Иммунология: структура и функции иммунной системы: учебное пособие / Р.М. Хаитов. - 2013. - 280 с.
|
|
|
|
Глава 9. ТОРМОЖЕНИЕ ИММУННОЙ СИСТЕМЫ
Для нормальной жизнедеятельности организма процессы торможения в иммунной системе так же необходимы, как и процессы её активации. Поясним на нескольких примерах: после уничтожения и выведения патогена из организма иммунный ответ естественным образом останавливается - развивается супрессия иммунного ответа, а иммунный ответ на собственные ткани организма предотвращает иммунная толерантность. Наконец, чрезвычайно важно торможение иммунной системы и при таких антропогенных вмешательствах, как трансплантация органов и тканей. Ключевые роли в торможении иммунной системы принадлежат двум механизмам: ингибирующим межклеточным взаимодействиям и апоптозу.
АПОПТОЗ
Апоптоз (от греч. apoptosis - листопад) - программированная (регулируемая) гибель клеток путём деградации её компонентов, включая конденсацию хроматина и фрагментацию ДНК, с последующим фагоцитозом макрофагами. Необходимые для апоптоза гены («программа смерти») есть в каждой клетке, но их транскрипция начинается только при получении клеткой сигнала к апоптозу.
• Апоптоз в иммунной системе
- В иммунной системе апоптоз развивается при удалении аутореактивных клонов иммунокомпетентных клеток, регуляции численности пролиферирующих клеточных популяций, повреждении генома клеток.
- Аномально повышенная устойчивость (резистентность) клеток к апоптозу играет важную роль в патогенезе аутоиммунных нарушений и злокачественных новообразований за счёт подавления процесса гибели дефектных и мутантных клеток: например, при аутоиммунном лимфопролиферативном синдроме угнетён
апоптоз лимфоцитов, что вызвано мутацией гена, кодирующего «рецептор смерти» - гликопротеин Fas.
- Аномально повышенная гибель клеток путём апоптоза сопровождает острые (инфекционные заболевания, ишемические повреждения), а также ряд хронических патологий (нейродегенеративные заболевания, синдром приобретённого иммунодефицита).
- В иммунной системе известны рецепторы, связывание которых с лигандами индуцирует апоптоз в клетке-носителе рецептора.
◊ Рецепторы DR (от Death Receptor), относящиеся к семейству рецепторов фактора некроза опухоли (TNFR). Известно 5 разновидностей молекул группы DR. Их лигандами являются мембранные или растворимые молекулы семейства фактора некроза опухоли:
- Fas (CD95, DR1), лиганд которого - FasL - экспрессирован на ДК тимуса и индуцирует апоптоз тимоцитов при негативной селекции. Тот же лиганд присутствует на мембране ЦТЛ и естественных киллеров и вызывает гибель клеток-мишеней;
- TNFRI (TNF Receptor-1, CD120a, DR2) - рецептор типа 1 для фактора некроза опухоли;
- DR3-DR6. Их лигандами являются: TRAIL (TNF-Related Apoptosis-Inducing Ligand) - для DR4 и DR5, TL1A и Tweak (TNF-related weak inducer of apoptosis) - для DR3 и N-APP -
для DR6.
◊ CD30 на тимоцитах или T-лимфоцитах. Эта молекула тоже участвует в негативной селекции, связываясь с лигандом CD30L на эпителии и ДК мозговой зоны тимуса.
◊ Ядерный рецептор для глюкокортикоидов, индуцирующих апоптоз тимоцитов при позитивной селекции в тимусе и, вероятно, апоптоз активированных лимфоцитов в периферических тканях.
• Факторы транскрипции, образующиеся при активации лимфоцитов, - АP-1, NFAT - способствуют экспрессии рецепторов, индуцирующих апоптоз, что приводит к гибели лимфоцитов после выполнения ими своих функций. Этот феномен получил название «индуцированной активацией смерти клеток» (AICD - ActivationInduced Cell Death).
• Гены, продукты которых предотвращают апоптоз: Bcl-2, Bcl-xL, Bcl-w, Mcl-1, ALG-3 и др. Транскрипция этих генов происходит при получении клеткой «сигнала на выживание». Для B-лимфоцитов таким сигналом служит связывание BCR с антигеном, для тимоцитов - удовлетворительное связывание TCR с MHC при позитивной селекции, для периферических T-лимфоцитов - постоянное узнавание эндогенных пептидов в комплексе с MHC.
• Макрофаги и ДК сорбируют и поглощают апоптозные тельца с помощью интегринов, молекул CD36 и рецепторов-«мусорщиков», а затем разрушают их содержимое до мелких метаболитов. При этом ДК способны экспрессировать на мембране комплексы этих метаболитов с молекулами MHC-I и MHC-II - это лежит в основе развития иммунного ответа на собственные повреждённые ткани и тканевые антигены чужеродных трансплантатов.
В зависимости от механизмов запуска выделяют митохондриальный (эндогенный) и рецепторный (экзогенный) апоптоз. Развитие или защита от апоптоза определяется балансом про- и противоапоптотических факторов семейства Вcl-2 в мембранах митохондрий (рис. 9-1). Вcl-2 и Bcl-xL являются противоапоптотическими факторами, постоянно связанными с мембранами митохондрий, а Bax, Bim, Bik, Bak и т.д. - проапоптотическими факторами, способными циркулировать в цитоплазме. Включение программы митохондриального апоптоза индуцируется определёнными сигналами из цитоскелета, в результате чего противоапоптотические факторы релокализуются, димеризуясь с молекулой Bcl-2 в митохондриальной мембране и нейтрализуют её антиапоптотический потенциал. Проапоптотические факторы формируют димеры, образующие пору в митохондриальной мембране, через которую из митохондрии в цитозоль поступает цитохром С. Apaf-1 (Apoptotic peptidase activating factor 1) связывает цитохром С, и к этому комплексу подсоединяется димер прокаспазы 9. Формирующаяся надмолекулярная структура называется апоптосомой. В составе апоптосомы прокаспаза 9 превращается в активную каспазу 9 путём аутокаталитического отщепления N-концевого участка.
Каспазы - это сериновые протеазы, разрывающие полипептидную связь после остатка Asp (отсюда их название). Выделяют инициаторные и эффекторные каспазы. Каспаза 9 относится к инициаторным каспазам. Она, как и другие инициаторные каспазы, отщепляет фрагменты эффекторных каспаз (чаще всего каспазы 3), переводя их в активную форму. Митохондриальная форма апоптоза играет основную роль в
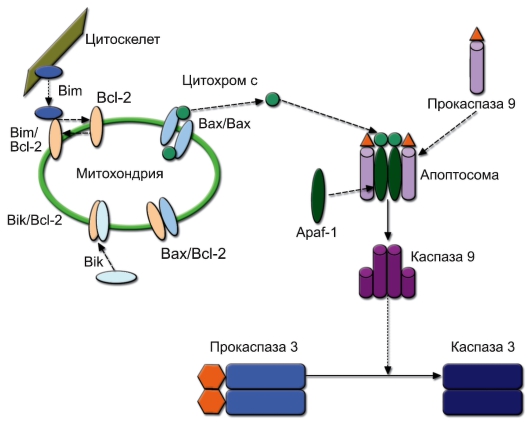
Рис. 9-1. Митохондриальный механизм запуска апоптоза. Пояснения см. в тексте
процессах морфогенеза, в том числе при положительной селекции Т-лимфоцитов.
Запуск апоптоза под влиянием внешних факторов (рецепторный апоптоз) обеспечивают рецепторы DR (от Death Receptor) - рис. 9-2. Следует отметить, что все рецепторы DR за исключением Fas могут также вызывать активацию NF-kB (индукция пролиферации и выживания). Рецепторы DR в цитоплазматической части содержат домен смерти DD (Death Domain), активация которого и запуск апоптотического сигнала происходят при тримеризации рецептора, вызываемой связыванием лигандов. Тримеризация рецепторов и их внутриклеточных DD придаёт последним способность образовывать так называемый «сигнальный комплекс». В случае рецепторов Fas и TRAIL это DISC (Death Initiating Signaling Complex), образуемый при взаимодействии с гомологичным DD доменом цитоплазматического адаптерного белка FADD (Fas-Associated
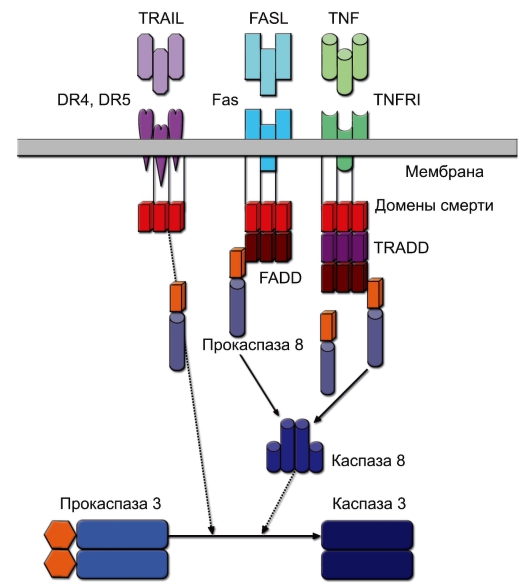
Рис. 9-2. Рецепторный механизм запуска апоптоза. Пояснения см. в тексте
Death Domain protein). В случае рецептора TNFRI, чтобы сформировать сигнальный комплекс 1 и обеспечить связывание с белком FADD, необходимо вначале рекрутировать белок TRADD (TNF-Related Death Domain protein). FADD кроме DD домена также содержит эффекторный DED-домен (Death Effector Domain), который приобретает сродство к гомологичному участку некоторых инициаторных прокаспаз, в типичном случае - прокаспазы 8 (а также каспазы 10). Связывание этих молекул приводит к аутокаталитическому отщеплению от каспазы 8 гомолога DED и формированию активной каспазы 8, способной активировать каспазу 3 и другие эффекторные каспазы. В некоторых клетках каспа-
за 8 может также запускать митохондриальный путь апоптоза через активацию фактора Bid.
Каспаза 3 и другие эффекторные каспазы имеют многочисленные молекулы-мишени, локализующиеся в ядре и цитоплазме. Расщепление этих мишеней определяет многообразные изменения морфологии и функции клетки, которые проявляются в процессе апоптоза. Основной мишенью эффекторных каспаз является Са2+Mg2+-зависимая эндонуклеаза - CAD (от Caspase-Activated DNAse). Этот фермент обусловливает разрывы ДНК между нуклеосомами. Конечным результатом многообразных изменений в клетке является её гибель, как полагают, наступающая в результате истощения энергетических ресурсов, которые тратятся на неэффективную репарацию разрывов ДНК. Гибель по механизму апоптоза проявляется в сморщивании клетки, конденсации хроматина, образовании отростков клеточной мембраны и, наконец, фрагментации ядра и всей клетки с образованием апоптотических телец.
Рецепторная форма апоптоза свойственна зрелым лимфоцитам при их взаимодействии с лигандами апоптоза, локализующимися на активированных клетках, а также с антигеном, цитокинами и т.д. Рецепторный апоптоз реализуется при отрицательной селекции тимоцитов и незрелых В-клеток.
СУПРЕССИЯ ИММУННОГО ОТВЕТА
Супрессия иммунного ответа в норме развивается по мере элиминации антигенов из организма. Элиминация антигенов означает устранение исходного причинного фактора активации лимфоцитов через TCR и BCR, в результате новые (наивные) лимфоциты становится «нечем» активировать.
Супрессия лимфоцитов
• Терминально дифференцированные лимфоциты имеют ограниченное время жизни и погибают по механизму апоптоза, «отработав» свою программу. В таких лимфоцитах снижается экспрессия генов, защищающих лимфоцит от апоптоза на время иммуногенеза, но экспрессируются индуцирующие апоптоз рецепторы, а именно: молекула Fas (CD95), рецепторы для глюкокортикоидов и ФНОα. Следовательно, глюкокортикоидные гормоны, ФНОα и FasL в определённое время от начала развития иммунного ответа становятся факторами физиологической иммуносупрессии.
• Механизмы торможения. Известно несколько конкретных механизмов торможения активности лимфоцитов.
- T-лимфоциты-регуляторы. В первую очередь это активность регуляторных T-лимфоцитов (Treg), продуцирующих значительные количества иммуносупрессорных цитокинов - ИЛ-10 и ТФРβ.
- Th1-лимфоциты подавляют активированные B-лимфоциты той же специфичности через взаимодействие FasL-Fas.
- ИЛ-4 и ИЛ-13, продуцируемые тучными клетками, CD4-/ CD8- T-лимфоцитами, а также дифференцированными Th2клетками, подавляют дифференцировку Th1 из Th0.
- ИФНγ - продукт дифференцированных Th1-лимфоцитов - угнетает дифференцировку Th2 из Th0.
- IgG-антитела, при достижении определённых концентраций в жидких средах организма через специальный ингибирующий рецептор FcγRIIB, экспрессированный на дифференцированных B-лимфоцитах, подавляют биосинтез иммуноглобулинов в данном лимфоците и его дифференцировку в плазматическую клетку. В клинической практике это явление используют для профилактики резус-конфликта: если резус-отрицательной женщине ввести антирезус-антитела до того, как эритроциты плода успеют попасть в кровь матери, иммунный ответ матери на резус-антиген будет подавлен.
• «Аутокиллеры». В организме образуются особые T-лимфоцитыкиллеры с признаками NK-клеток, на которых экспрессировано много Fas-лиганда. Связывая Fas на активированных T-лимфоцитах, эти «аутокиллеры» индуцируют апоптоз активированных T-лимфоцитов.
- Таких «аутокиллеров» много в печени. Вероятно, их природная роль - ликвидировать приносимые с кровью воротной вены лимфоциты, активированные в тканях кишечника пищевыми антигенами.
- Печень как иммуносупрессорный орган. В печени локализовано большинство всех естественных киллеров организма, причём преобладает одна из двух больших субпопуляций NK-клеток, а именно CD56high CD16-, тогда как в крови и красной пульпе селезёнки преобладают естественные киллеры с фенотипом CD56low CD16+.
- На NK-клетках печени экспрессировано много Fas-лиганда, а на клетках эндотелия синусоидов печени - особого лектина, на-
зываемого галектин-1, который, возможно, тоже служит индуктором апоптоза активированных лимфоцитов. Не исключено, что это объясняет неотторжение чужеродных трансплантатов печени.
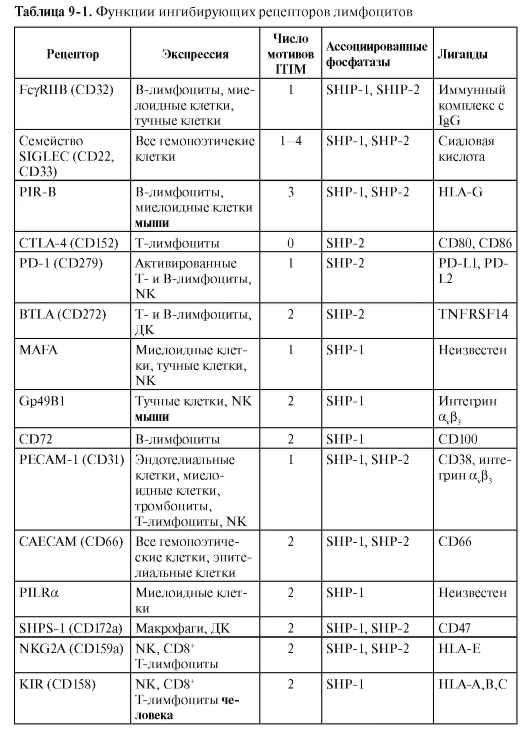
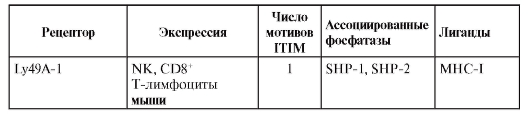
• Ингибирующие рецепторы. Известно не менее 22 мембранных молекул, функционирующих как негативные корецепторы, т.е. способных подавлять активацию клетки, вызванную через другие рецепторы. Ингибирующие рецепторы объединили в семейство молекул SIRP (Signal-Regulatory Proteins) суперсемейства иммуноглобулинов. Рецепторы семейства SIRP в цитоплазматической части содержат от 1 до 4 тирозинсодержащих аминокислотных ингибирующих последовательностей ITIM (Immunoreceptor Tyrosinebased Inhibitory Motif). Эти негативные корецепторы связаны со всеми рецепторами факторов роста, а также обнаружены при рецепторах гормонов (например, инсулина), взаимодействующих внутри клетки с тирозинкиназами.
- PIR-B (Paired Immunoglobulin-like Receptor - парные иммуноглобулиноподобные рецепторы B-лимфоцитов) на B-лимфоцитах мыши, PD-1 (Programmed cell Death - запрограммированная смерть клетки, CD279) и CTLA-4 (Cytotoxic T-lymphocyte Associated protein - белок, ассоциированный с ЦТЛ, CD152) на T-лимфоцитах связаны с ингибиторными фосфатазами SHP-1 и SHP-2, блокирующими активацию киназ семейства Tec и фосфолипазы PLCγ
- KIR (Killer cell Immunoglobulin-like Receptor - иммуноглобулиноподобные рецепторы клеток-киллеров). Рецепторы семейства KIR на ЦТЛ и NK-клетках человека распознают молекулы MHC-I и подавляют тот или иной тип цитотоксичности.
- FcγRIIB-1/2 на B-лимфоцитах и лейкоцитах миелоидного ряда распознают комплексы IgG с антигеном и подавляют образование антител и активацию лейкоцитов.
- На B-лимфоцитах есть ещё один ингибирующий рецептор - CD22. Это димерная молекула семейства лектинов, экспрессируемая только зрелыми B-лимфоцитами.
В табл. 9-1 приведена краткая характеристика некоторых ингибирующих рецепторов.
Супрессия лейкоцитов
Супрессия лейкоцитов достигается теми же двумя путями, что и в случае лимфоцитов: апоптозом и подавлением их активности при помощи сигналов через определённые рецепторы.
• Самые короткоживущие лейкоциты - нейтрофилы. Они погибают физиологическим апоптозом через 4-12 ч после выхода из костного мозга в циркуляцию. В очагах воспаления в тканях нейтрофилы погибают ещё быстрее.
• Эозинофилы и базофилы погибают вскоре после дегрануляции.
• Другие клетки, особенно тканевые макрофаги, живут несколько дольше. Именно поэтому для них (по крайней мере, макрофагов и тучных клеток) существуют биологические механизмы подавления активности. Однако после активного проявления деструктивной функции такие клетки тоже погибают и заменяются новыми, мигрировавшими из кровотока: в случае макрофагов - это моноциты, в случае тучных клеток - предшественники тучных клеток.
Известно несколько факторов и механизмов подавления активности лейкоцитов.
• ИЛ-10, продуцируемый дифференцированными естественными регуляторными Т-лимфоцитами, подавляет активность макрофагов.
• ИЛ-4/STAT6 индуцирует в макрофагах биосинтез антагониста рецептора для ИЛ-1.
• Ингибирующие рецепторы. На тучных клетках выявлено, по крайней мере, 3 ингибирующих рецептора. Один из них у мышей - gp49B1, лигандом которого служит интегрин Второй - уже известный по B-лимфоцитам FcyRIIB, связывающий иммунные комплексы антиген-IgG. Третий - MAFA (Mast cell-Associated Function Antigen) впервые идентифицирован на тучных клетках крысы. Лиганд для MAFA неизвестен, но этот рецептор конститутивно ассоциирован в мембране клетки с FcεRI - высокоаффинным активирующим рецептором для IgE.
ИММУННАЯ ТОЛЕРАНТНОСТЬ
Иммунная толерантность - отсутствие активации лимфоцитов (и, следовательно, выработки ими эффекторных молекул) при наличии доступного специфичного антигена. В природе толерантность лимфоцитов как отсутствие ответа на доступный антиген нужна только по отношению к антигенам собственных неповреждённых тканей организма.
• Следует отличать иммунную толерантность от супрессии уже состоявшегося иммунного ответа:
- супрессия - продуктивная активация клона начинается, реализуется, затем подавляется;
- толерантность - продуктивной активации антигенспецифичного клона лимфоцитов не происходит.
Механизмы супрессии и толерантности одинаковы - апоптоз и подавление внутриклеточного метаболизма сигналами с ингибирующих рецепторов, однако эти механизмы реализуются на разных этапах лимфопоэза и иммуногенеза лимфоцитов.
• Делеция клона. Делеция аутореактивных клонов на стадии лимфогенеза приводит к установлению центральной толерантности. По механизму делеции клона, т.е. путём апоптоза лимфоцитов, связавших антиген, происходит элиминация аутореактивных B-лимфоцитов в костном мозге и негативная селекция тимоцитов в тимусе.
• Анергия клона. Анергией клона называют отсутствие полной активации лимфоцитов, распознавших антиген, но не получивших при этом полноценных костимуляторных сигналов. Это один из основных механизмов развития периферической толерантности лимфоцитов после их выхода из центральных органов иммунной системы в периферические для прохождения иммуногенеза. Анергия, вероятно, имеет несколько разных механизмов реализации.
Продуктивная активация аутореактивного лимфоцита может развиться, если он распознаёт комплексы собственных пептидов со «своими» MHC и получает дополнительный сигнал от корецепторов при взаимодействии с активированной АПК. Сверхпороговый уровень экспрессии костимулирующих молекул достигается только при внешней стимуляции АПК, наиболее очевидный фактор которой - воспаление покровных тканей, повреждённых внедрившимся внешним патогеном (в естественных условиях - инфекционным). Таким образом, если организм не травмирован и инфекция не индуцировала развитие вос-
паления, иммунная толерантность лимфоцитов к собственным антигенам - единственно возможное их состояние.
Трансплантация
Трансплантацией называют пересадку тканей или органов, изъятых из одного организма (донора), во внутреннюю среду другого организма (реципиента). Если трансплантацию проводят между организмами одного вида, то это аллотрансплантация, а антигены трансплантата - аллоантигены, реакция иммунной системы - ответ на аллоантигены. Если же трансплантацию проводят между организмами разных видов, то это ксенотрансплантация.
ОТТОРЖЕНИЕ ТРАНСПЛАНТАТА
Трансплантация может быть успешной только при развитии иммунной толерантности организма-реципиента к антигенам трансплантата (на практике это достигается путём медикаментозной иммуносупрессии со всеми её побочными эффектами), в противном случае на тех или иных сроках после операции происходит отторжение пересаженных тканей.
• Сверхострое отторжение происходит во время или вскоре после операции. При этом развивается окклюзия кровеносных сосудов, связывающих трансплантат с организмом реципиента. Это происходит, если реципиент уже был иммунизирован антигенами донора (или антигенами, перекрёстно реагирующими с антигенами донора) и в крови реципиента есть достаточное количество антител к антигенам стенок сосудов или клеток крови донора. Эти антитела немедленно связываются со стенкой сосудов трансплантата, активируют комплемент и систему коагуляции крови, что приводит к быстрому тромбозу сосудов и выключению органа из кровотока.
• Острое отторжение - нормальный первичный иммунный ответ на трансплантат при отсутствии медикаментозной иммуносупрессии. В разрушение трансплантата могут быть вовлечены все известные эффекторные механизмы иммунного ответа - антителозависимые (антителозависимая клеточная цитотоксичность, активация комплемента иммунными комплексами и др.) и антителонезависимые (CD8+ ЦТЛ; Th1-клетки, активируют макрофаги, индуцируя ГЗТ; Тh2-клетки активируют эозинофилы посредством продуцируемого ими ИЛ-5).
• Отсроченное отторжение по эффекторным механизмам аналогично острому, однако в результате эффективной иммуносупрессии индукция иммунного ответа откладывается на несколько лет.
В отторжении аллогенного траснплантата участвуют практически все механизмы адаптивного иммунитета (рис. 9-3). Основными эффектора-
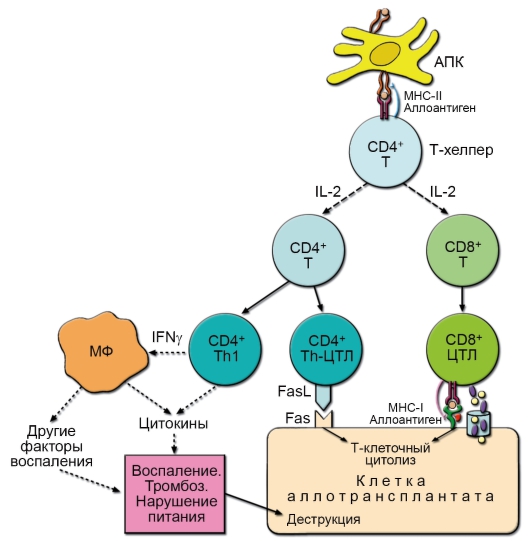
Рис. 9-3. Клеточные факторы и механизмы отторжения трансплантата. ThЦТЛ - это CD4+ T-клетки с цитотоксическим фенотипом. Сплошные стрелки обозначают последующую дифференциацию одной клетки в другую. Стрелки с пунктиром - секретируемые цитокины, воздействующие на другие клетки. Мелкая красная структура - β2-микроглобулин. А цилиндрическая структура рядом схематично показывает проникновение перфорина (мелкие фиолетовые овалы) c гранзимами (мелкие жёлтые кружочки)
ми отторжения являются клеточные факторы. CD8+ Т-клетки, которые обычным путём дифференцируются в цитотоксические Т-лимфоциты (ЦТЛ), вызывают гибель клеток трансплантата преимущественно по механизму перфоринзависимого и Fas-индуцированного цитолиза. CD4+ Т-клетки участвуют в отторжении с помощью двух групп механизмов. Одна из них включает индукцию гибели клеток трансплантата по механизму Fas- и ФНО-зависимого апоптоза. Вторая группа объединяет действие различных факторов воспаления, развивающегося в трансплантате вследствие развития Th1-клеток и активации с их участием макрофагов. Причиной гибели при этом в первую очередь является нарушение питания трансплантата, вызванное изменением микроциркуляции и развитием тромбозов, а также прямое действие цитокинов, ферментов и других факторов, выделяемых в очаге воспаления.
Роль антител в отторжении трансплантата заведомо второстепенна. Связываясь с антигенами трансплантата, антитела блокируют их, не давая возможности проявиться клеточным механизмам защиты. Привлечение в качестве эффекторных агентов факторов комплемента при этом невозможно в связи с активностью на аллогенных (как и на сингенных) клетках системы контроля комплемента, немедленно разрушающих связанные факторы комплемента. В то же время иммунные комплексы, образующиеся при соединении антител с мембранными антигенами трансплантата, могут привлечь клеточные эффекторные механизмы, основанные на распознавании Fc-частей молекул антител. В роли эффекторных клеток-киллеров могут выступать FcR+-клетки - NK-клетки и макрофаги. Такие реакции обозначают как антителозависимый клеточный цитолиз.
Трансплантация костного мозга. Особый случай - пересадка костного мозга или органов и тканей, содержащих много профессиональных АПК. На этих клетках присутствуют все костимуляторные молекулы, необходимые и достаточные для продуктивной активации T-лимфоцитов. Именно поэтому при пересадке кроветворных тканей отторжение MHC-совместимого трансплантата может произойти быстрее, чем MHC-несовместимого, поскольку T-лимфоциты реципиента будут эффективнее работать с АПК донорского происхождения (как с «родными» по MHC). Кроме того, лимфоциты донора могут начать атаковать клетки организма реципиента, вызывая реакцию «трансплантат против хозяина».
ИММУНОПРИВИЛЕГИРОВАННЫЕ ТКАНИ
В организме есть анатомические зоны, в которых аккуратно вживлённый трансплантат при определённых условиях не отторгается. Эти зоны называют иммунопривилегированными. У человека такими местами служат мозг, передняя камера глаза, беременная матка и семенники.
Первоначальное предположение о том, что антигены этих тканей не покидают своих мест и недоступны для распознавания T-лимфоцитами, не подтвердилось: антигены тканей из привилегированных мест покидают их, однако действительно не совсем так, как во всех остальных органах. А именно - антигены минуют классический лимфатический дренаж; иммунопривилегированные органы отграничены особыми барьерами, клетки которых продуцируют иммуносупрессорные цитокины (ТФРР) или экспрессируют много Fas-лиганда, убивающего проникающие в них активные лимфоциты.
С клинической точки зрения существенно, что именно ткани из иммунопривилегированных мест чаще прочих становятся объектом аутоиммунного повреждения (например, демиелинизирующие заболевания мозга, включая рассеянный склероз, симпатическая офтальмия).
ИММУННАЯ СИСТЕМА И ГЛЮКОКОРТИКОИДЫ
Не одно десятилетие глюкокортикоиды применяют в качестве противовоспалительных лекарственных средств, причём при патологиях с очевидным вовлечением в патогенез иммунной системы - ревматические, аутоиммунные, аллергические заболевания. Глюкокортикоиды облигатно вовлечены в лимфопоэз и иммуногенез.
Мишени глюкокортикоидов
• Тимус. Источником глюкокортикоидов, воздействующих на лимфоциты, служат не только надпочечниковые железы - глюкокортикоиды синтезируются и эпителиальными клетками тимуса. Другими словами, в тимусе создаётся нужная локальная концентрация глюкокортикоидов, необходимая для индукции апоптоза, - ≈95-99% тимоцитов при позитивной и негативной селекции.
• Лимфоидная периферия. Главный эффект физиологических концентраций системных глюкокортикоидов на лимфоциты в периферических тканях - индукция апоптоза активированных лимфоцитов:
глюкокортикоиды служат исполнителями индуцированной активацией клеточной смерти (AICD). Эффекты глюкокортикоидов. В фармакологических концентрациях глюкокортикоиды вызывают следующие эффекты:
• стимулируют в активированных лимфоцитах и эозинофилах эндонуклеазы, разрушающие ДНК в межнуклеосомных участках, что заканчивается апоптозом клеток;
• ингибируют биосинтез ИЛ-1, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-8, ФНОα, GM-CSF, что приводит к ослаблению воспалительных процессов, зависимых от этих цитокинов;
• подавляют NO-синтазу и, следовательно, снижают зависимую от оксида азота альтерацию тканей, в том числе стенки кровеносных сосудов;
• ингибируют фосфолипазу А2 и циклооксигеназу 2-го типа, необходимые для синтеза простагландинов и лейкотриенов. Как следствие, происходит угнетение воспалительных процессов и спазмов гладкой мускулатуры, зависящих от этих липидных медиаторов;
• угнетают экспрессию молекул межклеточной адгезии, что приводит к снижению экстравазации лейкоцитов и миграции их в очаги воспаления.
Побочные эффекты. Наиболее очевидные побочные эффекты применения терапевтических доз глюкокортикоидов состоят в задержке в организме натрия (а следовательно, и воды), увеличении массы тела, проявлении симптомов диабета, потере минеральных веществ из костей, истончении кожи (а следовательно, и ухудшении её барьерных свойств).