Биохимия: учебник для вузов/ под ред. Е.С.Северина - 5-е изд., - 2009. - 768 с.
|
|
|
|
РАЗДЕЛ 14 БИОХИМИЯ КРОВИ
Кровь - жидкая внутренняя среда организма. Общий объём крови взрослого человека составляет 5-6 л. Кровь состоит из жидкой части - плазмы, составляющей 55% её общего объёма, и форменных элементов, к которым относят эритроциты, лейкоциты и тромбоциты.
Благодаря работе сердца кровь циркулирует по замкнутой системе кровеносных сосудов и осуществляет транспорт различных химических веществ. Она переносит кислород из лёгких к тканям и углекислый газ из тканей в лёгкие в составе гемоглобина эритроцитов (дыхательная функция); доставляет продукты переваривания пищи из кишечника в ткани (трофическая функция); уносит конечные продукты обмена из тканей в выделительные органы (выделительная функция); перемещает промежуточные продукты обмена веществ, синтез и использование которых происходит в разных органах.
Кровь участвует в регуляции обмена веществ, доставляя сигнальные молекулы от органов внутренней секреции к тканям-мишеням.
Защитная функция крови имеет две стороны. Во-первых, в ней содержатся клеточные (лейкоциты) и гуморальные (антитела) элементы иммунного реагирования, которые защищают организм от любой чужеродной молекулы. Во-вторых, это способность крови свёртываться. При повреждении сосуда прерывается замкнутость циркуляции крови, а уменьшение количества крови может привести к серьёзным нарушениям функций клеток, вплоть до их гибели. Кровь здорового человека образует тромб в месте повреждения, который закупоривает просвет повреждённого сосуда и останавливает кровотечение.
Кровь поддерживает кислотно-щелочной и водный баланс организма. В норме рН крови составляет 7,36-7,4. Сохранение постоянства рН является важнейшей задачей, так как в кровь выделяется большое количество кислых (например, лактат, кетоновые тела, угольная кислота),
а также основных (аммиак) продуктов метаболизма. Регуляцию рН осуществляют буферные системы крови, которые подробно рассмотрены в курсе физиологии.
Выполняя терморегуляторную функцию, кровь поддерживает постоянство температуры тела в разных его частях.
Химический состав растворимых в плазме крови веществ относительно постоянен, так как существуют мощные нервные и гуморальные механизмы, поддерживающие гомеостаз (постоянство внутренней среды). Растворимые вещества плазмы составляют около 10% массы крови, из них на долю белков приходится около 7%, на долю неорганических солей - 0,9%, остальную часть образуют небелковые органические соединения. Диапазон концентраций разных веществ плазмы крови у здорового человека представлен в специальных биохимических справочниках и является важнейшим материалом для медицинской биохимии.
Кровь связана со всеми тканями организма, поэтому возникновение патологического процесса в каком-либо органе приводит к изменению биохимических показателей крови. Эта информация может быть ценной при постановке диагноза и оценке эффективности лечебных мероприятий.
I. МЕТАБОЛИЗМ ЭРИТРОЦИТОВ
Эритроциты - высокоспециализированные клетки, которые переносят кислород от лёгких к тканям и диоксид углерода, образующийся при метаболизме, из тканей к альвеолам лёгких. Транспорт О2 и СО2 в этих клетках осуществляет гемоглобин, составляющий 95% их сухого остатка. Организм взрослого человека содержит около 25х1012 эритроцитов, при этом каждые сутки обновляется примерно 1% этого количества клеток, т.е. в течение одной секунды в кровоток поступает около 2 млн эритроцитов.
А. ОСОБЕННОСТИ СТРОЕНИЯ И ДИФФЕРЕНЦИРОВКИ ЭРИТРОЦИТОВ
Эритроциты - единственные клетки, которые имеют только клеточную мембрану и цитоплазму. Дифференцировка стволовых клеток в специализированные происходит в клетках костного мозга и заканчивается в кровотоке. Особенности строения эритроцитов соответствуют их функциям: большая площадь поверхности обеспечивает эффективность газообмена, эластичная клеточная мембрана облегчает движение по узким капиллярам, специальная ферментативная система защищает эти клетки от активных форм кислорода.
Дифференцировка эритроцитов. Эритроциты, так же как и другие клетки крови, образуются из полипотентных стволовых клеток костного мозга (рис. 14-1).
Размножение и превращение начальной клетки эритроидного ряда в унипотентную стимулирует ростовой фактор интерлейкин-3. Интерлейкин-3 синтезируется Т-лимфоцитами, а также клетками костного мозга. Это низкомо-
лекулярный белок группы цитокинов - регуляторов роста и дифференцировки клеток.
Дальнейшую пролиферацию и дифференци-ровку унипотентной клетки эритроидного ряда регулирует синтезирующийся в почках гормон эритропоэтин. Скорость образования эритро-поэтина в почках зависит от парциального давления кислорода. При недостатке кислорода скорость образования гормона повышается и, соответственно, количество эритроцитов тоже увеличивается. Хроническая почечная недостаточность сопровождается снижением образования эритропоэтина в почках, что приводит к развитию анемии.
В процессе дифференцировки на стадии эритробласта происходят интенсивный синтез гемоглобина, конденсация хроматина, уменьшение размера ядра и его удаление. Образующийся ретикулоцит ещё содержит глобиновую мРНК и активно синтезирует гемоглобин. Циркулирующие в крови ретикулоциты лишаются рибосом, ЭР, митохондрий и в течение двух суток превращаются в эритроциты. Стволовая
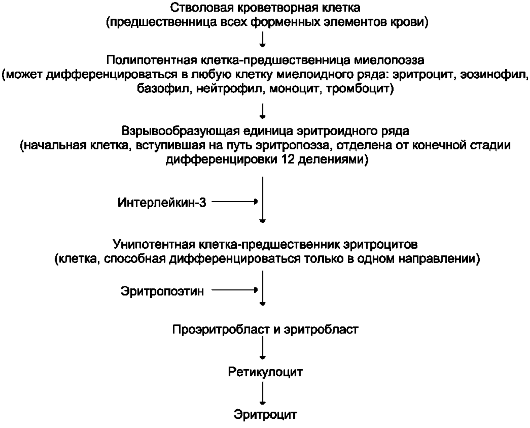
Рис. 14-1. Схема дифференцировки стволовых клеток костного мозга в зрелые эритроциты.
клетка превращается в эритроцит за две недели. Эритроциты не содержат ядра и поэтому не способны к самовоспроизведению и репарации возникающих в них повреждений. Эти клетки циркулируют в крови около 120 дней и потом разрушаются макрофагами в печени, селезёнке и костном мозге (см. раздел 13).
Строение эритроцитов. Двояковогнутая форма эритроцитов имеет большую площадь поверхности по сравнению с клетками сферической формы такого же размера. Это облегчает газообмен между клеткой и внеклеточной средой. Кроме того, такая форма, а также особенности строения мембраны и цитоскелета обеспечивают большую пластичность эритроцитов при прохождении ими мелких капилляров.
Важную роль в сохранении формы и способности к обратимой деформации эритроцитов играют липиды и белки плазматической мембраны. Липиды бислоя плазматической мембраны эритроцитов, так же, как плазматические мембраны других клеток, содержат глицеро-фосфолипиды, сфингофосфолипиды, гликоли-пиды и холестерол (см. раздел 5). Увеличение содержания холестерола в составе мембраны, которое может наблюдаться при некоторых заболеваниях, снижает её текучесть и эластичность, а следовательно, и способность к обратимой деформации. Это, в свою очередь, затрудняет движение эритроцитов через капилляры и может способствовать развитию гемостаза.
Методом электрофореза в мембране эритроцитов обнаруживают около 15 основных мембранных белков с молекулярной массой от 15 до 250 кД. Около 60% массы мембранных белков приходится на спектрин, гликофорин и белок полосы 3 (называется так по расположению этой белковой фракции на электрофореграмме относительно других белков). Интегральный гликопротеин гликофорин присутствует только в плазматической мембране эритроцитов (рис. 14-2). К N-концевой части белка, расположенной на наружной поверхности мембраны, присоединено около 20 олигосахаридных цепей (см. раздел 5). Олигосахариды гликофорина - антигенные детерминанты системы групп крови АВ0 (см. раздел 10).
Спектрин - периферический мембранный белок, нековалентно связанный с цитоплаз-матической поверхностью липидного бислоя мембраны. Он представляет собой длинную,
тонкую, гибкую фибриллу и является основным белком цитоскелета эритроцитов. Спектрин состоит из α- и β-полипептидных цепей, имеющих доменное строение; α- и β-цепи димера расположены антипараллельно, перекручены друг с другом и нековалентно взаимодействуют во многих точках. Спектрин может прикрепляться к мембране и с помощью белка анкирина. Этот крупный белок соединяется с β-цепью спектрина и цитоплазматическим доменом интегрального белка мембраны - белка полосы 3. Анкирин не только фиксирует спектрин на мембране, но и уменьшает скорость диффузии белка полосы 3 в липидном слое. Таким образом, на цитоплазматической поверхности эритроцитов образуется гибкая сетевидная структура, которая обеспечивает сохранение их формы при прохождении через узкие капилляры сосудов (рис. 14-2).
Интегральный белок полосы 3 - белок-переносчик ионов С1- и НСО3- через плазматическую мембрану эритроцитов по механизму пассивного антипорта. В разделе 1 подробно описана роль эритроцитов в газообмене. Поступающий из тканей в эритроциты СО2 под действием фермента карбоангидразы превращается в слабую угольную кислоту, которая распадается на Н+ и НСО3-. Образующиеся при этом протоны присоединяются к гемоглобину, уменьшая его сродство к О2, а бикарбонаты с помощью белка полосы 3 обмениваются на С1- и выходят в плазму крови.
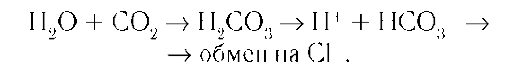
В лёгких увеличение парциального давления кислорода и взаимодействие его с гемоглобином приводят к вытеснению протонов из гемоглобина, обмену внутриклеточного С1- на НСО3- через белок полосы 3, образованию угольной кислоты и её разрушению на СО2 и Н2О.
Мембранный фермент Na+,К+-АТФ-аза обеспечивает поддержание градиента концентраций Na+ и K+ по обе стороны мембраны. При снижении активности Na+,К+-АТФ-азы концентрация Na+ в клетке повышается, так как небольшие ионы могут проходить через мембрану простой диффузией. Это приводит к увеличению осмотического давления, увеличению поступления воды в эритроцит и к его гибели в результате разрушения клеточной мембраны - гемолизу.
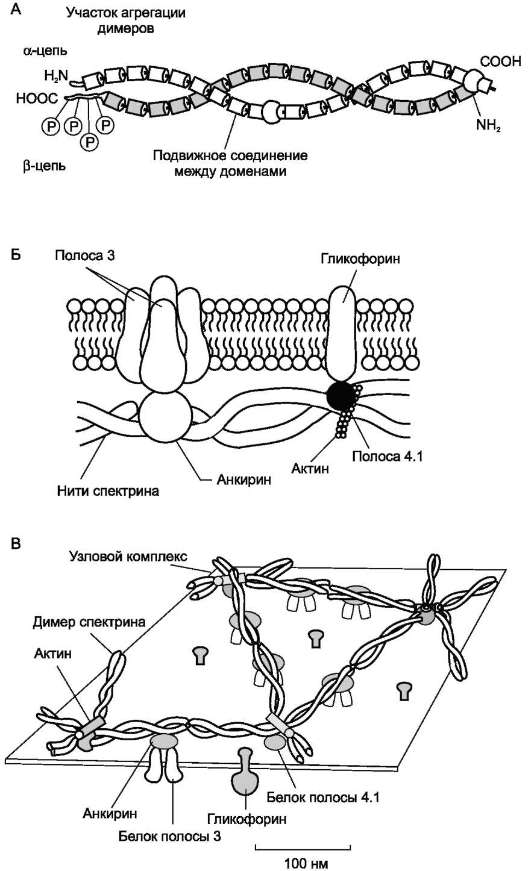
Рис. 14-2. Строение спектрина (А), околомембранного белкового комплекса (Б) и цитоскелета эритроцитов (В). Каждый димер спектрина состоит из двух антипараллельных, нековалентносвязанных между собой α- и β-полипептидных цепей (А). Белок полосы 4.1 образует со спетрином и актином «узловой комплекс», который посредством белка полосы 4.1 связывается с цитоплазматическим доменом гликофорина. Анкирин соединяет спектрин с основным интегральным белком плазматической мембраны - белком полосы 3 (Б). На цитоплаз-матической поверхности мембраны эритроцита имеется гибкая сетеобразная структура, состоящая из белков и обеспечивающая пластичность эритроцита при прохождении им через мелкие капилляры (В).
Са2+-АТФ-аза - ещё один мембранный фермент, осуществляющий выведение из эритроцитов ионов кальция и поддерживающий градиент концентрации этого иона по обе стороны мембраны.
Б. МЕТАБОЛИЗМ ГЛЮКОЗЫ В ЭРИТРОЦИТАХ
Эритроциты лишены митохондрий, поэтому в качестве энергетического материала они могут использовать только глюкозу. В эритроцитах катаболизм глюкозы обеспечивает сохранение структуры и функции гемоглобина, целостность мембран и образование энергии для работы ионных насосов. Глюкоза поступает в эритроциты путём облегчённой диффузии с помощью ГЛЮТ-2. Около 90% поступающей глюкозы используется в анаэробном гликолизе, а остальные 10% - в пентозофосфатном пути.
Конечный продукт анаэробного гликолиза лактат выходит в плазму крови и используется в других клетках, прежде всего гепатоцитах. АТФ, образующийся в анаэробном гликолизе, обеспечивает работу Na+,K+-ATФ-азы и поддержание самого гликолиза, требующего затраты АТФ в гексокиназной и фосфофруктокиназной реакциях (см. раздел 7).
Важная особенность анаэробного гликолиза в эритроцитах по сравнению с другими клетками - присутствие в них фермента бисфос-фоглицератмутазы. Бисфосфоглицератмутаза катализирует образование 2,3-бисфосфогли-церата из 1,3-бисфосфоглицерата (рис. 14-3). Образующийся только в эритроцитах 2,3-бис-фосфоглицерат служит важным аллостеричес-ким регулятором связывания кислорода гемоглобином (см. раздел 1).
Глюкоза в эритроцитах используется и в пентозофосфатном пути, окислительный этап которого обеспечивает образование кофермен-та NАDРН, необходимого для восстановления глутатиона (рис. 14-4).
В. ОБЕЗВРЕЖИВАНИЕ АКТИВНЫХ ФОРМ КИСЛОРОДА В ЭРИТРОЦИТАХ
• Большое содержание кислорода в эритроцитах определяет высокую скорость образования супероксидного анион-радикала (О2-), пероксида водорода (Н2О2) и гидроксил радикала (ОН ). Эритроциты содержат ферментативную систему, предотвращающую токсическое действие активных форм кислорода и разрушение мембран эритроцитов (рис. 14-4). Постоянный источник
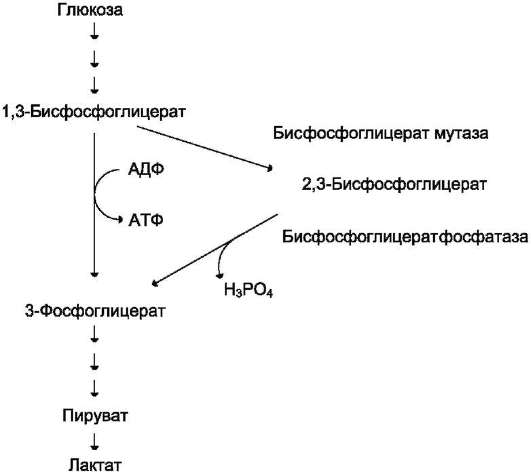
Рис. 14-3. Метаболизм 2,3-бисфосфоглицерата в эритроцитах.
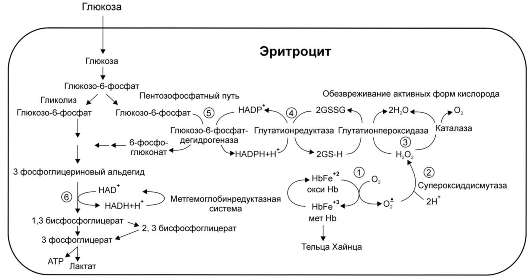
Рис. 14-4. Образование и обезвреживание активных форм кислорода в эритроците:
1 - спонтанное окисление Fe2+ в геме гемоглобина - источник супероксидного аниона в эритроцитах;
2 - супероксиддисмутаза превращает супероксидный анион в пероксид водорода и воду: O2- + O2- +2 H+ → H2O2 + O2;
3 - пероксид водорода расщепляется каталазой: 2 н202 → 2 н20 + 02 или глутатионпероксидазой: 2 gsh + н202 → gssg +2 н20;
4 - глутатионредуктаза восстанавливает окисленный глутатион: gssg + nadph + н+ → 2 gsh + nadp+;
5 - NADPH, необходимый для восстановления глутатиона, образуется на окислительном этапе пентозофосфатного пути превращения глюкозы;
6 - NADH, необходимый для восстановления гемоглобина метгемоглобинредуктазной системой, образуется в глицеральдегидфосфатдегид-рогеназной реакции гликолиза.
активных форм кислорода в эритроцитах - неферментативное окисление гемоглобина в метгемоглобин:
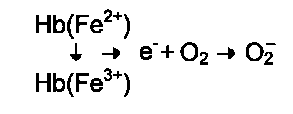
В течение суток до 3% гемоглобина может окисляться в метгемоглобин. Однако метгемогло-бинредуктазная система постоянно восстанавливает метгемоглобин в гемоглобин. Метгемогло-бинредуктазная система состоит из цитохрома b5 и флавопротеина цитохром b5 редуктазы, донором водорода для которой служит NADH, образующийся в глицеральдегиддегидрогеназной реакции гликолиза (рис. 14-4).
Цитохром b5 восстанавливает Fe3+ метгемог-лобина в Fe2+:
Hb-Fe3+ + Цит. b5 восст. → HbFe2+ + Цит. b5 ок.
Окисленный цитохром b5 далее восстанавливается цитохром b5 редуктазой:
Цит. b5 ок. + NADH → Цит. b5 восст. + NAD+
5 ок. 5 восст.
Супероксидный анион с помощью фермента супероксиддисмутазы превращается в пероксид водорода:
О2- + О2- +2 Н+ → Н2О2 + О2
Пероксид водорода разрушается каталазой и содержащим селен ферментом глутатионпе-роксидазой. Донором водорода в этой реакции служит глутатион - трипептид глутамилцисте-инилглицин (GSH) (см. раздел 12).
2Н2О → 2 Н2О +О2; 2GSH + H2O2 → GSSG+ +2 H2O
Окисленный глутатион (GSSG) восстанавливается NADPH-зависимой глутатионредуктазой. Восстановление NADP для этой реакции обеспечивают окислительные реакции пентозофос-фатного пути (см. раздел 7).
Г. НАРУШЕНИЯ МЕТАБОЛИЗМА ЭРИТРОЦИТОВ
Энзимопатии, обусловливающие гемолиз эритроцитов. Для эффективного обезвреживания активных форм кислорода, образующихся в эритроцитах, необходимы все перечисленные выше фермен-
тативные системы защиты. Однако у людей обнаружено около 3000 генетических дефектов глюкозо-6-фосфатдегидрогеназы. Этот фермент катализирует скорость-лимитирующую реакцию пентозофосфатного пути окисления глюкозы, которая обеспечивает образование NАDРН + Н+. Как известно, от количества NАDР + Н+ зависит активность глутатионредуктазы и глу-татионпероксидазы - ферментов, разрушающих пероксид водорода. Не менее 100 млн человек, у которых активность этого фермента снижена, являются носителями дефектных генов глюкозо-6-фосфатдегидрогеназы. При приёме некоторых лекарств, являющихся сильными окислителями (антималярийного препарата примахина, сульфаниламидов, парацетамола), у пациентов, имеющих генетические дефекты глюкозо-6-фосфатдегидрогеназы или глутатионредуктазы, глутатионовой защиты может оказаться недостаточно. Активные формы кислорода вызывают образование гидроперекисей ненасыщенных жирных кислот фосфолипидов, входящих в состав клеточных мембран, их разрушение и гемолиз эритроцитов.
Генетический дефект любого фермента гликолиза приводит к уменьшению образования АТФ и NАDН + Н+ в этих клетках. Вследствие снижения скорости синтеза АТФ падает активность Na+,K+-АТФ-азы, повышается осмотическое давление и возникает осмотический шок. Дефицит NАDН + Н+ приводит к накоплению метгемоглобина и увеличению образования активных форм кислорода, вызывающих окисление SH-групп в молекулах гемоглобина. Молекулы метгемоглобина образуют дисульфид-ные связи между протомерами и агрегируют с образованием телец Хайнца (рис. 14-5).
Гемоглобинопатии
Серповидноклеточная анемия - тяжёлое наследственное заболевание, обусловленное точечной мутацией гена, кодирующего структуру β-цепи гемоглобина (см. раздел 4). В результате в эритроцитах больных присутствует HbS, β-цепи которого в шестом положении вместо гидрофильной глутаминовой кислоты содержат гидрофобную аминокислоту валин. Появление гидрофобной аминокислоты недалеко от начала молекулы способствует возникновению нового центра связывания, поэтому при низком парциальном давлении кислорода
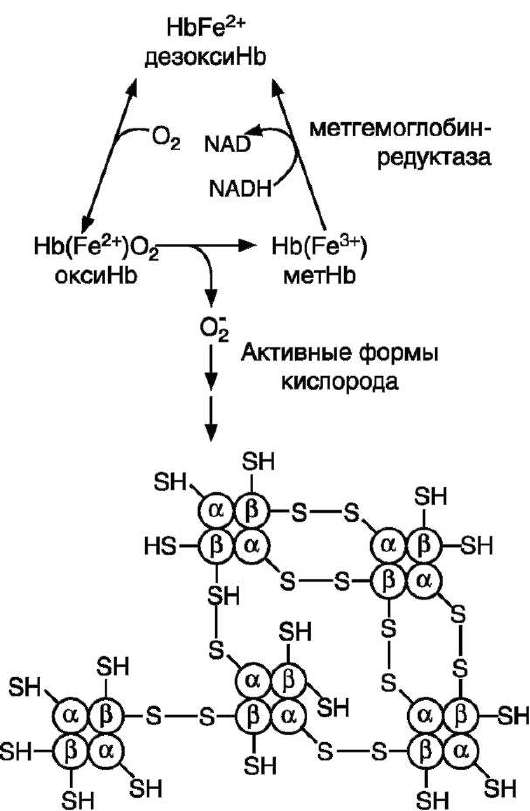
Рис. 14-5. Схема образования телец Хайнца - агрегация гемоглобина. В норме супероксиддисмутаза катализирует образование пероксида водорода, который под действием глутатионпероксидазы превращается в Н2О. При недостаточной активности ферментов обезвреживания активных форм кислорода между прото-мерами метгемоглобина образуются дисульфидные связи, и они агрегируют.
тетрамеры дезокси-HbS ассоциируют, образуя длинные микротрубчатые образования, которые полимеризуются внутри эритроцитов. Полимеризация приводит к нарушению структуры эритроцитов, они приобретают серповидную форму и легко разрушаются. При этом заболевании отмечают анемию, прогрессирующую слабость, отставание в развитии и желтуху.
Носители гена серповидноклеточной анемии чаще всего встречаются среди африканского населения, так как они приобретают некоторое преимущество при заболевании малярией, часто встречающейся в странах с тропическим
климатом. Причина сохранения гена серповид-ноклеточной анемии в популяции связана с тем, что в эритроцитах гетерозигот хуже развивается малярийный плазмодий, часть жизненного цикла которого проходит в эритроцитах человека. В связи с этим гетерозиготные носители дефектного гена выживали при эпидемиях малярии, однако четверть их потомства погибала от серповидно-клеточной анемии.
Талассемии - наследственные заболевания, обусловленные отсутствием или снижением скорости синтеза α- или β-цепей гемоглобина. В результате несбалансированного образования глобиновых цепей образуются тетрамеры гемоглобина, состоящие из одинаковых протомеров. Это приводит к нарушению основной функции гемоглобина - транспорту кислорода к тканям. Нарушение эритропоэза и ускоренный гемолиз эритроцитов и клеток-предшественников при талассемиях приводит к анемии.
При β-талассемии не синтезируются β-цепи гемоглобина. Это вызывает образование нестабильных тетрамеров, содержащих только α-цепи. При этом заболевании в костном мозге из-за преципитации нестабильных α-цепей усиливается разрушение эритробластов, а ускорение разрушения эритроцитов в циркулирующей крови приводит к внутрисосудистому гемолизу. Как известно, для образования фетального гемоглобина β-цепи не требуются (см. раздел 4), поэтому клинически β-талассемия не проявляется до рождения, после чего происходит переключение синтеза HbF на HbA.
В случае α-талассемии недостаток образования α-глобиновых цепей приводит к нарушению образования HbF у плода. Избыточные γ-цепи образуют тетрамеры, называемые гемоглобином Барта. Этот гемоглобин при физиологических условиях имеет повышенное сродство к кислороду и не проявляет кооперативных взаимодействий между протомерами. В результате гемоглобин Барта не обеспечивает развивающийся плод необходимым количеством кислорода, что приводит к тяжёлой гипоксии. При α-талассемии отмечают высокий процент внутриутробной гибели плода. Выжившие новорождённые при переключении с γ- на β-ген синтезируют β-тетрамеры или HbH, который, подобно гемоглобину Барта, имеет слишком высокое сродство к кислороду, менее стабилен,
чем HbA и быстро разрушается. Это ведёт к развитию у больных тканевой гипоксии и к смерти вскоре после рождения.
Наследственный сфероцитоз. Причиной этой патологии чаще всего является дефект белков цитоскелета эритроцитов - спектрина или анкирина, которые обеспечивают поддержание двояковогнутой формы клетки и эластичности мембраны. Эритроциты приобретают шарообразную форму, что приводит к уменьшению площади их поверхности и снижению скорости газообмена. Потеря эластичности клеточной мембраны приводит к повышению хрупкости и травматичности клеток и, как следствие, к ускорению их разрушения в сосудистом русле и селезёнке. Заболевание сопровождается анемией и желтухой. Удаление селезёнки (спленэктомия) при наследственном сфероцитозе улучшает состояние больных, так как предотвращает разрушение сфероцитов в селезёнке.
Мегалобластная (макроцитарная) анемия развивается при дефиците фолиевой кислоты или витамина В12.
Фолиевая кислота в виде кофермента (Н4-фо-лата) участвует в синтезе нуклеотидов. Недостаток фолиевой кислоты приводит к снижению скорости синтеза ДНК в быстроделящихся клетках, и в первую очередь в предшественниках эритроцитов. Клетки дольше пребывают в интерфазе, синтезируя гемоглобин, и становятся крупнее. Кроме того, из-за недостатка нуклеоти-дов они реже делятся, и количество эритроцитов снижается, а крупные мегалобласты быстрее разрушаются. Всё это в конечном итоге приводит к развитию анемии.
Аналогичная симптоматика развивается при недостатке в организме витамина В12. Этот витамин участвует в переносе метильной группы с N5-метил-Н4-фолата на гомоцистеин с образованием метионина и Н4-фолата (см. раздел 10). Недостаточность витамина В12 приводит к накоплению N5-метил-Н4-фолата в клетках. Дефицит Н4-фолата приводит к нарушению деления клеток и развитию анемии.
ii. особенности метаболизма фагоцитирующих клеток
Способность некоторых клеток крови к фагоцитозу - одна из защитных функций крови.
В фагоцитозе участвуют 2 типа лейкоцитов - нейтрофилы и моноциты. Нейтрофилы содержат многодольчатое ядро, поэтому их ещё называют полиморфноядерными лейкоцитами (ПЯЛ). Они поступают в кровоток из костного мозга и имеют продолжительность жизни около 8 сут. Взаимодействие белков интегринов (см. раздел 15) с рецепторами эндотелиальных клеток капилляров приводит к адгезии нейтрофилов, которые далее мигрируют в ткань.
Моноциты также могут выходить из кровяного русла, и тогда их называют макрофагами. Оба типа фагоцитов захватывают и разрушают бактерии. Макрофаги, кроме того, утилизируют старые повреждённые клетки и клеточные оболочки, в частности они поглощают около 1011 эритроцитов в сутки. Фагоцитоз - особая форма эндоцитоза, при которой образуются большие эндоцитозные пузырьки, размеры которых определяются размерами поглощаемых частиц.
Образование фагосомы начинается с взаимодействия специфических рецепторов фагоцитов с бактерией или комплексом антиген-антитело. Рецепторы, расположенные в тех участках плазматической мембраны, где локализован особый белок клатрин (см. раздел 5), «узнают» компоненты комплемента, олигосахариды на по-верхности микроорганизмов или Fc области комплекса антиген-антитело (см. раздел 1). Активация рецепторов, передающих сигнал в клетку с участием инозитолфосфатной системы, инициирует процессы, определяющие фагоцитарный ответ клетки. Он включает в себя формирование фагосомы, слияние её с лизо-сомой, образование фаголизосомы, активацию кислородзависимых бактерицидных механизмов уничтожения микробов и/или выработку клетками токсичного для микробов оксида азота, а также действие кислороднезависимых механизмов уничтожения микроорганизмов.
Формирование фагосомы. Взаимодействие микробной клетки с поверхностью фагоцита приводит к образованию на его мембране выростов - псевдоподий, окружающих микробную клетку. Фагосома, сформированная таким образом, вместе с захваченной бактерией погружается внутрь фагоцита.
Образование фаголизосомы. В цитозоле фаго-сомы сливаются с первичными лизосомами, образуя фаголизосомы. Первичные лизосомы, образованные аппаратом Гольджи, содержат ряд
заключённых в гранулы гидролаз, способных разрушать органические молекулы в кислой среде фаголизосом: протеиназы, фосфатазы, эстеразы, ДНК-азы, РНК-азы. Низкое значение рН внутри фагосом оказывает бактерицидное действие и создаёт оптимальную среду для активации лизосомальных гидролаз. В результате действия этих ферментов разрушаются полимерные молекулы микроорганизмов и образуются аминокислоты, моносахариды, нуклеотиды, которые поступают в цитозоль и могут использоваться клеткой. Большая часть мембранных компонентов и непереваренные субстраты локализуются в остаточных тельцах, которые путём экзоцитоза возвращаются на поверхность плазматической мембраны фагоцитов, при этом значительная часть мембранных компонентов может утилизироваться и в самой мембране (рис. 14-6).
Активация кислородзависимых бактерицидных механизмов уничтожения микробов. Ферментный комплекс мембраны фагосом - NАDРН-окси-даза восстанавливает О2, образуя супероксидный анион:
2 о2 + nadph → 2 o2- + nadp+ + h+.
Супероксидный анион спонтанно или при участии фермента супероксиддисмутазы превращается в пероксид водорода:
о2- + о2- + 2н+ → н2о2 + о2.
Под действием миелопероксидазы, проникающей в фагосому при её слиянии с лизосомой, из пероксидов в присутствии галогенов (йодидов и хлоридов) образуются дополнительные токсичные окислители - гипойодид и гипохлорид.
н2о2 + с1- +н+ → нос1 + н2о.
Все эти молекулы являются сильными окислителями и оказывают бактерицидное действие. Резкое увеличение потребления кислорода фагоцитирующей клеткой называется «респираторным взрывом» (рис. 14-7).
Активные формы кислорода инициируют свободнорадикальные реакции, разрушающие липиды клеточных мембран поглощённых фагоцитами бактерий.
Наследственная недостаточность NADP-оксида-зы, обусловленная дефектом одного из генов этого ферментного комплекса, приводит к хроническому гранулематозу. В результате дефекта фермента фагоциты больных не способны продуцировать супероксидный кислородный радикал и пероксид водорода и поэтому не могут быстро разрушать фагоцитированные клетки бактерий и грибов. Некоторые устойчивые микроорганизмы остаются жизнеспособными внутри фагоцитов, и их антигены
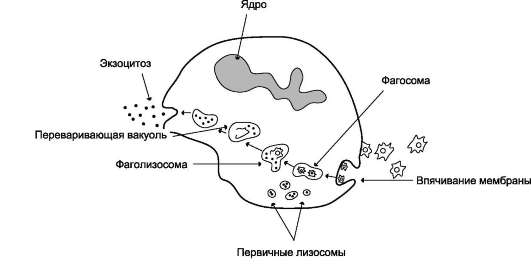
Рис. 14-6. Фагоцитоз в нейтрофилах.
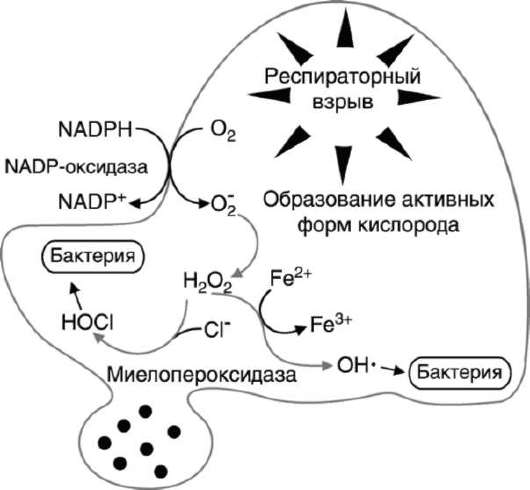
Рис. 14-7. Образование активных форм кислорода фагоцитирующими клетками при респираторном взрыве.
Активация NADPH оксидазы, локализованной в мембране клетки, вызывает образование супероксидного аниона. В результате впячивания мембраны супероксид вместе с бактериальной клеткой оказываются в фагосоме. Супероксидный анион генерирует образование других токсичных молекул, включая Н2О2 и ОН*. Миелопероксидаза, содержащаяся в гранулах фагоцитирующих клеток, секретируется в фагосому, где образует НОСl.
вызывают в месте скопления фагоцитов клеточный иммунный ответ и формирование гранулём. Наиболее часто встречается сцепленная с Х-хромосомой форма этого заболевания, связанная с дефектом гена одной из полипептидных цепей комплекса, локализованного на коротком плече Х-хромосомы.
Образование реакционноспособных метаболитов азота. Бактерицидное действие в макрофагах оказывает и оксид азота (NO). Оксид азота в этих клетках образуется, так же как и в других, под действием фермента NO син-тазы из аргинина (см. раздел 9). Активность NO синтазы в макрофагах заметно повышается при фагоцитозе в присутствии γ-интерферона и фактора некроза опухолей. Супероксид-анион образует с NO соединения, обладающие большими бактерицидными свойствами, чем сам NO:
• NO +O2- → ONOO- - OH +NO2.
Пероксинитрил (ONOO-), оксид азота, диоксид азота, радикал гидроксила вызывают окислительное повреждение белков, нуклеиновых кислот и липидов бактериальных клеток. Оксид азота может непосредственно взаимодействовать
с железосерными белками ЦПЭ, ингибируя дыхание и синтез АТФ в бактериях. При взаимодействии NO с О2 образуются нитриты, которые превращаются в нитраты, также обладающие токсическим действием (см. раздел 12).
Вспышка метаболической активности ней-трофила заканчивается его гибелью. Погибшие нейтрофилы, макрофаги, бактерии и тканевая жидкость входят в состав гноя.
Действие кислороднезависимых бактерицидных механизмов. Некоторые грамположительные бактерии погибают в фагосомах нейтрофилов под действием лизосомального фермента лизоцима. Этот фермент гидролизует связи между содержащимися в клеточной стенке N-ацетилмура-мовой кислотой и N-ацетил-D-глюкозамином и вызывает её разрушение.
В нейтрофилах человека обнаружены ка-тионные пептиды - дефензины , содержащие около 30 аминокислотных остатков и богатые цистеином и аргинином. Они составляют от 30 до 50% всех белков гранул. Дефензины вызывают образование ионных каналов в мембране микробной клетки сразу же после образования фаголизосомы, и тем самым способствуют её уничтожению. Дефензины действуют и на об-
ладающие оболочкой вирусы, например вирус простого герпеса.
Таким образом, огромное разнообразие микроорганизмов, атакующих клетки человека, определило многообразие бактерицидных механизмов, действующих как в аэробных, так и анаэробных условиях.
iii. свёртывающая система крови
При повреждении кровеносного сосуда инициируется каскад реакций, в результате которого образуется сгусток крови - тромб, предотвращающий кровотечение. Основную роль в свёртывании (коагуляции) крови играют тромбоциты и ряд белков плазмы крови.
В остановке кровотечения различают 3 этапа. На первом этапе происходит сокращение кровеносного сосуда. Затем к месту повреждения прикрепляются тромбоциты, которые, наслаиваясь друг на друга, образуют тромбоцитарную
пробку (белый тромб). Белый тромб является непрочным и может закупорить только небольшой кровеносный сосуд. На третьем этапе растворимый белок плазмы крови фибриноген превращается в нерастворимый белок фибрин, который откладывается между тромбоцитами, и формируется прочный фибриновый тромб. Такой тромб содержит эритроциты и поэтому называется красным тромбом.
Образованию фибринового тромба предшествует каскад протеолитических реакций, приводящий к активации фермента тромбина, который и превращает фибриноген в фибрин. Все белки, участвующие в свёртывании крови, называют факторами свёртывания. Они синтезируются в основном в печени и клетках крови в виде неактивных предшественников, обозначаются римскими цифрами, но имеют и тривиальные названия (табл. 14-1). Большинство этих белков активируется в каскаде ферментативных реакций свёртывания крови. Активные формы этих белков обозначают такими же римскими цифрами, но с добавлением буквы «а».
Таблица 14-1. Основные функции и содержание в плазме крови факторов свёртывания крови
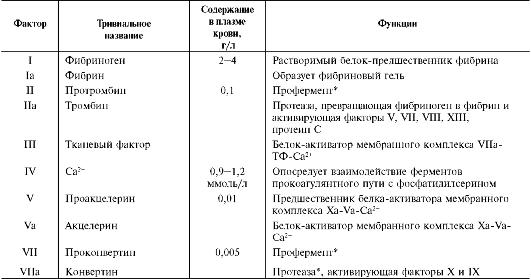

* Содержит остатки карбоксиглутаминовой кислоты, необходимые для образования мембранных ферментных комплексов прокоагулянтного пути свёртывания крови.
А. ОБРАЗОВАНИЕ ФИБРИНОВОГО ТРОМБА
Образование фибринового тромба начинается с превращения растворимого белка плазмы крови фибриногена в нерастворимый фибрин.
Фибриноген (фактор I) - гликопротеин с молекулярной массой 340 кД. Он синтезируется в печени и содержится в плазме крови в концентрации 8,02-12,9 мкмоль/л (2-4 г/л). Молекула фибриногена состоит из шести полипептидных цепей, которые связаны друг с другом дисуль-фидными связями. Состав полипептидных цепей молекулы фибриногена обозначают Аα2, Вβ2, γ2. Заглавные буквы соответствуют тем участкам, которые отщепляются под действием тромбина при превращении фибриногена в фибрин. Фрагменты А в цепях Аα и В в цепях Вβ содержат большое количество остатков аспартата и глутамата. Это создаёт сильный отрицательный заряд на N-концах молекул фибриногена и препятствует их агрегации.
Молекула фибриногена состоит из трех глобулярных доменов, по одному на каждом конце молекулы (домены Д) и один в середине (домен Е). Домены отделены друг от друга участками полипептидных цепей, имеющими стержнеоб-разную конфигурацию. Из центрального домена Е выступают N-концевые фрагменты А и В цепей Αα и Ββ (рис. 14-8).
В образовании фибринового тромба можно выделить 4 этапа.
1. Превращение фибриногена в мономер фибрина. Сначала молекулы фибриногена освобождаются от отрицательно заряженных фрагментов А и В, в результате чего образуются мономеры фибрина. Превращение фибриногена (фактор I) в фиб-
рин (фактор Ia) катализирует фермент тромбин (фактор На). В каждой молекуле фибриногена тромбин гидролизует четыре пептидные связи аргинил-глицил, две из которых соединяют фрагменты А с α-цепью, а две другие - В с β-цепью в Аα2- и Вβ2-цепях фибриногена. Мономер фибрина, образующийся из фибриногена, имеет состав (α, β, γ)2.
2. Образование нерастворимого геля фибрина. На втором этапе образуется нерастворимый полимерный фибриновый сгусток - гель фибрина. В результате превращения фибриногена в фибрин-мономер в домене Е открываются центры связывания с доменами D. Причём домен Е содержит центры агрегации, формирующиеся только после частичного протеолиза фибриногена под действием тромбина, а домен D является носителем постоянных центров агрегации. Первичная агрегация молекул фибрина происходит в результате взаимодействия центров связывания домена Е одной молекулы с комплементарными им участками на доменах D других молекул.Та-ким образом, между доменами молекул фибрина-мономера образуются нековалентные связи. При «самосборке» геля фибрина сначала образуются двунитчатые протофибриллы, в которых молекулы фибрина смещены друг относительно друга на 1/2 длины. После достижения протофибриллами определённой критической длины начинается их латеральная ассоциация, ведущая к образованию толстых фибриновых волокон (рис. 14-9). Образовавшийся гель фибрина непрочен, так как молекулы фибрина в нём связаны между собой нековалентными связями.
3. Стабилизация геля фибрина. Гель фибрина стабилизируется в результате образования амидных
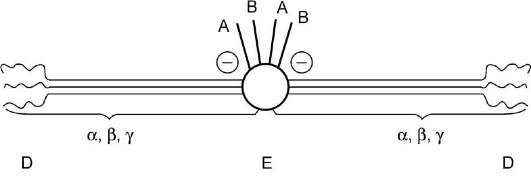
Рис. 14-8. Строение фибриногена. Фибриноген состоит из шести полипептидных цепей: Αα2, Ββ2 и γ2. А, В - отрицательнозаряженные фрагменты, благодаря которым молекулы фибриногена не агрегируют. Д, Е - глобулярные домены молекулы фибриногена. Домены отделены участками полипептидных цепей, имеющими стержнеобразную конфигурацию. Из центрального глобулярного домена Е выступают N-концевые участки фрагментов А и В цепей Αα2 и Ββ2.
связей между остатками лизина одной молекулы фибрина и остатками глутамина другой молекулы. Реакцию трансамидирования катализирует фермент трансглутамидаза (фактор XIIIа) (рис. 14-10). Фактор XIII активируется частичным протеолизом под действием тромбина.
Трансглутамидаза также образует амидные связи между фибрином и фибронектином - гли-копротеином межклеточного матрикса и плазмы крови (см. раздел 15). Таким образом, тромб фиксируется в месте повреждения сосуда.
4. Ретракция фибринового сгустка. Сжатие (ретракцию) геля обеспечивает актомиозин тромбоцитов - сократительный белок тромбос-тенин, обладающий АТФ-азной активностью. Тромбостенин участвует также в активации и агрегации тромбоцитов. Ретракция кровяного сгустка предупреждает полную закупорку сосудов, создавая возможность восстановления кровотока.
В механизме образования тромба есть три функционально разных этапа: прокоагулянтный путь, контактный путь и антикоагулянтная фаза, препятствующая распространению тромба.
Б. ПРОКОАГУЛЯНТНЫЙ ПУТЬ СВЕРТЫВАНИЯ КРОВИ
Для остановки кровотечения из капилляров и сосудов необходимо быстрое образование прочного тромба, препятствующего потере крови.
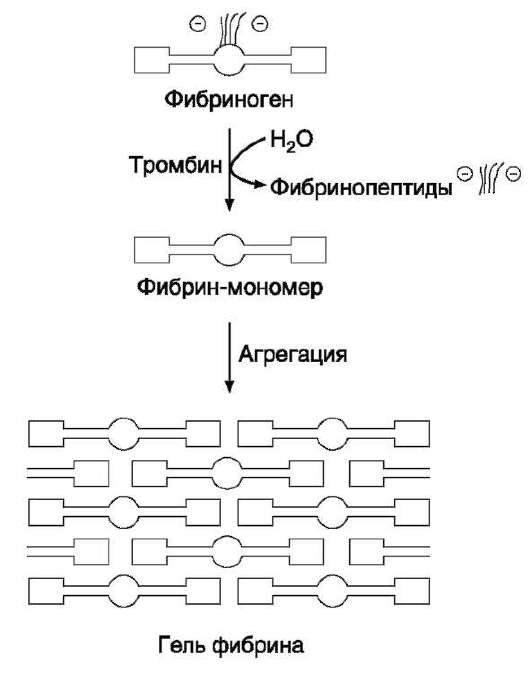
Рис. 14-9. Образование геля фибрина. Фибриноген, освобождаясь под действием тромбина от отрицательно заряженных фрагментов (фибринопептидов 2А и 2В), превращается в фибрин-мономер. В результате взаимодействия комплементарных участков Е- и D-доменов фибрина-мономера происходит сначала линейная, а затем латеральная полимеризация молекул с образованием геля фибрина.
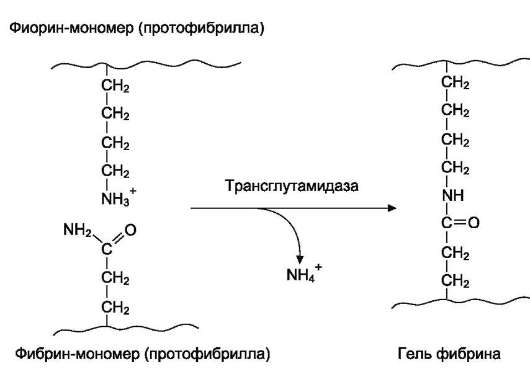
Рис. 14-10. Образование амидной связи между молекулами фибрина.

Это достигается каскадом ферментативных реакций с механизмами усиления на многих этапах.
Прокоагулянтный путь занимает центральное место в свёртывании крови (рис. 14-11).
В циркулирующей крови содержатся проферменты протеолитических ферментов: фактор II (протромбин), фактор VII (проконвертин), фактор IX (Кристмаса), фактор X (Стюарта). Находящиеся в крови факторы Vа (акцелерин) и VIIIa (антигемофильный фактор), а также мембранный белок - тканевый фактор (ТФ, фактор III) являются белками-активаторами этих ферментов (табл. 14-1).
При повреждении сосуда «включается» каскадный механизм активации ферментов с последовательным образованием трёх связанных с фосфолипидами клеточной мембраны ферментных комплексов. Каждый комплекс состоит из протеолитического фермента, белка-активатора и ионов Са2+: VIIa-TФ-Ca2+, IXa-VIIIa-Ca2+ (теназа), Xa-Va-Ca2+ (протромбиназа) (рис. 14-12). Комплекс Xa-Va-Са2+ (протром-биназный комплекс) активирует протромбин
(фактор II). Каскад ферментативных реакций завершается образованием мономеров фибрина и последующим формированием тромба.
В активации ферментов каскада выделяют три основных механизма: частичный проте-олиз, взаимодействие с белками-активаторами и взаимодействие с модифицированными клеточными мембранами.
Активация частичным протеолизом. Все ферменты прокоагулянтного пути являются сери-новыми протеазами, синтезируются в печени в виде неактивных проферментов и в такой форме циркулируют в крови. В процессе реализации тромбогенного сигнала проферменты (факторы VII, IX, X и II) частичным протеолизом превращаются в активные ферменты.
Тромбин (фактор IIa) - гликопротеин с молекулярной массой 39 кД. Он образуется в крови из неактивного предшественника протромбина. Протромбин синтезируется в печени, имеет молекулярную массу 70 кД и содержит остатки γ-карбоксиглутаминовой кислоты. Концентрация этого белка в крови в норме составляет 0,1 г/л.
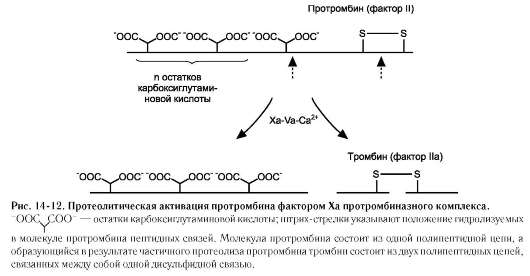
Он фиксируется на мембранном ферментном комплексе Xa-Va-Ca2+, взаимодействуя, с одной стороны, остатками γ-карбоксиглутамата с Са2+, а с другой - непосредственно с белком-активатором Va. Таким образом, создаются наилучшие стерические условия для протекания ферментативной реакции. Фактор Xa гидролизует две пептидные связи в молекуле протромбина. В результате этого образуется молекула тромбина, состоящая из двух цепей - лёгкой и тяжёлой, связанных между собой одной дисульфидной связью (рис. 14-12). Молекула тромбина не содержит остатков γ-карбоксиглутамата и освобождается из протромбиназного комплекса. Тромбин частичным протеолизом превращает фибриноген в фибрин и активирует факторы VII, VIII, V, XIII.
Тромбин выполняет ряд важных физиологических функций: является ферментом прокоа-гулянтного и контактного путей свёртывания крови, инициирует реакции антикоагулянтной фазы, вызывает агрегацию тромбоцитов и оказывает митогенное действие, участвуя в пролиферации и репарации клеток.
Частичным протеолизом активируются также факторы V и VIII, превращаясь, соответственно, в факторы Va и VIIIa. В результате активации этих факторов изменяется их конформация и
повышается сродство к фосфолипидам мембран и ферментам, которые они активируют.
Взаимодействие белков-активаторов с протеоли-тическими ферментами. Тканевый фактор, фактор Vа и фактор VIIIa имеют центры связывания с фосфолипидами мембран и ферментами VIIa, IXa и Xa, соответственно. При связывании с белками-активаторами в результате конформа-ционных изменений активность этих ферментов повышается.
Тканевый фактор (фактор III) представляет собой комплекс, состоящий из белка и фосфати-дилсерина. Белковая часть тканевого фактора (апопротеин III) экспонирована на поверхности многих клеток (мозга, лёгких, печени, селезёнки и др.) и связана с фосфатидилсерином плазматических мембран. Однако появление апопротеина III на поверхности клеток, соприкасающихся с кровью (эндотелиальных и моноцитов), происходит только при определённых условиях: при повреждении сосуда и/или нарушении нормальной асимметрии их плазматических мембран. Тканевый фактор в протеолитической активации не нуждается.
Фактор V и фактор VIII - доменные белки, циркулирующие в крови. Фактор V синтезируется в печени, а фактор VIII - эндотелиальными клетками. Оба фактора активируются частичным протеолизом под действием тромбина. Фактор
VIII в плазме крови находится в комплексе с белком - фактором тромбоцитов фон Вил-лебранда. Фактор фон Виллебранда в этом комплексе стабилизирует фактор VIII, препятствуя его разрушению протеолитическим ферментом антикоагулянтной фазы фактором Са.
Взаимодействие ферментных комплексов с клеточными мембранами происходит с участием ионов Са2+. Все проферменты прокоагулянтного пути (II, VII, IX, X) содержат остатки γ-карбоксиглу-таминовой кислоты, образующиеся в результате посттрансляционой модификации этих белков в ЭР гепатоцитов.
Остатки γ-карбоксиглутаминовой кислоты в факторах VIIa, IXа и Xа обеспечивают взаимодействие этих ферментов посредством Са2+ с отрицательно заряженными фосфолипидами клеточных мембран. В отсутствие ионов Са2+ кровь не свёртывается.
Роль витамина К в карбоксилировании остатков глутаминовой кислоты в проферментах прокоагулян-тного пути свёртывания крови. Карбоксилирование остатков глутаминовой кислоты в проферментах прокоагулянтного пути катализирует карбок-силаза, коферментом которой служит восстановленная форма витамина К (нафтохинона) - дигидрохинон витамина К (см. раздел 3).
Поступивший в организм витамин К (нафто-хинон) восстанавливается в печени NADPH-за-висимой витамин К редуктазой с образованием дигидрохинона витамина К. В ходе реакции карбоксилирования остатков глутаминовой кислоты в проферментах прокоагулянтного пути дигидрохинон окисляется и эпоксидиру-ется с образованием 2,3-эпоксида витамина К. Регенерация эпоксида в дигидрохинон витамина К происходит следующим образом: сначала 2,3-эпоксид витамина К восстанавливается в витамин К тиолзависимой эпоксидредукта-зой, коферментом которой является белок, подобный тиоредоксину. Затем образующийся в этой реакции витамин К восстанавливается ферментом витамин К тиолзависимой редук-тазой в дигидрохинон витамина К. Донором водорода в этой реакции, так же, как и в предыдущей, служит тиоредоксинподобный белок (рис. 14-13).
Недостаточность витамина К приводит к нарушению карбоксилирования проферментов прокоагулянтного пути и сопровождается
кровоточивостью, подкожными и внутренними кровоизлияниями.
Структурные аналоги витамина К дикумарол и варфарин ингибируют тиолзависимые ферменты витамин К 2,3-эпоксидредуктазу и витамин К редуктазу, вызывая торможение свёртывания крови (рис. 14-14). Эти препараты применяют в клинической практике для предупреждения тромбозов.
Инициация каскада реакций прокоагулянтного пути.
Ферментные мембранные комплексы прокоагу-лянтного пути образуются только при наличии на внешней поверхности плазматической мембраны клеток тканевого фактора и отрицательно заряженных фосфолипидов. Поперечная асимметрия плазматических мембран, в частности, определяется преобладанием в наружном слое нейтральных фосфолипидов (фосфатидилхолина и сфингомиелина), а во внутреннем - отрицательно заряженных (фосфатидилинозитолбис-фосфата и фосфатидилсерина) (см. раздел 5). Специальная ферментная система обеспечивает трансмембранный перенос и такое распределение фосфолипидов в клеточных мембранах, при котором в норме внешняя поверхность плазматических мембран клеток не заряжена (см. раздел 5).
При нарушении поперечной асимметрии мембран тромбоцитов и эндотелиальных клеток на их поверхности формируются отрицательно заряженные (тромбогенные) участки и экспонируется апопротеин III тканевого фактора. Такие нарушения могут возникнуть при физической травме. В этом случае тканевый фактор и внутренняя поверхность клеточной мембраны становятся доступными для плазменных факторов прокоагулянтного пути. Кроме того, взаимодействие сигнальных молекул, вызывающих тромбогенез, с рецепторами эндотелиальных клеток и тромбоцитов активирует Са2+-зависимые регуляторные системы (см. раздел 5). В конечном итоге это приводит к повышению содержания в цитоплазме Са2+, который ингибирует АТФ-зависимую амино-фосфолипидтранслоказу. Этот фермент играет важную роль в сохранении поперечной асимметрии мембран, так как переносит фосфа-тидилсерин из внешнего липидного слоя во внутренний. Снижение активности аминофос-фолипидтранслоказы приводит к увеличению содержания во внешнем слое клеточной мемб-
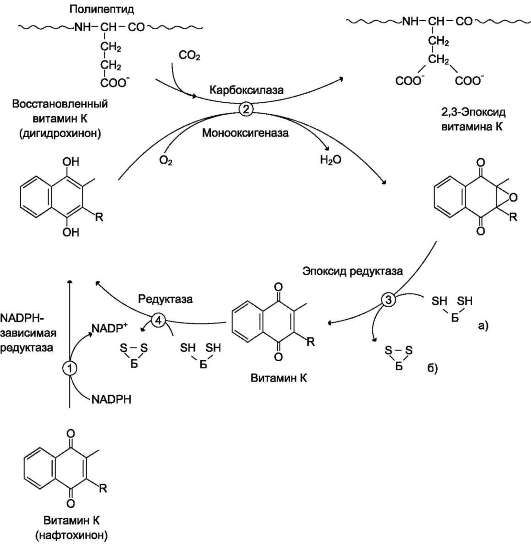
Рис. 14-13. Роль витамина К в посттрансляционном карбоксилировании глутаминовой кислоты. 1 - восстановление экзогенного витамина К NADPH-зависимой редуктазой; 2 - γ-карбоксилирование остатков глутаминовой кислоты в факторах II,VII, IX, X, протеине С витамин К зависимой карбоксилазой сопровождается окислением дигидрохинона с образованием 2,3-эпоксида витамина К; 3 - восстановление 2,3-эпоксида тиолзависимой витамин К редуктазой; 4 - восстановление витамина К тиолзависимой витамин К редуктазой; а) и б) - восстановленная и окисленная формы тиоредоксинподобного белка.
раны фосфатидилсерина и образованию отрицательно заряженных участков, необходимых для формирования мембранных ферментных комплексов. Кроме того, в результате такого нарушения структуры плазматической мембраны на её внешней поверхности экспонируется тканевый фактор и формируется первый
ферментный комплекс прокоагулянтного пути свёртывания крови VII-ТФ-Са2+.
Активация ферментов каждого комплекса - результат взаимодействия всех его компонентов. Если факторы IX, X и II требуют активации, то фактор VII обладает невысокой протеолитической активностью. Фактор VII

Рис. 14-14. Структурные аналоги витамина К дику-марол и варфарин.
мембранного комплекса VII-ТФ-Ca2+ частичным протеолизом активирует факторы IX и X. Активные факторы IXа и Xа включаются в образование мембранных комплексов IXa-VIIIa-Ca2+ и Xa-Va-Ca2+. При этом фактор Xa протеолити-чески активирует фактор V, а протромбиназный комплекс не только превращает протромбин в тромбин, но и активирует фактор VII, протеоли-тическая активность которого в комплексе VIIa-Тф-Ca2+ в 10 000 раз выше, чем в комплексе VII-Tф-Ca2+.
Образовавшийся в результате каскада реакций тромбин катализирует реакции частичного протеолиза фибриногена, фактора XIII и по принципу положительной обратной связи проте-олитически активирует факторы V, VII и VIII.
В процессе свёртывания действуют 2 механизма усиления сигнала: каскад реакций, в котором каждое ферментативное звено обеспечивает усиление сигнала, и положительные обратные связи.
В. КОНТАКТНЫЙ ПУТЬ СВЕРТЫВАНИЯ КРОВИ
Контактный путь свёртывания крови начинается с взаимодействия профермента фактора XII с повреждённой эндотелиальной поверхностью сосудистой стенки. Такое взаимодействие приводит к активации фактора XII и инициирует образование мембранных ферментных комплексов контактной фазы свёртывания. Они содержат ферменты калликреин, факторы XIa (плазменный предшественник тромбопластина) и XIIa (фактор Хагемана), а также белок-ак-
тиватор - высокомолекулярный кининоген (ВМК) (рис. 14-15).
Фактор XII - профермент, циркулирующий в крови. Он последовательно активируется двумя способами: сначала в результате изменения кон-формации при взаимодействии с отрицательно заряженной поверхностью повреждённого эндотелия, затем частичным протеолизом мембранным комплексом калликреин-ВМК.
Высокомолекулярный кининоген - белок-активатор в ферментных мембранных комплексах XIIa-ВМК, XIa-ВМК и калликреин-ВМК. ВМК - гликопротеин плазмы крови, который синтезируется в печени и имеет молекулярную массу 120 кД. Он опосредует взаимодействие протеолитических ферментов контактной фазы свёртывания крови с коллагеном субэндотелия и, кроме того, является компонентом каллик-реин-кининовой системы.
Калликреин - сериновая протеаза, субстратами которой являются, кроме фактора XII, белки плазмы крови плазминоген (профермент, участвующий в растворении фибрина) и кининогены с низкой (69 кД) и высокой (120 кД) молекулярной массой. При частичном протеолизе кининогенов образуются регуляторные пептиды кинины. В частности, мощный вазодилятатор брадикинин повышает проницаемость сосудов и вызывает разрушение клеточных мембран эндотелия.
В результате контакта фактора XII с субэндотелием сосудов он активируется. Активный фактор XIIа в комплексе с ВМК протеолити-чески превращает прекалликреин, связанный с мембраной посредством ВМК, в калликреин. Мембранный комплекс калликреин-ВМК по принципу положительной обратной связи частичным протеолизом активирует фактор XII. При этом фактор XII приобретает максимальную ферментативную активность и по принципу положительной обратной связи активирует связанный с ВМК прекалликреин. Кроме того, образовавшийся в результате частичного проте-олиза фактор XIIa протеолитически активирует фактор XI, а фактор XIa в составе ферментного комплекса XIa-ВМК активирует фактор IX. Фактор IXa мембранного комплекса IXa-VIIIa-Ca2+ активирует фактор X, который в составе протромбиназного комплекса активирует протромбин.
Каскад реакций, ведущий к образованию тромбина, может реализоваться двумя путями -

прокоагулянтным (внешним) и контактным (внутренним) (рис. 14-15). Для инициации реакций внешнего пути необходимо появление тканевого фактора на внешней поверхности плазматической мембраны клеток, соприкасающихся с кровью. Внутренний путь начинается с активации фактора XII при его контакте с повреждённой поверхностью эндотелия сосудов и взаимной активации ферментов прекалликреина и фактора XII.
Таким образом, в прокоагулянтном и контактном путях свёртывания крови последовательное образование мембранных ферментных комплексов приводит к активации фактора X и образованию протромбиназы. Этапы, одинаковые для обоих путей свёртывания крови, называют общим путём свёртывания крови. В настоящее время понятие
о внутреннем и внешнем путях свёртывания считают достаточно условным, так как стало ясно, что комплекс VIIa-ТФ-Са2+ более эффективно активирует фактор IX, чем фактор X, а фактор VII активируется фактором IXа, хотя и значительно медленнее по сравнению с активацией фактором Xа. Следовательно, можно полагать, что каскад реакций свёртывания крови идёт преимущественно в линейной последовательности, а не по двум относительно независимым путям. Контактный путь, очевидно, не является абсолютно необходимым для инициации свёртывания; по-видимому, он служит для сопряжения системы свёртывания крови с различными регуляторными системами организма, например калликреин-кининовой и системой ферментов фибринолиза, растворяющей тромб.
Кровь здорового человека in vitro свёртывается за 5-10 мин. При этом образование протромби-назного комплекса занимает 5-8 мин, активация протромбина - 2-5 с и превращение фибриногена в фибрин - 2-5 с.
Снижение свёртываемости крови. При снижении свёртываемости крови наблюдают заболевания, сопровождающиеся повторяющимися кровотечениями. Гемофилии - наследственные болезни, характеризующиеся повышенной кровоточивостью. Причиной этих кровотечений (спонтанных или вызванных травмой) является наследственная недостаточность белков свёртывающей системы крови.
Гемофилия А (классическая гемофилия) обусловлена мутацией гена фактора VIII, локализованного в Х хромосоме. Классическая гемофилия составляет 80% всех случаев заболевания гемофилией. Гемофилия В встречается реже и обусловлена генетическим дефектом фактора
IX.
Дефект гена фактора VIII проявляется как рецессивный признак, поэтому этой формой гемофилии болеют только мужчины. Это заболевание сопровождается подкожными, внутримышечными и внутрисуставными кровоизлияниями, иногда опасными для жизни. Дефект фактора VIII встречается примерно у одного из 10 000 новорождённых. Больных лечат препаратами, содержащими фактор VIII, получаемыми из донорской крови или методами генной инженерии.
Г. ПРОТИВОСВЕРТЫВАЮЩАЯ СИСТЕМА КРОВИ
Физиологические ингибиторы регуляции свёртывания крови играют важную роль в гемостазе, так как они сохраняют кровь в жидком состоянии и препятствуют распространению тромба за пределы повреждённого участка сосуда.
Тромбин, образующийся в результате реакций прокоагулянтного и контактного путей свёртывания крови, вымывается током крови из тромба. Он может инактивироваться при взаимодействии с ингибиторами ферментов свёртывания крови или активировать антикоагулянтную фазу, тормозящую образование тромба.
Антикоагулянтная фаза. Свёртывание крови должно быть ограничено не только в пространстве, но и во времени. Антикоагулянтная фаза ограничивает время существования актив-
ных факторов в крови и инициируется самим тромбином. Следовательно, тромбин, с одной стороны, ускоряет свёртывание крови, являясь последним ферментом каскада реакций коагуляции, а с другой - тормозит его, вызывая образование ферментных комплексов антикоа-гулянтной фазы на неповреждённом эндотелии сосудов. Этот этап представляет собой короткий каскад реакций, в котором кроме тромбина участвуют белок-активатор тромбомодулин (Тм), витамин К-зависимая сериновая протеаза протеин С, белок-активатор S и факторы Va и VIIIa (рис. 14-16).
В каскаде реакций антикоагулянтной фазы последовательно образуются 2 мембранных комплекса Па-Тм-Са2+ и Са-S-Са2+.
Тромбомодулин - интегральный белок мембран эндотелиальных клеток. Он не требует протеолитической активации и служит белком-активатором тромбина. Тромбин приобретает способность активировать протеин С только после взаимодействия с тромбомодулином, причём связанный с тромбомодулином тромбин не может превращать фибриноген в фибрин, не активирует фактор V и тромбоциты.
Протеин С - профермент, содержащий остатки γ-карбоксиглутамата. Тромбин в мембранном комплексеIIа-Тм-Са2+ активирует частичным
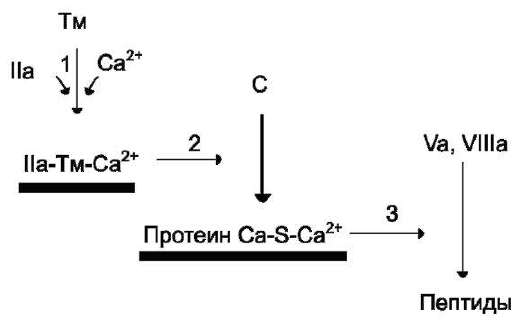
Рис. 14-16. Антикоагулянтная фаза. Тм - тромбо-модулин; С - протеин С; Са - активный протеин С; S - протеин S; жирные линии - мембранно-связан-ный комплекс. 1 - тромбин (IIa) образует мембранный комплекс с белком тромбомодулином (Тм); 2 - тромбин в составе мембранного комплекса IIа-Тм-Са2+ активирует протеин С; 3 - активированный протеин С в составе ферментного мембранного комплекса Са-S-Са2+ гидролизует по 2 пептидные связи в факторах Vа и VIIIа и превращает их в неактивные пептиды.
протеолизом протеин С. Активированный протеин С (Са) образует с белком-активатором S мембраносвязанный комплекс Са-S-Са2+. Протеин Са в составе этого комплекса гидролизует в факторах Va и VIIIa по две пептидные связи и инактивирует эти факторы. Под действием комплекса Са-S-Са2+ в течение 3 мин. теряется 80% активности факторов VIIIa и Va. Таким образом, тромбин по принципу положительной обратной связи не только ускоряет своё образование, но и, активируя протеин С, тормозит процесс свёртывания крови.
Наследственный дефицит протеина С и S ведёт к снижению скорости инактивации факторов VIIIa и Va и сопровождается тромботической болезнью. Мутация гена фактора V, при которой синтезируется фактор V, резистентный к протеину С, также приводит к тромбогенезу.
Антикоагулянтная фаза вызывает торможение каскада реакций свёртывания крови, а ингибиторы ферментов свёртывания инактивируют активные ферменты в кровяном русле.
Ингибиторы ферментов свёртывания крови. Физиологические ингибиторы ферментов свёртывания крови ограничивают распространение тромба местом повреждения сосуда. Белок плазмы крови антитромбин III - наиболее сильный ингибитор свёртывания крови; на его долю приходится около 80-90% антикоагулянтной активности крови. Он инактивирует ряд сери-новых протеаз крови: тромбин, факторы IXa, Xa, XIIa, калликреин, плазмин и урокиназу. Антитромбин III не ингибирует фактор VIIa и не влияет на факторы в составе мембранных комплексов, а устраняет ферменты, находящиеся в плазме крови, препятствуя распространению тромбообразования в кровотоке.
Взаимодействие антитромбина с ферментами свёртывания крови ускоряется в присутствии гепарина. Гепарин - гетерополисахарид, который синтезируется в тучных клетках. В результате взаимодействия с гепарином антитромбин III приобретает конформацию, при которой повышается его сродство к сериновым протеазам крови. После образования комплекса антитромбин III-гепарин-фермент гепарин освобождается из него и может присоединяться к другим молекулам антитромбина.
При наследственном дефиците антитромбина III в молодом возрасте наблюдают тромбозы и эмболии сосудов, опасные для жизни.
α2-Макроглобулин образует комплекс с серино-выми протеазами крови. В таком комплексе их активный центр полностью не блокируется, и они могут взаимодействовать с субстратами небольшого размера. Однако высокомолекулярные субстраты, например фибриноген, становятся недоступными для действия протеаз в комплексе α2-макроглобулин-тромбин.
Антиконвертин (тканевый ингибитор внешнего пути свёртывания) синтезируется в эндотелии сосудов. Он специфически соединяется с ферментным комплексом Тф-VIIa-Са2+, после чего улавливается печенью и разрушается в ней.
α,-Антитрипсин ингибирует тромбин, фактор ХIа, калликреин, однако он не рассматривается как важный ингибитор факторов свёртывания крови. α1-Антитрипсин в основном на тканевом уровне ингибирует панкреатические и лейкоцитарные протеазы, коллагеназу, ренин, урокиназу.
Пептиды, образующиеся в результате про-теолитической активации проферментов и профакторов, тоже обладают выраженными антикоагулянтными свойствами, но механизм их действия в настоящее время не выяснен.
Д. РОЛЬ ТРОМБОЦИТОВ В ГЕМОСТАЗЕ
Способность тромбоцитов прилипать к повреждённой поверхности стенки сосуда (адгезия) и друг к другу (агрегация), связываться с фибрином, образуя тромбоцитарный тромб, и секретировать в месте повреждения сосуда гемостатические факторы определяет их роль в гемостазе.
Циркулирующие в крови тромбоциты имеют дисковидную форму и не прилипают к неповреждённому эндотелию сосудов. Адгезию и агрегацию предотвращают взаимное отталкивание тромбоцитов и интактного эндотелия, а также простациклин (PG I2). Механизм действия некоторых индукторов и репрессора агрегации тромбоцитов рассмотрен на рис. 14-17.
Простациклин образуется из арахидоновой кислоты в эндотелиии сосудов и поступает в кровь (см. раздел 8). Синтез и секрецию про-стациклина эндотелиальными клетками стимулируют тромбин, гистамин, ангиотензин II и калликреин. Он реализует своё действие через аденилатциклазную систему передачи сигнала (см. раздел 5). Взаимодействие простациклина с
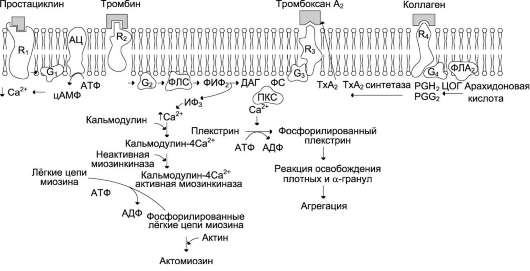
Рис. 14-17. Механизм действия некоторых индукторов и репрессора агрегации тромбоцитов. Взаимодействие простациклина с рецептором R1 вызывает активацию аденилатциклазы, повышение концентрации цАМФ и вследствие этого снижение концентрации Са2+ в цитозоле тромбоцитов. Взаимодействие коллагена с рецептором R4 приводит к активации фосфолипазы А2, которая гидролизует фосфолипиды клеточных мембран с образованием арахидоновой кислоты. Арахидоновая кислота ферментом циклооксигеназой превращается в простагландины PGG2 и PGH2, из которых под действием тромбоксансинтетазы образуется тромбоксан А2. Тромбоксан А2 секретируется активированными тромбоцитами. Тромбоксан и тромбин, соединяясь с соответствующими рецепторами R3 и R2, активируют фосфолипазу С. Фосфолипаза С гидролизует мембранный фосфо-липид фосфатидилинозитол 4,5-бисфосфат с образованием 1,4,5-инозитолтрифосфата и 1,2-диацилглицерола. Инозитолтрифосфат ускоряет поступление из плотной тубулярной системы в цитоплазму клетки Са2+, который соединяется с кальмодулином.
Активная миозинкиназа в комплексе кальмодулин-4Са2+ - миозинкиназа фосфорилирует миозин. Фосфорили-рованный миозин взаимодействует с актином с образованием сократительного белка актомиозина, вызывающего изменение формы тромбоцитов, их адгезию и агрегацию. Комплекс протеинкиназа С-фосфатидилсерин-ДАГСа2+ фосфорилирует белок плекстрин. Фосфорилированный плекстрин вызывает освобождение содержимого плотных гранул и а-гранул тромбоцитов: АДФ, ГДФ, Са2+, серотонина, фактора фон Виллебранда, β-тромбо-модулина. R1, R2, R3, R4 - специфические мембранные рецепторы;
АЦ - аденилатциклаза; G1, G2, G3, G4 - G-белки; ФЛС - фосфолипаза С; ФЛА2 - фосфолипаза А2; ФИФ2 - фосфатидилинозитол 4,5-бисфосфат; ИФ3 - 1,4,5-инозитол трифосфат; ДАГ - 1,2-диацилглицерол; ПКС - протеинкиназа С; ЦОГ - циклооксигеназа; ТхА2 - тромбоксан А2; ТхА2 синтетаза - тромбоксан-синтетаза.
рецептором вызывает активацию протеинкина-зы А. Активная протеинкиназа А фосфорилирует и таким образом активирует Са2+-АТФ-азу и Са2+-транслоказу. Это приводит к снижению уровня содержания Са2+ в цитоплазме тромбоцитов, сохранению ими дисковидной формы и снижению способности к агрегации.
Активация тромбоцитов сопровождается появлением на поверхности плазматической мембраны отрицательно заряженных участков, образованных фосфатидилсерином.
Основные индукторы активации и агрегации тромбоцитов - фактор фон Виллебранда, коллаген, тромбин, АДФ.
Фактор фон Виллебранда - гликопротеин, присутствующий в плазме крови, эндотелии сосудов и α-гранулах тромбоцитов. При повреждении стенки сосудов коллаген, базальная мембрана и миоциты субэндотелия взаимодействуют с тромбоцитами посредством фактора фон Вил-лебранда. Плазматическая мембрана тромбоцитов содержит несколько типов рецепторов этого фактора. Фактор фон Виллебранда, взаимодействуя с рецепторами, действует на тромбоциты через инозитолфосфатную систему передачи сигнала (см. раздел 5). В конечном итоге это приводит к повышению содержания Са2+ в цитоплазме тромбоцитов и образованию комплекса кальмодулин-4Са2+ - миозинкиназа. Фермент миозинкиназа в составе этого комплекса фос-форилирует сократительный белок миозин, который взаимодействует с актином с образованием актомиозина (тромбостенина). В результате этого тромбоциты приобретают шипо-видно-сферическую форму, облегчающую их взаимодействие друг с другом и с поверхностью повреждённого эндотелия.
Снижение концентрации фактора фон Вилле-бранда, уменьшение количества или изменение структуры его рецепторов ведут к нарушениям адгезии и агрегации тромбоцитов, что сопровождается кровоточивостью. Это наблюдают при синдроме Бернара-Сулье, обусловленном недостатком рецептора фактора фон Виллебранда гликопротеина Iа в тромбоцитах, и при болезни фон Виллебранда вследствие дефицита фактора фон Виллебранда.
Наиболее важные первичные индукторы активации тромбоцитов - тромбин и коллаген. Взаимодействие этих белков со специфическими рецепторами плазматической мембраны тромбоцитов приводит к мобилизации Са2+ из плотной тубулярной системы в цитоплазму, что в конечном итоге вызывает их адгезию и агрегацию.
Коллаген вызывает в тромбоцитах активацию фосфолипазы А2, которая освобождает арахидо-новую кислоту из фосфолипидов их мембраны. Арахидоновая кислота служит субстратом для фермента циклооксигеназы (ЦОГ). В результате реакции, катализируемой циклооксигеназой, образуются циклические эндоперекиси проста-гландин G2 (PG G2) и простагландин H2 (PG H2). Эти простагландины под действием тромбок-сансинтетазы превращаются в тромбоксан А2
(см. раздел 8). Тромбоксан А2 снижает уровень цАМФ и, активируя фосфолипазу С, ускоряет освобождение Са2+ из плотной тубулярной системы (рис. 14-17).
Тромбин взаимодействует со специфическим рецептором - интегральным белком, имеющим 7 трансмембранных доменов. Тромбин активирует рецептор частичным протеолизом, отщепляя от него N-концевой пептид, находящийся на внешней плазматической поверхности тромбоцита. Следовательно, тромбин, в отличие от других активаторов, действует каталитически, и одна молекула тромбина может активировать несколько рецепторов. Передача сигнала осуществляется через инозитолфосфатную систему, в результате чего в тромбоците повышается концентрация Са2+ и активируется протеинкина-за С. Образующийся комплекс кальмодулин- 4Са2+-миозинкиназа фосфорилирует миозин, взаимодействие которого с актином приводит к изменению формы тромбоцитов, к их адгезии и агрегации. Протеинкиназа С, кроме того, фосфорилирует белок тромбоцитов плекстрин. Фосфорилированный плекстрин вызывает «реакцию освобождения» содержащихся в гранулах тромбоцитов вторичных индукторов активации и агрегации тромбоцитов. К этим веществам относят содержащиеся в плотных гранулах тромбоцитов АДФ, Са2+, ГДФ, серотонин, гистамин и присутствующие в α-гранулах белок β-тромбоглобулин, фактор фон Виллебранда, белок фибронектин, тромбосподин и ВМК. Тромбосподин участвует во взаимодействии тромбоцитов друг с другом. β-Тромбоглобулин снижает секрецию простациклина и связывает гепарин. Фибронектин имеет центры связывания для коллагена, гепарина и тромбоцитов.
АДФ содержится в тромбоцитах, а также попадает в кровь при разрушении эритроцитов. АДФ взаимодействует со специфическими рецепторами и подавляет активность аденилатциклазы. Это вызывает увеличение мобилизации внутриклеточного Са2+ и в конечном итоге приводит к агрегации тромбоцитов.
Активация тромбоцитов, таким образом, сопровождается изменением их метаболизма и освобождением биологически активных веществ. Эти вещества вызывают морфологические изменения, адгезию, агрегацию тромбоцитов и участвуют в образовании тромба.
Нарушение функциональной активности рецепторов и системы вторичных посредников тромбоцитов приводит к изменению их функции и может явиться причиной ряда заболеваний, сопровождающихся тромбозами или кровотечениями.
Лекарственные препараты, нарушающие агрегацию тромбоцитов, используют для предупреждения возникновения тромбозов. Аспирин (ингибитор циклооксигеназы), никотиновая кислота (ингибитор тромбоксансинтетазы) и Са2+-блокаторы угнетают агрегацию тромбоцитов, влияя на разные этапы реализации тромбо-генного сигнала.
Е. ФИБРИНОЛИЗ
Тромб растворяется в течение нескольких дней после образования. Фибринолиз - ферментативное расщепление волокон фибрина с образованием растворимых пептидов, которые удаляются из сосудистого русла. Разрушение фибрина в составе тромба происходит под действием сериновой протеазы плазмина.
Плазмин образуется из плазминогена под действием активаторов. Неактивный профермент плазмина плазминоген синтезируется в печени, почках и костном мозге.
Тканевый активатор плазминогена (ТАП) -
протеолитический фермент, содержащийся в эндотелии сосудов всех тканей, кроме печени. Поступление этого активатора в кровь увеличивается при эмоциональном напряжении, боли, венозной тромбоэмболии, умеренной физической работе. ТАП частичным протеолизом превращает неактивный плазминоген в активный плазмин. Активаторами плазминогена также служат фактор XIIa и калликреин.
Растворение фибринового сгустка происходит при взаимодействии фибрина, плазминогена и ТАП (рис. 14-18).
Формирование сети фибриновых волокон при образовании тромба сопровождается сорбцией на ней плазминогена и его активаторов. В молекуле плазмина и плазминогена есть участки, комплементарные доменам фибрина, причём одна молекула плазмина может связывать несколько молекул фибрина. Молекулы ТАП тоже имеют центры связывания с фибрином. Образующийся из плазминогена под действием ТАП плазмин гидролизует фибрин с образованием пептидов X и Y, активирующих фибринолиз, и пептидов D и Е, его тормозящих. Растворимые пептиды X, Y, D, E поступают в кровоток и там фагоцитируются. Разрушение тромба приводит к освобождению

Рис. 14-18. Схема фибринолиза. 1 - абсорбированный на фибриновом сгустке плазминоген под действием активаторов (фактор XIIa, калликреин, ТАП) частичным протеолизом превращается в плазмин; 2 - плазмин гидролизует фибрин с образованием растворимых пептидов X, Y,D, E; 3 - в кровотоке ТАП инактивируется специфическими белками и-ТАП-1, и-ТАП-2; 4 - активность плазмина снижается под действием неспецифических ингибиторов сериновых протеаз (α2-антиплазмина, α2-макроглобулина, α1-антитрипсина, комплекса антитромбин-гепарин).
из него плазмина и ТАП. В кровяном русле последние быстро инактивируются специфическими ингибиторами и улавливаются печенью.
ТАП ингибируется ингибиторами тканевого активатора плазмина первого (и-ТАП-1) и второго (и-ТАП-2) типов, а плазмин - α2-антиплазмином или другими ингибиторами сериновых протеаз.
В почках синтезируется протеолитический активатор плазминогена урокиназа, которая, превращая плазминоген в плазмин, способствует освобождению почечных клубочков от фибри-новых волокон.
Из β-гемолитического стрептококка выделили белок стрептокиназу, образующий комплекс с плазминогеном, в котором плазминоген аутока-талитически превращается в плазмин.
Урокиназу, стрептокиназу и ТАП используют при тромболитической терапии инфаркта миокарда, тромбозах вен и артерий, гемодиализе.
Такие ингибиторы ферментов свёртывания крови, как α2-макроглобулин, α1-антитрипсин и комплекс антитромбин III-гепарин также обладают небольшой фибринолитической активностью.
Снижение фибринолитической активности крови сопровождается тромбозами. Нарушение разрушения фибринового сгустка может быть вызвано наследственным дефицитом плазмино-гена или генетическим дефектом его структуры, снижением поступления в кровь активаторов плазминогена, повышением содержания в крови ингибиторов фибринолиза (и-ТАП-1, и-ТАП-2, а2-антиплазмина).
Наследственные и приобретённые нарушения гемостаза могут привести как к геморрагическим заболеваниям, характеризующимся кровоточивостью, так и к тромботической болезни. Однако следует отметить, что повышенная склонность к тромбообразованию и внутрисосудистому свёртыванию (тромбофилии) встречается гораздо чаще, чем гемофилии. Например, частота разных форм гемофилий колеблется в разных странах от 6 до 18 на 100 000 мужчин, в то время как тромбофилии, вызванные дефицитом антитромбина III, встречаются у 1-2 больных на 5000, а при недостатке протеина С - у одного на 15 000 человек.
iv. белки плазмы крови
В плазме крови содержится 7% всех белков организма при концентрации 60-80 г/л. Белки
плазмы крови выполняют множество функций. Одна из них заключается в поддержании осмотического давления, так как белки связывают воду и удерживают её в кровеносном русле.
• Белки плазмы образуют важнейшую буферную систему крови и поддерживают рН крови в пределах 7,37-7,43.
• Альбумин, транстиретин, транскортин, трансферрин и некоторые другие белки (табл. 14-2) выполняют транспортную функцию.
• Белки плазмы определяют вязкость крови и, следовательно, играют важную роль в гемодинамике кровеносной системы.
• Белки плазмы крови являются резервом аминокислот для организма.
• Иммуноглобулины, белки свёртывающей системы крови, а1-антитрипсин и белки системы комплемента осуществляют защитную функцию.
Методом электрофореза на ацетилцеллюлозе или геле агарозы белки плазмы крови можно разделить на альбумины (55-65%), α1-глобулины (2-4%), α2-глобулины (6-12%), β-глобулины (8-12%) и γ-глобулины (12-22%) (рис. 14-19).
Применение других сред для электрофорети-ческого разделения белков позволяет обнаружить большее количество фракций. Например, при электрофорезе в полиакриламидном или крахмальном гелях в плазме крови выделяют 16-17 белковых фракций. Метод иммуноэлект-
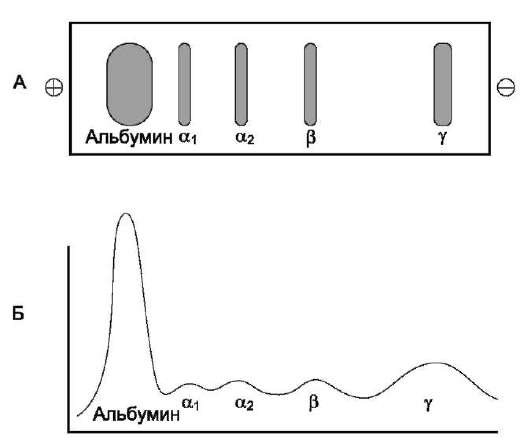
Рис. 14-19. Электрофореграмма (А) и денситограм-ма (Б) белков сыворотки крови.
рофореза, сочетающий электрофоретический и иммунологический способы анализа, позволяет разделить белки плазмы крови более чем на 30 фракций.
Большинство сывороточных белков синтезируется в печени, однако некоторые образуются и в других тканях. Например, γ-глобулины синтезируются В-лимфоцитами (см. раздел 4), пептидные гормоны в основном секретируют клетки эндокринных желёз, а пептидный гормон эритропоэтин - клетки почки.
Для многих белков плазмы, например альбумина, α1-антитрипсина, гаптоглобина, трансферрина, церулоплазмина, α2-макроглобулина и иммуноглобулинов, характерен полиморфизм (см. раздел 4).
Почти все белки плазмы, за исключением альбумина, являются гликопротеинами. Оли-госахариды присоединяются к белкам, образуя гликозидные связи с гидроксильной группой серина или треонина, или взаимодействуя с карбоксильной группой аспарагина. Концевой остаток олигосахаридов в большинстве случаев представляет собой N-ацетилнейраминовую
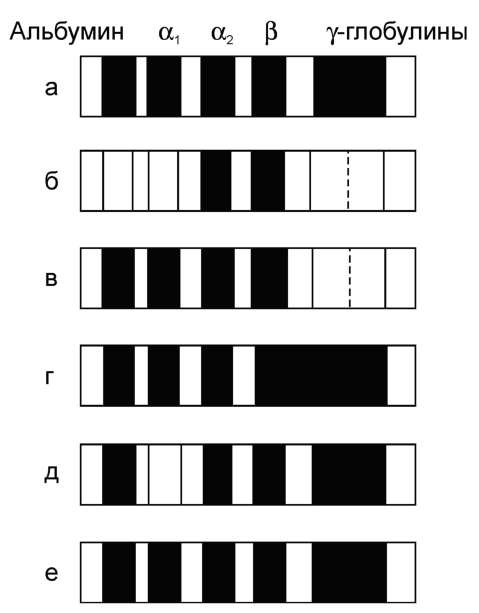
Рис. 14-20. Протеинограммы белков сыворотки крови.
а - в норме; б - при нефротическом синдроме; в - при гипогаммаглобулинемии; г - при циррозе печени; д - при недостатке а1-антитрипсина; е - при диффузной гипергамма-глобулинемии.
кислоту, соединённую с галактозой. Фермент эндотелия сосудов нейраминидаза гидролизу-ет связь между ними, и галактоза становится доступной для специфических рецепторов ге-патоцитов. Путём эндоцитоза «состарившиеся» белки поступают в клетки печени, где разрушаются. T1/2 белков плазмы крови составляет от нескольких часов до нескольких недель.
При ряде заболеваний происходит изменение соотношения распределения белковых фракций при электрофорезе по сравнению с нормой (рис. 14-20).
Такие изменения называют диспротеине-миями, однако их интерпретация часто имеет относительную диагностическую ценность. Например, характерное для нефротического синдрома снижение альбуминов, α1- и γ-глобулинов и увеличение α2- и β-глобулинов отмечают и при некоторых других заболеваниях, сопровождающихся потерей белков. При снижении гуморального иммунитета уменьшение фракции γ-глобулинов свидетельствует об уменьшении содержания основного компонента иммуноглобулинов - IgG, но не отражает динамику изменений IgA и IgM.
Содержание некоторых белков в плазме крови может резко увеличиваться при острых воспалительных процессах и некоторых других патологических состояниях (травмы, ожоги, инфаркт миокарда). Такие белки называют белками острой фазы, так как они принимают участие в развитии воспалительной реакции организма. Основной индуктор синтеза большинства белков острой фазы в гепатоцитах - полипептид интерлейкин-1, освобождающийся из мононуклеарных фагоцитов. К белкам острой фазы относят С-реактивный белок, называемый так, потому что он взаимодействует с С-по-лисахаридом пневмококков, а1-антитрипсин, гаптоглобин, кислый гликопротеин, фибриноген. Известно, что С-реактивный белок может стимулировать систему комплемента, и его концентрация в крови, например, при обострении ревматоидного артрита может возрастать в 30 раз по сравнению с нормой. Белок плазмы крови а1-антитрипсин может инактивировать некоторые протеазы, освобождающиеся в острой фазе воспаления.
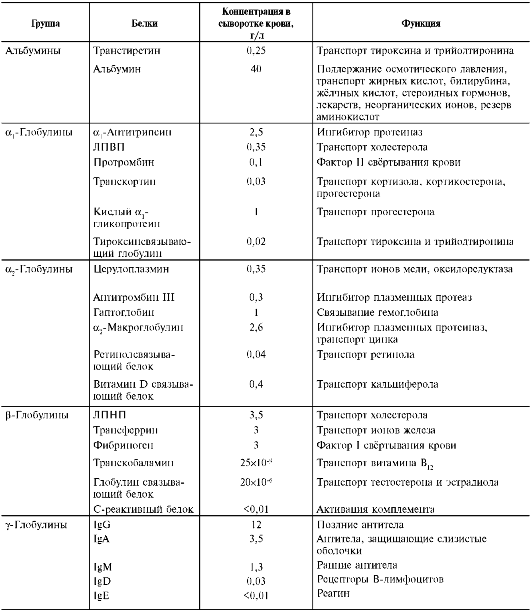
Содержание некоторых белков в плазме крови и их функции представлены в таблице 14-2.
Таблица 14-2. Содержание и функции некоторых белков плазмы крови
Альбумин. Концентрация альбумина в крови составляет 40-50 г/л. В сутки в печени синтезируется около
Благодаря относительно небольшой молекулярной массе и высокой концентрации альбумин обеспечивает до 80% осмотического давления плазмы. При гипоальбуминемии осмотическое давление плазмы крови снижается. Это приводит к нарушению равновесия в распределении внеклеточной жидкости между сосудистым руслом и межклеточным пространством. Клинически это проявляется как отёк. Относительное снижение объёма плазмы крови сопровождается снижением почечного кровотока, что вызывает стимуляцию системы ренин-ангиотензин-альдостерон, обеспечивающей восстановление объёма крови (см. раздел 11). Однако при недостатке альбумина, который должен удерживать Na+, другие катионы и воду, вода уходит в межклеточное пространство, усиливая отёки.
Гипоальбуминемия может наблюдаться и в результате снижения синтеза альбуминов при заболеваниях печени (цирроз), при повышении проницаемости капилляров, при потерях белка из-за обширных ожогов или катаболических состояний (тяжёлый сепсис, злокачественные новообразования), при нефротическом синдроме, сопровождающемся альбуминурией, и голодании. Нарушения кровообращения, характеризующиеся замедлением кровотока, приводят к увеличению поступления альбумина в межклеточное пространство и появлению отёков. Быстрое увеличение проницаемости капилляров сопровождается резким уменьшением объёма крови, что приводит к падению АД и клинически проявляется как шок.
Альбумин - важнейший транспортный белок. Он транспортирует свободные жирные кислоты (см. раздел 8), неконъюгированный билирубин (см. раздел 13), Са2+, Cu2+, триптофан, тироксин
и трийодтиронин (см. раздел 11). Многие лекарства (аспирин, дикумарол, сульфаниламиды) связываются в крови с альбумином. Этот факт необходимо учитывать при лечении заболеваний, сопровождающихся гипоальбуминемией, так как в этих случаях повышается концентрация свободного лекарства в крови. Кроме того, следует помнить, что некоторые лекарства могут конкурировать за центры связывания в молекуле альбумина с билирубином и между собой.
Транстиретин (преальбумин) называют тирок-синсвязывающим преальбумином. Это белок острой фазы. Транстиретин относят к фракции альбуминов. Он имеет тетрамерную структуру и способен присоединять в одном центре связывания ретинолсвязывающий белок, а в другом - до двух молекул тироксина и трийодтиронина. Соединение с этими лигандами происходит независимо друг от друга. В транспорте последних транстиретин играет существенно меньшую роль по сравнению с тироксинсвязывающим глобулином.
α1-Антитрипсин относят к α1-глобулинам. Он ингибирует ряд протеаз, в том числе фермент эластазу, освобождающийся из нейтрофилов и разрушающий эластин альвеол лёгких. При недостаточности α1-антитрипсина могут возникнуть эмфизема лёгких (см. раздел 15) и гепатит, приводящий к циррозу печени. Существует несколько полиморфных форм α1-антитрипси-на, одна из которых является патологической. У людей, гомозиготных по двум дефектным аллелям гена антитрипсина, в печени синтезируется α1-антитрипсин, который образует агрегаты, разрушающие гепатоциты. Это приводит к нарушению секреции такого белка гепатоцитами и к снижению содержания α1-антитрипсина в крови.
Гаптоглобин составляет примерно четверть всех α2-глобулинов. Гаптоглобин при внутрисосудис-том гемолизе эритроцитов образует комплекс с гемоглобином, который разрушается в клетках РЭС. Если свободный гемоглобин, имеющий молекулярную массу 65 кД, может фильтроваться через почечные клубочки или агрегировать в них, то комплекс гемоглобин-гаптоглобин имеет слишком большую молекулярную массу (155 кД), чтобы пройти через гломерулы. Следовательно, образование такого комплекса предотвращает потери организмом железа, содержащегося в гемоглобине. Определение со-
держания гаптоглобина имеет диагностическое значение, например, снижение концентрации гаптоглобина в крови наблюдают при гемолитической анемии. Это объясняют тем, что при T1/2 гаптоглобина, составляющем 5 дней, и T1/2 комплекса гемоглобин-гаптоглобин (около 90 мин) увеличение поступления свободного гемоглобина в кровь при гемолизе эритроцитов
вызовет резкое снижение содержания свободного гаптоглобина в крови.
Гаптоглобин относят к белкам острой фазы, его содержание в крови повышается при острых воспалительных заболеваниях.
Информация о некоторых других белках плазмы крови, представленных в табл. 14-2, имеется в соответствующих разделах учебника.