Патофизиология Новицкого, Е.Д. Гольдберга Тома 1 и 2 - 2009 г.
|
|
|
|
ГЛАВА 20 ПАТОФИЗИОЛОГИЯ ЭНДОКРИННОЙ СИСТЕМЫ
20.1. ОБЩАЯ ПАТОФИЗИОЛОГИЯ ЭНДОКРИННОЙ СИСТЕМЫ
Характер и локализация патологического процесса в эндокринной системе определяют особенности патогенеза и клинические проявления эндокринопатий. Выделяют следующие основные механизмы нарушения функции желез внутренней секреции: 1) нарушение центральных механизмов регуляции железы; 2) патологические процессы в самой железе и 3) периферические (внежелезистые) механизмы нарушения активности гормонов.
20.1.1. Нарушение центральных механизмов регуляции
Частыми причинами, приводящими к нарушению гипоталамической регуляции функции желез внутренней секреции, являются инфекционные и воспалительные процессы, сосудистые и травматические повреждения, опухоли. Патологические процессы, первично развивающиеся в гипоталамусе, ведут к нарушению: а) трансгипофизарного и б) парагипофизарного путей регуляции функции желез внутренней секреции. Деятельность гипоталамических центров может нарушаться и вторично в связи с нарушениями в лимбической системе (гиппокамп, миндалина, обонятельный мозг) и вышележащих этажах центральной нервной системы, которые тесно связаны с гипоталамусом. В этой связи необходимо указать на большую роль психических травм и других стрессовых состояний в развитии эндокринных нарушений. Так, например, под их влиянием угнетается функция половых желез, что может выражаться в снижении половой потенции у мужчин и расстройствах менструального цикла у женщин.
Нарушение трансгипофизарной регуляции. Трансгипофизарная регуляция является основной для щитовидной, половых и коры надпочечных желез. Она представляет собой трехступенчатый каскад усиления первичного регуляторного сигнала. Первая ступень включает образование в нейросекреторных клетках медиобазальной части подбугорья нанограммовых количеств олигопептидов, которые опускаются по аксонам до капилляров срединного возвышения и через венозные сосуды ножки гипофиза достигают аденогипофиза. Здесь они либо стимулируют, либо тормозят образование тропных гормонов. Стимулирующие олигопептиды получили название либеринов или рилизинг-факторов (от англ. release - освобождать). К их числу относятся тиреолиберин, гонадолиберины, соматолиберин и др. Тормозящие олигопептиды называют статинами, например тиростатин, соматостатин и др. Их соотношение между собой определяет образование соответствующего гормона.
Вторая ступень начинается с образования в аденогипофизе тропных гормонов (уже в микрограммовых количествах) - соматотропного (СТГ), или соматотропина, гонадотропных (ГТГ) и др. Эти тропные гормоны, действуя на соответствующие мишени, включают третью ступень. Из них тиреотропный, гонадотропные, адренокортикотропный гормоны стимулируют в соответствующих железах внутренней секреции образование гормонов, а СТГ вызывает в разных органах образование соматомединов - полипептидных гормонов, через которые и оказывает свое действие. Этих продуктов образуется уже намного больше. Они осуществляют генерализованное и относительно длительное влияние.
Избирательное нарушение образования в гипоталамусе того или иного либерина, а возможно и усиление образования статина, приводит к нарушению образования соответствующего тропного гормона в аденогипофизе. Так, например, недостаточное образование гонадолиберинов вызывает сокращение продукции гонадотропных гормонов, снижение выработки тиреолиберина, торможение продукции тиреотропного гормона и т.д.
Первичное поражение лимбических структур головного мозга с расстройством контроля секреции кортиколиберина и последующим вовлечением в патологический процесс аденогипофиза и коры надпочечников лежит в основе развития болезни Иценко-Кушинга и характеризуется усилением секреции кортизола с развитием синдрома гиперкортизолизма (см. раздел 20.2.2). Одновременно при
этом заболевании снижается чувствительность соответствующих центров гипоталамуса и аденогипофиза к кортизолу, что нарушает работу механизма обратной связи, в результате чего повышенная концентрация кортизола в крови не угнетает секреции кортиколиберина в гипоталамусе и продукции адренокортикотропного гормона (АКТГ) в гипофизе.
Важным фактором нарушения регуляции эндокринной системы являются сосудистые поражения. Так, например, иногда при поражении портальных сосудов срединного возвышения возникают ишемия гипофиза и его некроз. Это ведет к развитию гипопитуитаризма и выпадению второй ступени трансгипофизарной регуляции желез.
Нарушение парагипофизарной регуляции. Парагипофизарный путь является главным образом нервно-проводниковым. Через этот путь осуществляется секреторное, сосудистое и трофическое влияние центральной нервной системы на функцию желез внутренней секреции. Для мозгового слоя надпочечников, островков Лангерганса и паращитовидных желез это важнейший путь регуляции. В функции других желез играют важную роль оба пути регуляции. Так, например, функция щитовидной железы определяется не только выработкой тиреотропного гормона (ТТГ), но и симпатической импульсацией. Прямое раздражение симпатических нервов увеличивает поглощение йода железой, образование тиреоидных гормонов и их освобождение. Денервация яичников вызывает их атрофию и ослабляет реакцию на гонадотропные гормоны.
Нарушения транс- и парагипофизарной регуляции являются важнейшим механизмом дисфункции желез внутренней секреции. Выделяют различные варианты нарушений функции железы. Гипофункцией обозначают снижение образования гормонов данной железой, гиперфункцией - усиление их образования. При нарушении функции одной железы говорят о моногландулярном процессе, расстройство функций нескольких желез обозначают как плюригландулярный процесс. Нарушения функции железы могут быть парциальными, когда страдает образование какого-либо одного из нескольких секретируемых железой гормонов (например, в надпочечниках), либо тотальными, когда нарушается образование всех секретируемых железой гормонов. Нередко нарушение функций желез сопровождается вовлечением в патологический процесс центров вегетативной нервной системы. Примером последнего является адипозогенитальная дистрофия. При этом заболевании
находят изменения в паравентрикулярных и вентромедиальных ядрах гипоталамуса, что приводит к снижению образования гонадотропинов и развитию гипогонадизма, а также повышенного аппетита с развитием ожирения. Патогенез ожирения сложен. В нем играют роль: а) недостаточное образование в гипофизе (или освобождение) жиромобилизующих полипептидов или тех фрагментов молекул СТГ и АКТГ, которые активируют мобилизацию жира из жировых депо, повышают содержание жирных кислот в крови и стимулируют их окисление; б) поражение трофических центров гипоталамуса, что снижает активирующее действие симпатической нервной системы на мобилизацию жира из жировых депо; в) усиление образования или активности инсулина, который стимулирует переход углеводов в жиры.
Роль механизма обратной связи. Независимо от патогенетического пути нарушения функции желез внутренней секреции, как правило, в той или иной степени страдает механизм обратной связи, и это нарушение может стать причиной других расстройств. Механизм обратной связи является обязательным звеном в саморегуляции деятельности желез. Сущность регуляции заключается в том, что регулируемый параметр оказывает обратное влияние на активность железы. По характеру регулируемого параметра механизмы обратной связи можно разделить на два типа.
Первый тип - регулируемым параметром является концентрация гормона в крови. Механизм саморегуляции заключается в том, что повышение концентрации гормона в крови тормозит активность гипоталамического центра, секретирующего либерины. Это приводит к снижению образования тропного гормона и, следовательно, к уменьшению образования гормона. При уменьшении концентрации гормона возникает обратная ситуация. Так осуществляется регуляция секреции кортизола, тиреоидных и половых гормонов.
Второй тип - регулируемым параметром является содержание регулируемого вещества, например концентрация глюкозы в крови или ионов кальция. В этих случаях активность железы определяется концентрацией регулируемого вещества, которое действует непосредственно на данную железу. Знание типа механизма обратной связи важно для патофизиологического анализа нарушений и выяснения их механизмов. Допустим, при обследовании двух больных сахарным диабетом выявлены два вида изменений в механизме обратной связи. В первом случае в крови оказались увели-
ченными концентрация инсулина и глюкозы, а во втором - только глюкозы, а концентрация инсулина снижена. В обоих случаях увеличение концентрации глюкозы свидетельствует об инсулиновой недостаточности. Однако в первом случае концентрация инсулина увеличена. Следовательно, функция железы не нарушена, а действие инсулина блокируется где-то на периферии, вне железы, т.е. речь идет о внепанкреатическом, так называемом инсулинонезависимом сахарном диабете. Во втором же случае повышение концентрации глюкозы сопровождается снижением концентрации инсулина, что дает основание говорить о недостаточной функции островков Лангерганса и, следовательно, предполагать наличие инсулинозависимого сахарного диабета.
Механизм обратной связи включается и при лечении гормонами. При этом вводимый извне гормон тормозит функцию соответствующей железы и при длительном введении приводит к ее атрофии. Об этом очень важно помнить при лечении кортикостероидными гормонами. Они применяются с лечебной целью очень широко и нередко длительно, что приводит к атрофии коры надпочечников. Известно, что стрессовые состояния в связи с действием на организм различных повреждающих факторов (операционная или бытовая травма, холод, токсины, аллергическая альтерация и др.) сопровождаются активацией функции коры надпочечников и усилением секреции кортикостероидов. Это позволяет организму приспособиться к новым условиям. Больные, которые лечились кортикостероидами и прекратили это лечение, также могут попасть в такую ситуацию, когда под влиянием повреждающих факторов у них разовьется стрессовое состояние. Однако, в отличие от здоровых, у лечившихся кортикостероидами атрофированные надпочечники не отвечают адекватным усилением секреции кортикостероидов. В результате развивается острая надпочечниковая недостаточность, которая может закончиться гибелью больного.
При нарушении центральных механизмов регуляции также нарушается механизм обратной связи. Нередко он отключается, и изменение концентрации гормона в крови уже не изменяет секреции рилизинг-фактора. Выше, например, уже указывалось, что при болезни Иценко-Кушинга снижается чувствительность гипоталамических центров, воспринимающих колебания концентрации кортизола в крови. В этих случаях обычная концентрация кортизола не тормозит образования кортиколиберина, а это ведет к увеличению его образования и, соответственно, к увеличению
секреции АКТГ. Для оценки функции желез, имеющих трансгипофизарную регуляцию, важно определять концентрацию тропного гормона в крови. Это может помочь установить локализацию патологического процесса. Так, например, при гипотиреозе значительное увеличение концентрации ТТГ (в 4-10 раз) свидетельствует о поражении щитовидной железы, которая не реагирует на ТТГ, а снижение его концентрации до следовых количеств заставляет предполагать локализацию процесса в гипофизе или в центральной нервной системе.
20.1.2. Патологические процессы в самой железе
Различные патологические процессы могут развиваться в самой железе и тем самым вызывать нарушение ее функции.
Инфекционные процессы и интоксикации
Острые инфекционные заболевания могут приводить к нарушению функции желез внутренней секреции. Так, например, менингококковая инфекция может сопровождаться кровоизлиянием в надпочечники, что приводит к разрушению ткани железы и развитию острой надпочечниковой недостаточности. Подобная недостаточность может возникать при дифтерии в связи с коагуляционными некрозами в надпочечниках. Эпидемический паротит у взрослых мужчин часто вызывает орхит, который в 30-50% случаев заканчивается одноили двусторонней атрофией яичек. Тестикулы могут поражаться и при гонорее в связи с восходящей инфекцией уретры. Такие инфекционные заболевания, как туберкулез и сифилис, также поражают различные железы. При туберкулезе отмечается постепенное разрушение ткани железы в связи с творожистым некрозом туберкулезных бугорков, а при сифилисе - в связи с некрозом сифилитической гранулемы (гуммы). При локализации процесса в надпочечных железах развивается хроническая надпочечниковая недостаточность, которая называется аддисоновой болезнью по имени врача Аддисона, впервые описавшего это заболевание. При локализации процесса в тестикулах развивается гипогонадизм, характеризующийся снижением образования андрогенов и нарушением сперматогенеза. При локализации в паращитовидных железах развивается гипопаратиреоз и т.д.
Опухолевые процессы в железах
Это один из частых патологических процессов в железах внутренней секреции. Опухоль может развиваться в любой железе. Клиника заболевания будет определяться характером и количеством секретируемых гормонов и влиянием опухоли на окружающую ткань железы. Есть опухоли, которые не секретируют гормоны, а только сдавливают и приводят к атрофии нормальные участки железы. Клинически это будет выражаться в гипофункции соответствующей железы, как, например, при хромофобных аденомах гипофиза. Среди других опухолей гипофиза эта опухоль встречается чаще всего. Она не секретирует гормоны, но сдавливает гипофиз, вызывая его гипофункцию. Уменьшается секреция тропных гормонов, что приводит к гипофункции половых желез, щитовидной железы и надпочечников. Одновременно она может сдавливать зрительные нервы и хиазму. Это приводит к выпадениям полей зрения вплоть до полной слепоты.
Чаще всего развитие опухоли сопровождается избыточным образованием гормона и клиникой гиперфункции. Так, например, при эозинофильной аденоме гипофиза - опухоли, происходящей из эозинофильных клеток, продуцируется избыточное количество СТГ. В период роста организма это приводит к развитию гигантизма, а после окостенения эпифизарных хрящей - к акромегалии (от греч. akros - крайний, megas - большой). В последнем случае происходит непропорциональное увеличение и утолщение концевых частей скелета (кисти рук, стопы ног) и костей черепа вследствие периостального роста (рис. 20-1). Одновременно увеличиваются внутренние органы.
При базофильной аденоме гипофиза - опухоли из базофильных клеток, продуцируется избыточное количество АКТГ. Это приводит к увеличению секреции кортизола надпочечными железами и развитию синдрома гиперкортизолизма. Данный же синдром может быть вызван и опухолью пучковой зоны коры надпочечников, которая секретирует избыточные количества кортизола. Определенная роль в развитии указанных изменений при этом синдроме принадлежит механизму обратной связи. Если при базофильной аденоме избыточная секреция АКТГ вызывает гиперплазию обоих надпочечников, то при опухоли пучковой зоны одного надпочечника механизм обратной связи выключает секрецию АКТГ и это ведет к тому, что второй - нормальный - надпочечник атрофируется (рис. 20-2).
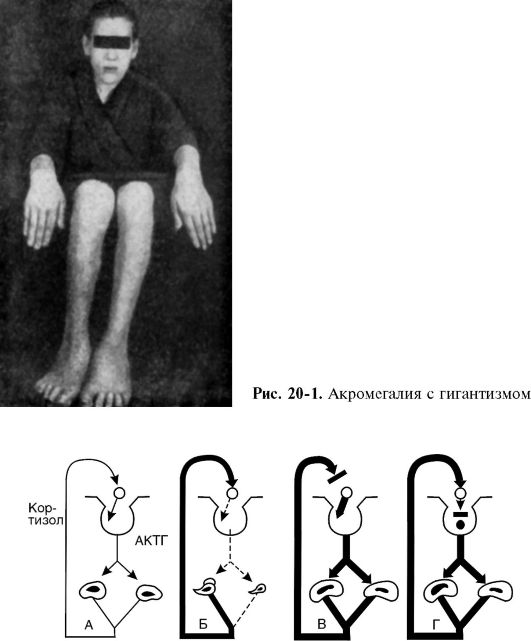 Рис. 20-2. Механизмы
развития гиперкортизолизма: А - саморегуляция продукции кортизола в
норме; Б - при аденоме пусковой зоны коры надпочечников; В - при
снижении чувствительности гипоталамических центров, регулирующих
образование фактора, освобождающего кортикотропин; Г - при базофильной
аденоме гипофиза; АКТГ - адренокортикотропный гормон
Рис. 20-2. Механизмы
развития гиперкортизолизма: А - саморегуляция продукции кортизола в
норме; Б - при аденоме пусковой зоны коры надпочечников; В - при
снижении чувствительности гипоталамических центров, регулирующих
образование фактора, освобождающего кортикотропин; Г - при базофильной
аденоме гипофиза; АКТГ - адренокортикотропный гормон
При опухолях тестикул, происходящих из клеток Лейдига, усиливается образование андрогенов. Если опухоль возникает у мальчиков до 9-летнего возраста, то это ведет к преждевременному половому созреванию, характеризующемуся быстрым ростом тела и развитием вторичных половых признаков. Однако опухолевый процесс не сопровождается сперматогенезом и непораженные участки железы остаются незрелыми.
Опухоли сетчатой зоны коры надпочечников продуцируют гормоны, обладающие андрогенными и эстрогенными свойствами, и приводят к развитию адреногенитальных синдромов (см. ниже).
Иногда опухоли поражают несколько эндокринных желез. Описаны аденомы, одновременно развивающиеся в аденогипофизе, паращитовидных железах и островках поджелудочной железы. Одна или все они могут быть гормонально-активными, и клиника будет зависеть от количества и вида секретируемых гормонов. Иногда этот синдром носит семейный характер и сопровождается развитием пептических язв. Примером является синдром Золлингера-Эллисона (синдром ульцерогенных аденом островков Лангерганса). Его развитие связано с наличием гастринсекретирующей опухоли поджелудочной железы, вызывающей высокую желудочную секрецию соляной кислоты, развитие пептических язв и диарею.
Железа внутренней секреции может быть не только источником опухоли, но и местом, куда метастазируют опухоли из других органов. В этих случаях растущая опухоль будет сдавливать железу, вызывать ее атрофию и гипофункцию. Так, при метастазе рака молочной железы в заднюю долю гипофиза нарушается выделение антидиуретического гормона (АДГ) и развивается несахарный диабет. Рак легкого, помимо костей, дает метастазы в надпочечники, а рак желудка - нередко в яичники (так называемый крукенбергский рак яичников).
Иногда опухоли эндокринных желез или даже неэндокринных органов начинают продуцировать гормоны, не свойственные данной железе или вообще клеткам данного органа. Например, опухоль щитовидной железы или бронхогенный рак начинает продуцировать АКТГ с развитием как следствие синдрома гиперкортизолизма. Такое изменение фенотипа клеток связано с их опухолевой трансформацией, при которой происходит дерепрессия не функционирующих в норме участков клеточного генома.
Генетически обусловленные дефекты биосинтеза гормонов
Биосинтез любого гормона представляет собой сложный многозвеньевой процесс, в котором принимает участие множество ферментов. При этом образование любого фермента, точнее, его апофермента, определяется активностью соответствующего гена. Мутация гена может привести к недостаточности образования апофермента или такому его изменению, при котором образующийся фермент теряет свою активность. В этом случае нарушается последовательный ход биосинтеза соответствующего гормона, что обусловливает: 1) гипофункцию железы; 2) накопление в железе промежуточных продуктов биосинтеза, образующиеся до места блокады, которые выделяются в кровь и оказывают специфический патофизиологический эффект; 3) нарушение механизма обратной связи и развитие дополнительных патологических процессов. Иллюстрацией к этому служат два примера.
Первый пример. На рис. 20-3 в общих чертах представлен биосинтез кортизола и участки его блокады. В настоящее время хорошо изучены два вида блокады образования кортизола в связи с дефицитом ферментов - 21-гидроксилазы (I) в одном случае и 11 β-гидроксилазы (II) - в другом. При дефиците 21-гидроксилазы (I) процесс биосинтеза заканчивается образованием прогестерона и 17а-оксипрогестерона. Кортизол не образуется. Это по механизму обратной связи растормаживает секрецию кортиколиберина в гипоталамусе, что, в свою очередь, ведет к усилению образования АКТГ. АКТГ стимулирует стероидогенез до места блокады, и так как кортизол не образуется, то вся эта стимуляция переключается на образование D4-андростен-3,17-диона, обладающего андроген-
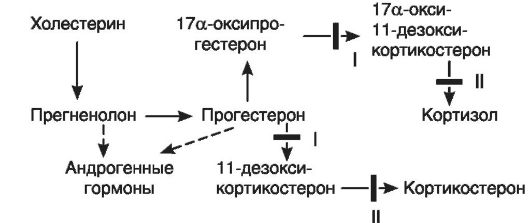 Рис. 20-3. Участки блокады биосинтеза кортизола
Рис. 20-3. Участки блокады биосинтеза кортизола
ными свойствами. Его поступление в кровь значительно увеличивается. Образующиеся в надпочечниках андрогены включаются в механизм обратной связи, регулирующей развитие половых желез, и приводят к выключению этой регуляции, что сопровождается атрофией половых желез как у мальчиков, так и у девочек. Дефект выявляется уже в период эмбрионального развития. У эмбриона женского пола к этому периоду внутренние половые органы уже заложены, поэтому избыток андрогенов вызывает их гипоплазию и развитие вирилизма. Маскулинизация продолжается и после рождения. У мальчиков же появляются признаки преждевременного полового созревания.
Подобный механизм включается и при дефекте фермента 11b-гидроксилазы (II). Кортизол также не образуется, но в этом случае (в отличие от предыдущего синдрома) накапливается избыточное количество 11-дезоксикортикостерона и 17а-окси-11- дезоксикортикостерона, первый из которых обладает выраженными минералокортикоидными свойствами. Это ведет к повышению кровяного давления. Всю эту патогенетическую цепь можно разорвать введением глюкокортикоидов (рис. 20-4). Они тормозят образование АКТГ и тем самым уменьшают образование андрогенов.
Второй пример. Биосинтез тиреоидных гормонов, происходящий в клетках фолликулярного эпителия щитовидной железы,
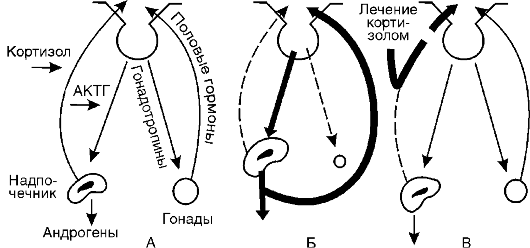 Рис. 20-4. Механизм
атрофии половых желез при врожденном адреногенитальном синдроме и
механизм лечебного действия кортизола: А - регуляторные механизмы в
норме; Б - адреногенитальный синдром; В - патогенетическая терапия
кортизолом (по Гоффу); АКТГ - адренокортикотропный гормон
Рис. 20-4. Механизм
атрофии половых желез при врожденном адреногенитальном синдроме и
механизм лечебного действия кортизола: А - регуляторные механизмы в
норме; Б - адреногенитальный синдром; В - патогенетическая терапия
кортизолом (по Гоффу); АКТГ - адренокортикотропный гормон
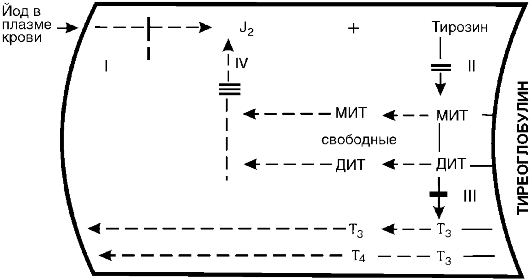 Рис. 20-5. Участки блокады биосинтеза тиреоидных гормонов. МИТ - монойодтирозин, ДИТ - дийодтирозин
Рис. 20-5. Участки блокады биосинтеза тиреоидных гормонов. МИТ - монойодтирозин, ДИТ - дийодтирозин
также является сложным многозвеньевым процессом. В общих чертах он представлен на рис. 20-5 и состоит из следующих основных процессов: 1) захват йода железой и окисление его пероксидазой в молекулярный йод или йодит; 2) йодирование тирозина тирозинйодиназой с образованием монойодтирозина (МИТ) и дийодтирозина (ДИТ); тирозин, как и МИТ и ДИТ, находится в составе тиреоглобулина; 3) конденсация молекул МИТ и ДИТ с образованием трийодтиронина (Т3) и тироксина (Т4); 4) образование свободных МИТ и ДИТ и их дегалогенизация; выделяющийся при этом йод снова идет на йодирование тирозина. В связи с дефектами соответствующих ферментов каждый из указанных этапов может блокироваться.
Установлена возможность блокады йодзахватывающей системы (I). Для этого случая характерна неспособность железы поглощать J131 при соответствующем исследовании. Исправление этого дефекта достигается введением в организм небольших доз йодистого калия, который в связи с повышением его концентрации в крови, в силу диффузии проникает в щитовидную железу и, таким образом, компенсирует дефект йодзахватывающей системы. II - блокада йодирования тирозина. Поглощенный йод сохраняется в железе в неорганической форме и не включается в тирозин. Этот дефект на данном этапе компенсируется введением готовых тиреоидных гормонов. III - дефект конденсации йодтирозинов. Характеризуется
накоплением промежуточных продуктов - МИТ и ДИТ и следовыми количествами Т3 и Т4. Компенсация дефекта проводится также введением гормонов. IV - дефект йодтирозин-дегалогеназы. Он характеризуется угнетением дегалогенизации МИТ и ДИТ. Эти продукты накапливаются, выделяются в кровь и выводятся из организма. Организм теряет йод, развивается йодная недостаточность. Компенсация дефекта может быть обеспечена введением в организм йодистого калия.
Каждый из указанных дефектов приводит к недостаточному образованию тиреоидных гормонов. В результате возникает гипофункция щитовидной железы, сопровождаемая развитием зоба (увеличением щитовидной железы) и кретинизма. Последнее объясняется тем, что эти дефекты возникают еще до рождения или в детском возрасте.
20.1.3. Периферические (внежелезистые) механизмы нарушения активности гормонов
Большую роль в развитии эндокринных и ряда других заболеваний играют периферические механизмы, определяющие активность уже выделившихся в кровь гормонов. Эта активность может изменяться либо в сторону ее повышения, либо снижения, что клинически проявляется гиперили гипофункцией соответствующей железы.
Очевидно, все выделившиеся из желез гормоны связываются в крови с определенными белками и циркулируют в двух формах - связанной и свободной. Из этих двух форм связанный гормон биологически неактивен. Активностью обладает только свободная форма гормона, которая и оказывает физиологическое действие в клетках-мишенях. Известно связывание белками тироксина, инсулина, гормона роста, стероидных гормонов. Так, например, в физиологических условиях в плазме крови кортизол и кортикостерон связаны белками более чем на 90%, и лишь незначительное количество этих кортикостероидов находится в свободном состоянии.
Общее количество циркулирующего тироксина в организме составляет: связанного - 1,0 мг; свободного - 0,001 мг при концентрации последнего в сыворотке крови 0,1 мкг/л. Таким образом, концентрация свободной формы гормона очень незначительна по отношению к связанной.
Механизм действия гормонов на уровне клеток-мишеней различен и сложен. В соответствии с современными представлениями
все гормоны по механизму их действия на клетки-мишени можно разделить на две группы. Одна группа гормонов управляет различными обменными процессами в клетке с ее поверхности, как бы на расстоянии, поэтому данную группу можно назвать гормонами «дистантного» (непрямого) действия. Сюда входят белковые и пептидные гормоны, факторы роста, катехоламины, а также ряд других лигандов. Эти гормоны связываются на поверхности клетки-мишени с соответствующим рецептором, что включает ряд биохимических процессов, приводящих к образованию вторичных посредников. Обычно это выражается в активации ферментовэффекторов (аденилатциклаза, гуанилатциклаза, фосфолипаза С) и накоплении цАМФ, цГМФ или диацилглицерола и инозинтрифосфата. Вторичные посредники, в свою очередь, запускают последующую цепь процессов, важнейшими звеньями которых являются активация протеинкиназ и фосфорилирование белковых субстратов. По такому механизму, в частности, катехоламины регулируют интенсивность гликогенолиза. Специфичность ответа клетки на тот или иной гормон определяется специфичностью рецептора, который связывается только со своим гормоном, а также природой специфических для клетки протеинкиназ и белковых субстратов.
Другая группа гормонов проникает в клетку, где оказывает свое действие. Эту группу можно обозначить как группу гормонов «непосредственного» (прямого) действия. Сюда входят андрогены, эстрогены, прогестины, кортикостероиды. Главным в действии стероидных гормонов является активация или торможение того или иного гена, что сопровождается усилением или угнетением образования соответствующего фермента. Однако ряд эффектов осуществляется другими путями, не связанными с влиянием на активность генов.
В механизме доставки стероида к генетическому локусу можно выделить три звена. Первое звено - связывание поступившего в клетку гормона с белком, находящимся в цитоплазме и выполняющим роль специфического рецептора для данного гормона. Второе звено - модификация комплекса «стероид + рецепторный белок». Эта модификация дает возможность осуществления третьего звена - проникновения стероида в комплексе с рецепторами в ядро клетки и избирательного соединения со специфическим участком хроматина.
Общий механизм влияния гормонов «непосредственного» действия можно проиллюстрировать на примере глюкокортикоидов
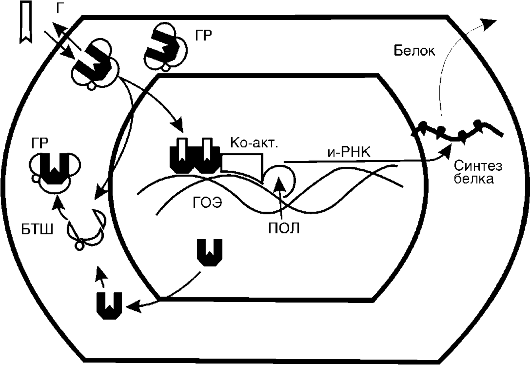 Рис. 20-6. Молекулярные
механизмы действия глюкокортикостероидов (Г): ГР - глюкокортикоидный
рецептор; БТШ - белок теплового шока; Ко-акт. - коактиваторы; ПОЛ -
полимераза; ГОЭ - гормонотвечающий элемент
Рис. 20-6. Молекулярные
механизмы действия глюкокортикостероидов (Г): ГР - глюкокортикоидный
рецептор; БТШ - белок теплового шока; Ко-акт. - коактиваторы; ПОЛ -
полимераза; ГОЭ - гормонотвечающий элемент
(рис. 20-6). Гормон свободно проникает в клетку и связывается со специфическими рецепторными белками цитоплазмы - глюкокортикоидными рецепторами (ГР). Очевидно, связывается неметаболизированный гормон, поскольку из стероидно-белкового комплекса удается выделить глюкокортикоид как таковой. Об этом свидетельствует и тот факт, что метаболиты кортизола не вызывают эффектов кортизола и конкурентно не угнетают его действия. Рецепторные белки обладают высоким сродством к стероиду, выраженной специфичностью и малой емкостью. Поэтому данный вид связывания называют специфическим. В зависимости от вида клеток количество рецепторов колеблется от 3000 до 5000 на одну клетку. Сравнение различных тканей одного вида животных показало, что связывание глюкокортикоида различно в разных тканях. Так, растворимая фракция клеток тимуса связывала в 3 раза больше триамсинолона, чем такие же фракции из коры головного мозга и тестикул.
Глюкокортикоидные рецепторы относятся к суперсемейству стероид/ядерных регуляторных протеинов, которые функционируют как лигандактивируемые факторы транскрипции. В цитоплаз-
ме ГР в несвязанном с гормоном состоянии представляют собой гетерогенные комплексы, состоящие из собственно рецептора и связанных с ним по крайней мере четырех белков теплового шока (БТШ). Роль последних заключается в поддержании конформации ГР в состоянии, подходящем для связывания гормона и предупреждения транслокации несвязанного с гормоном ГР в ядро. После связывания ГР с гормоном он освобождается из комплекса с белками теплового шока и перемещается в ядро. Здесь глюкокортикоидрецепторный комплекс превращается в димер и связывается в регуляторной части соответствующего гена с определенным участком ДНК, называемым гормонотвечающим элементом (ГОЭ). ГР-димер регулирует транскрипцию этого гена, вызывая либо активацию транскрипции, либо ее угнетение. При снижении уровня гормона и диссоциации гормонально-рецепторного комплекса в ядре ГР освобождается и возвращается обратно в цитоплазму, где снова образует комплекс с белками теплового шока.
Препараты глюкокортикоидных гормонов обычно применяют для подавления воспаления при многих заболеваниях (аутоиммунные процессы, бронхиальная астма и др.). Механизмы их антивоспалительного действия многообразны и реализуются через изменение регуляции активности многих генов, кодирующих образование провоспалительных цитокинов, ферментов и других продуктов, участвующих в развитии воспаления. Так, глюкокортикоиды:
1) усиливают экспрессию генов, которые кодируют образование ферментов, оказывающих угнетающее влияние на развитие воспаления (липокортин-1, нейтральная эндопептидаза в эпителиальных клетках слизистой дыхательных путей разрушают тахикинины, лейкоцитарный секретируемый ингибитор протеазы в слизистой дыхательных путей и др.);
2) угнетают экспрессию генов, кодирующих образование провоспалительных цитокинов (интерлейкины-1-6, гранулоцитарномакрофагальный колониестимулирующий фактор, фактор некроза опухоли и др.);
3) угнетают экспрессию генов, кодирующих образование энзимов, способствующих развитию воспаления (синтетаза оксида азота, индуцибельная изоформа циклоксигеназы-2);
4) угнетают экспрессию генов, кодирующих образование молекул адгезии (ICAM-I) и рецепторов для провоспалительных медиаторов (для вещества Р).
Одним из важных механизмов действия глюкокортикоидов является так называемое пермиссивное действие. Оно означает, что некоторые метаболические эффекты гормонов дистантного действия, о которых упоминалось выше, реализуются только в присутствии физиологических концентраций глюкокортикоидов.
Все гормоны, циркулирующие в организме, метаболизируются и выводятся из него. В основном метаболизм гормонов происходит в печени. Однако ряд гормонов метаболизируется и в других тканях.
В организме для каждого гормона существует равновесие между его секрецией, связыванием белками, действием в тканях-мишенях и метаболизмом в тканях. В поддержании такого равновесия большую роль играет механизм обратной связи. Нарушение любого из внежелезистых компонентов этого равновесия может приводить к таким изменениям, которые будут клинически проявляться как нарушение функции соответствующей железы.
Нарушение связывания гормонов белками
Связывание кортикостероидов белками плазмы крови при определенных условиях может нарушаться. Это может стать патогенетическим фактором либо сниженной, либо повышенной физиологической активности кортикостероидных гормонов. Клинические наблюдения указывают на такую возможность. Так, например, при синдроме Иценко-Кушинга выявляются случаи, сопровождаемые снижением связывания кортизола белками плазмы крови, что приводит к увеличению свободной фракции кортизола. При снижении способности белков плазмы крови связывать кортизол обнаруживали также признаки диабета или преддиабета, нарушения менструального цикла, гипертензию и др. Нарушение связывания тиреоидных гормонов может приводить к таким изменениям, которые определяются как гипоили гипертиреоз. Усиление связывания инсулина может способствовать возникновению инсулиновой недостаточности.
Блокада циркулирующего гормона
Этот вид изменений гормональной активности касается полипептидных гормонов и имитирует картину гипофункции соответствующей железы. Возможны следующие механизмы инактивации:
а) инактивация гормона в связи с образованием аутоантител к тому или иному гормону. Такая возможность хорошо известна при лечении экзогенными гормональными препаратами. Установлено образование антител к инсулину, СТГ, АКТГ у большинства лечившихся людей, что в ряде случаев сопровождается снижением лечебного эффекта препарата. Возможно образование аутоантител и к гормонам, образующимся в самом организме;
б) изменения в активном центре или конформации молекулы гормона в связи с мутацией и замещением в молекуле гормона одной аминокислоты на другую. Такие замещения обнаружены в активном центре инсулина. Можно предположить такую возможность в отношении других гормонов и, в частности, СТГ. Последнее предположение вытекает из клинических наблюдений. Так, есть группа больных карликовостью с очень высокой концентрацией СТГ в плазме крови, однако действия этот гормон на рост организма не оказывает. Гормон определяется иммунологически, что свидетельствует о сохранности его антигенных свойств. Больные отвечают увеличением роста на введение экзогенного СТГ, что указывает на наличие нормально функционирующих рецепторов к данному гормону. Сопоставление этих двух фактов дает основание сделать заключение о недостаточной активности эндогенного СТГ;
в) нарушение превращения прогормона в гормон. Установлено, что белковые гормоны секретируются вначале как прогормоны в составе более крупных полипептидных цепей, которые затем расщепляются. Так, например, плацента секретирует АКТГ, липотропин и β-эндорфин как общую молекулу. В некоторых случаях у больных сахарным диабетом обнаружен инсулин, у которого С-терминальный конец β-цепи связан с С-пептидом. В обычных условиях С-пептид соединяет α- и β-цепи инсулина, и вся молекула называется проинсулином. Это одноцепочный белок с м. м. 10 000 Да, физиологически неактивный. В островках Лангерганса или даже на периферии от проинсулина в результате протеолиза отщепляется С-пептид, и проинсулин превращается в активный инсулин. Нарушение отщепления С-пептида, очевидно, не дает инсулину возможности принять такую конформацию, в которой он наиболее активен.
Блокада гормонального рецептора
Очевидно, это довольно распространенный механизм, приводящий к гормональной недостаточности. Это происходит в тех
случаях, когда активный гормон не находит своего рецептора на клетке или в ней в связи с потерей рецептора либо в связи с фиксацией на его поверхности антагонистов, конформационными изменениями и другими факторами, препятствующими соединению с гормоном. Обычно концентрация гормона в таких случаях нормальна либо увеличена. Введение таким больным с лечебной целью гормонов не приводит к нужному результату. Для получения некоторого эффекта нужно вводить большие дозы препарата.
Описаны случаи вазопрессинрезистентных форм несахарного диабета, сопровождающиеся значительным увеличением содержания антидиуретического гормона в крови и отсутствием эффекта на его введение извне. В ряде случаев карликового роста концентрация СТГ в крови нормальна, и у больных отсутствует реакция на экзогенный СТГ. Введение СТГ не стимулируют (как в норме) образование соматомедина, через который СТГ оказывает свое влияние на рост. При псевдогипопаратиреозе развивается синдром, сходный с гипопаратиреозом, сопровождащийся гипокальциемией, гиперфосфатемией и даже развитием тетании. Такие больные не реагируют на введение экзогенного паратгормона. Аналогичные изменения выявлены и в отношении ГР. Обнаружена изоформа ГР, которая не связывала гормон, поэтому не было влияния на экспрессию генов. В других случаях определялся укороченный в карбоксильном конце глюкокортикоидный рецептор, который также оказался функционально неполноценным. В Т-лимфоцитах стероидрезистентных больных бронхиальной астмой выявлялось обратимое цитокинопосредованное снижение аффинности ГР к гормону, которое ассоциировалось с изменением функции этих клеток.
Нарушение пермиссивного действия глюкокортикоидов
Как указывалось выше, эффекты ряда гормонов «дистантного» действия, и, в частности, катехоламинов реализуются на фоне физиологических концентраций кортизола. Эту роль кортизола называют пермиссивной. Поэтому снижение концентрации кортизола ведет к уменьшению, а иногда и к извращению эффекта катехоламинов. Так, например, адреналин вызывает гликогенолиз в печени и липолиз в жировой ткани в присутствии кортизола. Поэтому у адреналэктомированных животных значительно снижены оба эти эффекта адреналина. Вызываемый адреналином гликогенолиз яв-
ляется сложным и многозвеньевым процессом. Он начинается с соединения адреналина с β-адренергическим рецептором на клеточной мембране. Это вызывает активацию аденилатциклазы и приводит к усилению образования циклического аденозинмонофосфата, который, в свою очередь, через ряд реакций приводит к активации фосфорилазы и гликогенолизу.
Механизм пермиссивного действия кортизола может реализоваться на разных уровнях в зависимости от характера стимулируемой обменной реакции и вида клеток. Он не влияет на связывание адреналина с его рецепторами на лейкоцитах, в том числе и на лимфоцитах, так как не обнаружено различий в их связывающей способности у больных бронхиальной астмой по сравнению со здоровыми. Однако число β-адренорецепторов на клетках тканей дыхательных путей и лейкоцитах этих больных оказывалось сниженным. Лечение глюкокортикоидами увеличивало экспрессию этих рецепторов. В определенных случаях кортизол в физиологических концентрациях оказывает непосредственное активирующее влияние на аденилатциклазу, что ведет к увеличению концентрации цАМФ. В других случаях при нормальной или повышенной под влиянием катехоламинов концентрации цАМФ в отсутствие глюкокортикоидов оказывались блокированными последующие звенья процесса. В подобных случаях находили блокаду одной из стадий активации фосфорилазы в связи с недостаточной мобилизацией ионов кальция. Увеличение концентрации этих ионов или добавление глюкокортикоидов восстанавливало ход процесса.
Нарушение метаболизма гормонов
При гепатитах и циррозах печени метаболизм гормонов угнетается. Замедление метаболизма кортизола приводит к задержке его в организме. Это включает механизм обратной связи и угнетает функцию коры надпочечников, что приводит к некоторой их атрофии (рис. 20-7). Снижение инактивации эстрадиола в печени у мужчин вызывает включение механизма обратной связи, в результате чего угнетается образование гонадотропных гормонов в гипофизе и как следствие снижается функция тестикул, развивается импотенция. Одновременно при циррозах печени тестостерон легче превращается в эстрогены.
Таким образом, причины и механизмы нарушения функции желез внутренней секреции многообразны. Они могут действовать
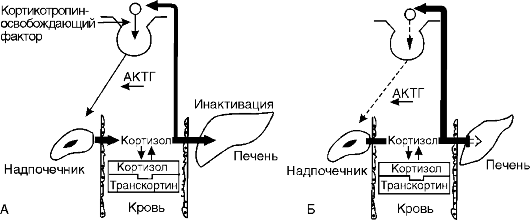 Рис. 20-7. Регуляция
образования кортизола в норме (А) и при гепатитах и циррозах (Б).
Угнетение метаболизма кортизола в печени по механизму обратной связи
тормозит его образование в надпочечниках. АКТГ - адренокортикотропный
гормон
Рис. 20-7. Регуляция
образования кортизола в норме (А) и при гепатитах и циррозах (Б).
Угнетение метаболизма кортизола в печени по механизму обратной связи
тормозит его образование в надпочечниках. АКТГ - адренокортикотропный
гормон
как изолированно, так и в различных комбинациях, приводя к сложному переплетению обменных функциональных и структурных нарушений.
20.1.4. Роль аутоаллергических (аутоиммунных) механизмов в развитии эндокринных нарушений
Все больше появляется данных о том, что наиболее частым механизмом нарушения функции эндокринной системы является образование аутоантител к различным ее компонентам. Эти аутоантитела гетерогенны по своему составу и свойствам и действуют на различных участках эндокринной регуляции. Описана группа аутоантител, повреждающих клетки желез внутренней секреции и приводящих к развитию недостаточности той или иной железы. Так, известны аутоиммунные формы недостаточности щитовидной, паращитовидных, надпочечных желез. Аналогичным образом развивается инсулинозависимая (1 тип) форма сахарного диабета.
Наиболее ярко аутоаллергический механизм повреждения выявляется при тиреоидите Хасимото. Это заболевание щитовидной железы было описано Хасимото в 1912 г. Оно встречается преимущественно у женщин в возрасте за 50 лет и сопровождается снижением функции железы - гипотиреоидизмом и увеличением ее объема, т.е. развитием зоба. Строение железы резко изменено. Она инфильтрирована главным образом лимфоцитами, поэтому
это заболевание иногда называют лимфоидным зобом. Инфильтрация носит диффузный и очаговый характер. Количество фолликулов постепенно уменьшается, и они заменяются соединительной тканью. Это приводит к постепенному снижению функции железы, иногда вплоть до развития микседемы. В железе имеется как минимум три антигена (естественные или изолированные). Они находятся в тиреоглобулине, в коллоиде фолликулярного эпителия. Аутоантитела могут образовываться ко всем трем антигенам. Одновременно в повреждении участвует и аллергическая реакция замедленного типа.
Инсулинозависимый тип сахарного диабета часто сочетается с образованием аутоантител к островкам. Состав этих аутоантител различен. Можно обнаружить антитела к α- и β-клеткам, причем они могут быть направлены к рецепторам для глюкозы, к участкам мембраны, ответственным за Са2+-опосредованный экзоцитоз глюкагона и/или инсулина. Это создает различные сочетания в нарушениях образования глюкагона и инсулина, что находит свое отражение в разнообразии клинических проявлений диабета.
Действие другой группы аутоантител направлено против полипептидных гормонов.
Наибольшее внимание привлекает третья группа аутоантител, действие которых направлено на рецепторы для гормонов на различных клетках-мишенях. Эти аутоантитела получили название антирецепторных. Рецептор представляет собой обычно сложный белок, состоящий из нескольких субъединиц, и выполняет, как правило, две функции: а) узнавания, в которой рецептор специфически связывает химический сигнал (гормон, медиатор, токсин, вирус), и б) передачи, в которой взаимодействие химического сигнала с рецептором трансформируется в определенный биохимический процесс. Антирецепторные антитела могут быть направлены к различным частям рецептора. Поэтому возможны различные последствия связывания аутоантител с рецепторами. Установлены следующие варианты:
1. Антитела блокируют место узнавания на рецепторе, поэтому естественный или экзогенный гормон полностью или частично связаться с ним не может. Развивается клиника недостаточности данной железы, хотя гормон в крови есть. Выявляется резистентность к экзогенному гормону.
2. Антитела связываются с активным местом рецептора. Возникает имитация действия гормона, развивается клиника гиперфунк-
ции данной железы. По механизму обратной связи образование естественного гормона снижается.
3. Образование комплекса «антитело + рецептор» в зависимости от вида антител может приводить к активации комплемента и повреждению рецепторов.
4. Образовавшиеся комплексы «антитело + рецептор» собираются в одном месте на поверхности клетки (кэппинг - образование шапки), после чего в этом месте происходит впячивание части мембраны внутрь клетки с образованием фагосомы, где происходит деградация комплексов. Взамен утраченных рецепторов клетка образует новые. При хроническом течении процесса может происходить истощение воспроизводящей функции клетки, и на ее поверхности уменьшается число рецепторов к данному гормону.
Процесс поглощения, деградации и воспроизведения рецепторов происходит и в норме. Так поглощаются и разрушаются гормонрецепторные комплексы. От избытка гормона клетка защищается, уменьшая образование рецепторов. Этот механизм, в частности, лежит в основе снижения чувствительности клеток-мишеней к инсулину у людей, употребляющих избыточное количество пищи. Последнее ведет к усилению образования инсулина. В ответ на избыток инсулина клетки-мишени снижают число рецепторов. Развивается один из видов инсулинорезистентности, который хорошо лечится ограничением приема пищи.
Характер функциональных нарушений будет определяться свойствами образовавшихся аутоантител и их соотношением. Чаще идет образование аутоантител одновременно к различным субъединицам рецептора. Так, например, при диффузном токсическом зобе (базедова болезнь, Гревса болезнь) примерно у 95% нелеченых больных выявляются аутоантитела к рецептору для ТТГ. Они получили разные названия (длительно действующий стимулятор - LATS; протектор длительно действующего стимулятора - LATS-P; тиреоидстимулирующие антитела - ТSАb; тиреотропинсвязывающий ингибитор ТSI и др.).
Более детальные исследования показали, что одни из них направлены к гликопротеиновой субъединице (место узнавания сигнала), другие - к ганглиозидной субъединице (функция передачи сигнала). Все они в той или иной степени блокируют связывание ТТГ, но при этом одни стимулируют образование цАМФ, синтез и освобождение Т3 и Т4, а другие - рост тиреоидных клеток без влияния на образование цАМФ. Отсюда первые приводят к раз-
витию клиники гипертиреоидизма, а вторые - к развитию зоба с небольшим увеличением содержания в крови Т3 и Т4. По ходу развития заболевания количество антител разных видов обычно меняется. Поэтому изменяются функция щитовидной железы и клиника заболевания. Аналогичным образом дело обстоит с функцией других клеток-мишеней.
Возникает вопрос: почему вырабатываются аутоантитела к рецепторам клеток? Считают, что это связано с дисбалансом в механизмах идиотип-антиидиотипического взаимодействия. Суть его сводится к тому (рис. 20-8), что на антигенную детерминанту гормона, которая может оказаться той частью, которой гормон связывается с рецептором клетки, образуются специфические антитела с уникальной конфигурацией на антигенсвязывающем конце. Эта специфическая, уникальная конфигурация получила название
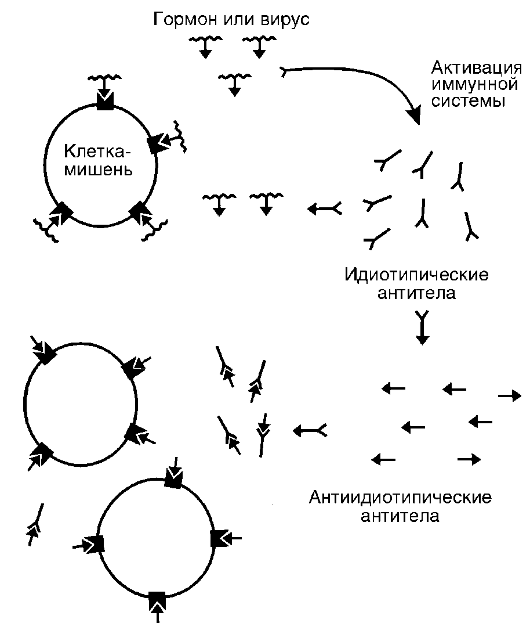 Рис. 20-8. Нарушение идиотип-антиидиотипического взаимодействия как причина образования антирецепторных аутоантител
Рис. 20-8. Нарушение идиотип-антиидиотипического взаимодействия как причина образования антирецепторных аутоантител
идиотипа. Идиотип является зеркальным отражением конфигурации антигенной детерминанты, поэтому и способен связываться с ней. Но сам идиотип, т.е. его конфигурация, является чужеродной для иммунной системы организма, и она начинает образовывать анти-антитела, специфичные к идиотипу и получившие название антиидиотипических антител. Последние, являясь зеркальным отражением специфичности идиотипических антител, становятся по конфигурации аналогичными антигенной детерминанте гормона. Поэтому они могут связываться как с идиотипическими антителами, так и с гормональными рецепторами клетки-мишени.
Полипептидные гормоны имеют обычно несколько антигенных детерминант. Некоторые из них могут оказаться теми участками, через которые гормон связывается с местом узнавания или передачи сигнала на рецепторе. Поэтому обычно образуются различные виды аутоантител со специфичностью к различным участкам рецептора со всеми вытекающими отсюда последствиями.
У подавляющего числа людей антирецепторные антитела не обнаруживаются, так как в физиологических условиях к собственным гормонам имеется иммунологическая толерантность, и иммунная реакция на них не включается. Что же должно произойти, чтобы этот механизм включился?
Во-первых, должны быть какие-то особенности реагирования самой иммунной системы. Установлено, что имеется связь между образованием антирецепторных аутоантител и определенными антигенами гистосовместимости. Они образуются обычно у людей, имеющих гаплотип HLA-B8-DW3-DR-3. Поскольку имеется особенность иммунного реагирования, то обычно образуются аутоантитела не к одному, а ко многим антигенам, что создает основу для развития плюригландулярных расстройств, например сочетание недостаточности надпочечников, диффузного тиреотоксического зоба, сахарного диабета и др.
Во-вторых, должен быть какой-то стимул, выводящий нейроэндокринную систему из равновесия и приводящий к избыточному образованию гормона. Таким стимулом может быть стресс. Уже указывалось, что клетки-мишени защищаются от избытка гормона тем, что усиливают поглощение и разрушение гормонально-рецепторных комплексов. Очевидно, при этом может включаться и иммунный механизм защиты. Можно также представить, что употребление больших количеств легко усваиваемых углеводов приведет к усиленному образованию инсулина и как следствие к включению иммунного механизма.
В-третьих, все больше накапливается данных, что причиной образования антирецепторных аутоантител может быть вирусная инфекция, вызванная, в частности, вирусами Коксаки В, паротита, краснухи, гепатита. Описывают развитие у детей инсулинозависимого сахарного диабета после этих вирусных заболеваний. В эксперименте у мышей ряд вирусных инфекций приводил к развитию расстройств, похожих на сахарный диабет, и даже к развитию полиэндокринопатий с появлением аутоантител. Известно, что вирус проникает в клетку после соединения с рецепторами на ее поверхности. Если таким рецептором окажется гормональный рецептор, то легко может быть запущен механизм идиотипантиидиотипического взаимодействия (см. рис. 20-8), и вирус спровоцирует образование антирецепторных аутоантител.
20.2. ПАТОФИЗИОЛОГИЯ ОТДЕЛЬНЫХ
ЭНДОКРИННЫХ ЖЕЛЕЗ
20.2.1. Патофизиология гипофиза Недостаточность функции гипофиза
Гипофизэктомия. Последствия гипофизэктомии зависят от вида и возраста животного. Возникающие нарушения связаны в основном с выпадением функции аденогипофиза.
Общими признаками гипофизэктомии являются: задержка роста, нарушение функции размножения, атрофия щитовидной и половых желез, коры надпочечников, астения, кахексия, полиурия. У рыб, рептилий и амфибий теряется способность приспосабливать окраску к окружающему фону. Нарушаются обмен веществ, утилизация основных компонентов пищи. Животные чувствительны к инсулину и резистентны к гипергликемическому действию адреналина. Повышается чувствительность к действию факторов внешней среды и снижается устойчивость к инфекции.
Пангипопитуитаризм. У человека полная недостаточность функции гипофиза выявляется при разрушении 90% его ткани. Этот синдром называют пангипопитуитаризмом, или синдромом Симмондса-Шиена. К развитию данного синдрома могут привести следующие причины: сосудистые нарушения в гипофизе и гипоталамусе (наиболее часто послеродовой длительный спазм сосудов мозга и гипофиза вследствие кровопотери на фоне гиперплазии
аденогипофиза - послеродовой гипопитуитаризм), травмы основания черепа, опухоли гипофиза и гипоталамуса, воспалительное повреждение (туберкулез, сепсис) гипофиза, врожденная аплазия и гипоплазия гипофиза и т.д.
Чаще всего в основе развития гипопитуитаризма лежит нарушение гонадотропной функции гипофиза и секреции СТГ с последующим присоединением недостаточности секреции ТТГ, АКТГ и пролактина. Клинически это проявляется нарушением половых функций, снижением полового влечения, уменьшением размера половых органов, выпадением волос на лобке и в подмышечных впадинах, бледностью кожных покровов, утомляемостью, мышечной слабостью.
В редких случаях наблюдается общее истощение. Смерть может наступить от гипогликемической комы (гипогликемия - как следствие снижения секреции контринсулярных гормонов - глюкокортикоидов и СТГ). При развитии синдрома у детей наблюдается отставание в росте, физическом (недостаток СТГ, ТТГ, АКТГ), психическом (недостаток ТТГ) и половом развитии (ГТГ).
Изолированная недостаточность гормонов передней доли гипофиза
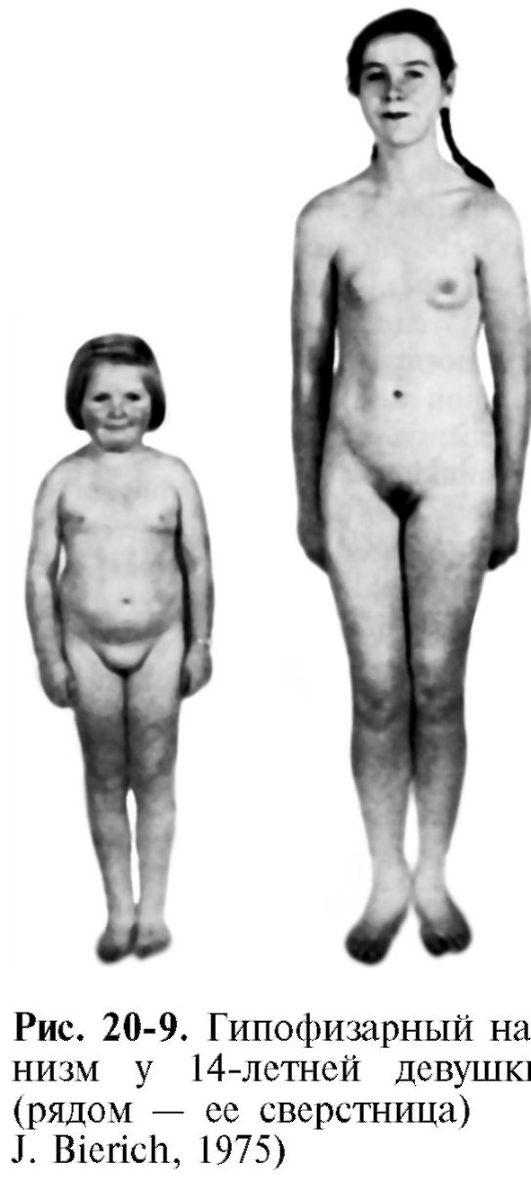
Недостаточность соматотропного гормона. Недостаточное образование СТГ приводит к развитию гипофизарной карликовости, или нанизма. В более чем половине случаев развитие заболевания связано с генетически обусловленным снижением секреции СТГ, которое проявляется двумя основными типами нарушений: врожденной аплазией гипофиза и семейным пангипопитуитаризмом или изолированной недостаточностью СТГ. При этом наследование может быть как аутосомным, так и сцепленным с полом.
У остальных больных причина болезни либо не установлена (идиопатический нанизм), либо причиной ее являются органические нарушения гипоталамо-гипофизарной области (травмы, опухоли, нарушение кровообращения, воспалительные изменения).
В результате недостаточного образования СТГ наблюдаются:
а) снижение интенсивности синтеза белка, что ведет к задержке и остановке роста (более чем на 30% от средних значений данной возрастной группы), развития костей, внутренних органов, мышц; нарушение синтеза белков соединительной ткани приводит к потере ее эластичности и развитию дряблости;
б) уменьшение ингибирующего действия СТГ на поглощение глюкозы и преобладание инсулинового эффекта, что выражается в развитии гипогликемии;
в) выпадение жиромобилизующего действия и тенденция к ожирению.
Обычно гипофизарный нанизм сопровождается половым недоразвитием, что связано с недостаточным образованием ГТГ и, следовательно, с недостаточным образованием половых гормонов. Отсюда у карликов детские черты лица, что наряду с дряблостью кожи придает им вид «старообразного юнца». Снижение интенсивности синтеза белка лежит и в основе некоторой недостаточности синтеза гормонов коры надпочечников и щитовидной железы (рис. 20-9). Это снижает выносливость таких больных при действии неблагоприятных факторов.
Недостаточность адренокортикотройного гормона. Недостаточное образование АКТГ ведет к вторичной частичной недостаточности коры надпочечников. Страдает в основном глюкокортикоидная функция. Минералокортикоидная функция практически не меняется, так как механизмы ее регуляции иные. Отличием от первичной гипофункции коры надпочечников является отсутствие развития гиперпигментации, связанное с тем, что уровень АКТГ снижен и его меланофорный эффект не проявляется.
Недостаточность тиреотропного гормона. Снижение образования ТТГ вызывает вторичное снижение функции щитовидной железы, что ведет к развитию симптоматики вторичного гипотиреоза. В отличие от первичной гипофункции щитовидной железы введение ТТГ может восстановить ее функцию. Содержание ТТГ в крови (по может снижаться и в связи с включением механизма обратной связи при
 первичной
гиперфункции щитовидной железы. Так, например, при диффузном
токсическом зобе в связи с гиперфункцией железы и избыточным
образованием Т3 и Т4 угнетается образование ТТГ.
первичной
гиперфункции щитовидной железы. Так, например, при диффузном
токсическом зобе в связи с гиперфункцией железы и избыточным
образованием Т3 и Т4 угнетается образование ТТГ.
Недостаточность гонадотропных гормонов. При недостаточном образовании ГТГ возникают различные расстройства, картина которых зависит от того, какие ГТГ не образуются и насколько их недостаточность сочетается с выпадением секреции других гормонов аденогипофиза. Недостаточное образование у мужчин фолликулостимулирующего гормона (ФСГ) приводит к снижению способности клеток Сертоли накапливать андрогены, что вызывает определенное угнетение сперматогенеза, а это ведет к снижению фертильности у мужчины, т.е. способности к оплодотворению. Во всех других отношениях эти лица здоровы. Клетки Лейдига при этом не страдают и продуцируют андрогены. Угнетение образования лютеинизирующего гормона (ЛГ) (у мужчин он обозначается ГСИК - гормон, стимулирующий интерстициальные клетки) при адекватном образовании ФСГ нарушает функцию клеток Лейдига. Иногда они даже полностью отсутствуют. В результате отсутствует образование андрогенов. Развивается евнухоидизм с сохранением частичной способности к оплодотворению, так как процесс созревания сперматозоидов полностью не прекращается. Одновременное снижение секреции ФСГ и ГСИК приводит к подавлению функциональной активности семенных канальцев и клеток Лейдига. Если этот процесс развивается до наступления полового созревания, появляются евнухоидизм с недоразвитием наружных половых органов и крипторхизм (задержка опущения яичек в мошонку).
Недостаточное образование ГТГ у девочек также приводит к недоразвитию половых органов и вторичных половых признаков.
Секреция ГТГ по механизму обратной связи тормозится половыми гормонами, причем эстрогены являются более мощными ингибиторами, чем андрогены. В физиологических условиях в половых железах мужчин образуется небольшое количество эстрогенов. При патологии это образование эстрогенов может увеличиваться, что приводит к угнетению образования ГТГ и тем самым к развитию гипогонадизма.
При поражении вентромедиальных ядер инфундибулотуберальной части гипоталамуса со вторичным вовлечением гипофиза преимущественно в виде недостаточной секреции ГТГ развивается так называемая адипозо-генитальная дистрофия. Она
проявляется в виде гипогенитализма и ожирения с преимущественным отложением жира в области нижней части живота, таза и верхней части бедер. Недостаточная секреция ГТГ вызывает задержку полового созревания.
Гиперфункция передней доли гипофиза
Избыточная секреция соматотропного гормона (гормон роста). Избыточная секреция этого гормона наблюдается чаще всего при эозинофильной аденоме гипофиза.
Клинически это проявляется развитием акромегалии и гигантизма (рис. 20-10). Акромегалия - заболевание у людей с закончившимся ростом, проявляющееся диспропорциями скелета, мягких тканей (увеличение размеров кистей, стоп, носа, ушей, нижней челюсти) (рис. 20-11), кифосколиозом, спланхномегалией (увеличение размера внутренних органов). Избыточная секреция СТГ в детском возрасте приводит к развитию гигантизма, сопровождающегося увеличением роста более 190 см в сочетании с признаками акромегалии. В 90% случаев развитие акромегалии и гигантизма связано с наличием гормонально-активной эозинофильной адено-
 Рис. 20-10. Акромегалический
гигантизм у больного 25 лет, рост 220 см (вокруг стоят люди нормального
роста). Случай Ланнуа и Роя (по Н.А. Шерешевскому)
Рис. 20-10. Акромегалический
гигантизм у больного 25 лет, рост 220 см (вокруг стоят люди нормального
роста). Случай Ланнуа и Роя (по Н.А. Шерешевскому)
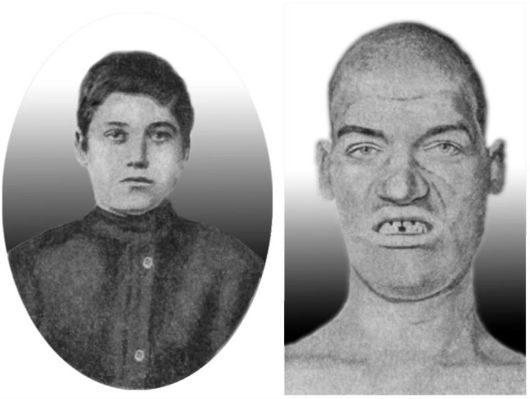 Рис. 20-11. Юноша 16 лет до заболевания акромегалией. Тот же человек в 20-летнем возрасте во время болезни (по W. Schultze, 1904)
Рис. 20-11. Юноша 16 лет до заболевания акромегалией. Тот же человек в 20-летнем возрасте во время болезни (по W. Schultze, 1904)
мы гипофиза. В ряде случаев опухоль не обнаруживается, а развитие гиперплазии гипофиза может быть объяснено, по-видимому, либо избыточной секрецией соматолиберина, либо недостаточной секрецией соматостатина, возникающей в результате повреждения гипоталамуса. Такими повреждениями могут быть травмы (в том числе родовые), инфекции (вирусные инфекции, скарлатина, сыпной тиф, туберкулез, сифилис), нарушения кровообращения. Увеличенное образование СТГ приводит к нарушению обмена белков, углеводов и жиров.
Нарушения белкового обмена. Усиление роста свидетельствует об активации синтеза белков или торможении их разрушения. Действительно, введение СТГ животным вызывает задержку азота в организме, положительный азотистый баланс и понижение распада белков. При этом установлено увеличение включения разных аминокислот в белки тканей и снижение отношения остаточного азота к белковому.
Считается, что действие СТГ опосредовано действием пептидных ростовых факторов - инсулиноподобных факторов роста (ИФР), синтезируемых в тканях и прежде всего в печени. Именно с их действием связывают такие анаболические эффекты, как:
1) стимуляция включения SO4 в протеогликаны;
2) стимуляция включения тимидина в ДНК;
3) стимуляция синтеза РНК;
4) стимуляция синтеза белка СТГ.
Анаболический эффект СТГ обусловливают два момента:
1. Наличие инсулина. На фоне экспериментального диабета у животных и сахарного диабета у людей СТГ обычно не усиливает синтеза белков. Очевидно, это связано с тем, что инсулин активирует обмен углеводов и стимулирует синтез белка.
2. Концентрация глюкокортикоидов. Малые их дозы способствуют реализации анаболического эффекта СТГ, а большие дозы, наоборот, тормозят анаболический эффект СТГ и задерживают рост, что может быть связано с тем, что кортизол в больших дозах угнетает образование ИФР. У больных с эозинофильной аденомой гипофиза часто усилена продукция глюкокортикоидов. Не исключено, что это один из компенсаторных процессов, направленных на ограничение эффекта избыточных количеств СТГ.
Нарушение углеводного обмена. Это нарушение имеет различную степень выраженности. В своей крайней форме проявляется в виде сахарного диабета. Механизм этих нарушений сложен и включает участие следующих факторов:
а) СТГ активирует выход глюкозы из печени за счет активации секреции глюкагона альфа-клетками островков поджелудочной железы, который усиливает гликогенолиз;
б) в поджелудочной железе СТГ стимулирует продукцию инсулина, что усиливает утилизацию глюкозы тканями, однако на уровне клеток тканей СТГ совместно с глюкокортикоидами выступает как антагонист инсулина, т.е. тормозит поглощение глюкозы. Механизм торможения связан с активацией ингибирующей активности β-липопротеиновой фракции сыворотки крови, которая угнетает гексокиназную реакцию, являющуюся пусковой в углеводном обмене;
в) СТГ активирует инсулиназу печени, расщепляющую инсулин. Конечный результат влияния СТГ на углеводный обмен зависит от всех указанных факторов.
Нарушение жирового обмена. СТГ активирует липолиз в жировой ткани, что ведет к увеличению содержания свободных неэстерифицированных жирных кислот в крови, их накоплению в печени и окислению. Усиление окисления выражается, в частности, в увеличении образования кетоновых тел. Этот катаболический эффект осуществляется в присутствии небольших концентраций глюкокортикоидов. Увеличение их количества тормозит мобилизацию жира и его окисление СТГ.
Избыточная секреция адренокортикотропного гормона. Повышенная секреция АКТГ гипофизом приводит к развитию болезни Иценко-Кушинга, которая проявляется двусторонней гиперплазией надпочечников и повышенной секрецией гормонов коры надпочечников. От болезни Иценко-Кушинга следует отличать синдром Иценко-Кушинга, имеющий аналогичную клиническую картину, но обусловленный гормонально-активной аденомой или аденокарциномой коры надпочечников, а также злокачественными опухолями вненадпочечниковой локализации, продуцирующими АКТГ-подобные пептиды (например, бронхогенный рак легких).
В 1924 г. Н.М. Иценко опубликовал наблюдение за больными со следующими признаками: изменение очертаний лица (ожирение его нижней части), перераспределение жира (ожирение туловища при отсутствии ожирения конечностей), мраморность кожных покровов, стрии (багрово-синюшные полосы) на передней стенке живота и бедрах, атрофия мышц конечностей и увеличение живота, повышение артериального давления, остеопороз, нарушение половых функций. Патологоанатомически Н.М. Иценко в ряде случаев определял изменения в гипоталамусе, что и позволило ему связать наблюдаемую клиническую картину с этими изменениями.
В 1932 г. Кушинг описал ту же клиническую картину, связав ее с базофильной аденомой гипофиза. В настоящее время вопрос этиологии болезни Иценко-Кушинга все еще не решен. Было установлено, что данное заболевание возникает на фоне развития стресса, нейроинфекций, травм мозга, абортов, родов, полового созревания и чрезмерной физической нагрузки. По-видимому, действие этих факторов опосредуется через центральные нейромедиаторы (ацетилхолин, серотонин, норадреналин и др.), которые, в свою очередь, регулируют секрецию кортиколиберина в гипоталамусе. В патогенезе болезни Иценко-Кушинга может иметь значение невосприимчивость нейронов мозга к ингибирующим влияниям механизма обратной связи, развивающаяся и закрепляющаяся в результате повреждения гипоталамуса и высших отделов ЦНС. Так или иначе продукция кортиколиберина в гипоталамусе увеличивается, что приводит к гиперплазии базофильных клеток гипофиза, вырабатывающих повышенные количества АКТГ. Если причина, вызвавшая повышение продукции кортиколиберина, сохраняется длительно, то гиперплазия превращается в микроаденому, а затем и в аденому. Повышенный уровень АКТГ при этом заболевании
сочетается с повышением уровня и других продуктов проопиомеланокортина.
Избыточно образующийся АКТГ оказывает свое действие двояко: а) через надпочечники и б) вненадпочечниковым путем.
В надпочечниках АКТГ стимулирует пучковую и (в меньшей степени) сетчатую зону, усиливая образование главным образом кортизола и кортикостерона, выражением чего является гиперкортизолизм.
Избыточная секреция глюкокортикоидов, в свою очередь, приводит к развитию гипергликемии, поскольку она угнетает утилизацию глюкозы на периферии и усиливает глюконеогенез. Следствием этого является повышенная секреция инсулина, чувствительность к которому в тканях снижается. Усиливая образование кортизола, АКТГ тем самым увеличивает катаболизм белка. С повышенным распадом белка связано развитие ряда симптомов заболевания, таких, как остеопороз (деградация белковой матрицы кости), мышечная слабость (атрофия мышц), стрии.
Избыток кортизола приводит помимо этого к задержке натрия и воды, а также синергически с катехоламинами кортизол действует на периферические сосуды, что вызывает их спазм. Все это приводит к развитию артериальной гипертонии. Повышенное выведение калия способствует развитию мышечной слабости. Избыток кортизола может быть в некоторой степени причиной развития гирсутизма (избыточного оволосенения) у больных женщин.
Вненадпочечниковое действие АКТГ на некоторые обменные процессы отличается от его действия на эти же процессы через усиление секреции кортизола. Так, АКТГ способен увеличивать активность тирозиназы в меланоцитах, что приводит к такому нередкому клиническому признаку заболевания, как гиперпигментация. На жировой обмен АКТГ воздействует следующим образом: добавление его непосредственно к жировой ткани стимулирует ее липолитическую активность (распад жира) и тем самым мобилизацию жира с образованием свободных высших неэстерифицированных жирных кислот. Однако усиливая образование кортизола, АКТГ оказывает следующее влияние: а) тормозит мобилизацию жира; б) активирует глюконеогенез и тем самым способствует образованию жира; в) тормозит действие СТГ, активирующее окисление жира. Очевидно, конечный результат зависит от соотношения надпочечникового и вненадпочечникового действия гормона.
Избыточная секреция тиреотропного гормона. Избыточное образование ТТГ стимулирует функцию щитовидной железы, что приводит к усиленному образованию тиреоидных гормонов, развитию так называемого вторичного гипертиреоза и тиреотоксикоза (см. ниже). Кроме того, ТТГ увеличивает содержание кислых мукополисахаридов в коже, мышцах и ретроорбитальной клетчатке как интактных, так и тиреоидэктомированных животных. Причиной данного нарушения могут выступать аденомы из базофильных клеток, секретирующих тиротропин. Они являются редкой формой опухолей передней доли гипофиза. В этом случае к симптоматике гипертиреоза и токсикоза прибавляются и офтальмологические нарушения (изменение полей зрения и глазного дна), возникающие вследствие сдавливания опухолью перекреста зрительного нерва при выходе ее за пределы турецкого седла.
Избыток кортизола приводит помимо этого к задержке натрия и воды, а также синергически с катехоламинами кортизол действует на периферические сосуды, что вызывает их спазм. Все это приводит к развитию артериальной гипертонии. Повышенное выведение калия способствует развитию мышечной слабости. Избыток кортизола может быть в некоторой степени причиной развития гирсутизма (избыточного оволосенения) у больных женщин.
Вненадпочечниковое действие АКТГ на некоторые обменные процессы отличается от его действия на эти же процессы через усиление секреции кортизола. Так, АКТГ способен увеличивать активность тирозиназы в меланоцитах, что приводит к такому нередкому клиническому признаку заболевания, как гиперпигментация. На жировой обмен АКТГ воздействует следующим образом: добавление его непосредственно к жировой ткани стимулирует ее липолитическую активность (распад жира) и тем самым мобилизацию жира с образованием свободных высших неэстерифицированных жирных кислот. Однако усиливая образование кортизола, АКТГ оказывает следующее влияние: а) тормозит мобилизацию жира; б) активирует глюконеогенез и тем самым способствует образованию жира; в) тормозит действие СТГ, активирующее окисление жира. Очевидно, конечный результат зависит от соотношения надпочечникового и вненадпочечникового действия гормона.
Избыточная секреция тиреотропного гормона. Избыточное образование ТТГ стимулирует функцию щитовидной железы, что приводит к усиленному образованию тиреоидных гормонов, развитию так называемого вторичного гипертиреоза и тиреотоксикоза (см. ниже). Кроме того, ТТГ увеличивает содержание кислых мукополисахаридов в коже, мышцах и ретроорбитальной клетчатке как интактных, так и тиреоидэктомированных животных. Причиной данного нарушения могут выступать аденомы из базофильных клеток, секретирующих тиротропин. Они являются редкой формой опухолей передней доли гипофиза. В этом случае к симптоматике гипертиреоза и токсикоза прибавляются и офтальмологические нарушения (изменение полей зрения и глазного дна), возникающие вследствие сдавливания опухолью перекреста зрительного нерва при выходе ее за пределы турецкого седла.
Избыточная секреция гонадотропных гормонов. К их числу относятся: а) ФСГ (фоллитропин); б) ЛГ (лютропин), или ГСИК; в) пролактин, или лактотропный гормон. Их секреция тесно связана с функцией гипоталамуса. В гипоталамусе выделяются соответствующие либерины, которые, спускаясь в гипофиз, стимулируют там образование ФСГ и ЛГ (ГСИК). Образование же пролактина при этом тормозится. Повреждение срединного возвышения, как и гипофизэктомия, ведет к уменьшению секреции ГТГ и к атрофии половых желез. Наоборот, повреждение задних образований гипоталамуса вызывает усиление секреции ГТГ и в детском возрасте приводит к преждевременному половому созреванию. Определенное значение в патогенезе одного из видов преждевременного полового созревания - макрогентосомии придают нарушению функции шишковидной железы, так как считают, что в физиологических условиях она до определенного возраста тормозит секрецию ГТГ, поскольку мелатонин, секретируемый эпифизом, угнетает секрецию гонадотропинов. Преждевременное угнетение функции этой железы (гипопинеализм) растормаживает секрецию ГТГ и приводит к раннему половому созреванию. Согласно другим представлениям, имеют значение опухоли подбугорья вообще, которые каким-то образом стимулируют секрецию ГТГ гипофизом. Секреция ГТГ увеличивается и при первичном выпадении инкреторной активности половых желез, однако это не ведет к повышению продукции половых гормонов.
Избыточное образование пролактина отмечено у больных синдромом лактореи - аменореи, возникающим в связи с первичным
повреждением гипоталамуса. При этом нередко находят опухоль гипоталамуса или хромофобную аденому гипофиза.
20.2.2. Патофизиология надпочечников
Кортикостероидная недостаточность
Кортикостероидная недостаточность может быть тотальной, когда выпадает действие всех гормонов, и частичной - при выпадении активности одного из гормонов коры надпочечников.
Тотальная кортикостероидная недостаточность в эксперименте вызывается адреналэктомией. После адреналэктомии животное неминуемо погибает при явлениях выраженной адинамии и гипотонии. Продолжительность жизни составляет от нескольких часов до нескольких суток.
У людей острая тотальная недостаточность надпочечников (синдром Уотерхауса-Фридрихсена) может возникать при некоторых инфекционных болезнях или нарушениях кровообращения. В связи с быстрым выпадением функции надпочечников развивается коллапс, и больные могут умереть в течение первых же суток.
Хроническая надпочечниковая недостаточность характерна для болезни Аддисона (или бронзовой болезни). Причиной развития болезни Аддисона чаще всего является туберкулезная инфекция или аутоиммунный процесс (аутоиммунный адреналит), лежащий, повидимому, в основе патогенеза так называемой идиопатической атрофии коры надпочечников. В основе патофизиологических изменений, возникающих в результате прогрессирующей гибели ткани коры надпочечника, лежит комбинация недостаточности всех гормонов его коры. При этом наблюдаются: 1) нарушения водного, минерального и углеводного обмена; 2) расстройство функции сердечно-сосудистой системы; 3) развитие адинамии (мышечная слабость); 4) пигментация кожных покровов и слизистых оболочек, в связи с чем это заболевание называют бронзовой болезнью.
Водный и минеральный обмен. В основе нарушения этого обмена лежит недостаток минералокортикоида - альдостерона и в меньшей степени глюкокортикоидов - кортизола и кортикостерона. Нарушение минерального обмена сводится к перераспределению ионов натрия и калия между клетками тканей и внеклеточным депо. Натрий начинает переходить из внеклеточного депо внутрь клетки, а калий - наоборот. Вслед за натрием в клетки устремля-
ется вода, что ведет к развитию водной интоксикации. Уменьшение количества воды в экстрацеллюлярном пространстве приводит к дегидратации организма и уменьшению объема крови. В канальцах почек снижается реабсорбция натрия, и он теряется с мочой. Ионы калия, наоборот, реабсорбируются более интенсивно, и калий начинает накапливаться в организме. В связи со снижением кровяного давления падает фильтрационное давление в клубочках почек, и в результате уменьшается образование первичной мочи. Одновременно увеличивается реабсорбция воды в канальцах. Это связано с нарастанием концентрации ионов калия, что повышает чувствительность канальцевого эпителия к АДГ.
Таким образом, уменьшение фильтрации и усиление реабсорбции воды ведут к понижению суточного диуреза. Потеря натрия обусловливает уменьшение активности симпатических окончаний, что является одним из механизмов развития адинамии и гипотонии. С другой стороны, снижение секреции кортизола, который совместно с катехоламинами регулирует тонус сосудистой стенки, является фактором, ведущим к развитию гипотонии. Задержка калия приводит к снижению сократительной способности скелетной и сердечной мускулатур и, следовательно, к брадикардии и аритмии.
Углеводный обмен. Недостаток глюкокортикоидов вызывает гипогликемию в результате: а) снижения глюконеогенеза из белка за счет уменьшения активности некоторых трансаминаз и активности «ключевого» фермента глюконеогенеза - фосфоэнолпируваткарбоксилазы; б) увеличения активности инсулина, по отношению к которому глюкокортикоиды являются антагонистами: поэтому больные с недостаточностью надпочечников очень чувствительны к инсулину, и введение его в обычных дозах всегда дает более выраженный эффект; в) уменьшения активации глюкозо-6-фосфатазы, что ведет к менее интенсивному поступлению в кровь глюкозы из клеток печени; г) снижения всасывания глюкозы в кишечнике в связи с нарушением соотношения между ионами натрия и калия. Проявляется гипогликемия приступами слабости, раздражительности, чувством голода, потливостью.
Сердечно-сосудистая система. Кортикостероидная недостаточность сопровождается снижением артериального давления. Это объясняется: а) уменьшением объема циркулирующей крови; б) брадикардией, являющейся одной из причин снижения минутного объема крови; в) снижением сосудистого тонуса, в основе которого
лежит падение чувствительности сосудистой стенки к адреналину и норадреналину и снижение тонуса сосудодвигательного центра в связи с общим уменьшением катаболизма белка, в частности, в ЦНС. Это приводит к менее интенсивному образованию аммиака, необходимого для поддержания нормального уровня возбудимости сосудодвигательного и дыхательного центров.
Адинамия. В основе мышечной слабости, кроме указанного выше нарушения сократительных свойств мускулатуры, лежит и дефицит андростендиона (гормона, секретируемого сетчатой зоной коры надпочечников) в связи с выпадением его анаболического действия в отношении мышечных белков.
Пигментация. При аддисоновой болезни пигментация возникает в связи с увеличением отложения меланина в коже и слизистых оболочках. При недостатке кортизола по механизму обратной связи усиливается секреция и β-липотропина, и АКТГ, который имеет в своей молекуле участок с такой же последовательностью аминокислот, какая прослеживается в молекуле меланофорного гормона. Поэтому большие количества АКТГ также оказывают некоторое меланофорное влияние.
Гиперкортикостероидизм
Гиперкортикостероидизмом (гиперкортицизмом) называются такие изменения в организме, которые соответствуют усилению функции коры надпочечников. Гиперкортикостероидизм может развиваться за счет избыточного образования (или повышения активности) одного или сразу нескольких гормонов. Наиболее часто встречаются следующие виды гиперкортикостероидизма: гиперкортизолизм, альдостеронизм и адреногенитальные синдромы.
Гиперкортизолизм - это комплекс таких изменений в организме, которые вызываются либо избыточным образованием кортизола в пучковой зоне коры надпочечников, либо повышением активности кортизола за счет уменьшения связывания его транскортином. Как указывалось выше, избыточное образование возможно при опухоли пучковой зоны одного из надпочечников, называемой глюкостеромой (первичный гиперкортизолизм). Возможно также нарушение центральных механизмов регуляции гипоталамогипофизарно-надпочечниковой системы. В этом случае усиливается образование кортиколиберина и, следовательно, секреция АКТГ (третичный гиперкортизолизм). Секреция АКТГ увеличи-
вается и при опухоли передней доли гипофиза - базофильной аденоме (вторичный гиперкортизолизм). Возникающие при этом изменения составляют картину синдрома Иценко-Кушинга. Он характеризуется нарушениями углеводного, белкового, жирового, водно-солевого обмена и функции сердечно-сосудистой системы. У больных на коже боковой поверхности живота, бедрах, груди появляются полосы с фиолетовым оттенком, похожие на полосы растяжения у беременных. Характерно отложение жира в области туловища и лица («лунообразное» лицо) (рис. 20-12).
Углеводный обмен. Гиперкортизолизм приводит к развитию гипергликемии за счет: а) усиления глюконеогенеза из глюкогенных аминокислот; б) торможения перехода глюкозы в жир; в) торможения декарбоксилирования пирувата, что увеличивает способность пирувата ресинтезироваться в глюкозу; г) повышения активности глюкозо-6-фосфатазы в печени, что способствует переходу глюкозы в кровь. Одновременно в связи с усиленным образованием глюкозы увеличивается образование в печени гликогена. В свою очередь, гипергликемия усиливает образование инсулина островковым аппаратом поджелудочной железы; в случаях функциональной неполноценности инсулярного аппарата его гиперфункция сменяется истощением и развитием сахарного диабета (так называемый стероидный диабет).
Белковый обмен. Усиливается катаболизм белков и тормозится их синтез преимущественно в мышцах и мезенхимальных элементах, что выражается в повышении выделения азота с мочой.
В костной ткани в связи с нарушением образования белкового каркаса тормозится отложение солей кальция и развивается остеопороз.
Жировой обмен. Избыточное отложение жира вызвано: а) гипергликемией, которая активирует синтез триацилглицеролов и увеличивает образование инсулина в поджелудочной железе и тем самым усиливает липогенез; б) уменьшением окисления жирных кислот в печени в связи с увеличением в ней гликогена, что тормозит действие СТГ, активирующее окисление жира.
Водно-солевой обмен. В связи с некоторыми минералокортикоидными свойствами кортизола и кортикостерона отмечаются изменения электролитного и водного обмена. В канальцах почек усиливается реабсорбция ионов натрия, что ведет к задержке этих ионов в организме и некоторому увеличению их концентрации в экстрацеллюлярной жидкости. Одновременно уменьшается реаб-
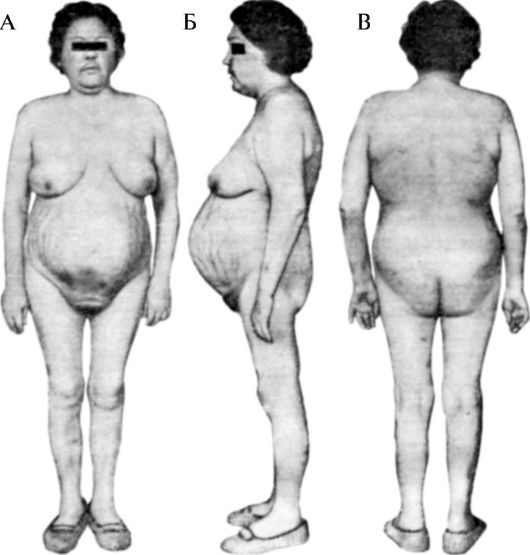
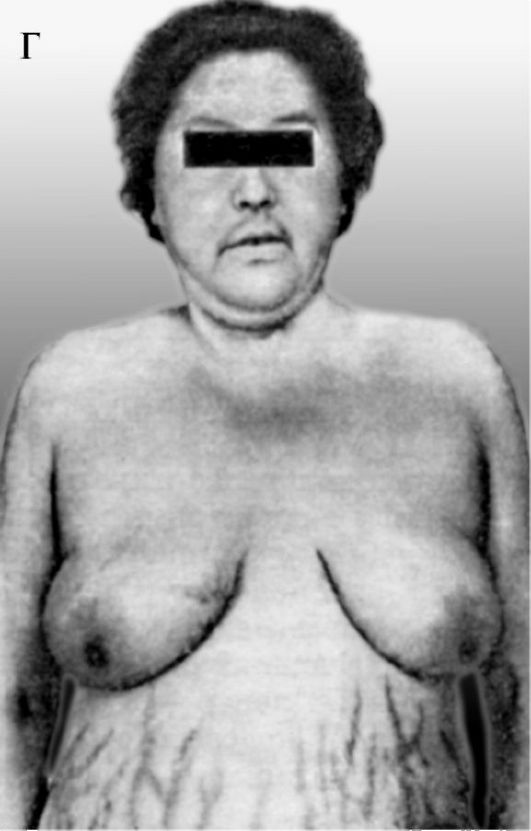 Рис. 20-12. Больная
с синдромом Иценко-Кушинга. Выглядит значительно старше своего возраста
(30 лет). Перераспределение подкожной жировой клетчатки: отложение жира
на животе (А, Б), истончение конечностей (А, Б, В), скошенность ягодиц
(В), лунообразное лицо (Г). На животе (Г) яркие полосы растяжения
(стрии) (по Г.С. Васильченко, 1983)
Рис. 20-12. Больная
с синдромом Иценко-Кушинга. Выглядит значительно старше своего возраста
(30 лет). Перераспределение подкожной жировой клетчатки: отложение жира
на животе (А, Б), истончение конечностей (А, Б, В), скошенность ягодиц
(В), лунообразное лицо (Г). На животе (Г) яркие полосы растяжения
(стрии) (по Г.С. Васильченко, 1983)
сорбция ионов калия в почках, вызывающая некоторую потерю калия в организме. В связи с этими изменениями повышается содержание воды в экстрацеллюлярном депо и увеличивается объем крови. Нарушается и обмен кальция. Тормозится его всасывание в кишечнике и усиливается экскреция с мочой. Это ведет к вторичному гиперпаратироидизму. Усиление секреции паратгормона активирует в кости переход стволовых костных клеток в остеокласты и тормозит превращение последних в остеобласты. В результате увеличивается количество остеокластов и как следствие происходит резорбция костной ткани, развивается остеопороз.
Сердечно-сосудистая система. Гиперкортизолизм приводит к повышению кровяного давления в результате: а) увеличения объема крови; б) повышения чувствительности сосудистой стенки к адреналину и норадреналину за счет как увеличения содержания натрия, так и пермиссивной (т.е. облегчающей действие других гормонов) активности глюкокортикоидов; в) усиления процесса возбуждения в ЦНС, по-видимому, вследствие повышения концентрации аммиака в головном мозгу. Это ведет к усилению тонуса сосудодвигательного центра. Таким образом, кровяное давление повышается в связи с действием различных механизмов. Однако однократное внутривенное введение глюкокортикоидов в эксперименте всегда вызывает снижение кровяного давления. Очевидно, только неоднократное их введение включает механизмы, приводящие к его повышению.
В связи с повышением кровяного давления увеличивается фильтрация в клубочках почек и одновременно тормозится реабсорбция воды за счет блокады действия АДГ, что ведет к повышению диуреза. Кортизол активирует развитие эритроцитов и нейтрофилов, но тормозит развитие лимфоцитов и эозинофилов и усиливает их апоптоз.
Альдостеронизм. Различают первичный и вторичный альдостеронизм. Первичный альдостеронизм (синдром Конна) чаще всего обусловлен гормонально-активной аденомой клубочковой зоны, называемой альдостеромой, которая секретирует избыточное количество альдостерона. Это приводит к усилению реабсорбции натрия в канальцах почек. Натрий задерживается в организме. Его концентрация в экстрацеллюлярных депо в большинстве случаев увеличивается. Одновременно в почках в связи с усилением реабсорбции натрия конкурентно тормозится реабсорбция калия, что ведет к значительной потере калия из клеток организма. Эта по-
теря компенсируется вхождением в клетки ионов натрия и водорода. Можно отметить следующие проявления альдостеронизма: а) повышение кровяного давления в связи с повышением тонуса артериол; это вызвано увеличением концентрации ионов натрия в клетках, что усиливает реакцию клеток на симпатические импульсы и потенцирует действие норадреналина; б) развитие мышечной слабости и временных параличей в связи с потерей калия; снижается сократимость мышц, и возникают парезы и параличи, которые могут длиться на протяжении многих суток; в) полиурия в связи со снижением концентрации калия в клетках, что уменьшает реакцию канальцевого эпителия почек на АДГ. Возможно, полиурия является одной из причин того, что при первичном альдостеронизме, несмотря на задержку натрия, не бывает отеков в отличие от вторичного альдостеронизма. Определенную роль играет и отсутствие застоя в венозной системе; г) гипокалиемический алкалоз; потеря ионов хлора (вместе с ионами калия) ведет к снижению их уровня в крови и компенсаторному увеличению в экстрацеллюлярном депо бикарбонатов (связывание избытка натрия); алкалоз может стать некомпенсированным и привести к развитию тетании; д) уменьшение в плазме крови концентрации ренина и ангиотензина-II; это связано с гиперволемией, которая тормозит секрецию ренина. Вторичный альдостеронизм развивается на фоне первичных процессов, протекающих вне надпочечников. К этим процессам относятся недостаточность правого сердца, циррозы печени, злокачественная гипертония и др.
Адреногенитальные синдромы - изменения в организме, которые развиваются при избыточной секреции андрогенов или эстрогенов сетчатой зоной коры надпочечников. Характер изменения зависит в значительной степени от пола, возраста больного и вида секретируемых гормонов. Различают два основных адреногенитальных синдрома: 1) гетеросексуальный - избыточное образование у данного пола половых гормонов противоположного пола; 2) изосексуальный - раннее или избыточное образование половых гормонов, присущих данному полу.
Избыточное образование андрогенных стероидов. К группе данных соединений относятся андростендион и адреностерон - слабые андрогены, способные в периферических тканях превращаться в тестостерон. Их гиперпродукция связана с опухолью сетчатой зоны коры надпочечников (андростерома) или ее гиперплазией. По механизму обратной связи они тормозят синтез гонадотроп-
ных гормонов, что приводит к атрофии половых желез. У женщин под действием андрогенов атрофируются женские первичные и вторичные половые признаки и развиваются мужские вторичные половые признаки - вирилизм (от лат. virilis - мужской, подобающий мужчине; синоним - маскулинизация, от лат. masculinus - мужской). Различают врожденный и постпубертатный вирильные синдромы. При врожденной форме синдрома у девочек действие андрогенов реализуется уже на этапе внутриутробного развития и при рождении проявляется формированием урогенитального синуса и гипертрофией клитора (ложный женский гермафродитизм). Период полового созревания начинается рано (в 6-7 лет) и протекает по гетеросексуальному типу (мужское телосложение, увеличение мышечной массы в результате анаболического действия гормонов, отсутствие молочных желез, аменорея, акне, низкий тембр голоса). Постпубертатная форма характеризуется олигоили аменореей, нередко бесплодием, атрофией молочных желез, уменьшением размеров матки и яичников, гирсутизмом. Гирсутизм - это избыточный рост терминальных (или стержневых) волос в андрогензависимых зонах (над верхней губой, на подбородке, щеках, верхней части груди, спины, живота) по мужскому типу (рис. 20-13). Гирсутизм следует дифференцировать с гипертрихозом - избыточным ростом пушковых и терминальных волос в тех местах, где
 Рис. 20-13. Рост волос на лице у жен- щины 32 лет (по Н.А. Шерешевскому)
Рис. 20-13. Рост волос на лице у жен- щины 32 лет (по Н.А. Шерешевскому)
 Рис. 20-14. Гипертрихоз у мужчины
Рис. 20-14. Гипертрихоз у мужчины
обычный их рост является нормой как у женщин, так и у мужчин, в том числе в андрогеннезависимых областях. Примерами гипертрихоза являются избыточный рост волос на спине, груди и лице у мужчин (рис. 20-14), на голенях у женщин.
У мужчин избыток андрогенов не сопровождается клиническими проявлениями, поскольку основной андрогенный эффект во взрослом мужском организме оказывает вырабатываемый яичками тестостерон. У мальчиков чрезмерная секреция андрогенов надпочечниками (изосексуальный тип адреногенитального синдрома) проявляется ускоренным ростом, преждевременным появлением вторичных половых признаков, полового влечения, эрекции, увеличением размеров полового члена и мошонки.
Избыточное образование эстрогенов. Реже опухоль сетчатой зоны продуцирует эстрогены (кортикоэстрома). У девочек это вызывает преждевременное половое и физическое развитие. У мужчин развивается феминизация, в процессе которой исчезают мужские
вторичные половые признаки и появляются женские. Отмечаются изменения телосложения, голоса, отложения жировой ткани по женскому типу.
Гиперфункция мозгового слоя надпочечников
Функция мозгового слоя усиливается, как правило, при попадании организма в экстремальные условия, действии ноцицептивных (от лат. посеге - вредить) раздражителей. В этих условиях происходит активация симпатоадреналовой системы, что является частью общего адаптивного синдрома. Иногда в основе гиперфункции лежит образование опухоли из клеток мозгового слоя надпочечника или вненадпочечниковой хромафинной ткани - хромаффиномы. Она чаще бывает доброкачественной (феохромоцитома) и реже злокачественной (феохромобластома). Опухоль встречается относительно редко - по данным вскрытий, в 0,04%. Однако среди больных артериальной гипертензией она встречается намного чаще. Размеры опухоли колеблются в широких пределах - от микроскопических до опухолей массой 3,5 кг. Клетки хромаффиномы секретируют катехоламины - адреналин, норадреналин, предшественник - дофамин и иногда серотонин. Количество и соотношение секретируемых продуктов резко варьируют, что создает большие различия в клинических проявлениях заболевания.
Сердечно-сосудистый синдром проявляется прежде всего пароксизмальным или постоянным повышением артериального давления. Наблюдаются различные изменения деятельности сердца: тахикардия или брадикардия, нарушения ритма типа экстрасистолии, блокады пучка Гиса, мерцания предсердий.
Нарушение обмена веществ характеризуется симптомами умеренного диабета, тиреотоксикоза, гиперхолестеринемии. Для больных с феохромоцитомой типично раннее развитие атеросклероза.
Нервно-психический синдром проявляется во время пароксизмов головокружением, головной болью, галлюцинациями, повышенной возбудимостью нервной системы, судорогами.
Реже феохромоцитома сопровождается желудочно-кишечным синдромом. Он выражается в тошноте, рвоте, запорах, иногда в изъязвлении стенки желудка кишечника с последующим развитием кровотечения.
Напротив, гипофункция мозгового слоя надпочечников может служить одним из патогенетических факторов гипотонических состояний.
20.2.3. Патофизиология щитовидной железы
Гипертиреоз
Гипертиреоз - синдром, вызываемый повышением функции щитовидной железы. Резко выраженный гипертиреоз называют тиреотоксикозом. Гипертиреоз, в зависимости от места, где произошло первичное нарушение, можно разделить на первичный, вторичный и третичный. Причинами первичного гипертиреоза может быть нарушение функции щитовидной железы, развивающееся при таких болезнях, как диффузный токсический зоб (базедова болезнь, болезнь Грейвса, болезнь Парри), тиреотоксическая аденома щитовидной железы. Причиной вторичного гипертиреоза может являться развитие ТТГ-секретирующей опухоли аденогипофиза, а причиной третичного гипертиреоза - нарушение в гипоталамусе.
В целом наиболее частой причиной развития гипертиреоза является диффузный токсический зоб. Считается, что при этом заболевании в организме вырабатываются тиреоидстимулирующие антитела, которые, подобно ТТГ, способны связываться с рецепторами на базальной мембране тиреоцита, что приводит к активации клетки. Одновременно уровень ТТГ в крови больных снижен по механизму обратной связи.
Гипертиреоз сопровождается нарушением энергетического и повышением основного обмена, усилением потребления кислорода, расстройством различных видов обмена, похуданием, нарушением функции ЦНС, сердечно-сосудистой системы и других органов.
Энергетический обмен. Трийодтиронин разобщает окисление и фосфорилирование в митохондриях клеток, в результате чего энергия окисления НАД2Н и НАДФ2Н не аккумулируется в АТФ и рассеивается. Уменьшение синтеза АТФ увеличивает концентрацию его предшественников - АДФ и неорганического фосфата, также изменяется перенос АДФ в митохондрии, поскольку трийодтиронин связывается с переносчиком АДФ транслоказой, что, в свою очередь, усиливает окислительные процессы и тем самым рассеивание энергии. Это ведет к увеличению основного обмена.
Углеводный обмен. При гипертиреозе усиливается обмен углеводов, увеличивается утилизация глюкозы тканями. Активируется фосфорилаза печени и мышц, следствием чего является усиление гликогенолиза и обеднение этих тканей гликогеном. Повышается активность гексокиназы и всасывание глюкозы в кишечнике, что сопровождается алиментарной гипергликемией. Активируется ин-
сулиназа печени. Это в совокупности с гипергликемией вызывает напряженное функционирование инсулярного аппарата и в случае его неполноценности может привести к развитию сахарного диабета. Кроме того, усиление пентозофосфатного пути обмена углеводов способствует повышенному образованию НАДФ2Н.
Белковый обмен. На белковый обмен тиреоидные гормоны в больших дозах оказывают главным образом катаболическое действие, что приводит к отрицательному азотистому балансу. Усиливается выделение азота, фосфора и калия с мочой, указывающее на клеточный распад. Увеличивается выделение аммиака. В крови повышается уровень остаточного азота и азота аминокислот. С повышенным катаболизмом белка связано развитие таких симптомов диффузного токсического зоба, как атрофия мышц и остеопороз.
Жировой обмен. В связи с усилением энергетического обмена больные тиреотоксикозом худеют главным образом за счет уменьшения запасов жира в жировых депо. Уменьшение запасов жиров происходит вследствие: а) мобилизации жиров из депо за счет сенсибилизации симпатических нервных окончаний в жировой ткани; б) ускорения окисления жиров в печени; в) торможения перехода углеводов в жиры. В связи с усилением окисления жира увеличивается образование кетоновых тел. При одновременном дефиците углеводов это приводит к нарушению их окисления и, следовательно, к гиперкетонемии и кетонурии. Повышенный распад жиров приводит к развитию общего похудания больных диффузным токсическим зобом.
Водный и минеральный обмен. Повышение в крови концентрации тиреоидных гормонов вызывает увеличение: а) относительного содержания воды в организме в связи с похуданием; б) объема плазмы; в) скорости фильтрации воды через капиллярные стенки; г) диуреза в связи с усилением почечного кровотока и клубочковой фильтрации; д) потоотделения; е) потери воды с выдыхаемым воздухом. При этом усиливается выведение кальция, фосфора и калия из организма.
Центральная нервная система и другие органы. Тиреоидные гормоны оказывают выраженное влияние на центральную нервную систему. Возбудимость коры головного мозга повышается. В клетках коры, ствола головного мозга и передних рогов спинного мозга развиваются токсически-дегенеративные изменения. Меняется возбудимость гипоталамических вегетативных центров, а в связи с этим и функция внутренних органов.
Со стороны сердечно-сосудистой системы отмечаются стойкая тахикардия, наклонность к мерцанию предсердий. В основе этого явления лежит повышение чувствительности миокарда к адреналину и норадреналину в связи с увеличением количества бета-адренергических рецепторов под влиянием тиреоидных гормонов. Возможно также, что при распаде тиреоидных гормонов образуются активные продукты, способные функционировать как псевдокатехоламины. Усиление работы сердца вызывает его гипертрофию и дистрофические изменения. Нарастание возбуждения симпатического отдела нервной системы приводит к повышению тонуса артериол и развитию гипертонии, развитию тремора. Снижение количества гликогена в печени уменьшает ее дезинтоксикационную функцию и способность синтезировать белки. Повышена влажность и температура кожи. Развивающийся в ряде случаев при диффузном токсическом зобе экзофтальм (пучеглазие) (рис. 20-15), так же как и изменение кожи голеней и кистей (акропатия), может быть следствием аутоиммунного повреждения тканей.
 Рис. 20-15. Тиреотоксикоз у женщины 33 лет (по Н.А. Шерешевскому)
Рис. 20-15. Тиреотоксикоз у женщины 33 лет (по Н.А. Шерешевскому)
Гипотиреоз
Гипотиреоз - состояние, возникающее при недостатке тиреоидных гормонов в организме. Так
же как и гипертиреоз, он может быть первичным, вторичным и третичным. Первичный гипотиреоз встречается при тиреоидите Хасимото, дефектах биосинтеза тиреоидных гормонов, тиреоидэктомии, лечении радиоактивным йодом, недостаточном поступлении йода в организм и других патологических процессах в железе. Вторичный и третичный гипотиреоз являются следствием выпадения регуляторных влияний (поражения гипофиза, дефицит тиролиберина). Наиболее выраженную форму гипотиреоза у взрослых
 Рис. 20-16. Кретинизм
у 8-детней девочки (рядом - ее сверстница) (А). Этот же ребенок через 2
месяца после лечения тиреоидными гормонами (Б) (по J. Bierich, 1975)
Рис. 20-16. Кретинизм
у 8-детней девочки (рядом - ее сверстница) (А). Этот же ребенок через 2
месяца после лечения тиреоидными гормонами (Б) (по J. Bierich, 1975)
называют микседемой. Синдром, который развивается у детей в связи с полной недостаточностью щитовидной железы, называют кретинизмом. Кретинизм характеризуется выраженной задержкой роста и своеобразной внешностью больного (рис. 20-16). В основе кретинизма лежит, как правило, аплазия щитовидной железы.
Тиреоидэктомия в эксперименте сопровождается отставанием в росте молодых животных, задержкой роста трубчатых костей и полового развития. Возникают отклонения от нормы во внешнем виде. Меняется конфигурация черепа - укорачивается передняя часть лица, а задняя приобретает шаровидную форму, останавливается развитие зубов. У собак конечности становятся толстыми, движения - неуклюжими, прекращается рост шерсти. Развивается слизистый отек подкожной клетчатки вследствие задержки воды, хлористого натрия и накопления в соединительной ткани мукопо-
лисахаридов, обладающих гидрофильными свойствами. При хорошем содержании животные могут жить месяцы и годы.
При гипотиреозе наблюдаются следующие нарушения обмена веществ и функций органов:
Энергетический обмен. Гипотиреоз сопровождается уменьшением интенсивности окислительных процессов, что приводит к снижению основного обмена.
Белковый обмен. При функциональной недостаточности щитовидной железы снижается интенсивность синтеза белка. Свидетельством этого является торможение скорости включения метионина в белки тканей. При этом усиливается катаболизм аминокислот, уменьшается содержание РНК в тканях.
Углеводный обмен. Интенсивность обмена углеводов падает. Повышается содержание гликогена в печени в связи со снижением активности фосфорилазы. В результате ослабления активности гексокиназы уменьшается всасывание глюкозы в кишечнике. Следствием замедления окислительных процессов в тканях может быть развитие гиперкетонемии.
Жировой обмен. Скорость синтеза холестерина в печени и надпочечниках снижается, однако еще более замедляется его распад, что ведет к гиперхолестеринемии и способствует развитию атеросклероза.
После тиреоидэктомии у собак снижается возбудимость центральной нервной системы. У людей при гипотиреозах отмечается замедление психических реакций, ослабление памяти, в тяжелых случаях - слабоумие.
Эндемический зоб. Особой формой гипотиреоза является эндемический зоб. Он развивается в определенных географических районах, в которых население не получает с пищей достаточного количества йода. Недостаток йода снижает синтез тиреоидных гормонов, что по механизму обратной связи усиливает секрецию ТТГ гипофизом. Это вызывает гиперплазию железы, что вначале компенсирует недостаток тиреоидных гормонов. Однако при продолжающемся дефиците йода эта компенсация оказывается недостаточной для образования тиреоидных гормонов, и развивается гипотиреоз, который в далеко зашедших случаях может перейти в микседему и кретинизм (рис. 20-17). Профилактическое введение йода препятствует возникновению этого заболевания. С этой целью к поваренной соли добавляют 0,002% йодистого натрия или калия. Потребление 6 г соли ежедневно означает прием 120 мкг йодида, что является оптимальной суточной дозой для взрослых.
 Рис. 20-17. Группа эндемических кретинов из местечка Аарау (Германия, 1908) (по W. Falta, 1913)
Рис. 20-17. Группа эндемических кретинов из местечка Аарау (Германия, 1908) (по W. Falta, 1913)
Нарушение секреции тиреокальцитонина
Тиреокальцитонин - ТКТ (он же кальцитонин) образуется в светлых клетках парафолликулярного эпителия щитовидной железы (так называемых С-клетках, обозначенных по названию гормона). Он оказывает эффект, противоположный действию паратгормона (ПГ): угнетает функцию остеокластов и усиливает их превращение в остеобласты; оказывает прямое действие на остеокласты через соответствующие рецепторы на этих клетках (рис. 20-18). В результате угнетается резорбция кости остеокластами. Помимо указанного действия, кальцитонин обладает кальцийуретическим и фосфоруретическим эффектом, а также увеличивает образование 1,25-дигидроксивитамина D3, что усиливает абсорбцию кальция в кишечнике.
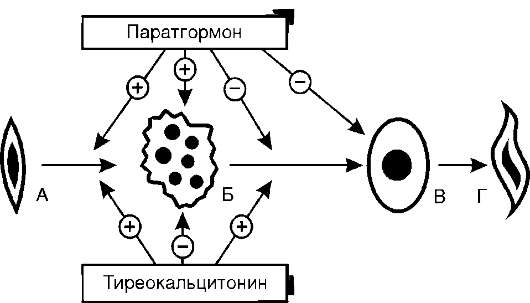 Рис. 20-18. Пути
развития ткани и точки приложения в действии паратгормона и
тиреокальцитонина: А - стволовая клетка; Б - остеокласт; В - остеобласт;
Г - остеоцит. Активация обозначена знаком «плюс», торможение - знаком
«минус»
Рис. 20-18. Пути
развития ткани и точки приложения в действии паратгормона и
тиреокальцитонина: А - стволовая клетка; Б - остеокласт; В - остеобласт;
Г - остеоцит. Активация обозначена знаком «плюс», торможение - знаком
«минус»
Увеличено образование ТКТ при аденомах и медуллярной аденокарциноме щитовидной железы, происходящих из С-клеток. Вторично нарушается образование ТКТ при гипер- и гипотиреозах. При гипертиреозах усиливается катаболизм белковой основы костной ткани, в связи с чем усиливается вымывание из кости кальция. Это включает механизмы обратной связи, которые, с одной стороны, тормозят образование ПГ, а с другой - усиливают секрецию ТКТ. Последний тормозит развитие остеопороза, но при длительном и тяжелом течении гипертиреоза истощается компенсаторное образование ТКТ и развивается остеопороз. При гипотиреозе кальций задерживается в организме и накапливается в костях.
Патофизиология околощитовидных желез
Гиперпаратиреоз - синдром, вызываемый усилением функции паращитовидных желез. Встречается при паратиреоидной дистрофии (первичный гиперпаратиреоз, болезнь Реклингхаузена). В основе этого заболевания лежит образование аденом в околощитовидных железах. Понижение уровня кальция в плазме крови также стимулирует функцию железы. Поэтому происходят вторичная гиперплазия и гиперфункция этих желез при первичном нарушении функции почек, недостатке кальция в пище, потеря его во время беременности и лактации, при поносах, авитаминозе D.
При выраженном гиперпаратиреозе костная ткань теряет кальций. Развивается остеопороз, костная ткань заменяется фиброзной, становится мягкой (остеомаляция).
В тканях лактат и цитрат кальция легко окисляются, поэтому кальций выпадает в осадок, образуя кальциевые отложения. Этот процесс идет и в почках. Увеличивается выведение кальция с мочой, что приводит к полиурии и гипотонии мочи. Одновременно происходит обызвествление клеток канальциевого эпителия и выпадение фосфорнокислых и углекислых солей кальция в просвете канальцев. Иногда это является основой образования камней в мочевом тракте. В резко выраженных случаях гиперпаратиреоза нарушение функции почек приводит к анурии и уремии.
Гипопаратиреоз - синдром, развивающийся при угнетении функции паращитовидных желез. Синдром, возникающий при резистентности органа-мишени к ПГ, обозначают как псевдогипопаратиреоз. Наиболее выраженные явления гипопаратиреоза развиваются при паратиреоидэктомии. При этом у собак, кошек, обезьян в эксперименте и у человека (при случайном удалении во время операции тиреоидэктомии) развиваются острые явления, обычно со смертельным исходом. Картина нарушений характеризуется повышением мышечной возбудимости вплоть до развития приступа тетании в виде периодически возникающих тонических и клонических судорог с нарушением дыхания, сердечно-сосудистой деятельности, усилением моторики желудочно-кишечного тракта, развитием пироло- и ларингоспазма.
20.2.4. Патофизиология половых желез Нарушение функций мужских половых желез
Гипогонадизм (гипофункция половых желез) проявляется либо угнетением функции семенных канальцев без нарушения продукции андрогенов, либо недостаточным образованием этих гормонов, либо сочетанием обоих процессов.
Кастрация. Наиболее полные проявления гипогонадизма развиваются после удаления половых желез. Кастрация в препубертатном периоде предупреждает развитие придаточных половых органов и вторичных половых признаков. Эта же операция после завершения развития сопровождается атрофией придаточных половых органов (семенных пузырьков, предстательной железы, пре-
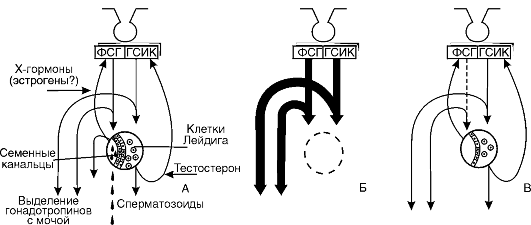 Рис. 20-19. Регуляция
функции семенных желез в норме (А), ее нарушение при кастрации (Б) и
при недостаточности секреции гонадотропинов гипофизом (В) (с изменениями
по Williams). ФСГ - фолликулостимулирующий гормон, ГСИК - гормон,
стимулирующий интерстициальные клетки
Рис. 20-19. Регуляция
функции семенных желез в норме (А), ее нарушение при кастрации (Б) и
при недостаточности секреции гонадотропинов гипофизом (В) (с изменениями
по Williams). ФСГ - фолликулостимулирующий гормон, ГСИК - гормон,
стимулирующий интерстициальные клетки
пуциальных желез и др.) и вторичных половых признаков, уменьшается масса мышц, и в них откладывается большое количество жира. Кости становятся более тонкими и длинными. Задерживается инволюция вилочковой железы. Гипофиз гипертрофируется, и в нем появляются так называемые клетки кастрации. В связи с выпадением тормозящего влияния андрогенов усиливается выделение гипофизом гонадотропных гормонов (рис. 20-19).
У лиц, кастрированных до наступления половой зрелости, развивается евнухоидизм. При этом происходит чрезмерный рост костей в длину с запаздыванием заращения эпифизарных поясков. Это ведет к относительному увеличению длины конечностей. Наружные половые органы недоразвиты. Наблюдается скудный рост волос на теле и лице с женским типом оволосения на лобке. Мышцы недостаточно развиты и слабы, тембр голоса высокий. Распределение жира и строение таза имеют черты, свойственные женскому организму (рис. 20-20, 20-21). Половое влечение (либидо) и способность к половому акту (потенция) отсутствуют. При кастрации зрелых мужчин изменения менее резки, так как рост, формирование скелета и половых органов уже закончились.
Гипергонадизм (усиление функции семенных желез) в препубертатном периоде приводит к преждевременному созреванию. Усиление функции семенников может быть вызвано: 1) повышением
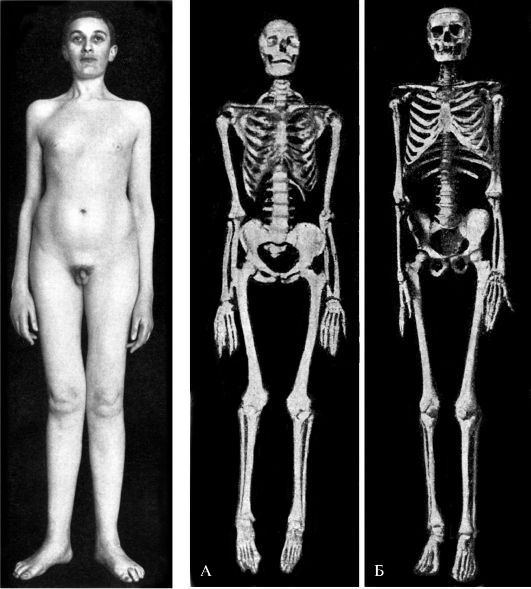 Рис. 20-20. Евнухоидизм Рис. 20-21. Скелет нормального мужчины (А) и
Рис. 20-20. Евнухоидизм Рис. 20-21. Скелет нормального мужчины (А) и
у мужчины (по Falta, евнухоида (Б) (по А. Вэйль, 1925)
1913)
секреции гонадотропинов, как правило, в связи с патологическими процессами в гипоталамусе (воспалительные процессы, опухоли области серого бугра); 2) опухолями, исходящими из клеток Лейдига.
Ранняя секреция андрогенов приводит к преждевременному развитию половых органов, лонного оволосения и полового влечения. Сначала мальчик быстро растет, а затем происходит задержка роста в результате преждевременного окостенения эпифизарных хрящей. В случаях преждевременного созревания, вызванного ранней секрецией гонадотропинов, стимулируется образование как андрогенов, так и сперматозоидов в семенных канальцах. При опухолях, исходящих из клеток Лейдига, образуются только андрогены. При этом угнетается сперматогенез, так как отсутствует секреция гонадотропинов и в первую очередь фолликулостимулирующего гормона.
Нарушение функций женских половых желез
Задержка полового созревания. В норме половое созревание у женщин происходит в возрасте 9-14 лет. Задержка наступления половой зрелости сопровождается недоразвитием вторичных половых органов. Матка, влагалище, фаллопиевы трубы, молочные железы остаются недоразвитыми. Во многих случаях недостаточность функции яичников сопровождается отставанием общего физического развития, что обозначается как инфантилизм. Инфантилизм обычно является следствием недостаточности гипофиза, который не продуцирует не только гонадотропины, но и другие тропные гормоны, в результате чего задерживается рост и отмечается гипофункция надпочечников и щитовидной железы. Если же недостаточность ограничивается только яичниками, недоразвитие касается главным образом половой системы и сопровождается преимущественно евнухоидизмом. В обоих случаях наблюдается аменорея. Недостаточность яичников может быть следствием недостаточности гонадотропинов, рефрактерности яичников к этим гормонам или разрушения ткани яичников (при аутоиммунном оофорите или облучении). В первом случае обнаруживают снижение, а во втором и третьем - повышение содержания гонадотропинов в моче.
Недостаток эстрогенов приводит к следующим изменениям: 1) снижается способность вызывать гипертрофию и гиперплазию эпителиальной, мышечной и соединительной тканей; 2) предупреждается развитие гиперемии и отека родовых путей, а также секреция слизистых желез; 3) понижается чувствительность мышечной оболочки матки к окситоцину, что уменьшает ее сократительную
способность; 4) уменьшается гиперплазия канальцев и интерстициальной соединительной ткани в молочных железах.
Недостаточность гормонов желтого тела предупреждает возникновение изменений, обеспечивающих имплантацию оплодотворенного яйца в эндометрий матки.
Гиперфункция яичников. Этиологическими факторами гиперфункции яичников являются: а) патологические процессы в мозгу (опухоль задней части подбугорья, водянка мозга, менингиты, энцефалиты, дефекты мозга), которые приводят к раздражению ядер подбугорья, стимулирующих гонадотропную функцию гипофиза и усиливающих неврогенным путем реакцию яичников на действие гонадотропинов. Предполагают, что несекретирующие опухоли эпифиза могут являться причиной преждевременного полового созревания, так как мелатонин эпифиза тормозит секрецию гонадотропинов; б) гормонально-активные опухоли яичников. К их числу относится гранулезоклеточная опухоль (фолликулома) из клеток гранулезы фолликула и текома из клеток, окружающих фолликул. Обычно эта опухоль продуцирует эстрогены, реже - андрогены. Поэтому их называют феминизирующими в первом случае и вирилизирующими во втором; в) опухоль надпочечников, секретирующая эстрогены. В этом случае функция яичников по механизму обратной связи угнетается. Однако изменения в организме соответствуют таковым при гиперфункции. Результат гормональных расстройств зависит от основного механизма и возраста больного. Усиление функции яичников в препубертатном периоде приводит к преждевременному половому созреванию, которое заключается в развитии вторичных половых органов и признаков до 9-летнего возраста. Рано появляются менструации. Усиливается рост, который впоследствии задерживается в результате преждевременного окостенения эпифизарных хрящей. Идет накопление жира по женскому типу. Развиваются молочные железы и половые органы. В репродуктивном периоде выявляются расстройства менструального цикла.
Расстройства менструального цикла. Состояние, в котором у половозрелой женщины в генеративном периоде жизни менструация не наступает, называют вторичной аменореей. Другие виды расстройств выражаются в том, что менструация может возникать чаще, чем обычно, или редко, быть слишком обильной или скудной, а также сопровождаться необычной болезненностью.
Можно выделить 4 основных патогенетических пути расстройства гормональной функции яичников, которые приводят к нарушениям менструального цикла: 1) увеличенное выделение эстрогенов (гиперэстрогенизм); 2) недостаточное выделение эстрогенов (гипоэстрогенизм); 3) увеличенное выделение прогестерона (гиперлютеинизм); 4) недостаточное выделение прогестерона (гиполютеинизм). Любое из этих изменений приводит к нарушению последовательности включения различных гонадотропных и овариальных гормонов, регулирующих последовательность стадий менструального цикла.