Биология: учебник: в 2 т. / под ред. В. Н. Ярыгина. - 2011. - Т. 1. - 736 с. : ил.
|
|
|
|
Глава 5. МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ И КЛЕТОЧНЫЕ МЕХАНИЗМЫ ОБЕСПЕЧЕНИЯ СВОЙСТВ НАСЛЕДСТВЕННОСТИ И ИЗМЕНЧИВОСТИ У ЛЮДЕЙ КАК ПРОЯВЛЕНИЕ БИОЛОГИЧЕСКОГО НАСЛЕДСТВА ЧЕЛОВЕКА. ВВЕДЕНИЕ В ГЕНЕТИКУ ЧЕЛОВЕКА
Человек как отдельный вид представляет собой продукт биологической эволюции. Возникновение вида Homo sapiens, его расселение по планете и длительное (люди современного типа появились порядка 40 тыс. лет назад; архаичные формы человека, к которым некоторые антропологи причисляют неандертальцев - Homo neandertalensis, возникли порядка 100-300 тыс. лет назад или раньше) существование на Земле обусловлены в том числе наличием таких универсальных свойств жизни, как наследственность и биологическая изменчивость.
Важнейшее доказательство эволюционного родства человека с другими организмами из мира земной жизни - идентичность генетического материала (ДНК), принципов его надмолекулярной (двойная спираль) и морфологической (хромосомы) организации, способов записи биоинформации (биологический код - нуклеотидный и аминокислотный) и ее использования (матричный синтез: репликация ДНК, транскрипция информационных или матричных РНК, трансляция полипептидов). Имея в виду высокую степень аргументированности гипотезы о происхождении человека и современных человекообразных обезьян от общего предка, не вызывает удивления большое (на 99%) сходство, наблюдаемое в спектре образуемых белков, а также в нуклеотидных последовательностях ДНК у человека и, в частности, у шимпанзе.
Кариотипы людей и шимпанзе различаются по числу пар хромосом - 23 и 24 соответственно. Вместе с тем характер исчерченности хромосом при их избирательном окрашивании у названных видов прак-
тически идентичен. Анализ результатов такого окрашивания дает повод к заключению, что крупная хромосома 2 человека - это слившиеся две мелкие хромосомы (12 и 13) шимпанзе.
Гены человека мутируют с частотой, соизмеримой с таковой у других организмов от прокариот до высших позвоночных животных - в среднем от 10-5 до 10-7 мутаций на 1 локус (сайт ДНК) или гамету за поколение (но: см. п. 4.3.1.3).
Соматические клетки, строящие тело и органы людей, по содержанию ДНК и по количеству наборов хромосом диплоидны, тогда как гаметы гаплоидны. Диплоидность соматических клеток в ряду поколений и гаплоидность гамет обеспечиваются тем, что у всех животных в основе образования новых клеток, исключая половые, лежит процесс митоза, а в гаметогенезе задействован процесс мейоза с включенными в него кроссинговером и рекомбинацией генетического материала в профазе и независимым расхождением негомологичных хромосом в анафазе первого деления. Напомним, что восстановление диплоидного количества ДНК (2с) и числа хромосом (2n) в клетках в индивидуальном развитии и у человека, и у представителей всех других видов животных, размножающихся половым путем, происходит в момент и вследствие оплодотворения гаплоидной (с; n) яйцеклетки гаплоидным (с; n) сперматозоидом. Напомним, что закономерная смена диплоидной и гаплоидной фаз жизненного цикла организмов, которым свойственно половое размножение, в сочетании со свойством аллельного состояния генов и правилом чистоты гамет раскрывает природу механизмов независимого наследования признаков, открытого Г. Менделем.
Таким образом, молекулярно-генетические и клеточные основы явлений наследственности и изменчивости у человека и представителей других видов животных полностью совпадают.
Следует, однако, помнить, что в силу социальности человек создает в процессе своей деятельности новую среду жизни (биосфера трансформируется в социотехносферу, ноосферу). С одной стороны, составляющие этой среды (отапливаемые и/или кондиционируемые жилища и производственные помещения, одежда и т.д.) избавляют организм человека от прямого действия на него факторов природной среды, среди которых немалое число относится к категории экстремальных, вредных для здоровья. С другой стороны, далеко не всегда параметры этой среды оптимальны для существования людей. В частности, растет концентрация и расширяется спектр мутагенов, что не может не сказаться на интенсивности мутационного процесса, происходящего в генетическом
материале (ДНК) как человека, так и организмов других видов. Специального внимания заслуживает факт снижения давления на гено(аллело) фонды популяций людей естественного отбора, активными факторами которого на протяжении большей части истории человечества служили особо опасные инфекционные и паразитарные болезни, туберкулез. Это приводит к сохранению в названных гено(аллело)фондах мутантных аллелей с неблагоприятным фенотипическим эффектом, что благодаря формированию так называемого генетического груза снижает приспособительный потенциал популяций и жизнеспособность отдельных индивидуумов. Вытекающая из этих обстоятельств опасность ухудшения здоровья и качества жизни осознается людьми, которые пытаются найти эффективные меры предупреждения неблагоприятных последствий.
5.1. наследственность и биологическая изменчивость у человека
Благодаря значительному объему (3,2 млрд п.н.) человеческого генома и сниженному в силу социальности людей давлению естественного отбора, утратившего относительно популяций людей функцию видообразования, в гено(аллело)фонде человечества за тысячелетия его существования вследствие постоянно происходящего мутационного процесса накоплено большое количество разнообразных (мутантных) аллелей многих генов. Это стало причиной формирования у людей разных вариантов фенотипических признаков и свойств на структурном, функциональном, биохимическом (метаболическом) и поведенческом уровне. Действительно, основу индивидуальных различий по многим белкам составляют изменения соответствующих генов. Анализ аминокислотного состава вариантов белков человеческого организма, интенсивности их синтеза и функциональной активности дает ценные сведения об организации и экспрессии (реализации биоинформации) генетического материала людей.
Наглядным примером действия универсальных молекулярно-гене-тических и клеточных механизмов наследственности и изменчивости у человека может служить гемоглобин - специфический белок эритроцитов, играющий ключевую роль в транспорте О2 и легко выделяемый для проведения исследований. Масштабы изменчивости гемоглобина поражают. На настоящее время известно около 400 вариантов белка. Одни из этих вариантов закономерно образуются в организме на определенных стадиях онтогенеза, являясь необходимой предпосылкой здоровья, тогда
как другие связаны с развитием определенных патологических состояний. При этом в ряде случаев известен тот позитивный момент, который «оплачивается» неблагоприятными последствиями сохранения «проблемных» аллелей в гено(аллело)фондах некоторых популяций людей (серповид-ноклеточная анемия и тропическая малярия, см. здесь же, ниже).
Функционально полноценный гемоглобин представляет собой гете-робелковый комплекс - тетрамер. У взрослого человека он представлен четырьмя полипептидными глобиновымим молекулами (двумя α и двумя β), соединенными с железосодержащим элементом гемом. Оба полипептида существуют в организме в виде нескольких вариантов, образование которых контролируют разные, но близкие нуклеотидные последовательности ДНК. Так, полипептиды α- (141 аминокислотный остаток) и β- (146 аминокислотных остатков) глобинов различаются по десяти аминокислотным остаткам. Гены, контролирующие синтез обоих полипептидов, характеризуются кластерной организацией. Кластер α-глобиновых генов располагается на коротком плече хромосомы 16, а β-глобиновых - на коротком плече хромосомы 11. Два варианта полипептида β фетального гемоглобина (гемоглобин плода, HbF) yG и γΑ различаются одним аминокислотным остатком - в 136-м положении находится либо глицин, либо аланин. Различные члены кластеров α- и β-генов за исключением так называемых псевдогенов (нуклеотидные последовательности, которые вследствие накопления в них точечных мутаций, не подхваченных естественным отбором, утратили биоинформационную функцию и не экспрессируются; псевдогены имеются в обоих кластерах), будучи необходимыми для нормального онтогенеза и жизнедеятельности, транскрибируются и транслируются в клетках различного типа, находящихся в разных органах (стенка желточного мешка, печень, красный костный мозг), в разные периоды индивидуального развития, образуя гемоглобины эмбриона, плода и родившегося человека (см. также п. 4.3.3.2).
Из многочисленных мутаций генов глобинов большинство редки (обычно не более 1% мутантных аллелей в аллелофонде популяции) и лишь немногие из них встречаются, особенно в некоторых человеческих популяциях, относительно часто, например HbS (Средиземноморье и жаркий влажный пояс Африки, доля гетерозигот HbA/HbS - 10-30%), HbC (Западная Африка, порядка 10% гетерозигот HbA/HbC), HbD (северо-запад полуострова Индостан, до 5% гетерозигот HbA/HbD), HbE (Таиланд, не менее 10% гетерозигот HbA/HbE). Большая часть вариантов гемоглобина различается единичными аминокислотными замена-
ми, причиной которых являются генные мутации, связанные с заменой нуклеотидов (изменением азотистых оснований) в нуклеотидных последовательностях членов α- или β-глобиновых генных семейств.
Замены аминокислот, нарушающие спиральную структуру полипептидов, нередко фенотипически проявляются в неустойчивости гемо-глобина-тетрамера. Замена аминокислот в участках, которыми α- и β-полипептиды контактируют друг с другом, влияют на сродство гемоглобина к кислороду. Нарушения функций гемоглобина, обусловленные отмеченными изменениями структуры α- и β-глобиновых генов, ведут к заболеваниям, которые принято делить на четыре группы.
• Гемолитические анемии. Проявляются в распаде эритроцитов в связи с неустойчивостью гемоглобина. Известно порядка 100 вариантов нестабильных гемоглобинов с мутациями в гене β-глобина.
• Метгемоглобинемии. Обусловлены ускоренным окислением двухвалентного железа до трехвалентного с образованием метгемогло-бина (гемоглобина М). Известны пять точечных мутаций генов α- и β-глобинов, фенотипически проявляющиеся в развитии указанного патологического состояния.
• Эритроцитоз. Заключается в образовании большего, чем обычно, количества эритроцитов, что обусловлено повышенным сродством гемоглобина к кислороду, который с трудом высвобождается в тканях. Известно порядка тридцати таких мутаций.
• Серповидноклеточная анемия. К указанному патологическому состоянию чаще всего ведет замена в эритроцитах HbA на HbS. В условиях гипоксии HbS склонен к кристаллизации, что приводит к изменению формы эритроцитов на серповидную, а фенотипиче-ски проявляется многообразием симптомов (см. рис. 4.3), важнейшее место среди которых занимает анемия.
Заболевания крови первых трех групп наследуются по доминантному типу, в связи с чем отклонения в здоровье наблюдаются не только у гомозигот, но и у гетерозигот по мутантному аллелю. В обычных условиях наследование серповидноклеточной анемии происходит по рецессивному типу. В условиях выраженной гипоксии, например, при нахождении человека на высоте свыше 3000 м над уровнем моря анемия развивается у гетерозигот HbA/HbS (кодоминирование). Накопление в гено(аллело) фондах некоторых популяций людей аллелей серповидноклеточности эритроцитов - HbS, HbC, HbD, HbE (см. здесь же, выше) связано с тем, что гетерозиготы по названным аллелям обеспечивают выживание соответствующих популяций в регионах Земли, где распространены воз-
будители тропической малярии и ряда других тяжелых паразитарных болезней.
Описанные выше мутантные формы гемоглобина, как уже отмечалось, возникают вследствие изменений структуры генов по типу замены азотистых оснований (нуклеотидов). Мутации иного характера приводят к появлению аллелей глобинов, обусловливающих другие виды патологии красной крови. В частности, нарушение процесса рекомбинации между аллельными генами в виде неравного кроссинговера дает изменение числа нуклеотидов в них, что приводит к сдвигу «рамки считывания». Нередким результатом таких мутаций бывает подавление образования соответствующего полипептида гемоглобина, приводящее к развитию патологических состояний, известных как талассемии. Так, делеция одного нуклеотида в 139-м триплете гена α-глобина (всего 141 триплет) вызывает сдвиг «рамки считывания» и, как следствие, «прочтение» 142-го кодона и далее, в результате чего мутантный α-полипептид становится длиннее на 5 аминокислотных остатков. Присутствие таких α-полипептидов характеризует один из редких вариантов - гемоглобин Vayne. Если делеция случается ближе к 5'-концу генов α-, β- или γ-глобинов, синтез соответствующего полипептида может блокироваться, вследствие чего развиваются различные клинические формы α-, β- и γ-талассемии.
Некоторые мутантные варианты гемоглобинов образуются вследствие дупликаций определенных участков глобиновых генов. Гемоглобин Grandy, например, отличается дупликацией аминокислотных остатков, занимающих в α-глобине 116-118 положение. β-Глобин му-тантного гемоглобина Cranston имеет длину не 146, а 158 аминокислотных остатков, что является результатом дупликации нуклеотидной последовательности АГ после 144-го триплета, сдвига «рамки считывания» и, как следствие, «прочтения» кодона-терминатора.
Различные по своему конкретному выражению (нуклеотидные замены, делеции, дупликации) изменения структуры глобиновых генов приводят к аминокислотным заменам в соответствующих полипептидах, к укорочению или удлинению последних или же к прекращению их синтеза. Они могут быть причиной развития заболеваний, объединяемых в семейство гемоглобинопатии.
Из сказанного по поводу гемоглобина следует, что образование те-трагетеробелкового комплекса, определяющего выполнение эритроцитами их главной функции, находится под генетическим контролем. При этом формирование тетрамера требует скоординированной экспрессии
двух неаллельных генов - α- и β-глобиновых. Генетический контроль распространяется также на небелковый элемент гемоглобина - гем, представляющий собой комплекс порфиринового кольца и двухвалентного железа. В первую очередь речь здесь идет о генах, контролирующих синтез гема, в частности, в эритроцитах. Мутации этих генов (локу-сы 10q25.2-q26.3, 18q21.3) фенотипически проявляются в различных клинических формах эритропоэтической порфирии (симптомы - фотосенсибилизация, светочувствительный дерматит, желтуха, гепато-спленомегалия, покраснение зубов, полиневропатии, задержка физического и психического развития).
Среди генных наследственных болезней человека ферменто(энзимо) патиям, развивающимся вследствие мутаций генов, контролирующих образование белков-ферментов, принадлежит заметное место. Так, нарушение структуры соответствующего гена приводит к функциональной недостаточности организма по ферменту фенилаланингидроксилазы (см. также п. 5.2.2.8). В такой ситуации аминокислота фенилаланин не превращается в аминокислоту тирозин (рис. 5.1) и накапливается в крови (до 0,5-0,6 г/л вместо обычных 0,03-0,04 г/л). Избыток фенила-ланина и некоторых продуктов его обмена оказывает токсическое действие на мозг ребенка, что приводит к задержке умственного развития. Одновременно нарушается образование пигмента меланина (слабая пигментация радужной оболочки глаз, волос). Повышенная концентрация фенилаланина, подавляя активность некоторых ферментных систем организма, может привести к развитию судорожного синдрома.
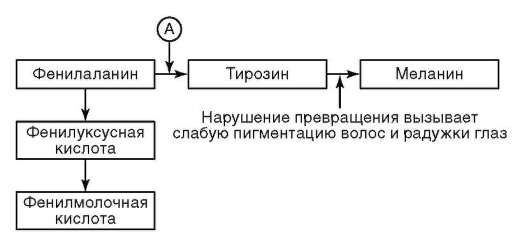
Рис. 5.1. Сокращенная схема обмена аминокислоты фенилаланина: А - фермент фенилаланингидроксилаза, наследственный дефект которого приводит к развитию фенилкетонурии
Описанный симптомокомплекс характеризует наследственное заболевание фенилкетонурию.
Генотипические изменения (мутации генов, контролирующих распад определенных веществ) составляют важное патогенетическое звено некоторых болезней накопления , например лизосомных (различные формы мукополисахаридозов - синдром Гурлера, синдром Хантера; сфинголипидозов - болезнь Нимана-Пика, болезнь Тея-Сакса).
Наследственные дефекты в виде генных мутаций - причина многих болезней обмена веществ , В зависимости от специфических фено-типических проявлений выделяют болезни углеводного (галактозе-мия , непереносимость фруктозы , дисахаридов), жирового (семейная гиперхолистеринемия, болезнь острова Танжер) метаболизма, обмена транспортных белков (цистинурия, цистиноз), аминокислот (фе-нилкетонурия, см. здесь же, выше) и органических кислот (метилма-лоновая ацидемия, пропионовая ацидемия), пуринов и пиримидинов (болезнь Леша-Найана, недостаточность пуриннуклеозидфосфорила-зы, наследственная оротовая ацидурия), металлов (меди - болезнь Вильсона-Коновалова, цинка - наследственный энтеропатический акродерматит ).
5.2. генетика человека как научно-практическая дисциплина
5.2.1. ЧЕЛОВЕК КАК ОБЪЕКТ ГЕНЕТИЧЕСКОГО АНАЛИЗА
Основные закономерности наследования и изменчивости признаков живых форм и вытекающие из этих закономерностей генетические законы были установлены классической (домолекулярной) генетикой благодаря применению гибридологического метода (метод скрещивания) генетического анализа (см. п. 4.3.5.3-в), основоположником которого является Г. Мендель. И в настоящее время в целях генетического анализа широко используют такие объекты классической генетики, как растения, одноклеточные эукариоты, дрожжи, среди многоклеточных животных - плодовая муха дрозофила, круглый червь Caenorhabditis elegans, мышь и другие виды, сравнительно легко скрещивающиеся в лабораторных условиях. Общие характеристики этих видов - достаточно высокая плодовитость (возможность использовать статистический подход при оценке потомства), непродолжительный жизненный цикл и, следовательно, быстрая смена поколений (открывает
перед генетиками перспективу в короткие отрезки времени наблюдать наследование и изменчивость признаков в ряду многих поколений), небольшое число групп сцепления (хромосом), умеренное влияние на состояние фенотипических признаков факторов среды (параметры нормы реакции - см. п. 4.1.1). Молекулярная генетика расширила список «привлекательных» объектов генетического анализа за счет микроорганизмов, вирусов и фагов, привлечение которых позволило получить информацию о химической природе вещества наследственности (ДНК), о структуре гена как функциональной генетической единице, о некоторых механизмах регуляции активности этой единицы.
С точки зрения приведенных выше характеристик, делающих объект удобным для проведения генетических исследований с использованием гибридологического метода генетического анализа, человек как вид обладает существенными ограничениями. Во-первых, среди людей не могут практиковаться заранее спланированные в интересах генетика-исследователя направленные скрещивания (браки). Во-вторых, сравнительно низкая плодовитость затрудняет эффективное применение статистического подхода. В-третьих, медленная (продолжительность существования поколения людей - 25 лет) смена поколений даже при относительно большой длительности жизни не позволяет генетику-исследователю наблюдать закономерности наследования и изменчивости признаков у представителей более чем двух-трех поколений. В-четвертых, генетический анализ людей затрудняет наличие большого числа групп сцепления (23 хромосомы у женщин и 24 хромосомы у мужчин). В-пятых, люди характеризуются выраженным фенотипическим полиморфизмом, что нередко обусловлено действием факторов среды.
Важная позитивная особенность человека, рассматриваемого в качестве объекта генетического анализа, - высокий в сравнении с большинством других живых форм (в частности, многоклеточных эукариот) уровень изученности его фенотипа - результат работы морфологов, физиологов, биохимиков, иммунологов, этологов и психологов, социологов, клиницистов и др.
Невозможность использовать в интересах генетического анализа гибридологический метод стимулировала поиск и применение других, нередко специфических методов (например, близнецовый метод), дающих сведения о закономерностях наследования и изменчивости признаков у человека.
Эффективность фундаментальных и биомедицинских прикладных, прежде всего диагностических и скрининговых (от англ. screening -
тестирование большого числа людей на наличие, например, болезни, «проблемных» аллелей и т.п.), исследований в области антропо-генетики и медицинской генетики существенно повысилась в связи с реализацией проекта «геном человека», а также разработкой, совершенствованием и применением в практическом здравоохранении новейших молекулярно-биологических и молекулярно-генетических геномных технологий.
5.2.2. МЕТОДЫ, ИСПОЛЬЗУЕМЫЕ В ГЕНЕТИКЕ ЧЕЛОВЕКА
Потребность в информативных методах генетического анализа людей вытекает в немалой степени из интересов медицинской и клинической генетики, а также здравоохранения в целом. Этим объясняется устойчивый интерес к соответствующим разработкам, выполняемым в рамках биомедицинского направления современной науки о жизни (см. предисловие). Некоторые особенности разработок такого рода диктует специфика человека как объекта генетического анализа (см. п. 5.2.1). Свой отпечаток накладывают типичные для настоящего времени смещение приоритетов в область молекулярной и клеточной биологии, биоинформатики, а также повышенный интерес к нанобиомедицинским профилактически-превентивным, диагностическим и терапевтическим технологиям. Это порождает проблему разумного сочетания методологии и наработок классической домолекулярной («обратной») и современной молекулярной («прямой») генетики. От оптимального решения названной проблемы уже сегодня зависит эффективность функционирования службы МГК.
С практической медицинской точки зрения важно возможно более раннее в онтогенезе обнаружение генетических дефектов, что стимулирует создание методов пренатальной и предимплантационной диагностики генетической конституции зачатого и начавшего свое индивидуальное развитие человека. Успешное развитие этой группы методов в немалой степени зависит от возможности получить биологический материал, необходимый для проведения современных диагностических молекулярно-биологических и клеточно-биологических медико-генетических исследований.
Для организации превентивных мер, направленных на профилактику рождения генотипически «проблемного» потомства и распространения мультифакторной патологии, а также для оценки ожидаемой материально-финансовой и кадровой нагрузки на здравоохранение
отдельных регионов и государства в исторической перспективе (в том числе ближайшей), необходимы популяционно-генетические скрининго-вые исследования, дающие информацию об особенностях гено(аллело) фондов групп людей (популяций, этнических, производственно-профессиональных, обитающих в определенных, в частности экстремальных, климатогеографических условиях). На уровне семей и людей активного детородного возраста, предполагающих вступить в брак, решение такого рода задач стоит в связи с получением информации о генетической конституции конкретных лиц. Законодательно предписываемое составление индивидуальных геномно-протеомных паспортов (портретов), о необходимости которого много говорится, способствовало бы радикальному решению соответствующих задач. В любом случае в арсенале антропогенетиков должны быть методы генетического анализа, делающие возможными как персонифицированные, так и популяционно-групповые исследования.
Один из подходов к решению названной задачи состоит в совершенствовании молекулярно-биологических геномных технологий биомедицинского диагностического и/или скринингового исследования образцов биоматериала, основанных на использовании панелей (ми-кро)чипов (англ. (micro)chip technology - предположительно от англ. chip - фишка, небольших размеров кусочек пластика, используемый для обозначения определенной суммы денег в некоторых азартных играх). В англоязычной научной литературе используется также термин (micro)array (предположительно от франц. arrai - набор объектов, расположенных в правильном порядке). В исходном биомедицинском понимании (микро)чипы - это серия коротких нуклеотидных последовательностей ДНК, отличающихся друг от друга по составу, которые зафиксированы в строго определенном порядке на твердой подложке (стекло, пластик). На 1 см2 поверхности удается разместить порядка 10 тыс. последовательностей. Такие панели путем проведения процедуры молекулярной гибридизации с ДНК-зондами (см. п. 5.2.2.3-а) или других приемов используются для изучения феномена группового (частота присутствия конкретных аллелей или мутаций в гено(аллело) фондах популяций, этносов и т.д.) и индивидуального (идентификация генных мутаций с целью уточнения диагноза генетической патологии или обнаружения факта гетерозиготного носительства неблагоприятного по фенотипическому эффекту аллеля, детекция кандидатного гена или генетического маркера предрасположенности к определенному муль-тифакторному заболеванию) наследственного полиморфизма. Биоин-
формационные технологии на основе ДНК-чипов, биоэкспрессионные технологии на основе РНК-чипов или белковых чипов, допускающие обработку результатов в автоматическом режиме, существенно повышают производительность и информативность молекулярно-биологических диагностических и скрининговых исследований.
Необходимой предпосылкой использования методов ДНК-диагностики (см. п. 5.2.2.3-б), в том числе в формате (micro)chip или (micro) array technologies, является идентификация возможно большего количества генов и их аллелей, а также картирование генов и генетических маркеров на хромосомах.
По моральным и этическим соображениям эксперименты над людьми недопустимы. Благодаря действию в природе закона гомологичных рядов генотипической изменчивости (Н.И. Вавилов) есть животные, несущие те же мутации, что и люди, страдающие определенными наследственными, в частности моногенными, заболеваниями. К примеру, пациенты, страдающие прогрессирующей мышечной дистрофией Дюшена, и мыши линии mdx имеют мутацию в 23-м экзоне гена, который контролирует и у человека, и у мыши образование важного с функциональной точки зрения белка скелетной мускулатуры и сердечной мышцы - дистрофина (у человека ген расположен на коротком плече хромосомы Х - Хр21.2). Таким образом, закон гомологичных рядов, работающий в живой природе планеты, способствует решению проблемы биологических (генетических) моделей ряда патологических состояний людей. На таких моделях изучаются патогенетические механизмы соответствующих заболеваний, проводятся испытания лекарственных средств.
К методам генетического анализа, применяемым для изучения закономерностей наследования и изменчивости признаков у людей (в том числе патологических), появившимся во времена домолекулярной генетики, относятся генеалогический, близнецовый, цитогенетический, биохимический, популяционно-статистический, генетики соматических клеток и ряд других. Благодаря прогрессу науки некоторые из названных методов были модифицированы (см. п. 5.2.2.3-а), тогда как возможности других были существенно расширены. Появились и принципиально новые методы генетического анализа.
5.2.2.1. Генеалогический метод (метод родословных) генетического анализа человека
В основе этого метода лежат составление и анализ родословных. Его, в сочетании с методом скрещивания, основанном на целенаправленном
подборе родительских пар, применяют с древних времен и до наших дней в коневодстве (см. рис. 1.2), селекции пород крупного рогатого скота и свиней, при получении чистопородных собак, выведении новых пород пушных животных. Родословные (генеалогические древа) составлялись на протяжении многих столетий в отношении членов царствующих семейств и знати в странах Европы (см. рис. 5.9) и Азии, в Древнем Египте.
В антропогенетике генеалогический метод стали активно использовать с начала ХХ в., когда выяснилось, что анализ родословных, в которых прослеживается передача в ряду поколений конкретного признака, например патологического, может заменить собой практически неприменимый в отношении людей гибридологический метод.
При составлении родословной исходным является человек - про-банд, родословную которого изучают. Обычно это либо больной, либо носитель определенного признака, характер наследования которого предполагается исследовать. Результаты генеалогического анализа оформляют в виде таблиц с использованием унифицированных обозначений, предложенных Г. Юстом в 1931 г. (рис. 5.2). В этих таблицах
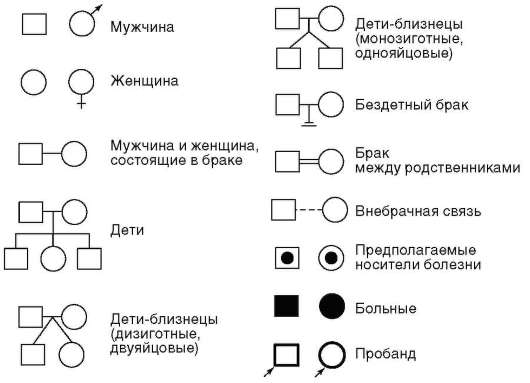
Рис. 5.2. Условные обозначения, используемые при составлении родословных
последовательные поколения обозначают римскими цифрами, а конкретных лиц в каждом поколении - арабскими.
С помощью генеалогического метода устанавливается наследственная обусловленность изучаемого признака, а также тип его наследования (аутосомно-доминантный и т.д., см. здесь же, ниже). При анализе родословных, составленных по нескольким признакам одновременно, может быть выявлен сцепленный характер их наследования, что используют для составления генетических карт хромосом человека. Этот метод дает возможность изучать интенсивность мутационного процесса, оценить экспрессивность и пенетрантность аллеля (признака). Его широко используют в практике МГК в решении таких задач, как планирование семьи и прогноз генетического здоровья потомства.
Определенность медико-генетического заключения на основании анализа родословных снижается в малодетных семьях, а также в связи с явлением фенокопирования (см. п. 4.3.1.1), при наличии в анамнезе матери контактов с вредными агентами (профессиональные вредности на рабочем месте, лучевые диагностические и/или терапевтические манипуляции), фактов приема определенных лекарственных препаратов.
5.2.2.1-а. Родословные при аутосомно-доминантном типе наследования
Для аутосомного типа наследования (ген, определяющий развитие признака, расположен на аутосоме) в целом характерна равная вероятность встречаемости признака среди мужчин и женщин. Это обусловливается наличием в генотипе человека вне зависимости от пола двух аллелей генов, локализованных в аутосомах. Напомним, что один из пары аллелей потомок получает через сперматозоид отца, другой - через яйцеклетку матери, а состояние соответствующего фенотипического признака у потомка зависит от характера взаимодействия аллелей гена, т.е. от того, является ли индивид доминантной гомозиготой, рецессивной гомозиготой или гетерозиготой (см. п. 4.3.4).
При доминировании признака у детей, рожденных родительской парой, в которой хотя бы один из родителей является его носителем, признак проявляется с большей или меньшей вероятностью в зависимости от генетической конституции родителей (рис. 5.3).
Если предмет генетического анализа путем составления родословной - доминантный признак, не оказывающий влияния на жизнеспособность организма, то его носителями среди детей будут и гомозиготы, и гетерозиготы. В случае аутосомно-доминантного наследования патологического признака (фактически генетического заболевания) дети, гомозиготные по соответствующему аллелю, нередко нежизнеспособ-
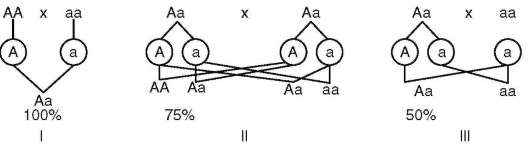
Рис. 5.3. Вероятность появления ребенка с аутосомным доминантным фено-типическим признаком при различной генетической конституции родителей
ны, и, следовательно, живые носители анализируемого признака по их генетической конституции являются гетерозиготами.
Таким образом, при аутосомно-доминантном типе наследования признак обнаруживается в равной мере среди мужчин и женщин и при достаточном числе потомков обнаруживается в каждом поколении.
При анализе родословных следует помнить о возможности неполной пенетрантности и варьирующей экспрессивности доминантного аллеля (см. п. 4.3.1.1). Известно также, что некоторые генные болезни дают клинические проявления не с рождения, а лишь по достижении определенного возраста. Так, тяжелое нейродегенеративное заболевание хорея Гентингтона (мутантный ген располагается на коротком плече хромосомы 4 - 4р16.3, содержит 67 экзонов, контролирует синтез белка «гентингтин» с неустановленными окончательно функциями; мутация состоит в увеличении в первом экзоне числа повторов ЦАГ до 36-180 при нормальном их количестве 6-32) диагностируется клинически обычно у лиц не моложе 35-40 лет. В связи с отмеченным, при прогнозировании по данным анализа родословных возможности появления у родительской пары детей с указанным заболеванием уже родившиеся братья и сестры, не достигшие «критического» возраста, в расчет не принимаются. Так как мутация, приводящая к развитию хореи Ген-тингтона, заключается в экспансии до критических цифр числа трину-клеотидных повторов, для заболевания типично явление антиципации, т.е. утяжеление клинических проявлений и более раннее начало заболевания из поколения в поколение в рамках одной родословной (см.
п. 4.3.1.3).
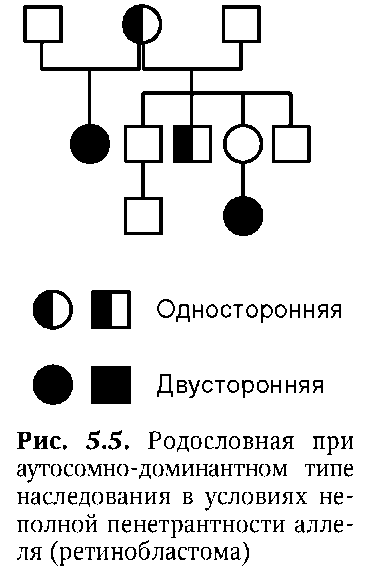
Первое описание и анализ родословной при аутосомно-доминант-ном типе наследования патологического фенотипа (брахидактилия -
рис. 5.4), дано в 1905 г. На рисунке 5.5 представлена родословная при аутосомно-доминантном типе наследования патологического фенотипа (ретинобластома) в условиях неполной пенетрантности.
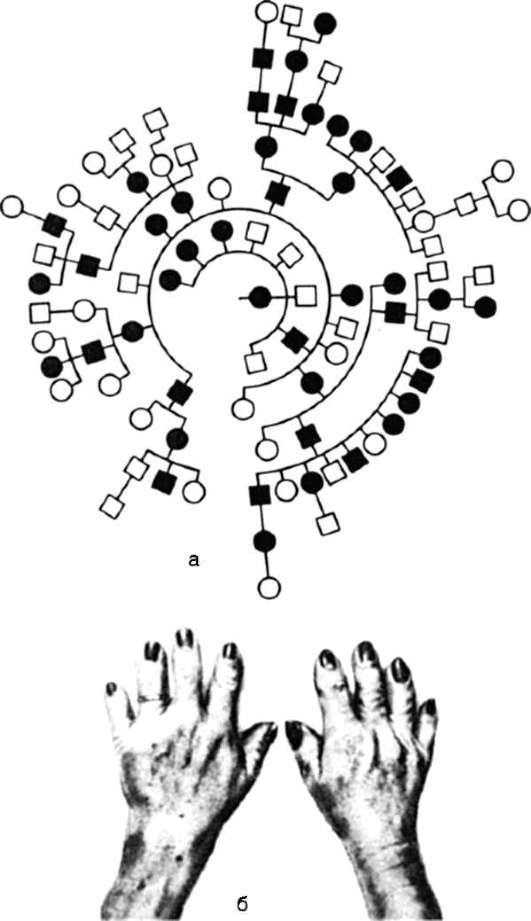
Рис. 5.4. Родословная (а) при аутосомно-доминантном типе наследования (бра-хидактилия - б)
5.2.2.1-6. Родословные при аутосомно-рецессивном типе наследования
Рецессивные признаки проявляются фенотипически лишь у гомозигот по рецессивным аллелям. Эти признаки, как правило, обнаруживаются у детей фенотипически нормальных родителей, которые, будучи гетерозиготами, являются носителями соответствующих аллелей и могут передать их потомкам. В таких случаях вероятность рождения потомков рецессивных гомозигот равна 25%. Вероятность рождения детей с указанной генетической конституцией выше в близкородственных браках. У родителей рецессивных гомозигот по анализируемому аллелю рецессивный признак будет воспроизведен в фенотипе всех (100%) рожденных ими детей (рис. 5.6).
Для родословных при аутосомно-рецессивном типе наследования характерно, что потомки-носители признака обнаруживаются не в каждом поколении.
В качестве примера аутосомно-рецессивного типа наследования патологического фенотипа приводится родословная пациента с псевдогипертрофической прогрессивной миопатией, в которой высока частота близкородственных браков (рис. 5.7).
5.2.2.1-в. Родословные при доминантном Х-сцепленном типе наследования
Гены, размещающиеся в хромосоме Х и не имеющие парных аллелей (гомологичных локусов) в хромосоме Y, представлены в генотипах
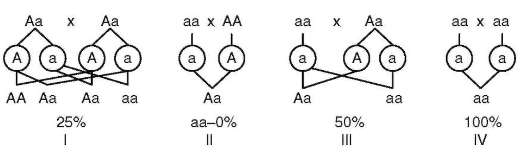
Рис. 5.6. Вероятность появления ребенка с аутосомным рецессивным признаком при различной генетической конституции родителей (I-IV)
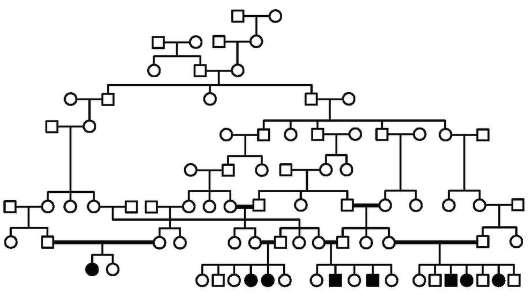
Рис. 5.7. Родословная при аутосомно-рецессивном типе наследования (псевдогипертрофическая прогрессирующая миопатия)
мужчин и женщин в разных дозах. Женщина (гомогаметный пол) получает две свои хромосомы Х и соответствующие гены как от отца, так и от матери, тогда как мужчина (гетерогаметный пол) получает свою единственную хромосому Х только от матери. Соответствующий фенотипи-ческий признак у мужчин, таким образом, определяется единственным аллелем, присутствующим в его генотипе, а у женщин он определяется характером взаимодействия двух аллельных генов. В связи с этим признаки, наследуемые по Х-сцепленному типу, воспроизводятся в фенотипах мужчин и женщин с разной вероятностью.
При доминантном Х-сцепленном типе наследования признак чаще обнаруживается у женщин, которые могут получить соответствующий аллель и от отца, и от матери. Мужчины наследуют доминантный Х-сцепленный признак (аллель) исключительно по материнской линии. Женщины передают доминантный Х-сцепленный признак (аллель) в равной степени сыновьям и дочерям, а мужчины - только дочерям.
В качестве примера доминантного Х-сцепленного типа наследования приводится родословная пациента с фолликулярным кератозом - кожным заболеванием, сопровождающимся потерей ресниц, бровей, волос на голове (рис. 5.8). Характерно более тяжелое течение заболевания у гемизиготных мужчин в сравнении с женщинами, большинство которых гетерозиготны (см. п. 4.3.4).
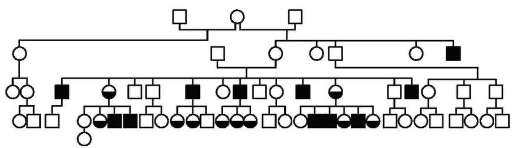
Рис. 5.8. Родословная при доминантном Х-сцепленном типе наследования (фолликулярный кератоз)
При ряде наследственных болезней (пигментный дерматоз), определяемых получением потомком доминантного Х-сцепленного аллеля, гемизиготные зародыши мужского пола нежизнеспособны и погибают на ранних стадиях онтогенеза. В таких случаях в родословных все пораженные лица имеют женский пол. Среди их детей отношение пораженных дочерей, здоровых дочерей и здоровых сыновей равно 1:1:1. Если гемизиготы мужского пола, унаследовавшие летальный доминантный Х-сцепленный аллель, переживают самые ранние стадии онтогенеза, то их смертью, случающейся в плодный период внутриутробного развития, объясняется часть самопроизвольных выкидышей (абортов) и мертворождений.
5.2.2.1-г. Родословные при рецессивном Х-сцепленном типе наследования
Характерная особенность родословных при рассматриваемом типе наследования - преимущественное проявление соответствующего признака в фенотипах гемизиготных мужчин, которые наследуют его от фе-нотипически нормальных матерей, носительниц рецессивных аллелей в гетерозиготном состоянии. Как правило, признак наследуется мужчинами через поколение (дед по материнской линии внук). У женщин проявление признака возможно, если они гомозиготны по рецессивному аллелю анализируемого гена, вероятность чего выше в близкородственных браках.
Классический пример рецессивного Х-сцепленного типа наследования - гемофилия А (рис. 5.9).
5.2.2.1-д. Родословные при Y-сцепленном типе наследования
Наличие хромосомы Y исключительно у представителей мужского пола объясняет главную особенность Y-сцепленного типа наследова-
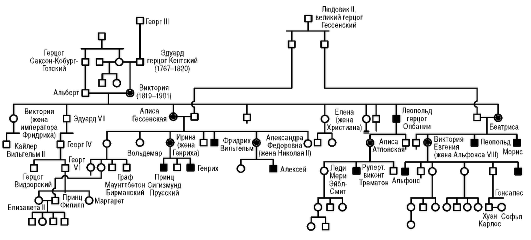
Рис. 5.9. Родословная последнего наследника российского престола царевича Алексея, страдавшего гемофилией. Рецессивный Х-сцепленный тип наследования
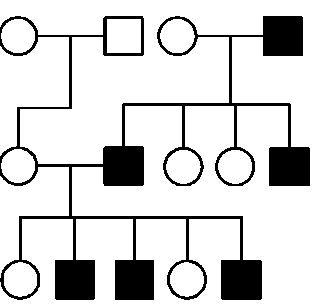
Рис. 5.10. Родословная при Y-сцепленном типе наследования
ния, заключающуюся в том, что соответствующий признак наследуется в ряду поколений только по мужской линии, т.е. передается от отца к сыну (рис. 5.10). Именно таким образом наследуется ряд патологических состояний, проявляющихся в нарушениях развития признаков мужского пола, важных с точки зрения выполнения генеративной функции, например дисгенезия гонад типа 3.
Первый и долгое время остававшийся единственным фенотипический признак, в отношении которого предполагалось голан-дрическое (Y-сцепленное) наследование, -
гипертрихоз ушной раковины (пучки жестких волос, растущие из ушных раковин). Y-сцепленный тип наследования этого признака и сейчас остается предметом обсуждения.
На настоящий момент в хромосоме Y идентифицировано несколько генов, контролирующих образование белков «домашнего хозяйства» (housekeeping proteins), имеющих гомологи в хромосоме Х, не инакти-вируемые при гиперспирализации хромосомы Х с образованием тельца полового хроматина. Тип наследования признаков, контролируемых этими генами, соответствует тому, который присущ признакам, контролируемым аутосомными генами.
Убедительно доказать факт голандрического наследования на основании анализа родословных объективно трудно. Всегда существует необходимость дифференцировать его от аутосомно-доминантного типа наследования.
5.2.2.2. Близнецовый метод генетического анализа человека
Близнецовый метод генетического анализа человека предложен Ф. Гальтоном (см. п. 4.2) в 1895 г. первоначально для оценки роли наследственности и среды в развитии психических свойств человека. Названный метод заключается в изучении закономерностей наследования признаков в парах одно- и двуяйцовых близнецов. В настоящее время он широко используется в изучении закономерностей наследования и изменчивости разнообразных нормальных и патологических признаков у человека для суждения о соотносительной роли генетических и сре-довых факторов (генофенотипические корреляции) в их формировании. Этот метод дает возможность выявить наследуемость признака,
определить пенетрантность аллеля (признака), а также оценить эффективность действия на организм некоторых внешних факторов, например лекарственных средств, воспитания, обучения.
Генетическая основа метода состоит в том, что сравнивается проявление фенотипического признака в разных группах детей-близнецов при учете большего или меньшего сходства их генотипов. Действительно, в отличие от двуяйцовых (дизиготных) близнецов (ДБ), однояйцовые (монозиготные) близнецы (ОБ), развивающиеся из одной оплодотворенной яйцеклетки (зиготы), генетически идентичны, т.е. имеют 100% общих генов (сайтов, нуклеотидных последовательностей ДНК). В силу отмеченного среди монозиготных близнецов наблюдается высокий процент конкордантных пар по наличию в фенотипе обоих сиб-сов - братьев и сестер (сибсы - все дети одной супружеской пары) и конкретному фенотипическому проявлению соответствующего признака.
Сравнение фенотипов монозиготных близнецов, разлученных вскоре после рождения и, следовательно, воспитывавшихся в разных условиях (например, в разных семьях), позволяет выявить признаки, в формировании которых существенная роль принадлежит факторам среды, в том числе социально-культурной. По названным фенотипи-ческим признакам между однояйцовыми близнецами (генетически идентичными) наблюдается дискордантность, т.е. либо отсутствие признака у одного из них, либо признак у близнецов находится в разном состоянии. Напротив, сходство (конкордантность) вплоть до уровня идентичности близнецов по состоянию определенного признака, несмотря на различия условий постнатального развития и существования сибсов, свидетельствует о наследственной обусловленности анализируемого признака.
Сопоставление данных, характеризующих конкордантность по конкретному признаку в парах генетически идентичных монозиготных близнецов (100% общих генов) и в парах дизиготных близнецов, которые имеют в среднем порядка 50% общих генов, дает возможность более объективно судить о роли генотипа в формировании соответствующего признака. Близость показателей конкордантности в парах монозиготных и дизиготных близнецов говорит о незначительном вкладе генетических факторов и об определяющей роли среды в формировании признака или в развитии заболевания. Достоверно различающиеся, но достаточно низкие показатели конкордантности в парах одно- и двуяй-цовых близнецов указывают на наличие наследственной предрасполо-
женности к формированию признака (в том числе патологического), развивающегося под очевидным влиянием факторов среды.
Трудности применения в целях генетического анализа человека близнецового метода связаны, во-первых, с относительно низкой частотой рождения близнецов в большинстве популяций людей (согласно данным мировой статистики, 1 роды двойней на 86-88 родов или 1-2% близнецов среди новорожденных; к сведению, 1 роды тройней приходятся на 10-15 тыс. родов), что осложняет подбор достаточного числа пар с анализируемым признаком, и, во-вторых, с надежной идентификацией (диагностикой) факта монозиготности близнецов, что имеет принципиальное значение для получения достоверных выводов.
Для подтверждения монозиготности близнецов используют ряд подходов:
• сравнение по многим, главным образом морфологическим признакам - пигментация глаз, волос и кожи, особенности волосяного покрова на голове и теле, а также форма волос, форма ушей, носа, губ и ногтей, пальцевые узоры (полисимптомный подход);
• сравнение по эритроцитарным антигенам - группы крови АВ0, резус, MN и др., по белкам сыворотки крови - γ-глобулин, гаплотипам HLA (от англ. Human Leukocyte Antigen - человеческие лейкоцитарные антигены, аналог главного комплекса гистосовместимости, или MHC (англ. Major Histocompatibility Complex), животных): все перечисленные маркеры относятся к категории моногенных мен-делирующих признаков, а контролирующие их гены отличаются узкой нормой реакции, см. п. 4.1.1 (иммунологический подход);
• сравнение данных ЭКГ и ЭГ - электрокардиограмм и энцефалограмм - близнецов (клинико-функциональный подход);
• трансплантационный тест, заключающийся в перекрестной пересадке кожи у близнецов (вариант иммунологического подхода, успешная перекрестная пересадка - наиболее достоверный критерий монозиготности).
Приведем несколько примеров применения близнецового метода из области клинической медицины. Так, конкордантность в парах монозиготных и дизиготных близнецов составляет по умственной отсталости 97 и 37%, по шизофрении 69 и 10%, по эпилепсии 67 и 30%, по кори 97 и 94%, по скарлатине 55 и 47%. Можно заключить, что роль генотипа в развитии первых 3 из названных патологических состояний представлена достаточно отчетливо, тогда как в развитии последних двух патологий (инфекционные заболевания) приоритетное значение имеет
контакт с возбудителем, т.е. с фактором, находящимся в среде. В отношении туберкулеза (конкордантность в монозиготных парах 53% и в дизиготных парах 21%) можно думать о наличии генетической предрасположенности. Вместе с тем важно помнить, что в качестве обязательной характеристики среды жизни, в которой при любой генетической конституции возможно заражение туберкулезом, является присутствие в ней возбудителя - бациллы Коха.
Близнецовым исследованиям принадлежит заметное место в изучении генетики поведения, в частности, таких характерологических (личностных) свойств людей, как агрессивность или склонность к действиям, направленным на отпор насилию, асоциальное (преступное) поведение и законопослушность, лживость и искренность, а также генетики конформизма и лидерства, интеллекта, гениальности. Близнецовый метод остается одним из активно используемых специалистами по общей и медицинской психологии.
5.2.2.3. Цитогенетический метод генетического анализа человека
В основе цитогенетического метода лежит изучение с помощью микроскопа хромосом клеток человека. Его стали широко применять в исследованиях по генетике человека (включая медицинскую генетику) с 1956 г., когда шведские ученые Дж. Тийо и А. Леван (см. п. 4.2), предложив оригинальную методику изучения метафазных хромосом, доказали, что в кариотипе человека 46 хромосом, а не 48, как считали ранее. Современный этап в применении цитогенетического метода связан с разработкой и введением в практику работы цитогенетиков дифференциальной или избирательной окраски хромосом (Т. Ка-сперсон, Швеция, 1969), что дало возможность точно идентифицировать каждую хромосому по характеру распределения окрашиваемых сегментов (см. п. 4.3.4, рис. 5.11). При отсутствии методов дифференциальной окраски идентификацию хромосом проводили по их размерам, положению центромеры (первичная перетяжка) и соотношению длин плеч (центромерный индекс), наличию вторичных перетяжек и спутников (рис. 5.12). В таких условиях не удавалось достичь абсолютной персонификации хромосом. Каждую хромосому относили к одной из 9 групп (А - 1, 2 и 3, В - 4 и 5, С- 6, 7 и Х, С" - 8 и 9, С" " " - 10, 11 и 12, D - 13, 14 и 15, Е - 16, 17 и 18, F - 19 и 20, G - 21, 22 и Y) в порядке убывания размеров. Разграничение между хромосомами в пределах групп проводили, используя дополнительные критерии, например присутствие спутников.
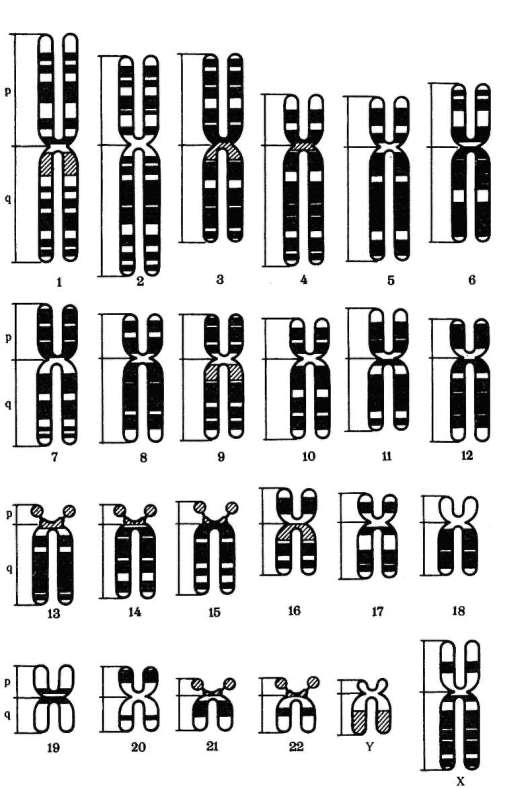
Рис. 5.11. Расположение полос в хромосомах человека при их избирательном окрашивании: p - короткое плечо, q - длинное плечо; 1-22 - порядковый номер хромосомы (аутосомы), - половые хромосомы
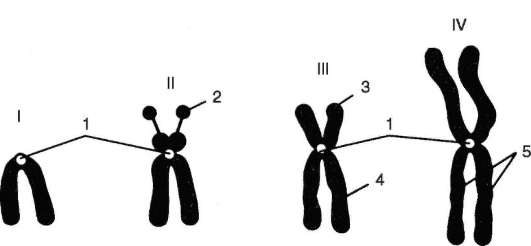
Рис. 5.12. Метафазные хромосомы при сплошной их окраске: I - телоцентри-ческая хромосома; II - акроцентрическая хромосома; III - субметацентриче-ская хромосома; IV - метацентрическая хромосома, основания идентификации: 1 - центромера, 2 - спутник, 3 - короткое плечо, 4 - длинное плечо, 5 - хро-матиды
Цитогенетический метод позволяет изучать нормальную морфологию хромосом (в том числе с учетом их полиморфизма) и кариотипа в целом, устанавливать генетический (хромосомный) пол особи, а также диагностировать хромосомные болезни, связанные с изменением числа отдельных хромосом и хромосомных наборов (анэуплоидии, гаплоидия и полиплоидия, см. п. 4.3.3.3) или с нарушением их структуры (хромосомные аберрации, см. п. 4.3.2.2). Цитогенетический метод широко применяется в МГК в целях пренатальной (в том числе предымпланта-ционной) диагностики хромосомных болезней, что дает возможность путем своевременного прерывания беременности избежать рождения потомства с грубыми нарушениями развития.
Материалом для цитогенетических исследований служат клетки из разных тканей и органов человека - лимфоциты периферической крови, клетки костного мозга, фибробласты кожи, клетки опухолей и эмбриональных тканей. Непременное условие применения цитогене-тического метода - наличие в материале делящихся клеток. Цитоге-нетики обычно используют относительно легкодоступный материал - лимфоциты периферической крови, которые в условиях in vitro путем обработки веществом фитогемагглютинином переводят в состояние митотического деления. Непосредственный объект цитогенетических исследований - метафазные хромосомы на гистологических препаратах так называемых метафазных пластинок. Находящиеся в
состоянии максимальной спирализации (см. п. 3.1.1.2) метафазные хромосомы хорошо видны в микроскоп. Для повышения количества мета-фазных клеток переход из метафазы в анафазу митоза блокируют путем обработки клеточной культуры колхицином или колцемидом, которые разрушают веретено деления.
Мазки, приготовленные из культуры, обогащенной клетками в ме-тафазе митоза, после обычной или избирательной окраски хромосом микроскопируют. Отдельные метафазные пластинки фотографируют. Фотографии используют для составления кариограмм, в которых гомологичные хромосомы выстроены парами, распределены по группам или, при избирательном окрашивании, располагаются попарно в соответствии с их порядковым номером в кариотипе (рис. 5.13, см. также рис. 5.11). В последнем случае хромосомный ряд кариограммы завершает пара половых хромосом.

Рис. 5.13. Нормальный кариотип человека: а - женщина, б - мужчина; вверху - хромосомные комплексы (метафазные пластинки); внизу - кариограм-
мы
5.2.2.3-а. Неинвазивные методы генетического анализа человека: научно-практическое наследие классической генетики
В медицине к категории неинвазивных методов относят такие, осуществление которых происходит при сохранности пограничных тканей и структур организма (методы лучевой диагностики - рентгенологическое и ультразвуковое исследования, различные варианты томографии), а также если требуемый для исследования биологический материал получают без нарушения целостности кожных покровов, слизистых оболочек, стенок полостей тела, кровеносных сосудов и т.п.
С целью диагностики изменения числа хромосом Х в цитогенетике применяют неинвазивный метод определения числа телец полового хроматина в неделящихся клетках слизистой оболочки щеки, которые получают путем соскоба. Тельце полового хроматина (тельце Барра) в ядрах клеток генотипически нормальных женщин присутствует в единственном экземпляре - в связи с функционально-генетической инактивацией одной из двух хромосом Х путем ее гетерохроматизации (см. п. 2.4.3.4-в). Такое тельце выглядит как интенсивно окрашенная красителем основного характера (например, гематоксилином), расположенная обычно вблизи ядерной оболочки с внутренней ее стороны структура (рис. 5.14). В случае увеличения в кариотипе числа хромосом Х сверх положенных двух растет и число выявляемых телец полового хроматина, которое всегда на единицу меньше числа хромосом Х. При уменьшении числа хромосом Х до одной (моносомия Х, синдром Шерешевского-Тернера, кариотип 45Х0) тельце Барра в ядрах соматических клеток отсутствует.
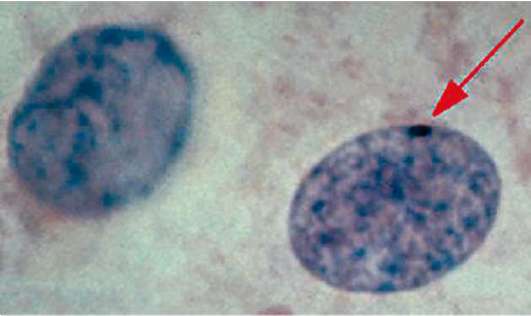
Рис. 5.14. Тельце Барра в ядре клетки
В кариотипе мужчин можно идентифицировать хромосому Y по более интенсивной в сравнении с другими хромосомами флюоресценции после обработки метафазных препаратов акрихинипритом и, следовательно, посчитать их количество (цитогенетическая диагностика синдрома Клейнфельтера, возможные кариотипы 47XYY, 48XYYY).
В антропогенетике используются методы дерматоглифики и паль-москопии, которые также неинвазивны. Они заключаются в изучении, в первом случае кожных гребешковых узоров пальцев и ладоней, во втором - сгибательных ладонных борозд. Основанием к применению названных методов в целях генетического анализа людей послужили результаты исследований, указывающие на то, что характерные изменения дерматоглифических рисунков кожи пальцев и ладоней, характера основных ладонных борозд наблюдаются при определенных хромосомных болезнях - синдромах Дауна, Клейнфельтера, Шерешевского- Тернера, синдроме «кошачьего крика» (делеция короткого плеча хромосомы 5; симптомы - низкая масса тела при рождении, отставание в развитии, лунообразное лицо с широко расставленными глазами, недоразвитие гортани, что проявляется в характерном плаче, напоминающем кошачье мяукание).
Дерматоглифические исследования проводят с целью установления отцовства и идентификации близнецов (см. также п. 5.2.2.2). В последнем случае заключение в пользу монозиготности следует, если из 10 гомологичных пальцев сходные узоры имеют не менее 7, а в пользу дизиготно-сти, если сходные узоры наблюдаются на 4-5 пальцах.
Методы дерматоглифики и пальмоскопии предложены Ф. Гальтоном в 1892 г., хотя основы для классификации кожных узоров были разработаны Я. Пуркинье в 1823 г. В современной антропогенетике и медицинской генетике дерматоглифика и пальмоскопия как методы генетического анализа людей утрачивают свои позиции, так как в сравнении с цитогенети-ческим, молекулярно-цитогенетическим и молекулярно-генетическими методами проигрывают в информативности и, следовательно, в определенности заключений. Тем не менее в некоторых ситуациях они могут быть использованы в скрининговых исследованиях или как вспомогательные врачами практического здравоохранения при решении вопроса о целесообразности медико-генетической консультации.
5.2.2.3-6. Молекулярно-цитогенетический метод генетического анализа человека
Молекулярно-цитогенетический метод анализа хромосом дает возможность преодолеть ограничения цитогенетического метода в его клас-
сическом формате. Так, в кариотипе обследуемого удается обнаружить хромосомные аберрации, даже если их несколько и они произошли в разных хромосомах, а также, если они захватывают участок хромосомы относительно небольших размеров.
Основу метода составляет процедура флуоресцентной гибридизации in situ (англ. FISH - fluorescent in situ hybridization). Речь идет о приготовлении ДНК-зондов, представляющих собой определенные по нуклеотидному составу фрагменты ДНК, помеченные флюорохромом (флюоресцирующий краситель). В указанной конструкции функцию зонда выполняет фрагмент ДНК, который находит в геноме (генотипе, кариотипе) обследуемого «свой» - т.е. комплементарный - участок ДНК и прикрепляется к нему («садится» на него). Благодаря наличию в конструкции флюорох-рома место «посадки» ДНК-зонда определяется по специфическому свечению при микроскопировании гистологических препаратов. При этом обычно используют люминесцентный микроскоп, допускающий работу в УФ части спектра. Объектом микроскопирования могут быть как метафаз-ные хромосомы (см. п. 5.2.2.3, цитогенетический метод), так и хроматин ядер неделящихся клеток (интерфазные хромосомы).
Молекулярно-цитогенетический метод (FlSH-метод в различных его модификациях, например, с одновременным использованием нескольких флюорохромов, дающих разную окраску; известна технология 24-цветная FISH для одномоментной идентификации и нумерации 22 ау-тосом, хромосом Х и У), в сравнении с классическим вариантом цитоге-нетического исследования, позволяет провести анализ количественных изменений и структурных перестроек хромосом быстрее и эффективнее. В силу этого ему отдают предпочтение в ситуациях, требующих проведения высокоинформативной экспресс-диагностики (пренатальное выявление хромосомных аберраций). При соблюдении ряда технических условий он дает возможность идентифицировать места хромосомных разрывов при транслокациях, инверсиях, делециях.
FISH молекулярно-цитогенетический анализ в силу того, что он позволяет локализовать ген (нуклеотидную последовательность ДНК) на хромосоме, нашел применение при составлении физических карт хромосом (см. п. 4.3.2.1) человека. В практике МГК он используется в целях уточнения характера генетического дефекта (ДНК-диагностика), вызвавшего патологические фенотипические проявления у пациента (про-банда), или же для подтверждения факта гетерозиготного носительства «проблемного» рецессивного аллеля (например, фенилкетонурии) потенциальными родителями (супругами).
5.2.2.3-в. Молекулярно-генетические методы генетического анализа человека (ДНК-диагностика)
В последнее время практическая медицина обогатилась значительным числом молекулярно-генетических диагностических методов, что связывают с осуществлением проекта «Геном человека» - геномные (постгенеомные) технологии. Интерес к методам молекулярно-генетического анализа людей (ДНК-диагностика) стимулируется осознанием того, что непременное условие повышения эффективности профилактических, превентивных и терапевтических медицинских (здравоохраненческих) мероприятий - информация о генетической конституции отдельных лиц (см. предисловие: геномное тестирование или портретирование) и об особенностях гено(аллело)фондов человеческих популяций или иных групп населения. Этим в немалой степени объясняется «бум» в области биомедицинских исследований, направленных на поиск диагностических и прогностических маркеров (в том числе генетических) распространенных и «грозных» патологических состояний, в частности онкологических.
Нельзя забывать о таких генетических явлениях, как генокопиро-вание и фенокопирование, неполная экспрессивность и пенетрант-ность, генетическая гетерогенность (см. п. 4.3.1.1), затрудняющих диагностику многих наследственных болезней. К примеру, моногенное наследственное заболевание «муковисцидоз» (кистозный фиброз поджелудочной железы), по распространенности занимающее первое место в группе аутосомно-рецессивных болезней человека, обусловлено мутациями в разных частях гена, картированного на длинном плече хромосомы 7 (7q31.2). У 50-70% пациентов мутация представлена делецией трех нуклеотидов в 10-м экзоне гена - delF508. Фенотипиче-ский эффект мутации состоит в отсутствии в кодируемом полипептиде (всего 1480 аминокислот) в 508-м положении аминокислоты фенила-ланина. Названный полипептид участвует в обеспечении функции хлор-натриевого чрезмембранного транспортного канала в железистых экзокринных клетках. К настоящему времени известно порядка 600 (по другим источникам, 800) мутаций гена, приводящих к муковисцидозу (часть пациентов несет одновременно две мутации - генетические компаунды). Из нескольких сотен мутаций клинический интерес представляют порядка шести, так как именно они являются причиной развития патологического фенотипа (т.е. заболевания) у 70% пациентов. При синтезе железистыми клетками бронхов, поджелудочной железы и эпителиальной выстилки кишечника, слизистой оболочки придаточных па-
зух носа и других органов мутантного полипептида повышается вязкость образуемого ими слизистого секрета. Это приводит к закупориванию путей оттока слизи и развитию воспаления. Диагноз может быть поставлен на основании клинической картины, в которой сочетаются симптомы поражения бронхолегочного аппарата, кишечные расстройства, признаки дисфункции поджелудочной железы, а также на основании данных лабораторных исследований, в частности, если в поте обнаруживается ненормально высокая (свыше 60 ммоль/л) концентрация хлоридов. Установлена определенная зависимость тяжести заболевания от типа мутации, которая его вызвала. Сейчас методами ДНК-диагностики тип мутации, приводящей к муковисцидозу, определяется с надежностью в 72%, т.е. диагностическая эффективность (информативность) не достигает желаемых 100%. Предположительно, одна из причин состоит в многочисленности мутаций гена, приводящих к соответствующему патологическому фенотипу (т.е. к болезни). Показатели эффективности ДНК-диагностики различны для разных заболеваний. В случае ахон-дроплазии и хореи Гентингтона, например, они достигают 100%, тогда как в случае миодистрофии Дюшена - 60%.
Различают прямые и косвенные методы ДНК-диагностики. Первые применяются, если известны: ген, ответственный за развитие соответствующего наследственного заболевания, основные типы его патологических (патогенных) мутаций - в случае муковисцидоза наиболее частая (мажорная) мутация - delF508 (см. здесь же, выше).
В настоящее время приоритетная роль в качестве основы ДНК-диагностики переходит к методу ПЦР (Полимеразная Цепная Реакция), или PCR (англ. Polymerase Chain Reaction). Названный метод дает возможность в условиях in vitro в течение 1 ч получить миллионы копий заданного (представляющего интерес для исследователя или врача) фрагмента молекулы (нуклеотидной последовательности) ДНК, использование которых благодаря их многочисленности существенно облегчает идентификацию в геноме пациента (пробанда) сайта ДНК, представляющего диагностический интерес. Популярность метода ПЦР в медицинской среде объясняется также тем, что он широко используется в целях высокоточной и надежной диагностики вирусных (СПИД, вирусные гепатиты) и инфекционных заболеваний по выявлению нуклеиновой кислоты возбудителя.
Альтернативным методу ПЦР, уступающим ему позиции в качестве основы ДНК-диагностики, является метод блот-гибридизации (от англ. blot - пятно, клякса; blotting paper - промокашка, фильтроваль-
ная бумага). В арсенале молекулярных и медицинских генетиков этот метод появился раньше метода ПЦР. Технически он более сложен, требует использования радиоактивных материалов. В настоящее время его модификации применяются для решения ряда конкретных задач - гибридизация ДНК-зондов с разделенными с помощью электрофореза молекулами РНК, а также с белковыми молекулами, фиксируемыми на фильтрах с мечеными антителами.
Косвенные методы ДНК-диагностики наследственных (прежде всего, моногенных) болезней (в общем виде молекулярно-генетические методы генетического анализа людей - см. здесь же, выше) основаны на идентификации не патологических (патогенных) мутаций генов непосредственно, а на анализе наличия в геноме и поведения (распределение аллелей генетического маркера в геномах родственников: у сибсов, в парах «дед-внук», «дядя-племянник») в семье обследуемого так называемых полиморфных генетических маркеров - участков (ло-кусов) ДНК, тесно сцепленных в соответствующем районе конкретной хромосомы с локусом, ответственным за генетическое заболевание. На настоящий момент на хромосомах человека картировано порядка 6 тыс. таких ДНК-маркеров (полиморфных генных локусов). Для некоторых из них доказана ассоциация с конкретным наследственным или мульти-факторным заболеванием. Так, аллель В27 генетической системы HLA ассоциирован с «анкилозирующим спондилитом» и «псориатическим спондилитом», аллель DR3 названной системы - с болезнью Аддисона и с рассеянным склерозом, аллель DR4 - с ревматоидным артритом, аллель CW6 - с псориазом.
Применение косвенных методов ДНК-диагностики возможно при отсутствии полной информации о «проблемном» гене (он не идентифицирован и поэтому не известен порядок следования в нем нуклеотидных последовательностей, т.е. ген не секвенирован). Необходимо, однако, знать, на какой хромосоме этот ген расположен.
5.2.2.3-г. Современные тенденции в ДНК-диагностике. Использование полиморфных генетических маркеров
Нередко полиморфные локусы ДНК, используемые в качестве генетических маркеров, представляют собой нейтральные мутации, не проявляющиеся фенотипически и не влияющие на жизнеспособность и репродуктивные свойства особей, концентрирующиеся в основном в некодирующих областях генома и характеризующиеся менделевским типом наследования.
Высоким уровнем полиморфизма отличаются минисателлитные (длина повторяющегося элемента от 11 до 500 п.н.) и микросателлитные (длина повторяющегося элемента от 2 до 10 п.н.) тандемные повторы (минисателлитный и микросателлитный генетический полиморфизм) с меняющимся от хромосомы к хромосоме или от участка к участку одной хромосомы числом повторов. Их число и распределение в геноме каждого человека индивидуализированы настолько, что могут быть использованы для идентификации личности (как отпечатки пальцев). Это обстоятельство послужило основой для разработки методов диагностики наличия родственных связей между людьми (геномная дактилоскопия), а также предрасположенности к определенным наследственным и мультифакторным заболеваниям в связи со сцеплением этих локусов с так называемыми кандидатными генами. Кандидатным называют ген (сайт, нуклеотидную последовательность ДНК), наличие которого указывает на предрасположенность к конкретной болезни. Ген, вызывающий моногенное наследственное заболевание, один. Кан-дидатных генов предрасположенности обычно несколько. Так, называют 9 кандидатных генов эссенциальной артериальной гипертензии, не менее 6 кандидатных генов атеросклероза. Число идентифицированных кандидатных генов распространенных мультифакторных болезней постоянно растет.
Для целей генодиагностики предрасположенности к мультифактор-ным заболеваниям по обнаружению соответствующего полиморфного генетического маркера в настоящее время в качестве наиболее перспективного называют анализ ОНП геномов обследуемых. ОНП также называют снипсами (англ. SNPs). Это самый распространенный вариант ДНК-полиморфизмов, многократно превосходящий по представленности в геноме минисателлитные и микросателлитные полиморфизмы. По своему происхождению ОНП - следствие точечных мутаций, затрагивающих всего одну пару нуклеотидов. Подсчитано, что в геноме человека количество таких вариабельных пар нуклеотидов составляет 3 млн (в среднем одна измененная пара нуклеотидов на каждую 1000 п.н.). Уже идентифицировано более 2,2 млн ОНП, причем около 99% их расположено в участках молекул ДНК, не кодирующих последовательности аминокислот в полипептидах, что указывает на возможную причину высокой сохранности ОНП в геноме. Принимая, что количество кодирующих (транскрибируемых и транслируемых, экспрессируемых) генов в геноме человека равно 30-35 тыс., а их размеры колеблются в диапазоне от 1 тыс. до 1 млн п.н., можно предположить нахождение внутри
каждого гена (в том числе «проблемного» - патогенного, кандидатно-го) или в непосредственной близости от него (в состоянии сильного сцепления с ним) одного или даже нескольких ОНП. Этим определяется перспективность анализа ОНП (SNP) в целях получения информации о «биологическом или генетически-биоинформационном и, таким образом, отчасти медицинском качестве» генома отдельно взятого лица. Для получения названной информации необходимо иметь «сводную карту» расположения ОНП в геноме человека. Работа по составлению такой карты начата в 1999 г., когда был запущен исследовательский проект идентификации и картирования исключительно ОНП. Наличие такой карты - одна из предпосылок к проведению широкомасштабной работы по геномной паспортизации людей, что даст персонифицированную информацию, полезную при выборе профессии, вида спорта, супруги или супруга, местожительства, при определении приоритетных действий, направленных на профилактику болезней, сохранение и/или даже преумножение здоровья, воплощение в практическом здравоохранении принципа «лечить не болезнь, а больного».
5.2.2.4. Метод генетики соматических клеток
Суть метода генетики соматических клеток сводится к использованию в целях генетического анализа человека культур клеток, получаемых из различных источников, - периферическая кровь, кожа, скелетная мускулатура, биопсийный материал (клетки плаценты и ворсин хориона плода, опухолей), амниотическая жидкость. В зависимости от задачи проводят простое культивирование клеток in vitro, клонирование (получение от одной клетки генетически идентичного клеточного потомства), селекцию (отбор из клеточной массы клеток с заданной характеристикой, например, несущих определенную мутацию), гибридизацию клеток, различающихся по некоторым характеристикам, полученных от разных людей или от человека и животного другого вида - мыши, крысы, курицы, хомячка, обезьяны, генетическую модификацию клеток с использованием генно-инженерных технологий knock out (инактивация конкретного гена, замена аллеля дикого типа на мутант-ный) и knock in (введение в клеточный геном определенного гена).
Культивирование клеток решает задачу увеличения массы биоматериала, получаемого, например, от эмбриона или плода, до уровня, позволяющего выполнить в полном объеме цитогенетические - см. п. 5.2.2.3, биохимические - см. п. 5.2.2.5, иммунологические - см. п. 5.2.2.6, иные молекулярно-биологические и клеточно-биологические, прежде всего
диагностические, исследования. Практикуемые в целях активной профилактики рождения детей с наследственной патологией плацентоби-опсии и хорионбиопсии (8-12-е недели беременности), амниоцентез (забор амниотической жидкости с находящимися в ней клетками, 15-18-е недели беременности), кордоцентез (забор пуповинной крови с находящимися в ней клетками, беременность более 18 нед), забор клеток из бластоцист, получаемых путем экстракорпорального оплодотворения или маточного лаважа (от франц. lavage - мытье, стирка, здесь - промывание полости органа жидкостью с целью извлечения зародыша; срок 90-130 ч после оплодотворения) дают недостаточное количество клеток. Без последующего наращивания объема биоматериала в условиях in vitro качественная дородовая (пренатальная) и предымплантационная диагностика генетических дефектов невозможна.
Селекция, клонирование, генетическая модификация клеток расширяют возможности научного анализа форм и степени генетического контроля развития различных (в том числе патологических) фе-нотипических признаков, повышают вероятность выявления стартового патогенетического звена заболевания, в частности, из числа наследственных болезней обмена веществ или наследственных иммунодефи-цитов (отсутствие синтеза или образование функционально дефектного продукта генной активности - фермента, рецептора, иммуноглобулина, транспортного или сигнального белка и т.п.). Названные выше манипуляции с клетками находят применение при создании терапевтических генно-инженерных конструкций, тканеинженерных конструкций для регенеративной медицины (см. п. 3.2).
Гибридизация соматических клеток в условиях культуры дает возможность исследовать сцепление генов и их локализацию на той или иной хромосоме (картирование). Особенность межвидовых клеточных гибридов состоит в том, что в последовательных делениях из кариотипа теряются хромосомы предпочтительно одного вида. Клетки-гибриды «человек-мышь», например, утрачивают, причем постепенно, все хромосомы человека, что дает возможность проследить с потерей каждой очередной хромосомы утрату определенных генов (сайтов, ну-клеотидных последовательностей ДНК).
Используя метод генетики соматических клеток, изучают характер межгенных взаимодействий, механизмы регуляции генной активности. Получаемые этим методом данные позволяют судить о генетической гетерогенности (см. п. 4.3.1.1) наследственных болезней, а также изучать их патогенез на молекулярном и клеточном уровнях.
5.2.2.5. Биохимический подход в генетическом анализе человека
В основе биохимического подхода, в рамках которого в целях генетического анализа человека используются лабораторно-биохи-мические (в том числе клинические) методы, лежит выявление в фенотипе обследуемого субъекта (пробанда) нормальных или измененных первичных продуктов функциональной активности конкретных генов, например, контролирующих образование α- и β-глобиновых полипептидов гемоглобина или ферментов (см. п. 5.1).
Использование названного подхода и, следовательно, лабораторно-биохимических методов оказалось эффективным в решении диагностических задач и выяснении существенных звеньев патогенеза обширной группы наследственных болезней обмена веществ: аминокислот (альбинизм, фенилкетонурия), углеводов (гликогенозы, глюкозурии, галактоземия), липидов (липидозы, семейная гиперхолистеринемия), стероидных гормонов (адреногенитальный синдром), эритрона (гемолитические анемии), пуринов и пиримидинов (синдром Криглера-Найяра), металлов (болезнь Вильсона-Коновалова), лизосомных болезней (мукополисахаридозы), пероксисомных болезней (синдром Цельвегера) и др.
Наиболее часто генетический дефект в виде мутации соответствующего гена дает фенотипический (в том числе патологический, клинически значимый) эффект из-за нарушения того или иного метаболического процесса в связи выпадением каталитической функции фермента (см. рис. 5.1). Вследствие такого выпадения могут страдать синтезы, утилизация, транспорт субстратов и/или продуктов соответствующих биохимических реакций. Функционально дефектными могут оказаться белки-клеточные рецепторы, что вызывает отклонения в процессах, требующих «правильно» организованного взаимодействия разных клеток, например морфогенезы (см. синдром тестикулярной феминизации Морриса).
5.2.2.6. Иммунохимический подход в генетическом анализе человека
В основе иммунохимического подхода, в рамках которого в целях генетического анализа человека используют иммуноаналитические методы, лежит выявление в фенотипе обследуемого субъекта (пробан-да) нормальных или измененных первичных продуктов функциональной активности конкретных генов путем постановки специфической реакции связывания антитела с антигеном.
Эффективность применения иммунохимических методов существенно повысилась в связи с разработкой на основе целенаправленной селекции и клонирования соматических клеток (см. п. 5.2.2.4) технологии получения гибридом. Так как гибридома представляет собой клеточный клон, полученный путем многократных последовательных делений одной клетки-родоначальницы (гибрид опухолевой клетки, способной к неограниченной пролиферации, и нормального иммунного лимфоцита, синтезирующего антитела), все ее клетки образуют определенное антитело или, другими словами, чистый препарат одинаковых иммуноглобулинов. Антитела, образуемые гибридомой, называются моноклональными и способны вступать в реакцию исключительно со своим антигеном. Этим обеспечивается высочайшая степень надежности детекции белка, являющегося таким антигеном и одновременно первичным продуктом исследуемого гена. В целях визуализации реакции «антиген-моноклональное антитело» антиген или антитело метятся радиоактивным изотопом (радиоиммунный вариант), ферментом, катализирующим превращение субстрата с образованием окрашенного продукта (иммуноферментный вариант), или флюорохромом (имму-нофлюоресцентный вариант). Таким образом, моноклональные антитела выполняют роль молекулярных зондов (см. п. 5.2.2.3-б).
5.2.2.7. Популяционно-статистический подход в генетическом анализе людей
Особенность популяционно-статистических методов генетического анализа человека состоит в статистической обработке результатов наблюдений.
Популяционно-статистические методы предназначены для изучения распределения избранных фенотипических признаков (в том числе патологических) в группах людей (этнических, региональных, популяциях, демах, изолятах) в одном или в ряду поколений. На основании данных, полученных этим методом, рассчитывают частоту встречаемости в исследуемой группе населения различных аллелей гена или разных генотипов по этим аллелям, степень гетерозиготности и полиморфизма, выясняют распространение в группе определенных наследуемых признаков, включая генетические и мультифакторные болезни, анализируют влияние факторов внешней среды на экспрессию (показатели экспрессивности и пенетрантности) генов, устанавливают факт наличия отбора (положительного или отрицательного) по отдельным аллелям соответствующих генов, а также природу факторов отбора. Популяционно-
статистический подход может быть использован для определения и/или подтверждения типа наследования заболевания; он дает ценные сведения для идентификации факторов, путей и генетических механизмов антропогенеза и расогенеза. Его широко применяют для определения степени межпопуляционного генетического разнообразия, в расчетах генетических расстояний между сравниваемыми популяциями, что дает возможность судить об уровне их генетического родства. Последнее может дать основания к заключению (предположению) об историческом и языковом (лингвистическом, культурном) родстве.
При статистической обработке материала, получаемого путем обследования членов избранной группы людей по интересующему исследователя или врача-генетика признаку, основой для суждений об особенностях генетической структуры группы (генетического состава или аллелофонда популяции) служит закон генетического равновесия Харди-Вайнберга. Он отражает закономерность, согласно которой при соблюдении ряда условий соотношение частот аллелей генов в гено(аллело)фонде, а также разных гено(аллело)типов в обследуемой популяции в ряду поколений остается неизменным. Имея данные о частоте встречаемости в популяции рецессивного фенотипа (генотип аа), легко рассчитать частоту встречаемости указанного аллеля (а) в гено(аллело)фонде обследуемого поколения людей. Распространив результаты расчетов на ближайшие поколения, можно предсказать появление в них людей - рецессивных гомозигот и гетерозиготных носителей соответствующего рецессивного аллеля. Данные такого рода важны в прогностическом плане, если этот аллель является патогенным, т.е. в гомозиготном состоянии приводит к развитию наследственной (моногенной) болезни, или же его наличие свидетельствует о генетической предрасположенности к мультифакторному заболеванию.
Математическим выражением закона Харди-Вайнберга служит формула:
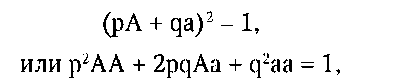
где р2 - доля гомозигот по аллелю а; р - частота аллеля а; q2 - доля гомозигот по альтернативному аллелю A; q - частота аллеля А; 2рq - доля гетерозигот.
Очевидно, что p + q = 1. Формула дает возможность рассчитать частоту встречаемости людей с разными генотипами и в первую очередь, что представляет непосредственный интерес для практической медицины, частоту встречаемости гетерозигот - носителей скрытого рецессивного
(нередко «проблемного», патогенного) аллеля. К примеру, альбинизм связан с отсутствием в организме синтеза черного пигмента меланина и является наследственным рецессивным признаком. Частота, с которой в большинстве популяций встречаются альбиносы (генотип аа), составляет 1:20 000. Если q2 = 1/20 000, то q = 1/141, а р = 140/141. В соответствии с законом Харди-Вайнберга частота встречаемости гетерозигот равна 2pq, т.е. в нашем примере 2(1/141)(140/141)=280/20 000 или 1/70. Заключаем, что в обследованной популяции гетерозиготные носители аллеля альбинизма встречаются с частотой один на 70 человек.
Факт соответствия частот встречаемости разных признаков среди членов обследуемой популяции людей закону Харди-Вайнберга дает основания утверждать, что развитие анализируемых признаков контролируют аллели одного гена. Так, путем изучения фенотипов было установлено, что среди белого населения США 29,16% имеют группу крови М, 49,58% - группу крови MN и 21,26% - группу крови N, что в точности отвечает формуле р2М + 2pqMN + q2N = 1. Был сделан вывод, что развитие трех вариантов признака обусловлено одним геном (I), имеющего два аллеля (Iм и IN) с формой взаимодействия в виде кодо-минирования: у лиц с группой крови М генотип IMIM, у лиц с группой крови N - ININ и у лиц с группой крови MN - IMIN.
5.2.2.8. Медико-генетическое консультирование
Медико-генетическое консультирование (МГК) - один из видов специализированной медицинской помощи. По своему содержанию это комплекс мероприятий, имеющих целью, во-первых, сообщить людям информацию о возможности появления или повторения у их детей наследственной патологии, т.е. осуществить медико-генетигеский прогноз, и, во-вторых, помочь им сформулировать отношение к сообщенной информации. При этом важно, чтобы люди, которые получают информацию, желали и были бы способны контролировать свою репродуктивную функцию. Медико-генетический прогноз может быть проспективным и ретроспективным. От варианта прогноза зависит выбор технологий, применяемых в практике МГК. В силу этого нередко говорят о проспективном и ретроспективном МГК.
Самостоятельная задача МГК состоит в содействии врачам в постановке точного диагноза заболеваний генетической природы. Патогенетическую основу таких заболеваний могут составлять:
• новая патогенная мутация (возникает непосредственно у пациента-пробанда);
• неблагоприятный («проблемный») патогенный аллель или неблагоприятная комбинация мутантных патогенных аллелей, достающихся пациенту от родителей (моногенные наследственные болезни с доминантным, рецессивным и сцепленным с полом типами наследования, см. пп. 5.2.2.1-а, б, в, г, д);
• структурные перестройки или изменение числа хромосом (хромосомные наследственные болезни, см. пп.4.3.2.2 и 4.3.3.3);
• особая генетическая конституция человека, при которой требуемый белок-фермент в организме либо не образуется вовсе, либо функционально неполноценен: контакт такого лица с конкретным «разрешающим» фактором внешней среды неизбежно приводит к развитию патологического состояния, тогда как при отсутствии этого фактора человек практически здоров (непереносимость молочного сахара лактозы - выявляется у 85-100% членов некоторых популяций Юго-восточноазиатского региона, у 70-75% представителей ряда популяций афроамериканцев и североамериканских индейцев);
• наличие во внешней среде патогенных факторов, вызывающих при «неблагоприятном» генетическом фоне развитие соответствующего заболевания (болезни с наследственной предрасположенностью).
В задачу проспективного МГК входит определение для конкретной пары родителей риска рождения генетически «проблемного» потомства до зачатия (на основании анализа кариотипов потенциальных родителей, выявления гетерозиготного носительства, в частности, рецессивных патогенных аллелей; этот вариант МГК настоятельно рекомендуется в ситуациях повышенного риска - близкородственные браки), до имплантации начавшего развитие зародыша в стенку матки (путем, например, экстракорпорального оплодотворения получают несколько зародышей, развитие которых до стадии бластоцисты происходит «в пробирке»; единичные клетки таких зародышей помещают в культуру с питательной средой, где их количество, так как они размножаются, увеличивается настолько, что, используя цитогенети-ческий, молекулярно-цитогенетический, биохимические, иммунологические методы, молекулярно-генетические и геномные технологии ДНК-диагностики - см. пп. 5.2.2.3, 5.2.2.3-б, в, г, 5.2.2.5, 5.2.2.6, проводят анализ кариотипа и генома (генотипа) на предмет выявления генотипически «полноценного» эмбриона, который и будет имплантирован), до родов - в период внутриутробного развития (получая для
медико-генетического исследования клеточный материал зародыша путем биопсии плаценты или ворсин хориона, пункции кровеносных сосудов пуповины - кордоцентез, из околоплодной жидкости путем амниоцентеза, см. п. 5.2.2.4) или же используя такие диагностические методы, как фетоскопия, УЗИ. На использовании ультразвуковой дородовой диагностики (УЗИ или ультразвуковое исследование плода) пороков развития остановимся особо. В настоящее время это единственный повсеместно применяемый в акушерской практике современный неинвазивный (см. п. 5.2.2.3-а) метод. Его можно осуществлять фактически на любом сроке беременности - с 6-8-й до 30-32-й недели. Попытки использовать такие неинвазивные методы лучевой диагностики, как радио- и рентгенография, имели место в прошлом, но не нашли широкого применения. Современные неинвазивные томографические технологии, например магнитно-резонансная томография (МРТ), в силу технических обстоятельств (относительно медленное формирование изображения при высокой двигательной активности плода) не гарантируют требуемой определенности результата. Метод фетоскопии (инвазивный) заключается в осмотре плода с помощью оптической системы - зонда, вводимого в амниотическую полость. В настоящее время применяется по особым показаниям, во-первых, в силу высокой вероятности осложнений (7-8% фетоскопий приводят к выкидышу) и, во-вторых, в силу того, что почти все врожденные пороки развития, выявляемые методом фетоскопии, диагностируются с помощью УЗИ. Дородовое МГК рекомендуется проводить не позднее 20-22-й недели беременности. В указанные сроки, если принимается решение о «нецелесообразности» продолжения беременности по медико-генетическим основаниям, плод нежизнеспособен, и осуществляемые в таких случаях преждевременные роды психологически воспринимаются более спокойно.
Практикуется также перинатальное МГК, заключающееся в проведении непосредственно после рождения или в раннем перинатальном периоде (первая неделя после рождения) просеивающей диагностики с целью выявления определенных наследственных заболеваний. Разработка и применение технологий перинатального МГК стимулируется возможностью при некоторых наследственных патологиях блокировать развитие патологического фенотипа (т.е. генетического заболевания) путем организации профилактических мероприятий.
Так, в целях ранней диагностики фенилкетонурии на 3-5-й день после рождения в роддоме готовят образцы пятен капиллярной крови
младенцев, высушенные на хроматографической или фильтровальной бумаге. Эти образцы доставляют в специализированную биохимическую лабораторию (их можно пересылать по почте), где определяют концентрацию аминокислоты фенилаланина (количественный высокоинформативный тест). В случае обнаружения гиперфенилаланинемии у врачей возникает настороженность. При последующем подтверждении диагноза фенилкетонурии и уточнении, в частности, с использованием методов ДНК-диагностики (см. п. 5.2.2.3-в) характера генетического дефекта (болеют рецессивные гомозиготы по мутациям гена PAH фермента фенилаланингидроксилазы - 12q22-q24.2; из более чем 200 известных мутаций в России и в восточноевропейских странах распространена R408W - изменение в 12 экзоне гена 1 п.н., что приводит к замене в 408-м положении аминокислоты аргинина на аминокислоту триптофан) ребенок переводится на искусственную безфенилаланиновую диету. Дело в том, что при отсутствии активности фенилаланингидроксилазы нарушается обмен поступающего с пищей фенилаланина, что приводит к накоплению аминокислоты и продуктов побочных реакций метаболизма с ее участием - фенилпировиноградная, фенилуксусная и фенил-молочная кислоты - прежде всего в головном мозге. В таких условиях страдает процесс умственного развития (еще одно название болезни - фенилпировиноградная олигофрения). Если перевод младенца на требуемую диету происходит в первые недели жизни, то болезнь не развивается. Перинатальное (т.е. раннее) МГК в описанном случае важно потому, что при отсутствии специфических профилактических мер (без-фенилаланиновая диета) первые клинические проявления заболевания обнаруживаются уже спустя 2-3 нед после рождения, а к 6-месячному возрасту дефицит умственного развития становится необратимым. По достижении ребенком возраста 11-12 лет диету расширяют и делают более разнообразной без угрозы нарушений в умственном развитии. О том, что новорожденный несет соответствующий генетический дефект и болен фенилкетонурией, говорит специфический (мышиный) запах мочи новорожденного или приобретение ею зеленого окрашивания при добавлении треххлорного железа - проба Фёллинга (оба теста качественные).
Ретроспективное МГК проводится в отношении семей, уже имеющих ребенка с заболеванием генетической природы (пробанд), с целью определения риска рождения детей с наследственной патологией в будущем.
Необходимость медико-генетического прогноза связана с тем, что гено(аллело)фонды популяций людей отягощены генетическим гру-
зом, который определяется как некоторая (обычно меньшая) часть членов популяции, имеющая измененную наследственность, в результате чего в каждом поколении рождаются люди с наследственной патологией. Такие люди отличаются пониженной жизнеспособностью и поэтому подвергаются избирательной гибели в процессе естественного (стабилизирующего) отбора. Вместе с тем некоторые из них доживают до репродуктивного возраста и оставляют потомство, как правило, также с измененной наследственностью. Свой вклад в суммарный объем генетического груза вносят гетерозиготы-носители «проблемных» патогенных аллелей. Такие гетерозиготы сами обычно генетически здоровы, так как в дополнение к рецессивному патогенному аллелю они имеют нормальный доминантный аллель дикого типа. Известны лица-гетерозиготы, у которых патогенный аллель доминантен. В силу дозированности действия генов при многих наследственных патологиях с доминантным аутосомным или доминантным Х-сцепленным типом наследования гетерозиготы тоже страдают генетическим заболеванием, но, в сравнении с доминантными гомозиготами (которые нередко нежизнеспособны), в легкой форме.
По степени нарушения здоровья различают несколько вариантов генетического груза. Генетический груз характеризуют как незначительный, если вызываемые им отклонения в здоровье снижают жизнеспособность индивида в малой степени - дальтонизм (цветовая слепота). В таких случаях определяемое генетическим грузом нарушение здоровья обычно проявляет себя на протяжении всей жизни человека. При некоторых наследственных заболеваниях вклад генетического груза в нарушение здоровья и снижение жизнеспособности оценивают как весомый, причем неблагоприятное влияние этого груза на состояние здоровья наблюдается на протяжении достаточно длительного отрезка жизни человека, делая его инвалидом - псевдогипертрофическая мышечная дистрофия Дюшена (моногенное наследственное заболевание, мутация в 23-м экзоне гена мышечного белка дистрофина - Хр21.2, фенотипически проявляется в неуклонно прогрессирующей мышечной слабости с распространением на все большее число скелетных мышц; болезнь начинается с неуверенной походки обычно в первые три года жизни, к 10-11 годам дети нередко уже прикованы к постели, средняя продолжительность жизни порядка 20 лет). Есть заболевания, при которых патогенетическое действие генетического груза является весомым, но появление клинически значимых фенотипических признаков и, следовательно, начало болезни как таковой отсрочены по возрасту- хорея Гентингтона (моногенное заболевание, мутация в гене белка гентингтина
с неустановленной функцией - 4р16.3; фенотипически проявляется в образовании амилоидподобных бляшек с исходом в гибель нервных клеток стриопаллидарной системы головного мозга; характерен комплекс клинических проявлений в виде гиперкинезов, слабоумия и психических нарушений, оформляющийся в типичных случаях на 4-5-м десятилетии жизни человека). Генетический груз оценивают как интенсивный, если он обусловливает появление признаков болезни в раннем детстве, тяжесть и быстрое нарастание степени выраженности клинических симптомов, раннюю смерть - амавротическая идиотия или болезнь Тея-Сакса (моногенное наследственное заболевание обмена веществ, мутация в локусе 15q22.4; фенотипически проявляется в функциональной недостаточности лизосомального фермента гексоаминидазы А, что приводит к накоплению, в частности, в головном мозге Gm2-ганглиозида с исходом в генерализованную гибель нервных клеток и демиелинизацию, в замещение нервных структур нейроглией; клинические проявления регистрируются обычно на 4-5-м месяце жизни, быстро прогрессируют и, достигнув максимальной степени выраженности в виде функциональных расстройств, связанных с поражением жизненно важных нервных центров продолговатого мозга и фактической декортикацией больного, глухоты, слепоты, полной обездвиженности, трофических нарушений, кахексии, приводят к смерти ребенка обычно в возрасте 3-4 лет). В таких случаях действие генетического груза относительно кратковременно.
Благодаря наличию генетического груза существует генетический риск развития у потомства с известной вероятностью определенной наследственной патологии. Величина генетического риска рассчитывается тем или иным из существующих методов. Выбор метода в немалой степени определяется типом наследования и рядом других характеристик наследственной патологии, по которой готовится медико-генетический прогноз. Так, учитывают, является ли заболевание моногенным, хромосомным или мультифакторным, а также информацию о пенетрантности и экспрессивности «проблемного» гена, о наличии генокопий, фенокопий, о генетической гетерогенности. Генетический риск в своем количественном выражении - величина переменная. Так, в браках гетерозигот-носителей патологических аллелей риск рождения ребенка с аутосомно-рецессивным или аутосомно-доминантным наследственным заболеванием равен 25 и 50% соответственно.
Риск рождения генетически «проблемного» по состоянию здоровья ребенка не выше 5% оценивается как низкий, до 10% - как повышенный, до 20% - как средний и свыше 20% - как высокий.
Исходя из величины генетического риска, формулируется медико-генетический прогноз, и путем соотносительной оценки «пользы»
(рождается здоровый ребенок) и «потерь» (рождается больной ребенок) определяется цена, которую должны будут заплатить родители в случае появления на свет генетически «проблемного» потомства. Исключительное право на окончательное решение принадлежит родителям, тогда как в функцию сотрудника медико-генетической консультации (здесь - медицинское учреждение) входит в доступной форме объяснить им медицинские последствия конкретной генетической ситуации.
В общении с консультируемыми врачу-генетику приходится учитывать целый ряд обстоятельств, различающихся от ситуации к ситуации. Так, в браках гетерозигот-носителей рецессивных патологических (патогенных) аллелей риск рождения ребенка с генетическим заболеванием в 25% соответствует максимальному. На самом деле у таких родительских пар, причем с вероятностью в 75%, рождаются здоровые дети. Необходимо, следовательно, говорить о диапазоне риска от 0 до 25%. Риск рождения генетически «проблемного» ребенка у гетерозиготных родителей-носителей патогенных рецессивных аллелей в диапазоне от 0% до 25% характеризует каждую отдельную беременность и не зависит от того, какой ребенок - больной или здоровый - был рожден ранее. Необходимо учитывать возраст родителей. Так, у женщин в возрасте 35 лет и старше увеличен риск рождения ребенка с болезнью Дауна (трисомия по хромосоме 21). Повышение риска рождения потомства с определенной наследственной патологией по мере увеличения возраста родителей, включая отцов, предполагается для ряда болезней, в том числе моногенной природы. По мере увеличения возраста отца растет, например, вероятность рождения ребенка с ахондроплазией (описаны разные формы заболевания с локализацией мутантных генов - 4p14-p16, Xq28, Xp22.32).
Причину роста генетического риска с увеличением возраста родителей многие врачи-генетики видят в явлении «перезревания гамет»: появление и накопление в гаметах (и женских, и мужских) изменений за отрезок времени между их созреванием и моментом оплодотворения. Предположительно чем длиннее названный отрезок, тем ниже вероятность физиологического оплодотворения и тем выше частота аномалий развития, в частности, генетической природы. Также предположительно факт «перезревания» половых клеток связывают с десинхронизаци-ей процессов овуляции (разрыв зрелого граафова фолликула в яичнике
женщины детородного возраста с выбросом в брюшную полость яйцеклетки) и оплодотворения. Причины такой десинхронизации разнообразны: гормональные изменения в преклимактерическом периоде жизни женщины, сниженная, возможно, вследствие имевшего место воспаления, проходимость маточных труб, приводящая к задержке продвижения яйцеклетки и сперматозоидов навстречу друг другу, пониженная двигательная активность спермиев.
Описаны случаи, когда наличие в семье больного (в частности, эпилепсией) ребенка повышает риск рождения в последующих беременностях потомства с другой генетической патологией - гемофилией.
При неблагоприятном медико-генетическом прогнозе, если риск появления на свет больного ребенка оценивается как высокий, а рождение ребенка рассматривается как безусловный семейный интерес, ставится вопрос о необходимости пренатальной (или даже предымплантацион-ной) оценки генетической конституции зачатого и/или осуществляющего развитие ребенка.
5.2.2.8-а. Генетический груз как биомедицинское явление: популяционный и индивидуально-семейный аспекты. Евгеника в исторический период молекулярно-генетических и геномных технологий
Выше необходимость МГК связывалась с наличием популяционно-го генетического груза. Такой груз в гено(аллело)фондах популяций (групп) людей есть. Вместе с тем выше в примерах наследственных болезней, иллюстрирующих неблагоприятное действие популяцион-ного генетического груза, речь шла о снижении жизнеспособности и нарушениях здоровья отдельных лиц, что требует пояснения. В своем конкретном выражении популяционный генетический груз проявляется в снижении уровня здоровья некоторого числа членов популяции в связи с функционально-генетической «дефектностью» их генотипов. Количество пораженных и характер наследственной патологии от популяции к популяции различаются. Так, в большинстве человеческих популяций один пациент, страдающий болезнью Тея-Сакса, приходится на 360 000 населения, тогда как среди евреев-ашкенази один такой пациент приходится в среднем на 3600 человек. Среди евреев-ашкенази родом из Польши или Литвы один пациент с болезнью Тея-Сакса приходится на 5000 человек. Известны человеческие популяции, в которых регистрируется сверхвысокая частота обнаружения лиц с определенным наследственным дефектом. Так, в некоторых этнических группах один мужчина с недостаточностью эритроцитарного фермента глюкозо-6-
фосфатдегидрогеназы, Г6ФД (признак Х-сцепленный, Kq28) приходится на каждые 4-20 лиц мужского пола. Таким образом, генетический груз можно определить как вероятность появления конкретной наследственной патологии у обратившегося за консультацией или у его потомков (индивидуально-семейный аспект). При этом человек, обращающийся за медико-генетической консультацией, является членом конкретной популяции людей (популяционный аспект), что обязательно принимается в расчет.
Наличие индивидуально-семейного и популяционного аспектов требует корректного отношения к определению стратегических задач, которые ставятся перед МГК как одним из видов специализированной медицинской помощи населению. С одной стороны, такая задача может состоять в снижении риска или недопущении рождения определенным лицом и/или в конкретной семье генетически «проблемного» ребенка. C другой стороны, речь может идти о генетическом оздоровлении населения страны или региона, отдельных популяций или групп населения, человечества в целом.
Первая из названных выше задач решаема. Конкретные меры, предпринимаемые для ее решения, различны. В одних случаях используют методы пассивной (первичной - предупреждение зачатия больного ребенка или вторичной - прерывание беременности по медико-генетическим показаниям) профилактики появления лиц с наследственной патологией, такие, как отказ от близкородственных браков или браков гетерозиготных носителей, исключение наступления нежелательной беременности путем отказа от деторождения вообще и/или ограничение деторождения возрастом 30-35 лет, прерывание беременности по результатам дородовой диагностики, исключение имплантации «проблемных» бластоцист по результатам доимплантацион-ной диагностики. Перечисленные выше методы могут быть сведены в группу под общим названием - планирование семьи. Первичная профилактика включает также мероприятия по улучшению среды жизни человека: жесткий контроль присутствия и концентрации в среде мутагенов и тератогенов. Необходимость такого контроля вытекает из данных расчетов, свидетельствующих о том, что порядка 20% наследственных болезней в каждом поколении людей обусловлены новыми мутациями.
В других случаях применяют методы активной (третичной или нормокопирования, если речь идет о заболеваниях строго генетической природы) профилактики появления лиц с наследственной
или мультифакторной патологией, такие, как исключение из среды жизни провоцирующих или «разрешающих» патогенных факторов с целью при «проблемном» генотипе получить нормальный фенотип, путем, например, перевода ребенка на безфенилаланиновую диету - см. здесь же, выше. Суть данного подхода, по мнению ряда генетиков, состоит в управлении экспрессией генов. Возможность активно формировать хорошие качества, исправлять или не допускать генетически обусловленных патологических проявлений у людей путем изменения условий среды жизни (диета, лекарства, воспитание и др.) в середине 20-х годов ХХ в. обосновывал Н.К. Кольцов, опираясь на открытия классической генетики своего времени (была установлена зависимость фенотипиче-ского проявления генов от характеристик генотипической и внешней среды, в профессиональный генетический обиход вошли понятия экспрессивности и пенетрантности, специфичности действия генов). Для обозначения указанного подхода он предложил термин «евфеника». Введением этого термина подчеркивалась принципиальная разница между двумя научно-практическими подходами к решению проблемы генетического оздоровления людей - евгеникой и евфеникой. Это было принципиально важным и своевременным, так как в некоторых странах, исходя из евгенической идеологии того исторического периода, предпринимались практические шаги (включая принятие соответствующих законов), ведущие к насильственному ограничению деторождения представителями некоторых категорий граждан, к примеру, страдающих наследственными, психическими и рядом других заболеваний или же принадлежащих к определенным социальным группам.
Перспективы решения второй из названных выше стратегических задач оцениваются в свете достижений молекулярной генетики, клеточной биологии и биотехнологии как реальные, но неопределенные по времени. Они зависят от успехов научно-практических разработок в таких областях, как генная и клеточно-тканевая инженерия, нанобио-медицинские технологии.
В случае генной инженерии речь идет о создании биотехнологических конструкций, содержащих необходимую для обеспечения генотерапевтического эффекта генетическую информацию (другими словами, требуемый ген, выделенный из клеток генетически здорового субъекта или синтезированный в лаборатории), о введении таких конструкций в организм пациента с наследственным заболеванием и о функциональной активации (актуализации) введенного генетического материала. Проблема введения биотехнологической конструкции ре-
шается разными способами. В одних протоколах используют так называемые векторы (проводники), функцию которых обычно выполняет специально приготовленная ДНК, в других протоколах - клетки пациента в условиях ex vivo (то есть вне организма) генетически модифицируют, удаляя патогенный аллель - технология «knock out» или вводя требуемый аллель - технология «knock in», после чего эти клетки возвращают в организм больного. На сегодняшний день в мире прошло апробацию (в испытаниях приняли участие порядка 750 пациентов; объекты генотерапии - тяжелые комбинированные иммунодефициты, му-ковисцидоз, миодистрофия Дюшена, семейная гиперхолистеринемия, онкологические заболевания) и утверждение более 150 генотерапев-тических протоколов. Поставлен, но еще ждет своего решения в отношении человека вопрос о «терапии зародышевого пути как такового», т.е. о технологиях, которые позволили бы исключить или резко снизить образование генетически «проблемных» половых клеток. Еще одно направление генной инженерии - получение антисмысловых олигонукле-отидов из 15-20 мономеров, способных благодаря комплементарному (высокоизбирательному) взаимодействию соединяться с ДНК патогенных аллелей человека и блокировать их экспрессию - антисмысловая генотерапия.
Клеточно-тканевая инженерия представляет собой основу регенеративной медицины (см. п. 3.2) и состоит в приготовлении из стволовых клеток и/или их производных клеточного трансплантата (клеточного продукта). Трансплантат (продукт) вводится в организм пациента, вызывая положительный терапевтический эффект. Названный эффект может быть следствием замещения собственных функционально «дефектных» клеток больного функционально полноценными клетками трансплантата (опыт гематологии: анемия Фанхони) либо вводимые пациенту стволовые и/или прогениторные (предшественники) клетки стимулируют собственные клетки, которые обеспечивают восстановление дефицита функции.
Используемые современной медицинской генетикой подходы и методы можно разделить на две группы. В одну из них включаются манипуляции, позитивный эффект которых сопряжен с удалением из гено(аллело)фонда «проблемного» патогенного аллеля (запрет на определенные категории браков и деторождение, прерывание беременности по медико-генетическим показаниям, зачатие в пробирке с выбором для имплантации генетически «беспроблемной» бластоцисты, некоторые варианты генетической модификации клеток), в другую - манипуля-
ции, позитивный эффект которых распространяется на фенотип, тогда как патогенный аллель остается в гено(аллело)фонде и, следовательно, может быть унаследован потомством (методы активной профилактики, направленные на изменение в требуемом направлении условий среды жизни, антисмысловая генотерапия). Методы, относящиеся к первой группе, в плане решения задачи генетического оздоровления людей более радикальны, так как действуют и на индивидуально-семейном, и на популяционном уровне. Вместе с тем именно они вызывают серьезные возражения по соображениям разной природы - этическим, религиозным, этническим и др. Узаконенное их применение многими рассматривается как действие, связанное с нарушением прав человека. Методы, относящиеся ко второй группе, вызывают меньше споров и критики, так как они не нарушают установленных природой генетических (биоинформационных) отношений между поколениями. Предложение о целесообразности их применения обычно исходит от врача.
Расширение круга манипуляций с наследственным материалом человека послужило основанием к оживлению евгенической идеи. Необходимость усилий, направленных на генетическое оздоровление человечества, евгеники ХХI в. связывают с ответственностью ныне живущих людей перед людьми будущих поколений. Особое внимание в этом плане привлекают такие технологии, как экстракорпоральное оплодотворение и клонирование, а также развитие функциональной геномики. Достижения последней помогут оптимизировать создание геномно-протеомных портретов (или паспортов) людей. Дело в том, что, хотя порядок следования пар нуклеотидов в ДНК всех хромосом человека установлен, знания о функционировании подавляющего большинства сайтов ДНК отсутствуют или недостаточны. Усовершенствованные технологии экстракорпорального оплодотворения и доимплантационной диагностики наследственной патологии, а также клонирования дадут, по мнению современных евгеников, возможность отказаться от главного постулата негативной евгеники начала минувшего столетия - добиваться генетического оздоровления людей путем регулируемого снижения рождаемости среди генетически менее удачных и ориентироваться на постулат позитивной евгеники - добиваться генетического оздоровления людей путем регулируемого повышения рождаемости у тех, кто наделен генетическими преимуществами. Одним из практических следствий таких представлений было, в частности, создание банка спермы нобелевских лауреатов. При этом замалчивается известный факт существования людей (иногда их называют гениями-идиотами),
которые с трудом справляются с элементарными каждодневными жизненными задачами, будучи в то же время выдающимися музыкантами, скульпторами или же обладая способностью со скоростью калькулятора перемножать в уме многозначные цифры. Можно заключить, что у этих людей отдельный дар не компенсирует недостатка остальных способностей. Вместе с тем в науке о человеке существует понятие «общее умственное развитие».
Вопросы для самоконтроля
1. В чем состоят особенности человека как объекта генетики?
2. Какие методы используются в антропогенетике, и в чем их суть?
3. Перечислите особенности родословных при различных типах моногенного наследования.
4. Что такое медико-генетическое консультирование, и каковы его задачи?