Наглядная детская гастроэнтерология и гепатология : учеб. пособ. - Кильдиярова Р. Р., Лобанов Ю. Ф. 2013. - 124 с. : ил.
|
|
|
|
Глава 8. Редкие синдромы
ГАСТРОЭНТЕРОЛОГИЯ
Врожденный изолированный трахеальнопищеводный свищ
Код по МКБ-10
Q39.2. Врожденный трахеально-пищеводный свищ без атрезии.
Врожденное изолированное соединение трахеи и пищевода - трахеально-пищеводный свищ (ТПС) - составляет 1-4% всех заболеваний пищевода в детском возрасте, чаще встречается у мальчиков.
Классификация
Встречаются три основных анатомических варианта ТПС (по Баирову Г.А., 1968):
• короткий и широкий (рис. 8-1, а);
• длинный и узкий (рис. 8-1, б);
• соединение трахеи и пищевода без свищевого канала (рис. 8-1, в).
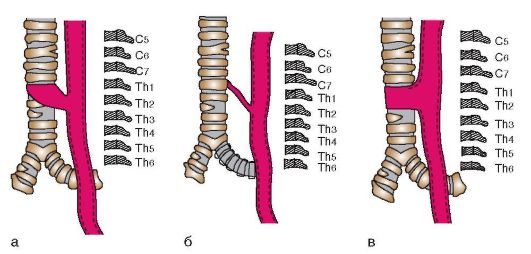
Рис. 8-1. Анатомические варианты ТПС (пояснения в тексте)
Ход свища обычно косой - от трахеи вниз, к пищеводу, его отверстие в пищеводе открывается на передней стенке также на достаточно высоком уровне, в шейном или верхнегрудном отделе, в пределах позвонков CVI-ТhIII.
Клиническая картина
У многих больных клиническая картина ТПС характеризуется классической триадой основных симптомов:
• кашлем и поперхиванием при приеме пищи;
• явлениями аспирационной пневмонии;
• вздутием живота, которое возникает в результате прямого попадания воздуха из трахеи в пищевод и желудок.
Про данное заболевание образно говорят: «Ребенок со свищом дышит в свой живот». Особенно вздутие увеличивается при кашле и плаче. Однако выраженность этих признаков может быть различной как по интенсивности, так и по сочетанию.
Диагностика
Для подтверждения диагноза необходимы рентгенологическое и эндоскопическое исследования больного. К рентгенологическим методам относятся обзорная рентгенография грудной и брюшной полостей, контрастная эзофагоскопия и эзофагография, контрастная эзофагоскопия с использованием электронно-оптического преобразователя.
Лечение
Лечение только оперативное.
Прогноз
Прогноз благоприятный при своевременном хирургическом лечении.
Врожденный стеноз пищевода
Код по МКБ-10
Q39.3. Врожденные стеноз и стриктура пищевода.
Среди всех заболеваний пищевода врожденные сужения составляют у детей от 2 до 6%.
Классификация
Врожденный стеноз пищевода (ВСП) подразделяется на две формы: циркулярные сужения, в большей или меньшей степени напоминающие песочные часы, и мембранозные сужения с центральным или эксцентрически расположенным отверстием. Его возникновение связано с задержкой процесса рассасывания эпителиальных масс, заполняющих просвет пищевода, что может привести к образованию стеноза.
Клиническая картина
Наиболее частыми симптомами ВСП у детей любого возраста являются рвота, срыгивания, регургитация, дисфагия, дефицит массы тела и расстройства дыхания (одышка, цианоз и др.), связанные с приемом пищи. В дальнейшем формируется хроническая пневмония, а также могут обнаруживаться инородные тела в пищеводе.
Диагностика
Рентгеноскопия пищевода является высокоинформативным методом при ВСП. Выделяют три наиболее часто встречаемых варианта рентгенологической картины ВСП:
• короткий циркулярный (рис. 8-2, а);
• длинный циркулярный (рис. 8-2, б);
• циркулярный в форме песочных часов (рис. 8-2, в).
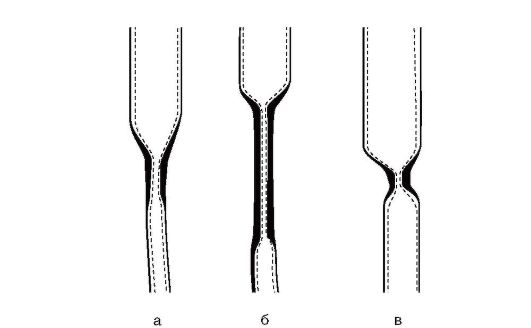
Рис. 8-2. Рентгенологические варианты ВСП (пояснения в тексте)
В связи с тем что многие дифференциально-диагностические вопросы решаются с помощью рентгенографии, а также тщательным выяснением анамнеза заболевания, эзофагоскопию применяют только в случаях, когда происхождение сужения остается неясным.
Лечение
Лечение ВСП возможно консервативными методами или с помощью операции. До настоящего времени не определены четкие показания к каждому из них.
Независимо от возраста больного и формы ВСП начинать лечение следует с бужирования. Операция показана только тем больным, у которых полностью отсутствовал эффект от консервативного лечения или наблюдалось временное улучшение после трех курсов бужирования. Разработаны новые методы оперативного вмешательства, что увеличило выживаемость детей с данной врожденной патологией.
Прогноз
Прогноз благоприятный при своевременных диагностике и лечении.
Синдром Маллори-Вейсса
Код по МКБ-10
K22.6. Желудочно-пищеводный разрывно-геморрагический синдром Мaллори-Вейссa.
Синдром Маллори-Вейсса - остро развивающийся синдром желудочного кровотечения в результате продольных разрывов слизистого и подслизистого слоев в области пищеводно-желудочного соустья (кардиального отдела), наступающий при рецидивирующей рвоте. Синдром назван по именам американских патологоанатома G. Mallory и врача S. Weiss, описавших его в 1932 г. Частота синдрома составляет около 1-2% среди всех больных с желудочно-кишечным кровотечением. Чаще синдром встречается у лиц мужского пола в возрасте после 30 лет (77%), у детей наблюдается редко.
Этиология и патогенез
Повышение внутрижелудочного давления, перерастяжение и разрыв слизистой оболочки возникают вследствие дискорреляции замыкательной функции кардиального и пилорического сфинктеров, а также выпадения слизистой оболочки желудка в просвет пищевода и ущемления ее в кардиальном жоме.
Предрасполагающими факторами разрывов считаются:
• малая растяжимость слизистой оболочки кардиальной части желудка вследствие фиксации ее внежелудочными связками и продольного расположения складок;
• наличие скользящей грыжи пищеводного отверстия диафрагмы, которую обнаруживают более чем у 50% больных;
• трофические изменения слизистой оболочки вследствие хронических заболеваний ЖКТ.
Классификация
Изолированная пищеводная локализация трещин встречается в 8% случаев, желудочная - в 44%, почти в половине всех случаев синдрома Маллори-Вейсса разрыв слизистой оболочки переходит с желудка на пищевод. Следует различать свежие трещины, когда от момента разрыва прошло не более 1-2 сут, и поздние.
По глубине разрывов выделяют три стадии:
• I - повреждение только слизистой оболочки, когда возможно самоизлечение или образование субкардиальной язвы;
• II - разрыв слизистого и подслизистого слоев, при котором повреждаются кровеносные сосуды и возникает кровотечение;
• III - разрыв всех слоев желудка или пищевода, при котором развиваются перитонит, медиастинит или пневмоторакс.
Клиническая картина
Синдром Маллори-Вейсса проявляется частой рвотой на фоне имеющихся заболеваний, таких как хронический гастрит, хронический холецистит и др. У детей эти заболевания могут сопровождаться лихорадкой, что, как известно, способствует гиперемии слизистых оболочек и повышенной хрупкости сосудов. Перед проявлением синдрома дети могут страдать респираторными заболеваниями, быть оперированы по поводу острого аппендицита; также синдром может развиться после лечения салицилатами в связи с ревматической инфекцией и может быть связан с нарушениями диеты, когда вначале возникает рвота пищей, а затем - кровью.
Распознать синдром Маллори-Вейсса при внешнем осмотре и пальпации желудка удается очень редко.
Диагностика
Диагностика синдрома Маллори-Вейсса остается сложной и проводится с учетом современных методов исследования.
Эзофагогастроскопию можно считать основным методом диагностики, так как ничто не может быть достовернее, чем визуальное обнаружение разрыва слизистой оболочки в типичном месте. Диагноз устанавливают в 94-97% случаев. Эндоскопическая картина при этом заболевании довольно характерна.
На высоте кровотечения в кардиальном отделе желудка или в пищеводе обнаруживают продольные одиночные или множественные трещины (рис. 8-3, а). Края разрывов отечны, пропитаны кровью, покрыты сгустками крови и фибрином. Глубину трещин определить трудно, дном их часто является мышечный слой. Со дна разрыва из-под сгустков крови иногда отмечается подтекание свежей крови. Однако даже при эндоскопии о природе обнаруженной патологии не всегда можно говорить с уверенностью, идет ли речь о кровоточащей трещине, язве или эрозии (см. рис. 8-3, а). В то же время эзофагогастродуоденоскопия позволяет выявить сопутствующие заболевания желудка и ДПК, которые имеют место почти у 50% больных с синдромом Маллори-Вейсса.
Значение рентгенологического исследования в диагностике синдрома Маллори-Вейсса незначительное, так как поверхностные разрывы не видны, а глубокие обычно заполняются сгустками крови и никак не проявляются.
В последнее время диагностическое значение при синдроме Маллори-Вейсса приобретает селективная ангиография, которая может превратиться и в лечебную процедуру за счет эмболизации сосудов желудка.
Дифференциальная диагностика
Дифференциальную диагностику проводят с варикозным расширением вен пищевода, язвенной болезнью желудка и ДПК, грыжей пищеводного отверстия диафрагмы с помощью рентгенологического исследования.
Лечение
Лечение синдрома Маллори-Вейсса с годами претерпело большие изменения. Вначале больных подвергали хирургическому лечению, летальность при этом составляла 50-59%. С внедрением в практику эндоскопии и более эффективных способов консервативной терапии хирургическое лечение (ушивание трещин, рис. 8-3, б) проводят у 30-35% больных, при этом летальность снизилась до 14%.
В настоящее время около 90% больных лечат консервативно, а летальность составляет 3%. Операционная летальность сохраняется на уровне 15%, что объясняется более тяжелым контингентом больных, у которых консервативное лечение оказалось неэффективным. Консервативное лечение: холод, антациды, стимуляторы свертывающей системы крови, ε-аминокапроновая кислота в/в и внутрь и т.д.
Прогноз
Отдаленные результаты лечения больных с синдромом Маллори-Вейсса зависят главным образом от сопутствующих заболеваний желудка, ДПК и пищевода. В отдельных случаях наблюдаются повторные разрывы слизистой оболочки пищеводно-кардиальной области и рецидивы кровотечения.
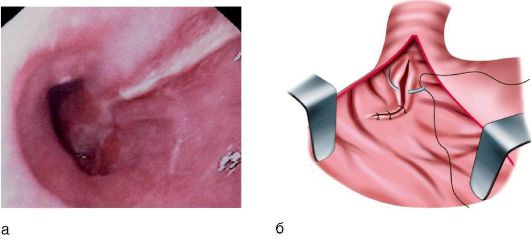
Рис. 8-3. Синдром Маллори-Вейсса: а - трещины слизистой оболочки пищеводно-желудочного соустья (эндоскопическая картина); б - схема ушивания трещин
Безоары желудка
и двенадцатиперстной кишки
Код по МКБ-10
К92.8. Другие уточненные болезни органов пищеварения.
Это группа заболеваний, близких к инородным телам. Возникают при попадании в желудок веществ, которые не перевариваются в нем, а накапливаются и формируются в инородные тела, с пищей или в результате вредных привычек.
Этиология и патогенез, классификация
Известно около 11 видов безоаров ЖКТ в зависимости от их состава.
Чаще других наблюдаются фитобезоары, которые составляют 70-75% всех безоаров. Формируются они из растительной клетчатки, кожицы, семян и косточек плодов и фруктов: хурмы, диких слив, винограда, инжира, черемухи и др. Скорость образования фитобезоаров зависит от их органической природы и колеблется от 1-5 дней до 16 лет. В зависимости от сроков формирования фитобезоары имеют консистенцию от мягкой до плотности природного камня. Они бывают единичными и множественными, темно-коричневого или зеленого цвета, издают зловонный запах. Их размеры колеблются от нескольких миллиметров до 20 см и более. Среди фитобезоаров наиболее часто сообщается о диоспиросбезоарах, формирующихся из хурмы, которая содержит большое количество смолистых веществ, претерпевающих коагуляцию под воздействием желудочного сока и способствующих слипанию частей хурмы в плотную массу.
Безоары эмбрионального происхождения образуются из экто- и энтодермы и представляют собой дермоидные кисты - тератомы желудка (рис. 8-4, а).
Трихобезоары образуются при попадании в желудок волос и встречаются чаще всего у людей с неуравновешенной психикой, которые страдают непреодолимым желанием кусать волосы. Описаны случаи, когда трихобезоары желудка, постепенно увеличиваясь в размерах (иногда достигают 3,5 кг), спускаются своей дистальной частью в двенадцатиперстную и тощую кишку (рис. 8-4, б).
Шеллачные камни (шеллакобезоары) встречаются реже, имеют массу 500 г и более. Причиной их образования является прием внутрь спиртового лака, нитролака и политуры. При добавлении к шеллачному лаку воды шеллачная смола выпадает в осадок, который вначале имеет вязкую консистенцию, склеивает частицы пищи и под действием перистальтики желудка формируется в конгломерат, затвердевает и превращается в камень. Эти безоары буровато-белого цвета, с гладкой или слегка шероховатой поверхностью, на разрезе имеют черно-коричневый цвет, горят, режутся ножом, крошатся.
Пиксобезоары образуются у лиц, имеющих привычку жевать смолу, вар. Формируются они медленно и достигают крупных размеров.
Себо(стибо)безоары образуются при уплотнении животного жира (козье, баранье и говяжье сало в растопленном виде) в результате кристаллизации триглицеридов.
Лактобезоары образуются у недоношенных детей, находящихся на высококалорийном искусственном вскармливании с высоким содержанием лактозы и казеина, в течение первых 2 нед жизни. Они самостоятельно распадаются после промывания желудка, коррекции диеты и применения адаптированных молочных смесей, близких по составу к грудному молоку.
Полибезоары (миксобезоары) могут включать клубок хлопчатобумажных и шерстяных ниток, спички и разволокненные кусочки древесины.
Гемобезоары встречаются очень редко и образуются при длительном заглатывании крови у больных портальной гипертензией, системной красной волчанкой и другими заболеваниями.
Кроме истинных безоаров, существует группа псевдобезоаров, образующихся из плохо разжеванной пищи, слизи.
Клиническая картина
Проявления безоаров желудка и ДПК зависят от природы, размера, массы, локализации и давности их образования, а также от нервно-психического состояния больного и осложнений, связанных с безоаром. В диагностике имеют значение анамнестические данные о вредных привычках пациента.
Наиболее частыми симптомами являются боли в эпигастральной области, тошнота, тухлая отрыжка, рвота. Боли чаще тупые, но могут быть схваткообразными, распространяясь на низ живота, что бывает обусловлено частичной или полной закупоркой тонкой кишки. Иногда больные ощущают «перекатывание мяча» в желудке.
Многие больные жалуются на общую слабость, быстрое насыщение, плохой аппетит, похудение. При исследовании крови выявляется анемия.
Заболевание может протекать волнообразно, периодически обостряясь по мере накопления безоаров в желудке и затихая после эвакуации их из желудка в ДПК или с рвотными массами наружу. При достаточной величине безоара в эпигастральной области можно прощупать опухолевидное образование, безболезненное и легко смещаемое.
Общее состояние и показатели лабораторных исследований у подростков меняются мало, в то время как у детей младшего возраста появляются отеки, гипопротеинемия,
анемия, обусловленные метаболическими нарушениями, недостаточной абсорбцией фолиевой кислоты и витамина B12, бурным размножением бактерий в верхнем отделе тонкой кишки.
Безоары могут способствовать образованию ЯБ желудка и ДПК, что может быть связано с трофическими нарушениями в слизистой оболочке и нарушениями желудочной секреции. Частота образования язв при фитобезоарах составляет 25%, при трихобезоарах - 10%. Безоары значительно отягощают течение ЯБ. Перфорация желудочной язвы чаще наблюдается при трихобезоарах. Имеются случаи малигнизации язвы желудка при безоарах, а также сочетания безоаров желудка с карциномой.
Безоары могут вызывать непроходимость желудка, двенадцатиперстной и тонкой кишки.
Диагностика
Большое место в обнаружении безоаров желудка и ДПК отводится рентгенологическому исследованию (рис. 8-4, в). При безоарах желудка и ДПК наиболее информативным является эндоскопическое исследование, которое позволяет не только поставить правильный диагноз, но и установить природу безоара и оценить состояние слизистой оболочки желудка и ДПК.
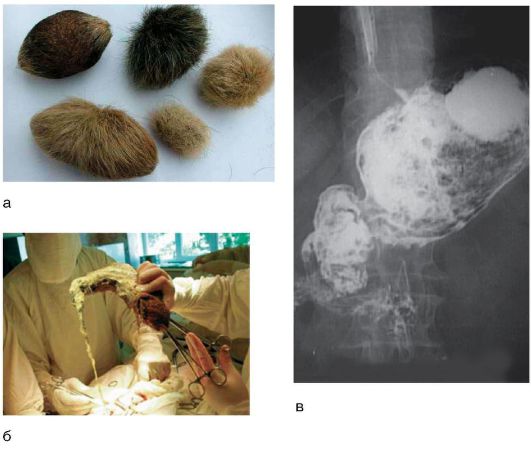
Рис. 8-4. Безоары желудка и ДПК: а - безоары эмбрионального происхождения; б - этап операции: выведение трихобезоара из желудка; в - рентгеноконтрастное исследование: безоар в желудке
Лечение
Лечение безоаров в каждом отдельном случае индивидуальное. При небольших и мягких безоарах, при отсутствии осложнений следует проводить консервативное лечение, включающее питье натощак теплых минеральных вод, молока и 10% раствора гидрокарбоната натрия, массаж живота, лечебную гимнастику, прием слабительных средств.
В последние годы получил распространение метод размельчения и удаления безоаров при эзофагогастродуоденоскопии с последующим вымыванием содовым раствором и удалением специальной петлей.
При трихо-, шеллако-, миксо-, пиксобезоарах, при крупных фитобезоарах, а также при отсутствии эффекта от консервативного лечения показано хирургическое вмешательство (см. рис. 8-4, б).
Прогноз
Непосредственные и отдаленные результаты хирургического лечения безоаров желудка и ДПК при отсутствии осложнений, как правило, хорошие.
Незавершенный поворот кишечника
Код по МКБ-10
Q40-Q43. Врожденные аномалии (пороки развития) органов пищеварения.
Незавершенный поворот кишечника - врожденная аномалия развития кишечника, связанная с нарушением нормального поворота первичной трубки плода, возникающая с 1-го по 12-й месяц гестации.
Незаконченный поворот кишечника - наиболее частая аномалия ЖКТ, наблюдается у 75 из 119 детей с пороками развития кишечника.
Этиология и патогенез
Возникновение аномалий кишечника во время вращения первичной трубки плода (рис. 8-5) связано с влиянием неблагоприятных факторов в конкретные сроки гестации. Периоды вращения первичной кишки плода
• I период вращения начинается на 5-й неделе гестации, когда первичная кишка висит в сагиттальной плоскости на брыжейке (рис. 8-5, а), а с 8-й недели петля средней кишки, находящаяся в пупочном канатике, поворачивается на 90° против часовой стрелки из сагиттальной в горизонтальную плоскость (рис. 8-5, б).
• II период вращения начинается на 10-й неделе внутриутробного развития. Происходит поворот кишки на следующие 180° и одновременное самопроизвольное вправление пупочной грыжи (рис. 8-5, в). В итоге на 11-й неделе слепая кишка оказывается в правом верхнем квадранте, кишечник повернулся на 270° (рис. 8-5, д).
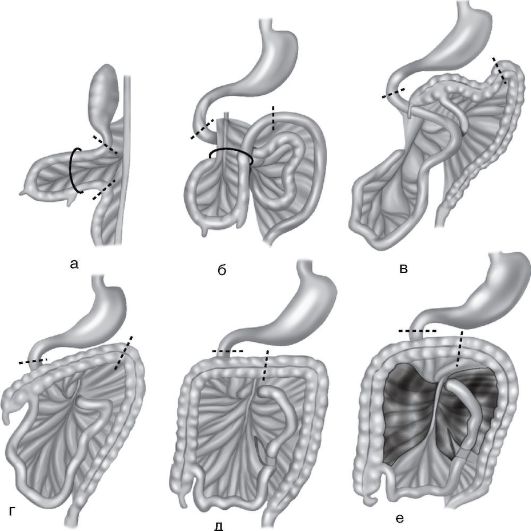
Рис. 8-5. Схематическое изображение нормального поворота первичной трубки плода: а, б - I период; в, г - II период; д, е - III период вращения
• III период вращения - слепая кишка опускается в свое обычное положение - правый нижний квадрант, но брыжейка еще не фиксирована на задней брюшной стенке (рис. 8-5, д). Окончательный поворот первичной кишки заканчивается фиксацией брыжейки на задней стенке брюшной полости (рис. 8-5, е).
Классификация
Расстройства поворотов первичной кишки плода:
- I периода вращения - грыжа пупочного канатика;
- II периода вращения - несостоявшийся поворот кишечника, непроходимость ДПК, внутренняя грыжа, поворот в обратном направлении, синдром Ледда;
- III периода вращения - высокое расположение, подвижная слепая кишка, ретроцекальное положение аппендикса.
Клиническая картина
Дуоденальная непроходимость у детей старшего возраста наблюдается редко и обычно не диагностируется, так как клиническая картина напоминает ЯБ, холецистит, панкреатит и другие заболевания этой зоны. Порок заключается во внутриутробном нарушении вращения средней кишки вокруг общей брыжейки, высоком расположении слепой кишки и наличии тяжей брюшины, которые сдавливают просвет ДПК. Сдавление происходит чаще в верхней половине нисходящей ее части. При нарушении проходимости ДПК появляются ощущение тяжести после еды, боли в эпигастральной области, тошнота, рвота с примесью желчи. В ряде случаев частичная закупорка ДПК сочетается с заворотом тонкой кишки, создавая клиническую картину острой высокой кишечной непроходимости. Этот синдром получил название «синдром Ледда».
Диагностика
При рентгенологическом исследовании в вертикальном положении обнаруживают неравномерное распределение газа по кишечнику, горизонтальные уровни в желудке и кишках. Отделы ДПК выше препятствия удлинены, расширены и могут образовывать дополнительные изгибы. Контур вдавления, как правило, четкий, протяженностью до 1 см. Дуоденоскопия в сочетании с рентгеноскопией позволяют исключить ЯБ ДПК. При непроходимости, вызванной нарушением поворота ДПК, переход ее в тонкую кишку находится справа от позвоночника. Для синдрома Ледда характерно смещение слепой кишки кверху, однако нормальное положение толстой кишки не исключает аномалии вращения ДПК. Дооперационная диагностика данной патологии затруднена.
Лечение
Лечение больных с незаконченным поворотом кишечника хирургическое.
Прогноз
Прогноз не всегда благоприятный из-за поздней диагностики и несвоевременного оперативного вмешательства.
Болезнь Гиршпрунга
Код по МКБ-10
Q43.1. Болезнь Гиршпрунга.
Болезнь Гиршпрунга - аномалия развития толстой кишки, приводящая к нарушению иннервации фрагмента кишки (врожденному аганглиозу) и проявляющаяся упорным запором. Болезнь названа по имени датского педиатра H. Hirshprung, описавшего ее в 1887 г. По данным разных авторов,
частота болезни Гиршпрунга колеблется от 1:1000 до 1:20 000. Мальчики болеют в 4-5 раз чаще девочек.
Этиология и патогенез
Болезнь возникает из-за врожденного нарушения иннервации кишечника, приводящего к снижению (вплоть до отсутствия) перистальтики аганглионарного сегмента. На определенном его участке, чаще ампулярной части прямой кишки или дистальном отделе сигмовидной кишки, отсутствует нормальная структура нервных мейсснеровых и ауэрбаховых сплетений. Возможно наличие аганглиоза и в вышележащих отделах толстой кишки. Крайне редко поражается подвздошная кишка. Чем выше начало зоны аганглиоза, тем тяжелее протекает болезнь, поскольку в вышележащих отделах скапливается кишечное содержимое и возникает упорный запор. Аганглиарный участок кишки сужен, а выше места сужения кишка растянута (рис. 8-5, а).
Классификация
Различают следующие анатомические формы болезни Гиршпрунга:
• ректальную, с поражением части прямой кишки (25% случаев);
• ректосигмоидальную, с поражением части прямой кишки и дистальной трети сигмовидной кишки (70%);
• сегментарную, с поражением одного сегмента сигмовидной кишки и более (1,5%);
• субтотальную, с поражением левой половины и распространением процесса на правую половину толстой кишки (3%);
• тотальную, с поражением всей толстой кишки и иногда части тонкой кишки (0,5%).
Клиническая картина
У новорожденных и детей грудного возраста клиническая картина своеобразна и разнообразна, что связано с протяженностью и высотой расположения зоны аганглиоза по отношению к анальному отверстию. Чем протяженнее зона аганглиоза и чем выше она расположена, тем острее и ярче проявляются симптомы заболевания. В раннем возрасте заболевание включает триаду симптомов:
• запор;
• увеличение живота;
• повторный илеус (кишечная непроходимость).
У детей старшего возраста характерны ранние и поздние проявления. Запор с первых дней (недель) жизни усиливается при введении плотной пищи; стул - только после клизмы, метеоризм - с первых дней жизни, отмечается «лягушачий» живот (рис. 8-5, б). Поздние проявления включают анемию, гипотрофию, рахитоподобную деформацию грудной клетки, каловые камни (рис. 8-5, в), каловую интоксикацию.
Выделяют следующие клинические стадии заболевания: компенсированную, субкомпенсированную и декомпенсированную (табл. 8-1).
Таблица 8-1. Стадии болезни Гиршпрунга

Окончание табл. 8-1
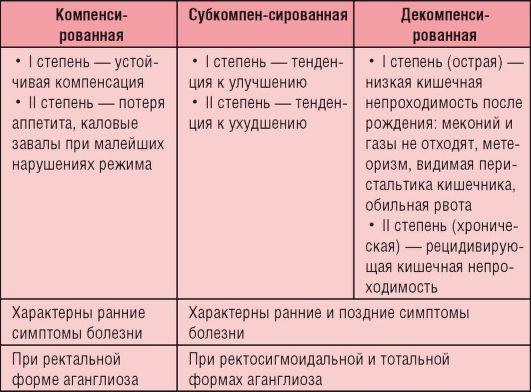
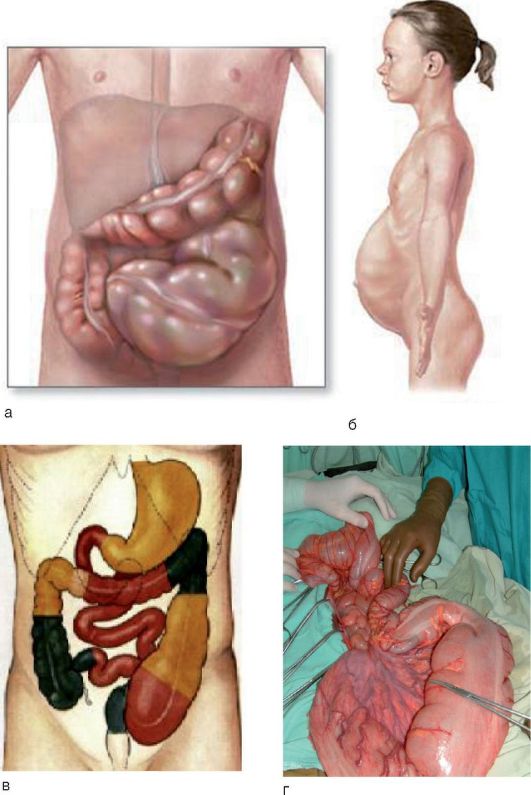
Рис. 8-5. Болезнь Гиршпрунга: а - схематическое изображение увеличенной сигмовидной кишки; б - внешний вид больной; в - преимущественная локализация каловых масс; г - этап оперативного вмешательства
Диагностика
Диагностика тем сложнее, чем младше ребенок, так как такие симптомы, как задержка стула и вздутие живота, часто встречаются у младенцев. Обязательные диагностические процедуры:
• УЗИ кишечника;
• обзорная рентгенография брюшной полости в вертикальном положении больного (наличие расширенных петель толстой кишки с горизонтальными уровнями или без них);
• ирригография;
• боковая рентгенограмма после опорожнения кишки (определение ширины ретроректального пространства);
• биопсия;
• аноректальная манометрия;
• гистохимический и морфометрический методы (в специализированных учреждениях).
Дифференциальная диагностика
Дифференциальную диагностику проводят с мекониевой пробкой, стенозом терминального отдела подвздошной кишки, динамической кишечной непроходимостью, мегаколоном, привычным запором, эндокринопатиями, гиповитаминозом В1.
Лечение
При явлениях непроходимости кишечника, связанных с энтероколитом, применяют антибактериальную терапию и ежедневные клизмы с изотоническим раствором. Купирование явлений энтероколита приводит к исчезновению непроходимости. Радикальной и обязательной операции (рис. 8-5, г) предшествует консервативное лечение: послабляющая диета, очистительные и сифонные клизмы, симптоматическая терапия.
Прогноз
Прогноз благоприятный при ректальной форме и своевременном оперативном вмешательстве.
Спаечная болезнь брюшины
Код по МКБ-10
K66.0. Брюшинные спайки абдоминальные (стенки), кишечника, брыжейки, спаечные тяжи.
Спаечная болезнь брюшины - синдром, обусловленный наличием спаек в брюшной полости и характеризуемый частыми приступами кишечной непроходимости, возникающий, как правило, вследствие хирургических вмешательств на органах брюшной полости и отличающийся многообразием клинических проявлений.
В последнее время отмечается рост распространенности спаечной болезни брюшины (СББ) и спаечной кишечной непроходимости (СКН) среди прочих видов илеуса у детей.
Этиология и патогенез
Хирургические вмешательства на брюшной полости являются непосредственной причиной образования спаек. Любой воспалительный процесс в брюшной полости как результат травмирования брюшины и воздействия инфекционного агента приводит к активации иммунного ответа с участием медиаторов воспаления, цитокинов, иммунных клеток, в результате чего образуются патологические иммунные комплексы. Эти комплексы прикрепляются к клеткам здоровых тканей и вызывают активацию системы комплемента. Цитотоксические реакции, вызванные их активацией, приводят к повреждению ткани и упорному течению воспалительного процесса, сопровождающегося пролиферацией соединительной ткани, способствуя тем самым усилению спаечного процесса в брюшной полости.
Классификация
Классификация СББ (Гатауллин Н.Г., 1978) основана на клиническом течении заболевания с учетом его тяжести и позволяет определить наиболее удобную тактику ведения больных.
В ней выделены 4 формы заболевания:
• латентная бессимптомная форма СББ;
• спаечная болезнь с преобладанием болевого синдрома;
• спаечная болезнь с преобладанием дискомфорта в ЖКТ;
• СКН (рис. 8-6).
Таблица 8-2. Основные формы и клинические проявления СББ
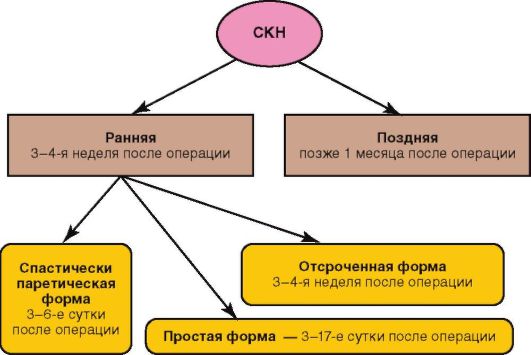
Рис. 8-6. Формы СКН у детей (по Баирову Г.А., 1983)
Клиническая картина
Клиническая картина спаечной болезни и спаечных сращений брюшной полости вариабельна. Она складывается из симптомов, обусловливающих тяжесть общего состояния больного, и местных проявлений (табл. 8-2). Правильно собранный анамнез, в котором особое значение отводится выявлению перенесенных ранее воспалительных процессов и оперативных вмешательств на органах брюшной полости, а также давность заболевания, являются ведущими в постановке диагноза.
Важно правильно провести пальпацию живота в целях выявления некоторых характерных симптомов, механизм которых основан на принципе висцеросенсорных рефлексов и связан с появлением болевых ощущений при растяжении спаек:
- симптом Блинова - боль в рубце при наклоне туловища вперед;
- симптом Андросова - боль в эпигастрии при глубокой пальпации гипогастральной области;
- симптом Хунафина - появление болей при искусственном надувании живота (при спайках любой локализации).
При развитии острой СКН, помимо острых, схваткообразных болей, рвоты застойным содержимым, вздутия живота, задержки стула и газов, при осмотре выявляют асимметрию живота, видимую перистальтику кишечных петель на передней брюшной стенке, шум плеска, шум падающей капли.
В поздние сроки присоединяются перитонеальные явления, лихорадка, нейтрофильный лейкоцитоз, резкое повышение СОЭ.
Диагностика
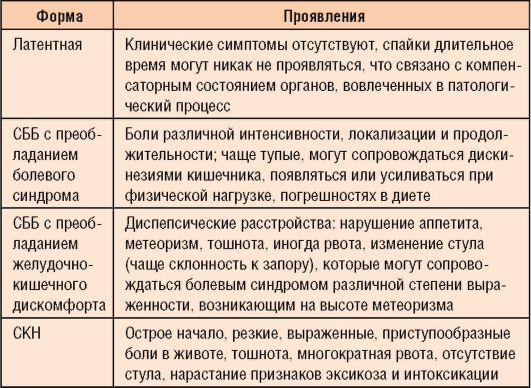
Ведущая роль в диагностике принадлежит рентгенологическому исследованию. Основные симптомы СББ, выявляемые при обзорной рентгенографии органов брюшной полости (рис. 8-7, а):
• неравномерное распредение газов в петлях кишечника;
• чаши Клойбера и арки, позволяющие определить уровень кишечной непроходимости.
Однако прямых признаков, позволяющих определить размеры, вид, локализацию спаек, при данном методе исследования нет.
Более информативной признана методика рентгенологического исследования ЖКТ с контрастированием сернокислым барием. Время появления контрастного вещества в слепой кишке в норме - 3-4 ч с момента приема бариевой взвеси per os. Выявляют косвенные признаки спаечного процесса (рис. 8-7, б):
• фиксацию петель кишечника к передней брюшной стенке, послеоперационному рубцу или другим органам;
• конгломераты петель тонкой кишки с неравномерной скоростью эвакуации контрастного вещества;
• сужение сегментов тонкой кишки, расширение и утолщение стенки кишки выше спаек, образование ложных дивертикулов в тонкой кишке за счет тракции ее спайками.
Перспективным и наиболее информативным методом диагностики СББ является диагностическая лапароскопия (рис. 8-7, в).
Лечение
Борьба с уже сформированными спайками брюшной полости до настоящего времени остается сложной и до конца не решенной задачей абдоминальной хирургии. На практике большую часть операций выполняют в экстренном порядке при возникновении ситуации, угрожающей жизни больного.

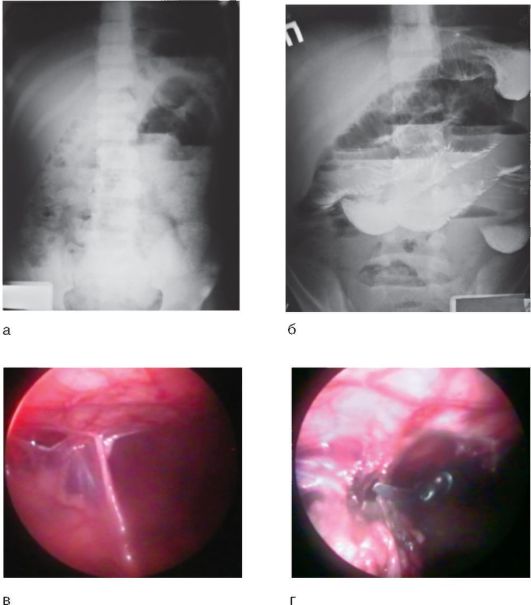
Рис. 8-7. СББ: а - обзорная рентгенограмма органов брюшной полости; б - рентгеноконтрастное исследование ЖКТ в прямых проекциях; в - лапароскопическая картина; г - рассечение коагулированной спайки
В зависимости от клинических проявлений заболевания (от изолированного болевого синдрома до выраженной картины СКН) применяют декомпрессию ЖКТ путем введения назогастрального зонда и промывания желудка, блокады, гипертонические компрессы на переднюю брюшную стенку, медикаментозную стимуляцию кишечника путем введения антихолинэстеразных средств, очистительные и гипертонические клизмы, снижение болевого синдрома путем введения спазмолитиков.
Данные мероприятия позволяют купировать кишечную непроходимость без оперативного вмешательства. Сроки консервативного лечения определяются в пределах 10-12 ч, а при улучшении общего состояния могут быть продлены до 24-36 ч.
При неэффективности консервативного лечения и ярко выраженной картине СКН вопрос о хирургическом вмешательстве решается однозначно. При диагностической лапароскопии она может перейти в лечебную при подтверждении наличия спаек в брюшной полости. Данная операция называется лапароскопическим адгезиолизисом. Она малоинвазивна, позволяет максимально снизить травматичность оперативного вмешательства и улучшить отдаленные результаты лечения СББ. Суть операции состоит в визуализации спаек брюшной полости и их коагуляции с последующим рассечением (рис. 8-7, г).
Профилактика
В ежедневной хирургической практике необходимо соблюдение принципов неспецифической интраоперационной профилактики спайкообразования, т.е. уменьшение инвазивности вмешательства, травмы, ишемии тканей.
Специальные (частные) методы профилактики предусматривают назначение препаратов, действующих в разных направлениях:
• фибринолитических ферментов (стрептокиназы, урокиназы, коллагеназы и др.);
• протеолитических ферментов (трипсина, химотрипсина и др.);
• антикоагулянтов (низкомоллекулярного гепарина);
• противовоспалительных препаратов (антибиотиков);
• неспецифических десенсибилизирующих средств (антигистаминных препаратов);
• препаратов гиалуронидазы (лидазы).
Кроме того, в послеоперационном периоде пациенты группы риска развития СББ нуждаются в назначении препаратов, замедляющих синтез коллагена и усиливающих его утилизацию. В комплексе с обычной противовоспалительной терапией им необходимо назначение пеницилламина (купренила*) внутрь с 3-х суток после операции в течение 10-14 дней в следующих дозировках: до 5 лет - 0,15 г; 5-12 лет - 0,3 г; старше 12 лет - 0,45 г. В целях разрушения и утилизации коллагеновых волокон одновременно проводят фонофорез коллализина со 2-3-го дня после операции по 10-15 сеансов.
Для предупреждения развития послеоперационного пареза кишечника используют электростимуляцию, иглорефлексотерапию, пролонгированные блокады рефлексогенных зон брюшной полости, гипербарическую оксигенацию.
Профилактика
Важным моментом является осведомленность больных и их родителей в отношении клинического течения СББ и необходимости врачебной консультации в целях ранней диагностики заболевания и предупреждения СКН и других осложнений.
После перенесенного оперативного лечения и выписки из стационара все больные должны быть взяты на диспансерный учет с регулярными осмотрами не реже 2 раз в год. Рекомендуют проведение реабилитационных мероприятий, направленных на повышение общей резистентности организма, на профилактику воспалительных заболеваний.
Прогноз
Прогноз неблагоприятный в случае позднего оперативного вмешательства при СКН, летальность при которой может составить 5-7%. В ходе длительного течения спаечного процесса с частыми рецидивами кишечной непроходимости может развиться стойкая инвалидизация больных.
ГЕПАТОЛОГИЯ Болезнь Вильсона-Коновалова
Код по МКБ-10
E83.0. Болезнь Вильсона-Коновалова.
Болезнь Вильсона-Коновалова (синонимы: гепатоцеребральная дистрофия, гепатолентикулярная дегенерация) - наследственное заболевание с аутосомно-рецессивным типом наследования, характеризуемое нарушением метаболизма меди, сочетанием цирроза печени с дистрофическим процессом в головном мозге. Болезнь описали в 1912 г. английский невролог S.A.K. Wilson и русский невропатолог Н.В. Коновалов.
Распространенность патологического аллеля гена болезни Вильсона-Коновалова колеблется от 1:200 до 1:500; гетеро-
зиготы составляют 1% клинически здоровых лиц, которые являются носителями и могут обнаруживать аномалии метаболизма меди. В скрытом гетерозиготном состоянии ген распространяется в популяции, не подвергаясь естественному отбору, поэтому наблюдается в популяции с частотой в среднем 1:200 000; частота выше среди народностей, где распространены близкородственные браки. Чаще болеют лица мужского пола, средний возраст дебюта - 11-25 лет.
Диагностируют у 5-10% больных циррозом печени дошкольного и школьного возраста.
Этиология и патогенез
Заболевание обусловлено низким или аномальным синтезом церулоплазмина - белка, транспортирующего медь (ATP7B); его структура представлена на рис. 8-8, а. Ген ATP7B, мутации которого вызывают заболевание, расположен на хромосоме 13.
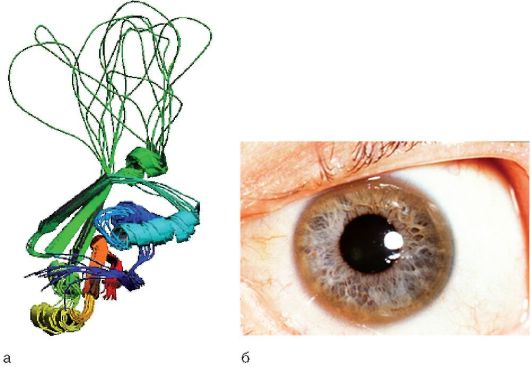
Рис. 8-8. Болезнь Вильсона-Коновалова: а - структура церулоплазмина (белка ATP7B); б - кольцо Кайзера-Флейшера
Основную роль в патогенезе играют нарушение обмена меди, ее накопление в нервной, почечной, печеночной ткани и роговице, а также токсическое повреждение медью данных органов. Нарушение метаболизма выражается в нарушении синтеза и снижении в крови концентрации церулоплазмина, участвующего в процессе выведения меди из организма.
Для проявления заболевания имеют значение экзогенные воздействия, поражающие печень, - интоксикация и инфекции.
Клиническая картина
Болезнь начинается в детском возрасте и имеет хроническое прогрессирующее течение. Во многих случаях появлению симптомов поражения нервной системы предшествуют висцеральные расстройства в виде нарушения деятельности печени и желудочно-кишечных расстройств.
ХГ с развитием цирроза, портальной гипертензии клинически проявляется гепатоспленомегалией, отеками, гемолитической анемией, тромбоцитопенией, лейкопенией.
Неврологическая патология: экстрапирамидные симптомы в виде мышечной ригидности, гиперкинезов, нарушений координации. Могут быть дизартрии, пирамидные симптомы, чувствительность обычно не нарушена.
Расстройства психики в виде депрессий, фобий у подростков; астенизация, повышенная утомляемость у детей младшего возраста.
Типичный симптом - кольцо Кайзера-Флейшера - желтовато-зеленая или зеленовато-коричневая пигментация по периферии роговицы, содержащая зеленовато-бурый пигмент меди. Более выражен при поздних формах заболевания, когда кольцо становится полным (рис. 8-8, б). Также могут отмечаться желтовато-коричневая пигментация кожи туловища и лица, геморрагические явления (кровоточивость десен, носовые кровотечения, положительные пробы щипка и жгута), мраморность кожи, акроцианоз. Редко отмечаются поражения сердца (кардиомиопатия, аритмии), костей (остеомаляция, остеопороз), почечный тубулярный ацидоз (глюкозурия, аминоацидурия, фосфатурия, уратурия, протеинурия).
Диагностика
При наличии признаков поражения печени, нервной системы и кольца Кайзера-Флейшера диагноз болезни Вильсона- Коновалова не вызывает сомнения. Для диагностики используют осмотр с помощью щелевой лампы, при котором обнаруживают зеленое кольцо Кайзера-Флейшера на роговице у лимба.
Диагноз подтверждается результатами следующих исследо ваний:
• содержанием меди в сыворотке крови (выше 80 мкг/100 мл, или 9,4 ммоль/л);
• концентрацией церулоплазмина (ниже 20 мг/100 мл, или 1 мкмоль/л);
• экскрецией меди с мочой (более 100 мк/сут, или 1,6 мкмоль);
• определением меди в биоптате печени - золотой стандарт (содержание меди более 50 мкг в 1 г сухого вещества печени).
Патоморфология
Печень вследствие формирования крупноузлового или смешанного цирроза бугристая, участки нормальной ткани чередуются с зонами дегенерации и некроза, островками регенерации; обильное новообразование сосудов приводит к появлению анастомозов между ветвями воротной и нижней полой вены.
В головном мозге поражаются чечевицеобразное ядро с образованием мелких кист, хвостатое ядро, глубокие слои коры, мозжечок, в частности зубчатые ядра, подбугорные ядра.
В почках поражаются в основном проксимальные канальцы.
Дифференциальная диагностика
Дифференциальную диагностику проводят с ХГ, первичным билиарным циррозом, наследственными (синдромом Жильбера и другими пигментными гепатозами), а также паразитарными (описторхозом, эхинококкозом) заболеваниями и др. В верификации заболевания используют данные биохимических проб печени, УЗИ печени, эзофагогастродуоденоскопии, КТ и других специальных методов исследования.
Лечение
Патогенетическое лечение направлено на увеличение выведения меди из организма.
Для выведения меди из организма применяют комплексоны (тиоловые соединения). Препаратом выбора является пеницилламин (купренил*) - комплексообразующее соединение, которое оказывает дезинтоксикационное и иммунодепрессивное действие, образует хелатные комплексы с ионами
меди. Начальная доза - 250 мг, затем ее постепенно увеличивают до 0,75-1,5 г/сут. Доза считается эффективной, если суточное выведение Cu2+ с мочой (после 1-й недели лечения) превышает 2 мг. В дальнейшем адекватность дозы определяют на основании измерения содержания свободной Cu2+ в сыворотке крови (оно должно быть <10 мкг/мл). В отдельных случаях суточная доза может составлять 2 г и более.

Лечение пеницилламином сопровождается заметным улучшением состояния больных или даже приводит к полной ликвидации симптомов. Вполне удовлетворительные результаты получены и при применении унитиола*.
Димеркаптопропансульфонат натрия (унитиол*) увеличивает выведение меди из металлосодержащих ферментов клеток; препарат выпускается в 5% растворе в ампулах по 2 и 5 мл № 5 и № 10, назначают детям старшего возраста по 250- 500 мг (5-10 мл 5% раствора) в/м ежедневно или через день. Курс лечения - 25-30 инъекций, при необходимости повторяют через 3-4 мес, далее назначают в таблетках по 0,25 и 0,50 г, в упаковках по 20 таблеток.
Пиридоксин (пиридоксина гидрохлорид* - витамин В6) в составе комплексной терапии назначают подкожно, в/м или в/в, если прием внутрь невозможен (при рвоте) и при нарушении всасывания в кишечнике, подросткам - по 50-100 мг/сут в 1-2 приема, детям - по 20 мг. Курс лечения - 1 мес.
По показаниям проводят трансплантацию печени.
Прогноз
Летальный исход от неврологических нарушений при отсутствии лечения возможен через 5-14 лет. Основными причинами служат интеркуррентные заболевания или желудочнокишечные кровотечения, портальная гипертензия.
Гемохроматоз
Код по МКБ-10
Е83.1. Гемохроматоз.
Гемохроматоз - наследственное, генетически обусловленное заболевание, проявляющееся нарушением обмена железа с накоплением его в печени и других тканях и органах.
Вероятность развития гемохроматоза, распространенного в странах Европы, составляет 0,01-0,07%; мужчины болеют в 10 раз чаще женщин. Потери железа во время менструаций и беременности, вероятно, предохраняют девушек и женщин от развития гемохроматоза.
Классификация
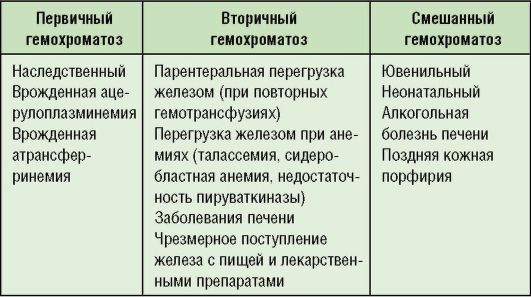
Различают первичный (классический) и вторичный гемохроматоз, связанный с другими заболеваниями.
Этиология и патогенез
Причины гемохроматоза представлены в табл. 8-3.
Таблица 8-3. Причины гемохроматоза
При наследственном (первичном) гемохроматозе ген расположен на хромосоме 6, заболевание наследуется по аутосомно-рецессивному типу.
В организме здорового человека содержится от 3-4 до 20-60 г. Железо является одним из важнейших микроэлементов и содержится во многих пищевых продуктах, входит в состав молекулы гемоглобина (протопорфирина-IX, присоединяющего железо), который в составе эритроцитов обеспечивает транспорт кислорода из легких по всему телу.
При гемохроматозе организм человека поглощает слишком много железа из пищи, которое накапливается в печени, сердце, поджелудочной железе и других органах. Опасна передозировка препаратами железа: на рис. 8-9 представлен аутопсийный материал больного, у которого возникли субтотальный некроз паренхимы печени, выраженное малокровие миокарда, почек.
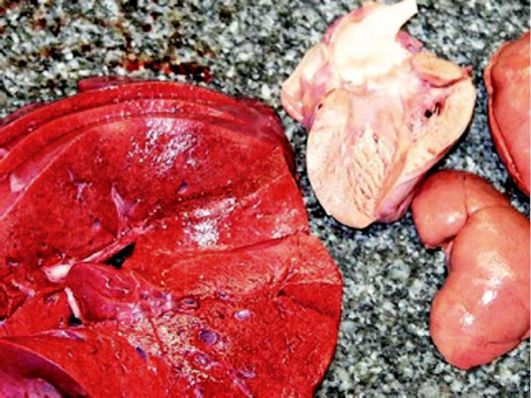
Рис. 8-9. Аутопсийный материал печени, сердца, почек: передозировка препаратами железа
Клиническая картина и диагностика
Диагностика не вызывает затруднений - помогает тщательный сбор семейного анамнеза в сочетании с лабораторной диагностикой. Симптомы гемохроматоза могут варьировать в широких пределах; 90% железа в организме поглощается печенью; артрит, нефропатия, пигментация кожи также могут быть следствием гемохроматоза. Клинические проявления:
• слабость и быстрая утомляемость;
• снижение давления;
• выраженное похудение;
• гиперпигментация кожного покрова (грифельно-серый цвет с коричневым оттенком), слизистых оболочек и сетчатки;
• сердечная недостаточность;
• отек и болезненность суставов.
Целесообразно обследовать всех родственников больного первой степени родства, лучше с помощью генетического скрининга в сочетании с определением содержания ферритина в сыворотке крови, железосвязывающей способности сыворотки.
Биопсия показана при изменениях биохимических показателей функциональных проб печени, превышении концентрации ферритина (более 1000 мкг/л), поскольку эти признаки ассоциируются с выраженным фиброзом печени.

Лечение
Для лечения гемохроматоза используют препарат, связывающий железо и способствующий его выведению, - дефероксамин (десферал*). Схему лечения (доза и способ введения) определяют индивидуально в зависимости от степени повышения содержания железа. Для оценки эффективности начатой терапии и подбора адекватной дозы необходим суточный контроль количества железа, выделяемого с мочой. Назначают наименьшую эффективную дозу, которая достаточна для установления отрицательного баланса железа. Средняя суточная доза - 20-40 мг/кг. Внутривенное введение эффективнее внутримышечного или подкожного способа введения.
Эффективный метод лечения - кровопускание (флеботомия или венесекция) до нормализации концентрации ферритина в сыворотке крови. Еженедельные кровопускания способствуют удалению железа из организма и приводят к улучшению общего состояния, уменьшению пигментации и размеров печени.
Прогноз
Прогноз зависит от длительности течения заболевания до момента диагностики.
Синдром Криглера-Найяра
Код по МКБ-10
E80.5. Синдром Криглера-Найяра.
Синдром Криглера Найяра - врожденная неконъюгированная гипербилирубинемия с аутосомно-доминантным (I типом синдрома Криглера-Найяра) и аутосомно-рецессивным (II типом синдрома Криглера-Найяра) типами наследования, характеризуемая желтухой и тяжелым поражением нервной системы.
Синдром описали американские педиатры J.F. Crigler и V.A. Najjar в 1952 г. Синдром Криглера-Найяра I типа наблюдается у представителей различных этнических групп, чаще у населения Азиатского региона. Заболеваемость составляет
1 случай на 1 млн новорожденных. С равной частотой встречается у мальчиков и девочек.
Этиология и патогенез
Гипербилирубинемия является следствием нарушения конъюгации в печени билирубина с глюкуроновой кислотой, обусловленного отсутствием или значительной недостаточностью фермента УДФГТ, переводящей свободный билирубин в связанный.
I тип характеризуется полным отсутствием активности УДФГТ. Обусловлен мутациями в кодирующей последовательности гена UGTIAI, что приводит к образованию неполноценного фермента УДФГТ, который разрушается, в связи с чем реакции глюкунизации билирубина не происходит, и свободный (непрямой, неконъюгированный) билирубин накапливается в организме, в том числе в ядрах серого вещества головного мозга, обусловливая тяжелое клиническое течение заболевания.
II тип - синдром Ариаса - активность фермента составляет менее 20% нормальной. Заболевание также обусловлено мутациями в той же кодирующей последовательности гена, но больные часто являются гетерозиготами. Гипербилирубинемия высокая, но лечение возможно.
Клиническая картина, классификация
Характерны симптомы желтухи (желтушность склер и кожного покрова) и неврологические нарушения (билирубиновая энцефалопатия).
I тип характеризуется злокачественным прогрессирующим течением. Манифестирует в первые часы жизни. При отсутствии лечебных мероприятий больные погибают в течение первого года жизни от ядерной желтухи.
В первой фазе билирубиновой энцефалопатии наблюдается угнетение безусловно-рефлекторной деятельности (апатия, вялость, сонливость), ребенок начинает плохо сосать, лежит в расслабленной позе, резко реагирует на слабые раздражители, дыхание становится редким, с длительными периодами остановки. Могут отмечаться монотонный крик, срыгивания, рвота, блуждающий взгляд, цианоз.
Вторая фаза ядерной желтухи продолжается от нескольких дней до нескольких недель. В этой фазе развивается клиническая картина поражения ядер головного мозга. Наблюдаются спастичность, ригидность затылочных мышц, вынужденное положение тела с опистотонусом, негнущимися конечностями и сжатыми в кулаки кистями. Ребенок пронзительно кричит, у него отмечаются выбухание большого родничка, подергивание мышц лица, крупноразмашистый тремор рук, исчезновение видимой реакции на звук, сосательного рефлекса. Наблюдаются нистагм, апноэ, брадикардия, летаргия, судороги.
Третья фаза билирубиновой энцефалопатии - период ложного благополучия. Явления спастичности полностью или частично исчезают.
В четвертой фазе (на 3-5-м месяце жизни) формируются стойкие неврологические нарушения: параличи, парезы, нистагм, атетоз. Наблюдается грубое отставание в физическом и психическом развитии: ребенок не держит голову, не реагирует на голос матери и другие звуковые раздражители, не следит за игрушкой.
Смерть пациентов при синдроме Криглера-Найяра I типа обусловлена развитием билирубиновой энцефалопатии и наступает в течение первых 2 лет жизни.
II тип занимает промежуточное положение по тяжести клинических проявлений между синдромом Криглера-
Найяра I типа и синдромом Жильбера. Манифестация наступает несколько позже, чем при I типе, - от нескольких месяцев до первых лет жизни.
У ряда больных желтуха может не проявляться до подросткового возраста, и неврологические осложнения наблюдаются редко; в некоторых случаях клиническая симптоматика отсутствует. Клинические проявления сходны с I типом, но менее тяжелые. Редко, при интеркуррентных инфекциях или в условиях стресса, у больных с синдромом Криглера-Найяра II типа может возникать билирубиновая энцефалопатия.
Диагностика
При I типе синдрома Криглера-Найяра основной биохимический показатель - уровень билирубина в крови выше 200 мкмоль/л. В желчи полностью отсутствует конъюгированный билирубин. На электроэнцефалограмме регистриру-
ются медленная активность в задних долях и пароксизмальная активность. При II типе уровень билирубина в крови менее 200 мкмоль/л, желчь пигментирована и содержит билирубин-глюкуронид.
ДНК-диагностика основана на поиске мутаций во всех экзонах гена UGTIAI, анализе промоторной области, а также косвенной ДНК-диагностики с использованием 3 полиморфных ДНК-маркеров, лежащих в районе локализации гена. Проба с фенобарбиталом позволяет определить фракции билирубина с помощью высокоэффективной жидкостной хроматографии.
Дифференциальная диагностика
Дифференциальная диагностика желтухи у детей первых месяцев жизни в значительной степени поддается логике, чем врачебной интуиции, и может быть представлена в виде алгоритма (рис. 8-10).
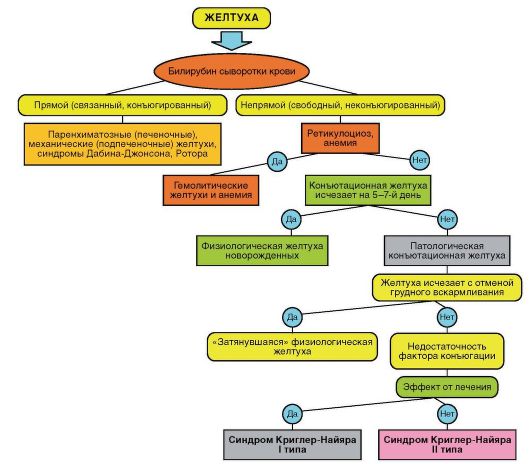
Рис. 8-10. Алгоритм дифференциальной диагностики различных типов желтухи и синдрома Криглера-Найяра I и II типа
Лечение
Рекомендуют соблюдение диеты № 5 с достаточной водной нагрузкой для профилактики синдрома сгущения желчи. Необходимы избегание провоцирующих факторов (инфекций, перегрузок), применение препаратов - конкурентов глюкуронирования или вытесняющих билирубин из связи с альбумином. Следует избегать ацидоза, сопровождаемого повышенной проницаемостью гематоэнцефалического барьера.
Медикаментозная терапия позволяет перевести неконъюгированный билирубин в конъюгированный, выведение, обеспечить связывание и разрушение билирубина.
Назначают индуктор микросомальных ферментов, способствующий переводу неконъюгированного билирубина в конъюгированный, - фенобарбитал в таблетках по 0,005 мг и растворе для приема внутрь до 5 мг/кг в сутки за 30-40 мин до еды 2 раза в день. У детей в возрасте до 6 мес РД - 5 мг,
6-12 мес - 10 мг, 1-2 лет - 20 мг, 3-4 лет - 30 мг, 5-6 лет - 40 мг, 7-9 лет - 50 мг, 10-14 лет - 75 мг. Детям старше 12 лет назначают глютетимид*.
Для выведения билирубина с помощью адсорбентов билирубина в кишечнике используют активированный уголь, лигнин гидролизный (полифепан*) и др. Полифепан* в виде пасты, порошка или гранул назначают детям до 1 года по 1 ч. л. на прием, 1-7 лет - по 1 десертной ложке, 7 лет и старше - по 1 ст. л., подросткам - по 0,5-1,0 г/кг 3-4 раза в день (1 ст. л. размешивают в 200 мл воды в течение 2 мин). В тяжелых случаях назначают плазмаферез, гемосорбцию.
Для связывания билирубина в крови используют введение альбумина в дозе 1 г/кг массы тела в течение 1 ч, особенно целесообразно введение альбумина перед заменным переливанием крови при синдроме Криглера-Найяра I типа.
Еще одна цель лечения - разрушение билирубина, фиксированного в тканях, тем самым происходит высвобождение периферических рецепторов, которые могут связать новые порции билирубина, предотвращая его проникновение через гематоэнцефалический барьер. Достигается это посредством фототерапии, показанием к которой считают концентрацию неконъюгированного билирубина в сыворотке крови более 256 мкмоль/л, у недоношенных детей - более 171 мкмоль/л. Максимальный эффект наблюдается при длине волны 450 нм. Фотоисточник (люминесцентные лампы синего света) помещают на высоте 40-45 см над туловищем (процедуру следует проводить только в кювезе под контролем температуры).
Необходимо экранирование глаз и половых органов (у мальчиков). Также можно использовать так называемые фотоодеяла. В последнем случае свет к коже ребенка передается от мощных галогеновых ламп с помощью оптоволоконных световодов.
Лечение
Лечебный эффект фототерапии основывается на способности молекул билирубина изменять свою химическую структуру под воздействием световой энергии. Билирубин поглощает световую энергию преимущественно в синей зоне видимого спектра (450-460 нм). Под воздействием света в коже происходят фотоизомеризация и фотоокисление билирубина. В процессе фотоизомеризации токсичная 4Z, 15Z форма билирубина (естественный изомер) превращается в менее токсичную 4Z, 15Е (фотоизомер). Экскреция фотоизомера, так же как и естественного изомера, осуществляется печенью, однако ее скорость не зависит от конъюгирующей способности гепатоцитов, поэтому происходит быстрее, чем обычное выведение билирубина. После попадания в тонкую кишку фотоизомер билирубина может частично всасываться обратно в кровь, поддерживая таким образом гипербилирубинемию.
При фотоокислении жирорастворимый неконъюгированный билирубин превращается в водорастворимый люмирубин, который выводится из организма с мочой. У детей, получающих фототерапию, концентрация люмирубина в крови может составлять до 15% общей концентрации билирубина.
Фотодеградацию билирубина усиливает рибофлавин. Фототерапия значительно эффективнее при одновременном проведении сеансов, так как кислород усиливает декомпозицию билирубина.
Частые сеансы фототерапии (до 16 ч в сутки) позволяют продлить жизнь больным; метод эффективен в 50% случаев, его можно проводить амбулаторно. Фототерапию следует
рассматривать как подготовку к трансплантации печени (при синдроме I типа).
Трансплантация печени принципиально улучшает прогноз заболевания, так как способствует нормализации обмена билирубина.
Прогноз
При синдроме Криглера-Найяра I типа прогноз неблагоприятный. При синдроме II типа желтуха сохраняется в той или иной степени в течение всей жизни, при своевременном лечении прогноз относительно благоприятный.
Синдром Дабина-Джонсона
Код по МКБ-10
K93.8. Поражение других уточненных органов пищеварения при болезнях, классифицированных в других рубриках.
Синдром Дубина-Джонсона (в отечественной литературе обозначается как синдром Дабина-Джонсона) - энзимопатическая желтуха, редкий пигментный гепатоз, характеризуемый нарушением экскреции связанного билирубина из гепатоцитов в желчные капилляры, что приводит к накоплению билирубина. Синдром описали американские врачи I.N. Dubin и F. Johnson в 1954 г.
Синдром Дабина-Джонсона распространен среди иранских евреев с частотой 1:1300 в ассоциации с недостаточностью VII фактора свертывания крови (60% случаев), приводящей к снижению активности протромбина. В 70% случаев синдром Дабина-Джонсона проявляется в молодом возрасте, преимущественно у лиц мужского пола.
Этиология и патогенез
Заболевание обусловлено наследственным дефектом с аутосомно-рецессивным типом наследования. Генетический дефект заключается в появлении мутации в гене, кодирующим белок, который является АТФ-зависимой транспортной системой канальцев гепатоцитов. В результате гепатобилиарный транспорт билирубина и органических анионов нарушается. Характерны следующие признаки:
• умеренное повышение в крови прямого (связанного) билирубина вследствие нарушения механизмов его транспорта из гепатоцитов в желчь;
• повышенное выделение с мочой желчных пигментов, билирубина;
• отложение в гепатоцитах темно-коричневого или бурооранжевого пигмента, который выявляется при окрашивании на липофусцин (устанавливают при пункционной биопсии печени).
Клиническая картина
Симптоматика выражена более ярко, чем при других формах гипербилирубинемии. Пациенты жалуются на повышенную утомляемость, плохой аппетит, боль в правом подреберье. Выраженность желтухи может варьировать, усиливаясь на фоне стрессов, инфекционных заболеваний, при приеме оральных контрацептивов и беременности. Во время ремиссии желтуха почти полностью исчезает.
Диагностика
Конъюгационная желтуха (конъюгированная гипербилирубинемия) диагностируется, если содержание прямой фракции билирубина превышает 5,2 мкмоль/л, а показатель общего сывороточного билирубина - более 34,2 мкмоль/л или если содержание прямой фракции билирубина составляет более 15% показателя общего сывороточного билирубина.
Для подтверждения диагноза проводят пробу с фенобарбиталом - снижение уровня билирубина на фоне приема фенобарбитала. Возможно умеренное повышение ферментов крови (АСТ, АЛТ, γ-глютамилтранспептидазы, ЩФ).
Обязательные инструментальные исследования:
• УЗИ органов брюшной полости (умеренное увеличение печени; размеры, форма, толщина стенок желчного пузыря и желчных протоков не изменены; конкременты отсутствуют; нередко увеличение размеров селезенки);
• пероральная или внутривенная холецистография (запаздывание или полное отсутствие контрастирования желчного пузыря и желчных протоков);
• пункционная биопсия печени (обнаружение в гепатоцитах печени характерного пигмента);
• диагностическая лапароскопия (характерное черное окрашивание печени).
Патоморфология
Особенностью синдрома Дабина-Джонсона является изменение цвета печени: она становится зеленовато-серой или коричневато-черной. Макроскопически в ткани печени определяются темные пятна («шоколадная печень», «черная печень»), появление которых связывают с нарушением секреции метаболитов тирозина, триптофана, фенилаланина (рис. 8-11, а). Структура печени остается нормальной. Отложение пигмента также происходит в селезенке. Гепатоциты и клетки Купфера заполнены темным пигментом, который выявляется при окрашивании на липофусцин, преимущественно в центре долек, а также желчью (рис. 8-11, б). При электронной микроскопии пигмент выявляется в плотных тельцах, связанных с лизосомами, выявляются нормальные желчные канальцы с интактными микроворсинками. Лизосомы имеют неровные контуры, увеличены, заполнены зернистым содержимым и часто - жировыми капельками, связанными с мембраной.
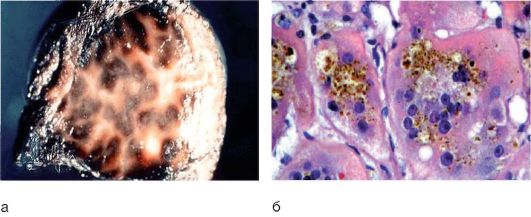
Рис. 8-11. Синдром Дабина-Джонсона: а - макропрепарат: «шоколадная печень»; б - скопление желчи и темного пигмента (окраска гематоксилин-эозином; χ 400)
Дифференциальная диагностика
Дифференциальную диагностику проводят с другими гипербилирубинемиями (синдромами Криглера-Найяра, Жильбера, Ротора; см. рис. 8-10), вирусным гепатитом, ХГ, проявляющимся холестатическим синдромом, механической желтухой, первичным билиарным циррозом.
Лечение
Больные должны стараться соблюдать щадящий режим - избегать провоцирующих факторов: инфекций, физических и психических нагрузок, инсоляции, употребления гепатотоксичных препаратов, алкоголя. Требуются санация хронических очагов инфекции и лечение имеющейся патологии желчевыводящих путей.
Рекомендуют диету с ограничением тугоплавких жиров и продуктов, содержащих консерванты.
Медикаментозного лечения не существует, хотя в определенной степени повышенный уровень билирубина поддается коррекции фенобарбиталом. Рекомендуются витамины группы В, желчегонные средства.
Критерии эффективности лечения - уменьшение интенсивности или устранение желтухи, нормализация (достоверное уменьшение) уровня билирубина в крови.
Продолжительность лечения - в течение всей жизни.
Прогноз
Прогноз благоприятный. Заболевание не влияет на продолжительность жизни пациентов.
Синдром Ротора
Код по МКБ-10
E80.6. Синдром Ротора.
Синдром Ротора - наследственный пигментный гепатоз с аутосомно-рецессивным типом наследования, напоминающий синдром Дабина-Джонсона, однако дефект экскреции билирубина менее выражен. Описал филиппинский терапевт A.B. Rotor в 1948 г.
Синдром Ротора - очень редкое состояние, первичный биохимический дефект, в отличие от других функциональных гипербилирубинемий, не идентифицирован. Как правило, заболевание возникает в детском возрасте.
Клиническая картина
Клиника проявляется интермиттирующей желтухой, нарастанием билирубина, повышением содержания копропорфирина в моче, задержкой бромсульфалеина. Желчный пузырь в данном случае контрастируется. У большинства болезнь протекает бессимптомно.
Диагностика
Наследственный синдром Ротора характеризуют следующие признаки:
• умеренное повышение в крови прямого (связанного) билирубина вследствие нарушения механизмов его транспорта из микросом гепатоцитов в желчь;
• неизмененная активность печеночных ферментов;
• повышенное выделение с мочой желчных пигментов;
• отсутствие темно-коричневого пигмента в гепатоцитах при пункционной биопсии;
• признаки жировой дистрофии гепатоцитов;
• визуализация желчного пузыря при холецистографии.
Дифференциальная диагностика
Алгоритм дифференциальной диагностики желтух представлен на рис. 8-10.
Лечение включает щадящий режим, исключение применения препаратов - конкурентов глюкуронирования или вытесняющих билирубин из связи с альбумином (сульфаниламидов, гепарина, салицилатов, оральных контрацептивов). Рекомендуют диету с ограничением тугоплавких жиров и продуктов, содержащих консерванты, с достаточной водной нагрузкой. Противопоказана инсоляция. Показаны санация хронических очагов инфекции и лечение имеющейся патологии желчевыводящих путей.
Прогноз
Прогноз благоприятный.
Недостаточность α-1-антитрипсина
Коды по МКБ-10
Q34.8. Врожденные аномалии органов дыхания. Q44.7. Врожденные аномалии органов печени.
Недостаточность α-1-антитрипсина - генетический дефект гликопротеина, ингибирующего активность протеолитических ферментов: трипсина, химотрипсина и эластазы, который может вызвать заболевания легких у взрослых и заболевания печени у взрослых и детей. Это наиболее распространенное генетическое заболевание печени, определяемое у младенцев. Впервые дефицит α-1-антитрипсина был описан в 1963 г.
При недостаточности а1-антитрипсина частота гомозиготного носительства составляет 1:600, гетерозиготного носительства - 1:2500. Около 1-3% взрослых больных с диагнозом «хроническая обструктивная болезнь легких» имеют недостаточность а1-антитрипсина.
Этиология и патогенез
α-1-Антитрипсин - сывороточный ингибитор сериновых протеаз, образующийся в печени. Формы α-1-антитрипсина детерминированы генетически. Около 90% здорового населения имеют генотип PiMM. У больных имеется генотип PiZZ, при котором α-1-антитрипсин не секретируется в кровь из гепатоцитов, так как полимеризуется в эндоплазматическом ретикулуме гепатоцитов. В мутантных вариантах синтезируемого белка ему не хватает возможностей для перехода с эндоплазматического ретикулума в зону Гольджи, и, следовательно, α1-антитрипсин накапливается в эндоплазме, как гиалиновые шарики (рис. 8-12). У лиц, гомозиготных по данному аллелю (PiZZ), активность α-1-антитрипсина в крови особенно низка, возможно развитие поражения легких и печени.
Клиническая картина
У детей с генотипом PiZZ заболевание может проявиться на первом году жизни: развивается тяжелая паренхиматозная желтуха, в дальнейшем - печеночно-клеточная недостаточность, быстро наступает летальный исход. Другой вариант поражения печени при недостаточности α-1-антитрипсина - длительно сохраняющаяся желтуха в первые недели жизни, обычно расцениваемая как неонатальный холестаз. В последующем развивается гепатоспленомегалия, однако даже при сформировавшемся циррозе печени дети могут чувствовать себя удовлетворительно.

Основные проявления касаются начала эмфиземы легких в возрасте от 20 до 50 лет. Медленнопрогрессирующая одышка - основной симптом, хотя многие пациенты изначально имеют кашель с выделением мокроты или свистящее дыхание, а также потерю массы тела. Курение или воздействия табачного дыма ускоряет появление симптомов и заболевания легких.
Около 10% детей и 15% взрослых имеют повреждения печени, ведущие к циррозу печени.
Признаки заболевания печени могут включать отеки нижних конечностей, желтуху и иктеричность склер, запор. В пубертатном периоде возможно развитие хронического АИГ и цирроза печени с гепатомегалией, портальной гипертензией, гиперспленизмом, печеночной энцефалопатией.
В редких случаях дефицит α-1-антитрипсина вызывает поражение кожных покровов.
Пример заболевания и смертельного исхода - легендарный поп-певец Майкл Джексон (см. рис. 8-12).

Рис. 8-12. Цирроз печени. Накопление α-1-антитрипсина в эндоплазме, подобно гиалиновым шарикам (указаны стрелками; окраска гематоксилин-эозином; х250). Фото и рисунок Майкла Джексона
Диагностика
Важны анамнестические данные о длительности желтухи или гепатита новорожденных, эмфиземы в юношеском возрасте.
На электрофореграмме белков в сыворотке крови: отсутствие α-1-глобулинов, в сыворотке крови снижена активность α-1-антитрипсина.
Индикатор повреждения печени - повышение ЩФ.
В генетическом анализе обнаруживают генотип PiZZ.
Патоморфология
В биоптатах печени: ШИК-положительные включения в перипортальных гепатоцитах, содержащие α-1-антитрипсин (см. рис. 8-12).
Дифференциальная диагностика
Дифференциальную диагностику проводят с жировой дистрофией печени, ХГ, включая АИГ, разнообразными видами цирроза печени. В раннем возрасте исключают атрезию желчных путей. При манифестации заболевания на первом году жизни его необходимо дифференцировать от неонатального гепатита, в дальнейшем - от вирусных гепатитов.
Лечение
Лечение симптоматическое, включает отказ от курения, физическую реабилитацию в программах, предназначенных для пациентов с хронической обструктивной болезнью легких. Назначают диетотерапию, гепатопротекторы, иммуносупрессивную терапию. Специфического лечения не существует.
Возможно применение донорского или генно-инженерного α-1-антитрипсина. При тяжелом течении цирроза рекомендуют трансплантацию печени.
Прогноз
Прогноз неблагоприятный. Неонатальный гепатит может разрешиться самопроизвольно в ХГ и цирроз печени у взрослых, в отдаленном периоде возможно развитие гепатоцеллюлярной карциномы.
Холестероз желчного пузыря
Код по МКБ-10
К82.4. Холестероз желчного пузыря.
Холестероз желчного пузыря - редкое и труднодиагностируемое заболевание, возникающее у лиц молодого возраста, связанное с абсорбцией и накоплением в стенке желчного пузыря липидов и сопровождаемое изменением его функции.
Холестероз чаще выявляется при инструментальных исследованиях желчного пузыря, а также при холецистэктомии (39%) и аутопсии (46%).
Этиология и патогенез
Причина заболевания в настоящее время не установлена. Решающую роль в развитии холестероза играют метаболические факторы. Часто холестероз сочетается с холелитиазом, что связывают с нарушением обмена липидов.
Большинство ученых считают, что при холестерозе липиды в стенку пузыря попадают из желчи. В норме стенка желчного пузыря абсорбирует определенное количество свободного холестерина и его циклических предшественников. Одна треть абсорбированного холестерина из желчи поступает в серозную оболочку пузыря, а затем - в лимфатические и кровеносные сосуды. Две трети холестерина возвращаются в желчь, и, таким образом, депонирования липидов в стенки желчного пузыря не происходит. Нарушение транспорта липидов в стенке желчного пузыря может быть обусловлено изменениями в системе лимфатических и кровеносных сосудов, релаксирующим действием гормонов (прогестерона), изменениями моторно-эвакуаторной функции вследствие раздражения нервных сплетений стенки, а также вследствие врожденной или приобретенной дисхолии. Нарушение обмена жиров сопровождается перенасыщением
желчи холестерином и способствует развитию холестероза желчного пузыря или холелитиаза.
Классификация
Существует несколько вариантов классификации холестероза. В настоящее время используют классификацию, в основу которой положены макроскопические изменения слизистой оболочки желчного пузыря.
Выделяют полипозную, сетчатую и смешанную формы холестероза.
Клиническая картина
Некоторые ученые считают, что холестероз не имеет собственных симптомов и клинически проявляется лишь при развитии холецистита или ЖКБ. Вместе с тем имеется точка зрения, указывающая на собственную симптоматику заболевания в виде приступообразных болей в правом подреберье, диспепсических явлений, обусловленных липидной инфильтрацией стенки пузыря, нарушением его сократительной функции, а также закупоркой пузырного протока слущивающимся эпителием. Не исключено, что в ряде случаев коликообразные боли обусловлены наличием конкрементов. Холестероз может быть причиной дисфункции органов билиарной системы.
Диагностика
Основными диагностическими методами являются УЗИ и рентгенография.
УЗИ проводят в условиях снижения режима работы аппарата. Холестероз желчного пузыря выявляют в основном на передне-боковой стенке, так как с помощью современной аппаратуры получить объективную информацию о строении задней стенки затруднительно. По форме холестероз желчного пузыря при УЗИ делят на очаговый и диффузносетчатый. Холестериновые отложения в виде пластинок чаще визуализируются на задней поверхности стенки пузыря (рис. 8-13, а), полипы - в виде образований округлой или овальной формы на внешней или внутренней поверхности стенки. При очаговой форме поражены отдельные участки его стенки, а при диффузно-сетчатой - значительная ее часть. Различают холестериновые полипы (рис. 8-13, б, в), которые отличаются от доброкачественных образований (папиллом и аденом) меньшей эхогенностью, их структура почти неотличима от паренхимы печени.
При холецистографии иногда выявляют маленькие фиксированные дефекты заполнения. Характерный рентгенологический признак заболевания - медленное появление пятнистого рисунка или исчерченности, стремительное опорожнение пузыря.
При исследовании желчи обнаруживают увеличение уровня холестерина, кристаллов билирубината кальция, снижение концентрации общих желчных кислот.

Патоморфология
Отложение жиров в подслизистой оболочке и эпителии проявляется множественными желтыми пятнами на розовой слизистой оболочке, что послужило причиной названия «земляничный желтый пузырь» (см. рис. 8-13, б).
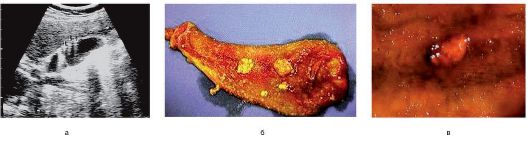
Рис. 8-13. Холестероз желчного пузыря: а - картина при УЗИ: стрелками указаны отложения холестерина в виде пластинок на задней стенке желчного пузыря; б - макропрепарат; в - холестериновый полип
Лечение
При отсутствии выраженного нарушения функций желчного пузыря проводят консервативную терапию. В нее входят:
• диета, обогащенная растительными волокнами, растительными маслами;
• желчегонные препараты, способствующие нормализации коллоидного состава желчи и моторно-эвакуаторной функции пузыря (лиобил*, холензим*, холагол* и др.);
• препараты желчных кислот (хенофальк*, урсофальк*, литофальк*), способствующие изменению соотношения желчные кислоты/холестерин желчи;
• антибактериальные препараты - при присоединении инфекции.
При нарушении моторно-эвакуаторной функции пузыря и наличия в нем множественных полипов основным методом лечения является холецистэктомия. Показаниями к операции являются:
• сопутствующий холелитиаз;
• нефункционирующий желчный пузырь;
• выраженные клинические проявления заболевания.
Прогноз
Прогноз в большинстве случаев благоприятный.