Эндокринная регуляция. Биохимические и физиологические аспекты : учеб. пособие / А.Н. Смирнов ; под ред. В.А. Ткачука - 2009. - 368 с.
|
|
|
|
ГЛАВА 10. ОБМЕН ЛИПИДОВ
Липиды - гетерогенный класс органических соединений, слабо окисленных по сравнению с углеводами, общим свойством которых является низкая растворимость в воде и высокая - в органических растворителях. Липиды обычно покрывают 40% потребности организма в энергии. Подавляющая часть липидов пищи (99%) и основная часть липидов в организме представлена жирными кислотами и их сложными эфирами с глицерином (жирами). Мембраны клеток включают фосфолипиды и холестерин. К липидам относятся также желчные кислоты, стероидные гормоны, эйкозаноиды и некоторые другие сигнальные соединения (рис. 10-1). Особую роль играют не образующиеся в организме полиненасыщенные жирные кислоты пищи, оказывающие регуляторное влияние на липидный обмен.
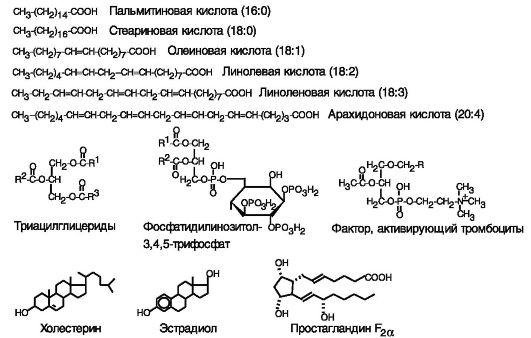 Рис. 10-1. Некоторые липиды
Рис. 10-1. Некоторые липиды
ЛИПИДЫ КРОВИ
Основными липидами крови являются холестерин, фосфолипиды, триацилглицериды и неэтерифицированные жирные кислоты, находящиеся в соотношении приблизительно 5:5:5:1 с суммарной концентрацией около 5 мг/мл, колеблющейся в весьма широких пределах (±2 мг/мл). Основная часть липидов крови находится в составе липопротеиновых частиц, служащих транспортной формой для водонерастворимых липидов. По соотношению липидов и белков, определяющему плавучую плотность частиц, различают несколько групп липопротеинов, которые отличаются друг от друга также по входящим в их состав белкам и соотношению различных липидов (рис. 10-2). В пределах этих групп выделяют отдельные подгруппы липопротеинов.
Аполипопротеин B (апо-B) синтезируется в печени и кишечнике. В кишечнике образуется укороченный вариант белка, апо-B48, в результате тканеспецифичного редактирования мРНК. Данный белок служит главным белковым компонентом хиломикронов (липопротеиновых частиц, образующихся в кишечнике), но лишен способности взаимодействовать с рецепторами. Взаимодействие ремнантов хиломикронов с рецепторами определяется другим белком этих частиц, апо-E. В печени образуется полноразмерный вариант апо- B, апо-B-100, являющийся одним из основных белков секретируемых печенью ЛОНП, узнаваемых рецептором липопротеинов низкой плотности (рецептором апо-B/E).
В состав липопротеинов могут входить минорные аполипопротеины, выполняющие вспомогательные функции. Так, апо-A-II, формируя с помощью дисульфидной связи димер с апо-D, способствует стабилизации липопротеинов высокой плотности (ЛВП). Aпо-C-II, обратимо взаимодействуя с липопротеинами очень высокой плотности (ЛОВП), активирует ряд триглицеридлипаз. Апо(а) с помощью дисульфидной связи образует комплексы с апо-B-100. Обладает протеиназной активностью, ингибируя, в частности, активатор 1 плазминогена тканевого типа. (Ауто)протеолитические фрагменты апо(а) конкурируют с плазминогеном за фибрин(оген) и, аккумулируясь в атеросклеротических повреждениях, стимулируют тромбогенез. Физиологическая функция апо(а) неизвестна. Апо-A-V в составе ЛВП и ЛОНП способствует стимулирующему действию апо-C-II на липопротеинлипазы и ингибирует образование ЛОНП, что ведет к снижению уровня триглицеридов плазмы крови. Является объектом тера-
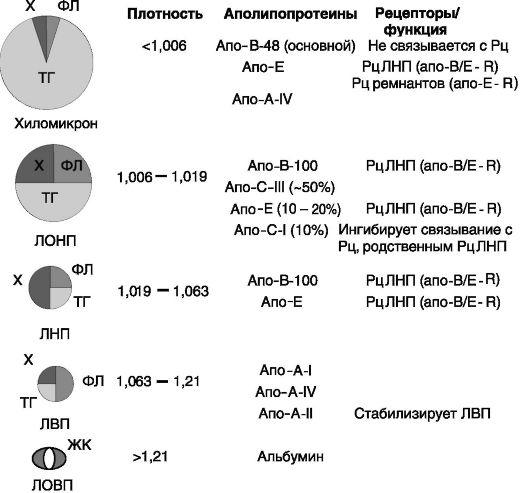 Рис. 10-2. Липопротеины крови:
Рис. 10-2. Липопротеины крови:
ТГ - триацилглицериды; Х - холестерин; ФЛ - фосфолипиды; ЖК - жирные кислоты
певтического действия антигиперлипидемических лигандов ядерного рецептора PPARa печени. Образующийся в печени апо-F в составе ЛНП ингибирует белок транспорта эфиров холестерина (CETP).
ПОСТУПЛЕНИЕ ЛИПИДОВ В КЛЕТКИ
Внеклеточные липазы
Внеклеточные липазы формируют семейство, включающее панкреатическую липазу, липопротеинлипазу, печеночную липазу, эндотелиальную липазу, фосфатидилсеринфосфолипазу A1, белки 1 и 2,
родственные панкреатической липазе. Липазы различаются по субстратной специфичности, тканевой локализации, регуляции.
Панкреатическая липаза в кишечнике гидролизует сложноэфирные связи между жирными кислотами и глицерином в положениях 1 и 3 триглицеридов. Белок 2, родственный панкреатической липазе, также секретируется поджелудочной железой, но гидролизует не только триглицериды, но и фосфолипиды. Остальные представители семейства участвуют в метаболизме липопротеинов крови.
Печеночная липаза синтезируется исключительно в печени и локализуется на микроворсинках поверхности гепатоцитов, обращенной в просвет синусоидов, а также на клетках эндотелия синусоидов путем заякоривания через гепарансульфатные протеогликаны. Печеночная липаза может быть перенесена током крови на клетки эндотелия коры надпочечников и яичников. Полагают, что основной функцией печеночной липазы является обеспечение захвата стероидогенными клетками и гепатоцитами эфиров холестерина, входящих в состав ЛВП. Активность фермента в стероидогенных тканях стимулируется соответствующими тропными гормонами гипофиза. Механизм стимуляции неизвестен; возможно, она связана с усилением захвата фермента из крови. Секреция фермента печенью стимулируется инсулином и гепарином и ингибируется адреналином, а также диетой с высоким содержанием холестерина. Печеночная липаза функционирует в тесной кооперации с мультилигандным рецептором-мусорщиком SR-B1, способствующим захвату клетками эфиров холестерина. В гене фермента выявлены регуляторные элементы, чувствительные к холестерину (SRE), эстрогенам, тиреоидным гормонам (Т-ЧЭ), глюкокортикоидам, цАМФ и, возможно, глюкозе и/или инсулину.
Липопротеинлипаза локализована на клетках эндотелия капилляров многих органов с наибольшей концентрацией в жировой ткани и мышцах. Основная функция фермента - снабжение соответствующих органов жирными кислотами из триглицеридов ЛОНП и хиломикронов. Субстратная специфичность липопротеинлипазы сходна с таковой панкреатической липазы. На поверхности клеток эндотелия липопротеинлипаза заякоривается благодаря взаимодействию с гепарансульфатными протеогликанами и с помощью ковалентно присоединенной фосфатидилинозитидгликановой группы. Помимо гидролиза эфиров липидов печеночная липаза и липопротеинлипаза могут стимулировать захват липидов неферментативным путем, пос-
редством интернализации комплексов липопротеинных частиц, их рецепторов и липазы.
Эндотелиальная липаза экспрессируется клетками эндотелия и локализуется на этих клетках во многих органах, а также на макрофагах. Данная липаза очень слабо гидролизует триацилглицериды, но эффективна в качестве фосфолипазы A1, гидролизуя связь жирной кислоты с глицерином фосфолипида в положении 1. Особенностью регуляции эндотелиальной липазы служит индукция ее экспрессии провоспалительными цитокинами, такими, как ИЛ-1β и ФНО-α, а также напряжением сдвига в сосуде. Физиологическая функция данной липазы не вполне ясна. Возможно, эндотелиальная липаза, локализованная в печени, предназначена для удаления фосфолипидов с поверхности ремнантов, образованных из липопротеинов очень низкой плотности. Допускается участие фермента в атеросклеротическом повреждении сосудов.
Фосфатидилсериновая фосфолипаза A1, продуцируемая тромбоцитами, обладает абсолютной субстратной специфичностью в отношении фосфатидилсерина и лизофосфатидилсерина. Активность фермента связана с высвобождением гистамина тучными клетками и воспалением.
Рецепторы липопротеинов
Липопротеиновые частицы плазмы крови узнаются, задерживаются, модифицируются, интернализуются и разрушаются с участием рецепторов липопротеинов, которых известно более 10. Структурнофункциональная организация таких рецепторов показана на рис. 10-3 на примере ЛНП-Рц и рецепторов ЛОНП (ЛОНП-Рц).
Данные рецепторы включают крупный внеклеточный домен, содержащий 7 - 8 обогащенных цистеином повторов, формирующих сайт связывания лиганда, область гомологии с предшественником эпидермального фактора роста, в том числе 3 обогащенных цистеином повтора, и примембранную область, обогащенную остатками серина и треонина; короткий трансмембранный домен и относительно короткий внутриклеточный домен. В состав внутриклеточного домена входит последовательность NPXY, обеспечивающая кластеризацию рецепторов в окаймленных ямках плазматической мембраны клетки.
ЛОНП-Рц экспрессируется в органах-потребителях жирных кислот: сердце, скелетной мышце, жировой ткани, мозгу, а ЛНП-Рц преимущественно в печени - органе элиминации обогащенных холестерином
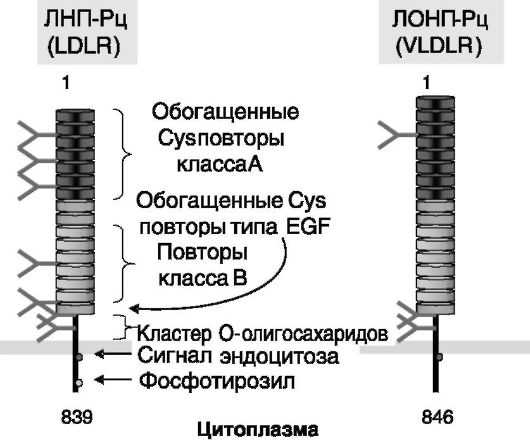 Рис. 10-3. Структурно-функциональная организация ЛНП-Рц и ЛОНП-Рц
Рис. 10-3. Структурно-функциональная организация ЛНП-Рц и ЛОНП-Рц
ЛНП. ЛОНП-Рц локализуется на клетках эндотелия капилляров, артериол, коронарной артерии, а также на макрофагах и гладкомышечных клетках сосудов, особенно при атеросклеротических повреждениях. В мозгу ЛОНП-Рц локализуется на клетках микроглии.
Лигандами ЛОНП-Рц служат содержащие апо-E липопротеиновые частицы: липопротеины очень низкой плотности, липопротеины промежуточной плотности, но не липопротеины низкой плотности. В то же время ЛНП-Рц связывает и апо-B-, и апо-E-содержащие частицы, включая липопротеины очень низкой, промежуточной и низкой плотности.
Рецепторы липопротеинов и внеклеточные липазы действуют в тесной кооперации: липопротеинлипаза, взаимодействуя с рецепторами, способна увеличивать сродство липопротеиновых частиц к рецепторам; она может способствовать перемещению липопротеиновых частиц к рецепторам за счет взаимодействия одновременно с гепарансульфатными протеогликанами клеточной поверхности и липопротеиновыми частицами; путем гидролиза триглицеридов липаза снижает размер липопротеиновых частиц и увеличивает концентрацию в них апопротеина, узнаваемого рецептором. В результате ЛОНП-Рц могут обеспечивать интернализацию ремнантов липопротеинов очень низкой плотности и хиломикронов.
Захват гепатоцитом липопротеиновой частицы низкой плотности с помощью рецепторного механизма сопровождается включением
механизма стабилизации уровня внутриклеточного холестерина: ингибированием экспрессии 3-гидрокси-3-метилглутарилредук- тазы (ключевого фермента биосинтеза холестерина) и ускорением деградации фермента; активацией ацил-CoA: холестеролацилтрансферазы (АХАТ), этерифицирующей холестерин жирными кислотами; подавлением экспрессии ЛНП-Рц. Последний эффект определяется наличием в гене ЛНП-Рц стеролчувствительного элемента (SRE) , Экспрессия ЛОНП-Рц может стимулироваться тиреоидными гормонами, эстрогенами, инсулином, гиполипидемическим агентом клофибратом и ингибируется стимуляторами цАМФ и липидной диетой.
Рецепторы липопротеинов могут играть роль в проведении сигналов внеклеточных факторов. Показано, что с ЛОНП-Рц и рецептором 2 апо-Е (apoER2) способна взаимодействовать сериновая протеиназа внеклеточного матрикса рилин, и что это взаимодействие сопровождается фосфорилированием остатков тирозина адапторного белка Dab-1, связывающегося с внутриклеточным доменом указанных рецепторов, а также ЛНП-Рц. Фосфорилирование Dab-1 обеспечивает активацию PI3K и ряда других внутриклеточных сигнальных белков, включая нерецепторные тирозинкиназы семейства Src, итогом чего является, в частности, позиционирование нейронов в развивающемся мозгу (рис. 10-4). Белок, родственный ЛНП-Рц (LRP), также способен взаимодействовать с адаптором Dab-1, а также адапторами Shc и CED-6/GULP, и это взаимодействие зависит от фосфорилирования рецептора ПК-Ca. LRP выполняет функцию вспомогательного рецептора в действии паракринных регуляторов групп Wnt и спондина-R.
ЛВП, обогащенные холестерином периферических тканей, захватываются гепатоцитами преимущественно с помощью рецепторамусорщика SR-B1. Рецепторы-мусорщики этого и ряда других классов (рис. 10-5) участвуют в захвате клетками модифицированных липопротеинов (включающих окисленные липиды, гликозилированные белки и т.д.), уровень которых возрастает при ряде патологических состояний, включая диабет, и которые, как полагают, связаны с повышенным риском атерогенеза. Помимо липопротеинов рецепторы-мусорщики способны взаимодействовать со многими другими соединениями, включая компоненты бактериальной стенки, остатки клеток, подвергшихся разрушению и т.д. Некоторые рецепторымусорщики кроме клиренса веществ из крови способны выполнять
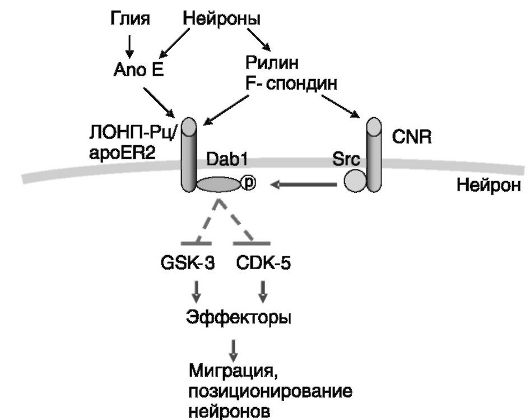 Рис. 10-4. ЛОНП-Рц
и рецептор 2 апо-Е (арoER2) способны проводить сигнал лигандов с
участием адаптора Dab 1 и тирозинкиназ семейства Src: CRN - родственные
кадгеринам нейрональные рецепторы; GSK - киназа гликогенсинтазы; CDK -
циклинзависимая киназа.
Рис. 10-4. ЛОНП-Рц
и рецептор 2 апо-Е (арoER2) способны проводить сигнал лигандов с
участием адаптора Dab 1 и тирозинкиназ семейства Src: CRN - родственные
кадгеринам нейрональные рецепторы; GSK - киназа гликогенсинтазы; CDK -
циклинзависимая киназа.
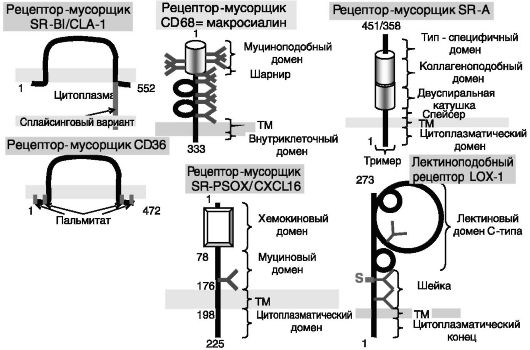 Рис. 10-5. Представители некоторых семейств рецепторов-мусорщиков. CD36 выполняет также функцию транслоказы жирных кислот (FAT)
Рис. 10-5. Представители некоторых семейств рецепторов-мусорщиков. CD36 выполняет также функцию транслоказы жирных кислот (FAT)
и другие функции, например обратный захват веществ из первичной мочи (в частности, 25-гидрокси-витамина D3), функцию хемоаттрактантов и др.
Транспортеры жирных кислот
Высвобождающиеся под действием внеклеточных липаз жирные кислоты поступают в клетки-потребители путем пассивной диффузии через плазматическую мембрану и за счет облегченной диффузии, определяемой ассоциированными с мембраной транспортерами: транслоказой жирных кислот FAT/CD36, белком транспорта жирных кислот FATP (рис. 10-5), белком плазматической мембраны, связывающим жирные кислоты (FABPpm). Помимо транспорта жирных кислот указанные белки могут, видимо, выполнять ряд других функций. Так, FAT/CD36 способен взаимодействовать с коллагеном, тромбоспондином, анионными фосфолипидами, окисленными ЛНП, может выполнять роль молекулы клеточной адгезии. Ряд представителей группы FATP обладает активностью ацил-CoA синтазы, т.е. могут обеспечивать и следующий после транспорта этап обмена жирных кислот.
Поступившие в клетки кишечника короткоцепочечные жирные кислоты (C <10) секретируются в воротную вену и транспортируются в печень. Длинноцепочечные жирные кислоты подвергаются повторной этерификации с глицерином и в форме триглицеридов поступают в кровь через грудной лимфатический проток в составе хиломикронов
и ЛОНП.
МОБИЛИЗАЦИЯ ЛИПИДОВ
Основной формой запасания липидов служит их хранение в виде триацилглицеридов в липидных каплях адипоцитов. Главным сигналом мобилизации липидов служит β-адренергическая стимуляция, эффективность которой контролируется рядом гормонов. Катехоламины стимулируют активность аденилатциклазы, продукт которой, цАМФ, активирует ПК-A, которая, в свою очередь, путем фосфорилирования активирует гормончувствительную липазу (HSL). Активность фермента при этом возрастает в 2 - 3 раза, что совершенно не соответствует степени индукции липолиза, скорость которого может возрастать в 100 раз. Причиной данного несоответствия служит то, что HSL является не единственным субстратом для ПК-A в адипоцитах, участвующим в липолизе. Важную роль в стимуляции
липолиза играет фосфорилирование липофильного белка перилипина, формирующего защитную пленку вокруг липидной капли. В результате фосфорилирования меняется конформация перилипина, что открывает доступ к субстрату HSL. При длительной стимуляции β-адренорецепторов фосфорилирование перилипина сопровождается фрагментацией липидной капли на множество мелких, что обеспечивает увеличение поверхности контакта HSL и/или других триглицеридлипаз с субстратом. У мышей с нокаутированным геном перилипина наблюдается конститутивная активация HSL, и жировые запасы у таких животных снижены. Каталитическая эффективность HSL усиливается взаимодействием фермента с белком, связывающим жирные кислоты (FABP). Этот белок обеспечивает отток продукта реакции от HSL, снимая возможность ингибирования реакции продуктом (рис. 10-6).
Помимо действия через ПК-A катехоламины могут стимулировать фосфорилирование и активацию HSL через MAPK-каскад. Липолитическое действие предсердного натриуретического пептида (ANP), по-видимому, включает фосфорилирование HSL и перили-
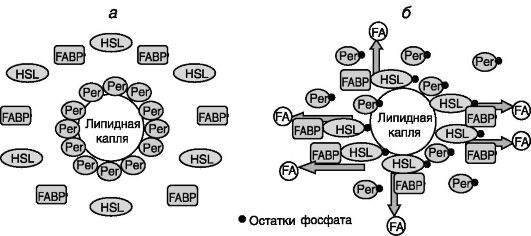 Рис. 10-6. Барьерная/транслокационная
модель стимуляции липолиза. В состоянии покоя (а) гормончувствительная
липаза (HSL) неактивна, а липиды защищены слоем молекул перилипина
(Per). β-Адренергическая стимуляция (б) через цАМФ и ПК-A вызывает
фосфорилирование, активацию и перемещение HSL к липидной капле.
Параллельное фосфорилирование Per ведет к его удалению с поверхности
липидной капли с открытием доступа субстрата для HSL. Белок, связывающий
жирные кислоты (FABP), обеспечивает отток образующихся жирных кислот
(FA), препятствуя ингибированию HSL продуктом.
Рис. 10-6. Барьерная/транслокационная
модель стимуляции липолиза. В состоянии покоя (а) гормончувствительная
липаза (HSL) неактивна, а липиды защищены слоем молекул перилипина
(Per). β-Адренергическая стимуляция (б) через цАМФ и ПК-A вызывает
фосфорилирование, активацию и перемещение HSL к липидной капле.
Параллельное фосфорилирование Per ведет к его удалению с поверхности
липидной капли с открытием доступа субстрата для HSL. Белок, связывающий
жирные кислоты (FABP), обеспечивает отток образующихся жирных кислот
(FA), препятствуя ингибированию HSL продуктом.
пина ПК-G, активируемой продуктом ферментативной активности рецептора ANP, цГМФ. Стимуляция липолиза гормоном роста включает путь с участием JAK/STAT, но механизм остается неизвестным. Липолиз под действием ФНО-α может быть связан с подавлением уровня перилипина в клетках. Липолитическое действие глюкокортикоидов может быть отчасти обусловлено индукцией экспрессии гена HSL. Хорошо известный факт усиления липолиза при гипертиреозе, по-видимому, определяется непрямым действием тиреоидных гормонов, опосредуемым усилением катехоламиновой стимуляции адипоцитов.
Инсулин препятствует липолизу. Один из механизмов включает активацию разрушающей цАМФ фосфодиэстеразы 3B (PDE3B) посредством ее фосфорилирования активируемой инсулином ПК-B. Известно, однако, что инсулин может действовать и через путь, не связанный с цАМФ. Не исключено, что этот путь включает стимуляцию протеинфосфатаз, дефосфорилирующих HSL и перилипин (рис. 10-7). Кроме того, инсулин способен индуцировать экспрессию перилипина через стимуляцию уровня PPARγ, повышающего транскрипционную активность гена перилипина.
HSL гидролизует сложноэфирные связи с жирными кислотами предпочтительно в положениях 1 и 3 глицерина. Образующийся 2-мо- ноацилглицерид служит субстратом для моноацилглицеридлипазы, которая не является лимитирующим ферментом липолиза.
HSL экспрессируется в нескольких тканеспецифичных изоформах, возникающих в результате использования нескольких промоторов,
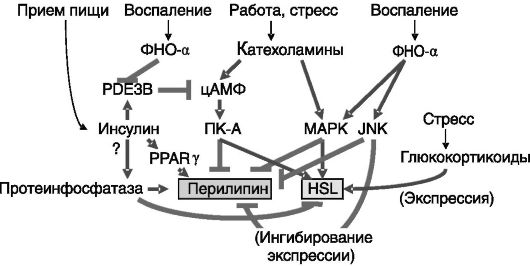 Рис. 10-7. Гормональная регуляция липолиза
Рис. 10-7. Гормональная регуляция липолиза
альтернативного сплайсинга и кодонов старта трансляции. Более длинная изоформа HSL характерна для стероидогенных тканей (кора надпочечников, семенники), где основной функцией HSL является гидролиз эфиров холестерина с жирными кислотами. Активаторами HSL в этих тканях служат соответствующие тропные гормоны гипофиза, стимулирующие фосфорилирование HSL через цАМФ-ПК-A. Самцы мышей с нокаутированным геном HSL бесплодны. В других тканях HSL может выполнять функцию регулятора уровня жирных кислот и диацилглицерола, обладающих сигнальными функциями. В частности, в β-клетках поджелудочной железы высокий уровень жирных кислот может нарушать стимулирующее действие глюкозы на секрецию инсулина.
ОБМЕН ЖИРНЫХ КИСЛОТ И КЕТОНОВЫХ ТЕЛ
Этапы окисления жирной кислоты. Перед окислением жирные кислоты подвергаются активации путем образования тиоэфиров с CoA с затратой энергии макроэрга. Последующее β-окисление длинноцепочечных жирных кислот отчасти проходит в пероксисомах. (Пероксисомы представляют собой органеллы округлой формы, берущие начало от эндоплазматического ретикулума. Свое название получили благодаря наличию в них пероксидаз, продуцирующих перекись водорода.) Укороченные фрагменты поступают в митохондрии, где β-окисление завершается. Образующийся ацетил-CoA поступает в цикл Кребса, где окисляется до CO2. Образующиеся при β-окислении и в цикле Кребса НАДH и ФАДH2 направляются в дыхательную цепь митохондрий для генерации АТФ.
Индукторами окисления жирных кислот служат прежде всего, сами жирные кислоты, действующие через свои внутриклеточные сенсоры, PPARs. PPARa экспрессируется в тканях с интенсивным β-окислением, таких, как печень, сердце. Роль PPARa особенно существенна в условиях голодания, когда высвобождающиеся из жировых депо жирные кислоты становятся преобладающим источником энергии для большинства тканей. Недостаточность PPARa в печени в условиях голодания ведет к гипокетонемии, гипогликемии и гипотермии. В мышечной ткани аналогичную PPARa функцию выполняет PPARδ. Среди разнообразных мишеней для PPARa находятся гены ферментов окисления жирных кислот в пероксисомах и митохондриях, ферментов обмена аминокислот и углеводов.
Кетогенез. Образование кетоновых тел (ацетоацетата, β-гидроксибутирата), или кетогенез, индуцируется при голодании, приеме жирной пищи, диабете и направлен на сохранение глюкозы для мозга путем обеспечения энергетических потребностей периферических тканей за счет синтезируемых из ацетил-CoA (продукта β-окисления жирных кислот) кетоновых тел. Кетогенез включает стадию образования гидроксиметилглутарил-CoA (HMG-CoA), являющегося общим предшественником кетоновых тел и холестерина (рис. 10-8), но катализ образования HMG-CoA, используемого для биосинтеза холестерина и кетоновых тел, осуществляют разные изозимы - цитозольная и митохондриальная HMG-CoA-син- тазы (HMGCS1 и 2) соответственно. Контролирующая кетогенез HMGCS2 быстро регулируется режимом питания и гормонами на транскрипционном уровне (рис. 10-9).
Физиологическое значение установленной регуляции экспрессии митохондриальной HMG-CoA-синтазы заключается в снятии избытка жирных кислот, возникающего при приеме жирной пищи, а также в условиях усиленного липолиза, вызванного голоданием, интенсивной работой, стрессом, с образованием легко усваиваемых кетоновых тел. Напротив, данный путь метаболизма ингибируется (через инсулин) в условиях избытка углеводов. Например, глюкокортикоиды эффективно повышают экспрессию HMG-CoA-синтазы 2 только у голодных животных, т.е. при низком уровне инсулина. Это напоминает ситуацию с гормональной регуляцией одного из ферментов глюконеогенеза - фосфоенолпируваткарбоксикиназы.
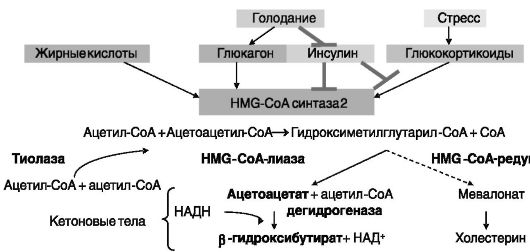 Рис. 10-8. HMG-CoA-синтаза 2 в кетогенезе
Рис. 10-8. HMG-CoA-синтаза 2 в кетогенезе
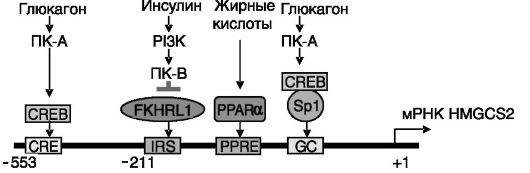 Рис. 10-9. Механизмы регуляции HMG-CoA-синтазы 2 на транскрипционном уровне
Рис. 10-9. Механизмы регуляции HMG-CoA-синтазы 2 на транскрипционном уровне
Помимо печени HMG-CoA-синтаза 2 интенсивно экспрессируется в ободочной кишке, где она, как предполагается, участвует в кетогенезе из образующихся в результате ферментации масляной и пропионовой кислот. Допускается также участие HMG-CoA-синтазы 2 в стероидогенезе в гонадах, где продукт катализируемой ей реакции может направляться на биосинтез холестерина. Значительная индукция фермента наблюдается у новорожденных.
Кетолиз. Утилизация кетоновых тел в периферических тканях, называющаяся кетолизом, включает стадию активации кетоновых тел, заключающуюся в образовании тиоэфиров кетоновых тел с CoA. К этому ведут два пути (рис. 10-10).
Считается, что главные потребители кетоновых тел (скелетная и сердечная мышцы) используют преимущественно первый путь. Второй путь катализируется двумя изозимами - митохондриальным и цитозольным. Образующийся в митохондриях ацетоацетил-CoA под действием тиолазы дает две молекулы ацетил-CoA, подвергающиеся затем окислению в цикле Кребса. Образующийся в цитозоле аце- тоацетил-CoA может быть направлен на синтез жирных кислот или холестерина (через образование HMG-CoA, см. выше) в липогенных
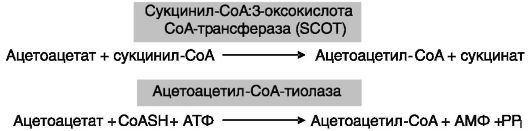 Рис. 10-10. Два пути активации кетоновых тел: SCOT (ранее) - тиофораза
Рис. 10-10. Два пути активации кетоновых тел: SCOT (ранее) - тиофораза
тканях, например в мозгу. Активность ферментов активации кетоновых тел стимулируется инсулином и (в мышце) физической нагрузкой.
Липосинтез. Синтез липидов происходит практически во всех тканях, но наиболее интенсивно - в печени и жировой ткани. Наиболее мощными индукторами липосинтеза служат углеводы пищи (преимущественно глюкоза) и инсулин, действующие синергично. Регуляция осуществляется преимущественно на уровне транскрипции ферментов синтеза липидов и в некоторых случаях - отчасти на уровне стабилизации их мРНК или белка.
Подготовительной к липосинтезу стадией является перенос образованного из углеводов или белков ацетил-CoA из митохондрий в цитоплазму. Стадия включает образование цитрата из ацетил-CoA и оксалоацетата, перемещение цитрата в цитоплазму и энергозависимый распад цитрата на ацетил-CoA и оксалоацетат. Последняя реакция катализируется АТФ-цитратлиазой.
Первой стадией собственно синтеза является карбоксилирование ацетил-CoA под действием ацетил-CoA-карбоксилазы с затратой энергии АТФ и образованием малонил-CoA. Последующие стадии катализируются синтазой жирных кислот - крупным белком, обладающим семью типами каталитической активности и способным выполнять функцию носителя промежуточных продуктов (АПБ), замещая собой коэнзим A.
Образующиеся жирные кислоты в активированной форме (ацил- CoA) взаимодействуют с α-глицерофосфатом с образованием триглицеридов и фосфолипидов.
РЕГУЛЯЦИЯ ОБМЕНА ЛИПИДОВ
Обмен липидов контролируется режимом питания и рядом гормонов, причем активность отдельных групп ферментов меняется координированно, что предполагает сходство регуляторных механизмов. Одними из наиболее универсальных проводников регуляторных стимулов на липидный обмен являются транскрипционные факторы группы SREBP.
SREBPs в липидном обмене
SREBPs (sterol regulatory element-binding proteins - белки, связывающие элементы регуляции стерином) представляют собой транскрипционные факторы семейства «спираль-петля-спираль- лейцино-
вая застежка» (bHLH-Zip). Соответствующие стеринчувствительные элементы (SREs, 10 п.н.) обнаружены в регуляторных областях многих генов, кодирующих ферменты и другие белки обмена жирных кислот и холестерина.
Клонировано 2 гена, кодирующих 3 изоформы SREBPs: SREBP1a, SREBP1c (сплайсинговые варианты) и SREBP2. SREBP2 предпочтительно активирует экспрессию генов, участвующих в биосинтезе холестерина и желчных кислот, а SREBP1c - жирных кислот и триглицеридов. SREBP1a стимулирует экспрессию генов обоих рядов, однако роль этой изоформы SREBP в регуляции, по-видимому, невелика, поскольку ее экспрессия (в отличие от SREBP1c и SREBP2) является конститутивной. На рис.10-11 показаны основные объекты регуляции изоформами SREBPs.
Регуляция активности SREBPs осуществляется на транскрипционном и посттранскрипционном уровне.
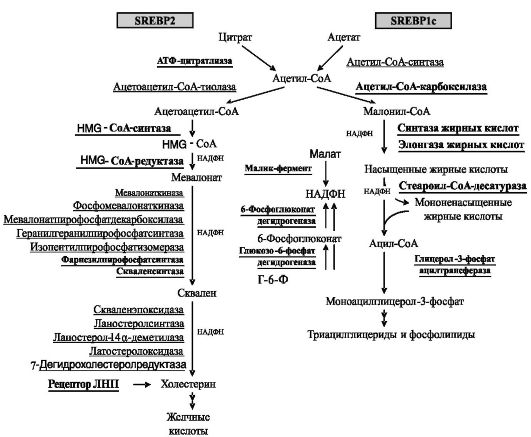 Рис. 10-11. Объекты регуляции изоформами SREBP (выделены шрифтом)
Рис. 10-11. Объекты регуляции изоформами SREBP (выделены шрифтом)
SREBPs экспрессируются в форме предшественников размером около 1150 а.к., которые являются интегральными белками мембран эндоплазматического ретикулума. Активация SREBPs происходит в комплексе Гольджи под действием двух протеиназ. Мембранносвязанная сериновая протеаза S1P разрезает молекулу SREBP пополам, после чего металлопротеиназа S2P отделяет домен bHLH-Zip от трансмембранного фрагмента половинки молекулы SREBP. Домен bHLH-Zip (ядерная форма SREBP, nSREBP) затем транслоцируется в ядро и взаимодействует с ДНК. Этап перехода SREBP из эндоплазматического ретикулума в комплекс Гольджи (и, следовательно, последующая активация) негативно контролируется липидами (оксистеринами, такими, как 27-гидроксихолестерин или 24(S),25- эпоксихолестерин). Этот контроль осуществляется через белок SCAP (SREBP cleavage-activating protein - белок расщепления-активации SREBP). Мембранный фрагмент SCAP служит сенсором липидов, а C-концевой цитоплазматический домен WD связан с регуляторной областью SREBP. Изменение конформации SCAP [или его взаимодействия с дополнительным регуляторным белком - продуктом индуцируемого инсулином гена 1 (INSIG-1)] при снижении уровня липидов в клетке приводит к переходу комплексов SCAP/SREBP в транспортные везикулы эндоплазматического ретикулума и транспортировке в комплекс Гольджи (рис. 10-12, а). Данный механизм служит одним из способов ауторегуляции биодинамики холестерина в клетках печени: повышенный уровень холестерина через оксистерины и SCAP блокирует активацию SREBP, что сдерживает поступление холестерина в клетки через рецептор ЛНП и биосинтез холестерина de novo. Конкурентные антагонисты SCAP, такие, как LY295427 (4а-аллилхолестан-3а-ол), снимают тормозное действие оксистеринов на экспрессию рецептора ЛНП, что сопровождается снижением уровня холестерина в крови
(рис. 10-12,б).
Регуляция экспрессии SREBPs на уровне транскрипции. SREBP1c и SREBP2 регулируются на транскрипционном уровне рядом факторов. Первый из них включает положительную ауторегуляцию через SRE генов SREBP1 и 2. Транскрипция SREBP1c избирательно стимулируется стеринами (включая оксистериновые промежуточные продукты биосинтеза холестерина) через ядерные рецепторы LXRa и β (рис. 10-13). Таким образом, действие оксистеринов на SREBP на транскрипционном и посттрансляционном уровнях является разнона-
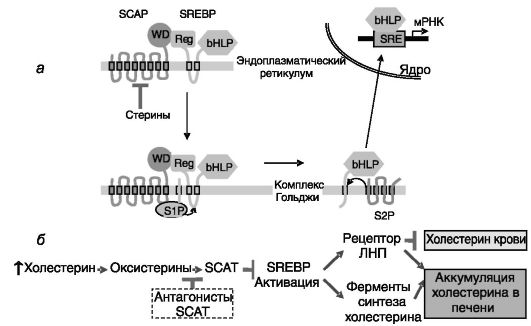 Рис. 10-12. Регуляция
активности SREBPs липидами на посттрансляционном уровне (а) и роль
этого механизма в ауторегуляции уровня холестерина в печени (б)
Рис. 10-12. Регуляция
активности SREBPs липидами на посттрансляционном уровне (а) и роль
этого механизма в ауторегуляции уровня холестерина в печени (б)
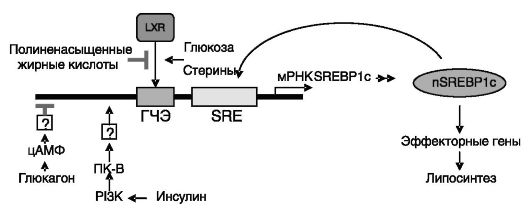 Рис. 10-13. Регуляция
экспрессии SREBP1 на транскрипционном уровне. Глюкагон через цАМФ
ингибирует, а инсулин через PI3K и ПК-B стимулирует экспрессию гена.
Экспрессия стимулируется также производными холестерина и ингибируется
полиненасыщенными жирными кислотами через ядерный рецептор LXR. SREBP1
положительно регулирует собственную продукцию.
Рис. 10-13. Регуляция
экспрессии SREBP1 на транскрипционном уровне. Глюкагон через цАМФ
ингибирует, а инсулин через PI3K и ПК-B стимулирует экспрессию гена.
Экспрессия стимулируется также производными холестерина и ингибируется
полиненасыщенными жирными кислотами через ядерный рецептор LXR. SREBP1
положительно регулирует собственную продукцию.
правленным. (Необходимо отметить, что лигандная специфичность SCAP и LXR перекрывается лишь частично.) Последствия нокаута генов SREBP1c и LXRa/β для обмена липидов сходны, поэтому
предполагается, что основная часть эффектов LXRs опосредована их влиянием на экспрессию SREBP1c. Индукция стеринами экспрессии SREBP1c одним из следствий имеет усиление синтеза олеата - предпочтительной жирной кислоты для образования эфиров холестерина как транспортной и запасаемой формы холестерина. Эффект LXRs на экспрессию SREBP1c может служить одной из форм связи между обменом жирных кислот и холестерина: полиненасыщенные жирные кислоты конкурентно ингибируют стимулирующее действие стеринов на LXR и тем самым (через снижение экспрессии SREBP1c) подавляют синтез жирных кислот и триглицеридов. Этот механизм обеспечивает отчасти известное благоприятное действие растительного масла (обогащенного полиненасыщенными жирными кислотами) на липидный обмен. Индукция экспрессии SREBP1c под действием LXR может быть еще одной формой связи липидного и углеводного обмена, поскольку оказалось, что LXR может служить сенсором не только стеринов, но и глюкозы или глюкозо-6-фосфата. В результате избыток поступающих в печень углеводов индуцирует направление собственного метаболизма на липосинтез. Стимулирующее действие инсулина и ингибирующее действие глюкагона на биосинтез жирных кислот в печени также связано с регуляцией экспрессии SREBP1c (см. рис. 10-13). Трансфекция клеток доминантной негативной формой SREBP1c снимает действие инсулина на липогенез. Реципрокная регуляция липосинтеза в печени и жировой ткани голоданием и поступлением пищи связана с указанным действием инсулина и глюкагона. Это действие гормонов дополняется их реципрокным влиянием на экспрессию коактиватора SREBP1c, PGC-1β. Регуляция данными гормонами экспрессии SREBP1c связана также с обменом углеводов: SREBP1c индуцирует экспрессию глюкокиназы и подавляет экспрессию фосфоенолпируваткарбоксикиназы - ключевых ферментов гликолиза и глюконеогенеза соответственно.
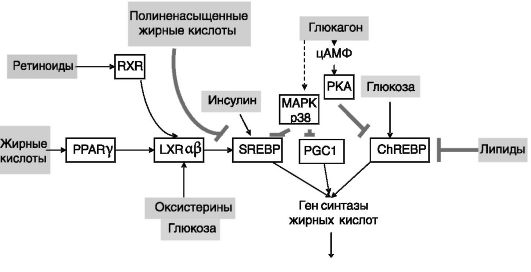
Примером участия SREBP1 в системе регуляции обмена липидов может служить контроль экспрессии гена синтазы жирных кислот (рис. 10-14).
ОБМЕН ХОЛЕСТЕРИНА В ПЕЧЕНИ
Холестерин попадает в печень двумя путями: за счет опосредованного рецепторами эндоцитоза липопротеинов (рецепторами хиломикронов, ЛНП-Рц и ЛВП-Рц - рецепторами-мусорщиками SR-B1)
 Липосинтез
Липосинтез
Рис. 10-14. Регуляция экспрессии синтазы жирных кислот
и синтеза de novo из ацетил-CoA. Часть холестерина подвергается этерификации активированными жирными кислотами в присутствии ацил-CoA-холестеролэфиртрансферазы (ACAT). Этерифицированный холестерин служит запасной формой холестерина и может быть вновь превращен в холестерин под действием холестерилэфиргидролазы (CEH) или экскретирован в желчь (рис. 10-15). У человека ~50% холестерина экскретируется в виде желчных кислот. Желчные кислоты, кроме того, служат в качестве эмульгаторов для плохорастворимого холестерина в желчи.
Баланс между различными направлениями обмена холестерина, его адаптация к режиму питания и координация с другими видами обменных процессов достигается с участием ряда внутриклеточных сенсоров производных холестерина, относящихся к классу ядерных рецепторов: рецептора фарнезоидов (FXR), рецептора прегнанов (PXR), рецептора печени (LXR), регулирующих активность ферментов и транспортеров на транскрипционном уровне.
Энтерогепатическая циркуляция желчных кислот
Желчь играет важную роль в кишечном пищеварении и абсорбции липидов, а также служит важным путем элиминации ксенобиотиков и конечных продуктов обмена эндогенных соединений (эндобиотиков), например холестерина, билирубина, стероидов. Приблизительно 75% желчи образуется путем секреции гепатоцитами в формируемые
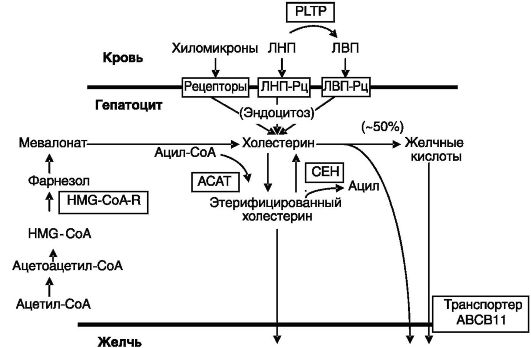 Рис. 10-15. Обмен холестерина в печени:
Рис. 10-15. Обмен холестерина в печени:
HMG-CoA-R - гидроксиметилглутарил-CoA-редуктаза; PLTP - белок переноса фосфолипидов плазмы
между ними желчные канальцы. Остальная часть желчи формируется при прохождении желчи по желчным протокам за счет секреции и абсорбции веществ выстилающими протоки холангиоцитами. В желчном пузыре желчь подвергается ~10-кратному концентрированию. Движущей силой образования первичной желчи и «зависимого от желчных кислот» тока желчи служит активный транспорт органических соединений (солей желчных кислот, фосфолипидов, холестерина) через апикальную мембрану гепатоцитов, сопровождаемый пассивным транспортом воды, электролитов и ряда других соединений. Клетки желчных протоков секретируют бикарбонат и восстановленный глутатион, которые обеспечивают «независимый от желчных кислот» ток желчи.
В кишечнике ряд органических соединений желчи (соли желчных кислот, холестерин, фосфолипиды) реабсорбируется и через портальную вену вновь поступает в печень. Размер пула солей желчных кислот в организме составляет 3 - 4 г, но за счет «энтерогепатической циркуляции» (6 - 10 оборотов за сутки) с желчью экскретируется
20 - 40 г солей желчных кислот. Часть желчных кислот в кишечнике подвергается метаболическим превращениям с образованием непригодных для дальнейшего использования продуктов (рис. 10-16).
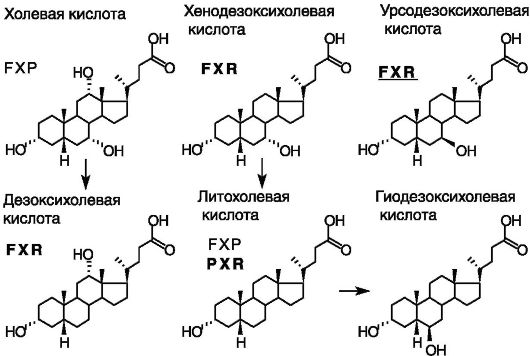 Рис. 10-16. Желчные
кислоты: превращения и сенсоры. Урсодезоксихолевая кислота (7β~эпимер
хенозезоксихолевой кислоты) является не агонистом, как другие желчные
кислоты, а антагонистом рецептора X фарнезоидов (FXR)
Рис. 10-16. Желчные
кислоты: превращения и сенсоры. Урсодезоксихолевая кислота (7β~эпимер
хенозезоксихолевой кислоты) является не агонистом, как другие желчные
кислоты, а антагонистом рецептора X фарнезоидов (FXR)
Ежесуточная потеря желчных кислот с фекальными массами, восполняемая биосинтезом de novo, составляет 0,5 г. В условиях холестаза существенное значение в биодинамике желчных кислот приобретает «холегепатический шунт», т.е. реабсорбция солей желчных кислот из желчных протоков холангиоцитами с возвращением в гепатоциты. Основная часть желчных кислот извлекается гепатоцитами из крови при первом прохождении через печень. Незахваченные желчные кислоты поступают в первичную мочу, откуда извлекаются с помощью транспортеров проксимальных нисходящих канальцев.
Транспорт желчных кислот в гепатоциты
Неконъюгированные желчные кислоты могут поступать из крови в гепатоциты путем пассивной диффузии, а конъюгированные с глицином или таурином - посредством активного (энергозависимого)
транспорта. В базальных условиях захват происходит почти исключительно гепатоцитами перипортальной области. Но после приема пищи и в условиях холестаза к захвату подключаются и гепатоциты перицентральной области печени. Различают Na+-зависимый захват, осуществляемый за счет работы транспортера желчных кислот NTCP (Na+/taurocholate cotransporting polypeptide), и независимый от Na+ захват, опосредуемый мультиспецифичными транспортерами органических анионов OATPs. Локализованный на базолатеральной мембране гепатоцитов NTCP функционирует как электрогенный (за счет котранспорта в клетку двух катионов Na+ вместе с одной молекулой органического аниона) котранспортер конъюгированных желчных кислот и Na+ (рис. 10-17). Энергетически его работа поддерживается Na+-K+-АТФазой, откачивающей Na+ из клеток. NTCP принимает также участие в транспорте некоторых других соединений, например конъюгированных стероидных и тиреоидных гормонов.
OATPs обеспечивают облегченную диффузию в гепатоциты небольшой части конъюгированных желчных кислот и большей части неконъюгированных в обмен на внутриклеточные анионы (глутатион, HCO3). У человека наиболее значим вклад OATP-A, OATP-C и OATP-8, особенно OATP-C (синонимы: OATP2, LST-1; номенклатурное наименование OATP1B1; см. рис. 10-17). Эти транспортеры участвуют в захвате гепатоцитами множества других органических соединений (эндобиотиков и ксенобиотиков), подлежащих экскреции с желчью.
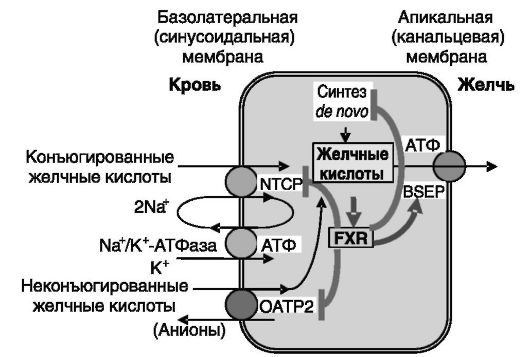 Рис. 10-17. Cистема ауторегуляции содержания желчных кислот в гепатоцитах с участием сенсора желчных кислот FXR
Рис. 10-17. Cистема ауторегуляции содержания желчных кислот в гепатоцитах с участием сенсора желчных кислот FXR
Экскреция желчных кислот гепатоцитами в желчь в нормальных условиях осуществляется локализованным на канальцевой мембране энергозависимым транспортером BSEP (bile salt export pump - насос экспорта солей желчных кислот) семейства АТФ-зависимых ABC-кас- сетных транспортеров. В условиях холестаза в гепатоцитах индуцируется экспрессия белков множественной резистентности к лекарствам (MRPs) того же семейства, откачивающих сульфаты и глюкурониды желчных кислот через базолатеральную мембрану в кровь с последующим их выведением с мочой (см. рис. 10-17).
Регуляция биосинтеза и транспорта желчных кислот
Желчные кислоты в высоких концентрациях оказывают токсичное действие на гепатоциты, поэтому процессы поступления желчных кислот из кровотока в гепатоциты, биосинтеза желчных кислот de novo и их экскреции в желчь строго сбалансированы. Основным координатором этих процессов является сенсор желчных кислот, рецептор X фарнезоидов (FXR) семейства ядерных рецепторов. Желчные кислоты через FXR ингибируют экспрессию в гепатоцитах белков, импортирующих желчные кислоты (OATP2, NTCP) и обеспечивающих их биосинтез (CYP7A1), и, наоборот, увеличивают экспрессию BSEP, обеспечивающего экспорт желчных кислот (см. рис. 10-17).
Превращение холестерина в желчные кислоты включает по меньшей мере 15 реакций и состоит из трех этапов: гидроксилирования стеролового ядра; восстановления Δ5 двойной связи в кольце B и эпимеризации 3β-гидроксильной группы; окисления боковой цепи и ее укорочения на 3 атома углерода. В зависимости от исходной реакции различают нейтральный путь (через 7а-гидроксилирование) и кислый - путь (через 27-гидроксилирование). У здорового человека преобладает нейтральный путь, у больных циррозом печени - кислый путь. У грызунов соотношение этих путей равно Соотношение новосинтезированных хенодезоксихолевой и холевой кислот у человека равно Участвующая в биосинтезе желчных кислот 3а- ГСД помимо каталитической выполняет функцию внутриклеточного транспортера желчных кислот.
CYP7A является лимитирующим ферментом в цепи превращений холестерина в желчные кислоты и поэтому служит объектом регуляции со стороны многих факторов. 85% мышей с нокаутированным геном CYP7A гибнут в первые 3 нед жизни из-за дефицита жира и жирорастворимых витаминов (недостаточность желчных кислот
- недостаточность всасывания пищевых липидов). У оставшихся в живых компенсаторно повышена экспрессия CYP7B (фермента, катализирующего 7а-гидроксилирование в кислом пути биосинтеза). мРНК CYP7A, благодаря мотивам AUUUA в 3'-нетранслируемой области, нестабильна (t1/2 1,5 ч), что обеспечивает адаптацию уровня фермента к режиму питания. Экспрессия CYP7A чутко реагирует на уровень желчных кислот: разрыв энтерогепатической циркуляции желчных кислот выведением желчного протока наружу приводит к быстрому повышению экспрессии CYP7A. Система отрицательной обратной связи между уровнем желчных кислот и экспрессией CYP7A по меньшей мере отчасти осуществляется через сиротский ядерный рецептор FXR, с которым связываются желчные кислоты. Действие FXR преимущественно направлено на повышение эффективности системы энтерогепатической системы транспорта желчных кислот. (Данный механизм используется для лечения детей с пороками ферментов синтеза желчных кислот. Промежуточные продукты этого синтеза у таких больных накапливаются и оказывают токсическое действие. Введение им холевой кислоты, с одной стороны, тормозит (через FXR) экспрессию CYP7A и соответствующих промежуточных продуктов, а с другой стороны, компенсирует недостаточность собственных желчных кислот.)
Ингибирующее действие желчных кислот на транскрипционную активность гена CYP7A через FXR является непрямым и включает индукцию экспрессии малого партнера гетеродимеризации SHP (семейство ядерных рецепторов). Этот транскрипционный репрессор, связываясь с гомологом рецептора печени (LRH-1), блокирует активирующее действие этого транскрипционного фактора на ген CYP7A. При этом подавляется и активирующее действие сенсора оксистеринов, рецептора X печени (LXR), сайт связывания которого также имеется в промоторе гена CYP7A (рис. 10-18). Сходный механизм с участием SHP действует при тормозном влиянии желчных кислот на экспрессию гена NTCP, основного транспортера конъюгированных желчных кислот в клетки печени.
Желчные кислоты экскретируются в желчь почти исключительно в форме амидов с глицином или таурином. Эти конъюгаты, с одной стороны, более эффективны для переваривания и всасывания липидов в кишечнике, а с другой стороны, менее токсичны для клеток печени по сравнению с неконъюгированными желчными кислотами. Образование таких конъюгатов последовательно катализирует-
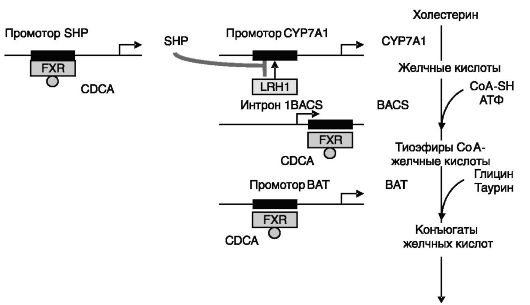 Рис. 10-18. Сенсор желчных кислот FXR в системе ауторегуляции биосинтеза желчных кислот:
Рис. 10-18. Сенсор желчных кислот FXR в системе ауторегуляции биосинтеза желчных кислот:
CDCA - хенодезоксихолевая кислота; SHP - малый партнер гетеродимеризации; LRH1 - гомолог 1 рецептора печени; CYP7A1 - 7α-гидроксилаза (p4507a); BACS - желчные кислоты-СоA-синтаза; BAT - желчная кислота- СоA:аминокислота N-ацетилтрансфераза
ся двумя ферментами: желчные кислоты-CоA-синтазой (BACS) и желчная кислота-CоA:аминокислота N-ацетилтрансферазой (BAT). Экспрессия обоих ферментов стимулируется желчными кислотами через FXR за счет прямого взаимодействия FXR с регуляторными областями генов этих ферментов (рис. 10-18). Сходный прямой механизм стимуляции желчными кислотами через FXR выявлен в отношении экспрессии гена BSEP, экспортирующего конъюгированные желчные кислоты в желчь.
Эффективными активаторами FXR являются первичная хенодезоксихолевая кислота и вторичная дезоксихолевая кислота, а также конъюгированные желчные кислоты (глико- и таурохолевая и хенодезоксихолевая кислоты). Вторичная литохолевая кислота и гидрофильная первичная холевая кислота являются более слабыми активаторами FXR. Урсодезоксихолевая кислота выступает в роли антагониста.
Биосинтез и транспорт желчных кислот являются важными компонентами биодинамики холестерина, которые в значительной мере определяют уровень и распределение холестерина в организме.
Избыточное скармливание холестерина нормальным мышам ведет к компенсаторному повышению образования желчных кислот и их элиминации с калом. Нокаут FXR у мышей ведет к повышению в крови желчных кислот, холестерина и триглицеридов, повышению в печени холестерина и триглицеридов, возникновению проатерогенного профиля липопротеинов. Общий пул желчных кислот у таких животных снижен, как и их экскреция с калом из-за сниженной экспрессии канальцевого белка транспорта солей желчных кислот (BSEP).
Еще одним объектом действия оккупированного желчной кислотой FXR служит экспрессия белка кишечника, связывающего желчные кислоты (IBABP), являющегося переносчиком желчных кислот из просвета кишечника в кровь, т.е. обеспечивающего энтерогепатическую циркуляцию желчных кислот. У человека 90 - 95% желчных кислот кишечника возвращается через кровь и печень в желчь для повторного использования. Под действием FXR экспрессия гена IBABP возрастает. Захват желчных кислот клетками кишечника осуществляется несколькими механизмами, включая пассивную диффузию, облегченный и активный транспорт. Последний обеспечивается мембранным транспортером желчных кислот кишечника (IBAT), использующим для работы градиент Na+. Этот транспортер относится к тому же семейству (10), что и NTCP печени. Экспрессия IBAT подавляется желчными кислотами через их сенсор FXR посредством индукции транскрипционного репрессора, малого партнера гетеродимеризации (SHP). Напротив, желчные кислоты повышают активность своего транспорта, транспортера органических соединений (OST), на базолатеральной мембране энтероцитов. Индукция осуществляется на транскрипционном уровне благодаря прямому взаимодействию комплексов FXR-желчная кислота с регуляторными участками генов обеих субъединиц (а и β) этого транспортера. Таким образом, в энтероцитах, как и в гепатоцитах, система ауторегуляции желчных кислот с участием FXR направлена на снижение уровня потенциально токсичных желчных кислот путем подавления их поступления в клетку и стимуляции экспорта из клетки.
В контроле обмена желчных кислот принимает участие также сиротский ядерный рецептор PXR, служащий сенсором литохолевой кислоты. Действие PXR направлено преимущественно на элиминацию этой кислоты. Гидрофобная литохолевая кислота (LCA) является вторичной желчной кислотой, образующейся путем дегидроксилирования хенодезоксихолевой кислоты в положении 7а под действием
кишечной флоры. LCA вызывает внутрипеченочный холестаз, сопровождающийся нарушением переваривания и всасывания липидов и аккумуляцией в печени токсичных продуктов. В качестве компенсации этих негативных процессов LCA и ее метаболит З-кето-LCA через PXR обеспечивают индукцию экспрессии дополнительных транспортеров желчных кислот канальцевой мембраны гепатоцитов (MRP4, семейство АТФ-связывающих кассетных транспортеров) и цитохромов CYP3A и CYP2B, обеспечивающих дополнительное гидроксилирование желчных кислот, снижающее их цитотоксичность, и подавляют синтез желчных кислот через ингибирование экспрессии CYP7A1 (рис. 10-19). Посредством взаимодействия с PXR аналогичные эффекты способен вызывать ряд ксенобиотиков.
Позитивным регулятором экспрессии CYP7A служит LXRa, функционирующий в форме гетеродимеров с RXRa. LXR служат сенсорами холестерина в форме его гидроксипроизводных. Действие LXR направлено преимущественно на снижение уровня холестерина. Существенно, что имеется межвидовая разница в чувствительности гена CYP7A к LXR: в клетках крысы и мыши LXR стимулировал, а в клетках человека и хомячка практически не влиял на экспрессию гена. Эти различия объясняют известную способность мышей и крыс справляться с гиперхолестеринемией за счет стимуляции превращения
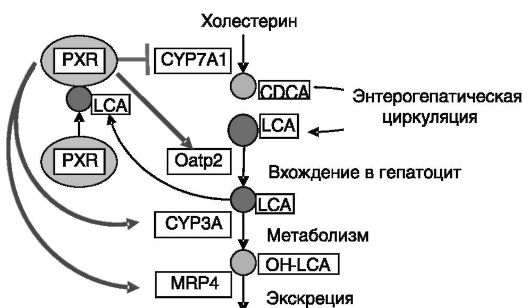 Рис. 10-19. Система ауторегуляции обмена желчных кислот с участием сенсора желчных кислот, рецептора прегнанов (PXR):
Рис. 10-19. Система ауторегуляции обмена желчных кислот с участием сенсора желчных кислот, рецептора прегнанов (PXR):
CDCA - хенодезоксихолевая кислота; LCA-литохолевая кислота; OH-LCA - гидроксилитохолевая кислота; Oatp2 - полипептидный транспортер 2 органических анионов; MRP - белок, связанный с резистентностью ко многим лекарствам
холестерина в желчные кислоты и отсутствие такой способности у других видов. Всасывание холестерина в кишечнике осуществляется с участием транспортеров ABCA1, ABCG5 и ABCG8, экспрессия которых стимулируется лигандами LXR. Интересно, что ABCA1 локализуется на базолатеральной мембране энтероцитов и обеспечивает поступление холестерина в кровь, а ABCG5 и ABCG8 - на апикальной поверхности и способствуют экскреции холестерина с фекальными массами. Суммарный результат приема обогащенной холестерином диеты или активаторов LXR (фитостерины, синтетические агонисты) заключается в подавлении всасывания холестерина. В печени ABCG5 и ABCG8 локализуются на канальцевой мембране гепатоцитов, обеспечивая усиленную экскрецию холестерина в желчь при избыточном поступлении холестерина. Агонисты LXR способны индуцировать экспрессию транспортера ABCA1 и в других типах клеток, в частности в макрофагах, что может быть использовано для профилактики и/или лечения атеросклероза за счет стимуляции выхода холестерина из клеток.
Связь обмена стеринов с другими видами обмена липидов
Обмен холестерина и желчных кислот связан с другими видами обмена. Показано, например, что FXR/RXR и их лиганды стимулируют экспрессию транспортного белка для фосфолипидов (PLTP). Этот белок переносит фосфолипиды и холестерин с липопротеинов очень низкой плотности на липопротеины высокой плотности и тем самым способствует доставке фосфолипидов и холестерина в печень из периферических тканей. (PLTP находится в плазме крови в свободной и связанной с ЛВП формах и экспрессируется во многих тканях.) Другим объектом действия FXR, связывающим обмен желчных кислот с обменом других липидов, служит апо-C-II. Скармливание мышам холевой кислоты приводило через активацию FXR к повышению экспресии апо-C-II и снижению уровня триглицеридов крови. Кроме предшественника холестерина фарнезола и желчных кислот активаторами FXR могут служить метаболиты андрогенов андростерон и этиохоланолон. Некоторые другие пути влияния желчных кислот на обмен липидов показаны на рис. 10-20.
Связь между обменом желчных кислот и жирных кислот и триглицеридов является двусторонней. Сенсором уровня жирных кислот служит ядерный рецептор PPAR. Одной из форм влияния жирных кислот на желчные кислоты служит ускорение элиминации желчных кислот путем их конъюгации с глюкуроновой кислотой (рис. 10-21).
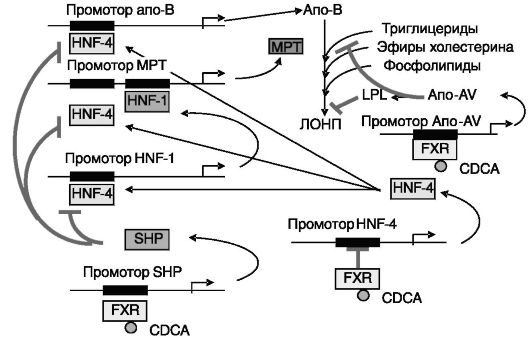 Рис. 10-20. Связь обмена холестерина и триглицеридов.
Рис. 10-20. Связь обмена холестерина и триглицеридов.
Желчные кислоты снижают секрецию ЛОНП клетками печени и кишечника посредством ингибирования экспрессии апо-B и микросомального белка транспорта триглицеридов (MTP). Желчные кислоты подавляют перенос триглицеридов на апо-B в печени и ускоряют утилизацию ЛОНП посредством активации липопротеинлипазы (LPL) через усиление экспрессии апо- AV. CDCA - хенодезоксихолевая кислота; FXR - ядерный рецептор фарнезоидов (сенсор желчных кислот); SHP - малый партнер гетеродимеризации ядерных рецепторов
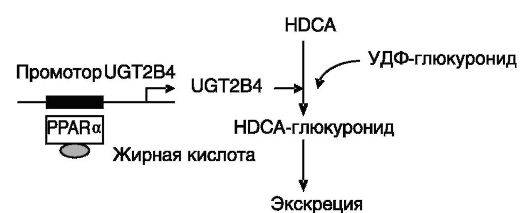 Рис. 10-21. Связь обмена холестерина и жирных кислот
Рис. 10-21. Связь обмена холестерина и жирных кислот
Жирные кислоты через PPARα повышают в печени экспрессию УДФ-глюкуронозилтрансферазы (UGT), которая за счет образования глюкуронидов желчных кислот обеспечивает их ускоренную экскрецию. HDCA - гиодезоксихолевая кислота (продукт литохолевой кислоты)
ПЕЧЕНЬ В ОБМЕНЕ ЛИПОПРОТЕИНОВ
Печень играет важнейшую роль в поддержании баланса между различными липидами и адаптации их обмена к режиму питания. В значительной мере эта функция печени связана с секрецией и захватом гепатоцитами липопротеинов. Печенью секретируются ЛОНП, транспортирующие преимущественно триглицериды в жировую ткань для запасания и ткани-потребители жирных кислот. Обедненные триглицеридами, но обогащенные холестерином и фосфолипидами ремнанты хиломикронов и образующихся из ЛОНП липопротеины низкой (и промежуточной) плотности захватываются печенью для последующей переработки и/или экскреции с желчью. Секретируемые печенью белковые компоненты ЛВН выполняют функцию мусорщиков, транспортируя в печень избыток холестерина из периферических тканей (рис. 10-22). Необходимость поддержания очень тонкого баланса между этими процессами подчеркивается
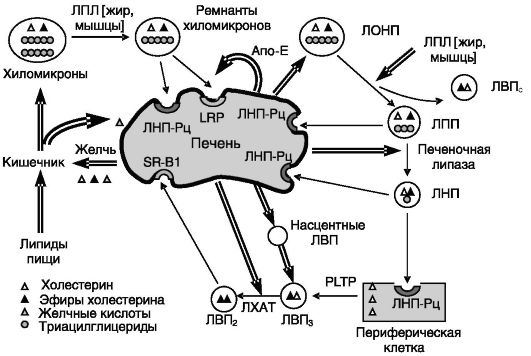 Рис. 10-22. Печень в обмене липопротеинов:
Рис. 10-22. Печень в обмене липопротеинов:
ЛПЛ - липопротеидлипаза; ЛХАТ - лецитин: холестеролацилтрансфераза; PLTP - белок транспорта фосфолипидов; LRP - белок, родственный ЛНП-Рц; SR-B1 - рецептор-мусорщик B1
существованием значительных половых различий в риске развития желчно-каменной болезни (точнее, ее наиболее распространенной формы - холестеринового холелитиаза) (у женщин риск выше, чем у мужчин) и атерогенеза (у мужчин риск выше, чем у женщин). Одной из причин этих различий служит большая эффективность системы доставки холестерина из периферических тканей в печень женского организма по сравнению с мужским при менее эффективной системе элиминации холестерина с желчью.
Более эффективная доставка холестерина в печень у женщин (эстрогены) определяется более высоким уровенем ЛВП за счет:
- усиленной продукции в печени основного аполипопротеина (апо-A-I) ЛВП;
- более медленной деградации ЛВП в печени под действием печеночной эндотелиальной липазы, активность которой у женщин ниже, чем у мужчин;
- более высокого уровня ЛНП-Рц в печени, способствующего захвату обогащенных холестерином липопротеиновых частиц.
Механизм менее эффективной элиминации холестерина из печени у женщин (эстрогены) обусловлен сниженной экспрессией 7а-гидрокси- лазы холестерина - ключевого фермента биосинтеза желчных кислот.
В результате усиленного притока холестерина в печень и его ослабленного превращения в желчные кислоты индекс литогенности желчи (т.е. отношение фактического содержания холестерина в желчи к максимальному количеству, которое может быть растворено при данном уровне желчных кислот и фосфолипидов) у женщин оказывается выше, чем у мужчин.
Меньшая эффективность системы выведения холестерина из кровотока и периферических тканей у мужчин способствует их предрасположенности к атерогенезу, включающему, в частности, аккумуляцию холестерина в атеросклеротических повреждениях сосудов. Антиатерогенное действие эстрогенов включает также внепеченочное действие, в частности ингибирование захвата окисленных ЛНП клетками сосудов.
Рекомендуемая литература
Розен В.Б., Матарадзе Т.Д., Смирнова О.В., Смирнов А.Н. Половая дифференцировка функций печени. - М.: Медицина, 1991. - 336 с.
Dean M., Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11(7):1156 - 1166.
Hegardt F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J. 1999;338 ( Pt 3):569 - 582.
Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125 - 1131.
Moore K.J., Freeman M.W. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26(8):1702 - 1711.
Trauner M., Boyer J.L. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83(2):633 - 671.
Wong H., Schotz M.C. The lipase gene family. J Lipid Res. 2002;43(7):
993 - 999.
Yeaman S.J. Hormone-sensitive lipase - new roles for an old enzyme. Biochem J. 2004;379(Pt - 22.