транскетолазы (по В.Б.
Спиричеву, Ю.И. Барашневу, 1977). ЦТК - цикл трикарбоновых кислот
и функций витамина: подострая некротизирующая энцефаломиелопатия, или болезнь Лея, при которой нарушено образование коферментной формы витамина в мозговой ткани; перемежающаяся атаксия, обусловленная врожденным дефектом пируватдегидрогеназного ферментативного комплекса; тиаминзависимая форма болезни «моча с запахом кленового сиропа», связанная с дефектом системы окислительного декарбоксилирования разветвленных кетокислот.
При авитаминозе В1 в крови и тканях происходит накопление α-кетокислот, с мочой в повышенных количествах выделяются аминокислоты и креатин. Нарушение окислительного декарбоксилирования приводит к накоплению пирувата и его недоокисленных метаболитов, оказывающих токсическое действие на ЦНС. Развиваются метаболический ацидоз, гипоэнергетические состояния в связи с нарушением метаболизма углеводов, нарушается работа ионных насосов нервных и мышечных клеток, в том числе кардиомиоцитов. В связи с тем, что для получения энергии организм начинает использовать больше белков и липидов, нарушаются синтез
жирных кислот, ацетилирование холина, превращение углеводов в липиды, развивается отрицательный азотистый баланс.
В токсичных дозах витамин В1 угнетает ферменты холинэстеразу и гистаминазу, вызывает дегрануляцию тучных клеток и острую аллергическую реакцию. Возможны кровоизлияния в кишечник, отек легких, судороги. Наиболее тяжелая реакция - анафилактический шок с потерей сознания, коллапсом и даже смертельным исходом. Развитие аллергических реакций может быть связано не только с величиной дозы витамина В1, но и с повышенной чувствительностью некоторых людей к тиамину.
Основное количество витамина В1 человек получает с растительной пищей. Много витамина В1 содержится в дрожжах, пшеничном хлебе из муки грубого помола, оболочке и зародышах семян хлебных злаков, сое, фасоли, горохе, меньше - в картофеле, моркови, капусте. Из продуктов животного происхождения наиболее богаты витамином В1 печень, почки, мозг. Некоторые бактерии, населяющие кишечник животных, способны синтезировать витамин В1. Норма суточного потребления тиамина для взрослого человека составляет от 1,2 до 2,2 мг.
Витамин В2
Дефицит витамина В2 (рибофлавина, витамина роста) может быть вызван как недостаточным поступлением его с пищей, так и нарушением всасывания витамина, связанным с ингибирующим действием на флавокиназу ряда лекарственных препаратов (адриамицин, антидепрессанты и др.).
Острый арибофлавиноз, развивающийся при полном отсутствии витамина в пище у человека и животных, характеризуется внезапным развитием коматозного состояния и быстро наступающей гибелью. Недостаток рибофлавина у животных вызывает поражения кожных покровов (облысение, дерматиты с шелушением кожи и появлением эрозий), глаз (кератиты, катаракты), развитие гипохромной анемии. У собак отмечаются нарушения координации движений, а также процессов высшей нервной деятельности со снижением условных и безусловных рефлексов. У человека дефицит рибофлавина проявляется прежде всего хейлозом с мацерацией и трещинами на губах и ангулярным стоматитом (рис. 12-12). Могут развиваться дерматиты на носогубной складке, крыльях носа, веках и ушах, волосистой части головы, мошонке и других частях
тела. Отмечаются глосситы, сопровождающиеся чувством жжения языка, а также поражения глаз (васкуляризация роговой оболочки, блефариты, конъюнктивиты и кератиты); развиваются анемия, нервные расстройства, проявляющиеся мышечной слабостью, жгучими болями в ногах, атаксией, гиперкинезами. Рибофлавин входит в состав флавиновых коферментов (ФМН - флавинмононуклеотид и ФАД - флавинадениндинуклеотид), являющихся, в свою очередь, простетическими группами ряда ферментов, катализирующих реакции дегидрирования исходного субстрата или промежуточного метаболита (оксидазы L- и D-аминокислот, ксантиноксидаза и др.); а также реакции, характеризующиеся переносом электронов и протонов не от исходного субстрата, а от восстановленных пиридиновых коферментов. Витамин В2, входящий в состав коферментов оксидоредуктаз, принимает участие в процессах окислительного фосфорилирования, окисления альдегидов, моноаминов, пуриновых оснований, углеводов и др. При гипорибофлавинозе вследствие недостатка флавопротеинов страдают в первую очередь высокоаэробные эпителии кожи и полости рта. Дефицит рибофлавина характеризуется снижением интенсивности
окислительно-восстановительных превращений ксантина, аминокислот, угнетением ферментов цикла трикарбоновых кислот, β-окисления жирных кислот, дыхательной цепи митохондрий. При гиповитаминозе В2 снижается детоксикационная активность ферментов печени в отношении лекарственных препаратов.
Из пищевых продуктов богаты рибофлавином хлеб (из муки грубого помола), семена злаков, яйца, молоко, мясо, печень, свежие овощи и др. Суточная потребность в витамине В2 составляет 1,7-3,0 мг.
Витамин РР
Недостаточность витамина РР
(никотиновая кислота и никотинамид; ниацин; антипеллагрический
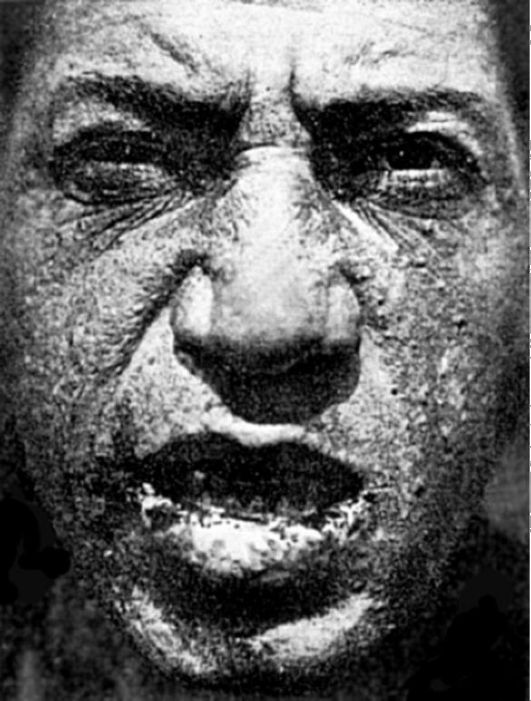 Рис. 12-12. Арибофлавиноз. Себорейные поражения кожи, ангулярный стоматит, хейлоз (по В.В. Ефремову, 1944)
Рис. 12-12. Арибофлавиноз. Себорейные поражения кожи, ангулярный стоматит, хейлоз (по В.В. Ефремову, 1944)
фактор) в организме может быть вызвана его дефицитом в пище, особенно в сочетании с неполноценным белковым питанием и/или недостатком витамина В6 (некоторое количество витамина РР синтезируется в организме человека и животных из триптофана через кинуренин и хинолиновую кислоту при участии пиридоксальфосфата), а также мальабсорбцией, алкоголизмом, длительным приемом ряда лекарственных препаратов (изониазид, цитостатики и др.). Синтез никотиновой кислоты из триптофана может быть нарушен при карциноидном синдроме, что объясняется усилением превращения триптофана в гидроксииндолы.
Дефицит в организме ниацина в сочетании с недостаточностью витаминов А, В1, В2, С вызывает развитие пеллагры, наиболее характерными признаками которой являются поражения кожи (дерматиты), пищеварительного тракта (диарея) и нарушения нервной деятельности (деменция). Развитию клинической картины пеллагры обычно предшествуют неспецифические симптомы гиповитаминоза: вялость, апатия, слабость в ногах, быстрая утомляемость, снижение аппетита, головокружение, раздражительность, бессонница, снижение сопротивляемости организма инфекциям.
Ранними клиническими симптомами пеллагры являются вызванная атрофией эпителия желудочно-кишечного тракта диарея, приводящая к обезвоживанию организма и нарушению всасывания ниацина и триптофана, воспаление и изъязвление слизистой оболочки толстой кишки и изменения в полости рта (фуксиноподобный язык со вздутиями и трещинами, стоматиты, гингивиты). Повышенная чувствительность кожных покровов к ультрафиолетовой части спектра, связанная со значительным усилением обмена порфиринов, которые обладают фотосенсибилизирующими свойствами, приводит к появлению на коже открытых частей тела симметричных красных пятен (пеллагрическая эритема). В дальнейшем развиваются гиперпигментация, гиперкератоз с отшелушиванием, трещинами, пиодермия (рис. 12-13). Со стороны сердечно-сосудистой системы отмечаются миокардиодистрофия и гипотония. Нарушения липидного обмена проявляются гипохолестеринемией. Гибель нейронов и дегенерация проводящих путей приводят к глубоким нарушениям функций центральной и периферической нервной системы, проявляющимся шумом и звоном в ушах, нарушением вкуса, нервно-мышечными и головными болями, болями в позвоночнике, конечностях, ощущением онемения, парестезиями в форме жжения, расстройствами брюшных кожных
и сухожильных рефлексов, тяжелой адинамией, артикуляционными нарушениями речи, парезами, мышечными атрофиями, нарушениями координации движений. Самое тяжелое в клинической картине пеллагры - нарушения психики (психозы с дезориентацией, бред, галлюцинации).
Классическим объектом для моделирования пеллагры в эксперименте являются собаки, у которых отсутствие никотиновой кислоты в пище приводит к болезни «черный язык».
Тяжелые последствия дефицита витамина РР в организме в значительной степени объясняются вхождением никотинамидных ко- ферментов (НАД+, НАДФ+) в со-
став большого числа ферментов (малатдегидрогеназа, глюкозо-6-фосфатдегидрогеназа, глутаматдегидрогеназа и др.), действующих на различных этапах окисления и синтеза углеводов, аминокислот, липидов и др. При участии никотинамидных коферментов специфические дегидрогеназы катализируют обратимые реакции дегидрирования спиртов, оксикислот и некоторых аминокислот в соответствующие альдегиды, кетоны и кетокислоты. В процессе биологического окисления НАД+ выполняет роль промежуточного переносчика электронов и протонов между окисляемым субстратом и флавиновыми ферментами.
Недостаток витамина РР приводит к нарушению биосинтеза стероидных гормонов, оказывающих разностороннее влияние на обмен веществ в различных тканях. У ниациндефицитных крыс возможно переключение окислительной ветви пентозофосфатного пути на неокислительную. Высказывается мнение, что РРнедостаточность нарушает биосинтез белка, а еще раньше - биосинтез нуклеиновых кислот, при этом гибель клеток, особенно высокоаэробных, не уравновешивается их пролиферацией.
Существенный недостаток никотиновой кислоты может наблюдаться при врожденных нарушениях ее биосинтеза из триптофана
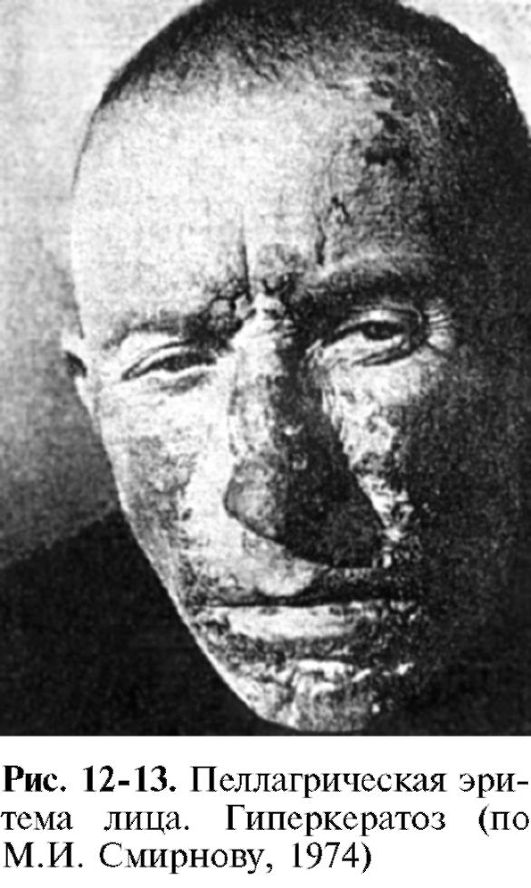 вследствие дефекта пиридоксальзависимого фермента кинурениназы (наследственная ксантуренурия - синдром Кнаппа-Комровера).
вследствие дефекта пиридоксальзависимого фермента кинурениназы (наследственная ксантуренурия - синдром Кнаппа-Комровера).
К редким наследственным нарушениям, ведущим к развитию пеллагроподобных симптомов, относится болезнь Хартнупа - нарушение реабсорбции в почках группы монокарбоновых моноаминокислот, в число которых входит и триптофан, сочетающееся с дефектом всасывания триптофана в тонком кишечнике.
Высокие дозы никотиновой кислоты, имеющие гистаминлиберирующий эффект, при приеме внутрь или инъекциях вызывают расширение кровеносных сосудов, гиперемию кожи; могут появляться зуд, сыпь, возможны диспепсия, гипотония, нарушения функций печени, снижение толерантности к глюкозе. Встречается индивидуальная чувствительность к никотиновой кислоте.
Для человека основными источниками никотиновой кислоты и ее амида являются рис, хлеб, картофель, мясо, печень, почки, морковь и другие продукты. В молоке и яйцах витамин РР почти не содержится, хотя эти продукты обладают антипеллагрическим действием за счет высокого содержания триптофана, из которого в организме человека может синтезироваться никотиновая кислота. Суточная потребность в витамине РР составляет 14-25 мг.
Витамин Вь
Витамин В6 (пиридоксин, антидерматитный витамин) синтезируется микрофлорой кишечника в количествах, частично покрывающих потребности организма. Гиповитаминоз В6 может развиваться при нарушении всасывания витамина в кишечнике, повышенном его распаде (например, при алкоголизме), при повышенном катаболизме белка (стресс, лихорадка, гипертиреоз), при применении ряда лекарственных препаратов (изониазид, фтивазид, L-ДОФА и др.). Признаком авитаминоза В6 у человека является развитие сухого себорейного пеллагроподобного дерматита (шелушение, эритема, гиперпигментация), хейлоза, стоматита и глоссита, что может быть отчасти связано с вторично развивающейся (из-за нарушения обмена триптофана) недостаточностью никотиновой кислоты. При дефиците витамина В6 развивается гипохромная микроцитарная анемия и появляются судороги. При недостаточном содержании пиридоксина в искусственной пище у грудных детей возникают дерматиты и поражения нервной системы, в том числе эпилептиформные судороги. Патогенез судорог, гипервоз-
будимости и повышенной чувствительности к шуму у новорожденных и некоторых животных (например, крыс) связан со снижением скорости образования в нервной ткани γ-аминомасляной кислоты (тормозного медиатора нервной системы) из глутамата в реакции, катализируемой глутаматдекарбоксилазой, в состав которой входит коферментная форма витамина В6 - пиридоксальфосфат. Витамин В6 участвует также в метаболизме триптофана, при нарушении которого образуются соединения типа ксантуреновой кислоты, способные препятствовать инсулиногенезу, вызывающие диабетоподобные состояния.
Проявления авитаминоза у крыс (весьма чувствительных к недостатку пиридоксина) выражаются в специфическом дерматите с преимущественным поражением кожи лапок, носа, ушей и хвоста (акродиния), выпадении шерсти на пораженных участках, шелушении кожи, в дальнейшем - изъязвлении кожи конечностей и гангрене. При авитаминозе В6 у крыс, собак, свиней, кур отмечаются гипохромная анемия, эпилептиформные припадки, дегенеративные изменения в ЦНС.
Пиридоксальфосфат является необходимым кофактором пиридоксалевых ферментов, катализирующих ключевые реакции белкового обмена (аминотрансфераз, осуществляющих обратимый перенос аминогруппы от аминокислот на кетокислоты, и декарбоксилаз аминокислот, катализирующих необратимое отщепление СО2 от аминокислот с образованием биогенных аминов).
Недостаток витамина В6 сопровождается нарушениями белкового обмена (отрицательный азотистый баланс), от которых особенно страдают быстро пролиферирующие ткани. Развиваются гипераминоацидемия и гипераминоацидурия. Нарушение биосинтеза гема гемоглобина на этапе синтеза δ-аминолевулиновой кислоты наряду с нарушением синтеза белка являются метаболической причиной развития анемии при В6-авитаминозе.
Большая численность пиридоксальзависимьгх ферментов объясняет существование широкого спектра энзимопатий: врожденной гомоцистинурии, связанной с дефектом цистатионинсинтазы, врожденной цистатионинурии, обусловленной нарушением активности цистатионазы, наследственной ксантуренурии (синдром Кнаппа-Комровера), связанной с дефектом кинурениназы и пиридоксинзависимого судорожного синдрома, в патогенезе которого существенную роль может играть снижение активности глутаматдекарбоксилазы.
В больших суточных дозах (250-500 мг) витамин В6 может вызвать кожные высыпания, головокружение и судороги, что вынуждает прерывать лечение. Развитие сенсорной нейропатии и нарушений вибрационной чувствительности возможно при парентеральном введении свыше 2 г пиридоксина в сутки. Длительное введение лечебных доз витамина может угнетать противосвертывающую систему крови.
Витамин В6 содержится в дрожжах, хлебе, горохе, фасоли, картофеле, мясе, печени, почках. Наиболее высока концентрация витамина в зародышевой части семян и зерен злаков. Суточная потребность установлена ориентировочно (2-4 мг в сутки). Она увеличивается при потреблении с пищей большого количества белка, физической нагрузке, охлаждении, облучении, беременности, при применении сульфаниламидов и антибиотиков, угнетающих микрофлору кишечника, а также с возрастом.
Витамин В12
Гипо- и авитаминоз В12 у человека может развиваться вследствие недостаточности витамина В12 (кобаламин, антианемический фактор) в пище (строгое вегетарианство), отсутствия или недостаточности вырабатывающегося в обкладочных клетках слизистой желудка антианемического фактора Касла, образующего с витамином В12 комплекс, который способен всасываться в кишечнике при взаимодействии с рецептором, а также вследствие расстройства всасывания витамина В12 в кишечнике (энтериты, в том числе спру, резекции кишечника) и абсорбции витамина кишечными паразитами (широкий лентец, анкилостома). Примером гастрогенного авитаминоза является болезнь Аддисона-Бирмера (злокачественная пернициозная анемия) - заболевание, связанное с генетически обусловленной секрецией слизистой желудка биологически неактивного внутреннего фактора Касла либо отсутствием его секреции (наследственная форма) или развивающееся в связи с образованием аутоантител к внутреннему фактору Касла или обкладочным клеткам слизистой желудка, а также при угнетении секретирующей функции желудка вследствие атрофии его слизистой оболочки, резекции, рака и др. (приобретенная форма).
Недостаточность витамина В12 в организме приводит к развитию злокачественной гиперхромной макроцитарной мегалобластической анемии, лейкопении, нейтропении и тромбоцитопении, па-
тологии органов пищеварения (глоссит, атрофия слизистой желудка и др.), а также дегенерации задних и боковых столбов спинного мозга (фуникулярный миелоз), проявляющейся парестезиями, судорогами, нарушениями кожной и вибрационной чувствительности, ахилловых и коленных рефлексов.
Известно, что витамин В12 является одним из важнейших факторов нормального кроветворения, обеспечивающих необходимое равновесие процессов пролиферации и дифференцировки клеток крови в физиологических условиях. Витамин В12, будучи синергистом фолиевой кислоты, играет важную роль в синтезе нуклеиновых кислот и белков. Дефицит витамина В12 нарушает деметилирование N5-метилтетрагидрофолиевой кислоты, что затрудняет использование в клетках тетрагидрофолиевой кислоты, необходимой для синтеза пуриновых и пиримидиновых нуклеотидов, в первую очередь в быстро пролиферирующих тканях (костный мозг, эпителий желудочно-кишечного тракта). Наряду с этим недостаток витамина В12 приводит к нарушению превращения метилмалонил-КоА в сукцинил-КоА, в связи с чем формируется избыток метилмалоновой и пропионовой кислот, которые, превращаясь в жирные кислоты с нечетным числом углеродных атомов в скелете, включаются в липиды нейронов, вызывая демиелинизацию нервных волокон и жировую дистрофию нервных клеток.
К врожденным нарушениям обмена витамина В12 у человека, проявляющимся мегалобластической анемией, наряду с болезнью Аддисона-Бирмера относятся болезнь Имерслунда-Грэсбека (нарушение всасывания витамина В12, связанное с дефектом рецепторов в клетках эпителия кишечника) и дефект транскобаламина II (нарушение его образования и транспорта витамина В12), для которого характерны также неврологические расстройства, задержка роста, диарея, неукротимая рвота. Врожденный дефект транскобаламина I (нарушение его образования) не сопровождается клинической симптоматикой. Известен ряд генетических нарушений, затрагивающих синтез ферментов, в состав которых входят кобамидные коферменты, а также энзимов, принимающих участие в образовании метил- и дезоксиаденозилкобаламина. Дефицит тетрагидроптероилглутаматметилтрансферазы проявляется снижением иммунитета, отставанием в росте и умственном развитии, анемией, а также повышенным выделением с мочой гомоцистеина и цистатионина. Дефекты синтеза метилмалонил-КоА-мутазы и
дезоксиаденозилкобаламинтрансферазы приводят к повышению содержания в крови и моче метилмалоновой кислоты и ацидозу.
Избыточное количество В12 нетоксично, но встречается индивидуальная чувствительность к витамину и в редких случаях - плохая переносимость.
Для человека основными источниками витамина В12 являются мясо, говяжья печень, почки, рыба, молоко, яйца. Суточная потребность в витамине В12 составляет 2-3 мкг.
Фолиевая кислота
При недостаточности фолиевой кислоты (витамин Вс, В9, фолацин) у экспериментальных животных отмечаются снижение аппетита, задержка роста, геморрагические гастроэнтериты, жировая инфильтрация печени, дерматиты, спинномозговые параличи, макроцитарная анемия, лейкопения, тромбоцитопения.
Дефицит фолиевой кислоты в организме человека, вызываемый подавлением лекарственными препаратами (сульфаниламиды) синтеза фолацина из парааминобензойной кислоты микрофлорой кишечника или нарушением всасывания витамина (при спру, алкоголизме, приеме барбитуратов), сопровождается развитием гиперхромной макроцитарной мегалобластической анемии. В тяжелых случаях авитаминоза отмечаются тромбоцитопения и лейкопения. Мегалобластическая фолиеводефицитная анемия может быть обусловлена недостаточностью только фолатов (монодефицит витамина В9), а также дефицитом витамина В12 и связанным с ним нарушением метаболизма фолиевой кислоты (см. выше) (сочетанный дефицит витаминов В12 и В9). Наряду с нарушениями кроветворения при фолиеводефицитной анемии развиваются хейлоз, глоссит, эзофагит, атрофический или эрозивный гастрит, энтерит, ахлоргидрия, диарея, стеаторея.
Коферментные функции тетрагидрофолиевой кислоты (биологически активной формы фолиевой кислоты) непосредственно связаны с переносом одноуглеродных групп (формильная, метильная, метиленовая, метенильная, оксиметильная, формиминовая). Путем включения фолиевых коферментов в обмен одноуглеродных соединений осуществляется их участие в биосинтезе предшественников нуклеиновых кислот - пуриновых и пиримидиновых нуклеотидов, а также в метаболизме ряда аминокислот: серина, глицина, гистидина, метионина, триптофана и др. При недостат-
ке фолиевой кислоты биосинтез белков может быть лимитирован недостатком метионина и серина, синтез ДНК тормозится дефицитом тимидиловых нуклеотидов (дезоксиуридиловая кислота не превращается в тимидиловую) и пуриновых нуклеотидов, что приводит к нарушениям клеточного цикла быстро пролиферирующих эпителиальных и гемопоэтических клеток и определяет клинику фолатдефицитных состояний.
Известен наследственный дефект НАДФН-зависимой дигидрофолатредуктазы, вызывающий нарушение обмена фолиевой кислоты и приводящий к развитию мегалобластической анемии.
Избыточное количество фолиевой кислоты может быть токсично и вызывать гистаминоподобный эффект.
Богатыми источниками фолиевой кислоты являются зеленые листья растений, дрожжи, капуста, томаты, шпинат, а также почки, печень, мясо и другие продукты. Фолацин синтезируется микрофлорой кишечника в количествах, достаточных для удовлетворения потребности организма взрослого человека, которая составляет от 0,5 до 2 мг в сутки.
Пантотеновая кислота
Апантотеноз у экспериментальных животных проявляется замедлением роста, потерей массы тела, дерматитами, дегенеративными изменениями миелиновой оболочки спинного мозга, седалищного нерва и связанными с ними параличами, дискоординацией движений; нарушениями со стороны желудочно-кишечного тракта (гастроэнтериты, колиты, профузная диарея), дегенеративными изменениями репродуктивных органов и гибелью плода, нарушениями биосинтеза стероидных гормонов, связанными с повреждениями надпочечников; замедлением процесса антителообразования; анемией; в далеко зашедших случаях наблюдается летальный исход. В связи с широким распространением пантотеновой кислоты (антидерматитный фактор) в продуктах питания у людей ее недостаточность встречается редко, однако при тяжелых нарушениях питания отмечались поражения кожных покровов, депигментация волос и потеря волосяного покрова; депрессия, апатия; инфекции верхних дыхательных путей; нарушения со стороны сердечнососудистой системы и желудочно-кишечного тракта; слабость мышечных групп разгибателей, онемение пальцев ног, ощущение жжения в стопах.
Пантотеновая кислота в организме человека и животных входит в состав кофермента А (коэнзима А), который принимает участие в осуществлении таких биохимических процессов, как окислительное декарбоксилирование α-кетокислот (пируват, α-кетоглутарат), β-окисление и биосинтез высших жирных кислот, синтез стероидных гормонов, триацилглицеролов, фосфолипидов, ацетилхолина, гиппуровой кислоты, гема гемоглобина и других, выступая в роли промежуточного акцептора и переносчика различных кислотных остатков (ацилов) и образуя так называемые ацилпроизводные кофермента А (в том числе ацетил-КоА - ключевой метаболит, посредством которого происходит взаимодействие белкового, углеводного и липидного обменов).
Развитие симптомов гиповитаминоза в основном обусловлено нарушениями синтетических процессов (в том числе синтеза стероидных гормонов), процессов энергообразования и усилением катаболизма белков, углеводов и липидов, вызванными недостатком кофермента А в организме. Нарушения со стороны нервной системы обусловлены снижением биосинтеза ацетилхолина и фосфолипидов; развитие анемии - нарушением синтеза гема гемоглобина. Развитие дерматитов может быть связано с нарушением обмена соединительной ткани (биосинтеза гликозаминогликанов, в том числе ацетилирования гексозаминов).
Пантотеновая кислота содержится практически во всех продуктах растительного и животного происхождения. Особенно богаты ею печень, почки, яичный желток, икра, мясо, дрожжи, цветная капуста, картофель, помидоры. Кроме того, пантотеновая кислота синтезируется микрофлорой кишечника. Суточная потребность для взрослого человека в пантотеновой кислоте - 3-10 мг.
Витамин Н
У человека экзогенный авитаминоз Н встречается редко в связи с тем, что витамин Н (биотин, антисеборейный фактор) в достаточных количествах синтезируется микрофлорой кишечника (эндогенный синтез может снижаться при приеме антибиотиков и сульфаниламидов). Дефицит биотина может проявляться при мальабсорбции, парентеральном питании, а также при употреблении в пищу большого количества сырого яичного белка, содержащего гликопротеин авидин, способный к необратимому связыванию с витамином, образуя комплекс, не подвергающийся расщепле-
нию в пищеварительном тракте. Биотиновый авитаминоз у животных характеризуется прекращением роста, снижением массы тела, дерматитом (себорея десквамационного типа), депигментацией производных кожи, атактической походкой, отеком конечностей, параличами, нарушениями нервно-трофических процессов и липидного обмена. У человека при недостаточности биотина развиваются себорейный дерматит носогубного треугольника и волосистой части головы, поражения ногтей, выпадение волос, конъюнктивит, миалгия, гиперестезии, атаксия, вялость, сонливость, анорексия, анемия.
Биотиновые ферменты катализируют реакции карбоксилирования (пируваткарбоксилаза, ацетил-КоА-карбоксилаза, пропионилКоА-карбоксилаза, β-метилкротоноил-КоА-карбоксилаза) и транскарбоксилирования (метилмалонилоксалоацетаттранскарбоксилаза), играющие важную роль в синтезе пуриновых нуклеотидов, белков, высших жирных кислот, превращении пирувата в оксалоацетат, что приводит к пополнению запаса оксалоацетата в ЦТК.
При биотиновой недостаточности нарушается обмен белков, липидов, углеводов, нуклеиновых кислот, образование АТФ, в том числе включение СО2 в пуриновые нуклеотиды и ацетоуксусную кислоту; карбоксилирование пропионил-КоА и β-метилкротоноил-КоА; карбоксилирование ацетил-КоА с образованием малонил-КоА (первая стадия синтеза высших жирных кислот). Недостаточность биотина влияет на синтез амилазы в поджелудочной железе и сывороточного альбумина в печени, что, возможно, связано с нарушением синтеза субстратов ЦТК и снижением энергообеспечения клеток.
К врожденным нарушениям обмена и функций биотина относятся врожденная пропионатацидемия, обусловленная дефектом пропионил-КоА-карбоксилазы, и врожденная β-метилкротоноилглицинурия, связанная со снижением активности β-метилкротоноил-КоА-карбоксилазы.
При недостаточности панкреатической биотинидазы, необходимой для усвоения витамина Н, у новорожденных возможно развитие десквамативной эритродермии Лейнера, сопровождающейся себореей и диареей.
Из продуктов животного и растительного происхождения витамином Н богаты почки, печень, молоко, желток яйца, картофель, лук. Суточная потребность - 10-30 мкг.
Витамин Р
Витамин Р (рутин, цитрин, фактор проницаемости) представлен группой веществ с общим названием биофлавоноиды. Наибольшей активностью обладают катехины, рутин, кверцетин.
При недостаточности витамина Р, развивающейся у лиц, не употребляющих растительной пищи, отмечается повышение проницаемости кровеносных сосудов, сопровождающееся кровоизлияниями и кровотечениями. У человека одной из причин общей слабости, быстрой утомляемости и болей в конечностях может являться дефицит биофлавоноидов. Механизм действия витамина Р на сосудистую проницаемость окончательно не изучен. Биофлавоноиды выступают синергистами витамина С в процессе посттрансляционной модификации коллагена - белка, определяющего механические свойства соединительной ткани и сосудистую проницаемость, регулируя активность пролилгидроксилазы. Недостаток витамина Р вызывает увеличение сосудистой проницаемости также за счет активации гиалуронидазы и расщепления гиалуроновой кислоты. Биофлавоноиды защищают от окисления катехоламины (предотвращают переход адреналина в адренохром).
При дефиците витаминов группы Р одной из причин возникающих нарушений могут быть изменения структуры и функции биологических мембран, в частности мембран лизосом, приводящие к выбросу протеолитических ферментов и росту проницаемости капилляров. Антиоксидантные свойства биофлавоноидов проявляются в лимитировании перекисного окисления липидов, восстановлении витамина С, токоферолов, SH-групп (в том числе в составе глутатиона) и т.д. Витамины группы Р оказывают влияние на коагуляционный гемостаз, ограничивая выход в кровь тканевых факторов свертывания; снижают адгезивность тромбоцитов за счет торможения активности фосфодиэстеразы, накопления циклических нуклеотидов и уменьшения продукции тромбоксанов; снижают агрегационную активность тромбоцитов и вовлечение их в реакцию высвобождения.
Обезболивающий, седативный и гипотензивный эффекты ряда биофлавоноидов связаны с их действием на бензодиазепиновые рецепторы мозга.
Витамин Р в больших количествах содержится в цитрусовых, винограде, красном перце и многих других овощах и фрук-
тах. Установленная суточная потребность в витамине Р от 50 до
100 мг.
12.4. ПАТОФИЗИОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА
Обмен углеводов включает в себя несколько этапов:
1. Переваривание и всасывание углеводов.
2. Депонирование углеводов.
3. Промежуточный обмен углеводов:
• анаэробное (гликолиз) расщепление глюкозы;
• аэробное (окислительное декарбоксилирование пирувата до ацетил-КоА, цикл трикарбоновых кислот (ЦТК), пентозофосфатный шунт (ПФШ)) расщепление глюкозы;
• процесс глюконеогенеза (синтез глюкозы из неуглеводных предшественников - лактата, глюкогенных аминокислот и глицерола);
• взаимопревращение гексоз;
• образование пентоз, необходимых для синтеза нуклеотидов;
• синтез жирных кислот (из ацетил-КоА и глицерина).
4. Выделение глюкозы почками и ее реабсорбция.
12.4.1. Нарушение углеводного обмена на этапе переваривания (расщепления) и всасывания
Гидролиз гликогена и крахмала пищи начинается в ротовой полости под влиянием α-амилазы (а-1,4-гликозидаза) слюны. Моносахариды способны всасываться уже в ротовой полости. В желудке нет ферментов, осуществляющих гидролиз углеводов. В полости тонкой кишки под влиянием α-амилазы сока поджелудочной железы они гидролизуются до декстринов и мальтозы (полостное пищеварение) (см. раздел 17.2.4). На поверхности микроворсинок энтероцитов локализованы ферменты: сахараза, мальтаза, лактаза, изомальтаза и др., расщепляющие декстрины и дисахариды до моносахаридов (пристеночное пищеварение) (см. раздел 17.2.4).
Причинами нарушения процессов переваривания (расщепления) и всасывания углеводов являются:
1. Интестинальные энзимопатии наследственного и приобретенного характера (см. раздел 17.2.4.3).
2. Нарушение выработки и выделения панкреатического сока при поражении ацинозной ткани поджелудочной железы (диффуз-
ный панкреатит, закупорка выводного протока камнем или опухолью).
3. Действие ферментных ядов, блокирующих процесс фосфорилирования и дефосфорилирования (флоридзин, монойодацетат), так как моносахариды всасываются только в фосфорилированном виде.
4. Недостаток Na+, например, при гипофункции коры надпочечников.
5. Нарушение кровоснабжения кишечной стенки. Нарушение расщепления (переваривания) углеводов чаще всего
отмечается при дефектах ферментов, участвующих в гидролизе углеводов в кишечнике (интестинальные энзимопатии). К числу наиболее типичных дефектов можно отнести врожденную и приобретенную недостаточность как отдельных ферментов-дисахаридаз - лактазы, сахаразы и изомальтазы, так и недостаточность всего сахаразо-изомальтазного ферментативного комплекса.
Чаще всего встречается врожденная недостаточность лактазы. Фермент гидролизует дисахарид лактозу до глюкозы и галактозы. Новорожденные дети обычно получают 50-60 г лактозы (с молоком) в день. Негидролизованная лактоза поступает в нижние отделы тонкого кишечника, где сбраживается кишечной микрофлорой с образованием газов (вызывает метеоризм) и органических (молочной и уксусной) кислот. Их осмотическое действие привлекает в полость кишечника большое количество воды, что вызывает диарею, поэтому наиболее характерное проявление недостаточности лактазы - диарея после приема молока. При этом кал имеет кислое значение рН и содержит лактозу, иногда регистрируется лактозурия. Симптомы развиваются сразу после рождения. Со временем у ребенка развиваются хронический дисбактериоз и нарушение физического развития. Этот синдром следует отличать от приобретенного дефицита лактазы, а также от недостаточности кишечной лактазы, встречающейся у взрослых людей вследствие снижения экспрессии гена фермента в онтогенезе.
Недостаточность сахаразо-изомальтазного ферментативного комплекса приводит к тому, что дисахариды сахароза и изомальтоза не расщепляются и не усваиваются организмом. Накапливающиеся при этом в просвете кишечника дисахариды осмотически связывают значительное количество воды, что становится причиной диареи. В этих условиях возможно также поглощение клетками эпителия некоторого количества дисахаридов. Однако они остают-
ся метаболически неактивными и в неизмененном виде довольно быстро выводятся с мочой. Например, при дефекте активности сахаразы нагрузка дисахаридами не вызывает гипергликемии в интервале 30-90 мин (плоские гликемические кривые), как это имеет место у здоровых людей. В то же время прием смеси глюкозы и фруктозы (моносахариды, из которых состоит сахароза) вызывает повышение уровня сахара в крови. Врожденный вариант этой патологии проявляется, когда в рацион детей добавляют сахарозу и крахмал, поэтому больные дети неохотно едят сладкое. Другие сахара (глюкоза, фруктоза, лактоза) усваиваются хорошо. Установлено, что наследственные формы этих энзимопатий передаются по аутосомно-рецессивному типу.
Нарушение всасывания углеводов. Всасывание углеводов происходит главным образом в двенадцатиперстной кишке и тонком кишечнике с участием микроворсинок кишечного эпителия только в виде моносахаридов.
Транспорт моносахаридов в энтероциты осуществляется двумя путями:
• по градиенту концентрации путем облегченной диффузии без затраты энергии с помощью специальных белков - транспортеров глюкозы (ГЛЮТ) - ГЛЮТ 2 и ГЛЮТ 5;
• против градиента концентрации путем вторично-активного транспорта с затратой энергии АТФ. По второму пути глюкоза и галактоза транспортируются по механизму активного симпорта (симпорт - перенос двух веществ в одном направлении) параллельно транспорту Na+, происходящему в клетку по градиенту концентрации. Градиент Na+ при этом создается работой Na+/К+-АТФазы. Ионы натрия и моносахаров связываются в разных активных центрах переносчика, и Na+ стремится войти в клетку по градиенту концентрации, «увлекая» за собой глюкозу (галактозу), поэтому при гипонатриемии, когда концентрация Na+ вне клетки уменьшается, транспорт этих сахаров в клетку снижается. Кроме энтероцитов, по механизму активного симпорта происходит натрийзависимая реабсорбция глюкозы и аминокислот из первичной мочи в проксимальных отделах почечных канальцев.
При разной концентрации глюкозы в просвете кишечника «работают» различные механизмы транспорта. Благодаря вторичноактивному транспорту энтероциты могут поглощать глюкозу при ее очень низкой концентрации. Если концентрация глюкозы в
просвете кишечника велика, то она транспортируется в клетку путем облегченной диффузии с помощью ГЛЮТ 2 и ГЛЮТ 5, таким же образом, используя ГЛЮТ 5, может всасываться и фруктоза.
Нарушение всасывания углеводов отмечается при синдроме мальабсорбции (см. раздел 17.2.4.5). У новорожденных и младенцев недостаточно активны как пищеварительные ферменты, так и энзиматические системы фосфорилирования и дефосфорилирования углеводов, вследствие чего их всасывание замедлено.
При нарушении всасывания углеводов происходит уменьшение массы тела, уменьшение выработки энергии, снижается специфическая функция углеводов (образование пентоз, необходимых для синтеза нуклеотидов) и др.
12.4.2. Нарушение углеводного обмена на этапе депонирования гликогена
Глюкоза может поступать в клетки экзогенным путем (из пищи) и эндогенно из депонированного гликогена (в результате гликогенолиза) или из других субстратов в результате глюконеогенеза.
Всосавшаяся в тонкой кишке экзогенная глюкоза поступает через воротную вену в печень и попадает в гепатоциты, где с помощью активированной инсулином глюкокиназы превращается в глюкозо-6-фосфат (Г-6-Ф) (рис. 12-14). Глюкокиназа гепатоцитов и панкреоцитов работает в абсорбтивном периоде, когда концентрация глюкозы крови может повышаться до 8 ммоль/л, а в воротной вене - до 10 ммоль/л. Особые свойства глюкокиназы, которая в отличие от гексокиназ клеток других тканей не игибируется Г-6-Ф (конечным продуктом этой реакции), позволяют, в первую очередь гепатоцитам, захватывать глюкозу из кровотока в абсорбтивном периоде. Это предотвращает чрезмерное повышение ее концентрации в крови после приема пищи и такие нежелательные последствия гипергликемии, как гликозилирование белков.
Процесс облегченной диффузии глюкозы в клетки осуществляется с помощью белков-транспортеров глюкозы (ГЛЮТ 1, 2, 3, 5, 6), поэтому скорость трансмембранного переноса глюкозы в этом случае зависит только от ее концентрации в крови (табл. 12-1). Исключение составляют клетки мышечной и жировой ткани, где облегченная диффузия регулируется инсулином (гормон поджелудочной железы) - инсулинзависимые ткани. При отсутствии инсулина цитоплазматическая мембрана этих клеток непроницае-
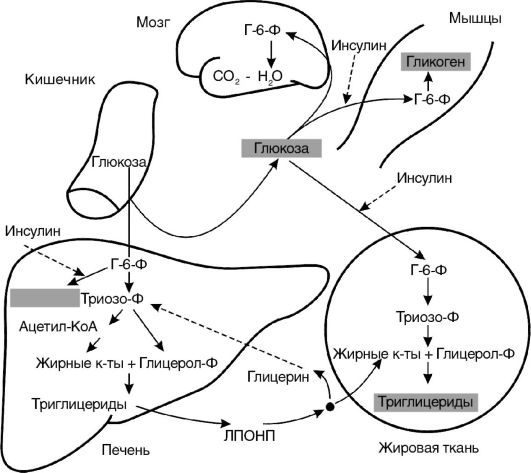 Рис.
12-14. Метаболизм глюкозы после еды. Всосавшаяся в кишечнике глюкоза
поступает в печень. Печень поддерживает постоянную доставку
энергетических субстратов для других органов, в частности мозга.
Поступление глюкозы в печень и мозг не зависит от инсулина, в мышцы и
жировую ткань - инсулинзависимое. Во всех клетках первый этап
метаболизма глюкозы - образование глюкозо-6-фосфата (Г-6-Ф). В печени
инсулин стимулирует фермент глюкокиназу, переводя глюкозу в Г-6-Ф и
далее в гликоген, избыток Г-6-Ф переводится в жирные кислоты с
последующим образованием триацилглицеролов (ТАГ), которые освобождаются
из печени в виде липопротеинов очень низкой плотности (ЛПОНП). В мышцах
глюкоза запасается в виде гликогена, в жировой ткани переходит в ТАГ, в
мозговой ткани глюкоза используется как энергетический субстрат
Рис.
12-14. Метаболизм глюкозы после еды. Всосавшаяся в кишечнике глюкоза
поступает в печень. Печень поддерживает постоянную доставку
энергетических субстратов для других органов, в частности мозга.
Поступление глюкозы в печень и мозг не зависит от инсулина, в мышцы и
жировую ткань - инсулинзависимое. Во всех клетках первый этап
метаболизма глюкозы - образование глюкозо-6-фосфата (Г-6-Ф). В печени
инсулин стимулирует фермент глюкокиназу, переводя глюкозу в Г-6-Ф и
далее в гликоген, избыток Г-6-Ф переводится в жирные кислоты с
последующим образованием триацилглицеролов (ТАГ), которые освобождаются
из печени в виде липопротеинов очень низкой плотности (ЛПОНП). В мышцах
глюкоза запасается в виде гликогена, в жировой ткани переходит в ТАГ, в
мозговой ткани глюкоза используется как энергетический субстрат
ма для глюкозы, так как она не содержит белки-транспортеры. В этих тканях ГЛЮТ 4 находятся в цитозольных везикулах, и их транслокация (перемещение) на мембрану клеток возможна лишь после взаимодействия инсулина с инсулиновым рецептором (рис. 12-15).
Таблица 12-1. Распределение и свойства белков-транспортеров глюкозы в организме человека
Типы ГЛЮТ | Локализация в органах, свойства ГЛЮТ |
ГЛЮТ 1 | На цитоплазматической мембране клеток мозга, почек, толстого кишечника, тканей глаза, плаценты, эритроцитов; обеспечивает стабильный поток глюкозы в клетку при нормогликемии |
ГЛЮТ 2 | На цитоплазматической мембране клеток печени, почек, тонкого кишечника, β-клеток островков Лангерганса; обеспечивает поток глюкозы в абсорбтивном периоде пищеварения (наследственный дефект ГЛЮТ 2 или нарушение его функции - один из механизмов снижения чувствительности β-клеток к глюкозе, приводящей к инсулинорезистентности при СД 2 типа) |
ГЛЮТ 3 | На цитоплазматической мембране клеток мозга, почек, плаценты, скелетных мыщц плода и др. тканей; обладает большим, чем ГЛЮТ 1, сродством к глюкозе, обеспечивает поток глюкозы при гипогликемии |
ГЛЮТ 4 | В цитоплазме клеток скелетных мышц, миокарда, жировой ткани в специальных везикулах; инсулинзависимый переносчик глюкозы, обеспечивает поток глюкозы в абсорбтивном периоде пищеварения (наследственный дефект ГЛЮТ 4 или нарушение его функции - один из механизмов инсулинорезистентности СД 2 типа) |
ГЛЮТ 5 | На цитоплазматической мембране энтероцитов, клеток почек; способен переносить и фруктозу |
ГЛЮТ 6 | Выделен в 1990 г. из тканей взрослого человека, на 80% по свойствам идентичен ГЛЮТ 3 |
Гликоген. Из Г-6-Ф в результате сочетанного действия гликогенсинтазы и «ветвящего» фермента (амило-1,4->1,6-глюкозил- транферазы) синтезируется гликоген - полисахаридный полимер человека и животных. Гликоген является депонированной формой глюкозы. Он содержится практически во всех тканях; в клетках нервной системы его количество минимально, а в печени и мышцах его особенно много. Гликоген содержит только два типа гликозиднъгх связей: α (1->4)-тип и α (1->6)-тип. Связь α (1->4)-тип формируется через каждые 8-10 остатков D-глюкозы (рис. 12-16). Основным гормоном, активирующим синтез гликогена, является инсулин.
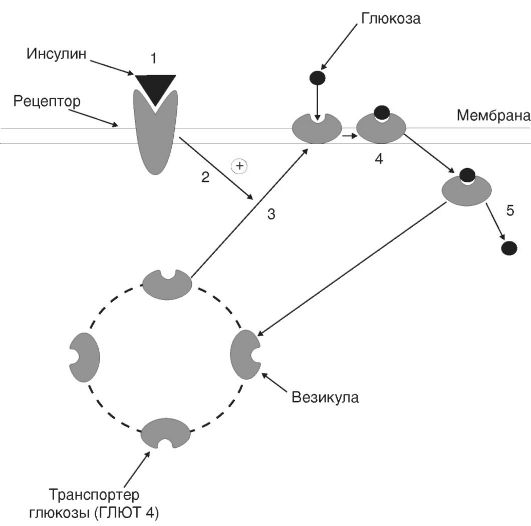 Рис.
12-15. Влияние инсулина на перемещение транспортера глюкозы (ГЛЮТ) 4 из
цитоплазмы в плазматическую мембрану клеток в инсулинзависимых тканях: 1
- связывание инсулина с рецептором; 2 - участок инсулинового рецептора,
обращенный внутрь клетки, который стимулирует перемещение ГЛЮТ 4; 3, 4 -
ГЛЮТ 4 в составе везикул перемещаются к плазматической мембране,
включаются в ее состав; 5 - ГЛЮТ 4 переносят глюкозу в клетку
Рис.
12-15. Влияние инсулина на перемещение транспортера глюкозы (ГЛЮТ) 4 из
цитоплазмы в плазматическую мембрану клеток в инсулинзависимых тканях: 1
- связывание инсулина с рецептором; 2 - участок инсулинового рецептора,
обращенный внутрь клетки, который стимулирует перемещение ГЛЮТ 4; 3, 4 -
ГЛЮТ 4 в составе везикул перемещаются к плазматической мембране,
включаются в ее состав; 5 - ГЛЮТ 4 переносят глюкозу в клетку
 Рис. 12-16. Структура гликогена
Рис. 12-16. Структура гликогена
Снижение синтеза гликогена и уменьшение его депонирования.
Синтез гликогена из глюкозы (гликогеногенез), как и любой анаболический процесс, происходит с затратой энергии макроэргов. Печень запасает глюкозу в виде гликогена не столько для собственных нужд, сколько для поддержания постоянной концентрации глюкозы в крови. В мышечной ткани гликоген депонируется для обеспечения энергией организма при физической нагрузке. Уменьшение депонирования гликогена может быть обусловлено повышенным его расщеплением в условиях недостатка ресинтеза или нарушением его синтеза.
Причинами снижения синтеза гликогена и его депонирования являются:
• снижение тонуса парасимпатической нервной системы;
• гипоксии различного генеза;
• поражение гепатоцитов (острые и хронические гепатиты, отравление гепатотропными ядами - фосфором, четыреххлористым углеродом, дихлорэтаном);
• гипоавитаминозы В1 и С;
• нарушение эндокринной регуляции (сахарный диабет, тиреотоксикоз, недостаточность коры надпочечников - болезнь Аддисона);
• наследственные болезни - агликогеноз или гликогеноз 0 (дефект гликогенсинтазы).
Гликогенолиз. Это путь расщепления гликогена. Основными гормонами, активирующими гликогенолиз, являются глюкагон, адреналин. Кортизол вызывает длительную активацию гликогенолиза в инсулинзависимых тканях при стрессе.
Причинами усиления распада гликогена и снижения его депонирования являются:
• возбуждение симпатической нервной системы (нервные импульсы проводятся к депо гликогена и активируют процесс его распада);
• повышение продукции гормонов, стимулирующих гликогенолиз (адреналина, глюкагона, тироксина и соматотропного гормона);
• интенсивная мышечная работа, что обусловливается увеличением потребления глюкозы мышцами;
• лихорадка, шок, эмоциональные нагрузки.
При недостаточности гликогена субстратом для энергетических процессов в клетке становятся жирные кислоты и белки. В ре-
зультате такого переключения метаболизма происходит избыточное образование кетоновых тел и развиваются гиперкетонемия и кетонурия. Использование клеткой белков как источника энергии обусловливает нарушения различных ферментативных и пластических процессов.
Нарушения обмена гликогена, связанные с его патологическим депонированием, проявляются гликогеновыми болезнями. Это группа наследственных нарушений, в основе которых лежит снижение или отсутствие активности ферментов, катализирующих реакции синтеза (агликогенозы) или распада гликогена (гликогенозы).
Гликогенозы - заболевания, обусловленные генетическими дефектами отдельных ферментов распада гликогена, при которых происходит его избыточное накопление в различных органах, прежде всего в печени и скелетных мышцах. При некоторых типах гликогенозов синтезируется гликоген с нарушенной структурой.
Известны 12 типов гликогенозов. В настоящее время выделяют 2 формы гликогенозов: печеночные и мышечные.
При печеночных формах в гепатоцитах нарушается гликогенолиз, обеспечивающий нормальный уровень глюкозы в крови, поэтому их общий симптом - это гипогликемия в постабсорбтивном периоде. К ним относятся гликогеноз I типа (болезнь Гирке), III типа (болезнь Кори и Форбса), IV типа (болезнь Андерсена), VI типа (болезнь Герса), IX типа (болезнь Хага) и X типа.
Мышечные формы гликогенозов характеризуются нарушением гликогенолиза, обеспечиваюшего энергоснабжение скелетных мыщц. Эти болезни проявляются при физических нагрузках и сопровождаются болями и судорогами в мышцах, слабостью и быстрой утомляемостью, гипогликемия не характерна. К ним относятся гликогенозы V (болезнь Мак-Ардла) и VIII (болезнь Таруи) типа.
1. Гликогеноз I типа, или болезнь Гирке (Гирке - Ван-Кревельда), или гепатонефромегальный. Впервые описан в 1929 г. Это наиболее часто регистрируемый тип гликогеноза. В основе данной патологии лежит наследственный дефицит фермента глюкозо- 6-фосфатазы в печени и почках. Под действием этого фермента происходит дефосфорилирование Г-6-Ф, что обеспечивает трансмембранный переход свободной глюкозы из гепатоцитов и клеток почек в кровь. Дефицит фермента вызывает накопление избытка гликогена в клетках печени и почек. Структура гликогена при этом не нарушается. У больных увеличен живот за счет существенного
увеличения размеров печени (гепатомегалия). Содержание гликогена в печени достигает 10-15% от массы органа (вместо 3-5% в норме). В связи с дефектом фермента поступление в кровь глюкозы значительно сокращается, что обусловливает тяжелую гипогликемию, являющуюся причиной приступов судорог. Дефицит глюкозы в крови приводит к торможению выделения инсулина из поджелудочной железы, что стимулирует липолиз в жировой ткани. Формируется характерная для данного типа гликогеноза гипертриацилглицеролемия. Интенсификация метаболизма липидов приводит к накоплению в крови молочной кислоты, кетоновых тел и к ацидозу. Дефицит инсулина сопровождается снижением интенсивности синтеза белка (нарушается транспорт аминокислот). Накопление в гепатоцитах Г-6-Ф увеличивает его использование в ПФШ с образованием рибозо-5-фосфата, из которого синтезируется большое количество пуриновых нуклеотидов, что приводит в конечном итоге к гиперурикемии. Больные имеют характерное выражение лица - «лицо китайской куклы». Пациенты, страдающие этим типом гликогеноза, как правило, рано умирают от интеркуррентных заболеваний или от ацидотической комы.
2. Гликогеноз III типа, или болезнь Кори (Кори и Форбса). Заболевание возникает по причине наследственного дефекта фермента амило-1,6-глюкозидазы («деветвящий» фермент) мышц, печени и миокарда. Молекула полимера при данном типе гликогеноза имеет патологическую структуру, для которой характерны многочисленные, но укороченные боковые ветви. Этот гликоген в избытке накапливается преимущественно в печени, мышечной ткани, эритроцитах и зернистых лейкоцитах. Затрудненный гидролиз гликогена с измененной структурой приводит к гипогликемии. Имеет место также гипертриацилглицеролемия, развивающаяся по механизму, аналогичному для гликогеноза I типа, но в более мягкой форме. Угрозы для жизни не представляет. Специфическим диагностическим признаком гликогеноза III типа является повышенный уровень гликогена в зернистых лейкоцитах.
3. Гликогеноз IV типа, или болезнь Андерсена. В основе заболевания лежит наследственный дефицит фермента амило-1,4->1,6- глюкозилтрансферазы, который обеспечивает ветвление в молекуле гликогена. Недостаточность фермента обусловливает накопление преимущественно в печени, мышцах и лейкоцитах гликогена, молекулы которого имеют аномально длинные цепи с очень малым количеством боковых ответвлений. Структура гликогена становит-
ся похожей на таковую у крахмала. Для клинической картины данного заболевания характерны: выраженная гипогликемия, гепатомегалия, прогрессирующий цирроз печени с летальным исходом в возрасте до 2 лет вследствие развития печеночной недостаточности.
4. Гликогеноз V типа, или болезнь Мак-Ардла (Мак-Ардла- Шмида-Пирсона болезнь). Болезнь с аутосомно-рецессивным наследованием, при которой в скелетных мышцах полностью отсутствует активность гликогенфосфорилазы, поэтому происходит накопление гликогена с нормальной структурой. Поскольку активность этого фермента в гепатоцитах нормальная, то гипогликемия не развивается (строение фермента в печени и мышцах кодируется разными генами). Больные плохо переносят тяжелые физические нагрузки, часто возникают судороги.
Помимо приведенных выше типов, описаны смешанные (генерализованные) гликогенозы, к ним относятся гликогеноз II типа (болезнь Помпе), гликогеноз VII типа (болезнь Томсона - когда недостаточность фосфоглюкомутазы имеет место в печени и в мышцах). Гликогеноз II типа, или болезнь Помпе развивается при наследственном дефекте фермента α-1,4-глюкозидазы, локализованного в лизосомах. Дефект фермента приводит к генерализованному накоплению гликогена с нормальной структурой в печени, селезенке, почках, скелетных и сердечной мышцах, нервной ткани, лейкоцитах и эритроцитах. Клинические симптомы заболевания проявляются уже на 2-6-м месяце жизни. Наиболее тяжелые нарушения развиваются в мышечной ткани: гипотония скелетных мышц и увеличение размеров сердца (кардиомегалия), что ведет к тяжелой кардиореспираторной недостаточности. Она является главной причиной летального исхода в возрасте до 2 лет.
12.4.3. Нарушения промежуточного обмена углеводов
Промежуточный обмен углеводов осуществляется на уровне клетки и включает в себя все превращения с момента их поступления в клетку (образование Г-6-Ф в глюкоили гексокиназной реакции) до образования конечных продуктов распада СО2 и Н2О. Основной путь обмена глюкозы - гликолиз. В физиологических условиях пищеварения роль гликолиза в жировой ткани, так же как и в печени, заключается в обеспечении источника субстратов для синтеза жирных кислот (рис. 12-17 - пути 1, 2, 3, 4). В тканях,
где нет или очень мало митохондрий (эритроциты, белые мышцы, клетки сетчатки глаза, мозгового слоя коры надпочечников), гликолиз является конечным энергетическим процессом, в результате которого образуется лактат. Избыток лактата образуется и в скелетных мышцах при интенсивной физической нагрузке. Благодаря реакции ресинтеза лактата в пируват в печени (см. рис. 12-17) - путь 2 (цикл Кори), избыток лактата возвращается в метаболический пул углеводов, так как из него вновь синтезируется глюкоза - путь 6 - (глюконеогенез), которая может снова возвращаться в кровь и поглощаться скелетными мышцами в условиях физической нагрузки - путь 7.
Когда поступление углеводов в составе пищи уменьшается, содержание глюкозы в крови поддерживается за счет гликогенолиза
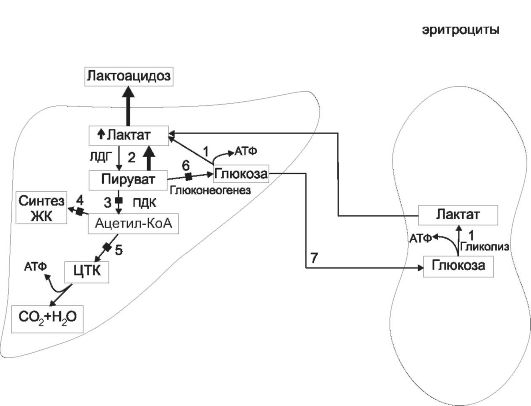 Рис. 12-17. Метаболизм
глюкозы, лактата и пирувата в норме и при гипоксии: 1 - основной путь
обмена глюкозы в печени, мышцах, эритроцитах; 2 - ресинтез пирувата; 3-7
- пути, обеспечивающие обмен глюкозы в норме; ■ - блокирование
ферментов метаболизма пирувата при гипоксии; - - усиление эффекта; ПДК -
пируватдекарбоксилаза; ЛДГ - лактатдегидрогиназа; ЖК - жирные кислоты,
ЦТК - цикл трикарбоновых кислот
Рис. 12-17. Метаболизм
глюкозы, лактата и пирувата в норме и при гипоксии: 1 - основной путь
обмена глюкозы в печени, мышцах, эритроцитах; 2 - ресинтез пирувата; 3-7
- пути, обеспечивающие обмен глюкозы в норме; ■ - блокирование
ферментов метаболизма пирувата при гипоксии; - - усиление эффекта; ПДК -
пируватдекарбоксилаза; ЛДГ - лактатдегидрогиназа; ЖК - жирные кислоты,
ЦТК - цикл трикарбоновых кислот
в печени, где запасы гликогена невелики, и они истощаются уже к 6-10 ч голодания. При продолжении голодания до 24 ч включается глюконеогенез в печени, энтероцитах и корковом веществе почек, обеспечивающий синтез 80-100 г глюкозы в сутки. На долю мозга при голодании приходится большая часть потребности организма в глюкозе.
Причины, механизмы и последствия нарушения промежуточного обмена углеводов:
1. Гипоксия, возникающая при снижении рО2 во вдыхаемом воздухе, недостаточности дыхания и кровообращения, анемиях, интоксикациях, инфекциях, переключает клеточный метаболизм с преимущественного аэробного окисления субстратов на анаэробный (рис. 12-17). При распаде глюкозы в процессе гликолиза образуется избыток лактата, что приводит к лактоацидозу.
2. Нарушения функции печени. Возникновению лактоацидоза способствует также нарушение глюконеогенеза в гепатоцитах (рис. 12-17), где часть молочной кислоты в норме ресинтезируется в глюкозу и гликоген. При острых и хронических гепатитах, циррозах, отравлениях гепатотропными ядами этот процесс нарушается, молочная кислота выходит в кровь, развивается лактоацидоз.
3. Гиповитаминоз В1. При недостаточности витамина В1 возникает дефицит кокарбоксилазы, что приводит к подавлению синтеза ацетил-КоА из пировиноградной кислоты. Последняя накапливается и частично переходит в молочную кислоту, содержание которой в связи с этим возрастает, и возникает лактоацидоз. Торможение окисления пировиноградной кислоты снижает синтез ацетилхолина и нарушает передачу нервных импульсов. При возрастании концентрации пировиноградной кислоты в 2-3 раза по сравнению с нормой возникают нарушения чувствительности, невриты, параличи.
4. Наследственные дефекты ферментов: дефект ферментов глюконеогенеза; недостаточность глюкозо-6-фосфатазы при гликогенозе I типа (болезнь Гирке) также приводят к выраженному лактоацидозу.
5. Ятрогенные факторы. Применение ряда лекарственных препаратов, например бигуанидов (блокаторы глюконеогенеза), при лечении сахарного диабета может привести к коме с лактатацидозом.
12.4.4. Нарушение выделения глюкозы почками
Нарушения выделения глюкозы почками связаны с уменьшением фильтрации глюкозы в клубочках или снижением ее реабсорбции в проксимальных канальцах нефрона. В клубочках почек глюкоза фильтруется, затем в эпителии проксимальных канальцев реабсорбируется путем вторично-активного транспорта с затратой энергии АТФ по градиенту концентрации Na+. При увеличении в сыворотке уровня глюкозы более 8,8-9,9 ммоль/л она начинает выделяться с мочой. Показатель гликемии, при котором появляется глюкозурия, называется почечным порогом. На выделение глюкозы с мочой влияет скорость клубочковой фильтрации, которая в норме составляет примерно 130 мл/мин. При снижении фильтрации глюкоза не определяется в моче даже при гликемии, значительно превышающей почечный порог, так как фильтруется меньше глюкозы и вся она успевает реабсорбироваться в проксимальных канальцах почек. В случае патологии канальцев (тубулопатии) при нарушении реабсорбции глюкоза может присутствовать в моче даже в условиях нормоили гипогликемии (почечный диабет). В связи с этим диагностика «сахарного диабета» только по одному показателю уровня глюкозы в моче не проводится.
12.4.5. Нарушение регуляции углеводного обмена
Уровень глюкозы в крови является жесткой гомеостатической константой организма и критерием адекватности углеводного обмена. Нормальный уровень гликемии поддерживается работой ЦНС, кишечника, печени, почек, поджелудочной железы, надпочечников, жировой ткани и других органов (см. рис. 12-14), в связи с чем выделяют нервную, гормональную, почечную и субстратную регуляцию углеводного обмена.
Субстратная регуляция. Основным фактором, определяющим метаболизм глюкозы, является уровень гликемии. Пограничная концентрация глюкозы, при которой продукция ее в печени равна потреблению периферическими тканями, составляет 5,5-5,8 ммоль/л. При уровне меньше указанного печень поставляет глюкозу в кровь; при большем уровне, напротив, доминирует синтез гликогена в печени и мышцах.
Нервная регуляция. Симпатическая импульсация приводит к освобождению адреналина из надпочечников, который стимули-
рует гликогенолиз, и развивается гипергликемия. Раздражение парасимпатических нервных волокон сопровождается усилением выделения инсулина поджелудочной железой, поступлением глюкозы в клетку и гипогликемическим эффектом.
Почечная регуляция. Нормальная работа почек поддерживает уровень глюкозы с помощью процессов фильтрации и реабсорбции (см. раздел 12.4.4).
Гормональная регуляция. На уровень глюкозы в крови влияет широкий спектр гормонов, при этом только инсулин вызывает гипогликемический эффект. Контринсулярным действием с повышением уровня глюкозы крови обладают следующие гормоны: глюкагон, адреналин, глюкокортикоиды, аденокортикотропный (АКТГ), соматотротгый (СТГ), тареотропный (ТТГ), тареоидные. Эффекты инсулина и контринсулярных гормонов в норме регулируют стабильный уровень глюкозы в крови. При низкой концентрации инсулина, в частности при голодании, усиливаются гипергликемические эффекты других гормонов, таких, как глюкагон, адреналин, глюкокортикоиды и гормон роста. Это происходит даже в том случае, если концентрация этих гормонов в крови не увеличивается.
В табл. 12-2 приведена характеристика действия гормонов на метаболизм глюкозы.
Таблица 12-2. Гормоны, контролирующие гомеостаз глюкозы
Название гормона | Место образования | Механизм действия и ткани-мишени |
Инсулин | β-клетки островков Лангерганса поджелудочной железы | Увеличивает: потребление глюкозы клетками (мышцы, жировая ткань); синтез гликогена и белков (печень, мышцы); липогенез (печень, жировая ткань) Снижает: липолиз (жировая ткань); глюконеогенез и кетогенез (печень); протеолиз (мышцы) |
Глюкагон | α-клетки островков Лангерганса поджелудочной железы | Увеличивает: гликогенолиз, глюконеогенез, кетогенез (печень), липолиз (жировая ткань) |
Окончание табл. 12-2
Адреналин | Мозговое вещество надпочечников | Увеличивает: гликогенолиз (печень, мышцы); липолиз (жировая ткань) |
СТГ (гормон роста) | Эозинофильные клетки аденогипофиза | Увеличивает: гликогенолиз (печень); липолиз (жировая ткань) |
АКТГ | Базофильные клетки аденогипофиза | Стимулирует освобождение глюкокортикоидов (надпочечники) Увеличивает липолиз (жировая ткань) |
Глюкокортикоиды | Пучковая зона коркового вещества надпочечников | Увеличивает: глюконеогенез, синтез гликогена (печень); протеолиз (мышцы) Снижает потребление глюкозы клетками (мышцы, жировая ткань) |
Гормоны щитовидной железы | Тироциты | Увеличивает: утилизацию глюкозы клетками, липолиз, протеолиз (опосредовано через усиление основного обмена) - все ткани Активирует инсулиназу (печень) |
В физиологических условиях в регуляции обмена глюкозы наиболее важны два гормона - инсулин и глюкагон.
Инсулин - видоспецифичный пептидный гормон (представляет собой полипептид, состоящий из двух аминокислотных цепей (А- и В-цепи), соединенных между собой двумя дисульфидными мостиками). Инсулин синтезируется в виде неактивной полипептидной цепи проинсулина, таким он сохраняется в гранулах β-клеток островков Лангерганса поджелудочной железы. Активация проинсулина заключается в частичном протеолизе пептида по Arg31 и Arg63 (рис. 12-18). В результате в эквимолярном количестве образуются инсулин и С-пептид, уровень которого в крови позволяет достаточно точно определить функциональное состояние β-клеток и является важным критерием в диагностике диабета. В сыворотке здоровых людей обнаруживается также небольшое количество проинсулина, его содержание значительно повышается у лиц с аденомой панкреатических β-клеток.
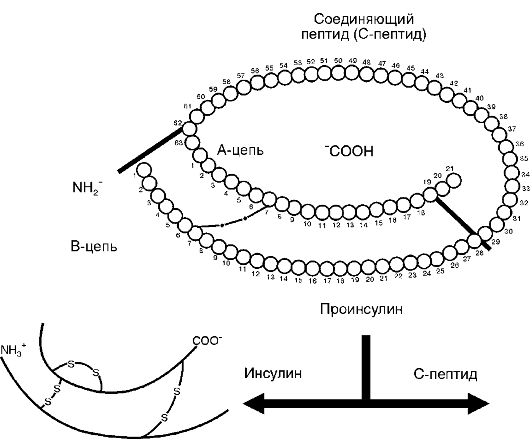 Рис.
12-18. Образование инсулина в поджелудочной железе. В результате
частичного протеолиза проинсулина формируются инсулин и С-пептид.
Инсулин состоит из двух полипептидных цепей, соединенных дисульфидными
мостиками
Рис.
12-18. Образование инсулина в поджелудочной железе. В результате
частичного протеолиза проинсулина формируются инсулин и С-пептид.
Инсулин состоит из двух полипептидных цепей, соединенных дисульфидными
мостиками
Характеризуя секрецию инсулина, выделяют базальную секрецию (утром, после ночного голодания), фазу 1 - ранний пик секреции инсулина (у человека выявляется в ходе внутривенного глюкозотолерантного теста (ГТТ) в первые 10 мин после поступления глюкозы в кровь), фазу 2 (глюкозо-стимулированная секреция) - постепенное повышение секреции инсулина (30-120 мин).
Известны 3 механизма регуляции секреции инсулина β-клеткой, включающие несколько сигнальных путей (рис. 12-19). Секреция инсулина стимулируется, помимо указанных на рис. 12-19 факторов, окситоцином, пролактином, эстрогенами, кортизолом, СТГ (в высоких концентрациях), вазопрессином, опиоидными пептидами, свободными жирными кислотами. Катехоламины и нейропептид Y, а также соматостатин и простагландины подавляют секрецию инсулина. Инсулин способен оказывать аутокринное ингибиторное влияние на свою секрецию через собственные рецепторы на
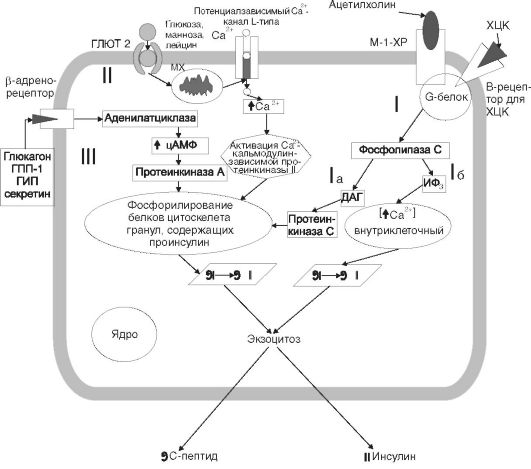 Рис. 12-19. Механизмы стимуляции секреции инсулина β-клеткой: I - стимуляция М1-холинорецепторов
(ХР) и В-рецепторов для холецистокинина (ХЦК) вызывает
G-белокопосредованную активацию фосфолипазы С, расщепляющей мембранные
фосфолипиды на два вторичных посредника - инозитолтрифосфат (ИФ3)
и диацилглицерол (ДАГ); Iа - ДАГ активирует протеинкиназу C, которая
фосфорилирует белки цитозоля и вызывает экзоцитоз секреторных гранул без
повышения уровня внутриклеточного Са2+; I6 - ИФ3 открывает Са2+-каналы в эндоплазматической сети и митохондриях (МХ) и увеличивает концентрацию внутриклеточного Са2+, что приводит к экзоцитозу секреторных гранул; II - активация секреции инсулина моносахаридами и аминокислотами Са2+-зависимый процесс; активация транспорта Са2+ происходит через усиление метаболизма этих субстратов в МХ и открытие Са2+-канала L-типа с последующей активацией Са2+-кальмодулинзависимой
протеинкиназы II, что приводит к экзоцитозу секреторных гранул; III -
стимуляция β-адренорецепторов активирует аденилатциклазу и повышает в
цитозоле уровень цАМФ, который активирует протеинкиназу А, вызывающую
фосфорилирование белков цитоскелета секреторных гранул и экзоцитоз. Примечание: ГПП-1 - глюкагоноподобный пептид 1; ГИП - гастринингибирующий пептид
Рис. 12-19. Механизмы стимуляции секреции инсулина β-клеткой: I - стимуляция М1-холинорецепторов
(ХР) и В-рецепторов для холецистокинина (ХЦК) вызывает
G-белокопосредованную активацию фосфолипазы С, расщепляющей мембранные
фосфолипиды на два вторичных посредника - инозитолтрифосфат (ИФ3)
и диацилглицерол (ДАГ); Iа - ДАГ активирует протеинкиназу C, которая
фосфорилирует белки цитозоля и вызывает экзоцитоз секреторных гранул без
повышения уровня внутриклеточного Са2+; I6 - ИФ3 открывает Са2+-каналы в эндоплазматической сети и митохондриях (МХ) и увеличивает концентрацию внутриклеточного Са2+, что приводит к экзоцитозу секреторных гранул; II - активация секреции инсулина моносахаридами и аминокислотами Са2+-зависимый процесс; активация транспорта Са2+ происходит через усиление метаболизма этих субстратов в МХ и открытие Са2+-канала L-типа с последующей активацией Са2+-кальмодулинзависимой
протеинкиназы II, что приводит к экзоцитозу секреторных гранул; III -
стимуляция β-адренорецепторов активирует аденилатциклазу и повышает в
цитозоле уровень цАМФ, который активирует протеинкиназу А, вызывающую
фосфорилирование белков цитоскелета секреторных гранул и экзоцитоз. Примечание: ГПП-1 - глюкагоноподобный пептид 1; ГИП - гастринингибирующий пептид
β-клетках. Особое значение для регуляции секреции инсулина придается лептину, повышение выработки которого адипоцитами ингибирует секрецию инсулина, а также экспрессию генов рецептора инсулина, субстрата инсулинового рецептора и ГЛЮТ 4 (см. раздел 12.5).
Нарушения секреции инсулина могут являться результатом:
• недостаточного питания плода, что приводит к нарушению внутриутробного развития поджелудочной железы;
• недостаточного питания в постнатальном периоде;
• действия глюкозотоксичности (при хронической гипергликемии);
• генетических дефектов в механизмах секреции инсулина (мутации генов инсулина, глюкокиназы, ГЛЮТ 2 и др.).
Нарушения секреции инсулина могут выражаться ее снижением в ответ на глюкозу и другие стимуляторы (аргинин, лейцин); нарушением пульсирующей секреции инсулина и превращения проинсулина в инсулин, что приводит к повышению содержания проинсулина в крови.
Процессы синтеза и секреции инсулина не являются строго сопряженными процессами. Основными стимуляторами синтеза инсулина являются глюкоза, манноза, аргинин и лейцин. Известны 2 пути регуляции глюкозой синтеза инсулина β-клеткой (рис. 12-20). Путь I, связанный с активацией трансляции уже существующей в цитозоле матричной РНК (мРНК) проинсулина, - быстрый, не требующий усиления транскрипции генов; поэтому за счет него осуществляется синтез инсулина в ответ на стимуляцию глюкозой, которая приходится на начало абсорбтивного периода. Глюкокортикоиды укорачивают время жизни проинсулиновой мРНК и таким образом могут снижать продукцию инсулина в β-клетках. Параллельно активируется путь II синтеза инсулина, обеспечивающий достаточное количество гормона в конце абсорбтивного периода (см. рис. 12-20).
Инсулин в крови находится в свободном и связанном с белками состоянии. Деградация инсулина происходит в печени (до 80%), почках и жировой ткани. С-пептид также подвергается деградации в печени, но значительно медленнее. Концентрация инсулина натощак составляет у здоровых лиц 36-180 пмоль/л. После пероральной нагрузки глюкозой уровень его через 1 ч повышается в 5-10 раз по сравнению с исходным.
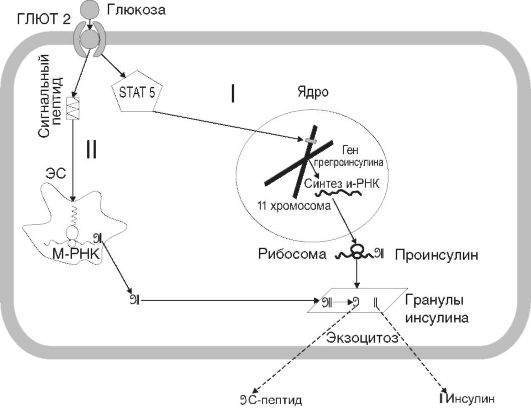 Рис. 12-20. Пути
регуляции глюкозой синтеза инсулина β-клеткой: I - путь, связанный с
активацией гена препроинсулина и транскрипцией м-РНК в ядре клетке; II -
путь, связанный с активацией цитозольной м-РНК препроинсулина на
рибосомах эндоплазматической сети; STAT 5 - активирующие
транскрипционные факторы
Рис. 12-20. Пути
регуляции глюкозой синтеза инсулина β-клеткой: I - путь, связанный с
активацией гена препроинсулина и транскрипцией м-РНК в ядре клетке; II -
путь, связанный с активацией цитозольной м-РНК препроинсулина на
рибосомах эндоплазматической сети; STAT 5 - активирующие
транскрипционные факторы
Инсулин - главный анаболический гормон, обладающий широким спектром действия на транспорт и обмен углеводов, аминокислот, ионов, липидов, а также на процессы репликации и транскрипции, клеточной дифференцировки, пролиферации и трансформации. Высокие концентрации инсулина в крови обладают анаболическим, а низкие - катаболическим действием на обмен веществ.
Метаболические эффекты инсулина:
1) увеличивают активность и количество ключевых ферментов гликолиза;
2) активируют фермент гексокиназу, фосфорилирующую глюкозу во всех тканях организма;
3) увеличивают проницаемость клеточных мембран в мышцах и жировой ткани для глюкозы, ионов калия, натрия, аминокислот; для кетоновых тел в мышцах;
4) активируют гликогенсинтазу, вызывая усиление гликогеногенеза в печени;
5) снижают гликогенолиз, подавляя активность гликогенфосфатазы и гликогенфосфорилазы;
6) уменьшают активность ферментов глюконеогенеза;
7) снижая процессы глюконеогенеза, опосредованно активируют синтез белка;
8) увеличивают липогенез, усиливая синтез триацилглицеролов из углеводов, активируя липопротеиновую липазу (ЛП-липазу) адипоцитов;
9) ускоряют использование глюкозы в ЦТК и ПФШ.
В то же время полипептидная молекула инсулина не способна проникать через клеточную мембрану, поэтому все эффекты инсулина осуществляются через специальные рецепторы на ее поверхности. Рецепторы инсулина обнаружены почти во всех типах клеток, но больше всего их в гепатоцитах и клетках жировой ткани. Клетки с разным содержанием рецепторов на мембране реагируют по-разному на одну и ту же концентрацию гормона. Инсулиновый рецептор относится к рецепторам с тирозинкиназной активностью, обеспечивающим фосфорилирование специфических внутриклеточных белков - субстратов инсулинового рецептора (IRS). Активированные IRS включают несколько сигнальных путей в клетке, что составляет основу многостороннего влияния инсулина на внутриклеточный метаболизм.
Глюкагон - одноцепочечный полипептид, состоящий из 29 аминокислотных остатков, его эффекты противоположны эффектам инсулина. Основные клетки-мишени для глюкагона - печень и жировая ткань. Связываясь с рецепторами клеток-мишеней, глюкагон ускоряет мобилизацию гликогена в печени и мобилизацию липидов в жировой ткани, активируя через аденилатциклазный каскад гормончувствительную ТАГ-липазу. В β-клетках поджелудочной железы глюкагон стимулирует секрецию инсулина из гранул в условиях высокой гликемии в течение абсорбтивного периода (см. рис. 12-19). Совместные эффекты инсулина и глюкагона в поджелудочной железе и в других органах представлены на рис. 12-21.
12.4.6. Нарушения углеводного обмена
При нарушении углеводного обмена могут развиваться состояния гипергликемии (концентрация глюкозы в крови более
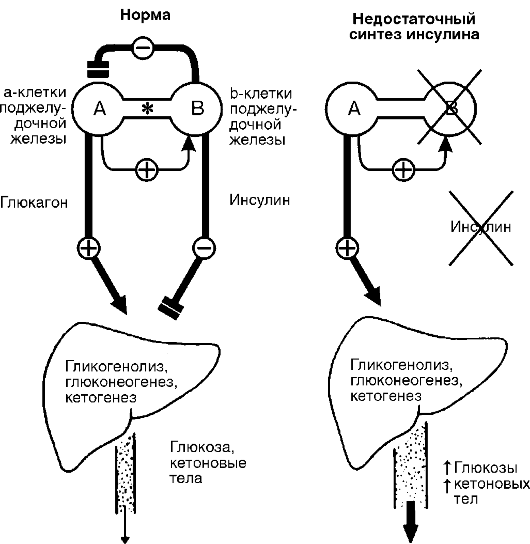 Рис. 12-21. Эффекты
инсулина и глюкагона в норме и при инсулиновой недостаточности. В норме
печень образует примерно 10 г/ч глюкозы, из них 65-75% в результате
действия глюкагона на гликогенолиз, глюконеогенез. Инсулин может
угнетать секрецию глюкагона α-клетками независимо от уровня глюкозы в
крови. В печени инсулин угнетает образование глюкозы и кетоновых тел.
При уменьшении отношения инсулин/глюка- гон увеличивается образование
глюкозы и кетоновых тел в печени
Рис. 12-21. Эффекты
инсулина и глюкагона в норме и при инсулиновой недостаточности. В норме
печень образует примерно 10 г/ч глюкозы, из них 65-75% в результате
действия глюкагона на гликогенолиз, глюконеогенез. Инсулин может
угнетать секрецию глюкагона α-клетками независимо от уровня глюкозы в
крови. В печени инсулин угнетает образование глюкозы и кетоновых тел.
При уменьшении отношения инсулин/глюка- гон увеличивается образование
глюкозы и кетоновых тел в печени
5,5 ммоль/л) и гипогликемии (менее 3,3 ммоль/л). Уровень глюкозы в крови зависит от вида пробы (цельная кровь, венозная или капиллярная, плазма) и от режима взятия пробы (натощак - уровень глюкозы утром после не менее чем 8-часового голодания; через 2 ч после глюкозотолерантного теста) (табл. 12-3). В клинике выделяют еще один показатель уровня глюкозы в крови - постпрандиальная гликемия - это уровень глюкозы через 2 ч после обычного приема пищи (не более 7,5 ммоль/л).
Таблица 12-3. Концентрация глюкозы в крови в норме (ммоль/л)
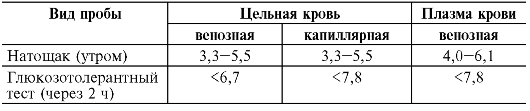 Патогенез гипогликемий может
быть связан с недостаточным поступлением глюкозы в кровь, ускоренным
выведением ее из крови либо комбинацией этих факторов. Различают
физиологическую и патологическую гипогликемию.
Патогенез гипогликемий может
быть связан с недостаточным поступлением глюкозы в кровь, ускоренным
выведением ее из крови либо комбинацией этих факторов. Различают
физиологическую и патологическую гипогликемию.
Физиологическая гипогликемия. Выявляется при тяжелой и длительной физической нагрузке; длительном умственном напряжении; у женщин в период лактации; развивается сразу вслед за алиментарной гипергликемией благодаря компенсаторному выбросу в кровь инсулина.
Патологическая гипогликемия (гиперинсулинизм). Чаще возникает у больных сахарным диабетом в связи с передозировкой инсулина при лечении. Причиной ее могут быть также: аденома островковых клеток поджелудочной железы (инсулома); синдром Золлингера- Эллисона (аденома или карцинома поджелудочной железы, которая, по-видимому, развивается из α-клеток островков Лангерганса, ответственных за выделение глюкагона и гастрина).
Патологическая гипогликемия (без гиперинсулинизма). Выявляется: при патологии почек, сопровождающейся снижением порога для глюкозы, что приводит к потере глюкозы с мочой; нарушении всасывания углеводов; заболеваниях печени, сопровождающихся торможением синтеза гликогена и глюконеогенеза (острые и хронические гепатиты); недостаточности надпочечников (дефицит глюкокортикоидов); гипоавитаминозе В1, галактоземиях и при печеночных формах гликогенозов; голодании или недостаточном питании (алиментарная гипогликемия); недостаточности механизмов регуляции углеводного обмена у новорожденных.
Гипергликемия у человека встречается чаще, чем гипогликемия. Различают следующие типы гипергликемий.
Физиологические гипергликемии. Это быстрообратимые состояния. Нормализация уровня глюкозы в крови происходит без каких-либо внешних корригирующих воздействий. К ним относятся:
1. Алиментарная гипергликемия. Обусловлена приемом пищи, содержащей углеводы. Концентрация глюкозы в крови нарастает вследствие ее быстрого всасывания из кишечника. Активация секреции гормона β-клетками островков Лангерганса поджелудочной железы начинается рефлекторно, сразу после попадания пищи в полость рта и достигает максимума при продвижении пищи в двенадцатиперстную кишку и тонкий кишечник. Пики концентраций инсулина и глюкозы в крови совпадают по времени. Таким образом, инсулин не только обеспечивает доступность углеводов пищи к клеткам организма, но и ограничивает повышение концентрации глюкозы в крови, не допуская потерю ее с мочой.
2. Нейрогенная гипергликемия. Развивается в ответ на эмоциональный стресс и обусловлена выбросом в кровь большого количества катехоламинов, образующихся в мозговом веществе надпочечников и реализующих свои гипергликемические эффекты (см. табл. 12-2). Освобождающаяся глюкоза быстро выходит в кровь, обусловливая гипергликемию. Физиологический смысл этого феномена состоит в обеспечении срочной мобилизации резерва углеводов для использования их в качестве источников энергии (окисления) в предстоящей повышенной двигательной активности в условиях стресса.
Патологические гипергликемии. Причинами их развития являются:
1) нейроэндокринные расстройства, когда нарушены соотношения уровня гормонов гипо- и гипергликемического действия. Например, при заболеваниях гипофиза, опухолях коры надпочечников, при феохромоцитоме, гиперфункции щитовидной железы; при недостаточной продукции инсулина, глюкагономе;
2) органические поражения центральной нервной системы, расстройства мозгового кровообращения различной этиологии;
3) нарушения функций печени при циррозе;
4) судорожные состояния, когда происходит расщепление гликогена мышц и образование лактата, из которого в печени синтезируется глюкоза;
5) действие наркотических веществ (морфин, эфир), возбуждающих симпатическую нервную систему и тем самым способствующих развитию гипергликемии.
Наиболее часто встречается гипергликемия при недостаточности инсулина и (или) его действия, которая лежит в основе сахарного диабета.
Диагностика нарушений углеводного обмена методом нагрузок
Исследование состояния углеводного обмена с диагностической целью в клинике начинают с определения натощак содержания глюкозы в крови и анализа мочи на присутствие в ней глюкозы и кетоновых тел. Если результаты анализов свидетельствуют о наличии гипергликемии, глюкозурии и кетонурии, то этого оказывается достаточно для подтверждения диагноза сахарного диабета. Доказано, что ни одно из других заболеваний внутренних органов не дает всей триады: гипергликемии, глюкозо- и кетонурии. Лишь при сахарном диабете имеют место существенные нарушения не только углеводного, но и жирового обмена.
Для углубленного исследования состояния углеводного обмена используют методы «сахарных» нагрузок.
Проба с однократной сахарной нагрузкой для получения гликемической («сахарной») кривой. У обследуемого утром натощак берут кровь из пальца для определения концентрации глюкозы, после чего дают сахарную нагрузку: прием внутрь 100 г глюкозы, растворенной в 200 мл кипяченой воды. Время, в течение которого раствор следует выпить, не должно превышать 5 мин. Повторные заборы проб крови из пальца ведут с интервалом в 30 или 60 мин. Длительность пробы (у взрослых) составляет 3 ч. На основании полученных данных строят кривую, откладывая по оси ординат концентрацию глюкозы, а по оси абсцисс - время. Типы гликемических кривых, присущих норме или сахарному диабету, представлены на рис. 12-22.
Для гликемической кривой у здоровых субъектов характерны следующие признаки. Уже через 15 мин после приема раствора
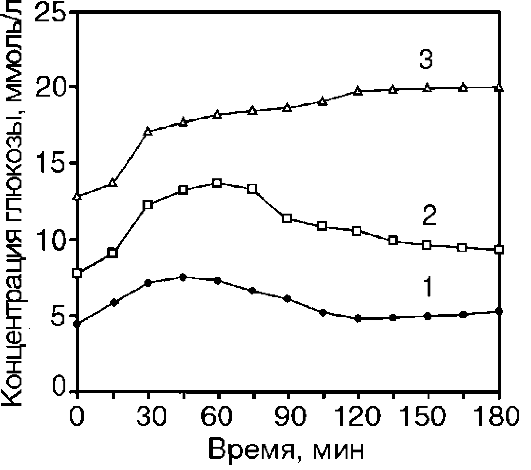 Рис. 12-22. Типы
гликемических кривых в норме и в состояниях, характеризующихся
пониженной толерантностью к глюкозе: 1 - норма; 2 - латентный сахарный
диабет (легкая форма заболевания); 3 - тяжелая форма сахарного диабета
Рис. 12-22. Типы
гликемических кривых в норме и в состояниях, характеризующихся
пониженной толерантностью к глюкозе: 1 - норма; 2 - латентный сахарный
диабет (легкая форма заболевания); 3 - тяжелая форма сахарного диабета
глюкозы внутрь в крови начинает расти концентрация глюкозы, достигая максимума к концу первого часа (в промежутке от 30-й до 60-й мин). При этом концентрация глюкозы превышает таковую натощак на 50-75%. Далее концентрация глюкозы в крови начинает снижаться, и к окончанию второго часа наблюдения она либо достигает исходного уровня (натощак), либо падает ниже исходного уровня (вариант физиологической гипогликемии), либо остается несколько повышенной, но не превышает 6,6 ммоль/л. К третьему часу во всех трех возможных вариантах концентрация глюкозы в крови не отличается от исходного значения (натощак).
У больных сахарным диабетом концентрация глюкозы натощак повышена, а нарастание гликемической кривой после сахарной нагрузки происходит медленнее. Максимальное значение показателя регистрируют только через 60-150 мин от начала наблюдения, при этом концентрация глюкозы может в 1,8 раза превышать ее исходное значение. Спад концентрации глюкозы крови (гипогликемическая фаза) также происходит чрезвычайно медленно (вплоть до отсутствия такового), что коррелирует со степенью тяжести заболевания. Если же понижение концентрации глюкозы все же происходит, то оно растягивается на 3-4 ч.
Тест толерантности к глюкозе (ГТТ). Кровь для проведения теста также берут из пальца дважды: натощак и спустя 120 мин после нагрузки глюкозой. Согласно критериям ВОЗ, у практически здорового человека концентрация глюкозы в крови натощак не должна превышать 5,5 ммоль/л. Спустя 120 мин после стандартной нагрузки глюкозой (одномоментный прием внутрь 75 г глюкозы в 300 мл воды) уровень глюкозы не должен превышать 6,7 ммоль/л. В случае если концентрация глюкозы в крови натощак превышает 6,1 ммоль/л, а тест свидетельствует о том, что спустя 120 мин уровень глюкозы остается выше 7,8 ммоль/л, то это веский аргумент в пользу если не диабета, то преддиабетического состояния у обследуемого.
12.4.7. Сахарный диабет
Сахарный диабет (СД) - это группа метаболических (обменных) заболеваний, характеризующихся гипергликемией, которая является результатом дефектов секреции инсулина, действия (активности) инсулина или обоих этих факторов. Хроническая гипергликемия при диабете сочетается с повреждением, дисфункцией и недоста-
точностью различных органов, особенно глаз, почек, нервной и сердечно-сосудистой системы.
Следствием выраженной гипергликемии могут быть полиурия, полидипсия, изменение массы тела, сочетающееся с полифагией, и снижение остроты зрения. Нарушение физического развития у детей и восприимчивость к инфекциям также могут сопровождать хроническую гипергликемию. Острые, угрожающие жизни осложнения диабета - гипергликемические - кетоацидотическая кома, а также гиперосмолярная кома без кетоацидоза.
Выделяют две наиболее распространенные патогенетические категории сахарного диабета. В основе развития СД 1 типа - абсолютный дефицит секреции инсулина. При СД 2 типа причина заключается в комбинации резистентности к инсулину (инсулинорезистентность) и неадекватного компенсаторного инсулинсекреторного ответа (гиперинсулинемия).
Моделирование сахарного диабета
Для углубленного изучения этиологии и патогенеза СД используются различные экспериментальные модели. Первая модель СД была получена в 1889 г. О. Минковским и Дж. Мерингом, которые вызвали диабет у собак путем удаления поджелудочной железы и установили, что необходимым фактором для развития СД является недостаточность секреции инсулина.
Парентеральное введение аллоксана крысам вызывает некроз β-клеток островков и через 24-48 ч приводит к развитию гипергликемии. При аллоксановом диабете снижено содержание инсулина в крови и поджелудочной железе, повышена чувствительность тканей к экзогенному инсулину, снижено содержание гликогена в печени, отмечается жировое истощение и снижается синтез белка, т.е. экспериментальная картина соответствует развитию СД 1 типа.
Экспериментальный диабет у кроликов получают путем внутривенного введения дитизона. При остром течении такого диабета развиваются высокая гликемия (>50 ммоль/л), глюкозурия, гиперкетонемия, полиурия, полидипсия, уменьшение массы тела. Животные погибают в течение 5-10 дней при состоянии, напоминающем диабетическую гипергликемическую кому.
Введение собакам СТГ в больших дозах вызывает соматотропный (гипофизарный) диабет с классическими симптомами - ги-
пергликемией, глюкозурией, полиурией и повышенным содержанием свободных жирных кислот (СЖК) в крови. В начальный период при использовании такой модели концентрация инсулина в 15-20 раз превышает нормальный уровень, затем нормализуется и в дальнейшем падает в результате истощения функциональных резервов инсулярного аппарата.
Введение глюкокортикоидов в больших дозах приводит к развитию стероидного диабета, для которого характерна начальная гиперинсулинемия, снижение чувствительности к инсулину (инсулинорезистентность) и последующее поражение инсулярного аппарата с возникновением классического синдрома инсулинодефицитного диабета.
В современной классификации сахарного диабета (ВОЗ, 1999) выделяют 4 группы: СД 1 типа (аутоиммунный и идиопатический), СД 2 типа, гестационный и другие специфические формы сахарного диабета.
Сахарный диабет I типа (деструкция β-клеток, обычно ведущая к абсолютному дефициту инсулина)
Иммуноопосредованный диабет. Основным звеном патогенеза СД 1 типа является воспаление в островках Лангерганса, приводящее к деструкции β-клеток и дисфункции остальных клеточных типов островка (это эндокриноциты энтодермального происхождения - α-, β-, δ- и PP-клетки; кровеносные капилляры, симпатические терминали, эфференты и афференты блуждающего нерва, нейроны, глиальные клетки и резидентные тканевые макрофаги). Это воспаление имеет аутоиммунный характер (аутоиммунный инсулит) и вызывает гибель β-клеток преимущественно путем апоптоза
(рис. 12-23).
СД 1 типа может быть результатом дефекта системы иммунологического надзора, связанного с наследованием определенной комбинации генов главного комплекса гистосовместимости. Показана четкая ассоциация заболевания с HLA-генами - В8 и В15 (гены I класса комплекса гистосовместимости); генами DR3 и DR4; DQ B1 и DQ B2 (гены II класса комплекса гистосовместимости). В связи с этим определение данных генов у больных СД 1 типа и у членов их семей может служить генетическими маркерами заболевания. Так, самый высокий титр антител к инсулину отмечен у лиц с антигеном В15, а у 95% больных потенциальный риск развития
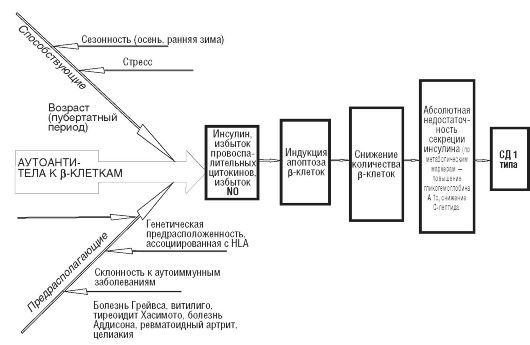 Рис. 12-23. Этиологические и патогенетические факторы развития сахарного диабета (СД) 1 типа.HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
Рис. 12-23. Этиологические и патогенетические факторы развития сахарного диабета (СД) 1 типа.HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
заболевания обусловлен сочетанием антигенов DR3+DR4. В пользу генетической предрасположенности этой формы диабета свидетельствует сочетание ее с другими аутоиммунными заболеваниями (болезнь Грейвса, тиреоидит Хасимото, целиакия, витилиго, болезнь Аддисона, ревматоидный артрит). Таким образом, в основе СД 1 типа лежит генетически обусловленный дисбаланс отдельных популяций иммунокомпетентных клеток.
Маркерами иммунной деструкции β-клеток являются аутоантитела к антигену β-клетки (ICA), аутоантитела к инсулину (IAA), аутоантитела к глутаматдекарбоксилазе (GADA) и аутоантитела к тирозин-фосфатазам IA-2 и IA2b. Один вид, а обычно и более, этих аутоантител присутствует у 85-90% индивидуумов при первоначальном обнаружении гипергликемии натощак.
Т-лимфоциты играют важную роль в инициации и развитии аутоиммунного процесса, приводящего к уничтожению инсулинсинтезирующих клеток. Основная роль принадлежит цитотоксическим Т-лимфоцитам. Роль Т-звена в формировании диабета показана в опытах на мышах линии NOD (Non-Obese Diabetic mice - диабетические мыши без ожирения со спонтанно развивающимся аутоиммунным диабетом), которых с помощью тимэктомии и введения антител к молекулам CD4+ защищали от развития спонтанного аутоиммунного диабета.
Метаболическими маркерами СД 1 типа является уровень инсулина и С-пептида в крови и моче (в норме соотношение концентраций инсулина и С-пептида равно 1/3); гликозилированного гемоглобина - HbA1c (более 5% от всего содержания гемоглобина); нарушение толерантности глюкозы в ГТТ.
Патогенез деструкции β-клеток поджелудочной железы достаточно сложен. В последние годы установлено, что существенную роль в этих механизмах играет свободный радикал - оксид азота (NO), который образуется из L-аргинина под действием нейрональной или эндотелиальной NO-синтазы и участвует в передаче межклеточных сигналов (снижает тонус периферических сосудов с помощью инсулинопосредованного эндотелийзависимого механизма вазодилатации; регулирует процессы в нервной ткани). Существует третья изоформа NO-синтазы - индуцибельная (iNOS), которая появляется в клетках при повышении содержания провоспалительных цитокинов и может выделяться непосредственно в β-клетку, вызывая цитотоксические и цитостатические эффекты.
Каскад иммунньгх реакций, приводящих к деструкции β-клеток, инициируется также резидентными макрофагами островков Лангерганса. Активированный диабетогенными факторами макрофаг продуцирует провоспалительные цитокины - интерлейкин (IL) 1, фактор некроза опухолей (TNF) α, интерферон γ, способные разрушать β-клетки. Примером одного из механизмов деструкции служит экспрессия гена iNOS в β-клетке с помощью IL-1, что увеличивает образование оксида азота. Важно отметить, что ген iNOS локализуется на 11-й хромосоме рядом с геном, кодирующим синтез инсулина, что позволяет предполагать вовлеченность этих двух областей в патогенез СД 1 типа.
Значимость NO как одного из ведущих патогенетических факторов деструкции β-клеток подтверждена применением никотинамида и аминогуанидина. Эти препараты ингибируют экспрессию iNOS, уменьшают образование NO и предотвращают деструкцию β-клеток и манифестацию СД 1 типа.
Начальное повреждение β-клеток уменьшает количество ГЛЮТ 2 на их цитоплазматической мембране, снижая чувствительность к глюкозе и тем самым уменьшает продукцию инсулина еще до их выраженной деструкции. Клинически СД 1 типа манифестирует (у детей чаще кетоацидозом), когда повреждены 80-90% островковых клеток. При раннем обнаружении деструкции β-клеток с помощью перечисленных выше маркеров и при адекватном лечении повреждение клеток можно предупредить и остановить. Лечение иммунодепрессантами (циклоспорин А) до развития гипергликемии может существенно снизить вероятность заболевания СД 1 типа.
Многие больные с СД 1 типа становятся зависимыми от инсулина и находятся в состоянии риска по кетоацидозу. На последней стадии заболевания секреция инсулина минимальна или отсутствует, что проявляется низким или неопределяемым уровнем С-пептида плазмы. Иммуноопосредованный СД обычно начинается в детском и подростковом возрасте, но может развиться в любой период жизни, даже у пожилых людей 80-90 лет.
Идиопатический диабет. Некоторые формы СД 1 типа не имеют известной этиологии. Больные с идиопатическим диабетом имеют постоянный дефицит инсулина и наклонность к кетоацидозу, но у них отсутствуют маркеры иммунной деструкции и нет связи с HLA- генами. Эта форма имеет четкое наследование, механизм которого еще неясен. Идиопатический диабет чаще всего развивается у лиц
африканского или азиатского происхождения. У пациентов с этой формой диабета эпизодически выявляются кетоацидоз и различные степени инсулинодефицита. Причиной инсулиновой недостаточности в этом случае может быть и частое употребление в пищу продуктов, содержащих цианиды (корни маниока, сорго, ямс). Цианиды в нормальных условиях обезвреживаются при участии серосодержащих аминокислот, но при белковой недостаточности, повсеместно распространенной у лиц, проживающих в странах Азии и Африки, создаются условия для накопления цианидов и повреждения β-клеток поджелудочной железы.
Актуальной задачей является диагностика предклинического периода развития деструкции инсулярного аппарата. Это достигается с помощью определения аутоантител (ICA, IAA, GADA) или по оценке инсулинового ответа на внутривенную нагрузку глюкозой (ГТТ). Уже на стадии доклинических проявлений деструкции исчезает ранний пик секреции инсулина на глюкозную нагрузку. Если эти метаболические и иммунные признаки сочетаются с наследственной предрасположенностью (HLA-типирование), то можно с большой вероятностью поставить диагноз деструкции инсулярного аппарата на доклинических этапах СД 1 типа.
Сахарный диабет 2 типа (от преобладающей инсулинорезистентности с относительным инсулинодефицитом до преобладающего дефекта секреции инсулина с инсулинорезистентностью)
СД 2 типа - общее собирательное название гетерогенных нарушений углеводного обмена, имеющих, в отличие от СД 1 типа, другую генетическую основу и разнообразные пусковые механизмы развития. Содержание инсулина в поджелудочной железе и крови может быть нормальным или даже повышенным, поэтому говорят об относительной инсулиновой недостаточности, подразумевая недостаточность метаболических эффектов инсулина в тканях. Это явление обозначают специальным термином - инсулинорезистентность. Инсулинорезистентность (ИР) - это снижение реакции инсулинчувствительных тканей на инсулин при его достаточной концентрации, в результате чего глюкоза не усваивается инсулинзависимыми тканями и развивается гипергликемия.
В основе патогенеза СД 2 типа лежат два ведущих фактора: ИР и дисфункция β-клеток. ИР бывает трех типов: пререцепторная, рецепторная и пострецепторная.
Пререцепторная ИР обусловлена следующими механизмами:
1) мутацией гена инсулина;
2) мутацией генов, контролирующих энергетический обмен в β-клетках и секрецию инсулина.
Рецепторная ИР связана с дефектом синтеза, ресинтеза или субстратной аффинности инсулиновых рецепторов на β-клетках и других клетках-мишенях, в основе которых лежат:
1) мутации гена инсулинового рецептора (в настоящее время выявлено более 30 точечных мутаций этого гена);
2) повышенное использование рецепторов инсулина;
3) уменьшение количества рецепторов инсулина на поверхности гипертрофированных адипоцитов;
4) увеличение количества висцеральной жировой ткани, имеющей низкий уровень экспрессии рецепторов к инсулину, что обусловливает ее изначальную ИР;
5) блокирование рецепторов к инсулину антителами. Пострецепторная ИР связана с патологией ассоциированных с
рецепторами инсулина тирозинкиназы и глюкозных транспортеров. К этому могут привести:
1) повреждение внутриклеточных посредников передачи инсулинового сигнала (например, мутации генов субстрата инсулинового рецептора - IRS-1);
2) снижение чувствительности ГЛЮТ 2 β-клеток поджелудочной железы к глюкозе;
3) снижение мембранной концентрации и активности ГЛЮТ 4 в мышечной и жировой ткани;
4) снижение чувствительности ГЛЮТ 2 к глюкозе в гепатоцитах;
5) нарушение обмена глюкозы в клетках-мишенях инсулина (например, при мутации гена глюкокиназы, гликогенсинтазы, митохондриального гена при MELAS-синдроме).
Наибольшее значение, вероятно, имеют рецепторная и пострецепторная ИР, которая по механизмам развития может быть первичной и вторичной. Первичная ИР определяется вышеперечисленными генетическими механизмами. Из-за этих нарушений возникает гиперинсулинемия, которая сначала имеет компенсаторный характер, так как необходима для преодоления ИР тканей и поддержания нормального транспорта глюкозы в клетки. Показано, что самым ранним признаком СД 2 типа является нарушение способности мышечных и жировых клеток реагировать на инсулин. До тех пор пока поджелудочная железа способна увеличивать
секрецию инсулина с тем, чтобы восполнить ИР этих тканей, толерантность к глюкозе остается в норме.
Однако с течением времени β-клеткам не удается поддерживать высокий уровень секреции инсулина, что снижает толерантность к глюкозе и постепенно приводит к развитию СД, т.е. возникает дисфункция β-клеток. Несмотря на то что у больных этой формой диабета нормальный или повышенный уровень инсулина, его все же недостаточно для компенсации высокой гликемии, характерной для СД 2 типа. Таким образом, секреция инсулина у этих больных неполноценна и недостаточна для того, чтобы компенсировать ИР, поэтому на определенных этапах лечения СД 2 типа пациентам назначают инсулинотерапию, несмотря на достаточно высокий уровень инсулина в крови.
Вторичная ИР связана: 1) с хронической гипергликемией, которая способствует десенситизации β-клеток, проявляющейся снижением их секреторной активности; с ожирением, в результате чего повышается продукция адипоцитами лептина, TNF-α, IL-1, IL-6; 2) с дислипопротеинемией, характеризующейся повышением содержания липопротеинов очень низкой плотности (ЛПОНП), липопротеинов низкой плотности (ЛПНП) и снижением уровня холестерина липопротеинов высокой плотности (ЛПВП), что способствует активации окислительного стресса в эндотелиоцитах и эндотелиальной дисфункции (см. раздел 12.5.2). При развитии вторичной ИР дисфункция β-клеток с преобладанием дефекта секреции инсулина выходит на первый план.
Несмотря на гетерогенность патогенетических факторов, обусловливающих ИР при развитии СД 2 типа, можно выделить 3 уровня нарушений гомеостаза глюкозы:
1. Нарушение механизма узнавания глюкозы β-клетками поджелудочной железы и вследствие этого потеря первой фазы секреции инсулина в ГТТ.
2. ИР инсулинозависимых периферических тканей, что приводит к недостаточному транспорту и метаболизму глюкозы в клетках и гипергликемии, результатом чего является нарушение толерантности к глюкозе в ГТТ.
3. Увеличение уровня глюкозы натощак свидетельствует о повышении продукции ее печенью (вследствие недостатка инсулина и/или избытка глюкагона и катехоламинов).
Таким образом, патогенез СД 2 типа может быть представлен следующей последовательностью событий: первичная ИР и дис-
функция β-клеток -> действие диабетогенных факторов -> хроническая гипергликемия -> гиперинсулинемия -> вторичная ИР -> нарастающий относительный дефицит инсулина -> атрофия поджелудочной железы -> абсолютный дефицит инсулина (рис. 12-24).
Из внешних факторов, неблагоприятно влияющих на чувствительность тканей к инсулину, наибольшее значение имеют гиподинамия и избыточное потребление жира (см. раздел 12.5.4). Гиподинамия сопровождается снижением транслокации ГЛЮТ 4 в мышечных клетках и усиливает ИР мышечной ткани (см. рис. 12- 15). Избыточное потребление животных жиров, содержащих насыщенные жирные кислоты, приводит к структурным изменениям мембранных фосфолипидов и нарушению экспрессии генов, контролирующих проведение инсулинового сигнала внутрь клетки (пострецепторные механизмы), что также усиливает ИР на уровне
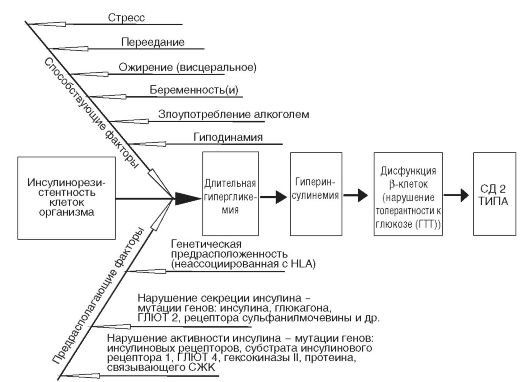 Рис. 12-24. Этиологические
и патогенетические факторы развития сахарного диабета (СД) 2 типа. ГЛЮТ
- глюкозный транспортер, ГТТ - глюкозотолерантный тест, СЖК - свободные
жирные кислоты, HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
Рис. 12-24. Этиологические
и патогенетические факторы развития сахарного диабета (СД) 2 типа. ГЛЮТ
- глюкозный транспортер, ГТТ - глюкозотолерантный тест, СЖК - свободные
жирные кислоты, HLA (human leukocytes antigen) - человеческий лейкоцитарный антиген
всего организма. Гипертриацилглицеролемия, в особенности постпрандиальная, часто наблюдаемая у пациентов с висцеральным типом ожирения, сопровождается избыточным отложением липидов в мышцах, которое нарушает активность ферментов, участвующих в метаболизме глюкозы.
Несмотря на то что транспорт глюкозы в клетки в условиях ИР нарушен, липогенетическое действие инсулина на клетки инсулинозависимых тканей может сохраняться и заключается в следующем:
• инсулин активирует ЛП-липазу адипоцитов, способствуя ее переносу на мембрану эндотелиоцитов кровеносных сосудов, где начинается распад хиломикронов и ЛПОНП на СЖК (они депонируются в жировой ткани в виде триацилглицеролов - ТАГ) и глицерол, поступающий в печень для синтеза новых
ТАГ и ЛПОНП;
• инсулин опосредованно тормозит липолиз в жировой ткани, угнетая гормончувствительную ТАГ-липазу, и ТАГ депонируются в адипоцитах. Именно поэтому СД 2 типа сопровождается ожирением.
Такое действие инсулина на ферменты жирового обмена объясняет развитие при СД 2 типа ретенционной гиперлипемии и дислипопротеинемии IV и V типов, когда в крови увеличивается содержание ЛПОНП и ТАГ (рис. 12-25).
При СД 1 типа отмечается транспортная гиперлипемия (см. раздел 12.5.3), а больные худеют. Кроме того, повышенный липолиз при СД 1 типа усугубляется усилением липолитического эффекта соматотропина, АКТГ, глюкагона, адреналина и тироксина в условиях абсолютной недостаточности инсулина.
Отмечая роль ожирения в патогенезе СД 2 типа, необходимо подчеркнуть наличие взаимосвязи между СД 2 типа и степенью ожирения. Риск развития данного вида диабета увеличивается в 2 раза при наличии ожирения I степени, в 5 раз - при средней степени ожирения и более чем в 10 раз при наличии III степени ожирения. При этом абдоминальное распределение жира тесно связано с развитием метаболических нарушений (см. раздел 12.5.4).
Представленные сложные взаимосвязи патогенетических факторов, участвующих в механизме нарушений регуляции углеводного и жирового обмена при СД 2 типа, позволили G. Reaven в 1988 г. сформулировать концепцию метаболического синдрома, основными компонентами которого является ИР тканей и гиперинсулинемия, нарушение толерантности к глюкозе и развитие СД 2 типа, абдо-
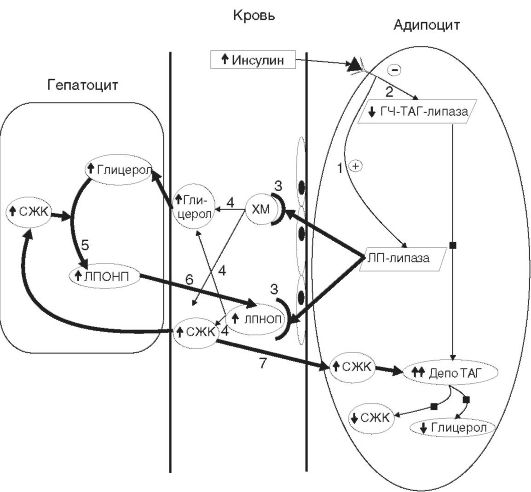 Рис. 12-25. Механизм
увеличения депонирования жиров в адипоцитах в условиях гиперинсулинемии
при СД 2 типа (липогенез > липолиз - увеличение депонирования жира):
1 - активация инсулином ЛП-липазы адипоцитов; 2 - угнетение
гч-ТАГ-липазы адипоцитов; 3 - перенос ферментативных комплексов
ЛП-липазы на мембрану эндотелиоцитов и связывание их с ХМ и ЛПОНП; 4 -
катаболизм ХМ и ЛПОНП; 5 - синтез в печени ЛПОНП из избытка глицерола и
СЖК; 6 - избыточное поступление ЛПОНП в кровь; 7 - избыточное
поступление СЖК в адипоциты; ТАГ - триацилглицеролы; СЖК - свободные
жирные кислоты; ХМ - хиломикроны; ЛПОНП - липопротеины очень низкой
плотности; ЛПлипаза - липопротеиновая липаза адипоцитов; гч-ТАГ-липаза -
гормончувствительная ТАГ-липаза, - - усиление эффекта; ■ - блокирование
эффекта; ® - активация фермента; θ - ингибирование фермента
Рис. 12-25. Механизм
увеличения депонирования жиров в адипоцитах в условиях гиперинсулинемии
при СД 2 типа (липогенез > липолиз - увеличение депонирования жира):
1 - активация инсулином ЛП-липазы адипоцитов; 2 - угнетение
гч-ТАГ-липазы адипоцитов; 3 - перенос ферментативных комплексов
ЛП-липазы на мембрану эндотелиоцитов и связывание их с ХМ и ЛПОНП; 4 -
катаболизм ХМ и ЛПОНП; 5 - синтез в печени ЛПОНП из избытка глицерола и
СЖК; 6 - избыточное поступление ЛПОНП в кровь; 7 - избыточное
поступление СЖК в адипоциты; ТАГ - триацилглицеролы; СЖК - свободные
жирные кислоты; ХМ - хиломикроны; ЛПОНП - липопротеины очень низкой
плотности; ЛПлипаза - липопротеиновая липаза адипоцитов; гч-ТАГ-липаза -
гормончувствительная ТАГ-липаза, - - усиление эффекта; ■ - блокирование
эффекта; ® - активация фермента; θ - ингибирование фермента
минальное (висцеральное) ожирение, дислипидемия, артериальная гипертензия и атеросклероз (рис. 12-26). Исследования последних лет расширили компоненты метаболического синдрома, добавив к ним гиперурикемию, гомоцистеинемию, гиперандрогению у женщин, нарушения гемостаза (увеличение содержания в крови инги-
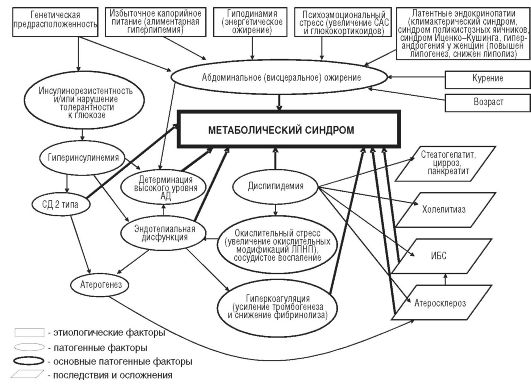 Рис. 12-26. Этиологические, патогенетические факторы и последствия метаболического синдрома
Рис. 12-26. Этиологические, патогенетические факторы и последствия метаболического синдрома
битора активатора плазминогена, гиперфибриногенемия). Наиболее часто в литературе используется термин «метаболический синдром» (метаболический синдром Х), хотя прилагательное «дисметаболический» более точно отражает сущность патогенеза этого синдрома.
В индустриалышх странах распространенность метаболического синдрома среди населения старше 30 лет составляет 10-20%. Метаболический синдром может быть генетически детерминирован, что подтверждается результатами близнецового метода и преобладанием заболеваемости в определенных этнических группах, например, у жителей Индии.
Необходимо особо отметить значение абдоминального (висцерального) ожирения в развитии ИР тканей, СД 2 типа и метаболического синдрома. Висцеральная жировая ткань, в отличие от жировой ткани другой локализации, непосредственно сообщается с портальной системой печени. При данном типе ожирения активация липолиза в жировой ткани сальника и брыжейки сопровождается избыточным поступлением СЖК в портальную циркуляцию и в печень (выше в 20-30 раз по сравнению с нормой). Висцеральные адипоциты характеризуются высокой плотностью β,,-адренорецепторов, кортикостероидных и андрогенных рецепторов и низким содержанием α2-адренорецепторов и рецепторов к инсулину. Эти особенности определяют высокую чувствительность висцеральной жировой ткани к липолитическому действию катехоламинов и других перечисленных выше факторов и низкую - к липогенетическому действию инсулина (см. раздел 12.5.4).
Таким образом, любые гормональные нарушения, сопровождающие стресс, возрастные гормональные перестройки, вызывают интенсивный липолиз в висцеральных адипоцитах и приводят к выделению большого количества СЖК в портальную циркуляцию и печень. В печени СЖК экранируют инсулиновые рецепторы и препятствуют связыванию инсулина гепатоцитами, обусловливая развитие ИР на уровне печени (снижается транспорт глюкозы в гепатоциты из-за нарушения глюкокиназного механизма, что приводит к гипергликемии), снижается экстракция инсулина гепатоцитами (инсулин становится недоступным для действия инсулиназы гепатоцитов), и развивается системная гиперинсулинемия. СЖК подавляют тормозящее действие инсулина на глюконеогенез, увеличивая продукцию глюкозы печенью. С другой стороны, СЖК в мышечной ткани конкурируют с глюкозой и обусловливают ИР на уровне мышечной ткани, усугубляя гипергликемию (рис. 12-27).
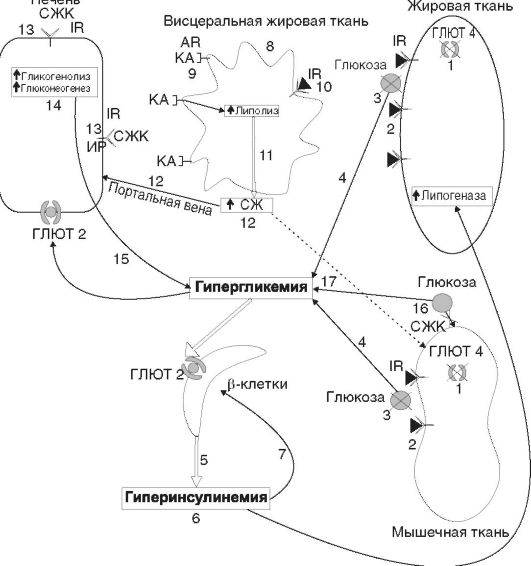 Рис. 12-27. Роль
нарушений ГЛЮТ 4 и висцерального ожирения в развитии
инсулинорезистентности: 1 - наследственный дефект ГЛЮТ 4; 2 -
инсулиновый сигнал не работает (ГЛЮТ 4 остаются в клетках
инсулинзависимых тканей); 3 - транспорт глюкозы в клетки
инсулинзависимых тканей прекращается; 4 - гипергликемия; 5 - увеличение
продукции инсулина β-клетками; 6 - гиперинсулинемия и усиление
липогенеза; 7 - истощение β-клеток; 8 - висцеральное ожирение; 9 -
увеличение количества β3-адренорецепторов; 10 - снижение
количества рецепторов к инсулину; 1l - усиление липолиза в результате
действия КА (высокие концентрации КА при действии психогенных факторов);
12 - повышение СЖК в портальной вене печени; 13 - экранирование
рецепторов к инсулину СЖК и ИР на уровне печени; 14 - отмена угнетающего
действия инсулина на гликогенолиз и глюконеогенез в печени; 15 -
гипергликемия натощак; 16 - конкуренция СЖК за поступление глюкозы в
мышечные клетки; 17 - гипергликемия
Рис. 12-27. Роль
нарушений ГЛЮТ 4 и висцерального ожирения в развитии
инсулинорезистентности: 1 - наследственный дефект ГЛЮТ 4; 2 -
инсулиновый сигнал не работает (ГЛЮТ 4 остаются в клетках
инсулинзависимых тканей); 3 - транспорт глюкозы в клетки
инсулинзависимых тканей прекращается; 4 - гипергликемия; 5 - увеличение
продукции инсулина β-клетками; 6 - гиперинсулинемия и усиление
липогенеза; 7 - истощение β-клеток; 8 - висцеральное ожирение; 9 -
увеличение количества β3-адренорецепторов; 10 - снижение
количества рецепторов к инсулину; 1l - усиление липолиза в результате
действия КА (высокие концентрации КА при действии психогенных факторов);
12 - повышение СЖК в портальной вене печени; 13 - экранирование
рецепторов к инсулину СЖК и ИР на уровне печени; 14 - отмена угнетающего
действия инсулина на гликогенолиз и глюконеогенез в печени; 15 -
гипергликемия натощак; 16 - конкуренция СЖК за поступление глюкозы в
мышечные клетки; 17 - гипергликемия
Примечание: IR - инсулиновый рецептор; ГЛЮТ 2 и ГЛЮТ 4 - глюкозные транспортеры; ИР - инсулинорезистентность; СЖК - свободные жирные кислоты; AR - адренорецепторы; КА - катехоламины
При рассмотрении взаимосвязи гиперинсулинемии и артериальной гипертензии выстраивается картина многофакторных взаимоотношений.
У здоровых людей инсулин в физиологической концентрации стимулирует эндотелиальную NO-синтазу через фосфатидилино- зитол-3-киназный путь. В результате увеличивается продукция NO, которая обеспечивает эндотелийзависимую вазодилатацию. В условиях ИР изменяются внутриклеточные сигнальные системы, и трансдукция сигналов через фосфатидилинозитол-3-киназный путь нарушается, с инсулиновых рецепторов сигнал передается через митогенактивируемую протеинкиназу. Активация этого сигнального пути приводит к увеличению продукции эндотелина и повышению уровня маркеров воспаления и тромбоза.
Кроме увеличения продукции эндотелина, гиперинсулинемия активирует целый ряд механизмов, повышающих тоническое напряжение сосудистой стенки:
• повышение активности симпатических центров головного мозга вследствие усиления метаболизма глюкозы в инсулиночувствительных клетках вентромедиального гипоталамуса;
• увеличение фильтрации глюкозы в почечных клубочках в условиях хронической гипергликемии сопровождается усилением обратной ее реабсорбции вместе с натрием в проксимальных канальцах, приводит к гиперволемии, повышению общего периферического сосудистого сопротивления и увеличению артериального давления;
• повышение уровня ангиотензиногена II, так как в условиях ИР отменяется инсулинопосредованное ингибирование экспрессии гена ангиотензиногена;
• увеличение чувствительности кардиомиоцитов и гладкомышечных клеток сосудов к прессорным воздействиям, так как при ИР и гиперинсулинемии подавляется активность Na+/K+- и Са2+-зависимой АТФаз, что сопровождается увеличением внутриклеточного содержания Na+ и Са2+ и уменьшением концентрации K+ и Mg2+ внутри клеток;
• развитие дислипидемии, которая, способствуя дисфункции эндотелия, приводит к нарушению физиологического механизма NO-опосредованной вазодилатации (см. рис. 12-26).
Рассмотренные выше нарушения углеводного и липидного обмена при метаболическом синдроме и СД 2 типа характеризуются изменениями, приводящими к нарушению микроциркуля-
ции, повышенному тромбообразованию, что является основой для развития осложнений этих заболеваний: раннего атеросклероза и сердечно-сосудистой патологии (ИБС, инфаркт миокарда, инсульт); нефро-, ретино- и нейропатиям; неалкогольному стеатогепатиту (гепатоспленомегалия, фиброз, цирроз); синдрому хиломикронемии (панкреатит, ксантома).
Таким образом, гиперинсулинемия, являясь компенсаторной ответной реакцией организма, направленной на обеспечение адекватного транспорта глюкозы в клетку, одновременно является и патологическим фактором, поскольку вызывает развитие «порочных кругов» в патогенезе метаболического синдрома и СД 2 типа.
Проявления СД 2 типа имеют свои особенности по сравнению с таковыми при СД 1 типа (табл. 12-4).
Таблица 12-4. Основные признаки сахарного диабета 1 и 2 типов
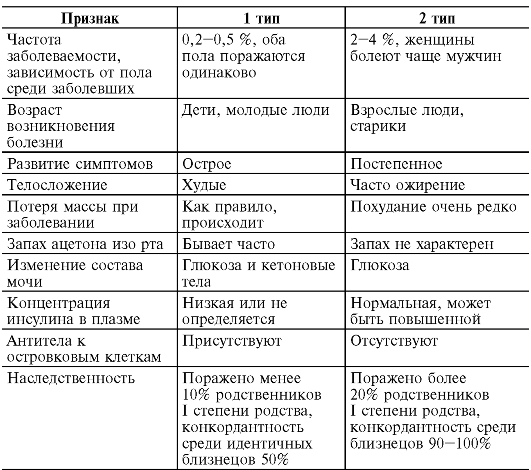 Окончание табл. 12-4
Окончание табл. 12-4
Ассоциация с HLA | В8, В15, DR3, DR4, DQ B1, DQ B2, Dw3, Dw4 | Нет ассоциации |
Лечение (основное) | Инсулин | Диета, сульфонилмочевинные препараты |
Гестационный сахарный диабет связан с ИР, развивающейся у женщины во время беременности (обычно во II триместре). Под этим названием объединяют любые нарушения толерантности к глюкозе, возникшие впервые во время беременности. Во второй половине беременности значительно возрастает уровень плацентарных гормонов, которые подавляют утилизацию глюкозы тканями матери, чтобы обеспечить поступление достаточного количества глюкозы в фетоплацентарную систему, поэтому у беременных уровень глюкозы в крови после приема пищи выше, чем у небеременных.
Постоянная легкая гипергликемия приводит к физиологической гиперинсулинемии. Во второй половине беременности возникает физиологическая инсулинорезистентность, обусловленная плацентарными гормонами - прогестероном, эстрогенами, пролактином и плацентарным лактогеном. ИР также способствует развитию гиперинсулинемии, поэтому, как правило, диабет беременных по патогенезу сходен с СД 2 типа и поддается диетотерапии. В большинстве случаев диабет беременных проходит после родов, но он существенно может повышать риск развития СД 2 типа у матери в будущем.
Другие специфические формы сахарного диабета включают в себя очень большую группу патологических процессов, имеющих как первичный, так и вторичный характер нарушений углеводного обмена. Основными причинами генетических дефектов функции β-клеток могут быть мутации гена глюкокиназы (все варианты MODY - от англ. Maturity onset type diabetes - «диабет взрослого типа у молодых лиц»), митохондриальных генов - MELAS- синдром; выработка антител к инсулину и к рецептору инсулина может привести к необычным формам иммуноопосредованного диабета.
Вторичный инсулинодефицит возникает в результате следующих заболеваний:
• экзокринного отдела поджелудочной железы (хронический панкреатит - алкогольный и тропический, травма, гемохроматоз, неоплазии и др.);
• эндокринопатиях (акромегалия, синдром Иценко-Кушинга, тиреотоксикоз, глюкагонома, феохромоцитома, синдром Конна, гиперандрогенемия и др.); болезнях печени (цирроз, хронический активный гепатит);
• генетических синдромах (синдром Дауна, Клайнфельтера, Шерешевского-Тернера, Прадера-Вилли, хорея Гентингтона и др.);
• вирусных инфекциях (цитомегаловирусная, краснуха и др.);
• при использовании лекарственных препаратов и химических веществ (кортикостероиды, оральные контрацептивы, тиазидовые диуретики, диазоксид, вакор, циклоспорин А, пентамидин и др.).
Алкогольный панкреатит является наиболее частой причиной хронического панкреатита. Заболеванию подвергаются люди в основном среднего возраста. Уровень глюкозы в крови весьма неустойчив в связи с нарушениями в диете, ухудшением всасывания и переваривания, снижением эндокринной функции поджелудочной железы; периодически могут возникать гипер- и гипогликемии; кетоацидоз - явление редкое. Лечение ферментами поджелудочной железы улучшает гликемический контроль.
Гемохроматоз сопровождается отложением железа в печени, поджелудочной железе, коже, половых органах. Подозрение на наличие гемохроматоза у больного сахарным диабетом должно возникнуть при сочетании бронзового оттенка кожи, гепатомегалии с аномальными функциональными печеночными тестами и импотенцией. Эффективное лечение гемохроматоза флеботомией и железосвязывающими препаратами улучшает толерантность к глюкозе.
Обширные поражения печени, такие как цирроз, снижают экстракцию инсулина печенью из портальной циркуляции, что приводит к периферической гиперинсулинемии и ИР. У предрасположенных пациентов на этом фоне возникает диабет. Нарушение толерантности к глюкозе и диабет средней тяжести отмечаются у 50-80% пациентов с установленным циррозом.
Повышенный уровень контринсулярных гормонов, особенно у предрасположенных лиц, снижает резервную функцию β-клеток и приводит к гипергликемии. Диагностика этих состояний обычно
не вызывает затруднений в связи с классической симптоматикой и клинической картиной, обусловленной избыточной продукцией гормонов.
Некоторые лекарственные препараты (см. выше) могут нарушать толерантность к глюкозе, вызывая либо ИР, либо дисфункцию β-клеток.
Часть редких генетических аномалий (см. выше) может также сопровождаться СД. Несмотря на разнообразие причин, к категории «вторичного» СД клинически может быть отнесена относительно небольшая его часть (<1%). Устранение или лечение основного заболевания у этих пациентов может привести к «излечению» СД.
12.4.8. Метаболические осложнения сахарного диабета
Основные патогенетические факторы метаболического повреждения включают следующие процессы:
1. Неферментативное гликозилирование белков - соединение глюкозы со свободными аминогруппами белков - коллагенов, кристаллинов, гемоглобина и др.
2. Ферментативное гликозилирование заключается в повышении скорости превращения глюкозы в сорбитол и маннитол, что приводит к их накоплению в клетках и межклеточном веществе сетчатки, хрусталика, клубочках почек, эндотелиоцитах, шванновских клетках, нейронах.
3. Резкие суточные колебания осмотического давления крови (из-за резкого повышения концентрации глюкозы после приема пищи).
4. Формирование внутриклеточной гиперосмолярности (за счет образования сорбитола).
5. Повреждение свободными радикалами («окислительный стресс»).
Патогенез диабетического кетоацидоза
Ведущие патогенетические звенья диабетического кетоацидоза, учитывающие нарушения всех видов обмена, представлены на интегральной схеме (рис. 12-28).
Среди важнейших метаболических нарушений при СД выделяются следующие:
1. Нарушения углеводного обмена при отсутствии инсулина (или его действия) характеризуются гипергликемией, глюкозурией и гиперлактатацидемией.
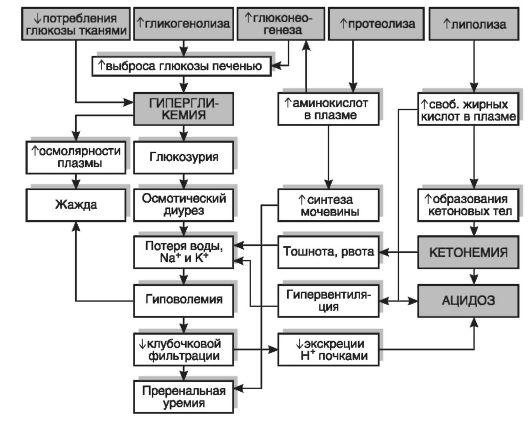 Рис. 12-28. Патогенез
диабетического кетоацидоза. Верхний ряд показывает основные
метаболические эффекты при недостатке инсулина, которые вызывают все
последующие изменения
Рис. 12-28. Патогенез
диабетического кетоацидоза. Верхний ряд показывает основные
метаболические эффекты при недостатке инсулина, которые вызывают все
последующие изменения
Гипергликемия обусловлена:
• нарушением поступления глюкозы из крови внутрь клеток;
• компенсаторным ускорением гликогенолиза;
• активацией глюконеогенеза вследствие снятия репрессивного действия инсулина на синтез ключевых ферментов этого метаболического пути;
• усилением секреции глюкокортикоидов, являющихся стимуляторами синтеза ключевых ферментов глюконеогенеза в печени и почках.
Глюкозурия и, как следствие, полиурия развиваются при достижении концентрации глюкозы в крови 9,9 ммоль/л, когда преодолевается почечный порог, и глюкоза появляется в моче.
Гиперлактатацидемия развивается вследствие торможения катаболизма лактата в ЦТК и нарушения ресинтеза пирувата из лактата (см. рис. 12-17).
2. Нарушения липидного обмена при отсутствии инсулина характеризуются усилением липолиза и снижением липогенеза. Дефицит инсулина при сахарном диабете приводит к тому, что:
а) контринсулярные гормоны (глюкагон, адреналин, глюкокортикоиды и др.) стимулируют мобилизацию липидов из жировых депо и доставку жирных кислот к органам, что является адаптивным механизмом, поставляющим альтернативный субстрат окисления в условиях снижения утилизации глюкозы клетками;
б) снижается активность ЛП-липазы адипоцитов, поэтому свободные жирные кислоты не поступают в жировую ткань;
в) начинает преобладать эффект глюкагона, стимулирующий кетогенез в печени и гормончувствительную ТАГ-липазу в адипоцитах;
г) в норме кетоновые тела стимулируют выход инсулина из поджелудочной железы, что угнетает липолиз и таким образом ограничивает доставку липидов в печень и соответственно кетогенез (при сахарном диабете этот регуляторный механизм нарушен: идет усиленная продукция кетоновых тел печенью благодаря интенсивному β-окислению жирных кислот);
д) при сахарном диабете в избыточном количестве образуется продукт β-окисления жирных кислот - ацетил-КоА, однако способность цикла Кребса утилизировать данный продукт существенно снижена. В результате этого избыток ацетил-КоА становится источником образования больших количеств кетоновых тел: β-оксимасляной, ацетоуксусной кислот и ацетона, что и приводит к кетонемии. Кетоновые тела начинают выделяться с мочой в виде натриевых солей (кетонурия), а ацетон - также и в составе выдыхаемого воздуха.
3. Нарушения белкового обмена при отсутствии инсулина характеризуются преобладанием процессов катаболизма вследствие активации глюконеогенеза из глюкогенных аминокислот и снижения проницаемости клеточных мембран для аминокислот, что приводит к недостатку в тканях свободных аминокислот и нарушению процесса синтеза белка. Стимулируется синтез мочевины, что характеризуется гиперазотемией и приводит к отрицательному азотистому балансу.
4. Нарушения кислотно-щелочного баланса организма развиваются в связи с накоплением кислых продуктов метаболизма в результате переключения аэробных путей утилизации глюкозы на анаэробный гликолиз с повышенной продукцией лактата и усиления ке-
тогенеза. По мере истощения емкости буферных систем организма формируется декомпенсированный метаболический ацидоз.
5. Водно-солевой обмен и дегидратация клеток. Нарушения водного обмена при СД проявляются полиурией (суточный диурез достигает 4-10 л) и полидипсией (до 9 л воды в сутки), обусловленной гипогидратацией организма и гиперосмией крови в связи с гипергликемией, гиперазотемией, кетонемией, гиперлактатацидемией, повышением содержания отдельных ионов. Именно эти явления дали основания для первоначального названия заболевания: diabetes mellitus (лат.) - сахарное мочеизнурение. Повышение осмотического давления крови сопровождается дегидратацией клеток, особенно чувствительны к этому клетки нервной ткани.
Необходимо особо отметить, что сочетание ацидоза и явлений дегидратации в эритроцитах приводит к снижению в этих клетках концентрации 2,3-дифосфоглицериновой кислоты - аллостерического модулятора функций гемоглобина. В этих условиях сродство гемоглобина к кислороду возрастает, но его способность отдавать кислород тканям уменьшается, вследствие чего гипоксия, вызванная глубокими метаболическими нарушениями при СД, усугубляется.
В результате перечисленных выше нарушений обмена веществ при СД могут развиваться серьезные осложнения, к которым относят комы и диабетические микро- и макроангиопатии (нефропатия, слепота, синдром «диабетической стопы»), вторичные инфекции из-за иммунодефицита.
Диабетическая кома. Критическая дегидратация тканей организма с поражением функций головного мозга ведет к развитию диабетической (гипергликемической) комы. Кома развивается при достижении концентрации глюкозы в крови от 19,4 до 33,3 ммоль/л и более. В этих условиях вследствие кетоацидоза ионы калия выходят во внеклеточное пространство (гиперкалиемия), что лежит в основе нарушения сократительной функции миокарда, а также дыхательной мускулатуры. Диабетическая кома может привести к летальному исходу, если больному не будет своевременно проведена специфическая противокоматозная терапия.
Различают следующие виды диабетической комы:
1. Гипергликемическая кетоацидотическая кома. Развивается чаще всего у больных СД 1 типа вследствие гипергликемии, гиперкетонемии и метаболического ацидоза. Глюкоза и кетоновые тела выводятся с мочой (глюкозурия и кетонурия), что способ-
ствует увеличению осмотического давления в первичной моче, потере ионов Na и сопровождается полиурией. При этом возникает обезвоживание, которое ведет к недостаточности периферического кровообращения и гипоксии тканей. Ацидоз вызывает дыхание Куссмауля, при котором теряется СО2 и как следствие усугубляются нарушения водно-электролитного баланса, кислотно-щелочного равновесия, возникает резкое нарушение метаболизма и функций клеток ЦНС, что приводит к расстройству высшей нервной деятельности. К клиническим проявлениям комы относятся: слабость, головная боль, адинамия, диспепсические расстройства (в 30-50% случаев - «абдоминальный синдром» - клиника «острого живота»), дыхание Куссмауля с запахом ацетона в выдыхаемом воздухе, снижение кровяного давления и частый слабый пульс, нарастающая сухость кожи и слизистых оболочек. Затем наступает полная потеря сознания, расслабление мышц, зрачки сужаются, отмечаются характерные признаки энцефалопатии. Содержание глюкозы в крови превышает 22 ммоль/л, кетоновых тел - 17 ммоль/л, повышено содержание остаточного азота, мочевины, холестерина, жирных кислот, уровень натрия чаще нормальный, реже - снижен, уровень калия чаще нормальный, у больных с почечной недостаточностью может быть повышен.
2. Гипергликемическая гиперосмолярная кома. Встречается реже, чем кетоацидотическая, и развивается у больных СД 2 типа старше 50 лет при дополнительном воздействии обезвоживающих факторов (рвота, понос, ограничение приема жидкости, ожоги, кровопотеря, полиурия, прием диуретиков). Основными звеньями патогенеза этого вида комы являются дегидратация организма и развитие гиперосмолярности плазмы, уровень гликемии может достигать 55 ммоль/л. У больных нет выраженной гиперкетонемии и кетонурии, отсутствует запах ацетона изо рта и, если не обратиться к врачу, нарастает уровень глюкозы в крови до крайне высокой степени, что способствует усилению диуреза (глюкозурический осмотический диурез). Возникающее обезвоживание приводит к гиповолемии, стимуляции секреции альдостерона и задержке ионов Na и Cl. Показатель осмолярности плазмы повышается в 1,5-2 раза (в норме около 300 мосмоль/л, при коме достигает 500 мосмоль/л), что приводит к резко выраженной внутриклеточной дегидратации, нарушению водного и электролитного равновесия в клетках мозга, гипоксии ЦНС с выраженной неврологической симптоматикой и потере сознания.
3. Гипергликемическая кома с лактат-ацидозом (лактацидотическая). Это относительно редкое, но опасное осложнение СД. В механизме ее развития важную роль играют следующие факторы:
а) снижение активности ферментативного пируватдегидрогеназного комплекса (выявляется при дефиците инсулина), превращающего пируват в ацетил-КоА. Пируват в обратимой реакции, катализируемой лактатдегдрогеназой, превращается в молочную кислоту (см. рис. 12-17);
б) применение лекарственных препаратов, стимулирующих анаэробный гликолиз и тем самым повышающих содержание лактата и пирувата в организме (например, бигуаниды, повышающие утилизацию глюкозы за счет ее анаэробного распада). При поражении печени или почек может иметь место кумуляция этих препаратов в организме, в результате чего развиваются лактоацидоз и кома;
в) гипоксическое состояние (при котором, как правило, стимулируется гликолиз), вызванное физическим переутомлением, сердечной или дыхательной недостаточностью.
Как следствие в крови накапливается молочная кислота (содержание лактата в плазме превышает 5 ммоль/л), что сопровождается развитием коллапса, нарушением сердечной деятельности и функций дыхательного центра (возникает патологическое дыхание Куссмауля), угнетением сознания, нарушением чувствительности, дисфункцией желудочно-кишечного тракта, резко выраженной дегидратацией тканей. Гиперкетонемия и кетонурия отсутствуют, могут выявляться незначительная гипергликемия и небольшая глюкозурия. Вследствие несвоевременной диагностики и трудности лечения прогноз может быть неблагоприятным.
4. Гипогликемическая кома. Связана с передозировкой инсулина, препаратов сульфонилмочевины, развитием вторичного гипопитуитаризма (следствие ангиопатии сосудов гипофиза), ослабляющего ответ на гипогликемию, и явлениями диабетического нефросклероза, что удлиняет время циркуляции инсулина и, кроме того, еще более снижает почечный порог для глюкозы, способствуя ее потере.
Причинами гипогликемии могут быть также гиперпродукция инсулина опухолью поджелудочной железы (инсулиномой), недостаточность контринсулярных гормонов, печеночные формы гликогенозов, заболевания печени, голодание, нарушение расщепления и всасывания углеводов в желудочно-кишечном тракте и др.
В механизме развития гипогликемической комы решающее значение имеет снижение доставки глюкозы к нервным клеткам, что ведет к их энергетическому истощению и нарушению функций ЦНС. При снижении уровня глюкозы менее 3 ммоль/л возникают потливость, тремор, чувство тревоги и голода, слабость. Затем развивается состояние, напоминающее алкогольное опьянение и сопровождающееся дезориентацией, агрессивностью, галлюцинациями. При дальнейшем падении содержания глюкозы (менее 2,5 ммоль/л) возникают клонические судороги и потеря сознания. В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
Микро- и макроангиопатии относят к более поздним осложнениям, развивающимся при СД обоих типов.
1. Микроангиопатия. Это осложнение выражается в повреждении сосудов микроциркуляции и развивается вследствие метаболических нарушений в сосудистой стенке (гликозилирование белков) и развития васкулита. Чаще всего поражаются сосуды почек и сетчатки глаза.
Поражение почек - диабетическая нефропатия из-за развития макро- и микроангиопатии в настоящее время является основной причиной ранней смертности у больных диабетом молодого возраста. При этом происходит избыточное гликозилирование коллагена базальных мембран почечных клубочков, приводящее к существенным нарушениям структуры и функций почечного фильтра.
Если в суточной моче концентрация альбумина превышает 30 мг (микроальбуминурия), и эти значения повторяются несколько раз, то необходимо проводить лечение, так как данные изменения характерны для начинающейся диабетической нефропатии. По мере прогрессирования поражения почек при диабете развивается выраженная протеинурия. Тщательный контроль за уровнем глюкозы в крови и лечение любых форм гипертонии могут приостановить микроальбуминурию и предупредить развитие манифестной почечной недостаточности.
Поражение сетчатки глаз при диабете - диабетическая ретинопатия относится к числу одной из наиболее частых причин развивающейся слепоты при этой патологии. Основную роль в патогенезе развития отечно-геморрагических форм диабетической ретинопатии играют максимальные суточные колебания уровня глюкозы в крови. Так, при повышении уровня глюкозы на 5,55
ммоль/л осмолярность сыворотки повышается на 5,5 мосмоль/л, что соответствует увеличению давления на 86 мм рт.ст., поэтому если увеличение концентрации сахара в крови идет быстро (в постабсорбтивном периоде), то в капиллярах сетчатки резко возрастает трансмуральное (разница между давлением внутри и снаружи сосуда) осмотическое давление. Жидкость «вытягивается» из ткани сетчатки в кровь, что ведет к резкому повышению трансмурального гидростатического давления в сосудах сетчатки. Повышение гидростатического давления за счет миогенной ауторегуляции приводит к сужению артериол вплоть до полного перекрытия кровотока (с развитием микроинфарктов сетчатки), а капилляры растягиваются с образованием микроаневризм и развития кровоизлияний.
Другой причиной, приводящей к потере зрения, является диабетическая катаракта, в патогенезе которой важную роль играет длительно существующая гипергликемия. Она вызывает, с одной стороны, усиление синтеза сорбитола и маннитола (ферментативное гликозилирование) и накопление этих углеводов в хрусталике глаза, приводящее по осмотическому градиенту к увеличению содержания в нем воды. С другой стороны, гликозилирование белков хрусталика и его капсулы (неферментативное) также вызывает необратимые нарушения его структуры.
2. Макроангиопатия. Диабетическая макроангиопатия проявляется атеросклерозом вследствие выраженного нарушения обмена липопротеинов. Особенностями атеросклероза при СД являются большая распространенность (захват многих сосудистых бассейнов), быстрое прогрессирование, начало в более молодом возрасте. Патологический процесс охватывает сосуды головного мозга, сердца, почек, а также сосуды конечностей, в особенности сосуды голени и стопы. Диабет, даже в условиях его лечения современными средствами, характеризуется ускоренными темпами старения организма. Наличием диабета обусловлены высокая частота инфарктов миокарда, инсультов и случаев гангрены пальцев ног или стопы.
В настоящее время считают, что диабет ускоряет развитие атеросклероза в результате:
• дислипопротенемии (см. раздел 12.5.2);
• гиперхолестеринемии (см. раздел 12.5.8);
• эндотелиальной дисфункции, возникающей из-за окислительного стресса, основным фактором которого являются модифицированные ЛПНП;
• активации неспецифического звена системы иммунитета, сопровождающейся выработкой провоспалительных цитокинов;
• эффекта гормона роста вследствие отсутствия противодействия со стороны инсулина в условиях его абсолютного или относительного дефицита, приводящего к усилению процесса пролиферации гладкомышечных клеток;
• усиленного синтеза тромбоксана А2.
Диабетическая нейропатия - нарушение функции нервов - развивается вследствие ангиопатии из-за нарушения кровоснабжения нервов, гликозилирования белков нейронов и миелиновых оболочек, гиперосмолярного поражения шванновских клеток (проявление гиперосмотической дегидратации при СД). Эти нарушения способны вызывать дисфункции любой системы организма, имитируя многочисленные неврологические заболевания. В патологический процесс могут быть вовлечены как чувствительные, так и двигательные или вегетативные нервные волокна. В качестве типичных примеров клинического проявления диабетических нейропатий можно назвать образование трофических язв на стопах - «диабетическую стопу», различные расстройства функций желудочно-кишечного тракта, мочевого пузыря, импотенцию. При исследовании структуры нервных волокон диабетиков с помощью световой микроскопии часто выявляются их демиелинизация, истончение и склероз эпиневрия, отек и дистрофии нервных волокон, глиальная клеточная реакция.
Патогенез диабетических нейропатий полностью не раскрыт, однако в настоящее время можно назвать ряд факторов, безусловно определяющих развитие этого осложнения диабета:
1) нарушение структуры миелина, когда патологический процесс обусловливает изменение как химической структуры, так и количественного соотношения между основными биохимическими компонентами (холестерин, ТАГ, фосфолипиды, гликолипиды и белки) миелина нервных волокон. Клинические наблюдения свидетельствуют о том, что под влиянием заместительной терапии инсулином происходит существенная коррекция многих из указанных сдвигов;
2) сорбитоловый путь окисления глюкозы - в результате данного процесса происходит ферментативное окисление глюкозы с образованием сначала сорбитола, а затем фруктозы, повышенное количество которых приводит к гиперосмотической дегидратации нейронов;
3) токсическое действие пирувата.
Следует отметить, что общая черта всех вышеперечисленных нарушений, по крайней мере до развития глубоких морфологических изменений, - их обратимость под влиянием терапии, приводящей к снижению гипергликемии.
12.5. ПАТОФИЗИОЛОГИЯ ОБМЕНА ЛИПИДОВ
Липиды - это химические соединения, нерастворимые в воде, но растворимые в хлороформе или спирте. К липидам относятся ненасыщенные и насыщенные жирные кислоты, моно-, ди-, триацилглицеролы, холестерин, фосфолипиды, гликолипиды, стерины и воски. По составу они делятся на простые, сложные липиды и стероиды.
Жирные кислоты - самые простые по строению липиды, их свыше 200 разновидностей, более 20 из которых представлено в тканях человека; полиненасыщенные жирные кислоты (линолевая, линоленовая, арахидоновая) относятся к незаменимым и условно названы витамином F.
Триацилглицеролы (ТАГ) - это эфиры трехатомного спирта глицерина и жирных кислот.
Сложные липиды - это фосфолипиды (производные фосфорной кислоты) и гликолипиды (содержат остатки сахаров).
Стероиды (производные циклопентанпергидрофенантрена): холестерин и его эфиры, желчные кислоты, стероидные гормоны - половые, глюко- и минералокортикоиды, гормоны и стероидные витамины (витамин D).
Холестерин относится к стероидным спиртам. Он является источником образования желчных кислот, стероидных гормонов, витамина D; входит в состав клеточных мембран и является важным компонентом (липопротеинов) плазмы крови.
Фосфолипиды - это сложные эфиры многоатомных спиртов с высшими жирными кислотами и фосфорной кислотой, в их состав входят азотсодержащие соединения: холин, этаноламин, серин. Они содержатся в мембранах клеток и клеточных органелл, регулируют их проницаемость и активность мембранных АТФ, аденилатциклазы и др.
Функции липидов в организме:
• пластическая, так как липиды являются основным компонентом биологических мембран, обеспечивают их проницаемость
и текучесть; входят в состав гликокаликса клеточной поверхности; участвуют в межклеточных взаимодействиях; служат рецепторами бактериальных токсинов, например, холерного токсина; определяют группу крови (система АВ0); являются основным элементом сурфактанта (поверхностно-активного вещества) легких, необходимого для расправления альвеол;
• энергетическая, поскольку в результате окисления жирных кислот образуется большое количество энергии. Так, при β-окислении одной молекулы пальмитиновой кислоты (из 16 С-атомов) образуется 131 молекула АТФ (при расщеплении одной молекулы глюкозы в гликолизе - всего лишь 2 молекулы АТФ, а в цикле трикарбоновых кислот и пентозофосфатном шунте - соответственно 38 и 36 молекул АТФ);
• регуляторная - как нейромедиаторы участвуют в передаче нервных импульсов (ацетилхолин), являются источником синтеза гормонов, жирорастворимых витаминов, биологически активных веществ (эйкозаноидов), фосфолипиды регулируют активность ионных насосов мембраны;
• защитная, механическая - термоизоляция организма, защита внутренних органов от механических воздействий.
Жировой обмен включает следующие этапы:
• переваривание липидов в кишечнике и всасывание их в кровь;
• транспорт липидов и переход их из крови в ткани и наоборот;
• депонирование жиров;
• промежуточный обмен липидов.
Патология обмена липидов связана с нарушением их расщепления, всасывания, транспорта, утилизации, депонирования и метаболизма.
12.5.1. Нарушение переваривания и всасывания липидов
Для нормального переваривания и всасывания липидов в кишечнике определяющее значение имеет взаимодействие таких факторов, как:
1) выработка поджелудочной железой липолитического фермента липазы;
2) поступление с желчью желчных кислот, эмульгирующих жиры и продукты их распада, активирующих панкреатическую ли-
пазу и участвующих во всасывании жирных кислот (всасывается комплекс жирных и желчных кислот);
3) захват продуктов переваривания липидов клетками слизистой оболочки тонкого кишечника;
4) превращение в стенке кишечника всосавшихся продуктов гидролиза липидов в частицы (хиломикроны) для дальнейшего транспорта их в лимфатические сосуды и далее в кровоток.
При нарушении любого из этих процессов развивается стеаторея - избыточное содержание жира в испражнениях.
Причинами нарушения переваривания и всасывания липидов являются:
1. Дефицит или низкая активность панкреатической липазы (поражение поджелудочной железы), что приводит к нарушению расщепления жиров.
2. Недостаточное поступление желчных кислот в кишечник (при гепатитах, циррозах, холециститах, обтурационной желтухе и др.) вызывает нарушение эмульгирования и расщепления жира, а также переноса продуктов его гидролиза к всасывающей поверхности эпителия кишечника.
3. Дефицит гормонов желудочно-кишечного тракта (холецистокинин, гастрин и др.), регулирующих сокращение стенок желчного пузыря, процессы эмульгирования и расщепления жиров, их транспорт через кишечную стенку.
4. Поражение эпителия тонкого кишечника различными ядами (флоридзин, монойодуксусная кислота) и инфекционными агентами, инактивирующими ферментные системы ресинтеза триацилглицеролов эпителия тонкого кишечника, а также процессы фосфорилирования и дефосфорилирования в стенке кишечника.
5. Авитаминозы А, В, С (поскольку эти витамины являются коферментами соответствующих биохимических реакций).
6. Избыточное потребление с пищей Са и Mg, что приводит к образованию нерастворимых в воде солей жирных кислот (мыла).
7. Дефицит холина в пище или недостаточное его образование из метионина при малобелковом питании тормозит реабсорбцию липидов.
8. Изменение деятельности нервной и эндокринной систем: перерезка блуждающего нерва ослабляет всасывание жиров из кишечника, аналогично действует наркоз; адренокортикотропный гормон (АКТГ) и тироксин усиливают всасывание жира. При недостатке гормонов коры надпочечников или избытке адреналина всасывание жира замедляется.
9. Усиленная перистальтика кишечника и диарея препятствуют реабсорбции большей части жира.
10. Нарушение метаболизма липидов в энтероцитах с образованием аномальных белково-липидных комплексов ухудшает всасывание жира и вызывает образование жировых скоплений в стенке тонкого кишечника и в мелких лимфатических протоках, что блокирует отток лимфы.
Дефицит липидов в организме может быть связан не только с нарушением их всасывания в кишечнике, но и с усилением их выведения. Организм может терять липиды с мочой (липидурия), что наблюдается при липоидном нефрозе. Возможны потеря липидов сальными железами (экзема, угревая сыпь) и выход липидов из депо при травматизации больших участков жировой ткани и костного мозга.
Последствиями недостатка липидов являются:
1) развитие гиповитаминозов (жирорастворимых витаминов
А, D, Е, К);
2) возникновение дефицита незаменимых полиненасыщенных жирных кислот с последующим нарушением синтеза биологически активных веществ (лейкотриены, простагландины и др.). Это, как правило, сопровождается выпадением волос, воспалительным поражением кожи, возникновением некротических очагов и экзематозных явлений, поражением почек, потерей способности к размножению;
3) развитие истощения.
Синтез и разрушение липидов происходят практически во всех тканях организма. Некоторые ткани выполняют специализированные функции. Так, поглощение экзогенных липидов происходит в стенках тонкого кишечника; накопление - в жировой ткани; выведение продуктов распада липидов - в кишечнике, почках, легких. Центральное место в липидном обмене занимает печень, где пересекаются пути метаболизма липидов, углеводов и белков. Здесь же синтезируются основная масса белков транспорта липидов и продукты деградации липидов, требующие выведения из организма. Окончательные продукты распада липидов выводятся из организма в форме солей желчных кислот, нейтральных стероидов и кетоновых тел.
12.5.2. Нарушение транспорта липидов
Всосавшиеся в кровь неполярные липидные молекулы циркулируют в крови и лимфе в комплексе с полярными соединениями
(белками). Существует большой спектр липопротеиновых частиц, отличающихся по размерам, плотности и составу. Липопротеины - это сферические частицы, состоящие из гидрофобной сердцевины и гидрофильной оболочки (в центре - неполярные липиды: триглицериды и эфиры холестерина; оболочка построена из полярных липидов - холестерина и фосфолипидов, причем их заряженные концы обращены наружу; также в составе оболочки - белки, нековалентно связанные с фосфолипидами и холестерином, апопротеины). Апопротеины поддерживают структуру частиц, обеспечивают их взаимодействие с рецепторами и служат «визитной карточкой» липопротеинов, поскольку рецепторы для липопротеинов на разных клетках распознают только определенные апопротеины. Липопротеины подразделяют на классы в зависимости от их плотности и подвижности при электрофорезе. Плотность липопротеиновой частицы определяется отношением апопротеины/ липиды: чем больше это отношение, тем выше плотность. Выделяют 4 основные группы липопротеинов (ЛП):
1. Липопротеины высокой плотности (ЛПВП, или α-ЛП). В состав ЛПВП входят 40-55% белка (процент общей массы частицы), 27-30% фосфолипидов, 3-8% триглицеридов, 2-3% свободного холестерина, 14-20% эфиров холестерина. Оболочка содержит апопротеины А, СП, Е. Они синтезируются паренхимой печени, в стенке тонкого кишечника и всегда присутствуют в плазме крови здоровых людей. Выполняют транспортную функцию, переводя избыток холестерина с поверхности сосудов в печень и выводя его излишек из клеток эндотелия (рис. 12-29).
Высокоспецифичные рецепторы ЛПВП обнаружены на гладкомышечных клетках и фибробластах. Количество этих рецепторов увеличивается при повышении концентрации холестерина в клетке. Связывание ЛПВП с рецепторами вызывает выброс холестерина из клеток. Первоначально холестерин встраивается в оболочку ЛПВП, но затем под действием лецитин-холестеринацетилтрансферазы (ЛХАТ) он этерифицируется и перемещается в сердцевину ЛПВП. В печени ЛПВП связываются с рецепторами и разрушаются. Таким образом, ЛПВП - это антиатерогенные липопротеины.
2. Липопротеины очень низкой плотности (ЛПОНП, или пре- β-ЛП). Представляют очень неоднородный класс частиц с различным содержанием компонентов: 8-12% - белок, 10-12% - свободный холестерин, 18-20% - фосфолипиды, 3-6% - эфиры
 Рис. 12-29. Обмен
липидов. ЛПНП - липопротеины низкой плотности, ЛПОНП - липопротеины
очень низкой плотности, ЛПВП - липопротеины высокой плотности, НЭЖК -
неэтерифицированные жирные кислоты
Рис. 12-29. Обмен
липидов. ЛПНП - липопротеины низкой плотности, ЛПОНП - липопротеины
очень низкой плотности, ЛПВП - липопротеины высокой плотности, НЭЖК -
неэтерифицированные жирные кислоты
холестерина, около 50% - триацилглицеролы (ТАГ). Они образуются в основном в гепатоцитах (и в меньшем количестве - в слизистой кишечника), являются главной транспортной формой эндогенных триацилглицеролов. В их составе апопротеины С, Е и В100. В плазме крови происходит трансформация ЛПОНП в β-ЛП (при участии ферментов - липопротеиновой липазы и ЛХАТ крови). В ходе их катаболизма размеры частиц уменьшаются, меняется их состав (теряются ТАГ и возрастает относительный процент холестерина).
3. Липопротеины низкой плотности (ЛПНП, или β-ЛП) имеют следующий состав: 24-31% - свободный холестерин, 16-28% - этерифицированный холестерин, 7-11% - триглицериды, около 30% - фосфолипиды, 20-25% - белок. Они образуются в плазме из ЛПОНП и являются самой атерогенной фракцией липопротеинов у человека. ЛПНП содержат только один апопротеин B100.
Около 70% ЛПНП удаляется из крови гепатоцитами с помощью высокоспецифичных рецепторов. Остальные ЛПНП захватываются клетками ретикулоэндотелиальной системы с помощью менее
специфичных нейтрализующих рецепторов. Синтез высокоспецифичных рецепторов в печени подавляется при повышении концентрации β-ЛП. Напротив, количество нейтрализующих рецепторов на клетках ретикулоэндотелиальной системы не зависит от уровня ЛПНП. Нарушение захвата ЛПНП в печени (вследствие дефекта апопротеина B100 или дефекта высокоспецифичньгх рецепторов) приводит к накоплению ЛПНП в других тканях и органах. Их атерогенный эффект опосредуется именно нейтрализующими рецепторами. Считается, что ЛПНП становятся атерогенными только после определенных превращений, например, при перекисном окислении (модифицированные ЛПНП). Окисленные ЛПНП захватываются макрофагами, которые после этого превращаются в ксантомные клетки, нагруженные эфирами холестерина. Нейтрализующие рецепторы обнаружены и в гладкомышечных клетках артерий.
4. Хиломикроны - самые крупные липопротеиновые частицы, поступающие в кровь из лимфы и представляющие собой транспортную форму пищевых жиров (экзогенных ТАГ). В их составе находятся: 3-8% фосфолипидов, 2-4% эфиров холестерина, около 2% свободного холестерина, 1-2% белка и 86-94% ТАГ. В оболочке имеются апопротеины B48, A, C, E. Хиломикроны образуются в стенке кишечника в процессе всасывания экзогенных ТАГ и холестерина, поступают в лимфатические сосуды и через грудной лимфатический проток попадают в большой круг кровообращения.
В крови хиломикроны встречаются с ЛПВП и обмениваются с ними апопротеинами: отдают часть апопротеинов A, получают C и E. В кровеносных капиллярах жировой ткани, миокарда, скелетных мышц и молочных желез хиломикроны расщепляются липопротеиновой липазой, расположенной на поверхности эндотелия и теряют значительное количество ТАГ (образуются свободные жирные кислоты (СЖК) и глицерин). При этом освобождается остаточный компонент хиломикрона (сердцевина), содержащий большое количество эфиров холестерина. Сами хиломикроны не обладают атерогенными свойствами, но остаточные компоненты хиломикронов, по-видимому, атерогенны.
Для ткани легких катаболизм хиломикронов особенно важен, поскольку играет ключевую роль в обеспечении высокой активности альвеолярных макрофагов и необходим для синтеза фосфолипидов сурфактанта (рис. 12-30). В связи с этим при заболеваниях легких положительный эффект дает жировая диета. Следует от-
 Рис. 12-30. Роль сурфактанта в предупреждении ателектаза
Рис. 12-30. Роль сурфактанта в предупреждении ателектаза
метить, что плазма крови здоровых людей натощак (через 12-14 ч после приема пищи) не содержит хиломикронов. В крови, взятой натощак, присутствуют только ЛПОНП, ЛПНП и ЛПВП, тогда как другие липопротеины (хиломикроны, остаточные компоненты хиломикронов, а также липопротеины промежуточной плотности) выявляются только после еды или при нарушениях обмена липидов. При ряде заболеваний липопротеиновый спектр сыворотки меняется и возникают гиперили гипо-, алипопротеинемии. При этом отмечаются увеличение или, наоборот, снижение содержания, вплоть до полного отсутствия одного или нескольких классов липопротеинов в крови, а также появление их определенных форм (дислипопротеинемии). Гипер- и дислипопротеинемии - один из главных факторов риска многих заболеваний, в первую очередь - атеросклероза. Их клинические проявления и тяжесть зависят от рациона и режима питания, а также от сопутствующих заболеваний. Дислипопротеинемии возникают или усиливаются при ожирении, сахарном диабете, гипотиреозе, болезнях почек и печени; их течение и прогноз зависят от тяжести основного заболевания. Различают 5 типов гиперлипопротеинемий:
I. Гиперхиломикронемия - характеризуется высоким содержанием хиломикронов в плазме натощак. Проявляется ксантоматозом - отложением холестерина и его эфиров в купферовских клетках печени, гистиоцитах подкожной клетчатки и сухожилиях с последующим разрастанием соединительной ткани в виде бляшек и узлов желтоватого цвета (рис. 12-31). У больных развивается гепатоспленомегалия, выявляются тромбоз и микронекрозы поджелудочной железы с последующим формированием хронического панкреатита, абдоминальные колики после принятия жирной пищи. На коже определяются ксантомы в виде желтоватых папул. Заболе-
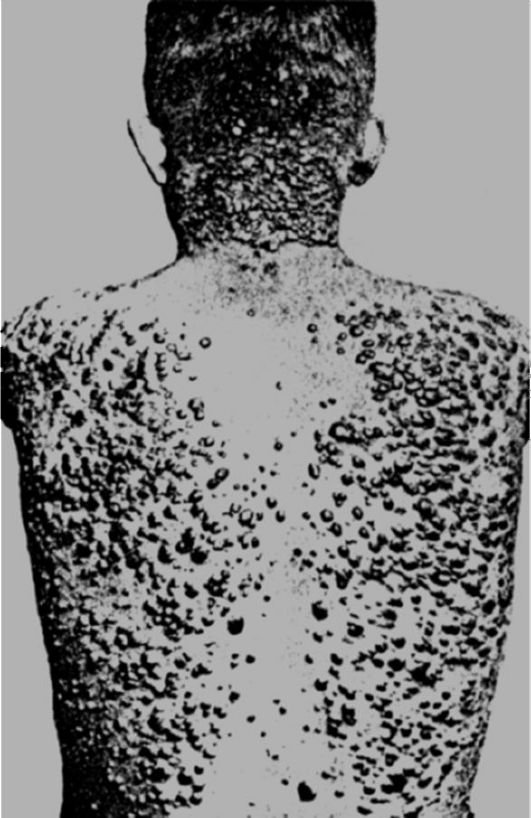 Рис. 12-31. Ксантоматоз кожи (по H.W. Siemens)
Рис. 12-31. Ксантоматоз кожи (по H.W. Siemens)
вание может быть вызвано наследственным аутосомно-рецессивным дефектом липопротеиновой липазы либо аутоиммунными заболеваниями соединительной ткани (при системной красной волчанке образуются антитела против гликозаминогликанов, что нарушает процесс гепариновой активации липопротеиновой липазы).
II. Гипер-b-липопротеинемия делится на 2 типа:
11а - увеличение содержания в крови β-ЛП при нормальном уровне пре- β-ЛП;
11б - увеличение содержания β-ЛП и пре- β-ЛП.
Для заболевания характерен выраженный ксантоматоз век, кожи, роговицы, развитие ишемической болезни сердца с инфарктом миокарда в очень раннем возрасте,
атеросклеротические поражения сосудов у детей. Предполагается, что в основе заболевания лежит аутосомно-доминантный дефект рецепторов ЛПОНП и ЛПНП (11б) либо изменение активности липопротеиновой липазы плазмы крови (11а).
III. «Флотирующая» гиперлипопротеинемия, или дис- β-липопротеинемия. В основе заболевания лежит наследственно обусловленное нарушение синтеза апопротеина Е (белок, входящий в состав хиломикронов и ЛПОНП). Заболевание характеризуется появлением в сыворотке флотирующих β-ЛП, которые называются промежуточными (липопротеины промежуточной плотности - ЛППП). Они обогащены холестерином, а содержание триглицеридов в них может быть снижено. Образуются эти частицы при нарушении катаболизма ЛПОНП и хиломикронов. Встречаются также приобретенные формы заболевания при гипотиреозе, танжерской болезни, некоторых аутоиммунных гаммапатиях. Этот вид гиперлипопротеинемии сопровождается ранними атеросклеротическими проявлениями (после 20 лет), развитием ишемической болезни сердца, ишемической энцефалопатии вплоть до инсультов, ксантоматозом, ожирением.
IV. Гипер-пре- β-липопротеинемия. Заболевание может быть наследственно обусловленным (аутосомно-доминантное) или приобретенным (при алкоголизме, остром гепатите, акромегалии, диабете и др.). Патогенез до конца не выяснен. Для этого типа гиперлипопротеинемии характерно нарастание уровня триглицеридов и ЛПОНП в крови. Содержание ЛПНП и ЛПВП варьирует от нормального до значительно сниженного. У больных развиваются ожирение и сахарный диабет 2 типа, появляются ксантомы, возможны атеросклеротическое поражение сосудов нижних конечностей, липидоз сетчатки и ухудшение зрения, проявления ишемической болезни сердца.
V. Гипер-пре- β-липопротеинемия и хиломикронемия. При этом заболевании в крови увеличивается содержание хиломикронов и ЛПОНП и снижается уровень ЛПНП и ЛПВП. У больных отмечаются гепато- и спленомегалия, ожирение, снижение толерантности к глюкозе (при сахарном диабете 2 типа), поражение миокарда. После приема жирной пищи могут наблюдаться внезапные приступы абдоминальной колики, ксантоматоз и атеросклероз слабо выражены. В патогенезе первичного заболевания главную роль играет наследственно обусловленное отсутствие кофактора липопротеинлипазы - апопротеина CII (аутосомно-рецессивное наследование), в результате два основных субстрата воздействия этого фермента накапливаются в крови. Фенокопия болезни развивается при алкоголизме, гликогенозе Гирке и некоторых других заболеваниях печени.
Гипо-(а)-липопротеинемии относятся к группе относительно редких аномалий спектра липопротеинов:
1. A-β-липопротеинемия. В основе заболевания лежит аутосомно-доминантный дефект синтеза апопротеина В, что приводит к аномалии строения хиломикронов, снижению содержания или полному отсутствию в плазме ЛПОНП и ЛПНП. Клинические проявления связаны с нарушением всасывания в кишечнике жиров и углеводов, гемолитической анемией, дегенерацией бокового и заднего канатиков спинного мозга, пигментной ретинопатией. Нарушение всасывания жиров проявляется сразу после рождения плохим аппетитом, рвотой, обильными испражнениями, стеатореей, развитием гипотрофии. Примерно у трети больных развивается умственная отсталость. С возрастом усиливаются неврологические расстройства, появляются скелетные деформации, сердечные аритмии, ухудшается зрение. В патогенезе заболевания решающее значение имеет снижение содержания холестерина в клеточных
мембранах и потеря жирорастворимых витаминов, особенно витамина Е, что ведет к утрате антиоксидантной защиты мембран.
2. Танжерская (или тэнжирская) болезнь. В основе заболевания лежит аутосомно-рецессивное нарушение синтеза апопротеина А, что, в свою очередь, нарушает продукцию ЛПВП. У больных нарушен транспорт эфиров холестерина, в результате эфиры захватываются макрофагами и откладываются в клетках ретикулоэндотелиальной системы селезенки, печени, лимфоидных органов. Выявляются лимфаденопатия, гепатоспленомегалия, неврологические нарушения - слабость, парестезии, снижение сухожильных рефлексов. Одним из ярких признаков заболевания является оранжево-желтый цвет увеличенных миндалин.
Существуют и другие формы гиполипопротеинемий: церебросухожильный ксантоматоз (наследственный дефект синтеза желчных кислот из холестерина), болезнь Вальмана (аутосомнорецессивный дефицит холинэстеразы), гипо-a-липопротеинемия (генетически детерминированное нарушение продукции апопротеина А и С) и др. Большинство из них связано с наследственной патологией синтеза белковой части липопротеинов либо с нарушением метаболизма холестерина.
12.5.3. Нарушение перехода липидов в ткани. Гиперлипемия
Гиперлипемия является одним из показателей нарушения жирового обмена и характеризуется повышенным содержанием в крови нейтральных жиров и триацилглицеролов.
Липиды поступают в лимфу, а затем в кровь из кишечника в виде самых крупных липопротеинов - хиломикронов, из печени в кровь выходят липопротеины очень низкой плотности. При липолизе из подкожной жировой клетчатки, легких, костного мозга освобождаются неэтерифицированные жирные кислоты (НЭЖК). Уровень липидов в плазме крови в норме не превышает 1-2 г/л.
Гиперлипемия может быть алиментарной, транспортной и ретенционной.
Алиментарная гиперлипемия - временное увеличение уровня хиломикронов в крови, вызванное приемом жирной пищи или проведением пробы с липидной нагрузкой. Она легко устраняется с помощью возросшей функциональной активности гепатоцитов, утилизирующих хиломикроны. Возможно также усиление депонирования липидов в жировой ткани (рис. 12-32).
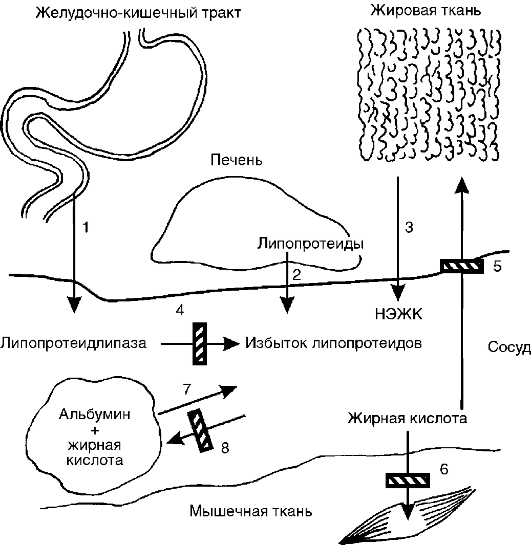 Рис. 12-32. Причины
гиперлипемии: 1 - усиленное поступление в кровь хиломикронов и жирных
кислот из кишечника; 2 - усиленное поступление в кровь липопротеинов из
печени; 3 - усиленное поступление в кровь неэтерифицированных жирных
кислот из жировой ткани; 4 - низкая активность липопротеиновой липазы;
5-6 - задержка поступления жирных кислот из крови в жировую ткань и
мышцы; 7 - усиленное расщепление комплекса альбумина с жирной кислотой; 8
- гипоальбуминемия и недостаточное образование комплекса альбумина с
жирной кислотой. Заштрихованные столбики - места нарушения процесса
Рис. 12-32. Причины
гиперлипемии: 1 - усиленное поступление в кровь хиломикронов и жирных
кислот из кишечника; 2 - усиленное поступление в кровь липопротеинов из
печени; 3 - усиленное поступление в кровь неэтерифицированных жирных
кислот из жировой ткани; 4 - низкая активность липопротеиновой липазы;
5-6 - задержка поступления жирных кислот из крови в жировую ткань и
мышцы; 7 - усиленное расщепление комплекса альбумина с жирной кислотой; 8
- гипоальбуминемия и недостаточное образование комплекса альбумина с
жирной кислотой. Заштрихованные столбики - места нарушения процесса
Транспортная гиперлипемия обусловлена либо усиленной мобилизацией из депо в виде неэтерифицированных жирных кислот при голодании, стрессе, сахарном диабете 1 типа, либо нарушением метаболизма циркулирующих в крови липопротеинов при различных формах семейной гипертриглицеролемии. Усилению мобилизации липидов из жировой ткани, костного мозга способствуют соматотропный и кортикотропный гормоны гипофиза, а также глюкагон, тироксин и адреналин, которые активируют тка-
невую липазу через аденилатциклазную систему. Из печени липопротеины (комплекс липидов с белками) поступают в кровь. Мобилизация жира из легких, приводящая к гиперлипемии, возникает также при длительной гипервентиляции легких, например, у пловцов и профессиональных певцов.
Ретенционная гиперлипемия (от лат. retentio - задерживать) развивается в результате задержки перехода нейтральных жиров из крови в ткани. Возникает при атеросклерозе, ишемической болезни сердца, нефрозе, сахарном диабете 2 типа, при механической желтухе, поступлении большого количества NaCl (ингибирует липопротеиновую липазу крови). В патогенезе этого вида гиперлипемии большое значение имеют следующие факторы:
1) снижение уровня гепарина, активирующего фактор просветления (липопротеиновая липаза) - при нефрозе, механической желтухе, атеросклерозе;
2) уменьшение содержания альбуминов в крови (осуществляют транспорт НЭЖК в клетки различных органов) - при нефротическом синдроме, заболеваниях печени и др.;
3) присутствие в сыворотке ингибитора липопротеиновой липазы - при нефротическом синдроме;
4) снижение активности липокаина, активирующего поступление в кровь липопротеиновой липазы, синтезируемой клетками многих тканей (жировой, мышечной, сердечной, тканей легких, селезенки) - при сахарном диабете.
12.5.4. Нарушение депонирования жиров
Нарушение нейроэндокринной регуляции жирового обмена. Масса тела человека находится под сложным контролем нервногуморальных влияний, определяющих пищевую мотивацию и уровень основного обмена. Центры регуляции пищевого поведения и основного обмена находятся в супраоптических ядрах гипоталамуса и контролируются таламусом, лимбической системой и корой. Центр голода локализован в вентролатеральных, а центр насыщения - в вентромедиальных ядрах подбугорья (связан с центром голода синапсами, передающими тормозные импульсы). Нейроны центра голода вырабатывают нейропептид Y, который активирует кортико-лимбические структуры мозга и стимулирует чувство голода, побуждая к поиску пищи. Эмоционально-поведенческие аспекты приема пищи регулируются отделами, расположенными
в кортикальной части лимбической системы (поясная извилина, гиппокамп, инфраорбитальная область) и в миндалине, разрушение которой вызывает безразличие к характеру предлагаемой пищи.
Подавление центра голода (при патологии вплоть до анорексии) обусловлено действием лептина1 (тормозит синтез нейропептида Y в центре голода и стимулирует образование в центре насыщения глюкагоноподобного пептида (ГПП) I, подавляющего аппетит; рис. 12-33); соматостатина, нейротензина, кортико- и тиреолиберина, меланокортинов гипофиза2; серотонина, вазопрессина, окситоцина; холецистокинина (сигналом для его выработки служит механическое растяжение) и других кишечных гормонов энтериновой системы (глюкагон, секретин, вазоактивный интестинальный пептид, гастрин, энтерогастрон); норадреналина, кальцитонина, инсулина; TNF-α (фактор некроза опухолей, или кахексин), выделяемого висцеральными адипоцитами в «сытом» состоянии. Перечисленные гормоны и нейротрансмиттеры связываются с рецепторами нейронов центра голода, снижают образование нейропептида Y и подавляют аппетит.
Активация центра голода и возникновение гиперрексии (булимии) может быть опосредовано нейропептидом Y (понижает симпатический и повышает парасимпатический тонус, а также угнетает половую функцию), эндорфинами и энкефалинами, соматолибери-
1 Лептин - основной гормон, контролирующий массу жировой ткани (обнаружен в 1994 г.). Механизмы действия лептина: усиление основного обмена, термогенеза и расхода энергии; стимуляция симпатического отдела ЦНС; повышение печеночного гликогенолиза и захвата глюкозы скелетными мышцами; активация липолиза в белой жировой ткани; усиление окисления жирных кислот в митохондриях гепатоцитов и скелетных мышц; подавление активности половых желез и репродуктивной функции; повышение продукции глюкокортикоидов. Лептин секретируется в основном адипоцитами белой жировой ткани. В норме его содержание в крови четко коррелирует с массой тела. Рецепторы к лептину обнаружены практически во всех клетках, но основной орган-мишень лептина - центральная нервная система. Действуя через специфические рецепторы гипоталамуса, лептин снижает аппетит (участвует в развитии кахексии, нервной анорексии и др.) и уменьшает запасы жира в жировых депо. Лептин является провоспалительным гормоном, способствующим преобладанию клеточного иммунитета, вследствие усиления продукции цитокинов ТЫ-типа. Лептин вырабатывается также остеобластами и тормозит образование остеокластов. Ген ожирения LEP (obese gene - ob) локализован на хромосоме 7q31.3, ген рецептора лептина - на хромосоме 1. Рецептор лептина человека имеет гомологичный участок с рецепторами интерлейкина (IL) 6 и других цитокинов.
2 Гипоталамическая система меланокортина и меланокортиновый рецептор MC-4R участвуют в передаче эффектов лептина.
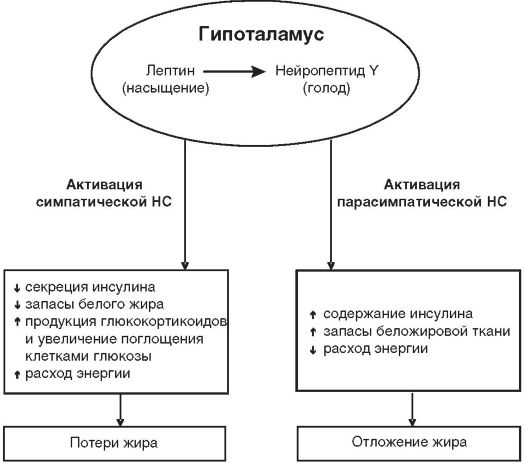 Рис. 12-33. Взаимосвязь между центральной нервной системой и лептином
Рис. 12-33. Взаимосвязь между центральной нервной системой и лептином
ном, дофамином, γ-аминомасляной кислотой (ГАМК), избытком инсулина.
Усиление липолиза имеет место при активации симпатоадреналовой системы (максимальный эффект через β,,-адренорецепторы, в буром жире адренергические терминали особенно обильны и образуют синапсы на самих адипоцитах, способствуя «залповому» липолизу); действии глюкагона (см. раздел 12.4.5), катехоламинов (высокие дозы стимулируют липолиз через β,,-адренорецепторы на адипоцитах, низкая концентрация адреналина действует через а2-рецепторы и увеличивает липогенез), тиреотропного гормона (ТТГ), тироксина, соматотропного гормона (СТГ), андрогенов, АКТГ (прямое действие на адипоциты), глюкокортикоидов (в физиологических концентрациях повышают использование жирных кислот в энергетическом обмене во всех тканях, кроме печени), TNF-α (снижает ответ мышечной и жировой ткани на инсулин и тормозит липогенез).
Усиление липогенеза может быть обусловлено парасимпатической импульсацией (парасимпатическая нервная система практически не иннервирует жировую ткань и потому не оказывает выраженного эффекта на ее метаболизм, однако вагусная импульсация уменьшает выделение норадреналина из пресинаптических окончаний, что косвенно может опосредовать преобладание липогенеза); действием инсулина (см. раздел 12.4), глюкокортикоидов (в высоких дозах усиливают липолиз в конечностях и липогенез в области лица и туловища; их эффект зависит от действия катехоламинов и различен в адипоцитах разной локализации, поскольку набор адренорецепторов на жировых клетках неодинаков и зависит от их анатомического расположения), АКТГ (см. раздел 12.4), пролактина и эстрогенов (через гиперинсулинемию; кроме того, пролактин стимулирует синтез фосфолипидов и ТАГ в молочных железах и других тканях).
Ожирение - избыточное отложение жира в жировой ткани. Ожирение значительно повышает риск развития артериальной гипертонии, сахарного диабета 2 типа, атеросклероза, поэтому очень важно следить за своим весом. Считается, что человек страдает ожирением, если его масса превышает нормальную более чем на 20% и продолжает увеличиваться далее. Этим недугом страдает более трети взрослого населения России. В 1998 г. Всемирная организация здравоохранения (ВОЗ) признала ожирение хроническим заболеванием. По статистике ВОЗ, в экономически развитых странах около 30% взрослых и до 10% детей имеют ту или иную форму и степень ожирения. В возрастных группах после 50 лет это заболевание встречается чаще. За последнее десятилетие число таких больных в мире увеличилось почти в два раза и по оценке специалистов в 2025 г. их количество составит 300 млн человек. Ситуация тем более сложная, что с каждым годом увеличивается число молодых людей, страдающих ожирением, снижается общая продолжительность жизни населения земного шара в связи с тяжелыми заболеваниями, сопутствующими ожирению (см. табл. 12-5).
У тучных людей старше 50 лет смертность увеличивается на 50% по сравнению с лицами, не имеющими ожирения. У женщин с ожирением чаще встречаются рак эндометрия, яичника, шейки матки, желчного пузыря и молочной железы, у мужчин - рак предстательной железы и толстой кишки, снижение потенции. Жировая ткань, составляющая в норме 15-20% от массы тела у мужчин и 20-29% у женщин, - это метаболически активное образование.
Таблица 12-5. Заболевания, сопутствующие ожирению (по С. Бутровой,
2001;
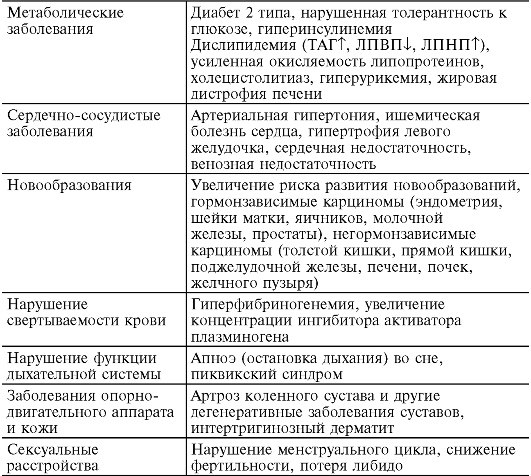 Адипоциты
секретируют гемопоэтины; выделяют цитокины - TNF-α, IL-6,
трансформирующий фактор роста (TGF) β и соответствующие им растворимые
рецепторы; синтезируют биоактивные вещества - ангиотензиноген, ингибитор
активатора плазминогена; ряд ферментов (липопротеиновая липаза,
индуцируемая NO-синтаза, аполипопротеин Е) и гормонов (лептин, резистин,
адипонектин, эстрогены); выделяют свободные жирные кислоты. Увеличение
массы жировой ткани влечет за собой повышение содержания лептина в
крови, причем его продукция в подкожной жировой клетчатке выше, чем в
висцеральных жировых депо. Уровень лептина отражает не только количество
накопленного жира, но и нарушения энергетического обмена: при голодании
он значи-
Адипоциты
секретируют гемопоэтины; выделяют цитокины - TNF-α, IL-6,
трансформирующий фактор роста (TGF) β и соответствующие им растворимые
рецепторы; синтезируют биоактивные вещества - ангиотензиноген, ингибитор
активатора плазминогена; ряд ферментов (липопротеиновая липаза,
индуцируемая NO-синтаза, аполипопротеин Е) и гормонов (лептин, резистин,
адипонектин, эстрогены); выделяют свободные жирные кислоты. Увеличение
массы жировой ткани влечет за собой повышение содержания лептина в
крови, причем его продукция в подкожной жировой клетчатке выше, чем в
висцеральных жировых депо. Уровень лептина отражает не только количество
накопленного жира, но и нарушения энергетического обмена: при голодании
он значи-
тельно снижается, при переедании - повышается. Избыток лептина вызывает инсулинорезистентность скелетных мышц и жировой ткани и подавляет действие инсулина на клетки печени (инсулин активирует адипоциты, повышая образование лептина, в свою очередь, лептин, воздействуя на собственные рецепторы, локализованные на поверхности β-клеток, тормозит секрецию инсулина).
У женщин наличие достаточно выраженной жировой ткани существенно для поддержания нормальной половой функции. Менструации у девочек, не достигших критической массы (около 48 кг), не начинаются, даже если пубертатный период пройден. При похудении на 10-15% нормы даже при сохранении цикла нет овуляции, возможна и полная аменорея. Эти изменения обратимы, при нормализации веса детородная функция восстанавливается. Вероятно, невозможность деторождения у женщин, не имеющих достаточных жировых запасов для успешного рождения и вскармливания ребенка, была выработана в процессе естественного отбора. У мужчин похудение и физические нагрузки приводят к уменьшению полового влечения, а если вес тела на 25% ниже нормы, выработка спермы угнетается. Следует отметить, что сильное развитие мускулатуры (например, у культуристов) действует на репродуктивные процессы так же, как и похудение. Избыток эстрогенов у полных мужчин приводит к снижению потенции, гинекомастии, гипогонадизму с понижением уровня тестостерона.
Классификация ожирения. Ожирение может возникать как самостоятельное заболевание - в этом случае говорят о первичном ожирении. Вторичное ожирение - это синдром, возникающий вследствие гормональных или других расстройств в организме.
Первичное ожирение возникает при нарушении гормональной связи между жировой тканью и гипоталамусом. Это генетически опосредованное нейроэндокринное заболевание, его главная черта - абсолютная или относительная лептиновая недостаточность. Около 20% больных имеют абсолютную лептиновую недостаточность, однако дефицит лептина1 не является основной причиной развития ожирения. Более 80% пациентов с первичным ожирени-
1 Генетически опосредованный дефицит лептина проявляется ранним ожирением, пониженным обменом веществ, гипогонадотропным гипогонадизмом, гиперинсулинемией, нарушением гипоталамо-питуитарных и тиреоидальных взаимодействий и нарушением количества и функции Т-лимфоцитов, что повышает восприимчивость больных к инфекциям. Известны 5 отдельных мутаций в гене лептина, вызывающих развитие первичного ожирения.
ем имеют выраженную гиперлептинемию, что свидетельствует о резистентности к гормону. Известны следующие механизмы резистентности:
• нарушение транспорта лептина через гематоэнцефалический барьер (введение гормона даже в больших дозах не дает результата);
• нарушение переноса гормона транспортными белками;
• мутации рецептора лептина1 (несмотря на продукцию лептина, центр голода продолжает секрецию нейропептида Y);
• мутации генов, кодирующих рецепторы к меланокортину2 (4% всех больных с ожирением). Следует отметить, что на фоне введения лептина у млекопитающих снижается только масса жировой ткани, в то время как при голодании снижается также масса других тканей.
Вторичное ожирение - синдром, возникающий при нарушении соотношения между процессами липолиза и липогенеза, носит симптоматический характер и порождается различными расстройствами (эндокринопатии, опухоли мозга, нарушения мозгового кровообращения и пр.).
По степени увеличения массы тела различают ожирение I степени (масса тела увеличена на 30%); II степени (на 30-50%); III степени (более чем на 50%).
Одним из наиболее распространенных показателей для оценки степени ожирения является индекс массы тела (ИМТ), рассчитываемый следующим образом:
ИМТ= Вес (кг) / [Рост (м)]2.
Больные с ИМТ 30 кг/м2 и более, а также пациенты с ИМТ 27 кг/м2 или более, ожирение которых связано с такими факторами риска, как диабет 2 типа или дислипопротеинемия, подлежат обязательному лечению (табл. 12-6).
1 Дети с дефектом рецептора лептина быстро набирают избыточную массу в течение первых месяцев жизни, отличаются гиперфагией и агрессивным поведением во время еды. Иногда дефект рецептора имеет более выраженные проявления (гипотиреоз), чем отсутствие лиганда.
2 Меланокортины (адренокортикотропный и меланоцитостимулирующий гормоны, а также и их фрагменты) образуются в гипофизе из проопиомеланокортина. Лептин стимулирует экспрессию гена проопиомеланокортина, образующийся проопиомеланокортин расщепляется до субстрата, который действует как супрессор пищевого поведения, возможно через MC4R. При снижении гипоталамического меланокортинэргетического сигнала через рецепторы MC4R наблюдаются гиперфагия и прибавка массы.
Таблица 12-6. Классификация избыточной массы у взрослых в зависимости от индекса массы тела (в соответствии с докладом ВОЗ 1998 г.)
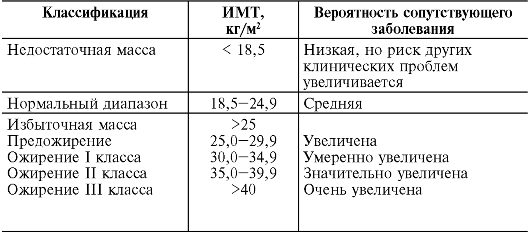 Наиболее
простым методом определения склонности к ожирению является измерение
окружности талии. В идеале окружность талии не должна превышать 94 см у
мужчин и 80 см у женщин. Если окружность талии у мужчин достигает 102
см, а у женщин - 88 см, возникает серьезная угроза увеличения риска
заболевания.
Наиболее
простым методом определения склонности к ожирению является измерение
окружности талии. В идеале окружность талии не должна превышать 94 см у
мужчин и 80 см у женщин. Если окружность талии у мужчин достигает 102
см, а у женщин - 88 см, возникает серьезная угроза увеличения риска
заболевания.
По особенностям морфологии жировой ткани выделяют гипертрофическое и гиперпластическое ожирение.
Гипертрофическое ожирение связано с увеличением размеров адипоцитов (это лабильный фактор, зависимый от питания), чаще встречается в зрелом возрасте. При этом виде ожирения масса тела может увеличиваться в 3-3,3 раза.
Гиперпластическое ожирение сопровождается увеличением количества адипоцитов. Начинается, как правило, в детском возрасте, так как дифференцировка фибробластических клеток-предшественниц в новые адипоциты во взрослом организме - явление довольно редкое (это происходит в период внутриутробного развития и в раннем грудном возрасте). В развитии гиперпластического ожирения огромное значение имеет наследственность, определяющая пролиферативные возможности этих клеток. Избыток массы тела при гиперпластическом ожирении может достигать гигантских величин (до 1000%). Следует отметить, что в подростковый и предклимактерический периоды повышается пролиферативная активность преадипоцитов. Кроме того, их деление индуцируют избыточная калорийность пищи, сахарный диабет или переедание у беременных. В этих случаях гиперпластическое ожирение развивается у взрослых.
Жир может располагаться в подкожно-жировой клетчатке (подкожный жир) и вокруг внутренних органов (висцеральный жир), вместе подкожно-жировая клетчатка в области живота и висцеральный жир брюшной полости составляют абдоминальный жир. Разная локализация жировых отложений при различных формах первичного и вторичного ожирения зависит от влияния мужских и женских половых гормонов на распределение и катехоламиновых рецепторов в разных отсеках жировой ткани. Жировая ткань, локализованная в различных частях тела, отличается по своей гормональной функции (см. раздел 12.4). У людей, склонных к первичному ожирению, уменьшена экспрессия β-адренорецепторов на адипоцитах.
В зависимости от характера распределения жировой ткани различают:
• андроидный (яблочный) тип
ожирения, когда избыточные отложения жира располагаются на животе и верхней части туловища (наиболее характерен для мужчин) (рис. 12-34);
• гиноидный (грушевидный) тип ожирения, когда избыточные отложения жира располагаются на бедрах, ягодицах и в нижней части туловища (наиболее характерен для женщин);
• смешанный тип ожирения - комбинирует признаки андроидного и гиноидного типов.
Гиноидное ожирение чаще носит гиперпластический характер, поэтому оно более резистентно к диетотерапии. Однако более патогенным считается андроидное, а более благоприятными - гиноидное, смешанное.
Отложение жировой клетчатки в абдоминальной области (яблочный или верхний тип ожирения) больше связано с заболеваемо-
 стью
и смертностью, чем гиноидный или нижний тип ожирения, и даже больше,
чем степень ожирения. Большое количество абдоминального жира
способствует развитию дислипидемии, сахарного диабета,
сердечно-сосудистых заболеваний, у женщин - возникновению саркомы. Эта
зависимость не связана с общим содержанием жира в организме. При
одинаковом ИМТ абдоминальное ожирение имеет более высокий риск развития
сопутствующих заболеваний, чем ожирение по нижнему типу, что увеличивает
смертность у людей.
стью
и смертностью, чем гиноидный или нижний тип ожирения, и даже больше,
чем степень ожирения. Большое количество абдоминального жира
способствует развитию дислипидемии, сахарного диабета,
сердечно-сосудистых заболеваний, у женщин - возникновению саркомы. Эта
зависимость не связана с общим содержанием жира в организме. При
одинаковом ИМТ абдоминальное ожирение имеет более высокий риск развития
сопутствующих заболеваний, чем ожирение по нижнему типу, что увеличивает
смертность у людей.
По этиологии ожирение классифицируют на экзогенно-конституциональное, гипоталамическое, гормональное (эндокринное).
Экзогенно-конституциональное ожирение (часто, но не всегда относится к первичной форме ожирения). Нарушение пищевого поведения (например, синдром ночной еды, повышенное потребление пищи в ответ на стресс) приводит к отложению избытка жира в организме в соответствии с формулой:
Отложение жира = Поступление энергии - Расход энергии.
Длительное повышение активности «пищевого центра» ведет к повышению аппетита (гиперфагии) и ожирению (рис. 12-35). Привычка переедать может быть приобретена в детстве. Так, установлено, что избыточное кормление ребенка первого года жизни
 Рис. 12-35. Ожирение
в связи с гиперфагией и перееданием. Матиас Галлас - полководец
Тридцатилетней войны (1618-1648). Гравюра неизвестного художника XVII в.
(из фонда кафедры патофизиологии
Рис. 12-35. Ожирение
в связи с гиперфагией и перееданием. Матиас Галлас - полководец
Тридцатилетней войны (1618-1648). Гравюра неизвестного художника XVII в.
(из фонда кафедры патофизиологии
СибГМУ)
способствует развитию гиперпластического ожирения, характеризующегося увеличением объема жировых клеток.
Гипоталамическое ожирение. Является следствием поражения области гипоталамуса. Причиной могут быть перенесенные травмы головного мозга, стойкая внутричерепная гипертензия, опухоли мозга, менингит, а также врожденные дегенеративные изменения гипоталамической области (например, синдром Фрелиха) (рис. 12-36).
Гормональное ожирение. Связано как с гипо-, так и с гиперфункцией желез внутренней секреции и развивается при гипотиреозе, гипофункции половых желез, а также при гиперинсулинизме и гиперкортицизме. В крови таких больных повышается содержание ЛПНП и ЛПОНП, НЭЖК. При гормональном ожирении рано развивается гипертриацилглицеролемия и несколько позже - гиперхолестеринемия. Нарушению обмена липидов сопутствует изменение углеводного обмена: развивается гипергликемия, стимулирующая секрецию инсулина и его предшественника. В свою очередь, секрецию проинсулина и инсулина стимулируют НЭЖК, ЛПОНП, ЛПНП. Усиленный выброс глюкокортикоидов, стимулирующих глюконеогенез, также повышает уровень инсулина в крови.
По патогенезу различают алиментарное, метаболическое и энергетическое ожирение.
Алиментарное ожирение - развивается при чрезмерном потреблении пищи, что может быть обусловлено:
а) нарушением деятельности гипоталамического пищевого центра (абсолютная или относительная лептиновая недостаточность, длительное возбуждение вентролатеральных ядер в результате травм, кровоизлияний, воспаления в диэнцефальной области (по этиологии это экзогенно-конституциональное или гипоталамическое ожирение);
б) афферентной импульсацией при частом возбуждении вкусовых рецепторов;
в) переходом от активного к малоподвижному образу жизни. При этом в некоторых случаях сохраняется высокий уровень возбудимости пищевого центра (характерный для лиц физического труда или спортсменов), что приводит к систематическому перееданию;
г) чрезмерным растяжением стенок желудка при его переполнении. Это снижает чувствительность нервных окончаний слизи-
 стой
оболочки, и тормозящие импульсы передаются в пищевой центр только при
очень большом скоплении пищи в желудке. В результате переедание
становится постоянным и возникает ожирение;
стой
оболочки, и тормозящие импульсы передаются в пищевой центр только при
очень большом скоплении пищи в желудке. В результате переедание
становится постоянным и возникает ожирение;
д) пожилым возрастом, что объясняется несоответствием между прежним уровнем возбудимости центра голода и меньшими энергозатратами (после 25 лет основной обмен снижается в каждые последующие 10 лет примерно на 7,5%). Интересно отметить, что в глубокой старости часто развивается исхудание, поскольку угнетается активность пищевого центра и снижается переход углеводов в жиры.
Метаболическое ожирение обусловлено повышенным синтезом жира из углеводов. В обычных условиях до 30% поступающей в организм глюкозы под действием инсулина превращается в жир. При гиперфункции инсулярного аппарата этот процент возрастает. Аналогичное изменение метаболизма развивается при повышенной продукции пролактина (гормона передней доли гипофиза), глюкокортикоидов (по этиологии это гормональное ожирение).
Энергетическое ожирение обусловлено недостаточным использованием жиров в качестве источника энергии. Развивается при гиподинамии в сочетании с хорошим аппетитом, при снижении тонуса симпатической нервной системы и недостаточной продукции жиромобилизующих гормонов (СТГ, тиреоидные гормоны, катехоламины), поскольку задерживается выход жира из депо и использование его в качестве энергетического субстрата (по этиологии соответствует экзогенно-конституциональному или гормональному ожирению).
Последствия ожирения. При ожирении постепенно изменяется белковый обмен, который характеризуется снижением уровня общего белка крови преимущественно за счет уменьшения концентрации альбуминов, увеличением содержания фибриногена, продуктов деградации фибрина, снижением уровня гепарина. Следствием этого является нарушение транспорта НЭЖК и других липидов, снижение фибринолитической активности и повышение тромбогенных свойств крови, возникновение тромбоэмболических осложнений. Эти изменения являются факторами риска атеросклероза, ишемической болезни сердца, инсульта, гипертонической болезни. Возникают нарушения функций ЦНС: отмечаются утомляемость, сонливость, ухудшение памяти; развивается преждевременное старение, возникают изменения во внутренних
 Рис. 12-37. Причины
жировой инфильтрации печени: 1 - усиленный выход неэтерифицированных
жирных кислот из жировой ткани; 2 - интенсивное длительное поступление
хиломикронов из кишечника в кровь, а затем в печень; 3 - задержка
окисления жирных кислот в печени до кетоновых тел; 4 - задержка выхода
из печени пре-β- и β-липопротеинов. Затемненные прямоугольники - места
нарушения процесса
Рис. 12-37. Причины
жировой инфильтрации печени: 1 - усиленный выход неэтерифицированных
жирных кислот из жировой ткани; 2 - интенсивное длительное поступление
хиломикронов из кишечника в кровь, а затем в печень; 3 - задержка
окисления жирных кислот в печени до кетоновых тел; 4 - задержка выхода
из печени пре-β- и β-липопротеинов. Затемненные прямоугольники - места
нарушения процесса
органах, например жировая инфильтрация (ожирение или жировая трансформация) печени (рис. 12-37).
При первичном ожирении многие из расстройств метаболизма после нормализации веса корректируются (уменьшается или совсем проходит инсулинорезистентность, гипер- и дислипопротеинемия). Тем не менее у больного сохраняется лептиновая недостаточность, повышена активность липопротеиновой липазы жировой ткани, снижена реакция центров насыщения на серотонин, а адипоцитов - на β-адреномиметики, нарушена рецепция инсулина в гипоталамусе, а при гиперпластическом и смешанном ожирении увеличено число адипоцитов и т.д.
При быстрой нормализации веса снижается продукция тиреотропина, ухудшается холодовая адаптация. При дальнейшем падении массы тела еще больше снижается основной обмен. Отмечается тенденция к лейкопении, брадикардии и гипотонии, снижается иммунитет. У женщин возможно нарушение овариально-менструального цикла, которое связано со снижением эстрогенпродуцирующей функции адипоцитов. Многие похудевшие пациенты испытывают дисфорию, отмечаются обсессивные неврозы в связи с понижением выработки опиатных пептидов. Не-
которые психосоматические особенности похудевших пациентов с первичным ожирением напоминают таковые при психогенной анорексии. При голодании, диетах отмечается недостаток выделения серотонина, норадреналина, β-эндорфина и других биологически активных веществ в кровь. Снижение уровня серотонина субъективно воспринимается организмом человека как состояние депрессии, уменьшение концентрации норадреналина - упадка сил, β-эндорфина - неудовольствия, дискомфорта. Напротив, выделение норадреналина после еды вызывает чувство прилива сил, энергии, увеличивает уровень основного обмена. У людей с нарушениями центральной серотонинергической системы особенно сильны негативные реакции на голод, выражающиеся в снижении продукции серотонина. Даже при незначительном голодании у них развивается выраженная депрессия.
Адекватное лечение больного ожирением возможно лишь под наблюдением врача и не должно быть только симптоматическим, т. е. сводиться к диетотерапии и лечебной гимнастике. После открытия лептина большие надежды связывали с его применением для лечения лептиновой недостаточности при первичном ожирении.
12.5.5. Ожирение и жировая инфильтрация печени
При нарушении расщепления и выведения жиров из клетки (гепатоцита) говорят о ее жировом пропитывании (инфильтрации). Сочетание инфильтрации с нарушением цитоплазматической структуры клетки и ее белковых компонентов называется жировой дистрофией (рис. 12-38). Причинами жировой инфильтрации печени являются: сахарный диабет; ожирение; гиперлипопротеинемия; алкоголизм; отравление фосфором, мышьяком, хлороформом и другими ядами гепатотропного действия; голодание и гиповитаминозы; инфекции и интоксикации; длительное стрессорное воздействие; недостаток в пище холина, метионина и других липотропных факторов, снижение синтеза липокаина в мелких протоках поджелудочной железы; беременность; наследственные дефекты окисления жирных кислот.
Патогенез ожирения печени связан с увеличением поступления липидов в гепатоциты и снижением их утилизации в результате торможения окисления свободных жирных кислот, нарушением образования ЛПОНП и их секреции в кровь. Например, гепато-
 Рис. 12-38. Компрессия
синусоидов и междольковых артерий при выраженной жировой инфильтрации
печени (ПВ - портальная вена; Ж - жиры в гепатоцитах): А - нормальная
печень; Б - гепатоциты, перегруженные липидами, это может сопровождаться
повышением давления в портальной вене (портальная гипертензия). При
нарушении междолькового кровотока в центре дольки возникают гипоксия,
клеточная атрофия и происходит гибель клеток
Рис. 12-38. Компрессия
синусоидов и междольковых артерий при выраженной жировой инфильтрации
печени (ПВ - портальная вена; Ж - жиры в гепатоцитах): А - нормальная
печень; Б - гепатоциты, перегруженные липидами, это может сопровождаться
повышением давления в портальной вене (портальная гипертензия). При
нарушении междолькового кровотока в центре дольки возникают гипоксия,
клеточная атрофия и происходит гибель клеток
тропные яды угнетают окисление СЖК в митохондриях печени, нарушают образование ЛПОНП, образуют активные кислородные радикалы, повреждающие гепатоциты.
Ожирение печени может заканчиваться гибелью гепатоцитов и формированием фиброза и цирроза органа. В то же время следует знать, что в принципе это процесс обратимый и в некоторых случаях он может протекать бессимптомно. Однако чаще при ожирении печени выявляются патологические печеночные пробы (см. главу 18), гиперкетонемия и ацидоз (в крови обнаруживаются ацетон,
ацетоуксусная и β-оксимасляная кислоты), появляются признаки печеночно-клеточной недостаточности и энцефалопатии.
12.5.6. Нарушение обмена липидов и ненасыщенных жирных кислот
Жирные кислоты окисляются в процессе β-окисления, образуя ацетил-КоА. В печени из двух молекул ацетил-КоА образуется ацетоацетил-КоА, который затем взаимодействует еще с одной молекулой ацетил-КоА. Образовавшийся β-окси-β-метилглутарилКоА способен под действием фермента оксиметилглутарил-КоАлиазы расщепляться на ацетоацетат и ацетил-КоА. При декарбоксилировании ацетоуксусной кислоты (ацетоацетат) образуется ацетон, при ее восстановлении - D-β-оксимасляная кислота (D-β- оксибутират). Существует второй путь синтеза кетоновых тел: при отщеплении КоА от ацетоацетил-КоА образуется свободная ацетоуксусная кислота. Процесс катализируется ферментом деацилазой, однако его активность в печени низкая, следовательно, этот путь не имеет существенного значения.
При жировой инфильтрации печени отмечается избыточное поступление НЭЖК, задержка их окисления и выведения липопротеинов из печени, происходит накопление кетоновых тел в крови (гиперкетонемия) и в моче (кетонурия). Возможно изменение рН крови.
Гиперкетонемия со сдвигом рН крови в кислую сторону (ацидоз) возможна при угнетении цикла Кребса (цикла трикарбоновых кислот), в котором происходит окисление кетоновых тел. Это может иметь место при голодании, сахарном диабете, стрессе различной этиологии, тяжелой мышечной работе. Однако следует помнить, что в экстремальных ситуациях из кетоновых тел путем глюконеогенеза может синтезироваться глюкоза, служащая источником энергии для работы центральной нервной системы.
При метаболизме липидов образуются полиненасыщенные высшие жирные кислоты (линолевая, линоленовая, арахидоновая). Они являются регуляторами жизненно важных функций и процессов гомеостаза: контролируют течение иммунных реакций, физиологических родов, репаративных процессов. Во время метаболизма арахидоновой кислоты по циклооксигеназному пути в организме образуются простагландины, простациклины, тромбоксаны, а при работе липооксигеназного метаболического пути из арахидоновой кислоты образуются лейкотриены (см. главу 10).
Таким образом, биологически активные вещества, являющиеся продуктами полиненасыщенных жирных кислот, в оптимальных условиях сохраняют гомеостаз организма. В случае изменения их количественного соотношения могут развиваться различные виды патологии.
12.5.7. Нарушение обмена фосфолипидов
Описаны некоторые наследственные заболевания и патологические состояния, связанные с избыточным отложением в тканях фосфолипидов.
Так, при болезни Гоше цереброзиды откладываются в макрофагальных клетках селезенки, печени, лимфатических узлах и в костном мозгу. При болезни Нимана-Пика в клетках различных органов происходит отложение фосфатида сфингомиелина. При амавротической (от греч. amauros - темный, слепой) семейной идиотии липиды отлагаются в нервных клетках, что сопровождается атрофией зрительных нервов и слабоумием.
12.5.8. Нарушение обмена холестерина
Холестерин1 - производное циклопентана и гидратированного фенантрена. Его название происходит от греческих слов «желчь» и «твердый», поскольку он впервые описан в XVIII столетии как компонент желчных камней.
1 Общее содержание холестерина в организме человека составляет от 100 до 300 г, при этом преобладает свободный холестерин (его почти в 3 раза больше, чем эфиров холестерина). Холестерин поступает в организм с пищей (яичный желток, печень, мясо, сливочное масло, сметана и сливки). Существует также эндогенный синтез холестерина в печени из ацетил-КоА. Кроме того, печень - это единственный орган, где образуются эфиры холестерина, поэтому снижение уровня этерифицированного холестерина служит одним из показателей недостаточности функции печени (см. главу 18). Суточное потребление холестерина варьирует от 0,2 до 0,5 г, в самом организме образуется около 1 г в день. Роль холестерина в организме огромна, он входит в состав клеточных мембран, изменяя их жидкие свойства и проницаемость, влияет на активность мембранных ферментов, может стимулировать пролиферацию способных к делению клеток (при его избытке в мембране). Холестерин является предшественником желчных кислот и стероидных гормонов: половых, глюкокортикоидов, минералкортикоидов, а также витамина D. Выведение холестерина из организма осуществляется несколькими путями - около 0,5 г холестерина в сутки превращается в желчные кислоты и удаляется с желчью через кишечник, примерно такое же его количество теряется ежесуточно с фекалиями (копростерин); около 0,1 г холестерина выделяют сальные железы.
В плазме крови здорового человека содержится 5,2-6,2 ммоль/л холестерина. Патогенными для организма являются как избыток, так и недостаток холестерина.
К возникновению гиперхолестеринемии могут привести:
1) возбуждение симпатической нервной системы (стресс), что способствует усиленной мобилизации жира из депо и синтезу эндогенного холестерина;
2) нарушение ресинтеза жирных кислот из ацетил-КоА (при сахарном диабете);
3) нарушение выведения холестерина из организма при угнетении перистальтики кишечника, дискинезии желчных путей, при механической желтухе;
4) эндокринные заболевания, нарушающие обмен липидов: гипотиреоз, гиперкортицизм;
5) беременность;
6) нефротический синдром (нарушения липидного обмена и снижение содержания альбуминов);
7) гиповитаминоз С, гипоксия, поскольку распад холестерина требует достаточного количества АТФ;
8) повышенное поступление с пищей (однако в этом случае угнетается синтез эндогенного холестерина);
9) усиленное поступление в организм животных жиров и рафинированных углеводов (повышает синтез эндогенного холестерина);
10) наследственно обусловленные дефекты ферментов обмена липидов (в том числе холестерина).
К последствиям гиперхолестеринемии следует отнести развитие атеросклероза, ксантоматоза, холестеатоза (отложение холестерина и его эфиров в паренхиматозных органах с последующим развитием цирроза), ожирения, ишемической болезни сердца, рассеянного склероза (наследственные формы накопления холестерина), ретинопатии и др.
Среди причин гипохолестеринемии следует указать на:
1) наследственно обусловленные α-β-липопротеинемии и ан- α-липопротеинемии;
2) заболевания печени с утратой способности к синтезу холестерина и его эфиров;
3) гипертиреоз;
4) неполное голодание (сниженное поступление продуктов, богатых холестерином, животных жиров, рафинированных углеводов);
5) некоторые виды анемий;
6) усиление выведения ХН при поносах. Последствиями гипохолестеринемии являются: 1) нарушение
барьерной функции клеточных мембран, повышение ее проницаемости и цитолиз (также гемолиз эритроцитов); 2) неврологические нарушения, связанные с нарушением структуры миелиновых нервных волокон и проведения нервного импульса (атаксия, гипорефлексия, парестезии); 3) снижение образования желчных кислот и, следовательно, нарушение пищеварения в кишечнике (потеря жиров и жирорастворимых витаминов); 4) гиповитаминоз D и соответствующие ему изменения; 5) гипопродукция стероидных гормонов.
Нарушение регуляции скорости биосинтеза холестерина. В клетке липопротеины расщепляются под влиянием ферментов лизосом с освобождением неэтерифицированного холестерина (рис. 12-39). По мере накопления холестерина в клетке ограничивается скорость его образования, угнетается активность гидроксиметилглютарилКоА-редуктазы (ГМГ-КоА-редуктазы), которая является ключевым ферментом, регулирующим скорость биосинтеза холестерина. Этот процесс возможен во всех клетках, кроме эритроцитов. Такой контроль предупреждает развитие атеросклероза. При недостаточности ЛПНП-рецепторов в тканях повышается уровень ЛПНП в крови, продлевается время их циркуляции, что способствует образованию модифицированных форм ЛПНП, обладающих высокой атерогенностью при неспецифическом эндоцитозе (избыточном неконтролируемом поступлении модифицированных ЛПНП в сосудистую стенку). Развивается атеросклероз.
Таким образом, бесконтрольное поступление липопротеинов в стенку сосудов, освобождение холестерина из молекулы ЛПНП и нарушение выведения его из клетки сосудистой мембраны, а также задержка утилизации холестерина в печени ведут к возникновению атеросклероза и ишемической болезни сердца (см. главу 15).
12.6. ПАТОФИЗИОЛОГИЯ БЕЛКОВОГО ОБМЕНА
Белки наряду с нуклеиновыми кислотами, при участии которых они образуются, являются основой всех жизненных процессов. Это важнейший структурный материал, необходимый для построения клеток и межклеточного вещества; белки входят как
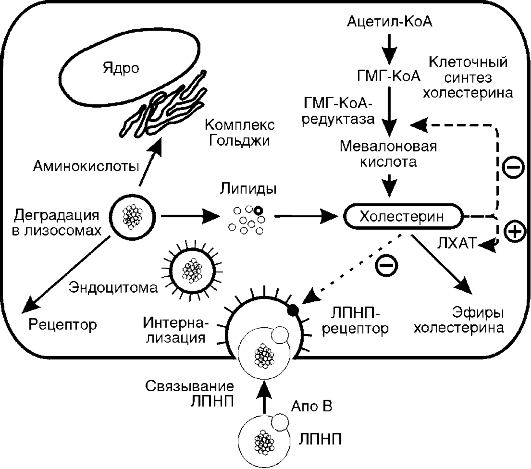 Рис. 12-39. Схема
внутриклеточного метаболизма холестерина. Липопротеины низкой плотности
(ЛПНП) могут захватываться клетками в области покрытых кластрином ямок
через связывание со специфическим ЛПНПрецептором, в результате
образуется эндоцитома, которая интернализуется внутрь клетки и
деградирует в лизосомах. Рецептор вновь способен вернуться на клеточную
мембрану. Белковая часть ЛПНП используется для внутриклеточного синтеза,
холестерин включается в клеточный метаболизм. Повышение уровня
свободного холестерина в клетке приводит к подавлению активности
ключевого фермента внутриклеточного синтеза холестерина
гидроксиметилглутарил-КоА (ГМГ-КоА-редуктазы), а также к увеличению
скорости образования эфиров холестерина с участием ЛХАТ
(лецитинхолестеринацетилтрансфераза)
Рис. 12-39. Схема
внутриклеточного метаболизма холестерина. Липопротеины низкой плотности
(ЛПНП) могут захватываться клетками в области покрытых кластрином ямок
через связывание со специфическим ЛПНПрецептором, в результате
образуется эндоцитома, которая интернализуется внутрь клетки и
деградирует в лизосомах. Рецептор вновь способен вернуться на клеточную
мембрану. Белковая часть ЛПНП используется для внутриклеточного синтеза,
холестерин включается в клеточный метаболизм. Повышение уровня
свободного холестерина в клетке приводит к подавлению активности
ключевого фермента внутриклеточного синтеза холестерина
гидроксиметилглутарил-КоА (ГМГ-КоА-редуктазы), а также к увеличению
скорости образования эфиров холестерина с участием ЛХАТ
(лецитинхолестеринацетилтрансфераза)
обязательная часть в состав всех ферментов, управляющих обменными процессами; значительная часть гормонов также являются белками (полипептидами). Белки играют важную роль в транспорте с током крови липидов, жирных кислот, гормонов, витаминов, неконъюгированного билирубина, микроэлементов и других веществ; при их участии осуществляются свертывание крови, иммунные реакции, поддержание кислотно-щелочного равновесия;
они создают онкотическое давление плазмы крови, что играет важную роль в осуществлении процессов микроциркуляции и водного обмена. Белки являются источником энергии - при полном распаде и окислении 1 г белка освобождается 4,1-4,3 ккал тепла, однако эта функция белков легко возмещается окислением углеводов и жиров.
Нарушение обмена белков может происходить на различных этапах:
1) при расщеплении и усвоении белков пищи;
2) их внутриклеточном синтезе и распаде;
3) межуточном обмене аминокислот;
4) образовании конечных продуктов и их экскреции.
12.6.1. Нарушение расщепления белков пищи и усвоения образующихся аминокислот
В желудке и кишечнике происходит гидролитическое расщепление белков пищи до пептидов и аминокислот под влиянием ферментов желудочного сока (пепсин, гастриксин), панкреатического (трипсин, химотрипсин, эластаза и карбоксипептидазы) и кишечного (аминопептидаза, дипептидазы) соков. Образующиеся при расщеплении белков аминокислоты всасываются стенкой тонкого кишечника в кровь и потребляются клетками различных органов. Нарушение этих процессов имеет место при заболеваниях желудка (воспалительные и язвенные процессы, опухоли), поджелудочной железы (панкреатиты, закупорка протоков, рак), тонкого кишечника (энтериты, диарея, атрофия слизистой, состояния мальдигестии и мальабсорбции). Такие обширные оперативные вмешательства, как удаление желудка или значительной части тонкого кишечника, также сопровождаются нарушением расщепления и усвоения белков пищи. Усвоение пищевых белков нарушается при лихорадке вследствие снижения секреции пищеварительных ферментов.
Недостаточность усвоения белков пищи сопровождается дефицитом аминокислот и нарушением синтеза собственных белков. Недостаток пищевых белков не может быть полностью компенсирован избыточным введением и усвоением каких-либо других веществ, так как белки являются основным источником азота для организма.
12.6.2. Нарушение процессов эндогенного синтеза и распада белка
Синтез белков происходит в организме непрерывно на протяжении всей жизни, но наиболее интенсивно совершается в период внутриутробного развития, в детском и юношеском возрасте.
Различают следующие виды синтеза белка в зависимости от его назначения:
• регенерационный, связанный с процессами физиологической и репаративной регенерации;
• синтез роста, сопровождающийся увеличением массы и размеров тела;
• стабилизирующий, связанный с возмещением структурных белков, утраченных в процессе диссимиляции, способствующий поддержанию структурной целостности организма;
• функциональный, связанный со специфической деятельностью различных органов (синтез гемоглобина, белков плазмы крови, антител, гормонов и ферментов).
Причинами нарушения синтеза белка являются:
• отсутствие достаточного количества аминокислот;
• дефицит энергии в клетках;
• расстройства нейроэндокринной регуляции;
• нарушение процессов транскрипции или трансляции информации о структуре того или иного белка, закодированной в геноме клетки.
Наиболее частой причиной нарушения синтеза белка является недостаток аминокислот в организме вследствие:
1) расстройств пищеварения и всасывания;
2) пониженного содержания белка в пище;
3) питания неполноценными белками, в которых отсутствуют или имеются в незначительном количестве незаменимые аминокислоты, не синтезирующиеся в организме (табл. 12-7).
Полный набор незаменимых аминокислот имеется в большинстве белков животного происхождения, тогда как растительные белки могут не содержать некоторые из них или содержат недостаточно (например, в белках кукурузы мало триптофана). Недостаток в организме хотя бы одной из незаменимых аминокислот (табл. 12-8) ведет к снижению синтеза того или иного белка даже при изобилии остальных.
Таблица 12-7. Незаменимые для человека аминокислоты (по И.П. Ашмарину, Е.П. Каразеевой, 1997)
Аминокислота | Примерная оптимальная норма, г/сутки | Примечание |
Триптофан | 1 | Наиболее дефицитные в растительной пище и в соединительной ткани животных |
Лизин | 3-5 | |
Метионин | 2-4 | |
Лейцин | 4-6 | |
Изолейцин | 3-4 | |
Валин | 3-4 | |
Фенилаланин | 2-4 | |
Треонин | 2-3 | |
Гистидин | 1,5-2 | Особенно важен для детей |
Аргинин | 6 | Незаменим для новорожденных |
Таблица 12-8. Проявления дефицита незаменимых аминокислот
Гистидин | Дерматит, анемия, снижение продукции гистамина, ухудшение умственной деятельности |
Изолейцин | Поражение почек, щитовидной железы, анемия, гипопротеинемия |
Лейцин | Поражение почек, щитовидной железы, гипопротеинемия |
Метионин (с цистеином) | Ожирение, некрозы печени, ускорение атерогенеза, надпочечниковая недостаточность, геморрагии почек, дефицит холина и адреналина |
Лизин | Анемия, миодистрофия, остеопороз, поражение печени и легких, головная боль, повышенная чувствительность к шуму |
Фенилаланин с тирозином | Гипотиреоз, недостаточность мозгового вещества надпочечников |
Аргинин | Нарушение сперматогенеза, цикла мочевины |
Окончание табл. 12-8
Триптофан | Пеллагра, катаракта, помутнение роговицы, анемия, облысение, гипопротеинемия, атрофия семенников, рассасывание плода |
Валин | Расстройство координации движений и гиперстезии |
Треонин | Отеки и падение массы тела |
Дефицит заменимых аминокислот в пище реже приводит к понижению синтеза белка, так как они могут образовываться в организме из кетокислот, являющихся продуктами метаболизма углеводов, жиров и белков.
Недостаток кетокислот возникает при сахарном диабете, нарушении процессов дезаминирования и трансаминирования аминокислот (гиповитаминоз В6).
Недостаток источников энергии имеет место при гипоксии, действии разобщающих факторов, сахарном диабете, гиповитаминозе В1, дефиците никотиновой кислоты и др. Синтез белка - энергозависимый процесс. Энергия макроэргов АТФ и ГТФ необходима для активации аминокислот и образования пептидных связей (21,9 кал на каждую пептидную связь).
Расстройства нейроэндокринной регуляции синтеза и расщепления белка. Нервная система оказывает на белковый обмен прямое и косвенное действие. При выпадении нервных влияний возникает расстройство трофики клетки1. Нарушения нервной трофики являются важным звеном патогенеза любого заболевания. Денервация тканей вызывает: прекращение их стимуляции вследствие нарушения выделения нейромедиаторов; нарушение секреции или действия комедиаторов, обеспечивающих регуляцию рецепторных, мембранных и метаболических процессов; нарушение выделения и действия трофогенов2. Подтверждением прямого трофического
1 Комплекс процессов, обеспечивающих жизнедеятельность клетки и поддержание генетически заложенных свойств. Нервные волокна регулируют в иннервируемых тканях не только кровообращение, но также метаболические, энергетические и пластические процессы в соответствии с текущими потребностями организма.
2 Трофогены - вещества преимущественно белковой природы, способствующие росту, дифференцировке и жизнедеятельности клеток, а также сохранению их гомеостаза. Они образуются в клетках периферических органов, в плазме крови; в нейронах, откуда поступают при помощи аксонального транспорта в иннервируемые ткани; в роли трофогенов могут выступать и анаболические гормоны.
влияния нервной системы на метаболизм белков в клетках является развитие атрофических и дистрофических изменений в денервированных тканях. Установлено, что в денервированных тканях процесс распада белка превалирует над синтезом. Косвенное влияние нервной системы на белковый обмен осуществляется путем изменения функции эндокринных желез.
Действие гормонов может быть анаболическим (усиливающим синтез белка) и катаболическим (повышающим распад белка в тканях).
Синтез белка увеличивается под действием:
• инсулина (обеспечивает активный транспорт в клетки многих аминокислот - особенно валина, лейцина, изолейцина; повышает скорость транскрипции ДНК в ядре; стимулирует сборку рибосом и трансляцию; тормозит использование аминокислот в глюконеогенезе, усиливает митотическую активность инсулинзависимых тканей, повышая синтез ДНК и РНК);
• соматотропного гормона (СТГ; ростовой эффект опосредуют соматомедины, вырабатываемые под его влиянием в печени). Другое название соматомединов - инсулиноподобные ростовые факторы - появилось в связи с их способностью снижать уровень глюкозы в крови. Основной из них - соматомедин С, который во всех клетках тела повышает скорость синтеза белка. Так стимулируется образование хрящевой и мышечной ткани. В хондроцитах имеются рецепторы и к самому гормону роста, что доказывает его прямое влияние на хрящевую и костную ткань;
• тиреоидных гормонов в физиологических дозах: трийодтиронин, связываясь с рецепторами в ядре клетки, действует на геном и вызывает усиление транскрипции и трансляции. Вследствие этого стимулируется синтез белка во всех клетках тела. Кроме того, тиреоидные гормоны стимулируют действие
СТГ;
• половых гормонов, оказывающих СТГ-зависимый анаболический эффект на синтез белка; андрогены стимулируют образование белков в мужских половых органах, мышцах, скелете, коже и ее производных, в меньшей степени - в почках и мозгу; действие эстрогенов направлено в основном на молочные железы и женские половые органы. Следует отметить, что анаболический эффект половых гормонов не касается синтеза белка в печени.
Распад белка повышается под влиянием:
• тиреоидных гормонов при повышенной их продукции (гипертиреоз);
• глюкагона (уменьшает поглощение аминокислот и повышает распад белков в мышцах; в печени активирует протеолиз, а также стимулирует глюконеогенез и кетогенез из аминокислот; тормозит анаболический эффект СТГ);
• катехоламинов (способствуют распаду мышечных белков с мобилизацией аминокислот и использованием их печенью);
• глюкокортикоидов (усиливают синтез белков и нуклеиновых кислот в печени и повышают распад белков в мышцах, коже, костях, лимфоидной и жировой ткани с высвобождением аминокислот и вовлечением их в глюконеогенез. Кроме того, они угнетают транспорт аминокислот в мышечные клетки, снижая синтез белка).
Анаболическое действие гормонов осуществляется в основном путем активации определенных генов и усилением образования различных видов РНК (информационная, транспортная, рибосомальная), что ускоряет синтез белков; механизм катаболического действия гормонов связан с повышением активности тканевых протеиназ.
Снижение синтеза гормонов анаболического действия, таких как СТГ и тиреоидные гормоны, в детском возрасте ведет к задержке роста.
Инактивацию тех или иных факторов, участвующих в биосинтезе белка, могут вызвать некоторые лекарственные препараты (например, антибиотики) и микробные токсины. Известно, что дифтерийный токсин тормозит присоединение аминокислот к синтезируемой полипептидной цепи; этот эффект устраняется анатоксином.
Стимулирующее или угнетающее действие на синтез белка могут оказать изменения концентрации различных ионов (прежде всего Mg2+), уменьшение или увеличение ионной силы.
Последствия нарушения общего синтеза белка
Длительное и значительное понижение синтеза белка приводит к развитию дистрофических и атрофических нарушений в различных органах и тканях вследствие недостаточного обновления структурных белков. Замедляются процессы регенерации. В детском возрасте тормозятся рост, физическое и умственное разви-
тие. Снижается синтез различных ферментов и гормонов (СТГ, антидиуретический и тиреоидный гормоны, инсулин и др.), что приводит к эндокринопатиям, нарушению других видов обмена (углеводного, водно-солевого, основного). Понижается содержание белков в сыворотке крови в связи со снижением их синтеза в гепатоцитах. Вследствие этого в крови уменьшается онкотическое давление, что способствует развитию отеков. Уменьшается продукция антител и других защитных белков и, как следствие, снижается иммунологическая реактивность организма. В наиболее выраженной степени эти расстройства возникают в результате длительного нарушения усвоения белков пищи при различных хронических заболеваниях органов пищеварения, а также при длительном белковом голодании, особенно если оно сочетается с дефицитом жиров и углеводов. В последнем случае повышается использование белка в качестве источника энергии.
Причины и механизм нарушения синтеза отдельных белков. В большинстве случаев эти нарушения имеют наследственную природу. В основе их лежит отсутствие в клетках информационной РНК (иРНК), специфической матрицы для синтеза какого-либо определенного белка, или нарушение ее структуры вследствие изменения структуры гена, на котором она синтезируется. Генетические нарушения, например замена или потеря одного нуклеотида в структурном гене, приводят к синтезу измененного белка, нередко лишенного биологической активности.
К образованию аномальных белков могут привести отклонения от нормы в структуре иРНК, мутации транспортной РНК (тРНК), вследствие чего к ней присоединяется несоответствующая аминокислота, которая и будет включаться в полипептидную цепь при ее сборке (например, при образовании гемоглобина).
Процесс трансляции является сложным, совершающимся при участии ряда ферментов, и нарушение функции какого-либо из них может привести к тому, что та или другая иРНК не передаст закодированную в ней информацию.
Нарушение синтеза отдельных белков-ферментов или структурных белков лежит в основе различных наследственных болезней (гемоглобинозы, альбинизм, фенилкетонурия, галактоземия, гемофилия и многие другие - см. раздел 5.1). Нарушение какой-либо ферментативной функции чаще всего связано не с отсутствием соответствующего белка - фермента, а с образованием патологически измененного неактивного продукта.
Причины, механизм и последствия повышенного распада тканевых белков. Наряду с синтезом в клетках организма постоянно происходит деградация белков под действием протеиназ. Обновление белков за сутки у взрослого человека составляет 1-2% общего количества белка в организме и связано преимущественно с деградацией мышечных белков, при этом 75-80% освободившихся аминокислот вновь используется для синтеза.
Азотистый баланс - интегральный показатель общего уровня белкового обмена, это суточная разница между поступающим и выделяющимся из организма азотом,
У здорового взрослого человека процессы распада и синтеза белка уравновешены, т.е. имеется азотистое равновесие. При этом суточная деградация белка составляет 30-40 г.
Азотистый баланс может быть положительным или отрицательным.
Положительный азотистый баланс: поступление в организм азота превышает его выведение, т.е. синтез белка преобладает над его распадом. Отмечается при регенерации тканей, в период выздоровления после тяжелых болезней, при беременности, в детском возрасте, при гиперпродукции СТГ, при полицитемии.
При патологии распад белка может превалировать над синтезом и азота поступает в организм меньше, чем выделяется (отрицательный азотистый баланс).
Причинами отрицательного азотистого баланса являются: инфекционная лихорадка; обширные травмы, ожоги и воспалительные процессы; прогрессирующий злокачественный опухолевый рост, эндокринные заболевания (сахарный диабет, гипертиреоз, гиперкортицизм); тяжелый эмоциональный стресс; обезвоживание, белковое голодание, лучевая болезнь; гиповитаминозы А, С, В1, В2, В6, РР, дефицит фолиевой кислоты. В механизме усиленного распада белков при многих из перечисленных состояний лежит повышенная продукция катаболических гормонов.
Следствием отрицательного азотистого баланса являются дистрофические изменения в органах, похудание, в детском возрасте - задержка роста и умственного развития.
12.6.3. Нарушение обмена аминокислот
Аминокислоты поступают в кровь и ткани из пищеварительного тракта; кроме того, они образуются при деструкции тканевых белков под действием внутриклеточных катепсинов (протеиназ).
Основная часть аминокислот используется в организме в качестве строительных блоков при синтезе белков. Кроме того, аминокислоты используются для синтеза пуриновых и пиримидиновых оснований, гормонов, гема, различных биологически активных пептидов (интерлейкины, факторы роста и т.д.), меланина, глюкозы, жирных кислот и ряда других веществ. Глицин и глутамат играют роль нейромедиаторов в ЦНС. Аминокислоты, не использованные для вышеупомянутых целей, подвергаются окислению до СО2 и Н2О с освобождением энергии.
В норме при окислении аминокислот освобождается 10-15% образующейся в организме энергии. Окисление аминокислот усиливается при избыточном поступлении их в организм, при голодании, сахарном диабете, гипертиреозе, снижении синтеза белков и некоторых других состояниях.
Катаболизм большинства аминокислот начинается с отщепления от них аминогруппы, что происходит в 2-х типах реакций: трансаминирования и дезаминирования.
Трансаминирование - перенос аминогруппы аминокислоты на α-кетокислоту, в результате образуются новая кетокислота и новая аминокислота.
Процесс трансаминирования легко обратим и катализируется трансаминазами, коферментом которых является пиридоксальфосфат - производное витамина В6 (служит переносчиком аминогрупп). Чаще всего в реакциях трансаминирования участвуют аминокислоты, содержание которых в тканях выше остальных, - глутамат, аланин, аспартат и соответствующие им кетокислоты - α-кетоглутарат, пируват, оксалацетат. Основным акцептором аминогрупп служит α-кетоглутарат. Принимая аминогруппу, он превращается в глутамат - донор аминогрупп, способный передавать их любым α-кетокислотам для образования новых аминокислот (рис. 12-40). Кетокислоты, образующиеся при трансаминировании (например, пировиноградная), также могут использоваться для синтеза глюкозы или окислиться до СО2 и Н2О подобно глюкозе и жирным кислотам. Трансаминирование - начальный этап катаболизма аминокислот, в результате которого аминный азот перераспределяется в тканях организма. Реакции трансаминирования играют роль в превращении аминокислот в кетокислоты или в образовании из кетокислот ряда заменимых аминокислот в том случае, если организм испытывает в них потребность. При трансаминировании общее количество аминокислот в клетке не меняется.
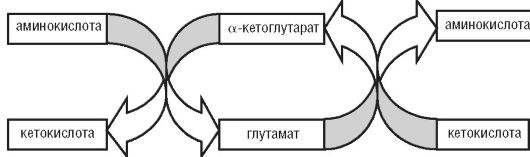 Рис. 12-40. Трансаминирование
Рис. 12-40. Трансаминирование
Нарушение процесса трансаминирования в целом организме происходит при гиповитаминозе В6, при недостатке α-кетокислот (голодание, сахарный диабет). Нарушение трансаминирования в отдельных органах, например в печени, происходит при некрозе клеток, что сопровождается выходом трансаминаз в кровь. Такое же явление имеет место при инфаркте миокарда. В поврежденных клетках может быть нарушен синтез белковой части трансаминаз.
Дезаминирование - реакция отщепления α-аминогруппы от аминокислоты, в результате чего образуется соответствующая α-кетокислота, которая может использоваться в реакциях трансаминирования, и выделяется молекула аммиака. Процесс окислительного дезаминирования снижается в связи с ослаблением трансаминирования, при гипоксии, гиповитаминозах В2, РР, С, белковом голодании.
Нарушение процессов трансаминирования и окислительного дезаминирования аминокислот ограничивает их использование для синтеза глюкозы, жирных кислот, заменимых аминокислот, а также их окисление с освобождением энергии. При этом повышается содержание свободных аминокислот в сыворотке крови и в моче (гипераминоацидемия и гипераминоацидурия), снижается синтез мочевины. Такие нарушения особенно выражены при обширных повреждениях гепатоцитов (вирусные и токсические гепатиты и др.), так как в этих клетках метаболизм аминокислот происходит наиболее интенсивно.
Наряду с внепочечной гипераминоацидурией, обусловленной усиленным поступлением аминокислот из крови в мочу, существует почечная форма гипераминоацидурии, связанная с нарушением реабсорбции аминокислот в почечных канальцах, при этом содержание аминокислот в сыворотке крови нормально или даже пони-
жено (см. главу 19). Гипераминоацидурия (физиологическая) обнаруживается у детей раннего возраста в связи с функциональной неполноценностью (незрелостью) эпителия почечных канальцев; у беременных повышается экскреция с мочой гистидина и ряда других аминокислот.
Одним из путей метаболизма аминокислот является их декарбоксилирование, которое состоит в отщеплении от аминокислоты α-карбоксильной группы. В результате образуются СО2 и биогенные амины: гистамин - из гистидина, серотонин - из 5-окситриптофана, тирамин - из тирозина, γ-аминомасляная кислота (ГАМК) - из глутаминовой, дофамин - из диоксифенилаланина и некоторые другие.
Эти процессы необратимы и катализируются декарбоксилазами, коферментом которых является пиридоксальфосфат (витамин В6); при его дефиците образование биогенных аминов снижается. В частности, уменьшается образование ГАМК, которая является основным тормозным нейромедиатором, как следствие этого наблюдается частое развитие судорог. Биогенные амины обладают высокой физиологической активностью. Наряду с ГАМК, серотонин и дофамин являются также нейромедиаторами в ЦНС, их повышенное или пониженное содержание в ткани мозга играет роль в патогенезе некоторых форм нейропатологии (нервной депрессии, паркинсонизма, шизофрении). Повышенное образование в организме серотонина (при опухоли, развивающейся из энтерохромафинных клеток кишечника) сопровождается спазмом мускулатуры бронхов и кишечника, диареей, усилением агрегации тромбоцитов; кроме того, серотонин является мощным вазоконстриктором. Хорошо известна роль гистамина в появлении болевых ощущений, развитии воспаления и аллергических реакций.
Устранение избытка биогенных аминов происходит при участии аминооксидаз, которые катализируют превращение их в альдегиды после отщепления аминогруппы в виде NH3. Серотонин превращается в оксииндолилуксусную кислоту, которая выделяется с мочой.
Наследственные нарушения обмена некоторых аминокислот (см. раздел 5.1).
12.6.4. Нарушение конечного этапа обмена белка и аминокислот
Конечные этапы белкового и нуклеинового обмена приводят к формированию экскретируемых из организма азотистых соедине-
ний - аммиака, мочевины, мочевой кислоты, креатинина, индикана. Основным показателем выведения и образования азотистых продуктов служит уровень остаточного азота, который в норме колеблется от 14,3 до 28,6 ммоль/л и состоит на 50% из азота мочевины; другие 50% составляет резидуальный (немочевинный) азот, входящий в состав аминокислот (25%), аммиака и других азотистых соединений.
Из всех компонентов остаточного азота наиболее токсичен аммиак. Он легко проникает через биологические мембраны, в том числе через гематоэнцефалический барьер. В норме его содержание в крови невелико и не превышает 0,07 ммоль/л. Он активно продуцируется органами, где идет интенсивный обмен аминокислот (мозг, печень, желудочно-кишечный тракт, почки).
Обезвреживание аммиака, образующегося в клетках различных органов, достигается путем реакции амидирования (главным образом в нервной ткани), т.е. присоединения его к аспарагиновой или глутаминовой кислотам с образованием амидов аспарагина и глутамина; путем образования солей аммония в почках и в результате синтеза мочевины.
Образование мочевины осуществляется гепатоцитами в орнитиновом цикле и имеет большое значение, так как благодаря этому процессу обезвреживается основная часть высокотоксичного аммиака, образующегося при дезаминировании аминокислот, а также поступающего в кровь из кишечника. Экскреция мочевины из организма осуществляется с мочой (более 70%), потом (1%) и около 25% продуцируемой мочевины диффундирует в кишечник, где разлагается бактериями с образованием аммиака и снова утилизируется.
Причинами понижения синтеза мочевины являются: 1) длительное белковое голодание (недостаток ферментов); 2) заболевания печени (циррозы, острые гепатиты с повреждением большого числа гепатоцитов, отравление печеночными ядами); 3) наследственные дефекты синтеза ферментов орнитинового цикла (карбамилфосфатсинтетазы, аргининсукцинатсинтетазы и аргининсукцинатлиазы).
При нарушении синтеза мочевины количество ее в крови и моче снижается, и нарастает содержание резидуального азота (продукционная гиперазотемия). Избыток аммиака может в некоторой степени устраняться за счет повышенного образования глутамина и присоединения к α-кетоглутаровой кислоте, которая при этом превращается в глутаминовую, и ее окисление в цикле трикарбо-
новых кислот резко снижается. Вследствие этого снижается образование АТФ.
Другой причиной накопления азотистых метаболитов в крови (креатинин, мочевина) является нарушение выделительной функции почек или нарушение проходимости мочевыводящих путей. Возникающая в данном случае гиперазотемия называется ретенционной. При этом концентрация остаточного азота в крови возрастает до 140-215 ммоль/л, а содержание небелковых азотистых продуктов в моче снижается. Ретенционная гиперазотемия является одним из факторов, играющих роль в развитии уремической комы.
Может возникать смешанная (комбинированная) форма гиперазотемии, при которой повышенный распад белка в тканях сочетается с недостаточным выведением азотистых продуктов с мочой. Такое сочетание возможно при острой почечной недостаточности, развившейся на почве септического аборта, или обширном сдавлении тканей (синдром раздавливания). К комбинированной форме гиперазотемии относится гипохлоремическая гиперазотемия, возникающая при неукротимой рвоте, стенозе привратника и профузных поносах.
12.6.5. Нарушение белкового состава плазмы крови
В плазме крови человека присутствуют более 200 белков, имеющих различную концентрацию. Количественно наиболее представлен альбумин. Содержание отдельных белков изменяется при многих физиологических и патологических состояниях.
Большинство белков плазмы (табл. 12-9) представлено гликопротеинами обычно с содержанием углеводов от 10 до 25%, исключение составляет альбумин, который не гликозилирован.
С клинической точки зрения удобно разделять белки по их функции, поэтому специфический индивидуальный белок может быть отнесен более чем к одной группе.
Таблица 12-9. Белки плазмы крови
Функциональные группы белков | Пример |
Транспортные белки | Трансферрин, тироксинсвязывающий белок |
Белки острой фазы | С-реактивный белок, фибриноген |
Комплемент | С3, С4 |
Окончание табл. 12-9
Факторы свертывания | Протромбин, фактор VIII, фибриноген |
Ферменты | Амилаза, ренин, холинэстераза |
Ингибиторы протеиназ | α,-антитрипсин, антитромбин III |
Гормоны | Инсулин, глюкагон, вазопрессин |
Иммуноглобулины | IgG, IgM, |
Белки, поддерживающие онкотическое давление | Все белки, особенно альбумин |
Белки, поддерживающие буферную емкость плазмы | Все белки |
Физиологические функции белков плазмы состоят в поддержании коллоидно-осмотического давления, буферной емкости плазмы, в осуществлении транспорта и депонировании молекул липидов, продуктов метаболизма, гормонов, лекарственных веществ и микроэлементов. Белки плазмы: ферменты, иммуноглобулины, компоненты комплемента и С-реактивный белок. Равновесие между белками - прокоагулянтами и ингибиторами свертывания обеспечивает жидкое состояние крови в норме и быстрое свертывание при повреждении. При непосредственном участии белков плазмы протекают все физиологические и патофизиологические реакции в организме.
Концентрация белка в плазме зависит от скорости синтеза, скорости удаления и объема распределения. Альбумины, α-глобулины и часть β-глобулинов синтезируются в печени, γ-глобулины и часть β-глобулинов - в клетках лимфоидной ткани. Концентрация белка в плазме может быстро меняться - через 30 мин в положении стоя после длительного лежания она может увеличиться на 10-20%, после венопункции может измениться в течение нескольких минут. В обоих случаях это связано с перераспределением жидкости между внутрисосудистым пространством и интерстицием.
На концентрацию белков в плазме влияют следующие факторы:
• возраст (у недоношенных детей содержание белка в крови составляет 36-60 г/л, у новорожденных - 46-70 г/л, у детей 2-12 лет - 50-75 г/л, у взрослых - 64-83 г/л);
• пол (мужские и женские половые гормоны влияют на концентрацию α-фетопротеина, ферритина, IgM и многих других белков);
• фенотипы, связанные с расовыми различиями;
• наследственный дефицит отдельных белков;
• окружающая среда (у жителей тропиков уровень иммуноглобулинов выше, чем у живущих в зоне с холодным климатом);
• физическая нагрузка (активная физическая работа повышает концентрацию белка в крови до 10%);
• сон;
• питание;
• беременность (влияет в первую очередь на концентрацию транспортных белков);
• прием лекарственных препаратов (оральные контрацептивы, тестостерон, фенотиазины, эстрогены).
К патологическим факторам, обусловливающим изменение концентрации белка в организме, относятся:
• потеря белка через поврежденный орган (при нефротическом синдроме, клубочковой и канальцевой протеинурии, патологии кишечника);
• нарушение синтеза белка (при заболеваниях печени, почек);
• изменение объема циркулирующей крови в результате гипер-, гипогидратации или перераспределения между водными пространствами организма;
• усиление катаболизма белка (при воспалении, опухолевых заболеваниях);
• изменение скорости утилизации белка (при воспалении, патологии почек).
Только существенные изменения уровня альбумина и иммуноглобулинов влияют на концентрацию общего белка в сыворотке. Концентрация общего белка в плазме быстро снижается при увеличении проницаемости капилляров, так как белок способен быстро диффундировать в интерстициальное пространство. Это может иметь место у больных с сепсисом или генерализованным воспалением. Причины увеличения и снижения концентрации общего белка в сыворотке представлены в табл. 12-10.
Гипопротеинемия - уменьшение концентрации белков в крови. Существуют различия между абсолютной гипопротеинемией, например после увеличения выделением почками альбумина при их патологии или в результате нарушения синтеза при хроническом циррозе печени, и относительной гипопротеинемией, например в результате избыточной инфузионной терапии или значительно уменьшенном количестве мочи (олигурия, анурия).
Таблица 12-10. Клинико-диагностическое значение изменения концентрации общего белка крови
Повышение концентрации выше 85 г/л | Снижение концентрации ниже 60 г/л |
Дегидратация: недостаточное питье; избыточные потери воды при потоотделении, профузных поносах, болезни Аддисона, диабетическом кетоацидозе Увеличение содержания одного или нескольких специфических белков: острые и хронические инфекции; аутоиммунные болезни; парапротеинемические гемобластозы: миеломная болезнь; болезнь Вальденстрема; болезнь тяжелых цепей; лимфогранулематоз; саркоидоз; активный хронический гепатит; цирроз печени без выраженной печеночно-клеточной недостаточности | Пониженный синтез белка: недостаток белка в пище, голодание; мальабсорбция, энтериты, панкреатиты; болезни печени (цирроз, атрофия и др.); длительный прием кортикостероидов Увеличенные потери белка: гломерулонефрит и другая патология почек; сахарный диабет; асцит, экссудаты и транссудаты; ожоги; кровотечения Повышенный распад белка: тиреотоксикоз; длительная физическая нагрузка; продолжительная лихорадка; травмы; опухоли Гипергидратация |
Как правило, основной причиной гипопротеинемии является гипоальбуминемия. Недостаточный синтез альбумина в печени может быть связан с уменьшенным поступлением аминокислот или с повреждением гепатоцитов. Нарушение всасывания в кишечнике (синдром мальабсорбции) может быть результатом бактериальной или паразитарной (лямблиоз) инфекции, муковисцидоза, колита, дисахаридазной недостаточности, энтеропатии с потерей белков или демпинг-синдрома. Поражение гепатоцитов может иметь место при циррозе, токсикозе, атрофии, метастазировании или первичном раке печени.
Потеря белка возникает при:
• нефротическом синдроме, гломерулонефрите (80%), диабете, системной красной волчанке и других аутоиммунных заболеваниях, амилоидозе, тромбозе почечных вен;
• энтеропатиях в результате заболеваний желудка или кишечника, колита, полипов;
• поражениях кожи (ожоги, дерматоз);
• образовании экссудатов и транссудатов (перитонит, плеврит, асцит);
• коагулопатиях;
• усиленном катаболизме белков (сепсис, лихорадки, множественные поражения, злокачественные опухоли).
Гиперпротеинемия - повышение концентрации общего белка в крови. Выделяют две основные причины повышения концентрации общего белка в сыворотке крови: уменьшение объема плазмы при дегидратации и повышение содержания в плазме одного или нескольких специфических белков. В связи с этим проводятся различия между абсолютной гиперпротеинемией, например повышение концентрации иммуноглобулинов (парапротеинемия), и относительной гиперпротеинемией при дегидратации. Гиперпротеинемия не может быть результатом усиленного синтеза альбумина, поэтому гиперальбуминемия указывает на дегидратацию.
Выраженное поликлональное увеличение концентрации иммуноглобулинов наблюдается при хроническом бактериальном воспалении, обострении вирусных инфекций (в частности, ВИЧинфекции), хронических заболеваниях печени (хронический и подострый гепатит), аутоиммунных болезнях (ревматоидный артрит, дерматомиозит), саркоидозе. Заподозрить гиперпротеинемию можно при изменении скорости оседания эритроцитов (СОЭ).
Диспротеинемия означает, что имеются количественные и качественные изменения концентрации нормальных белков плазмы, например при остром воспалении, циррозе печени, болезнях почек, парапротеинемии, опухолях. Диспротеинемия может быть обусловлена увеличением или уменьшением концентрации отдельных групп белков или продукцией новых белков, которые до этого не выявлялись. Диспротеинемия определяется путем электрофореза.
Гипоальбуминемия. Возможные причины ее представлены в табл. 12-11.
Известно более 20 генетических вариантов альбумина, что никак не связано со склонностью к заболеваниям. Этот эффект обозначается как бисальбуминемия. Наследственное отсутствие альбумина - анальбуминемия - асимптоматична, может проявляться лишь определенной склонностью к отекам. В клинической практике гипоальбуминемия чаще всего является следствием по-
Таблица 12-11. Клинико-диагностическое значение изменения содержания альбумина в плазме крови
Функция | Концентрация | ||
норма повышенная сниженная | |||
Связывание и транспорт катионов (Fe2+, Cu2+, Zn2+, Ca2+), малых и больших анионов, билирубина, жирных кислот, витаминов В12, С, лекарств, гормонов щитовидной железы. Нормализация коллоидноосмотического давления. Резерв белка (аминокислот) | 37-53 г/л | Острое обезвоживание Прием анаболических стероидов | Пониженный синтез: цирроз печени, недоедание, синдром мальабсорбции, анальбуминемия Повышенный катаболизм: травма, инфекция, сепсис, лихорадка, опухоли, гипоксия, синдром Кушинга, гипертиреоз, гиперкортицизм Аномальные потери: шок, кровотечение, энтероколиты, нефротический синдром Патологическое распределение: после операционного вмешательства, при ожогах, токсикозе, асците, плеврите |
тери альбумина при нефротическом синдроме, гастроэнтерите, активации катаболизма. При ожоговой болезни гипоальбуминемия развивается вследствие потери жидкости, изменения сосудистой проницаемости, угнетения синтеза. Выраженная гипоальбуминемия наблюдается при портальном циррозе и жировой дистрофии печени, амилоидозе, кахексии, тяжелых инфекциях, панкреатите, коллагенозах.
Гиперальбуминемия может быть либо артефактом (в частности, при взятии венозной крови в момент стаза), либо результатом чрезмерного внутривенного введения альбумина при инфузиях, либо связана с дегидратацией. При некоторых патологических состояниях отмечается повышенный синтез альбумина, однако это, как правило, не приводит к гиперальбуминемии.
Гипогаммаглобулинемия может быть физиологической и встречается у новорожденных (рис. 12-41). Контакт новорожденных с антигенами стимулирует В-лимфоциты, которые начинают активно продуцировать IgM. После трансформации в плазматические
 Рис. 12-41. Изменение концентрации иммуноглобулинов в сыворотке крови у новорожденных
Рис. 12-41. Изменение концентрации иммуноглобулинов в сыворотке крови у новорожденных
клетки начинается синтез и секреция IgG и IgA, при этом одновременно снижается содержание материнских IgG, поэтому у детей уровень IgG минимален в возрасте 3 месяцев. Наиболее подвержены инфекциям две группы детей: недоношенные, поскольку материнских IgG у них меньше, чем у доношенных, и дети, у которых происходит временная задержка синтеза IgG. В этом случае требуется вмешательство, направленное на активацию синтеза IgG. Патологическая гипогаммаглобулинемия как у детей, так и у взрослых может быть и врожденной, и приобретенной. В обоих случаях это сопровождается иммунодефицитом (см. главу 7).
Гипергаммаглобулинемия возможна при повышенном синтезе антител. Нарастает содержание иммуноглобулинов всех классов, но преобладают IgG. Концентрация иммуноглобулинов увеличивается при всех бактериальных и паразитарных заболеваниях, сепсисе, рожистом воспалении, инфекционном мононуклеозе, краснухе, бруцеллезе и др. Повышение уровня IgG имеет место при аутоиммунных заболеваниях; IgA - при инфекционных по-
ражениях кожи, желудка, дыхательных путей, почек; IgM - при первичной вирусной инфекции и паразитарных инфекциях с накоплением паразита в крови (малярия).
Парапротеинемия - появление в крови нехарактерных патологических белков.
Парапротеины (моноклональные иммуноглобулины) - это иммуноглобулины или их фрагменты, вырабатываемые плазматическими клетками. Парапротеины часто не способны выполнять функцию антител, обычно они структурно однородны, т.е. молекула состоит из тяжелых или легких цепей одного типа, иногда они состоят только из отдельных легких цепей (каппа или лямбда) или только из тяжелых цепей (фрагментов иммуноглобулинов). Класс и тип не меняются в течение болезни. Так как все молекулы идентичны, то парапротеины определяются при электрофорезе белков по наличию узкого пика (М-градиента). Часто при электрофорезе выявляется более чем одна полоса парапротеинов, что обусловлено присутствием фрагментов IgG или IgM, полимеризацией иммуноглобулинов или образованием комплексов иммуноглобулинов с другими белками плазмы.
Парапротеины (обычно IgG или IgM) встречаются наиболее часто при множественной миеломе, заболеваниях иммунной системы, как макроглобулинемия Вальденстрема, острый плазмобластный лейкоз, болезни тяжелых цепей, лимфомы с парапротеинемией и др.
Криоглобулины - патологические белки плазмы (10-80 мг/мл), обладающие свойством превращения в желеобразное состояние при температуре ниже 37 °С. Большинство криоглобулинов - это комплексы поликлональных иммуноглобулинов, в состав которых примерно наполовину входят моноклональные иммуноглобулины. Обычно это IgM. Криоглобулины могут появиться при макроглобулинемии Вальденстрема, миеломе, хроническом лимфолейкозе, инфекционных заболеваниях (мононуклеоз, сифилис, туберкулез, лепра), вирусных, аутоиммунных и паразитарных заболеваниях, циррозе печени, коллагенозах, при опухолевой трансформации клеток. С криоглобулинами связаны синдромы холодовой непереносимости, повышение вязкости крови, образование иммунных комплексов с факторами I, II, V, VII свертывания крови, что может сопровождаться кровоточивостью, активацией системы комплемента, вызывать поражение почек и гемолиз эритроцитов. Количество криоглобулинов определяют по отношению объема преципитированного глобулина к общему объему сыворотки.
12.7. ПАТОФИЗИОЛОГИЯ ОБМЕНА НУКЛЕИНОВЫХ КИСЛОТ
ДНК является главной составной частью хромосом. Специфика ее структуры определяет возможность передачи наследственной информации от родителей потомству и от исходной клетки к дочерним в процессе деления. На молекуле ДНК осуществляется синтез всех видов РНК (транскрипция), в том числе информационной РНК, которая является матрицей для синтеза специфических для данного организма белков.
В обмене нуклеиновых кислот можно выделить следующие этапы:
1) расщепление поступающих с пищей нуклеопротеидов в кишечнике с последующим всасыванием в кровь продуктов их гидролиза;
2) эндогенный синтез ДНК и РНК;
3) распад нуклеиновых кислот под действием внутриклеточных нуклеаз с образованием конечных продуктов их обмена и выведением из организма.
Нарушение усвоения поступающих с пищей нуклеиновых кислот и продуктов их гидролиза не имеет существенного значения, так как все высокоорганизованные существа способны синтезировать необходимые для них нуклеиновые кислоты из имеющихся в клетках метаболитов. Поступившие из кишечника в кровь нуклеотиды, пуриновые и пиримидиновые основания не включаются ни в синтезируемые нуклеиновые кислоты, ни в пуриновые и пиримидиновые коферменты, такие, как АТФ и НАД, а расщепляются в печени с образованием конечных продуктов - мочевой кислоты и мочевины. Но при парентеральном введении нуклеозидов и нуклеотидов они включаются в молекулы ДНК и РНК.
12.7.1. Нарушение эндогенного синтеза ДНК и РНК
Образование новых молекул ДНК и РНК происходит не только в растущем организме, но и у взрослого человека. Об этом свидетельствует включение введенного в организм радиоактивного изотопа фосфора (32Р) в их молекулы. Синтез ДНК наиболее интенсивно идет в постоянно регенерирующих тканях (костный мозг, слизистая желудочно-кишечного тракта и др.). Перед вступлением соматической клетки в митоз (в фазе S митотического цикла)
количество ДНК в ядре удваивается, что является необходимым условием удвоения числа хромосом. Синтез новых молекул РНК происходит во всех клетках, но наиболее интенсивно он протекает в органах, синтезирующих большое количество белков (костный мозг и лимфоидные органы, печень, слизистые желудка и кишечника, поджелудочная железа).
Для осуществления синтеза нуклеиновых кислот необходимо присутствие в клетках достаточного количества пуриновых и пиримидиновых оснований, рибозы и дезоксирибозы, а также макроэргических фосфорных соединений. Материалом для синтеза пуриновых и пиримидиновых оснований являются одноуглеродные фрагменты некоторых аминокислот и их производных (аспарагиновая кислота, глицин, серин, глутамин), а также аммиак и СО2 (рис. 12-42). Рибоза образуется из глюкозы в пентозном цикле, в дальнейшем она может превращаться в дезоксирибозу.
Причинами нарушения синтеза ДНК являются:
• действие мутагенов, в том числе ионизирующей радиации и радиотоксинов;
• повышение содержания свободных радикалов и продуктов перекисного окисления липидов;
• отравление ионами тяжелых металлов (хроническое отравление свинцом, соединениями мышьяка);
 Рис. 12-42. Происхождение атомов азота и углерода пуринового кольца
Рис. 12-42. Происхождение атомов азота и углерода пуринового кольца
• накопление внутриядерных липидов (нейтральные фосфолипиды инактивируют РНК-полимеразу, сфингомиелин активирует метилирование ДНК);
• недостаток железа, микроэлементов цинка и марганца, а также аминокислот метионина и гистидина;
• антибиотики и цитостатики - противоопухолевые антиметаболиты, структурные аналоги фолиевой кислоты, пуринов и пиримидинов (аналоги пуринов нарушают биосинтез пуриновых нуклеотидов; пиримидины превращаются в клетках в активные ингибиторы тимидин-синтетазы, участвующей в синтезе нуклеиновых кислот);
• избыток пуриновых нуклеотидов в клетке (приводит к торможению их синтеза);
• местное применение кортикостероидов (угнетает синтез ДНК в клетках базального слоя эпидермиса);
• оксид азота - NO (ингибирует синтез ДНК);
• действие высокой температуры;
• возрастные изменения;
• отравление хлорорганическими и фосфорорганическими соединениями (хлорофос, тетрахлорметан), нитритами, сернистыми и азотистыми соединениями, кислородным ипритом, этиленоксидом, этиленамином, гидразином и его производными, гидроксиламином, нитрозаминами, полициклическими углеводородами, метаболитами афлатоксинов и другими веществами (токсины образуют ковалентные связи с аминогруппами пуриновых и пиримидиновых оснований; измененные таким образом молекулы ДНК трансформируются и разрушаются эндонуклеазами);
• белковая недостаточность;
• дефицит цинка, так как ионы Zn входят в состав ДНК- и РНК-полимераз;
• сахарный диабет;
• дефицит витаминов (В1, B6, В9, В12), необходимых для синтеза азотистых оснований.
Наиболее выраженные нарушения синтеза ДНК имеют место при дефиците фолиевой кислоты и витамина В12.
При дефиците фолиевой кислоты нарушается использование одноуглеродных фрагментов аминокислот для синтеза пуриновых и пиримидиновых оснований.
Витамин В12 необходим для образования некоторых коферментных форм фолиевой кислоты, при дефиците которых нарушается синтез тимидина, что лимитирует образование новых молекул ДНК. Синтез РНК при дефиците витамина В12 и фолиевой кислоты не нарушается. Пониженное образование ДНК тормозит вступление клеток в митоз вследствие удлинения синтетической фазы митотического цикла. Задержка митозов ведет к замедлению клеточных делений, в результате тормозится процесс физиологической регенерации в костном мозгу и в других быстрообновляющихся тканях. Задержка митозов сопровождается увеличением размеров клеток, что, по-видимому, связано с удлинением интерфазы. Наиболее демонстративно эти изменения выражены в кроветворной ткани костного мозга: появляются гигантские эритробласты - мегалобласты, при созревании которых образуются эритроциты больших размеров - мегалоциты. Обнаруживаются также увеличенные в размерах миелоциты, метамиелоциты и более зрелые гранулоциты. Гигантские клетки появляются и в других тканях: слизистой языка, желудка и кишечника, влагалища.
Дефицит витамина В12 у человека возникает при длительной вегетарианской диете, при нарушении всасывания витамина в кишечнике в связи с прекращением продукции внутреннего фактора Касла в желудке, при атрофии его слизистой в результате повреждения аутоантителами. Другими причинами развития гиповитаминоза В12 могут быть гастрэктомия, инвазия широким лентецом, хроническое воспаление подвздошной кишки, отсутствие в слизистой кишечника специфических рецепторов, с которыми взаимодействует комплекс внутреннего фактора с витамином В12 (см. раздел 12.3).
Дефицит фолиевой кислоты возникает при длительном отсутствии в пище зеленых овощей и животных белков, у детей раннего возраста при вскармливании одним молоком (в нем содержание фолиевой кислоты незначительно). Эндогенный дефицит фолиевой кислоты может развиться при нарушении всасывания ее в кишечнике (заболевание спру), нарушении депонирования (заболевания печени), повышенном расходовании (беременность, в случае если исходные запасы витамина были понижены), при длительном лечении некоторыми лекарственными препаратами (сульфаниламиды), при алкоголизме (см. раздел 12.3).
Последствия угнетения синтеза нуклеиновых кислот. Вследствие замедления процессов регенерации развиваются тяжелая форма
малокровия (пернициозная анемия), лейкопения и тромбоцитопения, атрофия слизистых оболочек пищеварительного тракта и дыхательных путей. Отмечаются атрофические изменения во всех активно пролиферирующих тканях (семенниках, яичниках, волосяных фолликулах, ростковом слое кожи), в результате чего возникают бесплодие и облысение, кожа становится сухой и тонкой, на ее поверхности могут появляться эрозивные язвы. Негативные последствия касаются также тех тканей, где постоянно идет интенсивный функциональный синтез белка, требующий больших затрат РНК (печень, почки, железы внутренней и внешней секреции и др.).
12.7.2. Нарушения конечного этапа обмена нуклеиновых кислот
Наряду с синтезом в тканях постоянно происходит распад нуклеиновых кислот под действием клеточных нуклеаз, нуклеозидаз и ряда других ферментов. Освободившиеся из молекул ДНК и РНК пуриновые и пиримидиновые основания могут снова использоваться для синтеза нуклеиновых кислот и пуриновых и пиримидиновьгх коферментов (АТФ, НАД+). Большая часть оснований метаболизируется до конечных продуктов. Конечным продуктом превращений пиримидиновых оснований является мочевина, а пуриновых - мочевая кислота, синтезирующаяся под действием фермента ксантиноксидазы, который у человека имеется только в гепатоцитах и энтероцитах. Поскольку мочевая кислота не образуется при других видах обмена, она считается специфическим конечным продуктом обмена нуклеиновых кислот. Нормальное содержание мочевой кислоты в крови у женщин колеблется в пределах 0,12-0,38 ммоль/л, у мужчин - 0,12-0,46 ммоль/л; с мочой у здорового взрослого человека выделяется 0,6 г/сутки мочевой кислоты и ее солей.
Мочевая кислота не оказывает прямого токсического действия на клетки, негативные последствия ее повышенной плазменной концентрации связаны с низкой растворимостью уратов в жидкостях организма. Например, верхняя граница нормального содержания мочевой кислоты в плазме у мужчин находится примерно на уровне насыщения, а подобные насыщенные растворы могут легко выпадать в осадок. Кристаллизации уратов способствуют все виды ацидоза (соединительная ткань богата кислотами - глюкуроновой, хондроитинсерной; моча в норме имеет слабокислый рН;
причиной закисления могут быть также алкоголизация, сахарный диабет, физическое утомление, гипоксия), снижение температуры ниже 36-37 °С (наиболее холодными у человека считаются суставы стопы, коленный и голеностопный).
Повышенное содержание мочевой кислоты в крови - гиперурикемия может являться результатом:
1) генетически обусловленного нарушения активности ряда ферментов, участвующих в пуриновом обмене, что лежит в основе развития синдрома Леша-Нихана, первичной подагры, мочекаменной болезни, а также некоторых иммунодефицитных состояний - наследственная гиперурикемия (первичная);
2) избыточного поступления в организм продуктов с высоким содержанием нуклеиновых кислот (печень, почки, икра, молоки, консервированная рыба, темные сорта пива, красное вино, кофе, шоколад, бобовые и др.) - алиментарная гиперурикемия;
3) нарушения выведения мочевой кислоты и уратов с мочой (почечная недостаточность; ацидоз, поскольку органические кислоты конкурируют с мочевой за почечные переносчики; отравление свинцом или бериллием; гипотиреоз) - ретенционная гиперурикемия;
4) повышенной деградации нуклеиновых кислот или нуклеотидных макроэргов в тканях (действие радиации или цитостатиков, злокачественные новообразования, пневмония, псориаз, гемолитическая анемия новорожденных, гликогенозы, гипертиреоз, судорожные состояния, гипоксии любого генеза, ожирение, голодание, тяжелая мышечная работа и др.) - продукционная гиперурикемия;
5) сочетанного воздействия нескольких болезнетворных агентов - смешанная гиперурикемия.
Синдром Леша-Нихана (Х-сцепленная первичная гиперурикемия). Болеют только мальчики. Болезнь проявляется уже в раннем возрасте хореоатетозом и спастическим церебральным параличом, отставанием моторного и умственного развития. Характерным признаком синдрома Леша-Нихана является членовредительство (откусывание губ, пальцев). Кроме того, больные имеют симптомы подагры (гиперурикемия, повышенное выделение солей мочевой кислоты с мочой, образование камней в почках, отложение солей мочевой кислоты в суставы, острый артрит). В основе заболевания лежит полное отсутствие активности фермента гипоксантингуанин-фосфорибозил-трансферазы, что ведет к повышенной продукции уратов. Патогенез неврологических расстройств при этом заболевании невыяснен.
Подагра (от греч. podagra - капкан, боль в ногах) - заболевание, характеризующееся гиперурикемией и отложением солей мочевой кислоты (уратов) в различных тканях, преимущественно в области суставных хрящей, околосуставных тканей и в почках.
По этиологии различают подагру первичную и вторичную.
Первичная подагра является наследственным, сцепленным с Х-хромосомой рецессивным заболеванием; болеют в основном мужчины. Генетический дефект обусловливает резкое повышение активности фермента 5-фосфорибозил-1-пирофосфат-синтетазы (ФРПФ-синтетазы) или частичную потерю активности фермента гипоксантин-гуанин-фосфорибозил-трансферазы. В том и другом случае происходит избыточное образование и повышенная экскреция уратов.
Вторичная подагра обусловлена длительно существующей гиперурикемией приобретенного характера (главный этиологический фактор). Риск развития вторичной подагры увеличивается в регионах с повышенным содержанием молибдена в почве и воде (ФРПФсинтетаза - молибдензависимый фермент, при его активации повышается продукция мочевой кислоты), при лечении диуретиками, внеклеточной дегидратации, переохлаждении или воздействии факторов, снижающих растворимость мочевой кислоты в водных средах организма (см. выше). Комплексными способствующими факторами являются алкоголь (содержит пурины, подкисляет среду, ограничивает экскрецию уратов с мочой), стресс, травмы.
Болеют как мужчины, так и женщины, но у последних подагра возникает несколько реже, что связывают с защитной функцией эстрогенов, обеспечивающих более эффективную экскрецию уратов, а также стабилизирующих лизосомы нейтрофилов и других повреждаемых клеток. Защитную роль при подагре может играть и апопротеин В, входящий в состав липопротеинов очень низкой плотности (ЛПОНП) и липопротеинов низкой плотности (ЛПНП) (т.е. гиперлипопротеинемия при подагре может иметь компенсаторный характер). Адсорбируясь на поверхности кристаллов уратов, этот белок препятствует активации нейтрофилов и гуморальных медиаторов воспаления солями мочевой кислоты.
Патогенез подагры. Осаждающиеся соли мочевой кислоты активируют систему комплемента и фактор Хагемана, а через них - кининовую, свертывающую и фибринолитическую системы. Образующиеся хематтрактанты привлекают лейкоциты, фагоцитирующие кристаллы уратов. Гибель нейтрофилов в процессе фагоцитоза сопровождается выделением в окружающие ткани лизосомальных
ферментов и активных радикалов кислорода. Макрофаги, также принимающие активное участие в фагоцитозе, выделяют провоспалительные цитокины и другие медиаторы. Как следствие этих явлений возникает острое воспаление (острый подагрический артрит), в дальнейшем приобретающее хронический характер.
Главным патогенетическим фактором хронизации процесса является длительная активация макрофагов, внутри которых персистируют фагоцитированные кристаллы уратов, что приводит к формированию подагрической гранулемы (очага хронического пролиферативного воспаления). Наиболее часто поражаются мелкие суставы ног, реже - голеностопные и коленные, а также пальцы рук. Воспалительный процесс сопровождается (при обострении) резкими болями и постепенно приводит к образованию фиброзных подагрических узлов (подагрические шишки - tophi urici). Может развиться почечная недостаточность вследствие инфильтрации ткани почек уратами или развития мочекаменной болезни. Реже подагрические гранулемы могут возникать в костях, под кожей и в сердце. Известно также, что хроническая гиперурикемия может провоцировать аутоиммунный инсулит и таким образом служить диабетогенным фактором.
Очень интересными являются данные о влиянии гиперурикемии на формирование личности. Поскольку мочевая кислота (тригидроксиксантин) и кофеин (триметилксантин) являются близкими структурными аналогами, то гиперурикемия оказывает определенный стимулирующий эффект на умственную деятельность. В настоящее время можно считать доказанным наличие прямой корреляционной связи между уровнем мочевой кислоты в крови индивида и его интеллектуальными возможностями. Среди лиц, имевших аномалии пуринового обмена и страдавших подагрой, немало гениальных личностей, оставивших значительный след в истории человечества, - Микеланджело Буонарроти, Мартин Лютер, Петр Великий, Карл XII, Чарльз Диккенс и др.
Нарушение пиримидинового обмена проявляется в виде оротацидурии (повышенное выделение с мочой оротовой кислоты), возникающей в результате наследственного (аутосомно-рецессивного) дефекта ферментов, катализирующих синтез уридинтрифосфата. Избыток оротовой кислоты образует кристаллы, способные закупорить мочеточники и даже уретру. У больных детей наблюдается отставание в умственном и физическом развитии, возникают тяжелая мегалобластическая анемия и лейкопения.
12.8. РАССТРОЙСТВА ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА (ДИСГИДРИИ). ОБЕЗВОЖИВАНИЕ. OTEКИ
Вода является универсальной средой и непременным структурным компонентом любой живой системы. Она - уникальный растворитель биополимеров и необходимая среда для протекания всех биохимических реакций организма. В жидкой воде при температуре тела человека хаотически распределены ее свободные молекулы и структурированные «кристаллические» агрегаты, находящиеся в состоянии подвижного равновесия между собой. Степень структурированности воды в клетках отражает их функциональную активность при действии самых разнообразных факторов внешней и внутренней среды даже в сверхмалоинтенсивных дозах. Структурированность воды клеток определяет способность тканей насыщаться и отдавать воду (гидратационная способность тканей). Высокая частота расстройств водно-электролитного обмена и тяжесть течения процессов, вызванных этим расстройством, особенно в детском возрасте, делают проблему важной и актуальной.
12.8.1. Изменения распределения и объема воды в организме человека
Известны следующие нарушения водного баланса организма: отрицательный, сопровождающийся развитием обезвоживания организма со всеми наблюдающимися при этом последствиями, и положительный, приводящий к развитию отеков и водянок.
Общее содержание воды в организме (общая вода тела) зависит от возраста, массы тела и пола (табл. 12-12). Как видно из таблицы, содержание общей воды тела с возрастом непрерывно уменьшается, и этот процесс продолжается непрерывно до глубокой старости. При этом падает содержание воды в клетках, в то время как объем внеклеточной жидкости несколько увеличивается.
Таблица 12-12. Содержание воды в организме, % к массе тела
Возрастной период | Общая вода тела | Внеклеточная жидкость | Внутриклеточная жидкость |
Эмбрион 2 мес | 95 | - | - |
Плод 5 мес | 87 | - | - |
Окончание табл. 12-12
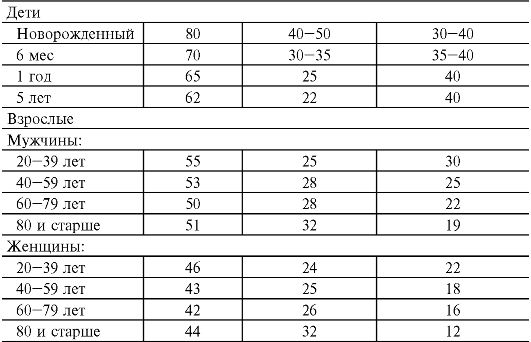 Общая
вода тела у взрослого мужчины в возрасте 25-30 лет составляет в среднем
примерно 60% массы его тела (около 42 л при массе тела 70 кг), у
взрослой женщины - 50%. Нормальные колебания от средних значений не
должны превышать 15%.
Общая
вода тела у взрослого мужчины в возрасте 25-30 лет составляет в среднем
примерно 60% массы его тела (около 42 л при массе тела 70 кг), у
взрослой женщины - 50%. Нормальные колебания от средних значений не
должны превышать 15%.
Внутриклеточный водный сектор организма (внутриклеточная вода тела). Значительная часть воды (30-35% массы тела) сосредоточена внутри клеток - внутриклеточный (интрацеллюлярный) водный сектор организма. Это вода клеточной массы организма. У мужчины 25 лет массой 70 кг внутри клеток сосредоточено примерно 25 л воды, у женщины того же возраста массой 60 кг - около 17 л (при объеме общей воды тела =32 л).
Внутриклеточная жидкость представлена в виде трех состояний:
1) связанная с гидрофильными структурами вода протоплазмы;
2) вода притяжения на поверхности коллоидных структур; 3) вода капиллярности - в лакунах протоплазмы - наиболее мобильная, относительно свободная вода клеток.
При различных патологических состояниях объем внутриклеточного водного сектора может меняться как в сторону его увеличения (например, при водной интоксикации), так и в сторону снижения (водное истощение). Эти изменения происходят чаще
за счет варьирования объема мобильной воды клеток. Как правило, изменение объема внутриклеточного сектора организма развивается медленнее и позже по сравнению с изменением объема внеклеточного водного сектора (особенно объема плазмы крови).
Внеклеточный (экстрацеллюлярный) водный сектор организма (внеклеточная вода тела). Объем его составляет 20-24% массы тела человека (около 17 л у мужчин массой тела 70 кг). Этот сектор включает в себя воду плазмы крови, интерстициальную и трансцеллюлярную жидкости.
Вода плазмы крови - часть внеклеточного водного сектора (интраваскулярный водный подсектор организма). Одной из важнейших функций плазмы крови является формирование среды для нормальной жизнедеятельности форменных элементов крови. Объем плазмы крови составляет 3,5-5% массы тела. Содержание белков в плазме крови у взрослого человека равно 70-80 г/л (это создает величину коллоидно-осмотического давления 3,25- 3,64 кПа, или 25-28 мм рт.ст.), что значительно превышает их содержание в интерстициальной жидкости (10-30 г/л). На долю чистой воды в плазме крови приходится 93% ее объема.
Интерстициальная жидкость представляет собой жидкость внеклеточного и внесосудистого пространств. Она непосредственно омывает клетки, близка по ионному и молярному составу к плазме крови (за исключением содержания белка) и вместе с лимфой составляет 15-18% массы тела. Эта жидкость находится в постоянном обмене с плазмой крови, так что за сутки из сосудов в ткани переходит приблизительно 20 л жидкости с растворенными в ней веществами, и такое же количество возвращается из тканей в общий кровоток, причем 3 л - через лимфатические сосуды.
Трансцеллюлярная жидкость - особая группа жидкостей организма. Она не состоит в простом диффузном равновесии с плазмой крови, а образуется в результате активной деятельности клеток, отчего занимает в организме особое положение. К этой группе жидкостей относятся пищеварительные соки, содержимое почечных канальцев, синовиальная, суставная и спинно-мозговая жидкости, камерная влага глаз и др. На их долю у взрослого человека приходится 1-1,5% массы его тела.
Изменения объемов водных секторов организма. Объем всех указанных жидкостей организма, входящих в состав внеклеточного водного сектора, как и жидкостей внутриклеточного водного сектора, может существенно изменяться и в сторону уменьшения, и
в сторону повышения. Эти изменения могут происходить вследствие: 1) первичного изменения электролитного состава жидкостных сред организма (уменьшение или увеличение электролитов); 2) первичного обезвоживания организма; 3) патологической задержки воды в организме. При этом в первую очередь меняют свои объемы мобильные жидкости организма - интраваскулярная и интерстициальная.
Жидкие среды организма обладают довольно постоянным электролитным составом (табл. 12-13), электронейтральны и находятся в состоянии осмотического равновесия. Однако электролитный состав внеклеточных жидкостей существенно отличается от электролитного состава внутриклеточных жидкостей. Клеточные жидкости содержат значительно больше ионов калия, магния, фосфатов, внеклеточные жидкости - ионов натрия, хлора, кальция, бикарбонатов. Содержание белков в клетках намного превышает их содержание в межтканевой жидкости.
Таблица 12-13. Молярный и ионный состав жидкостных сред человеческого организма*
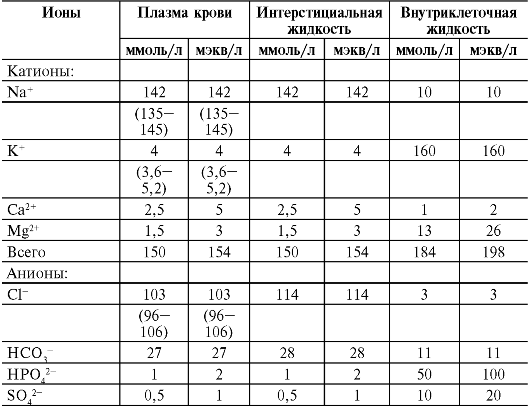
 * Не учтена концентрация недиссоциирующих соединений (глюкоза, мочевина и др.); она составляет приблизительно 7-9 ммоль/л.
* Не учтена концентрация недиссоциирующих соединений (глюкоза, мочевина и др.); она составляет приблизительно 7-9 ммоль/л.
Постоянство электролитного состава жидкостных сред организма поддерживает постоянство объемов этих жидкостей и определенное распределение их по водным секторам организма. И, напротив, постоянство объемов жидкостей организма поддерживает постоянство их электролитного состава.
Нейрогуморальная регуляция водно-электролитного обмена
Питьевое поведение человека обусловлено изменением состояния центра жажды в гипоталамусе, представляющего собой нейроны, чувствительные к ангиотензину-II, в меньшей степени к ангиотензину-III (эти вещества активируют центр жажды), к атриопептину - предсердному натрийуретическому фактору (он снижает активность центра жажды). Кроме этого, жажда бывает обусловлена как возрастанием осмотической концентрации внеклеточной жидкости и раздражением осморецепторов гипоталамуса (это так называемая гиперосмотическая жажда), так и сокращением объема циркулирующей крови (ОЦК) и воздействием образующегося при этом ангиотензина-II на центр жажды (гиповолемическая жажда).
Гуморальную регуляцию осуществляют антидиуретическая и антинатрийуретическая системы, главным исполнительным органом которых являются почки. Сенсорами антидиуретической системы служат осморецепторы, основная рефлексогенная зона которых заложена в переднем гипоталамусе. Кроме того, имеются и менее чувствительные осморецепторы печени (от печени по афферентным нервным путям сигналы достигают гипоталамуса). При повышении осмотического давления крови отмечается раздражение осморецепторов, что приводит к увеличению выделения антидиуретического гормона (АДГ) гипоталамусом. АДГ повышает реабсорбцию воды в дистальных канальцах нефрона, при этом диурез уменьшается.
Сенсорами антинатрийуретической системы являются волюморецепторы (рецепторы объема) предсердий, адекватным раздражителем для которых служит изменение объема жидкостных сред организма (главным образом ОЦК). При уменьшении ОЦК раздражение волюморецепторов сопровождается увеличением секреции надпочечниками альдостерона, который повышает реабсорбцию натрия в почечных канальцах и способствует его задержке в организме. Активация секреции альдостерона осуществляется также через ренин-ангиотензин-альдостероновую систему - РААС. Ренинстимулирующие факторы (уменьшение минутного объема сердца, гиповолемия, гипотонические состояния, ишемия почек, отрицательный натриевый баланс) обеспечивают выработку ренина клетками юкстагломерулярного аппарата почек. Под влиянием ренина из ангиотензиногена образуется ангиотензин-I. Ангиотензиноген продуцируется печенью и находится в а2-глобулиновой фракции плазмы. Под влиянием ангиотензинпревращающего фермента (АПФ) из ангиотензина-I преимущественно в сосудах легких образуется ангиотензин-II. Ангиотензин-II - ключевой эффектор в рениновой системе. Он является мощным вазоконстриктором. Кроме этого, действие ангиотензина-II на гипоталамус способствует активации симпатической нервной системы, что ведет к гипертензии. Ангиотензин-II стимулирует центр жажды; в гипоталамусе усиливает продукцию вазопрессина; в надпочечниках усиливает продукцию альдостерона; в больших концентрациях он стимулирует глюкокортикостероидогенез. На миоциты сосудов ангиотензин-II оказывает митогенное действие. Ангиотензин-II угнетает ренинообразование (это явление называют «короткой петлей обратной связи» РААС). Выработку ренина также снижают атриопептин и вазопрессин. Время жизни ангиотензина-II в кровотоке короткое - до 2 мин. При последовательном протеолизе он дает ряд производных, среди них - ангиотензин-III, который обладает слабой, сходной с ангиотензином-II биологической активностью. Ангиотензин-II и ангиотензин-III стимулируют синтез минералкортикоида - альдостерона. В почечных канальцах альдостерон способствует реабсорбции натрия. Под влиянием ангиотензинов задержка натрия происходит также в потовых и слюнных железах, тонком и толстом кишечнике. В результате в крови увеличивается содержание натрия - наблюдается гипернатриемия. Это сопровождается раздражением осморецепторов гипоталамуса и увеличением секреции антидиуретического гормона гипотала-
мусом. АДГ усиливает реабсорбцию воды в дистальных почечных канальцах. Таким образом, в результате активации РААС происходит задержка в организме воды и натрия, что может привести к появлению отеков и водянок.
Атриопептины (предсердный натрийуретический фактор - ПНУФ) синтезируются в секреторных клетках предсердий, ЦНС, легких. Секреция атриопептинов возрастает при гиперволемии, солевой нагрузке, растяжении предсердий. Атриопептины обеспечивают увеличение натрийуреза и диуреза в почках. Атриопептины подавляют функцию РААС, уменьшают секрецию ренина, альдостерона, АДГ, обладают вазодилататорным эффектом, угнетают центр жажды. Таким образом, они предохраняют организм от перегрузки солью и водой.
При различных патологических состояниях (сердечно-сосудистая и почечная недостаточность, голодание и др.) нейроэндокринная регуляция, направленная на поддержание водно-электролитного гомеостаза в здоровом организме, повреждается и может становиться важным патогенетическим звеном, приводящим к серьезным расстройствам обмена воды и электролитов.
12.8.2. Потери и потребность в воде организма человека в норме и при патологии
Человек за сутки должен потреблять такое количество жидкости, которое в состоянии возмещать суточные потери ее через почки и внепочечными путями. Оптимальный суточный диурез у здорового взрослого человека составляет 1200-1700 мл (при патологических состояниях он может увеличиваться до 20-30 л и понижаться до 50-100 мл за сутки). Выведение воды происходит также при испарении с поверхности альвеол и кожи - неощутимое пропотевание (от лат. perspiratio insensibilis). При нормальных температурных условиях и влажности воздуха взрослый человек таким путем за сутки теряет от 800 до 1000 мл воды. Эти потери при определенных условиях могут возрасти до 10-14 л. Наконец, незначительная часть жидкости (100-250 мл/сутки) теряется через желудочно-кишечный тракт. Однако суточные потери жидкости через желудочно-кишечный тракт при патологии могут достигать 5 л. Это происходит при тяжелых расстройствах деятельности пищеварительной системы. Таким образом, суточные потери жидкости у здоровых взрослых людей при выполнении умеренной
физической работы и местопребывании их в средней географической полосе могут колебаться от 1000 (1500) до 3000 мл. В тех же условиях нормальная сбалансированная суточная потребность взрослого человека в воде колеблется в этих же объемах - от 1000 (1500) до 3000 мл и зависит от массы тела, возраста, пола и т.п. (табл. 12-14).
Таблица 12-14. Суточные потери и потребность в воде у здоровых взрослых и детей, мл
Потери воды | Взрослый массой 70 кг | Ребенок массой до 10 кг | Поступление воды | Взрослый массой 70 кг | Ребенок массой до 10 кг |
С мочой | 800-1700 | 300-500 | С пищей | 600-1200 | 350-750 |
С калом | 100-250 | 25-50 | Питьевая вода | 800-1500 | - |
При дыхании и потоотделении | 650-1000 | 75-300 | Эндогенная вода* | 150-250 | 50-100 |
Всего | 1550-2950 | 400-850 | Всего Потребность на 1 кг массы | 1550-2950 30-50 | 400-850 120-150 |
* Эндогенная (метаболическая) вода, образующаяся в процессе обмена и утилизации белков, жиров и углеводов, составляет 8-10% суточной потребности воды организмом (120-250 мл). Этот объем может возрастать в 2-3 раза при некоторых патологических процессах (тяжелая травма, инфекция, лихорадка и др.)
При различных обстоятельствах и ситуациях, в которых может оказаться человек, и особенно при патологических состояниях суточные потери и потребление воды могут существенно отличаться от средненормальных. Это ведет к разбалансировке водного обмена и сопровождается развитием отрицательного или положительного водного баланса.
12.8.3. Виды обезвоживания и причины их развития
Обезвоживание (гипогидрия, дегидратация, эксикоз) развивается в тех случаях, когда потери воды превышают поступление ее в организм. При этом возникает абсолютный дефицит общей воды тела, сопровождающийся развитием отрицательного водного баланса. Этот дефицит может быть связан с уменьшением объема
внутриклеточной воды тела или с уменьшением объема внеклеточной воды тела, что на практике встречается наиболее часто, а также за счет одновременного снижения объемов внутриклеточной и внеклеточной воды тела. Виды обезвоживания:
1. Обезвоживание, вызываемое первичной абсолютной нехваткой воды (водное истощение, «десикация»). Этот вид обезвоживания развивается либо вследствие ограничения приема воды, либо вследствие избыточного выведения гипотонической или вовсе не содержащей электролитов жидкости из организма при недостаточной компенсации потерь.
2. Обезвоживание, вызываемое первичным недостатком минеральных солей в организме. Данный вид дегидратации развивается тогда, когда организм теряет и недостаточно восполняет запасы минеральных солей. Все формы этого обезвоживания характеризуются отрицательным балансом внеклеточных электролитов (в первую очередь ионов натрия и хлора) и не могут быть устранены только приемом чистой воды.
При развитии обезвоживания практически важно учитывать два момента: скорость потери жидкости (если дегидратация вызвана избыточной потерей воды) и каким путем теряется жидкость. Эти факторы во многом определяют характер формирующегося обезвоживания и принципы его терапии: при быстрой (в течение нескольких часов) потере жидкости (например, при острой высокой тонкокишечной непроходимости) в первую очередь уменьшаются объем внеклеточного водного сектора организма и содержание электролитов, входящих в его состав (прежде всего ионов натрия). Возмещать потерянную жидкость в этих случаях следует быстро. Основой переливаемых сред должны быть изотонические солевые растворы - в данном случае изотонический раствор хлорида натрия с добавлением небольшого количества белков (альбумина).
Медленно (в течение нескольких дней) развивающаяся дегидратация (например, при резком снижении или полном прекращении поступления воды в организм) сопровождается уменьшением диуреза и потерей значительных количеств внутриклеточной жидкости и ионов калия. Возмещение таких потерь должно быть медленным: в течение нескольких дней вводят жидкости, основным электролитным компонентом которых является хлорид калия (под контролем уровня диуреза, который должен быть близким к норме).
Таким образом, в зависимости от скорости потерь жидкости организмом выделяют острую и хроническую дегидратацию. В зависимости от преимущественной потери воды или электролитов выделяют гиперосмолярную и гипоосмолярную дегидратацию. При потере жидкости с эквивалентным количеством электролитов развивается изоосмолярная дегидратация.
Для правильной терапевтической коррекции различных видов обезвоживаний организма, помимо представления о причинах дегидратации, изменения осмотической концентрации жидкостей и объема водных пространств, за счет которых преимущественно происходит обезвоживание, необходимо знать и об изменении рН жидкости организма. С этой точки зрения различают дегидратации с изменением рН в кислую сторону (например, при хронических потерях кишечного содержимого, панкреатического сока или желчи), в щелочную сторону (например, многократная рвота при стенозе привратника сопровождается значительными потерями HCl и ионов калия и компенсаторным повышением содержания в крови HCO3-, что ведет к развитию алкалоза), а также дегидратацию без изменения рН жидкостных сред организма (например, обезвоживание, развивающееся при снижении поступления воды извне).
Обезвоживание в связи с первичной абсолютной нехваткой воды (водное истощение, «десикация»). К развитию обезвоживания в связи с первичной абсолютной нехваткой воды могут приводить: 1) алиментарное ограничение поступления воды; 2) избыточные потери воды через легкие, почки, кожу (с потом и через обширные обожженные и травмированные поверхности тела). В всех указанных случаях возникают гиперосмолярная или изоосмолярная дегидратации.
Ограничение поступления воды. У здоровых людей ограничение или полное прекращение поступления воды в организм происходит при чрезвычайных обстоятельствах: у заблудившихся в пустыне, у засыпанных при обвалах и землетрясениях, при кораблекрушениях и т.д. Однако значительно чаще водный дефицит наблюдается при различных патологических состояниях: 1) при затруднении глотания (сужение пищевода после отравления едкими щелочами, при опухолях, атрезии пищевода и др.); 2) у тяжелобольных и ослабленных лиц (коматозное состояние, тяжелые формы истощения и др.); 3) у недоношенных и тяжелобольных детей; 4) при некоторых формах заболеваний головного мозга, сопровождающихся отсутствием чувства жажды (идиотия, микроцефалия), а также в
результате кровоизлияния, ишемии, опухолевого роста, при сотрясении головного мозга.
При полном прекращении поступления питательных веществ и воды (абсолютное голодание) у здорового человека возникает суточный дефицит воды в 700 мл (табл. 12-15).
Таблица 12-15. Водный баланс здорового взрослого человека, мл, в состоянии абсолютного голодания (по Гемблу)
Потеря жидкости организмом, л | Образование жидкости в организме, л | ||
Минимальное количество мочи | 500 | Метаболическая вода | 200 |
Минимальная потеря через кожу и легкие (perspir, insensib) | 900 | Вода, освобождающаяся из депо | 500 |
Итого | 1400 | Итого 700 | |
При голодании без воды организм начинает использовать прежде всего мобильную жидкость внеклеточного водного сектора (вода плазмы, интерстициальная жидкость), позже используются мобильные водные резервы внутриклеточного сектора. У взрослого человека массой 70 кг таких резервов мобильной воды - до 14 л (при средней суточной потребности 2 л), у ребенка массой 7 кг - до 1,4 л (при средней суточной потребности 0,7 л).
Продолжительность жизни взрослого человека при полном прекращении поступления воды и питательных веществ (при обычных температурных условиях внешней среды) составляет 6-8 суток. Теоретически рассчитанная продолжительность жизни ребенка массой в 7 кг в тех же условиях в 2 раза меньше. Детский организм значительно тяжелее переносит обезвоживание по сравнению со взрослым. При одинаковых условиях грудные дети на единицу поверхности тела, приходящейся на 1 кг массы, теряют через кожу и легкие в 2-3 раза больше жидкости. Экономия воды почками у грудных детей выражена плохо (концентрационная способность почек низкая, в то время как способность разводить мочу формируется быстрее), а функциональные резервы воды (соотношение между резервом мобильной воды и суточной ее потребностью) у ребенка в 3,5 раза меньше, чем у взрослого. Интенсивность обменных процессов у детей намного выше. Следовательно, и потребность в воде (см. табл. 12-15), а также чувствительность к ее недостатку у детей существенно выше по сравнению со взрослым организмом.
Избыточные потери воды от гипервентиляции и усиленного потоотделения. У взрослых суточная потеря воды через легкие и кожу может повышаться до 10-14 л (в нормальных условиях это количество не превышает 1 л). В детском возрасте особенно большое количество жидкости может теряться через легкие при так называемом гипервентиляционном синдроме, нередко осложняющем инфекционные заболевания. При этом возникает частое глубокое дыхание, продолжающееся в течение значительного времени, что приводит к потере большого количества чистой (почти без электролитов) воды, газовому алкалозу.
При лихорадке через кожу (за счет пота с незначительным содержанием солей) и дыхательные пути может теряться значительное количество гипотонической жидкости. При искусственной вентиляции легких, которую проводят без достаточного увлажнения дыхательной смеси, также идет потеря гипотонической жидкости. В результате данной формы обезвоживания (когда потери воды превышают потери электролитов) повышается концентрация электролитов внеклеточных жидкостей организма и увеличивается их осмолярность - развивается гиперосмолярная дегидратация. Концентрация натрия в плазме крови, например, может достигать 160 ммоль/л (норма 135-145 ммоль/л) и более. Увеличивается показатель гематокрита, относительно возрастает содержание белка плазмы крови (рис. 12-43, 2). В результате повышения осмолярности плазмы развивается дефицит воды в клетках, внутриклеточная дегидратация, что проявляется возбуждением, беспокойством. Появляется мучительное чувство жажды, сухость кожных покровов, языка и слизистых оболочек, повышается температура тела, серьезно расстраиваются функции сердечно-сосудистой системы из-за сгущения крови, центральной нервной системы, почек. В тяжелых случаях возникает опасное для жизни коматозное состояние.
Избыточные потери воды через почки. Обезвоживание от полиурии может возникнуть, например, при несахарном диабете (недостаточной выработке или высвобождении АДГ). Чрезмерные потери воды через почки имеют место при врожденной форме полиурии (врожденно обусловленное снижение чувствительности дистальных канальцев и собирательных трубочек почек к АДГ), некоторых формах хронического нефрита и пиелонефрита и т.д. При несахарном диабете суточное количество мочи с низкой относительной плотностью у взрослых может достигать 20 л и более.
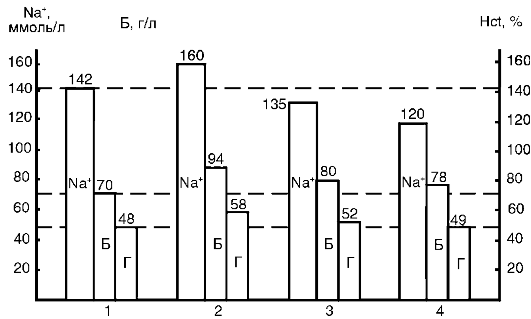 Рис. 12-43. Изменение
содержания натрия (Na, ммоль/л), белка плазмы крови (Б, г/л) и
показателя гематокрита (Hct, %) при различных видах дегидратации: 1 -
норма; 2 - гипертоническая дегидратация (водное истощение); 3 -
изотоническая дегидратация (острая потеря внеклеточной жидкости с
эквивалентным количеством солей); 4 - гипотоническая дегидратация
(хроническая дегидратация с потерей электролитов)
Рис. 12-43. Изменение
содержания натрия (Na, ммоль/л), белка плазмы крови (Б, г/л) и
показателя гематокрита (Hct, %) при различных видах дегидратации: 1 -
норма; 2 - гипертоническая дегидратация (водное истощение); 3 -
изотоническая дегидратация (острая потеря внеклеточной жидкости с
эквивалентным количеством солей); 4 - гипотоническая дегидратация
(хроническая дегидратация с потерей электролитов)
В результате развивается гиперосмолярная дегидратация. Если потеря жидкости компенсируется, то водный обмен остается в равновесии, обезвоживание и расстройства осмотической концентрации жидкостных сред организма не возникают. Если же потеря жидкости не компенсируется, то в течение нескольких часов развивается тяжелое обезвоживание с коллапсом и лихорадкой. Возникает прогрессирующее расстройство деятельности сердечно-сосудистой системы из-за сгущения крови.
Потери жидкости с обширных обожженных и травмированных поверхностей тела. Таким путем возможны значительные потери из организма воды с малым содержанием солей, т.е. потери гипотоничной жидкости. В этом случае вода из клеток и плазмы крови переходит в интерстициальный сектор, увеличивая его объем (см. рис. 12-43, 4). При этом содержание электролитов там может не измениться (см. рис. 12-43, 3) - развивается изоосмолярная дегидратация. Если же потеря воды из организма происходит относительно медленно, но достигает значительных размеров, то содержание электролитов в интерстициальной жидкости может повыситься - развивается гиперосмолярная дегидратация.
Обезвоживание от недостатка электролитов. К развитию обезвоживания от недостатка электролитов могут приводить: 1) потери преимущественно электролитов через желудочно-кишечный тракт, почки и кожу; 2) недостаточное поступление электролитов в организм.
Электролиты организма обладают способностью связывать и удерживать воду. Особенно активны в этом отношении ионы натрия, калия и хлора. Поэтому потеря и недостаточное пополнение электролитов сопровождается развитием обезвоживания. Этот вид обезвоживания продолжает развиваться при свободном приеме чистой воды и не может быть устранен одним только введением воды без восстановления нормального электролитного состава жидкостных сред организма. При потерях электролитов могут возникнуть гипоосмолярная или изоосмолярная дегидратации.
Потеря электролитов и воды через почки. Большое количество солей и воды может теряться при некоторых формах нефритов, при болезни Аддисона (недостаточности альдостерона), при полиурии с высокой осмотической плотностью мочи («осмотический» диурез при сахарном диабете) и т.д. (см. рис. 12-43, 4; рис. 12-44). Потери электролитов в этих случаях превышают потерю воды, и развивается гипоосмолярная дегидратация.
Потеря электролитов и воды через кожу. Содержание электролитов в поте относительно низкое. Средняя концентрация натрия - 42 ммоль/л, хлора - 15 ммоль/л. Однако при обильном потоотделении (тяжелая физическая нагрузка, работа в горячих цехах, длительные марши) потеря их может достигать значительных величин. Суточное количество пота у взрослого человека в зависимости от температурных факторов внешней среды и мышечной нагрузки колеблется от 800 мл до 10 л, при этом натрия может теряться более 420 ммоль/л, а хлора - более 150 ммоль/л. Поэтому при обильном потоотделении без соответствующего приема соли и воды наблюдается столь же тяжелое и быстрое обезвоживание, как при тяжелых гастроэнтеритах и неукротимой рвоте. Развивается гипоосмолярная дегидратация. Возникает внеклеточная гипоосмия и переход воды в клетки с последующим клеточным отеком. Если пытаться возместить потерянную воду бессолевой жидкостью, то внутриклеточный отек усугубляется.
Потеря электролитов и воды через желудочно-кишечный тракт. При хронических потерях жидкости, содержащей большое количество электролитов, возникает гипоосмолярная дегидратация (см.
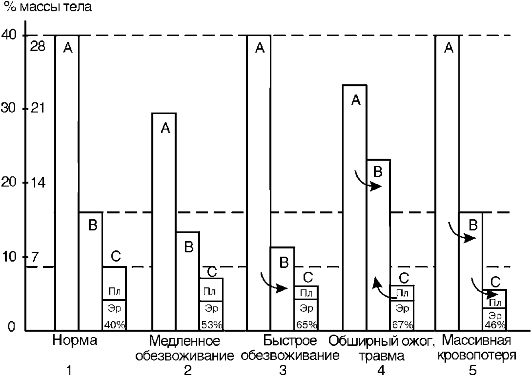 Рис. 12-44. Изменение
объема внутри- и внеклеточной жидкости организма, а также сдвиги воды
из одних пространств в другие при различных патологических состояниях у
взрослого человека: А - объем внутриклеточной жидкости; В - объем
интерстициальной жидкости; С - объем крови. Пл - плазма крови, Эр -
эритроциты
Рис. 12-44. Изменение
объема внутри- и внеклеточной жидкости организма, а также сдвиги воды
из одних пространств в другие при различных патологических состояниях у
взрослого человека: А - объем внутриклеточной жидкости; В - объем
интерстициальной жидкости; С - объем крови. Пл - плазма крови, Эр -
эритроциты
рис. 12-43, 4). Чаще других такие потери могут происходить через желудочно-кишечный тракт: многократная рвота и поносы при гастроэнтеритах, долго незаживающие свищи желудка, протока поджелудочной железы.
При острых стремительных потерях соков желудочно-кишечного тракта (при стенозе привратника, острой бактериальной дизентерии, холере, язвенном колите, высокой тонкокишечной непроходимости) изменения осмолярности и состава внеклеточной жидкости практически не происходят. При этом возникает солевой дефицит, осложненный потерей эквивалентного количества жидкости. Развивается острая изоосмолярная дегидратация (см. рис. 12- 43, 3). Изоосмолярная дегидратация может развиться также при обширной механической травме, массивных ожогах поверхности тела и др.
При данном виде обезвоживания (изоосмолярная дегидратация) потеря воды организмом происходит в основном за счет внеклеточной жидкости (до 90% объема потерянной жидкости), что крайне неблагоприятно сказывается на гемодинамике из-за бы-
стро наступающего сгущения крови. На рисунке 12-44 показаны изменения объема внутри- и внеклеточной жидкости организма, а также перемещение (сдвиги) воды из одних водных пространств в другие при острой потере внеклеточной жидкости (см. рис. 12-44,
3, 5).
При быстром обезвоживании организма теряются главным образом интерстициальная жидкость и вода плазмы крови. При этом имеет место сдвиг воды внутриклеточного сектора в интерстициальный. При обширных ожогах и травмах вода из клеток и плазмы крови перемещается в интерстициальный сектор, увеличивая его объем. После сильной кровопотери вода быстро (от 750 до 1000 мл за сутки) перемещается из интерстициального водного сектора в сосуды, восстанавливая объем циркулирующей крови. При неукротимой рвоте и поносах (гастроэнтериты, токсикоз беременности и др.) организм взрослого ежесуточно может терять до 15% от общего количества натрия, до 28% общего количества хлора и до 22% всей внеклеточной жидкости.
Нарушения функций органов и систем организма при изоосмолярном обезвоживании проявляются быстрее и протекают тяжелее, чем при гиперосмолярной дегидратации - прогрессивно уменьшается масса тела, падает артериальное и центральное венозное давление, уменьшается минутный объем сердца, расстраивается деятельность центральной нервной системы, нарушается выделительная функция почек. Быстро нарастают апатия и адинамия, расстраивается сознание и возникает коматозное состояние.
При медленном обезвоживании объем воды пропорционально уменьшается за счет всех водных пространств организма. Проявления его менее стремительны и опасны, чем при изоосмолярной дегидратации.
12.8.4. Влияние обезвоживания на организм
Нарушение деятельности сердечно-сосудистой системы. Значительное обезвоживание организма ведет к сгущению крови - ангидремии. Это состояние сопровождается расстройством ряда гемодинамических показателей.
Объем циркулирующей крови и плазмы при обезвоживании уменьшается. Так, при экспериментальном обезвоживании животных (собак) потеря воды, составляющая 10% от массы тела, вызывает снижение объема циркулирующей крови на 24% при умень-
шении количества плазмы на 36%. Происходит перераспределение крови. Жизненно важные органы (сердце, мозг, печень) за счет значительного снижения кровоснабжения почек и скелетной мускулатуры относительно лучше других снабжаются кровью.
При тяжелых формах обезвоживания систолическое артериальное давление падает до 70-60 мм рт.ст. и ниже. Венозное давление также понижается. Минутный объем сердца в тяжелых случаях обезвоживания снижается до 1/3 и даже до 1/4 нормальной величины. Время кругооборота крови удлиняется по мере снижения величины минутного объема сердца. У грудных детей при тяжелом обезвоживании оно может быть удлинено в 4-5 раз по сравнению с нормой.
Нарушение деятельности центральной нервной системы. В основе расстройств центральной нервной системы при обезвоживании (судороги, галлюцинации, коматозное состояние и т.д.) лежит нарушение кровоснабжения нервной ткани и интоксикация ее продуктами обмена. Это приводит к следующим явлениям: а) недостаточному поступлению питательных веществ (глюкозы) к нервной ткани; б) недостаточному снабжению нервной ткани кислородом; в) нарушению ферментативных процессов в нервных клетках. Величина парциального давления кислорода в венозной крови головного мозга человека достигает критических цифр, приводящих к коматозному состоянию (ниже 19 мм рт.ст.). Расстройству деятельности центральной нервной системы способствуют понижение артериального давления в большом круге кровообращения, нарушение осмотического равновесия жидкостных сред организма, ацидоз и гиперазотемия, развивающиеся при обезвоживании.
Нарушение деятельности почек. Главной причиной снижения выделительной способности почек является недостаточное кровоснабжение почечной паренхимы. Это может быстро привести к гиперазотемии с последующей уремией.
В тяжелых случаях обезвоживания могут наблюдаться и анатомические изменения почек (некротическое обызвествление канальцев с исчезновением активности фосфатазы их эпителия; тромбозы почечных вен, закупорка почечной артерии, симметричные кортикальные некрозы и др.). Возникновение гиперазотемии зависит как от понижения фильтрации, так и от повышения реабсорбции мочевины в канальцах. Непропорционально большая реабсорбция мочевины, видимо, связана с поражением канальцевого эпителия. Нагрузка на почки как на выделительный орган при
обезвоживании повышена. Почечная недостаточность является решающим фактором в механизме негазового ацидоза (накопление кислых продуктов белкового обмена, кетоновых тел, молочной, пировиноградной кислот и др.).
Расстройство деятельности желудочно-кишечного тракта. Вследствие торможения ферментативных процессов, а также из-за угнетения перистальтики желудка и кишечника при обезвоживании возникают растяжение желудка, парез кишечной мускулатуры, уменьшение всасывания и другие расстройства, вызывающие нарушение пищеварения. Ведущим фактором при этом является тяжелое ангидремическое расстройство кровообращения желудочнокишечного тракта.
12.8.5. Задержка воды в организме
Задержка воды в организме (гипергидратация, гипергидрия, обводнение) наблюдается при чрезмерном введении воды (водное отравление, водная интоксикация) либо при ограничении выделения жидкости из организма. При этом могут развиваться видимые и скрытые отеки и водянки.
Водное отравление (интоксикация). У человека и животных водное отравление возникает в том случае, если поступление воды в организм превосходит способность почек к ее выведению. От чрезмерной водной нагрузки увеличивается объем циркулирующей крови (олигоцитемическая гиперволемия), относительно уменьшается содержание белков и электролитов крови, гемоглобина, возникают гемолиз эритроцитов и гематурия. Такое состояние сопровождается развитием гипоосмолярной гипергидратации, переходом воды в клетки с последующим появлением признаков внутриклеточного отека.
В экспериментальных условиях у собак при повторном (до 10- 12 раз) введении в желудок воды по 50 мл на 1 кг массы с интервалом в 0,5 ч наступает острая водная интоксикация. При этом возникают тошнота, рвота, мышечные подергивания, судороги, коматозное состояние и нередко смертельный исход.
Диурез первоначально увеличивается, затем начинает относительно отставать от количества поступающей воды, а при развитии гемолиза и гематурии происходит истинное уменьшение мочеотделения. Особенно резко усугубляются эти явления при одновременном введении антидиуретического гормона или альдостерона.
У человека водное отравление может возникнуть при некоторых почечных заболеваниях (гидронефроз, во вторую стадию острой почечной недостаточности и др.), при состояниях, сопровождающихся острым уменьшением или прекращением отделения мочи (у больных в послеоперационном периоде, в шоковом состоянии при внутривенном капельном введении больших количеств жидкостей). Описано возникновение водного отравления у больных несахарным мочеизнурением, продолжавших принимать большое количество жидкостей на фоне лечения антидиуретическими гормональными препаратами, у детей, выпивающих на спор большое количество жидкости и др.
Виды гипергидратации. Увеличение общего содержания воды в организме может наблюдаться при сохранении ее нормальной осмотической концентрации (300-330 мосмоль/л). В этом случае возникает изоосмолярная гипергидратация. Такое состояние наблюдается, например, при повышении гидростатического давления в капиллярах и усилении процесса фильтрации жидкости из сосудов в интерстициальное пространство (например, при недостаточности правого сердца). Скопление изотонической жидкости в тканях имеет место при резком снижении онкотического давления в крови (потери белка через почки, цирроз печени, белковое голодание и т.д.), при повышении проницаемости капиллярной стенки (диффузный капиллярит при гломерулонефрите), при затруднении лимфооттока (закупорка лимфатических сосудов, например круглыми червями-филяриями, метастазы в лимфоузлы и лимфососуды - см. механизм развития отеков). Изоосмолярная гипергидратация может развиваться после введения избыточных количеств изотонических растворов (неправильная коррекция водно-электролитных нарушений).
В случае уменьшения осмотической концентрации жидкостных сред организма ниже 300 мосмоль/л (при увеличенной массе общей воды тела) говорят о гипоосмолярной гипергидратации. Такое состояние возникает при водном отравлении (поступление воды в организм превосходит способность почек к ее выведению), а также при гиперпродукции антидиуретического гормона (синдром Пархона). Гипоосмолярная гипергидратация сопровождается перемещением воды в клетки с последующим появлением признаков внутриклеточного отека.
Гиперосмолярная гипергидратация (при повышении осмотической концентрации жидкостных сред организма выше 330 мосмоль/л
на фоне увеличенной массы общей воды тела) может возникнуть, например, при вынужденном неограниченном употреблении морской воды, осмолярность которой намного превышает осмолярность плазмы крови. Подобное состояние имеет место также при избыточном введении различных гипертонических растворов с лечебной целью (неправильная коррекция водно-электролитных нарушений). При этом развиваются опасные для жизни нарушения деятельности органов и систем, обусловленные дегидратацией клеток. Особенно быстро возникают признаки клеточной дегидратации, если гипертонические растворы вводят в организм при нарушении функции почек в отношении выведения солей или на фоне избыточной продукции альдостерона (первичный, вторичный альдостеронизм).
12.8.6. Отеки и водянки
Отеком называется патологическое скопление жидкости в тканях и межтканевых пространствах вследствие нарушения обмена воды между кровью и тканями. Отек - типовой патологический процесс, встречающийся при многих заболеваниях.
Невоспалительная отечная жидкость называется транссудатом. По своим физико-химическим свойствам транссудат существенно отличается от воспалительного выпота - экссудата (см. главу 10).
Отеки различаются:
• по распространенности: местный и общий (генерализованный);
• по скорости развития: молниеносные (развиваются в течение нескольких секунд, например, после укуса насекомых, змей), острые (развиваются в течение часа, например, при острой сердечной недостаточности отек легких), хронические (в течение нескольких суток, недель, например, при голодании);
• по патогенезу: гидростатические (застойные), онкотические, мембраногенные, лимфатические (лимфогенные), осмотические;
• по этиологии: сердечные, почечные (нефротические и нефритические), печеночные, токсические, нейрогенные, аллергические, воспалительные, кахектические, голодные.
Патологическое скопление жидкости в серозных полостях организма называется водянкой (в брюшной полости - асцит, в плевральной полости - гидроторакс, в околосердечной сумке - гидроперикардиум, между листками серозной оболочки яичка -
гидроцеле, в желудочках мозга и в субарахноидальном пространстве - гидроцефалия и др.).
Механизмы возникновения отеков. Обмен жидкости между сосудами и тканями происходит через капиллярную стенку. Эта стенка представляет собой достаточно сложно устроенную биологическую структуру, через которую относительно легко транспортируются вода, электролиты, некоторые органические соединения (мочевина), но значительно труднее - белки. В результате этого концентрации белков в плазме крови (60-80 г/л) и тканевой жидкости (10-30 г/л) неодинаковы.
Согласно классической теории Э. Старлинга (1896), нарушение обмена воды между капиллярами и тканями определяется следующими факторами: 1) гидростатическим давлением крови в капиллярах и давлением межтканевой жидкости; 2) коллоидноосмотическим давлением плазмы крови и тканевой жидкости; 3) проницаемостью капиллярной стенки.
Кровь движется в капиллярах с определенной скоростью и под определенным давлением (рис. 12-45), в результате чего создаются гидростатические силы, стремящиеся вывести воду из капилляров в интерстициальное пространство. Эффект гидростатических сил будет тем больше, чем выше кровяное давление и чем меньше величина давления тканевой жидкости.
Гидростатическое давление крови в артериальном конце капилляра кожи человека составляет 30-32 мм рт.ст., а в венозном конце - 8-10 мм рт.ст.
Установлено, что давление тканевой жидкости является величиной отрицательной. Она на 6-7 мм рт.ст. ниже величины атмосферного давления и, следовательно, обладая присасывающим эффектом действия, способствует переходу воды из сосудов в межтканевое пространство.
Таким образом, в артериальном конце капилляров создается эффективное гидростатическое давление (ЭГД) - разность между гидростатическим давлением крови и гидростатическим давлением межклеточной жидкости, равное ~ 36 мм рт.ст. (30 - (-6)). В венозном конце капилляра величина ЭГД соответствует 14 мм рт.ст.
(8 - (-6)).
Удерживают воду в сосудах белки, концентрация которых в плазме крови (60-80 г/л) создает коллоидно-осмотическое давление, равное 25-28 мм рт.ст. Определенное количество белков содержится в межтканевых жидкостях. Коллоидно-осмотическое
 Рис. 12-45. Обмен
жидкости между различными частями капилляра и тканью (по Э. Старлингу):
pa - нормальный перепад гидростатического давления между артериальным
(30 мм рт.ст.) и венозным (8 мм рт.ст.) концом капилляра; bc -
нормальная величина онкотического давления крови (28 мм рт.ст.). Влево
от точки A (участок Ab) происходит выход жидкости из капилляра в
окружающие ткани, вправо от точки А (участок Ac) происходит ток жидкости
из ткани в капилляр (А1 - точка равновесия). При повышении
гидростатического давления (p'a') или снижении онкотического давления
(b'c') точка A смещается в положение А1 и А2. В этих случаях переход жидкости из ткани в капилляр затрудняется и возникает отек
Рис. 12-45. Обмен
жидкости между различными частями капилляра и тканью (по Э. Старлингу):
pa - нормальный перепад гидростатического давления между артериальным
(30 мм рт.ст.) и венозным (8 мм рт.ст.) концом капилляра; bc -
нормальная величина онкотического давления крови (28 мм рт.ст.). Влево
от точки A (участок Ab) происходит выход жидкости из капилляра в
окружающие ткани, вправо от точки А (участок Ac) происходит ток жидкости
из ткани в капилляр (А1 - точка равновесия). При повышении
гидростатического давления (p'a') или снижении онкотического давления
(b'c') точка A смещается в положение А1 и А2. В этих случаях переход жидкости из ткани в капилляр затрудняется и возникает отек
давление интерстициальной жидкости для большинства тканей составляет ~ 5 мм рт.ст. Белки плазмы крови удерживают воду в сосудах, белки тканевой жидкости - в тканях. Эффективная онкотическая всасывающая сила (ЭОВС) - разность между величиной коллоидно-осмотического давления крови и межтканевой жидкости. Она составляет ~ 23 мм рт. ст. (28-5). Если эта сила превышает величину эффективного гидростатического давления, то жидкость будет перемещаться из интерстициального пространства в сосуды. Если ЭОВС меньше ЭГД, обеспечивается процесс ультрафильтрации жидкости из сосуда в ткань. При выравнивании величин ЭОВС и ЭГД возникает точка равновесия А (см. рис. 12-45).
В артериальном конце капилляров (ЭГД = 36 мм рт.ст., а ЭОВС = 23 мм рт.ст.) сила фильтрации преобладает над эффективной онкотической всасывающей силой на 13 мм рт.ст. (36-23). В точке равновесия А эти силы выравниваются и составляют 23 мм рт.ст. В венозном конце капилляра ЭОВС превосходит эффективное гидростатическое давление на 9 мм рт.ст. (14 - 23 = -9), что определяет переход жидкости из межклеточного пространства в сосуд.
По Э. Старлингу, имеет место равновесие: количество жидкости, покидающей сосуд в артериальной части капилляра, должно быть равно количеству жидкости, возвращающейся в сосуд в венозном конце капилляра. Как показывают расчеты, такого равновесия не происходит: сила фильтрации в артериальном конце капилляра равна 13 мм рт.ст., а всасывающая сила в венозном конце капилляра -9 мм рт.ст. Это должно приводить к тому, что в каждую единицу времени через артериальную часть капилляра в окружающие ткани жидкости выходит больше, чем возвращается обратно. Так оно и происходит - за сутки из кровяного русла в межклеточное пространство переходит около 20 л жидкости, а обратно через сосудистую стенку возвращается только 17 л. Три литра транспортируется в общий кровоток через лимфатическую систему. Это довольно существенный механизм возврата жидкости в кровяное русло, при повреждении которого могут возникать так называемые лимфатические отеки.
Обмен жидкости между капилляром и тканью показан на рис.
12-45.
В развитии отеков играют роль следующие патогенетические факторы.
1. Гидростатический (гидродинамический) фактор. При возрастании гидростатического давления в сосудах (рис. 12-45, р, а) увеличивается сила фильтрации, а также поверхность сосуда (А1b, а не Аb, как в норме), через которую происходит фильтрация жидкости из сосуда в ткань. Поверхность же, через которую осуществляется обратный ток жидкости (А1с, а не Ас, как в норме), уменьшается. При значительном повышении гидростатического давления в сосудах может возникнуть такое состояние, когда через всю поверхность сосуда осуществляется ток жидкости только в одном направлении - из сосуда в ткань. Происходит накопление и задержка жидкости в тканях.
Возрастание гидростатического давления в сосудах отмечается при повышении венозного давления (из-за застоя крови при сер-
дечной недостаточности) и при увеличении ОЦК (из-за увеличения выработки АДГ при хронической сердечной недостаточности). Когда ведущим патогенетическим фактором в развитии отека является повышение гидростатического давления крови, развивается так называемый застойный отек. Этот механизм играет существенную роль при возникновении сердечных отеков (застой крови при сердечной недостаточности), при развитии асцита при циррозе печени (застой крови при портальной гипертензии). По такому механизму развиваются отеки при тромбофлебитах, отеки ног у беременных, т.к. повышается местное венозное давление из-за обтурации или сдавления вен.
2. Онкотический фактор. При уменьшении величины онкотического давления крови (рис. 12-45, Ъ, с) возникают отеки, механизм развития которых связан с падением величины эффективной онкотической всасывающей силы. Белки плазмы крови, обладая высокой гидрофильностью, удерживают воду в сосудах и, кроме этого, в силу значительно более высокой концентрации их в крови по сравнению с межтканевой жидкостью стремятся перевести воду из межтканевого пространства в кровь. Помимо этого увеличивается поверхность сосудистой площади (b1А2, а не ЪА, как в норме), через которую происходит процесс фильтрации жидкости при одновременном уменьшении резорбционной поверхности сосудов (А2с1, а не Ас, как в норме).
Таким образом, существенное уменьшение величины онкотического давления крови (не менее чем на 1/3) сопровождается выходом жидкости из сосудов в ткани в таких количествах, которые не успевают транспортироваться обратно в общий кровоток, даже несмотря на компенсаторное усиление лимфообращения. Происходит задержка жидкости в тканях и формирование отека.
Впервые экспериментальные доказательства значения онкотического фактора в развитии отеков были получены Э. Старлингом (1896). Оказалось, что изолированная лапа собаки, через сосуды которой перфузировали изотонический раствор поваренной соли, становилась отечной и прибавляла в массе. Масса лапы и отечность резко уменьшались при замене изотонического раствора поваренной соли на белковосодержащий раствор сыворотки крови.
Онкотический фактор играет важную роль в происхождении многих видов отеков: почечных (большие потери белка с мочой - протеинурия), печеночных (снижение синтеза белков-альбуминов в печени при ее заболеваниях - гипопротеинемия, уменьшение
альбумино-глобулинового коэффициента), голодных, кахектических. По механизму развития отеки, в возникновении которых ведущим является снижение онкотического давления крови, называются онкотическими.
3. Проницаемость сосудистой стенки. Увеличение проницаемости сосудистой стенки способствует возникновению и развитию отеков. Такие отеки по механизму развития называются мембраногенными. Однако повышение проницаемости сосудов может привести к усилению процессов как фильтрации в артериальном конце капилляра, так и резорбции в венозном конце. При этом равновесие между фильтрацией и резорбцией воды может и не нарушаться. Поэтому здесь большое значение имеет повышение проницаемости сосудистой стенки для белков плазмы крови, вследствие чего падает эффективная онкотическая всасывающая сила - в первую очередь за счет увеличения онкотического давления тканевой жидкости. Отчетливое повышение проницаемости капиллярной стенки для белков плазмы крови отмечается, например, при остром воспалении - воспалительном отеке. Содержание белков в тканевой жидкости при этом резко нарастает в первые 15-20 мин после действия патогенного фактора, стабилизируется в течение последующих 20 мин, а с 35-40-й мин начинается вторая волна увеличения концентрации белков в ткани, связанная, по-видимому, с нарушением лимфооттока и затруднением транспорта белков из очага воспаления (см. главу 10). Нарушение проницаемости сосудистых стенок при воспалении связано с накоплением медиаторов повреждения, а также с расстройством нервной регуляции тонуса сосудов.
Проницаемость сосудистой стенки может повышаться при действии некоторых экзогенных химических веществ (хлор, фосген, дифосген, люизит и др.), бактериальных токсинов (дифтерийный, сибиреязвенный и др.), а также ядов различных насекомых и пресмыкающихся (комары, пчелы, шершни, осы, змеи и др.). Под влиянием воздействия этих агентов, помимо повышения проницаемости сосудистой стенки, происходит нарушение тканевого обмена и образование продуктов, усиливающих набухание коллоидов и повышающих осмотическую концентрацию тканевой жидкости. Возникающие при этом отеки называются токсическими.
К мембраногенным отекам относятся также нейрогенные (вследствие нарушения нервной регуляции сосудистого тонуса, например при нейродистрофических процессах) и аллергические отеки
(вследствие воздействия медиаторов аллергии при аллергических заболеваниях). Например, велика роль медиаторов (гистамина, комплемента и др.) в развитии различных форм отека Квинке (немецкий врач-терапевт, описавший в 1882 г. острый локальный ангионевротический отек). Различают аллергический (см. главу 8) и неаллергический (наследственный, связанный с дефицитом ингибиторов протеаз, и в частности ингибитора С1-эстеразы системы комплемента) отеки Квинке. Чаще эти отеки развиваются на лице и в глотке, но могут затрагивать и внутренние органы (пищевод, желудок, кишечник, матку и другие органы и ткани).
Повышение проницаемости сосудистой стенки отмечается также при тромбоцитопениях, ацидозах.
4. Лимфообращение (лимфатический фактор). Затруднение транспорта жидкости и белков по лимфатической системе из интерстициального пространства в общий кровоток создает благоприятные условия для задержки воды в тканях и развития отеков. Так, например, при повышении давления в системе верхней полой вены (недостаточность правого сердца, сужение устья полых вен) возникает мощный прессорный рефлекс на лимфатические сосуды организма, вследствие чего затрудняется отток лимфы из тканей. Такое нарушение лимфообращения называется механической лимфатической недостаточностью и является одним из важных механизмов развития отека при сердечной недостаточности, а также при циррозе печени. Механическая лимфатическая недостаточность развивается также при закупорке лимфатических сосудов филяриями, при сдавлении лимфатических сосудов опухолью, экссудатом, рубцом, увеличенным соседним органом и др.
При значительном понижении содержания белков в крови (ниже 40 г/л), например, при нефротическом синдроме, линейная и объемная скорости лимфооттока возрастают в несколько раз. Однако, несмотря на это, вследствие чрезвычайно интенсивной фильтрации жидкости из сосудов в ткани (см. роль коллоидно-осмотического фактора) лимфатическая система не в состоянии возвращать в общий кровоток столь значительные объемы тканевой жидкости. В связи с перегрузкой транспортных возможностей лимфатических путей возникает так называемая динамическая лимфатическая недостаточность. Этот патогенетический фактор играет важную роль в формировании отеков при нефротическом синдроме.
Существует также резорбционная лимфатическая недостаточность, возникающая при увеличении концентрации белков в тка-
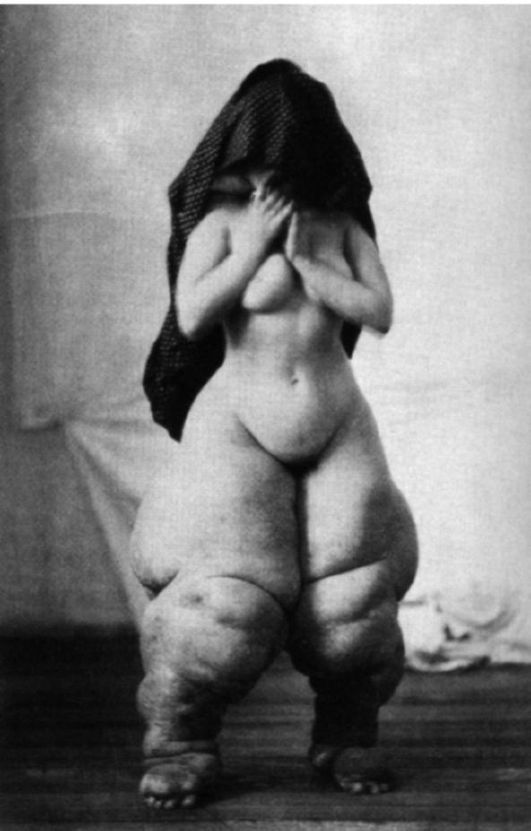 Рис. 12-46. Слоновость нижних конечностей (O.G. Mason, 1878)
Рис. 12-46. Слоновость нижних конечностей (O.G. Mason, 1878)
ни, например при воспалении. Молекулы белка удерживают воду в ткани, интенсивность лимфооттока уменьшается, и развивается отек.
В некоторых случаях роль лимфатического фактора в механизме развития отеков настолько непосредственна и велика, что выделяют так называемые лимфатические отеки. Примером может служить слоновость (elephantiasis) (рис. 12-46). Заболевание встречается преимущественно в тропических странах и возникает вследствие механической закупорки лимфатических сосудов круглыми паразитическими червями - филяриями. Развивающаяся при этом механическая лимфатическая недостаточность является ведущим патогенетическим механизмом формирования сильнейшей отечности конечностей (масса одной нижней конечности может достигать 50 кг и более), половых органов и других частей тела (по типу анасарки). Заболевание быстро приводит к инвалидности.
5. Активная задержка электролитов и воды (осмотический фактор). Важным звеном в развитии многих видов отеков (сердечные, почечные, печеночные и др.) является включение механизмов, активно задерживающих электролиты и воду в организме. Регуляцию постоянства электролитного состава жидкостных сред организма и их объема в норме осуществляют антидиуретическая и антинатрийуретическая системы. Раздражение осморецепторов гипоталамуса (при повышении осмотического давления крови) и волюморецепторов предсердий (при снижении объема циркулирующей крови) сопровождается увеличением секреции АДГ гипоталамусом, а также альдостерона надпочечниками (см. раздел 12.8.1, а также рис. 12-47, 12-48). Основным регулятором выработки альдостерона является РААС.
 Рис. 12-47. Автоматическая регуляция гомеостаза натрия и воды. АДГ антидиуретический гормон
Рис. 12-47. Автоматическая регуляция гомеостаза натрия и воды. АДГ антидиуретический гормон
Рис. 12-48. Нервно-гуморальное звено в механизме задержки воды и солей при сердечной недостаточности. АДГ - антидиуретический гормон
При сердечной недостаточности, циррозе печени, недостаточной функции почек, сопровождающейся активацией РААС, обнаруживается значительное повышение содержания альдостерона в крови (вторичный альдостеронизм). Секреция АДГ при этих состояниях также возрастает. Установлено, что стойкий гиперальдостеронизм при сердечной недостаточности и циррозе печени является результатом не только повышенной секреции клубочковой зоной коры надпочечников, но и пониженной инактивации альдостерона печенью. При этом наблюдается увеличение объема внеклеточных жидкостей организма, которое, казалось, должно было бы снизить секрецию альдостерона и АДГ и тем самым уменьшить задержку воды и солей в организме. Однако этого не происходит. При указанных патологических состояниях избыток альдостерона и АДГ уже не выполняет защитной роли, и механизмы, направленные на сохранение гомеостаза у здорового человека, в этих условиях «ошибаются», в результате чего задержка воды и солей усугубляется. В этом отношении отечные состояния могут рассматриваться, по Г. Селье, как «болезни адаптации».
В клинике наиболее часто встречаются сердечные, почечные (нефротические и нефритические), нейрогенные отеки, а также асцит при циррозе печени.
Сердечные отеки. Такие отеки возникают при развитии сердечной недостаточности (см. главу 15). Наибольшей выраженности они достигают в стадию сердечной декомпенсации, однако могут быть выявлены еще задолго до этой стадии, особенно при физической нагрузке.
В формировании сердечных отеков принимают участие многие патогенетические факторы, значимость и последовательность включения которых меняется по мере нарастания повреждения деятельности сердечно-сосудистой системы. Несомненно, что венозный застой крови и повышение гидростатического давления (см. роль гидростатического фактора) играют ведущую роль в развитии сердечных отеков. Поэтому эти отеки называют застойными.
Нарушение кровоснабжения почек из-за застойных явлений сопровождается активацией деятельности РААС. Увеличение в крови биологически активных пептидов (ангиотензина-II и ангиотензина-III) сопровождается усилением синтеза альдостерона и антидиуретического гормона (повышению концентрации этих гормонов в крови способствует также снижение инактивации их в печени) с последующей задержкой воды и натрия в организме и
тканях. Происходит активная задержка электролитов и воды (осмотический патогенетический фактор отека).
Нарушение кровоснабжения печени из-за застойных явлений сопровождается снижением синтеза белков в печени, снижается онкотическое давление крови, что также является одним из патогенетических факторов отека.
Повышение давления в полых венах (при недостаточности правых отделов сердца) вызывает рефлекторный спазм лимфатических сосудов, приводя к механической лимфатической недостаточности, что также является важным звеном формирования сердечных отеков.
Нарастающее расстройство общего кровообращения вызывает гипоксию тканей с последующим расстройством их трофики, развитием ацидоза и повышением проницаемости стенки сосудов (см. рис. 12-48).
Почечные отеки. При заболеваниях почек происходит нарушение водно-электролитного обмена, сопровождающееся задержкой воды в организме и возникновением отеков (см. главу 19). Характерной локализацией почечных отеков являются веки, лицо, при прогрессировании развивается отечность всего тела (анасарка), скопление жидкости в серозных полостях (асцит, гидроторакс, гидроперикардиум и др.).
Почечный нефротический отек (при нефротическом синдроме). При развитии нефротического синдрома ведущее место в формировании отека принадлежит резкому уменьшению содержания белков плазмы крови - гипопротеинемии. Это обусловлено большой потерей белков плазмы крови с мочой (главным образом альбуминов, но также теряются и другие белки: церулоплазмин, трансферрин, гаптоглобин, γ-глобулин и др.). Протеинурия связана с повышением проницаемости почечных клубочков для белков и нарушением реабсорбции белков в почечных канальцах. При этом концентрация белков в крови может падать до 30- 20 г/л и ниже, а суточная потеря белков с мочой достигает 30- 50 г (в норме не превышает 50 мг/сутки). Отсюда становится понятным значение онкотического патогенетического фактора в развитии нефротических отеков. Поэтому данный отек по патогенезу называется онкотическим.
Усиленная транссудация жидкости из капилляров в ткани и развитие из-за этого динамической лимфатической недостаточности могут способствовать появлению гиповолемии (уменьшению объ-
ема циркулирующей крови) с последующей мобилизацией ренинангиотензин-альдостеронового механизма задержки натрия и воды в организме (рис. 12-49).
При нефротическом синдроме отмечается увеличение эффективного гидростатического давления вследствие сдавления отечной тканью венул, включающее гидростатический патогенетический фактор отека.
Определенное значение в механизме отека при нефротическом синдроме может иметь повышение проницаемости сосудистой стен-
 Рис. 12-49. Патогенетические факторы, участвующие в развитии отеков при нефротическом синдроме
Рис. 12-49. Патогенетические факторы, участвующие в развитии отеков при нефротическом синдроме
ки вследствие интоксикации и ацидоза, которые развиваются при почечной недостаточности.
Почечный нефротический отек (при гломерулонефрите). В патогенезе отеков при хроническом гломерулонефрите большое значение имеет снижение клубочковой фильтрации, что уже само по себе может вести к задержке воды и солей в организме. Кроме того, в крови больных гломерулонефритом нередко отмечается повышенная концентрация альдостерона и АДГ. Это обусловлено нарушением внутрипочечной гемодинамики с последующим включением РААС, активной задержки электролитов и воды (т.е. работает осмотический патогенетический фактор отека). При гломерулонефрите часто отмечается повышение проницаемости обширной части капиллярной системы организма - развивается «генерализованный капиллярит». Имеются сведения и о повышении активности плазменного калликреина у больных с гломерулонефритом, что также ведет к увеличению сосудистой проницаемости. При гломерулонефритах развитие отеков обеспечивается также включением гидростатического патогенетического фактора отека с обширной задержкой воды в тканях вследствие увеличения ОЦК и развития артериальной гипертензии.
Асцит и отек при циррозе печени. При циррозе печени, наряду с местным скоплением жидкости в брюшной полости (асцитом), имеет место скопление ее в тканях и межтканевых пространствах организма (печеночные отеки). Первичным моментом возникновения асцита при циррозе печени является затруднение внутрипеченочного кровообращения с последующим повышением гидростатического давления в системе воротной вены (портальная гипертензия) и выходом жидкости в брюшную полость. Поэтому асцит по патогенезу называется застойным. Постепенно скапливающаяся внутри брюшной полости жидкость повышает внутрибрюшное давление до такой степени, что оно противодействует выходу жидкости из сосудов в полость и затрудняет дальнейшее развитие асцита.
Важная роль в механизме развития отеков и асцита при циррозе печени отводится активной задержке натрия и воды в организме. Отмечено, что концентрация натрия в моче, слюне и поте при асците низкая, а концентрация калия высокая. Все это указывает на повышение секреции в организме альдостерона, а также антидиуретического гормона. Происходит это из-за активации РААС вследствие недостаточности кровоснабжения почек при порталь-
ной гипертензии. Кроме этого имеется недостаточная инактивация альдостерона и АДГ в печени.
При нарушении способности печени синтезировать альбумины понижается онкотическое давление крови вследствие развивающейся гипоальбуминемии, и к перечисленным выше факторам, участвующим в механизме развития отека и водянки, присоединяется еще онкотический.
С повышением гидростатического давления в области воротной вены резко усиливается лимфоотток. При развитии асцита транссудация жидкости превосходит транспортную емкость лимфатических путей, и развивается динамическая лимфатическая недостаточность.
При заболеваниях печени (циррозах, гепатитах) отмечается увеличение содержания медиаторов воспаления, отмечаются гипоксия и ацидоз в тканях брюшной полости, это способствует повышению проницаемости сосудов (мембраногенный фактор отека).
Нейрогенные отеки. При некоторых видах отеков роль нервной системы выступает наиболее ярко и непосредственно, так как нервная система обеспечивает трофику сосудистой стенки и обменных процессов в тканях. Такие отеки называют нейрогенными. В их происхождении важную роль играют повышение проницаемости сосудистых стенок и нарушение обмена веществ в тканях с развитием метаболического ацидоза. Так, например, развиваются отеки конечностей при сирингомиелии (образование полостей в сером веществе спинного мозга) и сухотке спинного мозга (поражение задних рогов и столбов спинного мозга). Невралгия тройничного нерва нередко сопровождается развитием отека лица. К нейрогенным относятся отеки кожи при истерии, контузионные отеки и др.
Значение отека для организма. Как указывалось выше, в образовании различных видов отеков (сердечные, почечные, печеночные, кахектические, токсические, воспалительные, аллергические, нейрогенные и др.) участвуют многие общие механизмы: повышение гидростатического давления в сосудах, увеличение проницаемости сосудистой стенки для белков плазмы крови, повышение онкотического давления тканевой жидкости, уменьшение онкотического давления крови, недостаточность лимфообращения и возврата жидкости из тканей в кровь, включение механизмов, активно задерживающих натрий и воду в организме.
Можно выделить несколько фаз в образовании отеков. В первую фазу имеет место увеличение массы, связанной фиксирован-
ной с основным веществом соединительной ткани воды. Когда масса связанной воды выравнивает величину межтканевого давления до атмосферного (в норме эта величина на 6-7 мм рт.ст. ниже величины атмосферного давления), развивается вторая фаза обводнения, характеризующаяся накоплением свободной межтканевой жидкости, - видимый отек.
При формировании отеков включается ряд компенсаторных механизмов, направленных на предотвращение, а по возможности и на устранение отека, - резко увеличивается лимфоотток, активнее вымываются мелкодисперсные белки из межтканевой жидкости, что снижает ее коллоидно-осмотическое давление. Видимый отек не развивается до тех пор, пока отрицательное давление межтканевой жидкости не будет выравнено с атмосферным или превысит его. Если резервные возможности организма повышаются, а видимого отека еще не наблюдается, говорят о предотечном состоянии.
Однотипность механизмов формирования отека у животных и у человека позволяют отнести его к типовым патологическим процессам. Подобно любому типовому патологическому процессу, воздействие отека на организм является повреждающим, но при этом он имеет и защитно-приспособительное значение для организма.
Развитие отека приводит к механическому сдавлению тканей и нарушению в них кровообращения. Избыток межтканевой жидкости затрудняет обмен веществ между кровью и клетками. Вследствие нарушения трофики отечные ткани легче инфицируются, иногда отмечается развитие в них соединительной ткани. Если отечная жидкость является гиперосмолярной (например, у больных с сердечными отеками, которые нарушают солевой режим), наступает обезвоживание клеток с мучительным чувством жажды, повышением температуры, двигательным беспокойством и т.д. Если же отечная жидкость гипоосмолярная, развивается отек клеток с клиническими признаками водного отравления. Нарушение электролитного баланса при отеках может привести к нарушению кислотно-щелочного состояния жидких сред организма. Опасность отека в значительной мере определяется его локализацией. Скопление жидкости в полостях головного мозга, сердечной сумке, в плевральной полости нарушает функции этих важных органов и нередко угрожает жизни.
Из защитно-приспособительных реакций следует указать на следующие: переход жидкости из сосудов в ткани способствует освобождению крови от растворенных в ней (иногда токсических) веществ и сохранению изоосмолярности жидкостных сред орга-
низма; отечная жидкость способствует уменьшению концентрации различных химических и токсических веществ в крови, снижая их патогенное действие. При воспалительных, аллергических, токсических и некоторых других видах отеков вследствие затруднения оттока крови и лимфы из очага повреждения (отечная жидкость сдавливает кровеносные и лимфатические сосуды) происходит уменьшение всасывания и распространения по организму различных токсических веществ.
12.8.7. Принципы терапии водно-электролитных нарушений
Важнейшими звеньями терапии водно-электролитных нарушений являются: 1) восстановление и поддержание нормального объема циркулирующей крови и других водных пространств организма; 2) ликвидация наиболее опасных нарушений баланса электролитов и сдвигов кислотно-щелочного состояния; 3) восстановление и поддержание диуреза на уровне, обеспечивающем баланс воды и электролитов в организме; 4) обеспечение нормального распределения жидкостных объемов и электролитов по секторам.
Осмысленное осуществление перечисленных мероприятий невозможно без установления причин развития водно-электролитных нарушений, правильной интерпретации клинических симптомов и данных лабораторных исследований.
Важное место в коррекции водно-электролитных нарушений принадлежит инфузионной терапии (метод лечения, направленный на восстановление и поддержание нормального объема и электролитного состава жидких сред организма), медикаментозной терапии тех патологических состояний, которые привели к нарушению водно-электролитного обмена, и соответствующей диетотерапии.
12.9. ПАТОФИЗИОЛОГИЯ МИНЕРАЛЬНОГО ОБМЕНА
12.9.1. Нарушения обмена макроэлементов
Белки, липиды, углеводы, нуклеиновые кислоты организма человека и животных состоят в основном из углерода, кислорода, водорода и азота. Необходимым для существования организма является также ряд других химических элементов, которые по суточной потребности и содержанию в организме могут быть условно разделены на макроэлементы (Ca, P, K, Na, S, Cl, Mg) и микроэлементы (Fe, Cu, Zn, Mn, Se, Mo, Cr, I, Co, F).
Нарушения обмена натрия
На долю натрия - основного катиона внеклеточной жидкости, определяющего ее осмолярность, приходится более 90% всех катионов плазмы крови. Третья часть натрия связана в костной ткани, остальные ионы свободно обмениваются. Нормальная концентрация натрия в сыворотке крови у взрослых составляет 130-145 ммоль/л, в эритроцитах - 13-22 ммоль/л.
Na+/К+-АТФаза, входящая в состав мембран клеток и осуществляющая активное выведение ионов натрия из цитоплазмы во внеклеточную среду, поддерживает градиент натрия. Он образует на клеточной мембране электрохимический потенциал, обеспечивающий функции возбудимости и проводимости нервной и мышечной тканей. Натрий играет ведущую роль в поддержании осмотического давления внеклеточной жидкости и ее объема, участвует в регуляции кислотно-основного состояния, в транспорте углекислого газа, в активном переносе глюкозы и аминокислот в клетку, в поддержании пространственной конфигурации биомолекул.
Ведущую роль в поддержании гомеостаза натрия, который содержится в обычном рационе в количествах, в несколько раз превышающих потребность организма, играют почки. Основными факторами регуляции почечной экскреции натрия являются ангиотензин-II и альдостерон, снижающие выведение иона в периоды ограниченного поступления его с пищей, а также предсердный натрийуретический фактор (секретируемый предсердиями при их растяжении, вызванном повышением артериального давления, в том числе связанном с увеличением объема плазмы), стимулирующий экскрецию натрия. Стимуляция центра жажды, а также высвобождение вазопрессина (антидиуретического гормона - АДГ) из секреторных гранул в ответ на повышение осмотического давления межклеточной жидкости позволяют восстановить нормальную концентрацию натрия за счет увеличения объема воды в организме.
Гипонатриемия - концентрация натрия в сыворотке крови ниже 130 ммоль/л. Различают относительную и абсолютную гипонатриемию.
Абсолютная гипонатриемия имеет место при общем дефиците натрия в организме. Дефицит натрия в организме, объясняющийся недостаточным его поступлением, может быть вызван неадекватным парентеральным питанием, а также встречается у больных с сердечно-сосудистой недостаточностью, вынужденных длительное
время соблюдать бессолевую диету. Чрезмерные потери макроэлемента через почки наблюдаются при полиурической фазе острой почечной недостаточности, лечении диуретиками, недостаточной выработке минералокортикоидов (болезнь Аддисона и др.) и некоторых других заболеваниях. Натрий может теряться через кожу при обильном потоотделении, ожогах, генерализованном дерматите и через желудочно-кишечный тракт при продолжительной рвоте, диарее и кровотечениях.
Относительная гипонатриемия развивается при нормальном или даже повышенном общем запасе натрия в организме за счет его разведения в увеличенном объеме экстрацеллюлярной жидкости, что, например, имеет место при задержке воды в организме под действием АДГ, при нефротическом синдроме.
По скорости развития различают острую и хроническую гипонатриемию. Острая гипонатриемия развивается быстро, в сроки не более 48 ч (при введении больным больших количеств не содержащих натрий растворов, например до 15 литров 5% раствора глюкозы в сутки и более; при психогенной полидипсии).
Хроническая гипонатриемия встречается чаще, чем острая. Это состояние можно наблюдать у больных с отеками (сердечная недостаточность, цирроз печени, нефротический синдром и др.)
Гипонатриемия может сочетаться с повышенной, нормальной и пониженной осмолярностью внеклеточной жидкости. На этом основании различают гипертоническую, изотоническую и гипотоническую гипонатриемию.
Гипертоническая (транслокационная) гипонатриемия возникает при перемещении воды из клеток во внеклеточное пространство. Может быть вызвана быстрым возрастанием содержания в плазме крови глюкозы или другого осмотически активного вещества, наблюдается чаще всего при сахарном диабете или при внутривенном введении значительных количеств глюкозы или маннитола за короткое время. При этом концентрация содержащегося в плазме крови натрия снижается из-за разведения. Для этого состояния характерна дегидратация клеток.
Изотоническая гипонатриемия возникает вследствие разведения крови при задержке в экстрацеллюлярном пространстве изотонических жидкостей, не содержащих натрий (например, при внутривенной инфузии изотонической глюкозы).
Гипотоническая (гипоосмолярная) гипонатриемия характеризуется избытком воды по отношению к имеющемуся в организме
запасу натрия, подразделяется на гиперволемическую, нормоволемическую и гиповолемическую.
Гиперволемическая гипотоническая гипонатриемия развивается при неадекватной продукции АДГ (увеличение выработки гормона в гипоталамусе отмечается при тошноте, рвоте, эмоциональных расстройствах, кровоизлияниях в мозг, воспалении мозга; эктопическая секреция гормона возможна в ткани опухоли - раке бронхов, тимуса, средостения, предстательной железы, лимфомах и др.), при застойной сердечной недостаточности (в случае выраженной гиперсекреции АДГ в ответ на уменьшение объема сердечного выброса), олигурической фазе острой почечной недостаточности, первичной психогенной полидипсии. Проявляется головной болью, апатией, спутанностью сознания, отеками, увеличением массы тела, артериального давления, спазмами мышц, судорогами.
Нормоволемическая гипотоническая гипонатриемия может развиться при выраженном гипотирозе, отмене больших доз глюкокортикоидов, выраженном снижении в организме содержания калия, приеме тиазидных диуретиков и др.
Гиповолемическая гипотоническая гипонатриемия может развиваться при потерях содержащей натрий жидкости при ожогах, патологии желудочно-кишечного тракта, через почки: при передозировке диуретиков, первичном гипокортицизме, нефропатиях с потерей солей (в том числе при поликистозе, хроническом пиелонефрите и др.), а также в условиях удаления жидкости, выходящей в полости тела: выпот в плевру, быстрое развитие асцита (во всех этих случаях потери натрия обычно несколько преобладают над потерями воды). Это же состояние может развиваться при потерях изотонической жидкости через желудочно-кишечный тракт, почки или кожу в условиях частичного возмещения только воды, но не натрия.
Наиболее частыми причинами гипонатриемии у взрослых являются лечение тиазидами, неадекватная продукция АДГ в послеоперационном периоде, полидипсия у психических больных; у новорожденных и детей - потеря желудочно-кишечных соков при рвоте, диарее, мнократных клизмах водопроводной водой.
У ослабленных пациентов нередко отмечается повышение проницаемости клеточных мембран для натрия при нормальной или сниженной активности натриевого насоса («синдром больной клетки»). Ионы натрия при этом тяжелом метаболическом нарушении
по градиенту концентраций перемещаются внутрь клеток, ионы калия выходят в межклеточное пространство. Неадекватный ионный состав содержимого клеток приводит к еще более глубоким нарушениям метаболизма, что формирует «порочный круг». Наряду с этим у некоторых больных может наблюдаться неадекватная секреция вазопрессина, вызванная влиянием стресса.
Клинические проявления гипонатриемии в основном касаются нарушений функций ЦНС - головная боль, тошнота, рвота, спазм мышц, сонливость, дезориентация, беспокойство и торможение рефлексов. Неврологическая симптоматика гипонатриемии проявляется при снижении уровня натрия в сыворотке крови до 125-120 ммоль/л.
При гиповолемической гипонатриемии клинические проявления связаны в основном со снижением объема внеклеточной жидкости: отмечаются слабость, апатия, раздражительность, головокружение, гипотензия, тахикардия, обмороки при переходе из горизонтального в вертикальное положение, сухость слизистых оболочек. При быстро развивающейся гипоосмолярной гипонатриемии могут возникать судороги, кома, остановка дыхания, мозговая грыжа, может наступить летальный исход. В основе этих тяжелых нарушений лежит усиленное поступление воды в клетки мозга, так как в клетках осмотическое давление будет выше, чем в экстрацеллюлярной жидкости. Развивается отек мозга, возрастает внутричерепное давление. При медленном развитии гипонатриемии в организме включаются адаптивные реакции - осмотически активные вещества начинают покидать ткань мозга, что сопровождается выходом из клеток воды. Этот механизм препятствует развитию отека мозга.
Гипернатриемия всегда вызывает перераспределение воды между внутриклеточной и внеклеточной средами в пользу последней и клеточную дегидратацию. Гипернатриемия соответствует дефициту воды по отношению к запасам натрия в организме. Повышение концентрации натрия в сыворотке крови на 3-4 ммоль/л соответствует дефициту 1 л воды в организме.
Причинами гипернатриемии могут стать обезвоживание (профузное потение с низким содержанием натрия в выделяемой жидкости, наблюдающееся при тяжелой физической нагрузке и высокой температуре окружающего воздуха); несахарный диабет (избыточная потеря воды без натрия); нарушение механизма жажды; прием внутрь морской воды, недоступность питьевой воды и
избыток натрия при парентеральном введении гипертонического солевого раствора или бикарбоната натрия, а также при нарушении выведения макроэлемента (почечная недостаточность; избыток минералокортикоидов при застойной сердечной недостаточности, нефротическом синдроме, стенозе почечной артерии, декомпенсированном циррозе печени, синдромах Кушинга и Конна).
Симптомы гипернатриемии связаны с дегидратацией в первую очередь нервных клеток - происходит их сморщивание. Возможны разрывы субарахноидальных сосудов и кровоизлияния, что вызывает стойкое повреждение мозга и летальный исход. При медленном развитии гипернатриемии включается приспособительная реакция клеток мозга - происходит захват электролитов из экстрацеллюлярной жидкости, вслед за ними поступает вода, что нормализует объем мозга.
Клинические проявления гипернатриемии - жажда, одышка, отеки, венозный застой, гипертензия, избыточная масса тела, потливость, усталость, беспокойство, возбуждение, усиление мышечного тонуса, кома.
Суточная потребность организма взрослого человека в хлориде натрия составляет около 10 г.
Нарушения обмена калия
Калий является основным внутриклеточным катионом. Концентрация этого минерала в клетках в десятки раз превышает его содержание во внеклеточной жидкости (в сыворотке крови калия содержится 3,7-5,2 ммоль/л, в эритроцитах - 77-96 ммоль/л). В клетках организма калий находится в свободном и связанном с белками, глюкозой, фосфатами и другими соединениями виде; внеклеточная жидкость содержит только 2% от общего его количества, преимущественно в виде ионов, концентрация которых поддерживается в узком диапазоне. Уровень калия в плазме крови не является показателем его общего содержания в организме. Главная биологическая роль калия - создание трансмембранного потенциала, обеспечивающего в том числе возбудимость и проводимость нервных и мышечных клеток. Основным механизмом его поддержания является работа Nа+/К+-АТФазы. Распределение калия между внеклеточной жидкостью и внутриклеточной средой зависит от рН (ацидоз способствует развитию гиперкалиемии, алкалоз - гипокалиемии), некоторых гормонов (инсулин, адрена-
лин, альдостерон), а также от концентрации катионов (например, магний влияет на активность Nа+/К+-насоса). Общее количество калия в организме и его содержание в плазме крови регулируются изменением количества экскретируемого минерала почками, которые в то же время не способны так же эффективно удерживать калий, как натрий, и поэтому выведение калия наблюдается даже при существенном недостатке его в организме.
Калий участвует в поддержании кислотно-основного состояния, осмотического давления, транспорте углекислого газа кровью, способствует всасыванию глюкозы и кальция в кишечнике, оказывает влияние на многие стороны внутриклеточного обмена веществ (например, активирует ферменты гликолиза енолазу и пируваткиназу, участвуя в синтезе АТФ).
Гипокалиемия может развиваться вследствие сниженного поступления (нерациональное парентеральное введение, редко дефицит в пище) и/или повышенного выведения калия из организма через почки (полиурическая фаза острой почечной недостаточности, избыток минералокортикоидов: первичный и вторичный альдостеронизм, синдром Кушинга и др.), желудочно-кишечный тракт (диарея, рвота, стеноз пилорического отдела желудка и др.), в результате усиленного потоотделения. Гипокалиемия может быть вызвана перемещением калия внутрь клеток (алкалоз, избыток инсулина, быстрая клеточная пролиферация после ожогов, травм или голодания, лечение β-адренергическими агонистами или повышение β-адренергической активности при стрессе, ишемии миокарда и др.). При гипокалиемии наблюдаются нарушения сердечных, нейромышечных, почечных функций: гипотония, мышечная слабость, спазмы мышц ног, сниженные рефлексы, запоры, рвота, нарушение концентрационной способности почек, ведущее к полиурии; на ЭКГ отмечаются тахикардия, аритмии, депрессия сегмента ST, депрессия или инверсия зубца Т, увеличение интервалов PR и QT, выраженный зубец U. Действие гипокалиемии на почки объясняется повышением синтеза простагландинов - антагонистов вазопрессина, нейромышечные и сердечные проявления - нарушением возбудимости и проводимости клеток. Тяжелая гипокалиемия (менее 2 ммоль/л) вызывает остановку сердца в систоле или паралич дыхательных мышц и летальный исход.
Гиперкалиемия развивается при избыточном поступлении калия в организм (внутривенное введение препаратов калия, переливание консервированной крови); сниженном выведении калия
(прием сохраняющих калий диуретиков или ингибиторов ангиотензинпревращающего фермента, острая и хроническая почечная недостаточность, недостаточность минералокортикоидов, например, при болезни Аддисона); выходе калия из клеток (ацидоз, недостаток инсулина, катаболические состояния при лихорадке, травме, сепсисе и др.). К неврологическим симптомам гиперкалиемии относятся раздражительность, беспокойство, парестезии, слабость, вялый паралич. На ЭКГ больных отмечаются прежде всего увеличение амплитуды и заострение зубца Т, затем уплощение зубца Р вплоть до его исчезновения, расширение комплекса QRS, удлинение интервала PQ, брадиаритмия. Тяжелая гиперкалиемия (13 ммоль/л) вызывает остановку сердца в диастоле.
Суточная потребность организма взрослого человека в калии составляет 2-5 г.
Нарушения обмена кальция и фосфора
Организм взрослого человека содержит около 1000 г кальция и 600 г фосфора, 99% кальция и 85% фосфора депонировано в скелете в виде кристаллов гидроксиапатита. В сыворотке крови кальций и фосфат содержатся в связанной с белками и другими соединениями форме, а также в виде ионов. Ионизированные формы кальция и фосфата являются физиологически активными. Содержание общего кальция в сыворотке крови в норме составляет 2,3-2,7 ммоль/л, ионизированного - 1,1-1,4 ммоль/л, неорганического фосфора - 0,7-1,4 ммоль/л.
На гомеостаз кальция и фосфора оказывают влияние паратиреоидный гормон, кальцитонин и кальцитриол, основными органамимишенями которых являются кости, почки и кишечник. Паратиреоидный гормон стимулирует освобождение кальция и фосфата из костной ткани и почечный синтез кальцитриола, а также усиливает реабсорбцию кальция и подавляет реабсорбцию фосфата в почках. Кальцитриол активирует резорбцию кости и усиливает почечную реабсорбцию кальция и фосфата и абсорбцию кальция и фосфата в кишечнике. Кальцитонин тормозит остеокластическую резорбцию, тем самым снижая высвобождение кальция и фосфата, а также способствует поступлению фосфата в клетки кости и периостальную жидкость.
Функции фосфора в организме не сводятся только к формированию минерального компонента кости. Он входит в состав ну-
клеиновых кислот, фосфопротеинов и фосфолипидов, участвует в метаболизме белков, липидов и углеводов, образовании макроэргических соединений (АТФ, креатинфосфата и др.), поддержании кислотно-основного равновесия, необходим для активации ряда ферментов и нормального функционирования нервов и мышц.
Ионы кальция принимают участие в мышечном сокращении, выделении нейромедиаторов, контроле возбудимости, внутриклеточном метаболизме, свертывании крови, поддержании целостности мембран, трансмембранном транспорте, высвобождении веществ, синтезируемых в клетке (в том числе гормонов), поступлении в клетку веществ путем фаго- и пиноцитоза, минерализации костей; являются кофакторами многих ферментов и неферментных белков. Кальций воздействует на разнообразные обменные процессы, выполняя функцию внутриклеточного мессенджера (в том числе в комплексе с кальмодулином служит посредником в передаче регуляторных сигналов). Концентрация свободных ионов кальция в цитозоле, поддерживаемая в пределах 0,1-10 мкмоль/л, зависит от активности Са2+-АТФазы, кальциевых каналов и от концентрации кальция во внеклеточной жидкости. Ряд гормонов (а1-адренергические катехоламины, вазопрессин и др.) изменяют проницаемость мембран для кальция, тем самым регулируя его вход в клетку, воздействуя на Nа+/Са2+-обменник. Возможны также как мобилизация кальция из митохондрий и эндоплазматического ретикулума, так и накопление его в этих органеллах.
В физиологических условиях происходит постоянное обновление костной ткани - ее резорбция протекает параллельно с образованием остеоида и его минерализацией. При различных нарушениях гормональной регуляции (избыток или недостаток кальцитриола, паратиреоидного гормона, кальцитонин, глюкокортикоидов, эстрогенов и андрогенов, тиреоидных гормонов, инсулина, глюкагона, соматотропина), а также при нарушениях обмена кальция и фосфата сдвигается динамическое равновесие, обеспечивающее стабильное состояние скелета.
К заболеваниям с нарушениями метаболизма кальция и фосфора относятся: рахит и остеомаляция, остеопороз, гиперпаратиреоз (генерализованная фиброзная остеодистрофия, болезнь Реклингхаузена), гипопаратиреоз (паратиреопривная тетания), почечная остеодистофия, паранеопластическая гиперкальциемия. Кроме того, выделяют наследственные формы нарушения кальциевого и фосфорного обмена (семейный гипофосфатемический рахит).
Повышение концентрации внутриклеточного кальция, регистрируемое при патологии, может служить причиной нарушения функций и даже гибели клеток различных органов и тканей организма. Вытеснение кальция из комплекса с кальмодулином ионами двухвалентных металлов (Cu, Zn, Co, Mn) вызывает угнетение регулируемых Са2+-кальмодулином клеточных процессов и играет важную роль в патогенезе интоксикации этими металлами.
Гипокальциемия обычно бывает вызвана возрастанием потерь кальция или нарушением регуляции его обмена между межклеточной жидкостью и костной тканью, а также затруднением абсорбции минерала в кишечнике большими количествами жиров, оксалатов или фитиновой кислоты. Основным механизмом развития гипокальциемии при недостатке витамина D в пище и/или нарушении его метаболизма у больных хронической печеночной и почечной недостаточностью, при нефротическом синдроме, мальабсорбции, лечении противосудорожными препаратами (фенобарбитал), наследственной недостаточности 1-а-гидроксилазы является снижение количества кальцитриола и нарушение абсорбции Са2+ в кишечнике.
Снижение концентрации ионизированного кальция в сыворотке крови может быть вызвано также врожденным или приобретенным гипопаратиреозом; псевдогипопаратиреозом I типа (вследствие генетического дефекта нарушен процесс активации аденилатциклазы, поэтому связывание паратиреоидного гормона с рецептором не приводит к образованию цАМФ) и II типа (нарушена реакция клетки на цАМФ); недостатком магния (снижение секреции и эффекта паратиреоидного гормона); гиперфосфатемией или введением больших количеств цитрата (связывание кальция анионами); алкалозом (увеличение связывания кальция альбуминами); опухолевым процессом (обладающие остеобластической активностью клеточные элементы опухоли могут задерживать кальций), хроническим алкоголизмом, острым панкреатитом и др.
Клинические проявления гипокальциемии в значительной степени связаны с повышением возбудимости нейронов и миоцитов и судорожным синдромом. У больных наблюдаются парестезии, судороги, спазмы мышц, тетания, ларингоспазм, положительные симптомы Труссо (I) (тоническая судорога кисти, придающая ей форму руки акушера, в ответ на сдавление в области плеча) и Хвостека (одностороннее сокращение мышц лица при перкуссии в области прохождения лицевого нерва). Судороги наблюдаются
преимущественно в сгибательных мышцах. При тетании сгибательных мышц нижних конечностей стопа сгибается внутрь, пальцы подгибаются к подошве («конская стопа»). Судороги лицевой мускулатуры сопровождаются тризмом и образованием «рыбьего рта». Может развиться пилороспазм с рвотой, тошнотой, спазмы мускулатуры кишечника, мочевого пузыря. Спазм венечных сосудов сердца сопровождается резкими болями в области сердца. Понижение сократимости сердечной мышцы приводит к развитию застойной сердечной недостаточности, гипотензии, увеличению интервала QT. При тяжелой гипокальциемии тетанус дыхательных мышц приводит к летальному исходу. Приступы тетании могут провоцироваться различными раздражителями: болевыми, механическими, термическими, гипервентиляцией. При длительной гипокальциемии могут развиться гипокальциемическая катаракта, ломкость ногтей, ломкость и кариес зубов. Наблюдаются изменения психики: снижение интеллекта, нарушение памяти, неврозы.
Повышение концентрации кальция в крови наблюдается при первичном или вторичном гиперпаратиреозе, развивающемся на фоне длительно существующей гипокальциемии (полиурическая фаза острой почечной недостаточности - период восстановления диуреза или поражение желудочно-кишечного тракта), гранулематозных заболеваний (повышенное образование кальцитриола мононуклеарными фагоцитами), отравления витамином D, лечения тиазидсодержащими диуретиками (снижение экскреции кальция с мочой), акромегалии, синдрома Иценко-Кушинга, тиреотоксикоза, идиопатической гиперкальциемии в раннем детском возрасте и др. Причинами развития гиперкальциемии при опухолевом росте могут являться секреция клетками опухоли пептида, подобного паратиреоидному гормону, избыточное образование простагландина Е, фактора некроза опухоли α или интерлейкина-1, активирующих остеокласты, а также резорбция кости при наличии в ней метастазов. Увеличение концентрации ионов кальция в сыворотке обнаруживается при ацидозе.
Гиперкальциемия проявляется слабостью, утомляемостью, гипотонией мышц нижних и верхних конечностей, болями в стопах, жаждой, расшатыванием и выпадением зубов, снижением массы тела. Основными симптомами при почечной форме гиперкальциемии являются: полидипсия, полиурия, гипоизостенурия, щелочная реакция мочи. Развивается двусторонний нефрокальциноз, иногда гидронефроз, что при длительном течении может привести к уре-
мии. Больных беспокоят диспептические расстройства (анорексия, тошнота, рвота, запоры), частые приступы почечной колики, повышение артериального давления. При висцеропатической форме гиперкальциемии часто развиваются пептические язвы двенадцатиперстной кишки, желудка, кишечника, которые склонны к рецидивированию и развитию кровотечений. Могут наблюдаться нарушение сна, снижение памяти, депрессия, спутанность сознания. В тяжелых случаях наблюдаются изменение личности, ступор, кома. На ЭКГ больных регистрируются укорочение сегмента ST, интервала QT, желудочковые аритмии.
Откладывание в органах (почки, роговица, сосуды, желчный пузырь и др.) фосфата кальция наблюдается в случае, если гиперкальциемия сопровождается нормальным или повышенным уровнем фосфата в сыворотке крови.
Восстановление уровня кальция в сыворотке крови в случае его повышения или снижения осуществляется гормональной регуляцией процессов абсорбции кальция в кишечнике, реабсорбции его в почках и резорбции кости (рис. 12-50, 12-51).
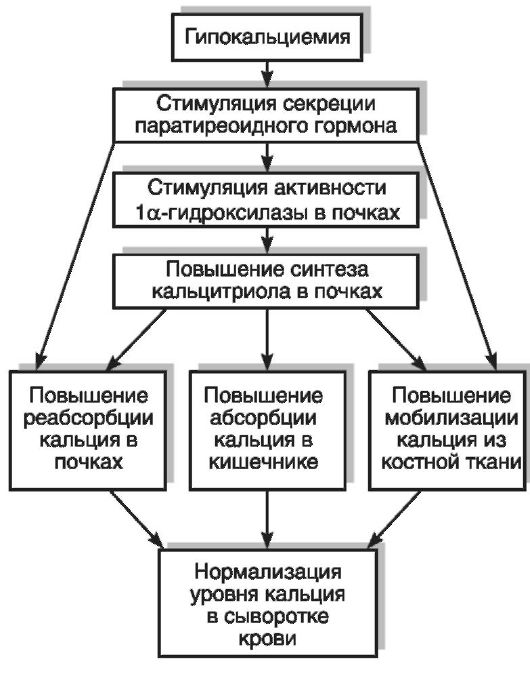 Рис. 12-50. Механизм коррекции гипокальциемии (по В.К. Бауману, 1989)
Рис. 12-50. Механизм коррекции гипокальциемии (по В.К. Бауману, 1989)
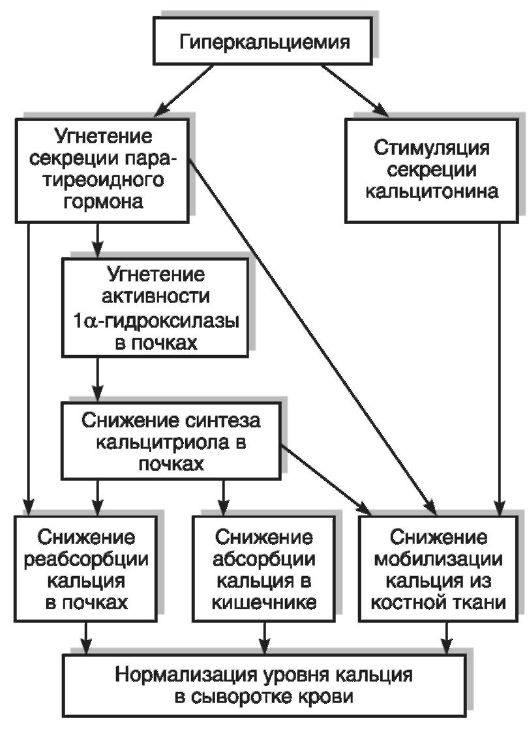 Рис. 12-51. Механизм коррекции гиперкальциемии (по В.К. Бауману, 1989)
Рис. 12-51. Механизм коррекции гиперкальциемии (по В.К. Бауману, 1989)
Гипофосфатемия может быть результатом неадекватного парентерального питания либо сниженной абсорбции фосфата из желудочно-кишечного тракта или его повышенных потерь через желудочно-кишечный тракт или почки при рвоте, диарее, мальабсорбции, дефиците витамина D, гиперпаратиреозе, использовании тиазидных диуретиков, гипомагниемии, семейном гипофосфатемическом рахите, алкоголизме и др. Концентрация фосфата в сыворотке крови может снижаться также при повышенном использовании его клетками (при заживлении ран и после голодания), а также в результате перехода фосфата в клетки при алкалозе.
Нарушения функций ЦНС при гипофосфатемии (ухудшение памяти, спутанность сознания, дискоординация движений, летаргия) объясняются в том числе и снижением образования макроэргических фосфатсодержащих соединений. Развивающиеся гипоксемия и гипоксия связаны со снижением содержания 2,3- дифосфоглицерата в эритроцитах. Наряду с этим хроническая ги-
пофосфатемия приводит к развитию признаков рахита и остеомаляции, проявляющихся болями в костях и переломами. У больных развивается мышечная слабость, в тяжелых случаях - острый рабдомиолиз (распад поперечно-полосатой мышечной ткани), а также уменьшение сократительной способности миокарда со снижением сердечного выброса и артериального давления. Снижение уровня АТФ и других фосфатсодержащих соединений в лейкоцитах и тромбоцитах крови ведет к развитию инфекций и возникновению кровотечений.
Гиперфосфатемия наиболее часто развивается при острой и хронической почечной недостаточности, а также при повышенном потреблении фосфата (при кормлении грудных детей неразбавленным коровьим молоком) или витамина D, гипопаратиреозе и псевдогипопаратиреозе, при перемещении фосфата из клеток во внеклеточную жидкость при дыхательном ацидозе и диабетическом кетоацидозе без лечения, высвобождении фосфата при катаболических состояниях (опухолевый лизис, рабдомиолиз). Выраженная гиперфосфатемия, приводящая к подавлению гидроксилирования 25-гидроксихолекальциферола в почках и нарушению образования кальцитриола, а также к образованию метастатических отложений гидроксиапатита, за счет этих двух механизмов вызывает гипокальциемию. Большинство клинических проявлений гиперфосфатемии связано с развитием гипокальциемии и отложением фосфата кальция в тканях (роговица, кровеносные сосуды, почки, легкие и др.). Кальцификация сердечной мышцы вызывает нарушение проводимости и аритмии, суставов - артралгию и ограничение их подвижности. В тяжелых случаях развивается гипокальциемическая тетания.
Суточная потребность организма взрослого человека в кальции составляет 0,8-1 г, в фосфоре - 1-1,5 г.
Нарушения обмена магния
Из 25 г магния, содержащихся в организме взрослого человека, 50-60% сосредоточено в костях, 1% - во внеклеточной жидкости, остальная часть - в клетках тканей (среди катионов по концентрации в клетках магний уступает только калию). 25-35% магния сыворотки крови связано с белками (в основном с альбуминами), небольшая часть присутствует в комплексных соединениях. Физиологически значимым является ионизированный (свободный)
магний. Гипоальбуминемия вызывает снижение общего содержания магния в сыворотке, количество ионизированного магния при этом остается в норме.
Магний является кофактором либо активатором более чем 300 ферментов, участвующих в обмене белков и нуклеиновых кислот (пептидгидролазы, аргиназа, аминоацил-тРНК-синтетаза, ДНК- и РНК-полимеразы, полинуклеотидфосфорилаза, ДНКаза, РНКаза и др.), углеводов (большинство ферментов гликолиза); принимает участие в трансмембранном транспорте ионов (поддерживает уровень ионов калия в клетке, принимая участие в работе Na+/ К+-насоса). Активируя ферменты цикла Кребса и участвуя в сопряжении окисления и фосфорилирования (АТФ-синтазной реакции), Mg2+ оказывает влияние на энергетический потенциал клетки. Взаимодействуя с кальцием, магний влияет на проницаемость мембран клеток и их электрические свойства, являясь регулятором механизма проводимости нейронов и волокон миокарда.
Гипомагаиемия (наблюдающаяся примерно у 10% госпитальных больных) может быть вызвана повышенными потерями магния из желудочно-кишечного тракта (рвота, диарея, дренирование желудка); нарушением всасывания - синдромом мальабсорбции (опухоли и резекции желудочно-кишечного тракта, энтериты, панкреатит); приемом лекарственных препаратов, повышающих почечную экскрецию (петлевые диуретики, гентамицин, цисплатин, циклоспорин и др.); нарушением функции почечных канальцев; хроническим избытком минералокортикоидов; хроническим алкоголизмом (сниженное поступление с пищей, нарушение абсорбции, повышенная экскреция); белково-калорийным голоданием (гипоальбуминемии сопутствует снижение содержания в сыворотке крови магния, связанного с белками).
Значительное уменьшение содержания в организме магния, нарушающее цАМФ-зависимые процессы секреции паратиреоидного гормона и пострецепторной передачи гормонального сигнала в клетках тканей-мишеней, влечет за собой развитие гипокальциемии. Гипомагниемия сопровождается гипофосфатемией. Развитие дефицита калия при гипомагниемии связано с повышенной его экскрецией почками и одновременно с нарушением регуляции активности Na+/К+-насоса (Mg2+ является кофактором Na+/К+-АТФазы). Клинические проявления гипомагниемии (при снижении концентрации ионов ниже 0,5 ммоль/л) зачастую обусловлены присоединившимися недостатками калия и кальция:
тошнота, рвота, анорексия, бессонница, изменения настроения, психические расстройства, бред, галлюцинации, атаксия, тремор, хорееподобные движения, судороги, мышечная слабость, тетания, положительные симптомы Хвостека и Труссо, парестезии. На ЭКГ регистрируются увеличение длительности интервалов PR и QT, депрессия сегмента ST, уплощение зубца Т; возможны тахиаритмии, фибрилляция предсердий.
Причинами гипермагниемии (приобретающей клиническое значение при концентрации ионов магния не менее 2,0 ммоль/л) могут стать употребление магнийсодержащих лекарственных препаратов (антациды, сульфат магния и др.) и снижение экскреции магния, встречающееся у больных с почечной недостаточностью и недостаточностью коры надпочечников (болезнь Аддисона), повышенное поступление магния в кровь при усиленном клеточном катаболизме. Избыток в организме магния проявляется тошнотой, рвотой, потливостью, сонливостью, мышечной слабостью, гипотензией вследствие периферической вазодилатации, снижением глубоких сухожильных рефлексов, кальцификацией мягких тканей. Магний в концентрации не менее 5,0 ммоль/л в сыворотке крови, воздействуя на проводимость сердечной мышцы, вызывает брадикардию, атриовентрикулярную блокаду. Тяжелая гипермагниемия (концентрация ионов превышает 7,5 ммоль/л), развивающаяся только при почечной недостаточности, вызывает паралич дыхания и остановку сердца.
Содержание магния в сыворотке крови в норме составляет 0,7- 1,1 ммоль/л. Суточная потребность организма взрослого человека в магнии - 0,3-0,5 г, из которых абсорбируется 30-40%.
12.9.2. Нарушения обмена микроэлементов
Эссенциальными (незаменимыми) микроэлементами на современном этапе развития науки считаются Fe, Cu, Zn, Mn, Se, Mo, Cr,
I, Co, F, для которых доказаны проявления синдромов истинного дефицита у человека. Наряду с этими микроэлементами для некоторых животных незаменимыми являются As, B, Br, Li, Ni, Si, V. А.П. Авцын (1983) разработал концепцию и классификацию патологических процессов, вызванных дефицитом, избытком или дисбалансом микроэлементов в организме, и предложил для них объединяющее название «микроэлементозы».
Нарушения обмена железа
В организме взрослого мужчины содержится 3-5 г железа, женщины - 3-4 г, из них 65-70% железа входит в состав эритроцитов и эритрокариоцитов; железо, связанное с ферритином и гемосидерином, составляет 20% его общего количества; 15% входит в состав миоглобина; около 1% - в состав гемовых ферментов и белков, содержащих негемовое железо; на долю транспортного железа, связанного с трансферрином, приходится 0,1-0,2%.
Железо входит в состав простетических групп окислительновосстановительных ферментов (оксидоредуктаз), обеспечивает транспорт электронов цитохромами и железосеропротеинами, транспорт и депонирование О2 и СО2 гемоглобином и миоглобином.
Железо участвует не только в процессах окислительного фосфорилирования и свободного окисления, но и в поддержании вторичной и третичной структуры ДНК и РНК, превращениях аминокислот по радикалу (фенилаланина в тирозин и др.), активации β-окисления ацил-КоА, повышении активности пептидгидролаз.
Во внеклеточных жидкостях железо содержится в виде трансферрина и лактоферрина. Внутриклеточные соединения железа можно подразделить на гемопротеины, структурным элементом которых является гем (гемоглобин, миоглобин, цитохромы, каталаза, пероксидазы и др.); негемовые железосодержащие ферменты (ацил-КоА-дегидрогеназа, пролилоксидаза, ксантиноксидаза и др.); ферритин и гемосидерин внутренних органов; железо, рыхло связанное с белками и другими органическими веществами.
Железодефицит (гипосидероз) - один из наиболее распространенных микроэлементозов человека. Гипосидероз может развиться при недостаточном поступлении железа с пищей, а также в результате нарушения кислотообразующей функции желудка (атрофический гастрит, тотальная и субтотальная гастроэктомия), нарушения его всасывания в кишечнике (обширная резекция тонкой кишки, хронический энтерит, конкурентная абсорбция цинка и меди, недостаток аскорбиновой кислоты, способствующей переводу железа в двухвалентую форму). Прием нестероидных противоревматических препаратов и некоторых антибиотиков также может стать причиной развития железодефицитных состояний. Распространенная причина дефицита железа - хронические кровотечения из мочеполовой системы и желудочно-кишечного тракта (в
том числе при язвенной болезни желудка, злокачественных опухолях, полименорее и др.). Потребность в железе возрастает при беременности, лактации, росте организма.
Первым следствием превышения расхода железа над поступлением его в организм является истощение его депо в печени, селезенке и других органах и тканях. Дальнейшее развитие гипосидероза приводит к разнообразным тканевым и органным повреждениям, многие из которых проявляются еще до наступления железодефицитной анемии.
Неспецифичные признаки железодефицита выражаются в легкой утомляемости, головокружении, головных болях, повышенной возбудимости или, напротив, депрессии. У детей ухудшаются внимание, память, замедляется умственное развитие. У некоторых больных наблюдается отсутствие аппетита или извращение вкуса (геофагия, пагофагия, амилофагия). Хронический недостаток железа вызывает поражения кожи и ее производных (сухость и трещины кожи, ломкость ногтей, выпадение волос), слизистых оболочек рта, глотки, пищевода, желудка, верхних дыхательных путей (ангулярный стоматит, глоссит, эзофагит, ларинго- и фаринготрахеит, гастрит). При анемизации проявляются симптомы, связанные с недостаточным обеспечением тканей кислородом: мышечная слабость (причиной которой является также нарушение метаболизма миоглобина), одышка, сердцебиение, обмороки. Дефицит железа вызывает угнетение иммунитета.
Железодефицитная гипохромная микроцитарная анемия является наиболее тяжелым следствием гипосидероза, приводящим к развитию гипоксии, поражению всех органов и тканей, стойкой утрате трудоспособности, вплоть до гибели организма. Прежде всего от гипоксии страдают чувствительные к кислородному голоданию ткани - нервная и эпителиальная. Причинами развития гипоэнергетических состояний в тканях, наряду с недостатком кислорода, являются нарушения в митохондриальной цепи переноса электронов, связанные с дефицитом железа, входящего в состав цитохромов и железосерных белков.
Предполагается, что нарушения со стороны нервной системы при дефиците железа связаны также с изменениями активности дофаминергических рецепторов и метаболизма γ-аминомасляной кислоты.
Гиперсидероз может возникать при избыточном содержании железа в пище, повышенном всасывании железа в кишечнике, при
неадекватном парентеральном введении препаратов железа при лечении рефрактерных анемий или многочисленных трансфузиях крови (трансфузионный сидероз), при хроническом усиленном гемолизе при талассемиях. Профессиональный сидероз (отложения железа в легких) нередко наблюдается у шахтеров и металлургов. Гиперсидероз может иметь местный (например, сидероз глазного яблока) и генерализованный характер.
Избыточное железо накапливается в основном в виде гемосидерина в клетках ретикулоэндотелиальной системы печени и селезенки, что со временем может привести к фиброзу печени. Повреждение миокарда при гиперсидерозе способствует развитию сердечной недостаточности. Токсическое действие избыточных концентраций железа во многом объясняется его участием в свободнорадикальных процессах (реакции Фентона). Длительная перегрузка организма железом ведет к накоплению в клетках ферритина и гемосидерина, способствующих нарушению целостности лизосомальных мембран и выходу протеолитических ферментов, повреждающих клеточные структуры.
Острое отравление железом может стать причиной некротического гастроэнтерита, некроза печени, почечной недостаточности, вплоть до летального исхода.
У человека известны два наследственных заболевания, затрагивающих обмен железа, - атрансферринемия, механизм возникновения которой до конца не изучен, и первичный гемохроматоз, основной причиной которого является избыточное всасывание железа в кишечнике. Отложения железа в клетках печени, селезенки, поджелудочной железы, сердца, надпочечников, развивающиеся при наследственном гемохроматозе, приводят к нарушению структуры и функций этих органов (цирроз печени, сахарный диабет, гепатоспленомегалия, сердечная недостаточность и т.д.).
У животных известны следующие генетические дефекты обмена железа: «анемия, сцепленная с полом» у мышей (sla); микроцитарная анемия у мышей (mk) и белградская анемия у крыс (b) - аутосомные рецессивные мутации, ведущие к развитию тяжелой гипохромной анемии.
Как избыток, так и дефицит железа оказывают эмбриотоксическое действие, проявляющееся в нарушении развития нервной и иммунной систем.
При нормальном питании в организм человека поступает около 15 мг железа в сутки, из них в кишечнике всасывается 1-1,5 мг,
при некоторых видах анемий - до 2-3 мг в день. Из организма железо выделяется с желчью, через почки и потовые железы, а также с менструальной кровью. При распаде гемоглобина 90% железа в организме реутилизируется, а 10% должно пополняться за счет пищи.
В норме концентрация сывороточного железа у мужчин составляет 14-25 мкмоль/л, у женщин - 11-22 мкмоль/л.
Нарушения обмена меди
Медь - один из основных незаменимых микроэлементов, входящих в состав важнейших ферментов, опосредующих в организме жизненно важные процессы, например тканевое дыхание и эритропоэз. Медь, принимая участие в обмене веществ и энергии (в том числе окислительном фосфорилировании и свободном окислении), оказывает влияние на воспроизведение, рост и развитие организма. Медь необходима для мобилизации железа из резервов, она также способствует его включению в структуру гема цитохромоксидазы и гемоглобина, являясь соответственно важным фактором эритро- и гранулоцитопоэза. Входя в состав активного центра дофамин-р-гидроксилазы, медь участвует в синтезе нейромедиаторов (превращении дофамина в норадреналин). Медь необходима также для нормального течения процессов кератинизации и пигментации кожи и волос, формирования миелина, синтеза различных производных соединительной ткани и др.
Ионы меди активируют аскорбатоксидазу, ингибируют ксантиноксидазу, принимают участие в поддержании вторичной и третичной структуры ДНК и РНК. Медь входит в состав цитохромоксидазы (компонента цепи переноса электронов в митохондриях), тирозиназы (катализирующей окисление тирозина, превращение ряда фенолов в хиноны, из которых в результате дальнейшего окисления образуются меланины). Переносчиком меди является церулоплазмин (ферроксидаза), имеющий окислительную активность. Медь наряду с цинком входит в состав субъединиц цитозольной супероксиддисмутазы, прерывающей свободнорадикальные процессы (защищающей клеточные структуры от повреждающего действия супероксидных анион-радикалов). Медьсодержащие аминоксидазы принимают участие в катаболизме многих аминов, таких как гистамин, тирамин, путресцин, спермин и другие, в окислении адреналина.
Причинами гипокупреоза у человека могут являться пищевой недостаток меди при нерациональном искусственном вскармливании новорожденных, неадекватном парентеральном питании; конкурентная абсорбция избыточного количества цинка в кишечнике. Модель пищевого дефицита меди, полученная на крысах и свиньях, характеризуется нарушением овуляции и прерыванием беременности.
Дефицит меди приводит к развитию микроцитарной анемии и лейкопении. Гипокупреоз у человека вызывает депигментацию кожи и ее производных, у грызунов - ахромотрихию, что связано с нарушением синтеза тирозиназы. Недостаток меди, входящей в состав лизилоксидазы - фермента, необходимого для образования ковалентных сшивок между полипептидными цепями коллагена и эластина, может стать причиной дефектов формирования соединительной ткани, в том числе сердечно-сосудистой системы (вазопатии, расслаивающая аневризма аорты) и скелета (остеопатии с изменениями в костях). Нарушением синтеза катехоламинов и демиелинизацией при гипокупреозе, связанными с недостаточностью цитохромоксидазы и дофамин-β-гидроксилазы, объясняются поражения нервной системы (спинальная демиелинизирующая нейропатия у овец в бедных медью районах Австралии, дискоординация движений у человека).
Недостаток меди в организме матери вызывает тяжелые нарушения развития нервной системы, дефекты соединительной ткани, снижение иммунитета у плода, возможна его гибель.
К наследственным формам гипокупреоза относится синдром Менкеса («болезнь курчавых волос» с тяжелым поражением ЦНС), при котором нарушаются функции ряда медьсодержащих ферментов - тирозиназы (депигментация волос), сульфидоксидазы (нарушение процесса кератинизации), лизилоксидазы (поражения соединительной ткани: аневризмы, эмфизема, остеопатии), дофамин-р-гидроксилазы и цитохромоксидазы (нейродегенеративные явления).
Значительная часть меди в клетках находится в связанном виде с металлотионеином - белком, содержащим много остатков цистеина и играющим важную роль в нейтрализации потенциально токсичных ионов тяжелых металлов. Избыточное накопление меди в клетках печени, происходящее при некоторых наследственных заболеваниях, а также под воздействием токсических веществ, может быть связано с нарушением регуляции синтеза металлотионеина и, в свою очередь, приводить к индукции биосинтеза этого
белка: создается «порочный круг». Избыток меди приводит к повреждению цитоскелета и мембран, в том числе лизосомальных, что также способствует дальнейшему накоплению меди в клетках в связи с нарушением выделительной функции лизосом.
Повышенное накопление меди в печени, базальных ганглиях головного мозга, роговице глаз и других тканях при наследственном нарушении ее метаболизма, известном как болезнь ВильсонаКоновалова, или гепатоцеребральная дистрофия (снижение экскреции меди с желчью, уменьшение концентрации церулоплазмина в плазме и включения в него меди, гипераминоацидурия), становится причиной развития цирроза печени, артритов, катаракты, моторных неврологических нарушений, гемолитической анемии. Механизм нарушения синтеза церулоплазмина при гепатоцеребральной дистрофии до конца не изучен. Наряду с генетически обусловленным дефектом лизосом гепатоцитов, определяющим нарушение важнейшего механизма регуляции баланса меди в организме - экскреции ее с желчью, возможно образование комплексов меди с аминокислотами, которые не всасываются в почечных канальцах. Дегенерация паренхимы печени, разрастание соединительной ткани обусловлены повреждением ферментных систем гепатоцитов накапливающимися ионами меди. Накопление меди в тканях связано с ее высвобождением из поврежденных гепатоцитов. Развитие внутрисосудистого гемолиза обусловлено ингибированием медью ферментных систем эритроцитов.
Генетические дефекты обмена меди у животных сходны с таковыми у человека и могут рассматриваться как их модели.
Известны также профессиональный (у работников медных рудников и химических предприятий, сварщиков цветных металлов) и гемодиализный гиперкупреоз. При выраженном гемолизе (гемоглобин в моче) могут развиться острая почечная недостаточность с анурией и уремией, гемолитическая желтуха, анемия. При попадании высокодисперсной пыли меди в верхние дыхательные пути возникает острая литейная лихорадка (озноб, сухой кашель, головная боль, слабость, одышка, повышение температуры). Возможны аллергические реакции кожи. Избыток меди может оказывать эмбриотоксическое действие.
В суточной диете взрослых людей должно содержаться от 2 до 5 мг меди, из которых усваивается около 30%. Нормальное содержание меди в сыворотке крови у женщин составляет 13-24 мкмоль/л, у мужчин - 11-22 мкмоль/л.
Нарушения обмена цинка
Цинксодержащие и активируемые цинком ферменты (их известно более 200) принимают участие в синтезе и распаде углеводов, липидов, белков и нуклеиновых кислот, делении клеток, тканевом дыхании, играя важную роль в процессах размножения и роста организма, в фотохимическом акте зрения, остеогенезе, кератинизации, иммунном ответе и др. Эти ферменты относятся ко всем шести известным классам (карбоксипептидазы А и В, щелочная фосфатаза, дипептидазы, карбоангидраза, малат-, глутамат-, алкогольдегидрогеназа и др.), наибольшее их число принадлежит к классу гидролаз. Микроэлемент может либо непосредственно выполнять каталитическую функцию, входя в состав активного центра фермента, либо стабилизировать его третичную или четвертичную структуру, а также являться регулятором активности фермента.
Биосинтез белков и нуклеиновых кислот осуществляется при разностороннем участии цинка. Микроэлемент необходим для функционирования всех нуклеотидилтрансфераз, ДНК- и РНКполимераз, тимидинкиназ, обратных транскриптаз, для стабилизации спиральной структуры ДНК и РНК. Принимая участие в образовании полисом и входя в состав аминоацил-тРНК-синтетаз и факторов элонгации полипептидной цепи у млекопитающих (например, еEF-1), цинк играет важную роль в процессе трансляции.
Цинк участвует в формировании активной формы инсулина. Входя в состав цитозольной супероксиддисмутазы, микроэлемент тормозит свободнорадикальное окисление в клетках. Рецептором глюкокортикоидов - гормонов, влияющих на все виды обмена веществ и играющих важную роль в адаптации к стрессам, является цинксодержащий белок. Цинк принимает участие в конформационных изменениях, происходящих с ретинолом в сетчатке.
Всасывание цинка в тонком кишечнике тормозится медью, фосфатом, кальцием, фитатом (в большом количестве содержащимся в бездрожжевом хлебе из неочищенной муки, чем объясняется широкое распространение гипоцинкоза среди сельских жителей Ирана); затруднено при энтеритах, опухолях кишечника и циррозе печени. Дефицит цинка может возникать при катаболических состояниях, в частности при тяжелых травмах и хронических гемолитических анемиях. Ятрогенный дефицит возможен при
парентеральном питании, лечении цитостатиками и некоторыми другими препаратами.
Гипоцинкоз может проявляться тяжелой анемией, гепатоспленомегалией, задержкой физического развития, карликовостью, гипогонадизмом, бесплодием, гипокератическим дерматитом, нарушением нормального оволосения, изменением или утратой восприятия вкуса и запаха, угнетением иммунных реакций (снижение пролиферации и дифференцировки лимфоцитов), ухудшением сумеречного зрения. Недостаток цинка в организме матери может являться причиной слабости родовой деятельности, преждевременных родов, а также гидроцефалии, микро- и анофтальмии, искривления позвоночника, пороков сердца у новорожденных. Наследственное заболевание, связанное с недостаточностью цинка - наследственный энтеропатический акродерматит, вызываемый нарушением синтеза цинксвязывающего протеина, который необходим для нормальной кишечной абсорбции цинка. Он проявляется с момента прекращения вскармливания материнским молоком, содержащим цинксвязывающий белок, характеризуется иммунодефицитом, дерматитом, неврологическими нарушениями, замедлением заживления ран, симптомами квашиоркора, прогрессирующей потерей зрения и другими проявлениями цинкдефицита.
Болезнь Адема у крупного рогатого скота, аналогичная энтеропатическому акродерматиту; мутация «летальное молоко» у мышей (lm), ведущая к гибели их потомства; синдром тестикулярной феминизации у крыс (Tfm), характеризующийся нечувствительностью к андрогенам и крипторхизмом; мутация SM (супермыши) относятся к рецессивным аутосомным мутациям, затрагивающим обмен цинка.
Отравление парами цинка, наблюдающееся у электросварщиков, работающих в закрытых помещениях, проявляется головной болью, кашлем, гиперсаливацией, повышением температуры тела и лейкоцитозом. Избыток цинка оказывает эмбриотоксическое действие. Избыток цинка тормозит всасывание меди в желудочнокишечном тракте. На этом основано применение фармакологических доз цинка для лечения болезни Вильсона-Коновалова.
Суточная потребность организма в цинке составляет 10-15 мг, из которых усваивается 30%. В норме содержание цинка в сыворотке крови составляет у взрослых людей 11-18 мкмоль/л.
Нарушения обмена марганца
Марганец является активатором или входит в состав ряда ферментов, среди которых известны гидролазы, оксидоредуктазы, трансферазы, лиазы, лигазы (пируваткарбоксилаза, аргиназа, пептидгидролазы, декарбоксилазы аминокислот, фосфотрансферазы, изоцитратдегидрогеназы и малатдегидрогеназы). Микроэлемент необходим для глюконеогенеза и регуляции уровня глюкозы в крови, синтеза гликопротеинов и гемоглобина, он также стимулирует синтез холестерина и жирных кислот, участвует в формировании спиральной структуры нуклеиновых кислот, обеспечивает пептидилтрансферазную реакцию при сборке полипептидных цепей.
При дефиците марганца (гипоманганоз) отмечены снижение активности гликозилтрансфераз, играющих важную роль в синтезе гликозаминогликанов, в том числе в костной матрице, и нарушение включения сульфата в хрящевую ткань, обусловливающие аномалии развития скелета с замедлением оссификации и задержкой роста у человека и животных. Недостаток микроэлемента нарушает формирование скелета как во внутриутробном, так и в постнатальном периоде жизни.
Гипоманганоз приводит к развитию гипохолестеринемии, связанной с нарушением активации диметилаллилтрансферазы, и анемии, вызванной нарушением синтеза гемоглобина. С нарушением синтеза холестерина - предшественника половых гормонов связано воздействие дефицита микроэлемента на репродуктивную функцию у человека и животных.
Влияние недостатка марганца на углеводный обмен проявляется ухудшением усвоения глюкозы, связанным с гибелью β-клеток островков Лангерганса под действием супероксидного радикала (снижение активности Мn2+-супероксиддисмутазы).
У человека генетические дефекты обмена марганца неизвестны. Мыши с аутосомной рецессивной мутацией «pallid» (гомозиготы) приносят потомство с врожденными нарушениями координации движений, что объясняется аномальным развитием отолитов при недостатке марганца.
Избыточное поступление марганца в организм ведет к формированию в костях рахитоподобных изменений («марганцевый рахит»), затрудняет всасывание железа и меди в желудочно-кишечном тракте, вызывая анемию. Профессиональный манганоз у шахтеров, регулярно вдыхающих марганцевую пыль, проявляется пар-
кинсоноподобным синдромом, выражающимся в расстройстве двигательной активности (нарушение письма, «петушиная походка»), психическими нарушениями (эйфория, благодушие); у больных также развивается астеновегетативный синдром с угнетением функции гонад и пневмокониоз. Повышенное содержание марганца в организме беременных может привести к гибели плода.
Суточная потребность взрослого человека в марганце - 2-7 мг. В норме в цельной крови содержится 30-50 мкг/л марганца.
Нарушения обмена хрома
Хром обеспечивает толерантность организма к глюкозе, усиливая действие инсулина во всех метаболических процессах, регулируемых этим гормоном. Механизм этого воздействия, возможно, связан с влиянием Сг на рецепцию инсулина. Микроэлемент принимает участие в формировании спиральной структуры нуклеиновых кислот; при недостатке магния может активировать фосфоглюкомутазу, осуществляющую обратимое превращение глюкозо-1-фосфата в глюкозо-6-фосфат.
Дефицит хрома, развивающийся у человека при длительном нерациональном парентеральном питании, проявляется нарушениями обмена углеводов (гипергликемия, глюкозурия) и липидов (повышение концентрации триацилглицеролов и холестерина, снижение уровня липопротеинов высокой плотности в сыворотке крови, увеличение атеросклеротических бляшек).
При профессиональном гиперхромозе могут развиваться изъязвление слизистой оболочки носа, дерматит, гепатоз. В высоких концентрациях соединения хрома могут оказывать мутагенное и канцерогенное действие.
Содержание Сr в цельной крови - 1,4-3,1 нмоль/л. Снижение содержания хрома в крови отмечается при гастрогенной железодефицитной и апластической анемиях, повышение - при лейкозах. Нормальным для взрослого человека считается ежесуточное поступление хрома с пищей в количестве 5-200 мкг.
Нарушения обмена селена
Селен один или вместе с железом и молибденом присутствует в ряде ферментов, таких, как некоторые оксидоредуктазы, в том числе глутатионпероксидаза, трансферазы и др. Глутатионпероксидаза является частью антиоксидантной системы клеток,
предохраняющей мембраны от токсического действия перекиси водорода и гидропероксидов липидов. Синергистом антиокислительного действия глутатионпероксидазы является витамин Е, который, возможно, принимает также участие в метаболизме селена, защищая его от окисления. Микроэлемент, избирательно ингибируя транскрипцию генов и принимая участие в окислительновосстановительных процессах, оказывает влияние на обмен белков, липидов и углеводов. Селен оказывает модифицирующее воздействие на ферменты биотрансформации ксенобиотиков. Относительно высокая концентрация селена в сетчатке глаза позволяет предположить его участие в фотохимических реакциях светоощущения. Селенсодержащие белки обнаружены в селезенке, семенниках и других органах.
Недостаток селена в организме у людей, проживающих в селенодефицитном поясе Китая, в Забайкалье, некоторых других регионах, является причиной эндемической кардиомиопатии - болезни Кешана (многоочаговой некротической миокардиодистрофии) и фактором риска ишемической болезни сердца и инфаркта миокарда. Дефицит селена может развиваться также при нерациональном парентеральном питании или белково-калорийной недостаточности. Селенодефицит приводит к угнетению иммунных реакций, снижению противовирусной и противоопухолевой резистентности организма. В тяжелых случаях возможно развитие дилатационной кардиомегалии и застойной сердечной недостаточности.
У животных селенодефицит характеризуется задержкой роста и развития, азоспермией, алиментарной мышечной дистрофией (беломышечная болезнь сельскохозяйственных животных), гепатозом, экссудативным диатезом, некрозом и фиброзом поджелудочной железы.
К генетическим дефектам обмена селена относятся наследственные селенодефицитные ферментопатии (например, дефицит глутатионпероксидазы эритроцитов и тромбоцитов), наследственный кистозный фиброз поджелудочной железы (муковисцидоз), наследственная миотоническая дистрофия.
Распространенный в некоторых регионах (штат Юта, США; Новая Зеландия и др.) эндемический селеноз, развивающийся при хроническом превышении рекомендуемой суточной дозы селена в 5-6 раз, проявляется дерматитом, повреждением эмали зубов, анемией и нервными расстройствами, дегенерацией печени, увеличением селезенки, поражением ногтей и волос. Селеноз у круп-
ного рогатого скота в эндемических районах приводит к развитию алкалоза, очагового некроза и цирроза печени, а также пороков внутриутробного развития.
Суточная потребность организма взрослого человека в селене составляет в среднем 0,2 мг. Нормальное содержание селена в сыворотке крови - 53-105 мкг/л.
Нарушения обмена молибдена
Недостаток в организме молибдена, возникающий чаще всего при парентеральном питании, характеризуется снижением активности молибденсодержащих ферментов: ксантиноксидазы, катализирующей окисление гипоксантина и ксантина в мочевую кислоту; сульфитоксидазы, превращающей сульфит в сульфат; альдегидоксидазы, окисляющей альдегиды до органических кислот.
Генетический дефект ксантиноксидазы у человека вызывает развитие ксантинурии с одновременным снижением содержания мочевой кислоты в сыворотке крови и моче. Наследственный дефект сульфитоксидазы характеризуется выраженными нарушениями развития нервной системы, умственной отсталостью, эктопией хрусталика. В моче повышено содержание сульфитов, сульфо-L-цистеина при практическом отсутствии сульфатов. Можно предположить, что выявленные изменения наступают как в связи с накоплением токсических количеств сульфитов в органах и тканях, так и из-за отсутствия сульфатов, необходимых для синтеза сложных белков, сульфогалактозилцерамидов и других молекул. Страдающие этим нарушением дети погибают в первые годы жизни.
Накопление избыточных количеств молибдена в организме человека и животных приводит к диарее, нарушениям кальциевофосфорного обмена и обмена меди, деформации костей, нарушению функций опорно-двигательного аппарата, бесплодию. Повышенная активность ксантиноксидазы приводит к ускоренному распаду пуриновых нуклеотидов в организме, вызывая возрастание концентрации мочевой кислоты в сыворотке крови и ее накопление в виде солей в суставах и сухожилиях - развивается молибденовая подагра (болезнь Ковальского).
Хронический профессиональный молибденоз характеризуется полиартралгиями, артрозами (накопление солей мочевой кислоты в суставах), гипотонией, анемией, лейкопенией.
Суточная потребность организма взрослого человека в молибдене составляет 0,5 мг. Содержание молибдена в плазме крови в норме - 13,5-15,2 мкг/л, при анемиях различного генеза может снижаться.
Нарушения обмена йода
Йодсодержащие тиреоидные гормоны тироксин и трийодтиронин регулируют деятельность центральной и периферической нервной системы, рост и дифференцировку тканей, обмен белков, углеводов и липидов, водно-электролитный и энергетический обмен, оказывают влияние на функции сердечно-сосудистой системы и пищеварительного тракта, гемопоэз и т.д.
Эндемический зоб, развивающийся при недостатке йода в организме, характеризуется компенсаторным увеличением щитовидной железы. Хронический недостаток йода, являющийся причиной снижения синтеза гормонов щитовидной железы, у детей приводит к кретинизму (умственная отсталость, карликовость, недоразвитие костной системы), у взрослых гипойодоз вызывает микседему (снижение основного обмена, отечность лица и конечностей, ожирение, сухость кожных покровов, быстрая утомляемость, артралгии, брадикардия). Поступление в организм блокирующих утилизацию йода щитовидной железой веществ, поражения печени и желудочно-кишечного тракта или нарушения интратиреоидного обмена йода могут вызывать развитие спорадического зоба у людей, проживающих в благополучных по йоду районах.
Известно несколько генетических дефектов обмена йода, являющихся причиной «семейного зоба»: нарушение синтеза тироксина из монойодтирозина и дийодтирозина; циркуляция в крови атипичного белка, прочно связывающего йод; неспособность железы концентрировать йод. Нарушение дейодирования монойодтирозина и дийодтирозина вызывается дефектом синтеза специфической дейодиназы, приводящим к повышенным потерям йода из организма. При синдроме Пендреда нарушение синтеза тиреоидных гормонов связано с дефектом тиреопероксидазы (одним из симптомов является тугоухость).
При повышенной чувствительности к йоду могут возникнуть аллергические реакции (отек Квинке, крапивница). Контакт с йодом может вызвать дерматит. При вдыхании паров йода поражаются верхние дыхательные пути. При попадании концентрированных
растворов йода внутрь развиваются тяжелые ожоги пищеварительного тракта, слизистые оболочки приобретают характерный желтый цвет.
Минимальная суточная потребность в йоде у взрослых - 100- 150 мкг в сутки, у беременных и кормящих женщин - 230-260 мкг в сутки. В норме в сыворотке крови содержится 45-90 ммоль/л йода.
Нарушения обмена кобальта
Кобальт в составе витамина В12 и соответственно кобамидных коферментов (метил- и дезоксиаденозилкобаламина) влияет на кроветворение, обмен белков, липидов и углеводов и нуклеиновых кислот, репродуктивную функцию и рост организма. Ионы кобальта повышают активность пептидгидролаз, аргиназы, альдолазы, фосфоглюкомутазы и других ферментов, участвуют в стабилизации вторичной и третичной структуры ДНК и РНК.
Недостаточное потребление кобальта с пищей в составе витамина В12 сопровождается клиническими проявлениями (пернициозная анемия, атрофия слизистой оболочки желудочно-кишечного тракта, фуникулярный миелоз и др.), обусловленными недостаточностью кобамидных коферментов (см. раздел 12.3.2).
Крупный рогатый скот в местностях с пониженным содержанием кобальта в почве страдает эндемическим заболеванием, известным как «кустарниковая болезнь», характеризующимся истощением, анемией, стеатозом печени, остеодистрофией.
Пребывание в производственных условиях в контакте с порошкообразными соединениями кобальта вызывает поражения органов дыхания (хронический бронхит, пневмония и пневмосклероз), кроветворения, сердечно-сосудистой и нервной систем, а также развитие аллергического дерматита.
Превышение поступления кобальта в организм над его выведением, нарушающее в том числе окислительное декарбоксилирование пирувата (кобальт в высоких концентрациях может взаимодействовать с липоевой кислотой), приводит к миокардиодистрофии, поражению нервной системы, полицитемии. Нарушение метаболизма йода при избытке кобальта в организме приводит к гиперплазии щитовидной железы. Совокупность вышеперечисленных симптомов получила название «болезнь любителей пива» в те годы, когда для стабилизации пены в пиво добавлялся хлорид кобальта.
Нормальное содержание кобальта в цельной крови составляет 34-48 нмоль/л.
Нарушения обмена фтора
Почти весь фтор в организме сосредоточен в костях и зубах, наиболее насыщен фтором поверхностный слой зубной эмали. Микроэлемент входит в состав фторапатита, необходимого для придания костной ткани прочности и кислотоустойчивости.
Дефицит фтора (гипофтороз) у экспериментальных животных вызывает задержку роста, связанную с нарушением минерализации костной ткани, снижение плодовитости и продолжительности жизни. При недостатке фтора в костной ткани отмечается снижение активности щелочной и кислой фосфатаз.
Дефицит фтора у людей, проживающих в эндемических зонах с низким содержанием фтора в питьевой воде, приводит к поражению зубов кариесом (эмаль и дентин кариозных зубов дефторированы), а в старческом возрасте также к развитию фторзависимого остеопороза, являющегося причиной частых переломов, особенно у женщин.
Острое отравление фторидами (например, входящими в состав инсектицидов) проявляется рвотой и поносом, возбуждением, неврологическими нарушениями, тетанией. При тяжелом отравлении паралич дыхательной мускулатуры может вызвать гибель организма.
Эндемический флюороз проявляется поражением зубов, связанным с избыточным накоплением фторидов (пятнистость или крапчатость зубной эмали), печени, почек, центральной нервной системы и эндокринной системы. Отмечаются слабость мышц, ломкость костей, кальцификация сухожилий. При профессиональном флюорозе наблюдаются фторный ринит с носовыми кровотечениями, язвенно-некротический фаринголарингит, атрофический гастрит, фторный гепатоз и гиперпаратиреоз, гипогонадизм, миокардиодистрофия. Нарушения углеводного, липидного и белкового обменов при избытке фтора связаны в том числе с образованием его комплексных соединений с кальцием, магнием и другими ионами - активаторами многочисленных ферментов.
Нормальным считается поступление фтора в организм в количестве 1,5-4 мг в сутки. Содержание фторидов в плазме крови в норме составляет 0,5-10,5 мкмоль/л.
12.10. НАРУШЕНИЯ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
Под кислотно-основным состоянием (КОС) подразумевается соотношение концентраций водородных (Н+) и гидроксильных (ОН) ионов в биологических средах. Необходимым условием существования живого организма является поддержание постоянства этого параметра внутренней среды. От величины рН зависят стабильность мембран, функции ферментов, диссоциация электролитов, нервно-мышечная возбудимость и проводимость, комплексообразование и другие процессы.
Белковый, липидный, углеводный обмены являются источниками образования летучих (угольная) и нелетучих кислот (фосфорная, серная, пировиноградная, молочная и др.), часть из которых претерпевает дальнейшее окисление; небольшое количество кислых эквивалентов удаляется из организма в свободном состоянии или в виде солей. Основные соединения (ОН-, креатинин и др.) образуются в организме в значительно меньших количествах.
Тенденция к увеличению концентрации Н+ (и соответственно снижению рН) традиционно называется ацидозом; тенденция к снижению концентрации Н+ (повышению рН) получила название «алкалоз». Значения рН крови ниже 6,8 и выше 8,0 считаются несовместимыми с жизнью и в клинике практически не встречаются.
Механизмы регуляции КОС весьма эффективны и способны компенсировать значительные сдвиги рН.
КОС в организме характеризуется такими основными показателями, как:
1. Актуальный рН - отрицательный десятичный логарифм концентрации водородных ионов - является интегральным показателем кислотно-основного состояния. В норме рН артериальной крови составляет 7,35-7,45, венозной - 7,26-7,36.
2. Парциальное давление (напряжение) углекислого газа в крови (рСО2) отражает концентрацию углекислоты (под термином «углекислота» подразумеваются различные соединения двуокиси углерода в крови). Напряжение углекислого газа в артериальной крови (раСО2) в норме составляет 4,7-6,0 кПа, в венозной - 6,1-7,7 кПа.
3. Парциальное давление (напряжение) кислорода в крови (рО2)
отражает концентрацию растворенного в крови кислорода. Напряжение О2 в артериальной крови (раО2) составляет в норме 12,0- 12,6 кПа, в венозной - 4,6-6,0 кПа.
4. Стандартный бикарбонат плазмы крови (SB) - концентрация бикарбоната в плазме крови, уравновешенной при 37 °С со стандартной газовой смесью при рСО2=5,33 кПа и рО2 >13 кПа, - в норме составляет 21,3-21,8 ммоль/л.
5. Буферные основания крови (ВВ) - сумма анионов буферных систем, в основном ионов бикарбоната и анионов белков, - в норме составляет 40-60 ммоль/л.
6. Нормальные буферные основания крови (NBB) - показатель, определяемый при рН=7,38 и рСО2=5,33 кПа.
7. Избыток (или дефицит) оснований (ВЕ) - показатель избытка (или недостатка) буферных мощностей (ВВ-NBB) - в норме колеблется от +2,3 до -2,3 ммоль/л.
Стабилизация КОС организма обеспечивается буферными системами (бикарбонатная, фосфатная, белковая и гемоглобиновая), а также функционированием специфических физиологических механизмов компенсации КОС в некоторых органах (легкие, почки, печень, желудочно-кишечный тракт, костная ткань).
1. Бикарбонатная буферная система (10% от буферной емкости крови) представляет собой сопряженную кислотно-основную пару, состоящую из молекул угольной кислоты Н2СО3, играющей роль донора протона, и бикарбонат-ионов НСО3- (во внеклеточной жидкости в виде натриевой соли NaНСО3, во внутриклеточной - КНСО3). Концентрация недиссоциированных молекул Н2СО3 в крови незначительна и находится в прямой зависимости от концентрации растворенного СО2, поэтому известное уравнение Гендерсона-Хассельбаха1 для бикарбонатной системы представимо в следующем виде:
рН=рКа + lg(НСО3- ] / [СО2]).
Отношение концентраций Н2СО3 и НСО3- в крови в норме составляет 1:20. Эта буферная система эффективно функционирует при значениях рН около 7,4.
2. Фосфатная буферная система (только 1% от буферной емкости крови, при этом ее роль в тканях, особенно в почках, весьма существенна). В ее состав входят однозамещенный фосфат Н2РО4- (донор протона) и двузамещенный фосфат НРО42- (акцептор протона), соотношение которых в норме - 1:4. Фосфатный буфер спо-
1 Уравнение Гендерсона-Хассельбаха выражает рН буферного раствора через константу диссоциации Ка и соотношение концентраций акцептора и донора протонов: рН = рКа + ^([акцептор протонов] / [донор протонов]).
собен оказывать влияние на концентрацию протонов в растворе в диапазоне рН от 6,1 до 7,7, наиболее эффективен при рН = 7,2.
3. Белковая буферная система наиболее эффективна в области значений рН от 7,2 до 7,4. Белки, являясь амфотерными электролитами за счет наличия в составе их молекул свободных кислотных и основных групп, в кислой среде связывают ионы водорода, в щелочной - отдают.
4. Гемоглобиновая буферная система является наиболее мощной (около 70% буферной емкости крови). Она состоит из ННb и ННbО2 (слабые органические кислоты, доноры протонов) и КНb и КНbО2 (сопряженные основания, акцепторы протонов). Система, состоящая из взаимопревращающихся гемоглобина и оксигемоглобина, функционирует как единое целое.
Буферные системы оказывают компенсаторное воздействие на изменения КОС непосредственно в момент их возникновения, влияние легких сказывается в течение нескольких минут, время восстановления почками физиологического соотношения концентраций компонентов буферных систем и нарушенного КОС измеряется часами.
Выделение СО2 регулируется изменением скорости и объема легочной вентиляции. Увеличение альвеолярной вентиляции приводит к снижению рСО2 в артериальной крови, уменьшение - к увеличению рСО2. В организме человека присутствуют два типа хеморецепторов, принимающих участие в регуляции этого процесса: рецепторы рН в каротидных тельцах и рецепторы, чувствительные к СО2 в продолговатом мозгу, аортальном и каротидных тельцах.
С легочным механизмом регуляции КОС непосредственно связана бикарбонатная буферная система крови, находящаяся в равновесии с газообразным СО2. Накопление в организме угольной кислоты вызывает компенсаторную гипервентиляцию (одышку), приводящую к удалению избытка СО2 с выдыхаемым воздухом. Компенсаторная гиповентиляция при алкалозе приводит к сохранению СО2 и восстановлению запасов Н2СО3 в крови.
Участие гемоглобиновой буферной системы в регуляции КОС связано с кислородтранспортной функцией гемоглобина. Освобождение протона при оксигенировании гемоглобина компенсирует подщелачивание крови в капиллярах легких, обусловленное снижением концентрации СО2. Дезоксигенированный гемоглобин в капиллярах тканей связывает протон и предотвращает понижение рН крови (рис. 12-52).
 Рис. 12-52. Участие
гемоглобиновой буферной системы в поддержании кислотно-основного
равновесия: А - реакции в капиллярах тканей; Б - реакции в легких (по В.
Эллиот, Д. Эллиот, 1999)
Рис. 12-52. Участие
гемоглобиновой буферной системы в поддержании кислотно-основного
равновесия: А - реакции в капиллярах тканей; Б - реакции в легких (по В.
Эллиот, Д. Эллиот, 1999)
Почки участвуют в поддержании КОС, осуществляя регулируемый процесс реабсорбции натрия и секреции протонов. Поддержанию в крови концентрации бикарбоната натрия и выведению избыточного количества протонов способствуют превращения в канальцах почек двузамещенных фосфатов в однозамещенные, бикарбонатов в угольную кислоту, экскреция слабых органических кислот, образование в почках аммиака и использование его для нейтрализации и выведения кислых эквивалентов с мочой (рис. 12-53). Ацидоз увеличивает синтез и экскрецию NН4+ в почках, алкалоз оказывает обратное действие. К факторам регуляции секреции протонов почками относятся напряжение СО2 в артериальной крови (СО2 легко проникает в клетки канальцев и вызывает в них снижение рН, приводящее к повышению секреции Н+), активность карбоангидразы, рН артериальной крови (частично определяющий
 Рис. 12-53. Роль почек в компенсации нарушений кислотно-основного гомеостаза (А- - анион)
Рис. 12-53. Роль почек в компенсации нарушений кислотно-основного гомеостаза (А- - анион)
рН клеток канальцев), паратиреоидный гормон (снижающий активность ?+/Н+-обменника), альдостерон. Минералокортикоиды, стимулируя реабсорбцию натрия, облегчают секрецию протонов, кроме того, альдостерон непосредственно активирует Н+-АТФазу, осуществляющую перемещение Н+ в просвет канальцев.
Роль печени в поддержании КОС связана с синтезом в ее клетках белков, относящихся к буферной системе, окислением органических кислот до СО2 и воды, преобразованием лактата в глюкозу и в дальнейшем в гликоген, а также выведением вместе с желчью из организма кислых и щелочных продуктов обмена.
Влияние желудочно-кишечного тракта на КОС организма связано с выделением соляной кислоты в полость желудка и бикарбоната натрия в проток поджелудочной железы (рис. 12-54, 12-55).
Ионы натрия, калия, кальция, магния, содержащиеся в костной ткани, могут обмениваться на ионы водорода, компенсируя аци-
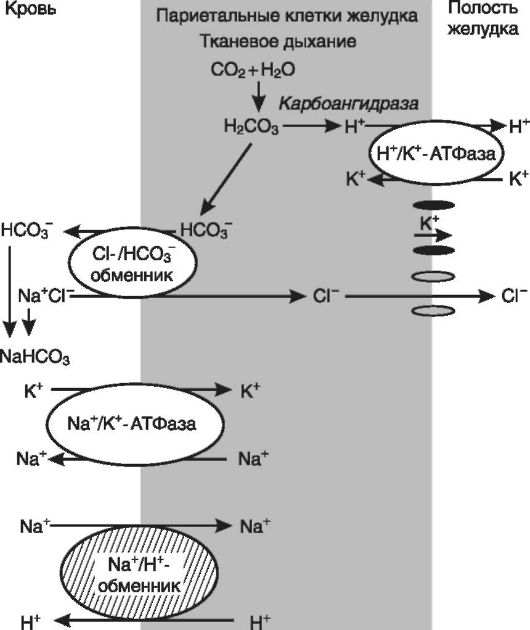 Рис. 12-54. Участие париетальных клеток желудка в поддержании кислотно-основного равновесия
Рис. 12-54. Участие париетальных клеток желудка в поддержании кислотно-основного равновесия
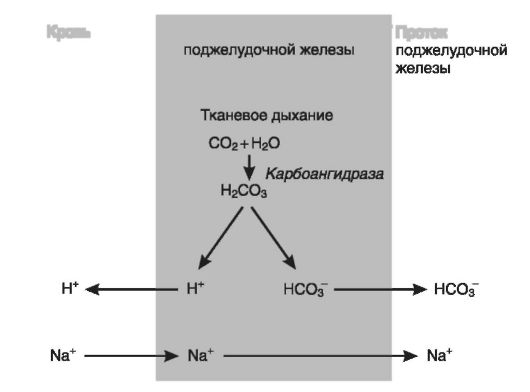
Рис. 12-55. Секреция НСО- клетками поджелудочной железы
доз. В тяжелых случаях этот процесс может приводить к декальцификации скелета.
Различают острые и хронические нарушения КОС. Для компенсации острых нарушений бывают задействованы зачастую только буферные системы организма, в регуляции хронических (установившихся) отклонений рН принимают участие легкие, почки, другие органы и ткани. Ацидозы и алкалозы могут быть компенсированными (компенсаторные механизмы способны поддерживать рН крови в пределах нормы за счет химических и функциональных сдвигов) и декомпенсированными (рН крови соответственно 7,24 и ниже и 7,55 и выше). Промежуточные состояния принято называть субкомпенсированными.
В зависимости от механизмов развития нарушений КОС выделяют газовые (дыхательные, респираторные) и негазовые (метаболические, обменные) ацидозы и алкалозы.
12.10.1. Газовый ацидоз
Газовый (дыхательный, респираторный) ацидоз характеризуется накоплением в крови углекислоты. Причинами острого дыхательного ацидоза могут стать:
1) высокая концентрация СО2 во вдыхаемом воздухе;
2) повышение сопротивления дыхательных путей (бронхоспазм, ларингоспазм, аспирация);
3) нарушения дыхательной функции легких (тяжелая пневмония, пневмоторакс, гемоторакс, отек легких);
4) угнетение дыхательного центра (анестетики, седативные препараты, черепно-мозговая травма, инфаркт головного мозга);
5) нервно-мышечные расстройства (приступ миастении, курареподобные вещества, токсины);
6) системные нарушения кровообращения (сердечная недостаточность, массивная тромбоэмболия);
7) ятрогенные воздействия: неадекватная механическая вентиляция легких, избыточное введение в организм СО2 (карбонаркоз) и др.
Хронический дыхательный ацидоз может быть обусловлен:
1) угнетением дыхательного центра (опухоль мозга, хроническая передозировка седативных препаратов);
2) нарушениями нервно-мышечной передачи (рассеянный склероз, полиомиелит, мышечная дистрофия, повреждения спинного мозга);
3) хроническими обструктивными заболеваниями легких (эмфизема, хронический бронхит);
4) ограничением движений, вызывающим снижение вентиляции (кифосколиоз, ожирение).
При гиперкапнии рН может некоторое время оставаться в границах физиологической нормы за счет действия компенсаторных механизмов. Возрастание парциального давления СО2 ведет к повышению возбудимости дыхательного центра, развитию одышки и выведению из организма избытка углекислого газа в более или менее достаточной степени. Особенностью функционирования буферных систем в условиях газового ацидоза является повышенная емкость бикарбонатного буфера, обусловленная высокой концентрацией в крови СО2. Возрастание концентрации протонов в плазме частично компенсируется белковой и фосфатной буферными системами, часть ионов водорода связывается гемоглобиновым буфером.
В почках ацидозом стимулируется образование и секреция NH4+, а также выведение Н+ в свободном виде. Увеличение выделения почками ионов водорода приводит к реабсорбции больших количеств НСО3-. Задержка почками бикарбоната при хроническом газовом ацидозе вызывает еще большее возрастание его концентрации в плазме, что способствует поддерживанию
нормального или близкого к нормальному значения рН (отмечается компенсаторный метаболический алкалоз). Развитие декомпенсации сопровождается общими нарушениями газообмена, вызванными в том числе снижением сродства гемоглобина к кислороду. Буферная емкость гемоглобиновой системы уменьшается (табл. 12-16, 12-17).
Таблица 12-16. Клинические признаки и лабораторные показатели при нарушениях кислотно-основного состояния (по М. Горн и соавт., 2000)
Вариант нарушений | рН | рСО2 | [НСО3] | Клинические признаки и симптомы |
Острый дыхательный ацидоз | Снижен | Повышено | Слабо повышена или норма | Тахикардия, тахипноэ, потливость, головная боль; беспокойство, переходящее в летаргию и кому; цианоз, аритмии |
Хронический дыхательный ацидоз | Снижен | Повышено | Повышена (компенсаторная реакция) | Диспноэ или тахипноэ с повышением уровня СО2, который превышает компенсаторные возможности; прогрессирование нарушений ЦНС до летаргии, спутанности сознания и комы |
Острый дыхательный алкалоз | Повышен | Снижено | Без изменений (снижение отмечается при длительном процессе и адекватной функции почек) | Парестезии, особенно в пальцах, головокружение |
Хронический дыхательный алкалоз | Повышен | Снижено | Снижена (компенсаторная реакция) | Симптомы отсутствуют |
Окончание табл. 12-16
Острый метаболический ацидоз | Снижен | Снижено (компенсаторная реакция) | Снижена | Тахипноэ, ведущее к дыханию типа Куссмауля; гипотензия, влажная холодная кожа, кома, аритмии |
Хронический метаболический ацидоз | Снижен | Снижено (но не так сильно, как при остром метаболическом ацидозе) | Снижена | Слабость, анорексия, недомогание (может быть отнесено к проявлению хронического заболевания так же, как и к проявлению ацидоза), декальцификация костей |
Острый метаболический алкалоз | Повышен | Повышено (почти до 60 - компенсаторная реакция) | Повышена | Мышечная слабость и гипорефлексия (обусловленная выраженной гипокалиемией), аритмии, апатия, спутанность сознания и ступор |
Хронический метаболический алкалоз | Повышен | Повышено (компенсаторная реакция) | Повышена | Обычно симптомы отсутствуют |
Таблица 12-17. Основные механизмы коррекции нарушений кислотноосновного состояния (по М. Горн и соавт., 2000)

 Нарушение
кислородтранспортной функции гемоглобина при респираторном ацидозе
приводит к усилению гипоксии, сопровождающейся в дальнейшем
присоединением явлений метаболического ацидоза вследствие нарушений
процесса тканевого дыхания и накопления недоокисленных продуктов в
клетках.
Нарушение
кислородтранспортной функции гемоглобина при респираторном ацидозе
приводит к усилению гипоксии, сопровождающейся в дальнейшем
присоединением явлений метаболического ацидоза вследствие нарушений
процесса тканевого дыхания и накопления недоокисленных продуктов в
клетках.
При хроническом дыхательном ацидозе приобретает клиническое значение обмен избыточных ионов водорода внеклеточной жидкости на ионы натрия и кальция костной ткани, приводящий к развитию остеопороза.
Гиперкапния приводит к повышению артериального давления за счет возбуждения сосудодвигательного центра - развивается спазм артериол, в первую очередь легочных. Весьма неблагоприятным следствием избытка углекислого газа в крови является спазм бронхиол и выделение больших количеств вязкой слизи, что в еще большей степени ухудшает газообмен и создает дополнительную нагрузку на дыхательную мускулатуру. Ухудшение вентиляции и интенсивная работа дыхательных мышц, увеличивающая продукцию углекислого газа, создают «порочный круг».
При хроническом газовом ацидозе снижение активности адренорецепторов вызывает ослабление сердечной деятельности и падение артериального давления. Большой избыток СО2 может привести к развитию брадикардии, вплоть до остановки сердца, вследствие повышения тонуса блуждающего нерва.
Компенсаторные изменения при респираторном ацидозе служат причиной возрастания показателей ВВ и SB. ВЕ в норме или повышен.
12.10.2. Газовый алкалоз
Газовый (дыхательный, респираторный) алкалоз является результатом альвеолярной гипервентиляции. Синдромом гипервентиляции называется острая альвеолярная гипервентиляция в результа-
те психического возбуждения. К причинам дыхательного алкалоза относятся:
1) гипервентиляция при гипоксии (пневмония, пребывание на высокогорье, выраженная анемия, застойная сердечная недостаточность);
2) центральная стимуляция дыхательного центра (заболевания центральной нервной системы - инсульт, опухоль; прием лекарственных препаратов - салицилаты, агонисты адренорецепторов);
3) легочные расстройства (тромбоэмболия легочной артерии, астма, интерстициальный фиброз);
4) механическая гипервентиляция.
Важнейшим механизмом компенсации гипокапнии является снижение возбудимости дыхательного центра, приводящее к задержке СО2 в организме. Самопроизвольная гипервентиляция не может продолжаться очень долго, поэтому причинами истинного дыхательного алкалоза могут стать только поражения мозга и искусственная вентиляция легких без контроля концентрации СО2 в крови.
Компенсация роста рН осуществляется преимущественно за счет высвобождения протонов из тканевых небикарбонатных буферов. Ионы Н+ перемещаются из клеток во внеклеточное пространство в обмен на ионы калия (возможно развитие гипокалиемии) и образуют при взаимодействии с НСО3- угольную кислоту. Выход протонов из клеток может вызвать развитие внутриклеточного алкалоза.
Уменьшение содержания углекислого газа в эритроцитах повышает сродство гемоглобина к кислороду, затрудняя переход кислорода в ткани, и таким образом способствует развитию гипоксии. Следствием гипоксии при устоявшейся гипервентиляции является развитие метаболического ацидоза, компенсирующего смещение рН.
Хроническая адаптация к развивающемуся алкалозу связана с деятельностью почек: секреция протонов снижается, что выражается уменьшением выведения органических кислот и аммиака. Наряду с этим угнетается реабсорбция и стимулируется секреция бикарбоната, что приводит к уменьшению его уровня в плазме крови и возвращению величины рН к норме (см. табл. 12-16, 12-17).
Снижение рСО2 при газовом алкалозе, воздействуя на рецепторы сосудодвигательного центра, ведет к падению кровяного давления. При длительной гипервентиляции могут наблюдаться явления коллапса с нарушениями со стороны центральной нервной
системы. Развивающаяся в условиях алкалоза гипокальциемия становится причиной повышения нервно-мышечной возбудимости и может приводить к судорожным явлениям (тетании). У пациентов могут отмечаться беспокойство, головокружение, парестезии, сердечные аритмии, в тяжелых случаях наблюдаются спутанность сознания, обмороки.
Показатели ВВ и SВ снижаются при компенсации газового алкалоза, ВЕ обычно в пределах нормы, может быть снижен.
12.10.3. Негазовый ацидоз
Негазовый (метаболический) ацидоз - наиболее часто встречающееся в клинической практике изменение КОС, которое может быть вызвано следующими причинами:
1) нарушениями обмена веществ, приводящими к накоплению кислых продуктов (ацетоуксусная, молочная, β-гидроксимасляная и другие кислоты): кетоацидоз при сахарном диабете, нарушении функций печени, голодании, гипоксии и пр.; лактатацидоз при гипоксии, инфекциях, нарушениях функций печени; накопление органических и неорганических кислот при катаболических состояниях: травмах, ожогах, воспалительных процессах;
2) задержкой кислот или повышенным выведением щелочей при заболеваниях почек (почечный канальцевый ацидоз тип II, диффузный нефрит, обессоливающий нефрит, уремия, интоксикация сульфаниламидами);
3) потерей бикарбоната через фистулы, при диарее, дренировании поджелудочной железы;
4) длительным приемом кислот с пищей или отравлением кислотами, а также приемом некоторых лекарственных препаратов.
Повышение концентрации СО2, обусловленное сдвигом в бикарбонатной буферной системе при изменении pH, вызывает усиление легочной вентиляции (развивается компенсаторный газовый алкалоз). Снижение рН также стимулирует центральные хеморецепторы и способствует гипервентиляции. Значение гипервентиляции, кроме выведения СО2, состоит в насыщении крови и тканей кислородом, необходимым для окисления недоокисленных продуктов.
Избыток Н+ внеклеточной жидкости обменивается на ионы калия эритроцитов и тканевых клеток, концентрация калия в плазме
крови возрастает. Часть протонов переходит в костную ткань в обмен на кальций и натрий, поэтому длительный негазовый ацидоз может приводить к декальцификации костей, особенно если выведение органических кислот и аммонийных солей с мочой ограничено из-за почечной патологии. Истощение бикарбонатной буферной системы уменьшает обмен НСО3- эритроцитов на ионы хлора в венозной крови, приводя к развитию гиперхлоремии.
В почках активно реабсорбируются основания, и в повышенных количествах выделяются кислые эквиваленты, вследствие чего в моче повышается содержание кислот (рН мочи может понижаться до 4,5) и их аммонийных солей, так как в клетках почечных канальцев усилено образование аммиака. Усиленный аммониогенез (приобретающий важное значение при продолжительном негазовом ацидозе) способствует сохранению в организме натрия, калия, кальция и других катионов, в избытке фильтрующихся в клубочках. При ацидозе возрастает распад белков с увеличением содержания свободных аминокислот в крови (см. табл. 12-16, 12-17).
Клинические проявления негазового ацидоза зависят от основного патологического процесса и тяжести нарушения кислотноосновного состояния. Снижение рСО2 крови вследствие гипервентиляции приводит к снижению возбудимости дыхательного центра, возможно появление дыхания Куссмауля, характерного для диабетической, печеночной или уремической комы. Неизбежны нарушения водно-электролитного баланса, связанные с потерей катионов с мочой. При остром ацидозе отмечаются изменения сознания, снижение артериального давления, аритмии и шоковые состояния. В случае значительного повышения концентрации ионов калия в крови при низком содержании их в миокарде возможно развитие фибрилляции желудочков, чему способствует усиленная секреция катехоламинов надпочечниками, стимулируемая снижением рН. Хронический негазовый ацидоз может проявляться слабостью, недомоганием и анорексией, связанными с основным заболеванием.
При метаболическом ацидозе показатели ВВ, SB, ВЕ снижены.
12.10.4. Негазовый алкалоз
Негазовый (метаболический) алкалоз развивается в случае накопления в организме избытка оснований в результате потери ионов водорода или чрезмерного потребления щелочных веществ. К причинам метаболического алкалоза относятся:
1) нарушение выделения НСО3- (молочно-щелочной синдром);
2) введение больших количеств НСО3- (терапия раствором бикарбоната натрия, введение щелочных минеральных вод);
3) накопление в организме НСО3- в результате окисления избыточных количеств солей органических кислот, поступающих с пищей;
4) потеря большого количества соляной кислоты из желудка при неукротимой рвоте;
5) выделение избыточного количества протонов почками при приеме диуретиков, особенно петлевых и тиазидных;
6) хлордиарея - врожденный метаболический алкалоз: патологические изменения в кишечнике, приводящие к потерям хлора и калия;
7) уменьшение содержания калия в организме, вызывающее переход Н+ в клетку и повышение выделения их с мочой.
Увеличение рН при негазовом алкалозе снижает возбудимость дыхательного центра. Гиповентиляция, как и затруднение диссоциации оксигемоглобина в щелочной среде, способствует гипоксии; накопление недоокисленных продуктов наряду с повышенным рСО2 частично компенсирует избыток оснований. Нейтрализация повышенных количеств щелочных эквивалентов частично осуществляется их взаимодействием с угольной кислотой. Увеличение содержания СО2 стимулирует дыхание, и он удаляется из организма, поэтому дыхательная компенсация метаболического алкалоза недостаточна. В почках повышается выделение НСО3-, двузамещенных фосфатов, может развиваться потеря калия, а в тяжелых случаях и натрия. Кислотность мочи и содержание в ней аммиака снижены. Выведению калия почками способствует избыточное образование альдостерона. Развивающаяся гипокалиемия усугубляет алкалоз за счет перемещения части протонов внутрь клеток в обмен на выходящий в межклеточную среду ион калия - параллельно в части тканей развивается внутриклеточный ацидоз (см. табл.
12-16, 12-17).
Сопутствующая гипокалиемия проявляется предсердножелудочковыми аритмиями, мышечной слабостью, гипорефлексией, полиурией и полидипсией, ослаблением перистальтики кишечника вплоть до динамической непроходимости. Наряду с этим наблюдаются признаки повышенной нервно-мышечной возбудимости, спутанности сознания и ступора, а также гиповоле-
мии. Если при алкалозе снижается уровень Са2+, могут развиваться спазмы, симптомы тетании, гипокальциемические судороги.
Недостаток калия в организме может увеличивать потери Н+ с мочой, частично обеспечивающие сохранение калия в плазме. Таким образом, при гипокалиемии наряду с развитием алкалоза у больных может выделяться кислая моча - «парадоксальная ацидурия».
При метаболическом алкалозе показатели ВВ, SB, ВЕ повышены.
12.10.5. Сочетанные нарушения кислотно-основного состояния
Зачастую у больного имеется больше одного типа нарушений КОС (сочетанные нарушения), проявления которых зависят от тяжести отдельных простых нарушений и от их специфики. Наличие однонаправленных нарушений увеличивает вероятность изменения рН до опасной для жизни величины, тогда как разнонаправленные отклонения, оказывающие взаимно компенсирующее воздействие, зачастую делают возможным поддержание концентрации Н+ в границах физиологической нормы.
В качестве распространенного примера однонаправленных нарушений можно привести сочетание негазового алкалоза, вызываемого тяжелой рвотой у женщин в первом триместре беременности, и дыхательного алкалоза, наблюдающегося и при нормально протекающей беременности. Повышение концентрации бикарбоната и снижение рСО2 приводят в совокупности к значительному увеличению рН.
У больных с хроническими обструктивными заболеваниями легких, получающих в период обострения болезни инъекции диуретиков для снижения объема внеклеточной жидкости, явления гипоксии и накопление СО2 могут стать еще более выраженными за счет угнетения дыхательного центра, вызванного развитием метаболического алкалоза.
Больные тяжелым гастроэнтеритом, испытывающие как рвоту, вызывающую потерю НС1, так и понос, приводящий к развитию метаболического ацидоза, нередко имеют близкое к нормальному значение рН крови, что может существенно затруднять диагностику.