Эндокринная регуляция. Биохимические и физиологические аспекты : учеб. пособие / А.Н. Смирнов ; под ред. В.А. Ткачука - 2009. - 368 с.
|
|
|
|
ГЛАВА 6 СТРЕСС
В середине 1930-х годов канадский исследователь Ганс Селье занимался поиском нового гормона яичников. Введение экстрактов яичников крысам вызывало воспроизводимую «триаду» эффектов: гипертрофию коры надпочечников, инволюцию тимуса и изъязвление желудка и двенадцатиперстной кишки. Удивительно было то, что чем чище был экстракт, тем слабее он воспроизводил указанные эффекты. Оказалось, что любое достаточно сильное повреждающее воздействие (холод, перегрев, рентгеновское облучение и т.д.) способно индуцировать указанную реакцию организма. Оказалось также, что умеренное воздействие повреждающего агента может повышать защитные функции организма в ответ на действие не только данного, но и любого другого повреждающего агента. Эти факты послужили основой для создания концепции общего адаптационного синдрома: неспецифической реакции организма на любой достаточно сильный раздражитель - стрессор. По мнению Селье, значительная часть стрессорной реакции опосредуется гормонами надпочечников глюкокортикоидами.
В современной физиологии интерпретация значения участия глюкокортикоидов в стрессорной реакции выходит за рамки первоначально высказанной Селье идеи о том, что индуцированный стрессом выброс глюкокортикоидов усиливает и опосредует стрессорный ответ. Уже базальный уровень глюкокортикоидов служит пермиссивным фактором для стрессорной реакции, но они также ограничивают стрессорную реакцию и стимулируют ее прекращение (рис. 6-1).
ДИНАМИКА РАЗВИТИЯ СТРЕССОРНОЙ РЕАКЦИИ
В соответствии с концепцией Селье, ответ организма на стрессорное воздействие включает стадии тревоги, резистентности, а при продолжающемся воздействии стрессора - и истощения.
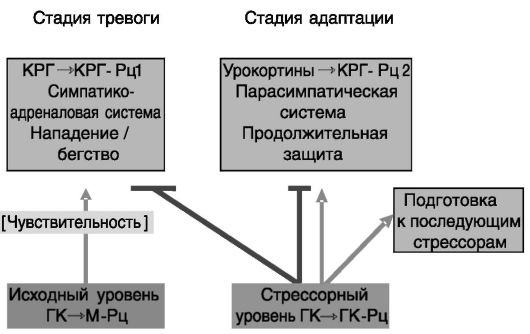 Рис. 6-1. Множественность функций глюкокортикоидов в развитии стрессорной реакции
Рис. 6-1. Множественность функций глюкокортикоидов в развитии стрессорной реакции
В стрессорной реакции принимают участие нервные, нейроэндокринные и эндокринные факторы, среди которых важнейшую роль играют симпатико-адреналовая и гипоталамо-гипофизарнонадпочечниковая системы. Динамика индукции секреции сигнальных соединений, принимающих участие в стрессорном ответе, начала их действия и развития физиологического ответа колеблется в широком диапазоне, от нескольких секунд до многих часов и даже дней (рис. 6-2). Поэтому, хотя набор таких сигнальных соединений и является стереотипным, их индивидуальный вклад в стрессорную реакцию может зависеть от продолжительности и интенсивности воздействия стрессора. При хроническом стрессе из-за постоянного дисбаланса сигнальных соединений могут развиваться так называемые болезни адаптации, включая, в частности, гипертензию и другие нарушения сердечно-сосудистой системы.
При остром стрессе 1-я волна, развивающаяся в течение нескольких секунд, включает выброс катехоламинов, кортикотропина рилизинг-гормона (КРГ), АКТГ, снижение секреции гонадотропинов, выброс глюкагона, ПРЛ и (у приматов) СТГ. При геморрагии массивно секретируются аргинин-вазопрессин и ренин. 2-я волна развивается через минуты и включает стероидные гормоны. Гормоны 1-й волны
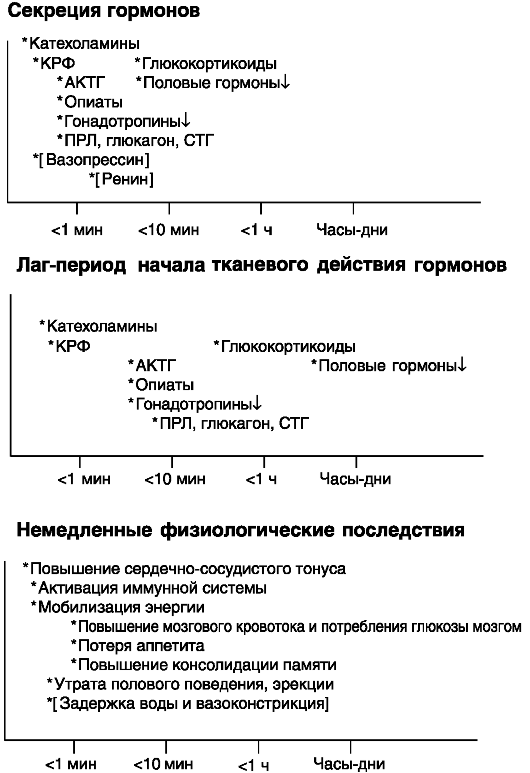 Рис. 6-2. Динамика секреции и инициации эффектов сигнальных соединений при остром стрессе
Рис. 6-2. Динамика секреции и инициации эффектов сигнальных соединений при остром стрессе
оказывают быстрые (секунды-минуты) эффекты через вторичные посредники. Эффекты гормонов 2-й волны за редкими исключениями развиваются медленно (1 ч и более).
ФУНКЦИИ ГЛЮКОКОРТИКОИДОВ ПРИ СТРЕССЕ
Модулирующие эффекты глюкокортикоидов, меняющие ответ организма на стрессовое воздействие, следующие:
• пермиссивные эффекты на включение защитных механизмов организма оказывает базальный уровень глюкокортикоидов до воздействия стрессора и проявляются немедленно после воздействия стрессора;
• супрессорные эффекты глюкокортикоидов проявляются за счет индуцированного стрессором повышения их концентрации, начинаются не ранее чем через 1 ч после воздействия стрессора и предназначены для сдерживания активированных стрессором защитных реакций от чрезмерного развития;
• стимулирующие эффекты глюкокортикоидов также проявляются
за счет индуцированного стрессором повышения их концентрации, начинаются не ранее чем через 1 ч после воздействия стрессора и предназначены для усиления эффектов гормонов 1-й волны;
• подготовительные эффекты глюкокортикоидов не влияют на немедленные ответы на стрессор, но модулируют ответы на повторное действие стрессора.
Сердечно-сосудистая система. В ответ на стрессор повышаются артериальное давление, частота сердцебиений и сердечный выброс, увеличивается поступление крови в мышцы за счет вазоконстрикции в мезентерии и почках и вазодилятации в мышцах. Эти изменения связаны с катехоламинами и КРГ, который через ЦНС вызывает активацию симпатической системы. Глюкокортикоиды также повышают давление и сердечный выброс. Эти эффекты пермиссивны, поскольку обеспечивают полноту действия катехоламинов и других вазоконстрикторов.
Антагонист глюкокортикоидов RU486 снижает эффективность катехоламинов и ангиотензина II. Действие глюкокортикоидов определяется несколькими механизмами: стимуляцией фенилаланин-^метил- трансферазы, обеспечивающей синтез адреналина из норадреналина в надпочечниках; торможением обратного захвата катехоламинов и снижением на периферии уровня ферментов метаболизма катехоламинов катехол-O-метилтрансферазы и моноаминоксидазы; повышением количества и сродства к агонистам β-адренорецепторов в гладкомышечных клетках сосудов; повышением сопряжения этих рецепторов
с G-белками и индуцированного синтеза цАМФ; торможением синтеза простагландинов, оказывающих вазодилататорное действие (это основной механизм гипертензии при болезни Кушинга). При адреналэктомии и болезни Адиссона развивается гипотония, которая может приводить к острому кризису при воздействии стрессора.
Объем крови и геморрагия. Геморрагия - сильный стрессор, вызывающий дополнительно выброс вазопрессина и ренина. Глюкокортикоиды опосредованно тормозят выброс вазопрессина, повышают клубочковую фильтрацию и увеличивают секрецию и эффективность действия натриуретического фактора предсердий, что способствует выведению воды.
У голодных адреналэктомированных животных геморрагия вызывает гибель, по-видимому, из-за гиперпродукции вазопрессина, который вызывает мощную вазоконстрикцию в печени и сердце и соответственно глубокую гипогликемию. Заместительная терапия глюкокортикоидами была эффективна лишь при дозировках, обеспечивающих уровень глюкокортикоидов, соответствующий циркадному пику.
Таким образом, при геморрагии глюкокортикоиды выступают в роли супрессоров ответа поддержания объема крови. Значение - защита от избыточного действия защитных механизмов.
Иммунитет и воспаление. Гормоны 1-й волны оказывают разнообразное, часто противоположное действие на иммунную систему. Например, внутрижелудочковое введение антител против КРГ блокирует снижение пролиферации T-клеток и цитотоксичности естественных киллеров, вызываемое КРГ в ответ на стрессор. В очень высоких концентрациях КРГ оказывает противовоспалительное и противоотечное действие. Но, с другой стороны, КРГ повышает пролиферацию B-клеток и пролиферативный ответ лимфоцитов на митогены.
Общие стрессоры вызывают быстрый иммунный ответ, сходный с ответом на действие инфекционных стрессоров. Этот ответ развивается еще до выброса глюкокортикоидов. Более того, ряд цитокинов, продуцируемых активированной иммунной системой, может стимулировать адренокортикальную ось. Например, ИЛ-1, действуя на гипоталамус и гипофиз, стимулирует секрецию КРГ и АКТГ.
Глюкокортикоиды тормозят иммунные и воспалительные реакции, снижая синтез, секрецию и/или эффективность цитокинов
(ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-12, фактора, стимулирующего колонии гранулоцитов-моноцитов (GM-CSF), ИФН-γ, ФНО-α, цитокина, регулируемого активацией экспрессии и секреции нор-
мальных T-клеток (RANTES), воспалительного белка 1α макрофагов), а также посредников воспаления (гистамина, брадикинина, эйкозаноидов, NO, коллагеназы, эластазы, активатора плазминогена). Глюкокортикоиды тормозят стимулирующее действие ФНО-α на молекулу межклеточной адгезии 1 (ICAM-1), подавляют презентацию антигена и экспрессию белков главного комплекса гистосовместимости (MHC) класса II, снижают активацию и пролиферацию T- и B-клеток, смещают ответ с Th1 клеток (продуцирующих ИЛ-2 и ИФН-γ) на Th2-клетки (продуцирующие противовоспалительный ИЛ-10). Глюкокортикоиды повышают активность трансформирующего фактора роста β (ТФР-β), подавляющего активацию T-клеток и макрофагов, и могут индуцировать экспрессию липокортина-1, оказывающего влияние на иммунные реакции.
Глюкокортикоиды влияют на передвижение клеток иммунной системы: быстро снижают в крови уровень лимфоцитов (T-клетки > B-клетки, хелперы CD4 > цитотоксичные CD8 и естественные киллеры), эозинофилов, базофилов, макрофагов и моноцитов, но повышают уровень нейтрофилов; подавляют хемотаксис лимфоцитов, моноцитов, гранулоцитов; в месте воспаления снижают аккумуляцию фагоцитирующих клеток; вызывают атрофию тимуса и других лимфоидных тканей, стимулируя апоптоз предшественников T- и B-клеток и зрелых T-клеток. Физиологический смысл этих эффектов может заключаться в облегчении селекции T-клеток и удалении потенциально токсических клеток.
Вместе с тем описано и усиление иммунных реакций под действием глюкокортикоидов. Например, у адреналэктомированных животных через 1 нед наблюдалось значительное снижение ответа T-клеток на митоген. Заместительная терапия кортикостероном в низких дозах восстанавливала ответ, а в высоких дозах (на уровне стресса) - подавляла. Предполагается, что стимулирующее действие низких доз глюкокортикоидов реализуется через рецепторы минералокортикоидов (М-Рц), а супрессорное действие высоких доз - через ГК-Рц.
Сходное пермиссивное или супрессивное действие глюкокортикоиды оказывают при ответе острой фазы на травму и инфекцию. Секретируемые иммунными клетками цитокины (ИЛ-1, ФНО-α) индуцируют в печени синтез белков острой фазы (амилоид A, С-реактивный белок, компоненты комплемента). Глюкокортикоиды повышают чувствительность печени к цитокинам, но снижают продукцию последних.
Физиологическая роль стрессорного выброса глюкокортикоидов заключается, по-видимому, в отсечении возможности перерастания иммунного ответа в аутоиммунную реакцию. Это достигается за счет предпочтительного подавления глюкокортикоидами малоактивных клеток или клеток, продуцирующих низкоаффинные антитела.
Метаболизм. Стрессор через катехоламины, глюкагон, СТГ вызывает быстрое повышение глюкозы крови за счет ее высвобождения из депо и блокировки повторного хранения из-за развивающейся резистентности к инсулину. Глюкокортикоиды также повышают уровень глюкозы, а в низких дозах - усиливают аппетит. Базальный уровень глюкокортикоидов стимулирует гликогенолиз и глюконеогенез под действием глюкагона и катехоламинов, повышает глюконеогенез в печени и образование гликогена, тормозит утилизацию глюкозы на периферии. Кроме того, глюкокортикоиды стимулируют липолиз в жировых депо и высвобождение аминокислот из мышц за счет подавления синтеза белка. Таким образом, они действуют пермиссивно и синергично по отношению к гормонам 1-й волны, усиливают и продлевают действие адреналина или глюкагона. Стимулирующее действие глюкокортикоидов на синтез гликогена можно рассматривать как подготовительное к повторному стрессу.
Действие глюкокортикоидов и инсулина в большинстве случаев антагонистично, но в синтезе гликогена они действуют синергично.
Центральная нервная система. Стрессор усиливает утилизацию глюкозы в мозгу уже через несколько секунд, что связано с активацией симпатической нервной системой сердечно-сосудистого тонуса и поступления крови в мозг. Адреналэктомия повышает утилизацию глюкозы в мозгу. Глюкокортикоиды в стрессовых концентрациях через ГК-Рц тормозят утилизацию глюкозы в мозгу, снижают транспорт глюкозы в нейроны и клетки глии за счет удаления транспортера глюкозы с клеточной поверхности в места хранения (минуты-часы) и снижения уровня мРНК транспортера глюкозы (часы-дни). Таким образом, глюкокортикоиды оказывают супрессорное действие на данный показатель.
Стрессор подавляет пищевое поведение в пределах 1 ч, что, видимо, связано с действием КРГ, который является анорексигенным агентом: антагонисты КРГ снимают тормозное действие стресса на потребление пищи; эффект КРГ сохраняется у гипофизэктомированных животных и наблюдается при внутрижелудочковом введении. Глюкокортикоиды, напротив, стимулируют аппетит в течение нескольких дней; адреналэктомия подавляет пищевое поведение.
Эффект глюкокортикоидов, видимо, реализуется через паравентрикулярное ядро гипоталамуса; их стрессорные концентрации, однако, подавляют аппетит, но это ингибирование является опосредованным, через стимуляцию секреции инсулина, который обладает анорексигенным действием. Альдостерон и глюкокортикоиды в малых концентрациях повышают потребление как углеводов, так и жиров, а избирательные агонисты глюкокортикоидов - только углеводов. В целом глюкокортикоиды оказывают супрессорное действие, помогают выйти из-под анорексигенного действия стресса и оказывают подготовительное действие к повторному стрессу.
Стрессоры усиливают запоминание, что происходит с участием катехоламинов: антагонисты β-адренорецепторов снимают этот эффект стрессора. Эффект, возможно, связан с повышением утилизации глюкозы. Основным объектом действия глюкокортикоидов на память служит гиппокамп, в котором много М-Рц и ГК-Рц. Базальный уровень глюкокортикоидов через М-Рц повышает возбудимость (за счет укорочения фазы рефрактерности после потенциала действия) и синаптическую пластичность, служащую основой обучения. Адреналэктомия и антагонисты М-Рц ухудшают процессы запоминания. Глюкокортикоиды в стрессорных концентрациях через ГК-Рц оказывают противоположное действие; при длительном их действии высокие концентрации вызывают атрофические изменения в гиппокампе. Таким образом, базальный уровень глюкокортикоидов оказывает пермиссивное действие, а стрессорный уровень - супрессивное действие. Физиологическая роль этой супрессии непонятна.
Репродуктивная система. Стрессор угнетает половое поведение и репродуктивную сферу в целом. В пределах нескольких минут происходит падение секреции ЛГРГ и гонадотропинов, быстро пропадает эрекция. Эффекты обусловлены гормонами 1-й волны.
Введение (внутрижелудочковое, но не периферическое) антагонистов КРГ частично снимает индуцированное стрессом падение ЛГ через подавление секреции ЛГРГ. Эффект, по-видимому, опосредован опиатами. Активация симпатической нервной системы блокирует опосредуемую парасимпатической системой инициацию эрекции. β-Адреноблокаторы снижают ингибирующее действие стресса на гонадотропины.
Глюкокортикоиды в стрессовых концентрациях действуют сходно с гормонами 1-й волны: они снижают секрецию ЛГРГ, прямо подавляют секрецию ЛГ, снижают чувствительность гонад к ЛГ. Базальный уровень глюкокортикоидов, по-видимому, либо слабо подавляет, либо
не влияет на репродуктивную сферу (отсутствие или слабое влияние адреналэктомии и низких доз глюкокортикоидов). Таким образом, глюкокортикоиды действуют синергично с гормонами 1-й волны и могут играть роль в подготовке к повторному стрессу.
Влияние стресса на овуляторный выброс гонадолиберина. Стрессорные воздействия могут задерживать и ослаблять овуляторный выброс гонадолиберина, что отрицательно сказывается на фертильности.
Тела гонадолиберинергических нейронов находятся в медиальной преоптической области гипоталамуса (MPOA; рис. 6-3). Стимулом для секреции ЛГРГ служат сигналы, поступающие в MPOA по аксонам от норадреналинергических нейронов ствола мозга, расположенных в областях A1 (ventrolateral medulla, VLM) или A2 (n. of the tractus solitarius, NTS). Большинство этих нейронов содержит Э-Рц. Норадреналинергические аксоны из этих областей также прямо или опосредованно стимулируют активность ГАМК-ергических нейронов в MPOA, которые, в свою очередь, тормозят активность ЛГРГергических нейронов. Таким образом, существует сбалансированное влияние ствола мозга на продукцию ЛГРГ. Сами ГАМК-ергические нейроны в MPOA чувствительны к действию эстрогенов и прогестерона. В лютеиновую фазу цикла прогестерон, а в раннюю фолли-
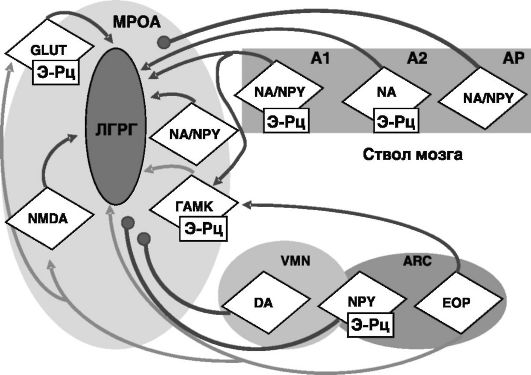 Рис. 6-3. Нейроны, участвующие в регуляции продукции ЛГРГ
Рис. 6-3. Нейроны, участвующие в регуляции продукции ЛГРГ
кулярную фазу эстрадиол повышают активность ГАМК-ергических нейронов, поддерживая низкую частоту прямоугольных импульсов ЛГРГ. В середине и конце фолликулярной фазы возрастающий уровень эстрогенов стимулирует секрецию норадреналина в MPOA. При этом происходит снятие стимулирующего действия норадреналина на ГАМК-ергические нейроны. Соответственно происходит растормаживание ЛГРГ-ергических нейронов. Продукция гонадолиберина контролируется также сигналами из аркуатного ядра (ARC) и вентромедиального гипоталамического ядра (VMN), формирующих медиобазальный гипоталамус (MBH). Аксоны опиоидергических нейронов ARC заканчиваются синапсами непосредственно на ЛГРГергических нейронах, а также на промежуточных нейронах в MPOA. Активность опиоидергических нейронов снижается непосредственно перед овуляторным выбросом ЛГРГ. Опиоиды подавляют стимулирующее действие катехоламинов, глутамина и NO на продукцию ЛГРГ и усиливают ингибирующее действие ГАМК. Стимулирующее действие на ЛГРГ оказывают нейропептид Y-ергические нейроны MBH. Изменения характера секреции ЛГРГ, гормонов и нейромедиаторов в ходе полового цикла показаны на рис. 6-4. Полагают, что в раннюю фолликулярную фазу эстрогены через активацию ГАМК-ергических
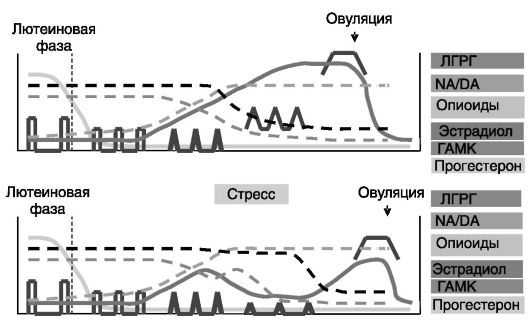 Рис. 6-4. Изменения продукции ЛГРГ, гормонов и нейромедиаторов при воздействии стрессора в половом цикле
Рис. 6-4. Изменения продукции ЛГРГ, гормонов и нейромедиаторов при воздействии стрессора в половом цикле
нейронов снижают амплитуду, но повышают частоту пульсов ЛГРГ. Последующее торможение активности ГАМК-ергических нейронов ведет к изменению формы импульсов ЛГРГ (из прямоугольных в треугольные). Последующее снижение действия опиоидов сопровождается уширением импульсов и увеличением секреции ЛГРГ между импульсами. Помимо действия на гипоталамические структуры эстрогены повышают чувствительность гипофиза к ЛГРГ, стимулируя экспрессию рецептора ЛГРГ в гипофизе.
Стрессор снижает пульсацию ЛГРГ уже через 5-10 мин. Стрессорное воздействие повышает продукцию кортиколиберина и вазопрессина в паравентрикулярном ядре (PVN) гипоталамуса, причем при усилении стрессора соотношение двух пептидов меняется в сторону вазопрессина. Роль последнего в подавлении репродуктивной функции стрессором доказывается тем, что антагонисты вазопрессина подавляют ингибирующее действие стресса на секрецию ЛГ. Предполагается, что КРГ через ГАМК-норадреналинергические нейроны снижает частоту импульсов ЛГРГ, а вазопрессин через опиоидергические нейроны подавляет амплитуду импульсов ЛГРГ. Показано, что аксоны КРГергических нейронов PVN образуют синапсы на ГАМК-ергических нейронах в MPOA. Кроме того, на самих ЛГРГ-ергических нейронах выявлены синапсы, содержащие КРГ или вазопрессин. Нейропептид Y, по-видимому, участвует в ингибировании ЛГРГ опосредованно, через стимуляцию продукции КРГ. Уровень самого нейропептида Y в ARC и PVN возрастает при стрессе. КРГ может подавлять секрецию ЛГРГ еще одним путем - через ингибирование активности серотонинергических нейронов ствола мозга (ядро Raphe), иннервирующих MPOA и стимулирующих секрецию ЛГРГ. Конечным итогом этих воздействий являются задержка овуляторного выброса ЛГРГ, снижение объема выброса и неполноценная овуляция. Стрессор, кроме того, снижает чувствительность гипофиза к действию ЛГРГ. Половые стероиды на разных уровнях повышают чувствительность репродуктивной системы к ингибирующему действию стрессора.
Стресс и желудочно-кишечный тракт. На протяжении многих десятилетий полагали, что вызываемому стрессорным агентом язвообразованию способствует стрессорный выброс глюкокортикоидов. В пользу этой точки зрения свидетельствовал факт ульцерогенного действия фармакологических доз глюкокортикоидов. Результаты исследований последних лет, однако, свидетельствуют о защитной функции глюкокортикоидов. Действительно, блокада биосинтеза или действия
глюкокортикоидов значительно повышает язвообразование у крыс, подвергаемых холодовому или плавательному стрессу, а заместительная терапия гормоном восстанавливает степень язвообразования. Предположительно, механизм защитного действия глюкокортикоидов включает подавление моторики желудка и кишечника, стимуляцию кровотока в слизистой и образования слизи. На модели вызываемого индометацином язвообразования показано, что глюкокортикоиды действуют в кооперации с простагландинами, NO и чувствительной к капсаицину нервной афферентацией, причем глюкокортикоиды эффективны в концентрациях, соответствующих и их базальному и стрессорному уровню. Таким образом, в данной системе глюкокортикоиды действуют в качестве супрессорного фактора в отношении индуцируемого стрессом язвообразования и в качестве пермиссивного фактора в отношении других защитных механизмов. Более того, глюкокортикоиды способствуют заживлению уже образовавшихся язв, о чем свидетельствует замедление заживления после адреналэктомии.
Стрессор может индуцировать быстрое опорожнение кишечника, включая стимуляцию подвижности ободочной кишки, секрецию в просвет кишечника слизи и воды, повышение проницаемости слизистой для ионов, а при длительном воздействии - развитие диареи. Ведущую роль в этих процессах играет КРГ. Об этом свидетельствуют следующие факты: введение КРГ или его гомолога урокортина в желудочек мозга или непосредственно в структуры мозга, чувствительные к КРГ (PVN, структуры моста - комплекс locus coeruleus/ ядро Баррингтона), но не в передний гипоталамус, воспроизводит эффект стрессора на ободочную кишку; введение в указанные отделы мозга или желудочек мозга антагонистов рецептора 1 КРГ - (КРГ- Рц1) блокирует действие стрессора на ободочную кишку; нокаут гена КРГ-Рц1 у мыши снижает индуцированную стрессором дефекацию. Эфферентный путь влияния стрессора на ободочную кишку включает проекции из PVN гипоталамуса и структур моста в крестцовый отдел спинного мозга, обеспечивающий парасимпатическую иннервацию ободочной кишки (рис. 6-5). Ваготомия и введение ганглиоблокатора атропина блокируют эффект центрального введения КРГ. Конечное звено эфферентации включает высвобождение вещества P и серотонина.
Стрессор с участием КРГ повышает также болевую чувствительность ободочной кишки к растяжению, что, очевидно, служит дополнительным фактором стимулирующего действия стрессора на
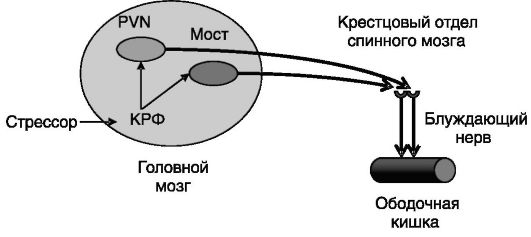 Рис. 6-5. Эфферентный путь влияния стрессора через КРГ на ободочную кишку
Рис. 6-5. Эфферентный путь влияния стрессора через КРГ на ободочную кишку
дефекацию. Центральное введение антагониста КРГ-Рц1 подавляет вызванную стрессором висцеральную гипералгезию. Высказывается предположение, что между хроническим стрессом и синдромом раздраженного кишечника у человека возникает порочная связь с участием КРГ, которую можно разорвать с помощью его антагонистов.
Парадоксально, но влияние стрессора на ободочную кишку может быть отчасти подавлено не только центральным, но и периферическим (внутрибрюшинным или внутривенным) введением антагонистов КРГ-Рц1, которые, по-видимому, не проникают из системного кровотока в мозг. Данный эффект антагонистов, возможно, связан с выявленной экспрессией нейронами мышечно-кишечного сплетения ободочной кишки КРГ-Рц1, а также урокортина. Можно предположить, что центральное действие стрессора каким-то образом дублируется на периферии с участием одного и того же КРГ-Рц.
Соотношение форм участия глюкокортикоидов в стрессорном ответе. Среднее время погони хищников за травоядными (острый стресс) составляет приблизительно 1 мин. Это значительно меньше, чем требуется для развития эффектов стрессорных концентраций глюкокортикоидов, поэтому трудно переоценить их пермиссивную функцию, обеспечивающую адекватность действия гормонов 1-й волны.
Стрессорный выброс глюкокортикоидов может иметь важное значение для подготовки организма к действию следующего стрессора.
Пермиссивные эффекты глюкокортикоидов, как полагают, в ряде случаев осуществляются через М-Рц, а супрессивные и подготовительные - через ГК-Рц.
Вероятный механизм двойного влияния глюкокортикоидов на развитие стрессорного ответа. Когнитивные способности, в значительной мере определяющие успешность реакции на стрессор, зависят от уровня глюкокортикоидов: как их недостаточность (например, при адреналэктомии), так и их избыток (например, при болезни Кушинга) оказывают неблагоприятное влияние на возбудимость и другие показатели эффективности функционирования мозга (рис. 6-6). В качестве примера можно привести гиперполяризующее действие серотонина на нейроны гиппокампа и зубчатой извилины, которое является минимальным при нормальном уровне глюкокортикоидов и возрастает у адреналэктомированных животных и в присутствии высоких концентраций глюкокортикоидов.
В ряде областей мозга, связанных со стрессорной реакцией и играющих важную роль в формировании эмоций и когнитивной функции (область CA1 гиппокампа, зубчатая извилина, ядра миндалины, медиальная префронтальная кора), выявлена совместная локализация двух типов ядерных рецепторов М-Рц и ГК-Рц, способных опосредовать эффекты глюкокортикоидов. М-Рц обладает приблизительно в 10 раз более высоким сродством к глюкокортикоидам по сравнению с ГК-Рц, и поэтому в начале развития стрессорного ответа глюкокортикоиды (еще до начала подъема их концентрации) действуют преимущественно через М-Рц. Спустя 10-15 мин после начала воздействия стрессора уровень глюкокортикоидов в крови значительно возрастает, и глюкокортикоиды начинают взаимодействовать не только с М-Рц, но и с ГК-Рц. Комплексы глюкокортикоидов с диме-
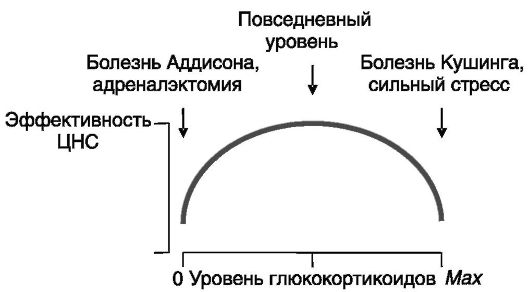 Рис. 6-6. Зависимость эффективности функционирования ЦНС от уровня глюкокортикоидов
Рис. 6-6. Зависимость эффективности функционирования ЦНС от уровня глюкокортикоидов
рами М-Рц и ГК-Рц узнают одни и те же ГЧЭ в регуляторных областях компетентных генов, и поэтому общая направленность действия низких и высоких концентраций глюкокортикоидов, реализуемых соответственно через М-Рц и ГК-Рц в качестве транскрипционных факторов, является сходной. Однако мономерный ГК-Рц, в отличие от М-Рц, способен действовать еще и в качестве трансрепрессора. Данная функция реализуется за счет взаимодействия ГК-Рц с другими транскрипционными факторами, такими, как AP-1 и NF-κΒ, без связывания ГК-Рц с ДНК. В результате глюкокортикоиды в высокой концентрации оказывают ингибирующее действие на транскрипционные факторы, активируемые катехоламинами и другими нейромедиаторами, ограничивая дальнейшее развитие стрессорного ответа (рис. 6-7). Преобладание трансрепрессорной функции ГК-Рц над его действием в качестве транскрипционного фактора может, вероятно, служить одной из причин развития болезней адаптации. Так, мыши с мутацией ГК-Рц, блокирующей его взаимодействие с ДНК, но не влияющей на его связывание с другими транскрипционными факторами, обладают сниженной способностью к обучению при повышенной активности гипоталамо-гипофизарно-надпочечниковой системы.
Подавление высокими концентрациями глюкокортикоидов иммунного ответа также, видимо, включает трансрепрессорную функцию
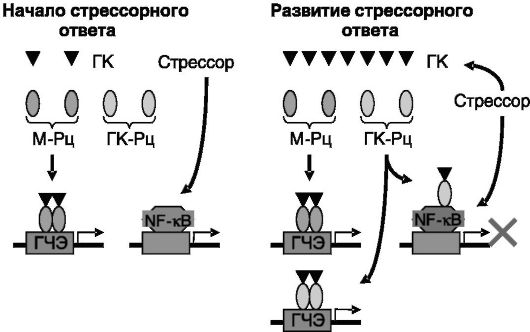 Рис. 6-7. В
низкой концентрации глюкокортикоиды (ГК) действуют преимущественно
через М-Рц. В высокой концентрации они действуют и через М-Рц, и через
ГК-Рц, причем мономерная форма ГК-Рц может выполнять функцию
трансрепрессора
Рис. 6-7. В
низкой концентрации глюкокортикоиды (ГК) действуют преимущественно
через М-Рц. В высокой концентрации они действуют и через М-Рц, и через
ГК-Рц, причем мономерная форма ГК-Рц может выполнять функцию
трансрепрессора
ГК-Рц. Благодаря стимуляции взаимодействия ГК-Рц с c-Jun и p65, являющимися субъединицами транскрипционных факторов AP-1 и NFkB соответственно, стрессорный уровень глюкокортикоидов подавляет активность этих факторов, направленную, в частности, на продукцию провоспалительных цитокинов. Данный эффект развивается независимо от способности ГК-Рц взаимодействовать с ДНК. Кроме того, в моноцитах и лимфоцитах глюкокортикоиды через ГК-Рц (уже как транскрипционный фактор) повышают экспрессию ингибитора NF-kB - IkB. Стимуляция высокими концентрациями глюкокортикоидов апоптоза клеток иммунной и нервной систем, как и подавление ими продукции цитокинов в ряде типов клеток, не зависит от трансактивационной функции ГК-Рц. Базальный же уровень глюкокортикоидов, напротив, повышает жизнеспособность клеток, и этот эффект реализуется преимущественно через М-Рц. Предполагается, что данные эффекты глюкокортикоидов реализуются с участием транскрипционного фактора p53, регулирующего экспрессию про- и антиапоптотических белков Bax, Bcl-2 и Bcl-xL. Необходимо отметить, что один из компонентов триады Селье, атрофия тимуса, не наблюдается у мышей с мутантным ГК-Рц, неспособным взаимодействовать с ДНК. Этот факт, с одной стороны, подтверждает участие глюкокортикоидов в действии стрессора на тимус, а с другой стороны, свидетельствует о функционировании ГКРц в клетках тимуса в качестве транскрипционного фактора.
Участие М-Рц в действии глюкокортикоидов осуществляется в клетках (например, гиппокампа), лишенных защиты от глюкокортикоидов. Клетки ряда других тканей и органов (например, дистальных отделов нефрона) включают систему, позволяющую им дифференцированно отвечать на минералокортикоиды и не реагировать на глюкокортикоиды. Эта система представляет собой 11β-ГСД2, окисляющую 11β-гидроксигруппу глюкокортикоидов до кетогруппы, наличие которой блокирует их взаимодействие и с ГК-Рц, и с М-Рц.
ДРУГИЕ ЭФФЕКТОРЫ СТРЕССОРНОГО ОТВЕТА
Вазопрессин относится к стрессорным гормонам 1-й волны. Секретируемый нейрогипофизом вазопрессин способствует задержке воды (через стимуляцию встраивания аквапоринов в мембраны клеток концевой части нефрона), повышению артериального давления (посредством стимуляции вазоконстрикции, прямо и через индукцию
секреции эндотелина), развитию гипергликемии (за счет активации фосфорилазы гликогена в печени). Продуцируемый мелкоклеточными ядрами гипоталамуса вазопрессин попадает в портальную систему переднего гипофиза, где он синергично с КРГ стимулирует секрецию АКТГ. В ЦНС вазопрессин выполняет функции нейромедиатора и таким образом участвует в формировании стрессорного ответа, в частности его поведенческого компонента. Показано, например, что селективный антагонист изоформы VIb рецептора вазопрессина оказывает анксиолитическое и антидепрессантное действие. Гиперпродукция вазопрессина может служить одной из причин патологической реакции на стрессорные воздействия.
Адреналин, выбрасываемый надпочечниками под действием стрессора, стимулирует сердечно-сосудистую систему, вызывает гипергликемию. Попадая из кровотока в гипоталамические структуры, адреналин ускоряет принятие и реализацию решения о форме стрессорного ответа (атака или бегство).
Бомбезин и родственные пептиды участвуют в интеграции стрессорных ответов, поскольку их введение имитирует эндокринные, автономные и поведенческие эффекты стрессора, а блокировка действия бомбезина и родственных пептидов тормозит развитие стрессорной реакции. Полагают, что эти пептиды активируют кортиколиберин- и/или вазопрессинергические нейроны.
Серотонин, по-видимому, служит одним из эффекторов стрессорной реакции. При этом доминантные особи отличаются от подчиненных более мощным и быстрым выбросом серотонина. Подчиненные особи характеризуются хронической активацией серотонинергической системы. КРГ, вазопрессин и другие гормоны 1-й волны, а также глюкокортикоиды служат модуляторами серотонинергической системы.
Пролактин, относящийся к гормонам 1-й волны стрессорного ответа, оказывает множественные влияния на ЦНС и периферические ткани, усиливающие или ослабляющие стрессорную реакцию. Так, ПРЛ оказывает анксиолитическое действие, подавляет реактивность гипоталамо-гипофизарно-адреналовой системы и выброс окситоцина нейрогипофизом (супрессорное действие). Особенно ярко эти эффекты проявляются в период до и после родов. ПРЛ является стимулятором гематопоэза и иммунной системы (стимулирующее действие), хотя он и не служит необходимым фактором для активации иммунной системы при стрессе, так же как ГР и ИФР-I. ПРЛ прямо и опосредованно (через катехоламины и КРГ) подавляет секрецию
гонадолиберина гипоталамусом (т.е. действует на репродуктивную функцию сходно с другими гормонами 1-й волны).
Стресс, КРГ ослабляют гематоэнцефалический барьер, антагонист рецептора КРГ-Рц блокирует действие стресса на него.
Центральную роль в организации ответа организма на стрессорное воздействие играет кортиколиберин. КРГ действует преимущественно через рецептор КРГ-Рц1 (надсемейство рецепторов, сопряженных с G-белками), экспрессируемый в коре мозга, мозжечке, медиальной перегородке, переднем гипофизе. Второй рецептор, КРГ-Рц2, предпочтительно связывает родственный КРГ пептид урокортин, продукция которого также возрастает при стрессе. КРГ-Рц2 экспрессируется в латеральной перегородке, вентромедиальном гипоталамусе, хориоидном сплетении, а также в сердце, скелетной мускулатуре, сосудах, желудочно-кишечном тракте. Нокаут гена КРГ-Рц1 снижает вызываемую стрессором тревожность, подавляет ответ гипоталамо-гипофизарно-надпочечниковой системы. Напротив, нокаут гена КРГ-Рц2 повышает чувствительность к стрессорному воздействию по скорости и величине реакции. Одной из причин служит значительное увеличение экспрессии вазопрессина, усиливающего действие КРГ. Полученные результаты позволяют предполагать, что КРГ через КРГ-Рц1 проводит стрессорный сигнал на поведенческий и эндокринный компоненты, а более медленно действующий урокортин через КРГ-Рц2 ограничивает данные эффекты и обеспечивает адаптацию поведения (см. рис. 6-1).
ХРОНИЧЕСКИЙ СТРЕСС
К хроническому стрессу могут приводить болезненные состояния, нервные потрясения, часто повторяющиеся или постоянные неблагоприятные социальные факторы. Хроническая активация нервных, нейроэндокринных и эндокринных компонентов стрессорной реакции может вести к сбоям на различных уровнях с развитием или обострением болезней адаптации (связанных со стрессом нарушений). К таким заболеваниям относятся некоторые психические нарушения (глубокая депрессия, посттравматическое стрессорное нарушение и т.д.), гипертензия и другие нарушения сердечно-сосудистой системы, резистентность к инсулину и связанные с этим метаболические нарушения (гипергликемия, дислипидемия, абдоминальное ожирение). В значительной мере эти нарушения ассоциированы с аномальным
функционированием системы гипоталамус-гипофиз-кора надпочечников. Доминирующей парадигмой, объясняющей данный феномен, служит так называемая гипотеза глюкокортикоидного каскада, согласно которой индуцированная стрессорным воздействием длительная избыточная продукция глюкокортикоидов вызывает повреждение структур мозга (прежде всего гиппокампа), участвующих в системе отрицательной обратной связи в продукции глюкокортикоидов. Это соответственно сопровождается дальнейшей несдерживаемой секрецией глюкокортикоидов, оказывающих неблагоприятное действие и на периферии. Действительно, при многих состояниях, связанных с хроническим стрессом, уровень глюкокортикоидов повышен.
Известны, однако, связанные с хроническим стрессом нарушения (посттравматическое стрессорное нарушение, синдром хронической усталости, фибромиалгия), при которых уровень глюкокортикоидов снижен. Поэтому выдвигается альтернативная гипотеза, согласно которой при хроническом стрессе может развиваться относительная глюкокортикоидная недостаточность либо за счет сниженной секреции гормонов, либо вследствие пониженной эффективности их действия. Тот факт, что лечение глюкокортикоидами предотвращает развитие некоторых связанных со стрессом заболеваний, подтверждает эту гипотезу. В пользу этой гипотезы свидетельствуют также данные о сниженной чувствительности к глюкокортикоидам системы отрицательной обратной связи регуляции продукции этих гормонов (тест подавления кортизола введением синтетического глюкокортикоида дексаметазона) и клеток иммунной системы на фоне повышенного общего уровня глюкокортикоидов при глубокой депрессии. По-видимому, из-за сниженной эффективности глюкокортикоидов данное нарушение связано с обострением заболеваний, включающих воспалительный компонент (ревматоидный артрит, рассеянный склероз). Несдерживаемая глюкокортикоидами продукция провоспалительных цитокинов может приводить к обострению патологии в периферических тканях, таких, как сердце и сосуды. Избыточная продукция провоспалительных цитокинов может также служить одной из причин развития резистентности к инсулину при хроническом стрессе (известно, например, что ФНО-α ингибирует тирозинкиназную активность рецептора инсулина, подавляет экспрессию генов, связанных с транспортом глюкозы). Кроме того, провоспалительные цитокины могут индуцировать ряд психических симптомов, характерных для состояний хронического стресса. Провоспалительные цитокины могут и прямо блокировать эффекты
глюкокортикоидов, в частности, путем индукции ингибирующего фосфорилирования ГК-Рц активируемой митогенами протеинкиназой (MAPK). Относительная или абсолютная недостаточность глюкокортикоидов при хроническом стрессе может служить также причиной несдерживаемого роста КРГ и катехоламинов и связанных с этим симптомов возбуждения и тревожности, нарушений аппетита и сна.
Возникающий при хроническом стрессе порочный круг может быть разорван, в частности, лечением антидепрессантами. Предполагается, что эти лекарства через активацию аденилатциклазы и ПК-A индуцируют активирующее фосфорилирование ГК-Рц, восстанавливая тем самым чувствительность клеток к глюкокортикоидам. Парадоксально, но лечебное действие при нервно-психических заболеваниях, связанных с хроническим стрессом, могут оказывать и глюкокортикоиды, и их антагонисты. Возможно, этот парадокс обусловлен функционированием мономерной формы ГК-Рц в качестве транскрипционного репрессора (см. выше); данный тип активности стимулируется и гормоном, и антигормоном.
Высказывается предположение, что нарушения при хроническом стрессе, связанные с резистентностью к глюкокортикоидам, могут иметь эволюционное оправдание: подчиненные особи имеют повышенный риск получения травм от доминантных, более агрессивных и сильных сородичей, и ослабленный контроль со стороны глюкокортикоидов за воспалительными процессами, являясь адаптивным, позволяет эффективнее залечивать раны (преимущественно благодаря врожденному иммунитету). Ослабление глюкокортикоидного контроля за продукцией КРГ и катехоламинов в ЦНС также может способствовать выживанию подчиненных особей, обеспечивая постоянную готовность (состояние тревожности, облегчение консолидации памяти) к адекватной поведенческой реакции на возможную агрессию.
ФЕТАЛЬНЫЙ СТРЕСС
Фетальный стресс в конце беременности является нормальной физиологической реакцией плода на непропорционально слабый рост матки в данный период, ее спорадические сокращения и сопутствующую этому гипоксию. Считается, что активация гипоталамо-гипофизарно-надпочечниковой системы плода служит решающим фактором в инициации родовой деятельности. Глюкокортикоиды плода, кроме
того, обеспечивают подготовку плода к внеутробной жизни, ускоряя созревание легких и систем, связанных с пищеварением, и в акушерской практике глюкокортикоиды могут применяться для подготовки плода к рождению в случаях риска рождения ребенка малого размера и/или с нарушениями развития легких.
Вместе с тем избыточный стресс матери и/или плода может вести к необратимым неблагоприятным последствиям в будущей жизни новорожденного, что позволяет говорить об импринтинге. Считается, что данный импринтинг реализуется за счет действия глюкокортикоидов. Отдаленными последствиями служат повышенная активность гипоталамо-гипофизарно-надпочечниковой системы (возможно, изза ослабления системы отрицательной обратной связи вследствие снижения экспрессии ГК-Рц); гипертензия или гипотензия (в зависимости от срока беременности при стрессе); склонность к развитию метаболического синдрома с относительной резистентностью к инсулину и гипергликемией; депрессивный тип поведения, включая состояние тревожности; склонность к приему наркотиков. Предполагается, что поведенческие аспекты связаны с повышенной активностью КРГ в миндалине. У макак-резусов снижаются объем гиппокампа и нарушается нейрогенез в зубчатой извилине.
К сходным последствиям ведет неонатальный стресс. Например, ежедневный отъем крысят от матери на 3 ч в первые 3 нед жизни ведет к развитию впоследствии у взрослых животных депрессивного типа поведения, сниженной способности к обучению, повышенной реакции гипоталамо-гипофизарно-надпочечниковой системы на стрессор, усиленной экспрессии КРГ в паравентрикулярном ядре гипоталамуса. Способность антагониста ГК-Рц нормализовать эти показатели позволяет предполагать формирование в результате неонатального стресса порочного круга из-за ослабления системы отрицательной обратной связи в регуляции продукции глюкокортикоидов, вероятно, на уровне ГК-Рц гиппокампа.
ХИМИЧЕСКИЙ СТРЕСС
Для усиления защитных функций организма на практике давно используется длительный прием небольших доз ксенобиотиков (кататоксических соединений), таких, как яды. В результате возрастает устойчивость не только к принимаемому, но и ко многим другим ксенобиотикам. Эта ситуация близка к общему адаптационному синдрому не только формально, но и отчасти по механизму развития адаптации.
Общим компонентом развития толерантности к ксенобиотикам служит индукция ферментов и других белков, способствующих инактивации ксенобиотиков и их выведению из организма. Эти ферменты условно делят на три группы, обеспечивающие соответственно: оксидоредукцию ксенобиотиков (фаза 1; рис. 6-8); конъюгацию исходных или модифицированных в фазу 1 ксенобиотиков с гидрофильными или гидрофобными фрагментами, а также синтез антиоксидантов и их восстанавливающее действие на компоненты клетки (фаза 2); транспортировку неконъюгированных или конъюгированных ксенобиотиков из клетки и экскрецию (фаза 3).
К ферментам фазы 1 относят цитохромы P-450 (CYP), действие которых преимущественно направлено на введение в структуру субстратов гидроксильных групп; флавиновые монооксигеназы (FMO), катализирующие присоединение кислорода к азоту, сере и другим нуклеофильным гетероатомам ксенобиотиков; аминооксидазы; гидролазы, такие, как альдегидоксидаза и ксантиноксидаза (которые, в отличие от CYP, присоединяют к субстратам кислород воды, а не воздуха). Ферменты фазы 2 включают ариламин-N-ацетилтрансферазу, УДФглюкуронозилтрансферазы (UGT), глютатион-S-трансферазы (GST),
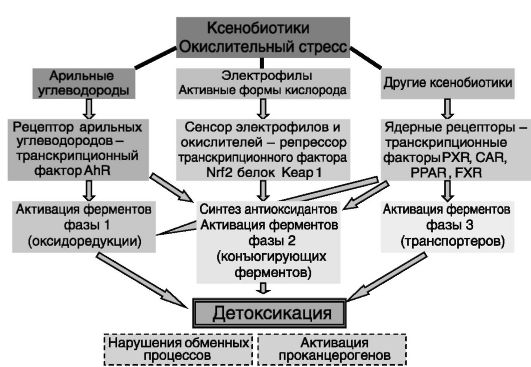 Рис. 6-8. Развитие неспецифической адаптации к токсичным агентам химический стресс
Рис. 6-8. Развитие неспецифической адаптации к токсичным агентам химический стресс
сульфотрансферазы (ST), O-, S- и N-метилтрансферазы. Сюда же относят глутамат-цистеинлигазу (синтез антиоксиданта глутатиона), глутатионпероксидазу и пероксиредоксин (восстановление перекиси водорода до воды), глутатионредуктазу (восстановление глутатиона), НАД(Ф)Н:хиноноксидоредуктазу (восстановление хинонов перед их конъюгацией), ферритин-L (запасание железа в растворимой, нетоксичной форме), металлотионеин-1 (хелатор тяжелых металлов). Белки фазы 3 включают белки, связанные с устойчивостью к многим лекарствам (MRP), полипептиды транспорта органических анионов (OATP) и транспортеры органических катионов (OCT), белки пероксисом.
Выявлено несколько сенсоров, опосредующих действие ксенобиотиков и оксидантов на указанные ферменты. Эти сенсоры либо сами являются транскрипционными факторами (рецептор арильных углеводородов (AhR), ядерные рецепторы PXR, CAR, PPAR, FXR), либо непосредственно регулируют активность транскрипционных факторов (Keap1). При этом активность AhR и ядерных рецепторов модулируется нековалентным связыванием лигандов, а активность Keap1 (и соответственно ассоциированного с ним транскрипционного фактора Nrf2) регулируется ковалентной модификацией его сульфгидрильных групп ксенобиотиками или оксидантами. Регулируемые ксенобиотиками (оксидантами) транскрипционные факторы (AhR, Nrf2 и ядерные рецепторы) взаимодействуют с регуляторными элементами генов ферментов фаз 1-3: ксенобиотикчувствительными элементами (XRE), антиоксидантчувствительными элементами (ARE) и ГЧЭ, соответственно, влияя тем самым на транскрипцию этих генов. Группа ядерных рецепторов способна влиять на ферменты всех трех фаз. Действие AhR ограничивается в основном ферментами фаз 1 и 2, а Nrf2 регулирует преимущественно ферменты фазы 2 (см. рис. 6-8).
Неспецифическая адаптация к действию ксенобиотиков, выполняющая, безусловно, защитную функцию, может иметь и негативные последствия. Например, индукция лекарственными препаратами детоксицирующих ферментов может вести к ускоренной инактивации эндобиотиков, таких, как половые стероиды, с соответствующими последствиями для репродуктивной функции. Используемые при химиотерапии опухолей агенты в ходе лечения могут терять свою эффективность из-за индукции ферментов собственной детоксикации. Для нейтрализации подобных нежелательных эффектов предлагается использование антагонистов сенсоров ксенобиотиков, в частности веществ, присутствующих в пищевых продуктах.
Другим нежелательным последствием индукции детоксицирующих агентов служит усиление метаболической активации проканцерогенных соединений в канцерогены (примером такой активации может служить гидроксилирование имеющегося в табачном дыме 7Н-дибензо[с,g]карбазола в положении 4, катализируемое преимущественно CYP1B1; образующийся продукт образует мутагенные аддукты с ДНК).
Прямое действие ксенобиотиков на инактивирующие их клетки может дополняться и усиливаться эндокринным компонентом. Так, активация рецептора X прегнанов (PXR) его лигандом, антибиотиком рифампицином или путем введения активирующей мутации ведет к повышению биосинтеза в надпочечниках и концентрации в крови кортикостероидов. Глюкокортикоиды, в свою очередь, индуцируют экспрессию в клетках печени PXR и конститутивно активного рецептора (CAR), которые стимулируют транскрипцию ряда цитохромов P-450. Эффект глюкокортикоидов на цитохромы может быть двуфазным, что, возможно, отражает изменение формы действия гормонов при переходе от базальных к стрессорным концентрациям.
Рекомендуемая литература
Селье Г. Очерки об адаптационном синдроме. - М., 1960.
De KloetE.R. Hormones, brain and stress. Endocr Regul. 2003;37(2):51- 68.
Dobson H., Ghuman S., Prabhakar S, Smith R. A conceptual model of the influence of stress on female reproduction. Reproduction. 2003;125(2):151-163.
Filaretova L., Podvigina T., Bagaeva T. et al. Gastroprotective role of glucocorticoid hormones. J Pharmacol Sci. 2007;104(3):195-201.
Liu H., Colavitti R., Rovira Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97(10):967-974.
Raison C.L., Miller A.H. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554 - 1565.
Sapolsky R.M., Romero L.M., Munck A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative
actions. Endocr Rev. 2000;21(1):55-89.
Tache Y., Martinez V, Wang L., Million M. CRF1 receptor signaling pathways are involved in stress-related alterations of colonic function and viscerosensitivity: implications for irritable bowel syndrome. Br J Pharmacol. 2004; 141(8):1321-1330.
Vrzal R., Ulrichova J., Dvorak Z. Aromatic hydrocarbon receptor status in the metabolism of xenobiotics under normal and pathophysiological conditions. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2004;148(1):3-10.