Биоорганическая химия : учебник / Н. А. Тюкавкина, Ю. И. Бауков, С. Э. Зурабян. - 2010. - 416 с.
|
|
|
|
ГЛАВА 4. РЕАКЦИОННАЯ СПОСОБНОСТЬ СПИРТОВ, ТИОЛОВ И АМИНОВ
4.1. Общая характеристика
4.1.1. Одноатомные спирты, фенолы и их производные
Производные алифатических углеводородов, в которых один или несколько атомов водорода замещены гидроксильной группой, называют спиртами; аналогичные производные ароматических углеводородов называют фенолами.
В зависимости от числа гидроксильных групп спирты и фенолы бывают одно-, двух-, трехатомными и т. д. Спирты, содержащие две гидроксильные группы или более, называют многоатомными.
Одноатомные спирты и фенолы имеют общие формулы:
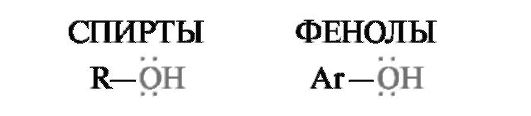
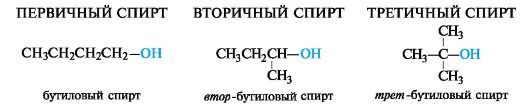
В зависимости от того, у какого атома углерода находится функциональная группа, спирты называют первичными, вторичными или третичными.
Спирты имеют аномально высокие температуры кипения (табл. 4.1), что объясняется их межмолекулярной ассоциацией с помощью водородных связей (см. 2.2.3).
В результате образования водородных связей с молекулами воды низшие спирты (метанол, этанол, пропанолы) смешиваются с водой в любых соотношениях. С увеличением молекулярной массы растворимость спиртов в воде уменьшается.
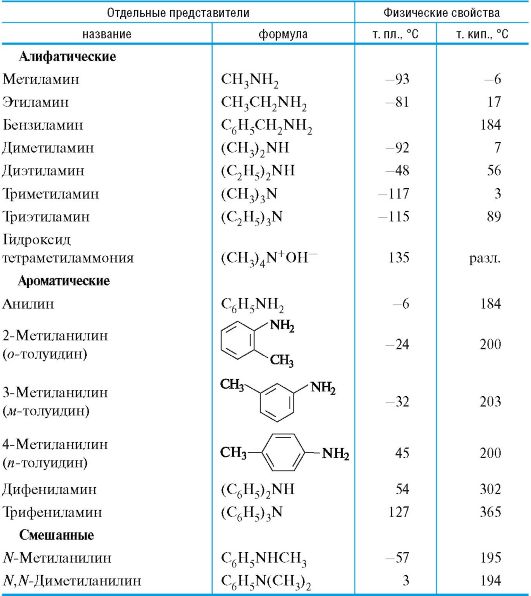
Метанол (метиловый спирт) является сильным ядом.
Этанол (этиловый спирт) используется для приготовления настоек и в качестве антисептического средства.
Таблица 4.1. Одноатомные спирты и фенолы
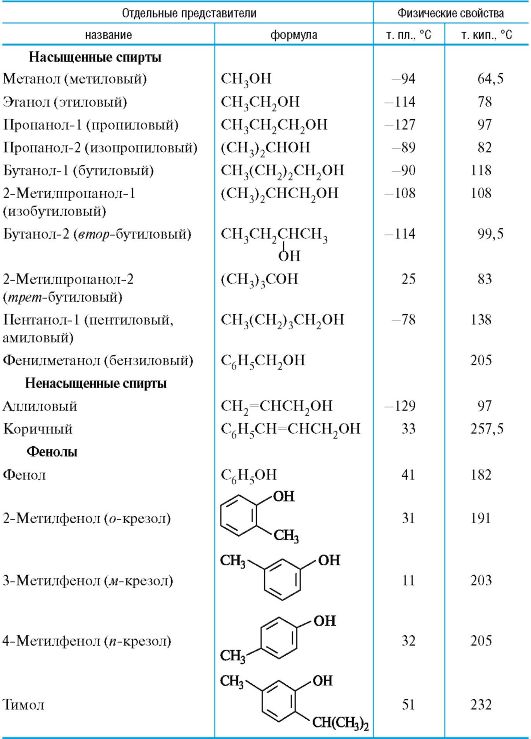
Бутиловые и амиловые спирты известны как основные компоненты сивушных масел.
Фенол (карболовая кислота) был первым антисептиком, введенным в хирургию. Токсичен и может вызвать ожог кожи.
Тимол содержится во многих эфирных маслах и оказывает антисептическое действие.
Соединения, в которых атом кислорода связан с двумя углеводородными радикалами, называют простыми эфирами.
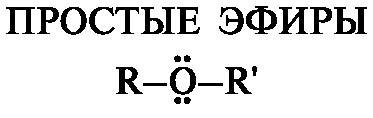
Простые эфиры можно рассматривать как производные спиртов или фенолов, в которых атом водорода гидроксильной группы замещен алкильным или арильным радикалом. Простые эфиры бывают также ненасыщенными (например, виниловые) и циклическими. Виниловые эфиры CH2=CH-OR являются производными винилового спирта. Виниловый спирт CH2=CH-OH - простейший представитель соединений, называемых енолами, поскольку в их составе у атома углерода двойной связи (-ен) находится гидроксильная группа (-ол). Енолы - крайне неустойчивые соединения, например виниловый спирт в момент его образования изомеризуется в уксусный альдегид.
Простейший из циклических эфиров - этиленоксид - образуется при каталитическом окислении этилена.
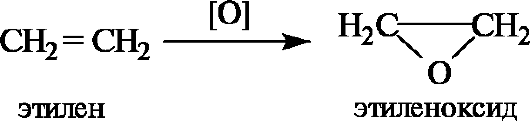
Для простых эфиров несложного строения (с относительно простыми радикалами) удобна радикально-функциональная номенклатура (табл. 4.2). Названия эфиров формулы ROR' по заместительной номен- клатуре строятся добавлением составного префикса алкокси- (сокраще- ние от алкил + окси) или арилокси-, обозначающего заместитель R'O, к названию углеводорода, соответствующего радикалу R родоначальной структуры, например 1-метоксипропан для эфира СН3ОСН2СН2СН3.
Простые эфиры практически не смешиваются с водой, хорошо растворяют многие органические вещества и поэтому часто исполь- зуются как растворители, но они весьма огнеопасны. В частности, диэтиловый эфир легко воспламеняется, если рядом находится открытый огонь или просто сильно нагретый предмет.
Таблица 4.2. Простые эфиры
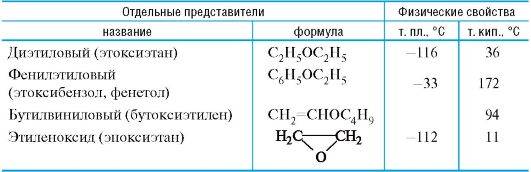
Диэтиловый эфир применяют для ингаляционного наркоза, а также как растворитель веществ животного и растительного происхож- дения. При хранении на воздухе диэтиловый эфир легко образует взрывчатый гидропероксид (см. 3.2.1).
Бутилвиниловый эфир используют для получения полимера, применяемого в качестве ранозаживляющего средства, под названием винилин («бальзам Шостаковского»).
Структурные фрагменты виниловых эфиров также встречаются в некоторых природных соединениях, в частности в сложных липидах плазмалогенах (см. 10.4.1).
4.1.2. Тиолы и их производные
Серосодержащие аналоги спиртов называют тиолами, фенолов - тиофенолами.
Тиолы и тиофенолы содержат функциональную группу SH.
Серосодержащие аналоги простых эфиров и органических пероксидов называют сульфидами и дисульфидами соответственно.

Тиолы и сульфиды можно также рассматривать как производные сероводорода H2S, у которого один или оба атома водорода замещены органическим радикалом (табл. 4.3).
В отличие от спиртов, тиолы не склонны к образованию межмолекулярных водородных связей, так как связь S-H практически неполярна из-за близких значений электроотрицательности серы и водорода (см. 2.2.1).
Таблица 4.3. Тиолы, сульфиды и дисульфиды
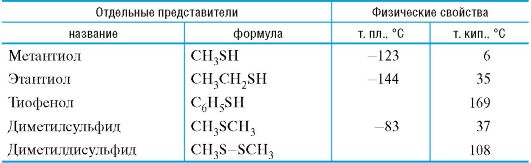
Тиолы и их производные обладают крайне неприятным запахом. Низшие тиолы используют в качестве пахучей добавки к природному газу, не имеющему собственного запаха.
4.1.3. Амины
Производные аммиака, в котором один, два или три атома водорода замещены органическими радикалами, называют аминами.
В зависимости от числа замещенных атомов водорода различают первичные, вторичные и третичные амины. Обратим внимание на иное использование этих понятий в применении к аминам, где они означают число органических радикалов, с которыми связан атом азота.
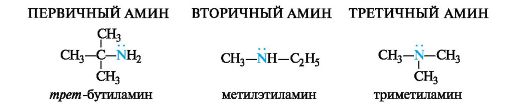
В зависимости от природы органических радикалов амины относят к алифатическому или ароматическому ряду, также они могут быть и смешанными (табл. 4.4).
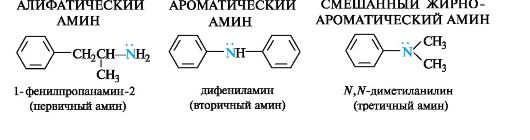
Таблица 4.4. Амины
Существуют и соли четырехзамещенного аммония, например хлорид тетраметиламмония (CH3)4N+C- . Соответствующее ему основание, гидроксид тетраметиламмония(CH3)4N+OH , представляет собой сильное основание, аналогичное гидроксидам щелочных металлов, так как связь с гидроксильной группой здесь ионная.
Аминогруппа при физиологических значениях рН чаще всего протонирована. В организме образуются так называемые биогенные амины путем декарбоксилирования α-аминокислот (см. 12.1.5).
Многие амины довольно токсичны. Анилин и другие ароматические амины являются кровяными и нервными ядами, легко проникают в организм человека через кожу или при вдыхании паров.
4.1.4. Реакционные центры в спиртах, тиолах и аминах
В спиртах, тиолах и аминах алифатического ряда sp3-гибридизованный атом углерода связан σ-связью с гетероатомом (O, S или N) соответствующей функциональной группы. Поскольку согласно шкале Полинга (см. 2.2.1) гетероатомы, как правило, имеют более высокую электроотрицательность, чем углерод, то электроны σ-связи смещены в их направлении, т. е. функциональные группы проявляют отрицательный индуктивный эффект. Каждый из гетероатомов имеет по крайней мере одну пару n-электронов, обусловливающую его основные свойства. Распределение электронной плотности в рассматриваемых соединениях с учетом -/-эффекта гетероатома и наличия у него неподеленной пары электронов представлено на схеме 4.1.
Схема 4.1. Реакционные центры в молекулах спиртов, тиолов и аминов
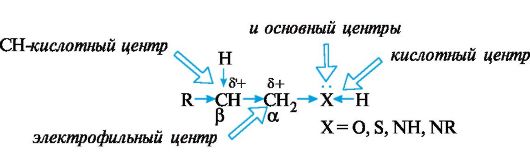
Такое распределение электронной плотности обусловливают несколько реакционных центров:
• кислотный центр в составе группы ХН;
• основный центр - гетероатом Х (кислород, сера или азот с неподеленной парой электронов);
• электрофильный центр - α-атом углерода, непосредственно связанный с гетероатомом Х;
• нуклеофильный центр - гетероатом Х;
• СН-кислотный центр - с участием β-атома углерода, связанного с электрофильным центром.
4.2. Кислотные и основные свойства
По теории Брёнстеда (протолитической теории), кислотность и основность соединений обусловливаются переносом протона Н+.
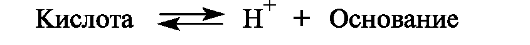
Кислота и основание образуют сопряженную* кислотно-основную пару, в которой чем сильнее кислота, тем слабее сопряженное ей основание, и напротив, чем сильнее основание, тем слабее сопряженная ему кислота.
Кислоты Брёнстеда (протонные кислоты) - нейтральные молекулы или ионы, способные отдавать протон (доноры протонов).
Основания Брёнстеда - нейтральные молекулы или ионы, способные присоединять протон (акцепторы протонов).
Теоретически любое соединение, в состав которого входит хотя бы один атом водорода, может отдавать его в виде протона и, следовательно, проявлять свойства кислоты. Основаниями могут быть и нейтральные молекулы, в состав которых входит гетероатом (обычно атом кислорода, азота или серы), имеющий неподеленную пару электронов, например спирты ROH, амины RNH2, тиолы RSH.
В органической химии часто используется путь сравнительного (качественного) сопоставления свойств одного соединения с другим или внутри какой-либо группы соединений.
4.2.1. Кислотные свойства
В зависимости от природы элемента, с которым связан протон, различают следующие кислоты Брёнстеда (табл. 4.5):
• OH-кислоты (спирты, фенолы, карбоновые кислоты);
• SH-кислоты (тиолы, тиофенолы);
• NH-кислоты (амины);
• CH-кислоты (углеводороды и их производные).
* Этот термин не имеет ничего общего с понятием «сопряжение», которое было введено ранее (см. 2.3). «Сопряженный» означает здесь «дополняющий».
Таблица 4.5. Органические кислоты по Брёнстеду
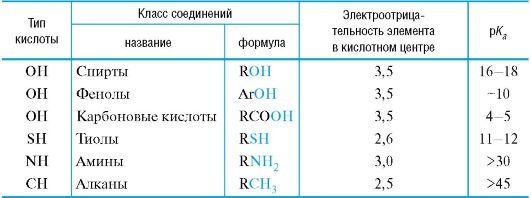
Элемент и связанный с ним потенциально способный к отщеплению атом водорода называют кислотным центром.
Большинство органических соединений, за исключением карбоновых кислот, проявляют чрезвычайно низкую кислотность, которую нельзя обнаружить с помощью индикаторов.
Для прогнозирования кислотных свойств используют качественный подход, основанный на оценке возможности отщепления протона, которая возрастает с увеличением электроотрицательности элемента в кислотном центре. Так, по силе кислотности однотипные соединения располагаются в следующий ряд: OH-кислоты > NH-кис- лоты > CH-кислоты.
Другим, зачастую более важным критерием, позволяющим оценивать кислотные свойства близких по строению соединений, является стабильность аниона, образующегося при ионизации кислоты.
Чем стабильнее анион, тем сильнее сопряженная кислота.
Стабильность аниона определяется степенью делокализации в нем отрицательного заряда. В общем случае она зависит от:
• природы атома в исходном кислотном центре;
• возможности стабилизации аниона путем сопряжения;
• характера органического радикала, связанного с исходным кислотным центром.
Эти факторы могут действовать в одинаковом или противоположном направлении, и в каждом случае их нужно рассматривать в совокупности.
Одноатомные спирты являются нейтральными веществами. Они практически не взаимодействуют со щелочами, но вступают в реакцию со щелочными металлами, образуя соли - алкоксиды металлов.
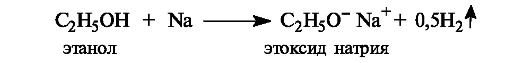
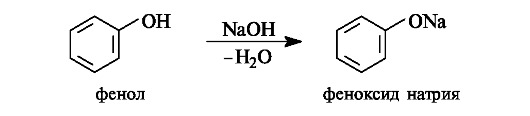
Фенолы по сравнению со спиртами проявляют более заметные кислотные свойства (см. табл. 4.5). Так,
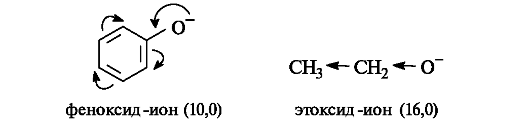
Таким образом, различие в кислотных свойствах спиртов и фенолов определяется типом радикала, связанного с гидроксильной группой.
Обладая относительно невысокой кислотностью, фенолы образуют соли только с сильными основаниями, например гидроксидом натрия, а со слабыми основаниями, например гидрокарбонатом натрия NaHCO3, в реакцию не вступают.
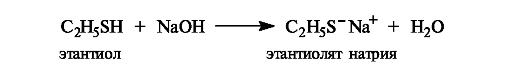
Тиолы обладают большей кислотностью, чем соответствующие спирты (см. табл. 4.5), поскольку тиолят-ионы стабильнее алкоксидионов вследствие большего атомного радиуса серы по сравнению с радиусом кислорода (фактор поляризуемости) и, следовательно, более эффективной делокализации отрицательного заряда на атоме серы.
В соответствии с достаточно высокой кислотностью тиолы при обработке водным раствором щелочи превращаются в соли.
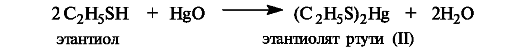
При действии солей тяжелых металлов, в частности ртути или свинца, тиолы образуют труднорастворимые соли.
Кислотность аминов и тем более углеводородов значительно ниже кислотности спиртов (см. табл. 4.5). Причиной этого является меньшая электроотрицательность атомов азота и углерода по сравнению с атомом кислорода. Это снижает возможность отщепления протона и в то же время приводит к меньшей стабильности анионов (сопряженных оснований) с зарядом на атомах азота и углерода.
Совокупность всех перечисленных факторов определяет силу кислоты в каждом конкретном случае. С некоторым допущением можно сказать, что кислотность органических соединений возрастает в ряду: CH-кислоты < NH-кислоты < ОН-кислоты < SH-кислоты.
4.2.2. Основные свойства
Для образования ковалентной связи с протоном основания Брёнстеда должны предоставлять либо неподеленную пару электронов, либо электроны π-связи. В соответствии с этим различают п-основания (табл. 4.6) и π-основания.
п-Основания могут быть нейтральными веществами или отрицательно заряженными частицами. Как правило, анионы обладают более сильно выраженными основными свойствами, чем нейтральные молекулы. Отсюда амид-ион NH2-, алкоксид-ион RO-, тиолят-ион RS-,
Таблица 4.6. Органические я-основания по Брёнстеду
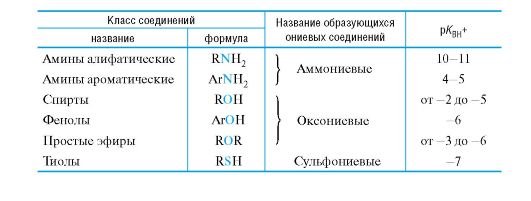
гидроксид-ион HO- по основности превосходят аммиак NH3, спирты ROH, тиолы RSH, воду H2O соответственно.
В π-основаниях, к которым относятся алкены и арены, центром основности, т. е. местом присоединения протона, являются электроны π-связи. Это очень слабые основания, так как протонируемые пары электронов несвободны.
В результате присоединения протона к нейтральной молекуле я-основания образуются солеобразные ониевые соединения.
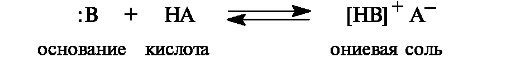
В зависимости от природы гетероатома могут образовываться соли:
• аммониевые (центр основности - атом азота)

• оксониевые (центр основности - атом кислорода)
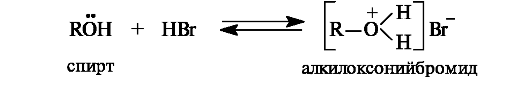
• сульфониевые (центр основности - атом серы). Атом серы является очень слабым центром основности, поэтому тиолы практически не проявляют основных свойств. Для качественной оценки основности органических соединений привлекаются те же факторы, что и для оценки кислотности, с той лишь разницей, что влияние этих факторов на основность противоположно тому влиянию, которое они оказывали на кислотность. Основность соединений с разными гетероатомами зависит от электроотрицательности и поляризуемости элемента в основном центре.
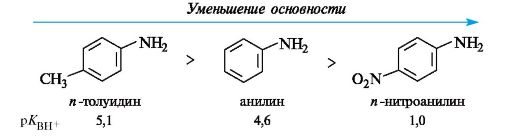
Наибольшей основностью среди органических соединений обладают амины (см. табл. 4.6). При сравнении атомов азота и кислорода можно отметить, что более электроотрицательный кислород прочнее удерживает неподеленную пару электронов и менее склонен присоединять протон, чем азот. Несмотря на еще меньшую электроотрицательность серы, электронная плотность этого атома рассредоточена
в большем объеме и поэтому атом серы слабее связывает протон. Таким образом, тиолы - более слабые основания, чем амины и даже чем спирты. Основность рассматриваемых классов соединений с одинаковыми заместителями при гетероатоме увеличивается в ряду:
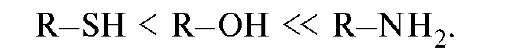
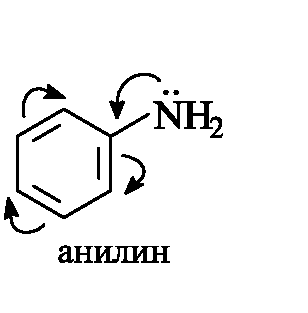
Ароматические амины значительно менее основны, чем алифатические, что связано с делокализацией неподеленной пары электронов азота благодаря р,π-сопряжению с ароматической системой и, следовательно, с меньшей доступностью для атаки протоном. Тем не менее, анилин легко образует соли с минеральными кислотами.
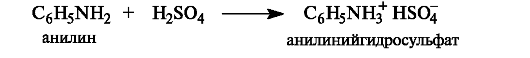
Влияние заместителей в углеводородной части молекулы на основность противоположно рассмотренному выше влиянию их на кис- лотность. Электронодонорные заместители увеличивают основность, электроноакцепторные - уменьшают.
Спирты и простые эфиры как очень слабые основания могут протонироваться только сильными кислотами. Образующиеся соли легко гидролизуются.
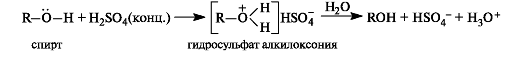
4.3. Нуклеофильное замещение у насыщенного атома углерода
Как следует из распределения электронной плотности (см. схему 4.1), в молекулах спиртов, тиолов и аминов одновременно сосуществуют заметно выраженные электрофильный и нуклеофильный
центры. Такая особенность электронного строения, в свою очередь, влечет за собой двойственный характер их участия в реакциях нуклеофильного замещения SN.
В одном случае это будет замещение у sp3-гибридизованного атома углерода в составе молекул спирта, тиола или амина, выступающих в роли субстратов и предоставляющих для участия в реакции «свой» электрофильный центр.
В другом случае это будет нуклеофильное замещение у sp3--гибридизованного атома углерода в составе реагента, который атакуется молекулами спиртов, тиолов или аминов за счет участия их нуклеофильных центров.
Для прогнозирования результата реакции важное значение имеет качественная оценка нуклеофильности участников процесса.
Нуклеофильность можно определить как способность к взаимодействию с атомом углерода, несущим частичный или полный положительный заряд, т. е. как сродство к углероду.
Оценочными критериями нуклеофильности служат обобщения:
• нуклеофильность заряженного нуклеофила выше, чем соответствующей нейтральной молекулы, например:
HO- > H2O; RO- > ROH; RS- > RSH; RCOO- > RCOOH;
• нуклеофильность уменьшается с увеличением электроотрицательности элемента в пределах одного периода Периодической системы, например, NH3 >H2O > HF. В первом приближении нуклеофильность изменяется параллельно основности (см. 4.2.2); исключение составляют серосодержащие нуклеофилы.
Таким образом, в ряду нейтральных нуклеофилов наиболее сильными являются амины. Спирты, вода и особенно фенолы обладают гораздо более низкой нуклеофильностью и в качестве нуклеофилов используются обычно в виде соответствующих анионов.
Общее описание механизма реакции. Электрофильный центр предопределяет возможность нуклеофильной атаки (см. схему 4.1).
В ходе этих реакций атакующий реагент (нуклеофил) предоставляет субстрату свою пару электронов, за счет которой образуется связь между атомом углерода субстрата и нуклеофилом. Группа Х (нуклеофуг), называемая также уходящей группой, отщепляется с парой электронов. По отношению к реагенту это превращение можно рассматривать как алкилирование нуклеофила.
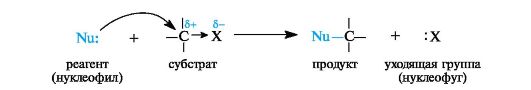
В схеме реакции намеренно не обозначены заряды: нуклеофил и нуклеофуг могут быть заряжены отрицательно или быть нейтральными, а субстрат и продукт - либо нейтральными, либо заряженными положительно.
Реакция нуклеофильного замещения может быть обратимой, так как уходящая группа сама может проявлять нуклеофильные свойства.
Для успешного протекания реакции нуклеофильного замещения необходимо, чтобы уходящая группа была более стабильной и проявляла более слабую нуклеофильность по сравнению с атакующим нуклеофилом.
Лучшие уходящие группы - наиболее слабые основания (а соответствующие им сопряженные кислоты - наиболее сильные). К хорошим уходящим группам относятся галогенид-ионы.
В отличие от галогенид-ионов сильные основания, например гидроксид-ион НО , алкоксид-ион RO и амид-ион NH2 , являются плохими уходящими группами, поэтому их прямое нуклеофильное замещение осуществить не удается. В таких случаях используют преобразование плохой уходящей группы в хорошую уходящую группу. Для этого обычно в субстрате переводят уходящую группу в ониевую, чтобы в дальнейшем она отщепилась в виде нейтральной молекулы.
Вместе с тем спирты, амины, тиолы и сульфиды в реакциях нуклеофильного замещения сами могут быть нуклеофильными реагентами в результате либо присутствия в их молекулах нуклеофильного центра, либо образования анионов при разрыве связи гетероатом-водород.
Спирты как субстраты в реакциях нуклеофильного замещения. В спиртах превращение группы ОН в оксониевую достигается проведением реакции в условиях кислотного катализа. Уходящей группой в этой реакции является молекула воды, а в качестве субстрата выступает протонированная по кислороду молекула спирта.
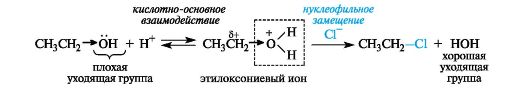
Спирты как реагенты в реакциях нуклеофильного замещения.
Спирты могут участвовать в реакциях в роли нуклеофильных реагентов, например, по отношению к наиболее активным третичным галогенопроизводным углеводородов.
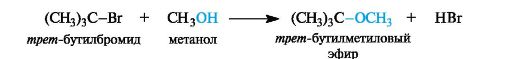
Со вторичными и особенно с первичными галогенопроизводными необходимы более сильные нуклеофилы - анионы спиртов (алкоксиды).

Амины как реагенты в реакциях нуклеофильного замещения. Следует отметить некоторые особенности реакций, в которых нуклеофильным реагентом является аммиак или амины. Рассмотрим это на примере алкилирования аммиака.
Первоначальным продуктом реакции должна быть замещенная аммониевая соль.
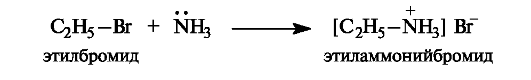
Аммиак проявляет не только нуклеофильные, но и основные свойства, заключающиеся в способности отщеплять протон от образовавшейся аммониевой соли, которая при этом превращается в первичный амин (этиламин). Этот амин в свою очередь также является нуклеофилом (даже более сильным, чем аммиак) и способен подвергаться алкилированию. При этом сначала образуется соль вторичного амина, а затем и сам амин (диэтиламин) и т. д. В итоге реакция галогеноалканов с аммиаком или аминами приводит к сложной смеси первичных, вторичных и третичных аминов и их солей.
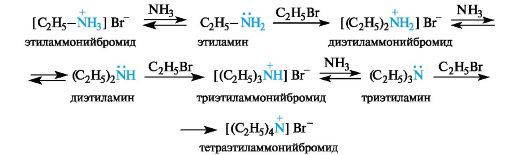
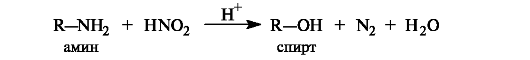
Группы NH2, NHR, NR2 представляют собой чрезвычайно плохие уходящие группы. Их замещение даже после превращения в ониевые ионы осуществить не удается. Однако нуклеофильное замещение группы NH2, например на группу ОН, в первичных аминах можно осуществить путем их взаимодействия с азотистой кислотой. Такая реакция называется дезаминированием.
Обзор реакций алкилирования как нуклеофильного замещения представлен на схеме 4.2.
Схема 4.2. Реакции алкилирования с участием спиртов, тиолов и аминов
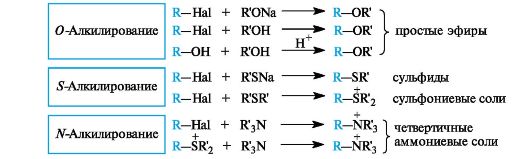
Как видно из схемы 4.2, в большинстве приведенных примеров реакций нуклеофильного замещения в качестве субстратов принимают участие галогенопроизводные углеводородов. В классической органической химии этим соединениям уделяется достаточно большое внимание. В биоорганической химии они по мере необходимости будут использоваться в качестве модельных соединений.
4.4. Реакции отщепления
Слабый СН-кислотный центр в производных углеводородов типа H-С-С-X (где Х - галоген, ОН, NR3+ и другие электроноакцепторные группы) предопределяет возможность его атаки основанием.
Поскольку каждый нуклеофил в то же время является и основанием, в субстратах, содержащих β-атомы водорода, с реакцией нуклеофильного замещения конкурирует реакция отщепления (элиминирования).
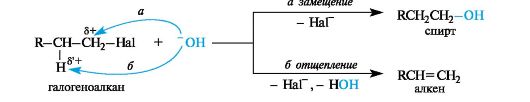
В определенных условиях такая реакция может стать основной и приводить к образованию алкенов. Наиболее часто это происходит при отщеплении галогеноводородов от алкилгалогенидов и воды от спиртов.
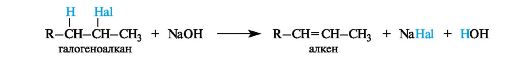
В реакциях отщепления галогеноводорода от алкилгалогенида или воды от спирта наблюдается преимущественное отщепление протона от атома углерода, содержащего минимальное число атомов водорода, т. е. от наименее гидрогенизированного атома углерода (правило Зайцева).
Так, дегидрогалогенирование алкилгалогенидов протекает под действием сильных оснований - концентрированного раствора гидроксида щелочного металла в спирте (спиртовая щелочь) или алкоксида (алкоголята) щелочного металла.
Дегидратация спиртов осуществляется при их нагревании в силь- нокислой среде, например в присутствии концентрированной серной или фосфорной кислоты.
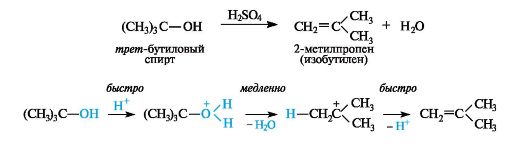
Легче всего элиминирование проходит от третичных алкилгалогенидов и спиртов. В случае вторичных и особенно первичных производных реакция протекает в значительно более жестких условиях. Первичные спирты при этом легче подвергаются конкурентной межмолекулярной реакции с образованием простых эфиров.
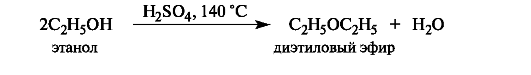
4.5. Окисление
Спирты (первичные и вторичные) по сравнению с углеводородами окисляются в значительно более мягких условиях. Альдегид, образующийся при этом из первичного спирта, легко окисляется далее в карбоновую кислоту.
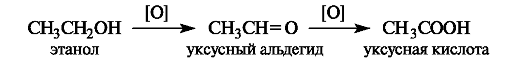
Особым случаем окисления является дегидрирование, когда субстрат теряет два атома водорода, что эквивалентно потере двух протонов и двух электронов (2Н+ и 2е-) или протона и гидрид-иона (Н и Н ).
Тиолы при окислении образуют ряд продуктов последовательного окисления - сульфеновые, сульфиновые и сульфоновые кислоты. В этом состоит их отличие от спиртов, у которых окислению подвергается атом углерода.
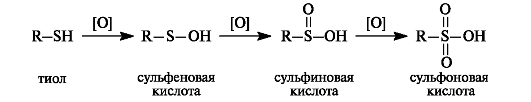
Использование мягких окислителей (пероксид водорода, кислород воздуха) приводит к образованию дисульфидов. Реакция окисления тиолов и обратный ей процесс восстановления имеют важное значение в биологических системах.
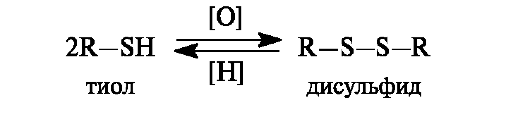
Примером дисульфида, участвующего в биохимическом окислении, может служить липоевая кислота, имеющая в составе пятичленное кольцо с дисульфидной группировкой. Восстановленная форма - дигидролипоевая кислота - представляет собой дитиол.
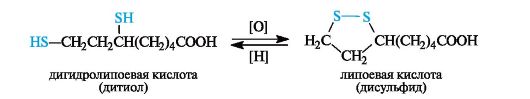
Сульфиды, как и тиолы, окисляются достаточно легко. Первичными продуктами окисления являются сульфоксиды, которые далее могут быть окислены в сульфоны.
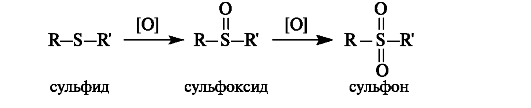
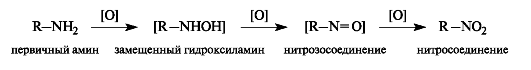
Амины легко окисляются; конечными продуктами при окислении первичных аминов RNH2 являются нитросоединения RNO2. На промежуточных стадиях образуются замещенный гидроксиламин RNHOH и нитрозосоединение RN=O.