Биохимия: учебник для вузов/ под ред. Е.С.Северина - 5-е изд., - 2009. - 768 с.
|
|
|
|
РАЗДЕЛ 2 ЭНЗИМОЛОГИЯ
Основу жизнедеятельности любого организма составляют химические процессы. Практически все реакции в живом организме протекают с участием природных биокатализаторов, называемых ферментами, или энзимами. Среди множества энергетически возможных реакций ферменты избирательно преобразуют реагенты, называемые субстратами, по физиологически полезному пути. Таким образом, ферменты управляют всеми метаболическими процессами организма.
В научной литературе на русском языке утвердились оба термина: «ферменты» и «энзимы», но предпочтение отдают термину «фермент», хотя наука о ферментах называется энзимология. Слово «фермент» происходит от лат. fermentum - закваска, а «энзим» - от греч. en - в, внутри и zyme - дрожжи. Данная терминология возникла исторически при изучении ферментативных процессов спиртового брожения.
Становление энзимологии как науки произошло в начале XIX века. Активное её развитие продолжается до настоящего времени. В задачи этой науки входят определение роли отдельных ферментов в ускорении химических реакций, протекающих в организме, выделение и очистка ферментов, установление их структуры, исследование механизма действия, изучение кинетических характеристик и особенностей регуляции активности in vivo.
Для практической медицины важность энзимологии обусловлена тем, что она даёт фармакологам инструмент направленного изменения метаболизма клетки путём воздействия определёнными химическими веществами на активность ферментов. Огромное количество фармацевтических препаратов - ингибиторы ферментов. Другая, не менее важная задача энзимологии для практической медицины - использование методов определения активности ферментов в биологических жидкостях для диагностики заболеваний. Кроме того, выделенные и очищенные ферменты могут использоваться в качестве терапевтических средств.
I. ОБЩАЯ ХАРАКТЕРИСТИКА ФЕРМЕНТОВ КАК БИОЛОГИЧЕСКИХ
КАТАЛИЗАТОРОВ
Ферменты, как было установлено ещё в
В роли биокатализаторов могут выступать и небелковые соединения. Например, некоторые типы РНК вызывают гидролиз фосфодиэфирных связей нуклеиновых кислот. Такие молекулы РНК с каталитической активностью называют рибозимами, однако их значение в химическом превращении соединений намного меньше, чем у ферментов.
Поскольку ферменты - белковые молекулы, следовательно, они обладают всеми свойствами, характерными для белков. В то же время они имеют особенности строения, характеризующие их как катализаторы. Рассмотрим основные свойства ферментов как биологических катализаторов.
А. СПЕЦИФИЧНОСТЬ
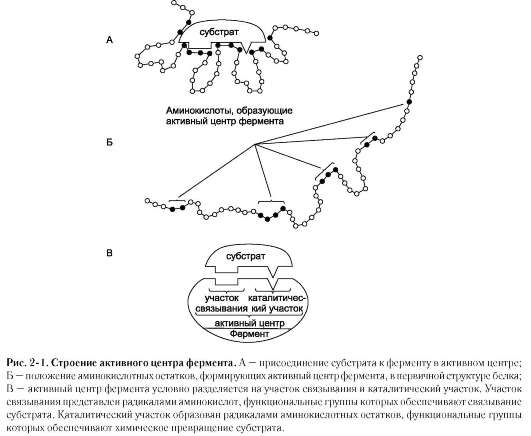
Биологическая функция фермента, как и любого белка, обусловлена наличием в его структуре активного центра. Лиганд, взаимодействующий с активным центром фермента, называют субстратом. В активном центре фермента есть аминокислотные остатки, функциональные группы которых обеспечивают связывание субстрата, и аминокислотные остатки, функциональные группы которых осуществляют химическое превращение субстрата. Условно
эти группы обозначают как участок связывания субстрата и каталитический участок, однако следует помнить, что не всегда эти участки имеют чёткое пространственное разделение и иногда могут «перекрываться» (рис. 2-1).
В участке связывания субстрат при помощи нековалентных связей взаимодействует (связывается) с ферментом, формируя фермент-субстратный комплекс. В каталитическом участке субстрат претерпевает химическое превращение в продукт, который затем высвобождается из активного центра фермента. Схематично процесс катализа можно представить следующим уравнением:
E + S ↔ ES ↔ ЕР ↔ Е + P,
где E - фермент (энзим), S - субстрат, P - продукт. Данные обозначения общеприняты и происходят от английских слов enzyme, substrat, product.
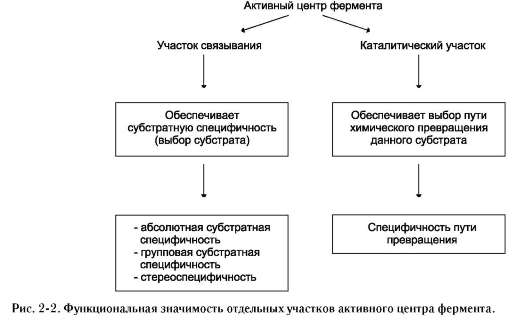
Специфичность - наиболее важное свойство ферментов, определяющее биологическую значимость этих молекул. Различают субстратную и каталитическую специфичности фермента, определяемые строением активного центра (рис. 2-2).
1. Субстратная специфичность
Под субстратной специфичностью понимают способность каждого фермента взаимодействовать лишь с одним или несколькими определёнными субстратами. Различают:
• абсолютную субстратную специфичность;
• групповую субстратную специфичность;
• стереоспецифичность.
Абсолютная субстратная специфичность
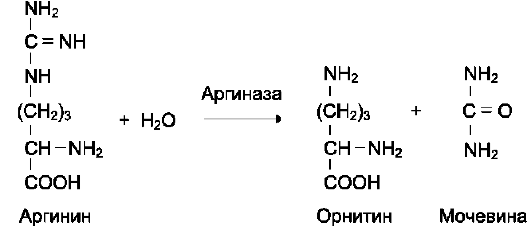
Активный центр ферментов, обладающих абсолютной субстратной специфичностью, комплементарен только одному субстрату. Следует отметить, что таких ферментов в живых организмах мало.
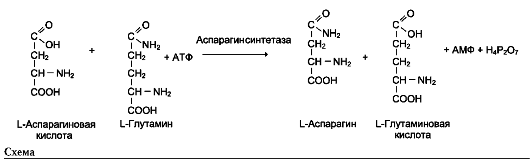
Пример фермента с абсолютной субстратной специфичностью - аргиназа, катализирующая реакцию расщепления аргинина до мочевины и орнитина:
Другой пример фермента с абсолютной субстратной специфичностью - уреаза, катализирующая гидролиз мочевины до диоксида углерода и аммиака.
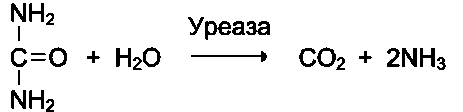
Групповая субстратная специфичность
Большинство ферментов катализирует однотипные реакции с небольшим количеством (группой) структурно похожих субстратов.
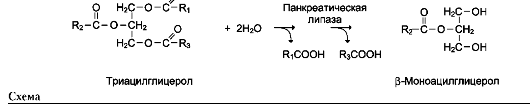
Так, фермент панкреатическая липаза катализирует гидролиз жиров в двенадцатиперстной кишке человека, катализируя превращение любой молекулы жира (триацилглицерола) до молекулы моноацилглицерола и двух молекул высших жирных кислот. Панкреатическая липаза гидролизует эфирную связь у α-атомов углерода глицерола, независимо от того, какие жирные кислоты входят в состав молекулы жира (см. схему на след. стр.).
Большинство протеолитических ферментов, осуществляющих гидролиз белков, имеет групповую субстратную специфичность, гидролизуя пептидные связи, образованные разными аминокислотами.
Стереоспецифичность
При наличии у субстрата нескольких сте-реоизомеров фермент проявляет абсолютную
специфичность к одному из них. В организме человека наблюдают специфичность ферментов к следующим стереоизомерам.
Стереоспецифичность к D-сахарам. Большинство моносахаридов и продуктов их обмена в организме человека и других млекопитающих относят к D-стереоизомерам. Ферменты, осуществляющие их метаболизм, имеют специфичность к D-, а не к L-сахарам.
Стереоспецифичность к L-аминокислотам. Белки человека состоят из аминокислот L-ряда. Большинство ферментов, обеспечивающих превращение аминокислот, имеет стереоспе-цифичность к L-аминокислотам.
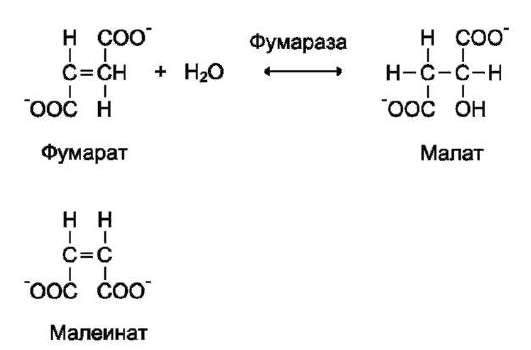
Стереоспецифичность к цис-транс-изомерам. Фермент фумараза оказывает действие только на фумарат. Малеинат (цис-изомер фумарата) не является субстратом фумаразы.
Исключение составляют только ферменты эпимеразы (рацемазы), катализирующие превращение оптических изомеров.
Стереоспецифичность к α- и β-гликозидным связям.
Фермент амилаза действует только на α-гли-козидные связи, что позволяет гидролизо-вать крахмал и гликоген (полимеры глюкозы), остатки глюкозы в которых соединены α-гликозидными связями. Целлюлоза - также полимер глюкозы, однако остатки глюкозы в нём связаны β-гликозидными связями. В результате отсутствия у человека ферментов, специфичных к β-гликозид-ной связи, целлюлоза не гидролизуется в кишечнике человека и не может служить источником глюкозы.
2. Каталитическая специфичность
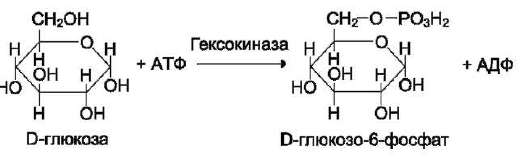
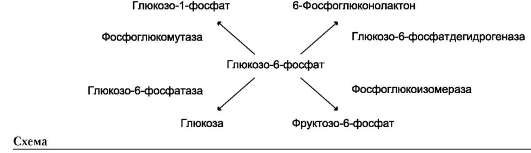
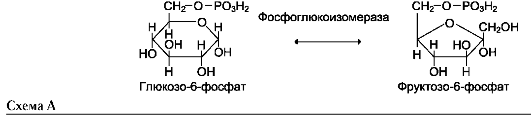
Фермент катализирует превращение присоединённого субстрата по одному из возможных путей его превращения. Это свойство обеспечивается строением каталитического участка активного центра фермента и называется каталитической специфичностью, или специ-фич-ностью пути превращения субстрата. Так, молекула глюкозо-6-фосфата в клетках печени человека - субстрат 4 различных ферментов: фосфоглюкомутазы, глюкозо-6-фосфатфосфа-тазы, фосфоглюкоизомеразы и глюкозо-6-фос-фатдегидрогеназы. Однако из-за особенностей строения каталитических участков этих ферментов происходит различное превращение этого соединения с образованием 4 различных продуктов (см. схему на след. стр.).
Б. КАТАЛИТИЧЕСКАЯ ЭФФЕКТИВНОСТЬ
Большинство катализируемых ферментами реакций высокоэффективны, они протекают в 108-1014 раз быстрее, чем некатализируемые реакции. Каждая молекула фермента способна за секунду трансформировать от 100 до 1000 молекул субстрата в продукт.
Количество молекул субстрата, превращённых в продукт с помощью одной молекулы фермента за 1 с, называют числом оборотов фермента, или молярной активностью.
в. лабильность ферментов
Каталитическая эффективность фермента, как и любой белковой молекулы, зависит от его конформации, и в частности от конформации активного центра.
Для ферментов характерна конформацион-ная лабильность - способность к небольшим изменениям нативной конформации вследствие разрыва слабых связей. Поэтому воздействие денатурирующих агентов, способных изменять конформацию молекулы фермента, приводит к изменению конформации активного центра и снижению способности присоединять субстрат. В результате этого уменьшается каталитическая эффективность фермента.
г. способность ферментов к регуляции
Активность ферментов в клетке зависит от количества молекул субстрата, продукта, наличия кофакторов и коферментов. Действие ферментов в клетке, как правило, строго упорядочено: продукт одной ферментативной реакции является субстратом другой, образуя таким образом «метаболические пути». Среди множества ферментов практически каждого метаболического пути различают ключевые, или регуляторные, ферменты, активность которых может изменяться в зависимости от потребности клетки в конечном продукте метаболического пути. Регуляторные ферменты расположены, как правило, в начале и/или в месте разветвления метаболического пути. Они катализируют либо самые медленные (скорость-лимитирующие реакции), либо
необратимые реакции. Подробно о том, как осуществляется контроль метаболизма путём регуляции активности ферментов, описано в подразделе VII.
II. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ФЕРМЕНТОВ
Каждый фермент имеет 2 названия. Первое - короткое, так называемое рабочее, удобное для повседневного использования. Второе (более полное) - систематическое, применяемое для однозначной идентификации фермента.
а. рабочее название
В названии большинства ферментов содержится суффикс «аза», присоединённый к названию субстрата реакции, например уреаза, сахараза, липаза, нуклеаза или к названию химического превращения определённого субстрата, например лактатдегидрогеназа, аденилатциклаза, фос-фо-глюкомутаза, пируваткарбоксилаза. Согласно российской классификации ферментов (КФ), названия ферментов пишутся слитно. Однако в употреблении сохранился ряд тривиальных, исторически закреплённых названий ферментов, которые не дают представления ни о субстрате, ни о типе химического превращения, например трипсин, пепсин, ренин, тромбин.
б. классы ферментов
Международный союз биохимии и молекулярной биологии в
преобразуемой химической группы субстрата, донора и акцептора преобразуемых группировок, наличия дополнительных молекул и т.д. Каждый из 6 классов имеет свой порядковый номер, строго закреплённый за ним.
1. Оксидоредуктазы
Катализируют различные окислительно-восстановительные реакции с участием 2 субстратов (перенос е- или атомов водорода с одного субстрата на другой).
Систематическое наименование ферментов составляют по формуле «донор: акцептор-ок-сидоредуктаза», рабочее - субстрат-подкласс оксидоредуктаз.
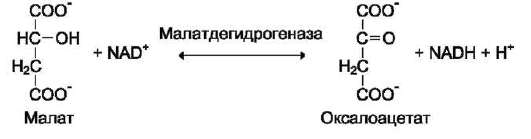
Дегидрогеназы. В этот подкласс входят ферменты, катализирующие реакции дегидрирования (отщепления водорода). В качестве акцепторов электронов используются ко-ферменты NAD+, NADP+, FAD, FMN (см. ниже). Все ферменты этой группы обладают высокой субстратной специфичностью. Пример реакции:
Оксидазы. Акцептором электрона служит молекулярный кислород. Пример реакции, катализируемой цитохромоксидазой:
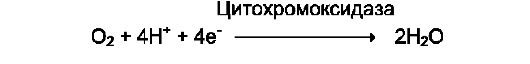
Оксигеназы (гидроксилазы) - атом кислорода из молекулы кислорода присоединяется к субстрату. Пример реакции:
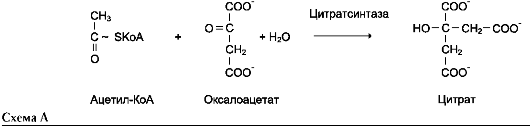
2. Трансферазы
Катализируют перенос функциональных групп от одного соединения к другому. Подразделяют в зависимости от переносимой группы.
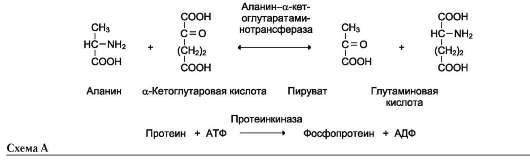
Название этих ферментов составляют по формуле «донор: акцептор-транспортируемая груп-па-трансфераза». К классу трансфераз относят аминотрансферазы, ацилтрансферазы, метил-транс-феразы, гликозилтрансферазы, киназы (фосфо-трансферазы). Примеры реакций (см. схему А на след. стр.).
3. Гидролазы
Катализируют реакции гидролиза (расщепления ковалентной связи с присоединением молекулы воды по месту разрыва). Подразделяют в зависимости от расщепляемой связи.
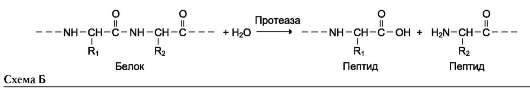
Наименование ферментов составляют по формуле «субстрат-гидролаза» или прямым присоединением к названию субстрата суффикса «аза», например протеаза, липаза, фосфолипаза, рибонуклеаза. Пример реакции (см. схему Б на след. стр.).
Для отдельных классов гидролаз применимы специальные термины, характеризующие гидролиз определённой химической связи: эстеразы, фосфатазы и др.
4. Лиазы
К лиазам относят ферменты, отщепляющие от субстратов негидролитическим путём определённую группу (при этом могут отщепляться СО2, Н2О, NH2, SH2 и др.) или присоединяющие чаще всего молекулу воды по двойной связи.
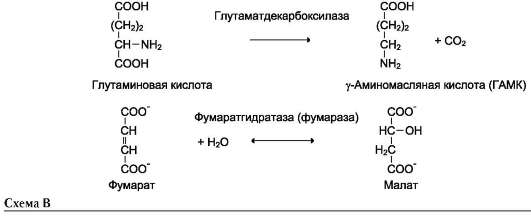
Наименование ферментов составляют по формуле «субстрат-отщепляемая или присоединяемая группировка». Примеры реакций (см. схему В на след. стр.).
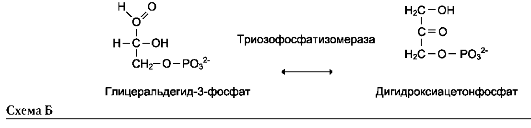
5. Изомеразы
Катализируют различные внутримолекулярные превращения. Подразделяют в зависимости от типа реакции изомеризации.
Как общее название ферментов этого класса применяют термин «изомеразы», например (см. схему А на след. стр.).
Изомеразы могут катализировать внутримолекулярные окислительно-восстановительные реакции, осуществляя взаимопревращения альдоз и кетоз, кетонных и енольных групп, перемещения двойных связей внутри молекулы (см. схему Б на след. стр.).
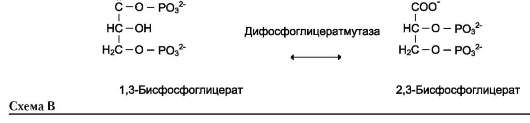
Когда изомеризация состоит во внутримолекулярном переносе группы, фермент называют
«мутазой», например (см. схему В на след. стр.).
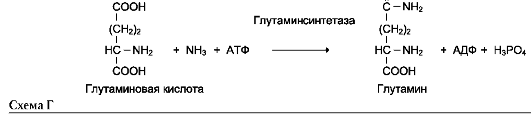
6. Лигазы (синтетазы)
Катализируют реакции присоединения друг к другу двух молекул с образованием ковалент-ной связи. Этот процесс сопряжён с разрывом фосфоэфирной связи в молекуле АТФ (или других нуклеозидтрифосфатов) или с разрывом макроэргических связей других соединений. В первом случае (при использовании энергии гидролиза АТФ) такие ферменты называют лигазами, или синтетазами (см. схему Г на след. стр.).
В случае, когда источником энергии служит любое другое макроэргическое соединение (не
АТФ), ферменты называют синтазами (см. схему А на след. стр.).
в. систематическое название
В соответствии с классификацией каждый фермент получил систематическое название, однозначно характеризующее катализируемую им химическую реакцию. Например, D-гли-церальдегид-3-фосфат: NAD-оксидоредуктаза (рабочее название - глицеральдегидфосфат дегидрогеназа). Из названия фермента следует, что субстратом этого фермента служит D-глице-ральдегид-3-фосфат, тип катализируемой реак-
ции - окислительно-восстановительная в присутствии кофермента NAD+.
В
малатдегидрогеназа имеет систематическое название L-малат: NAD-оксидоредуктаза и кодовый шифр 1.1.1.38. Шифр означает, что этот фермент относят к первому классу ферментов - оксидоредуктаз, окисляемая группа - гидро-ксильная группировка (1) в присутствии кофер-мента NAD+ (1) и порядковый номер фермента в этой подгруппе - 38. Кодовую номенклатуру ферментов в основном используют в научной литературе.
III. КОФАКТОРЫ И КОФЕРМЕНТЫ
Большинство ферментов для проявления ферментативной активности нуждается в низкомолекулярных органических соединениях небелковой природы (коферментах) и/или в ионах металлов (кофакторах).
Термин «кофермент» был введён в начале XX века и обозначал часть некоторых ферментов, которая легко отделялась от белковой молекулы фермента и удалялась через полупроницаемую мембрану при диализе. Несколько позже было выяснено, что большинство ферментов состоит из термолабильной белковой части и термостабильного небелкового фактора - кофермента. Белковая часть получила название «апофер-мент», который в отсутствие кофермента не обладает каталитической активностью. Кофермент с белковой молекулой (апоферментом) фор-
мируют молекулу холофермента, обладающую каталитической активностью.
А. КОФАКТОРЫ
Более 25% всех ферментов для проявления полной каталитической активности нуждается в ионах металлов. Рассмотрим роль кофакторов в ферментативном катализе.
1. Роль металлов в присоединении субстрата в активном центре фермента
Ионы металла выполняют функцию стабилизаторов молекулы субстрата, активного центра фермента и конформации белковой молекулы фермента, а именно третичной и четвертичной структур.
Ионы металлов - стабилизаторы молекулы субстрата
Для некоторых ферментов субстратом служит комплекс превращаемого вещества с ионом металла. Например, для большинства киназ в качестве одного из субстратов выступает не молекула АТФ, а комплекс Mg2+-АТФ. В этом случае ион Mg2+ не взаимодействует непосредственно с ферментом, а участвует в стабилизации молекулы АТФ и нейтрализации отрицательного заряда субстрата, что облегчает его присоединение к активному центру фермента (см. схему Б).
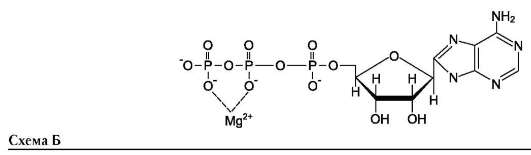
Схематично роль кофактора при взаимодействии фермента и субстрата можно представить как комплекс E-S-Me, где Е - фермент, S - субстрат, Ме - ион металла.
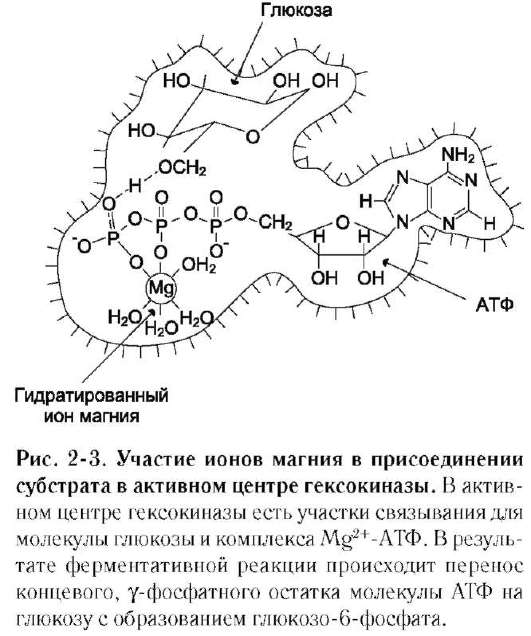
В качестве примера можно привести расположение субстратов в активном центре гексо-киназы (рис. 2-3).
Гексокиназа катализирует перенос концевого, γ-фосфатного остатка молекулы АТФ на глюкозу с образованием глюкозо-6-фосфата:
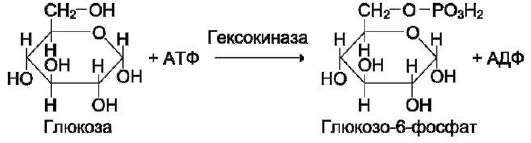
Ион Mg2+ участвует в присоединении и «правильной» ориентации молекулы АТФ в активном центре фермента, ослабляя фосфоэфирную связь и облегчая перенос фосфата на глюкозу.
Ионы металла - стабилизаторы активного центра фермента
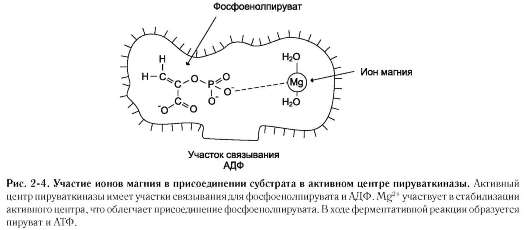
В некоторых случаях ионы металла служат «мостиком» между ферментом и субстратом.
Они выполняют функцию стабилизаторов активного центра, облегчая присоединение к нему субстрата и протекание химической реакции. В ряде случаев ион металла может способствовать присоединению кофермента. Перечисленные выше функции выполняют такие металлы, как Mg2+, Mn2+, Zn2+, Co2+, Mo2+. В отсутствие металла эти ферменты активностью не обладают. Такие ферменты получили название «металлоэнзимы». Схематично данный процесс взаимодействия фермента, субстрата и металла можно представить следующим образом:
E-Me-S
К металлоэнзимам относят, например, фермент пируват киназу (рис. 2-4), катализирующий реакцию:

2. Роль металлов в стабилизации третичной и четвертичной структуры фермента
Ионы металлов обеспечивают сохранение вторичной, третичной, четвертичной структуры молекулы фермента. Такие ферменты в отсутствие ионов металлов способны к химическому катализу, однако они нестабильны. Их активность снижается и даже полностью исчезает при небольших изменениях рН, температуры и других незначительных изменениях внешнего окружения. Таким образом, ионы металлов выполняют функцию стабилизаторов оптимальной конформации белковой молекулы.
Иногда в стабилизации вторичной и третичной структуры принимают участие ионы щёлочно-земельных металлов. Так, для поддержания третичной конформации пируваткиназы необходимы ионы К+.
Для стабилизации четвертичной структуры алкогольдегидрогеназы, катализирующей реакцию окисления этанола, необходимы ионы цинка. Алкогольдегидрогеназа состоит из 4 субъединиц с молекулярной массой 151 кД. В состав фермента входят 4 атома Zn2+. Удаление Zn2+ приводит к потере активности фермента за
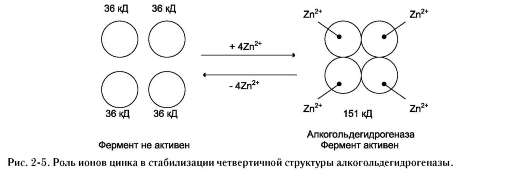
счёт диссоциации на 4 неактивные субъединицы с молекулярной массой 36 кД (рис. 2-5).
3. Роль металлов в ферментативном катализе
Не менее важную роль отводят ионам металлов в осуществлении ферментативного катализа.
Участие в электрофильном катализе
Наиболее часто эту функцию выполняют ионы металлов с переменной валентностью, имеющие свободную d-орбиталь и выступающие в качестве электрофилов. Это, в первую очередь, такие металлы, как Zn2+, Fe2+, Mn2+, Cu2+. Ионы щёлочно-земельных металлов, такие как Na+ и К+, не обладают этим свойством. В качестве примера можно рассмотреть
функционирование фермента карбоангидразы. Карбоангидраза - цинксодержащий фермент, катализирующий реакцию образования угольной кислоты:
СО2 + Н2О ↔ Н2СО3.
Ион Zn2+ в результате электрофильной атаки участвует в образовании Н+ и ОН- ионов из молекулы воды:
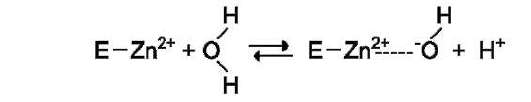
Протон и гидроксильная группа последовательно присоединяются к диоксиду углерода с образованием угольной кислоты (см. схему А на след. стр.).
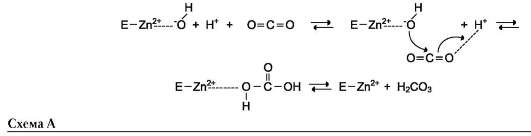
В ходе электрофильного катализа ионы металлов часто участвуют в стабилизации промежуточных соединений.
Участие в окислительно-восстановительных реакциях
Ионы металлов с переменной валентностью могут также участвовать в переносе электронов. Например, в цитохромах (гемсодержащих белках) ион железа способен присоединять и отдавать один электрон:
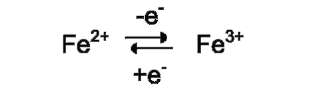
Благодаря этому свойству цитохромы участвуют в окислительно-восстановительных реакциях.
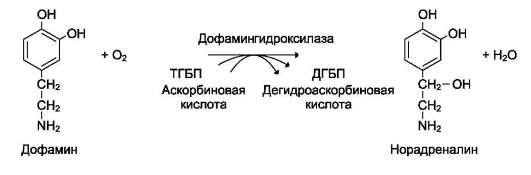
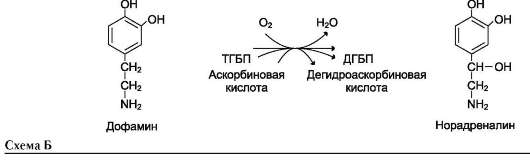
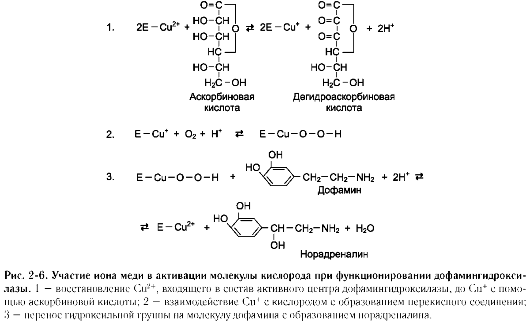
Другой пример участия ионов металлов в окислительно-восстановительных реакциях - работа фермента дофамингидроксилазы, катализирующего реакцию образования норадреналина при участии витамина С (см. схему Б).
За окислительно-восстановительные свойства у дофамингидроксилазы отвечает ион меди (рис. 2-6).
Фермент, содержащий ион Cu2+, не вступает в реакцию с молекулой кислорода. При восстановлении Cu2+до Cu+ с помощью аскорбиновой
кислоты образуется ион меди, способный взаимодействовать с кислородом с образованием перекисного соединения. Далее гидроксильная группа переносится на молекулу дофамина с образованием норадреналина.
4. Роль металлов в регуляции активности ферментов
Иногда ионы металлов выступают в роли регуляторных молекул. Например, ионы Са2+ служат активаторами фермента протеинкиназы С, катализирующего реакции фосфорилиро-вания белков (см. раздел 5). Ионы Са2+ также изменяют активность ряда кальций-кальмоду-линзависимых ферментов (см. подраздел V).
Б. КОФЕРМЕНТЫ
Как уже было сказано, для проявления каталитической активности большинству ферментов необходимо наличие кофермента. Исключение составляют гидролитические ферменты (например, протеазы, липазы, рибонуклеаза), выполняющие свою функцию в отсутствие кофермента.
Кофермент, локализуясь в каталитическом участке активного центра, принимает непосредственное участие в химической реакции, выступая в качестве акцептора и донора химических группировок, атомов, электронов. Кофермент
может быть связан с белковой частью молекулы ковалентными и нековалентными связями. В первом случае он называется простетической группой (например, FAD, FMN, биотин, липо-евая кислота). Вместе с тем известны примеры, когда кофермент присоединяется к ферменту нековалентными связями настолько прочно, что не диссоциирует от белковой молекулы, например тиаминдифосфат.
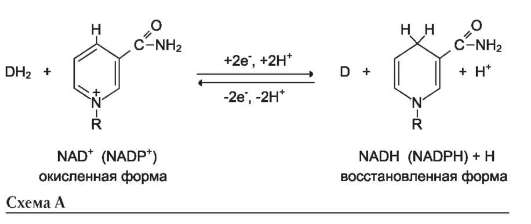
Во втором случае кофермент взаимодействует с ферментом только на время химической реакции и может рассматриваться в качестве второго субстрата. Примеры - NAD+, NADP+.
Апофермент обеспечивает специфичность действия и отвечает за выбор типа химического превращения субстрата. Один и тот же кофермент, взаимодействуя с различными апоферментами, может участвовать в разных химических превращениях субстрата. Например, пиридоксальфосфат в зависимости от того, с каким апоферментом взаимодействует, участвует в реакциях трансаминирования или декарбок-силирования аминокислот.
Химическая природа коферментов, их функции в ферментативных реакциях чрезвычайно
разнообразны. Традиционно к коферментам относят производные витаминов, хотя помимо них есть значительный класс небелковых соединений, принимающих участие в проявлении каталитической функции ферментов.
К коферментам относят следующие соединения:
• производные витаминов;
• гемы, входящие в состав цитохромов, ка-талазы, пероксидазы, гуанилатциклазы, NO-синтазы и являющиеся простетической группой ферментов;
• нуклеотиды - доноры и акцепторы остатка фосфорной кислоты;
• убихинон, или кофермент Q, участвующий в переносе электронов и протонов в ЦПЭ;
• фосфоаденозилфосфосульфат, участвующий в переносе сульфата;
• S-аденозилметионин (SAM) - донор ме-тильной группы;
• глутатион, участвующий в окислительно-восстановительных реакциях.
Строение и функции этих коферментов подробно рассмотрены в соответствующих разделах учебника.
В. МУЛЬТИСУБСТРАТНЫЕ РЕАКЦИИ
Большинство ферментов катализирует реакции, в которых участвует более чем один субстрат. В случае если кофермент не является простетической группой, его также можно рассматривать как ещё один субстрат. Следовательно, участников ферментативной реакции может быть несколько: непосредственно фермент, несколько субстратов и кофермент.
В этих случаях механизм ферментативной реакции, как правило, может идти по одному из двух путей: по механизму «пинг-понг» (механизму двойного замещения) или последовательному. Рассмотрим оба механизма.
1. Механизм «пинг-понг»
Схематично механизм «пинг-понг» может быть представлен следующим образом:
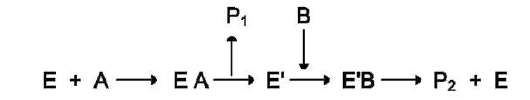
Субстрат А, взаимодействуя с ферментом (Е), превращается в продукт (Р1). Фермент остаётся в результате этого преобразования не в нативной форме, а в изменённой (Е') в результате модификации кофермента. Далее к активному центру Е' присоединяется субстрат В, подвергающийся преобразованию в продукт (Р2) с высвобождением нативной формы фермента (Е).
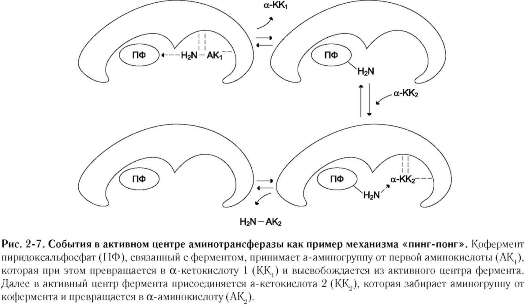
Хороший пример механизма «пинг-понг» - реакции трансаминирования с участием ферментов аминотрансфераз (кофермент пиридок-сальфосфат). Аминотрансферазы, открытые отечественным учёным А.Е. Браунштейном, катализируют обратимые реакции переноса аминогруппы с аминокислоты на кетокислоту. Механизм «пинг-понг» данной реакции схематично представлен на рис. 2-7.
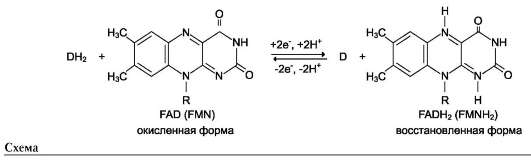
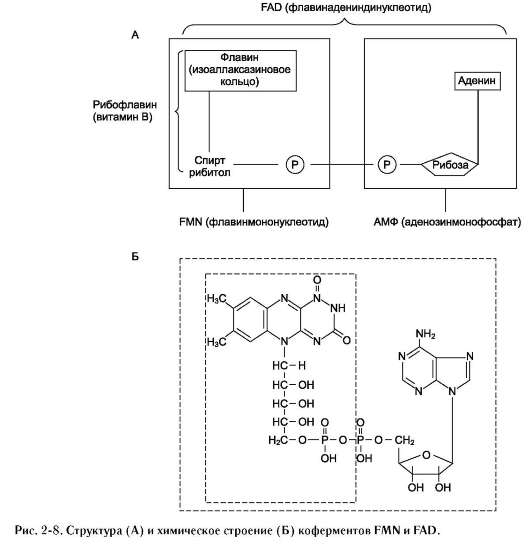
Другой пример механизма «пинг-понг» - реакции дегидрирования с участием кофермента FAD (флавинадениндинуклеотид) или FMN (флавинмононуклеотид), которые прочно связаны с ферментом и, следовательно, не могут рассматриваться в качестве второго субстрата.
Схематично структура этих коферментов и соответствующие им химические формулы представлены на рис. 2-8.
FMN и FAD участвуют в окислительно-восстановительных реакциях, акцептируя 2 е- и 2 Н+ в изоаллаксазиновом кольце (см. схему ниже).
Схему реакции дегидрирования (как пример механизма «пинг-понг» с участием коферментов FMN и FAD) можно представить в следующем виде:
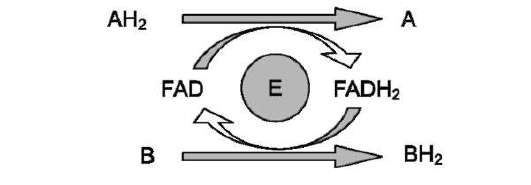
где АН2 - донор водорода, окисляемый субстрат 1; А - окисленная форма субстрата 1; В - акцептор водорода - субстрат 2; ВН2 - восстановленная форма субстрата 2; Е (FAD), Е (FADH2) - окисленная и восстановленная формы кофермен-та FAD, входящего в состав фермента Е.
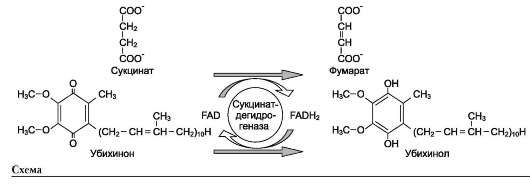
В качестве примера FAD-зависимой реакции можно привести сукцинатдегидрогеназную реакцию. В этой реакции в качестве второго субстрата участвует убихинон - один из посредников ЦПЭ (см. схему на след. стр.).
2. Последовательный механизм
В случае последовательного механизма для протекания ферментной реакции требуется
одновременно взаимодействие двух субстратов. В этом случае возможно присоединение субстратов двумя различными путями:
Механизм упорядоченного взаимодействия субстрата с активным центром фермента:
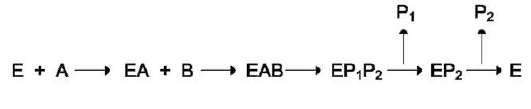
Первым в активный центр фермента присоединяется субстрат А, облегчая присо-
единение субстрата В. После химической модификации также наблюдают определённый порядок высвобождения продуктов реакции. Механизм случайного взаимодействия субстрата с активным центром фермента:
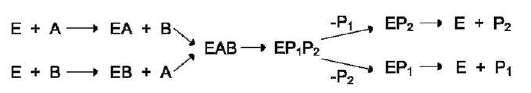
Приоритетности за взаимодействие субстратов А и В в активном центре фермента
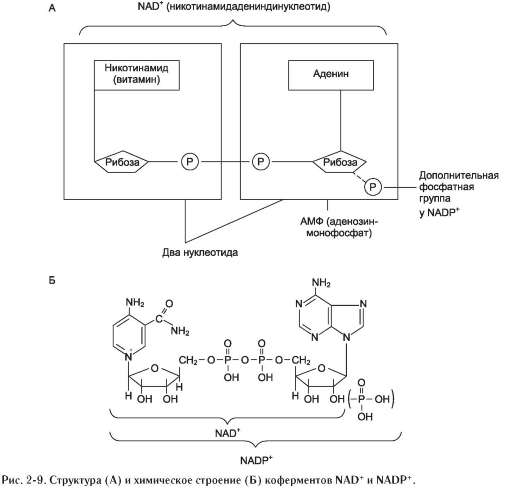
нет (каждый субстрат имеет свой центр связывания в активном центре). Также нет строгой закономерности высвобождения продуктов реакции. Примером последовательного упорядоченного механизма может быть реакция дегидрирования с участием коферментов NAD+, NADP+.
Схематично структура и химические формулы этих коферментов представлены на рис. 2-9.
Оба кофермента функционируют как посредники переноса двух электронов и одного протона от донора к акцептору, другого протона - в среду (см. схему А на след. стр.).
Донор и акцептор не обязательно участвуют в одном метаболическом пути. Другими словами, восстановленная форма этих нуклеотидов действует как общий пул электронов, образованный в результате окислительных реакций, и может быть использована в различных восстановительных реакциях. Такие реакции называют сопряжёнными (см. схему Б на след. стр.). где АН2 - донор водорода, восстановленная форма субстрата 1; А - окисленная форма субстрата 1; В - акцептор водорода - второй субстрат; ВН2 - восстановленная форма субстрата 2; NAD+, NADH - окисленная и вос-
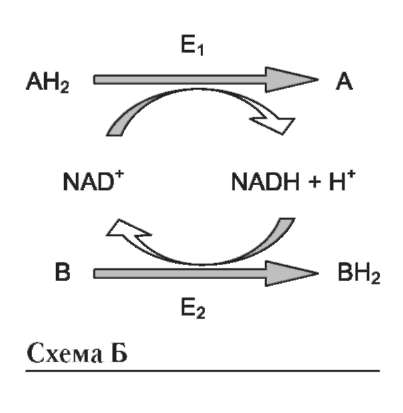
становленная формы кофермента; Е1 и Е2 - ферменты.
Две ферментативные реакции, катализируемые ферментами Е1 и Е2, сопряжены друг с другом посредством кофермента NAD+, служащего в каждом из этих случаев субстратом. Для первого фермента субстратом служит окисленная форма NAD, в качестве второго субстрата выступает донор водорода - пример последовательных реакций, продуктом - восстановленная форма NAD, для фермента Е2 - наоборот.
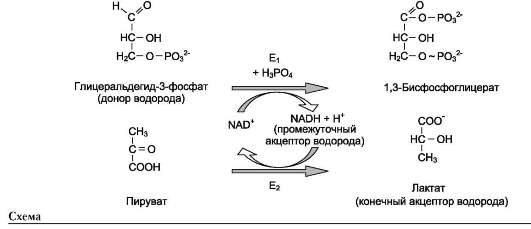
В качестве примера можно рассмотреть следующие сопряжённые реакции (см. схему выше), где E1 - глицеральдегидфосфат дегидрогеназа; Е2 - лактатдегидрогеназа.
IV.МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ
Механизм действия ферментов может быть рассмотрен с двух позиций: с точки зрения изменения энергетики химических реакций и с точки зрения событий в активном центре.
а. энергетические изменения при химических реакциях
Любые химические реакции протекают, подчиняясь двум основным законам термодинамики: закону сохранения энергии и закону энтропии. Согласно этим законам, общая энергия химической системы и её окружения остаётся постоянной, при этом химическая система стремится к снижению упорядоченности (увеличению энтропии). Для понимания энер-
гетики химической реакции недостаточно знать энергетический баланс входящих и выходящих из реакции реагентов, необходимо учитывать изменения энергии в процессе данной химической реакции и роль ферментов в динамике этого процесса. Рассмотрим реакцию разложения угольной кислоты:
H2CO3 → H2O + CO2.
Угольная кислота слабая; реакция её разложения пойдёт при обычных условиях, если молекулы угольной кислоты имеют энергию, превышающую определённый уровень, называемый энергией активации Еа (рис. 2-10).
Энергией активации называют дополнительное количество кинетической энергии, необходимое молекулам вещества, чтобы они вступили в реакцию.
При достижении этого энергетического барьера в молекуле происходят изменения, вызывающие перераспределение химических связей и образование новых соединений. Говорят, что молекулы, обладающие Еа, находятся в переходном состоянии. Разницу энергий между исходным реагентом Н2СО3 и конечными соединениями Н2О и СО2 называют изменением свободной энергии реакции DG. Молекулы Н2О и СО2 - более стабильные вещества, чем Н2СО3, т.е. обладают меньшей энергией и при обычных условиях практически не реагируют. Выделившаяся энергия в результате этой реакции рассеивается в виде тепла в окружающую среду.
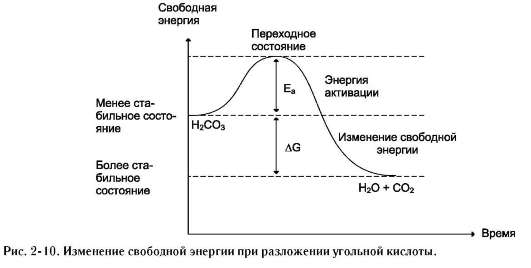
Чем больше молекул обладает энергией, превышающей уровень Еа, тем выше скорость химической реакции. Повысить скорость химической реакции можно нагреванием. При этом увеличивается энергия реагирующих молекул. Однако для живых организмов высокие температуры губительны, поэтому в клетке для ускорения химических реакций используются ферменты. Ферменты обеспечивают высокую скорость реакций при оптимальных условиях, существующих в клетке, путём понижения уровня Еа. Таким образом, ферменты снижают высоту энергетического барьера, в результате возрастает количество реакционно-способных молекул, следовательно, увеличивается скорость реакции.
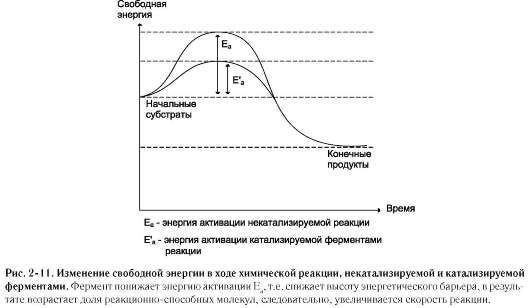
В механизме ферментативного катализа решающее значение имеет образование нестойких промежуточных соединений - фермент-субстратный комплекс ES, подвергающийся превращению в нестабильный переходный комплекс ЕР, который почти мгновенно распадается на свободный фермент и продукт реакции.
Таким образом, биологические катализаторы (ферменты) не изменяют свободную энергию субстратов и продуктов и поэтому не меняют равновесие реакции (рис. 2-11).
Фермент, выполняя функцию катализатора химической реакции, подчиняется общим законам катализа и обладает всеми свойствами, характерными для небиологических катализаторов, однако имеет и отличительные свойства, связанные с особенностями строения ферментов.
Сходство ферментов с небиологическими катализаторами заключается в том, что:
• ферменты катализируют энергетически возможные реакции;
• энергия химической системы остаётся постоянной;
• в ходе катализа направление реакции не изменяется;
• ферменты не расходуются в процессе реакции.
Отличия ферментов от небиологических катализаторов заключаются в том, что:
• скорость ферментативных реакций выше, чем реакций, катализируемых небелковыми катализаторами;
• ферменты обладают высокой специфичностью;
• ферментативная реакция проходит в клетке, т.е. при температуре 37 ?С, постоянном атмосферном давлении и физиологическом значении рН;
• скорость ферментативной реакции может регулироваться.
Б. ЭТАПЫ ФЕРМЕНТАТИВНОГО КАТАЛИЗА
1. Формирование фермент-субстратного комплекса
Тот факт, что ферменты обладают высокой специфичностью, позволил в
ветствует ему как «ключ замку». После взаимодействия субстрата («ключ») с активным центром («замок») происходят химические превращения субстрата в продукт. Активный центр при этом рассматривался как стабильная, жёстко детерминированная структура.
В
2. Последовательность событий в ходе ферментативного катализа
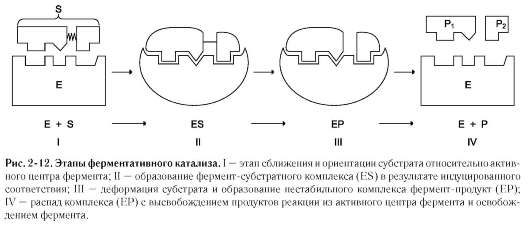
Процесс ферментативного катализа условно можно разделить на следующие этапы (рис. 2-12).
Первый, второй и четвёртый этапы катализа непродолжительны и зависят от концентрации субстрата (для первого этапа) и констант связывания лигандов в активном центре фермента (для первого и третьего этапов). Изменения энергетики химической реакции на этих стадиях незначительны.
Третий этап наиболее медленный; длительность его зависит от энергии активации химической реакции. На этой стадии происходят разрыв связей в молекуле субстрата, образование новых связей и формирование молекулы продукта.
3. Роль активного центра в ферментативном катализе
В результате исследований было показано, что молекула фермента, как правило, во много раз больше молекулы субстрата, подвергающегося химическому превращению этим ферментом. В контакт с субстратом вступает лишь небольшая часть молекулы фермента, обычно от 5 до 10 аминокислотных остатков, формирующих активный центр фермента. Роль остальных аминокислотных остатков состоит в обеспечении правильной конформации молекулы фермента
для оптимального протекания химической реакции.
Активный центр на всех этапах ферментативного катализа нельзя рассматривать как пассивный участок для связывания субстрата. Это комплексная молекулярная «машина», использующая разнообразные химические механизмы, способствующие превращению субстрата в продукт.
В активном центре фермента субстраты располагаются таким образом, чтобы участвующие в реакции функциональные группы субстратов находились в непосредственной близости друг к другу. Это свойство активного центра называют эффектом сближения и ориентации реагентов. Такое упорядоченное расположение субстратов вызывает уменьшение энтропии и, как следствие, снижение энергии активации (Еа), что определяет каталитическую эффективность ферментов.
Активный центр фермента также способствует дестабилизации межатомных связей в молекуле субстрата, что облегчает протекание химической реакции и образование продуктов. Это свойство активного центра называют эффектом деформации субстрата (рис. 2-12).
В. МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ ФЕРМЕНТАТИВНОГО КАТАЛИЗА
Механизмы ферментативного катализа определяются ролью функциональных групп активного центра фермента в химической реакции превращения субстрата в продукт. Выделяют 2 основных
механизма ферментативного катализа: кислотно-основной катализ и ковалентный катализ.
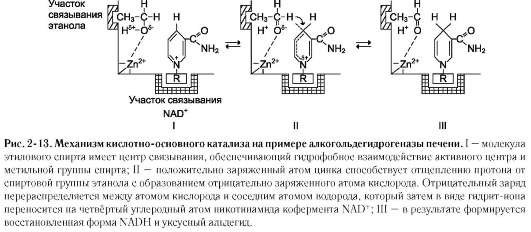
1. Кислотно-основной катализ
Концепция кислотно-основного катализа объясняет ферментативную активность участием в химической реакции кислотных групп (доноры протонов) и/или основных групп (акцепторы протонов). Кислотно-основной катализ - часто встречающееся явление. Аминокислотные остатки, входящие в состав активного центра, имеют функциональные группы, проявляющие свойства как кислот, так и оснований.
К аминокислотам, участвующим в кислотно-основном катализе, в первую очередь относят Цис, Тир, Сер, Лиз, Глу, Асп и Гис. Радикалы этих аминокислот в протонированной форме - кислоты (доноры протона), в депротониро-ванной - основания (акцепторы протона). Благодаря этому свойству функциональных групп активного центра ферменты становятся уникальными биологическими катализаторами, в отличие от небиологических катализаторов, способных проявлять либо кислотные, либо основные свойства.
Примером кислотно-основного катализа, в котором кофакторами являются ионы Zn2+, а в качестве кофермента используется молекула NAD+, можно привести фермент алкогольде-гидрогеназу печени, катализирующую реакцию окисления спирта (рис. 2-13):
2. Ковалентный катализ
Ковалентный катализ основан на атаке нук-леофильных (отрицательно заряженных) или электрофильных (положительно заряженных) групп активного центра фермента молекулами субстрата с формированием ковалентной связи между субстратом и коферментом или функциональной группой аминокислотного остатка (как правило, одной) активного центра фермента.
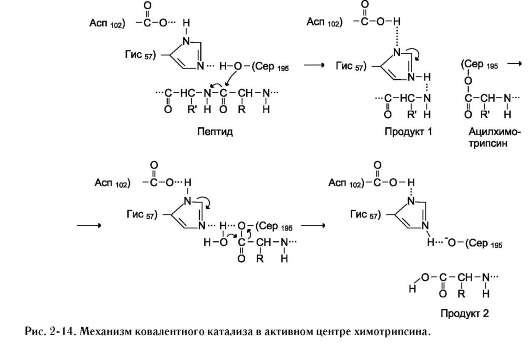
Действие сериновых протеаз, таких как трипсин, химотрипсин и тромбин, - пример механизма ковалентного катализа, когда ковалентная связь образуется между субстратом и аминокислотным остатком серина активного центра фермента. Термин «сериновые протеазы» связан с тем, что аминокислотный остаток серина входит в состав активного центра всех этих ферментов и участвует непосредственно в катализе. Рассмотрим механизм ковалентного катализа на примере химотрипсина, осуществляющего гидролиз пептидных связей при переваривании белков в двенадцатиперстной кишке (см. раздел 9). Субстратами химотрипсина служат пептиды, содержащие аминокислоты с ароматическими и циклическими гидрофобными радикалами (Фен, Тир, Три), что указывает на участие гидрофобных сил в формировании фермент-субстратного комплекса. Механизм ковалентного катализа химотрипсина рассмотрен на рис. 2-14.
Радикалы Асп102, Гис57 и Сер195 участвуют непосредственно в акте катализа. Вследствие
нуклеофильной атаки пептидной связи субстрата происходит разрыв этой связи с образованием ковалентно-модифицированного серина - ацил-химотрипсина. Другой пептидный фрагмент высвобождается в результате разрыва водородной связи между пептидным фрагментом и Гис57 активного центра химотрипсина. Заключительный этап гидролиза пептидной связи белков - деацилирование химотрипсина в присутствии молекулы воды с высвобождением второго фрагмента гидролизуемого белка и исходной формы фермента.
v. основы кинетики ферментативных реакций
Кинетика ферментативных реакций - раздел энзимологии, изучающий зависимость скорости химических реакций, катализируемых ферментами, от химической природы реагирующих веществ, а также от факторов окружающей среды.
Для измерения каталитической активности ферментов используют такие показатели, как скорость реакции или активность фермента. Скорость ферментативной реакции определяется изменением количества молекул субстрата или продукта за единицу времени. Скорость ферментативной реакции - мера каталитической активности фермента, её обозначают как активность фермента.
Математически скорость ферментативной реакции выражается в изменении концентрации субстрата (уменьшение) или продукта (увеличение) за единицу времени:
V= D[S]/t = D[P]/t.
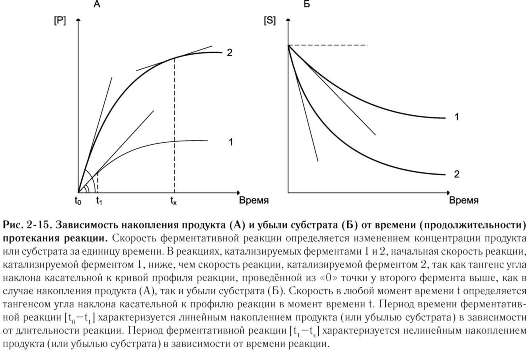
На начальном этапе [0 - t0] скорость реакции прямо пропорциональна времени и имеет линейную зависимость. Графически изменение скорости ферментативной реакции определяется тангенсом угла наклона касательной к кривой профиля реакции. Чем больше угол наклона, тем больше изменение скорости реакции (рис. 2-15).
С течением времени изменение скорости ферментативной реакции в экспериментальных условиях уменьшается, об этом свидетельствует уменьшение угла наклона касательной в момент времени t. Снижение скорости ферментативной реакции может происходить за счёт ряда факторов: уменьшения концентрации субстрата, увеличения концентрации продукта, который может оказывать ингибирующее действие, могут происходить изменения рН раствора, инактивация фермента и т.д.
На этапе [t1 - tx] скорость реакции изменяется нелинейно в зависимости от времени. Поэтому для определения скорости ферментативной реакции чаще всего исследуют изменение скорости на начальном этапе [t0 - t1], где наблюдают линейное изменение концентрации продукта (или субстрата).
Скорость ферментативной реакции зависит от ряда факторов, таких как количество и активность ферментов, концентрация субстрата, температура среды, рН раствора, присутствие регуляторных молекул (активаторов и ингибиторов). Рассмотрим влияние этих факторов на скорость ферментативной реакции.
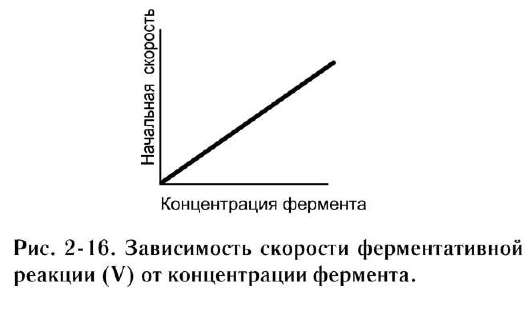
а. зависимость скорости ферментативной реакции от количества ферментов
При проведении ферментативной реакции в условиях избытка субстрата скорость реакции будет зависеть от концентрации фермента. Графическая зависимость такой реакции имеет вид прямой линии (рис. 2-16). Однако количество фермента часто невозможно определить в абсолютных величинах, поэтому на
практике пользуются условными величинами, характеризующими активность фермента: одна международная единица активности (МЕ) соответствует такому количеству фермента, которое катализирует превращение 1 мкмоль субстрата за 1 мин при оптимальных условиях проведения ферментативной реакции. Оптимальные условия индивидуальны для каждого фермента и зависят от температуры среды, рН раствора, при отсутствии активаторов и ингибиторов.

Количество единиц активности nME определяют по формуле:

В

Международная единица ферментативной активности МЕ связана с каталом следующими равенствами:
1 кат = 1 моль S/с = 60 моль S/мин = 60х106 мкмоль/мин = 6х107 МЕ,
1 МЕ = 1 мкмоль/мин = 1/60 мкмоль/с 1 60 мккат = 16,67 нкат.
В медицинской и фармацевтической практике для оценки активности ферментов часто используют международные единицы активности - МЕ. Для оценки количества молекул фермента среди других белков данной ткани определяют удельную активность (уд. ак.) фермента, численно равную количеству единиц активности фермента (nME) в образце ткани, делённому на массу (мг) белка в этой ткани:
По удельной активности судят об очистке фермента: чем меньше посторонних белков, тем выше удельная активность.
Б. ЗАВИСИМОСТЬ СКОРОСТИ
ФЕРМЕНТАТИВНОЙ РЕАКЦИИ
ОТ ТЕМПЕРАТУРЫ СРЕДЫ
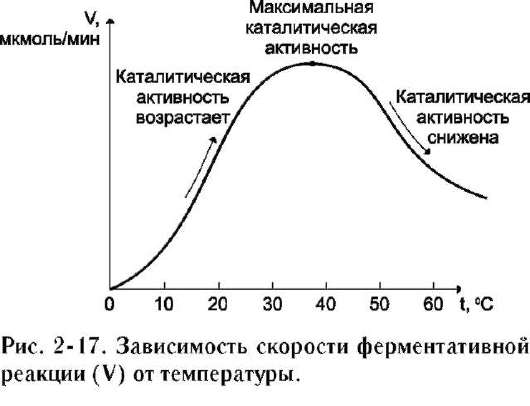
Повышение температуры до определённых пределов оказывает влияние на скорость ферментативной реакции, подобно влиянию температуры на любую химическую реакцию. С повышением температуры ускоряется движение молекул, что приводит к повышению вероятности взаимодействия реагирующих веществ. Кроме того, температура может повышать энергию реагирующих молекул, что также приводит к ускорению реакции. Однако скорость химической реакции, катализируемая ферментами, имеет свой температурный оптимум, превышение которого сопровождается понижением ферментативной активности, возникающим из-за термической денатурации белковой молекулы (рис. 2-17).
Для большинства ферментов человека оптимальна температура 37-38 ?С. Однако в природе существуют и термостабильные ферменты. Например, Taq-полимераза, выделенная из микроорганизмов, живущих в горячих источниках, не инактивируется при повышении температуры до 95 ?С. Этот фермент используют в научно-практической медицине для молекулярной диагностики заболеваний с использованием метода полимеразной цепной реакции (ПЦР).
В. ЗАВИСИМОСТЬ СКОРОСТИ ФЕРМЕНТАТИВНОЙ РЕАКЦИИ
ОТ РН СРЕДЫ
Активность ферментов зависит от рН раствора, в котором протекает ферментативная реак-
ция. Для каждого фермента существует значение рН, при котором наблюдается его максимальная активность. Отклонение от оптимального значения рН приводит к понижению ферментативной активности.
Влияние рН на активность ферментов связано с ионизацией функциональных групп аминокислотных остатков данного белка, обеспечивающих оптимальную конформацию активного центра фермента. При изменении рН от оптимальных значений происходит изменение ионизации функциональных групп молекулы белка. Например, при закислении среды происходит протонирование свободных аминогрупп (NH3+), а при защелачивании происходит отщепление протона от карбоксильных групп (СОО-). Это приводит к изменению конформации молекулы фермента и конформации активного центра; следовательно, нарушается присоединение субстрата, кофакторов и коферментов к активному центру. Кроме того, рН среды может влиять на степень ионизации или пространственную организацию субстрата, что также влияет на сродство субстрата к активному центру. При значительном отклонении от оптимального значения рН может происходить денатурация белковой молекулы с полной потерей ферментативной активности.
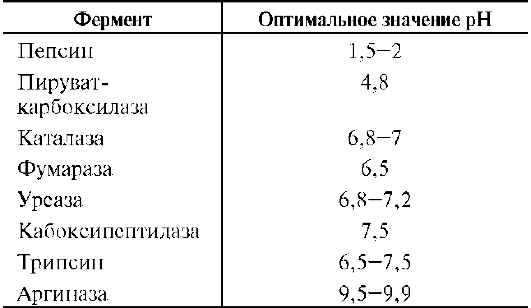
Оптимум значения рН у разных ферментов различный (рис. 2-18). Ферменты, работающие в кислых условиях среды (например, пепсин в желудке или лизосомальные ферменты), эволю-ционно приобретают конформацию, обеспечивающую работу фермента при кислых значениях рН. Однако большая часть ферментов организма
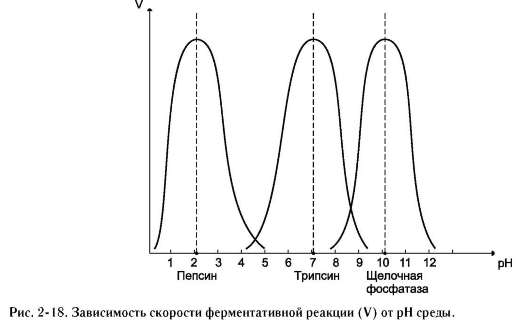
человека имеет оптимум рН, близкий к нейтральному, совпадающий с физиологическим значением рН (табл. 2-1).
Г. ЗАВИСИМОСТЬ СКОРОСТИ ФЕРМЕНТАТИВНОЙ РЕАКЦИИ
ОТ КОЛИЧЕСТВА СУБСТРАТА
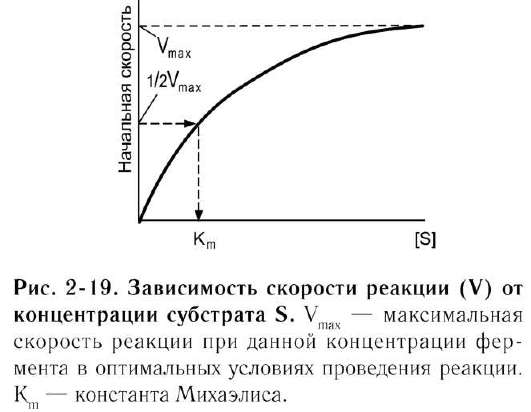
Если концентрацию ферментов оставить постоянной, изменяя только количество субстрата, то график скорости ферментативной реакции описывают гиперболой (рис. 2-19).
При увеличении количества субстрата начальная скорость возрастает. Когда фермент
Таблица 2-1. Оптимальные значения рН для некоторых ферментов
становится полностью насыщенным субстратом, т.е. происходит максимально возможное при данной концентрации фермента формирование фермент-субстратного комплекса, наблюдают наибольшую скорость образования продукта. Дальнейшее повышение концентрации субстрата не приводит к увеличению образования продукта, т.е. скорость реакции не возрастает. Данное состояние соответствует максимальной скорости реакции Vmax.
Таким
образом, концентрация фермента - лимитирующий фактор в образовании
продукта. Это наблюдение легло в основу ферментативной кинетики,
разработанной учёными Л. Михаэли-сом и М. Ментен в
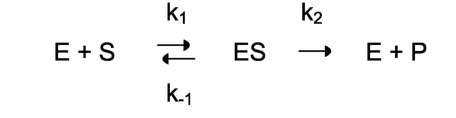
Ферментативный процесс можно выразить следующим уравнением:

где k1 - константа скорости образования фермент-субстратного комплекса; - константа скорости обратной реакции, распада фермент-субстратного комплекса; k2 - константа скорости образования продукта реакции.
Следующее соотношение констант скоростей (k-1 + k2)/k1 называют константой Михаэлиса и обозначают K .
Скорость реакции пропорциональна концентрации фермент-субстратного комплекса ES, а скорость образования ES зависит от концентрации субстрата и концентрации свободного фермента. На концентрацию ES влияет скорость формирования и распада ES.
Наибольшая скорость реакции наблюдается в том случае, когда все молекулы фермента находятся в комплексе с субстратом, т.е. в фермент-субстратном комплексе ES, т.е. [E] = [ES].
Зависимость скорости ферментативной реакции от концентрации субстрата выражается следующим уравнением (математическое выведение этой формулы можно найти в пособиях по ферментативной кинетике):
Это уравнение получило название уравнения Михаэлиса-Ментен .
В случае, когда скорость реакции равна половине максимальной, Km = [S] (рис. 2-19). Таким образом, константа Михаэлиса численно равна концентрации субстрата, при которой достигается половина максимальной скорости.
Уравнение Михаэлиса-Ментен - основное уравнение ферментативной кинетики, описывающее зависимость скорости ферментативной реакции от концентрации субстрата.
Если концентрация субстрата значительно больше Кт (S>>Km), то увеличение концентра-
ции субстрата на величину Km практически не влияет на сумму (Km + S) и её можно считать равной концентрации субстрата. Следовательно, скорость реакции становится равной максимальной скорости: V = Vmax. В этих условиях реакция имеет нулевой порядок, т.е. не зависит от концентрации субстрата. Можно сделать
вывод, что Vmax - величина постоянная для
данной концентрации фермента, не зависящая от концентрации субстрата.
Если концентрация субстрата значительно меньше Km (S<<Km), то сумма (Km + S) примерно равна Km, следовательно, V = Vmax[S]/Km, т.е. в данном случае скорость реакции прямо пропорциональна концентрации субстрата (реакция имеет первый порядок).
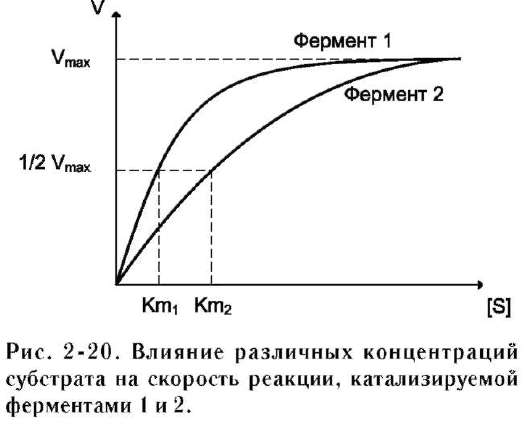
Vmax и Km - кинетические характеристики эффективности фермента. Vmax даёт характеристику каталитической активности фермента и имеет размерность скорости ферментативной реакции моль/л, т.е. определяет максимальную возможность образования продукта при данной концентрации фермента и в условиях избытка субстрата. Km характеризует сродство данного фермента к данному субстрату и является величиной постоянной, не зависящей от концентрации фермента. Чем меньше Km, тем больше сродство фермента к данному субстрату, тем выше начальная скорость реакции и наоборот, чем больше Km, тем меньше начальная скорость реакции, тем меньше сродство фермента к субстрату.
На рис. 2-20 представлена зависимость скорости двух ферментативных реакций (1 и 2) от концентрации субстрата. Константа Михаэлиса первого фермента меньше константы Михаэлиса второго фермента (Km1<Km2). Следовательно, сродство первого фермента к субстрату выше, чем у второго фермента, и начальная скорость реакции, катализируемой первым ферментом, выше в сравнении со вторым ферментом.
VI. ИНГИБИРОВАНИЕ ФЕРМЕНТАТИВНОЙ АКТИВНОСТИ
Под термином «ингибирование ферментативной активности» понимают снижение каталитической активности в присутствии определённых веществ - ингибиторов. К ингибиторам следует относить вещества, вызывающие снижение ак-
тивности фермента. Следует отметить, что все денатурирующие агенты также вызывают уменьшение скорости любой ферментативной реакции, вследствие неспецифической денатурации белковой молекулы, поэтому денатурирующие агенты к ингибиторам не относят.
Ингибиторы вызывают большой интерес для выяснения механизмов ферментативного катализа, помогают установить роль отдельных ферментов в метаболических путях организма. В основе действия многих лекарственных препаратов и ядов лежит ингибирование активности ферментов, поэтому знание механизмов этого процесса крайне важно для молекулярной фармакологии и токсикологии.
Ингибиторы способны взаимодействовать с ферментами с разной степенью прочности. На основании этого различают обратимое и необратимое ингибирование. По механизму действия ингибиторы подразделяют на конкурентные и неконкурентные.
А. ОБРАТИМОЕ ИНГИБИРОВАНИЕ
Обратимые ингибиторы связываются с ферментом слабыми нековалентными связями и при определённых условиях легко отделяются от фермента. Обратимые ингибиторы бывают конкурентными и неконкурентными.
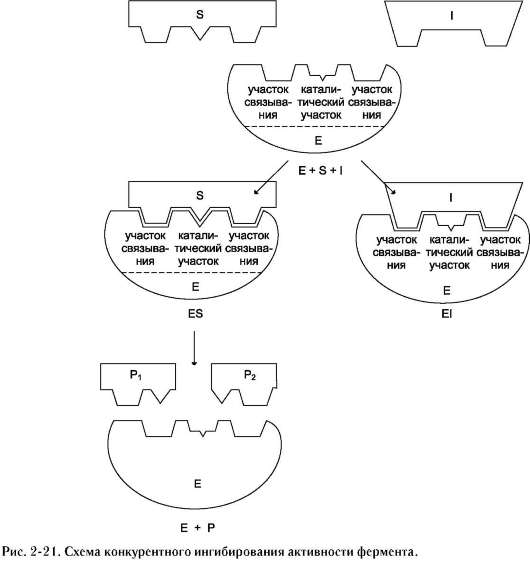
1. Конкурентное ингибирование
К конкурентному ингибированию относят обратимое снижение скорости ферментативной реакции, вызванное ингибитором, связы-
вающимся с активным центром фермента и препятствующим образованию фермент-субстратного комплекса. Такой тип ингибирования наблюдают, когда ингибитор - структурный аналог субстрата, в результате возникает конкуренция молекул субстрата и ингибитора за место в активном центре фермента. В этом случае с ферментом взаимодействует либо субстрат, либо ингибитор, образуя комплексы фермент-субстрат (ES) или фермент-ингибитор (EI). При формировании комплекса фермента и ингибитора (EI) продукт реакции не образуется (рис. 2-21).
Для конкурентного типа ингибирования справедливы следующие уравнения:
E + S <=> ES → E + P,
E + I <=> EI.
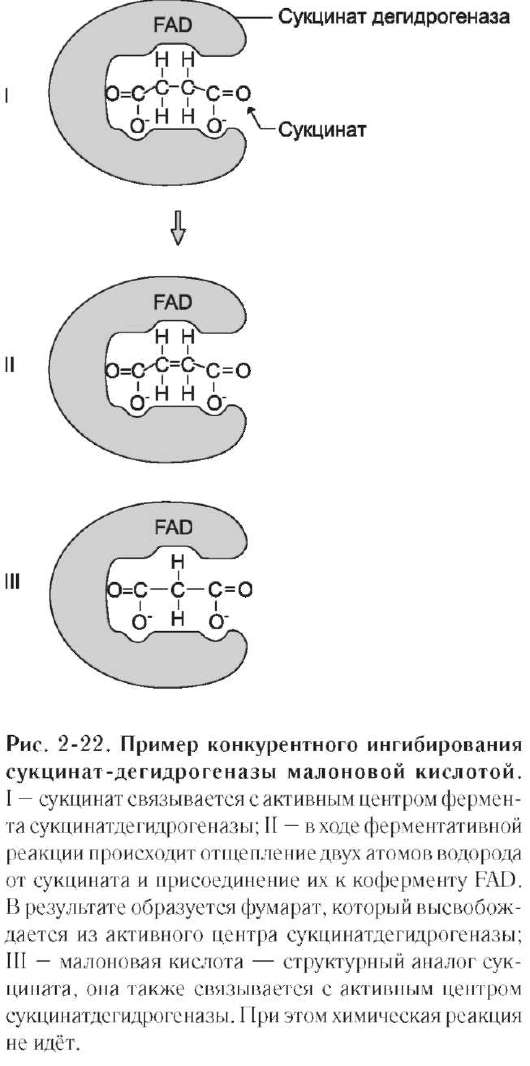
Классический пример конкурентного инги-бирования - ингибирование сукцинатдегид-рогеназной реакции малоновой кислотой (рис. 2-22). Малоновая кислота - структурный аналог сукцината (наличие двух карбоксильных групп) и может также взаимодействовать с активным центром сукцинат дегидрогеназы. Однако отщепление двух атомов водорода от малоновой кислоты невозможно; следовательно, скорость реакции снижается.
Кинетические зависимости
Конкурентные ингибиторы уменьшают скорость химической реакции. Конкурентный ингибитор повышает Кm для данного субстрата (уменьшает сродство субстрата к ферменту). Это означает, что в присутствии конкурентного ингибитора необходима большая концентрация субстрата для достижения 1/2 Vmax.
Увеличение соотношения концентрации субстрата и ингибитора снижает степень ин-гибирования. При значительно более высоких концентрациях субстрата ингибирование полностью исчезает, потому что активные центры всех молекул фермента будут находиться преимущественно в комплексе с субстратом.
Лекарственные препараты как конкурентные ингибиторы
Многие лекарственные препараты оказывают своё терапевтическое действие по механизму
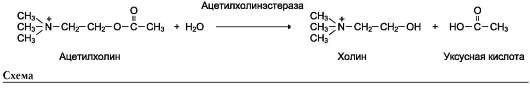
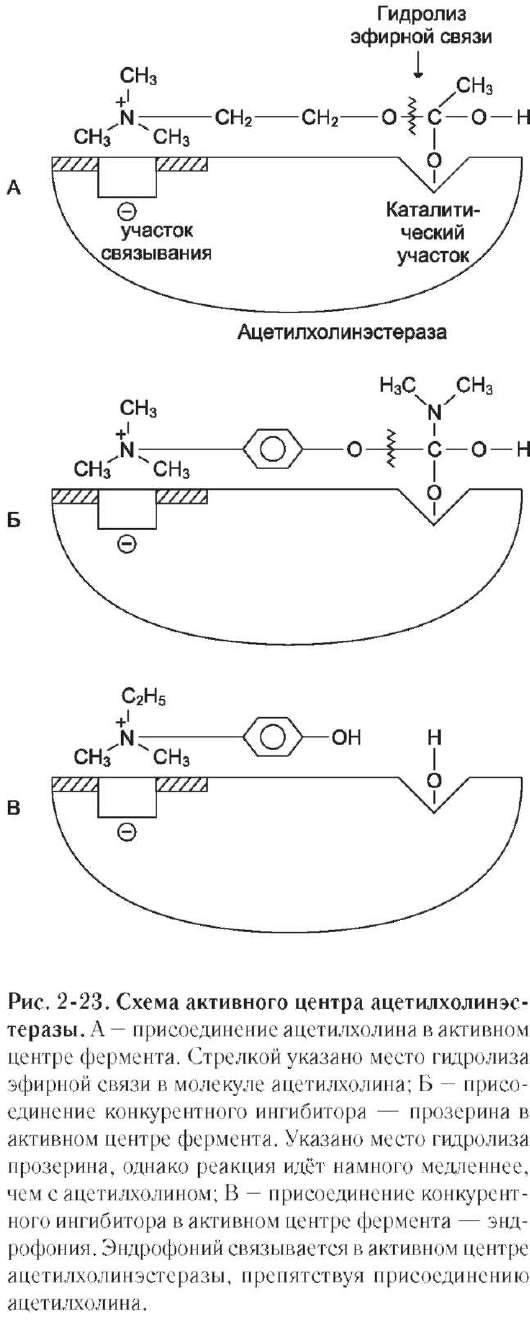
конкурентного ингибирования. Например, четвертичные аммониевые основания ингибируют ацетилхолинэстеразу, катализирующую реакцию гидролиза ацетилхолина на холин и уксусную кислоту (см. схему ниже)
При добавлении ингибиторов активность аце-тилхолинэстеразы уменьшается, концентрация
ацетилхолина (субстрата) увеличивается, что сопровождается усилением проведения нервного импульса. Ингибиторы холинэстеразы используют при лечении мышечных дистрофий. Эффективные антихолинэстеразные препараты - прозерин, эндрофоний и др. (рис. 2-23).
Антиметаболиты как лекарственные препараты
В качестве ингибиторов ферментов по конкурентному механизму в медицинской практике используют вещества, называемые антиметаболитами. Эти соединения, будучи структурными аналогами природных субстратов, вызывают конкурентное ингибирование ферментов, с одной стороны, и, с другой - могут использоваться этими же ферментами в качестве псевдосубстратов, что приводит к синтезу аномальных продуктов. Аномальные продукты не обладают функциональной активностью; в результате наблюдают снижение скорости определённых метаболических путей.
В качестве лекарственных препаратов используют следующие антиметаболиты: сульфаниламидные препараты (аналоги пара-аминобен-зойной кислоты), применяемые для лечения инфекционных заболеваний (см. раздел 9), аналоги нуклеотидов для лечения онкологических заболеваний (см. раздел 10).
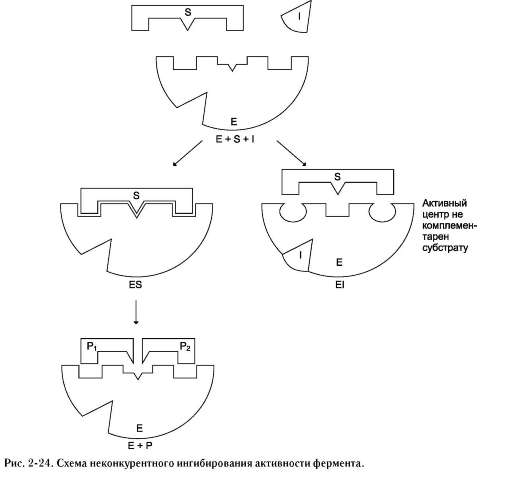
2. Неконкурентное ингибирование
Неконкурентным называют такое ингибиро-вание ферментативной реакции, при котором ингибитор взаимодействует с ферментом в участке, отличном от активного центра (рис. 224). Неконкурентные ингибиторы не являются структурными аналогами субстрата.
Неконкурентный ингибитор может связываться либо с ферментом, либо с фермент-субстратным комплексом, образуя неактивный комплекс. Присоединение неконкурентного ингибитора вызывает изменение конформации молекулы фермента таким образом, что нарушается взаимодействие субстрата с активным центром фермента, что приводит к снижению скорости ферментативной реакции.
Кинетические зависимости
Кинетическая зависимость неконкурентного ингибирования представлена на рис. 2-25. Этот тип ингибирования характеризуется снижением Vmax ферментативной реакции и уменьшением сродства субстрата к ферменту, т.е. увеличением Кm.
Б. НЕОБРАТИМОЕ ИНГИБИРОВАНИЕ
Необратимое ингибирование наблюдают в случае образования ковалентных стабильных
связей между молекулой ингибитора и фермента. Чаще всего модификации подвергается активный центр фермента. В результате фермент не может выполнять каталитическую функцию.
К необратимым ингибиторам относят ионы тяжёлых металлов, например ртути (Hg2+), серебра (Ag+) и мышьяка (As3+), которые в малых концентрациях блокируют сульфгидрильные группы активного центра. Субстрат при этом не может подвергаться химическому превращению (рис. 2-26). При наличии реактиваторов ферментативная функция восстанавливается. В больших концентрациях ионы тяжёлых металлов вызывают денатурацию белковой молекулы фермента, т.е. приводят к полной инактивации фермента.
1. Специфические и неспецифические ингибиторы
Использование необратимых ингибиторов представляет большой интерес для выяснения механизма действия ферментов. С этой целью применяют вещества, блокирующие определённые группы активного центра ферментов. Такие ингибиторы называют специфическими. Ряд соединений легко вступает в реакции с определёнными химическими группами. Если эти группы участвуют в катализе, то происходит полная инактивация фермента.
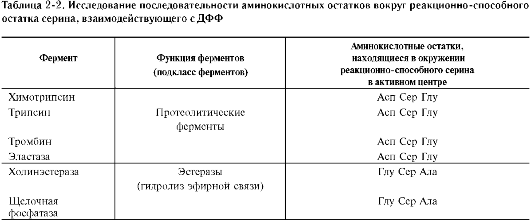
Роль гидроксильных групп серина в механизме катализа исследуют с помощью фтор-фосфатов, например диизопропилфторфосфата. Диизопропилфторфосфат (ДФФ) специфически реагирует лишь с одним из многих остатков серина в активном центре фермента. Остаток Сер, способный реагировать с ДФФ, имеет идентичное или очень сходное аминокислотное окружение (табл. 2-2). Высокая реакционная способность этого остатка по сравнению с другими остатками Сер обусловлена аминокислотными остатками, также входящими в активный центр ферментов.
ДФФ относят к специфическим необратимым ингибиторам «сериновых» ферментов, так как он образует ковалентную связь с гидроксиль-ной группой серина, находящегося в активном центре и играющего ключевую роль в процессе катализа (рис. 2-27).
Ацетат йода, п-хлормеркурибензоат легко вступают в реакции с SH-группами остатков
цистеина белков (рис. 2-28). Эти ингибиторы не относят к специфичным, так как они реагируют с любыми свободными SH-группами белков и называются неспецифическими ингибиторами. Если SH-группы принимают участие непосредственно в катализе, то с помощью этих ингибиторов представляется возможным выявление роли SH-групп фермента в катализе.
2. Необратимые ингибиторы ферментов как лекарственные препараты
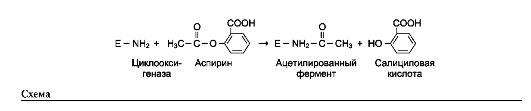
Пример лекарственного препарата, действие которого основано на необратимом ингиби-
ровании ферментов, - широко используемый препарат аспирин. Противовоспалительный нестероидный препарат аспирин обеспечивает фармакологическое действие за счёт ингибиро-вания фермента циклооксигеназы, катализирующего реакцию образования простагландинов из арахидоновой кислоты. В результате химической реакции ацетильный остаток аспирина присоединяется к свободной концевой NH2-группе одной из субъединиц циклооксигеназы (см. схему на след. стр.).
Это вызывает снижение образования продуктов реакции простагландинов (см. раздел 8), которые обладают широким спектром био-
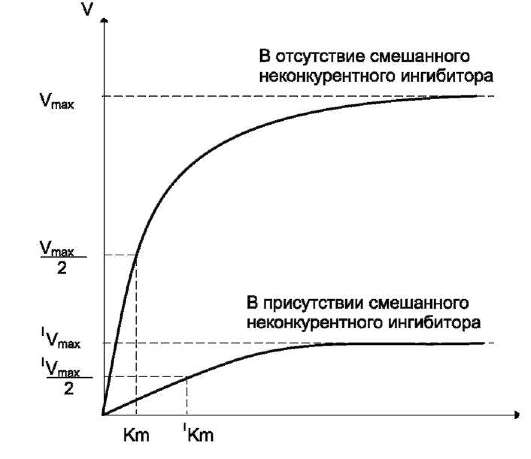
Рис. 2-25. Влияние неконкурентного ингибитора на скорость ферментативной реакции в зависимости от концентрации субстрата. Vmax - максимальная скорость реакции в отсутствие ингибитора; IVmax - максимальная скорость реакции в присутствии ингибитора; Кm - константа Михаэлиса в отсутствие ингибитора; IKm - константа Михаэлиса в присутствии ингибитора.
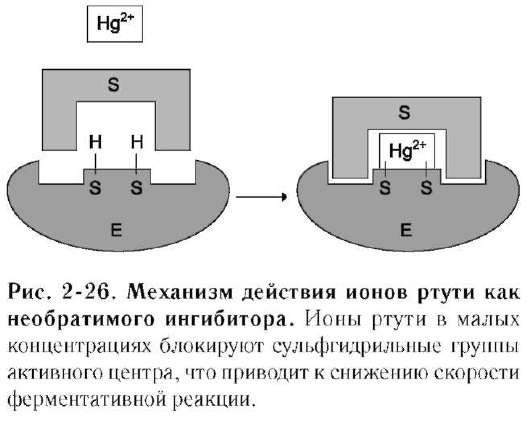
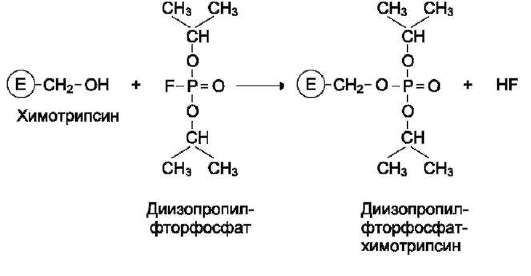
Рис. 2-27. Ингибирование активности химотрипсина с помощью диизопропилфторфосфата.
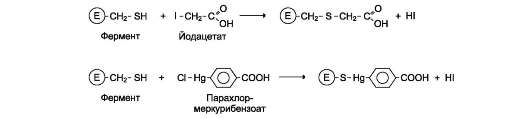
Рис. 2-28. Ингибирование активности ферментов вследствие ковалентной модификации остатков цистеина.
логических функций, в том числе являются медиаторами воспаления.
VII. РЕГУЛЯЦИЯ МЕТАБОЛИЧЕСКИХ ПРОЦЕССОВ
Живая клетка - открытая система, постоянно обменивающаяся с внешней средой веществами и энергией: в неё поступают питательные вещества, которые подвергаются превращениям и используются в качестве строительного и энергетического материала, из клетки выводятся конечные продукты метаболизма. В многоклеточном организме клетка реагирует не только на изменение окружающей среды, но и на функциональную активность соседних клеток. При этом она стремится сохранить неизменным свой внутренний состав. Это состояние называют стационарным или клеточным гомеостазом.
В клетке постоянно происходит большое количество разнообразных химических реакций, которые формируют метаболические пути - последовательное превращение одних соединений в другие. Метаболизм - совокупность всех метаболических путей, протекающих в клетках организма.
Среди всех метаболических путей, протекающих в организме, выделяют противоположно направленные процессы: катаболизм и анаболизм. Катаболизм - распад сложных веществ до простых с высвобождением энергии. Анаболизм - синтез из простых более сложных веществ. Ме-
таболические пути согласованы между собой по месту, времени и интенсивности протекания. Эта согласованность протекания всех процессов обеспечивается сложными и многообразными механизмами регуляции.
А. ОРГАНИЗАЦИЯ ХИМИЧЕСКИХ РЕАКЦИЙ В МЕТАБОЛИЧЕСКИЕ ПУТИ
Оптимальная активность ферментов, катализирующих реакции одного метаболического пути, достигается благодаря определённой пространственной организации в клетке.
1. Пространственная локализация ферментов
Большинство ферментов имеет внутриклеточную локализацию и распределены в организме неравномерно. Все ферменты одного метаболического пути, как правило, находятся в одном отделе клетки. Особенно разделение метаболических путей важно для противоположно направленных катаболических и анаболических процессов. Например, синтез жирных кислот происходит в цитоплазме, а их распад в митохондриях. Если бы такого разделения не существовало, образовывались бы бесполезные с функциональной и энергетической точки зрения пути.
В метаболических путях продукт первой ферментативной реакции служит субстратом второй и так далее до формирования конечного продукта. Промежуточные продукты метаболического пути могут высвобождаться из последо-
вательности реакций и использоваться в других метаболических путях, т.е. метаболические пути связаны между собой промежуточными продуктами.
В ряде случаев пространственная организация ферментов настолько сильно выражена, что продукт реакции ни при каких условиях не может быть вычленен из метаболического пути и обязательно служит субстратом следующей реакции. Такая организация метаболического пути носит название мультиферментного комплекса и возникает в результате структурно-функциональной организации ферментов. Обычно такие комплексы связаны с мембранами. В качестве примеров мультиферментных комплексов можно привести пируватдегидрогеназный комплекс, под действием которого происходит окислительное декарбоксилирование пировиноград-ной кислоты (пирувата) (см. раздел 6), синтазу
жирных кислот, катализирующую синтез пальмитиновой кислоты (см. раздел 8).
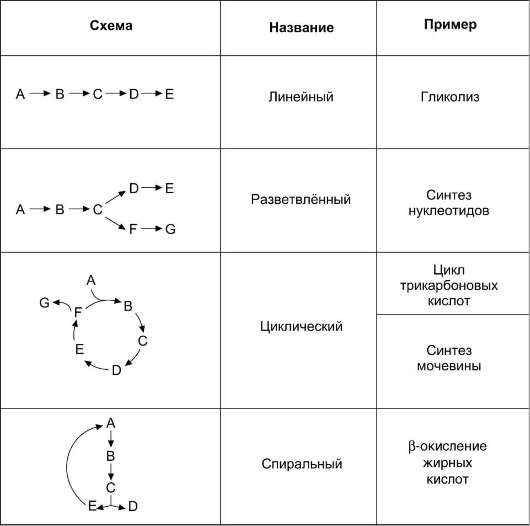
2. Структура метаболических путей
Структура метаболических путей в клетке крайне разнообразна (см. табл. 2-3). В случае, когда субстрат в результате ряда ферментативных процессов превращается в один продукт, такой путь носит название линейного метаболического пути. Часто встречаются разветвлённые метаболические пути, приводящие к синтезу различных конечных продуктов в зависимости от потребности клетки. В процессе изучения курса биологической химии вы также познакомитесь с циклическими и спиральными метаболическими путями.
Органоспецифичность
Ферментный состав различных клеток неодинаков. Ферменты, выполняющие функцию
Таблица 2-3. Типы метаболических путей
жизнеобеспечения клетки, находятся во всех клетках организма. В процессе дифференци-ровки клеток происходит изменение ферментного состава клеток. Так, фермент аргиназа, участвующий в синтезе мочевины, находится только в клетках печени, а кислая фосфатаза, участвующая в гидролизе моноэфиров ортофос-форной кислоты, - в клетках простаты. Это так называемые органоспецифичные ферменты.
Если говорить об узко специализированных клетках, то ферментов, выполняющих функции в этих клетках, находится больше, чем в других клетках. Например, в клетках сердечной мышцы имеется повышенное количество ферментов креатинкиназы и аспартатаминотрансферазы, в клетках печени - аланинаминотрансферазы и аспартатаминотрансферазы, в остеобластах - щелочной фосфатазы и т.д.
Компартментализация
Клетка - сложнофункциональная система, регулирующая своё жизнеобеспечение. Многообразие функций клетки обеспечивается пространственной и временной (в первую очередь, в зависимости от ритма питания) регуляцией определённых метаболических путей.
Пространственная регуляция связана со строгой локализацией определённых ферментов в различных органеллах. Так, в ядре находятся ферменты, связанные с синтезом молекул ДНК и РНК, в цитоплазме - ферменты гликолиза, в лизосомах - гидролитические ферменты, в матриксе митохондрий - ферменты ЦТК, во внутренней мембране митохондрий - ферменты цепи переноса электронов и т.д. (рис. 2-29). Такая субклеточная локализация ферментов способствует упорядоченности биохимических процессов и увеличивает скорость обмена веществ.
Б. ПРИНЦИПЫ РЕГУЛЯЦИИ МЕТАБОЛИЧЕСКИХ ПУТЕЙ
Все химические реакции в клетке протекают при участии ферментов. Поэтому, чтобы воздействовать на скорость протекания метаболического пути, достаточно регулировать количество или активность ферментов. Обычно в метаболических путях есть ключевые ферменты, благодаря которым происходит регуляция скорости всего пути. Эти ферменты (один или несколько в метаболическом пути) называются регуляторными ферментами; они катализируют,
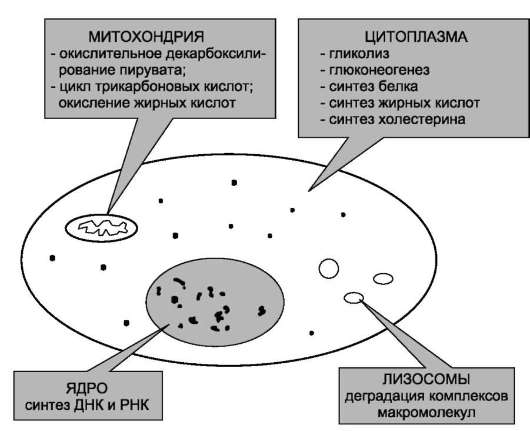
Рис. 2-29. Внутриклеточная локализация ферментов.
как правило, начальные реакции метаболического пути, необратимые реакции, скорость-ли-митирующие реакции (самые медленные) или реакции в месте переключения метаболического пути (точки ветвления).
Регуляция скорости ферментативных реакций осуществляется на 3 независимых уровнях:
• изменением количества молекул фермента;
• доступностью молекул субстрата и кофер-мента;
• изменением каталитической активности молекулы фермента.
1. Регуляция количества молекул фермента в клетке
Известно, что белки в клетке постоянно обновляются. Количество молекул фермента в клетке определяется соотношением 2 процессов - синтеза и распада белковой молекулы фермента:
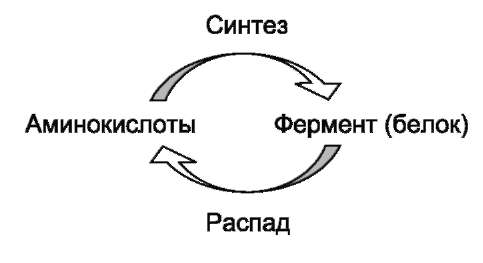
Синтез и фолдинг белка - многостадийный процесс. Регуляция синтеза белка может происходить на любой стадии формирования белковой молекулы. Наиболее изучен механизм регуляции синтеза белковой молекулы на уровне транскрипции, который осуществляется определёнными метаболитами, гормонами и рядом биологически активных молекул (см. раздел 4).
Что касается распада ферментов, то регуляция этого процесса менее изучена. Можно только предполагать, что это не просто процесс протеолиза (разрушения белковой молекулы), а сложный механизм, возможно, определяемый на генетическом уровне.
2. Регуляция скорости ферментативной реакции доступностью молекул субстрата и коферментов
Важный параметр, контролирующий протекание метаболического пути, - наличие суб-
стратов, и главным образом - наличие первого субстрата. Чем больше концентрация исходного субстрата, тем выше скорость метаболического пути.
Другой параметр, лимитирующий протекание метаболического пути, - наличие регенерированных коферментов. Например, в реакциях дегидрирования коферментом дегидрогеназ служат окисленные формы NAD+, FAD, FMN, которые восстанавливаются в ходе реакции. Чтобы коферменты вновь участвовали в реакции, необходима их регенерация, т.е. превращение в окисленную форму.
3. Регуляция каталитической активности ферментов
Важнейшее значение в изменении скорости метаболических путей играет регуляция каталитической активности одного или нескольких ключевых ферментов данного метаболического пути. Это высокоэффективный и быстрый способ регуляции метаболизма.
Основные способы регуляции каталитической активности ферментов:
• аллостерическая регуляция;
• регуляция с помощью белок-белковых взаимодействий;
• регуляция путём фосфорилирования/дефос-форилирования молекулы фермента;
• регуляция частичным (ограниченным) про-теолизом.
• Аллостерическая регуляция. Аллостерическими ферментами называют ферменты, активность которых регулируется не только количеством молекул субстрата, но и другими веществами, называемыми эффекторами. Участвующие в аллостерической регуляции эффекторы - клеточные метаболиты часто именно того пути, регуляцию которого они осуществляют. Роль аллостерических ферментов в метаболизме клетки. Аллостерические ферменты играют важную роль в метаболизме, так как они чрезвычайно быстро реагируют на малейшие изменения внутреннего состояния клетки. Аллостерическая регуляция имеет большое значение в следующих ситуациях:
- при анаболических процессах. Ингиби-рование конечным продуктом метаболи-
ческого пути и активация начальными метаболитами позволяют осуществлять регуляцию синтеза этих соединений;
- при катаболических процессах. В случае накопления АТФ в клетке происходит ин-гибирование метаболических путей, обеспечивающих синтез энергии. Субстраты при этом расходуются на реакции запасания резервных питательных веществ;
- для координации анаболических и ката-болических путей. АТФ и АДФ - аллос-терические эффекторы, действующие как антагонисты;
- для координации параллельно протекающих и взаимосвязанных метаболических путей (например, синтез пуриновых и пиримидиновых нуклеотидов, используемых для синтеза нуклеиновых кислот). Таким образом, конечные продукты одного метаболического пути могут быть аллостерическими эффекторами другого метаболического пути.
Аллостерические эффекторы. Эффектор, вызывающий снижение (ингибирование) активности фермента, называют отрицательным эффектором, или ингибитором. Эффектор, вызывающий повышение (активацию) активности ферментов, называют положительным эффектором, или активатором. Аллостерическими эффекторами часто служат различные метаболиты. Конечные продукты метаболического пути - часто ингибиторы аллостерических ферментов, а исходные вещества - активаторы. Это так называемая гетеротропная регуляция. Такой вид аллос-терической регуляции очень распространён в биологических системах. Более редкий случай аллостерической регуляции, когда сам субстрат может выступать в качестве положительного эффектора. Такая регуляция называется гомотроп-ной (эффектор и субстрат - одно и то же вещество). Эти ферменты имеют несколько центров связывания для субстрата, которые могут выполнять двойную функцию: каталитическую и регуляторную. Аллостерические ферменты такого типа используются в ситуации, когда субстрат накапливается в избытке и должен быстро преобразоваться в продукт. Выявить ферменты с аллостерической регуляцией можно, изучая кинетику этих
ферментов. Эти ферменты не подчиняются законам Михаэлиса-Ментен, они имеют характерную S-образную кривую зависимости скорости реакции от концентрации субстрата.
Особенности строения и функционирования аллос-терических ферментов:
- обычно это олигомерные белки, состоящие из нескольких протомеров или имеющие доменное строение;
- они имеют аллостерический центр, пространственно удалённый от каталитического активного центра;
- эффекторы присоединяются к ферменту нековалентно в аллостерических (регуля-торных) центрах;
- аллостерические центры, так же, как и каталитические, могут проявлять различную специфичность по отношению к лигандам: она может быть абсолютной и групповой. Некоторые ферменты имеют несколько аллостерических центров, одни из которых специфичны к активаторам, другие - к ингибиторам.
- протомер, на котором находится ал-лостерический центр, - регуляторный протомер, в отличие от каталитического протомера, содержащего активный центр, в котором проходит химическая реакция;
- аллостерические ферменты обладают свойством кооперативности: взаимодействие аллостерического эффектора с аллостери-ческим центром вызывает последовательное кооперативное изменение конформации всех субъединиц, приводящее к изменению конформации активного центра и изменению сродства фермента к субстрату, что снижает или увеличивает каталитическую активность фермента (рис. 2-30);
- регуляция аллостерических ферментов обратима: отсоединение эффектора от регуляторной субъединицы восстанавливает исходную каталитическую активность фермента;
- аллостерические ферменты катализируют ключевые реакции данного метаболического пути.
Локализация аллостерических ферментов в метаболическом пути. Скорость метаболических процессов зависит от концентрации веществ, использующихся и образующихся в данной
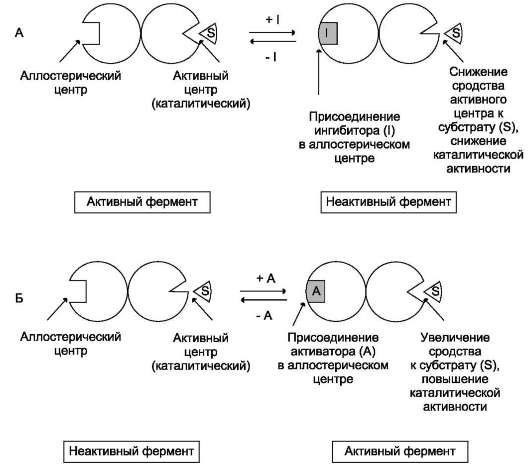
Рис. 2-30. Схема, поясняющая работу аллостерического фермента. А - действие отрицательного эффектора (ингибитора); Б - действие положительного эффектора (активатора).
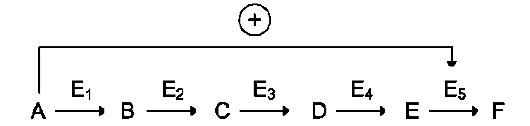
цепи реакций. Такая регуляция представляется логичной, так как при накоплении конечного продукта он (конечный продукт) может действовать как аллостерический ингибитор фермента, катализирующего чаще всего начальный этап данного метаболического пути:
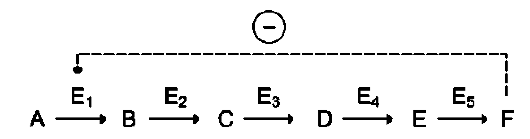
Фермент, катализирующий превращение субстрата А в продукт В, имеет аллостерический центр для отрицательного эффектора, которым служит конечный продукт метаболического пути F. Если концентрация F увеличивается (т.е. вещество F синтезируется быстрее, чем расходуется), ингибируется активность одного
из начальных ферментов. Такую регуляцию называют отрицательной обратной связью, или ретроингибированием. Отрицательная обратная связь - часто встречающийся механизм регуляции метаболизма в клетке. В центральных метаболических путях исходные вещества могут быть активаторами ключевых ферментов метаболического пути. Как правило, при этом аллостерической активации подвергаются ферменты, катализирующие ключевые реакции заключительных этапов метаболического пути:
В качестве примера можно рассмотреть принципы регуляции гликолиза - специфи-
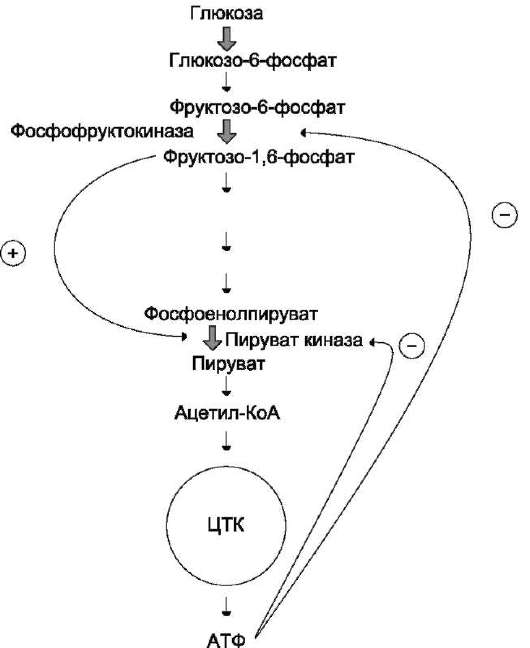
Рис. 2-31. Схема положительной и отрицательной регуляции катаболизма глюкозы. Молекула АТФ участвует в ретроингибировании аллостерических ферментов фосфо-фруктокиназы и пируваткиназы. Фруктозо-1,6-бисфосфат - активатор метаболического пути распада глюкозы. Плюсами отмечена активация, минусами - ингибирование ферментов - цикл трикарбоновых кислот.
ческого (начального) пути распада глюкозы (рис. 2-31). Один из конечных продуктов распада глюкозы - молекула АТФ. При избытке в клетке АТФ происходит ретро-ингибирование аллостерических ферментов
• фосфофруктокиназы и пируваткиназы. При образовании большого количества фруктозо-1,6-бисфосфата наблюдают аллостеричес-кую активацию фермента пируваткиназы. Благодаря такой регуляции осуществляется слаженность протекания метаболического пути распада глюкозы. Регуляция каталитической активности ферментов белок-белковыми взаимодействиями. Некоторые ферменты изменяют свою каталитическую активность в результате белок-белковых взаимодействий. Рассмотрим 2 механизма активации ферментов с помощью белок-белковых взаимодействий:
- активация ферментов в результате присоединения регуляторных белков;
- изменение каталитической активности ферментов вследствие ассоциации или диссоциации протомеров фермента.
Активация ферментов в результате присоединения регуляторных белков. Этот тип регуляции можно рассмотреть на примере активации фермента аденилатциклазы, локализованной в плазматической мембране клетки.
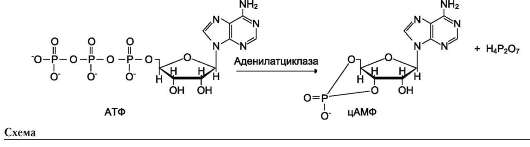
Активный центр аденилатциклазы локализован на цитоплазматической стороне плазматической мембраны. Активированная аденилат-циклаза катализирует реакцию образования из АТФ циклического 3',5'-АМФ (цАМФ) - вторичного, внутриклеточного посредника действия гормонов (см. схему ниже).
В мембране аденилатциклаза функционирует в комплексе с другими белками:
- рецептором гормона, выступающего во внеклеточную среду и взаимодействующего с гормонами;
- с G-белком, занимающим промежуточное положение между рецептором и ферментом аденилатциклазой. G-белок - олигомер-
ный белок, состоящий из 3 субъединиц - α, β, γ. α-Субъединица имеет центр связывания и расщепления ГТФ. Поэтому этот белок называется ГТФ-связывающим белком, или G-белком;
- в результате связывания гормона с рецептором происходит изменение конфор-мации G-белка, уменьшение его сродства к молекуле ГДФ, с которой он связан в отсутствие гормонального сигнала, и увеличение сродства к ГТФ. Присоединение ГТФ вызывает конформационные изменения в G-белке и диссоциацию его на субъединицы: субъединицу α, связанную с ГТФ (α-ГТФ), димер βγ;
- α-ГТФ имеет высокое сродство к адени-латциклазе, его присоединение приводит к активации последней, поэтому α-ГТФ - регуляторный белок, а данный механизм активации аденилатциклазы называют активацией ферментов в результате присоединения регуляторных белков (рис. 2-32).
Регуляция каталитической активности ферментов
ассоциацией/диссоциацией протомеров Протеинкиназы - группа ферментов, катализирующих перенос остатка фосфорной кислоты с АТФ на специфические ОН-группы аминокислотных остатков белков (вызывают фосфорилирование белков). Механизмы активации различных протеинкиназ неодинаковы. В качестве примера регуляции каталитической активности ферментов ассоциацией или диссоциацией протомеров можно привести регуляцию активности фермента протеинкиназы А. Протеинкиназа А (цАМФ-зависимая) состоит из 4 субъединиц 2 типов: 2 регуля-торных (R) и 2 каталитических (С). Такой тетрамер не обладает каталитической активностью. Регуляторные субъединицы имеют участки связывания для цик-ли-ческого 3',5'-АМФ (цАМФ), по 2 на каждую субъединицу. Присоединение 4 молекул цАМФ к 2 регуляторным субъединицам приводит к изменению кон-формации регуляторных протомеров и к диссоциации тетрамерного комплекса, при этом высвобождаются 2 активные каталитические субъединицы (рис. 2-32). Такой механизм регуляции обратим. Отщепление молекул цАМФ от регуляторных
субъединиц приведёт к ассоциации регуля-торных и каталитических субъединиц про-теинкиназы А с образованием неактивного комплекса.
• Регуляция каталитической активности ферментов путём фосфорилирования/дефосфорилирования
В биологических системах часто встречается механизм регуляции активности ферментов с помощью ковалентной модификации аминокислотных остатков. Быстрый и широко распространённый способ химической модификации ферментов - фосфорилирова-ние/дефосфорилирование. Модификации подвергаются ОН-группы фермента. Фос-форилирование осуществляется ферментами протеинкиназами, а дефосфорилирование - фосфопротеинфосфатазами. Присоединение остатка фосфорной кислоты приводит к изменению конформации активного центра и его каталитической активности. При этом результат может быть двояким: одни ферменты при фосфорилировании активируются, другие, напротив, становятся менее активными (рис. 2-33).
Изменение активности фермента, вызванное фосфорилированием, обратимо. Отщепление остатка фосфорной кислоты осуществляется ферментами фосфопротеинфосфатазами. Активность протеинкиназ и фосфопроте-инфосфатаз регулируется гормонами, что позволяет быстро изменять активность клю-чевых ферментов метаболических путей в зависимости от условий внешней среды. Антагонистичные по функции гормоны противоположным образом влияют на фосфорилирование/дефосфорилирование ферментов, вызывая противоположные эффекты изменения метаболизма клетки.
Например, под действием глюкагона (в период между приёмами пищи) в клетках происходит уменьшение синтеза энергетического материала - жира, гликогена и усиление его распада (мобилизация), вызванного фосфорилированием ключевых ферментов этих процессов. А под действием инсулина (во время пищеварения), наоборот, активируется синтез гликогена и ингибируется его распад, так как взаимодействие инсулина с рецептором активирует сигнальный путь, приводящий к дефосфорилированию тех же ключевых ферментов.
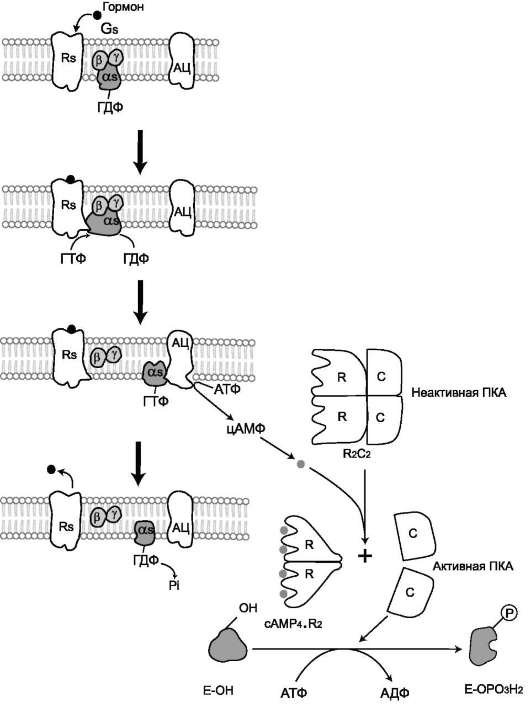
Рис. 2-32. Регуляция активности аденилатциклазы. Гормон (Г), взаимодействуя с рецептором (R) на поверхности клеток, приводит к уменьшению сродства ГТФ-связывающего белка (G-белка, состоящего из протомеров α, β, γ) к ГТФ и увеличению сродства к ГТФ. Присоединение молекулы ГТФ к активному центру G-белка вызывает диссоциацию комплекса на субъединицы α-ГТФ и димер βγ. Комплекс α-ГТФ активирует аденилатциклазу, что способствует синтезу из АТФ внутриклеточных регуляторных молекул цАМФ. АЦ - аденилатциклаза, ПКА - протеинкиназа А, P. - Н3РО4.
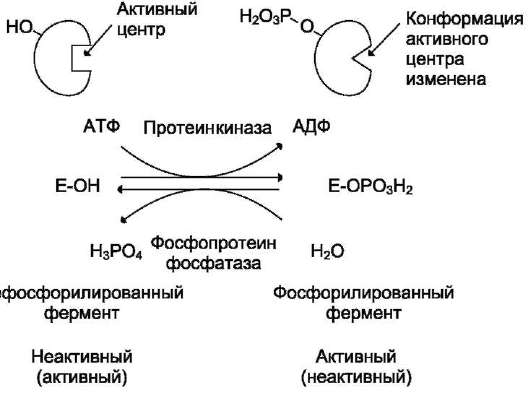
Рис. 2-33. Регуляция активности ферментов фосфорилированием/дефосфорилированием.
• Регуляция каталитической активности ферментов частичным (ограниченным) протеолизом
Некоторые ферменты, функционирующие вне клеток (в ЖКТ или в плазме крови), синтезируются в виде неактивных предшественников и активируются только в результате гидролиза одной или нескольких определённых петидных связей, что приводит к отщеплению части белковой молекулы предшественника. В результате в оставшейся части белковой молекулы происходит кон-формационная перестройка и формируется активный центр фермента.
Рассмотрим механизм частичного протеолиза на примере активации протеолитичес-кого фермента трипсина (рис. 2-34). Трип-синоген, синтезируемый в поджелудочной железе, при пищеварении по протокам поджелудочной железы поступает в двенадцатиперстную кишку, где и активируется путём частичного протеолиза под действием фермента кишечника энтеропептидазы. В результате отщепления гексапептида с N-конца формируется активный центр в оставшейся части молекулы. Следует напомнить, что трипсин относят к семейству «сериновых» протеаз - активный центр фермента содержит функционально важный остаток Сер.
Частичный протеолиз - пример регуляции, когда активность фермента изменяется необратимо. Такие ферменты функционируют, как правило, в течение короткого времени,
определяемого временем жизни белковой молекулы. Частичньгй протеолиз лежит в основе активации протеолитических ферментов, белков свёртывающей системы крови и фибринолиза, белков системы комплемента, а также пептидных гормонов.
VIII. ЭНЗИМОПАТИИ
В основе многих заболеваний лежат нарушения функционирования ферментов в клетке - энзимопатии. Различают первичные (наследственные) и вторичные (приобретённые) энзимопатии. Приобретённые энзимопатии, как и вообще протеинопатии, по-видимому, наблюдают при всех болезнях.
При первичных энзимопатиях дефектные ферменты наследуются, в основном, по аутосомно-рецессивному типу. Гетерозиготы, чаще всего, не имеют фенотипических отклонений. Первичные энзимопатии обычно относят к метаболическим болезням, так как происходит нарушение определённых метаболических путей. При этом развитие заболевания может протекать по одному из ниже перечисленных «сценариев». Рассмотрим условную схему метаболического пути:
Вещество А в результате последовательных ферментативных реакций превращается в про-

дукт Р. При наследственной недостаточности какого-либо фермента, например фермента Е3, возможны разные нарушения метаболических путей:
Нарушение образования конечных продуктов. Недостаток конечного продукта этого метаболического пути (Р) (при отсутствии альтернативных путей синтеза) может приводить к развитию клинических симптомов, характерных для данного заболевания:
можно рассмотреть альбинизм. При альбинизме нарушен синтез в меланоцитах пигментов - меланинов. Меланин находится в коже, волосах, радужке, пигментном эпителии сетчатки глаза и влияет на их окраску. При альбинизме наблюдают слабую пигментацию кожи, светлые волосы, красноватый цвет радужки глаза из-за просвечивающих капилляров. Проявление альбинизма связано с недостаточностью фермента тирозингидроксилазы (тирозиназы) - одного из ферментов, катализирующего метаболический путь образования меланинов (см. раздел 9). Накопление субстратов-предшественников. При недостаточности фермента Е3 будут накапли-
ваться вещество С, а также во многих случаях и предшествующие соединения. Увеличение субстратов-предшественников дефектного фермента - ведущее звено развития многих заболеваний:
алкаптонурия, при котором нарушено окисление гомогентизиновой кислоты в тканях (гомогентизиновая кислота - промежуточный метаболит катаболизма тирозина). У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты - диоксигеназы гомогентизиновой кислоты, приводящей к развитию заболевания. В результате увеличиваются концентрация го-могентизиновой кислоты и выведение её с мочой. В присутствии кислорода гомогенти-зиновая кислота превращается в соединение чёрного цвета - алкаптон. Поэтому моча таких больных на воздухе окрашивается в чёрный цвет. Алкаптон также образуется и в биологических жидкостях, оседая в тканях, коже, сухожилиях, суставах. При значительных отложениях алкаптона в суставах нарушается их подвижность.
Нарушение образования конечных продуктов и накопление субстратов предшественников. Отмечают заболевания, когда одновременно недостаток продукта и накопление исходного субстрата вызывают клинические проявления.
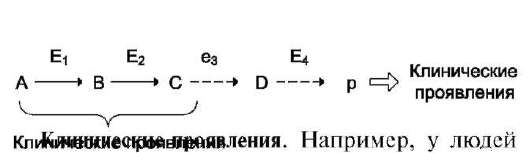
с болезнью Гирке (гликогеноз I типа) наблюдают снижение концентрации глюкозы в крови (гипогликемия) в перерывах между приёмами пищи. Это связано с нарушением распада гликогена в печени и выходом из неё глюкозы вследствие дефекта фермента глюкозо-6-фосфатфосфатазы (см. раздел 7). Одновременно у таких людей увеличиваются
размеры печени (гепатомегалия) вследствие накопления в ней не используемого гликогена.
IX. ПРИМЕНЕНИЕ ФЕРМЕНТОВ В МЕДИЦИНЕ
Ферментные препараты широко используют в медицине. Ферменты в медицинской практике находят применение в качестве диагностических (энзимодиагностика) и терапевтических (энзимотерапия) средств. В этом разделе мы остановимся на основных принципах энзимо-диагностики и энзимотерапии.
Кроме того, ферменты используют в качестве специфических реактивов для определения ряда веществ. Так, глюкозооксидазу применяют для количественного определения глюкозы в моче и крови. Фермент уреазу используют для определения содержания количества мочевины в крови и моче. С помощью различных дегидрогеназ обнаруживают соответствующие субстраты, например пируват, лактат, этиловый спирт и др.
A. ЭНЗИМОДИАГНОСТИКА
Энзимодиагностика заключается в постановке диагноза заболевания (или синдрома) на основе определения активности ферментов в биологических жидкостях человека. Принципы энзимодиагностики основаны на следующих позициях:
• при повреждении клеток в крови или других биологических жидкостях (например, в моче) увеличивается концентрация внутриклеточных ферментов повреждённых клеток;
• количество высвобождаемого фермента достаточно для его обнаружения;
• активность ферментов в биологических жидкостях, обнаруживаемых при повреждении клеток, стабильна в течение достаточно длительного времени и отличается от нормальных значений;
• ряд ферментов имеет преимущественную или абсолютную локализацию в определённых органах (органоспецифичность);
• существуют различия во внутриклеточной локализации ряда ферментов.
1. Причины, приводящие к увеличению количества ферментов в крови
Ферменты плазмы крови можно разделить на 2 группы. Первая, относительно небольшая группа ферментов активно секретируется в плазму крови определёнными органами. Например, печень синтезирует неактивные предшественники ферментов свёртывающей системы крови. Ко второй относят большую группу ферментов, высвобождающихся из клеток во время их нормального функционирования. Обычно эти ферменты выполняют свою функцию внутри клетки и не имеют физиологического значения в плазме крови. У здорового человека активность этих ферментов в плазме низкая и достаточно постоянная, так как постоянно соотношение скоростей высвобождения их из клеток и скоростей разрушения.
При многих заболеваниях происходит повреждение клеток, и их содержимое, в том числе и ферменты, высвобождаются в кровь. К причинам, вызывающим высвобождение внутриклеточного содержимого в кровь, относят нарушение проницаемости мембраны клеток (при воспалительных процессах) или нарушение целостности клеток (при некрозе). Определение в крови активности ряда ферментов хорошо налажено в биохимических лабораториях, что используют для диагностики заболеваний сердца, печени, скелетной мускулатуры и других тканей. Уровень активности ферментов в плазме коррелирует со степенью повреждения клеток.
Для энзимодиагностики имеют большое значение знания о субклеточной локализации ферментов. Так, появление в плазме крови ферментов, имеющих только цитозольную локализацию, свидетельствует о воспалительном процессе; при обнаружении митохондриальных или ядерных ферментов можно говорить о более глубоких повреждениях клетки, например о некрозе.
Однако повышение концентрации ферментов не всегда связано с повреждением тканей. При избыточной клеточной пролиферации, например при онкопролиферативных процессах, при повышенной скорости синтеза некоторых ферментов в клетках или при нарушенном клиренсе (способности выводиться почками) наблюдают повышение концентрации в крови определённых ферментов. Врачам следует учитывать, что нормальные значения активности ферментов в
крови детей и беременных женщин отличаются от показателей, характерных для взрослых здоровых людей.
2. Изоферменты
Ферменты, катализирующие одну и ту же химическую реакцию, но отличающиеся по первичной структуре белка, называют изофер-ментами, или изоэнзимами. Они катализируют один и тот же тип реакции с принципиально одинаковым механизмом, но отличаются друг от друга кинетическими параметрами, условиями активации, особенностями связи апофермента и кофермента.
Природа появления изоферментов разнообразна, но чаще всего обусловлена различиями в структуре генов, кодирующих эти изофермен-ты. Следовательно, изоферменты различаются по первичной структуре белковой молекулы и, соответственно, по физико-химическим свойствам. На различиях в физико-химических свойствах основаны методы определения изо-ферментов.
По своей структуре изоферменты в основном являются олигомерными белками. Причём та или иная ткань преимущественно синтезирует определённые виды протомеров. В результате определённой комбинации этих протомеров формируются ферменты с различной структурой - изомерные формы. Обнаружение определённых изоферментных форм ферментов позволяет использовать их для диагностики заболеваний.
Изоформы лактатдегидрогеназы. Фермент лак-татдегидрогеназа (ЛДГ) катализирует обратимую реакцию окисления лактата (молочной кислоты) до пирувата (пировиноградной кислоты) (см. раздел 7).
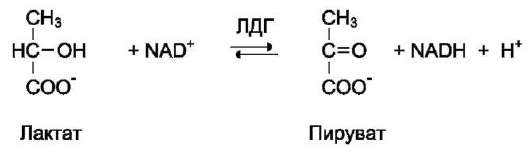
Лактатдегидрогеназа - олигомерный белок с молекулярной массой 134 000 Д, состоящий из 4 субъединиц 2 типов: М (от англ. muscle - мышца) и Н (от англ. heart - сердце). Комбинация этих субъединиц лежит в основе формирования 5 изоформ лактатдегидрогеназы (рис. 2-35, А). ЛДГ1 и ЛДГ2 наиболее активны в сердечной
мышце и почках, ЛДГ4 и ЛДГ5 - в скелетных мышцах и печени. В остальных тканях имеются различные формы этого фермента.
• Изоформы ЛДГ отличаются электрофо-ретической подвижностью, что позволяет устанавливать тканевую принадлежность изоформ ЛДГ (рис. 2-35, Б).
• Появление в эволюции различных изоформ ЛДГ обусловлено особенностями окислительного метаболизма тканей. Изоферменты
ЛДГ4 и ЛДГ5 (М-типы ЛДГ) работают эффективно в анаэробных условиях, ЛДГ1 и ЛДГ2 (Н-типы) - в аэробных, когда пируват быстро окисляется до СО2 и Н2О, а не восстанавливается до молочной кислоты.
• При ряде заболеваний исследуют активность ЛДГ в плазме крови. В норме активность ЛДГ составляет 170-520 ЕД/л. Повышение активности наблюдают при острых поражениях сердца, печени, почек, а также при мегалобластных и гемолитических анемиях. Однако это указывает на повреждение лишь одной из перечисленных тканей.
• Для постановки диагноза необходимо исследование изоформ ЛДГ в плазме крови методом электрофореза. На рис. 2-35, B представлены электрофореграммы плазмы крови здорового человека, больного инфарктом миокарда и больного гепатитом. Выявление в плазме крови тканеспецифи-ческих изоформ ЛДГ используют в качестве диагностического теста повреждения данной ткани.
Изоформы креатинкиназы. Креатинкиназа (КК) катализирует реакцию образования креатин-фосфата:
Молекула КК - димер, состоящий из субъединиц двух типов: М (от англ. muscle - мышца) и В (от англ. brain - мозг). Из этих субъединиц образуются 3 изофермента - ВВ, МВ, ММ. Изофермент ВВ находится преимущественно в головном мозге, ММ - в скелетных мышцах и
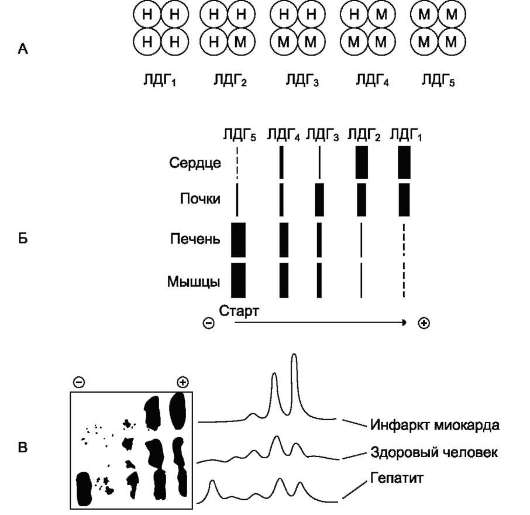
Рис. 2-35. Изоформы лактатдегидрогеназы. А - строение различных изоформ ЛДГ; Б - распределение на электрофореграмме и относительные количества изоформ ЛДГ в различных органах; В - содержание изоформ ЛДГ в плазме крови в норме и при патологии (электрофореграммы - слева и фотометрическое сканирование - справа).
МВ - в сердечной мышце. Изоформы КК имеют разную электрофоретическую подвижность (рис. 2-36).
Активность КК в норме не должна превышать 90 МЕ/л. Определение активности КК в плазме крови имеет диагностическое значение при инфаркте миокарда (происходит повышение уровня МВ-изоформы). Количество изоформы ММ может повышаться при травмах и повреждениях скелетных мышц. Изоформа ВВ не может проникнуть через гематоэнцефалический барьер, поэтому в крови практически не определяется даже при инсультах и диагностического значения не имеет.
3. Энзимодиагностика при инфаркте миокарда
Примерно 30% больных инфарктом миокарда имеют атипичную клиническую картину этого
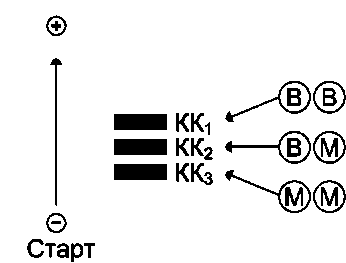
Рис. 2-36. Структура и электрофоретическая подвижность различных изоформ креатинкиназы.
заболевания. Поэтому необходимо проводить дополнительные методы исследования для подтверждения повреждения сердечной мышцы.
При инфаркте миокарда наблюдают достоверные изменения в крови активности ферментов КК, ЛДГ и аспартатаминотрансферазы - АСТ,
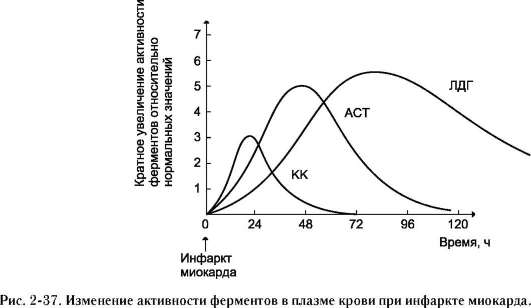
которые зависят от времени, прошедшего от начала развития инфаркта и от зоны тканевого повреждения. Типичную кривую изменения активности этих ферментов можно видеть на рис. 2-37. После закупорки (окклюзии) коронарного сосуда в крови вначале отмечают повышение активности КК изоформы МВ, однако фермент быстро удаляется из кровотока. Обнаружение повышенной активности КК в плазме крови - основной энзимодиагностический критерий инфаркта миокарда. Если у пациента с загру-динными болями не обнаружено изменения в активности КК, диагноз инфаркта миокарда маловероятен.
Дополнительным подтверждением диагноза инфаркта миокарда служит обнаружение активностей ферментов АСТ и ЛДГ в крови больных. Динамика изменений этих активностей также представлена на этом рисунке. Активность АСТ в норме составляет 5-40 МЕ/л. При инфаркте миокарда активность АСТ повышается через 4- 6 ч; максимум активности наблюдают в течение 2-3 дней. Уровень ЛДГ также увеличивается в плазме крови через несколько часов после закупорки кровеносного сосуда; максимум активности наблюдают на 3-4-й день, затем наступает постепенная нормализация активности. Уровень повышения активности ЛДГ коррелирует с размерами повреждения сердечной мышцы.
Б. ПРИМЕНЕНИЕ ФЕРМЕНТОВ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Использование ферментов в качестве терапевтических средств имеет много ограничений вследствие их высокой иммуногенности. Тем не менее энзимотерапию активно развивают в следующих направлениях:
• заместительная терапия - использование ферментов в случае их недостаточности;
• элементы комплексной терапии - применение ферментов в сочетании с другой терапией.
Заместительная энзимотерапия эффективна при желудочно-кишечных заболеваниях, связанных с недостаточностью секреции пищеварительных соков. Например, пепсин используют при ахилии, гипо- и анацидных гастритах. Дефицит панкреатических ферментов также в значительной степени может быть компенсирован приёмом внутрь препаратов, содержащих основные ферменты поджелудочной железы (фестал, энзистал, мезим-форте и др.).
В качестве дополнительных терапевтических средств ферменты используют при ряде заболеваний. Протеолитические ферменты (трипсин, химотрипсин) применяют при местном воздействии для обработки гнойных ран с целью расщепления белков погибших клеток, для удаления сгустков
крови или вязких секретов при воспалительных заболеваниях дыхательных путей. Ферментные препараты рибонуклеазу и дезоксирибонуклеазу используют в качестве противовирусных препаратов при лечении аденовирусных конъюнктивитов, герпетических кератитов.
Ферментные препараты стали широко применять при тромбозах и тромбоэмболиях. С этой целью используют препараты фибринолизина, стрептолиазы, стрептодеказы, урокиназы.
Фермент гиалуронидазу (лидазу), катализирующий расщепление гиалуроновой кислоты, используют подкожно и внутримышечно для рассасывания контрактур рубцов после ожогов и операций (гиалуроновая кислота образует сшивки в соединительной ткани) (см. раздел 8).
Ферментные препараты используют при онкологических заболеваниях. Аспарагиназа, катализирующая реакцию катаболизма ас-парагина, нашла применение для лечения лейкозов:
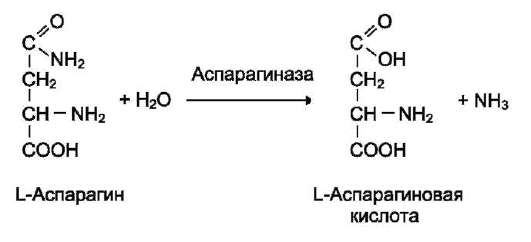
Предпосылкой антилейкемического действия аспарагиназы послужило обнаружение в лейкозных клетках дефектного фермента аспарагинсинтетазы, катализирующего реакцию синтеза аспарагина (см. схему ниже).
Лейкозные клетки не могут синтезировать аспарагин и получают его из плазмы крови. Если имеющийся в плазме аспарагин разрушать введением аспарагиназы, то в лейкоз-ных клетках наступит дефицит аспарагина и в результате - нарушение метаболизма клетки.