Биохимия: учебник для вузов/ под ред. Е.С.Северина - 5-е изд., - 2009. - 768 с.
|
|
|
|
РАЗДЕЛ 11 ГОРМОНАЛЬНАЯ РЕГУЛЯЦИЯ ОБМЕНА ВЕЩЕСТВ И ФУНКЦИЙ ОРГАНИЗМА
I. ОСНОВНЫЕ СИСТЕМЫ РЕГУЛЯЦИИ МЕТАБОЛИЗМА И МЕЖКЛЕТОЧНОЙ
КОММУНИКАЦИИ
Для нормального функционирования многоклеточного организма необходима взаимосвязь между отдельными клетками, тканями и органами. Эту взаимосвязь осуществляют 4 основные системы регуляции (рис. 11-1).
• Центральная и периферическая нервные системы через нервные импульсы и нейро-медиаторы;
• Эндокринная система через эндокринные железы и гормоны, которые секретируются в кровь и влияют на метаболизм различных клеток-мишеней;
• Паракринная и аутокринная системы посредством различных соединений, которые секретируются в межклеточное пространство и взаимодействуют с рецепторами либо близлежащих клеток, либо той же клетки (простагландины, гормоны ЖКТ, гистамин и др.);
• Иммунная система через специфические белки (цитокины, антитела).
А. ИЕРАРХИЯ РЕГУЛЯТОРНЫХ СИСТЕМ
Системы регуляции обмена веществ и функций организма образуют 3 иерархических уровня.
Первый уровень - ЦНС. Нервные клетки получают сигналы, поступающие из внешней и внутренней среды, преобразуют их в форму нервного импульса и передают через синапсы, используя химические сигналы - медиаторы. Медиаторы вызывают изменения метаболизма в эффекторных клетках.
Второй уровень - эндокринная система. Включает гипоталамус, гипофиз, периферические эндокринные железы (а также отдельные клетки), синтезирующие гормоны и высвобождающие их в кровь при действии соответствующего стимула.
Третий уровень - внутриклеточный. Его составляют изменения метаболизма в пределах клетки или отдельного метаболического пути, происходящие в результате:
• изменения активности ферментов путём активации или ингибирования;
• изменения количества ферментов по механизму индукции или репрессии синтеза белков или изменения скорости их разрушения;
• изменения скорости транспорта веществ через мембраны клеток.
Б. РОЛЬ ГОРМОНОВ В РЕГУЛЯЦИИ ОБМЕНА
ВЕЩЕСТВ И ФУНКЦИЙ
Интегрирующими регуляторами, связывающими различные регуляторные механизмы и метаболизм в разных органах, являются гормоны. Они функционируют как химические посредники, переносящие сигналы, возникающие в различных органах и ЦНС. Ответная реакция клетки на действие гормона очень разнообразна и определяется как химическим строением гормона, так и типом клетки, на которую направлено действие гормона.
В крови гормоны присутствуют в очень низкой концентрации. Для того чтобы передавать сигналы в клетки, гормоны должны распознаваться и связываться особыми белками клетки - рецепторами, обладающими высокой специфичностью.
Физиологический эффект гормона определяется разными факторами, например концентрацией гормона (которая определяется скоростью инактивации в результате распада гормонов, протекающего в основном в печени, и скоростью выведения гормонов и его метаболитов из организма), его сродством к белкам-переносчикам (стероидные и тиреоидные гормоны транспортируются по кровеносному руслу в комплексе с белками), количеством и типом рецепторов на поверхности клеток-мишеней.
Синтез и секреция гормонов стимулируются внешними и внутренними сигналами, поступающими в ЦНС (рис. 11-2).
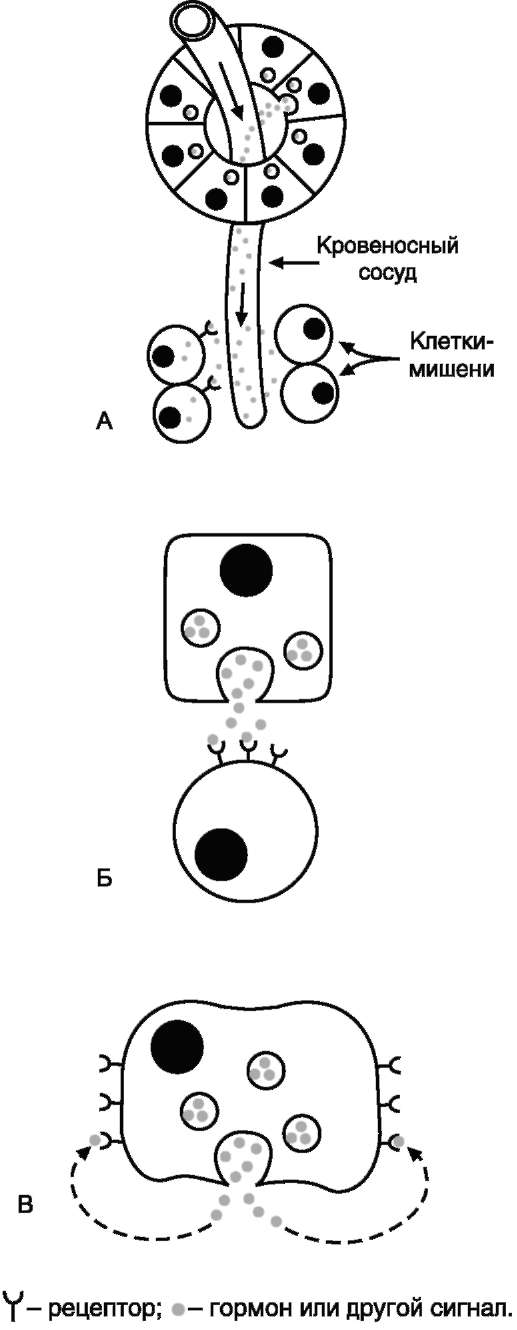
Рис. 11-1. Системы регуляции метаболизма.
А - эндокринная - гормоны секретируются железами в кровь, транспортируются по кровеносному руслу и связываются с рецепторами клеток-мишеней; Б - паракринная - гормоны секретируются во внеклеточное пространство и связываются с мембранными рецепторами соседних клеток; В - аутокринная - гормоны секретируются во внеклеточное пространство и связываются с мембранными рецепторами клетки, секретирующей гормон.
Эти сигналы по нейронам поступают в гипоталамус, где стимулируют синтез пептидных рилизинг-гормонов (от англ. release - освобождать) - либеринов и статинов, которые, со-ответственно, стимулируют или ингибируют синтез и секрецию гормонов передней доли гипофиза. Гормоны передней доли гипофиза, называемые тропными гормонами, стимулируют образование и секрецию гормонов периферических эндокринных желёз, которые поступают в общий кровоток и взаимодействуют с клетками-мишенями.
Поддержание уровня гормонов в организме обеспечивает механизм отрицательной обратной связи. Изменение концентрации метаболитов в клетках-мишенях по механизму отрицательной обратной связи подавляет синтез гормонов, действуя либо на эндокринные железы, либо на гипоталамус. Синтез и секреция тропных гормонов подавляется гормонами эндокринных периферических желёз. Такие петли обратной связи действуют в системах регуляции гормонов надпочечников, щитовидной железы, половых желёз.
Не все эндокринные железы регулируются подобным образом. Гормоны задней доли гипофиза (вазопрессин и окситоцин) синтезируются в гипоталамусе в виде предшественников и хранятся в гранулах терминальных аксонов ней-рогипофиза. Секреция гормонов поджелудочной железы (инсулина и глюкагона) напрямую зависит от концентрации глюкозы в крови.
В регуляции межклеточных взаимодействий участвуют также низкомолекулярные белковые соединения - цитокины. Влияние цитокинов на различные функции клеток обусловлено их взаимодействием с мембранными рецепторами. Через образование внутриклеточных посредников сигналы передаются в ядро, где происходят активация определённых генов и индукция синтеза белков. Все цитокины объединяются следующими общими свойствами:
• синтезируются в процессе иммунного ответа организма, служат медиаторами иммунной и воспалительной реакций и обладают в основном аутокринной, в некоторых случаях паракринной и эндокринной активностью;
• действуют как факторы роста и факторы диф-ференцировки клеток (при этом вызывают преимущественно медленные клеточные реакции, требующие синтеза новых белков);
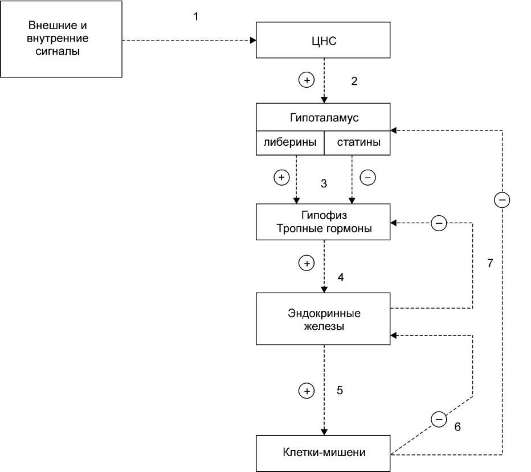
Рис. 11-2. Схема взаимосвязи регуляторных систем организма. 1 - синтез и секреция гормонов стимулируется внешними и внутренними сигналами; 2 - сигналы по нейронам поступают в гипоталамус, где стимулируют синтез и секрецию рилизинг-гормонов; 3 - рилизинг-гормоны стимулируют (либерины) или ингибируют (статины) синтез и секрецию тропных гормонов гипофиза; 4 - тропные гормоны стимулируют синтез и секрецию гормонов периферических эндокринных желез; 5 - гормоны эндокринных желез поступают в кровоток и взаимодействуют с клетками-мишенями; 6 - изменение концентрации метаболитов в клетках-мишенях по механизму отрицательной обратной связи подавляет синтез гормонов эндокринных желез и гипоталамуса; 7 - синтез и секреция тропных гормонов подавляется гормонами эндокринных желез; Е - стимуляция синтеза и секреции гормонов; Е - подавление синтеза и секреции гормонов (отрицательная обратная связь).
• обладают плейотропной (полифункциональной) активностью.
В. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ГОРМОНОВ
Все гормоны классифицируют по химическому строению, биологическим функциям и механизму действия.
1. Классификация гормонов по химическому строению
По химическому строению гормоны делят на 3 группы: пептидные (или белковые), стероидные и непептидные производные аминокислот (табл. 11-1).
2. Классификация гормонов по биологическим функциям
По биологическим функциям гормоны можно разделить на несколько групп (табл. 11-2). Эта классификация условна, поскольку одни и те же гормоны могут выполнять разные функции. Например, адреналин участвует в регуляции обмена жиров и углеводов и, кроме этого, регулирует частоту сердечных сокращений, АД, сокращение гладких мышц. Кортизол не только стимулирует глюконеогенез, но и вызывает задержку NaCl.
Таблица 11-1. Классификация гормонов по химическому строению

II. ВЗАИМОДЕЙСТВИЕ ГОРМОНОВ С РЕЦЕПТОРАМИ И МЕХАНИЗМЫ ПЕРЕДАЧИ ГОРМОНАЛЬНЫХ СИГНАЛОВ В КЛЕТКИ
Биологическое действие гормонов проявляется через их взаимодействие с рецепторами клеток-мишеней. Для проявления биологической активности связывание гормона с рецептором должно приводить к образованию химического сигнала внутри клетки, который вызывает специфический биологический ответ, например изменение скорости синтеза ферментов и других белков или изменение их активности (см. раздел 5). Мишенью для гормона могут служить клетки одной или нескольких тканей. Воздействуя на клетку-мишень, гормон вызывает специфическую ответную реакцию. Например, щитовидная железа - специфическая мишень для тиреотропина, под действием которого увеличивается количество ацинарных клеток щитовидной железы, повышается скорость биосинтеза тиреоидных гормонов. Глюкагон, воздействуя на адипоциты, активирует липо-лиз, в печени стимулирует мобилизацию гликогена и глюконеогенез. Характерный признак клетки-мишени - способность воспринимать информацию, закодированную в химической структуре гормона.
А. РЕЦЕПТОРЫ ГОРМОНОВ
Начальный этап в действии гормона на клетку-мишень - взаимодействие гормона с рецептором клетки. Концентрация гормонов во внеклеточной жидкости очень низка и обычно колеблется в пределах 10-6-10-11 ммоль/л. Клетки-мишени отличают соответствующий гормон от множества других молекул и гормонов благодаря наличию на клетке-мишени соответствующего рецептора со специфическим центром связывания с гормоном.
1. Общая характеристика рецепторов
Рецепторы пептидных гормонов и адреналина располагаются на поверхности клеточной мембраны. Рецепторы стероидных и тиреоидных гормонов находятся внутри клетки. Причём внутриклеточные рецепторы для одних гормонов, например глюкокортикоидов, локализованы в цитозоле, для других, таких как андрогены,
эстрогены, тиреоидные гормоны, расположены в ядре клетки (см. раздел 5).
Рецепторы по своей химической природе являются белками и, как правило, состоят из нескольких доменов.
В структуре мембранных рецепторов можно выделить 3 функционально разных участка. Первый домен (домен узнавания) расположен в N-концевой части полипептидной цепи на внешней стороне клеточной мембраны; он содержит гликозилированные участки и обеспечивает узнавание и связывание гормона. Второй домен - трансмембранньгй. У рецепторов одного типа, сопряжённых с G-белками, он состоит из 7 плотно упакованных α-спиральных полипептидных последовательностей. У рецепторов другого типа трансмембранный домен включает только одну α-спирализованную полипептидную цепь (например, обе β-субъединицы гетеротетра-мерного рецептора инсулина α2β2). Третий (ци-топлазматический) домен создаёт химический сигнал в клетке, который сопрягает узнавание и связывание гормона с определённым внутриклеточным ответом. Цитоплазматический участок рецептора таких гормонов, как инсулин, фактор роста эпидермиса и инсулиноподобный фактор роста-1 на внутренней стороне мембраны обладает тирозинкиназной активностью, а цитоплазматические участки рецепторов гормона роста, пролактина и цитокинов сами не проявляют тирозинкиназную активность, а ассоциируются с другими цитоплазматическими протеинкиназами, которые их фосфорилируют и активируют.
Рецепторы стероидных и тиреоидных гормонов содержат 3 функциональные области. На С-концевом участке полипептидной цепи рецептора находится домен узнавания и связывания гормона. Центральная часть рецептора включает домен связывания ДНК. На N-концевом участке полипептидной цепи располагается домен, называемый вариабельной областью рецептора, отвечающий за связывание с другими белками, вместе с которыми участвует в регуляции транскрипции.
2. Регуляция количества и активности рецепторов
Концентрация рецепторов внутри клетки или на её поверхности и их сродство к данному гормону в норме регулируются различными способа-
ми, а также могут меняться при заболеваниях или при использовании гормонов или их агонистов в качестве лекарственных средств. Например, при воздействии β-адренергических агонистов на клетки в течение нескольких минут в ответ на новое добавление агониста прекращается активация аденилатциклазы, и биологический ответ исчезает. Такое снижение чувствительности рецептора к гормону (десенситизация) может происходить в результате изменения количества рецепторов по механизму понижающей регуляции. Гормон связывается с рецептором, комплекс гормон-рецептор путём эндоцитоза проникает в клетку (интернализуется), где часть рецепторов подвергается протеолитическому расщеплению под действием ферментов лизо-сом, а часть инактивируется, отделяясь от других мембранных компонентов. Это приводит к уменьшению количества рецепторов на плазматической мембране. Например, в случае инсулина, глюкагона, катехоламинов это происходит в течение нескольких минут или часов. При снижении концентрации гормона рецепторы возвращаются на поверхность клетки, и чувствительность к гормону восстанавливается. Активность рецептора, т.е. его сродство к гормону, может изменяться также в результате ковалентной модификации, главным образом путём фосфо-рилирования. Концентрация внутриклеточных рецепторов может также регулироваться по механизму индукции и репрессии.
Б. МЕХАНИЗМЫ ПЕРЕДАЧИ ГОРМОНАЛЬНЫХ СИГНАЛОВ В КЛЕТКИ
По механизму действия гормоны можно разделить на 2 группы. К первой группе относят гормоны, взаимодействующие с мембранными рецепторами (пептидные гормоны, адреналин, а также гормоны местного действия - цитокины, эйкозаноиды). Вторая группа включает гормоны, взаимодействующие с внутриклеточными рецепторами.
Связывание гормона (первичного посредника) с рецептором приводит к изменению конфор-мации рецептора. Это изменение улавливается другими макромолекулами, т.е. связывание гормона с рецептором приводит к сопряжению одних молекул с другими (трансдукция сигнала). Таким образом, генерируется сигнал, который регулирует клеточный ответ путём изменения
активности или количества ферментов и других белков. В зависимости от способа передачи гормонального сигнала в клетках меняется скорость реакций метаболизма:
• в результате изменения активности ферментов;
• в результате изменения количества ферментов (табл. 11-3).
1. Передача гормональных сигналов через мембранные рецепторы
Гормоны (первичные посредники), связываясь с рецепторами на поверхности клеточной мембраны, образуют комплекс гормон-рецептор, который трансформирует сигнал первичного посредника в изменение концентрации особых молекул внутри клетки - вторичных посредников. Вторичными посредниками могут быть следующие молекулы: цАМФ, цГМФ, ИФ3, ДАГ, Ca2+, NO.
Гормоны, взаимодействие которых с рецептором клетки-мишени приводит к образованию цАМФ, действуют через трёхкомпонентную систему, которая включает белок-рецептор, G-белок и фермент аденилатциклазу. Образующийся под действием аденилатциклазы цАМФ активирует протеинкиназу А, фосфорилиру-ющую ферменты и другие белки (см. раздел 5). Известно более 200 различных G-белков, в структуре которых обнаружены 3 субъединицы α, β и γ (см. раздел 5). В отсутствие гормона α-субъединица G-белка связана с ГДФ. Образование комплекса гормон-рецептора приводит к конформационным изменениям α-субъедини-цы, замене ГДФ на ГТФ и отщеплению димера βγ от α-ГТФ. В случае рецепторов, сопряжённых с Gs-белком, субъединица αs-ГТФ активирует аденилатциклазу (рис. 11-3).
В случае рецепторов, сопряжённых с Gi белком, субъединица αi-ГТФ ингибирует аденилатциклазу. В таблице 11-4 приведены примеры гормонов, взаимодействие которых с соответствующим рецептором активирует или ингибирует аденилатциклазу.
Другая система, генерирующая цГМФ как вторичный посредник, сопряжена с гуанилат-циклазой. Цитоплазматический домен такого типа рецепторов обладает активностью гуа-нилатциклазы, которая катализирует реакцию образования цГМФ из ГТФ (подобно аденилат-
Таблица 11-3. Основные этапы передачи гормональных сигналов
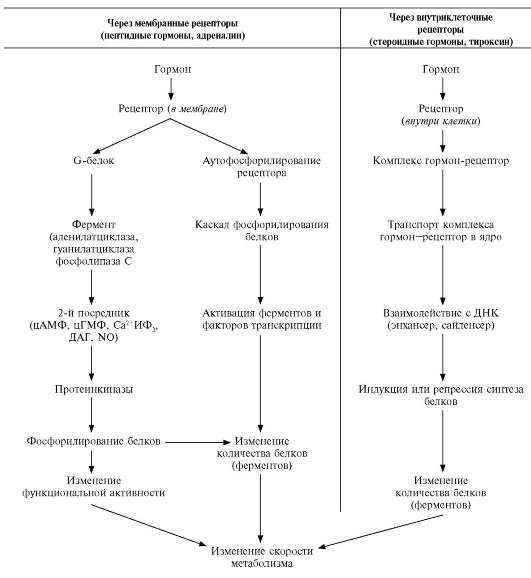
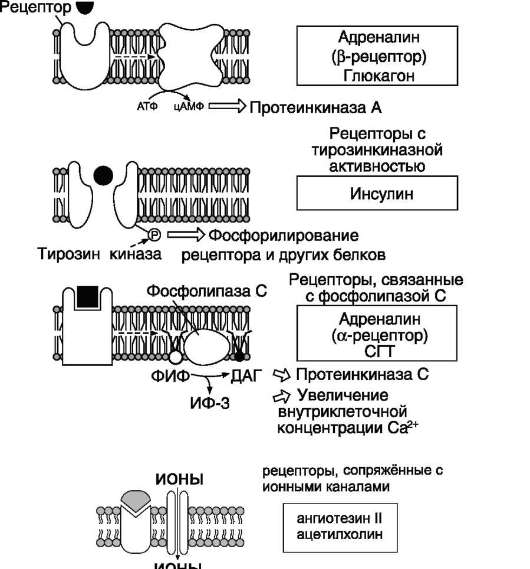
Рис. 11-3. Передача гормональных сигналов через мембранные рецепторы. ИФ3 - инозитол-3-фосфат; ДАГ - диацилглицерол; ФИФ2 - фосфоинозитолбисфосфат; СТГ - соматотропный гормон.
Таблица 11-4. Активация и ингибирование адени-
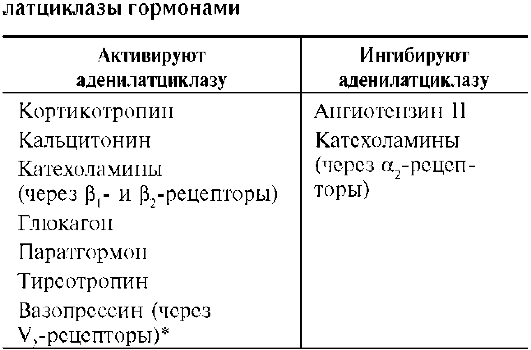
* V1- и V2-рецепторы рассмотены ниже (IV, A).
циклазе). Молекулы цГМФ могут активировать ионные каналы либо активировать цГМФ-зави-симую протеинкиназу G, участвующую в фосфо-рилировании других белков в клетке. Например, фосфодиэстераза, которая гидролизует цАМФ до АМФ, активируется в результате фосфорили-рования цГМФ-зависимой протеинкиназой.
Некоторые гормоны (например, вазопрес-син или адреналин), образуя комплекс с соответствующими рецепторами (рецептор V1 для вазопрессина и α1-рецептор для адреналина), через активацию соответствующих G-белков активируют фосфолипазу С, в результате чего в клетке появляются вторичные посредники ИФ3, ДАГ. Молекула ИФ3 стимулирует высвобождение Ca2+ из ЭР. Кальций связывается с белком кальмодулином. Этот комплекс активирует
Са2+-кальмодулинзависимую протеинкиназу. Ионы кальция и ДАГ участвуют в активации протеинкиназы С (см. раздел 5).
Многие гормоны передают сигнал в клетку через рецепторы, которые либо обладают тиро-зинкиназной активностью, либо связываются с цитоплазматическими белками, проявляющими активность тирозинкиназы. Связывание инсулина с мембранным рецептором, который является тирозинкиназой и имеет центр фосфорилиро-вания, инициирует аутофосфорилирование и последующее фосфорилирование субстратов рецептора инсулина и других белков (см. разделы 5 и ниже подраздел III, Ж).
В случае взаимодействия, например, эпидер-мального фактора роста или инсулиноподобно-го фактора роста -1 с мембранным рецептором сначала происходят димеризация рецептора и его активация. Активированный таким образом гомодимер рецептора, участок которого на внутренней стороне мембраны обладает активностью тирозинкиназы, фосфорилиру-ется сам (аутофосфорилирование) и вызывает фосфорилирование других белков и ферментов, которые участвуют в активации факторов транскрипции генов.
Некоторые гормоны (например, гормон роста, пролактин, интерферон, цитокины) взаимодействуют с мембранными рецепторами, ассоциированными с цитоплазматическими протеин-киназами (так называемыми «Янус-киназами», или киназами семейства JAK). Присоединение гормона вызывает димеризацию рецептора, присоединение Янус-киназ, их аутофосфорилирова-ние и активацию. Янус-киназы, в свою очередь, фосфорилируют рецептор по остаткам тирозина, в результате чего рецептор связывается с другими белками, например, особыми белками - переносчиками сигнала и активаторами транскрипции (ПСАТ, или STAT - от англ. signal transducer and activator of transcription - переносчик сигнала и активатор транскрипции). Далее следует инициируемый тирозинкиназой каскад реакций фос-форилирования. Белки STAT фосфорилируются, образуют димеры, транспортируются в ядро, где, связываясь со специфическими участками ДНК, участвуют в регуляции транскрипции (рис. 11-4).
Сигнальной молекулой в клетке может служить также оксид азота NO, образующийся в организме из аргинина при участии фермента
NO-синтазы, присутствующего в нервной ткани, эндотелии сосудов, тромбоцитах и других тканях (см. раздел 9). Молекула NO может быстро диффундировать через мембрану эн-дотелиальных клеток, где она синтезируется, в соседние клетки. Действие оксида азота кратковременно, так как T1/2 NO колеблется в пределах 5-10 с. В крови молекула существует примерно 100 мс, поскольку быстро взаимодействует с молекулярным кислородом, образуя нитрит, который далее превращается в нитрат и экскрети-руется с мочой. В клетках-мишенях, например, эндотелиальных клетках NO взаимодействует с входящим в активный центр гуанилатциклазы ионом железа (см. раздел 5), способствуя тем самым быстрому образованию цГМФ. Увеличение концентрации цГМФ в клетках гладких мышц вызывает активацию киназ, что в конечном итоге приводит к расслаблению ГМК сосудов и последующему их расширению. Механизм действия оксида азота объясняет использование нитроглицерина в качестве лекарственного препарата для снятия острых болей в сердце, поскольку нитроглицерин - источник образующихся молекул NO, которые и вызывают расслабление кровеносных сосудов и увеличение притока крови в миокард.
2. Передача сигналов через внутриклеточные рецепторы
Стероидные и тиреоидные гормоны связываются с рецепторами внутри клетки и регулируют скорость транскрипции специфических генов (рис. 11-5).
В отсутствие гормона внутриклеточные рецепторы связаны обычно с другими белками в ци-тозоле или ядре. Например, рецепторы глю-ко-кортикоидов образуют в цитозоле комплекс с шапероном, что препятствует связыванию рецептора с молекулой ДНК (рис. 11-6).
Взаимодействие гормона с центром связывания на С-концевом участке полипептидной цепи рецептора вызывает конформационные изменения и освобождение рецептора от шаперона. Происходит объединение 2 молекул рецептора с образованием гомодимера. Димер рецептора узнаёт специфическую последовательность нуклеотидов, которая расположена в промоторной области гена. Взаимодействие со специфическим участком ДНК НРЕ (от англ. hormone response element,
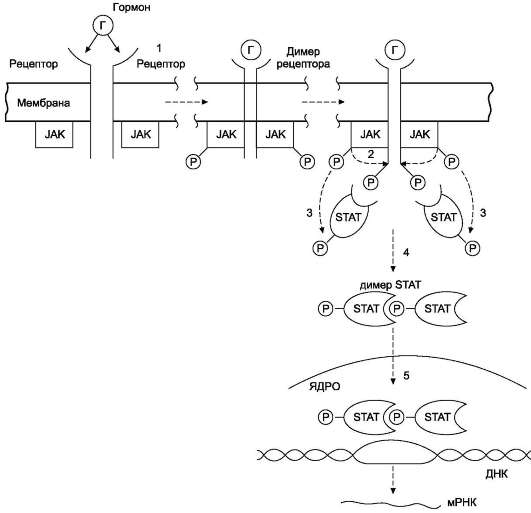
Рис. 11-4. Механизм передачи сигнала через мембранные рецепторы, ассоциированные с Янус-киназами (JAK). 1 - гормон взаимодействует с мембранным рецептором и вызывает димеризацию рецептора. Янус-ки-назы (цитоплазматические тирозинкиназы, имеющие два активных центра) связываются с димером мембранного рецептора, что приводит к их активации и аутофосфорилированию; 2 - янус-киназы (JAK) фосфорилируют димер рецептора по остаткам тирозина; 3 - комплекс фосфорилированного димера рецептора с Янус-киназами связывает особые цитоплазматические белки (STAT), которые фосфорилируются Янус-киназами; 4 - фосфо-рилированные белки STAT активируются, образуя димер; 5 - димер STAT перемещается из цитозоля в ядро, связывается с промоторным участком ДНК и индуцирует транскрипцию генов.
элемент, реагирующий на воздействие гормона) обеспечивает центральный домен рецептора. Этот домен содержит аминокислотную последовательность, образующую 2 «цинковых пальца». В каждом «цинковом пальце» атом цинка связан с 4 остатками цистеина (рис. 11-7).
В структуре одного «цинкового пальца» имеется последовательность аминокислот, отвечающая за связывание с ДНК, а второй «цинковый палец» содержит последовательность аминокислот, участвующую в димеризации рецепторов. Взаимодействие комплекса гормон-рецептор
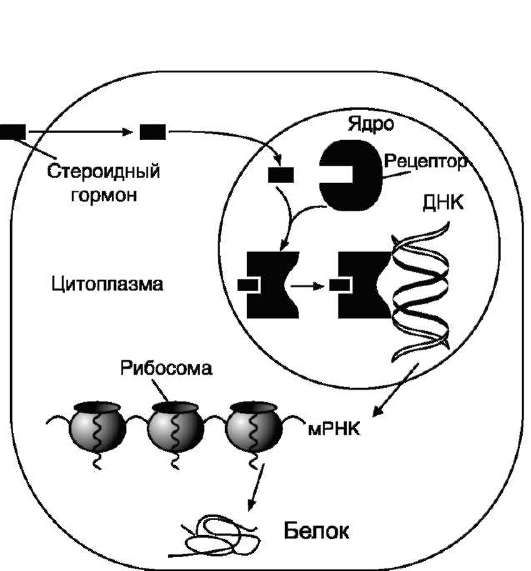
Рис. 11-5. Передача гормональных сигналов через внутриклеточные рецепторы (рецепторы стероидных гормонов могут находиться в цитоплазме и ядре).
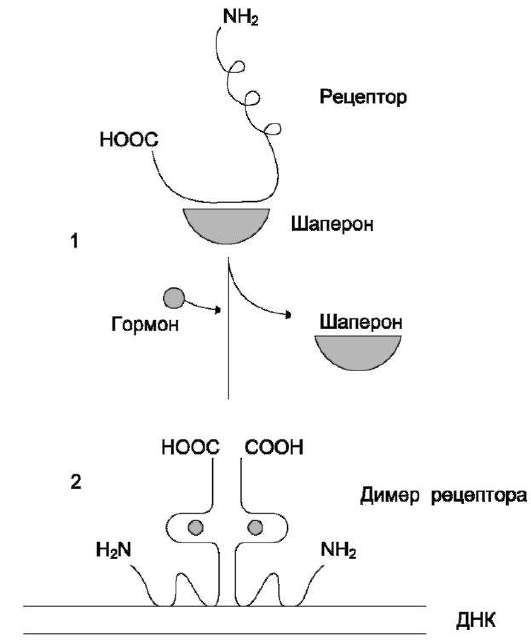
Рис. 11-6. Регуляция активности рецептора стероидных гормонов. 1 - в отсутствие гормона рецептор через гормонсвязывающий домен образует комплекс с шапероном, что препятствует связыванию рецептора с молекулой ДНК; 2 - в присутствии гормона рецептор освобождается от шаперона, образуется димер рецептора, который присоединяется к молекуле ДНК и вызывает активацию транскрипции.
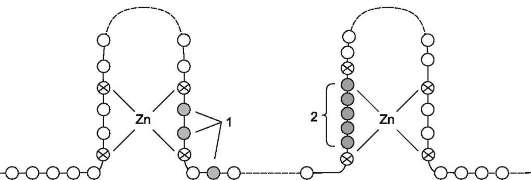
Рис. 11-7. Структура центрального домена стероидного гормона. 1 - аминокислотные остатки, участвующие в связывании ДНК; 2 - область димеризации. Центральный ДНК-связывающий домен содержит 2 «цинковых пальца». Атомы цинка связаны с аминокислотной последовательностью через остатки цистеина. Функциональные области 1 и 2 отвечают соответственно за связывание ДНК и димеризацию рецептора.
с определённой последовательностью нуклеотидов в промоторной части ДНК приводит к активации транскрипции.
Рецепторы тиреоидных гормонов всегда связаны с ДНК. В отсутствие гормонов соответствующие рецепторы ингибируют экспрессию генов. Напротив, взаимодействие с гормоном превращает их в активаторы транскрипции.
3. Передача сигналов через рецепторы, сопряжённые с ионными каналами
Рецепторы, сопряжённые с ионными каналами, являются интегральными мембранными белками, состоящими из нескольких субъединиц. Они действуют одновременно как ионные каналы и как рецепторы, которые способны специфически связывать с внешней стороны эффектор, изменяющий их ионную проводимость. Эффекторами такого типа могут быть гормоны и нейромедиаторы (см. рис. 11-3).
Известны рецепторы для ряда гормонов, ассоциированных с ионными каналами, и большинства медиаторов, среди которых наиболее изучен рецептор ацетилхолина. Рецептор ацетилхолина
состоит из пяти цилиндрообразных субъединиц, расположенных в мембране параллельно друг другу: α2, β, γ, δ. Между ними вдоль оси цилиндров находится заполненный молекулами воды канал. Каждая субъединица рецептора состоит из большого количества гидрофобных аминокислотных остатков. Кроме этого, все субъединицы содержат один спирализованный трансмембранный фрагмент, аминокислотные радикалы которого (полярные незаряженные аминокислотные остатки, в основном серин и треонин) выстилают центральный канал рецептора изнутри. В средней части субъединиц, обращённой к каналу, локализованы остатки лейцина. В присутствии ацетилхолина боковые взаимодействия между субъединицами поддерживают канал в открытом состоянии и создают возможность для транспорта ионов. В отсутствие ацетилхолина в результате изменения ориентации субъединиц относительно друг друга канал закрывается, так как выступающие внутрь канала остатки лейцина образуют плотное гидрофобное кольцо, блокируя движение гидра-тированных ионов в этой области (рис. 11-8).
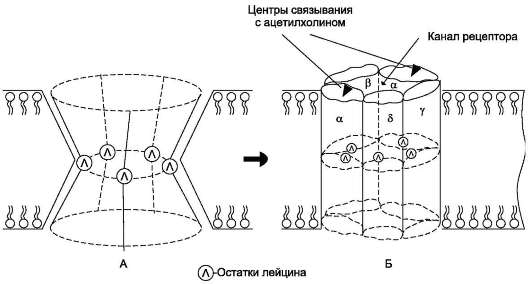
Рис. 11-8. Схема строения рецептора ацетилхолина. А - закрытый канал рецептора в отсутствие ацетилхо-лина; Б - открытый канал рецептора в присутствии ацетилхолина. Трансмембранные спирализованные участки всех 5 субъединиц содержат полярные незаряженные радикалы аминокислот; гидрофобные остатки лейцина (Л), локализованные в середине каждого спирализованного гидрофильного участка, выступают в центральную часть канала и препятствуют движению ионов.
iii. строение, биосинтез и биологическое действие гормонов
Гормоны образуются специализированными клетками, многие из них собраны в железы и секретируют гормоны непосредственно в кровоток (гипоталамус, гипофиз, островковые клетки поджелудочной железы, щитовидная и паращито-видные железы, надпочечники, половые железы). Многие эндокринные железы вырабатывают несколько гормонов, имеющих различное строение и осуществляющих различные функции.
Избыточная продукция или дефицит гормона могут быть причиной эндокринных заболеваний. Среди причин гиперсекреции гормонов первое место занимают гормонально-активные опухоли. Причинами гипосекреции часто являются генетические нарушения структуры и функции участвующих в синтезе гормонов ферментов, повреждение клеток, продуцирующих гормон, в результате инфекции, опухоли или аутоиммунных реакций. Клиническую картину гипер- и гипосекреции гормонов может вызывать и применение гормонов с лечебной целью. В некоторых случаях введение гормона приводит
к подавлению его секреции железами, поэтому резкая отмена гормонотерапии вызывает гипофункцию эндокринных желёз.
Причинами эндокринных заболеваний могут также быть дефекты структуры самих гормонов или их рецепторов, нарушения метаболизма гормонов и механизмов передачи гормональных сигналов в клетки-мишени.
А. ГОРМОНЫ ГИПОТАЛАМУСА
Гипоталамус занимает важнейшее место в иерархической системе, объединяя высшие отделы ЦНС и эндокринные железы. В клетках нейронов гипоталамуса синтезируются пептидные гормоны 2 типов. Одни через систему гипоталамо-гипофизарных сосудов поступают в переднюю долю гипофиза, где стимулируют или ингибируют синтез тропных гормонов; другие, как окситоцин и вазопрессин, поступают через аксоны нервных клеток в заднюю долю гипофиза, где они хранятся в везикулах и секретируются в кровь в ответ на соответствующие сигналы.
В настоящее время известно несколько гипо-таламических гормонов, регулирующих синтез и секрецию гормонов гипофиза (табл. 11-5).
Таблица 11-5. Строение и функции гормонов гипоталамуса
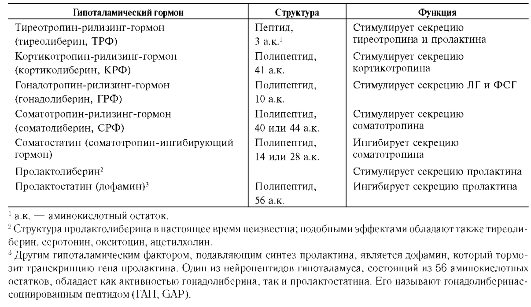
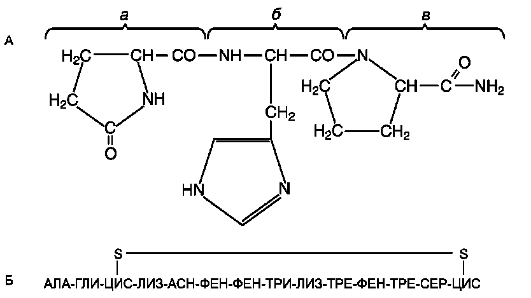
Рис. 11-9. Структура некоторых гормонов гипоталамуса. А. Структура тиреолиберина: а - пироглутаминовая кислота; б - гистидин; в - пролинамид. Б. Структура соматостатина.
1. Тиреолиберин - трипептид, состоящий из пироглутаминовой кислоты, гистидина и про-линамида (рис. 11-9).
Синтез тиреолиберина происходит в различных участках гипоталамуса, но в большей степени в паравентрикулярном ядре, а также в других областях ЦНС, где он выполняет функцию нейромедиатора, повышающего двигательную активность и АД. Предшественник тиреолибе-рина препротиреолиберин человека включает 242 аминокислотных остатка. Образование активного гормона происходит по механизму частичного протеолиза. В передней доле гипофиза тиреолиберин стимулирует синтез и секрецию тиреотропина, а также оказывает стимулирующее влияние на синтез многих других гормонов. В результате взаимодействия тиреолиберина с рецепторами плазматической мембраны клеток гипофиза происходит повышение концентрации внутриклеточного цАМФ и Са2+. Трансдукция сигнала происходит как через аденилатциклазную, так и через инози-толфосфатную системы.
Тиреолиберин разрушается в клетках-мишенях и в крови под действием специфических протеаз. T1/2 в крови составляет 3-4 мин.
2. Кортиколиберин
Кортиколиберин - полипептид, содержащий 41 аминокислотный остаток. Как и другие пептидные гормоны, кортиколиберин синтезируется в виде прогормона. T1/2 кортиколиберина в плазме крови составляет 60 мин. Основное
количество кортиколиберина образуется в гипоталамусе, однако он обнаруживается и в других отделах ЦНС, где выполняет роль медиатора, участвуя в ответной реакции на различные стрессовые ситуации.
В передней доле гипофиза кортиколиберин увеличивает синтез и секрецию проопиоме-ланокортина и образование кортикотропина. Рецепторы кортиколиберина находятся в плазматической мембране клеток в составе адени-латциклазного комплекса. Стимуляция секреции АКТГ требует присутствия ионов Са2+. Увеличение уровня внутриклеточного кальция, вероятно, является результатом фосфорилирования белков кальциевых каналов.
3. Гонадолиберин
Гонадолиберин - декапептид. Предшественник гонадолиберина человека состоит из 92 аминокислотных остатков и имеет молекулярную массу около 10 кД. Гонадолиберин стимулирует синтез и секрецию 2 гормонов гипофиза - ЛГ и ФСГ. Помимо гипоталамуса, нейроны, содержащие гонадолиберин, находятся и в других областях ЦНС, контролирующих эмоциональное и половое поведение. Рецептор гонадолиберина в плазматической мембране входит в состав инозитолфосфат-ного комплекса, активация которого стимулирует фосфорилирование белков и мобилизацию Са2+, что приводит к освобождению гормонов. T1/2 гонадолиберина в плазме крови составляет 5- 7 мин. Инактивация гонадолиберина происходит при участии специфических протеаз.
4. Соматолиберин
Соматолиберин - полипептид, состоящий из 44 аминокислотньгх остатков. В передней доле гипофиза соматолиберин стимулирует синтез и секрецию соматотропина. Трансдукция сигнала сопровождается повышением концентрации как цАМФ, так и ионов кальция. T1/2 соматолиберина в крови составляет около 7 мин. Соматолиберин применяют в клинической практике для диагностики нарушений функции гипофиза.
5. Соматостатин
Соматостатин первично был выделен из гипоталамуса, но впоследствии оказалось, что он синтезируется во многих клетках, расположенных вне гипоталамуса: в желудке, кишечнике, поджелудочной железе, в области периферических нервных окончаний, в плаценте, надпочечниках и в сетчатке глаза. Соматостатин выполняет функции гормона и медиатора, вызывая торможение секреторных процессов, снижение активности гладкой мускулатуры и нейронов. Соматостатин состоит из 14 аминокислотных остатков и имеет циклическую структуру, образованную дисульфидной связью между двумя остатками цистеина (рис. 11-10).
Биологической активностью обладает и ациклическая восстановленная форма пептида. В тканях соматостатин присутствует в форме пептида, содержащего 28 аминокислотных остатков и может служить предшественником пептида, состоящего из 14 аминокислотных остатков. Обе формы проявляют биологическую активность, но в разной степени. Соматостатин-14 находится в основном в ЦНС, а соматостатин-28 преимущественно в кишечнике.
Подобно другим пептидным гормонам, сома-тостатин взаимодействует с рецепторами плазматической мембраны клеток. Различают 5 типов рецепторов соматостатина, ассоциированных с G-белками. Все типы рецепторов экспрессиру-ются в передней доле гипофиза и гипоталамусе и обладают различной степенью сродства к разным структурным формам соматостатина. Рецепторы к соматостатину присутствуют во многих опухолевых клетках, секретирующих гормоны. Это обстоятельство используется для разработки методов ранней диагностики опухолей поджелудочной железы, феохромоцитомы, рака щитовидной железы, рака почек и молочной железы.
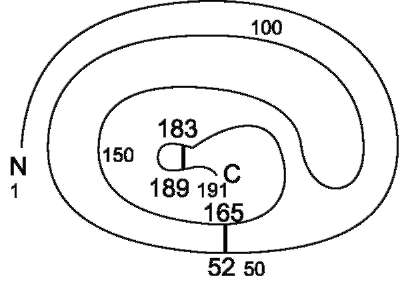
Рис. 11-10. Гормон роста человека. Полипептидная цепь включает 191 аминокислотный остаток. Две дисульфидные связи образованы между остатками цистеина в положениях 183-189 и 53-165.
Результат трансдукции сигнала соматостатина - снижение уровня внутриклеточной концентрации цАМФ и Са2+ в цитозоле клеток. Соматостатин тормозит секрецию гормона роста, глюкагона, инсулина, гастрина, секретина, вазоактивного интестинального пептида (ВИП, VIP), холецистокинина, кальцитонина, паратгормона, иммуноглобулинов, ренина; он также ингибирует секрецию бикарбонатов и ферментов поджелудочной железы, уменьшает кровоток на всём протяжении ЖКТ, снижает секрецию жёлчи.
Б. ГОРМОНЫ ГИПОФИЗА
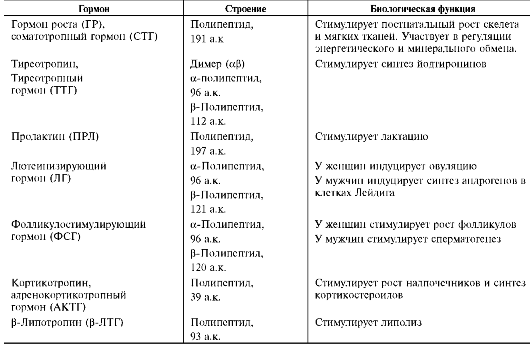
Гипофиз секретирует большое количество гормонов, участвующих в регуляции различных биохимических процессов и физиологических функций. В передней доле гипофиза (аденоги-пофизе) синтезируются так называемые тропные гормоны, стимулирующие синтез и секрецию гормонов других эндокринных желёз или оказывающие влияние на метаболические реакции в других тканях-мишенях (табл. 11-6).
Задняя доля гипофиза, или нейрогипофиз, секретирует гормоны, регулирующие в основном водный баланс и лактацию.
Секреция гормонов гипофиза обусловлена сочетанием нервных и гуморальных сигналов. При этом один и тот же агонист (например, но-радреналин) может вызывать противоположные изменения в секреции гипофизарных гормонов. С другой стороны, секреция каждого гормона может контролироваться многочисленными факторами.
Таблица 11-6. Строение и биологические функции гормонов передней доли гипофиза
Синтез и секреция гормонов передней доли гипофиза регулируются гормонами гипоталамуса, которые поступают в гипофиз через портальную систему кровеносных сосудов, связывающих гипоталамус и переднюю долю гипофиза. Кроме того, секреция гормонов гипоталамуса и гипофиза регулируется по механизму обратной связи гормонами, продукцию которых они стимулируют в органах-мишенях.
В передней доле гипофиза синтезируются гормоны, которые по химическому строению являются пептидами и гликопротеинами.
По механизму их синтеза и биологическим функциям эти гормоны объединяют в 3 группы.
1. Гормон роста, пролактин
Гормон
роста синтезируется в соматотрофных клетках, наиболее многочисленных в
передней доле гипофиза. Содержание гормона роста составляет 5-16 мг в
числяется в мкг/г. T1/2 гормона в плазме крови составляет около 50 мин.
Гормон роста у всех видов млекопитающих представляет собой одноцепочечный пептид с молекулярной массой 22 кД, состоящий из 191 аминокислотного остатка и имеющий 2 внутримолекулярные дисульфидные связи (рис. 11-10).
Гормон роста образуется из прогормона с молекулярной массой 28 кД, не обладающего гормональной активностью. Уровень гормона роста в плазме крови не превышает 3 нг/мл. Секреция гормона роста носит пульсирующий характер с интервалами в 20-30 мин. Один из самых больших пиков отмечается вскоре после засыпания.
Под влиянием различных стимулов (стресс, физические упражнения, гипогликемия, голодание, белковая пища, аминокислота аргинин) даже у нерастущих взрослых людей уровень гормона роста в крови может возрастать до 30-100 нг/мл.
Регуляция синтеза и секреции гормона роста осуществляется множеством факторов. Основной стимулирующий эффект оказывает соматолиберин, основной тормозящий - ги-поталамический соматостатин.
Рецепторы гормона роста находятся в плазматической мембране клеток печени, жировой ткани, яичках, жёлтом теле, скелетных мышцах, хрящевой ткани, мозге, лёгких, поджелудочной железе, кишечнике, сердце, почках, лимфоцитах. Рецептор гормона роста - белок с одним внутримембранным доменом и молекулярной массой 70 кД. Связывание рецептора с гормоном роста вызывает димеризацию 2 рецепторов, что приводит к активации связанных с рецептором Янус-киназ и фосфорилированию Янус-киназ и рецептора по остаткам тирозина. Активация рецептора гормона роста сопровождается повышением активности тирозинкиназ и фосфо-липазы С с последующим повышением уровня ДАГ и ИФ3 и активацией протеинкиназы С (см. раздел 5).
Первичные эффекты гормона роста кратковре-менны и инсулиноподобны. Они проявляются в основном в отношении обмена жиров и углеводов. В жировой ткани усиливается потребление
глюкозы и липогенез, вследствие чего происходит снижение концентрации глюкозы в крови. Однако в дальнейшем проявляются более медленные (в основном, противоположные инсулину) эффекты: усиливается липолиз в жировой ткани, увеличивается концентрация жирных кислот в крови, а в случае недостаточности инсулина увеличивается содержание кетоновых тел в крови. Энергия, образующаяся при повышенном распаде жиров, используется на анаболические процессы. В то же время использование глюкозы жировыми и мышечными клетками снижается, а в печени ускоряется глюконеогенез, следствием чего может быть гипергликемия, особенно при недостатке инсулина (рис. 11-11).
Основное действие гормона роста направлено на регуляцию обмена белков и процессов, связанных с ростом и развитием организма. Под влиянием гормона роста усиливаются транспорт аминокислот в клетки мышц, синтез белка в костях, хрящах, мышцах, печени и других внутренних органах, увеличивается общее количество РНК, ДНК и общее число клеток.
Влияние гормона роста на рост скелета и мягких тканей требует участия веществ, которые синтезируются в ответ на взаимодействие
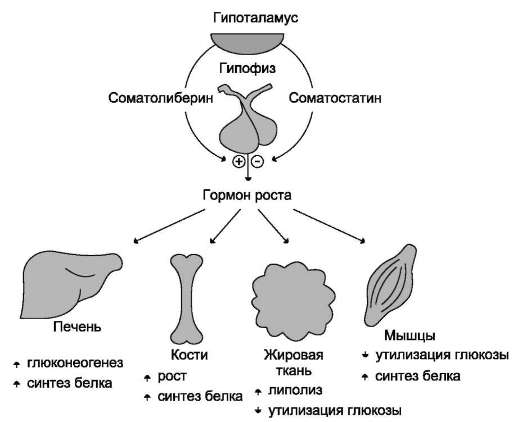
Рис. 11-11. Биологическое действие гормона роста.
гормона роста с рецепторами плазматической мембраны клеток различных тканей, в основном печени, и носят название соматомединов. Поскольку эти молекулы отличаются высокой гомологичностью друг к другу, а также к проинсулину и обладают инсулиноподобной активностью и мощным ростстимулирующим действием, они называются инсулиноподобны-ми факторами роста (ИФР-1, или соматомедин С; ИФР-2, или соматомедин А). ИФР-1 - од-ноцепочечный полипептид основного характера, содержащий 70 аминокислотных остатков, а полипептид ИФР-2 носит кислотный характер и состоит из 67 аминокислотных остатков. В крови примерно 95% соматомединов циркулирует в комплексе с белками. Синтез ИФР-1 в большей степени зависит от концентрации гормона роста в крови, чем синтез ИФР-2. В то же время ИФР-1, образующийся в печени, ингибирует синтез и секрецию гормона роста
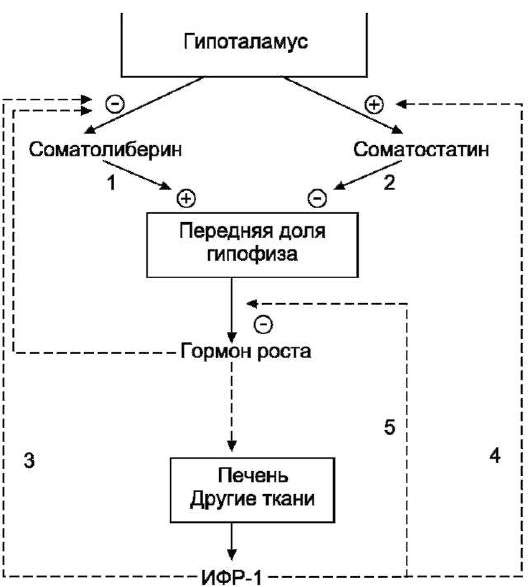
Рис. 11-12. Регуляция секреции гормона роста.
Соматолиберин стимулирует (1), а соматостатин ингибирует (2) освобождение гормона роста (ГР) из передней доли гипофиза. ИФР-1 ингибирует секрецию соматолиберина (3) и стимулирует секрецию сома-тостатина (4). ИФР-1 ингибирует секрецию гормона роста также на уровне гипофиза (5).
по механизму ретроингибирования, действуя на уровне гипофиза и гипоталамуса (рис. 11-12).
Инсулиноподобные факторы роста оказывают своё действие различными путями: эндокринным, паракринным и аутокринным (рис. 11-13).
Подобно рецептору инсулина, рецептор ИФР-1 обладает тирозинкиназной активностью и инициирует каскад реакций фосфорилирования других белков, участвующих в различных внутриклеточных процессах, включая активацию транскрипции генов. В большинстве случаев ИФР-1, как и инсулин, инициирует клеточное развитие, однако при значительно меньших, почти физиологических концентрациях. Это указывает на то, что инсулиноподобные факторы роста более активны в отношении их действия на рост и развитие клеток.
Под влиянием гормона роста увеличивается ширина и толщина костей, и одновременно с этим ускоряется рост других тканей, включая соединительную ткань, мышцы и внутренние органы.
Пролактин синтезируется лактотрофными клетками передней доли гипофиза в виде про-гормона с молекулярной массой 40 кД. Число этих клеток резко возрастает при беременности под влиянием эстрогенов. Пролактин близок по химическому строению гормону роста. Он состоит из 199 аминокислотных остатков, образующих одну полипептидную цепь с тремя дисульфидными связями. 35% аминокислотной последовательности пролактина идентично последовательностям гормона роста. Оба гормона имеют общие антигенные детерминанты, сходное строение рецепторов и пути трансдукции сигналов в клетки.
Рецепторы пролактина присутствуют в клетках многих тканей: в печени, почках, надпочечниках, яичках, яичниках, матке и других тканях.
Основная физиологическая функция про-лактина - стимуляция лактации. Пролактин индуцирует синтез α-лактальбумина и казеина, активирует синтез фосфолипидов и ТАГ.
На процессы роста пролактин влияет в значительно меньшей степени, чем гормон роста.
У мужчин пролактин повышает чувствительность клеток Лейдига к лютеинизирующему гормону, поддерживая таким образом необходимый уровень синтеза тестостерона; в почках пролак-
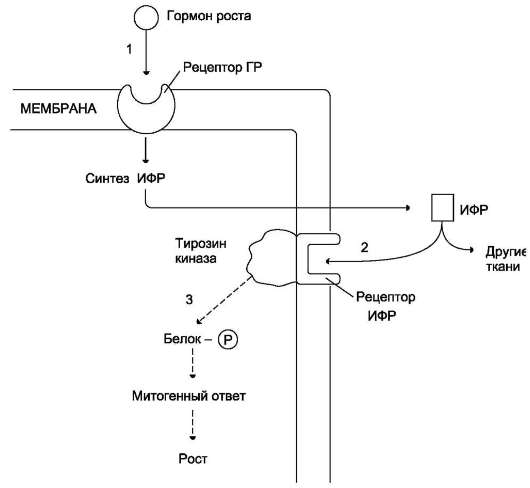
Рис. 11-13. Действие гормона роста через ИФР. Гормон роста взаимодействует с рецептором плазматической мембраны клеток, стимулируя синтез ИФР (1). ИФР, в свою очередь, взаимодействуют со специфическими рецепторами клеток той же или других тканей (2) и стимулируют фосфорилирование белков, участвующих в митозе и росте (3).
тин снижает экскрецию воды, влияет на реабсор-бцию ионов Na+ и К+; пролактин также повышает гуморальный и клеточный иммунитет.
Синтез и секрецию пролактина стимулируют тиреолиберин, серотонин, окситоцин, ацетил-холин, ингибирующий эффект оказывает дофамин.
Подобно большинству гормонов, пролактин секретируется в кровь эпизодически с интервалами 30-90 мин. Максимум секреции отмечается через 6-8 ч после начала сна. Концентрация пролактина в плазме крови женщин составляет 8-10 нг/мл, а мужчин - 5-8 нг/мл. T1/2 пролактина составляет 15-20 мин.
Плацента продуцирует гормон (плацентарный лактоген), гомологичный по аминокислотному составу гормону роста и пролактину. Все 3 гормона имеют общие антигенные детерминанты
и обладают рост-стимулирующей и лактогенной активностью. Существует гипотеза, согласно которой гены этих гормонов возникли в результате дупликации одного гена-предшественника.
2. Тиреотропин, лютеинизирующий гормон и фолликулостимулирующий гормон
Тиреотропин, ЛГ и ФСГ - гликопротеины. Тиреотропин (ТТГ) с молекулярной массой около 30 кД синтезируется в тиреотрофных клетках передней доли гипофиза.
Стимуляция секреции тиреотропина происходит под влиянием тиреолиберина, а основное ингибирующее действие оказывает повышение уровня тиреоидных гормонов. Пик секреции ТТГ отмечается в часы, непосредственно предшествующие сну, с последующим снижением в течение ночи.
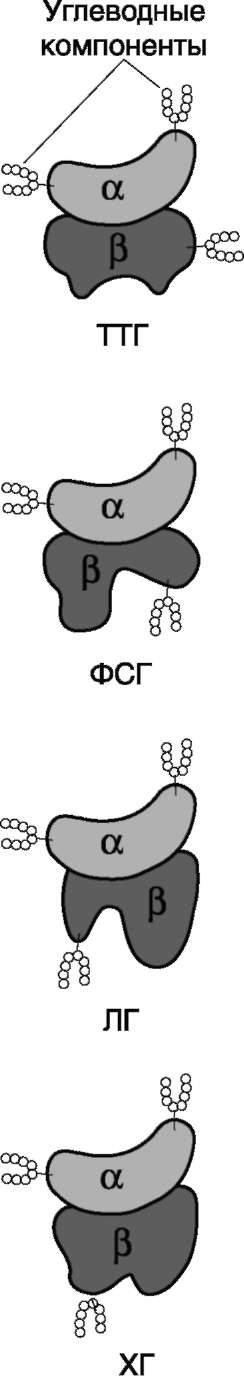
Рис. 11-14. Строение гормонов передней доли гипофиза и хорионического гонадотропина. ТТГ, ФСГ, ЛГ и ХГ - гликопротеины, состоящие из 2 субъединиц; α-субъединицы всех 4 гормонов идентичны; β-субъединицы различаются первичной структурой, строением олигосахаридных фрагментов и участков гликозилирования и определяют биологическую активность; α- и β-субъединицы содержат олигосахаридные фрагменты.
Основная биологическая функция тиреотро-пина - стимуляция синтеза и секреции йод-тиронинов (Т3 и Т4) в щитовидной железе. Трансдукция сигнала тиреотропина в клетки щитовидной железы происходит через рецепторы плазматической мембраны и активацию аденилатциклазы.
Рецептор тиреотропина состоит из 2 доменов, один из которых представляет собой гликопро-теин, а второй - ганглиозид (гликолипид, содержащий сиаловую кислоту). Для проявления биологического действия необходимо связывание тиреотропина с обоими доменами рецептора.
Тиреотропин оказывает на щитовидную железу 2 типа эффектов. Одни проявляются быстро (в течение нескольких минут) и включают стимуляцию всех стадий синтеза и секреции йодтиро-нинов (см. ниже подраздел III, В). Проявление других требует нескольких дней. К ним относят стимуляцию синтеза белков, фосфолипидов, нуклеиновых кислот, увеличение размеров и количества тиреоидных клеток.
Некоторые иммуноглобулины класса G, взаимодействуя с рецепторами тиреотропина, имитируют эффекты гормона. Подобные иммуноглобулины обнаруживаются у большинства больных гипертиреозом (см. ниже подраздел III, В). Помимо стимулирующих, обнаруживаются и антитела, вызывающие разрушение клеток щитовидной железы. Образование антител, имитирующих эффекты тиреотропина, - одна из частых причин нарушений функций щитовидной железы.
В группу гормонов, относящихся к глико-про-теинам, входят также гонадотропные гормоны гипофиза ЛГ и ФСГ и хорионический гонадо-тропин (ХГ) (рис. 11-14).
3. Группа гормонов, образующихся из проопиомеланокортина
Проопиомеланокортин (ПОМК) с молекулярной массой 28,5 кД синтезируется в передней и промежуточной долях гипофиза и в некоторых других тканях (кишечнике, плаценте). Полипептидная цепь ПОМК состоит из 265 аминокислотных остатков (рис. 11-15).
После отщепления сигнального пептида происходит частичный протеолиз оставшейся полипептидной цепи с образованием АКТГ и β-липот-
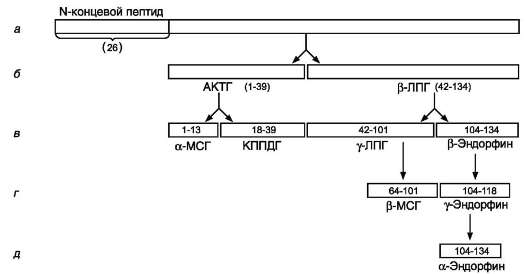
Рис. 11-15. Пептидные гормоны, образующиеся из ПОМК. А - ПОМК состоит из 265 аминокислотных остатков (а.к.), включая N-концевой сигнальный пептид из 26 аминокислот; Б - после отщепления сигнального пептида полипептидная цепь расщепляется на 2 фрагмента: АКТГ (39 а.к.) и β-липотропин (42-134 а.к); В, Г, Д - при дальнейшем протеолизе происходит образование α- и β-МСГ и эндорфинов. КППДГ - кор-тикотропиноподобный гормон промежуточной доли гипофиза.
ропина (β-ЛП). В разных клетках в результате избирательного протеолиза образуется разный набор пептидов: α- и β-меланоцитстимулиру-ющих гормонов (α- и β-МСГ) и эндорфинов. β-МСГ и кортикотропиноподобный гормон промежуточной доли у человека практически не образуются, так как у взрослых людей промежуточная доля не развита. В гипофизе человека найдены β-липотропин, γ-липотропин и β-эн-дорфин. Функции всех продуктов разрушения ПОМК недостаточно изучены.
Кортикотропин (АКТГ) - пептидный гормон; состоит из 39 аминокислотных остатков; синтезируется в клетках передней доли гипофиза под влиянием кортиколиберина.
Кортикотропин секретируется в импульсивном режиме. Скорость секреции составляет 5-25 мкг/сут. При стрессе (травма, ожог, хирургическое вмешательство, интоксикация химическими веществами, кровотечение, боль, психическая травма) концентрация АКТГ в крови возрастает во много раз. У здоровых людей наименьший уровень АКТГ в крови отмечается в конце дня и непосредственно перед сном, наибольший - в 6-8 ч утра, в момент пробуждения. T1/2 в крови составляет 15-25 мин.
Механизм действия АКТГ включает взаимодействие с рецептором плазматической мембраны клеток, активацию аденилатциклазы и фос-форилирование белков, участвующих в синтезе кортикостероидов (см. ниже подраздел III, Д). Эти эффекты усиливаются в присутствии ионов Са2+. В клетках коры надпочечников АКТГ стимулирует гидролиз эфиров холестерола, увеличивает поступление в клетки холестерола в составе ЛПНП; стимулирует превращение холестерола в прегненолон; индуцирует синтез митохондриальных и микросомальных ферментов, участвующих в синтезе кортикостероидов. Подробнее этапы синтеза кортикостероидов рассматриваются в подразделе III, Д.
4. Гормоны задней доли гипофиза
Задняя доля гипофиза, или нейрогипофиз, секретирует 2 активных гормона - вазопрессин, или антидиуретический гормон (АДГ), и оксито-цин. Окситоцин и вазопрессин - нонапептиды со сходной первичной структурой (рис. 11-16).
Оба гормона образуются в гипоталамусе в нейронах разных гипоталамических ядер в форме прогормонов, из которых в результате
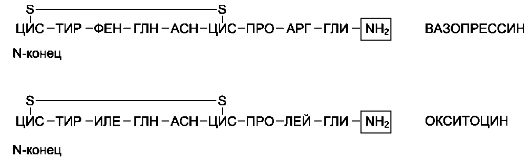
Рис. 11-16. Структура вазопрессина и окситоцина. Каждый нонапептид содержит остатки цистеина в положениях 1 и 6, связанные дисульфидными связями. У большинства животных и человека в положении 8 вазопрессина находится аргинин вместо лизина, в связи с чем он обозначается как аргинин вазопрессин.
посттрансляционной модификации образуются гормон и транспортный пептид нейрофизин (окситоцин+нейрофизин I и вазопрессин+нейро-физин II). В процессе транспорта в клетки задней доли гипофиза гормоны остаются нековалентно связанными со своими транспортными пептидами. В крови гормоны не связаны с нейрофизи-ном. T1/2 составляет 2-4 мин.
Основные биологические эффекты вазопрессина проявляются через взаимодействие с 2 типами рецепторов. V1-рецепторы расположены в клетках гладкой мускулатуры сосудов в комплексе с фосфолипазой С. Результат трансдукции сигнала в эти клетки - сокращение сосудов. V2-рецепторы расположены в клетках почечных канальцев. Взаимодействие вазопрессина с V2-рецепторами активирует аденилатциклазную систему, увеличивая в клетках концентрацию цАМФ и активность протеинкиназы А. В результате этой активации происходит фосфо-рилирование белков, стимулирующих экспрессию генов белков, которые образуют каналы, обеспечивающие реабсорбцию воды (см. ниже подраздел VI, A).
Окситоцин стимулирует сокращение гладкой мускулатуры матки, а также играет важную роль в стимуляции лактации. Он вызывает сокращение миоэпителиальных клеток молочных желёз, в результате чего происходит перераспределение молока из альвеолярных протоков в область соска.
Акт сосания материнской груди стимулирует секрецию пролактина, обеспечивая образование и секрецию молока.
В. НАРУШЕНИЯ ФУНКЦИЙ ГИПОТАЛАМО-ГИПОФИЗАРНОЙ СИСТЕМЫ
Нарушения функций гипоталамо-гипофи-зарной системы характеризуются разнообразными клиническими проявлениями.
Гипофункция может быть следствием уменьшения или полного подавления продукции тропных гормонов (пангипопитуитаризм) или частичного, при котором происходит нарушение синтеза и секреции одного или нескольких гормонов. Недостаток тропных гормонов гипофиза ведёт к резкому снижению функции периферических эндокринных желёз.
Выпадение гонадотропной функции гипофиза приводит к недостаточности яичников, аменорее, атрофии матки, молочных желёз. Вследствие снижения продукции кортикотро-пина развивается хроническая недостаточность коры надпочечников.
Дефицит гормона роста особенно опасен у детей. Известно несколько типов нарушений способности к нормальному росту вследствие абсолютного или относительного дефицита СТГ.
Гипофизарный нанизм, или карликовость (от греч. nanos - карлик). Причина нарушения роста и физического развития - дефицит гормона роста. Большинство форм гипофизарного нанизма развивается вследствие мутаций гена гормона роста. У большинства больных гипо-физарным нанизмом нарушение роста сочетается с другими эндокринными нарушениями. В некоторых случаях гипосекреция гормона роста может быть результатом аутоиммунного повреждения соматотрофных клеток гипофиза, черепно-мозговой травмы или радиации.
Нанизм Ларона возникает вследствие дефекта рецепторов гормона роста гепатоцитов и снижения синтеза ИФР-1 и ИФР-2. Концентрация СТГ в крови при этом повышена. Карликовость африканских пигмеев - результат нарушения пострецепторной передачи гормонального сигнала СТГ. При этой форме карликовости концентрация гормона роста в плазме нормальная, а концентрация ИФР-1 значительно снижена. Гиперфункция гормона роста обычно возникает в результате образования гормон-про-дуцирующей опухоли соматотрофных клеток гипофиза, что приводит к повышению ростовой активности. Если гиперсекреция гормона роста возникает у детей и подростков с незакончив-шимся процессом окостенения эпифизарных хрящей, но продолжающимся ростом длинных костей, развивается гигантизм (от греч. gigantos - великан). При гигантизме увеличение костей, мягких тканей и органов происходит сравнительно пропорционально. Гиперсекреция гормона роста у взрослых людей приводит к развитию акромегалии (от греч. akros - крайний, megas - большой), при которой рост тела ускоряется, но не в длину, а в ширину с диспропорциональным увеличением размеров лица, кистей рук, стоп, черепа, увеличением размеров внутренних органов.
У многих (~40%) больных акромегалией обнаруживается мутация в αs-субъединице G-бел-ка плазматической мембраны соматотрофных клеток, в результате которой αs-субъединица теряет ГТФ-азную активность. Вследствие этого развиваются продолжительная активация адени-латциклазы, избыточное образование цАМФ и избыточная секреция соматотропного гормона.
Г. ГОРМОНЫ ЩИТОВИДНОЙ ЖЕЛЕЗЫ
В щитовидной железе синтезируются гормоны - йодированные производные тирозина. Они объединены общим названием йодтирони-ны. К ним относят 3,5,3'-трийодтиронин (три-йодтиронин, Т3) и 3,5,3',5'-тетрайодтиронин (Т4), или тироксин (рис. 11-17).
Йодтаронины участвуют в регуляции многих процессов метаболизма, развития, клеточной диф-ференцировки, в регуляции экспрессии генов.
Заболевания, возникающие в результате нарушений синтеза, секреции и функций йодтирони-
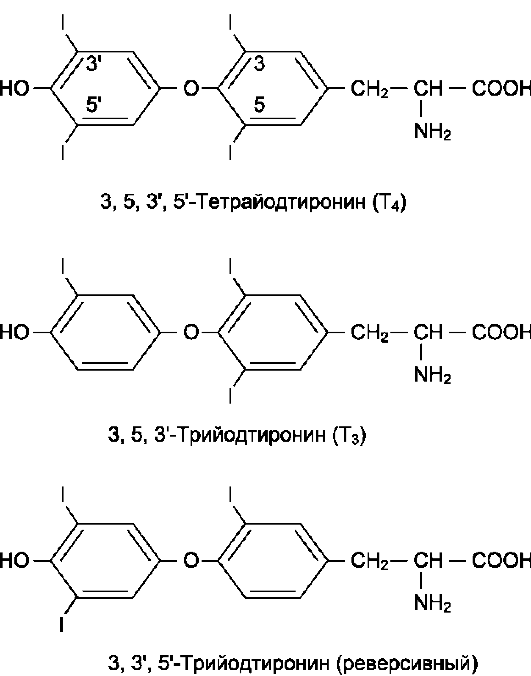
Рис. 11-17. Структура гормонов щитовидной железы.
нов, - наиболее распространённые заболевания эндокринной системы.
1. Биосинтез йодтиронинов
Йодтиронины синтезируются в составе белка тиреоглобулина (Тг) (рис. 11-18) в фолликулах, которые представляют собой морфологическую и функциональную единицу щитовидной железы.
Тиреоглобулин - гликопротеин с молекулярной массой 660 кД, содержащий 115 остатков тирозина. 8-10% массы тиреоглобулина представлено углеводами. Содержание йодида в организме составляет 0,2-1%.
Тиреоглобулин синтезируется на рибосомах шероховатого ЭР в виде претиреоглобулина, затем переносится в цистерны ЭР, где происходит формирование вторичной и третичной структуры, включая процессы гликозилирования. Из цистерн ЭР тиреоглобулин поступает в аппарат Гольджи, включается в состав секреторных гранул и секретируется во внеклеточный коллоид, где происходит йодирование остатков тирозина и образование йодтиронинов.
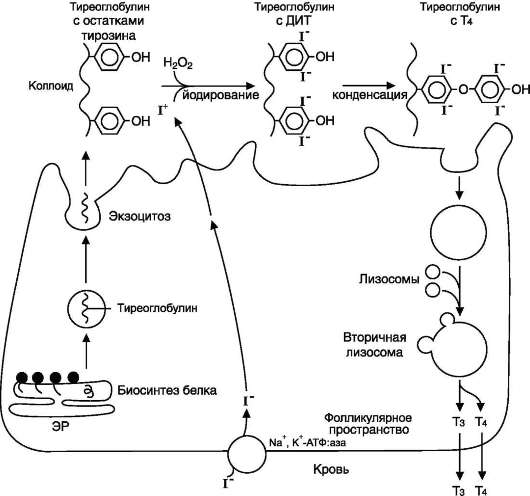
Рис. 11-18. Схема синтеза йодтиронинов. Тиреоглобулин синтезируется на рибосомах, далее поступает в аппарат Гольджи, а затем во внеклеточный коллоид, где он хранится и где происходит йодирование остатков тирозина. Образование йодтиронинов происходит в несколько этапов: транспорт йода в клетки щитовидной железы; окисление йода; йодирование остатков тирозина; образование йодтиронинов; транспорт йодтиронинов в кровь. ЭР - эндоплазматический ретикулум; ДИТ - дийодтиронин; Тг - тиреоглобулин; Т3 - трийодтиронин, Т4 - тироксин.
Йодирование тиреоглобулина и образование йодтиронинов осуществляется в несколько этапов (рис. 11-18).
Транспорт йода в клетки щитовидной железы. Йод в виде органических и неорганических соединений поступает в ЖКТ с пищей и питьевой водой. Суточная потребность в йоде составляет 150-200 мкг. 25-30% этого количества йодидов захватывается щитовидной железой. Транспорт йодида в клетки щитовидной железы - энергозависимый процесс и происходит при участии специального транспортного белка против
электрохимического градиента (соотношение концентраций I- в железе к концентрации I- в сыворотке крови в норме составляет 25:1). Работа этого йодид-переносящего белка сопряжена с Nа+,К+-АТФ-азой.
Окисление йода. Окисление I- в I+ происходит при участии гемсодержащей тиреопероксидазы и Н2О2 в качестве окислителя.
Йодирование тирозина. Окисленный йод взаимодействует с остатками тирозина в молекуле тиреоглобулина. Эта реакция также катализируется тиреопероксидазой.
Образование йодтиронинов. Под действием ти-реопероксидазы окисленный йод реагирует с остатками тирозина с образованием монойод-тирозинов (МИТ) и дийодтирозинов (ДИТ). Две молекулы ДИТ конденсируются с образованием йодтиронина Т4, а МИТ и ДИТ - с образованием йодтиронина Т3. Йодтиреоглобулин транспортируется из коллоида в фолликулярную клетку путём эндоцитоза и гидролизуется ферментами лизосом с освобождением Т3 и Т4. В нормальных условиях щитовидная железа секретирует 80-100 мкг Т4 и 5 мкг Т3 в сутки. Ещё 22-25 мкг Т3 образуется в результате дейодирования Т4 в периферических тканях по 5'-углеродному атому.
Транспорт и метаболизм йодтиронинов. От половины до двух третей Т3 и Т4 находятся в организме вне щитовидной железы. Большая часть их циркулирует в крови в связанной форме в комплексе с белками: тироксинсвязывающим глобулином (ТСГ) и тироксинсвязывающим преальбумином (ТСПА). ТСГ служит основным транспортным белком йодтиронинов, а также формой их депонирования. Он обладает более высоким сродством к Т3 и Т4 и в нормальных условиях связывает почти всё количество этих гормонов. Только 0,03% Т4 и 0,3% Т3 находятся в крови в свободной форме.
T1/2 Т4 в плазме в 4-5 раз больше, чем Т3. Для Т4 этот период составляет около 7 дней, а для Т3 - 1-1,5 дня. Биологическая активность йодтиро-нинов обусловлена несвязанной фракцией. Т3 - основная биологически активная форма йодтиро-нинов; его сродство к рецептору клеток-мишеней в 10 раз выше, чем у Т4. В периферических тканях в результате дейодирования части Т4 по пятому углеродному атому образуется так называемая «реверсивная» форма Т3, которая почти полностью лишена биологической активности.
Другие пути метаболизма йодтиронинов включают полное дейодирование, дезаминирование или декарбоксилирование. Йодированные продукты катаболизма йодтиронинов конъюгируют-ся в печени с глюкуроновой или серной кислотами (см. раздел 12), секретируются с жёлчью, в кишечнике вновь всасываются, дейодируются в почках и выделяются с мочой.
2. Регуляция синтеза и секреции йодтиронинов
Скорость синтеза и секреции йодтиронинов регулируются гипоталамо-гипофизарной
системой по механизму обратной связи (рис. 11-19).
Стимулом для повышения секреции тире-олиберина и тиреотропина служит снижение концентрации йодтиронинов в крови.
3. Механизм действия и биологические функции йодтиронинов
Клетки-мишени йодтиронинов имеют 2 типа рецепторов к этим гормонам. Основные эффекты йодтиронинов - результат их взаимодействия с высокоспецифичными рецепторами, которые в комплексе с гормонами постоянно находятся в ядре и взаимодействуют с определёнными последовательностями ДНК, участвуя в регуляции экспрессии генов.
Другие рецепторы расположены в плазматической мембране клеток, но это не те же самые белки, что в ядре. Они обладают более низким сродством к йодтиронинам и, вероятно, обеспе-
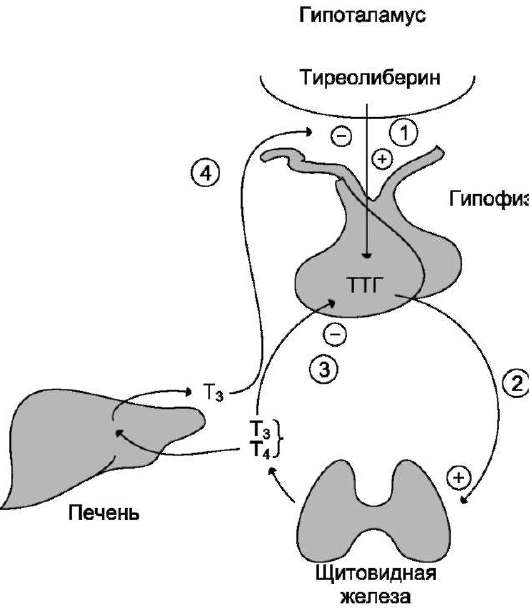
Рис. 11-19. Регуляция синтеза и секреции йодти-ронинов.
1 - тиреолиберин стимулирует освобождение ТТГ;
2 - ТТГ стимулирует синтез и секрецию йодтирони-нов; 3, 4 - йодтиронины тормозят синтез и секрецию ТТГ и тиреолиберина.
чивают связывание гормонов для удержания их в непосредственной близости к клетке.
При физиологической концентрации йодти-ронинов их действие проявляется в ускорении белкового синтеза, стимуляции процессов роста и клеточной дифференцировки. В этом отношении йодтиронины - синергисты гормона роста. Кроме того, Т3 ускоряет транскрипцию гена гормона роста. У животных при дефиците Т3 клетки гипофиза теряют способность к синтезу гормона роста.
Очень высокие концентрации Т3 тормозят синтез белков и стимулируют катаболические процессы, показателем чего служит отрицательный азотистый баланс.
Метаболические эффекты йодтиронинов относят в основном к энергетическому метаболизму, что проявляется в повышении поглощения клетками кислорода. Этот эффект проявляется во всех органах, кроме мозга, РЭС и гонад.
В разных клетках Т3 стимулирует работу Na+,К+-АТФ-азы, на что затрачивается значительная часть энергии, утилизируемой клеткой.
В печени йодтиронины ускоряют гликолиз, синтез холестерола и синтез жёлчных кислот. В печени и жировой ткани Т3 повышает чувствительность клеток к действию адреналина и косвенно стимулирует липолиз в жировой ткани и мобилизацию гликогена в печени. В физиологических концентрациях Т3 увеличивает в мышцах потребление глюкозы, стимулирует синтез белков и увеличение мышечной массы, повышает чувствительность мышечных клеток к действию адреналина.
Йодтиронины также участвуют в формировании ответной реакции на охлаждение увеличением теплопродукции, повышая чувствительность симпатической нервной системы к норадреналину и стимулируя секрецию но-радреналина (см. раздел 6).
4. Заболевания щитовидной железы
Гормоны щитовидной железы необходимы для нормального развития человека.
Гипотиреоз у новорождённых приводит к развитию кретинизма, который проявляется множественными врождёнными нарушениями и тяжёлой необратимой задержкой умственного развития.
Гипотиреоз развивается вследствие недостаточности йодтиронинов. Обычно гипотиреоз связан
с недостаточностью функции щитовидной железы, но может возникать и при заболеваниях гипофиза и гипоталамуса.
Наиболее тяжёлые формы гипотиреоза, сопровождающиеся слизистым отёком кожи и подкожной клетчатки, обозначают термином «микседема» (от греч. myxa - слизь, oedema - отёк). Отёчность обусловлена избыточным накоплением гликозаминогликанов и воды. В подкожной клетчатке накапливается глюку-роновая и в меньшей степени хондроитинсер-ная кислоты. Избыток гликозаминогликанов вызывает изменения коллоидной структуры межклеточного матрикса, усиливает его гид-рофильность и связывает ионы натрия, что приводит к задержке воды.
Характерные проявления заболевания: снижение частоты сердечных сокращений, вялость, сонливость, непереносимость холода, сухость кожи. Эти симптомы развиваются вследствие снижения основного обмена, скорости гликолиза, мобилизации гликогена и жиров, потребления глюкозы мышцами, уменьшения мышечной массы и снижения теплопродукции. При возникновении гипотиреоза у детей старшего возраста наблюдают отставание в росте без задержки умственного развития.
В настоящее время у взрослых людей частой причиной гипотиреоза является хронический аутоиммунный тиреоидит, приводящий к нарушению синтеза йодтиронинов (зоб Хашимото).
Гипотиреоз может быть также результатом недостаточного поступления йода в организм - эндемический зоб. Эндемический зоб (нетоксический зоб) часто встречается у людей, живущих в районах, где содержание йода в воде и почве недостаточно. Если поступление йода в организм снижается (ниже 100 мкг/сут), то уменьшается продукция йодтиронинов, что приводит к усилению секреции ТТГ (из-за ослабления действия йодтиронинов на гипофиз по механизму отрицательной обратной связи), под влиянием которого происходит компенсаторное увеличение размеров щитовидной железы (гиперплазия), но продукция йодтиронинов при этом не увеличивается.
Гипертиреоз возникает вследствие повышенной продукции йодтиронинов. Диффузный токсический зоб (базедова болезнь, болезнь Грейвса) - наиболее распространённое заболевание щитовидной железы. При этом заболевании отме-
чают увеличение размеров щитовидной железы (зоб), повышение концентрации йодтиронинов в 2-5 раз и развитие тиреотоксикоза.
Характерные признаки тиреотоксикоза: увеличение основного обмена, учащение сердцебиений, мышечная слабость, снижение массы тела (несмотря на повышенный аппетит), потливость, повышение температуры тела, тремор и экзофтальм (пучеглазие). Эти симптомы отражают одновременную стимуляцию йодтиронинами как анаболических (рост и дифференцировка тканей), так и катаболических (катаболизм углеводов, липидов и белков) процессов. В большей мере усиливаются процессы катаболизма, о чём свидетельствует отрицательный азотистый баланс.
Гипертиреоз может возникать в результате различных причин: развитие опухоли, тиреои-дит, избыточное поступление йода и йодсодер-жащих препаратов, аутоиммунные реакции.
Болезнь Грейвса возникает в результате образования антител к тиреоидным антигенам. Один из них, иммуноглобулин (IgG), имитирует действие тиреотропина, взаимодействуя с рецепторами тиреотропина на мембране клеток щитовидной железы. Это приводит к диффузному разрастанию щитовидной железы и избыточной неконтролируемой продукции Т3 и Т4, поскольку образование IgG не регулируется по механизму обратной связи. Уровень ТТГ при этом заболевании снижен вследствие подавления функции гипофиза высокими концентрациями йодтиронинов.
Д. ГОРМОНЫ КОРЫ НАДПОЧЕЧНИКОВ (КОРТИКОСТЕРОИДЫ)
В коре надпочечников синтезируется более 40 различных стероидов, различающихся по структуре и биологической активности. Биологически активные кортикостероиды объединяют в 3 основные класса в зависимости от их преобладающего действия.
Глюкокортикоиды, С21-стероиды, играют важную роль в адаптации к стрессу. Они оказывают разнообразные эффекты, но наиболее важный - стимуляция глюконеогенеза (см. раздел 7). Основной глюкокортикоид человека - кортизол.
Минералокортикоиды, С21-стероиды, необходимы для поддержания уровня Na+ и К+. Самый активный гормон этого класса - альдостерон (см. ниже подраздел VI).
Андрогены - С19-стероиды. В коре надпочечников образуются предшественники андрогенов, из которых наиболее активный - дегидроэпи-андростерон (ДЭА) и слабый - андростендион. Самый мощный андроген надпочечников тестостерон синтезируется в надпочечниках в небольшом количестве. Эти стероиды превращаются в более активные андрогены вне надпочечников. Тестостерон в незначительных количествах может превращаться в надпочечниках в эстрадиол. Но в норме продукция этих гормонов надпочечниками не играет существенной роли.
1. Биосинтез и метаболизм кортикостероидов
Общим предшественником кортикостероидов служит холестерол (рис. 11-20).
В митохондриях холестерол превращается в прегненолон при участии гидроксилазы, относящейся к группе цитохромов Р450. Цитохром Р450, отщепляющий боковую цепь, локализован во внутренней мембране митохондрий. Отщепление боковой цепи холестерола включает 2 реакции гидроксилирования: одна - по атому С22, другая - по С20. Последующее отщепление шестиуглеродного фрагмента приводит к образованию С21-стероида - прегненолона. Дальнейшее превращение прегненолона происходит под действием различных гидроксилаз с участием молекулярного кислорода и NADPH, а также дегидрогеназ, изомераз и лиаз. Эти ферменты имеют различную внутри- и межклеточную локализацию. В коре надпочечников различают 3 типа клеток, образующих 3 слоя, или зоны: клубочковую, пучковую и сетчатую. Каким именно стероидом окажется конечный продукт, зависит от набора ферментов в клетке и последовательности реакций гидроксилиро-вания. Например, ферменты, необходимые для синтеза альдостерона, присутствуют только в клетках клубочковой зоны, а ферменты синтеза глюкокортикоидов и андрогенов локализованы в пучковой и сетчатой зонах.
Путь биосинтеза кортизола. Кортизол синтезируется из холестерола, который в основном поступает из крови в составе ЛПНП или синтезируется в клетках из ацетил-КоА. Значительная часть эфиров холестерола накапливается в цитозоле клеток в липидных каплях. Под влиянием АКТГ происходит активация специфической эстеразы,
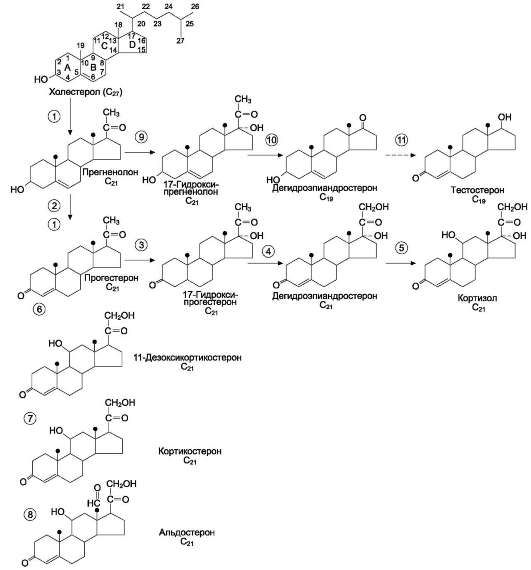
Рис. 11-20. Строение и основные этапы синтеза кортикостероидов. 1 - превращение холестерола в прегненолон (гидроксилаза, отщепляющая боковую цепь); 2 - образование прогестерона (3-Ь-гидроксис-тероиддегидрогеназа); 3, 4, 5 - реакции синтеза кортизола (3 - 17-гидроксилаза, 4 - 21-гидроксилаза, 5 - 11-гидроксилаза); 6, 7, 8 - путь синтеза альдостерона
(6 - 21-гидроксилаза, 7 - 11-гидроксилаза, 8 - 18-гидроксилаза, 18-гидроксидегидрогеназа); 9, 10, 11 - путь синтеза тестостерона (9 - 17-гидроксилаза, 10 - 17,20-лиаза, 11 - дегидрогеназа).
и свободный холестерол транспортируется в митохондрии (рис. 11-21).
Синтез кортизола начинается с превращения прегненолона в прогестерон. Эта реакция протекает в цитозоле клеток пучковой зоны коры надпочечников, куда прегненолон транспортируется из митохондрий. Реакцию катализирует 3-β-гидроксистероиддегидрогеназа.
В мембранах ЭР при участии 17-α-гидрок-силазы происходит гидроксилирование про-гес-терона по С17 с образованием 17-гидрокси-прогестерона. Этот же фермент катализирует превращение прегненолона в 17-гидроксип-регненолон, от которого далее при участии
17,20-лиазы может отщепляться двухуглеродная боковая цепь с образованием С19-стероида - дегидроэпиандростерона. 17-гидроксипрогес-терон служит предшественником кортизола, а дегидроэпиандростерон - предшественником андрогенов. Далее 17-ОН-прогестерон гидрок-силируется 21-гидроксилазой (Р450-С21), локализованной в мембране ЭР, и превращается в 11-дезоксикортизол, который переносится во внутреннюю мембрану митохондрий, где гид-роксилируется при участии цитохрома Р450-С11 с образованием кортизола.
Скорость синтеза и секреции кортизола стимулируются в ответ на стресс, травму, ин-
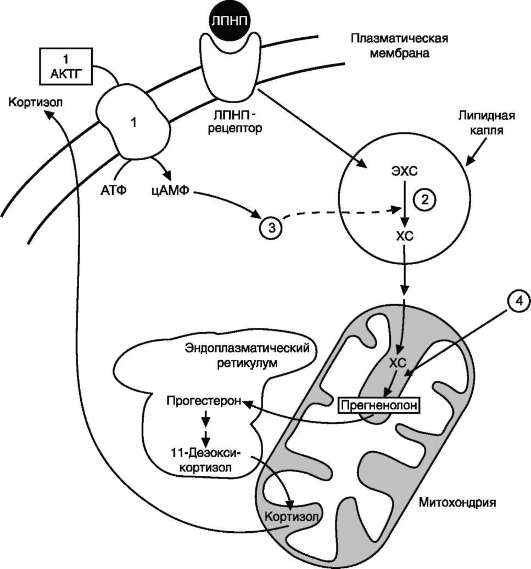
Рис. 11-21. Внутриклеточная локализация синтеза кортизола. 1 - аденилатциклазный комплекс; 2 - хо-лестеролэстераза; 3 - протеинкиназа А; 4 - холестеролдесмолаза отщепляет боковую цепь холестерола. ХС - холестерол; ЭХС - эфиры холестерола.
фекцию, понижение концентрации глюкозы в крови. Повышение концентрации кортизола подавляет синтез кортиколиберина и АКТГ по механизму отрицательной обратной связи.
Синтез минералокортикоидов в клетках клу-бочковой зоны коры надпочечников также начинается с превращения холестерола в пре-гненолон, а затем в прогестерон. Прогестерон гидроксилируется вначале по С21 с образованием 11-дезоксикортикостерона. Следующее гидроксилирование происходит по С11, что приводит к образованию кортикостерона, обладающего слабовыраженной глюкокортикоидной и минералокортикоидной активностью.
В клетках клубочковой зоны 17-а-гидроксилаза отсутствует, но есть митохондриальная 18-гид-роксилаза, при участии которой кортикостерон гидроксилируется, а затем дегидрируется с образованием альдегидной группы у С18.
Главным стимулом для синтеза альдостерона служит ангиотензин II (см. ниже подраздел V).
Транспорт кортикостероидов. Кортизол в плазме крови находится в комплексе с α-глобулином транскортином и в небольшом количестве в свободной форме. Синтез транскортина протекает в печени и стимулируется эстрогенами.
T1/2 кортизола составляет 1,5-2 ч. Несвязанный, или свободный кортизол, составляет около 8% от общего количества гормона в плазме и является биологически активной фракцией.
Альдостерон не имеет специфического транспортного белка, но образует слабые связи с альбумином.
Катаболизм гормонов коры надпочечников происходит прежде всего в печени. Здесь протекают реакции гидроксилирования, окисления и восстановления гормонов. Продукты катаболизма кортикостероидов (кроме кортикостерона и альдостерона) выводятся с мочой в форме 17-кетостероидов, образующихся в результате отщепления боковой цепи. Эти продукты метаболизма выделяются преимущественно в виде конъюгатов с глюкуроновой и серной кислотами. 17-Окси- и 17-кетостероиды образуются также при катаболизме половых гормонов, которые имеют у С17 гидроксиили кетогруппы. У мужчин 2/3 кетостероидов образуется за счёт кортикостероидов и 1/3 за счёт тестостерона (всего 12-17 мг/сут). У женщин 17-кетосте-роиды образуются преимущественно за счёт кортикостероидов (7-12 мг/сут). Определение
17-кетостероидов в моче позволяет оценить как количество глюкокортикоидов, секретируемых корой надпочечников, так и функцию надпочечников.
2. Биологические функции кортикостероидов отличаются широким спектром влияний на процессы метаболизма и подробно рассматриваются в соответствующих разделах.
Важнейший фактор в механизме действия кортикостероидов - взаимодействие их со специфическими рецепторами, расположенными в цитозоле клетки или в ядре. Регуляция внутриклеточных процессов под влиянием кортико-стероидных гормонов проявляется в изменении количества белков, обычно ключевых ферментов метаболизма, путём регуляции транскрипции генов в клетках-мишенях.
Влияние глюкокортикоидов на промежуточный метаболизм связано с их способностью коор-динированно воздействовать на разные ткани и разные процессы, как анаболические, так и катаболические.
Кортизол стимулирует образование глюкозы в печени, усиливая глюконеогенез и одновременно увеличивая скорость освобождения аминокислот - субстратов глюконеогенеза из периферических тканей. В печени кортизол индуцирует синтез ферментов катаболизма аминокислот (аланинаминотрансферазы, триптофанпирро-лазы и тирозинаминотрансферазы и ключевого фермента глюконеогенеза - фосфоенолпиру-ваткарбоксикиназы). Кроме того, кортизол стимулирует синтез гликогена в печени и тормозит потребление глюкозы периферическими тканями. Это действие кортизола проявляется в основном при голодании и недостаточности инсулина (см. ниже подраздел V). У здоровых людей эти эффекты кортизола уравновешиваются инсулином.
Избыточное количество кортизола стимулирует липолиз в конечностях и липогенез в других частях тела (лицо и туловище). Кроме того, глюкокортикоиды усиливают липолитическое действие катехоламинов и гормона роста.
Влияние глюкокортикоидов на обмен белков и нуклеиновых кислот проявляется двояко: в печени кортизол в основном оказывает анаболический эффект (стимулирует синтез белков и нуклеиновых кислот). В мышцах, лимфоид-ной и жировой ткани, коже и костях кортизол тормозит синтез белков, РНК и ДНК и стимулирует распад РНК и белков.
При высокой концентрации глюкокорти-коиды подавляют иммунные реакции, вызывая гибель лимфоцитов и инволюцию лимфоидной ткани; подавляют воспалительную реакцию, снижая число циркулирующих лейкоцитов, а также индуцируя синтез липокортинов, которые инги-бируют фосфолипазу А2, снижая таким образом синтез медиаторов воспаления - простагланди-нов и лейкотриенов (см. раздел 8).
Высокая концентрация глюкокортикоидов вызывает торможение роста и деления фибро-бластов, а также синтез коллагена и фибро-нектина (см. раздел 15). Для гиперсекреции глюкокортикоидов типичны истончение кожи, плохое заживление ран, мышечная слабость и атрофия мышц.
Глюкокортикоиды участвуют в физиологическом ответе на стресс, связанный с травмой, инфекцией или хирургическим вмешательством. В этом ответе в первую очередь участвуют кате-холамины, но во многих случаях для проявления их максимальной активности необходимо участие глюкокортикоидов.
Минералокортикоиды стимулируют реабсорб-цию Na+ в дистальных извитых канальцах и собирательных трубочках почек. Кроме того, они способствуют секреции К+, NH4+ в почках, а также в других эпителиальных тканях: потовых железах, слизистой оболочке кишечника и слюнных железах. В организме человека альдостерон - наиболее активный минералокортикоид.
Механизм действия и биологические эффекты альдостерона подробно рассмотрены в подразделе VI этого раздела.
3. Изменения метаболизма при гипо-и гиперфункции коры надпочечников
Заболевания коры надпочечников могут проявиться симптомами как гипо-, так и гиперпродукции гормонов.
Большинство клинических проявлений над-почечниковой недостаточности обусловлено дефицитом глюкокортикоидов и минералокор-тикоидов.
Острая надпочечниковая недостаточность представляет большую угрозу для жизни, так как сопровождается декомпенсацией всех видов обмена и процессов адаптации. Она проявляется сосудистым коллапсом, резкой адинамией, потерей сознания. Такое состояние возникает вследствие нарушения обмена электролитов,
которое приводит к потере ионов Na+ и Cl- с мочой, обезвоживанию за счёт потери внеклеточной жидкости, повышению уровня К+ в сыворотке крови, в межклеточной жидкости и клетках, в результате чего может нарушаться сократительная способность миокарда. Изменение углеводного обмена проявляется в снижении уровня сахара в крови, уменьшении запаса гликогена в печени и скелетных мышцах.
Острая недостаточность функции коры надпочечников может быть следствием декомпенсации хронических заболеваний, а также развивается у больных, лечившихся длительное время глюкокортикоидными препаратами по поводу неэндокринных заболеваний, например инфекционно-аллергических заболеваний.
В результате длительного приёма глюко-кортикоидов подавляется функция гипотала-мо-гипофизарно-надпочечниковой системы и развивается атрофия клеток коры надпочечников. Резкая отмена гормональных препаратов может сопровождаться острой надпочечниковой недостаточностью (так называемый синдром «отмены»).
Первичная недостаточность надпочечников (болезнь Аддисона) развивается в результате поражения коры надпочечников туберкулёзным или аутоиммунным процессом. Основные клинические проявления выражаются в снижении массы тела, общей слабости, снижении аппетита, тошноте, рвоте, снижении АД и типичной для первичной надпочечниковой недостаточности гиперпигментации кожи («бронзовая болезнь») . Причина гиперпигментации - повышение продукции ПОМК - предшественника АКТГ и меланоцитстимулирующего гормона.
Вторичная недостаточность надпочечников может развиться при дефиците АКТГ, что, в свою очередь, может быть следствием опухоли или инфекционного поражения гипофиза. При вторичной недостаточности надпочечников, в отличие от болезни Аддисона, отсутствует гиперпигментация.
При врождённой гиперплазии надпочечников нарушается синтез кортизола. В 95% случаев при этой патологии обнаруживается дефект 21-гид-роксилазы (реже 11-гидроксилазы). Снижение продукции кортизола сопровождается увеличением секреции АКТГ, накоплением промежуточных продуктов синтеза кортикостероидов, в частности, предшественников андрогенов.
Избыток андрогенов ведёт к усилению роста тела, раннему половому созреванию у мальчиков и развитию мужских половых признаков у девочек (адреногенитальный синдром).
При частичной недостаточности 21-гидрок-силазы у женщин может нарушаться менструальный цикл.
Гиперпродукция глюкокортикоидов (гиперкор-тицизм) может быть следствием повышения уровня АКТГ при опухолях гипофиза (болезнь Иценко-Кушинга) и опухолях других клеток (бронхов, тимуса, поджелудочной железы), вырабатывающих кортикотропинподобные вещества, или избыточного синтеза кортизола при гормонально-активных опухолях коры надпочечников (синдром Иценко-Кушинга).
При гиперкортицизме наблюдаются гипергликемия и снижение толерантности к глюкозе, обусловленные стимуляцией глюконеогенеза («стероидный диабет»), усиление катаболизма белков, уменьшение мышечной массы, истончение кожи, остеопороз, инволюция лимфоидной ткани. Характерно своеобразное перераспределение отложений жира («лунообразное лицо», выступающий живот). Гипернатриемия, гипер-тензия, гипокалиемия обусловлены некоторой минералокортикоидной активностью кортизола, которая проявляется при его избытке.
Для выявления первичной причины гипер-кортицизма, помимо определения концентрации АКТГ в плазме крови, используют тесты с применением высоких доз синтетического глюкокорти-коида дексаметазона (структурного аналога корти-зола). Дексаметазон подавляет секрецию АКТГ по механизму отрицательной обратной связи.
Для болезни Иценко-Кушинга характерно снижение концентрации кортизола после применения дексаметазона более чем на 50%. Отсутствие реакции на введение дексаметазона может указывать на наличие опухоли надпочечников или внегипофизарной секреции АКТГ.
Е. ГОРМОНЫ МОЗГОВОГО СЛОЯ НАДПОЧЕЧНИКОВ
Подобно задней доле гипофиза, мозговой слой надпочечников - производное нервной ткани. Его можно рассматривать как продолжение симпатической нервной системы, так как преганглионарные волокна чревного нерва оканчиваются на хромаффинных клетках мозгового слоя надпочечников.
Своё название эти клетки получили потому, что они содержат гранулы, окрашивающиеся бихроматом калия в красный цвет. Такие клетки находятся также в сердце, печени, почках, половых железах, постганглионарных нейронах симпатической нервной системы и в ЦНС.
При стимуляции преганглионарного нейрона хромаффинные клетки продуцируют катехол-амины - дофамин, адреналин и норадрена-лин.
У большинства видов животных хромаффин-ные клетки секретируют в основном адреналин (~ 80%) и в меньшей степени норадреналин.
По химическому строению катехоламины - 3,4-дигидроксипроизводные фенилэтиламина. Непосредственным предшественником гормонов служит тирозин (см. раздел 9).
1. Синтез и секреция катехоламинов
Синтез катехоламинов происходит в цитоплазме и гранулах клеток мозгового слоя надпочечников (рис. 11-22). В гранулах происходит также запасание катехоламинов.
Катехоламины поступают в гранулы путём АТФ-зависимого транспорта и хранятся в них в комплексе с АТФ в соотношении 4:1 (гормон-АТФ). Разные гранулы содержат разные катехо-ламины: некоторые только адреналин, другие - норадреналин, третьи - оба гормона.
Секреция гормонов из гранул происходит путём экзоцитоза. Катехоламины и АТФ освобождаются из гранул в том же соотношении, в каком они сохраняются в гранулах. В отличие от симпатических нервов, клетки мозгового слоя надпочечников лишены механизма обратного захвата выделившихся катехоламинов.
В плазме крови катехоламины образуют непрочный комплекс с альбумином. Адреналин транспортируется в основном к печени и скелетным мышцам. Норадреналин образуется в основном в органах, иннервируемых симпатическими нервами (80% от общего количества). Норадреналин лишь в незначительных количествах достигает периферических тканей. T1/2 катехоламинов - 10-30 с. Основная часть катехоламинов быстро метаболизируется в различных тканях при участии специфических ферментов (см. раздел 9). Лишь небольшая часть адреналина (~ 5%) выделяется с мочой.
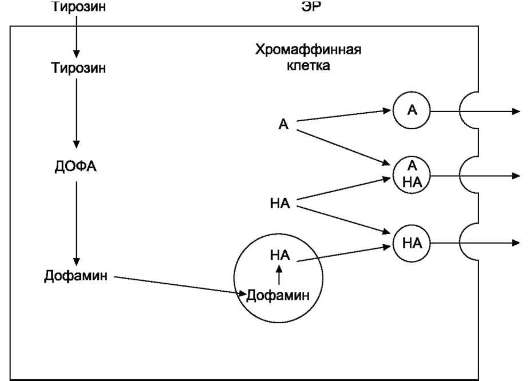
Рис. 11-22. Синтез и секреция катехоламинов. Биосинтез катехоламинов происходит в цитоплазме и гранулах клеток мозгового слоя надпочечников. В одних гранулах содержится адреналин, в других норадреналин, а в некоторых - оба гормона. При стимуляции содержимое гранул высвобождается во внеклеточную жидкость. А - адреналин; НА - норадреналин.
2. Механизм действия и биологические функции катехоламинов
Катехоламины действуют на клетки-мишени через рецепторы, локализованные в плазматической мембране. Выделяют 2 главных класса таких рецепторов: α-адренергические и β-адре-нергические. Все рецепторы катехоламинов - гликопротеины, которые являются продуктами разных генов, различаются сродством к агонис-там и антагонистам и передают сигналы в клетки с помощью разных вторичных посредников. Это определяет характер их влияния на метаболизм клеток-мишеней.
Адреналин взаимодействует как с α-, так и с β-рецепторами; норадреналин в физиологических концентрациях главным образом взаимодействует с α-рецепторами.
Взаимодействие гормона с β-рецепторами активирует аденилатциклазу, тогда как связывание с α2-рецептором её ингибирует. При взаимодействии гормона с α1-рецептором происходит активация фосфолипазы С и стимулируется инозитолфосфатный путь передачи сигнала (см. раздел 5).
Биологические эффекты адреналина и но-радреналина затрагивают практически все функции организма и рассматриваются в соответствующих разделах. Общее во всех этих эффектах заключается в стимуляции процессов, необходимых для противостояния организма чрезвычайным ситуациям.
3. Патология мозгового вещества надпочечников
Основная патология мозгового вещества надпочечников - феохромоцитома, опухоль, образованная хромаффинными клетками и продуцирующая катехоламины. Клинически феохромоцитома проявляется повторяющимися приступами головной боли, сердцебиения, потливости, повышением АД и сопровождается характерными изменениями метаболизма (см. разделы 7, 8).
Ж. ГОРМОНЫ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ И ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА
Поджелудочная железа выполняет в организме две важнейшие функции: экзокрин-ную и эндокринную. Экзокринная функция
обеспечивает синтез и секрецию ферментов и ионов, необходимых для процессов пищеварения. Эндокринную функцию выполняют клетки островкового аппарата поджелудочной железы, которые секретируют гормоны, участвующие в регуляции многих процессов в организме.
В островковой части поджелудочной железы (островки Лангерханса) выделяют 4 типа клеток, секретирующих разные гормоны: А- (или α-) клетки секретируют глюкагон, В- (или β-) - инсулин, D- (или δ-) - соматостатин, F-клетки секретируют панкреатический полипептид.
1. Инсулин. Строение, синтез и секреция
Инсулин - полипептид, состоящий из двух полипептидных цепей. Цепь А содержит 21 аминокислотный остаток, цепь В - 30 аминокислотных остатков. Обе цепи соединены
между собой двумя дисульфидными мостиками (рис. 11-23). Инсулин может существовать в нескольких формах: мономера, димера и гек-самера. Гексамерная структура инсулина стабилизируется ионами цинка, который связывается остатками Гис в положении 10 В-цепи всех 6 субъединиц.
Молекула инсулина содержит также внутримолекулярный дисульфидный мостик, соединяющий шестой и одиннадцатый остатки в А-цепи. Инсулины некоторых животных имеют значительное сходство по первичной структуре с инсулином человека.
Бычий инсулин отличается от инсулина человека по трём аминокислотным остаткам, а инсулин свиньи отличается только на одну аминокислоту, которая представлена алани-ном вместо треонина на карбоксильном конце В-цепи.
Рис. 11-23. Структура инсулина человека. А. Первичная структура инсулина. Б. Модель третичной структуры инсулина (мономер): 1 - А-цепь; 2 - В-цепь; 3 - участок связывания с рецептором.
В обеих цепях во многих положениях встречаются замены, не оказывающие влияния на биологическую активность гормона. Наиболее часто эти замены обнаруживаются в положениях 8, 9 и 10 цепи А.
В то же время в положениях дисульфидных связей, остатков гидрофобных аминокислот в С-концевых участках В-цепи и С- и N-кон-цевых остатков А-цепи замены встречаются очень редко, что свидетельствует о важности этих участков для проявления биологической активности инсулина. Использование химических модификаций и замен аминокислот в этих участках позволили установить структуру активного центра инсулина, в формировании которого принимают участие остатки фенила-ланина В-цепи в положениях 24 и 25 и N- и С-концевые остатки цепи А.
Биосинтез инсулина включает образование двух неактивных предшественников, препроинсулина и проинсулина, которые в результате последовательного протеолиза превращаются в активный гормон. Биосинтез препроинсулина начинается с образования сигнального пептида на полирибосомах, связанных с ЭР. Сигналыный пептид проникает в просвет ЭР и направляет поступление в просвет ЭР растущей полипептидной цепи. После окончания синтеза препроинсулина сигнальный пептид, включающий 24 аминокислотных остатка, отщепляется (рис. 11-24).
Проинсулин (86 аминокислотных остатков) поступает в аппарат Гольджи, где под действием специфических протеаз расщепляется в нескольких участках с образованием инсулина (51 аминокислотный остаток) и С-пептида, состоящего из 31 аминокислотного остатка.
Рис. 11-24. Схема биосинтеза инсулина в β-клетках островков Лангерханса. ЭР - эндоплазматический ретикулум. 1 - образование сигнального пептида; 2 - синтез препроинсулина; 3 - отщепление сигнального пептида; 4 - транспорт проинсулина в аппарат Гольджи; 5 - превращение проинсулина в инсулин и С-пептид и включение инсулина и С-пептида в секреторные гранулы; 6 - секреция инсулина и С-пептида.
Инсулин и С-пептид в эквимолярных количествах включаются в секреторные гранулы. В гранулах инсулин соединяется с цинком, образуя димеры и гексамеры. Зрелые гранулы сливаются с плазматической мембраной, и инсулин и С-пептид секретируются во внеклеточную жидкость в результате экзоцитоза. После секреции в кровь олигомеры инсулина распадаются. T1/2 инсулина в плазме крови составляет 3-10 мин, С-пептида - около 30 мин.
Разрушение инсулина происходит под действием фермента инсулиназы в основном в печени и в меньшей степени в почках.
Регуляция синтеза и секреции инсулина. Глюкоза - главный регулятор секреции инсулина, а β-клетки - наиболее важные глюкозо-чувстви-тельные клетки в организме. Глюкоза регулирует экспрессию гена инсулина, а также генов других белков, участвующих в обмене основных энергоносителей. Действие глюкозы на скорость экспрессии генов может быть прямым, когда глюкоза непосредственно взаимодействует с транскрипционными факторами, или вторичным, через влияние на секрецию инсулина и глюкагона. При стимуляции глюкозой инсулин быстро освобождается из секреторных гранул, что сопровождается активацией транскрипции мРНК инсулина.
Синтез и секреция инсулина не являются строго сопряжёнными процессами. Синтез гормона стимулируется глюкозой, а секреция его является Са2+-зависимым процессом и при дефиците Са2+ снижается даже в условиях высокой концентрации глюкозы, которая стимулирует синтез инсулина.
Потребление глюкозы β-клетками происходит в основном при участии ГЛЮТ-1 и ГЛЮТ-2, и концентрация глюкозы в клетках быстро уравнивается с концентрацией глюкозы в крови. В β-клетках глюкоза превращается в глюкозо-6-фосфат глюкокиназой, имеющей высокую Кт, вследствие чего скорость её фосфорилирования почти линейно зависит от концентрации глюкозы в крови. Фермент глюкокиназа - один из важнейших компонентов глюкозо-чувстви-тельного аппарата β-клеток, в который, помимо глюкозы, вероятно, входят промежуточные продукты метаболизма глюкозы, цитратного цикла и, возможно, АТФ. Мутации глюкокиназы приводят к развитию одной из форм сахарного диабета.
На секрецию инсулина влияют другие гормоны. Адреналин через α2-рецепторы тормозит секрецию инсулина даже на фоне стимуляции глюкозой, β-адренергические агонисты её стимулируют, вероятно, в результате повышения концентрации цАМФ. Этот механизм, полагают, лежит в основе действия гормонов ЖКТ, таких как секретин, холецистокинин и желудочный ингибирующий пептид (GIP), которые повышают секрецию инсулина. Высокие концентрации гормона роста, кортизола, эстрогенов также стимулируют секрецию инсулина.
2. Биологические функции инсулина
Инсулин - главный анаболический гормон. Он
участвует в регуляции метаболизма, транспорта глюкозы, аминокислот, ионов, в синтезе белков. Инсулин влияет также на процессы репликации и транскрипции, участвуя таким образом в регуляции клеточной дифференцировки, пролиферации и трансформации клеток. Участие инсулина в регуляции метаболизма рассмотрено в соответствующих разделах (см. разделы 7, 8, 9). Влияние инсулина на ключевые ферменты метаболизма представлено в табл. 11-7.
Транспорт глюкозы в клетки происходит при участии специальных белков-переносчиков (см. раздел 7). Переносчик, регулируемый инсулином (ГЛЮТ-4), содержится только в мышцах и жировой ткани (инсулинзависимые ткани). В отсутствие инсулина ГЛЮТ-4 находятся в цитозольных везикулах. Под влиянием инсулина происходит транслокация везикул в плазматическую мембрану; при снижении концентрации гормона глюкотранспортёры возвращаются в цитозоль, и транспорт глюкозы прекращается.
В клетках печени инсулин индуцирует синтез глюкокиназы. В результате фосфорилирования концентрация свободной глюкозы в клетках поддерживается на низком уровне, что способствует её транспорту из крови по градиенту концентрации.
Влияние инсулина на метаболизм глюкозы. Инсулин стимулирует утилизацию глюкозы в клетках разными путями. Около 50% глюкозы используется в процессе гликолиза, 30-40% превращается в жиры и около 10% накапливается в форме гликогена. Общий результат стимуляции этих процессов - снижение концентрации глюкозы в крови.
Таблица 11-7. Влияние инсулина на ключевые ферменты метаболизма

Влияние инсулина на метаболизм глюкозы осуществляется путём повышения активности и количества ключевых ферментов гликолиза: глюкокиназы, фосфофруктокиназы, пируват-киназы (см. раздел 7). В мышцах инсулин активирует гексокиназу II. В печени и мышцах под влиянием инсулина снижается концентрация цАМФ в результате активации фосфодиэсте-разы. Кроме того, инсулин активирует фосфа-тазы, дефосфорилирующие гликогенсинтазу, в результате чего происходит активация синтеза гликогена и тормозится его распад.
Эффекты инсулина, обусловленные фосфори-лированием и дефосфорилированием ферментов, проявляются очень быстро, в течение нескольких секунд и минут. Параллельно с активацией ферментов гликолиза инсулин тормозит глюконе-огенез, репрессируя синтез ключевого фермента глюконеогенеза - фосфоенолпируваткарбокси-киназы (ФЕП карбоксикиназы).
Влияние инсулина на метаболизм жиров. В печени и жировой ткани инсулин стимулирует синтез жиров, обеспечивая получение для этого процесса необходимых субстратов (ацетил-КоА, α-глицерофосфат и NADPH) из глюкозы. В адипоцитах инсулин активирует ацетил КоА-карбоксилазу и ЛП-липазу и индуцирует синтез синтазы жирных кислот, ацетил-КоА-карбокси-
лазы и ЛП-липазы (см. раздел 8 и табл. 11-7). Инсулин в жировой ткани тормозит мобилизацию жиров. Он активирует фосфатазу, которая дефосфорилирует и тем самым инактивирует гормончувствительную ТАГ-липазу. Таким образом, под влиянием инсулина снижается концентрация жирных кислот, циркулирующих в крови (см. раздел 8). Инсулин стимулирует потребление нейтральных аминокислот в мышцах и синтез белков в печени, мышцах и сердце.
Инсулин стимулирует пролиферацию большого количества клеток в культуре тканей, а также, вероятно, может участвовать в регуляции роста in vivo. Для изучения регуляции роста чаще всего используют культуры фиброб-ластов. В таких клетках инсулин усиливает способность фактора роста фибробластов (FGF), тромбоцитарного фактора роста (PDGF), фактора роста эпидермиса (EGF), простагландина (PGF2α), вазопрессина и аналогов цАМФ активировать размножение клеток, остановленных в фазе G.
3. Механизм действия инсулина
Действие инсулина начинается с его связывания со специфическим гликопротеиновым рецептором на поверхности клетки-мишени (см. раздел 5). Рецепторы инсулина обнару-
жены почти во всех типах клеток, но
больше всего их в гепатоцитах и клетках жировой ткани. Так как
концентрация инсулина в крови составляет ~10-
Инсулиновый рецептор (IR) постоянно синтезируется и разрушается. T1/2 рецептора составляет 7-12 ч. При высокой концентрации инсулина в плазме крови, например, при ожирении, число инсулиновых рецепторов может уменьшаться, и клетки-мишени становятся менее чувствительными к инсулину, что может быть одной из причин сахарного диабета II типа (см. ниже подраздел V).
Снижение чувствительности клеток к гормону (десенситизация) опосредуется 2 механизмами. Первый включает утрату рецепторов путём их интернализации. Комплекс инсулин-рецептор захватывается внутрь клетки эндоцитозом. В результате интернализации часть рецепторов подвергается разрушению в лизосомах, а часть возвращается в плазматическую мембрану. Второй механизм десенситизации - кова-лентная модификация рецептора в результате фосфорилирования. Так, фосфорилирование IR по остаткам серина и треонина снижает его сродство к инсулину.
Рецептор инсулина относят к типу рецепторов, обладающих тирозинкиназной активностью (см. раздел 5). Стимулированное инсулином аутофосфорилирование β-субъединицы IR по остаткам тирозина приводит к фосфорилиро-ванию других внутриклеточных белков - субстратов инсулинового рецептора (IRS). Известно несколько таких субстратов: IRS-1, IRS-2, а также некоторые белки семейства STAT.
Главную роль в формировании ответной реакции клетки на инсулиновый сигнал играет IRS-1. IRS-1 - фосфопротеин, состоящий из более чем 1200 аминокислотных остатков. Часть остатков серина, тирозина и треонина фосфори-лирована. При стимуляции инсулином степень фосфорилирования IRS-1 увеличивается и придаёт ему способность соединяться с другими цитозольными белками. Это приводит к активации нескольких сигнальных путей, представляющих каскад реакций активации специфических протеинкиназ. В результате активации
протеинкиназ происходит фосфорилирование ферментов и факторов транскрипции, что составляет основу многочисленных эффектов инсулина.
Активация инсулином сигнального пути Ras.
Белок, известный как Ras-белок, относят к семейству малых ГТФ-связывающих белков. В неактивном состоянии Ras-белок прикреплён к внутренней поверхности плазматической мембраны и связан с ГДФ. Стимуляция инсулином приводит к образованию активной ГТФ-связан-ной формы Ras (рис. 11-25).
Превращение Ras-белка в активную форму происходит при участии семейства белков, являющихся активаторами протеинкиназ и протеинкиназами и, так же, как Ras-белок, получившие свои названия от онкогенов (см. раздел 16). Один из субстратов инсулинового рецептора Shc участвует в образовании комплекса с небольшим цитозольным белком Grb. Образовавшийся комплекс взаимодействует с Ras-белком. В этот комплекс включаются другие белки: GAP (от англ. GTP-ase activating factor - фактор, активирующий ГТФ:азу), GEF (от англ. GTP exchange factor - фактор обмена ГТФ) и SOS (от англ. son of sevenless, названный по мутации гена у мушки дрозофилы). Два последних белка способствуют отделению ГДФ от Ras-белка и присоединению ГТФ. Активированный Ras соединяется с протеинкиназой Raf-1. Raf-1 в неактивном состоянии находится в цитозоле в соединении с шаперонами. Активация Raf-1 происходит в результате многоэтапного процесса, включающего присоединение белка к плазматической мембране, фосфорилирование и взаимодействие с рецептором инсулина. Активированная Raf-киназа стимулирует каскад реакций фосфорилирования и активации других протеинкиназ, в частности, митогенактиви-руемых протеинкиназ (МАПК). При участии Raf-1 сначала фосфорилируется и активируется киназа МАПК, которая, в свою очередь, фос-форилирует МАПК.
МАПК фосфорилирует многие цитоплаз-матические белки: протеинкиназу рр90S6, белки рибосом, фосфолипазу А2, активаторы транскрипции (ПСАТ). Путь Ras активируется не только инсулином, но и многими другими гормонами и факторами роста. Многие компоненты этого пути являются продуктами протоонкогенов, мутации которых приводят к
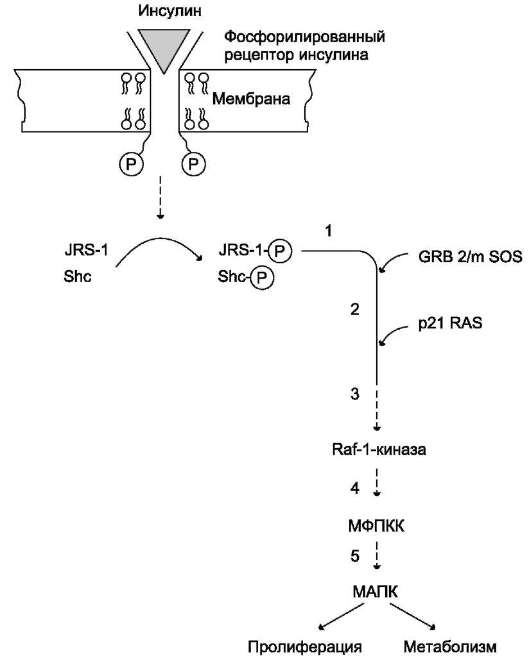
Рис. 11-25. Активация Ras-пути инсулином. 1 - GRB-2/mSOS - цитозольный белок нековалентно присоединяется к фосфорилированному рецептору инсулина при участии одного из субстратов инсулинововго рецептора - Shc; 2 - образовавшийся комплекс взаимодействует с белком Ras; в этот комплекс включаются также белки, которые обеспечивают отделение от Ras ГДФ и присоединение ГТФ; 3 - активированный Ras соединяется с протеинкиназой Raf-1, вследствие чего происходит активация Raf-1-киназы; 4, 5 - активированная Raf-1-киназа стимулирует каскад реакций фосфорилирования и активации других протеинкиназ, в частности, МАПКК и МАПК. МАПК фосфорилируют многие цитоплазматические белки и факторы транскрипции. МАПК - митогенактивируемые протеинкиназы.
злокачественной трансформации клеток (см. раздел 16).
Эффекты инсулина могут проявляться в течение секунд и минут (транспорт веществ, фосфо-рилирование и дефосфорилирование белков, активация и ингибирование ферментов, синтез РНК) или через несколько часов (синтез ДНК, белков, рост клеток).
Активация фосфоинозитол-3-киназы (ФИ-3-кина-зы). Этот фермент катализирует фосфорилирова-
ние ФИ, ФИ-4-фосфата и ФИ-4,5-бисфосфата в положении 3, образуя полифосфоинозитиды: ФИ-3-фосфат, ФИ-3,4-бисфосфат, ФИ-3,4,5-трифос-фат, которые в разных клетках стимулируют мобилизацию Са2+ и активацию специфических протеинкиназ (см. раздел 5). Активация ФИ-3-киназы стимулирует транслокацию ГЛЮТ-4 в плазматическую мембрану и таким образом ускоряет трансмембранный перенос глюкозы в клетки жировой и мышечной ткани. В жиро-
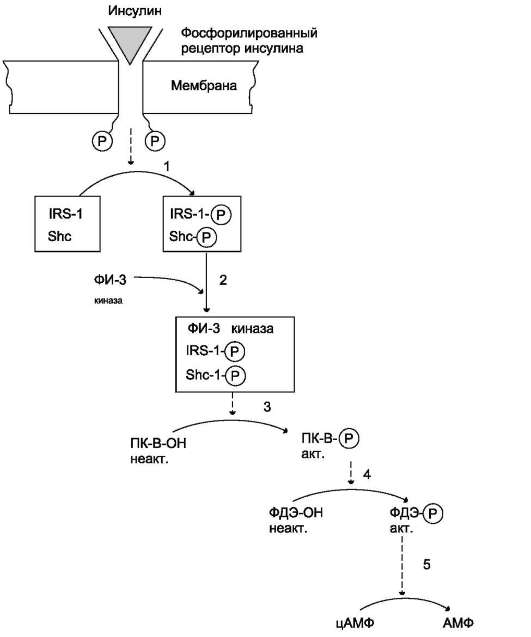
Рис. 11-26. Активация фосфодиэстеразы адипоцитов инсулином. 1 - фосфорилированный рецептор инсулина фосфорилирует субстраты инсулинового рецептора; 2 - образование комплекса фосфоинозитол-3-киназы (ФИ-3-киназы) с активированными субстратами инсулинового рецептора; 3 - активация протеинкиназы В (ПК-В); 4 - протеинкиназа В активирует фосфодиэстеразу (ФДЭ) путём фосфорилирования; 5 - ФДЭ катализирует реакцию превращения цАМФ в АМФ.
вой ткани активация ФИ-3-киназы приводит к торможению липолиза. Снижение скорости липолиза происходит в результате активации фосфодиэстеразы и уменьшения внутриклеточной концентрации цАМФ (рис. 11-26).
Активация гликогенсинтазы инсулином. Одной из протеинкиназ, активируемых через путь Ras, является протеинкиназа pp90S6. Этот фермент фосфорилирует протеинфосфатазу, связанную с
гранулами гликогена. При фосфорилировании протеинфосфатаза активируется и дефосфори-лирует киназу гликогенфосфорилазы, глико-генфосфорилазу и гликогенсинтазу. Дефосфо-рилированные формы киназыфосфорилазы и гликогенфосфорилазы неактивны, вследствие чего мобилизация гликогена замедляется. Гли-когенсинтаза, напротив, активируется, и синтез гликогена ускоряется (рис. 11-27).
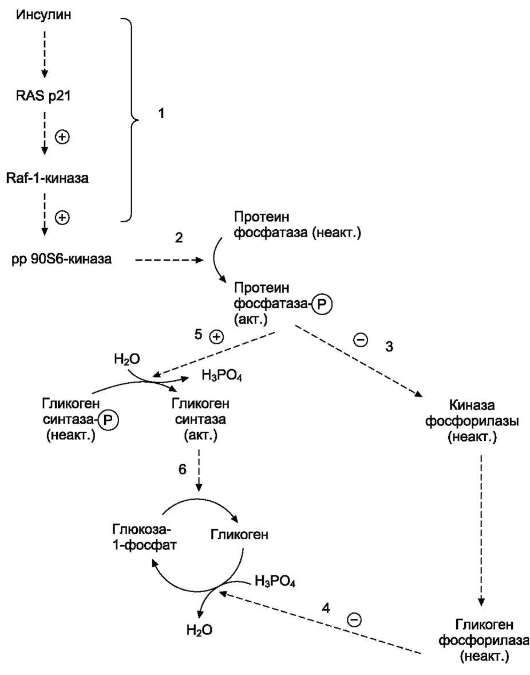
Рис. 11-27. Активация гликогенсинтазы инсулином. 1 - активация пути RAS; 2 - протеинкиназа рр90S6, активируемая инсулином через путь RAS (рис. 11-25), фосфорилирует протеинфосфатазу гранул гликогена, которая включает каскад реакций дефосфорилирования; 3 - инактивация киназыфосфорилазы и гликоген-фосфорилазы; 4 - торможение мобилизации гликогена; 5 - активация гликогенсинтазы; 6 - стимуляция синтеза гликогена.
Инсулин влияет на скорость транскрипции более, чем 100 специфических мРНК в печени, жировой ткани, скелетных мышцах и сердце. Впервые влияние инсулина на транскрипцию генов было показано на примере фосфоенол-пируваткарбоксикиназы - ключевого фермента глюконеогенеза, скорость синтеза которого в культуре клеток гепатомы снижалась в течение нескольких минут.
4. Глюкагон
Глюкагон - одноцепочечный полипептид, состоящий из 29 аминокислотных остатков. Биосинтез глюкагона происходит в α-клетках островков Лангерханса, в нейроэндокринных клетках кишечника и в некоторых отделах ЦНС. Неактивный предшественник проглюкагон в результате частичного протеолиза превращается в несколько пептидов. В клетках поджелудочной
железы главный пептид - глюкагон; в клетках кишечника образуются глюкагоноподобные пептиды (от англ. GLP - glucagon like peptide): GLP-1, GLP-2, глицентин и другие. GLP-1 ингибирует секрецию глюкагона и стимулирует синтез и секрецию инсулина. Стимулятором секреции GLP-1 служит другой гормон - желудочный ингибирующий полипептид (от англ. GIP - gastrial inhibitor peptide), который синтезируется в клетках слизистой оболочки верхних отделов тонкого кишечника. Секреция GIP стимулируется при приёме пищи; наиболее сильным стимулятором служит глюкоза. На секрецию глюкагона влияют и многие другие соединения, включая аминокислоты, жирные кислоты, кетоновые тела и нейромедиаторы. При приёме пищи, богатой углеводами, секреция глюкагона снижается. Белковая пища стимулирует секрецию инсулина и глюкагона; однако некоторые аминокислоты в большей степени влияют на секрецию одного из них. Например, аланин стимулирует секрецию глю-кагона, но не инсулина.
В плазме крови глюкагон не связан с каким-либо транспортным белком. T1/2 гормона составляет ~5 мин. В печени глюкагон быстро разрушается под действием специфических протеаз.
Эффекты глюкагона в основном противоположны эффектам инсулина. Основные клетки-мишени глюкагона - печень и жировая ткань. Связываясь с рецепторами на плазматической мембране клеток-мишеней, глюкагон повышает содержание цАМФ (см. раздел 5). В гепатоци-тах это приводит к активации фосфорилазы гликогена и к снижению активности гликоген-синтазы. В результате ускоряется мобилизация гликогена. Фосфорилирование пируваткиназы и БИФ вызывает торможение гликолиза и ускорение глюконеогенеза. Кроме того, глюкагон стимулирует глюконеогенез, индуцируя синтез ферментов: глюкозо-6-фосфатазы, фосфоенол-пируваткарбоксикиназы, фруктозо-1,6-бис-фосфатазы (см. раздел 7). В клетках жировой ткани глюкагон через аденилатциклазный каскад активирует гормончувствительную ТАГ-липазу и стимулирует липолиз (см. раздел 8). Таким образом, в противоположность инсулину глюкагон стимулирует мобилизацию основных энергоносителей - углеводов и жиров.
5. Другие гормоны желудочно-кишечного тракта
Из тканей ЖКТ выделено более 10 биологически активных пептидов. Многие из них по механизму действия могут быть причислены к истинным гормонам, проявляющим эндокринный эффект. К ним относят гастрин, секретин, GIP, холецистокинин, мотилин, панкреатический полипептид и энтероглюкагон. Другие желудочно-кишечные пептиды обладают па-ракринным или нейроэндокринным действием: вазоактивный интестинальный пептид (VIP), соматостатин.
К особенностям эндокринной системы ЖКТ относят то, что её клетки рассеяны по разным отделам, а не собраны в отдельном органе. Многие желудочно-кишечные пептиды найдены в нервах ЖКТ, а также в клетках ЦНС. Кроме того, для пептидов ЖКТ характерно наличие множественных форм, имеющих структурное и функциональное сходство, что ограничивает возможности изучения их структуры и функции. Из главных желудочно-кишечных гормонов только секретин существует в единственной форме. Многие гормоны ЖКТ по сходству первичной структуры и функции могут быть отнесены к одному из 2 семейств.
Семейство гастрина объединяет гастрин и хо-лецистокинин.
Семейство секретина объединяет секретин, глюкагон, GIP, VIP и глицентин.
Сравнительно мало известно о механизмах действия гормонов ЖКТ. На ацинарных клетках поджелудочной железы идентифицировано шесть разных классов рецепторов. Один из них для семейства гастрина функционирует при участии инозитолфосфатной системы; рецепторы секретина и VIP - компоненты адени-латциклазной системы. Биологическая роль основных гормонов ЖКТ рассматривается в курсе физиологии и в разделе 9.
IV. РЕГУЛЯЦИЯ ОБМЕНА ОСНОВНЫХ ЭНЕРГОНОСИТЕЛЕЙ
Основные пищевые вещества (углеводы, жиры, белки) окисляются в организме с освобождением свободной энергии, которая используется в анаболических процессах и при осуществлении физиологических функций.
Энергетическая ценность основных пищевых веществ выражается в килокалориях и составляет: для углеводов - 4 ккал/г, для жиров - 9 ккал/г, для белков - 4 ккал/г. Взрослому здоровому человеку в сутки требуется 2000-3000 ккал (8000-12 000 кДж) энергии.
При обычном ритме питания промежутки между приёмами пищи составляют 4-5 ч с 8-12-часовым ночным перерывом. Во время пищеварения и абсорбтивного периода (2-4 ч) основные энергоносители, используемые тканями (глюкоза, жирные кислоты, аминокислоты), могут поступать непосредственно из пищеварительного тракта. В постабсорбтивном периоде и при голодании энергетические субстраты образуются в процессе катаболизма депонированных энергоносителей.
Изменения в потреблении энергоносителей и энергетических затратах координируются путём чёткой регуляции метаболических процессов в разных органах и системах организма, обеспечивающей энергетический гомеостаз.
Основную роль в поддержании энергетического гомеостаза играют гормоны инсулин и глюкагон, а также другие контринсулярные гормоны - адреналин, кортизол, йодтиронины и сома-тотропин. Инсулин и глюкагон играют главную роль в регуляции метаболизма при смене абсор-бтивного и постабсорбтивного периодов и при голодании.
А. АБСОРБТИВНЫЙ ПЕРИОД
Абсорбтивный период характеризуется временным повышением концентрации глюкозы, аминокислот и жиров в плазме крови. Клетки поджелудочной железы отвечают на это повышение усилением секреции инсулина и снижением секреции глюкагона. Увеличение отношения ин-сулин/глюкагон вызывает ускорение использования метаболитов для запасания энергоносителей: происходит синтез гликогена, жиров и белков. Режим запасания включается после приёма пищи и сменяется режимом мобилизации запасов после завершения пищеварения. Тип метаболитов, которые потребляются, депонируются и экспортируются, зависит от типа ткани. Главные органы, связанные с изменениями потока метаболитов при смене режимов мобилизации и запасания энергоносителей, - печень, жировая ткань и мышцы (рис. 11-28).
1. Изменения метаболизма в печени в абсорбтивном периоде
После приёма пищи печень становится главным потребителем глюкозы, поступающей из пищеварительного тракта. Почти 60 из каждых
При повышении концентрации глюкозы в гепатоцитах происходит активация глюкокиназы, превращающей глюкозу в глюкозо-6-фосфат. Глюкокиназа имеет высокое значение Km для глюкозы, что обеспечивает высокую скорость фосфорилирования при высоких концентрациях глюкозы. Кроме того, глюкокиназа не ингиби-руется глюкозо-6-фосфатом (см. раздел 7). Инсулин индуцирует синтез мРНК глюкокиназы. Повышение концентрации глюкозо-6-фосфата в гепатоцитах обусловливает ускорение синтеза гликогена. Этому способствуют одновременная инактивация гликогенфосфорилазы и активация гликогенсинтазы. Под влиянием инсулина в гепатоцитах ускоряется гликолиз в результате повышения активности и количества ключевых ферментов: глюкокиназы, фосфофруктокиназы и пируваткиназы. В то же время происходит торможение глюконеогенеза в результате инактивации фруктозо-1,6-бисфосфатазы и снижения количества фосфоенолпируваткарбоксикина-зы - ключевых ферментов глюконеогенеза. Повышение концентрации глюкозо-6-фосфата в гепатоцитах в абсорбтивном периоде сочетается с активным использованием NADPH для синтеза жирных кислот, что способствует стимуляции пентозофосфатного пути.
Ускорение синтеза жирных кислот обеспечивается доступностью субстратов (ацетил-КоА и NADPH), образующихся при метаболизме глюкозы, а также активацией и индукцией ключевых ферментов синтеза жирных кислот (см. раздел 8 и табл. 11-7).
В абсорбтивном периоде в печени ускоряется синтез белков. Однако количество аминокислот, поступающих в печень из пищеварительного тракта, превышает возможности их использова-
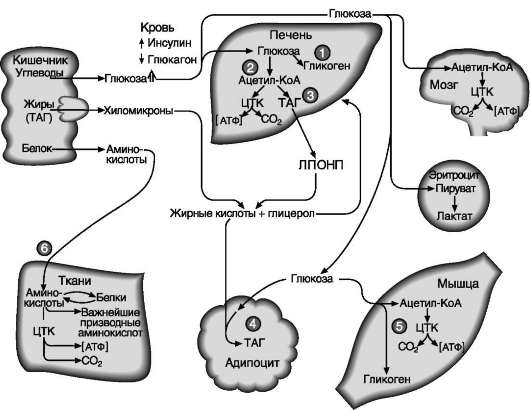
Рис. 11-28. Пути использования основных энергоносителей в абсорбтивном периоде. 1 - биосинтез гликогена в печени; 2 - гликолиз; 3 - биосинтез ТАГ в печени; 4 - биосинтез ТАГ в жировой ткани; 5 - биосинтез гликогена в мышцах; 6 - биосинтез белков в разных тканях, в том числе в печени.
ния для синтеза белков и других азотсодержащих соединений. Излишек аминокислот либо поступает в кровь и транспортируется в другие ткани, либо дезаминируется с последующим включением безазотистых остатков в общий путь катаболизма (см. раздел 9).
2. Изменения метаболизма в адипоцитах
Основная функция жировой ткани - запасание энергоносителей в форме триацилгли-церолов. Под влиянием инсулина ускоряется транспорт глюкозы в адипоциты. Повышение внутриклеточной концентрации глюкозы и активация ключевых ферментов гликолиза обеспечивают образование ацетил-КоА и глице-рол-3-фосфата, необходимых для синтеза ТАГ. Стимуляция пентозофосфатного пути обеспечивает образование NADPH, необходимого для синтеза жирных кислот. Однако биосинтез жирных кислот de novo в жировой ткани человека
протекает с высокой скоростью только после предшествующего голодания. При нормальном ритме питания для синтеза ТАГ используются в основном жирные кислоты, поступающие из ХМ и ЛПОНП под действием ЛП-липазы (см. раздел 8). Вместе с тем при увеличении отношения инсулин/глюкагон гормончувс-твительная ТАГ-липаза находится в дефосфо-рилированной неактивной форме, и процесс липолиза тормозится.
3. Изменение метаболизма в мышцах в абсорбтивном периоде
В абсорбтивном периоде под влиянием инсулина ускоряется транспорт глюкозы в клетки мышечной ткани. Глюкоза фосфорилируется и окисляется для обеспечения клетки энергией, а также используется для синтеза гликогена. Жирные кислоты, поступающие из ХМ и ЛПОНП, в этот период играют незначительную роль в
энергетическом обмене мышц. Поток аминокислот в мышцы и биосинтез белков также увеличиваются под влиянием инсулина, особенно после приёма белковой пищи.
Б. ПОСТАБСОРБТИВНЫЙ ПЕРИОД
Постабсорбтивным состоянием называют период после завершения пищеварения до следующего приёма пищи. Если пища не принимается в течение суток и более, то это состояние определяют как голодание. Типичным постаб-сорбтивным периодом считают состояние после 12-часового ночного перерыва в приёме пищи. В начале постабсорбтивного периода концентрация глюкозы в крови снижается, вследствие чего снижается секреция инсулина и повышается концентрация глюкагона. При снижении индекса инсулин/глюкагон ускоряются процессы мобилизации депонированных энергоносителей
(рис. 11-29).
В постабсорбтивном периоде изменения метаболизма направлены, главным образом, на поддержание концентрации в крови глюкозы, которая служит основным энергетическим субстратом для мозга и единственным источником энергии для эритроцитов. Основные изменения метаболизма в этот период происходят в печени и жировой ткани.
1. Изменения метаболизма в печени
В печени прежде всего ускоряется мобилизация гликогена (см. раздел 7). Однако запасы гликогена в печени истощаются в течение 18- 24 ч голодания. Главным источником глюкозы по мере исчерпания запасов гликогена становится глюконеогенез, который начинает ускоряться через 4-6 ч после последнего приёма пищи. Субстратами для синтеза глюкозы служат глице-рол, аминокислоты и лактат. При высокой концентрации глюкагона скорость синтеза жирных кислот снижается вследствие фосфорилирова-
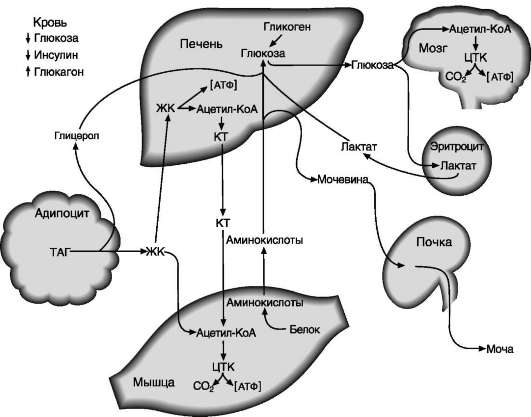
Рис. 11-29. Изменения метаболизма основных энергоносителей при смене абсорбтивного состояния на постабсорбтивное. КТ - кетоновые тела; ЖК - жирные кислоты.
ния и инактивации ацетил-КоА-карбоксилазы, а скорость β-окисления возрастает. Вместе с тем увеличивается снабжение печени жирными кислотами, которые транспортируются из жировых депо. Ацетил-КоА, образующийся при окислении жирных кислот, используется в печени для синтеза кетоновых тел.
2. Изменения метаболизма в жировой ткани
В жировой ткани при повышении концентрации глюкагона снижается скорость синтеза ТАГ и стимулируется липолиз. Стимуляция ли-полиза - результат активации гормончувстви-тельной ТАГ-липазы адипоцитов под влиянием глюкагона. Жирные кислоты становятся важными источниками энергии в печени, мышцах и жировой ткани.
Таким образом, в постабсорбтивном периоде концентрация глюкозы в крови поддерживается на уровне 80-100 мг/дл, а уровень жирных кислот и кетоновых тел возрастает.
В. ИЗМЕНЕНИЕ ГОРМОНАЛЬНОГО СТАТУСА И МЕТАБОЛИЗМА ПРИ ГОЛОДАНИИ
Голодание может быть кратковременным, в течение суток (I фаза), продолжаться в течение недели (II фаза) или нескольких недель (III фаза).
В отсутствие пищи в крови снижается уровень глюкозы, аминокислот и триацилглицеролов. Инсулин-глюкагоновый индекс снижается, и повышается концентрация контринсулярных гормонов, в первую очередь кортизола. В этих условиях возникает состояние, для которого характерно преобладание процессов катаболизма жиров, гликогена и белков на фоне общего снижения скорости метаболизма. Под влиянием контринсулярных гормонов в этот период происходит обмен субстратами между печенью, жировой тканью, мышцами и мозгом. Этот обмен служит двум целям: 1) поддержанию концентрации глюкозы в крови для обеспечения глюкозозависимых тканей (мозга, эритроцитов); 2) мобилизации других источников энергии, в первую очередь жиров, для обеспечения энергией всех других тканей. Вследствие переключения метаболизма на режим мобилизации энергоносителей даже после 5-6 нед голодания концентрация глюкозы в крови составляет не менее 60 мг/дл.
1. Обмен углеводов
Так как за счёт мобилизации гликогена обеспечивается только кратковременное голодание, основным источником глюкозы при длительном голодании служит глюконеогенез, а основными субстратами глюконеогенеза - аминокислоты, лактат и глицерол. При низкой концентрации инсулина глюкоза используется только инсу-линнезависимыми тканями, в основном мозгом, эритроцитами. Обеспечение энергетических потребностей других тканей происходит за счёт жирных кислот и кетоновых тел.
2. Обмен жиров
Жирные кислоты, образующиеся в процессе мобилизации жиров в жировых депо, становятся основными источниками энергии для большинства органов в первый период голодания. Во II фазе мобилизация жиров продолжается, и концентрация жирных кислот в крови увеличивается в 3-4 раза по сравнению с постабсорбтивным состоянием. Синтез кетоновых тел начинается в первые дни голодания. Во II фазе голодания скорость синтеза кетоновых тел значительно возрастает. Концентрация кетоновых тел в крови в этот период может достигать 20-30 мг/дл (в норме 1-3 мг/дл). Используются кетоновые тела, в основном, в мышцах. В этот период голодания часть энергетических потребностей мозга обеспечивается кетоновыми телами, а скорость окисления кетоновых тел в мышцах снижается.
3. Обмен белков
В
течение нескольких первых дней голодания быстро распадаются мышечные
белки - основной источник субстратов для глюконеогенеза. При голодании
более 3 нед скорость катаболизма белков стабилизируется и составляет
примерно
которых происходит потеря значительного количества белков. В теле человека массой
V. ИЗМЕНЕНИЯ ГОРМОНАЛЬНОГО СТАТУСА И МЕТАБОЛИЗМА ПРИ САХАРНОМ ДИАБЕТЕ
Сахарный диабет - заболевание, возникающее вследствие абсолютного или относительного дефицита инсулина.
А. ОСНОВНЫЕ КЛИНИЧЕСКИЕ ФОРМЫ САХАРНОГО ДИАБЕТА
Согласно данным Всемирной организации здравоохранения, сахарный диабет классифицируют с учётом различия генетических факторов и клинического течения на две основные формы: диабет I типа - инсулинзависимый (ИЗСД), и диабет II типа - инсулиннезависимый (ИНСД).
1. Инсулинзависимый сахарный диабет
Инсулинзависимый сахарный диабет - заболевание, вызываемое разрушением β-клеток островков Лангерханса поджелудочной железы.
Деструкция β-клеток - результат аутоиммунных реакций. В аутоиммунной реакции принимают участие лимфоциты и макрофаги (моноциты). Эти клетки продуцируют цитоки-ны, которые либо непосредственно повреждают β-клетки, либо опосредуют клеточные реакции против β-клеток.
Провоцировать возникновение диабета I типа может вирусная инфекция, вызывающая деструкцию b-клеток. К таким вирусам, называемым β-цитотропными, относят вирусы оспы, краснухи, кори, цитомегаловирус, эпидемического паротита, Коксаки, аденовирус. Некоторые β-цитот-ропные вирусы вызывают лизис β-клеток.
Известны некоторые токсические вещества, например, такие как производные нитрозомо-чевины и другие нитроили аминосодержащие соединения, избирательно поражающие β-клет-ки и индуцирующие аутоиммунную реакцию. Кроме того, ИЗСД может быть результатом частичного генетически обусловленного дефекта системы иммунологического надзора и сочетаться с другими аутоиммунными заболеваниями.
На долю ИЗСД приходится примерно 25-30% всех случаев сахарного диабета. Как правило, разрушение β-клеток происходит медленно, и начало заболевания не сопровождается нарушениями метаболизма. Когда погибает 80-95% клеток, возникает абсолютный дефицит инсулина, и развиваются тяжёлые метаболические нарушения. ИЗСД поражает в большинстве случаев детей, подростков и молодых людей, но может проявиться в любом возрасте (начиная с годовалого).
2. Инсулинонезависимый сахарный диабет
Инсулинонезависимый сахарный диабет - общее название нескольких заболеваний, развивающихся в результате относительного дефицита инсулина, возникающего вследствие нарушения секреции инсулина, нарушения превращения проинсулина в инсулин, повышения скорости катаболизма инсулина, а также повреждения механизмов передачи инсулинового сигнала в клетки-мишени (например, дефекта рецептора инсулина, повреждения внутриклеточных посредников инсулинового сигнала и др.). ИНСД поражает людей, как правило, старше 40 лет. Сахарный диабет II типа характеризуется высокой частотой семейных форм. Риск ИНСД у ближайших родственников больного достигает 50%, тогда как при ИЗСД он не превышает 10%. Заболевание поражает преимущественно жителей развитых стран, особенно горожан.
Возможными причинами ИНСД могут быть: образование антител к рецепторам инсулина; генетический дефект пострецепторного аппарата инсулинзависимых тканей; нарушения регуляции секреции инсулина. К факторам, определяющим развитие и клиническое течение болезни, относят ожирение, неправильный режим питания, малоподвижный образ жизни, стресс.
Мутации генов, контролирующих секрецию инсулина, энергетический обмен в β-клетках и обмен глюкозы в клетках-мишенях инсулина, приводят к возникновению нескольких форм ИНСД с аутосомно-доминантным наследованием.
Основным провоцирующим фактором инсу-линонезависимого диабета служит ожирение. Этот тип диабета часто сочетается с гиперинсу-линемией, что способствует ожирению. Таким
образом, ожирение, с одной стороны, важнейший фактор риска, а с другой - одно из ранних проявлений сахарного диабета.
Б. ИЗМЕНЕНИЯ МЕТАБОЛИЗМА ПРИ
САХАРНОМ ДИАБЕТЕ
При сахарном диабете, как правило, соотношение инсулин/глюкагон снижено. При этом ослабевает стимуляция процессов депонирования гликогена и жиров, и усиливается мобилизация запасов энергоносителей. Печень, мышцы и жировая ткань даже после приёма пищи функционируют в режиме постабсорб-тивного состояния.
1. Симптомы сахарного диабета
Для всех форм диабета характерно повышение концентрации глюкозы в крови - гипер-глике-мия. После приёма пищи концентрация глюкозы может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе. Снижение толерантности к глюкозе наблюдают в случаях скрытой (латентной) формы сахарного
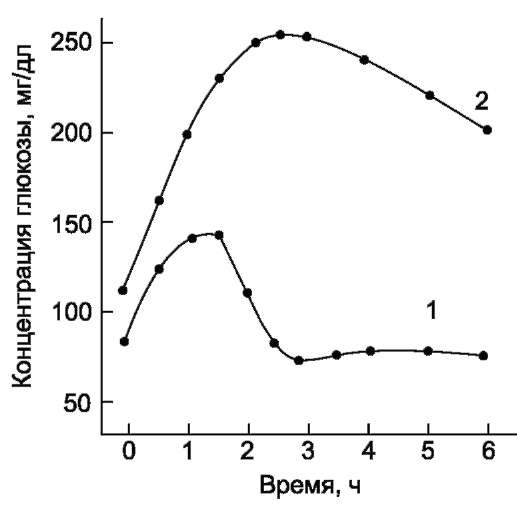
Рис. 11-30. Изменение толерантности к глюкозе у больных скрытой формой сахарного диабета. Определение
толерантности к глюкозе используют для диагностики сахарного диабета.
Обследуемый принимает раствор глюкозы из расчёта
диабета. В этих случаях у людей отсутствуют жалобы и клинические симптомы, характерные для сахарного диабета, а концентрация глюкозы в крови натощак соответствует норме. Однако использование провокационных проб (например, сахарной нагрузки) выявляет снижение толерантности к глюкозе (рис. 11-30).
Повышение концентрации глюкозы в плазме крови обусловлено снижением скорости использования глюкозы тканями вследствие недостатка инсулина или снижения биологического действия инсулина в тканях-мишенях.
При дефиците инсулина уменьшается количество белков-переносчиков глюкозы (ГЛЮТ-4) на мембранах инсулинзависимых клеток (жировой ткани и мышц). В мышцах и печени глюкоза не депонируется в виде гликогена, в жировой ткани уменьшается скорость синтеза и депонирования жиров. Кроме того, при снижении инсулин-глюкагонового индекса активируется глюконеогенез из аминокислот, глицерола и лактата. Повышение концентрации глюкозы в крови при сахарном диабете превышает концентрационный почечный порог, что становится причиной выделения глюкозы с мочой (глюко-зурия). В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу, если её уровень не превышает 8,9 ммоль/л (160 мг/дл).
К характерным признакам сахарного диабета относят также повышение концентрации в крови кетоновых тел - кетонемия. При низком соотношении инсулин/глюкагон жиры не депонируются, а ускоряется их катаболизм, так как гормончувствительная липаза в жировой ткани находится в фосфорилированной активной форме. Концентрация неэтерифицированных жирных кислот в крови повышается. Печень захватывает жирные кислоты, окисляет их до ацетил-КоА, который, в свою очередь, превращается в β-гидроксимасляную и ацетоуксусную кислоты. В тканях ацетоацетат частично декар-боксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии. Увеличение концентрации кетоновых тел в крови (выше 20 мг/дл, иногда до 100 мг/дл) приводит к кетонурии. Накопление кетоновых тел снижает буферную ёмкость крови и вызывает ацидоз.
Ещё один характерный признак сахарного диабета - повышенный уровень в крови ли-
попротеинов (в основном, ЛПОНП) - гипер-липопротеинемия. Пищевые жиры не депонируются в жировой ткани вследствие ослабления процессов запасания, а поступают в печень, где частично превращаются в триацилглицеролы, которые транспортируются из печени в составе
ЛПОНП.
При сахарном диабете дефицит инсулина приводит к снижению скорости синтеза белков в организме и усилению распада белков. Это вызывает повышение концентрации аминокислот в крови. Аминокислоты поступают в печень и дезаминируются. Безазотистые остатки глико-генных аминокислот включаются в глюконеоге-нез, что ещё более усиливает гипергликемию. Образующийся при этом аммиак вступает в орнитиновый цикл, что приводит к увеличению концентрации мочевины в крови и, соответственно, в моче - азотемия и азотурия.
Высокие концентрации глюкозы, кетоновых тел, мочевины требуют усиленной экскреции их из организма. Поскольку концентрационная способность почек ограничена, резко увеличивается выделение большого количества воды, в результате чего может наступить обезвоживание организма. Выделение мочи у больных возрастает в несколько раз и в некоторых случаях достигает 8-9 л в сутки, но чаще не превышает 3-4 л - полиурия. Потеря воды вызывает постоянную жажду - полидипсия.
2. Острые осложнения сахарного диабета. Механизмы развития диабетической комы
Нарушения обмена углеводов, жиров и белков при сахарном диабете могут приводить к развитию коматозных состояний (острые осложнения). Диабетическая кома проявляется в резком нарушении всех функций организма с потерей сознания. Основные предшественники диабетической комы - ацидоз и дегидратация тканей (рис. 11-31).
Параллельно кетоацидозу при декомпенсации диабета развивается нарушение водно-электролитного обмена. В его основе лежит гипергликемия, сопровождающаяся повышением осмотического давления в сосудистом русле. Для сохранения осмолярности начинается компенсаторное перемещение жидкости из клеток и внеклеточного пространства в сосудистое русло. Это ведёт к потере тканями воды и электролитов, прежде всего ионов Na+, K+, Cl-, HCO3.
В результате развиваются тяжёлая клеточная дегидратация и дефицит внутриклеточных ионов (прежде всего К+), затем возникает общая дегидратация. Это приводит к снижению периферического кровообращения, уменьшению мозгового и почечного кровотока и гипоксии. Диабетическая кома развивается медленно, в течение нескольких дней, но иногда может возникнуть и в течение нескольких часов. Первыми признаками могут быть тошнота, рвота, заторможенность. АД у больных снижено.
Коматозные состояния при сахарном диабете могут проявляться в трёх основных формах: кетоацидотической, гиперосмолярной и лакто-ацидотической. Для кетоацидотической комы характерны выраженный дефицит инсулина, кетоацидоз, полиурия, полидипсия. Гипергликемия (20-30 ммоль/л), обусловленная ин-сулиновой недостаточностью, сопровождается большими потерями жидкости и электролитов, дегидратацией и гиперосмоляльностью плазмы. Общая концентрация кетоновых тел достигает 100 мг/дл и выше.
При гиперосмолярной коме наблюдают чрезвычайно высокие уровни глюкозы в плазме крови, полиурию, полидипсию, всегда проявляется тяжёлая дегидратация. Предполагают, что у большинства больных гипергликемия обусловлена сопутствующим нарушением функции почек. Кетоновые тела в сыворотке крови обычно не определяются.
При лактоацидотической коме преобладают гипотония, снижение периферического кровообращения, гипоксия тканей, приводящая к смещению метаболизма в сторону анаэробного гликолиза, что обусловливает повышение концентрации молочной кислоты в крови (лакто-ацидоз).
Разные варианты диабетической комы в чистом виде практически не встречаются. Их возникновение может быть обусловлено разными факторами, например инфекционными заболеваниями, травмами, хирургическими вмешательствами, токсическими соединениями и др.
3. Поздние осложнения сахарного диабета
Главная причина поздних осложнений сахарного диабета - гипергликемия. Гипергликемия приводит к повреждению кровеносных сосудов и нарушению функций различных тканей и органов.
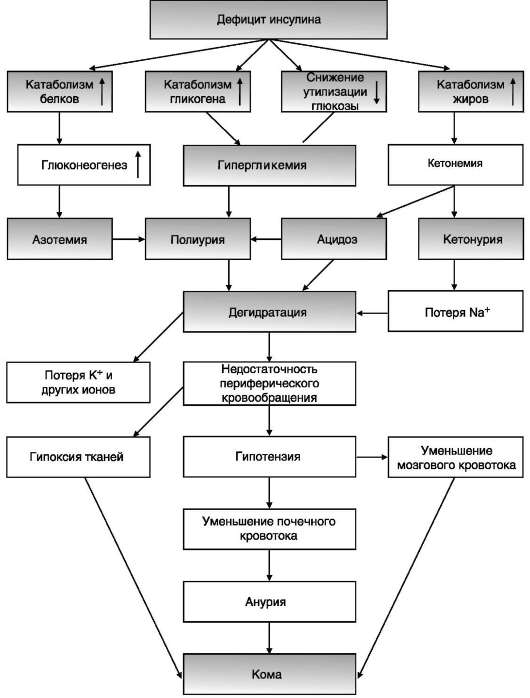
Рис. 11-31. Изменение метаболизма при сахарном диабете и причины диабетической комы.
Одним из основных механизмов повреждения тканей при сахарном диабете является гликози-лирование белков, приводящее к изменению их конформации и функций. Некоторые белки в норме содержат углеводные компоненты, причём образование таких гликопротеинов протекает ферментативно (например, образование гликопротеиновых гормонов аденогипофиза). Однако в организме человека может происходить и неферментативное взаимодействие глюкозы со свободными аминогруппами белков - неферментативное гликозилирование белков. В тканях здоровых людей эта реакция протекает медленно. При гипергликемии процесс глико-зилирования ускоряется. Степень гликозили-рования белков зависит от скорости их обновления. В медленно обменивающихся белках накапливается больше изменений. К одним из первых признаков сахарного диабета относят увеличение в 2-3 раза количества гликозили-рованного гемоглобина (норма HbA1C 5,8-7,2%). Другим примером медленно обменивающихся белков служат кристаллины - белки хрусталика. При гликозилировании кристаллины образуют многомолекулярные агрегаты, увеличивающие преломляющую способность хрусталика. Прозрачность хрусталика уменьшается, возникает его помутнение, или катаракта.
К медленно обменивающимся белкам относятся белки межклеточного матрикса, базаль-ных мембран. Утолщение базальных мембран, одно из характерных осложнений сахарного диабета, приводит к развитию диабетических ангиопатий.
Причиной многих поздних осложнений сахарного диабета также служит повышение скорости превращения глюкозы в сорбитол (см. раздел 7).
• Реакция превращения глюкозы в шестиатомный спирт (сорбитол) катализируется ферментом альдозоредуктазой. Сорбитол не используется в других метаболических путях, а скорость его диффузии из клеток невелика. У больных сахарным диабетом сорбитол накапливается в сетчатке и хрусталике глаза, клетках клубочков почек, шванновских клетках, в эндотелии.
• Сорбитол в высоких концентрациях токсичен для клеток. Его накопление в нейронах приводит к увеличению осмотического давления, набуханию клеток и отёку тканей. Так, например, помутнение хрусталика
может развиться вследствие вызванного накоплением сорбитола набухания хрусталика и нарушения упорядоченной структуры кристаллинов. Диабетические ангиопатии. Диабетические анги-опатии обусловлены прежде всего поражением базальных мембран сосудов. При высокой концентрации глюкозы в плазме крови протеогли-каны, коллагены, гликопротеины гликозилиру-ются, нарушается обмен и соотношение между компонентами базальных мембран, нарушается их структурная организация.
Макроангиопатии проявляются в поражениях крупных и средних сосудов сердца, мозга, нижних конечностей. Патологические изменения во внутренней оболочке артерий и повреждения артериальной стенки в средних и наружных слоях - следствие гликозили-рования базальных мембран и белков межклеточного матрикса (коллагена и эластина), что приводит к снижению эластичности артерий. В сочетании с гиперлипидемией это может быть причиной развития атеросклероза. При сахарном диабете атеросклероз встречается чаще, развивается в более раннем возрасте и прогрессирует значительно быстрее, чем в отсутствие диабета. Микроангиопатии - результат повреждения капилляров и мелких сосудов. Проявляются в форме нефро-, нейро- и ретинопатии. Нефропатия развивается примерно у трети больных сахарным диабетом. Электронно-микроскопические изменения базальной мембраны в почечных клубочках можно обнаружить уже на первом году после установления диагноза. Однако у большинства больных клинические признаки диабетической нефропатии проявляются через 10-15 лет существования диабета. Признаком ранних стадий нефропатии служит микроальбуминурия (в пределах 30- 300 мг/сут), которая в дальнейшем развивается до классического нефротического синдрома, характеризующегося высокой протеинурией, гипоальбуминемией и отёками.
Ретинопатия, самое серьёзное осложнение сахарного диабета и наиболее частая причина слепоты, развивается у 60-80% больных сахарным диабетом. На ранних стадиях развивается базальная ретинопатия, которая проявляется в кровоизлияниях в сетчатку, расширении сосудов сетчатки, отёках. Если изменения не затраги-
вают жёлтого пятна, потеря зрения обычно не происходит. В дальнейшем может развиться пролиферативная ретинопатия, проявляющаяся в новообразовании сосудов сетчатки и стекловидного тела. Ломкость и высокая проницаемость новообразованных сосудов определяют частые кровоизлияния в сетчатку или стекловидное тело. На месте тромбов развивается фиброз, приводящий к отслойке сетчатки и потере зрения.
в. диагностика сахарного диабета
Обычно диагноз сахарного диабета можно поставить на основе классических симптомов сахарного диабета - гипергликемии, полиурии, полидипсии, полифагии, ощущения сухости во рту. Важнейшие биохимические признаки ИЗСД выявляют на основе:
• теста толерантности к глюкозе (см. рис. 1130). Уровень глюкозы в плазме крови выше 10 ммоль/л через 2 ч после сахарной нагрузки свидетельствует о сахарном диабете;
• определения гликозилированного гемоглобина. При сахарном диабете уровень HbA1c, в норме составляющий около 5% от всего содержания гемоглобина, увеличивается в 2-3 раза;
• отсутствия или низкого уровня инсулина и С-пептида в крови и моче. В норме инсулин и С-пептид секретируются в эквимолярных концентрациях. Поскольку печенью задерживается примерно 2/3 инсулина, соотношение инсулин/С-пептид в воротной вене и периферических сосудах в норме составляет 1/3. Величина уровня С-пептида в сыворотке или моче позволяет достаточно точно оценить функциональное состояние β-клеток;
• альбуминурии. При сахарном диабете суточное выведение альбумина составляет примерно 30-300 мг - микроальбуминурия (в норме около 8 мг).
Поскольку ИНСД развивается значительно медленнее, классические клинические симптомы, гипергликемию и дефицит инсулина диагностируют позднее, часто в сочетании с симптомами поздних осложнений сахарного диабета.
г. подходы к лечению
сахарного диабета
Лечение сахарного диабета зависит от его типа (I или II), является комплексным и включает
диету, применение сахаропонижающих средств, инсулинотерапию, а также профилактику и лечение осложнений.
Современные сахаропонижающие препараты делят на две основные группы: производные сульфонилмочевины и бигуаниды. К препаратам, действие которых направлено на стимуляцию секреции инсулина, относят производные суль-фонилмочевины (например, манинил). Механизм действия препаратов сульфонилмочевины объясняют их влиянием на функцию АТФ-чувстви-тельных К+-каналов. Повышение внутриклеточной концентрации К+ приводит к деполяризации мембраны и ускорению транспорта ионов кальция в клетку, вследствие чего стимулируется секреция инсулина.
Другую основную группу сахаропонижающих препаратов составляют бигуаниды. По данным некоторых исследований, бигуаниды увеличивают количество переносчиков глюкозы ГЛЮТ-4 на поверхности мембран клеток жировой ткани и мышц.
К перспективным методам лечения сахарного диабета относят следующие: трансплантация островков поджелудочной железы или изолированных β-клеток, трансплантация генетически реконструированных клеток, а также стимуляция регенерации панкреатических островков.
При сахарном диабете обоих типов важнейшее значение имеет диетотерапия. Рекомендуют хорошо сбалансированную диету: на долю углеводов должно приходиться 50-60% общей калорийности пищи (исключение должны составлять легкоусвояемые углеводы, пиво, спиртные напитки, сиропы, пирожные и др.); на долю белков - 15-20%; на долю всех жиров - не более 25-30%. Пищу следует принимать 5-6 раз в течение суток.
vi. регуляция водно-солевого обмена
Важнейшие параметры водно-солевого го-меостаза - осмотическое давление, рН и объём внутриклеточной и внеклеточной жидкости. Изменение этих параметров может привести к изменению АД, ацидозу или алкалозу, дегидратации и отёкам тканей. Основные гормоны, участвующие в тонкой регуляции водно-солевого баланса и действующие на дистальные извитые канальцы и собирательные трубочки почек: ан-
тидиуретический гормон (АДГ), альдостерон и предсердный натриуретический фактор (ПНФ).
А. АНТИДИУРЕТИЧЕСКИЙ ГОРМОН
Антидиуретический гормон (АДГ), или вазо-прессин - пептид с молекулярной массой около 1100 Д, содержащий 9 аминокислот, соединённых одним дисульфидным мостиком.
1. Синтез и секреция антидиуретического гормона
АДГ синтезируется в нейронах гипоталамуса в виде предшественника препрогормона, который поступает в аппарат Гольджи и превращается в прогормон. В составе нейросекреторных гранул прогормон переносится в нервные окончания задней доли гипофиза (нейрогипофиз). Во время транспорта гранул происходит процессинг прогормона, в результате чего он расщепляется на зрелый гормон и транспортный белок - ней-рофизин. Гранулы, содержащие зрелый антидиуретический гормон и нейрофизин, хранятся в терминальных расширениях аксонов в задней доле гипофиза, из которых секретируются в кровоток при соответствующей стимуляции.
Стимулом, вызывающим секрецию АДГ, служит повышение концентрации ионов натрия и увеличение осмотического давления внеклеточной жидкости. При недостаточном потреблении воды, сильном потоотделении или после приёма большого количества соли осморецепторы гипоталамуса, чувствительные к колебаниям осмолярности, регистрируют повышение осмотического давления крови. Возникают нервные импульсы, которые передаются в заднюю долю гипофиза и вызывают высвобождение АДГ. Секреция АДГ происходит также в ответ на сигналы от барорецепторов предсердий. Изменение осмолярности всего на 1% приводит к заметным изменениям секреции АДГ.
2. Механизм действия
Для АДГ существуют 2 типа рецепторов: V1 и V2. Рецепторы V2, опосредующие главный физиологический эффект гормона, обнаружены на базолатеральной мембране клеток собирательных трубочек и дистальных канальцев - наиболее важных клеток-мишеней для АДГ, которые относительно непроницаемы для молекул воды. В отсутствие АДГ моча не концентрируется и может выделяться в количествах, превышаю-
щих
Рецепторы типа V1 локализованы в мембранах ГМК сосудов. Взаимодействие АДГ с рецептором V1 приводит к активации фосфолипазы С, которая гидролизует фосфатидилинозитол-4,5-бисфосфат с образованием инозитолтрифосфата и диацилглицерола. Инозитолтрифосфат вызывает высвобождение Са2+ из ЭР. Результатом действия гормона через рецепторы V1 является сокращение гладкомышечного слоя сосудов. Сосудосуживающий эффект АДГ проявляется при высоких концентрациях гормона. Поскольку сродство АДГ к рецептору V2 выше, чем к рецептору V1, при физиологической концентрации гормона в основном проявляется его антидиуретическое действие.
3. Несахарный диабет
Дефицит АДГ, вызванный дисфункцией задней доли гипофиза, а также нарушениями в системе передачи гормонального сигнала, приводит к развитию несахарного диабета. При этом происходит нерегулируемая экскреция воды, а наиболее опасным последствием является дегидратация организма.
Под названием «несахарный диабет» объединяют заболевания с разной этиологией. Так, основными причинами центрального несахарного диабета могут быть генетические дефекты синтеза препро-АДГ в гипоталамусе, дефекты процессинга и транспорта проАДГ (наследственная форма), а также повреждения гипоталамуса или нейрогипофиза (например, в результате черепно-мозговой травмы, опухоли, ишемии).
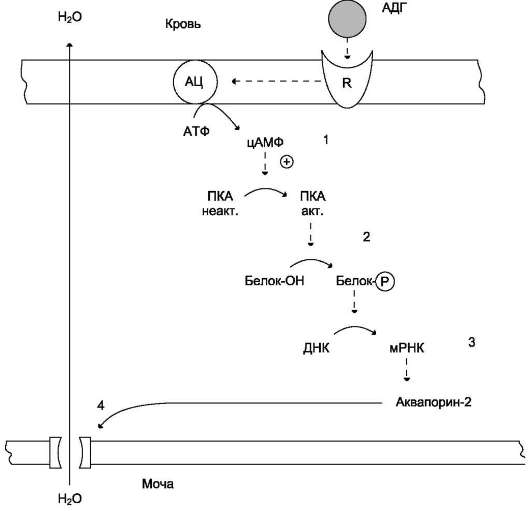
Рис. 11-32. Биологическое действие АДГ в клетках почечных канальцев. 1 - АДГ связывается с мембранным рецептором V2, вызывая активацию аденилатциклазы (АЦ) и образование цАМФ; 2 - цАМФ активирует протеинкиназу, фосфорилирующую белки; 3 - фосфорилированные белки индуцируют транскрипцию гена белка аквапорина; 4 - аквапорин встраивается в мембрану клетки почечного канальца.
Нефрогенный несахарный диабет возникает вследствие мутации гена рецептора АДГ типа V2 (наследственная
форма), следствием которого является неспособность почек реагировать
на гормон. Основное проявление несахарного диабета - гипотоническая
полиурия, т.е. выделение большого количества мочи низкой плотности.
Снижение секреции АДГ приводит также к усиленному потреблению воды.
Диагностические критерии несахарного диабета: выраженная по-лиурия (до
Б. АЛЬДОСТЕРОН
Альдостерон - наиболее активный минера-локортикостероид, синтезирующийся в коре надпочечников из холестерола.
Синтез и секреция альдостерона клетками клу-бочковой зоны непосредственно стимулируются низкой концентрацией Na+ и высокой концентрацией К+ в плазме крови. На секрецию альдо-стерона влияют также простагландины, АКТГ. Однако наиболее важное влияние на секрецию альдостерона оказывает ренин-ангиотензиновая система.
Альдостерон не имеет специфических транспортных белков, но за счёт слабых взаимодействий может образовывать комплексы с альбумином. Гормон очень быстро захватывается печенью, где превращается в тетрагидро-альдостерон-3-глюкуронид и экскретируется с мочой.
1. Механизм действия альдостерона
В клетках-мишенях гормон взаимодействует с рецепторами, которые могут быть локализованы как в ядре, так и в цитозоле клетки. Образовавшийся комплекс гормон-рецептор взаимодействует с определённым участком ДНК и изменяет скорость транскрипции специфических генов. Результат действия альдостерона - индукция синтеза: а) белков-транспортёров
Na+ из просвета канальца в эпителиальную клетку почечного канальца; б) Na+,Κ+,-ΑΤΦ-азы, обеспечивающей удаление ионов натрия из клетки почечного канальца в межклеточное пространство и переносящей ионы калия из межклеточного пространства в клетку почечного канальца; в) белков-транспортёров ионов калия из клеток почечного канальца в первичную мочу; г) митохондриальных ферментов ЦТК, в частности цитратсинтазы, стимулирующих образование молекул АТФ, необходимых для активного транспорта ионов (рис. 11-33).
Суммарным биологическим эффектом индуцируемых альдостероном белков является увеличение реабсорбции ионов натрия в канальцах нефронов, что вызывает задержку NaCl в организме, и возрастание экскреции калия.
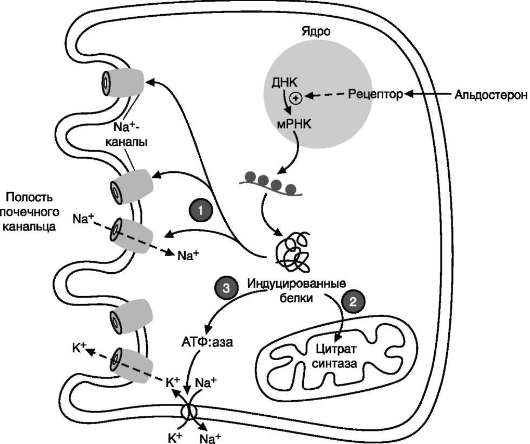
Рис. 11-33. Механизм действия альдостерона. Альдостерон, взаимодействуя с внутриклеточными рецепторами и стимулируя синтез белков: 1 - увеличивает реабсорбцию Nа+ из мочи; 2 - индуцирует синтез ферментов ЦТК, активность которых обеспечивает продукцию АТФ; 3 - активирует Nа+,К+,-АТФ-азу, которая поддерживает низкую внутриклеточную концентрацию ионов натрия и высокую концентрацию ионов калия.
2. Роль системы ренин-ангиотензин-альдостерон в регуляции водно-солевого обмена
Главным механизмом регуляции синтеза и секреции альдостерона служит система ренин-ангиотензин.
Ренин - протеолитический фермент, продуцируемый юкстагломерулярными клетками, расположенными вдоль конечной части афферентных (приносящих) артериол, входящих в почечные клубочки (рис. 11-34).
Юкстагломерулярные клетки особенно чувствительны к снижению перфузионного давления в почках. Уменьшение АД (кровотечение, потеря жидкости, снижение концентрации NaCl) сопровождается падением перфузионного
давления в приносящих артериолах клубочка и соответствующей стимуляцией высвобождения ренина.
Субстратом для ренина служит ангиотензино-ген. Ангиотензиноген - α2-глобулин, содержащий более чем 400 аминокислотных остатков. Образование ангиотензиногена происходит в печени и стимулируется глюкокортикоидами и эстрогенами. Ренин гидролизует пептидную связь в молекуле ангиотензиногена и отщепляет N-концевой декапептид (ангиотензин I), не имеющий биологической активности.
Под действием карбоксидипептидилпептида-зы, или антиотензин-превращающего фермента (АПФ), выявленного в эндотелиальных клетках, лёгких и плазме крови, с С-конца ангиотензина
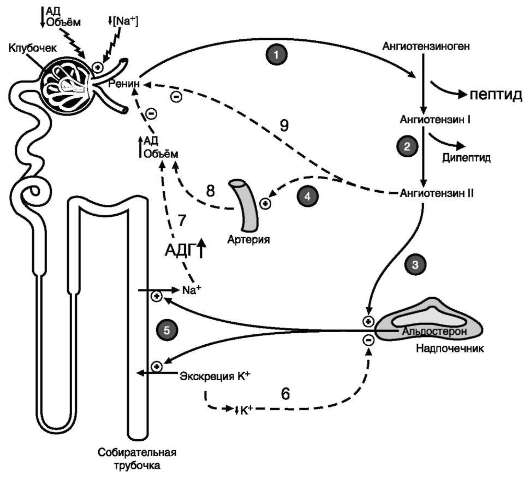
Рис. 11-34. Система ренин-ангиотензин-альдостерон. Ренин, протеолитический фермент, катализирует превращение ангиотензиногена (гликопротеина) в ангиотензин I (декапептид). 1 - ренин, протеолитический фермент, катализирует превращение ангиотензиногена (гликопротеина) в ангиотензин I; 2 - ангиотензин I превращается в ангиотензин II под действием АПФ, отщепляющего два аминокислотных остатка от декапеп-тида; 3 - ангиотензин II стимулирует синтез и секрецию альдостерона; 4 - ангиотензин II вызывает сужение сосудов периферических артерий; 5 - альдостерон стимулирует реабсорбцию Na+ и экскрецию К+; 6, 7, 8, 9 - торможение секреции ренина и альдостерона по механизму отрицательной обратной связи. Пунктирные линии - регуляция по принципу обратной связи.
I удаляются 2 аминокислоты и образуется окта-пептид - ангиотензин II.
Ангиотензин II, связываясь со специфическими рецепторами, локализованными на поверхности клеток клубочковой зоны коры надпочечников и ГМК, вызывает изменение внутриклеточной концентрации диацилглицерола и инозитолтри-фосфата. Инозитолтрифосфат стимулирует высвобождение из ЭР ионов кальция, совместно с которым активирует протеинкиназу С, опосредуя тем самым специфический биологический ответ клетки на действие ангиотензина II.
При участии аминопептидаз ангиотензин II превращается в ангиотензин III - гептапептид, проявляющий активность ангиотензина II. Однако концентрация гептапептида в плазме крови в 4 раза меньше концентрации октапептида, и поэтому большинство эффектов являются результатом действия ангиотензина II. Дальнейшее расщепление ангиотензина II и ангиотензина III протекает при участии специфических протеаз (ангиотензиназ).
Ангиотензин II оказывает стимулирующее действие на продукцию и секрецию альдостерона клетками клубочковой зоны коры надпочечников, который, в свою очередь, вызывает задержку ионов натрия и воды, в результате чего объём жидкости в организме восстанавливается. Кроме этого, ангиотензин II, присутствуя в крови в высоких концентрациях, оказывает мощное сосудосуживающее действие и тем самым повышает АД.
3. Восстановление объёма крови при обезвоживании организма
Уменьшение общего объёма жидкости, например в результате кровопотери, при обильной рвоте, диарее вызывает высвобождение ренина. Этому способствует также снижение импульса-ции от барорецепторов предсердий и артерий в результате уменьшения внутрисосудистого объёма жидкости. В результате увеличивается продукция ангиотензина II, наиболее мощного стимулятора секреции альдостерона. Повышение концентрации альдостерона в крови вызывает задержку ионов натрия, что является сигналом для осморецепторов гипоталамуса и секреции из нервных окончаний задней доли гипофиза АДГ, стимулирующего реабсорбцию воды из собирательных трубочек. Ангиотензин II, оказывая сильное сосудосуживающее действие, повышает АД и, кроме этого, усиливает жажду. Поступа-
ющая с питьём вода в большей мере, чем это происходит в норме, задерживается в организме. Увеличение объёма жидкости а, также повышение АД приводят к устранению стимула, который вызвал активацию ренин-ангиотензиновой системы, секрецию альдостерона и восстановление объёма крови (рис. 11-35).
4. Гиперальдостеронизм
Гиперальдостеронизм - заболевание, вызванное гиперсекрецией альдостерона надпочечниками. Причиной первичного гиперальдостеро-низма (синдром Конна) примерно у 80% больных является аденома надпочечников, в остальных случаях - диффузная гипертрофия клеток клу-бочковой зоны, вырабатывающих альдостерон. При первичном гиперальдостеронизме избыток альдостерона усиливает реабсорбцию натрия в почечных канальцах. Увеличение концентрации Na+ в плазме служит стимулом к секреции АДГ и задержке воды почками. Кроме того, усиливается выведение ионов калия, магния и протонов. В результате развиваются гипернатриемия, вызывающая, в частности, гипертонию, гиперволе-мию и отёки, а также гипокалиемия, ведущая к мышечной слабости, возникают дефицит магния и лёгкий метаболический алкалоз.
Вторичный гиперальдостеронизм встречается гораздо чаще, чем первичный, и может быть связан с рядом состояний (например, сердечная недостаточность, хронические заболевания почек, а также сопровождающиеся нарушением кровоснабжения опухоли, секретирующие ренин). При вторичном гиперальдостеронизме у больных наблюдают повышенный уровень ренина и ангиотензина II, что стимулирует кору надпочечников продуцировать и секретировать избыточное количество альдостерона. Клинические симптомы менее выражены, чем при первичном альдостеронизе. Одновременное определение концентрации альдостерона и активности ренина в плазме позволяет окончательно дифференцировать первичный (активность ренина в плазме снижена) и вторичный (активность ренина в плазме повышена) гиперальдостеронизм.
В. ПРЕДСЕРДНЫЙ НАТРИУРЕТИЧЕСКИЙ ФАКТОР (ПНФ)
Это пептид, содержащий 28 аминокислот с единственным дисульфидным мостиком. ПНФ
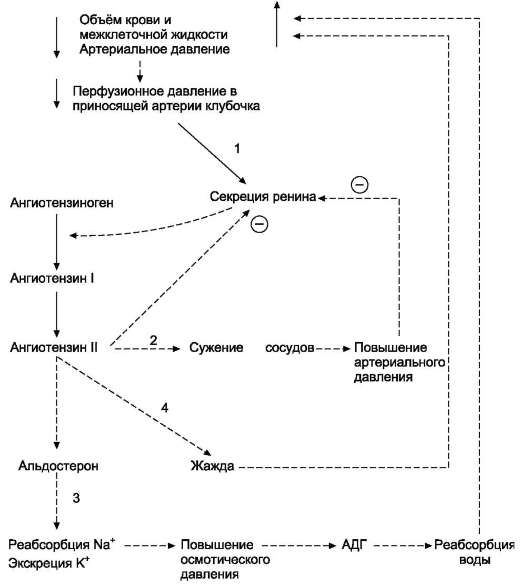
Рис. 11-35. Схема восстановления объёма крови при кровопотере и обезвоживании организма. 1 - уменьшение объёма жидкости и снижение АД активируют систему ренин-ангиотензин-альдостерон; 2 - ангиотензин II вызывает сужение сосудов, что является экстренной мерой для поддержания АД; 3 - альдостерон стимулирует задержку натрия, вследствие чего происходит высвобождение вазопрессина и усиливается реабсорбция воды; 4 - ангиотензин II вызывает также чувство жажды, что способствует увеличению жидкости в организме.
синтезируется, главным образом, в кардиоми-оцитах предсердий, и хранится в виде препро-гормона, состоящего из 126 аминокислотных остатков.
Основным фактором, регулирующим секрецию предсердного натрийуретического фактора, является увеличение АД. Другие стимулы секреции - увеличение осмолярности плазмы, повышение частоты сердцебиений, повышенный
уровень катехоламинов и глюкокортикоидов в крови.
Основные клетки-мишени ПНФ - почки, периферические артерии. В почках ПНФ стимулирует расширение приносящих артериол, усиление почечного кровотока, увеличение скорости фильтрации и экскреции ионов натрия. В периферических артериях ПНФ снижает тонус гладких мышц и соответственно расширяет арте-
риолы (рис. 11-36). Таким образом, суммарным действием ПНФ является увеличение экскреции Na+ и понижение АД.
Механизм передачи сигнала ПНФ не включает актвивацию G-белка. Рецептор ПНФ имеет доменное строение: домен связывания с лигандом, локализованный во внеклеточном пространстве, и один домен, пронизывающий мембрану и обладающий активностью гуанилатциклазы. В отсутствие ПНФ его рецептор находится в фосфорилированном состоянии и неактивен. Связывание ПНФ с рецептором вызывает конформационные изменения и возрастание гуанилатциклазной активности рецептора. В результате ГТФ превращается в циклический ГМФ (цГМФ), который активирует протеинкиназу G (см. раздел 5).
ПНФ обычно рассматривают как физиологический антагонист ангиотензина II, поскольку под его влиянием возникают не сужение просвета сосудов и задержка натрия, а, наоборот, расширение сосудов и увеличение почечной экскреции соли.
vii. регуляция обмена ионов кальция и фосфатов
В организме взрослого человека содержится в среднем
Кальций - не только структурный компонент костной ткани. Ионы кальция играют ключевую роль в мышечном сокращении, увеличивают проницаемость мембраны клеток для ионов калия, влияют на натриевую проводимость клеток, на работу ионных насосов, способствуют секреции гормонов, участвуют в каскадном механизме свёртывания крови. Кроме этого, они служат
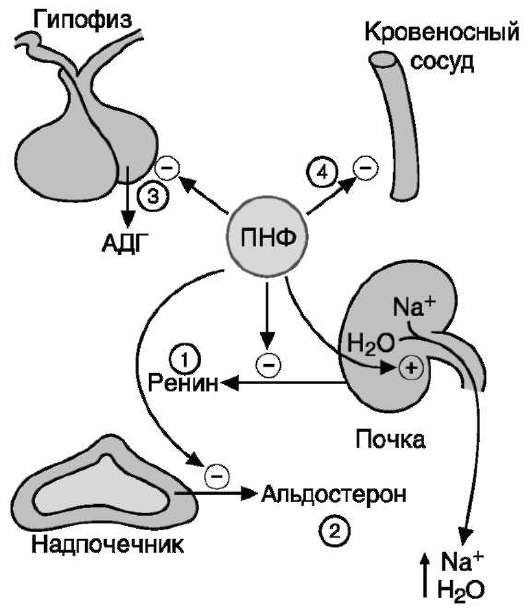
Рис. 11-36. Биологическое действие ПНФ. 1 - ин-
гибирует выделение ренина; 2 - ингибирует секрецию альдостерона; 3 - ингибирует секрецию АДГ; 4 - вызывает релаксацию сосудов.
важнейшими посредниками во внутриклеточной передаче сигналов.
Концентрация кальция внутри клеток зависит от его концентрации во внеклеточной жидкости. Пределы колебаний общей концентрации Са2+ в плазме крови здоровых людей составляют 2,12-2,6 ммоль/л, или 9-11 мг/дл. Кальций плазмы крови представлен в виде:
• несвязанного, ионизированного кальция (около 50%);
• ионов кальция, соединённых с белками, главным образом, с альбумином (45%);
• недиссоциирующих комплексов с цитратом, сульфатом, фосфатом и карбонатом (5%).
Биологически активной фракцией является ионизированный кальций, концентрация которого поддерживается в пределах 1,1-1,3 ммоль/л.
Изменение уровня кальция может привести к нарушению многих процессов: изменению порога возбудимости нервных и мышечных клеток, нарушению функционирования кальциевого насоса, снижению активности ферментов и нарушению гормональной регуляции метаболизма. Концентрация Са2+ в плазме регулируется с высокой точностью: изменение её всего на 1%
приводит в действие гомеостатические механизмы, восстанавливающие равновесие.
Основными регуляторами обмена Са2+ в крови являются паратгормон, кальцитриол и кальцитонин.
А. ПАРАТГОРМОН
Паратгормон (ПТГ) - одноцепочечный полипептид, состоящий из 84 аминокислотных остатков (около 9,5 кД), действие которого направлено на повышение концентрации ионов кальция и снижение концентрации фосфатов в плазме крови.
1. Синтез и секреция ПТГ
ПТГ синтезируется в паращитовидных железах в виде предшественника - препрогормона, содержащего 115 аминокислотных остатков. Во время переноса в ЭР от препрогормона отщепляется сигнальный пептид, содержащий 25 аминокислотных остатков. Образующийся прогормон транспортируется в аппарат Гольджи, где происходит превращение предшественника в зрелый гормон, включающий 84 аминокислотных остатка (ПТГ1-84). Паратгормон упаковывается и хранится в секреторных гранулах (везикулах). Интактный паратгормон может расщепляться на короткие пептиды: N-концевые, С-концевые и срединные фрагменты. N-кон-цевые пептиды, содержащие 34 аминокислотных остатка, обладают полной биологической активностью и секретируются железами наряду со зрелым паратгормоном. Именно N-концевой пептид отвечает за связывание с рецепторами на клетках-мишенях. Роль С-концевого фрагмента точно не установлена. Скорость распада гормона уменьшается при низкой концентрации ионов кальция и увеличивается, если концентрация ионов кальция высока.
Секреция ПТГ регулируется уровнем ионов кальция в плазме: гормон секретируется в ответ на снижение концентрации кальция в крови.
2. Роль паратгормона в регуляции обмена кальция и фосфатов
Органы-мишени для ПТГ - кости и почки. В клетках почек и костной ткани локализованы специфические рецепторы, которые взаимодействуют с паратгормоном, в результате чего инициируется каскад событий, приводящий к
активации аденилатциклазы. Внутри клетки возрастает концентрация молекул цАМФ, действие которых стимулирует мобилизацию ионов кальция из внутриклеточных запасов. Ионы кальция активируют киназы, которые фосфо-рилируют особые белки, индуцирующие транскрипцию специфических генов.
В костной ткани рецепторы ПТГ локализованы на остеобластах и остеоцитах, но не обнаружены на остеокластах. При связывании паратгормона с рецепторами клеток-мишеней остеобласты начинают усиленно секретировать инсулиноподобный фактор роста 1 и цитокины. Эти вещества стимулируют метаболическую активность остеокластов. В частности, ускоряется образование ферментов, таких как щелочная фосфатаза и коллагеназа, которые воздействуют на компоненты костного матрикса, вызывают его распад, в результате чего происходит мобилизация Са2+ и фосфатов из кости во внеклеточную жидкость (рис. 11-37).
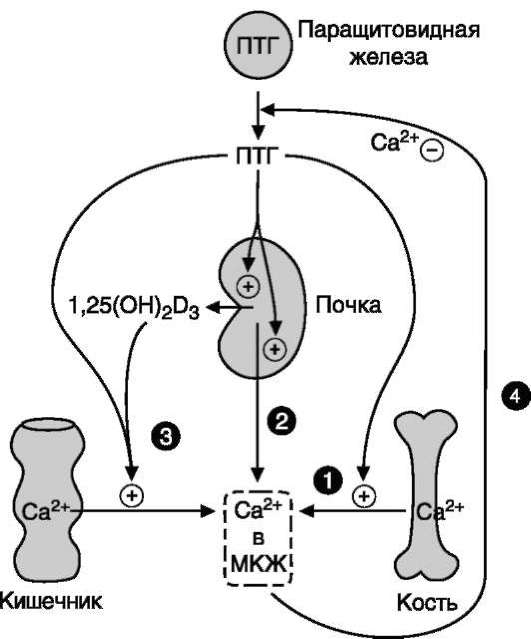
Рис. 11-37. Биологическое действие паратгормона.
1 - стимулирует мобилизацию кальция из кости;
2 - стимулирует реабсорбцию ионов кальция в дистальных канальцах почек; 3 - активирует образование кальцитриола, 1,25(ОН)2D3 в почках, что приводит к стимуляции всасывания Са2+ в кишечнике; 4 - повышает концентрацию кальция в межклеточной жидкости, тормозит секрецию ПТГ. МКЖ - межклеточная жидкость.
В почках ПТГ стимулирует реабсорбцию кальция в дистальных извитых канальцах и тем самым снижает экскрецию кальция с мочой, уменьшает реабсорбцию фосфатов.
Кроме того, паратгормон индуцирует синтез кальцитриола (1,25(OH)2D3), который усиливает всасывание кальция в кишечнике.
Таким образом, паратгормон восстанавливает нормальный уровень ионов кальция во внеклеточной жидкости как путём прямого воздействия на кости и почки, так и действуя опосредованно (через стимуляцию синтеза кальцитриола) на слизистую оболочку кишечника, увеличивая в этом случае эффективность всасывания Са2+ в кишечнике. Снижая реабсорбцию фосфатов из почек, паратгормон способствует уменьшению концентрации фосфатов во внеклеточной жидкости.
3. Гиперпаратиреоз
При первичном гиперпаратиреозе нарушается механизм подавления секреции паратгормона в ответ на гиперкальциемию. Это заболевание встречается с частотой 1:1000. Причинами могут быть опухоль околощитовидной железы (80%) или диффузная гиперплазия желёз, в некоторых случаях рак паращитовидной железы (менее 2%). Избыточная секреция паратгормона приводит к повышению мобилизации кальция и фосфатов из костной ткани, усилению ре-абсорбции кальция и выведению фосфатов в почках. Вследствие этого возникает гиперкаль-циемия, которая может приводить к снижению нервно-мышечной возбудимости и мышечной гипотонии. У больных появляются общая и мышечная слабость, быстрая утомляемость и боли в отдельных группах мышц, увеличивается риск переломов позвоночника, бедренных костей и костей предплечья. Увеличение концентрации фосфата и ионов кальция в почечных канальцах может служить причиной образования в почках камней и приводит к гиперфосфатурии и гипо-фосфатемии.
Вторичный гиперпаратиреоз встречается при хронической почечной недостаточности и дефиците витамина D3 и сопровождается гипокальциемией, связанной в основном с нарушением всасывания кальция в кишечнике из-за угнетения образования кальцитриола поражёнными почками. В этом случае секреция паратгормона увеличивается. Однако повышенный уровень паратгормона не
может нормализовать концентрацию ионов кальция в плазме крови вследствие нарушения синтеза кальцитриола и снижения всасывания кальция в кишечнике. Наряду с гипокальцие-мией, нередко наблюдают гиперфостатемию. У больных развивается повреждение скелета (остеопороз) вследствие повышения мобилизации кальция из костной ткани. В некоторых случаях (при развитии аденомы или гиперплазии околощитовидных желёз) автономная гиперсекреция паратгормона компенсирует гипокальциемию и приводит к гиперкальциемии (третичный гиперпаратиреоз).
4. Гипопаратиреоз
Основной симптом гипопаратиреоза, обусловленный недостаточностью паращитовидных желёз, - гипокальциемия. Понижение концентрации ионов кальция в крови может вызвать неврологические, офтальмологические нарушения и нарушения ССС, а также поражения соединительной ткани. У больного гипопарати-реозом отмечают повышение нервно-мышечной проводимости, приступы тонических судорог, судороги дыхательных мышц и диафрагмы, ларингоспазм.
Б. КАЛЬЦИТРИОЛ
Как и другие стероидные гормоны, кальци-триол синтезируется из холестерола.
Действие гормона направлено на повышение концентрации кальция в плазме крови.
1. Строение и синтез кальцитриола
В коже 7-дегидрохолестерол (провитамин D3) превращается в непосредственного предшественника кальцитриола - холекальциферол (витамин D3). В ходе этой неферментативной реакции под влиянием УФ-излучения связь между девятым и десятым атомами углерода в молекуле холестерола разрывается, раскрывается кольцо B, и образуется холекальциферол (рис. 11-38). Так образуется в организме человека большая часть витамина D3, однако небольшое его количество поступает с пищей и всасывается в тонком кишечнике вместе с другими жирорастворимыми витаминами.
В эпидермисе холекальциферол связывается со специфическим витамин D-связывающим белком (транскальциферином), поступает в
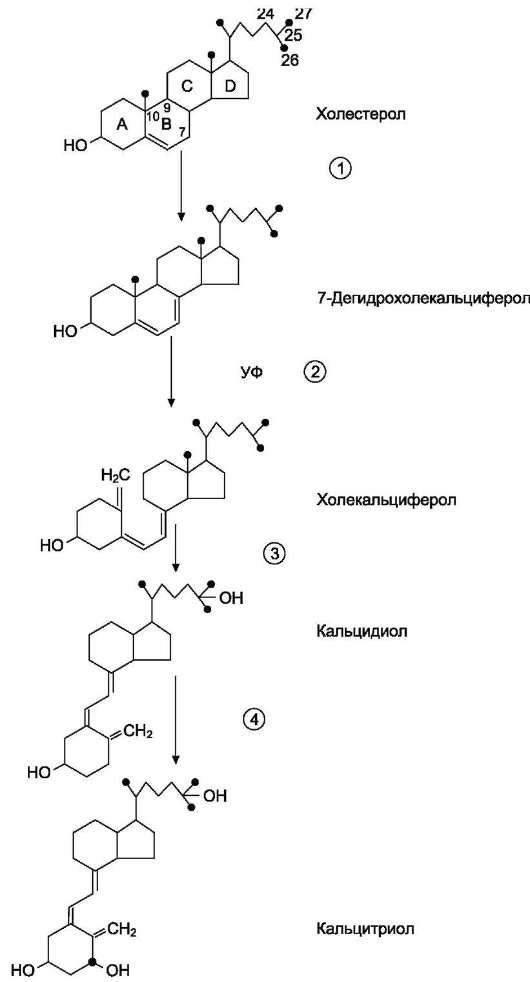
Рис. 11-38. Схема синтеза кальцитриола. 1 - холестерол является предшественником кальцитриола; 2 - в коже 7-дегидрохолестерол неферментативно превращается в холекальциферол; 3 - в печени 25-гидроксилаза превращает холекальциферол в кальцидиол; 4 - в почках образование кальцитриола катализируется 1а-гид-роксилазой.
кровь и переносится в печень, где происходит гидроксилирование по 25-му атому углерода с образованием кальцидиола [25-гидрокси-хо-лекальциферол, 25(OH)D3]. В комплексе с витамин D-связьгаающим белком кальцидиол транспортируется в почки и гидроксилируется по первому углеродному атому с образованием кальцитриола [1,25(OH)2D3]. Именно 1,25(OH)2D3 представляет собой активную форму витамина D3.
Гидроксилирование, протекающее в почках, является скорость-лимитирующей стадией. Эта реакция катализируется митохондриальным ферментом 1α-гидроксилазой. Паратгормон индуцирует 1α-гидроксилазу, тем самым стимулируя синтез 1,25(OH)2D3. Низкая концентрация фосфатов и ионов Са2+ в крови также ускоряет синтез кальцитриола, причём ионы кальция действуют опосредованно через паратгормон.
При гиперкальциемии активность 1 а-гидрок-силазы снижается, но повышается активность 24а-гидроксилазы. В этом случае увеличивается продукция метаболита 24,25(OH)2D3, который, возможно, и обладает биологической активностью, но роль его окончательно не выяснена.
2. Механизм действия кальцитриола
Кальцитриол оказывает воздействие на тонкий кишечник, почки и кости. Подобно другим стероидным гормонам, кальцитриол связывается с внутриклеточным рецептором клетки-мишени. Образуется комплекс гормон-рецептор, который взаимодействует с хроматином и индуцирует транскрипцию структурных генов, в результате чего синтезируются белки, опосредующие действие кальцитриола. Так, например, в клетках кишечника кальцитриол индуцирует синтез Са2+-переносящих белков, которые обеспечивают всасывание ионов кальция и фосфатов из полости кишечника в эпителиальную клетку кишечника и далее транспорт из клетки в кровь, благодаря чему концентрация ионов кальция во внеклеточной жидкости поддерживается на уровне, необходимом для минерализации органического матрикса костной ткани. В почках кальцитриол стимулирует реабсорбцию ионов кальция и фосфатов. При недостатке каль-цитриола нарушается образование аморфного фосфата кальция и кристаллов гидроксиапати-тов в органическом матриксе костной ткани,
что приводит к развитию рахита и остеомаляции. Обнаружено также, что при низкой концентрации ионов кальция кальцитриол способствует мобилизации кальция из костной ткани.
3. Рахит
Рахит - заболевание детского возраста, связанное с недостаточной минерализацией костной ткани. Нарушение минерализации кости - следствие дефицита кальция. Рахит может быть обусловлен следующими причинами: недостатком витамина D3 в пищевом рационе, нарушением всасывания витамина D3 в тонком кишечнике, снижением синтеза предшественников кальцитриола из-за недостаточного времени пребывания на солнце, дефектом 1а-гидрокси-лазы, дефектом рецепторов кальцитриола в клетках-мишенях. Всё это вызывает снижение всасывания кальция в кишечнике и снижение его концентрации в крови, стимуляцию секреции паратгормона и вследствие этого мобилизацию ионов кальция из кости. При рахите поражаются кости черепа; грудная клетка вместе с грудиной выступает вперёд; деформируются трубчатые кости и суставы рук и ног; увеличивается и выпячивается живот; задерживается моторное развитие. Основные способы предупреждения рахита - правильное питание и достаточная инсоляция.
В. РОЛЬ КАЛЬЦИТОНИНА В РЕГУЛЯЦИИ ОБМЕНА КАЛЬЦИЯ
Кальцитонин - полипептид, состоящий из 32 аминокислотных остатков с одной дисуль-фидной связью. Гормон секретируется парафол-ликулярными К-клетками щитовидной железы или С-клетками паращитовидных желёз в виде высокомолекулярного белка-предшественника. Секреция кальцитонина возрастает при увеличении концентрации Са2+ и уменьшается при понижении концентрации Са2+ в крови. Кальцитонин - антагонист паратгормона. Он ингибирует высвобождение Са2+ из кости, снижая активность остеокластов. Кроме того, каль-цитонин подавляет канальцевую реабсорбцию ионов кальция в почках, тем самым стимулируя их экскрецию почками с мочой. Скорость секреции кальцитонина у женщин сильно зависит от уровня эстрогенов. При недостатке эстрогенов
секреция кальцитонина снижается. Это вызывает ускорение мобилизации кальция из костной ткани, что приводит к развитию остеопороза.
viii. роль гормонов в регуляции репродуктивной функции организма
Репродуктивные функции организма регулируются половыми гормонами: у мужчин - тестостероном, у женщин - эстрогенами и прогестинами. Синтез и секреция половых гормонов, в свою очередь, находятся под контролем фолликулостимулирующего и лютеинизирую-щего гормонов.
А. ГОНАДОТРОПНЫЕ ГОРМОНЫ ГИПОФИЗА, СТИМУЛИРУЮЩИЕ СИНТЕЗ И СЕКРЕЦИЮ ПОЛОВЫХ ГОРМОНОВ
Фолликулостимулирующий гормон (ФСГ) и лютеинизирующий гормон (ЛГ) - гонадотроп-ные гормоны гипофиза. Представляют собой гликопротеины с молекулярной массой около 30 кД, состоящие из а- и β-субъединиц. а-Субъ-единицы содержат 92 аминокислоты и две боковые углеводные цепи и идентичны а-субъ-единице тиреотропина. β-Субъединицы индивидуальны для каждого гормона.
1. Регуляция секреции ФСГ и ЛГ
Образование и освобождение обоих гормонов стимулируется гипоталамическим декапепти-дом - гонадотропин-рилизинг-гормоном, секреция которого происходит эпизодически, что в основном и определяет импульсный характер секреции ЛГ и ФСГ.
У женщин эстрогены и прогестерон по механизму обратной связи влияют на секрецию ЛГ и ФСГ как на гипоталамическом, так и на гипофизарном уровне.
У мужчин тестостерон и эстроген, образованный в клетках Лейдига и в процессе метаболизма тестостерона, блокируют по механизму обратной связи синтез и секрецию гонадолиберина и го-надотропных гормонов гипофиза. Кроме этого, клетками гранулёзы фолликулов и клетками Сертоли вырабатывается белок ингибин, который тормозит гипофизарную секрецию ФСГ.
T1/2 ФСГ составляет примерно 150 мин, а T1/2 ЛГ - 30 мин.
2. Механизм действия и эффекты ФСГ и ЛГ
Гонадотропные гормоны ЛГ и ФСГ связываются с рецепторами на мембранах своих клеток-мишеней в яичниках и яичках, в результате чего происходит активация аденилатциклазной системы. Образующийся цАМФ активирует протеинкиназу, которая фосфорилирует белки, опосредующие эффекты ЛГ и ФСГ.
У женщин лютеинизирующий гормон стимулирует образование прогестерона клетками жёлтого тела, у мужчин - синтез тестостерона интерстициальными клетками Лейдига. ФСГ ускоряет развитие фолликулов в яичниках и образование эстрогенов, а действуя на клетки Сертоли, запускает процесс сперматогенеза.
Б. МУЖСКИЕ ПОЛОВЫЕ ГОРМОНЫ
Мужские половые гормоны (рис. 11-39) вырабатываются в основном в мужских половых железах - в интерстициальных клетках Лейдига семенников (95%). Небольшое количество анд-рогенов образуется в коре надпочечников.
1. Синтез андрогенов
Путь биосинтеза андрогенов в яичках и коре надпочечников одинаков. Предшественником андрогенов, как и других стероидных гормонов, служит холестерол (рис. 11-40), который либо поступает из плазмы в составе ЛПНП, либо синтезируется в самих железах из ацетил-КоА.
Отщепление боковой цепи холестерола и образование прегненолона -скорость-лимити-рующая реакция. Однако, в отличие от аналогичной реакции, протекающей в надпочечниках, эта стадия стимулируется ЛГ (а не АКТГ). ЛГ, связываясь с рецептором плазматической мембраны клеток Лейдига, активирует адени-латциклазу, увеличивая тем самым внутриклеточную концентрацию цАМФ, что в конечном итоге вызывает активацию фермента, который расщепляет боковую цепь холестерола между
С-20 и С-22.
Тестостерон. Превращение прегненолона в тестостерон катализируется пятью микро-сомальными ферментами и может протекать двумя путями: через образование дегидроэпиан-дростерона или через образование прогестерона (что, по-видимому, преобладает в семенниках человека).
Рис. 11-39. Мужские половые гормоны.
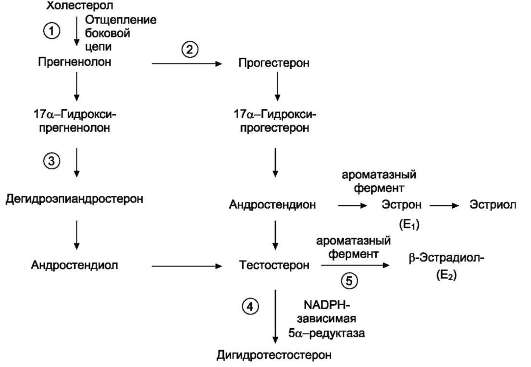
Рис. 11-40. Схема синтеза половых гормонов. Предшественником половых гормонов служит холестерол. Образование прегненолона происходит в результате отщепления боковой цепи холестерола (1). Превращение прегненолона в тестостерон может протекать двумя путями: через образование прогестерона (2) или дегидроэпиандростерона (3). Тестостерон служит предшественником дигидротестостерона (4). В некоторых периферических тканях небольшое количество тестостерона превращается в эстрадиол (5). В яичниках синтезируются женские половые гормоны, эстрогены и прогестины, среди которых наиболее активными являются 17β-эстрадиол и прогестерон. Ароматизация андрогенов протекает под действием ароматазного комплекса, содержащего цитохром Р450-оксидазу, и включает 3 реакции гидроксилирования с участием О2 и NADPH.
Суточная секреция тестостерона у мужчин составляет в норме примерно 5 мг и сохраняется на протяжении всей жизни организма. Гормон циркулирует в крови в связанном с белками плазмы состоянии: альбумином (40%) и специфически связывающим половые гормоны β-глобулином (называемым секс-гормонсвязывающим глобулином, СГСГ). Лишь 2% от общего количества гормона в крови транспортируется в свободном виде, и именно такие молекулы проявляют биологическую активность.
Дигидротестостерон. В семенных канальцах, предстательной железе, коже, наружных по-
ловых органах тестостерон служит предшественником более активного андрогена - ди-гидротестостерона (рис. 11-41, 11-42). Это превра-щение, в котором участвует примерно 4% тестостерона, происходит в результате восстановления двойной связи кольца А и 3-кето-группы при участии цитоплазматического фермента - NADPH-зависимой 5а-редуктазы. Семенники человека секретируют в сутки до 50-100 мкг дигидротестостерона. Однако большее количество гормона - следствие периферических превращений, и суммарная суточная секреция дигидротестостерона составляет
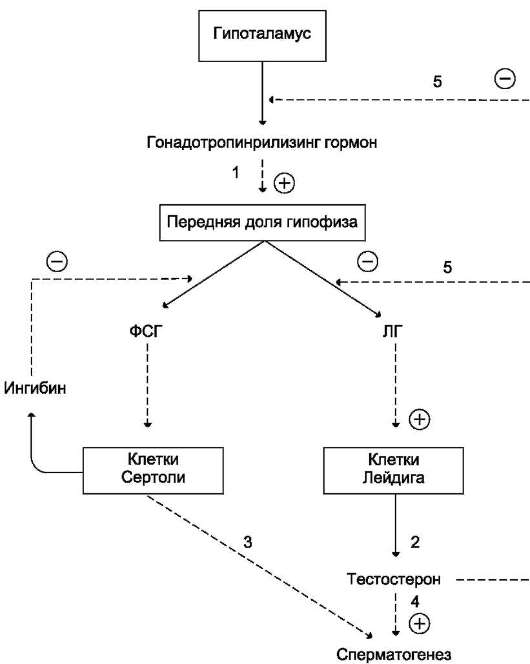
Рис. 11-41. Регуляция синтеза и секреции мужских половых гормонов. Синтез и секреция мужских половых гормонов регулируется гипоталамо-гипофизарной системой по механизму отрицательной обратной связи. Секреция ЛГ и ФСГ стимулируется гонадотропин-рилизинг гормоном. ЛГ ускоряет синтез и секрецию тестостерона клетками Лейдига, ФСГ стимулирует сперматогенез. Тестостерон стимулирует сперматогенез, ингибирует синтез и секрецию гонадотропин-рилизинг гормона и ЛГ.
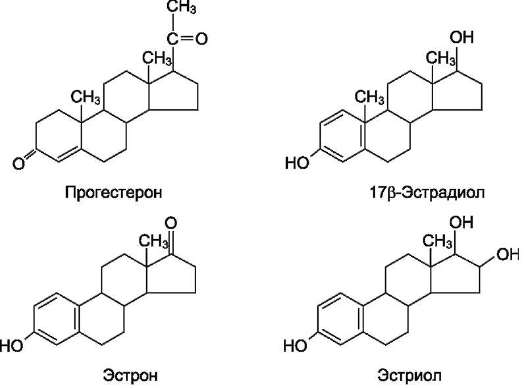
Рис. 11-42. Женские половые гормоны.
400 мкг, что почти в 10 раз меньше уровня секреции тестостерона.
В некоторых периферических тканях небольшое количество тестостерона превращается в эс-традиол. В качестве побочных продуктов клетки Лейдига также постоянно секретируют эстра-диол и прогестерон, хотя роль этих гормонов в развитии и поддержании функций размножения и формирования полового поведения у мужчин до настоящего времени не выяснена.
2. Регуляция синтеза и секреции андрогенов
В препубертатный период секреция андро-генов подавляет по механизму отрицательной обратной связи секрецию гонадотропина до начала пубертатного периода, когда гипофизарные клетки становятся менее чувствительными к ингибирующему действию циркулирующих в крови андрогенов. Эта потеря чувствительности приводит к циклически импульсному освобождению ЛГ и ФСГ. ЛГ, связываясь с рецепторами клеток Лейдига, стимулирует образование тестостерона интерстициальными клетками Лейдига, а ФСГ, связываясь с рецепторами клеток Сертоли в семенниках, стимулирует сперматогенез (рис.
11-41).
Тестостерон замыкает отрицательную обратную связь на уровне гипофиза и гипоталамуса, уменьшая частоту секреторных импульсов ЛГ. Торможение секреции ФСГ аденогипофизом
происходит под действием белка ингибина, вырабатываемого клетками Сертоли. ФСГ стимулирует синтез этого белка, который по механизму отрицательной обратной связи тормозит дальнейшую секрецию ФСГ.
3. Мишени для андрогенов
К мишеням тестостерона относят эмбриональные вольфовы структуры, сперматогонии, мьшцы, кости, почки, мозг. Подобно другим стероидным гормонам, андрогены образуют внутри клетки комплекс с рецептором, который связывается с определённым участком хроматина, активируя специфические гены, белковые продукты которых опосредуют биологические эффекты андрогенов.
4. Эффекты андрогенов
Физиологическое действие андрогенов различно в разные периоды жизни организма. У эмбриона под действием андрогенов из воль-фова протока образуются придаток яичка (эпи-дидимис), семявыносящий проток и семенной пузырёк. У плода мужского пола происходит маскулинизация мозга. Поскольку андрогены в организме обладают мощным анаболическим действием и стимулируют клеточное деление, повышенный уровень андрогенов в препубер-татный период приводит к скачкообразному
увеличению линейных размеров тела, увеличению скелетных мышц, росту костей, но одновременно способствуют и остановке роста, так как стимулируют сращение эпифизов длинных костей с их стволами. Андрогены вызывают изменение структуры кожи и волос, снижение тембра голоса вследствие утолщения голосовых связок и увеличения объёма гортани, стимулируют секрецию сальных желёз.
В. ЖЕНСКИЕ ПОЛОВЫЕ ГОРМОНЫ
В яичниках синтезируются женские половые гормоны - эстрогены и прогестины, среди которых наиболее активны 17β-эстрадиол и прогестерон (рис. 11-42).
1. Образование эстрогенов
Согласно современным представлениям, образование эстрогенов яичников предполагает выработку андрогенов (андростендиона) в клетках теки фолликулов с последующей ароматизацией андрогенов в клетках гранулёзы. В клетках теки синтезируются рецепторы ЛГ. Рецепторы ФСГ образуются в клетках гранулёзы. ЛГ, связываясь с рецепторами клеток теки и активируя фермент, который катализирует отщепление боковой цепи холестерола и превращение его в прегненолон, тем самым стимулирует образование основного андрогена яичников - андро-стендиона. ФСГ, взаимодействуя с рецепторами клеток гранулёзы, активирует содержащийся в этих клетках ароматазный ферментативный комплекс и стимулирует превращение андрогенов, вырабатываемых клетками теки, в эстрогены. Ароматизация андрогенов под действием аро-матазного комплекса, содержащего цитохром Р450-оксидазу, включает 3 реакции гидрокси-лирования, которые протекают с участием О2
и NADPH.
Непосредственно в клетках теки синтезируется очень небольшое количество эстрогенов. Значительная часть эстрогенов продуцируется путём периферической ароматизации андроге-нов в жёлтом теле, фетоплацентарном комплексе (во время беременности), корой надпочечников, в жировых клетках, печени, коже и других тканях, где обнаружена повышенная ароматазная активность.
В клетках гранулёзы может синтезироваться менее активный эстроген - эстрон, а ещё ме-
нее активный эстриол образуется из эстрона в крови. В печени β-эстрадиол инактивируется в результате гидроксилирования ароматического кольца по атому углерода С2 и образования конъюгатов с серной или глюкуроновой кислотами, которые и выводятся из организма с жёлчью или мочой.
Примерно 95% циркулирующих в крови эстрогенов связано с транспортными белками - СГСБ (секс-гормонсвязывающий белок) и альбумином. Биологической активностью обладает только свободная форма эстрогенов.
2. Регуляция секреции эстрогенов
В детском возрасте незрелые яичники вырабатывают небольшое количество гормонов, поэтому концентрация эстрогенов в крови низкая. В пубертатный период чувствительность гипоталамо-гипофизарной системы к действию ЛГ и ФСГ снижается. Импульсная секреция гонадотропин-рилизинг-гормона устанавливает суточный ритм секреции ЛГ и ФСГ. В начале каждого менструального цикла секреция ФСГ и ЛГ вызывает развитие первичных фолликулов. Созревающий фолликул в результате совместного действия ЛГ, стимулирующего продукцию андрогенов клетками теки, и ФСГ, стимулирующего ароматизацию андрогенов, секретирует эстрогены, которые по механизму отрицательной обратной связи угнетают секрецию ФСГ. Концентрация ФСГ в крови остаётся низкой ещё и в результате торможения секреции этого гормона белком ингибином, выделяемым яичниками (рис. 11-43).
По мере созревания фолликула (фолликулярная фаза) концентрация эстрадиола повышается, чувствительность гипофизарных клеток к гонадолиберину возрастает, и эстрадиол по механизму положительной обратной связи повышает секрецию ЛГ и ФСГ.
Повышение секреции ЛГ приводит к овуляции - освобождению яйцеклетки из лопнувшего фолликула. После овуляции клетки гранулёзы превращаются в жёлтое тело, которое, помимо эстрадиола, начинает вырабатывать всё большее количество основного гормона лютеиновой фазы - прогестерона (прогестина). Если возникает беременность, жёлтое тело продолжает функционировать и секретировать прогестерон, однако на более
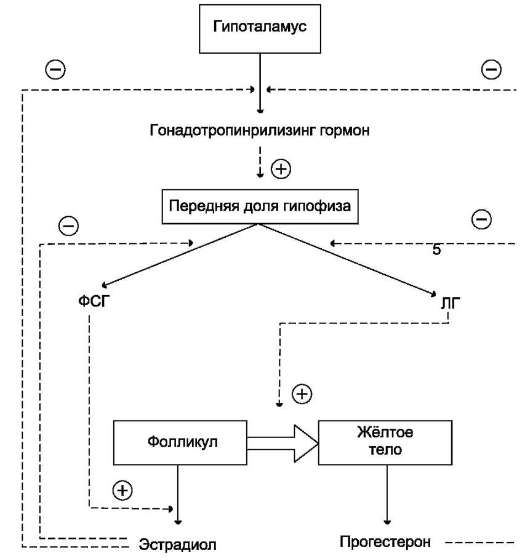
Рис. 11-43. Регуляция секреции женских половых гормонов. Гонадотропин-рилизинг гормон стимулирует секрецию ЛГ и ФСГ, которые совместно с эстрогеном и прогестероном регулируют половой цикл у женщин. Эстрадиол и прогестерон по механизму отрицательной обратной связи регулируют синтез и секрецию ЛГ и ФСГ.
поздних этапах беременности прогестерон в основном продуцируется плацентой. Если оплодотворение не происходит, высокая концентрация прогестерона в плазме крови по механизму отрицательной обратной связи угнетает активность гипоталамо-гипофизарной системы, тормозится секреция ЛГ и ФСГ, жёлтое тело разрушается, и снижается продукция стероидов яичниками. Наступает менструация, которая длится примерно 5 дней, после чего начинает формироваться новый поверхностный слой эндометрия, и возникает новый цикл.
3. Механизм действия и биологические эффекты эстрогенов
Эстрогены связываются с внутриклеточными рецепторами и, подобно другим стероидным гормонам, регулируют транскрипцию струк-
турных генов. Предполагается, что эстрогены индуцируют синтез свыше 50 различных белков, участвующих в проявлении физиологических эффектов эстрогенов.
Эстрогены стимулируют развитие тканей, участвующих в размножении, определяют развитие многих женских вторичных половых признаков, регулируют транскрипцию гена рецептора прогестина. В лютеиновой фазе под действием эстрогенов вместе с прогестинами пролиферативный эндометрий (эпителий матки) превращается в секреторный, подготавливая его к имплантации оплодотворённой яйце-клетки. Совместно с простагландином F2α эстрогены увеличивают чувствительность миометрия к действию окситоцина во время родов. Эстрогены оказывают анаболическое действие на кости и хрящи. Другие метаболические эффекты
эстрогенов включают поддержание нормальной структуры кожи и кровеносных сосудов у женщин, способствуют образованию оксида азота в сосудах гладких мышц, что вызывает их расширение и усиливает теплоотдачу. Эстрогены стимулируют синтез транспортных белков тире-оидных и половых гормонов. Эстрогены могут индуцировать синтез факторов свёртывания крови II, VII, IX и X, уменьшать концентрацию антитромбина III.
Эстрогены оказывают влияние на обмен ли-пидов. Так, увеличение скорости синтеза ЛПВП и торможение образования ЛПНП, вызываемое эстрогенами, приводит к снижению содержания холестерола в крови.
4. Образование прогестерона
Прогестерон, образующийся главным образом жёлтым телом во время менструации в лютеи-новую фазу, секретируется также фетоплацен-тарным комплексом во время беременности. В небольших количествах он вырабатывается у женщин и мужчин корой надпочечников. В фолликулярной фазе менструального цикла концентрация прогестерона в плазме обычно не превышает 5 нмоль/л, а в лютеиновой фазе увеличивается до 40-50 нмоль/л. В крови прогестерон связывается с транспортным глобу-
лином транскортином и альбумином, и только 2% гормона находится в свободной биологически активной форме. Диффундируя в клетки-мишени, прогестерон связывается со специфическим ядерным рецептором. Образующийся комплекс гормон-рецептор взаимодействует с промотор-ным участком ДНК и активирует транскрипцию генов. T1/2 прогестерона в крови составляет 5 мин. В печени гормон конъюгируется с глю-куроновой кислотой и выводится с мочой.
5. Биологические эффекты прогестерона
Действие прогестерона в основном направлено на репродуктивную функцию организма. Образование прогестерона отвечает за увеличение базальной температуры тела на 0,2- 0,5 ?С, которое происходит сразу после овуляции и сохраняется на протяжении лютеиновой фазы менструального цикла. При высоких концентрациях прогестерон взаимодействует с рецепторами, локализованными в клетках почечных канальцев, конкурируя таким образом с альдостероном. В результате конкурентного игибирования альдостерон теряет возможность стимулировать реабсорбцию натрия.
Прогестерон может также оказывать действие и на ЦНС, в частности вызывать некоторые особенности поведения в предменструальный период.