Общая неврология А.С. Никифоров, Е.И. Гусев - 2007г. - 720 с
|
|
|
|
ГЛАВА 7 МОЗЖЕЧОК И ПРИЗНАКИ ЕГО ПОРАЖЕНИЯ
7.1. СТРОЕНИЕ, СВЯЗИ И ФУНКЦИИ МОЗЖЕЧКА
Мозжечок (cerebellum) располагается под дубликатурой твердой мозговой оболочки, известной как намет мозжечка (tentorium cerebelli), который разделяет полость черепа на два неравных пространства - супратенториальное и субтенториальное. В субтенториальном пространстве, дном которого является задняя черепная ямка, помимо мозжечка, находится ствол мозга. Объем мозжечка составляет в среднем 162 см3. Масса его варьирует в пределах 136-169 г.
Мозжечок находится над мостом и продолговатым мозгом. Вместе с верхним и нижним мозговыми парусами он составляет крышу IV желудочка мозга, дном которого является так называемая ромбовидная ямка (см. главу 9). Над мозжечком находятся затылочные доли большого мозга, отделенные от него наметом мозжечка.
В мозжечке различают два полушария (hemispherum cerebelli). Между ними в сагиттальной плоскости над IV желудочком мозга располагается филогенетически наиболее древняя часть мозжечка - его червь (vermis cerebelli). Червь и полушария мозжечка фрагментируются на дольки глубокими поперечными бороздами.
Мозжечок состоит из серого и белого веществ. Серое вещество формирует кору мозжечка и находящиеся в его глубине парные ядра nuclei cerebelli (рис. 7.1). Самые крупные из них - зубчатые ядра (nucleus dentatus) - расположены в полушариях. В центральной части червя имеются ядра шатра (nuclei
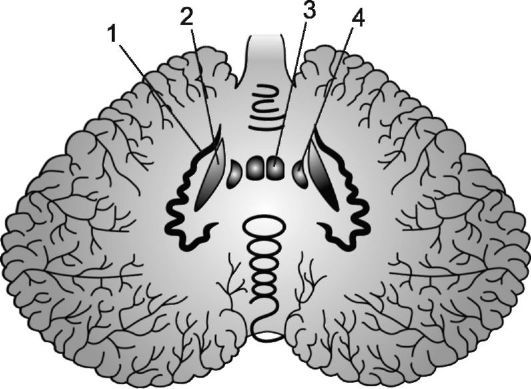
Рис. 7.1. Ядра мозжечка.
1 - зубчатое ядро; 2 - пробковидное ядро; 3 - ядро шатра; 4 - шаровидное ядро.
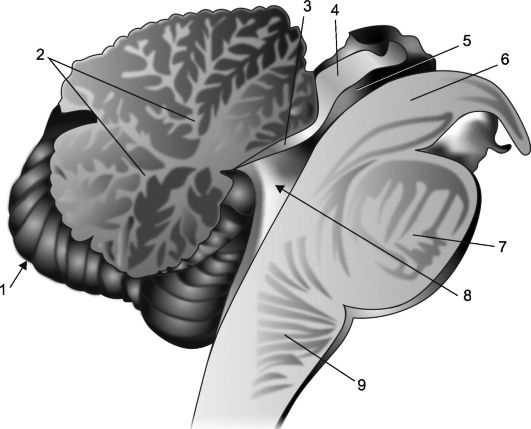
Рис. 7.2. Сагиттальный срез мозжечка и ствола мозга.
1 - мозжечок; 2 - «древо жизни»; 3 - передний мозговой парус; 4 - пластинка четверохолмия; 5 - водопровод мозга; 6 - ножка мозга; 7 - мост; 8 - IV желудочек, его сосудистое сплетение и шатер; 9 - продолговатый мозг.
fastigii), между ними и зубчатыми ядрами находятся шаровидные и пробковидные ядра (nuctei. globosus et emboliformis).
Ввиду того, что кора покрывает всю поверхность мозжечка и проникает в глубину его борозд, на сагиттальном разрезе мозжечка ткань его имеет рисунок листа, прожилки которого образованы белым веществом (рис. 7.2), составляющим так называемое древо жизни мозжечка (arbor vitae cerebelli). В основании древа жизни находится клиновидная выемка, являющаяся верхней частью полости IV желудочка; края этой выемки образуют его шатер. Крышей шатра служит червь мозжечка, а переднюю и заднюю его стенки составляют тонкие мозговые пластинки, известные под названием переднего и заднего мозговых парусов (vella medullare anterior et posterior).
Представляют интерес некоторые сведения об архитектонике мозжечка, дающие основания для суждения о функции его компонентов. У коры мозжечка есть два клеточных слоя: внутренний - зернистый, состоящий из мелких клеток-зерен, и наружный - молекулярный. Между ними расположен ряд крупных грушевидных клеток, носящих имя описавшего их чешского ученого И. Пуркинье (Purkinje I., 1787-1869).
В кору мозжечка импульсы поступают по проникающим в нее из белого вещества мшистым и ползучим волокнам, составляющим афферентные пути мозжечка. По мшистым волокнам импульсы, поступающие из спинного мозга,
вестибулярных ядер и ядер моста, передаются на клетки зернистого слоя коры. Аксоны этих клеток вместе с ползучими волокнами, проходящими через зернистый слой транзитом и несущими в мозжечок импульсы от нижних олив, доходят до поверхностного, молекулярного слоя мозжечка. Здесь аксоны клеток зернистого слоя и ползучие волокна Т-образно делятся, причем в молекулярном слое их разветвления принимают направление, продольное поверхности мозжечка. Импульсы, достигшие молекулярного слоя коры, пройдя через синаптические контакты, попадают на располагающиеся здесь же разветвления дендритов клеток Пуркинье. Далее они следуют по дендритам клеток Пуркинье к их телам, расположенным на границе молекулярного и зернистого слоев. Затем по аксонам тех же клеток, пересекающих зернистый слой, проникают в глубину белого вещества. Заканчиваются аксоны клеток Пуркинье в ядрах мозжечка. Главным образом в зубчатом ядре. Эфферентные импульсы, идущие от мозжечка по аксонам клеток, составляющих его ядра и принимающих участие в формировании мозжечковых ножек, покидают мозжечок.
Мозжечок имеет три пары ножек: нижнюю, среднюю и верхнюю. Нижняя ножка связывает его с продолговатым мозгом, средняя - с мостом, верхняя - со средним мозгом. Ножки мозга составляют проводящие пути, несущие импульсы к мозжечку и от него.
Червь мозжечка обеспечивает стабилизацию центра тяжести тела, его равновесие, устойчивость, регуляцию тонуса реципрокных мышечных групп, главным образом шеи и туловища, и возникновение при этом физиологических мозжечковых синергий, стабилизирующих равновесие тела.
Для успешного поддержания равновесия тела мозжечок постоянно получает информацию, проходящую по спиноцеребеллярным путям от проприоцепторов различных частей тела, а также от вестибулярных ядер, нижних олив, ретикулярной формации и других образований, участвующих в контроле за положением частей тела в пространстве. Большинство афферентных путей, идущих к мозжечку, проходит через нижнюю мозжечковую ножку, часть их расположена в верхней мозжечковой ножке.
Импульсы проприоцептивной чувствительности, идущие к мозжечку, как и другие чувствительные импульсы, следуя по дендритам первых чувствительных нейронов, достигают их тел, расположенных в спинномозговых узлах. В дальнейшем импульсы, идущие к мозжечку по аксонам тех же нейронов, направляются к телам вторых нейронов, которые располагаются во внутренних отделах основания задних рогов, формируя так называемые столбы Кларка. Аксоны их попадают в латеральные отделы боковых канатиков спинного мозга, где и образуют спиномозжечковые проводящие пути, при этом часть аксонов попадает в боковой столб той же стороны и формирует там задний спиномозжечковый путь Флексига (tractus spinocerebellaris posterior). Другая часть аксонов клеток задних рогов переходит на другую сторону спинного мозга и попадает в противоположный боковой канатик, образуя в нем передний спиномозжечковый путь Говерса (tractus spinocerebellaris anterior). Спиномозжечковые пути, увеличиваясь в объеме на уровне каждого спинального сегмента, поднимаются до продолговатого мозга.
В продолговатом мозге задний спиномозжечковый путь отклоняется в ла- теральном направлении и, пройдя через нижнюю мозжечковую ножку, проникает в мозжечок. Передний спиномозжечковый путь проходит транзитом через продолговатый мозг, мост мозга и достигает среднего мозга, на уровне которого совершает свой второй перекрест в переднем мозговом парусе и проходит в мозжечок через верхнюю мозжечковую ножку.
Таким образом, из двух спинномозговых путей один ни разу не подвергается перекресту (неперекрещенный путь Флексига), а другой переходит на противоположную сторону дважды (дважды перекрещенный путь Говерса). В результате оба проводят импульсы от каждой половины тела, преимущественно к гомолатеральной половине мозжечка.
Кроме спиномозжечковых путей Флексига, через нижнюю мозжечковую ножку импульсы к мозжечку проходят по вестибуломозжечковому пути (tractus vestibulocerebellaris), начинающемуся главным образом в верхнем вестибулярном ядре Бехтерева, и по оливомозжечковому пути (tractus olivocerebellaris), идущему от нижней оливы. Часть аксонов клеток тонкого и клиновидного ядер, не принимающих участие в формировании бульботаламического тракта, в виде наружных дугообразных волокон (fibre arcuatae externae) также попадает в мозжечок через нижнюю мозжечковую ножку.
Через свои средние ножки мозжечок получает импульсы из коры больших полушарий мозга. Эти импульсы проходят по корково-мостомозжечковым пу- тям, состоящим из двух нейронов. Тела первых нейронов располагаются в коре больших полушарий, главным образом в коре задних отделов лобных долей. Аксоны их проходят в составе лучистого венца, передней ножки внутренней капсулы и заканчиваются в ядрах моста. Аксоны клеток вторых нейронов, тела которых расположены в собственных ядрах моста, переходят на его противоположную сторону и составляют после перекреста среднюю мозжечковую ножку,
заканчивающуюся в противоположном полушарии мозжечка.
Часть импульсов, возникших в коре больших полушарий мозга, достигает противоположного полушария мозжечка, принося информацию не о произведенном, а лишь о намечаемом к выполнению активном движении. Получив такую информацию, мозжечок моментально высылает импульсы, корригирующие произвольные движения, главным образом, путем погашения инерции и наиболее рациональной регуляции тонуса реципрокных мышц - мышц-агонистов и антагонистов. В результате создается своеобразная эйметрия, делающая произвольные движения четкими, отточенными, лишенными нецелесообразных компонентов.
Пути, выходящие из мозжечка, состоят из аксонов клеток, тела которых формируют его ядра. Большинство эфферентных путей, в том числе пути, идущие от зубчатых ядер, покидают мозжечок через его верхнюю ножку. На уровне нижних бугров четверохолмия совершается перекрест эфферентных мозжечко- вых путей (перекрест верхних мозжечковых ножек Вернекинга). После перекреста каждый из них достигает красных ядер противоположной стороны среднего мозга. В красных ядрах мозжечковые импульсы переключаются на следующий нейрон и дальше движутся по аксонам клеток, тела которых заложены в красных ядрах. Эти аксоны формируются в красноядерно-спинномозговые проводящие пути (tracti rubro spinalis), пути Монакова, которые вскоре после выхода из красных ядер подвергаются перекресту (перекрест покрышки или перекрест Фореля), после чего спускаются в спинной мозг. В спинном мозге красноядерноспинномозговые пути располагаются в боковых канатиках; составляющие их волокна заканчиваются у клеток передних рогов спинного мозга.
Весь эфферентный путь от мозжечка до клеток передних рогов спинного мозга можно назвать мозжечково-красноядерно-спинномозговым (tractus cerebello-rubrospinalis). Он дважды совершает перекрест (перекрест верхних мозжечковых ножек и перекрест покрышки) и в итоге связывает каждое полушарие мозжечка с периферическими мотонейронами, находящимися в передних рогах гомолатеральной половины спинного мозга.
Из ядер червя мозжечка эфферентные пути идут в основном через нижнюю мозжечковую ножку к ретикулярной формации ствола мозга и вестибулярным ядрам. Отсюда по ретикулоспинномозговым и вестибулоспинномозговым путям, проходящим по передним канатикам спинного мозга, они также достигают клеток передних рогов. Часть импульсов, идущих от мозжечка, пройдя через вестибулярные ядра, попадает в медиальный продольный пучок, доходит до ядер III, IV и VI черепных нервов, обеспечивающих движения глазных яблок, и оказывает влияние на их функцию.
Подводя итог, необходимо подчеркнуть следующее:
1. Каждая половина мозжечка получает импульсы в основном а) из гомолатеральной половины тела, б) из противоположного полушария мозга, имеющего кортико-спинальные связи с той же половиной тела.
2. От каждой половины мозжечка эфферентные импульсы направляются к клеткам передних рогов гомолатеральной половины спинного мозга и к ядрам черепных нервов, обеспечивающих движения глазных яблок.
Такой характер мозжечковых связей позволяет понять, почему при поражении одной половины мозжечка мозжечковые расстройства возникают преимущественно в той же, т.е. гомолатеральной, половине тела. Это особенно отчетливо проявляется при поражении полушарий мозжечка.
7.2. ИССЛЕДОВАНИЕ ФУНКЦИЙ МОЗЖЕЧКА
И КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ЕГО ПОРАЖЕНИЯ
При поражении мозжечка характерны расстройства статики и координации движений, мышечная гипотония и нистагм.
Поражение мозжечка, прежде всего его червя, ведет к нарушениям статики - возможности поддержания стабильного положения центра тяжести тела человека, равновесия, устойчивости. При расстройстве указанной функции возникает статическая атаксия (от греч. ataxia - беспорядок, неустойчивость). Отмечается неустойчивость больного. Поэтому в положении стоя он широко расставляет ноги, балансирует руками. Особенно четко статическая атаксия выявляется при искусственном уменьшении площади опоры, в частности в позе Ромберга. Больному предлагается встать, плотно сдвинув ступни и слегка приподняв голову. При наличии мозжечковых расстройств отмечается неус- тойчивость больного в этой позе, тело его раскачивается, иногда его «тянет» в какую-то определенную сторону, при этом, если больного не поддержать, он может упасть. В случае поражения червя мозжечка больной обычно раскачивается из стороны в сторону и чаще падает назад. При патологии полушария мозжечка возникает тенденция к падению преимущественно в сторону патологического очага. Если расстройство статики выражено умеренно, его легче выявить в так называемой усложненной или сенсибилизированной позе Ромберга. Больному предлагается поставить ступни на одну линию, чтобы носок одной ступни упирался в пятку другой. Оценка устойчивости та же, что и в обычной позе Ромберга.
В норме, когда человек стоит, мышцы его ног напряжены (реакция опоры), при угрозе падения в сторону нога его на этой стороне перемещается в том же направлении, а другая нога отрывается от пола (реакция прыжка). При поражении мозжечка (главным образом червя) у больного нарушаются реакции
опоры и прыжка. Нарушение реакции опоры проявляется неустойчивостью больного в положении стоя, особенно в позе Ромберга. Нарушение реакции прыжка приводит к тому, что если врач, встав позади больного и подстраховывая его, толкает больного в ту или иную сторону, то больной падает при небольшом толчке (симптом толкания).
При поражении мозжечка походка больного обычно изменена в связи с развитием статолокомоторной атаксии. «Мозжечковая» походка во многом напоминает походку пьяного человека, поэтому ее иногда называют «походкой пьяного». Больной из-за неустойчивости идет неуверенно, широко расставляя ноги, при этом его «бросает» из стороны в сторону. А при поражении полушария мозжечка он отклоняется при ходьбе от заданного направления в сторону патологического очага. Особенно отчетлива неустойчивость при поворотах. Если атаксия оказывается резко выраженной, то больные полностью теряют способность владеть своим телом и не могут не только стоять и ходить, но даже сидеть.
Преимущественное поражение полушарий мозжечка ведет к расстройству его противоинерционных влияний, в частности к возникновению кинетической атаксии. Она проявляется неловкостью движений и особенно выражена при движениях, требующих точности. Для выявления кинетической атаксии проводятся пробы на координацию движений. Далее приводится описание некоторых из них.
Проба на диадохокинез (от греч. diadochos - последовательность). Больному предлагается закрыть глаза, вытянуть вперед руки и быстро, ритмично супинировать и пронировать кисти рук. В случае поражения полушария мозжечка движения кисти на стороне патологического процесса оказываются более размашистыми (следствие дисметрии, точнее - гиперметрии), в результате кисть начинает отставать. Это свидетельствует о наличии адиадохокинеза.
Пальценосовая проба. Больной с закрытыми глазами должен отвести руку, а затем, не торопясь, указательным пальцем дотронуться до кончика носа. В случае мозжечковой патологии рука на стороне патологического очага совершает избыточное по объему движение (гиперметрия), в результате чего больной промахивается. При пальценосовой пробе выявляется характерный для мозжечковой патологии мозжечковый (интенционный) тремор, амплитуда которого нарастает по мере приближения пальца к цели. Эта проба позволяет выявить и так называемую брадителекинезию (симптом узды): недалеко от цели движение пальца замедляется, иногда даже приостанавливается, а затем возобновляется вновь.
Пальце-пальцевая проба. Больному с закрытыми глазами предлагается широко развести руки и затем сближать указательные пальцы, стремясь попасть пальцем в палец, при этом, как и при пальценосовой пробе, выявляются интенционное дрожание и симптом узды.
Пяточно-коленная проба (рис. 7.3). Больному, лежащему на спине с закрытыми глазами, предлагают высоко поднять одну ногу и затем ее пяткой попасть в колено другой ноги. При мозжечковой патологии больной не может или ему трудно попасть пяткой в колено другой ноги, особенно выполняя пробу ногой, гомолатеральной пораженному полушарию мозжечка. Если все-таки пятка достигает колена, то предлагается провести ею, слегка касаясь передней поверхности голени, вниз, к голеностопному суставу, при этом в случае мозжечковой патологии пятка все время соскальзывает с голени то в одну, то в другую сторону.

Рис. 7.3. Пяточно-коленная проба.
Указательная проба: Больному предлагается несколько раз указательным пальцем попасть в резиновый наконечник молоточка, находящегося в руке обследующего. В случае мозжечковой патологии в руке пациента на стороне пораженного полушария мозжечка отмечается мимопопадание вследствие дисметрии.
Симптом Тома-Жюменти: Если пациент берет предмет, например стакан, он при этом чрезмерно раздвигает пальцы.
Мозжечковый нистагм. Подергивание глазных яблок при взгляде в стороны (горизонтальный нистагм) рассматривается как следствие интенционного дрожания глазных яблок (см. главу 30).
Расстройство речи: Речь теряет плавность, становится взрывчатой, фраг- ментированной, скандированной по типу мозжечковой дизартрии (см. главу 25).
Изменение почерка: В связи с расстройством координации движений руки почерк становится неровным, буквы деформированы, чрезмерно крупные (мегалография).
Пронаторный феномен: Больному предлагается удерживать вытянутые вперед руки в положении супинации, при этом на стороне пораженного полушария мозжечка вскоре происходит спонтанная пронация.
Симптом Гоффа-Шильдера: Если больной держит руки вытянутыми вперед, то на стороне пораженного полушария рука вскоре отводится кнаружи.
Имитационный феномен. Больной с закрытыми глазами должен быстро придать руке положение, аналогичное тому, которое обследующий перед этим придал другой его руке. При поражении полушария мозжечка гомолатеральная ему рука совершает движение, избыточное по амплитуде.
Феномен Дойникова. Пальцевой феномен. Сидящему пациенту предлагается супинированные кисти с разведенными пальцами положить на свои бедра и закрыть глаза. В случае поражения мозжечка на стороне патологического очага вскоре возникает спонтанное сгибание пальцев и пронация кисти и предплечья.
Симптом Стюарта-Холмса. Исследующий просит сидящего на стуле пациента сгибать супинированные предплечья и в то же время, взяв его руки за запястья, оказывает ему сопротивление. Если при этом неожиданно отпустить руки пациента, то рука на стороне поражения, сгибаясь по инерции, с силой ударит его в грудь.
Гипотония мышц. Поражение червя мозжечка ведет обычно к диффузной мышечной гипотонии. При поражении полушария мозжечка пассивные движения выявляют снижение мышечного тонуса на стороне патологического процесса. Гипотония мышц ведет к возможности переразгибания предплечья и голени (симптом Ольшанского) при пассивных движениях, к появлению симптомов «болтающейся» кисти или стопы при их пассивном встряхивании.
Патологические мозжечковые асинергии. Нарушения физиологических синергий при сложных двигательных актах выявляются, в частности, при следующих пробах (рис. 7.4).
1. Асинергия по Бабинскому в положении стоя. Если стоящий со сдвинутыми ногами пациент пытается прогнуться назад, запрокинув при этом голову, то в норме в таком случае происходит сгибание коленных суставов. При мозжечковой патологии в связи с асинергией это содружественное движение отсутствует, и больной, теряя равновесие, падает назад.
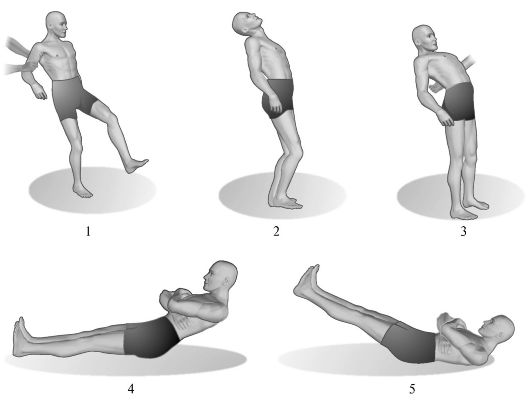
Рис. 7.4. Мозжечковая асинергия.
1 - походка больного с выраженной мозжечковой атаксией; 2 - наклон туловища назад в норме; 3 - при поражении мозжечка больной, наклоняясь назад, не может сохранить равновесия; 4 - выполнение пробы на мозжечковую асинергию по Бабинскому здоровым человеком; 5 - выполнение той же пробы больным с поражением мозжечка.
2. Асинергия по Бабинскому в положении лежа. Больному, лежащему на твердой плоскости с вытянутыми ногами, разведенными на ширину надплечий, предлагается скрестить руки на груди и затем сесть. При наличии мозжечковой патологии в связи с отсутствием содружественного сокращения ягодичных мышц (проявление асинергии) больной не может фиксировать на площади опоры ноги и таз, в результате ноги поднимаются и сесть ему не удается. Не следует переоценивать значимость этого симптома у пожилых пациентов, у людей с дряблой или ожиревшей брюшной стенкой.
Резюмируя изложенное, следует подчеркнуть многообразие и важность выполняемых мозжечком функций. Являясь частью комплексного регуляторного механизма с обратной связью, мозжечок выполняет роль координационного центра, обеспечивающего равновесие тела и поддержание мышечного тонуса. Как отмечает P. Duus (1995), мозжечок обеспечивает возможность выполнения дискретных и точных движений, при этом автор обоснованно считает, что мозжечок работает подобно компьютеру, отслеживая и координируя сенсорную информацию на входе и моделируя моторные сигналы на выходе.
7.3. МУЛЬТИСИСТЕМНЫЕ ДЕГЕНЕРАЦИИ
С ПРИЗНАКАМИ МОЗЖЕЧКОВОЙ ПАТОЛОГИИ
Мультисистемные дегенерации представляют собой группу нейродегенеративных заболеваний, общей особенностью которых является мультифокальный характер поражения с вовлечением в патологический процесс различных функциональных и нейромедиаторных систем мозга и в связи с этим полисистемность клинических проявлений.
7.3.1. Спиномозжечковые атаксии
К спиномозжечковым атаксиям относятся прогрессирующие наследственные дегенеративные заболевания, при которых в основном страдают структуры мозжечка, ствола головного мозга и проводящие пути спинного мозга, относящиеся главным образом к экстрапирамидной системе.
7.3.1.1. Наследственная атаксия Фридрейха
Наследственная болезнь, описанная в
Патогенез заболевания не уточнен. Отсутствует, в частности, представление о составляющем его основу первичном биохимическом дефекте.
Патоморфология. При патологоанатомических исследованиях выявляется выраженное истончение спинного мозга, обусловленное атрофическими процессами в его задних и боковых канатиках. Страдают, как правило, клиновидный (Бурдаха) и нежный (Голля) проводящие пути и спиномозжечковые пути Говерса и Флексига, а также перекрещенный пирамидный путь, содержащий
множество волокон, относящихся к экстрапирамидной системе. Дегенеративные процессы выражены также в мозжечке, в его белом веществе и ядерном аппарате.
Клинические проявления. Болезнь проявляется у детей или молодых людей в возрасте до 25 лет. С.Н. Давиденков (1880-1961) отмечал, что чаще клинические признаки болезни возникают у детей 6-10-летнего возраста. Первым признаком болезни обычно является атаксия. У больных возникают неуверенность, пошатывание при ходьбе, меняется походка (при ходьбе широко расставляют ноги). Походку при болезни Фридрейха можно назвать табетически- церебеллярной, так как ее изменения обусловлены сочетанием сенситивной и мозжечковой атаксии, а также обычно выраженным снижением мышечного тонуса. Характерны и расстройства статики, дискоординация в руках, интенционный тремор, дизартрия. Возможны нистагм, снижение слуха, элементы скандированности речи, признаки пирамидной недостаточности (сухожильная гиперрефлексия, стопные патологические рефлексы, иногда некоторое повышение мышечного тонуса), императивные позывы на мочеиспускание, снижение половой потенции. Иногда появляются гиперкинезы атетоидного характера.
Рано возникающее расстройство глубокой чувствительности ведет к прогрессирующему снижению сухожильных рефлексов: сначала на ногах, а затем на руках. Со временем формируется гипотрофия мышц дистальных отделов ног. Характерно наличие аномалий развития скелета. Прежде всего это проявляется наличием стопы Фридрейха: стопа укорочена, «полая», с очень высоким сводом. Основные фаланги ее пальцев разогнуты, остальные согнуты (рис. 7.5). Возможна деформация позвоночника, грудной клетки. Часто имеются проявления кардиопатии. Болезнь прогрессирует медленно, но неуклонно ведет к инвалидизации больных, которые со временем оказываются прикованными к постели.
Лечение. Патогенетическое лечение не раз- работано. Назначают препараты, улучшающие метаболизм в структурах нервной системы, общеукрепляющие средства. При выраженной деформации стоп показана ортопедическая обувь.
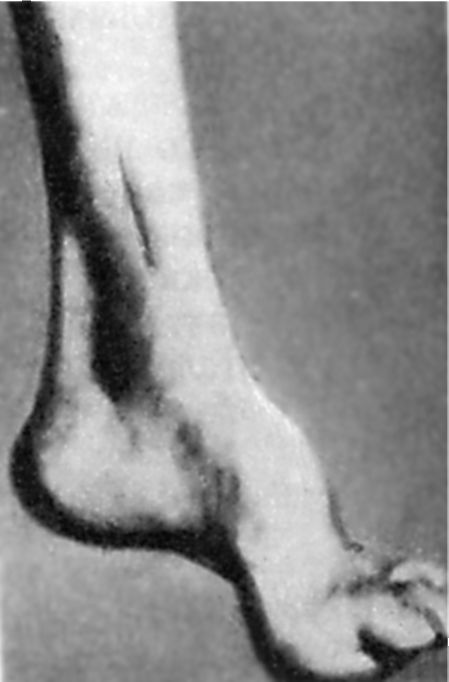
Рис. 7.5. Стопа Фридрейха.
7.3.1.2. Наследственная мозжечковая атаксия (болезнь Пьера Мари)
Это хроническое прогрессирующее наследственное заболевание, проявляющееся в возрасте 30-45 лет, с медленно нарастающими мозжечковыми расстройствами в сочетании с признаками пирамидной недостаточности, при этом характерны статическая и динамическая мозжечковая атаксия, интенционное дрожание, скандированная речь, сухожильная гиперрефлексия. Возможны клонусы, патологические пирамидные рефлексы, косоглазие, снижение зрения, сужение полей зрения в связи с первичной атрофией зрительных нервов и пигментной дегенерацией сетчатки. Течение болезни медленно прогрессирующее. Отмечаются уменьшение размеров мозжечка, дегенерация клеток
Пуркинье, нижних олив, спиномозжечковых путей. Наследуется по аутосомно-доминантному типу. Описал болезнь в
В настоящее время в понимании термина «болезнь Пьера Мари» нет единодушия, и вопрос о возможности выделения ее в самостоятельную нозологическую форму дискутабелен.
Лечение не разработано. Обычно применяются метаболически активные и общеукрепляющие, а также симптоматические средства.
7.3.2. Оливопонтоцеребеллярная дистрофия (болезнь Дежерина-Тома)
Это группа хронических прогрессирующих наследственных заболеваний, при которых развиваются дистрофические изменения главным образом в мозжечке, нижних оливах, в собственных ядрах моста и в связанных с ними структурах мозга.
При развитии заболевания в молодом возрасте около половины случаев наследуется по доминантному или рецессивному типу, остальные являются спорадическими. В спорадических случаях заболевания чаще встречаются проявления акинетико-ригидного синдрома и прогрессирующей вегетативной недостаточности. Средний возраст больного при проявлении в фенотипе наследственной формы заболевания - 28 лет, при спорадической - 49 лет, средняя продолжительность жизни - соответственно 14,9 и 6,3 года. При спорадической форме, кроме атрофии олив, моста и мозжечка, чаще обнару- живается поражение боковых канатиков спинного мозга, черного вещества и полосатого тела, голубоватого пятна в ромбовидной ямке IV желудочка мозга.
Характерны
симптомы нарастающего мозжечкового синдрома. Возможны расстройства
чувствительности, элементы бульбарного и акинетико-ригидного синдромов,
гиперкинезы, в частности миоритмии в язычке и мягком нёбе,
офтальмопарез, снижение остроты зрения, интеллектуальные расстройства.
Болезнь описали в
Заболевание чаще дебютирует нарушениями при ходьбе - неустойчивостью, дискоординацией, возможны неожиданные падения. Эти нарушения могут быть единственным проявлением заболевания в течение 1-2 лет. В дальнейшем возникают и нарастают координаторные расстройства в руках: затруднены манипуляции с мелкими предметами, нарушается почерк, возникает интенционный тремор. Речь становится прерывистой, смазанной, с носовым оттенком и не соответствующим построению речи ритмом дыхания (пациент говорит так, как будто его душат). В этой стадии заболевания присоединяются проявления прогрессирующей вегетативной недостаточности, появляются признаки акинетико-ригидного синдрома. Иногда доминирующими для больного симптомами становятся дисфагия, приступы ночного удушья. Они развиваются в связи со смешанным парезом бульбарной мускулатуры и могут представлять угрозу для жизни.
В
I тип (тип Менцеля). В
возрасте 14-70 (чаще 30-40) лет проявляется атак- сией, дизартрией,
дисфонией, гипотонией мышц, в поздней стадии - грубым тремором головы,
туловища, рук, мышц, признаками акинетико-ригидного синдрома. Возможны
патологические пирамидные знаки, парезы взора, наружная и внутренняя
офтальмоплегия, расстройства чувствительности, деменция. Наследуется по
аутосомно-доминантному типу. Как самостоятельную форму ее выделил в
II тип (тип Фиклера-Винклера). В возрасте 20-80 лет проявляется атаксией, снижением мышечного тонуса и сухожильных рефлексов. Наследуется по аутосомно-рецессивному типу. Возможны спорадические случаи.
III тип с ретинальной дегенерацией. Проявляется в детском или молодом (до 35 лет) возрасте атаксией, тремором головы и конечностей, дизартрией, признаками пирамидной недостаточности, прогрессирующим снижением зрения с исходом в слепоту; возможны нистагм, офтальмоплегия, иногда диссоциированные расстройства чувствительности. Наследуется по аутосомно-доминантному типу.
IV тип (тип Шута-Хаймакера). В возрасте 17-30 лет дебютирует мозжечковой атаксией или признаками нижнего спастического парапареза, в том и другом случае уже в ранней стадии болезни формируется сочетание этих проявлений, к которым в последующем присоединяются элементы бульбарного синдрома, пареза мимических мышц, расстройства глубокой чувствительности. Наследуется по доминантному типу.
V тип. Проявляется в возрасте 7-45 лет атаксией, дизартрией, признаками акинетико-ригидного синдрома и другими экстрапирамидными расстройствами, возможны прогрессирующие офтальмоплегия и деменция. Наследуется по доминантному типу.
7.3.3. Оливоруброцеребеллярная дегенерация (синдром Лежонна-Лермитта, болезнь Лермитта)
Заболевание
характеризуется прогрессирующей атрофией мозжечка, преимущественно его
коры, зубчатых ядер и верхних ножек мозжечка, нижних олив, красных
ядер. Проявляется прежде всего статической и динамической атаксией, в
дальнейшем возможны и другие признаки мозжечкового синдрома и поражения
ствола мозга. Описали болезнь французские невропатологи Ж. Лермитт
(Lhermitte J.J., 1877-1959) и Ж. Лежон (Lejonne J., род. в
7.3.4. Мультисистемная атрофия
В последние десятилетия в самостоятельную форму выделено спорадическое, прогрессирующее нейродегенеративное заболевание, названное мультисистемной атрофией. Оно характеризуется сочетанным поражением базальных ганглиев, мозжечка, ствола мозга, спинного мозга. Основные клинические проявления: паркинсонизм, мозжечковая атаксия, признаки пирамидной и вегетативной недостаточности (Левин О.С., 2002). В зависимости от преобладания тех или иных особенностей клинической картины выделяются три типа мультисистемной атрофии.
1) оливопонтоцеребеллярный тип, характеризующийся преобладанием признаков мозжечковой атаки;
2) стрионигральный тип, при котором доминируют признаки паркинсонизма;
3) синдром Шая-Дрейджера, характеризующийся преобладанием в клинической картине признаков прогрессирующей вегетативной недостаточности с явлениями ортостатической артериальной гипотонии.
В основе мультисистемной атрофии лежит избирательная дегенерация определенных участков преимущественно серого вещества мозга с поражением нейронов и глиальных элементов. Причины дегенеративных проявлений в ткани мозга и сегодня остаются неизвестными. Проявления мультисистемной атрофии по оливопонтоцеребеллярному типу связаны с поражением клеток Пуркинье в коре мозжечка, а также нейронов нижних олив, ядер моста мозга, демиелинизацией и дегенерацией главным образом понтоцеребеллярных про- водящих путей.
Мозжечковые расстройства обычно представлены статической и динамической атаксией с нарушением локомоторных движений. Характерны неустойчивость в позе Ромберга, атаксия при ходьбе, дисметрия, адиадохокинез, интенционный тремор, могут быть нистагм (горизонтальный вертикальный, бьющий вниз), прерывистость и замедленность следящих движений взора, нарушение конвергенции глаз, скандированная речь.
Мультисистемная атрофия обычно возникает в зрелом возрасте и быстро прогрессирует. Диагностика основывается на клинических данных и характеризуется сочетанием признаков паркинсонизма, мозжечковой недостаточности и вегетативными расстройствами. Лечение заболевания не разработано. Длительность заболевания - в пределах 10 лет, завершается летальными исходом.
7.4. ДРУГИЕ ЗАБОЛЕВАНИЯ, СОПРОВОЖДАЮЩИЕСЯ ПРИЗНАКАМИ ПОРАЖЕНИЯ МОЗЖЕЧКА
Если у пациента выявляются признаки поражения мозжечка, то в большинстве случаев прежде всего надо думать о возможности опухоли мозжечка (астроцитома, ангиобластома, медуллобластома, метастатические опухоли) или рассеянного склероза. При опухоли мозжечка рано проявляются признаки внутричерепной гипертензии. При рассеянном склерозе обычно удается выявить, кроме мозжечковой патологии, клинические проявления поражения и других структур ЦНС, прежде всего зрительной и пирамидной систем. В классической неврологии обычно упоминаются характерная для рассеянного склероза триада Шарко: нистагм, интенционное дрожание и скандированная речь, а также синдром Нонне: расстройство координации движений, дисметрия, скандированная речь и мозжечковые асинергии.
Мозжечковые нарушения являются основными и при посттравматическом синдроме Манна, для которого характерны атаксия, дискоординации, асинергии, нистагм. Травма или инфекционные поражения могут обусловить мозжечковый синдром Гольдштейна-Райхмана : расстройства статики и координации движений, асинергия, интенционное дрожание, снижение мышечного тонуса, гиперметрия, мегалография, нарушение восприятия массы (веса) предмета, находящегося в руках.
Расстройства функции мозжечка могут иметь и врожденный характер, проявляясь, в частности, синдромом Зеемана: атаксией, задержкой развития речи, а в последующем мозжечковой дизартрией.
Врожденная мозжечковая атаксия проявляется задержкой развития двигательных функций ребенка (в возрасте 6 мес он не может сидеть, поздно начинает ходить, при этом походка атактическая), а также задержкой речи, длительным сохранением дизартрии, иногда отставанием психического развития, нередки проявления микрокрании. На КТ уменьшены полушария мозжечка. Примерно к 10 годам обычно наступает компенсация мозговых функций, которая, однако, может нарушаться под влиянием вредных экзогенных воздействий. Возможны и прогредиентные формы заболевания.
Проявлением врожденной гипоплазии мозжечка является и синдром Фан- кони-Тернера. Он характеризуется нарушениями статики и координации движений, нистагмом, которые обычно сопровождаются и задержкой умственного развития.
К врожденным относится и наследуемая по аутосомно-рецессивному типу редко встречающаяся болезнь Беттена : Для нее характерна врожденная мозжечковая атаксия, проявляющаяся на первом году жизни нарушениями статики и координации движений, нистагмом, расстройством координации взора, умеренной мышечной гипотонией. Возможны диспластические признаки. Ребенок поздно, иногда только на 2-3 году жизни, начинает держать голову, еще позже - стоять, ходить, говорить. Речь его изменена по типу мозжечковой дизартрии. Возможны вегетативно-висцеральные расстройства, проявления иммунодепрессии. Через несколько лет клиническая картина обычно стабилизируется, больной в какой-то степени адаптируется к имеющимся дефектам.
Спастической атаксией по предложению A. Bell и E. Carmichel (1939) названа наследуемая по аутосомно-доминантному типу мозжечковая атаксия, которая характеризуется дебютом заболевания в 3-4-летнем возрасте и проявляется со- четанием мозжечковой атаксии с дизартрией, сухожильной гиперрефлексией и повышением мышечного тонуса по спастическому типу, при этом возможны (но не являются облигатными признаками болезни) атрофия зрительных нервов, дегенерация сетчатки, нистагм, глазодвигательные расстройства.
По аутосомно-доминантному типу наследуется синдром Фельдмана (описал немецкий врач H. Feldmann, род. в
Поздняя мозжечковая атрофия, или синдром Тома, описанный в
Сочетанием мозжечковых расстройств с миоклонией характеризуется миоклоническая мозжечковая диссинергия Ханта, или миоклонус-атаксия, при этом симптомокомплексе в клинической картине проявляются интенционный тремор, миоклонии, возникающие в руках, а в дальнейшем приобретающие генерализованный характер, атаксия и диссинергия, нистагм, скандированная речь, снижение мышечного тонуса. Является следствием дегенерации ядер мозжечка, красных ядер и их связей, а также корково-подкорковых структур.
В далеко
зашедшей стадии болезни возможны эпилептические припадки и деменция.
Прогноз плохой. Относится к редким формам прогрессирующих
наследственных атаксий. Наследуется по аутосомно-рецессивному типу.
Проявляется обычно в молодом возрасте. Нозологическая самостоятельность
симптомокомплекса оспаривается. Описал болезнь в
Среди дегенеративных процессов определенное место занимает мозжечковая дегенерация Холмса, или семейная церебеллооливарная атрофия, или
прогрессирующая атрофия системы мозжечка, преимущественно зубчатых
ядер, а также красных ядер, при этом в верхней ножке мозжечка выражены
проявления демиелинизации. Характерны статическая и динамическая
атаксия, асинергии, нистагм, дизартрия, снижение мышечного тонуса,
мышечная дистония, тремор головы, миоклонии. Почти одновременно
появляются эпилептические припадки. Интеллект обычно сохранен. На ЭЭГ
отмечается пароксимальная дизритмия. Заболевание признается
наследственным, но тип его наследования не уточнен. Описал болезнь в
(1876-1965).
Алкогольная мозжечковая дегенерация - следствие хронической алкогольной интоксикации. Происходит поражение преимущественно червя мозжечка, при этом прежде всего проявляются мозжечковая атаксия и нарушение координации движений ног, тогда как движения рук, глазодвигательные и речевые функции оказываются нарушенными в значительно меньшей степени. Обычно это заболевание сопровождается выраженным снижением памяти в сочетании с полиневропатией.
Паранеопластическая мозжечковая дегенерация проявляется мозжечковой атаксией, которая иногда может быть единственным клиническим симптомом, обусловленным злокачественной опухолью, без локальных признаков, указывающих на место ее возникновения. Паранеопластическая мозжечковая дегенерация может быть, в частности, вторичным проявлением рака грудной железы или яичников.
Синдром Барракера-Бордаса-Руиса-Лара проявляется
мозжечковыми расстройствами, возникающими в связи с быстро
прогрессирующей атрофией мозжечка. Описан синдром у больных раком
бронхов, сопровождающимся общей интоксикацией, современным испанским
врачом L. Barraquer-Bordas (род. в
Редко встречается рецессивная Х-хромосомная атаксия - наследственная болезнь, проявляющаяся практически только у мужчин медленно прогрессирующей мозжечковой недостаточностью. Передается по рецессивному, сцепленному с полом типу.
Заслуживает внимания и семейная пароксизмальная атаксия, или периодическая атаксия. Дебютирует чаще в детском возрасте, но может проявляться и позже - до 60 лет. Клиническая картина сводится к приступообразным проявлениям нистагма, дизартрии и атаксии, снижению мышечного тонуса, головокружению, тошноте, рвоте, головной боли, длительностью от нескольких минут до 4 нед.
Приступы семейной пароксизмальной атаксии могут быть спровоцированы эмоциональным стрессом, физическим переутомлением, лихорадочным состоянием, приемом алкоголя, при этом между приступами очаговая неврологическая симптоматика в большинстве случаев не выявляется, но иногда возможны нистагм и легкие мозжечковые симптомы.
Морфологическим
субстратом болезни признается атрофический процесс преимущественно в
передней части червя мозжечка. Впервые описал заболевание в
Острая мозжечковая атаксия, или синдром Лейдена-Вестфаля, представляет собой хорошо очерченный симптомокомплекс, являющийся параинфекционным осложнением. Возникает чаще у детей через 1-2 нед после перенесенной общей инфекции (грипп, сыпной тиф, сальмонеллез и др.). Характерны грубая статическая и динамическая атаксия, интенционное дрожание, гиперметрия, асинергии, нистагм, скандированная речь, снижение мышечного тонуса. В цереброспинальной жидкости выявляется лимфоцитарный плеоцитоз, умеренное повышение белка. В начале заболевания возможны головокружение, расстройства сознания, судороги. На КТ и МРТ патологии не выявляется. Течение доброкачественное. В большинстве случаев через несколько недель или месяцев - полное выздоровление, иногда - резидуальные расстройства в виде легкой мозжечковой недостаточности.
Болезнь Мари-Фуа-Алажуанина -
поздняя симметричная корковая атрофия мозжечка с преимущественным
поражением грушевидных нейронов (клетки Пуркинье) и зернистого слоя
коры, а также орального отдела червя мозжечка и дегенерацией олив.
Проявляется у лиц 40-75 лет расстройством равновесия, атаксией,
нарушением походки, координаторными расстройствами и снижением
мышечного тонуса, главным образом в ногах; интенционное дрожание в
руках при этом выражено незначительно. Нарушения речи возможны, но не
относятся к облигатным признакам заболевания. Болезнь описали в