Биохимия: учебник для вузов/ под ред. Е.С.Северина - 5-е изд., - 2009. - 768 с.
|
|
|
|
РАЗДЕЛ 8 ОБМЕН ЛИПИДОВ
Термин «липиды» объединяет вещества, обладающие общим физическим свойством - гид-рофобностью, т.е. нерастворимостью в воде. По структуре липиды настолько разнообразны, что у них отсутствует общий признак химического строения. Липиды разделяют на классы, в которые объединяют молекулы, имеющие сходное химическое строение и общие биологические свойства.
Основную массу липидов в организме составляют жиры - триацилглицеролы, служащие формой депонирования энергии. Жиры располагаются преимущественно в подкожной жировой ткани и выполняют также функции теплоизоляционной и механической защиты.
Фосфолипиды - большой класс липидов, получивший своё название из-за остатка фосфорной кислоты, придающего им свойства амфифильности. Благодаря этому свойству фосфолипиды формируют бислойную структуру мембран, в которую погружены белки. Клетки или отделы клеток, окружённые мембранами, отличаются по составу и набору молекул от окружающей среды, поэтому химические процессы в клетке разделены и ориентированы в пространстве, что необходимо для регуляции метаболизма.
Стероиды,
представленные в животном мире холестеролом и его производными,
выполняют разнообразные функции. Холестерол - важный компонент мембран и
регулятор свойств гидрофобного слоя. Производные холестерола (жёлчные
кислоты) необходимы для переваривания жиров. Стероидные гормоны,
синтезируемые из холестерола, участвуют в регуляции энергетического,
водно-солевого обменов, половых функций. Кроме стероидных гормонов,
многие производные липидов выполняют регуляторные функции и действуют,
как и гормоны, в очень низких концентрациях. Например,
тромбоци-тактивирующий фактор - фосфолипид особой структуры - оказывает
сильное влияние на агрегацию тромбоцитов в концентрации 10-
кислот, вырабатываемые почти всеми типами клеток, вызывают разнообразные биологические эффекты в концентрациях не более 10-
В тканях человека количество разных классов липидов существенно различается. В жировой ткани жиры составляют до 75% сухого веса. В нервной ткани липидов содержится до 50% сухого веса, основные из них фосфолипиды и сфингомиелины (30%), холестерол (10%), ган-глиозиды и цереброзиды (7%). В печени общее количество липидов в норме не превышает 10-13%.
Нарушения обмена липидов приводят к развитию многих заболеваний, но среди людей наиболее распространены два из них - ожирение и атеросклероз.
I. СТРУКТУРА, КЛАССИФИКАЦИЯ И СВОЙСТВА ОСНОВНЫХ ЛИПИДОВ ОРГАНИЗМА ЧЕЛОВЕКА
Липиды разных классов существенно отличаются по структуре и функциям. Большинство липидов имеют в своём составе жирные кислоты, связанные сложноэфирной связью c глицеролом, холестеролом или амидной связью с аминоспиртом сфингозином.
а. структура, состав и свойства жирных кислот и ацилглицеролов
Жирные кислоты в организме человека имеют чётное число атомов углерода, что связано с особенностями их биосинтеза, при котором к углеводородному радикалу жирной кислоты последовательно добавляются двухуглеродные фрагменты.
Жирные кислоты - структурные компоненты различных липидов. В составе триацилглице-ролов жирные кислоты выполняют функцию депонирования энергии, так как их радикалы
содержат богатые энергией СН2-группы. При окислении СН-связей энергии выделяется больше, чем при окислении углеводов, в которых атомы углерода уже частично окислены (-НСОН-). В составе фосфолипидов и сфинго-липидов жирные кислоты образуют внутренний гидрофобный слой мембран, определяя его свойства. Жиры и фосфолипиды организма при нормальной температуре тела имеют жидкую консистенцию, так как количество ненасыщенных жирных кислот преобладает над насыщенными. В фосфолипидах мембран ненасыщенных кислот может быть до 80-85%, а в составе жиров подкожного жира - до 60%.
В свободном, неэтерифицированном состоянии жирные кислоты в организме содержатся в
небольшом количестве, например в крови, где они транспортируются в комплексе с белком альбумином.
Жирные кислоты липидов человека представляют собой углеводородную неразветвлён-ную цепь, на одном конце которой находится карбоксильная группа, а на другом - метиль-ная группа (ω-углеродный атом). Большинство жирных кислот в организме содержат чётное число атомов углерода - от 16 до 20 (табл. 8-1 и 8-2). Жирные кислоты, не содержащие двойных связей, называют насыщенными. Основной насыщенной жирной кислотой в липидах человека является пальмитиновая (до 30-35%). Жирные кислоты, содержащие двойные связи, называют ненасыщенными. Ненасыщенные
Таблица 8-1. Строение жирных кислот
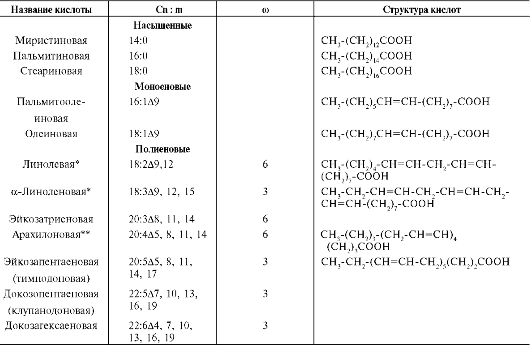
Примечания: Сn:m - число атомов углерода (n) и число двойных связей (m) в молекуле жирной кислоты; ω (6, 3) - номер углеродного атома, у которого находится первая двойная связь, считая от ω- (метильного) атома углерода; Δ - позиция двойной связи, считая с первого, карбоксильного атома углерода; * - жирные кислоты, которые не синтезируются в организме (незаменимые); ** - арахидоновая кислота может синтезироваться из линолевой кислоты.
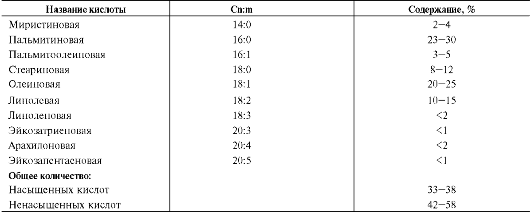
Таблица 8-2. Состав жирных кислот подкожного жира человека
жирные кислоты представлены моноеновыми (с одной двойной связью) и полиеновыми (с двумя и большим числом двойных связей). Если в составе жирной кислоты содержатся две и более двойных связей, то они располагаются через СН2-группу. Имеется несколько способов изображения структуры жирных кислот. При обозначении жирной кислоты цифровым символом (табл. 8-1, вторая графа) общее количество атомов углерода представлено цифрой до двоеточия, после двоеточия указывают число двойных связей. Позицию двойной связи обозначают знаком Δ, после которого указывают номер атома углерода, ближайшего к карбоксилу, у которого находится двойная связь. Например, C18:1Δ9 означает, что жирная кислота содержит 18 атомов углерода и одну двойную связь у 9-го атома углерода, считая от углеродного атома карбоксильной группы.
Позиция двойной связи может быть указана и другим способом - по расположению первой двойной связи, считая от метильного ω-атома углерода жирной кислоты. Например, линолевая кислота может быть обозначена как C18:2Δ9,12 или С18:2ω-6. По положению первой двойной связи от метильного углерода полиеновые жирные кислоты делят на семейства ω-3 и ω-6.
Двойные связи в жирных кислотах в организме человека имеют цис-конфигурацию. Это означает, что ацильные фрагменты находятся по одну сторону двойной связи. Цис-конфи-гурация двойной связи делает алифатическую цепь жирной кислоты изогнутой, что нарушает упорядоченное расположение насыщенных радикалов жирных кислот в фосфолипидах мембран (рис. 8-1) и снижает температуру плавления. Чем больше двойных связей в жирных кислотах липидов, тем ниже температура их плавления.
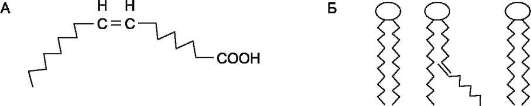
Рис. 8-1. Конфигурации радикалов жирных кислот. А - излом радикала жирной кислоты при двойной связи в цис-конфигурации; Б - нарушение упорядоченного расположения радикалов насыщенных жирных кислот в гидрофобном слое мембран ненасыщенной кислотой с цис-конфигурацией двойной связи.
Таблица 8-3. Состав жирных кислот и температура плавления некоторых пищевых жиров
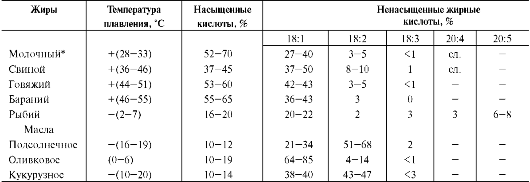
Примечания: сл. - кислоты, присутствующие в незначительных (следовых) количествах. В рыбьем жире, кроме указанных кислот, присутствуют 22:5 жирная кислота (клупанодоновая) - до 10% и 22:6 (цервоновая) - до 10%, которые необходимы для формирования структур фосфолипидов нервной системы человека. В других типах природных жиров они практически отсутствуют; * - жирные кислоты с числом атомов углерода от 4 до 10 содержатся в основном в липидах молока.
В таблице 8-1 выделены основные жирные кислоты в липидах человека.
Жирные кислоты с транс-конфигурацией двойной связи могут поступать в организм с пищей, например в составе маргарина. В этих кислотах отсутствует излом, характерный для цис-связи, поэтому жиры, содержащие такие ненасыщенные кислоты, имеют более высокую температуру плавления, т.е. более твёрдые по консистенции.
Большинство жирных кислот синтезируется в организме человека, однако полиеновые кислоты (линолевая и α-линоленовая) не синтезируются и должны поступать с пищей. Эти жирные кислоты называют незаменимыми, или эссенциальными. Основные источники поли-еновых жирных кислот для человека - жидкие растительные масла и рыбий жир, в котором содержится много кислот семейства ω-3 (табл. 8-1, 8-3).
Ацилглицеролы -
сложные эфиры трёхатомно-го спирта глицерола и жирных кислот. Глицерол
может быть связан с одной, двумя или тремя жирными кислотами,
соответственно образуя моно-, диили триацилглицеролы (МАГ, ДАГ, ТАГ).
Основную массу липидов в организме человека составляют триацилглицеролы -
жиры. У человека с массой тела
жится до
Моно- и диацилглицеролы образуются на промежуточных этапах распада и синтеза триацил-глицеролов. Атомы углерода в глицероле по-разному ориентированы в пространстве (рис. 8-2), поэтому ферменты различают их и специфически присоединяют жирные кислоты у первого, второго и третьего атомов углерода.
Номенклатура и состав природных триацил-глицеролов. В молекуле природного жира содержатся разные жирные кислоты. Как правило, в позициях 1 и 3 находятся более насыщенные
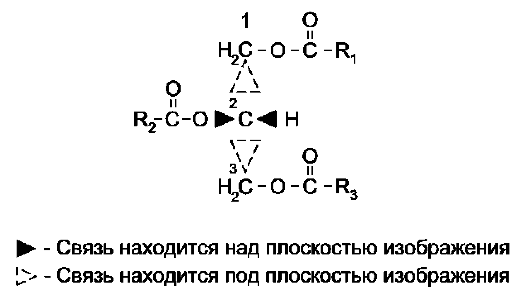
Рис. 8-2. Пространственное расположение углеродных атомов глицерола.
жирные кислоты, а во второй позиции - поли-еновая кислота. В названии триацилтлицерола перечисляются названия радикалов жирных кислот, начиная с первого углеродного атома глицерола, например пальмитоил-линоленоил-олеоилглицерол.
Жиры, содержащие преимущественно на-сы-щенные кислоты, являются твёрдыми (говяжий, бараний жиры), а содержащие большое количество ненасыщенных кислот - жидкими. Жидкие жиры или масла обычно имеют растительное происхождение (табл. 8-3).
Из животных пищевых жиров наиболее насыщен бараний жир, который практически не содержит незаменимых кислот. Ценными пищевыми жирами являются рыбий жир и растительные масла, содержащие незаменимые жирные кислоты. В организме рыб полиеновые жирные кислоты ω-3 и ω-6 также не синтезируются, рыбы получают их с пищей (водоросли, планктон).
Б. СТРУКТУРА И КЛАССИФИКАЦИЯ ФОСФОЛИПИДОВ И СФИНГОЛИПИДОВ
Фосфолипиды - разнообразная группа ли-пидов, содержащих в своём составе остаток фосфорной кислоты. Фосфолипиды делят на глицерофосфолипиды, основу которых составляет трёхатомный спирт глицерол, и сфинго-фосфолипиды - производные аминоспирта сфингозина. Фосфолипиды имеют амфифиль-ные свойства, так как содержат алифатические радикалы жирных кислот и различные полярные группы. Благодаря своим свойствам
фосфолипиды не только являются основой всех клеточных мембран, но и выполняют другие функции: образуют поверхностный гидрофильный слой липопротеинов крови, выстилают поверхность альвеол, предотвращая слипание стенок во время выдоха. Некоторые фосфо-липиды участвуют в передаче гормонального сигнала в клетки. Сфингомиелины являются фосфолипидами, формирующими структуру миелиновых оболочек и других мембранных структур нервных клеток.
Глицерофосфолипиды. Структурная основа глицерофосфолипидов - глицерол. Глицеро-фосфолипиды (ранее используемые названия - фосфоглицериды или фосфоацилглицеролы) представляют собой молекулы, в которых две жирные кислоты связаны сложноэфирной связью с глицеролом в первой и второй позициях; в третьей позиции находится остаток фосфорной кислоты, к которому, в свою очередь, могут быть присоединены различные заместители, чаще всего аминоспирты (табл. 8-4, рис. 8-3). Если в третьем положении имеется только фосфорная кислота, то глицерофосфолипид называется фосфатидной кислотой. Её остаток называют «фосфатидил»; он входит в название остальных глицерофосфолипидов, после которого указывают название заместителя атома водорода в фосфорной кислоте, например фосфатидилэта-ноламин, фосфатидилхолин и т.д.
Фосфатидная кислота в свободном состоянии в организме содержится в небольшом количестве (см. раздел 5, табл. 5-1), но является
Таблица 8-4. Классификация глицерофосфолипидов и сфинголипидов
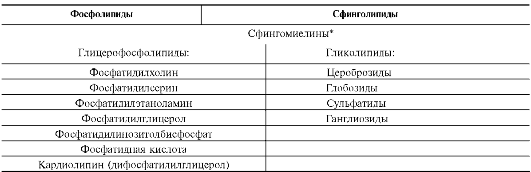
* Сфингомиелины относят как к фосфолипидам, так и сфинголипидам.
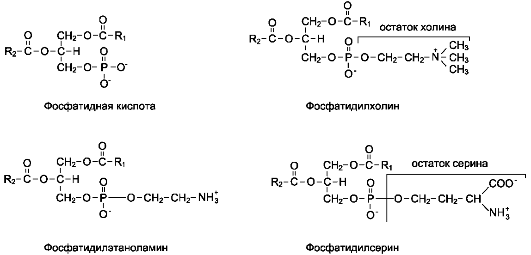
Рис. 8-3. Основные глицерофосфолипиды в организме человека.
промежуточным продуктом на пути синтеза как триацилглицеролов, так и глицерофосфолипидов. У глицерофосфолипидов, как и у триацилглицеролов, во второй позиции находятся преимущественно полиеновые кислоты; в молекуле фосфатидилхолина, входящего в структуру мембран, это чаще всего арахидоно-вая кислота. Жирные кислоты фосфолипидов мембран отличаются от других липидов человека преобладанием полиеновых кислот (до 80-85%), что обеспечивает жидкое состояние гидрофобного слоя, необходимое для функционирования белков, входящих в структуру мембран.
Плазмалогены. Плазмалогены - фосфолипи-ды, у которых в первом положении глицерола находится не жирная кислота, а остаток спирта с длинной алифатической цепью, связанный простой эфирной связью.
Характерный признак плазмалогенов - двойная связь между первым и вторым атомами
углерода в алкильной группе (рис. 8-4). Плазмалогены бывают 3 видов: фосфатидальэтано-ламины, фосфатидальхолины и фосфатидаль-серины. Плазмалогены составляют до 10% фос-фолипидов мембран нервной ткани; особенно много их в миелиновых оболочках нервных клеток.
Некоторые типы плазмалогенов вызывают очень сильные биологические эффекты, действуя как медиаторы. Например, тромбоцитак-тивирующий фактор (ТАФ) стимулирует агрегацию тромбоцитов. ТАФ отличается от других плазмалогенов отсутствием двойной связи в алкильном радикале и наличием ацетильной группы во втором положении глицерола вместо жирной кислоты.
ТАФ выделяется из фагоцитирующих клеток крови в ответ на раздражение и стимулирует агрегацию тромбоцитов, участвуя таким образом в свёртывании крови. Этот фактор обусловливает
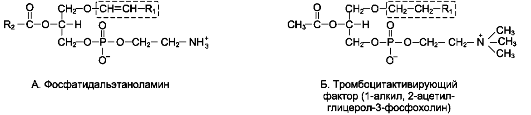
Рис. 8-4. Плазмалогены.
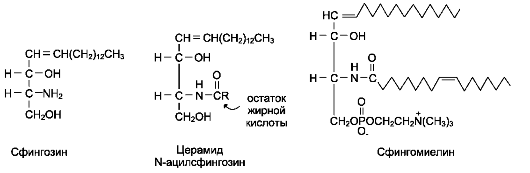
Рис. 8-5. Производные сфингозина: церамид и сфингомиелин.
также развитие некоторых признаков воспаления и аллергических реакций.
Сфинголипиды
Аминоспирт сфингозин, состоящий из 18 атомов углерода, содержит гидроксильные группы и аминогруппу. Сфингозин образует большую группу липидов, в которых жирная кислота связана с ним через аминогруппу. Продукт взаимодействия сфингозина и жирной кислоты называют «церамид» (рис. 8-5). В церамидах жирные кислоты связаны необычной (амидной) связью, а гидроксильные группы способны взаимодействовать с другими радикалами. Церамиды отличаются радикалами жирных кислот, входящих в их состав. Обычно это жирные кислоты с большой длиной цепи - от 18 до 26 атомов углерода.
Сфингомиелины. В результате присоединения к ОН-группе церамида фосфорной кислоты, связанной с холином, образуется сфингомие-лин (рис. 8-5). Сфингомиелины - основные компоненты миелина и мембран клеток мозга и нервной ткани. Сфингомиелины, как и глицеро-фосфолипиды, имеют амфифильные свойства, обусловленные, с одной стороны, радикалом жирной кислоты и алифатической цепью самого сфингозина, а с другой - полярной областью фосфорилхолина.
Гликолипиды. Церамиды - основа большой группы липидов - гликолипидов (см. выше табл. 8-4). Водород в гидроксильной группе церамида может быть замещён на разные углеводные фрагменты, что определяет принадлежность гликолипида к определённому классу. Гликоли-пиды находятся в основном в мембранах кле-
ток нервной ткани. Названия «цереброзиды» и «ганглиозиды» указывают на ткани, откуда они впервые были выделены.
Цереброзиды. Цереброзиды имеют в своём составе моносахариды. Наиболее распространены цереброзиды, имеющие в своём составе галактозу (галактоцереброзид), реже - глюкозу (глюкоцереброзид). Цереброзиды содержат необычные жирные кислоты, например, галакто-цереброзид френозин содержит цереброновую кислоту - 2-гидроксикислоту, содержащую 24 атома углерода (рис. 8-6).
Глобозиды. Глобозиды отличаются от церебро-зидов тем, что имеют в своём составе несколько углеводных остатков, связанных с церамидом:
церамид-глюкоза-галактоза-галактоза-N-ацети-
лгалактоза
Цереброзиды и глобозиды относят к нейтральным сфинголипидам, так как они не содержат заряженных групп.
Сульфатиды. Гидроксил у третьего углеродного атома моносахарида, входящего в состав цереброзида, может связывать остаток серной кислоты, т.е. сульфатироваться. В этом случае образуются сульфатиды, обладающие свойствами кислот и поэтому называемые кислыми сфинголипидами (рис. 8-7). При физиологических значениях рН сульфатированный углеводный остаток имеет отрицательный заряд. Около 25% цереброзидов мозга представляют собой сульфатированные производные. Сульфатиды в значительных количествах находят в белом веществе мозга.
Ганглиозиды - наиболее сложные по составу липиды. Они содержат несколько углеводных остатков, среди которых присутствует N-ацетил-
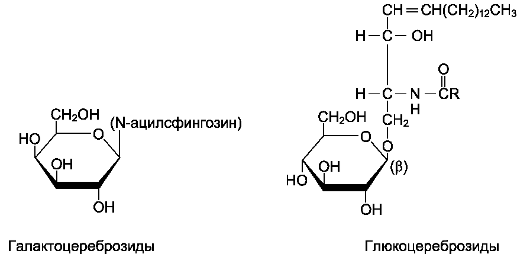
Рис. 8-6. Цереброзиды.
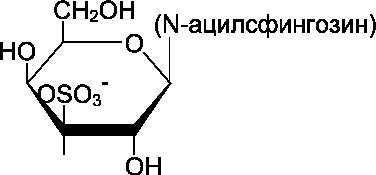
Рис. 8-7. Сульфатиды.
нейраминовая кислота. Нейраминовая кислота представляет собой углевод, состоящий из 9 атомов углерода и входящий в группу сиаловых кислот.
Строение ганглиозида Gm2 может быть представлено следующей схемой:
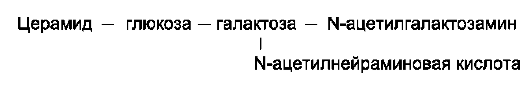
Номенклатура ганглиозидов. Ганглиозиды обозначают буквой G, например Gm2. Нижний индекс в виде букв M, D, T и Q означает, что молекула ганглиозида содержит 1, 2, 3 или 4 остатка сиаловых кислот. Цифра у нижнего индекса обозначает специфическую последовательность углеводов в ганглиозиде (рис. 8-8).
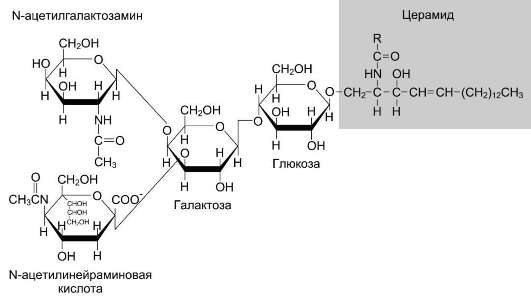
Рис. 8-8. Ганглиозид Gm2.
Ганглиозиды содержатся в основном в ганг-лиозных клетках нервной ткани, откуда они и получили своё название. Однако ганглиозиды находятся и в плазматических мембранах многих клеток - эритроцитов, гепатоцитов, клеток селезёнки и других органов. Главная роль ганг-лиозидов определяется их участием в осуществлении межклеточных контактов. Некоторые ганглиозиды служат своеобразными рецепторами для ряда бактериальных токсинов.
в. стероиды
Стероиды - производные восстановленных конденсированных циклических систем - цик-лопентанпергидрофенантренов.
В организме человека основной стероид - холестерол, остальные стероиды - его производные. Растения, грибы и дрожжи не синтезируют холестерол, но образуют разнообразные фитостеролы и микостеролы, не усваиваемые организмом человека. Бактерии не способны синтезировать стероиды.
Холестерол входит в состав мембран и влияет на структуру бислоя, увеличивая её жёсткость. Из холестерола синтезируются жёлчные кислоты, стероидные гормоны и витамин D3. Нарушение обмена холестерола приводит к развитию атеросклероза.
Холестерол представляет собой молекулу, содержащую 4 конденсированных кольца, обозначаемые латинскими буквами А, B, C, D, разветвлённую боковую цепь из 8 углеродных атомов в положении 17, 2 «ангулярные» метальные группы (18 и 19) и гидроксильную группу в положении 3. Наличие гидроксильной группы позволяет относить холестерол к спиртам, поэтому его правильное химическое название «холестерол», однако в медицинской литературе часто используют термин «холестерин».
Присоединение жирных кислот сложноэфир-ной связью к гидроксильной группе приводит к образованию эфиров холестерола (рис. 8-9).
В неэтерифицированной форме холестерол входит в состав мембран различных клеток. Гидроксильная группа холестерола обращена к водному слою, а жёсткая гидрофобная часть молекулы погружена во внутренний гидрофобный слой мембраны (см. рис. 5-6).
В крови 2/3 холестерола находится в этерифи-цированной форме и 1/3 - в виде свободного
холестерола. Эфиры холестерола служат формой его депонирования в некоторых клетках (например, печени, коры надпочечников, половых желёз). Из этих депо холестерол используется для синтеза жёлчных кислот и стероидных гормонов.
Жёлчные кислоты. Жёлчные кислоты обладают поверхностно-активными свойствами и участвуют в переваривании жиров, эмульгируя их и делая доступными для действия панкреатической липазы.
Жёлчные кислоты - производные холес-терола с пятиуглеродной боковой цепью в положении 17, которая заканчивается карбоксильной группой. В организме человека синтезируются две жёлчные кислоты: холевая, которая содержит три гидроксильные группы в положениях 3, 7, 12 (рис. 8-10), и хеноде-зоксихолевая, содержащая две гидроксильные группы в положениях 3 и 7. Так как карбоксильные группы этих жёлчных кислот имеют рК~6, они не полностью диссоциированы при физиологических значениях рН в кишечнике и не являются эффективными эмульгаторами. В печени эмульгирующие свойства жёлчных кислот увеличиваются за счёт реакции конъюгации, в которой к карбоксильной группе жёлчных кислот присоединяются таурин или глицин, полностью ионизированные при рН кишечного сока. Эти производные - конъ-югированные жёлчные кислоты - находятся в ионизированной форме и поэтому называются солями жёлчных кислот. Именно они служат главными эмульгаторами жиров в кишечнике.
ii. переваривание и всасывание пищевых липидов
С пищей в организм ежедневно поступает от 80 до
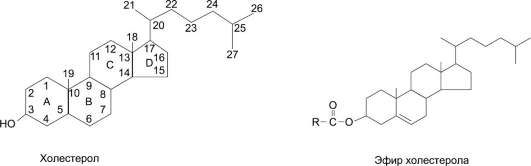
Рис. 8-9. Холестерол и его эфиры.
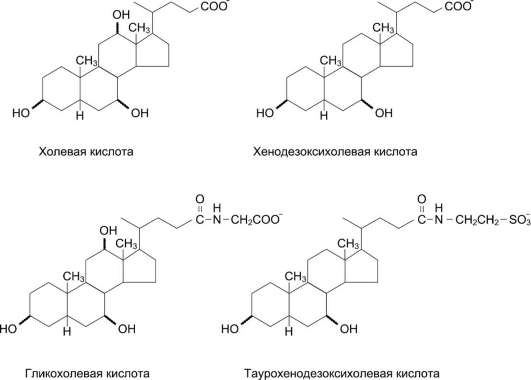
Рис. 8-10. Жёлчные кислоты.
пают и жирорастворимые витамины А, D, Е, К. Переваривание липидов пищи происходит в кишечнике. Основные продукты гидролиза (жирные кислоты и 2-моноацилглицеролы) после всасывания подвергаются ресинтезу и последующей упаковке в хиломикроны (ХМ) в клетках слизистой оболочки кишечника.
А. ЭМУЛЬГИРОВАНИЕ ЖИРОВ
Жиры составляют до 90% липидов, поступающих с пищей. Переваривание жиров происходит в тонком кишечнике, однако уже в желудке небольшая часть жиров гидролизуется под действием «липазы языка» . Этот фермент синтезируется железами на дорсальной поверх-
ности языка и относительно устойчив при кислых значениях рН желудочного сока. Поэтому он действует в течение 1-2 ч на жиры пищи в желудке. Однако вклад этой липазы в переваривание жиров у взрослых людей незначителен. Основной процесс переваривания происходит в тонкой кишке.
Так как жиры - нерастворимые в воде соединения, то они могут подвергаться действию ферментов, растворённых в воде только на границе раздела фаз вода/жир. Поэтому действию панкреатической липазы, гидролизующей жиры, предшествует эмульгирование жиров. Эмульгирование (смешивание жира с водой) происходит в тонком кишечнике под действием солей жёлчных кислот (рис. 8-11). Жёлчные кислоты синтезируются в печени из холес-терола и секретируются в жёлчный пузырь. Содержимое жёлчного пузыря - жёлчь. Это вязкая жёлто-зелёная жидкость, содержащая главным образом жёлчные кислоты; в небольшом количестве имеются фосфолипиды и холестерол. Жёлчные кислоты представляют собой в основном конъюгированные жёлчные кислоты: таурохолевую, гликохолевую и другие (см. выше рис. 8-10). После приёма жирной пищи жёлчный пузырь сокращается и жёлчь изливается в просвет двенадцатиперстной кишки. Жёлчные кислоты действуют как детергенты, располагаясь на поверхности капель жира и снижая поверхностное натяжение. В результате крупные капли жира распадаются на множество мелких, т.е. происходит эмульгирование жира. Эмульгирование приводит к увеличению площади поверхности раздела фаз жир/вода, что ускоряет гидролиз жира панкреатической липазой. Эмульгированию способствует и перистальтика кишечника.
Б. ГОРМОНЫ, АКТИВИРУЮЩИЕ
ПЕРЕВАРИВАНИЕ ЖИРОВ
При поступлении пищи в желудок, а затем в кишечник клетки слизистой оболочки тонкого кишечника начинают секретировать в кровь пеп-ттидный гормон холецистокинин (панкреозимин). Этот гормон действует на жёлчный пузырь, стимулируя его сокращение, и на экзокринные клетки поджелудочной железы, стимулируя секрецию пищеварительных ферментов, в том числе панкреатической липазы. Другие клетки
слизистой оболочки тонкого кишечника в ответ на поступление из желудка кислого содержимого выделяют гормон секретин. Секретин - гормон пептидной природы, стимулирующий секрецию бикарбоната (НСО3-) в сок поджелудочной железы.
В. ПЕРЕВАРИВАНИЕ ЖИРОВ
ПАНКРЕАТИЧЕСКОЙ ЛИПАЗОЙ
Переваривание жиров - гидролиз жиров панкреатической липазой. Оптимальное значение рН для панкреатической липазы =8 достигается путём нейтрализации кислого содержимого, поступающего из желудка, бикарбонатом, выделяющимся в составе сока поджелудочной железы:
Н+ + НСО3- → н2со3 → Н2О + СО2 ↑.
Выделяющийся углекислый газ способствует дополнительному перемешиванию содержимого тонкой кишки.
Панкреатическая липаза выделяется в полость тонкой кишки из поджелудочной железы вместе с белком колипазой. Колипаза попадает в полость кишечника в неактивном виде и частичным протеолизом под действием трипсина превращается в активную форму. Колипаза своим гидрофобным доменом связывается с поверхностью мицеллы эмульгированного жира. Другая часть молекулы способствует формированию такой конформации панкреатической липазы, при которой активный центр фермента максимально приближен к своим субстратам - молекулам жиров (рис. 8-12), поэтому скорость реакции гидролиза жира резко возрастает.
Панкреатическая липаза гидролизует жиры преимущественно в положениях 1 и 3 (рис. 813), поэтому основными продуктами гидролиза являются свободные жирные кислоты и 2-моно-ацилглицеролы (β-моноацилглицеролы).
Молекулы 2-моноацилглицеролов также обладают детергентными свойствами и способствуют эмульгированию жира.
Г. ПЕРЕВАРИВАНИЕ ДРУГИХ ЛИПИДОВ
Кроме жиров, с пищей поступают фосфоли-пиды, эфиры холестерола, однако количество этих липидов в составе пищи значительно меньше, чем жиров (=10%).
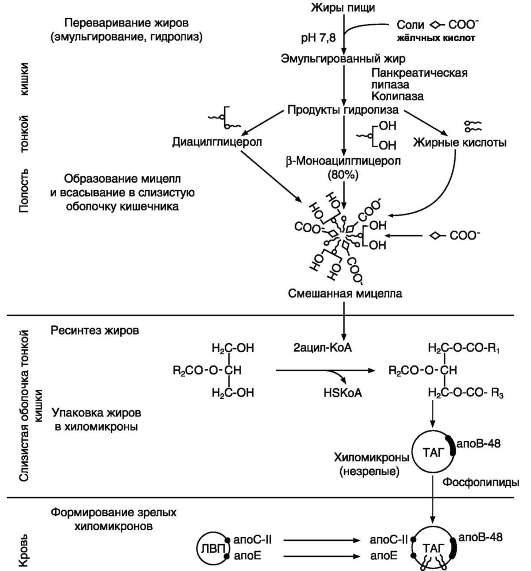
Рис. 8-11. Этапы поступления экзогенных жиров в организм.
Переваривание глицерофосфолипидов
В переваривании глицерофосфолипидов участвуют несколько ферментов, синтезирующихся в поджелудочной железе. Фосфолипаза А2 гид-ролизует сложноэфирную связь у второго атома углерода глицерола, превращая глицерофосфо-липиды в соответствующие лизофосфолипиды.
На рисунке 8-14 представлен пример гидролиза фосфатидилхолинов при переваривании.
Фосфолипаза А2 секретируется в кишечник в виде профермента и активируется уже в полости кишечника путём частичного протеолиза. Для проявления активности фосфолипазы А2 необходимы ионы кальция.
Жирная кислота в положении 1 отщепляется под действием лизофосфолипазы, а глицеро-фосфохолин гидролизуется далее до глицерола, холина и фосфорной кислоты, которые всасываются. Лизофосфолипиды - эффективные эмульгаторы жира, ускоряющие его переваривание.
Переваривание эфиров холестерола
В составе пищи холестерол находится в основном в виде эфиров. Гидролиз эфиров холестерола происходит под действием холесте-ролэстеразы - фермента, который также синтезируется в поджелудочной железе и секретирует-ся в кишечник (рис. 8-15). Продукты гидролиза (холестерол и жирные кислоты) всасываются в составе смешанных мицелл.
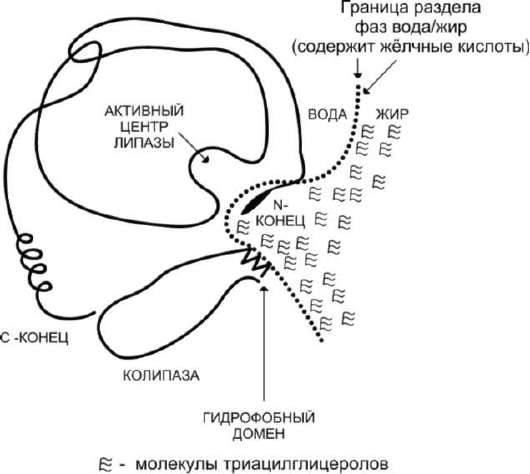
Рис. 8-12. Расположение панкреатической липазы и колипазы на границе раздела фаз вода/жир.
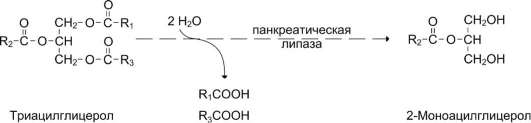
Рис. 8-13. Гидролиз триацилглицеролов панкреатической липазой.
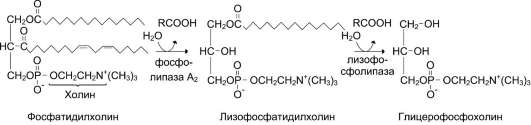
Рис. 8-14. Переваривание фосфатидилхолинов.
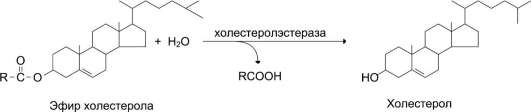
Рис. 8-15. Гидролиз эфиров холестерола в тонкой кишке.
Д. ПЕРЕВАРИВАНИЕ ЖИРА У ГРУДНЫХ ДЕТЕЙ
У грудных детей и детей младшего возраста основной пищей служит молоко. Молоко содержит жиры, в состав которых входят в основном жирные кислоты с короткой и средней длиной алифатических цепей (4-12 атомов углерода). Жиры в составе молока находятся уже в эмульгированном, смешанном с водой виде, поэтому они сразу же доступны для гидролиза ферментами. На жиры молока в желудке детей действует липаза, которая синтезируется в железах языка (липаза языка). Кроме того, в желудке детей грудного и младшего возраста вырабатывается желудочная липаза, которая активна при нейтральном значении рН, характерном для желудочного сока детей, и не активна у взрослых (рН желудочного сока ~1,5). Эта липаза гидролизует жиры, отщепляя, в основном, жирные кислоты у третьего атома углерода глицерола. Далее гидролиз жиров молока продолжается в кишечнике под действием панкреатической липазы. Жирные кислоты с короткой цепью, как водорастворимые, всасываются частично уже в желудке. Остальные жирные кислоты всасываются в тонком кишечнике. Для детей грудного возраста основным источником энергии являются жиры, в то время как у взрослых людей при нормальном питании основным источником энергии служит глюкоза.
Вследствие этого нарушение переваривания и всасывания жиров у детей более опасно, чем у взрослых.
Е. ВСАСЫВАНИЕ ПРОДУКТОВ ГИДРОЛИЗА ЛИПИДОВ В ТОНКОМ кишечнике. РЕСИНТЕЗ ЖИРОВ
Образование смешанных мицелл и всасывание продуктов гидролиза
Продукты гидролиза липидов - жирные кислоты с длинным углеводородным радикалом, 2-моноацилглицеролы, холестерол, а также соли жёлчных кислот образуют в просвете кишечника структуры, называемые смешанными мицеллами. Смешанные мицеллы построены таким образом, что гидрофобные части молекул обращены внутрь мицеллы, а гидрофильные - наружу, поэтому мицеллы хорошо растворяются в водной фазе содержимого тонкой кишки. Ста-
бильность мицелл обеспечивается в основном солями жёлчных кислот. Мицеллы сближаются со щёточной каймой клеток слизистой оболочки тонкого кишечника, и липидные компоненты мицелл диффундируют через мембраны внутрь клеток. Вместе с продуктами гидролиза липидов всасываются жирорастворимые витамины А, D, Е, К и соли жёлчных кислот. Наиболее активно соли жёлчных кислот всасываются в подвздошной кишке. Жёлчные кислоты далее попадают через воротную вену в печень, из печени вновь секретируются в жёлчный пузырь и далее опять участвуют в эмульгировании жиров. Этот путь жёлчных кислот называют «энтерогепатическая циркуляция». Каждая молекула жёлчных кислот за сутки проходит 5-8 циклов, и около 5% жёлчных кислот выделяется с фекалиями.
Всасывание жирных кислот со средней длиной цепи, образующихся, например, при переваривании липидов молока, происходит без участия смешанных мицелл. Эти жирные кислоты из клеток слизистой оболочки тонкого кишечника попадают в кровь, связываются с белком альбумином и транспортируются в печень.
Ресинтез жиров в слизистой оболочке тонкого кишечника
После всасывания продуктов гидролиза жиров жирные кислоты и 2-моноацилглицеролы в клетках слизистой оболочки тонкого кишечника включаются в процесс ресинтеза с образованием триацилглицеролов (рис. 8-16). Жирные кислоты вступают в реакцию этерификации только в активной форме в виде производных коэнзима А, поэтому первая стадия ресинтеза жиров - реакция активации жирных кислот: HS КоА + RCOOH + АТФ → R-CO ~ KoA +
АМФ + Н4Р2О7.
Реакция катализируется ферментом ацил-КоА-синтетазой (тиокиназой). Затем ацил~КоА участвует в реакции этерификации 2-моноаци-лглицерола с образованием сначала диацилг-лицерола, а затем триацилглицерола. Реакции ресинтеза жиров катализируют ацилтрансфе-разы.
В реакциях ресинтеза жиров участвуют, как правило, только жирные кислоты с длинной углеводородной цепью. В ресинтезе жиров участвуют не только жирные кислоты, всосавшиеся из кишечника, но и жирные кислоты, синте-
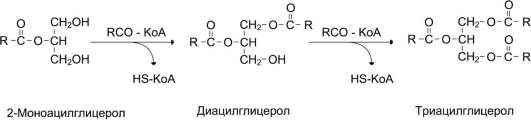
Рис. 8-16. Ресинтез жиров в клетках слизистой оболочки тонкой кишки.
зированные в организме, поэтому по составу ресинтезированные жиры отличаются от жиров, полученных с пищей. Однако возможности «адаптировать» в процессе ресинтеза состав пищевых жиров к составу жиров организма человека ограничены, поэтому при поступлении с пищей жиров с необычными жирными кислотами, например бараньего жира, в адипоцитах появляются жиры, содержащие кислоты, характерные для бараньего жира (насыщенные разветвлённые жирные кислоты). В клетках слизистой оболочки кишечника происходит активный синтез глицерофосфолипидов, необходимых для формирования структуры липопротеинов - транспортных форм липидов в крови.
Образование эфиров холестерола
В клетках слизистой оболочки тонкой кишки всосавшиеся молекулы холестерола также превращаются в эфиры путём взаимодействия с ацил-КоА (рис. 8-17). Эту реакцию катализирует ацилхолестеролацилтрансфераза (АХАТ). От активности этого фермента зависит скорость поступления экзогенного холестерола в организм.
В клетках эпителия тонкой кишки из жиров, образовавшихся в результате ресинтеза, а также из эфиров холестерола, жирорастворимых витаминов, поступивших с пищей, формируются липопротеиновые комплексы - хиломикроны
(ХМ). ХМ далее доставляют жиры в периферические ткани.
Нарушения переваривания и всасывания жиров. Стеаторея
Нарушение переваривания жиров может быть следствием нескольких причин. Одна из них - нарушение секреции жёлчи из жёлчного пузыря при механическом препятствии оттоку жёлчи. Это состояние может быть результатом сужения просвета жёлчного протока камнями, образующимися в жёлчном пузыре, или сдавлением жёлчного протока опухолью, развивающейся в окружающих тканях. Уменьшение секреции жёлчи приводит к нарушению эмульгирования пищевых жиров и, следовательно, к снижению способности панкреатической липазы гидролизовать жиры.
Нарушение секреции сока поджелудочной железы и, следовательно, недостаточная секреция панкреатической липазы также приводят к снижению скорости гидролиза жиров. В обоих случаях нарушение переваривания и всасывания жиров приводит к увеличению количества жиров в фекалиях - возникает стеаторея (жирный стул). В норме содержание жиров в фекалиях составляет не более 5%. При стеаторее нарушается всасывание жирорастворимых витаминов (А, D, Е, К) и незаменимых жирных кислот, поэтому при длительно текущей
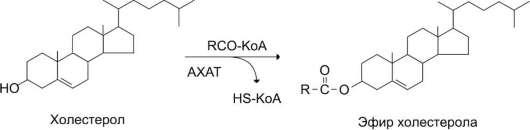
Рис. 8-17. Реакция этерификации холестерола в клетках слизистой оболочки тонкой кишки. АХАТ - ацил-холестерол-ацилтрансфераза.
стеаторее развивается недостаточность этих незаменимых факторов питания с соответствующими клиническими симптомами (см. раздел 3). При нарушении переваривания жиров плохо перевариваются и вещества нелипидной природы, так как жир обволакивает частицы пищи и препятствует действию на них ферментов.
iii. транспорт жиров из кишечника хиломикронами
Липиды в водной среде (а значит, и в крови) нерастворимы, поэтому для транспорта липидов кровью в организме образуются комплексы ли-пидов с белками - липопротеины.
А. ОБЩАЯ ХАРАКТЕРИСТИКА
ЛИПОПРОТЕИНОВ
Все типы липопротеинов имеют сходное строение - гидрофобное ядро и гидрофильный слой на поверхности (рис. 8-18). Гидрофильный слой образован белками, которые называют
апопротеинами, и амфифильными молекулами липидов - фосфолипидами и холестеролом. Гидрофильные группы этих молекул обращены к водной фазе, а гидрофобные части - к гидрофобному ядру липопротеина, в котором находятся транспортируемые липиды. Некоторые апопротеины интегральные и не могут быть отделены от липопротеина, а другие могут свободно переноситься от одного типа липоп-ротеина к другому. Апопротеины выполняют несколько функций:
• формируют структуру липопротеинов;
• взаимодействуют с рецепторами на поверхности клеток и таким образом определяют, какими тканями будет захватываться данный тип липопротеинов;
• служат ферментами или активаторами ферментов, действующих на липопротеины.
В организме синтезируются следующие типы липопротеинов (см. ниже табл. 8-5): хиломик-роны (ХМ), липопротеины очень низкой плотности (ЛПОНП), липопротеины промежуточной плотности (ЛППП), липопротеины низкой
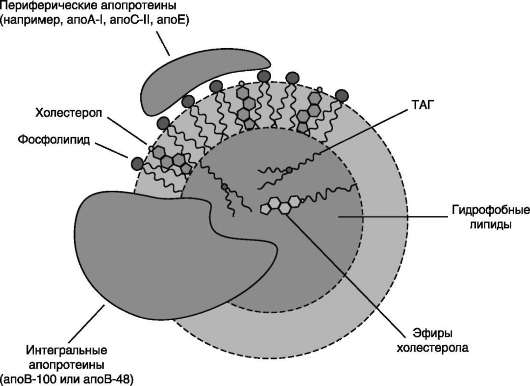
Рис. 8-18. Липопротеины плазмы крови.
плотности (ЛПНП) и липопротеины высокой плотности (ЛПВП).
Каждый из типов ЛП образуется в разных тканях и транспортирует определённые липиды. Например, ХМ транспортируют экзогенные (пищевые жиры) из кишечника в ткани, поэтому триацилглицеролы составляют до 85% массы этих частиц.
ЛП хорошо растворимы в крови, не коалес-цируют, так как имеют небольшой размер и отрицательный заряд на поверхности. Некоторые ЛП легко проходят через стенки капилляров кровеносных сосудов и доставляют липиды к клеткам.
Большой размер ХМ не позволяет им проникать через стенки капилляров, поэтому из клеток кишечника они сначала попадают в лимфатическую систему и потом через главный грудной проток вливаются в кровь вместе с лимфой.
Методы исследования. Состав ЛП крови можно исследовать разными методами (рис. 8-19). Метод ультрацентрифугирования позволяет разделить ЛП, используя их различие в плотности, которая зависит от соотношения количества липидов и белков в частице. Так как жир имеет меньшую, чем вода, плотность, то ХМ,
содержащие более 85% жиров, располагаются на поверхности сыворотки крови, а ЛПВП, содержащие наибольшее количество белков, имеют самую большую плотность и при центрифугировании располагаются в нижней части центрифужной пробирки. Так как ЛП впервые были выделены из сыворотки крови методом ультрацентрифугирования, то в названии указывают плотность частиц. Однако метод ультрацентрифугирования непригоден для широкого использования, поэтому в клинических лабораториях обычно применяют метод электрофореза. Скорость движения частиц при электрофорезе зависит от их заряда и размера. Заряд, в свою очередь, зависит от количества белков на поверхности ЛП (табл. 8-5). При электрофорезе в геле все типы ЛП движутся к положительному полюсу; ближе к старту располагаются ХМ, а ЛПВП, имеющие наибольшее количество белков и наименьший размер, удаляются от старта дальше других частиц.
Состав ЛП крови значительно изменяется в течение суток. В абсорбтивный период (особенно при употреблении жирной пищи) в крови появляются ХМ. Богатая углеводами пища способствует образованию ЛПОНП, так как эти ЛП транспортируют жиры, синтезированные
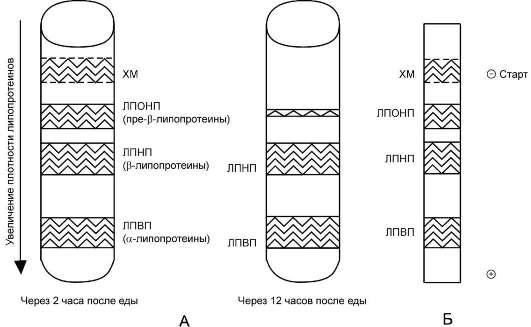
Рис. 8-19. Разделение липопротеинов сыворотки крови. А - метод ультрацентрифугирования. Б - метод электрофореза в полиакриламидном геле через 2 ч после еды.
Таблица 8-5. Липопротеины - транспортные формы липидов
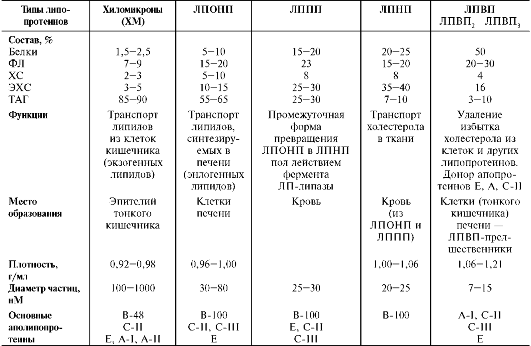
Примечания: ФЛ - фосфолипиды; ХС - холестерол; ЭХС - эфиры холестерола; ТАГ - триацилглицеролы. Функции апопротеинов
• В-48 - основной белок ХМ;
• В-100 - основной белок ЛПОНП, ЛПНП, ЛППП, взаимодействует с рецепторами ЛПНП;
• C-II - активатор ЛП-липазы, переносится с ЛПВП на ХМ и ЛПОНП в крови;
• C-III - ингибитор ЛП-липазы;
• Е - обеспечивает связывание нескольких типов липопротеинов с рецепторами ЛПНП и др. рецепторами;
• А-I - активатор фермента лецитин:холестеролацилтрансферазы (ЛХАТ).
в печени из углеводов. В постабсорбтивный период и при голодании в крови присутствуют ЛПНП, ЛПВП и в небольшом количестве ЛПОНП, основная функция которых заключается в транспорте холестерола.
Б. ОБРАЗОВАНИЕ ХИЛОМИКРОНОВ
Жиры, образовавшиеся в результате ресин-теза в клетках слизистой оболочки кишечника, упаковываются в ХМ. Основной апопротеин в составе ХМ - белок апоВ-48. Этот белок закодирован в том же гене, что и белок ЛПОНП - В-100 (табл. 8-5), который синтезируется в
печени. В кишечнике в результате посттранскрипционных превращений «считывается» последовательность мРНК, которая кодирует только 48% от длины белка В-100, поэтому этот белок называется апоВ-48. Белок апоВ-48 синтезируется в шероховатом ЭР и там же гликозилируется. Затем в аппарате Гольджи происходит формирование ХМ, называемых «незрелыми». По механизму экзоцитоза они выделяются в хилус, образующийся в лимфатической системе кишечных ворсинок, и через главный грудной лимфатический проток попадают в кровь. В лимфе и крови с ЛПВП на ХМ переносятся апопротеины Е (апоЕ) и С-II
(апоС-II); ХМ превращаются в «зрелые». ХМ имеют довольно большой размер, поэтому после приёма жирной пищи они придают плазме крови опалесцирующий, похожий на молоко, вид. ХМ транспортируют жир к различным тканям, где он утилизируется, поэтому концентрация ХМ в крови постепенно снижается, и плазма опять становится прозрачной. ХМ исчезают из крови в течение нескольких часов.
При редком наследственном заболевании - дефекте гена апопротеина В - нарушается синтез белков апоВ-100 в печени и апоВ-48 в кишечнике. В результате в клетках слизистой оболочки кишечника не формируются ХМ, а в печени - ЛПОНП. В клетках этих органов накапливаются капельки жира. Такое заболевание называется абеталипопротеинемия, так как второе название ЛПОНП - пре-р-липоп-ротеины.
в. использование экзогенных
жиров тканями
Действие липопротеинлипазы на ХМ. В крови триацилглицеролы, входящие в состав зрелых ХМ, гидролизуются ферментом липопротеин-липазой, или ЛП-липазой (рис. 8-20). ЛП-липаза связана с гепарансульфатом (гетерополисахари-дом), находящимся на поверхности эндотелиаль-ных клеток, выстилающих стенки капилляров кровеносных сосудов. ЛП-липаза гидролизует молекулы жиров до глицерола и 3 молекул жирных кислот. На поверхности ХМ различают 2 фактора, необходимых для активности ЛП-ли-пазы - апоС-II и фосфолипиды. АпоС-II активирует этот фермент, а фосфолипиды участвуют в связывании фермента с поверхностью ХМ.
ЛП-липаза синтезируется в клетках многих тканей: жировой, мышечной, в лёгких, селезёнке,
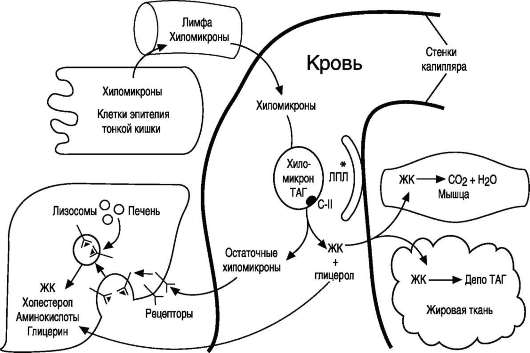
Рис. 8-20. Путь экзогенных жиров и хиломикронов. *ЛПЛ - липопротеинлипаза, ЖК - жирные кислоты.
клетках лактирующей молочной железы. Изофер-менты ЛП-липазы в разных тканях отличаются по значению Km: ЛП-липаза жировой ткани имеет в 10 раз более высокое значение Km, чем, например, ЛП-липаза сердца, поэтому гидролиз жиров ХМ в жировой ткани происходит в абсор-бтивный период. Жирные кислоты поступают в адипоциты и используются для синтеза жиров. В постабсорбтивном состоянии, когда количество жиров в крови снижается, ЛП-липаза сердечной мышцы продолжает гидролизовать жиры в составе ЛПОНП, которые присутствуют в крови в небольшом количестве, и жирные кислоты используются этой тканью как источники энергии, даже при низкой концентрации жиров в крови. ЛП-липазы нет в печени, но на поверхности клеток этого органа имеется другой фермент - печёночная липаза, не действующая на зрелые ХМ, но гидролизующая жиры в ЛППП, которые образуются из ЛПОНП.
Судьба жирных кислот, глицерола и остаточных хиломикронов. В результате действия ЛП-липа-зы на жиры ХМ образуются жирные кислоты и глицерол. Основная масса жирных кислот проникает в ткани (рис. 8-20). В жировой ткани в абсорбтивный период жирные кислоты депонируются в виде триацилглицеролов, в сердечной мышце и работающих скелетных мышцах используются как источник энергии. Другой продукт гидролиза жиров, глицерол, растворим в крови, транспортируется в печень, где в абсорбтивный период может быть использован для синтеза жиров.
В результате действия ЛП-липазы на ХМ количество жиров в них снижается на 90%, уменьшаются размеры частиц, апопротеин C-II переносится обратно на ЛПВП. Образовавшиеся частицы называются остаточными ХМ. Они содержат в себе фосфолипиды, холестерол, жирорастворимые витамины и апопротеины В-48 и Е. Остаточные ХМ захватываются гепатоцитами, которые имеют рецепторы, взаимодействующие с этими апопротеинами. Путём эндоцитоза остаточные ХМ попадают внутрь клеток, и ферментами лизосом белки и липиды гидролизуются, а затем утилизируются. Жирорастворимые витамины и экзогенный холестерол используются в печени или транспортируются в другие ткани.
Гиперхиломикронемия, гипертриглицеролемия. После приёма пищи, содержащей жиры, развивается физиологическая гипертриглицероле-
мия и, соответственно, гиперхиломикронемия, которая может продолжаться до нескольких часов.
Скорость удаления ХМ из кровотока зависит от:
• активности ЛП-липазы;
• присутствия ЛПВП, поставляющих апо-протеины С-II и Е для ХМ;
• активности переноса апоС-II и апоЕ на
ХМ.
Генетические дефекты любого из белков, участвующих в метаболизме ХМ, приводят к развитию семейной гиперхиломикронемии - гиперлипопротеинемии типа I. У таких больных в постабсорбтивном периоде концентрация три-ацилглицеролов повышена (более 200 мг/дл), плазма крови по виду напоминает молоко и при оставлении на холоде (+4 ?С) в ней всплывают белые жирные хлопья, что характерно для гипер-триглицеролемии и гиперхиломикронемии.
В тяжёлых случаях при этом заболевании происходит отложение триацилглицеролов в коже и сухожилиях в виде ксантом, у пациентов рано нарушается память, появляются боли в животе из-за сужения просвета сосудов и уменьшения кровотока, нарушается функция поджелудочной железы, что часто бывает причиной смерти больных. Если концентрация триацилглицеролов в крови превышает 4000 мг/дл, то липиды откладываются в сетчатке глаза, однако это не всегда влияет на зрительную функцию. При лечении гиперхиломикронемий необходимо прежде всего снизить потребление жиров с пищей, так как ХМ транспортируют экзогенные жиры.
iv. обмен триацилглицеролов
Приём
пищи человеком происходит иногда со значительными интервалами, поэтому
в организме выработались механизмы депонирования источников энергии.
Жиры - наиболее выгодная и основная форма депонирования энергии. Запасы
гликогена в организме не превышают
важную роль превращения части углеводов, поступающих с пищей, в жиры, которые затем секретируются в кровь в составе ЛПОНП и доставляются в другие ткани (в первую очередь, в жировую). Синтез жиров в печени и жировой ткани стимулируется инсулином. Мобилизация жира активируется в тех случаях, когда глюкозы недостаточно для обеспечения энергетических потребностей организма: в постабсорбтивный период, при голодании и физической работе под действием гормонов глюкагона, адреналина, соматотропина. Жирные кислоты поступают в кровь и используются тканями как источники энергии.
А. СИНТЕЗ ЖИРОВ В ЖИРОВОЙ ТКАНИ И ПЕЧЕНИ
Синтез жиров происходит в абсорбтивный период в печени и жировой ткани. Непосредственными субстратами в синтезе жиров являются ацил-КоА и глицерол-3-фосфат. Метаболический путь синтеза жиров в печени и жировой ткани одинаков, за исключением разных путей образования глицерол-3-фосфата.
Образование глицерол-3-фосфата
Синтез жиров в печени и жировой ткани идёт через образование промежуточного продукта - фосфатидной кислоты (рис. 8-21).
Предшественник фосфатидной кислоты - глицерол-3-фосфат, образующийся в печени двумя путями:
• восстановлением дигидроксиацетонфосфа-та - промежуточного метаболита гликолиза;
• фосфорилированием глицеролкиназой свободного глицерола, поступающего в печень из крови (продукт действия ЛП-липазы на
жиры ХМ и ЛПОНП).
В жировой ткани глицеролкиназа отсутствует, и восстановление дигидроксиацетонфосфа-та - единственный путь образования глицерол-3-фосфата. Следовательно, синтез жиров в жировой ткани может происходить только в абсорбтивный период, когда глюкоза поступает в адипоциты с помощью белка-переносчика глюкозы ГЛЮТ-4, активного только в присутствии инсулина, и распадается по пути гликолиза.
Синтез жиров в жировой ткани
В жировой ткани для синтеза жиров используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП (рис. 8-22). Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом, образуя сначала лизофосфатидную кислоту, а затем фосфатидную. Фосфатидная кислота после дефосфорилирования превращается в диацил-глицерол, который ацилируется с образованием триацил-глицерола.
Кроме жирных кислот, поступающих в ади-поциты из крови, в этих клетках идёт и синтез жирных кислот из продуктов распада глюкозы. В адипоцитах для обеспечения реакций синтеза жира распад глюкозы идёт по двум путям: гликолиз, обеспечивающий образование глицерол-3-фосфата и ацетил-КоА, и пентозофосфатный путь, окислительные реакции которого обеспечивают образование NADPH, служащего донором водорода в реакциях синтеза жирных кислот.
Молекулы жиров в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул. Подсчитано, что, если бы энергия, запасаемая в жирах, хранилась в форме сильно гидратированных молекул гликогена, то масса тела человека увеличилась бы на 14-15 кг.
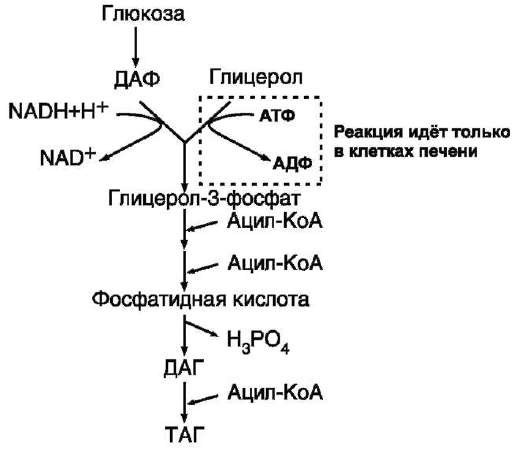
Рис. 8-21. Синтез жиров в печени и жировой ткани.
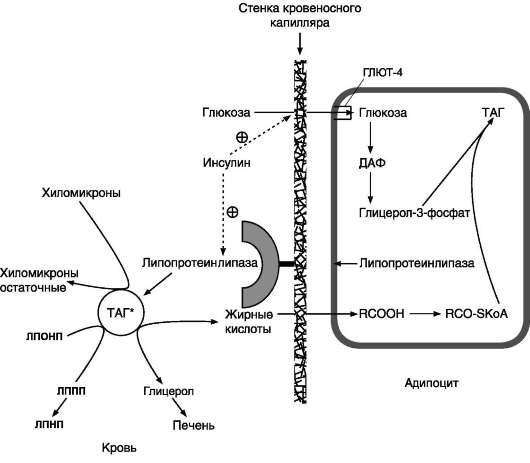
Рис. 8-22. Депонирование жира в адипоцитах в абсорбтивном периоде. После еды при повышении концентрации глюкозы в крови увеличивается секреция инсулина. Инсулин активирует транспорт глюкозы внутрь адипоцитов, действуя на ГЛЮТ-4, синтез ЛП-липазы в адипоцитах и её экспонирование на поверхности стенки капилляров. ЛП-липаза, связанная с эндотелием сосудов, гидролизует жиры в составе ХМ и ЛПОНП. АпоС-II на поверхности ХМ и ЛПОНП активирует ЛП-липазу. Жирные кислоты проникают в адипоцит, а глицерол транспортируется в печень. Так как в адипоцитах нет фермента глицеролкиназы, то свободный глицерол не может использоваться для синтеза ТАГ в этой ткани. Активированные жирные кислоты взаимодействуют с гли-церол-3-фосфатом, образующимся из дигидроксиацетонфосфата, и через фосфатидную кислоту превращаются в ТАГ, которые депонируются в адипоцитах. Сокращения: ТАГ* - триацилглицеролы в составе ХМ и ЛПОНП; ДАФ - дигидроксиацетонфосфат.
Синтез ТАГ в печени. Образование ЛПОНП в печени и транспорт жиров в другие ткани
Печень - основной орган, где идёт синтез жирных кислот из продуктов гликолиза. В гладком ЭР гепатоцитов жирные кислоты активируются и сразу же используются для синтеза жиров, взаимодействуя с глицерол-
3-фосфатом. Как и в жировой ткани, синтез жиров идёт через образование фосфатидной кислоты. Синтезированные в печени жиры упаковываются в ЛПОНП и секретируются в кровь (рис. 8-23).
В состав ЛПОНП, кроме жиров, входят хо-лестерол, фосфолипиды и белок - апоВ-100. Это очень длинный белок - одна молекула
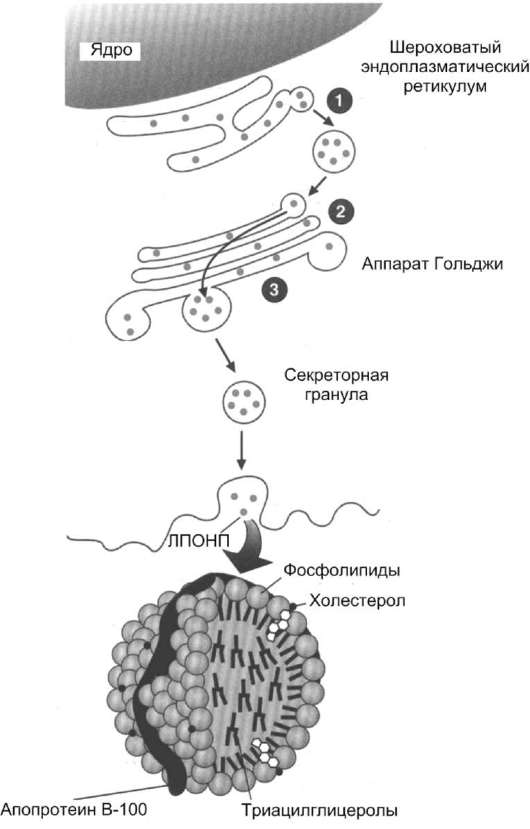
Рис. 8-23. Синтез и секреция ЛПОНП в печени. Белки, синтезированные в шероховатом ЭР (1), в аппарате Гольджи (2), формируют комплекс с ТАГ, называемый ЛПОНП. ЛПОНП комплектуются в секреторных гранулах (3), транспортируются к клеточной мембране и секретируются в кровь.
апоВ-100 покрывает поверхность всего липоп-ротеина.
ЛПОНП из печени секретируются в кровь (рис. 8-23), где на них, как и на ХМ, действует ЛП-липаза. Жирные кислоты поступают в ткани, в частности в адипоциты, и используются для синтеза жиров. В процессе удаления жиров из ЛПОНП под действием ЛП-липазы ЛПОНП сначала превращаются в ЛППП, а затем в ЛПНП. В ЛПНП основными липидными компонентами
служат холестерол и его эфиры, поэтому ЛПНП являются липопротеинами, доставляющими холестерол в периферические ткани. Глицерол, освободившийся из липопротеинов, кровью транспортируется в печень, где опять может использоваться для синтеза жиров.
Скорость синтеза жирных кислот и жиров в печени существенно зависит от состава пищи. Если в пище содержится более 10% жиров, то скорость синтеза жиров в печени снижается.
Б. МОБИЛИЗАЦИЯ ЖИРОВ ИЗ ЖИРОВОЙ ТКАНИ
Адипоциты (место депонирования жиров) располагаются в основном под кожей, образуя подкожный жировой слой, и в брюшной полости, образуя большой и малый сальники. Мобилизация жиров, т.е. гидролиз до глицерола и жирных кислот, происходит в постабсорбтивный период, при голодании и активной физической работе. Гидролиз внутриклеточного жира осуществляется под действием фермента гормончувствительной липазы - ТАГ-липазы. Этот фермент отщепляет одну жирную кислоту у первого углеродного атома глицерола с образованием диацилглицерола, а затем другие липазы гидролизуют его до глицерола и жирных кислот, которые поступают в кровь. Глицерол как водорастворимое вещество транспортируется кровью в свободном виде, а жирные кислоты (гидрофобные молекулы) в комплексе с белком плазмы - альбумином.
В. ГОРМОНАЛЬНАЯ РЕГУЛЯЦИЯ СИНТЕЗА
И МОБИЛИЗАЦИИ ЖИРОВ
Какой процесс будет преобладать в организме - синтез жиров (липогенез) или их распад (ли-полиз), зависит от поступления пищи и физической активности. В абсорбтивном состоянии под действием инсулина происходит липогенез, в постабсорбтивном состоянии - липолиз, активируемый глюкагоном. Адреналин, секреция которого увеличивается при физической активности, также стимулирует липолиз.
Регуляция синтеза жиров. В абсорбтивный период при увеличении соотношения инсулин/ глюкагон в печени активируется синтез жиров. В жировой ткани индуцируется синтез ЛП-липа-зы в адипоцитах и осуществляется её экспонирование на поверхность эндотелия; следовательно, в этот период увеличивается поступление жирных кислот в адипоциты. Одновременно инсулин активирует белки-переносчики глюкозы -
ГЛЮТ-4. Поступление глюкозы в адипоциты и гликолиз также активируются. В результате образуются все необходимые компоненты для синтеза жиров: глицерол-3-фосфат и активные формы жирных кислот. В печени инсулин, действуя через различные механизмы, активирует ферменты путём дефосфорилирования и индуцирует их синтез. В результате увеличиваются активность и синтез ферментов, участвующих в превращении части глюкозы, поступающей с пищей, в жиры. Это - регуляторные ферменты гликолиза, пируватдегидрогеназный комплекс и ферменты, участвующие в синтезе жирных кислот из ацетил-КоА. Результат действия инсулина на обмен углеводов и жиров в печени - увеличение синтеза жиров и секреция их в кровь в составе ЛПОНП. ЛПОНП доставляют жиры в капилляры жировой ткани, где действие ЛП-ли-пазы обеспечивает быстрое поступление жирных кислот в адипоциты, где они депонируются в составе триацилглицеринов.
Запасание
жиров в жировой ткани - основная форма депонирования источников
энергии в организме человека (табл. 8-6). Запасы жиров в организме
человека массой
Жиры образуют в адипоцитах жировые вакуоли. Жировые вакуоли иногда заполняют значительную часть цитоплазмы. Скорость синтеза и мобилизации подкожного жира происходит неравномерно в разных частях организма, что связано с неодинаковым распределением рецепторов гормонов на адипоцитах.
Регуляция мобилизации жиров. Мобилизация депонированных жиров стимулируется глюка-гоном и адреналином и, в меньшей степени, некоторыми другими гормонами (соматот-ропным, кортизолом). В постабсорбтивный период и при голодании глюкагон, действуя на адипоциты через аденилатциклазную систему, активирует протеинкиназу А, которая
Таблица 8-6. Запасы энергии в организме человека (масса

фосфорилирует и, таким образом, активирует гормончувствительную липазу, что инициирует липолиз и выделение жирных кислот и глицерина в кровь. При физической активности увеличивается секреция адреналина, который действует через β-адренергические рецепторы адипоцитов, активирующие аденилатциклазную систему (рис. 8-24). В настоящее время обнаружено 3 типа β-рецепторов: β1, β2, β3, активация которых приводит к липолитическому действию. К наибольшему липолитическому действию приводит активация β3-рецепторов. Адреналин одновременно действует и на α2-рецепторы адипоцитов, связанные с ингибирующим G-белком, что инактивирует аденилатциклазную систему. Вероятно, действие адреналина двояко: при низ-
ких концентрациях в крови преобладает его ан-тилиполитическое действие через α2-рецепторы, а при высокой - преобладает липолитическое действие через β-рецепторы.
Для мышц, сердца, почек, печени при голодании или физической работе жирные кислоты становятся важным источником энергии. Печень перерабатывает часть жирных кислот в кетоновые тела, используемые мозгом, нервной тканью и некоторыми другими тканями как источники энергии.
В результате мобилизации жиров концентрация жирных кислот в крови увеличивается приблизительно в 2 раза (рис. 8-25), однако абсолютная концентрация жирных кислот в крови невелика даже в этот период. T1/2 жирных кислот в крови
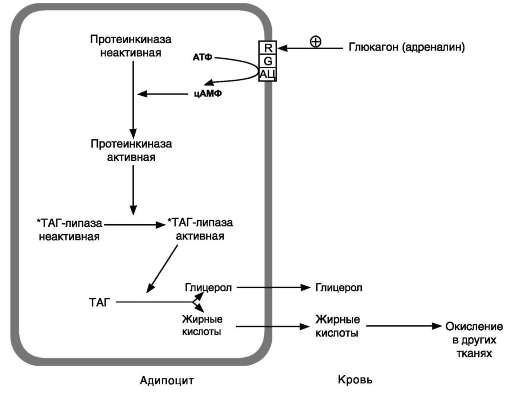
Рис. 8-24. Гормональная регуляция мобилизации жиров в постабсорбтивном периоде, при голодании и физической работе. При голодании увеличивается секреция глюкагона, при физической работе - адреналина. R - рецептор, G - G-белок, АЦ - аденилатциклаза Эти гормоны, действуя через аденилатциклазную систему, стимулируют мобилизацию жиров. *ТАГ-липаза имеет и другие названия: гормончувствительная липаза, тканевая липаза.
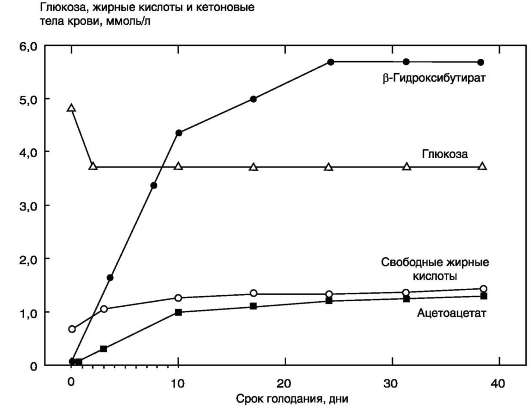
Рис. 8-25. Изменение концентрации жирных кислот, кетоновых тел и глюкозы в крови при голодании.
тоже очень мал (менее 5 мин), что означает существование быстрого потока жирных кислот из жировой ткани к другим органам. Когда пос-табсорбтивный период сменяется абсорбтивным, инсулин активирует специфическую фосфатазу, которая дефосфорилирует гормончувствительную липазу, и распад жиров останавливается.
Г. НАРУШЕНИЯ ЖИРОВОГО ОБМЕНА.
ОЖИРЕНИЕ
Жировая ткань составляет 20-25% от общей массы тела у женщин и 15-20% у мужчин. Однако избыточное накопление жира в ади-поцитах (ожирение) широко распространено. Среди взрослого населения некоторых стран около 50% людей страдает ожирением. Ожирение - важнейший фактор риска развития инфаркта миокарда, инсульта, сахарного диабета,
артериальной гипертензии и желчнокаменной болезни.
Ожирением считают состояние, когда масса тела превышает 20% от «идеальной» для данного индивидуума. Образование адипоцитов происходит ещё во внутриутробном состоянии, начиная с последнего триместра беременности, и заканчивается в препубертатный период. После этого жировые клетки могут увеличиваться в размерах при ожирении или уменьшаться при похудании, но их количество существенно не изменяется в течение жизни, за исключением случаев гиперпластического ожирения.
Первичное ожирение
Первичное ожирение характеризуется множеством гормональных и метаболических особенностей у лиц, страдающих этим заболеванием. В самом общем виде можно сказать, что пер-
вичное ожирение развивается в результате алиментарного дисбаланса - избыточной калорийности питания по сравнению с расходами энергии.
Суточные потребности организма в энергии складываются из:
• основного обмена - энергии, необходимой для поддержания жизни; основной обмен измеряют по поглощению кислорода или выделению тепла человеком в состоянии покоя утром, после 12-часового перерыва в еде;
• энергии, необходимой для физической активности.
Затраты энергии, необходимые для физической активности, разделяют на 3 уровня:
I - 30% энергии от основного обмена (у людей, ведущих сидячий образ жизни);
II - 60-70% от энергии основного обмена (у людей, которые 2 ч в день имеют умеренную физическую нагрузку);
III - 100% и более от энергии основного обмена (у людей, которые в течение нескольких часов в день занимаются тяжёлой физической работой).
В зависимости от интенсивности нагрузки и возраста суточная потребность в энергии колеблется у женщин от 2000 до 3000 ккал в день, а у мужчин - от 2300 до 4000 ккал.
Количество потребляемой пищи определяется многими факторами, в том числе и химическими регуляторами чувства голода и насыщения. Эти чувства определяются концентрацией в крови глюкозы и гормонов, которые инициируют чувство насыщения: холецистокинина, нейро-тензина, бомбезина, лептина. Причины первичного ожирения:
• генетические нарушения (до 80% случаев ожирения - результат генетических нарушений);
• состав и количество потребляемой пищи, метод питания в семье;
• уровень физической активности;
• психологические факторы. Генетические факторы в развитии ожирения. Метаболические различия между тучными и худыми людьми до настоящего времени не могут быть определены однозначно. Существует несколько теорий, объясняющих эти различия:
• генетически детерминированная разница в функционировании «бесполезных» циклов
(субстратных циклов, раздел 7). Эти циклы состоят из пары метаболитов, превращаемых друг в друга с помощью двух ферментов. Одна из этих реакций идёт с затратой АТФ. Например:
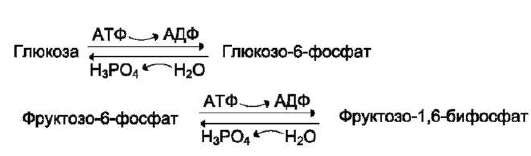
• если эти субстраты превращаются друг в друга с одинаковой скоростью, то происходит «бесполезный» расход АТФ и, соответственно, источников энергии, например жиров;
• у людей, склонных к ожирению, вероятно, имеется более прочное сопряжение дыхания и окислительного фосфорилирования, т.е. более эффективный метаболизм;
• возможно, разное соотношение аэробного и анаэробного гликолиза. Анаэробный гликолиз (как менее эффективный) «сжигает» гораздо больше глюкозы, в результате снижается её переработка в жиры;
• у отдельных индивидуумов имеется различие в активности Na+/К+-АТФ:азы, работа которой требует до 30% энергии, потребляемой клетками.
Роль лептина в регуляции массы жировой ткани
У человека и животных имеется «ген ожирения» - obese gene (ob). Продуктом экспрессии этого гена служит белок лептин, состоящий из 167 аминокислот, который синтезируется и секретируется адипоцитами и взаимодействует с рецепторами гипоталамуса. В результате его действия снижается секреция нейропептида Y. Нейропептид Y стимулирует пищевое поведение, поиск и потребление пищи у животных. Другие пептиды, участвующие в регуляции чувства сытости, например холецистокинин, также влияют на секрецию нейропептида Y. Таким опосредованным путём лептин выступает регулятором жировой массы, необходимой для роста и репродукции. Уровень лептина у больных ожирением может быть различным.
У 80% больных концентрация лептина в крови тучных людей больше в 4 раза, чем у людей с нормальной массой тела. В этих случаях имеется генетический дефект рецепторов лептина в
гипоталамусе, поэтому, несмотря на продукцию лептина, центр голода в гипоталамусе продолжает секрецию нейропептида Y.
20% больных имеют изменения в первичной структуре лептина. К настоящему времени описаны 5 одиночных мутаций в гене лептина, которые приводят к развитию ожирения. У этих больных наблюдают повышение отложения жиров в жировой ткани, чрезмерное потребление пищи, низкую физическую активность и развитие сахарного диабета типа II. Патогенез ожирения при дефекте гена ob может быть следующим: низкий уровень лептина в крови служит сигналом недостаточного количества запаса жиров в организме; этот сигнал включает механизмы, приводящие к увеличению аппетита и в результате к увеличению массы тела.
Следовательно, можно сделать вывод о том, что первичное ожирение - не просто следствие переедания, а результат действия многих факторов, т.е. ожирение - полигенное заболевание.
Вторичное ожирение - ожирение, развивающееся в результате какого-либо основного заболевания, чаще всего эндокринного. Например, к развитию ожирения приводят гипотиреоз, синдром Иценко-Кушинга, гипогонадизм и многие другие заболевания (см. раздел 11).
v. обмен жирных кислот и кетоновых тел
Жирные кислоты поступают с пищей или синтезируются в организме (кроме полиеновых кислот). Субстраты, необходимые для синтеза жирных кислот, образуются при катаболизме глюкозы и таким образом, часть глюкозы превращается сначала в жирные кислоты, а затем в жиры. Хотя специфический путь катаболизма жирных кислот заканчивается образованием ацетил-КоА, служащим исходным субстратом для синтеза жирных кислот, процессы синтеза и окисления жирных кислот необратимы. Они происходят в разных компартментах клеток (биосинтез протекает в цитозоле, а окисление - в митохондриях) и катализируются разными ферментами. Окисление жирных кислот как источников энергии увеличивается в постабсорбтивный период, при голодании и физической работе. В этих состояниях их концентрация в крови увеличивается в результате мобилизации из жировых депо, и они активно
окисляются печенью, мышцами и другими тканями. При голодании часть жирных кислот в печени превращается в другие «топливные» молекулы - кетоновые тела. Они, в отличие от жирных кислот, могут использоваться нервной тканью как источник энергии. При голодании и длительной физической работе кетоновые тела служат источником энергии для мышц и некоторых других тканей.
А. β-ОКИСЛЕНИЕ ЖИРНЫХ КИСЛОТ
β-окисление - специфический путь катаболизма жирных кислот, при котором от карбоксильного конца жирной кислоты последовательно отделяется по 2 атома углерода в виде ацетил-КоА. Метаболический путь - β-окисление - назван так потому, что реакции окисления жирной кислоты происходят у β-углеродного атома. Реакции β-окисления и последующего окисления ацетил-КоА в ЦТК служат одним из основных источников энергии для синтеза АТФ по механизму окислительного фосфорилирования. β-окисление жирных кислот происходит только в аэробных условиях.
Активация жирных кислот
Перед тем, как вступить в различные реакции, жирные кислоты должны быть активированы, т.е. связаны макроэргической связью с кофер-ментом А:
RCOOH + HSKoA + АТФ → RCO ~ КоА + АМФ + PPi.
Реакцию катализирует фермент ацил-КоА синтетаза. Выделившийся в ходе реакции пи-рофосфат гидролизуется ферментом пирофос-фатазой:
Н4Р2О7 + H2O → 2 Н3РО4.
Выделение энергии при гидролизе макроэрги-ческой связи пирофосфата смещает равновесие реакции вправо и обеспечивает полноту протекания реакции активации.
Ацил-КоА синтетазы находятся как в цитозоле, так и в матриксе митохондрий. Эти ферменты отличаются по специфичности к жирным кислотам с различной длиной углеводородной цепи. Жирные кислоты с короткой и средней длиной цепи (от 4 до 12 атомов углерода) могут проникать в матрикс митохондрий путём диффузии. Активация этих жирных кислот происходит
в матриксе митохондрий. Жирные кислоты с длинной цепью, которые преобладают в организме человека (от 12 до 20 атомов углерода), активируются ацил-КоА синтетазами, расположенными на внешней мембране митохондрий.
Транспорт жирных кислот с длинной углеводородной цепью в митохондриях
β-окисление жирных кислот происходит в матриксе митохондрий, поэтому после активации жирные кислоты должны транспортироваться внутрь митохондрий. Жирные кислоты с длинной углеводородной цепью переносятся через плотную внутреннюю мембрану митохондрий с помощью карнитина. Карнитин поступает с пищей или синтезируется из незаменимых аминокислот лизина и метионина. В реакциях синтеза карнитина участвует витамин С (аскорбиновая кислота).
В наружной мембране митохондрий находится фермент карнитинацилтрансфераза I (карни-тинпальмитоилтрансфераза I), катализирующий реакцию с образованием ацилкарнитина.
Образовавшийся ацилкарнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется
с помощью карнитинацилкарнитинтранслоказы на внутреннюю поверхность внутренней мембраны митохондрий, где фермент карнитина-цилтрансфераза II катализирует перенос ацила на внутримитохондриальный KoA (рис. 8-26). Таким образом, ацил-КоА становится доступным для ферментов β-окисления. Свободный карнитин возвращается на цитозольную сторону внутренней мембраны митохондрий той же транслоказой.
На внутренней поверхности внутренней мембраны находится фермент карнитинацил транс-фераза II, катализирующий обратный перенос ацила с карнитина на внутримитохондриальный КоА. После этого ацил-КоА включается в реакции β-окисления.
β-окисление жирных кислот - специфический путь катаболизма жирных кислот, протекающий в матриксе митохондрий только в аэробных условиях и заканчивающийся образованием аце-тил-КоА. Водород из реакций β-окисления поступает в ЦПЭ, а ацетил-КоА окисляется в цит-ратном цикле, также поставляющем водород для ЦПЭ. Поэтому β-окисление жирных кислот - важнейший метаболический путь, обеспечивающий синтез АТФ в дыхательной цепи.
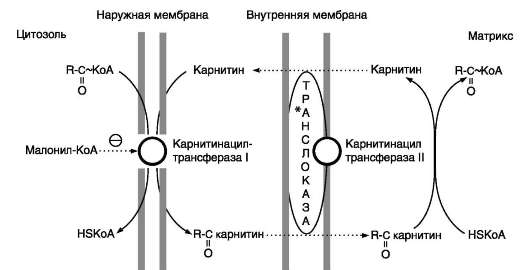
Рис. 8-26. Перенос жирных кислот с длинным углеводородным радикалом через мембраны митохондрий.
Фермент карнитинацилтрансфераза I - регуляторный фермент β-окисления; ингибируется малонил-КоА - промежуточным метаболитом, образующимся при биосинтезе жирных кислот. * - карнитинацилкарнитин-транслоказа.
β-окисление начинается с дегидрирования ацил-КоА FAD-зависимой ацил-КоА дегидро-геназой с образованием двойной связи между α- и β-атомами углерода в продукте реакции - еноил-КоА. Восстановленный в этой реакции кофермент FADH2 передаёт атомы водорода в ЦПЭ на кофермент Q. В результате синтезируются 2 молекулы АТФ (рис. 8-27). В следующей реакции β-окисления по месту двойной связи присоединяется молекула воды таким образом, что ОН-группа находится у β-углеродного атома ацила, образуя β-гидроксиацил-КоА. Затем β-гидроксиацил-КоА окисляется NAD+-зависи-мой дегидрогеназой. Восстановленный NADH,
окисляясь в ЦПЭ, обеспечивает энергией синтез 3 молекул АТФ. Образовавшийся β-кетоацил-КоА подвергается тиолитическому расщеплению ферментом тиолазой, так как по месту разрыва связи С-С через атом серы присоединяется молекула кофермента А. В результате этой последовательности из 4 реакций от ацил-КоА отделяется двухуглеродный остаток - ацетил-КоА. Жирная кислота, укороченная на 2 атома углерода, опять проходит реакции дегидрирования, гидратации, дегидрирования, отщепления ацетил-КоА. Эту последовательность реакций обычно называют «циклом β-окисления», имея в виду, что одни и те же реакции повторяются с
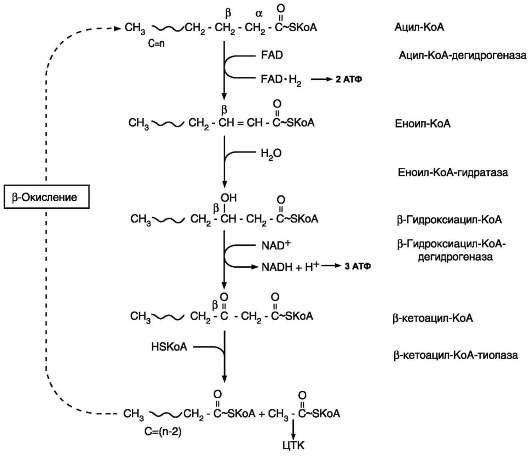
Рис. 8-27. β-окисление жирных кислот.
радикалом жирной кислоты до тех пор, пока вся кислота не превратится в ацетильные остатки.
Продуктами каждого цикла β-окисления являются FADH2, NADH и ацетил-КоА. Хотя реакции в каждом «цикле» одни и те же, остаток кислоты, который входит в каждый последующий цикл, короче на 2 углеродных атома. В последнем цикле окисляется жирная кислота из 4 атомов углерода, поэтому образуются 2 молекулы аце-тил-КоА, а не 1, как в предыдущих. Суммарное уравнение β-окисления, например пальмитоил-КоА может быть представлено таким образом: С15Н31СО-КоА + 7 FAD + 7 NAD+ + 7 HSKoA →
8 CH3-CO-KoA + 7 FADH2 + 7 (NADH + H+).
Если рассчитывать выход АТФ при окислении пальмитиновой кислоты (табл. 8-7), то из общей суммы молекул АТФ необходимо вычесть 2 молекулы, так как на активацию жирной кислоты тратится энергия 2 макроэргических связей (см. реакцию активации жирной кислоты).
Во многих тканях окисление жирных кислот - важный источник энергии. Это ткани с высокой активностью ферментов ЦТК и дыхательной цепи - клетки красных скелетных мышц, сердечная мышца, почки. Эритроциты, в которых отсутствуют митохондрии, не могут окислять жирные кислоты. Жирные кислоты не служат источником энергии для мозга и других нервных тканей, так как жирные кислоты не проходят через гематоэнцефалический барьер, как и другие гидрофобные вещества. В экспериментах показано, что скорость обмена жирных кислот в нервной ткани существенно меньше, чем в других тканях.
Регуляция скорости β-окисления
β-окисление - метаболический путь, прочно связанный с работой ЦПЭ и общего пути катаболизма. Поэтому его скорость регулируется потребностью клетки в энергии, т.е. соотношениями АТФ/AДФ и NADH/NAD+, так же, как и скорость реакций ЦПЭ и общего пути катаболизма (см. раздел 6). Скорость β-окисления в тканях зависит от доступности субстрата, т.е. от количества жирных кислот, поступающих в митохондрии. Концентрация свободных жирных кислот в крови повышается при активации липолиза в жировой ткани при голодании под действием глюкагона и при физической работе под действием адреналина. В этих условиях жирные кислоты становятся преимущественным источником энергии для мышц и печени, так как в результате β-окисления образуются NADH и ацетил-КоА, ингибирующие пируватдегидрогеназный комплекс. Превращение пирувата, образующегося из глюкозы, в ацетил-КоА замедляется. Накапливаются промежуточные метаболиты гликолиза и, в частности, глюкозо-6-фосфат. Глюкозо-6-фосфат ингибирует гексокиназу и, следовательно, препятствует использованию глюкозы в процессе гликолиза. Таким образом, преимущественное использование жирных кислот как основного источника энергии в мышечной ткани и печени сберегает глюкозу для нервной ткани и эритроцитов.
Скорость β-окисления зависит также от активности фермента карнитинацилтрансфе-

разы I. В печени этот фермент ингибируется малонил-КоА, веществом, образующимся при биосинтезе жирных кислот. В абсорбтивный период в печени активируется гликолиз и увеличивается образование ацетил-КоА из пирувата. Первая реакция синтеза жирных кислот - превращение ацетил-КоА в малонил-КоА. Малонил-КоА ингибирует β-окисление жирных кислот, которые могут использоваться для синтеза жира.
Окисление ненасыщенных жирных кислот
Около половины жирных кислот в организме человека ненасыщенные. β-окисление этих кислот идёт обычным путём до тех пор, пока двойная связь не окажется между третьим и четвёртым атомами углерода (рис. 8-28). Затем фермент еноил-КоА изомераза перемещает двойную связь из положения 3-4 в положение 2-3 и изменяет цис-конформацию двойной связи на транс-, которая требуется для β-окисления. В этом цикле β-окисления первая реакция дегидрирования не происходит, так как двойная
связь в радикале жирной кислоты уже имеется. Далее циклы β-окисления продолжаются, не отличаясь от обычного пути.
α-окисление жирных кислот. В липидах мозга и других отделах нервной ткани преобладают жирные кислоты с очень длинной цепью - более 20 углеродных атомов. Они окисляются по типу α-окисления, при котором от жирной кислоты отщепляется по одному атому углерода, выделяющемуся в виде СО2 (рис. 8-29).
Этот путь катаболизма жирных кислот не связан с синтезом АТФ. α-окислению подвергаются также жирные кислоты с разветвлённой углеводородной цепью, например фитановая, поступающая в организм с растительной пищей (рис. 8-30). Фитановая кислота образуется из фитола, который входит в состав хлорофилла. В этой кислоте у каждого третьего атома углерода находится метильная группа, что делает невозможным β-окисление данной кислоты. При α-окислении фитановой кислоты вначале удаляется метильная группа, а затем происходит цикл β-окисления.
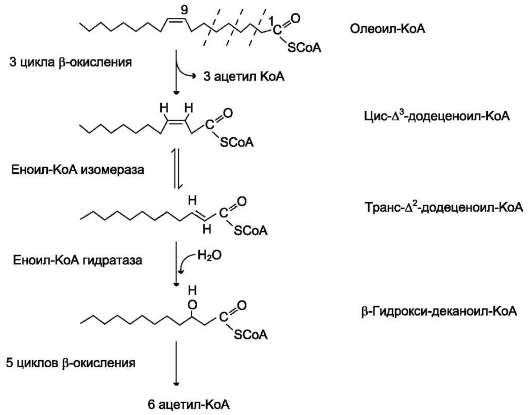
Рис. 8-28. Окисление жирных кислот с одной двойной связью.
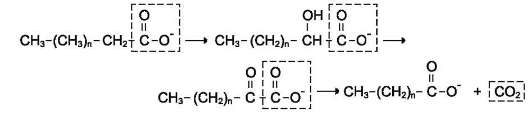
Рис. 8-29. α-Окисление жирных кислот.
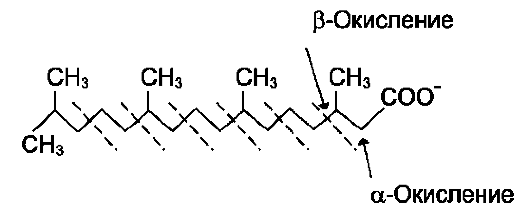
Рис. 8-30. Окисление фитановой кислоты.
Б. НАРУШЕНИЯ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Нарушение переноса жирных кислот в митохондрии. Скорость переноса жирных кислот внутрь митохондрий, а следовательно и скорость процесса β-окисления, зависит от доступности карнитина и скорости работы фермента кар-нитинацилтрансферазы I. β-Окисление могут нарушать следующие факторы:
• длительный гемодиализ, в ходе которого организм теряет карнитин;
• длительная ацидурия, при которой карнитин выводится как основание с органическими кислотами;
• лечение больных сахарным диабетом препаратами сульфонилмочевины, ингибирую-щими карнитинацилтрансферазу I;
• низкая активность ферментов, синтезирующих карнитин;
• наследственные дефекты карнитинацил-трансферазы I.
У людей с наследственными дефектами карнитинацилтрансферазы I или ферментов синтеза карнитина в скелетных мышцах снижается скорость поступления жирных кислот в матрикс митохондрий и, соответственно, скорость β-окисления. В этих случаях жирные кислоты с длинной цепью не используются как источники энергии. У таких людей снижена способность к физической активности; в мышечных клетках могут накапливаться жиры, образуя вакуоли.
Генетический дефект дегидрогеназы жирных кислот со средней длиной углеводородной цепи
В митохондриях имеется несколько ацил-КоА-дегидрогеназ, окисляющих жирные кислоты с длинной, средней или короткой цепью радикала. Жирные кислоты по мере укорочения радикала в процессе β-окисления могут последовательно окисляться этими ферментами. Генетический дефект дегидрогеназы жирных кислот со средней длиной радикала наиболее распространён по сравнению с другими наследственными заболеваниями - 1:15 000. Частота дефектного гена среди европейской популяции - 1:40. Это аутосомно-рецессивное заболевание, возникающее в результате замены Т на А в 985-й позиции гена. Активность этой дегидрогеназы особенно важна для грудных детей, у которых жиры молока служат основным источником энергии, а в триацилгли-церолах молока преобладают жирные кислоты со средней длиной цепи. Невозможность использовать жирные кислоты как источники энергии приводит к увеличению скорости окисления глюкозы. В результате у детей развивается гипогликемия - причина внезапной детской смертности (10% от общего числа умерших новорождённых). Если такие дети выживают, то после голодания в течение 6-8 ч у них развиваются гипогликемические приступы (слабость, головокружение, рвота, потеря сознания). Введение глюкозы приводит к исчезновению симптомов.
Во всех случаях, когда нарушается β-окисление, жирные кислоты накапливаются в клетках и распадаются по пути ω-окисления, которое в норме идёт с очень низкой скоростью. Окисление происходит по метильному ω-атому углерода (рис. 8-31), и в результате образуются дикарбоновые кислоты, выделяющиеся с мочой. Определение этих кислот в моче может служить диагностическим признаком нарушения β-окисления.
Нарушение окисления фитановой кислоты. При редком наследственном заболевании - болезни
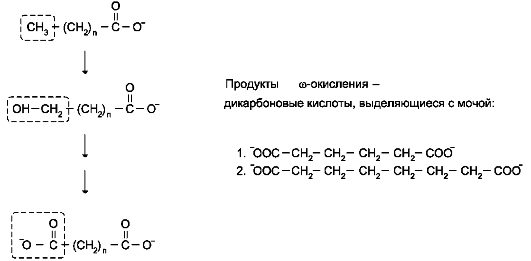
Рис. 8-31. ω-Окисление жирных кислот. ω-Окисление жирных кислот активируется в тех случаях, когда активность β-окисления жирных кислот снижена. 1 - адипиновая кислота; 2 - субериновая кислота.
Рефсума, развивающейся вследствие генетического дефекта одного из ферментов, участвующих в α-окислении, фитановая кислота, поступающая с пищей, не окисляется и накапливается в организме, в основном в нервной ткани. Это приводит к нарушению структуры нервной ткани и развитию многих неврологических симптомов.
В. ОБМЕН КЕТОНОВЫХ ТЕЛ
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислоты используются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновым телам относят β-гид-роксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления (рис. 8-32). Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.
Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА (рис. 8-33). С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА-синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА.
Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело - β-гидроксибутират путём восстановления.
В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови - именно β-гид-
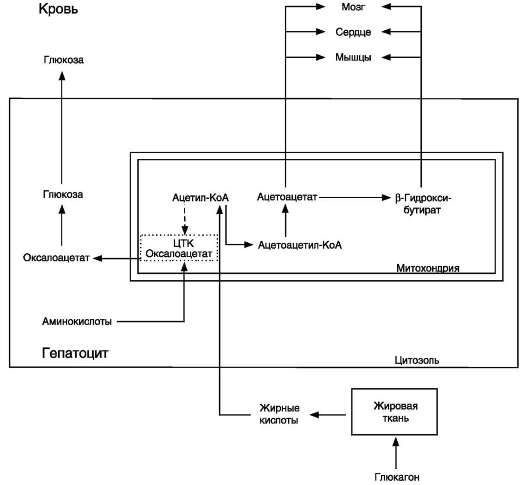
Рис. 8-32. Активация синтеза кетоновых тел при голодании. Точечные линии - скорость метаболических путей снижена; сплошные линии - скорость метаболических путей повышена. При голодании в результате действия глюкагона активируются липолиз в жировой ткани и β-окисление в печени. Количество оксалоацетата в митохондриях уменьшается, так как он, восстановившись до малата, выходит в цитозоль, где опять превращается в оксалоацетат и используется в глюконеогенезе. В результате скорость реакций ЦТК снижается и, соответственно, замедляется окисление ацетил-КоА. Концентрация ацетил-КоА в митохондриях увеличивается, и активируется синтез кетоновых тел. Синтез кетоновых тел увеличивается также при сахарном диабете (см. раздел 11).
роксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами.
При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется
тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз.
Регуляция синтеза кетоновых тел. Регуляторный фермент синтеза кетоновых тел - ГМГ-КоА синтаза.
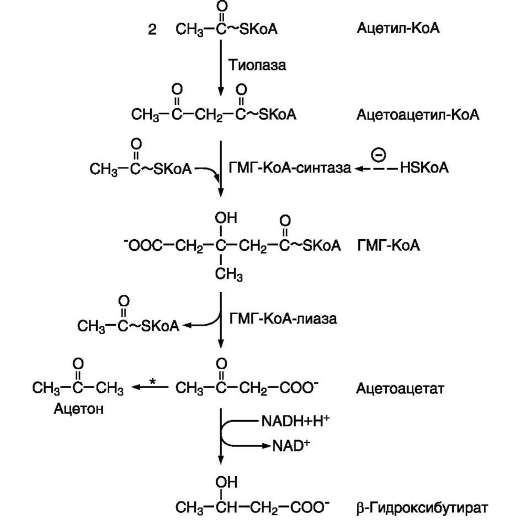
Рис. 8-33. Синтез кетоновых тел в митохондриях гепатоцитов. Регуляторный фермент синтеза кетоновых тел (ГМГ-КоАсинтаза) ингибируется свободным КоА. * - реакция идёт неферментативно при высокой концентрации кетоновых тел в крови.
• ГМГ-КоА-синтаза - индуцируемый фермент; его синтез увеличивается при повышении концентрации жирных кислот в крови. Концентрация жирных кислот в крови увеличивается при мобилизации жиров из жировой ткани под действием глюкагона, адреналина, т.е. при голодании или физической работе.
• ГМГ-КоА-синтаза ингибируется высокими концентрациями свободного кофермента А.
• Когда поступление жирных кислот в клетки печени увеличивается, КоА связывается с ними, концентрация свободного КоА снижается, и фермент становится активным.
• Если поступление жирных кислот в клетки печени уменьшается, то, соответственно, увеличивается концентрация свободного КоА,
ингибирующего фермент. Следовательно, скорость синтеза кетоновых тел в печени зависит от поступления жирных кислот.
Окисление кетоновых тел в периферических тканях
При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе.
β-Гидроксибутират (рис. 8-34), попадая в клетки, дегидрируется NAD-зависимой де-
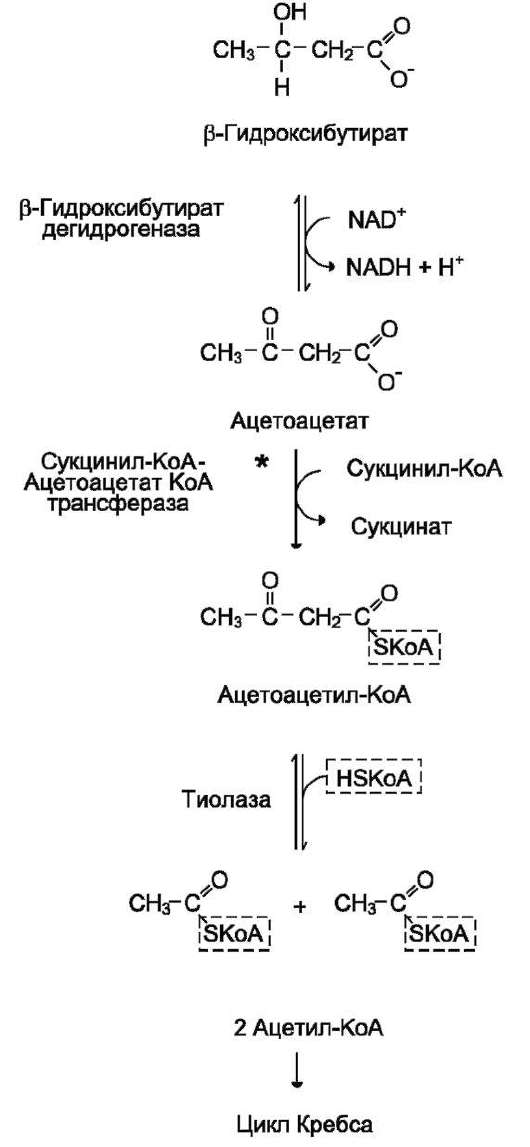
Рис. 8-34. Окисление кетоновых тел в тканях. * - реакция активации ацетоацетата.
гидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сукцинил-КоА - донором КоА: Ацетоацетат + Сукцинил-КоА → Ацетоацетил-КоА + Сукцинат.
Реакцию катализирует сукцинил-КоА-аце-тоацетат-КоА-трансфераза. Этот фермент не синтезируется в печени, поэтому печень не ис-
пользует кетоновые тела как источники энергии, а производит их «на экспорт». Кетоновые тела - хорошие топливные молекулы; окисление одной молекулы β-гидроксибутирата до СО2 и Н2О обеспечивает синтез 27 молекул АТФ. Эквивалент одной макроэргической связи АТФ (в молекуле сукцинил-КоА) используется на активацию ацетоацетата, поэтому суммарный выход АТФ при окислении одной молекулы β-гидроксибутирата - 26 молекул.
Кетоацидоз. В норме концентрация кетоновых тел в крови составляет 1-3 мг/дл (до 0,2 мМ/л), но при голодании значительно увеличивается. Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой - кетону-рией. Накопление кетоновых тел в организме приводит к кетоацидозу: уменьшению щелочного резерва (компенсированному ацидозу), а в тяжёлых случаях - к сдвигу рН (некомпенсированному ацидозу), так как кетоновые тела (кроме ацетона) являются водорастворимыми органическими кислотами (рК~3,5), способными к диссоциации:
СН3-СО-СН2-СООН ↔ СН3-СО-СН2-СОО- + Н+.
Ацидоз достигает опасных величин при сахарном диабете, так как концентрация кетоновых тел при этом заболевании может доходить до 400-500 мг/дл. Тяжёлая форма ацидоза - одна из основных причин смерти при сахарном диабете. Накопление протонов в крови нарушает связывание кислорода гемоглобином, влияет на ионизацию функциональных групп белков, нарушая их конформацию и функцию.
г. биосинтез жирных кислот
С пищей в организм поступают разнообразные жирные кислоты, в том числе и незаменимые. Значительная часть заменимых жирных кислот синтезируется в печени, в меньшей степени - в жировой ткани и лактирующей молочной железе. Источником углерода для синтеза жирных кислот служит ацетил-КоА, образующийся при распаде глюкозы в абсорбтивном периоде. Таким образом, избыток углеводов, поступающих в организм, трансформируется в жирные кислоты, а затем в жиры.
1. Синтез пальмитиновой кислоты
Образование ацетил-КоА и его транспорт в цитозоль
Синтез жирных кислот происходит в абсорб-тивный период. Активный гликолиз и последующее окислительное декарбоксилирование пи-рувата способствуют увеличению концентрации ацетил-КоА в матриксе митохондрий. Так как синтез жирных кислот происходит в цитозоле клеток, то ацетил-КоА должен быть транспортирован через внутреннюю мембрану митохондрий в цитозоль. Однако внутренняя мембрана митохондрий непроницаема для ацетил-КоА, поэтому в матриксе митохондрий ацетил-КоА конденсируется с оксалоацетатом с образованием цитрата при участии цитратсинтазы: Ацетил-КоА + Оксалоацетат → Цитрат + HS-KoA.
Затем транслоказа переносит цитрат в цитоплазму (рис. 8-35).
Перенос цитрата в цитоплазму происходит только при увеличении количества цитрата в митохондриях, когда изоцитратдегидрогеназа и α-ке-тоглутаратдегидрогеназа ингибированы высокими концентрациями NADH и АТФ. Эта ситуация создаётся в абсорбтивном периоде, когда клетка печени получает достаточное количество источников энергии. В цитоплазме цитрат расщепляется под действием фермента цитратлиазы: Цитрат + HSKoA + АТФ → Ацетил-КоА + АДФ
+ Pi + Оксалоацетат.
Ацетил-КоА в цитоплазме служит исходным субстратом для синтеза жирных кислот, а ок-салоацетат в цитозоле подвергается следующим превращениям (см. схему ниже).
Пируват транспортируется обратно в матрикс митохондрий. Восстановленный в результате действия малик-фермента NADPH используется как донор водорода для последующих реакций синтеза жирных кислот. Другой источник NADPH - окислительные стадии пентозофос-фатного пути катаболизма глюкозы.
Образование малонил-КоА из ацетил-КоА - регуляторная реакция в биосинтезе жирных кислот.
Первая реакция синтеза жирных кислот - превращение ацетил-КоА в малонил-КоА. Фермент, катализирующий эту реакцию (ацетил-КоА-кар-боксилаза), относят к классу лигаз. Он содержит ковалентно связанный биотин (рис. 8-36). В первой стадии реакции СО2 ковалентно связывается с биотином за счёт энергии АТФ, во второй стадии СОО- переносится на ацетил-КоА с образованием малонил-КоА. Активность фермента ацетил-КоА-карбоксилазы определяет скорость всех последующих реакций синтеза жирных кислот.
Реакции, катализируемые синтазой жирных кислот, - ферментным комплексом, катализирующим реакции синтеза пальмитиновой кислоты, описываются ниже.
После образования малонил-КоА синтез жирных кислот продолжается на мультифер-ментном комплексе - синтазе жирных кислот (пальмитоилсинтетазе). Этот фермент состоит из 2 идентичных протомеров, каждый из которых имеет доменное строение и, соответственно, 7 центров, обладающих разными каталитическими активностями (рис. 8-37). Этот комплекс последовательно удлиняет радикал жирной кислоты на 2 углеродных атома, донором которых служит малонил-КоА. Конечный продукт работы этого комплекса - пальмитиновая кислота, поэтому прежнее название этого фермента - пальмито-илсинтетаза.
Первая реакция - перенос ацетильной группы ацетил-КоА на тиоловую группу цистеина аце-тилтрансацилазным центром (рис. 8-38). Затем от малонил-КоА остаток малонила переносится на сульфгидрильную группу ацилпереносящего белка малонилтрансацилазным центром. После этого комплекс готов к первому циклу синтеза.
Ацетильная группа конденсируется с остатком малонила по месту отделившегося СО2. Реакция катализируется кетоацилсинтазным центром.
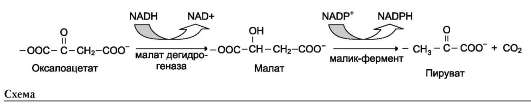
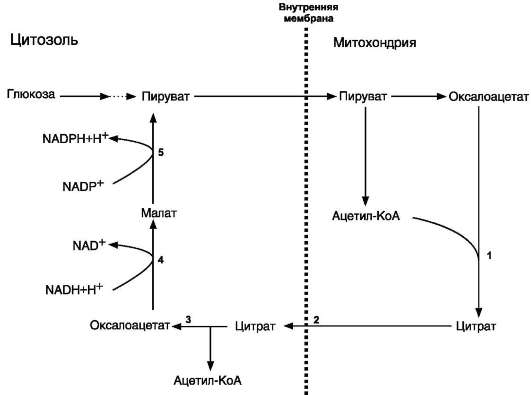
Рис. 8-35. Перенос ацетильных остатков из митохондрий в цитозоль. Действующие ферменты: 1 - цитрат-синтаза; 2 - транслоказа; 3 - цитратлиаза; 4 - малатдегидрогеназа; 5 - малик-фермент.
Рис. 8-36. Роль биотина в реакции карбоксилирования ацетил-КоА.
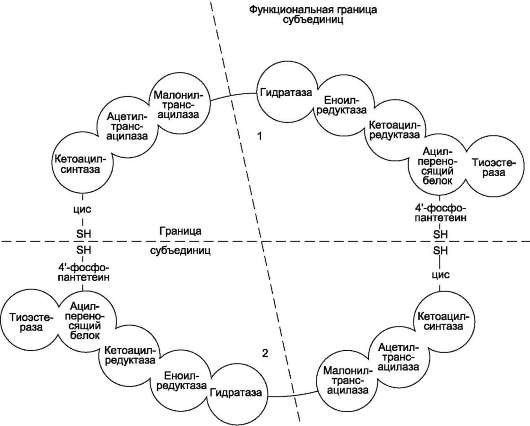
Рис. 8-37. Строение мультиферментного комплекса - синтазы жирных кислот. Комплекс - димер из двух идентичных полипептидных цепей, каждый из которых имеет 7 активных центров и ацилпереносящий белок (АПБ). SH-группы протомеров принадлежат различным радикалам. Одна SH-группа принадлежит цистеину, другая - остатку фосфопантетеиновой кислоты. SH-группа цистеина одного мономера расположена рядом с SH-группой 4-фосфопантетеината другого протомера. Таким образом, протомеры фермента расположены «голова к хвосту». Хотя каждый мономер содержит все каталитические центры, функционально активен комплекс из 2 протомеров. Поэтому реально синтезируются одновременно 2 жирных кислоты. Для упрощения в схемах обычно изображают последовательность реакций при синтезе одной молекулы кислоты.
Образовавшийся радикал ацетоацетила последовательно восстанавливается кетоацилредуктазой, затем дегидратируется и опять восстанавливается еноилредуктазой - активными центрами комплекса. В результате первого цикла реакций образуется радикал бутирила, связанный с субъединицей синтазы жирных кислот.
Перед вторым циклом радикал бутирила переносится из позиции 2 в позицию 1 (где находился ацетил в начале первого цикла реакций). Затем остаток бутирила подвергается тем же превращениям и удлиняется на 2 углеродных атома, происходящих из малонил-КоА.
Аналогичные циклы реакций повторяются до тех пор, пока не образуется радикал пальмитиновой кислоты, который под действием тиоэстеразного центра гидролитически отделяется от ферментного комплекса, превращаясь в свободную пальмитиновую кислоту (пальмитат, рис. 8-38, 8-39).
Суммарное уравнение синтеза пальмитиновой кислоты из ацетил-КоА и малонил-КоА имеет следующий вид:
CH3-CO-SKoA + 7 HOOC-CH2-CO-SKoA + 14 (NADPH + H+) → C15H31COOH + 7 CO2 + 6 H2O + 8 HSKoA + 14 NADP+.
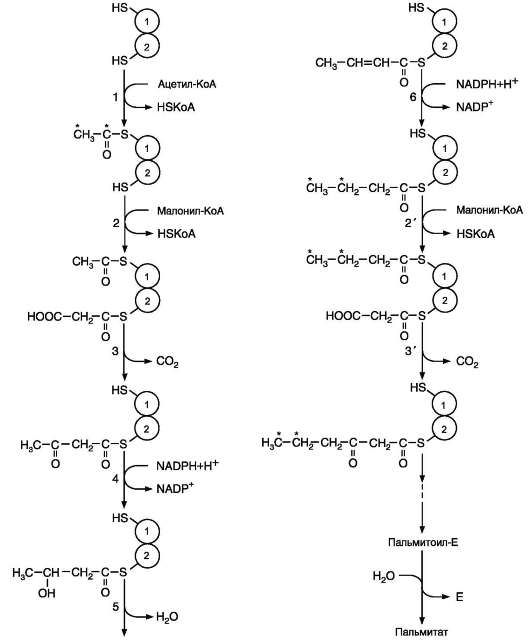
Рис. 8-38. Синтез пальмитиновой кислоты. Синтаза жирных кислот: в первом протомере SH-группа принадлежит цистеину, во втором - фосфопантетеину. После окончания первого цикла радикал бутирила переносится на SH-группу первого протомера. Затем повторяется та же последовательность реакций, что и в первом цикле. Пальмитоил-Е - остаток пальмитиновой кислоты, связанный с синтазой жирных кислот. В синтезированной жирной кислоте только 2 дистальных атома углерода, обозначенные *, происходят из ацетил-КоА, остальные - из малонил-КоА.
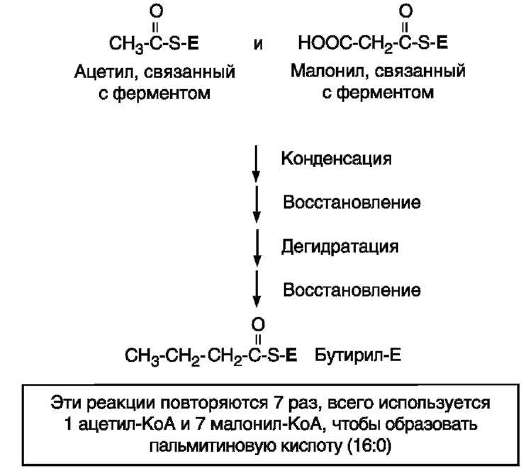
Рис. 8-39. Общая схема реакций синтеза пальмитиновой кислоты.
Основные источники водорода для синтеза жирных кислот
В каждом цикле биосинтеза пальмитиновой кислоты проходят 2 реакции восстановления, донором водорода в которых служит кофермент NADPH. Восстановление NADP+ происходит в реакциях:
• дегидрирования в окислительных стадиях пентозофосфатного пути катаболизма глюкозы;
• дегидрирования малата малик-ферментом;
• дегидрирования изоцитрата цитозольной NADP-зависимой дегидрогеназой.
2. Регуляция синтеза жирных кислот
Регуляторный фермент синтеза жирных кислот - ацетил-КоА-карбоксилаза. Этот фермент регулируется несколькими способами.
Ассоциация/диссоциация комплексов субъединиц фермента. В неактивной форме ацетил-КоА-карбоксилаза представляет собой отдельные комплексы, каждый из которых состоит из 4 субъединиц. Активатор фермента - цитрат; он стимулирует объединение комплексов, в результате чего активность фермента увеличивается. Ингибитор - пальмитоил-КоА; он
вызывает диссоциацию комплекса и снижение активности фермента (рис. 8-40). Фосфорилирование/дефосфорилирование аце-тил-КоА-карбоксилазы. В постабсорбтивном состоянии или при физической работе глюкагон или адреналин через аденилат-циклазную систему активируют протеинки-назу А и стимулируют фосфорилирование субъединиц ацетил-КоА карбоксилазы. Фосфорилированный фермент неактивен, и синтез жирных кислот останавливается. В абсорбтивный период инсулин активирует фосфатазу, и ацетил-КоА карбоксилаза переходит в дефосфорилированное состояние (рис. 8-41). Затем под действием цитрата происходит полимеризация протомеров фермента, и он становится активным. Кроме активации фермента, цитрат выполняет и другую функцию в синтезе жирных кислот. В абсорбтивный период в митохондриях клеток печени накапливается цитрат, в составе которого остаток ацетила транспортируется в цитозоль.
Индукция синтеза ферментов. Длительное потребление богатой углеводами и бедной жирами пищи приводит к увеличению секреции инсулина, который стимулирует
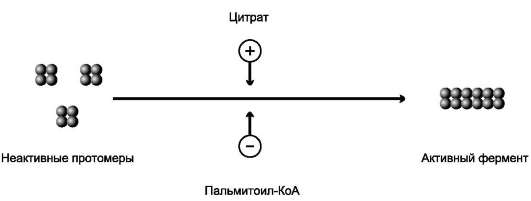
Рис. 8-40. Ассоциация/диссоциация комплексов ацетил-КоА-карбоксилазы.
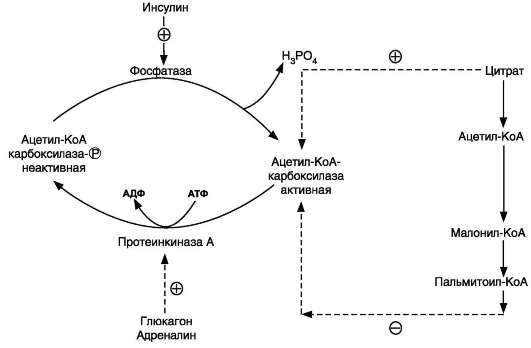
Рис. 8-41. Регуляция ацетил-КоА-карбоксилазы.
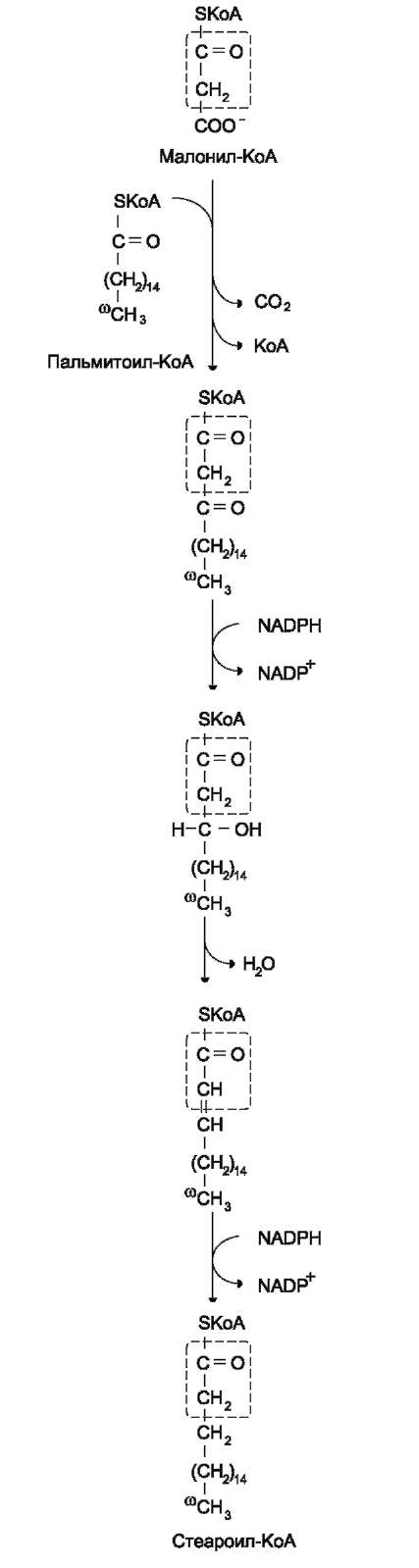
Рис. 8-42. Удлинение пальмитиновой кислоты в ЭР.
Радикал пальмитиновой кислоты удлиняется на 2 углеродных атома, донором которых служит малонил-КоА.
индукцию синтеза ферментов: ацетил-КоА-карбоксилазы, синтазы жирных кислот, цит-ратлиазы, глюкозо-6-фосфатдегидрогеназы. Следовательно, избыточное потребление углеводов приводит к ускорению превращения продуктов катаболизма глюкозы в жиры. Голодание или богатая жирами пища приводит к снижению синтеза ферментов и, соответственно, жиров.
3. Синтез жирных кислот из пальмитиновой кислоты
Удлинение жирных кислот. В ЭР происходит удлинение пальмитиновой кислоты с участием малонил-КоА. Последовательность реакций сходна с той, что происходит при синтезе пальмитиновой кислоты, однако в данном случае жирные кислоты связаны не с син-тазой жирных кислот, а с КоА. Ферменты, участвующие в элонгации, могут использовать в качестве субстратов не только пальмитиновую, но и другие жирные кислоты (рис. 8-42), поэтому в организме могут синтезироваться не только стеариновая кислота, но и жирные кислоты с большим числом атомов углерода.
Основной продукт элонгации в печени - стеариновая кислота (С18:0), однако в ткани мозга образуется большое количество жирных кислот с более длинной цепью - от С20 до С24, которые необходимы для образования сфинголипидов и гликолипидов.
В нервной ткани происходит синтез и других жирных кислот - α-гидроксикислот. Окси-дазы со смешанными функциями гидрок-силируют С22 и С24 кислоты с образованием лигноцериновой и цереброновой кислот, обнаруживаемых только в липидах мозга.
Образование двойных связей в радикалах жирных кислот. Включение двойных связей в радикалы жирных кислот называется деса-турацией. Основные жирные кислоты, образующиеся в организме человека в результате десатурации (рис. 8-43), - пальмитоолеино-вая (С16:19) и олеиновая (С18:1Δ9).
Образование двойных связей в радикалах жирных кислот происходит в ЭР в реакциях с участием молекулярного кислорода, NADН и цитохрома b5. Ферменты десатура-зы жирных кислот, имеющиеся в организме человека, не могут образовывать двойные
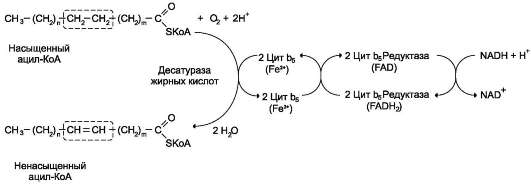
Рис. 8-43. Образование ненасыщенных жирных кислот.
связи в радикалах жирных кислот дисталь-нее девятого атома углерода, т.е. между девятым и метильным атомами углерода. Поэтому жирные кислоты семейства ω-3 и ω-6 не синтезируются в организме, являются незаменимыми и обязательно должны поступать с пищей, так как выполняют важные регуляторные функции. Для образования двойной связи в радикале жирной кислоты требуется молекулярный кислород, NADH, цитохром b5 и FAD-зави-симая редуктаза цитохрома b5. Атомы водорода, отщепляемые от насыщенной кислоты, выделяются в виде воды. Один атом молекулярного кислорода включается в молекулу воды, а другой также восстанавливается до воды с участием электронов NADH, которые передаются через FADH2 и цитохром b5.
vi. эйкозаноиды
Эйкозаноиды - биологически активные вещества, синтезируемые большинством клеток из полиеновых жирных кислот, содержащих 20 углеродных атомов (слово «эйкоза» по гречески означает 20).
Эйкозаноиды, включающие в себя простаглан-дины, тромбоксаны, лейкотриены и ряд других веществ, - высокоактивные регуляторы клеточных функций. Они имеют очень короткий T1/2, поэтому оказывают эффекты как «гормоны местного действия», влияя на метаболизм продуцирующей их клетки по аутокринному механизму, и на окружающие клетки - по паракринному
механизму. Эйкозаноиды участвуют во многих процессах: регулируют тонус ГМК и вследствие этого влияют на АД, состояние бронхов, кишечника, матки. Эйкозаноиды регулируют секрецию воды и натрия почками, влияют на образование тромбов. Разные типы эйкозаноидов участвуют в развитии воспалительного процесса, происходящего после повреждения тканей или инфекции. Такие признаки воспаления, как боль, отёк, лихорадка, в значительной мере обусловлены действием эйкозаноидов. Избыточная секреция эйкозаноидов приводит к ряду заболеваний, например бронхиальной астме и аллергическим реакциям.
А. СУБСТРАТЫ ДЛЯ СИНТЕЗА ЭЙКОЗАНОИДОВ
Главный субстрат для синтеза эйкозаноидов у человека - арахидоновая кислота (20:4, ω-6), так как её содержание в организме человека значительно больше остальных полиеновых кислот-предшественников эйкозаноидов (см. выше табл. 8-1).
В меньшем количестве для синтеза эйкозанои-дов используются эйкозапентаеновая (20:5, ω-3) и эйкозатриеновая (20:3, ω-6) жирные кислоты.
Полиеновые кислоты с 20 атомами углерода поступают в организм человека с пищей или образуются из незаменимых (эссенциальных) жирных кислот с 18 атомами углерода, также поступающими с пищей (рис. 8-44).
Полиеновые жирные кислоты, которые могут служить субстратами для синтеза эйкозаноидов, входят в состав глицерофосфолипидов мембран.
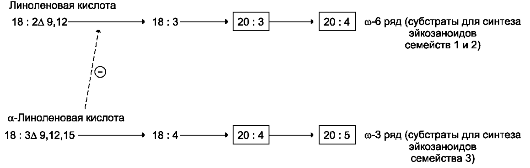
Рис. 8-44. Синтез полиеновых жирных кислот с 20 углеродными атомами в организме человека.
Под действием ассоциированной с мембраной фосфолипазы А2 жирная кислота отщепляется от глицерофосфолипида и используется для синтеза эйкозаноидов.
Б. СТРУКТУРА, НОМЕНКЛАТУРА И БИОСИНТЕЗ ПРОСТАГЛАНДИНОВ И ТРОМБОКСАНОВ
Хотя субстраты для синтеза эйкозаноидов имеют довольно простую структуру (полиеновые жирные кислоты), из них образуется большая и разнообразная группа веществ. Наиболее распространены в организме человека проста-гландины, которые впервые были выделены из предстательной железы, откуда и получили свое название. Позже было показано, что и другие ткани организма синтезируют простагландины и другие эйкозаноиды.
1. Структура и номенклатура простагландинов и тромбоксанов
Простагландины (рис. 8-45) обозначают символами, например PG A, где PG обозначает слово «простагландин», а буква А обозначает заместитель в пятичленном кольце в молекуле эйкозаноида.
Каждая из указанных групп простагландинов состоит из 3 типов молекул, отличающихся по числу двойных связей в боковых цепях. Число двойных связей обозначают нижним цифровым индексом, например, PG E2.
Число двойных связей в боковых цепях про-стагландинов зависит от структуры предшественника - полиеновой кислоты, из которой образовались простагландины. Две двойные
связи полиеновой кислоты используются при образовании кольца в молекуле простагланди-на, а количество оставшихся двойных связей в радикалах, связанных с кольцом, определяет серию простагландина: 1 - если одна двойная связь, 2 - если две двойные связи и 3 - если в радикалах имеются три двойных связи.
PG I - простациклины. Имеют 2 кольца в своей структуре: одно пятичленное, как и другие простагландины, а другое - с участием атома кислорода. Их также подразделяют в зависимости от количества двойных связей в радикалах
(PG I2, PG I3).
Тромбоксаны. В отличие от простагландинов, тромбоксаны синтезируются только в тромбоцитах, откуда и происходит их название, и стимулируют их агрегацию при образовании тромба.
Тромбоксаны имеют шестичленное кольцо, включающее атом кислорода (рис. 8-46). Так же, как и другие эйкозаноиды, тромбоксаны могут содержать различное число двойных связей в боковых цепях, образуя ТХ А2 или ТХ А3, отличающиеся по активности. ТХ В2 - продукт катаболизма ТХ А2 и активностью не обладает.
2. Циклооксигеназный путь: синтез простаглан-динов и тромбоксанов
Активация фосфолипаз. Синтез простагланди-нов начинается только после отделения поли-еновых кислот от фосфолипида мембраны под действием ферментов (рис. 8-47). Активация фосфолипаз, ассоциированных с мембранами, происходит под действием многих факторов: гормонов, гистамина, цитокинов, механического воздействия.
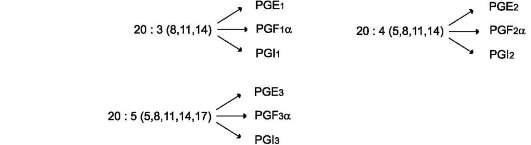
Рис. 8-45. Семейства простагландинов.
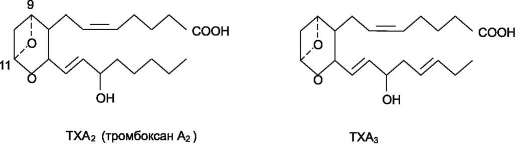
Рис. 8-46. Структура тромбоксанов. ТХ А2 синтезируется из арахидоновой кислоты; ТХ А3 синтезируется из эйкозапентаеновой кислоты.
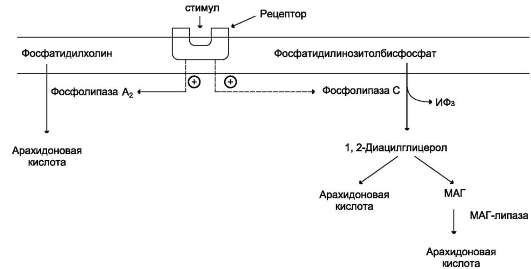
Рис. 8-47. Отделение арахидоновой кислоты от глицерофосфолипидов. МАГ - моноацилглицерол; ИФ3 - инозитолтри-фосфат.
Связывание стимулирующего агента с рецептором может активировать или фосфолипазу А2 или фосфолипазу С. Это зависит от типа клетки и типа рецепторов.
После отделения арахидоновой кислоты от фосфолипида она выходит в цитозоль и в различных типах клеток превращается в разные эйкозаноиды. В клетках имеется 2 основных пути превращения арахидоновой кислоты: циклооксигеназный, приводящий к синтезу простагландинов, простациклинов и тромбо-ксанов, и липоксигеназный, заканчивающийся образованием лейкотриенов или других эйкоза-ноидов (рис. 8-48).
Синтез простагландинов. Фермент, катализирующий первый этап синтеза простагландинов, называется PG Н2 синтазой и имеет 2 каталитических центра. Один из них называют цик-лооксигеназой, другой - пероксидазой. Этот фермент представляет собой димер гликопроте-инов, состоящий из идентичных полипептидных цепей. Фермент имеет гидрофобный домен, погружённый в липидный слой мембран ЭР, и каталитический домен, обращённый в полость ЭР. В активном центре циклооксигеназы находится тирозин (385), в активном центре пероксидазы - простетическая группа - гем. В организме имеются 2 типа циклооксигеназ (PG Н2 синтаз). Циклооксигеназа 1 - конститутивный фермент, синтезирующийся с постоянной скоростью. Синтез циклооксигеназы 2 увеличивается при воспалении и индуцируется соответствующими медиаторами - цитокинами.
Оба типа циклооксигеназ катализируют включение 4 атомов кислорода в арахидоновую кислоту и формирование пятичленного кольца. В результате образуется нестабильное гид-ропероксидпроизводное, называемое PG G2. Гидропероксид у 15-го атома углерода быстро восстанавливается до гидроксильной группы пероксидазой с образованием PG H2. До образования PG H2 путь синтеза разных типов проста-гландинов одинаков. Дальнейшие превращения PG H2 специфичны для каждого типа клеток.
Например, PG H2 в клетках ГМК может быть восстановлен под действием PG E синтазы с образованием PG E2 или под действием PG D синтазы с образованием PG D2. В тромбоцитах содержится фермент тромбоксансинтаза, превращающий тот же исходный PG H2 в ТХ А2, обладающий сильным сосудосуживающим
действием. В клетках эндотелия под действием фермента простациклинсинтазы из PG H2 синтезируется PG I2 (простациклин), имеющий сосудорасширяющее действие.
В. СТРУКТУРА И СИНТЕЗ ЛЕЙКОТРИЕНОВ, ГЭТЕ, ЛИПОКСИНОВ
Лейкотриены также образуются из эйкозано-евых кислот, однако в их структуре отсутствуют циклы, как у простагландинов, и они имеют 3 сопряжённые двойные связи, хотя общее число двойных связей в молекуле больше (рис. 8-49). Лейкотриены С4, D4 и Е4 имеют заместители в виде трипептида глутатиона, дипептида глицил-цистеина или цистеина, соответственно.
Липоксигеназный путь синтеза, приводящий к образованию большого количества разных эйкозаноидов, начинается с присоединения молекулы кислорода к одному из атомов углерода у двойной связи, с образованием гидропе-рокси-дов - гидропероксидэйкозатетраеноатов (ГПЭТЕ). Далее гидропероксиды превращаются в соответствующие гидроксиэйкозатетроеноаты
(ГЭТЕ).
Структура и синтез лейкотриенов и ГЭТЕ
Синтез лейкотриенов идёт по пути, отличному от пути синтеза простагландинов, и начинается с образования гидроксипероксидов - гидро-пероксидэйкозатетраеноатов (ГПЭТЕ). Эти вещества или восстанавливаются с образованием гидроксиэйкозатетроеноатов (ГЭТЕ) или превращаются в лейкотриены или липоксины. ГЭТЕ отличаются по положению гидроксильной группы у 5-го, 12-го или 15-го атома углерода, например: 5-ГЭТЕ, 12-ГЭТЕ.
Липоксигеназы действуют в 5-й, 12-й или 15-й позиции арахидоновой кислоты в зависимости от типа ткани. Например, в ПЯЛ содержится в основном 5-липоксигеназа, в тромбоцитах - 12-липок-сигеназа, в эозинофилах - 15-липоксигеназа.
В лейкоцитах и тучных клетках 5-ГПЭТЕ превращается в эпоксид-лейкотриен А4 (LT A4), где нижний индекс 4 обозначает общее количество двойных связей. Наличие 3 сопряжённых двойных связей обусловливает название «лей-котриен».
Другие типы лейкотриенов образуются из LТ А4. LТ В4 образуется под действием эпоксид-гидролазы в лейкоцитах и клетках эпителия
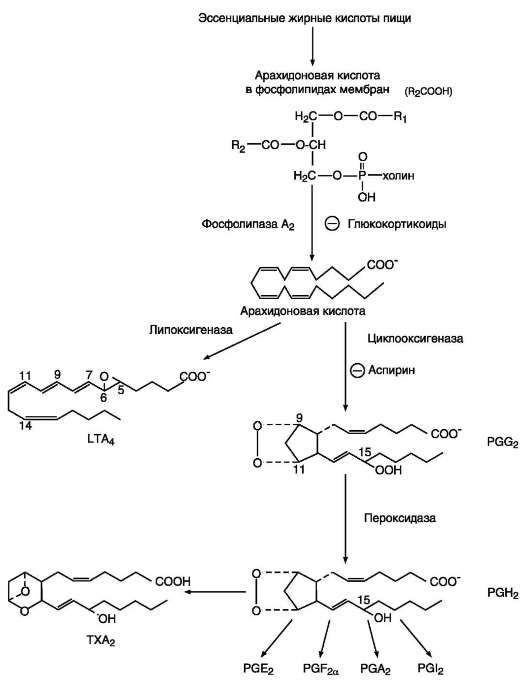
Рис. 8-48. Синтез эйкозаноидов из арахидоновой кислоты. Глюкокортикоиды ингибируют синтез всех типов эйкозаноидов, так как ингибируют фосфолипазу А2, и таким образом уменьшают количество субстрата для их синтеза. Аспирин и другие противовоспалительные препараты нестероидного действия ингибируют только циклооксигеназный путь.
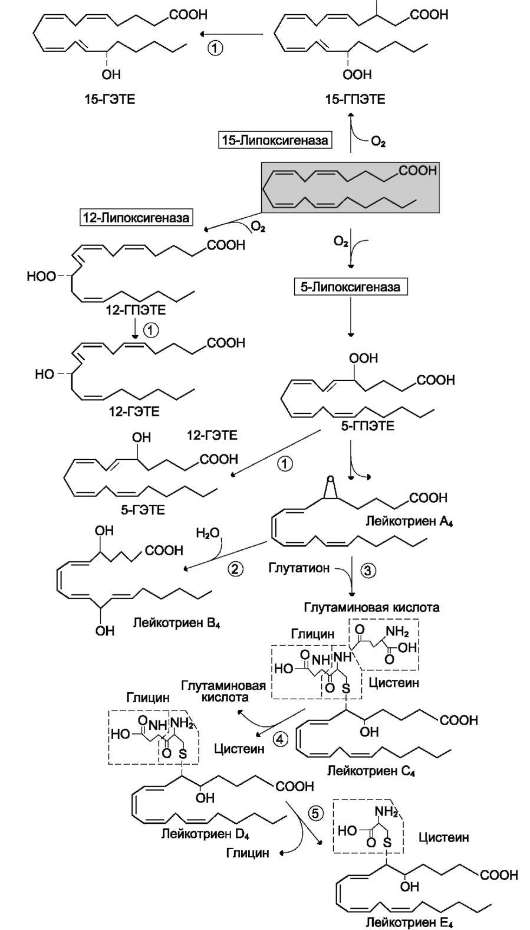
Рис. 8-49. Липоксигеназный путь синтеза эйкозаноидов.
сосудов. Другой путь приводит к образованию группы лейкотриенов: LT C4, LT D4, LT E4. Их синтез начинается с присоединения трипептида глутатиона к 6-му атому углерода с образованием LT С4 в реакции, катализируемой глутатион-S-трансферазой. В следующей реакции удаляется глутамат, и LT D4 содержит дипептид глицил-цистеин. На последней стадии отщепляется глицин, и LT Е4 содержит только цистеин.
Липоксины (например, основной липоксин А4) включают 4 сопряжённых двойных связи и 3 гидроксильных группы.
Синтез липоксинов начинается с действия на арахидоновую кислоту 15-липоксигеназы, затем происходит ряд реакций, приводящих к образованию липоксина А4 (рис. 8-50).
Г. МЕХАНИЗМЫ ДЕЙСТВИЯ ЭЙКОЗАНОИДОВ, ОСНОВНЫЕ БИОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Эйкозаноиды - гормоны местного действия по ряду признаков:
• образуются в различных тканях и органах, а не только в эндокринных железах;
• действуют по аутокринному или паракрин-ному механизмам;
• концентрация эйкозаноидов в крови меньше, чем необходимо, чтобы вызвать ответ в клетках-мишенях.
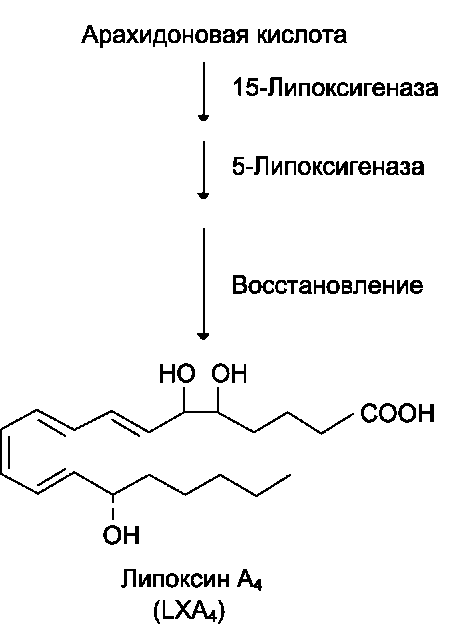
Рис. 8-50. Строение и синтез липоксина А4
Только при некоторых патологических состояниях эйкозаноиды могут оказывать системное действие, если их концентрация в крови увеличивается до количеств, когда они могут оказать действие на ГМК всего органа, например кишечника, лёгких, кровеносных сосудов.
Механизмы действия эйкозаноидов
Один и тот же тип эйкозаноида может действовать по паракринному и по аутокринному механизму. Например, ТХ А2, продуцируемый тромбоцитами при их активации, действует на сами тромбоциты, увеличивая их способность к агрегации, и в то же время действует на окружающие ГМК кровеносных сосудов, способствуя их сокращению. Таким образом создаются условия для образования тромба и предотвращения кровотечения в области повреждения сосудов.
Эйкозаноиды действуют на клетки через специальные рецепторы. Некоторые рецепторы эйкозаноидов связаны с аденилатциклазной системой и протеинкиназой А - это рецепторы PGE, PG D, PC I. PG F2α, ТХ А2, эндопереки-си (ГПЭТЕ) и лейкотриены действуют через механизмы, увеличивающие уровень кальция в цитозоле клеток-мишеней. Во многих клетках эйкозаноиды влияют на степень активации аденилатциклазной системы в ответ на действие других факторов, например гормонов. В этих случаях эйкозаноиды влияют на конформацию G-белков в плазматической мембране клеток. Если эйкозаноид связывается со стимулирующими Gs-белками, то эффект основного стимулирующего агента увеличивается; если с Gi-ингибируюигими - эффект снижается. Эйко-заноиды действуют на клетки почти всех тканей организма. Избыточная продукция эйкозанои-дов наблюдается при многих заболеваниях.
Роль эйкозаноидов в развитии воспаления
Воспаление - реакция организма на повреждение или инфекцию, направленная на уничтожение инфекционного агента и восстановление повреждённых тканей. Продукция медиаторов воспаления - эйкозаноидов, гистамина, кини-нов (пептидных гормонов местного действия) - активируется каскадами реакций, запускающимися при внедрении инфекционных агентов или повреждении тканей. Фактором, лимитирующим скорость синтеза эйкозаноидов, служит освобождение жирной кислоты под действием
фосфолипазы А2. Фосфолипаза А2 связана с мембранами клеток и активируется многими факторами: гистамином, кининами, механическим воздействием на клетку, контактом комплекса антиген-антитело с поверхностью клетки. Активация фосфолипазы А2 приводит к увеличению синтеза эйкозаноидов.
Многие эйкозаноиды выполняют функцию медиаторов воспаления и действуют на всех этапах воспаления. В результате увеличивается проницаемость капилляров, транссудат и лейкоциты проходят через сосудистую стенку. Лейкотриен В4 и липоксин А4 являются мощными факторами хемотаксиса; взаимодействуя с рецепторами, стимулируют движение лейкоцитов в область воспаления и секрецию ими лизосомальных ферментов и фагоцитоз чужеродных частиц.
Симптомы воспаления - покраснение, жар, отёк и боль. Покраснение и жар вызываются факторами, увеличивающими приток крови к
месту повреждения. Отёк - результат увеличения притока жидкости из капилляров и движения клеток белой крови в область воспаления. Боль вызывается химическими компонентами (продуктами распада тканей, протонами) и сдавлением нервных окончаний. В развитии этих признаков воспаления участвуют разные типы эйкозаноидов (табл. 8-8).
Роль эйкозаноидов в тромбообразовании
Свёртывание крови можно рассматривать как процесс, который поддерживается в состоянии равновесия противодействующими системами: свёртывания и противосвёртывания. В условиях патологии или при действии фармакологических средств это равновесие может смещаться в ту или другую сторону. В норме клетки эндотелия сосудов продуцируют простациклин I2, который препятствует агрегации тромбоцитов и сужению сосудов (рис. 8-51). При разрушении клеток эндотелия (например, в результате образова-
Таблица 8-8. Характеристика биологического действия основных типов эйкозаноидов
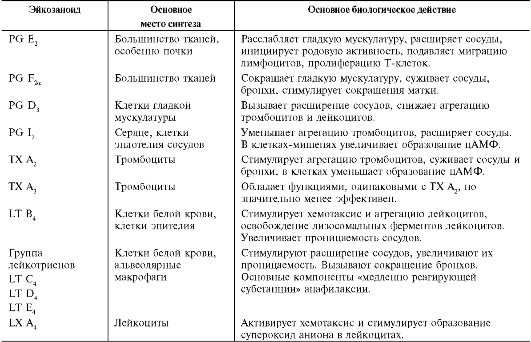
ния атеросклеротической бляшки) синтез PGI2 снижается. Тромбоциты контактируют с повреждённой стенкой сосуда, в результате чего активируется фосфолипаза А2. Это приводит к увеличению секреции ТХ стимулирующего агрегацию тромбоцитов и образование тромба в области повреждения сосуда (рис. 8-52), что часто приводит к развитию инфаркта.
При изучении факторов риска инфаркта миокарда было показано, что люди, потребляющие большое количество рыбьего жира, значительно меньше подвержены этому заболеванию, так как у них реже образуются тромбы в сосудах сердца. Оказалось, что на семейства эйкозаноидов, синтезируемых в организме, влияет состав жирных кислот пищи (см. выше табл. 8-3). Если с пищей поступает больше эйкозапентаеновой кислоты (20:5, ω-3), в большом количестве содержащейся в рыбьем жире, то эта кислота включается преимущественно в фосфолипиды мембран (вместо арахидоновой) и после действия фосфолипазы А2 служит основным субстратом для синтеза эйкозаноидов. Это имеет существенное влияние на свёртывание крови.
При обычной диете с преобладанием арахидо-новой кислоты (20:4, ω-6) над эйкозапентаено-вой действие ТХ А2 уравновешено действием PG I2 (рис. 8-53) и другими простагландинами. В случае диеты с преобладанием ω-3 кислот в клетках эндотелия образуются более сильные ингибиторы тромбообразования (PG I3, PG E3, PG D3), что снижает риск образования тромба и развития инфаркта миокарда.
Инактивация эйкозаноидов
Все типы эйкозаноидов быстро инактивируют-ся. T1/2 эйкозаноидов составляет от нескольких секунд до нескольких минут. Простагландины инактивируются путём окисления гидроксиль-ной группы в положении 15, важнейшей для их активности, до кетогруппы. Двойная связь в положении 13 восстанавливается. Затем происходит β-окисление боковой цепи, а после него - ω-окисление. Конечные продукты (дикарбоно-вые кислоты) выделяются с мочой. Активный ТХ А2 быстро превращается в биологически неактивный ТХ В2 путём разрыва кислородного мостика между 9-м и 11-м атомами углерода с образованием гидроксильных групп.
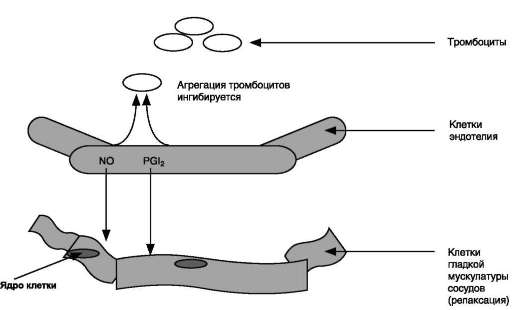
Рис. 8-51. Роль простациклинов в регуляции тонуса клеток гладкой мускулатуры стенок сосудов и агрегации тромбоцитов. В норме клетки эндотелия продуцируют PG I2, который вызывает релаксацию ГМК и ингибирует агрегацию тромбоцитов. Тромбоциты в неактивном состоянии не продуцируют тромбоксаны. N0 (оксид азота) - вазодилататор.
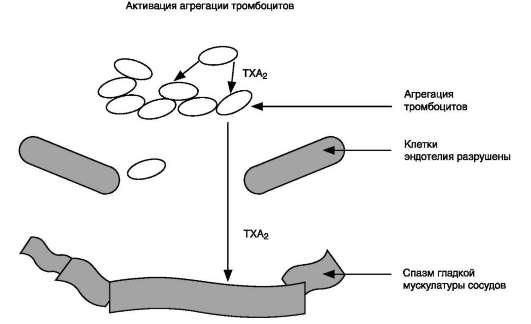
Рис. 8-52. Нарушение синтеза эйкозаноидов в области повреждения эндотелия. В области повреждения стенки сосуда преобладает действие ТХ А2, стимулирующего агрегацию тромбоцитов и сокращение стенок сосуда. В результате на повреждённом участке образуется тромб, происходит резкое сужение просвета сосуда. В миокарде это может привести к развитию инфаркта миокарда.
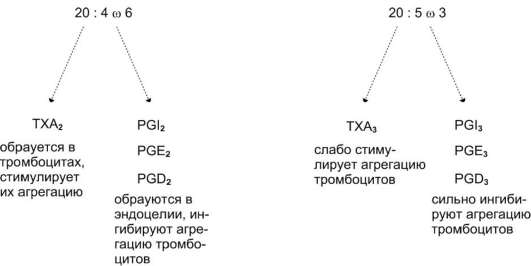
Рис. 8-53. Синтез тромбоксанов и простагландинов из арахидоновой и эйкозапентаеновой кислот.
Д. ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ - ИНГИБИТОРЫ СИНТЕЗА ЭЙКОЗАНОИДОВ
Механизм действия аспирина и других противовоспалительных препаратов нестероидного действия
Аспирин - препарат, подавляющий основные признаки воспаления. Механизм противовоспалительного действия аспирина стал понятен,
когда обнаружили, что он ингибирует циклоок-сигеназу. Следовательно, он уменьшает синтез медиаторов воспаления и, таким образом, уменьшает воспалительную реакцию. Циклооксигеназа необратимо ингибируется путём ацетилирования серина в положении 530 в активном центре (рис. 8-54). Однако эффект действия аспирина не очень продолжителен, так как экспрессия гена этого фермента не нарушается и продуцируются
Рис. 8-54. Механизм инактивации циклооксигеназы аспирином. Ацетильный остаток переносится с молекулы аспирина на ОН-группу фермента и необратимо ингибирует его.
новые молекулы фермента. Другие нестероидные противовоспалительные препараты (например, ибупрофен и ацетаминофен) действуют по конкурентному механизму, связываясь в активном центре фермента, и также снижают синтез про-стагландинов.
Механизм действия стероидных противоспали-тельных препаратов на синтез эйкозаноидов
Стероидные препараты обладают гораздо более сильным противовоспалительным действием, чем препараты нестероидного ряда. Механизм их действия заключается в индукции синтеза белков - липокортинов (или макро-кортинов), которые ингибируют активность фосфолипазы А2 и уменьшают синтез всех типов эйкозаноидов, так как препятствуют освобождению субстрата для синтеза эйкозаноидов - арахидоновой кислоты (или её аналога).
Использование стероидных противовоспалительных препаратов особенно важно для больных, страдающих бронхиальной астмой. Развитие симптомов этого заболевания (бронхоспазм и экссудация слизи в просвет бронхов) обусловлено, в частности, избыточной продукцией
лейкотриенов тучными клетками, лейкоцитами и клетками эпителия бронхов. Приём аспирина у больных, имеющих изоформу липоксигеназы с высокой активностью, может вызвать приступ бронхиальной астмы. Причина «аспириновой» бронхиальной астмы заключается в том, что аспирин и другие нестероидные противовоспалительные препараты ингибируют только цикло-оксигеназный путь превращений арахидоновой кислоты и, таким образом, увеличивают доступность субстрата для действия липоксигеназы и, соответственно, синтеза лейкотриенов. Стероидные препараты ингибируют использование арахидоновой кислоты и по липоксигеназному и по циклооксигеназному пути, поэтому они не могут вызывать бронхоспазма.
Использование производных эйкозаноидов в качестве лекарств
Хотя действие всех типов эйкозаноидов до конца не изучено, имеются примеры успешного использования лекарств - аналогов эйкоза-ноидов для лечения различных заболеваний. Например, аналоги PG E1 и PG E2 подавляют секрецию соляной кислоты в желудке, блоки-
руя гистаминовые рецепторы II типа в клетках слизистой оболочки желудка. Эти лекарства, известные как Н2-блокаторы, ускоряют заживление язв желудка и двенадцатиперстной кишки. Способность PG E2 и PG F2α стимулировать сокращение мускулатуры матки используют для стимуляции родовой деятельности.
vii. перекисное окисление липидов, роль в патогенезе повреждений клетки
Кислород, необходимый организму для функционирования ЦПЭ и многих других реакций, является одновременно и токсическим веществом, если из него образуются так называемые активные формы.
К активным формам кислорода относят:
ОН* - гидроксильный радикал;
О2- - супероксидный анион;
И2О2 - пероксид водорода.
Активные формы кислорода образуются во многих клетках в результате последовательного одноэлектронного присоединения 4 электронов к 1 молекуле кислорода. Конечный продукт этих реакций - вода, но по ходу реакций образуются химически активные формы кислорода. Наиболее активен гидроксильный радикал, взаимодействующий с большинством органических молекул. Он отнимает от них электрон и инициирует таким образом цепные реакции окисления. Эти свободнорадикальные реакции окисления могут выполнять полезные функции, например, когда клетки белой крови с участием активных форм кислорода разрушают фагоцитированные клетки бактерий. Но в остальных клетках свободнорадикальное окисление приводит к разрушению органических молекул, в первую очередь липидов, и, соответственно, мембранных структур клеток, что часто заканчивается их гибелью. Поэтому в организме функционирует эффективная система ингибирования перекисного окисления липидов (ПОЛ).
А. ИСТОЧНИКИ АКТИВНЫХ ФОРМ КИСЛОРОДА
ЦПЭ как источник активных форм кислорода
Утечка электронов из ЦПЭ и непосредственное их взаимодействие с кислородом - основ-
ной путь образования активных форм кислорода в большинстве клеток.
Кофермент Q в ЦПЭ принимает от доноров последовательно по одному электрону, превращаясь в форму семихинона (рис. 8-55) - KoQH' (см. раздел 6).
Этот радикал может непосредственно взаимодействовать с кислородом, образуя суперок-сидный анион О2-, который, в свою очередь, может превращаться в другие активные формы кислорода:
Реакции, катализируемые оксидазами и оксигеназами
Многие оксидазы - ферменты, непосредственно восстанавливающие кислород, образуют пероксид водорода - Н2О2. Оксидазы образуют пероксид водорода по схеме:
О2 + SH2 → S + H2O2,
где SH2 - окисляемый субстрат.
Примеры таких оксидаз - оксидазы аминокислот, супероксид дисмутаза, оксидазы, локализованные в пероксисомах. Оксидазы пероксисом окисляют, в частности, жирные кислоты с очень длинной углеродной цепью (более 20 углеродных атомов) до более коротких, которые далее подвергаются β-окислению в митохондриях.
Монооксигеназы, например цитохром Р450, включающий один атом кислорода в окисляемую молекулу, и диоксигеназы, включающие оба атома кислорода, также служат источниками активных форм кислорода.
Пероксид водорода химически не очень активен, но способствует образованию наиболее токсичной формы кислорода - гидроксильного радикала (ОН*) по следующей реакции:
Fe2+ + H2O2 → Fe3+ + OH- + OH*.
Наличие в клетках Fe2+ или ионов других переходных металлов увеличивает скорость образования гидроксильных радикалов и других активных форм кислорода. Например, в эритроцитах окисление иона железа гемоглобина способствует образованию супероксидного аниона.
Б. ПЕРЕКИСНОЕ ОКИСЛЕНИЕ ЛИПИДОВ
Реакции перекисного окисления липидов (ПОЛ) являются свободнорадикальными и постоянно
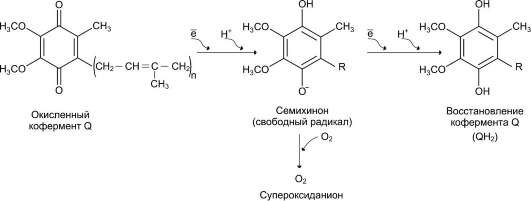
Рис. 8-55. Реакции последовательного восстановления убихинона в дыхательной цепи.
происходят в организме. Свободнорадикальное окисление нарушает структуру многих молекул. В белках окисляются некоторые аминокислоты. В результате разрушается структура белков, между ними образуются ковалентные «сшивки», всё это активирует протеолитические ферменты в клетке, гидролизующие повреждённые белки. Активные формы кислорода легко нарушают и структуру ДНК. Неспецифическое связывание Fe2+ молекулой ДНК облегчает образование гидроксиль-ных радикалов, которые разрушают структуру азотистых оснований. Но наиболее подвержены действию активных форм кислорода жирные кислоты, содержащие двойные связи, расположенные через СН2-группу. Именно от этой СН2-группы свободный радикал (инициатор окисления) легко отнимает электрон, превращая липид, содержащий эту кислоту, в свободный радикал.
ПОЛ - цепные реакции, обеспечивающие расширенное воспроизводство свободных радикалов, частиц, имеющих неспаренный электрон, которые инициируют дальнейшее распространение перекисного окисления.
Стадии перекисного окисления липидов
1) Инициация: образование свободного радикала (L*)
Инициирует реакцию чаще всего гидроксиль-ный радикал, отнимающий водород от СН2-групп полиеновой кислоты, что приводит к образованию липидного радикала.
2) Развитие цепи:
L ? + O2 → LOO ?
LOO? + LH → LOOH + L?
Развитие цепи происходит при присоединении О2, в результате чего образуется липо-пероксирадикал LOO» или пероксид липида LOOH.
ПОЛ представляет собой свободнорадикаль-ные цепные реакции, т.е. каждый образовавшийся радикал инициирует образование нескольких других.
3) Разрушение структуры липидов
Конечные продукты перекисного окисления полиеновых кислот - малоновый диальдегид и гидропероксид кислоты.
4) Обрыв цепи - взаимодействие радикалов между собой:
LOO? + L? → LOOH + LH L? + vit E → LH + vit Ε? vit Ε? + L? → LH + vit Εокисл.
окисл.
Развитие цепи может останавливаться при взаимодействии свободных радикалов между
собой или при взаимодействии с различными антиоксидантами, например, витамином Е, который отдаёт электроны, превращаясь при этом в стабильную окисленную форму.
В. ПОВРЕЖДЕНИЕ КЛЕТОК В РЕЗУЛЬТАТЕ ПЕРЕКИСНОГО ОКИСЛЕНИЯ ЛИПИДОВ
Активные формы кислорода повреждают структуру ДНК, белков и различные мембранные структуры клеток. В результате появления в гидрофобном слое мембран гидрофильных зон за счёт образования гидропероксидов жирных кислот в клетки могут проникать вода, ионы натрия, кальция, что приводит к набуханию клеток, органелл и их разрушению. Активация перекисного окисления характерна для многих заболеваний: дистрофии мышц (болезнь Дю-шенна), болезни Паркинсона, при которых ПОЛ разрушает нервные клетки в стволовой части мозга, при атеросклерозе, развитии опухолей. Перекисное окисление активируется также в тканях, подвергшихся сначала ишемии, а затем реоксигенации, что происходит, например, при спазме коронарных артерий и последующем их расширении.
Такая же ситуация возникает при образовании тромба в сосуде, питающем миокард. Формирование тромба приводит к окклюзии просвета сосуда и развитию ишемии в соответствующем участке миокарда (гипоксия ткани). Если принять быстрые лечебные меры по разрушению тромба, то в ткани восстанавливается снабжение кислородом (реоксигенация). Показано, что в момент реоксигенации резко возрастает образование активных форм кислорода, которые могут повреждать клетку. Таким образом, даже несмотря на быстрое восстановление кровообращения, в соответствующем участке миокарда происходит повреждение клеток за счёт активации перекисного окисления.
Изменение структуры тканей в результате ПОЛ можно наблюдать на коже: с возрастом увеличивается количество пигментных пятен на коже, особенно на дорсальной поверхности ладоней. Этот пигмент называют липофусцин, представляющий собой смесь липидов и белков, связанных между собой поперечными ковалентными связями и денатурированными в результате взаимодействия с химически активными группами продуктов ПОЛ. Этот
пигмент фагоцитируется, но не гидролизуется ферментами лизосом, и поэтому накапливается в клетках, нарушая их функции.
ПОЛ происходит не только в живых организмах, но и в продуктах питания, особенно при неправильном приготовлении и хранении пищи. Прогоркание жиров, образование более тёмного слоя на поверхности сливочного масла, появление специфического запаха у молочных продуктов - всё это признаки ПОЛ. В продукты питания, содержащие ненасыщенные липиды, обычно добавляют антиоксиданты - вещества, ингибирующие ПОЛ и сохраняющие структуру компонентов пищи.
Г. СИСТЕМЫ ЗАЩИТЫ КЛЕТОК ОТ АКТИВНЫХ
ФОРМ КИСЛОРОДА
Ферменты антиоксидантного действия
К ферментам, защищающим клетки от действия активных форм кислорода, относят супероксиддисмутазу, каталазу и глутатионпе-роксидазу. Наиболее активны эти ферменты в печени, надпочечниках и почках, где содержание митохондрий, цитохрома Р450 и пероксисом особенно велико. Супероксиддисмутаза (СОД) превращает супероксидные анионы в пероксид водорода:
2 О2- + 2 Н+ → Н2О2 + О2.
Изоферменты СОД находятся и в цитозоле и в митохондриях и являются как бы первой линией защиты, потому что супероксидный анион образуется обычно первым из активных форм кислорода при утечке электронов из дыхательной цепи.
СОД - индуцируемый фермент, т.е. синтез его увеличивается, если в клетках активируется перекисное окисление.
• Пероксид водорода, который может инициировать образование самой активной формы ОН , разрушается ферментом каталазой: 2 Н2О2 → 2 Н2О + О2.
Каталаза находится в основном в перокси-сомах, где образуется наибольшее количество пероксида водорода, а также в лейкоцитах, где она защищает клетки от последствий «респираторного взрыва» (см. раздел 6).
Глутатионпероксидаза - важнейший фермент, обеспечивающий инактивацию активных форм кислорода, так как он разрушает и пероксид водорода и гидропероксиды липидов.
Он катализирует восстановление пероксидов с помощью трипептида глутатиона (γ-глутамил-цистеинилглицин). Сульфгидрильная группа глутатиона (GSH) служит донором электронов и, окисляясь, образует дисульфидную форму глутатиона, в которой 2 молекулы глутатиона связаны через дисульфидную группу. Н2О2 + 2 GSH → 2 H2O + G-S-S-G.
Окисленный глутатион восстанавливается глутатионредуктазой:
GS-SG + NADPH + H+ → 2 GSH + NADP+.
Глутатионпероксидаза, которая восстанавливает гидропероксиды липидов в составе мембран, в качестве кофермента использует селен (необходимый микроэлемент пищи). При его недостатке активность антиоксидантной защиты снижается.
Витамины, обладающие антиоксидантным действием
Витамин Е (α-токоферол) - наиболее распространённый антиоксидант в природе - является липофильной молекулой, способной инактиви-ровать свободные радикалы непосредственно в гидрофобном слое мембран и таким образом предотвращать развитие цепи перекисного окисления. Различают 8 типов токоферолов, но α-токоферол наиболее активен.
• Витамин Е отдаёт атом водорода свободному радикалу пероксида липида (LOO ), восстанавливая его до гидропероксида (LOOH) и таким образом останавливает развитие ПОЛ (рис. 8-56).
Свободный радикал витамина Е, образовавшийся в результате реакции, стабилен и не способен участвовать в развитии цепи. Наоборот, радикал витамина Е непосредственно взаимодействует с радикалами липидных перекисей, восстанавливая их, а сам превращается в стабильную окисленную форму - токоферолхинон.
• Витамин С (аскорбиновая кислота) также является антиоксидантом и участвует с помощью двух различных механизмов в ингибировании ПОЛ. Во-первых, витамин С восстанавливает окисленную форму витамина Е и таким образом поддерживает необходимую концентрацию этого антиоксиданта непосредственно в мембранах клеток. Во-вторых, витамин С, будучи водорастворимым витамином и сильным восстановителем, взаимодействует с водорастворимыми активными формами кислорода - О2-, Н2О2, ОН и инактивирует их.
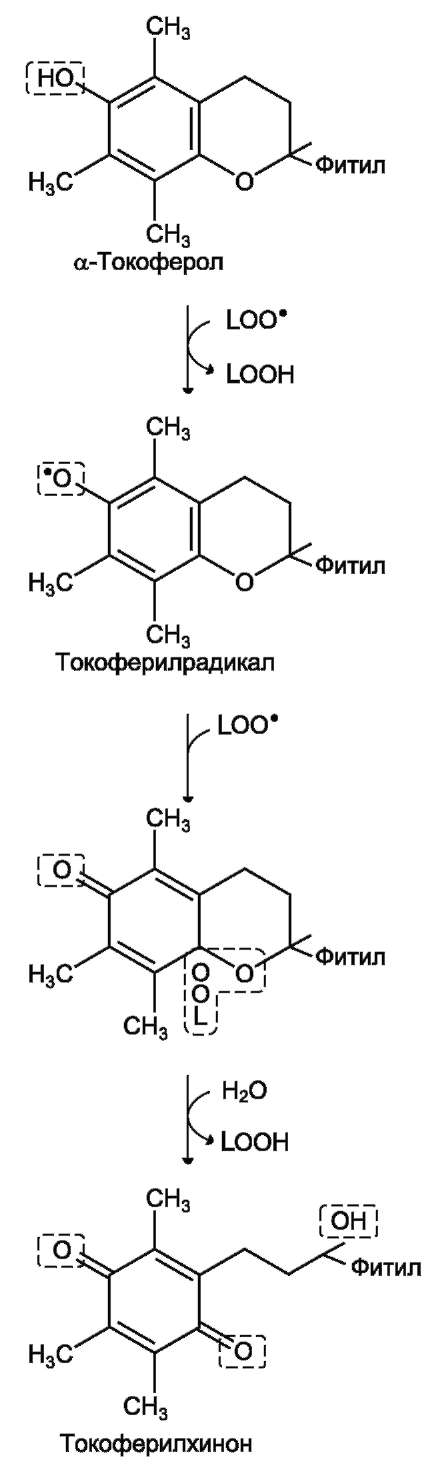
Рис. 8-56. Механизм антиоксидантного действия витамина Е. Витамин Е (a-токоферол) ингибирует свободнорадикальное окисление путём отдачи электрона, что приводит к инактивации радикала липида, а витамин Е превращается в стабильный, полностью окисленный токоферолхинон.
β-Каротин, предшественник витамина А, также обладает антиоксидантным действием и ингибирует ПОЛ. Показано, что растительная диета, обогащённая витаминами Е, С, ка-ротиноидами, существенно уменьшает риск развития атеросклероза и заболеваний ССС, подавляет развитие катаракты - помутнения хрусталика глаза, обладает антиканцерогенным действием. Имеется много доказательств в пользу того, что положительное действие этих компонентов пищи связано с ингибировани-ем ПОЛ и других молекул и, следовательно, с поддержанием нормальной структуры компонентов клеток.
viii. обмен и функции фосфолипидов
Метаболизм фосфолипидов тесно связан со многими процессами в организме: образованием и разрушением мембранных структур клеток, формированием ЛП, мицелл жёлчи, образованием в альвеолах лёгких поверхностного слоя, предотвращающего слипание альвеол во время выдоха. Нарушения обмена фосфолипидов - причина многих заболеваний, в частности, респираторного дистресс-синдрома новорождённых, жирового гепатоза, наследственных заболеваний, связанных с накоплением глико-липидов, - лизосомных болезней. При лизосом-ных болезнях снижается активность гидролаз, локализованных в лизосомах и участвующих в расщеплении гликолипидов.
А. ОБМЕН ГЛИЦЕРОФОСФОЛИПИДОВ
Синтез фосфатидилхолинов, фосфатидилэтанола-минов и фосфатидилсеринов
Начальные этапы синтеза глицерофосфоли-пидов и жиров происходят одинаково до образования фосфатидной кислоты. Фосфатидная кислота может синтезироваться двумя разными путями: через глицеральдегид-3-фосфат и через дигидроксиацетонфосфат (рис. 8-57).
На следующем этапе фосфатидаза отщепляет от фосфатидной кислоты фосфатный остаток, в результате чего образуется диацилглицерол. Дальнейшие превращения диацилглицерола также могут идти разными путями. Один из вариантов - образование активной формы «полярной головки» фосфолипида: холин, серин
или этаноламин превращаются в ЦДФ-холин, ЦДФ-серин (рис. 8-58) или ЦДФ-этаноламин.
Далее диацилглицерол взаимодействует c ЦДФ-производными, при этом выделяется ЦМФ, и образуется соответствующий фосфолипид, например фосфатидилхолин. Между глицерофосфолипида-ми возможны различные взаимопревращения. Фосфатидилхолин может образовываться и другим путём: из фосфатидилэтаноламина, получая последовательно 3 метильные группы от SAM. Фосфатидилсерин может превращаться в фосфа-тидилэтаноламин путём декарбоксилирования. Фосфатидилэтаноламин может превращаться в фосфатидилсерин путём обмена этаноламина на серин.
Дипальмитоилфосфатидилхолин - основной компонент сурфактанта лёгких
Сурфактант - внеклеточный липидный слой с небольшим количеством гидрофобных белков, выстилающий поверхность лёгочных альвеол и предотвращающий слипание стенок альвеол во время выдоха (рис. 8-59). Основной компонент сурфактанта - дипальмитоилфос-фатидилхолин, составляющий до 80% от всех фосфолипидов, входящих в состав сурфактанта. Кроме того, в сурфактант входят гидрофобные белки, общее количество которых не превышает 10-20%.
Синтез дипальмитоилфосфатидилхолина (лецитина) в пневмоцитах II типа происходит в процессе эмбрионального развития и резко увеличивается в период от 32 до 36 нед беременности.
Важным показателем нормального формирования сурфактанта служит соотношение фос-фатидилхолин/сфингомиелин >4 (рис. 8-60). Это соотношение можно определять, исследуя состав амниотической жидкости. Недостаточное формирование сурфактанта у недоношенных детей после рождения приводит к развитию респираторного дистресс-синдрома - основной причины смерти у этой группы новорождённых. Соотношение фосфатидилхолин/сфингомие-лин <2 указывает на высокий риск развития респираторного дистресс-синдрома. В случае необходимости лечение беременных кортикос-тероидами стимулирует синтез сурфактанта в лёгких плода и уменьшает риск развития респираторного дистресс-синдрома.
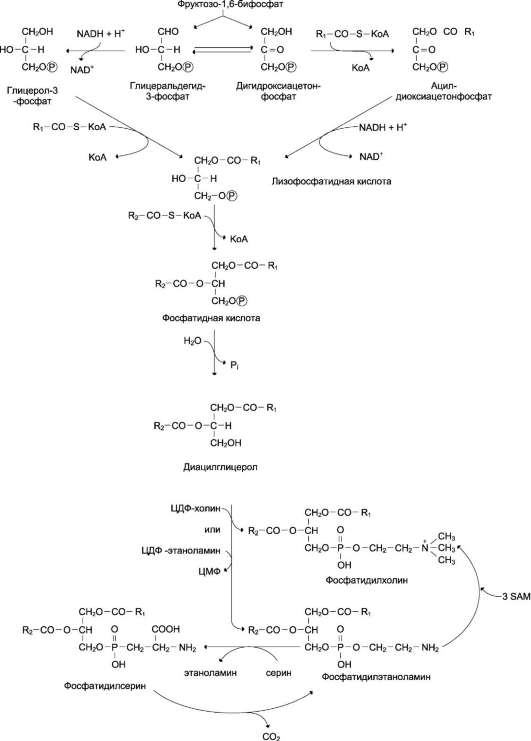
Рис. 8-57. Схема синтеза глицерофосфолипидов. R1 - радикал насыщенной жирной кислоты; R2 - радикал полиеновой жирной кислоты; SAM - S-аденозилметионин.
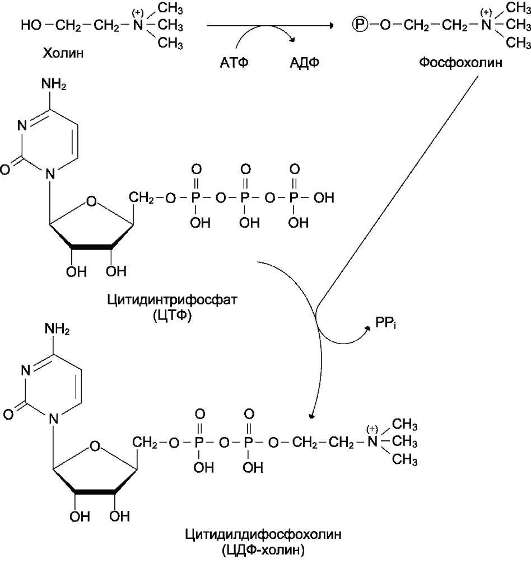
Рис. 8-58. Синтез ЦДФ-холина. «Полярная головка» фосфатидилхолина превращается за счёт энергии АТФ в активную форму - фосфохолин, который затем присоединяется к ЦТФ с одновременным удалением PPi, что сдвигает равновесие реакции вправо. Образовавшийся ЦДФ-холин - донор холина для синтеза молекул фосфатидилхолинов. ЦДФ-холин - цитидилдифосфохолин; ЦМФ - цитидилмонофосфат; Р - остаток фосфорной кислоты.
Синтез фосфатидилинозитола и кардиолипина
Другой путь превращений диацилглицерола, при котором образуется активная форма - ЦДФ-диацилглицерол приводит к образованию фосфатидилинозитола и кардиолипина (см. схему ниже).
Фосфатидилинозитол далее может фосфо-рилироваться с образованием фосфатидили-нозитол-4,5-бисфосфата, фосфолипида, располагающегося в наружной мембране клеток и участвующего в передаче гормональных сигналов внутрь клетки (см. раздел 5). Кардиолипин находится, главным образом, во внутренней
мембране митохондрий и в небольшом количестве в сурфактанте лёгких.
Катаболизм глицерофосфолипидов
Различные типы фосфолипаз, локализованных в клеточных мембранах или в лизосомах, катализируют гидролиз глицерофосфолипидов (см. раздел 5). Гидролиз некоторых глицерофос-фолипидов под действием фосфолипаз имеет значение не только как путь катаболизма, но и как путь образования вторичных посредников или предшественников в синтезе биологически активных веществ - эйкозаноидов. Кроме того,
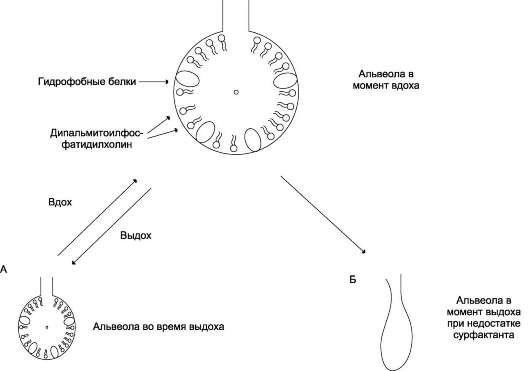
Рис. 8-59. Влияние сурфактанта на функцию альвеол. А - сурфактант уменьшает поверхностное натяжение жидкости, выстилающей поверхность альвеол, и предотвращает слипание стенок альвеол во время выдоха. Меньшее давление воздуха необходимо, чтобы наполнить альвеолы воздухом; Б - в отсутствие сурфактанта или при его недостаточном образовании (у недоношенных детей) стенки альвеол во время выдоха спадаются, и требуется давление воздуха в 10 раз большее, чтобы наполнить альвеолы.
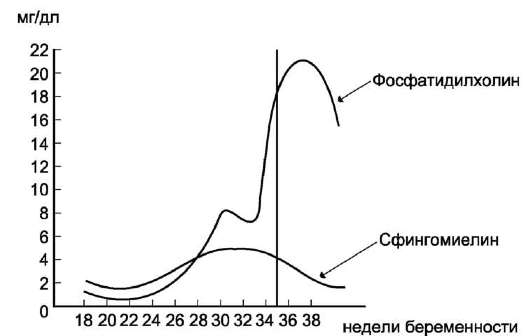
Рис. 8-60. Изменение соотношения фосфатидилхолин/сфингомиелин в амниотической жидкости в различные периоды беременности. На 35-й неделе беременности концентрация фосфатидилхолина увеличивается по отношению к сфингомиелину в 4 раза, что характеризует нормальное развитие лёгких.
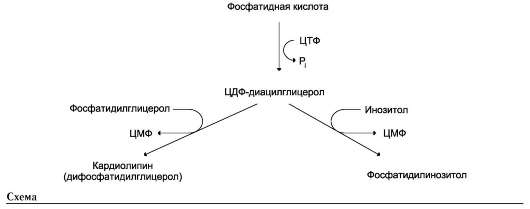
фосфолипазы А1 и А2 участвуют в изменении состава жирных кислот в глицерофосфолипидах, например при синтезе в эмбриональном периоде развития дипальмитоилфосфатидилхолина - компонента сурфактанта.
Б. ФУНКЦИИ И ОБМЕН СФИНГОЛИПИДОВ
Сфинголипиды - производные церамида, образующегося в результате соединения ами-носпирта сфингозина и жирной кислоты. В группу сфинголипидов входят сфингомиелины и гликос-финголипиды (см. табл. 8-4, рис. 8-61).
Сфингомиелины находятся в мембранах клеток различных тканей, но наибольшее их количество содержится в нервной ткани. Сфин-гомиелины миелиновых оболочек содержат в основном жирные кислоты с длинной цепью: лигноцериновую (24:0) и нервоновую (24:1) кислоты, а сфингомиелин серого вещества мозга содержит преимущественно стеариновую кислоту.
Гликосфинголипиды - гликолипиды, в состав которых входят церамид и один или несколько остатков углеводов, и сиаловая (N-ацетилнейра-миновая) кислота (см. рис. 8-5, 8-8, 8-61).

Рис. 8-61. Структуры гликосфинголипидов. А - общая схема строения гликосфинголипидов; Б - структура радикалов, связанных с церамидом, и названия отдельных гликосфинголипидов. Обозначения: цер - церамид; глк - глюкоза; гал - галактоза; гал-Naц - N-ацетилгалактозамин; N-АНК - N-ацетилнейраминовая кислота.
Гликосфинголипиды локализованы в плазматических мембранах клеток таким образом, что углеводная часть молекулы располагается на поверхности клеток и часто обладает антигенными свойствами. Эта часть молекул обеспечивает взаимное узнавание клеток и их взаимодействие. Интересно, что углеводная часть структуры антигенов на поверхности эритроцитов (по системе АВ0) может быть связана как с церамидом, так и с белками. В последнем случае структура антигена является не гликолипидом, а глико-протеином.
Некоторые ганглиозиды - рецепторы бактериальных токсинов. Например, GM1, находящийся на поверхности клеток кишечного эпителия, является местом прикрепления холерного токсина - белка, секретируемого возбудителями холеры.
Функции гликосфинголипидов можно суммировать следующим образом: Взаимодействие между:
• клетками;
• клетками и межклеточным матриксом;
• клетками и микробами. Модуляция:
• активности протеинкиназ;
• активности рецептора фактора роста;
• антипролиферативного действия (апопто-за, клеточного цикла).
Обеспечение:
• структурной жёсткости мембран;
• конформации белков мембран.
Синтез церамида и его производных. Синтез сфин-голипидов начинается с образования церамида. Серин конденсируется с пальмитоил-КоА. Продукт их взаимодействия сначала восстанавливается коферментом NADPH, затем к аминогруппе дигидросфингозина амидной связью присоединяется жирная кислота, содержащая, как правило, 24 атома углерода. После окисления FAD-зависимой дегидрогеназой образуется церамид. Церамид служит предшественником в синтезе большой группы сфинголипидов: сфингомиелинов, не содержащих углеводов, и гликосфинголипидов (рис. 8-62). Последующие реакции синтеза катализируются специфическими трансферазами, набор которых отличается в разных тканях. Соединение фосфорилхолина с церамидом сфингомиелин-синтазой приводит к образованию сфингомиелина. Присоединение углеводных компонентов катализируется специфическими гликозилтрансферазами. Донорами
углеводных компонентов служат активированные сахара: УДФ-галактоза и УДФ-глюкоза. Галактоцереброзид - главный липид миелино-вых оболочек; глюкоцереброзид входит в состав мембран многих клеток и служит предшественником в синтезе более сложных гликолипидов или продуктом на пути их катаболизма.
Катаболизм сфингомиелина и его нарушения. В лизосомах находятся ферменты, способные гидролизовать любые компоненты клеток. Эти ферменты называют кислыми гидролазами, так как они активны в кислой среде. Значение рН = 5, оптимальное для работы ферментов, создаётся протонным насосом, который, используя энергию АТФ, накачивает ионы водорода в лизосо-мы. Катаболизм сфингомиелинов и гликоли-пидов происходит в лизосомах. В распаде сфин-гомиелинов участвуют 2 фермента - сфингоми-елиназа, отщепляющая фосфорилхолин, и цера-мидаза, продуктами действия которой являются сфингозин и жирная кислота (рис. 8-63).
Генетический дефект сфингомиелиназы - причина болезни Ниманна-Пика. Дети с таким дефектом погибают в раннем возрасте. Симптомы заболевания: увеличение печени и селезёнки (гепатоспленомегалия), в лизосомах которых накапливается сфингомиелин; умственная отсталость. Генетический дефект другого фермента (церамидазы) приводит к развитию болезни Фарбера, симптомами которой также являются гепато- и спленомегалия, а также поражение суставов (болезненность, отёчность).
Катаболизм гликосфинголипидов. Катаболизм гликосфинголипидов начинается с перемещения их с поверхности клетки в цитоплазму по механизму эндоцитоза. В результате молекулы, расположенные на поверхности мембран, оказываются в эндоцитозных везикулах в цито-плазме и сливаются с лизосомами. В лизосомах находятся все ферменты, необходимые для гидролиза сложных молекул гликосфинголипидов: α- и β-галактозидазы, β-глюкозидазы, нейрамини-даза (сиалидаза) и церамидаза. В результате последовательных реакций гидролиза сложные молекулы гликосфинголипидов распадаются до мономеров: глюкозы, галактозы, жирной кислоты, сфингозина и других метаболитов.
Генетические дефекты лизосомных ферментов катаболизма гликосфинголипидов. В норме синтез и катаболизм гликосфинголипидов сбалансированы таким образом, что количество этих
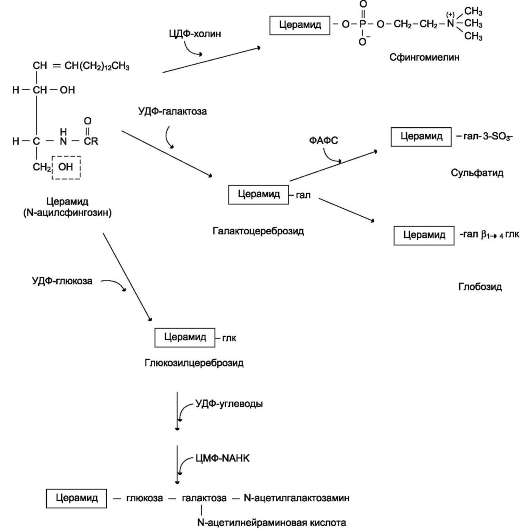
Рис. 8-62. Синтез сфинголипидов из церамида. Обозначения: гал - галактоза; глк - глюкоза; гал - Naц-N-ацетилгалактозамин; N-АНК - N-ацетилнейраминовая кислота; УДФ-галактоза, УДФ-глюкоза - активные формы углеводов, присоединяемые специфическими гликозилтрансферазами; ЦМФ-N-AHK - активная форма N-ацетилнейраминовой кислоты; ФАФС - фосфоаденозилфосфосульфат - активная форма серной кислоты. Фосфохолин или углеводы присоединяются по месту гидроксиметильной группы церамида (выделена пунктиром). Каждый остаток углевода присоединяется специфической гликозилтрансферазой в цистернах шероховатого ЭР и аппарате Гольджи.
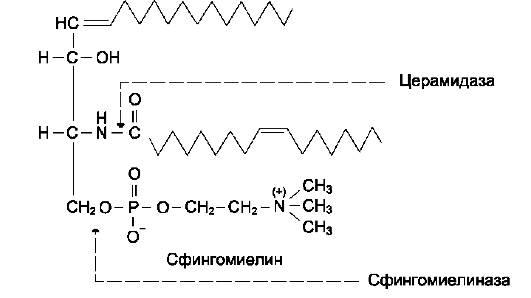
Рис. 8-63. Гидролиз сфингомиелина.
компонентов в мембранах постоянно. Если имеется генетический дефект какого-либо лизосомного фермента, участвующего в катаболизме гликосфинголипида, то в лизосомах
накапливается недеполимеризованный субстрат, так называемые «остаточные тельца», размеры лизосом увеличиваются, их мембрана может разрушаться, ферменты выходят в цитозоль, и функции клеток нарушаются. Генетические заболевания вследствие дефекта какого-либо из ферментов катаболизма гликосфинголипидов называют сфинголипидозами, или лизосомными болезнями. Эти заболевания редки, но среди некоторых популяций людей их частота очень высока. Так, болезнь Гоше вследствие дефекта фермента β-глюкозидазы (рис. 8-64) у евреев встречается с частотой 166:100 000, болезнь Тея-Сакса (дефект фермента β-гексозаминида-зы) - с частотой 33:100 000. Сфинголипидозы обычно приводят к смерти в раннем возрасте, так как происходит поражение клеток нервной ткани, где сконцентрированы гликосфинго-липиды. Однако при болезнях Гоше и Фабри больные живут относительно долго.
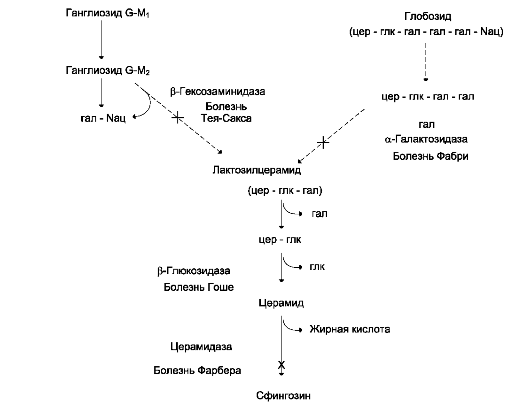
Рис. 8-64. Катаболизм гликосфинголипидов. На схеме указаны ферменты, генетические дефекты которых являются причиной наследственных заболеваний - сфинголипидозов.
ix. холестерол: функции, обмен
Холестерол
- стероид, характерный только для животных организмов. Он
синтезируется во многих тканях человека, но основное место синтеза -
печень. В печени синтезируется более 50% холестерола, в тонком
кишечнике - 15-20%, остальной холестерол синтезируется в коже, коре
надпочечников, половых железах. В сутки в организме синтезируется около
ми. Этерифицированный холестерол преобладает в крови и запасается в небольших количествах в некоторых типах клеток, использующих его как субстрат для синтеза других веществ. Холестерол и его эфиры - гидрофобные молекулы, поэтому они транспортируются кровью только в составе разных типов ЛП. Обмен холестерола чрезвычайно сложен - только для его синтеза необходимо осуществление около 100 последовательных реакций. Всего в обмене холестерола участвует около 300 разных белков. Нарушения обмена холестерола приводят к одному из наиболее распространённых заболеваний - атеросклерозу. Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения. Атеросклероз - «полигенное заболевание», т.е. в его развитии участвуют многие факторы, важнейшие из которых наследственные. Накопление холестерола в организме приводит к развитию и другого распространённого заболевания - желчнокаменной болезни.
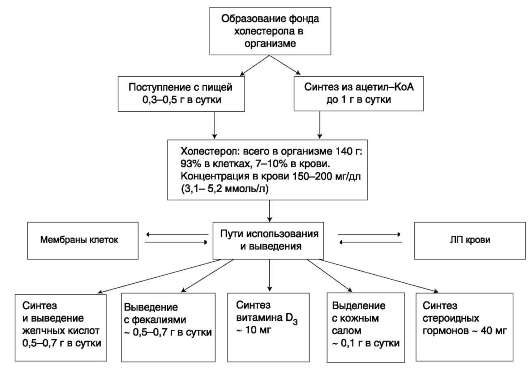
Рис. 8-65. Фонд холестерола в организме, пути его использования и выведения.
А. СИНТЕЗ ХОЛЕСТЕРОЛА И ЕГО РЕГУЛЯЦИЯ
Реакции синтеза холестерола происходят в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека.
Образование мевалоната
Сложный путь синтеза холестерола можно разделить на 3 этапа (рис. 8-66). Первый этап заканчивается образованием мевалоната (мева-лоновой кислоты). Две молекулы ацетил-КоА конденсируются ферментом тиолазой с образованием ацетоацетил-КоА. Фермент гидроксиме-тилглутарил-КоА-синтаза присоединяет третий ацетильный остаток с образованием ГМГ-КоА
(3-гидрокси-3-метилглутарил-КоА). Эта последовательность реакций сходна с начальными стадиями синтеза кетоновых тел (см. рис. 8-33). Однако реакции синтеза кетоновых тел происходят в митохондриях печени, а реакции синтеза холестерола - в цитозоле клеток.
Следующая реакция, катализируемая ГМГ-КоА-редуктазой, является регуляторной в метаболическом пути синтеза холестерола. В этой реакции происходит восстановление ГМГ-КоА до мевалоната с использованием 2 молекул NADPH. Фермент ГМГ-КоА-редуктаза - гликопротеин, пронизывающий мембрану ЭР, активный центр которого выступает в цито-золь.
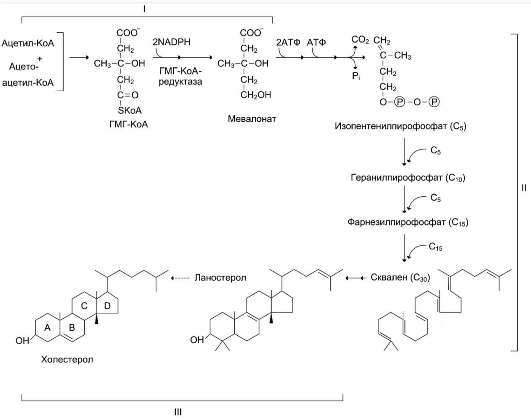
Рис. 8-66. Синтез холестерола. С5 - изопентенилпирофосфат; С15 - фарнезилпирофосфат. Все атомы углерода холестерола происходят из ацетил-КоА. Сквален - углеводород линейной структуры - превращается ферментом циклазой в ланостерол, содержащий 4 конденсированных кольца и гидроксильную группу. Ланостерол через ряд последовательных реакций превращается в холестерол (I, II, III - этапы синтеза).
Образование сквалена
На втором этапе синтеза мевалонат превращается в пятиуглеродную изопреноидную структуру, содержащую пирофосфат - изо-пентенилпирофосфат. Продукт конденсации
2 изопреновых единиц - геранилпирофосфат. Присоединение ещё 1 изопреновой единицы приводит к образованию фарнезилпирофосфа-та - соединения, состоящего из 15 углеродных атомов. Две молекулы фарнезилпирофосфата конденсируются с образованием сквалена - углеводорода линейной структуры, состоящего из 30 углеродных атомов.
Образование холестерола
На третьем этапе синтеза холестерола сква-лен через стадию образования эпоксида ферментом циклазой превращается в молекулу ланостерола, содержащую 4 конденсированных цикла и 30 атомов углерода. Далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол. На последних этапах синтеза от ланостерола отделяется
3 атома углерода, поэтому холестерол содержит 27 углеродных атомов. У холестерола имеется насыщенная разветвлённая боковая цепь из 8 углеродных атомов в положении 17, двойная связь в кольце В между атомами углерода в положениях 5 и 6, а также гидроксильная группа в положении 3.
В организме человека изопентенилпирофос-фат также служит предшественником убихинона (КоQ) и долихола, участвующего в синтезе гли-копротеинов.
Этерификация холестерола
В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием более гидрофобных молекул - эфиров холесте-рола. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролацил-трансферазой).
Реакция этерификации происходит также в крови в ЛПВП, где находится фермент ЛХАТ (лецитин:холестеролацилтрансфераза). Эфиры холестерола - форма, в которой они депонируются в клетках или транспортируются кровью. В крови около 75% холестерола находится в виде эфиров.
Регуляция синтеза холестерола
Регуляция ключевого фермента синтеза хо-лестерола (ГМГ-КоА-редуктазы) происходит разными способами.
Фосфорилирование/дефосфорилирование ГМГ-КоА-редуктазы (рис. 8-67). При увеличении соотношения инсулин/глюкагон этот фермент дефосфорилируется и переходит в активное состояние. Действие инсулина осуществляется через 2 фермента: фосфатазу киназы ГМГ-КоА-редуктазы, которая превращает киназу в неактивное дефосфо-рилированное состояние; фосфатазу ГМГ-КоА-редуктазы путём превращения её в дефосфорилированное активное состояние. Результатом этих реакций служит образование дефосфорилированной активной формы ГМГ-КоА-редуктазы. Следовательно, в абсорбтивный период синтез холестерола увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза холестерола - ацетил-КоА (в результате приёма пищи, содержащей углеводы, так как ацетил-КоА образуется в основном при распаде глюкозы). В постабсорбтивном состоянии глюкагон через протеинкиназу А стимулирует фосфо-рилирование ГМГ-КоА-редуктазы, переводя её в неактивное состояние. Это действие усиливается тем, что одновременно глю-кагон стимулирует фосфорилирование и инактивацию фосфатазы ГМГ-КоА-редук-тазы и фосфорилирование киназы ГМГ-КоА-редуктазы, удерживая, таким образом, ГМГ-КоАредуктазу в фосфорилированном неактивном состоянии. В результате синтез холестерола в постабсорбтивном периоде и при голодании ингибируется. Ингибирование синтеза ГМГ-КоА-редуктазы. Конечный продукт метаболического пути (холестерол) снижает скорость транскрипции гена ГМГ-КоА-редуктазы, подавляя таким образом собственный синтез. В печени активно идёт синтез жёлчных кислот из холестерола, поэтому и жёлчные кислоты (как конечные продукты синтеза) подавляют активность гена ГМГ-КоА-редуктазы (рис. 8-67). Так как молекула ГМГ-КоА-редук-тазы существует около 3 ч после синтеза, то ингибирование синтеза этого фермента
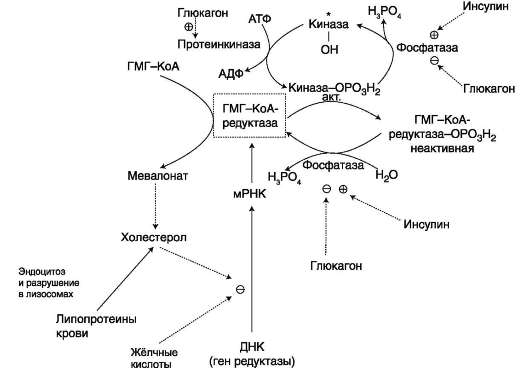
Рис. 8-67. Регуляция активности ГМГ-КоА-редуктазы в печени. Холестерол и жёлчные кислоты снижают скорость транскрипции и, таким образом, синтез фермента. Инсулин стимулирует дефосфорилирование, а глюкагон - фосфорилирование ГМГ-КоА-редуктазы. Инсулин активирует 2 фосфатазы: киназы ГМГ-КоА-редуктазы* и фосфатазу, дефосфорилирующую непосредственно ГМГ-КоА-редуктазу. Глюкагон стимулирует фосфорили-рование и инактивацию 2 фосфатаз и фосфорилирование и активацию киназы ГМГ-КоА-редуктазы.
конечным продуктом метаболического пути (холестеролом) является эффективной регуляцией.
Б. ТРАНСПОРТ ХОЛЕСТЕРОЛА ЛИПОПРОТЕИНАМИ КРОВИ
Холестерол транспортируется кровью только в составе ЛП. ЛП обеспечивают поступление в ткани экзогенного холестерола, определяют потоки холестерола между органами и выведение избытка холестерола из организма.
Транспорт экзогенного холестерола
Холестерол поступает с пищей в количестве 300-500 мг/сут, в основном в виде эфиров. После гидролиза, всасывания в составе мицелл,
этерификации в клетках слизистой оболочки кишечника эфиры холестерола и небольшое количество свободного холестерола включаются в состав ХМ и поступают в кровь. После удаления жиров из ХМ под действием ЛП-липазы холестерол в составе остаточных ХМ доставляется в печень. Остаточные ХМ взаимодействуют с рецепторами клеток печени и захватываются по механизму эндоцитоза. Затем ферменты лизосом гидролизуют компоненты остаточных ХМ, и в результате образуется свободный холестерол. Экзогенный холестерол, поступающий таким образом в клетки печени, может ингибировать синтез эндогенного холестерола, замедляя скорость синтеза ГМГ-КоА-редуктазы.
Транспорт эндогенного холестерола в составе ЛПОНП (пре-β-липопротеинов)
Печень - основное место синтеза холестеро-ла. Эндогенный холестерол, синтезированный из исходного субстрата ацетил-КоА, и экзогенный, поступивший в составе остаточных ХМ, образуют в печени общий фонд холестерола. В гепатоцитах триацилглицеролы и холестерол упаковываются в ЛПОНП. В их состав входят, кроме того, апопротеин В-100 и фосфолипиды. ЛПОНП секретируются в кровь, где получают от ЛПВП апопротеины Е и С-II. В крови на ЛПОНП действует ЛП-липаза, которая, как и в ХМ, активируется апоС-II и гидролизует жиры до глицерола и жирных кислот. По мере уменьшения количества ТАГ в составе ЛПОНП они превращаются в ЛППП. Когда количество жиров в ЛППП уменьшается, апопротеины С-II переносятся обратно на ЛПВП. Содержание хо-лестерола и его эфиров в ЛППП достигает 40%; часть этих липопротеинов захватывается клетками печени через рецепторы ЛПНП, которые взаимодействуют и с апоЕ и с апоВ-100.
Транспорт холестерола в составе ЛПНП. Рецепторы ЛПНП
На ЛППП, оставшиеся в крови, продолжает действовать ЛП-липаза, и они превращаются в ЛПНП, содержащие до 50% холестерола и его эфиров. Апопротеины Е и С-II переносятся обратно в ЛПВП. Поэтому основным апопро-теином в ЛПНП служит апоВ-100, Апопротеин В-100 взаимодействует с рецепторами ЛПНП и таким образом определяет дальнейший путь холестерола. ЛПНП - основная транспортная форма холестерола, в которой он доставляется в ткани. Около 70% холестерола и его эфиров в крови находится в составе ЛПНП. Из крови ЛПНП поступают в печень (до 75%) и другие ткани, которые имеют на своей поверхности рецепторы ЛПНП.
Рецептор ЛПНП - сложный белок, состоящий из 5 доменов и содержащий углеводную часть (рис. 8-68).
Рецепторы ЛПНП синтезируются в ЭР и аппарате Гольджи, а затем экспонируются на поверхности клетки, в специальных углублениях, выстланных белком клатрином. Эти углубления называют окаймлёнными ямками (рис. 8-69). Выступающий на поверхность N-концевой
домен рецептора взаимодействует с белками апоВ-100 и апоЕ; поэтому он может связывать
не только ЛПНП, но и ЛППП, ЛПОНП, остаточные ХМ, содержащие эти апопротеины. Клетки тканей содержат большое количество рецепторов ЛПНП на своей поверхности: например, на одной клетке фибробласта имеется от 20 000 до 50 000 рецепторов. Из этого следует, что холестерол поступает в клетки из крови в основном в составе ЛПНП.
Если количество холестерола, поступающего в клетку, превышает её потребность, то синтез рецепторов ЛПНП подавляется, что уменьшает
Рис. 8-68. Структура рецептора ЛПНП. Белок-рецептор состоит из 5 доменов. N - концевой домен, непосредственно связывает ЛПНП. Два других домена (связаны с олигосахаридами), выступающих на поверхности клетки, обеспечивают необходимую конформацию N-концевого домена ЛПНП.
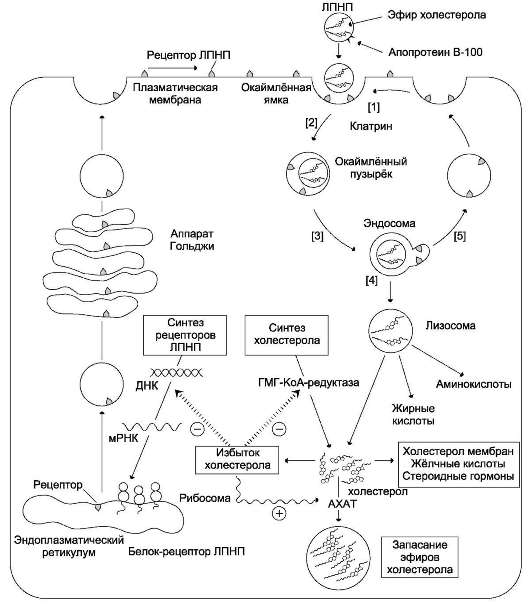
Рис. 8-69. Синтез рецепторов ЛПНП и их последующие превращения. После взаимодействия ЛПНП с рецептором (1) окаймлённые ямки вместе с рецептором и ЛПНП поглощаются по механизму эндоцитоза (2). В образовавшейся эндосоме снижается значение рН за счёт работы протонного насоса, использующего энергию АТФ. При снижении рН рецепторы ЛПНП отделяются от ЛПНП (3), и большая часть рецепторов возвращается в плазматическую мембрану (5). Таким образом, рецепторы ЛПНП могут многократно использоваться клеткой. После удаления рецептора ЛПНП эндосомы сливаются с лизосомами, и гидролитические ферменты лизосом расщепляют компоненты эндосом (4). В результате освобождается холестерол, который может быть использован для формирования структуры мембран, в клетках печени для синтеза жёлчных кислот, в клетках эндокринной системы для синтеза стероидных гормонов.
поток холестерола из крови в клетки. При снижении концентрации свободного холестерола в клетке, наоборот, активируется синтез ГМГ-КоА-редуктазы и рецепторов ЛПНП.
В регуляции синтеза рецепторов ЛПНП участвуют гормоны: инсулин и трийодтиронин (Т3), половые гормоны. Они увеличивают образование рецепторов ЛПНП, а глюкокортикоиды (в основном кортизол) уменьшают. Эффекты инсулина и Т3, вероятно, могут объяснить механизм гиперхолестеролемии и увеличение риска атеросклероза при сахарном диабете или гипотиреозе.
Другие пути поступления холестерола в клетки
Кроме рецепторов ЛПНП, на поверхности клеток многих органов (печени, мозга, плаценты) имеется другой тип рецептора, называемый «белком, сходным с рецептором ЛПНП». Этот рецептор взаимодействует с апоЕ и захватывает ремнантные (остаточные) ХМ и ЛППП. Основной функцией этих рецепторов, вероятно, является «очистка» плазмы крови от ремнантных частиц. Так как ремнантные частицы содержат холестерол, этот тип рецепторов также обеспечивает поступление его в ткани.
Кроме поступления холестерола в ткани путём эндоцитоза ЛП, некоторое количество холестерола поступает в клетки путём диффузии из ЛПНП и других ЛП при их контакте с мембранами клеток.
Роль ЛПВП в обмене холестерола
ЛПВП выполняют 2 основные функции: они поставляют апопротеины другим ЛП в крови и участвуют в так называемом «обратном транспорте холестерола». ЛПВП синтезируются в печени и в небольшом количестве в тонком кишечнике в виде «незрелых липопротеинов» - предшественников ЛПВП. Они имеют диско-видную форму, небольшой размер и содержат высокий процент белков и фосфолипидов. В печени в ЛПВП включаются апопротеины А, Е, С-II, фермент ЛХАТ. В крови апоС-II и апоЕ переносятся с ЛПВП на ХМ и ЛПОНП. Предшественники ЛПВП практически не содержат холестерола и ТАГ и в крови обогащаются холестеролом, получая его из других ЛП и мембран клеток.
Для переноса холестерола в ЛПВП существует сложный механизм. На поверхности ЛПВП находится фермент ЛХАТ - лецитин:холесте-рол-ацилтрансфераза. Этот фермент превращает холестерол, имеющий гидроксильную группу, выступающую на поверхность липопротеинов или мембран клеток, в эфиры холестерола. Радикал жирной кислоты переносится от фосфатидилхо-лина (лецитина) на гидроксильную группу холестерола. Реакция активируется апопротеином А-I, входящим в состав ЛПВП.
Гидрофобная молекула эфира холестерола перемещается внутрь ЛПВП. Таким образом, частицы ЛПВП обогащаются эфирами холес-терола. ЛПВП увеличиваются в размерах, из дисковидных небольших частиц превращаются в частицы сферической формы, которые называют ЛПВП3, или «зрелые ЛПВП». ЛПВП3 частично обменивают эфиры холестерола на триацилг-лицеролы, содержащиеся в ЛПОНП, ЛППП и ХМ (рис. 8-70). В этом переносе участвует «белок, переносящий эфиры холестерола» (он также называется апоD). Таким образом, часть эфиров холестерола переносится на ЛПОНП, ЛППП, а ЛПВП3 за счёт накопления триацилглицеролов увеличиваются в размерах и превращаются в ЛПВП2. ЛПОНП под действием ЛП-липазы превращаются сначала в ЛППП, а затем в ЛПНП. ЛПНП и ЛППП захватываются клетками через рецепторы ЛПНП.
Таким образом, холестерол из всех тканей возвращается в печень в основном в составе ЛПНП, но в этом участвуют также ЛППП и ЛПВП2. Практически весь холестерол, который должен быть выведен из организма, поступает в печень и уже из этого органа выделяется в виде производных с фекалиями. Путь возвращения холестерола в печень называют «обратным транспортом» холестерола.
Выведение холестерола из организма
Структурная основа холестерола - кольца циклопентанпергидрофенантрена - не может быть расщеплена до СО2 и воды, как другие органические компоненты, поступающие с пищей или синтезированные в организме. Поэтому основное количество холестерола выводится в виде жёлчных кислот.
Некоторое количество жёлчных кислот выделяется в неизменённом виде, а часть подвергается действию ферментов бактерий в кишечнике.
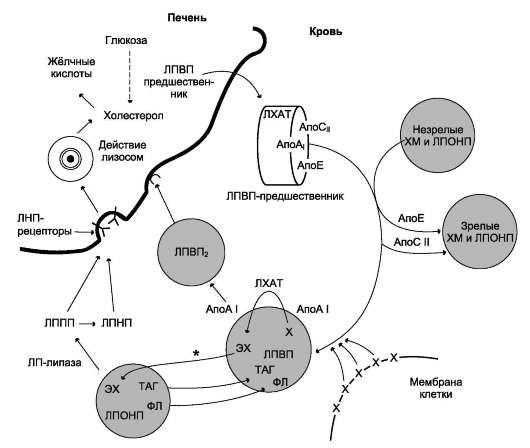
Рис. 8-70. Роль ЛПВП и ЛПНП в обратном транспорте холестерола в печень. Незрелые ЛПВП-предшествен-ники обогащаются холестеролом, который поступает в ЛПВП при участии фермента ЛХАТ с поверхности клеток и других липопротеинов, содержащих холестерол. Незрелые ЛПВП, обогащаясь холестеролом, превращаются в ЛПВП3 - частицы сферической формы и большего размера. ЛПВП3 обменивают эфиры холестерола на триацилглицеролы, содержащиеся в ЛПОНП, ЛППП при участии «белка, переносящего эфиры холестерола»*. ЛПВП3 превращается в ЛПВП2, размер которых увеличивается за счёт накопления триацилглицеролов. ЛПОНП и ЛППП под действием ЛП-липазы превращаются в ЛПНП, которые доставляют холестерол в печень. Часть ЛПВП захватывается клетками печени, взаимодействуя со специфическими для ЛПВП рецепторами к апоА-I. На поверхности клеток печени фосфолипиды и триацилглицеролы ЛППП, ЛПВП2 гидролизуются печёночной ЛП-липазой (Ω), что дестабилизирует структуру поверхности ЛП и способствует диффузии холестерола в гепатоциты. ЛПВП2 в результате этого опять превращаются в ЛПВП3 и возвращаются в кровоток. Х - холестерол, ЭХ - эфиры холестерола, ФЛ - фосфолипиды, ЛХАТ - лецитин-холестеролацилтрансфераза, А-I - апопротеин, активатор ЛХАТ.
Продукты их разрушения (в основном, вторичные жёлчные кислоты) выводятся из организма.
Часть
молекул холестерола в кишечнике под действием ферментов бактерий
восстанавливается по двойной связи в кольце В, в результате чего
образуются 2 типа молекул - холестанол и копростанол, выводимые с
фекалиями. В сутки из организма выводится от
В. СИНТЕЗ ЖЕЛЧНЫХ КИСЛОТ ИЗ ХОЛЕСТЕРОЛА И ЕГО РЕГУЛЯЦИЯ
Жёлчные кислоты синтезируются в печени из холестерола. Часть жёлчных кислот в печени подвергается реакции конъюгации - соединения с гидрофильными молекулами (глицином и таурином). Жёлчные кислоты обеспечивают эмульгирование жиров, всасывание продуктов их переваривания и некоторых гидрофобных веществ, поступающих с пищей, например жирорастворимых витаминов и холестерола. Жёлчные кислоты также всасываются, через воротную вену попадают опять в печень и многократно используются для эмульгирования жиров. Этот путь называют энте-рогепатической циркуляцией жёлчных кислот.
Синтез жёлчных кислот
В организме за сутки синтезируется 200600 мг жёлчных кислот. Первая реакция синтеза - образование 7-а-гидроксихолестерола - является регуляторной. Фермент 7-а-гидро-ксилаза, катализирующий эту реакцию, инги-бируется конечным продуктом - жёлчными кислотами. 7-а-Гидроксилаза представляет собой одну из форм цитохрома Р450 и использует кислород как один из субстратов. Один атом кислорода из О2 включается в гидроксильную группу в положении 7, а другой восстанавливается до воды. Последующие реакции синтеза приводят к формированию 2 видов жёлчных кислот: холевой и хенодезоксихолевой (рис. 8-71), которые называют «первичными жёлчными кислотами».
Конъюгирование жёлчных кислот
Конъюгирование - присоединение ионизированных молекул глицина или таурина к карбоксильной группе жёлчных кислот; усиливает их детергентные свойства, так как увеличивает амфифильность молекул.
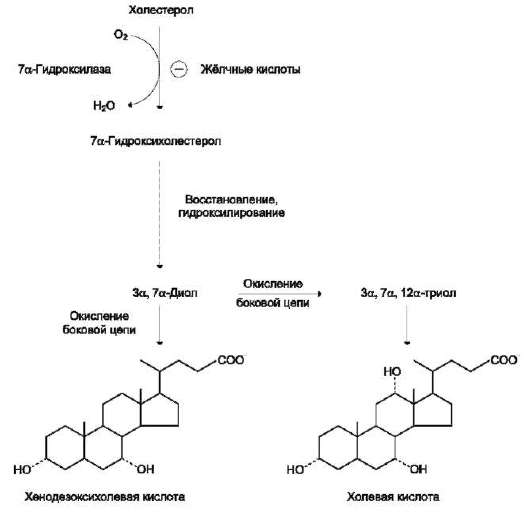
Рис. 8-71. Синтез первичных жёлчных кислот и его регуляция. В процессе синтеза жёлчных кислот холес-терол подвергается гидроксилированию, восстановлению двойной связи в положениях 5 и 6 и окислению боковой цепи. Образуется 2 типа жёлчных кислот: одна с гидроксильными группами в положениях 3 и 7, другая - с гидроксильными группами в положениях 3, 7, 12.
Конъюгация происходит в клетках печени и начинается с образования активной формы жёлчных кислот - производных КоА (рис. 8-72). Затем присоединяется таурин или глицин, и в результате образуется 4 варианта конъюгатов: таурохолевая и таурохенодезоксихолевая, глико-холевая или гликохенодезоксихолевая кислоты (они значительно более сильные эмульгаторы, чем исходные жёлчные кислоты).
Конъюгатов с глицином образуется в 3 раза больше, чем с таурином, так как количество таурина ограничено.
Энтерогепатическая циркуляция жёлчных кислот. Превращения жёлчных кислот в кишечнике
Продукты гидролиза жиров всасываются в основном в верхнем отделе тонкого кишечника, а соли жёлчных кислот - в подвздошной кишке. Около 95% жёлчных кислот, попавших в кишечник, возвращается в печень через ворот-
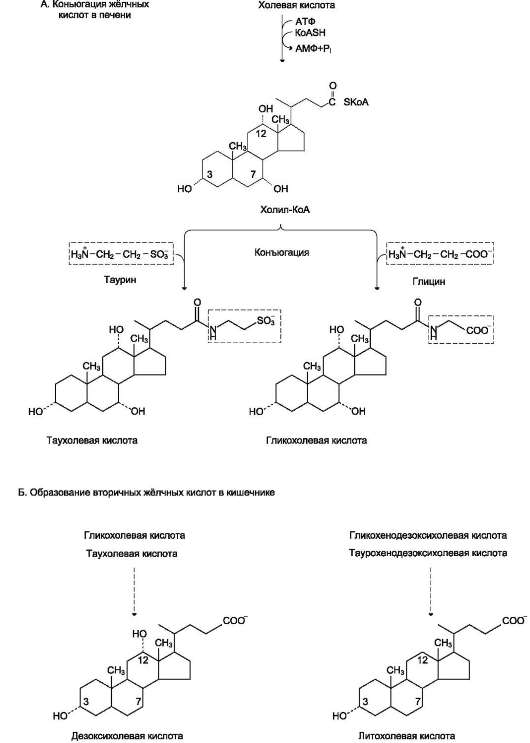
Рис. 8-72. Конъюгация жёлчных кислот в печени и разрушение в кишечнике. А - продукты конъюгации обладают лучшими детергентными свойствами, так как снижается константа диссоциации, и молекулы полностью диссоциированы при рН 6 в кишечнике. Конъюгации подвергаются холевая и хенодезоксихолевая кислоты; Б - в кишечнике небольшое количество жёлчных кислот под действием ферментов бактерий превращаются в литохолевую и дезоксихолевую кислоты.
ную вену, затем опять секретируются в жёлчь и повторно используются в эмульгировании жиров (рис. 8-73). Этот путь жёлчных кислот называют энтерогепатической циркуляцией. В сутки всего реабсорбируется 12-32 г солей жёлчных кислот, так как в организме имеется 2-4 г жёлчных кислот, и каждая молекула жёлчной кислоты проходит этот круг 6-8 раз.
Часть жёлчных кислот в кишечнике подвергается действию ферментов бактерий, которые
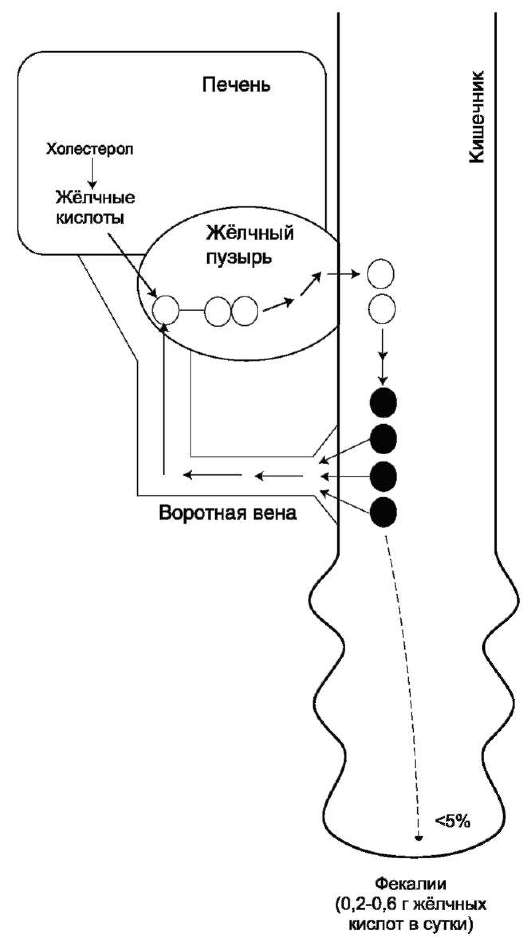
Рис. 8-73. Энтерогепатическая циркуляция жёлчных кислот. Светлые кружки - мицеллы жёлчи; тёмные кружки - смешанные мицеллы жёлчи и продуктов гидролиза триацилглицеролов.
отщепляют глицин и таурин, а также гидрок-сильную группу в положении 7 жёлчных кислот. Жёлчные кислоты, лишённые этой гидроксиль-ной группы, называют вторичными. Вторичные жёлчные кислоты: дезоксихолевая, образующаяся из холевой, и литохолевая, образующаяся из дезоксихолевой, хуже растворимы, медленнее всасываются в кишечнике, чем первичные жёлчные кислоты. Поэтому с фекалиями в основном удаляются вторичные жёлчные кислоты. Однако реабсорбированные вторичные жёлчные кислоты в печени опять превращаются в первичные и участвуют в эмульгировании жиров. За сутки из организма выводится 500-600 мг жёлчных кислот. Путь выведения жёлчных кислот одновременно служит и основным путём выведения холестерола из организма. Для восполнения потери жёлчных кислот с фекалиями в печени постоянно происходит синтез жёлчных кислот из холестерола в количестве, эквивалентном выведенным жёлчным кислотам. В результате пул жёлчных кислот (2-4 г) остаётся постоянным.
Регуляция синтеза жёлчных кислот
Регуляторные ферменты синтеза жёлчных кислот (7-а-гидроксилаза) и холестерола (ГМГ-КоА-редуктаза) ингибируются жёлчными кислотами. В течение суток активность обоих ферментов меняется сходным образом, т.е. увеличение количества жёлчных кислот в печени приводит к снижению синтеза как жёлчных кислот, так и холестерола. Возвращение жёлчных кислот в печень в процессе энтерогепатической циркуляции оказывает важное регуляторное действие; прерывание циркуляции приводит к активации 7-а-гидроксилазы и увеличению захвата холестерола из крови. Этот механизм лежит в основе одного из способов снижения концентрации холестеро-ла в крови при лечении гиперхолестеролемии. В этом случае используют препараты, адсорбирующие в кишечнике холестерол и жёлчные кислоты и препятствующие их всасыванию.
Регуляция 7-а-гидроксилазы осуществляется и другими механизмами:
фосфорилированием/дефосфорилированием, причём активна фосфорилированная форма, в отличие от ГМГ-КоА-редуктазы; изменением количества фермента; холестерол индуцирует транскрипцию гена, а жёлчные кислоты репрессируют.
На синтез 7-а-гидроксилазы влияют гормоны: тиреоидные гормоны индуцируют синтез, а эстрогены - репрессируют. Такое влияние эстрогенов на синтез жёлчных кислот объясняет, почему желчнокаменная болезнь встречается у женщин в 3-4 раза чаще, чем у мужчин.
Г. ЖЕЛЧНОКАМЕННАЯ БОЛЕЗНЬ
Желчнокаменная болезнь - патологический процесс, при котором в жёлчном пузыре образуются камни, основу которых составляет холестерол.
Выделение холестерола в жёлчь должно сопровождаться пропорциональным выделением жёлчных кислот и фосфолипидов, удерживающих гидрофобные молекулы холестерола в жёлчи в мицеллярном состоянии (табл. 8-9).
У большинства больных желчнокаменной болезнью активность ГМГ-КоА-редуктазы повышена, следовательно увеличен синтез холестерола, а активность 7-а-гидроксилазы, участвующей в синтезе жёлчных кислот, снижена. В результате синтез холестерола увеличен, а синтез жёлчных кислот из него замедлен, что приводит к диспропорции количества холестерола и жёлчных кислот, секретируемых в жёлчь.
Если эти пропорции нарушены, то холесте-рол начинает осаждаться в жёлчном пузыре, образуя вначале вязкий осадок, который постепенно становится более твёрдым. Иногда он пропитывается билирубином - продуктом распада гема, белками и солями кальция. Камни, образующиеся в жёлчном пузыре, могут состоять только из холестерола (холестериновые камни) или из смеси холестерола, билирубина, белков и кальция. Холестериновые камни обычно белого цвета, а смешанные камни - коричневого цвета разных оттенков. Причин, приводящих к изменению соотношения жёлчных кислот и холестерола,
Таблица 8-9. Компоненты жёлчи
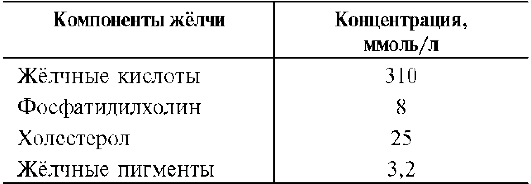
в жёлчи много: пища, богатая холестеролом, гиперкалорийное питание, застой жёлчи в жёлчном пузыре, нарушение энтерогепатической циркуляции, нарушение синтеза жёлчных кислот, инфекции жёлчного пузыря.
Если камни начинают перемещаться из жёлчного пузыря в жёлчные протоки, то они вызывают спазм жёлчного пузыря и протоков, что больной ощущает как приступ сильной боли. Если камень перекрывает проток некоторое время, то нарушается поступление жёлчи в кишечник, жёлчные пигменты проходят через мембраны гепатоцитов в сторону синусоидов и попадают в кровь, что приводит к развитию обтурационной (подпечёночной желтухи).
Лечение желчнокаменной болезни. В начальной стадии образования камней можно применять в качестве лекарства хенодезоксихолевую кислоту. Попадая в жёлчный пузырь, эта жёлчная кислота постепенно растворяет осадок холестерола (холестериновые камни), однако это медленный процесс, требующий нескольких месяцев.
Д. ДИСЛИПОПРОТЕИНЕМИИ. ГИПЕРХОЛЕСТЕ-РОЛЕМИЯ И РАЗВИТИЕ АТЕРОСКЛЕРОЗА
Дислипопротеинемии - нарушения обмена ЛП крови и, соответственно, нарушения обмена липидов, транспортируемых ЛП. Дислипопро-теинемии проявляются чаще всего повышением концентрации либо одного типа ЛП, либо со-четанным увеличением содержания нескольких типов ЛП. В настоящее время имеется несколько классификаций дислипопротеинемий. Основная классификация представлена в табл. 8-10.
Наиболее распространены нарушения обмена холестерола и триацилглицеролов.
Нарушения обмена холестерола чаще всего приводят к гиперхолестеролемии и последующему развитию атеросклероза. При атеросклерозе происходит образование на стенках артерий так называемых атеросклеротических бляшек, представляющих собой в основном отложения холес-терола. Атеросклеротические бляшки разрушают клетки эндотелия сосудов, и в таких местах часто образуются тромбы. Атеросклероз - полигенное заболевание. Одна из основных причин развития атеросклероза - нарушение баланса между поступлением холестерола с пищей, его синтезом и выведением из организма. Выведение холестерола ограничено, не превышает 1,2-1,5 г/сут, а
Таблица 8-10. Дислипопротеинемии
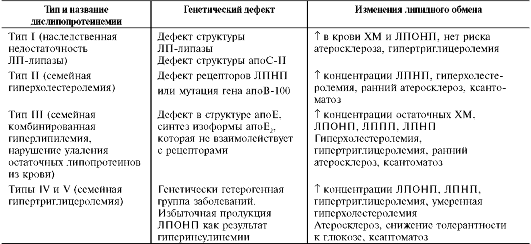
поступление с пищей при неправильном питании может превысить этот барьер, поэтому с возрастом постепенно происходит накопление холес-терола в организме. Важным фактором развития атеросклероза являются генетические дефекты белков и ферментов, участвующих в обмене холестерола.
Гиперхолестеролемия. Роль алиментарных факторов в развитии гиперхолестеролемии
Концентрация холестерола в крови взрослых людей должна быть <200 мг/дл (<5,2 ммоль/л) и, часто, увеличивается с возрастом. Превышение нормальной концентрации холестерола в крови называют гиперхолестеролемией.
Гиперхолестеролемия обычно развивается вследствие избыточного поступления холестерола с пищей, а также углеводов и жиров. Гиперкалорийное питание - один из распространённых факторов развития гиперхолестеролемии, так как для синтеза холестерола необходимы только ацетил-КоА, АТФ и NADРН. Все эти субстраты образуются при окислении глюкозы и жирных кислот, поэтому избыточное поступление этих компонентов пищи способствует развитию ги-перхолестеролемии. В норме поступление холе-стерола с пищей снижает синтез собственного холестерола в печени, однако с возрастом эффективность регуляции у многих людей снижается.
Правильное питание в течение всей жизни важнейший фактор профилактики гиперхолесте-ролемии. Доказана корреляция между увеличением концентрации холестерола в плазме крови и смертностью от заболеваний ССС - инфаркта миокарда и инсульта, развивающихся в результате атеросклероза (рис. 8-74).
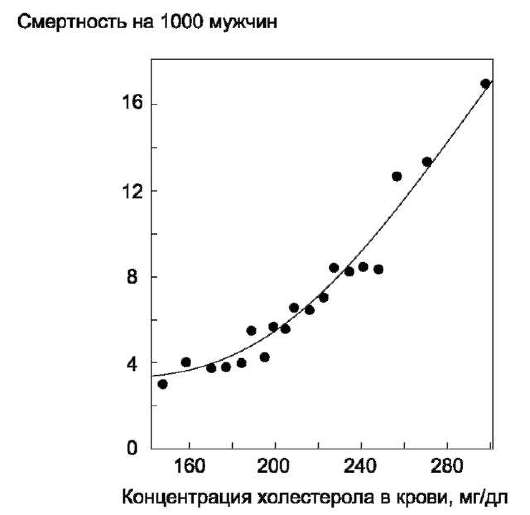
Рис. 8-74. Корреляция между концентрацией холес-терола в крови и смертностью от заболеваний ССС на 1000 мужчин.
Ген рецептора ЛПНП: структура и типы мутаций
Наследственные факторы играют важную роль в предрасположенности к развитию атеросклероза. Наиболее часто встречаются мутации в структуре гена рецептора ЛПНП.
Ген рецептора ЛПНП находится в хромосоме 19 и состоит из 18 экзонов (рис. 8-75). Различные группы экзонов кодируют различные домены в составе этого белка. Мутации в этом гене подробно изучены и разделены на 4 класса.
Первый класс мутаций, наиболее распространённый, приводит к полному отсутствию рецептора; второй класс мутаций характеризуется тем, что рецептор синтезируется, но не может транспортироваться на поверхность клетки; третий класс мутаций соответствует ситуации, когда рецептор транспортируется на поверхность клеток, но не связывает ЛПНП; четвёртый класс мутаций - рецептор связывает ЛПНП, но не происходит эндоцитоз. Изменения структуры рецепторов ЛПНП в результате всех типов мутаций приводит к гиперхолестеролемии, так как ЛПНП не захватываются клетками, и холестерол в составе ЛПНП накапливается в крови.
Семейная гиперхолестеролемия
Любой дефект рецептора ЛПНП или белка апоВ-100, взаимодействующего с ним, приводит к развитию наиболее распространённого наследственного заболевания - семейной гиперхолесте-ролемии. Причиной этого аутосомно-доминан-тного заболевания выступают указанные выше мутации в гене рецептора ЛПНП. Гетерозиготы, имеющие один нормальный ген, а другой де-
фектный, встречаются с частотой 1:500 человек, у некоторых народностей Африки - даже 1:100 человек. Количество рецепторов ЛПНП на поверхности клеток у гетерозигот снижено вдвое, а концентрация холестерола в плазме, соответственно, вдвое повышается. У гетерозигот концентрация холестерола в крови в 35-40 лет достигает 400-500 мг/дл, что приводит к выраженному атеросклерозу и ранней смерти в результате инфаркта миокарда или инсульта. Гомозиготы встречаются редко - 1:1 000 000 человек. Концентрации холестерола и ЛПНП в крови таких больных уже в раннем детском возрасте увеличены в 5-6 раз. ЛПНП захватываются макрофагами путём фагоцитоза. Макрофаги, нагруженные избытком холестерола и других липидов, содержащихся в ЛПНП, откладываются в коже и даже сухожилиях, образуя так называемые ксантомы. Холестерол откладывается также и в стенках артерий, образуя атеросклеро-тические бляшки. Такие дети без экстренных мер лечения погибают в возрасте 5-6 лет. Лечение данной формы заболевания проводят путём удаления ЛПНП из крови с помощью плазмафереза, но наиболее радикальный метод лечения - трансплантация печени. Печень донора с нормальным количеством рецепторов ЛПНП существенно понижает концентрацию холестерола в крови и предотвращает раннюю смерть от атеросклероза.
Кроме генетических дефектов рецептора ЛПНП, причинами гиперхолестеролемии и, следовательно, атеросклероза являются наследственные дефекты в структуре апоВ-100, а также повышенные синтез или секреция апоВ-100 в случае семейной комбинированной гиперлипидемии, при которой в крови повы-
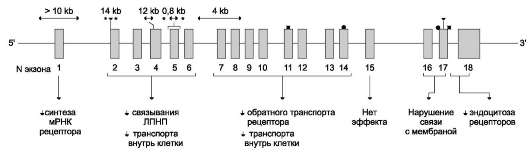
• Рис. 8-75. Типы мутаций в гене белка-рецептора ЛПНП. Экзоны представлены тёмными прямоугольниками, интроны - линиями, соединяющими экзоны. На рисунке показаны типы мутаций: ↔ - делеция, - нонсенс-мутация, ■ - миссенс-мутация; kb - тысячи оснований.
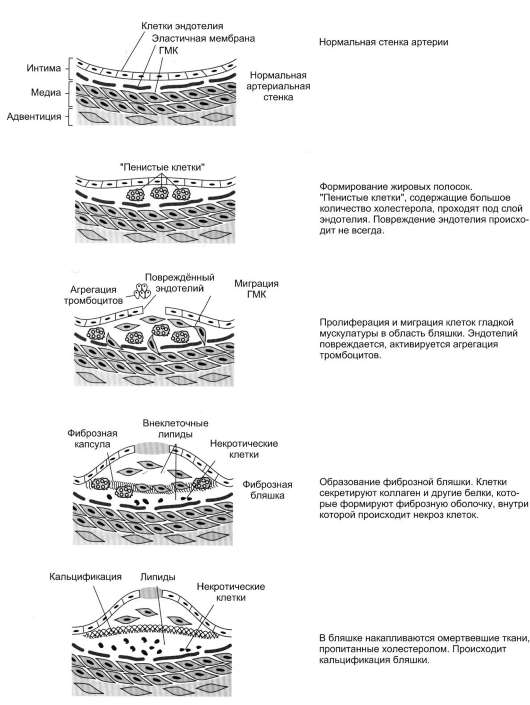
Рис. 8-76. Развитие атеросклеротической бляшки в клетках эндотелия кровеносных сосудов.
шены концентрации и холестерола и триаци-лглицеролов.
Химическая модификация липидов и белков ЛПНП и рецепторов ЛПНП
Изменение нормальной структуры липидов и белков в составе ЛПНП делает их чужеродными для организма и поэтому более доступными для захвата фагоцитами. Например, активация свободнорадикального окисления липидов приводит к изменению не только структуры липидов в липопротеинах, но и структуры апоВ-100. Другим фактором, изменяющим структуру как ЛПНП, так и рецептора ЛПНП, является неферментативное гликозилирование белков, происходящее при увеличении концентрации глюкозы в крови при сахарном диабете. Модифицированные ЛПНП поглощаются макрофагами с участием «скевенджер-рецепторов» (рецепторов-мусорщиков).
Молекулярные механизмы патогенеза атеросклероза
Развитие атеросклероза проходит несколько стадий (рис. 8-76).
Процесс начинается с повреждения эндотелия сосудов, причём повреждение может иметь различные механизмы. Важнейший механизм - повреждение эндотелия за счёт изменённой структуры ЛПНП, например в результате активации свободнорадикального ПОЛ в составе ЛПНП; повреждение провоцируется свободными радикалами, образующимися в процессе метаболизма или поступающими извне. В ходе ПОЛ в ЛПНП изменяется не только структура самих липидов, но и нарушается структура апопротеинов. Окисленные ЛПНП захватываются макрофагами через скевенджер-рецепто-ры. Этот процесс не регулируется количеством поглощённого холестерола, как в случае его поступления в клетки через специфические рецепторы, поэтому макрофаги перегружаются холестеролом и превращаются в «пенистые клетки», которые проникают в субэндотелиальное пространство. Это приводит к образованию жировых полосок в стенке кровеносных сосудов. На этой стадии эндотелий сосудов может сохранять свою структуру. При увеличении количества «пенистых клеток» происходит повреждение эндотелия сосудов. В норме клетки эндотелия
секретируют простагландин I2 (простациклин I2), который ингибирует агрегацию тромбоцитов. При повреждении клеток эндотелия тромбоциты активируются. Во-первых, они секретируют тромбоксан А2 (ТХ А2), который стимулирует агрегацию тромбоцитов, что может привести к образованию тромба в области атеросклероти-ческой бляшки; во-вторых, тромбоциты начинают продуцировать пептид - тромбоцитарный фактор роста, стимулирующий пролиферацию ГМК. ГМК мигрируют из медиального слоя во внутренний слой артериальной стенки и способствуют таким образом росту бляшки. Далее происходит прорастание бляшки фиброзной тканью (коллагеном, эластином); клетки под фиброзной оболочкой некротизируются, а холестерол откладывается в межклеточном пространстве. На этой стадии в центре бляшки образуются даже холестериновые кристаллы. На последних стадиях развития бляшка пропитывается солями кальция и становится очень плотной. В области бляшки часто образуются тромбы, перекрывающие просвет сосуда, что приводит к острому нарушению кровообращения в соответствующем участке ткани и развитию инфаркта. Чаще всего атеросклеротические бляшки развиваются в артериях миокарда, поэтому наиболее распространённое заболевание, развивающееся в результате атеросклероза, - инфаркт миокарда.
Биохимические основы лечения атеросклероза и предупреждения развития инфаркта миокарда
Важным лечебным фактором, снижающим риск развития гиперхолестеролемии и атеросклероза, является гипокалорийная и гипохолес-териновая диета. Поступление холестерола с пищей не должно превышать 300 мг/сут (табл.
8-11).
Холестерол - стероид животного происхождения, поэтому он поступает в организм при употреблении животных жиров и жирного мяса. Растительная пища не содержит холестерола, поэтому у людей среднего и старшего возраста она должна составлять основу рациона.
К лечебным и профилактическим факторам относят обогащение пищи полиеновыми жирными кислотами семейства ω-3, уменьшающими риск тромбообразования. Ненасыщенные жирные кислоты способствуют более быстрому
Таблица 8-11. Основы диеты, снижающей количество холестерола и жиров в организме человека
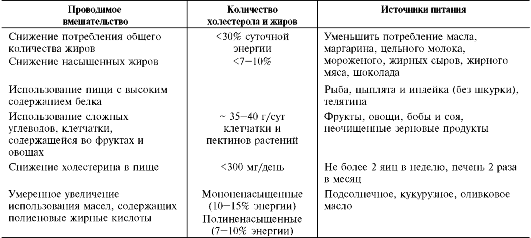
выведению холестерола из организма, хотя механизм этого явления до конца не выяснен. В то же время доказано, что полиеновые кислоты подавляют синтез тромбоцитарного фактора роста и таким образом замедляют развитие ате-росклеротической бляшки.
Витамины С, Е, А, обладающие антиокси-дантными свойствами, ингибируют перекисное (свободнорадикальное) окисление липидов в ЛПНП и поддерживают нормальную структуру липидов ЛПНП и их метаболизм.
Однако меры по исправлению диеты недостаточны при лечении выраженной гиперхолесте-
ролемии и атеросклерозе. Лечение гиперхолес-теролемии, как правило, комплексное.
Один из принципов лечения - «размыкание» цикла энтерогепатической циркуляции жёлчных кислот. Для этого используют лекарства типа холестирамина - полимера, который в кишечнике адсорбирует жёлчные кислоты, выделяется с фекалиями и таким образом уменьшает возврат жёлчных кислот в печень. В печени увеличивается захват холестерола из крови для синтеза новых жёлчных кислот. Препараты типа холестирамина называют секвестрантами жёлчных кислот.
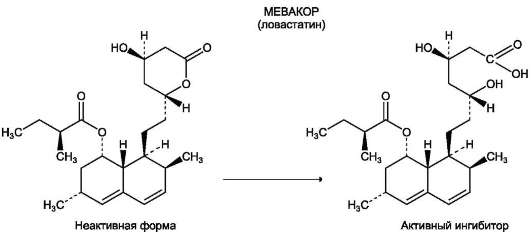
Рис. 8-77. Образование активной формы мевакора.
Наиболее эффективные препараты, применяемые при лечении атеросклероза, - ингибиторы ГМГ-КоА-редуктазы. Эти препараты - антибиотики, например мевакор, в печени трансфор-мируются в активную форму (рис. 8-77) и эффективно ингибируют регуляторный фермент биосинтеза холестерола. Такие препараты могут практически полностью подавить синтез собственного холестерола в организме. В этих условиях печень увеличивает захват хо-лестерола из крови. Для этого в клетках печени почти вдвое увеличивается синтез белков-рецепторов ЛПНП и, соответственно, увеличивается захват ЛПНП из крови. Таким образом концентрация холестерола в крови даже у больных с гетерозиготной формой семейной
гиперхолестеролемии может быть доведена практически до нормы.
Лекарственные препараты - фибраты (кло-фибрат, фенофибрат) - ускоряют катаболизм ЛПОНП, активируя ЛП-липазу. Эти препараты также активируют окисление жирных кислот в печени, уменьшая тем самым синтез триацилг-лицеролов и эфиров холестерола и, как следствие, секрецию ЛПОНП печенью. Клофибрат индуцирует синтез ферментов пероксисом, способных окислять жирные кислоты. Фибраты обычно применяют при сочетании гипертриг-лицеролемии и гиперхолестеролемии. Для эффективного лечения атеросклероза применяют, как правило, комбинированное воздействие нескольких лекарственных препаратов.