Эндокринная регуляция. Биохимические и физиологические аспекты : учеб. пособие / А.Н. Смирнов ; под ред. В.А. Ткачука - 2009. - 368 с.
|
|
|
|
ГЛАВА 1 РОСТ И ОБЩЕЕ РАЗВИТИЕ
Рост и общее развитие составляют количественную и качественную стороны онтогенеза. В многоклеточном организме процессы роста и развития в значительной мере контролируются сигнальными соединениями, включая гормональные, и тесно связаны между собой также с участием сигнальных соединений.
РОСТ
В общем случае под ростом понимают координированное увеличение размеров и числа клеток разного типа при параллельном увеличении скелета. Однако нередко термином «рост» обозначают лишь один тип изменений клеток, связанный либо с их пролиферацией (гиперплазией), либо с увеличением их размеров (гипертрофией). Например, рост злокачественной опухоли связан с пролиферацией клеток, а рост жировой или мышечной ткани у взрослых обусловлен гипертрофией клеток. Оба типа изменений связаны с биосинтезом белков, интегральным показателем которого служит азотистый баланс. Противоположностью роста являются программированная гибель клеток (апоптоз) и гипотрофия клеток. Данные процессы обычно активируются при неблагоприятных условиях, таких, как голодание или тяжелое заболевание, но могут происходить и в нормальных условиях. Так, апоптоз части иммунных клеток подавляет возможные аутоиммунные и аллергические реакции, а апоптоз альвеолярных клеток молочной железы при прекращении вскармливания обеспечивает прекращение уже ненужного отделения молока. Апоптоз и гипотрофия, как и процессы роста, контролируются разнообразными сигнальными соединениями, в том числе гормонами. Ведущую роль в ансамблях сигнальных соединений, контролирующих рост, играет ГР.
Система гормон роста - инсулиноподобный фактор роста I
Одной из причин недостаточности СТГ, или ГР, сопровождающейся карликовостью, служит мутация гена транскрипционного фак-
тора Pit-1, регулирующего активность гена СТГ и дифференцировку соматотрофов аденогипофиза. (Pit-1 экспрессируется исключительно в переднем гипофизе. В эмбриогенезе экспрессия Pit-1 коррелирует с клеточной пролиферацией и дифференцировкой сомато- и лактотрофов. Pit-1 обнаружен также в тиреотрофах, в которых он необходим для транскрипции гена β-субъединицы тиреотропина - ТТГ. Мутации Pit-1 могут приводить к недостаточности СТГ, пролактина - ПРЛ и ТТГ.) Другой известной причиной недостаточности СТГ и карликовости (линия lit/lit мышей) является мутация рецептора соматолиберина (ГРРГ-Рц), опосредующего стимулирующее действие соматолиберина, или ГГРГ, на секрецию и биосинтез СТГ, а также дифференцировку соматотрофов посредством сопряженного с Gs- белком каскада аденилатциклаза - циклический аденозинмонофосфат (цАМФ) - ПК-A - активация Pit-1. Карликовость развивается также в результате утраты чувствительности периферических тканей к СТГ из-за мутаций рецептора СТГ (ГР-Рц) - синдром Ларона. Избыточность СТГ, возникающая при образовании аденом соматотрофов из-за соматических мутаций Gs-белка (в частности, при конститутивной активации белка из-за нарушения ГТФазной активности Gαs субъединицы, приводящей к конститутивной активации аденилатциклазы), ведет к развитию гигантизма (если возникает до закрытия зон роста трубчатых костей) или акромегалии (если развивается у взрослого). Симптомы акромегалии - результат избыточной стимуляции СТГ продукции ИФР-I прежде всего в печени. Эффекты СТГ на клетки могут быть прямыми или опосредованными ИФР-I и ИФР-II, а также смешанными. В отсутствие ИФР-I (в результате нокаута гена) СТГ теряет способность оказывать ростовое действие, что может быть объяснено как посреднической, так и пермиссивной функцией ИФР-I в действии СТГ.
Контроль секреции СТГ
Соматолиберин, соматостатин, грелин и ИФР-I. Главными гипоталамическими регуляторами секреции ГР служат соматолиберин и соматостатин (SST, или SRIF - somatotropin release-inhibiting factor). Основная часть секретирующих соматостатин нервных терминалей, проецируемых на срединное возвышение, берет начало в передней перивентрикулярной области гипоталамуса, а терминалей ГРРГ - в аркуатном ядре. Секреция СТГ носит выраженный импульсный характер (паттерн), имеющий важное значение для отдельных пара-
метров гормонального действия. Считается, что этот паттерн определяется взаимодействием гипоталамических пептидергических систем секреции соматолиберина и соматостатина, которые секретируются также эпизодически. ГРРГ и соматостатин соответственно стимулируют и ингибируют и синтез, и секрецию СТГ. ГРРГ, кроме того, увеличивает количество соматотрофов в гипофизе. При хронической активации каскада ГРРГ-аденилатциклаза в аденомах гипофиза наблюдается гиперплазия соматотрофов и развивается акромегалия. ГРРГ-Рц относится к надсемейству рецепторов, сопряженных с G-белками. Обнаружено по меньшей мере 5 подтипов рецептора соматостатина (sstr1-5), ассоциированных с рядом G-белков. Рецептор соматостатина обнаружен не только в гипофизе, но и на ГРРГ-нейронах. Возможно, импульсы СТГ связаны со снятием блокирующего действия соматостатина на ГРРГ-нейроны, обладающие внутренней ритмикой.
Ритмичность секреции СТГ также определяется режимом питания. Голодание стимулирует секрецию желудком гормона грелина, стимулирующего продукцию СТГ. Через рецептор грелина могут действовать синтетические стимуляторы секреции СТГ, СТГ-рилизинг пептиды (GHRPs). Эти пептиды, используемые в клинике при недостаточности СТГ, потенцируют действие и увеличивают секрецию ГРРГ, снижают действие и секрецию соматостатина.
Третий уровень ритмичности секреции СТГ связан со сменами сна и бодрствования. Особенно выраженные ночные выбросы СТГ наблюдаются в период полового созревания, сопровождающегося ускоренным ростом.
Секреция СТГ контролируется системами обратной связи. Один из механизмов (короткая обратная связь) заключается в подавлении под действием циркулирующего СТГ секреции ГРРГ и стимуляции секреции соматостатина. ГР-Рц обнаружен в гипоталамических ядрах, контролирующих секрецию СТГ (в аркуатном ядре, содержащем основную часть ГРРГ-нейронов, в перивентрикулярном ядре на соматостатиновых нейронах или рядом с ними), а также в других отделах мозга. Экспрессия ГР-Рц в аркуатном ядре контролируется самим СТГ (стимулируется), гормонами гонад и надпочечников.
Важную роль в системе регуляции секреции СТГ (длинная обратная связь) играет ИФР-I, продуцируемый печенью в ответ на действие СТГ. ИФР-I действует и на гипоталамическом уровне, подавляя секрецию ГРРГ и стимулируя секрецию соматостатина, и на гипо-
физарном уровне, ингибируя транскрипцию гена СТГ. Кроме того, ИФР-I подавляет экспрессию рецептора грелина и рецептора ГРРГ в гипофизе. Эксперименты с избирательным нокаутом гена ИФР-I в печени мыши показали, что основная функция продуцируемого печенью ИФР-I - не стимуляция ростовых процессов, как это предполагалось ранее, а ингибирование секреции СТГ (рост таких животных почти не отличается от нормального; рис. 1-1). Основные элементы системы саморегуляции секреции СТГ показаны на рис. 1-2.
Другие регуляторы секреции СТГ. Многие гормоны способны действовать на биосинтез и секрецию СТГ прямо и/или опосредованно, причем эффекты, реализуемые разными путями, могут быть как однонаправленными, так и противоположными.
Глюкокортикоиды. Глюкокортикоиды служат одним из факторов, стимулирующих дифференцировку соматотрофов. При лече-
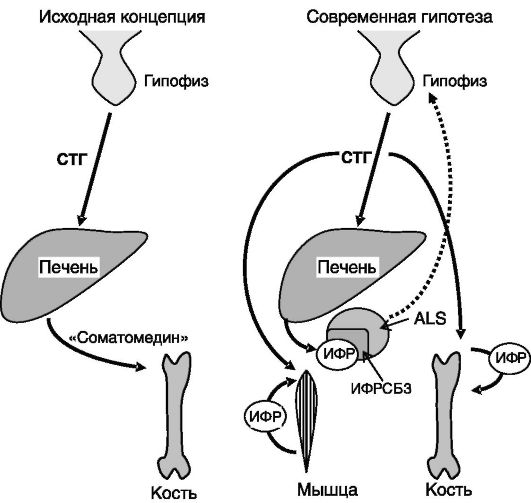 Рис. 1-1. Эволюция концепций соматомедина:
Рис. 1-1. Эволюция концепций соматомедина:
ИФРСБ3 - белок 3, связывающий ИФР; ALS - кислотолабильная субъединица. Пунктиром показано ингибирующее действие ИФР-I из печени на секрецию СТГ
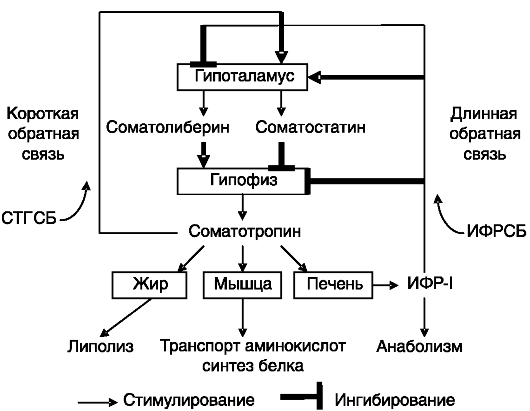 Рис. 1-2. Система саморегуляции секреции ГР:
Рис. 1-2. Система саморегуляции секреции ГР:
СТГСБ - белок, связывающий СТГ; ИФРСБ - белок, связывающий ИФР
нии адиссоновой болезни (первичной гипофункции надпочечников) глюкокортикоидами происходит стимуляция секреции СТГ. С другой стороны, гиперкортицизм часто сопровождается сниженными ростом и уровнем СТГ, а гипокортицизм (например, при пониженной чувствительности надпочечников к АКТГ) - повышенными ростом и уровнем СТГ. Подавление секреции СТГ глюкокортикоидами может быть связано с ингибированием стимулирующего действия ГРРГ и грелина. Действительно, в промоторе гена рецептора грелина выявлен глюкокортикоидчувствительный элемент (ГКЧЭ), опосредующий ингибирующее действие глюкокортикоидов на экспрессию гена. В то же время глюкокортикоиды повышают экспрессию гена рецептора ГРРГ, и остается неясным, за счет чего глюкокортикоиды снижают эффективность действия ГРРГ. Глюкокортикоиды вызывают быстрое высвобождение гипоталамическими нейронами соматостатина, и это должно вести к ингибированию секреции СТГ. Но на уровне гипофиза глюкокортикоиды подавляют экспрессию ряда рецепторов соматостатина, что должно способствовать секреции СТГ. Кроме того, действуя на печень, глюкокортикоиды подавляют секрецию ИФР, что также должно вести к увеличению секреции СТГ.
Экспрессия гена самого СТГ может прямо стимулироваться глюкокортикоидами. Можно предполагать, что в разных физиологических и патологических ситуациях может преобладать тот или иной путь действия глюкокортикоидов с противоположным конечным действием на секрецию СТГ.
Тиреоидные гормоны. Гипотиреоз разной этиологии (йодная недостаточность, мутация тироглобулина и т.д.) сопровождается низкорослостью при низком уровне СТГ, а лечение гипотиреоза тиреоидными гормонами восстанавливает ростовые процессы и уровень СТГ. Как и глюкокортикоиды, тиреоидные гормоны способствуют дифференцировке соматотрофов гипофиза. Как и глюкокортикоиды, тиреоидные гормоны прямо активируют экспрессию гена СТГ и гена рецептора ГРРГ. В отличие от глюкокортикоидов, тиреоидные гормоны увеличивают также экспрессию рецептора грелина. Следует отметить, однако, что избыток тиреоидных гормонов при тиреотоксикозе подавляет секрецию СТГ в ответ на ГРРГ. Тиреоидные гормоны снижают также секрецию ГРРГ гипоталамусом, что ограничивает их стимулирующее действие на СТГ.
Половые гормоны. Хорошо известны половые различия в скорости ростовых процессов. Эти различия в определенной мере связаны с действием половых гормонов на паттерн секреции СТГ. Для мужских особей характерны редкие, высокоамплитудные колебания концентрации СТГ при низком межпиковом уровне гормона, а для женских особей - более частые, низкоамплитудные колебания при высоком межпиковом уровне СТГ. Половые гормоны оказывают влияние на продукцию СТГ несколькими путями, включая неонатальное (у грызунов) программирование андрогенами количества соматотрофов в гипофизе и ГРРГ-ергических нейронов в аркуатном ядре гипоталамуса и способности гипоталамуса взрослых самцов к повышенному биосинтезу ГРРГ и соматостатина. У взрослых животных андрогены усиливают действие этой программы. Кроме того, андрогены у взрослых повышают чувствительность соматотрофов к действию ГРРГ и грелина, стимулируя экспрессию соответствующих рецепторов, и подавляют ингибирующее действие соматостатина. Половые гормоны, кроме того, способны прямо влиять на уровень ИФР-I, ингибирующего секрецию СТГ (андрогены стимулируют, а эстрогены подавляют секрецию печенью ИФР-I).
Лептин. Лептин способен действовать прямо на соматотрофы гипофиза, увеличивая синтез и секрецию СТГ. Кроме того, лептин
подавляет высвобождение соматостатина и стимулирует высвобождение ГРРГ гипоталамусом, что также должно способствовать секреции СТГ. Лептин служит также пермиссивным фактором для активации репродуктивной системы, которая через половые гормоны стимулирует секрецию СТГ. Физиологическое значение стимулирующего действия лептина на продукцию СТГ, по-видимому, заключается в поддержании массы тела на постоянном уровне через систему обратной связи: наполнение адипоцитов жиром - повышение секреции лептина - увеличение уровня СТГ - стимуляция липолиза, ингибирование липосинтеза.
Ретиноиды. Ретиноевые кислоты способствуют дифференцировке соматотрофов гипофиза. Транс-ретиноевая и 9-цис-ретиноевая кислоты, сами по себе слабо влияющие на экспрессию рецептора ГРРГ, значительно потенцируют стимулирующий эффект глюкокортикоидов. Недостаточность ретиноевой кислоты проявляется в нарушении паттерна секреции СТГ со значительным снижением амплитуды пиков гормона, что позволяет предполагать преимущественное влияние на гипоталамические регуляторы секреции СТГ.
Тиролиберин. Эндогенный тиролиберин (ТРГ) оказывает стимулирующее действие на продукцию и секрецию СТГ, о чем свидетельствуют задержка роста и пониженный уровень мРНК СТГ в гипофизе мышей с нокаутированным геном рецептора ТРГ. Напротив, введение ТРГ животным или больным людям повышает уровень СТГ.
Интермедин. Интермедин - член семейства адреномедуллина - экспрессируется в промежуточной и передней доле гипофиза. Подавляет стимулирующее действие ГРРГ на соматотрофы.
Нейропептид Y. Нейропептид Y (NPY) - важнейший стимулятор потребления пищи и ингибитор энергозатрат - оказывает модулирующее действие на СТГ, направленность которого зависит от концентрации самого NPY, а также других регуляторов продукции и секреции СТГ. Система регуляции СТГ с участием NPY включает прямое действие NPY на соматотрофы гипофиза, а также влияние на активность гипоталамических нейронов, секретирующих ГРРГ и соматостатин. NPY может также влиять на чувствительность клеток гипофиза к грелину.
Аминокислоты, углеводы, липиды. Возбуждающие аминокислоты (глутаминовая и аспарагиновая) стимулируют секрецию СТГ, действуя преимущественно на гипоталамическом уровне. Интересно, что данный эффект опосредуется ионотропными рецепторами аминокислот, тогда как активация метаботропных рецепторов оказывает противо-
положное действие. Глюкоза и свободные жирные кислоты, напротив, ингибируют секрецию СТГ. Перечисленные эффекты питательных веществ, по-видимому, составляют систему обратной связи: СТГ стимулирует утилизацию аминокислот, но оказывает гипергликемическое и гиперлипидемическое действие (рис. 1-3). Ингибирующее
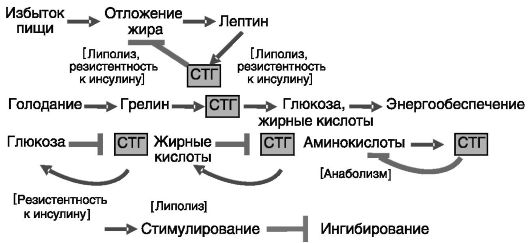 Рис. 1-3. Участие СТГ в системах ауторегуляции обменных процессов
Рис. 1-3. Участие СТГ в системах ауторегуляции обменных процессов
действие глюкозы на секрецию СТГ используется в провоцирующем тесте при подозрении на недостаточность СТГ: у здорового человека вызываемая введением инсулина гипогликемия сопровождается ростом концентрации СТГ в крови.
Транспорт СТГ и ИФР-I
В кровяном русле значительная часть СТГ находится в виде комплексов с СТГ-связывающим белком (СТГСБ).
Этот белок у разных видов имеет разное происхождение. У человека он возникает в результате «слущивания» внеклеточного фрагмента рецептора СТГ под действием специфической протеазы. У крысы СТГСБ возникает в результате альтернативного сплайсинга продукта транскрипции гена ГР-Рц, при котором трансмембранный и внутриклеточный домены ГР-Рц замещаются коротким гидрофильным фрагментом. Уровень СТГСБ у человека дифференцирован по полу (женщины - 1307 пмоль/л, мужчины - 944 пмоль/л) за счет действия эстрогенов, феминизирующих паттерн секреции СТГ и таким образом индуцирующих экспрессию ГР-Рц. СТГСБ, как и ГР-Рц, взаимодействует с СТГ в соотношении 2:1.
СТГСБ снижает эффективность действия СТГ на компетентные клетки и замедляет клиренс СТГ из кровяного русла.
Основная часть ИФР-I в кровяном русле находится в виде комплексов с ИФР-1-связывающими белками (ИФРСБ). Обнаружено по меньшей мере 6 таких белков - продуктов разных генов. ИФРСБ ингибируют действие ИФР-I, причем этот эффект может быть избирательным.
Так, ИФРСБ3 снимает гипогликемическое, но не анаболическое действие ИФР-I. Синтез и деградация ИФРСБ находятся под контролем ряда систем. Опухолевые клетки высвобождают протеазы, разрушающие ИФРСБ, обеспечивая тем самым проявление антиапоптотической активности ИФР-I. СТГ стимулирует синтез кислотолабильной субъединицы (ALS) ИФРСБ3 - основного транспортера ИФР-I крови. Тройной комплекс ИФР-I/ИФРСБ3/ALS оказывается чрезвычайно стабильным, обеспечивая длительное пребывание ИФР в крови. Уровень ИФР-I регулируется инсулином, что отчасти определяет ростовое действие инсулина.
Действие СТГ и ИФР
Рецептор СТГ относится к надсемейству рецепторов, ассоциированных с тирозинкиназами («рецепторы цитокинов»). Необходимым условием проведения сигнала СТГ является димеризация ГР-Рц с образованием комплекса СТГ/ГР-Рц2. При высоких концентрациях СТГ образуются комплексы с ГР-Рц в соотношении 1:1, неэффективные в проведении сигнала. В результате кривая «доза - эффект» для СТГ имеет колоколообразную форму. Эффективность СТГ зависит также от паттерна его секреции или введения и реагирующей системы. Так, уровень ГР-Рц увеличивается при продолжительной инфузии, но не при болюсном введении СТГ, а ростовой эффект СТГ при этих способах введения одинаков. Видимо, разные эффекты СТГ опосредуются разными каналами проведения сигнала, различающимися по динамике десенситизации и восстановления. Еще один пример роли импульсного типа секреции СТГ - влияние на жировую ткань. Известно, что СТГ стимулирует распад липидов у человека и сельскохозяйственных животных. У мышей линии dw/dw с недостаточностью СТГ при пищевом ожирении инфузия СТГ приводила к липолизу, а импульсное введение было неэффективно. В то же время рост костей и мышц у этих животных в одинаковой мере стимулировался постоянной инфузией и прерывистым введением СТГ.
Структурно-функциональная организация СТГ. СТГ экспрессируется в виде нескольких сплайсинговых вариантов, способных формировать олигомерные комплексы. Полноразмерный полипептид СТГ человека построен из 191 а.к. Благодаря наличию двух дисульфидных связей молекула СТГ формирует глобулу, в которой N- и С-концевые фрагменты контактируют между собой. Связывание с рецептором является упорядоченным во времени двустадийным процессом. Первая молекула рецептора с высоким сродством взаимодействует с поверхностью гормона (сайтом 1), включающей α-спирали 1 и 4 (рис. 1-4). Затем с сайтом 2 гормона, включающим участки α-спиралей 1 и 3, связывается вторая молекула рецептора, с уже меньшим сродством. После этого примембранные области двух молекул рецептора начинают взаимодействовать между собой, инициируя проведение сигнала. Возможно, составной характер сайтов взаимодействия СТГ с рецептором лежит в основе давно установленного экспериментально факта проявления отдельными фрагментами гормона форм активности, противоположных биологической активности целой молекулы гормона.
Соотношение прямых и опосредованных ИФР эффектов СТГ. Многие клетки чувствительны одновременно и к самому СТГ, и к ИФР. Дифференцировка прямых и опосредованных ИФР эффектов СТГ является достаточно сложной проблемой, поскольку часто СТГ вызывает локальное образование ИФР-I. К настоящему времени установлено, что СТГ и ИФР-I оказывают независимое действие на ростовые процессы, а при комбинированном введении действуют аддитивно. Что касается тканевой специфичности, то СТГ наиболее эффективен в стимуляции роста кости и печени, а ИФР-I - других тканей, включая почки,
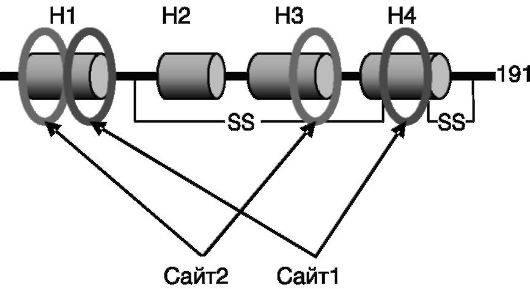 Рис. 1-4. Фрагментированная локализация сайтов взаимодействия с рецептором в молекуле СТГ:
Рис. 1-4. Фрагментированная локализация сайтов взаимодействия с рецептором в молекуле СТГ:
H1-H4 - α-спирали 1-4; S-S - дисульфидные внутримолекулярные связи
кишечник, лимфоидную ткань. Недублируемое ИФР-I (т.е. прямое) действие СТГ наблюдается в отношении индукции ALS комплекса ИФР-I/ ИФРСБ3: при гипофизэктомии концентрация ALS снижается в 100 раз и восстанавливается введением СТГ, но не ИФР-I. Роль ИФР-I в ростовых процессах видоспецифична. У сельскохозяйственных животных ИФР-I не стимулировал роста; у детей с нечувствительностью к СТГ ИФР-I вызывал неравномерный рост, особенно костей лица. ИФР-I сам по себе не стимулировал липолиз, но усиливал данный эффект СТГ. При использовании в качестве антикатаболитной терапии (например, при СПИДе, мышечной дистрофии, острой ишемии) лучшие результаты дает комбинированное введение СТГ и ИФР-I.
ИФР-I и его рецептор. ИФР-I - полипептид семейства инсулина, экспрессируемый в виде нескольких сплайсинговых вариантов. В отличие от инсулина процессинг пропептида ИФР-I не включает протеолитическое вырезание вставочного С-пептида центральной части молекулы. Рецептор ИФР-I относится к классу рецепторных тирозинкиназ, подклассу тетрамерных рецепторов. Может образовывать гибридные рецепторы с рецептором инсулина. ИФР-I экспрессируется практически повсеместно, и его экспрессия контролируется не только ГР, но и рядом других сигнальных соединений. Поэтому ИФР-I следует рассматривать не только в качестве посредника действия СТГ, но и как пара/эндокринный фактор с самостоятельными функциями.
У мыши с повреждением гена ИФР-I оказываются нарушенными процессы миелинизации, что сопровождается задержкой роста плодов. ИФР-I - важнейший антиапоптотический фактор. В отсутствие рецептора ИФР-I (ИФР-IРц) или самого ИФР-I многие клетки теряют способность к трансформации онкогенами, а уже трансформированные клетки подвергаются апоптозу. При этом они начинают экспрессировать p53 супрессор опухолей. В лимфоидной ткани ИФР-I является важнейшим ростовым фактором, стимулируя развитие В-клеток в костном мозгу и созревание T-клеток в тимусе (антиапоптотическое действие). Концентрация ИФР-I и ИФР-II в крови достигает 200 - 400 нг/мл. Введение ИФР-I приводит к гипогликемии, стимулируя транспорт глюкозы в клетки, т.е. ИФР-I дублирует эффект инсулина, тогда как СТГ оказывает диабетогенное (гипергликемическое) действие.
Мышечная ткань. Количество и размер миофибрилл в скелетных мышцах, а значит - мышечная масса и сила мышц зависят от физической активности, характера питания, общего состояния здоровья, возраста и разнообразных ауто-, пара- и эндокринных факторов.
Негативными регуляторами мышечной массы служат глюкокортикоиды, эффект которых усиливается трийодтиронином (Т3), сопровождающий многие заболевания провоспалительный цитокин ФНО-α, ангиотензин II. Стимуляторами мышечной массы выступают ГР, ИФР-I, андрогены. В раннем развитии и при регенерации мышцы объектами регуляции являются и количество, и размер мышечных волокон, а во взрослом организме - преимущественно размер волокон. Регуляция, по-видимому, включает действие механических и химических факторов как на собственно сократительные элементы мышцы, так и на их предшественники, часть которых представлена так называемыми сателлитными клетками. Внешние стимулирующие и ингибирующие влияния в значительной мере замыкаются на негативном ауто/паракринном регуляторе мышечной ткани миостатине, или факторе роста и дифференцировки 8 (GDF-8), являющемся членом надсемейства белков ТФР-β. Инактивирующие мутации гена миостатина у скота и человека, а также нокаут гена миостатина у мыши вызывают значительное увеличение («удвоение») мышечной массы (при параллельном снижении массы жира). Сходные изменения можно вызвать введением животным антител против миостатина. И наоборот, избыточная экспрессия трансгена вызывает атрофию мышц и дифференцировку клеток-предшественников мышечных волокон в адипоциты с увеличением жировых запасов. Соответственно СТГ и ИФР-I, а также физическая нагрузка подавляют продукцию миостатина, а глюкокортикоиды - стимулируют (регуляторная область гена миостатина включает ряд глюкокортикоидчувствительных элементов). Более того, нокаут гена миостатина у мыши полностью блокирует негативное действие глюкокортикоидов на мышечную массу.
Взаимное усиление эффектов СТГ и физической активности на мышечную массу в определенной мере определяется их совместным стимулирующим влиянием на экспрессию гена ИФР-I в мышце. В мышечной ткани функционируют два промотора этого гена, один из которых (дистальный) активируется механическими стимулами, а другой (проксимальный) - эндокринными факторами, включая СТГ.
При кратковременном действии на мышцу СТГ и ИФР-I оказывают сходные анаболические эффекты, повышая интенсивность биосинтеза белка. В отличие от СТГ и ИФР-I анаболическое действие инсулина реализуется преимущественно за счет подавления деградации белка в мышце.
Функции СТГ в онтогенезе. Максимальный линейный рост у человека наблюдается на 18-й неделе внутриутробного развития (макси-
мальная прибавка массы тела - на 34-й неделе), после чего скорость роста замедляется.
В 1-й год жизни рост увеличивается на 25 см, на 2-й - на 12,5 см, на 3 - 4-й годы прибавка составляет по 8 см, на 5 - 6-й годы - по 6 см. В последующие несколько лет до полового созревания прирост составляет 5,5 см. При этом во внутриутробный период и 1-й год жизни мальчики растут быстрее девочек, со 2-го по 4-й год, наоборот, девочки растут быстрее мальчиков, а в период 5 - 9 лет скорость роста у мальчиков и девочек выравнивается.
С 9 лет у девочек начинается пубертатный ростовой спурт, достигающий максимума в возрасте 11,5 года (8,3 см/год). У мальчиков пубертатный спурт начинается с 11 лет и достигает максимума в 13,5 года (9,5 см/год). После этого скорость роста быстро снижается. Половые различия в росте взрослых (около 13 см) складываются из роста мальчиков в течение дополнительных двух лет перед половым созреванием (8 - 11 см) и более интенсивного пубертатного спурта (3 - 5 см; рис. 1-5).
Динамика массы тела и его состава в онтогенезе также различны у мальчиков и девочек (рис. 1-6). Можно видеть, что уже с момента
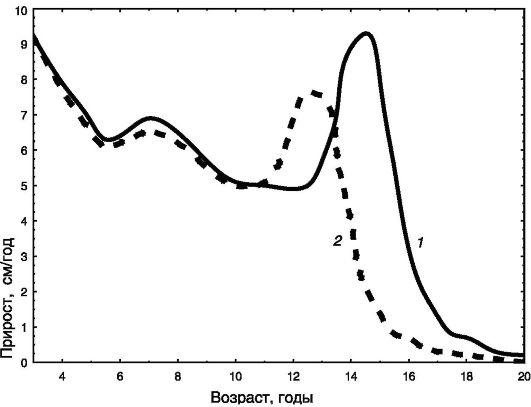 Рис. 1-5. Динамика скорости линейного роста мальчиков (1) и девочек (2)
Рис. 1-5. Динамика скорости линейного роста мальчиков (1) и девочек (2)
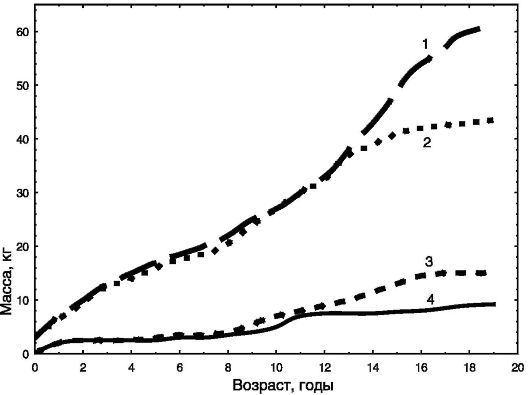 Рис. 1-6. Динамика массы тела за вычетом жира (1, 2) и массы жира (3, 4) в онтогенезе мальчиков (1, 4) и девочек (2, 3)
Рис. 1-6. Динамика массы тела за вычетом жира (1, 2) и массы жира (3, 4) в онтогенезе мальчиков (1, 4) и девочек (2, 3)
рождения имеются половые различия в общей массе тела и доле жира с преобладанием жира у девочек. При половом созревании эти различия резко усиливаются, и к 20 годам при преобладании общей массы тела у мужчин доля жира у них оказывается в 2 раза ниже, чем у женщин.
У человека СТГ начинает секретироваться с 12-й недели внутриутробного развития, достигает максимума между 16-й и 32-й неделями (40-кратное превышение уровня взрослых) и значительно снижается к моменту рождения. Ростовая активность СТГ во внутриутробный период развития и в первые месяцы жизни, однако, низка: ростовые процессы зависят от СТГ лишь на 20%. Полагают, что СТГ в эти периоды выполняет преимущественно адаптивные функции. Роль СТГ как стимулятора роста проявляется в фазе вторичного подъема ростовых процессов.
Аналогичная ситуация имеет место у лабораторных животных. До приблизительно 3-недельного возраста крысята и мышата с нарушениями продукции (например, в результате разрушения гипоталамических ГРРГ-нейронов неонатальным введением глутамата) или
действия (при нокауте гена ГР-Рц) СТГ не отстают в росте, но затем рост замедляется, и животные остаются карликами. Аналогичным образом, приблизительно 1000-кратное увеличение уровня СТГ у трансгенных мышей начинает сказываться на ускорении роста лишь после 3-й недели жизни.
Резкий подъем секреции СТГ в период полового созревания под действием половых гормонов служит причиной бурного роста (часто называемого спуртом) в этот период.
Самостоятельное ростовое действие других гормонов
Многие из гормонов, регулирующих секрецию СТГ, способны оказывать влияние на ростовые процессы не только через систему СТГ/ИФР-I, но и самостоятельно. Так, глюкокортикоиды в большинстве тканей (исключая печень) обусловливают отрицательный азотистый баланс, ингибируя биосинтез белка. Андрогены оказывают противоположное действие. При этом нередко, например в печени, СТГ может оказывать пермиссивное влияние на эффекты андрогенов. Периферическое действие половых гормонов служит важным фактором половой дифференцировки ростовых процессов. В целом андрогены повышают эффективность действия СТГ, а эстрогены - снижают, что отражается, в частности, в большей пропорции жир/ мышечная масса в женском организме по сравнению с мужским (см. выше). Обусловленные половыми гормонами половые различия в стимулирующем рост действии СТГ отмечаются и при терапевтическом применении СТГ у больных с гипопитуитаризмом. Эстрогены помимо анаболического действия на жировую ткань, участвуют в регуляции роста и минерализации скелета. Именно благодаря закрытию точек роста костей под действием эстрогенов (вероятно, за счет стимуляции эстрогенами апоптоза хондроцитов) девушки прекращают расти раньше своих сверстников-юношей.
Недостаточность рецепторов эстрогенов может вести к задержке закрытия точек роста кости и соответственно высокорослости. Считается, что действие андрогенов на костную ткань у мужчин на 70% опосредуется ароматизацией андрогенов в эстрогены. Стимулирующее действие экзогенных андрогенов на развитие кости при терапии мужского гипогонадизма снимается одновременным введением ингибиторов ароматазы. Остальные 30% эффекта андрогенов на костный аппарат реализуются собственно андрогенами через А-Рц, имеющийся, в частности, в остеобластах. Лица с инактивирующими
мутациями А-Рц (синдром тестикулярной феминизации) имеют рост, промежуточный между нормальными мужчинами и женщинами.
У взрослых эстрогены стимулируют минерализацию кости, активируя остеобласты и тормозя активность остеокластов. Тиреоидные гормоны прямо стимулируют пролиферацию нейронов в ЦНС, активируют глиальные клетки к биосинтезу миелиновой оболочки. Самостоятельное ростовое действие оказывает также ПРЛ. Этот гормон даже называют ювенильным, поскольку он продлевает фазу роста и ингибирует дифференцировку.
АПОПТОЗ
Регуляция апоптоза белками семейства Bcl2
Непосредственными исполнителями программы гибели клеток (апоптоза) служат сериновые протеазы, называемые каспазами. Их активация осуществляется за счет протеолиза предшественников - прокаспаз. Основные пути активации каспаз представлены на рис. 1-7.
Путь, индуцируемый внутриклеточными стрессорными сигналами (внутренний путь), является более древним по сравнению с путем, активируемым через рецепторы смерти (внешний путь), и регулируется белками семейства Bcl2. Передача сигнала происходит с участием фактора Apaf1 после его взаимодействия с Cyt c, выделяющимся из поврежденных митохондрий. Активность каспаз 9 и 3 ограничивается белками IAPs (inhibitors of apoptosis proteins, ингибиторы апоптотических белков). В свою очередь, эффект IAPs может быть блокирован белками Diablo/SMAC (second mitochondria-derived activator of caspase, второй активатор каспазы из митохондрий) и Omi, выделяющимися из поврежденных митохондрий. Эволюционно более молодой путь регуляции апоптоза, определяющий взаимовлияния клеток, опосредуется «рецепторами смерти» через адапторные белки, в частности белок FADD.
Непосредственное исполнение программы гибели клетки запускается через формирование в цитозоле клетки олигомерных белковых комплексов, апоптосом, включающих Apaf 1, Cyt c и латентные протеиназы в форме прокаспаз 9 и 3(7) (рис. 1-8). На ключевую роль Apaf 1 в инициации апоптоза указывают данные об устойчивости клеток, лишенных Apaf 1, к действию разнообразных апоптотических стимулов.
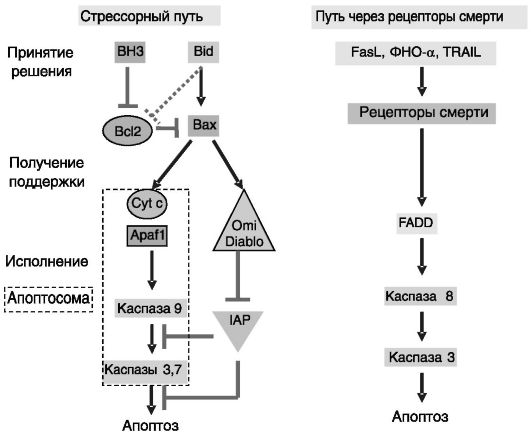 Рис. 1-7. Основные пути, ведущие к апоптозу:
Рис. 1-7. Основные пути, ведущие к апоптозу:
Bcl2 - белок 2 B-клеточной лимфомы; Apaf1 - фактор 1, активирующий апоптотическую протеазу; Cyt c - цитохром c; IAP - ингибитор апоптотических белков; FADD - ассоциированный с Fas белок с доменом смерти; Diablo - белок с низким pI, прямо связывающий IAP; Omi - один из белков, специфически взаимодействующих с Mxi2; Bax - ассоциированный с Bcl2 белок X; Bid - агонист смерти, взаимодействующий с доменом BH3 (BH3 interacting domain death agonist); BH3 - белок семейства BH3-only
Ингибирование инициирующей апоптоз каспазы-9 и исполнительных каспаз 3 и 7 белками группы IAP осуществляется путем взаимодействия повторов BIR (бакуловирусных IAP повторов) этих белков с каталитическим доменом каспаз. Снятие данного ингибирования под действием митохондриального белка Diablo зависит от связывания короткого фрагмента N-концевой области этого белка с теми же повторами BIR белков IAP, что, вероятно, создает стерическое препятствие для взаимодействия IAP с каспазами (рис. 1-9). Аналогично может действовать белок Omi. Этот белок, кроме того,
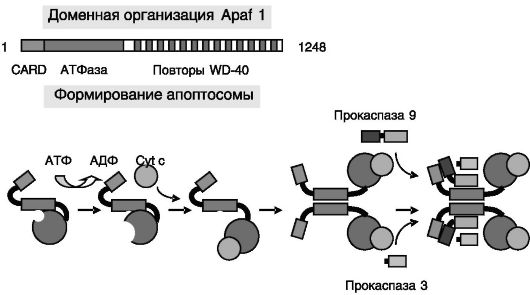 Рис. 1-8. Модель формирования апоптосомы
Рис. 1-8. Модель формирования апоптосомы
Связывание и гидролиз АТФ АТФазным доменом Apaf 1 ведут к изменениям конформации этого белка, обеспечивающим связывание Cyt c с последующей олигомеризацией комплексов. Домены CARD Apaf 1 в этих комплексах рекрутируют (гомофильное связывание) прокаспазу 9 через аналогичные домены последней. Прокаспаза 9 при этом аутоактивируется протеолизом, рекрутирует в комплексы прокаспазу 3 и активирует ее. CARD - домен рекрутирования каспаз.
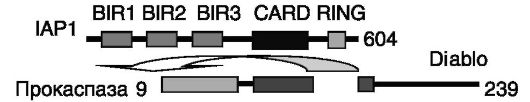 Рис. 1-9. Diablo блокирует ингибирование каспаз белком IAP, взаимодействуя с доменом BIR3 IAP
Рис. 1-9. Diablo блокирует ингибирование каспаз белком IAP, взаимодействуя с доменом BIR3 IAP
обладает протеиназной активностью, которая способствует апоптозу не только посредством активации каспаз.
Вышележащие эффекторы включают регуляторы целостности митохондрий. У млекопитающих имеется по меньшей мере 20 членов семейства Bcl2, каждый из которых содержит хотя бы одну область гомологии с Bcl2 (BH). В семейство входят как антиапоптотические белки (Bcl2, Bcl-xL, Bcl-w, A1 и Mcl1), так и проапоптотические факторы подсемейств Bax (Bcl2-associated X protein, ассоциированный с Bcl2 белок X) и «BH3-only» («только BH3»). Члены подсемейства
Bax содержат области гомологии BH1, BH2 и BH3, а члены подсемейства BH3-only - лишь короткий мотив BH3, необходимый и достаточный для индукции апоптоза. Белки группы BH3-only действуют как сенсоры повреждения и прямые антагонисты белков, обеспечивающих выживание клеток, а белки группы Bax действуют позднее, возможно, на уровне разрушения митохондрий. Структурная организация основных представителей семейства представлена на рис. 1-10.
В Bcl2 и его гомологах гидрофобный C-концевой трансмембранный домен обеспечивает заякоривание этих белков на цитоплазматической поверхности внутриклеточных мембран: митохондриальной, эндоплазматического ретикулума и ядерной. Домены BH1, BH2 и BH3 формируют гидрофобную бороздку, способную взаимодействовать с доменом BH3 белков семейства BH3-only.
Выживание клеток разных тканей обеспечивается разными представителями семейства Bcl2: сам Bcl2 способствует выживанию стволовых клеток почек и меланоцитов, а также зрелых лимфоцитов, Bcl-xL - нейрональных и эритроидных клеток, Bcl-w - предшественников сперматозоидов, A1 - нейтрофилов, Mcl1 обеспечивает имплантацию зиготы.
Исключая, возможно, Bid, белки семейства BH3-only действуют путем связывания и нейтрализации белков семейства Bcl2. Активность
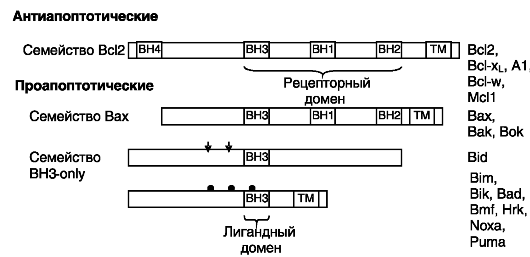 Рис. 1-10. Структурная
организация представителей семейства Bcl2. Стрелками показаны сайты
активирующего протеолиза Bid; черными кружочками отмечены положения
фосфорилируемых остатков в Bad. ТМ - трансмембранный домен
Рис. 1-10. Структурная
организация представителей семейства Bcl2. Стрелками показаны сайты
активирующего протеолиза Bid; черными кружочками отмечены положения
фосфорилируемых остатков в Bad. ТМ - трансмембранный домен
белков BH3-only контролируется рядом механизмов, включая фосфорилирование, протеолиз, интенсивность транскрипции.
В здоровой клетке Bim и Bmf находятся в неактивном состоянии за счет связывания с легкими цепямидинеина, ассоциированными смикротрубочками или актиновым цитоскелетом. Фосфорилированный киназой Akt в положениях Ser75 или Ser99 Bad (Bcl2 antagonist of cell death, антагонист Bcl2 в клеточной гибели) связывается белками «лесов» 14-3-3. Это связывание, в свою очередь, способствует дополнительному фосфорилированию Bad в области BH3 киназой ПК-A, что блокирует возможность связывания Bad с Bcl2. Bid неактивен до его протеолитического расщепления. Активность Noxa, Puma и Hrk контролируется на транскрипционном уровне. В действии белков семейства BH3-only имеется определенная тканевая специализация: Bid стимулирует гибель гепатоцитов, Bim - гематопоэтических и нейрональных клеток.
Помимо инактивации антиапоптотических белков семейства Bcl2 Bid стимулирует апоптоз путем активации Bax и Bak (Bcl2 antagonist/ killer, антагонист Bcl2/киллер).
Активация самого Bid, как отмечалось, происходит за счет его протеолитического расщепления. К полученному полипептидному фрагменту присоединяется миристат, который обеспечивает заякоривание молекулы на митохондриях. При этом в присутствии Bak или Bax происходит быстрое выделение Cyt c из митохондрий. Предполагается, что Bid стимулирует олигомеризацию Bax и Bak, формирующих поры в митохондриальной мембране.
В здоровой клетке Bax в виде гетеродимера с Bcl локализован в цитоплазме. Апоптотический сигнал ведет к его отделению от Bcl и встраиванию в наружную мембрану митохондрий и олигомеризации. В отличие от Bax Bak является интегральным белком митохондриальной мембраны и в здоровой клетке. Но апоптотический сигнал повышает его олигомеризацию.
Полагают, что функция Bcl2-подобных белков заключается в сохранении целостности митохондрий и тем самым в сохранении «в запертом» состоянии активаторов гибели: цитохрома c (который, связываясь с повторяющимся доменом WD40 белка Apaf1, активирует этот белок и соответственно каспазу 9), белков Diablo и Omi (которые блокируют белок IAP, тормозящий активность каспаз), флавопротеинового индуцирующего апоптоз фактора AIF (apoptosis-inducing factor), который участвует в стимуляции конденсации хроматина и
деградации ДНК, эндонуклеазы G, которая участвует в нуклеосомальной фрагментации ДНК (рис. 1-11).
Данный механизм не исчерпывает всех вариантов индукции апоптоза. В некоторых случаях разрушение митохондрий является не причиной, а следствием активации каспаз в ответ на апоптотический стимул. К числу каспаз, активируемых независимо от повреждения митохондрий, могут относиться каспазы 2, X, 12.
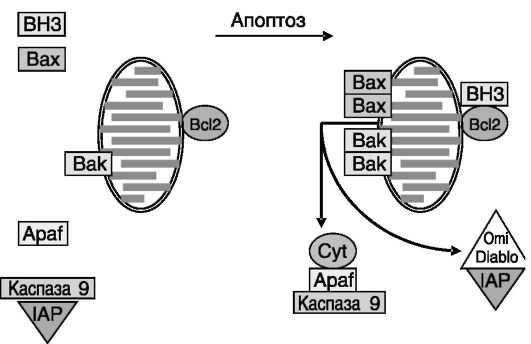 Рис. 1-11. Модель стимуляции апоптоза с участием белков семейства Bcl2 и факторов митохондрий
Рис. 1-11. Модель стимуляции апоптоза с участием белков семейства Bcl2 и факторов митохондрий
Функционирование членов семейства Bcl2 тесно связано с канцерогенезом, причем антиапоптотические члены семейства выступают в роли онкогенов, а проапоптотические - в роли супрессоров опухолей. Создание препаратов, активирующих стимуляторы апоптоза, может открыть новые возможности в лечении раковых заболеваний. Примером успешной реализации знания механизмов апоптоза может служить применение 7-членного N-концевого пептида Diablo для активации каспаз.
Гормональная регуляция апоптоза
Процессы апоптоза контролируются разнообразными сигнальными соединениями. При этом могут быть задействованы оба пути апоптоза (внутренний и внешний). Клинически значимым примером может служить проапоптотическое действие глюкокортикоидов на клетки иммунной системы: массированное введение глюкокортико-
идов больным лейкозом или рядом других форм рака крови за счет стимуляции апоптоза может вести к излечению или ремиссии заболевания. К сожалению, отдельные раковые клетки избегают гибели за счет нарушения проведения апоптотического сигнала глюкокортикоидов и становятся родоначальниками нечувствительных к глюкокортикоидам популяций раковых клеток. Механизмы апоптотического действия глюкокортикоидов остаются неясными. Установлено, что индуцируемый глюкокортикоидами апоптоз включает эффекты гормона на транскрипционном уровне и зависит от рецептора глюкокортикоидов. Наиболее вероятно, что глюкокортикоиды запускают митохондриальный путь апоптоза, но действуют сразу на несколько объектов. В частности, показано, что индуцируемый глюкокортикоидами апоптоз подавляется в отсутствие белков группы BH3-only, Puma (p53 up-regulated modulator of apoptosis, увеличиваемый p53 модулятор апоптоза) и Bim (Bcl-2 interacting mediator of cell death, взаимодействующий с Bcl-2 посредник клеточной гибели), и стимуляция экспрессии Bim является одним из ранних эффектов глюкокортикоидов. Глюкокортикоиды стимулируют также экспрессию каспазы 9 (возможно, через индукцию транскрипционного фактора AP-4), а ингибирование этой каспазы подавляет и индуцируемый гормоном апоптоз. Атрофия скелетных мышц под действием глюкокортикоидов, включающая активацию каспаз, как отмечалось выше, может опосредоваться ауто/паракринным фактором миостатином. Необходимо отметить, однако, что в отношении ряда типов клеток глюкокортикоиды могут выступать в качестве антиапоптотических факторов.
Примером роли гормонов в контроле физиологически значимого процесса апоптоза может служить инволюция молочной железы в конце периода лактации. Ведущую роль в индукции апоптоза альвеолярного эпителия молочной железы при прекращении лактации играет утрата поддерживающего действия ПРЛ вследствие утраты его рефлекторного выброса в ответ на акты сосания (доения у домашнего скота). ПРЛ, с одной стороны, стимулирует локальное образование антиапоптотического ИФР-I, а с другой - подавляет экспрессию его ингибитора ИФРСБ5. Роль ИФРСБ5 как посредника прекращения действия ПРЛ продемонстрирована в экспериментах с локальной гиперэкспрессией трансгена ИФРСБ5 у мыши. Напротив, гиперэкспрессия трансгена ИФР-I в молочной железе у мыши замедляет инволюцию железы и продлевает лактацию. Антиапоптотическое действие ИФР-I включает активацию PI3K - Akt пути, одним из следствий
чего является ингибирование локальной экспрессии ТФР-β, способствующего инициации апоптоза. Путь PI3K - Akt (и, возможно, путь Ras-MAPK) обеспечивает фосфорилирование проапоптотического белка Bad, что ведет к его инактивации. Снятие этого блока апоптоза в результате ослабления фосфорилирования Bad при снижении экспрессии и биологической доступности ИФР-I ведет к вытеснению активированным Bad проапоптотического Bax из комплексов с Bcl2. В результате Bax становится способным к формированию пор в наружной мембране митохондрий, обеспечивающих выход в цитоплазму посредников апоптоза (Cyt c, Diablo и т.д.; рис. 1-12).
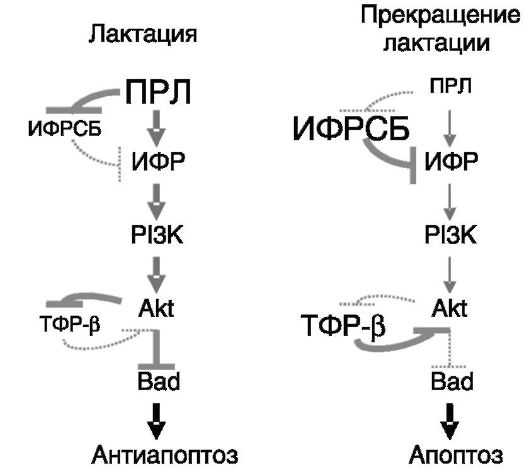 Рис. 1-12. Снижение
секреции ПРЛ при прекращении лактации сопровождается повышением
продукции контррегуляторов (ИФРСБ5, ТФР-β) и снятием ингибирования с
апоптотического пути (прекращением фосфорилирования Bad)
Рис. 1-12. Снижение
секреции ПРЛ при прекращении лактации сопровождается повышением
продукции контррегуляторов (ИФРСБ5, ТФР-β) и снятием ингибирования с
апоптотического пути (прекращением фосфорилирования Bad)
ОБЩЕЕ РАЗВИТИЕ
Начиная с первых делений оплодотворенной яйцеклетки процессы дифференцировки клеток эмбриона, а в последующие периоды развития - плода и новорожденного животного оказываются под контролем многочисленных сигнальных соединений, спектр и специализация действия которых в ходе онтогенеза постоянно расширяются. Например, первоначальная дифференцировка клеток
прилегающей к сердцу эмбриона эндодермы в печень происходит под контролем секретируемых сердцем ФРФ-1 и ФРФ-2, а последующий рост - под контролем ФРФ-8, также секретируемого сердцем. После формирования эндокринных желез к пара/аутокринным регуляторам добавляются гормоны, оказывающие мощное системное действие по координации процессов развития разных физиологических систем, отдельных органов и тканей. Роль и механизмы действия гормонов в индивидуальном развитии будут проиллюстрированы на примерах влияния тиреоидных гормонов на метаморфоз головастиков и становление морфологии и функций мозга у млекопитающих.
Метаморфоз амфибий
Тиреоидные гормоны являются абсолютно необходимыми факторами для метаморфоза личиночных форм (головастиков) бесхвостых амфибий в формы взрослых особей. При редком врожденном отсутствии щитовидной железы, тиреоидэктомии, блокировке функций щитовидной железы тиреостатиком или при повышенной инактивации тиреоидных гормонов гиперэкспрессируемой йодтирониндейодиназой D3 метаморфоз не наступает, и головастики достигают гигантских размеров с морфологически зрелыми гонадами (хотя неотения у бесхвостых амфибий не описана). И наоборот, введение головастикам трийодтиронина индуцирует преждевременный метаморфоз. Метаморфоз обеспечивает животным возможность смены образа жизни, характера питания, способа передвижения, поведения и т.д. и затрагивает практически все физиологические системы и органы (рис. 1-13).
 Рис. 1-13. Некоторые морфофункциональные изменения при метаморфозе головастиков
Рис. 1-13. Некоторые морфофункциональные изменения при метаморфозе головастиков
Изменениям подвергаются морфофункциональная организация мозга, форма головы и, в частности, нижней челюсти, кишечник (сокращение длины на 75%, замена личиночного эпителия, появление соединительнотканного слоя, разрушение части радиальных мышечных волокон), кожа (замена личиночного эпидермиса, в котором делятся все клетки, эпидермисом взрослых, обновляемым за счет деления только базальных клеток), органы дыхания (резорбция жабр, развитие легких), конечности (развитие и рост конечностей, деградация хвоста). Наряду с морфологическими изменениями у животных при метаморфозе происходит глубокая перестройка обменных процессов. В качестве адаптации к смене способа дыхания происходит замена личиночных изоформ гемоглобина изоформами, характерными для взрослых животных. При этом разрушение эритроцитов с личиночным гемоглобином индуцируется тиреоидными гормонами, а их замена эритроцитами с гемоглобином взрослых стимулируется уже другим гормоном - гидрокортизоном. Бóльшая степень независимости взрослых животных от водной среды определяется, в частности, заменой в сыворотке крови личиночного альбумина (фетопротеина) альбумином взрослых, обеспечивающим более эффективную регуляцию электролитного и водного обмена. При метаморфозе в печени начинают экспрессироваться ферменты синтеза мочевины, и азотистый обмен меняется с аммониотелического на уротелический, что позволяет избежать избыточной аккумуляции электролитов в крови в условиях редукции жабр и отсутствия постоянного контакта с водой.
Перестройка органов и тканей происходит с активацией тиреоидными гормонами процессов программированной гибели клеток - апоптоза. Деградирующие клетки элиминируются фагоцитами, активность которых также стимулируется тиреоидными гормонами.
Физиологическими антагонистами тиреоидных гормонов являются ПРЛ и, возможно, ГР. Гиперэкспрессия ПРЛ у трансгенных головастиков стимулирует дальнейший рост животных и тормозит их метаморфоз. В результате образуются хвостатые лягушки. Одним из механизмов антиметаморфного действия ПРЛ является индукция экспрессии инактивирующей тиреоидные гормоны дейодиназы 3. В данной модели, а также в ряде других случаев, когда метаморфоз останавливается на стадии хвостатых лягушек, животные гибнут.
Сведения о молекулярных механизмах, лежащих в основе метаморфоза, остаются весьма фрагментарными. Принято считать, что тиреоидные гормоны непосредственно влияют на активность так
называемых ранних генов, к которым, в частности, относятся гены ряда транскрипционных факторов (например, белка, связывающего основной транскрипционный элемент, BTEB). Продукты этих ранних генов, в свою очередь, индуцируют экспрессию поздних генов, кодирующих, например, факторы пролиферации или апоптоза клеток (например, каспазу 9) или протеиназы, разрушающие внеклеточный матрикс, что способствует перестройке ткани.
Рецепторы тиреоидных гормонов. Эксперименты с трансгенезом головастиков доминантно негативными и доминантно позитивными вариантами рецепторов тиреоидных гормонов (Т-Рц) показали абсолютную зависимость метаморфного действия тиреоидных гормонов от функционирования Т-Рц, относящихся к классу ядерных рецепторов. Эти рецепторы экспрессируются в большинстве подвергаемых метаморфозу тканей, что обеспечивает так называемую тканевую автономность метаморфной реакции. Например, рост хрящей конечностей, мышц конечностей и их иннервация спинным мозгом под действием тиреоидных гормонов происходят независимо друг от друга. В то же время реакция некоторых типов клеток, например клеток медленных скелетных мышц и хорды хвоста, на тиреоидные гормоны, по-видимому, является непрямой и опосредуется находящимися рядом фибробластами. Даже деградация быстрых мышц хвоста, прямо контролируемая тиреоидными гормонами, усиливается теми же фибробластами, стимулированными тиреоидными гормонами. В опосредуемой другими клетками перестройке тканей могут принимать участие местно действующие паракринные регуляторы, такие, как PDGF или аналог морфогенного белка кости (BMP). Таким образом, автономность метаморфной реакции не является абсолютной.
Как и у других позвоночных, Т-Рц у амфибий представлены двумя изоформами, Т-Рца и Т-Рцβ. Т-Рца начинает экспрессироваться в большинстве тканей конститутивно еще до начала секреции тиреоидных гормонов у головастиков. Промоторная область гена Т-Рцβ содержит элементы, чувствительные к тиреоидным гормонам (ТЧЭ), и экспрессия гена прямо стимулируется возрастающим в ходе метаморфоза уровнем тиреоидных гормонов. Роль двух изоформ рецепторов в индукции метаморфоза разных органов различна. Формирование и рост конечностей, являющиеся одними из наиболее ранних проявлений метаморфного действия тиреоидных гормонов, происходят еще до индукции в них Т-Рцβ и, следовательно, реализуются через Т-Рца. В то же время резорбция хвоста начинается лишь после индукции
в нем значительного уровня Т-Рц β, при соотношении Т-Рцβ/Т-Рцα приблизительно 5:1, т.е. здесь действие тиреоидных гормонов опосредуется преимущественно Т-Рцβ. Аналогичная ситуация имеет место в гипофизе при формировании системы отрицательной обратной связи в конце метаморфоза. Вывод о преобладающей роли Т-Рц β в редукции хвоста подтверждается применением избирательного агониста Т-Рц β, GC-1, который воспроизводит действие T3 на хвост, а также жабры головастика, но не индуцирует рост конечностей.
Корегуляторы рецепторов. И в отсутствие, и в присутствии тиреоидных гормонов их рецепторы конститутивно связаны с ТЧЭ. Но в отсутствие гормонов Т-Рц оказывают репрессорное действие на транскрипцию компетентных генов, а в присутствии гормонов - стимулирующее. Противоположность данных эффектов отчасти связана с рекрутированием и активацией рецепторами гистондеацетилаз и гистонацетилтрансфераз, формирующих непермиссивную и пермиссивную среду для транскрипции соответственно. Для нормального метаморфоза необходимы оба типа взаимодействий. Например, ингибитор гистондеацетилаз тормозит метаморфоз как развивающихся de novo, так и подвергающихся регрессии органов. Рекрутирование осуществляется с участием корепрессоров и коактиваторов. Как правило, гормон индуцирует отделение от рецептора корепрессора и присоединение коактиватора. Это, однако, происходит не на всех гормонкомпетентных генах и зависит от типа корегулятора. Предполагается, что тканевая специфичность реакции на гормон может в значительной мере определяться типом экспрессируемых в данной ткани корегуляторов. Экспрессия некоторых из корегуляторов (например, коактиватора 3 рецепторов стероидов, SRC3 или белка 7, взаимодействующего с Т-Рц TRIP7) может контролироваться тиреоидными гормонами. Интересно, что клетки даже одного типа, но с разной локализацией, могут реагировать на гормон противоположным образом. Примером могут служить мышечные клетки формируемых конечностей и деградирующего хвоста. Сравнение профилей индуцируемых гормоном генов в этих клетках показало их полное несовпадение, что может служить косвенным подтверждением тканеспецифичной роли корегуляторов. Установлено, что для индукции метаморфоза важно как снятие ингибирующего действия корепрессоров, так и проявление стимулирующего действия коактиваторов.
Роль локального метаболизма. Упомянутые выше морфологические изменения тканей и органов в процессе метаморфоза происходят не одновременно, а упорядоченно во времени. Наиболее ранним резуль-
татом повышения секреции тиреоидных гормонов являются рост конечностей, а также пролиферация клеток, выстилающих желудочки мозга и полость спинного мозга. Затем происходит укорочение кишечника и резорбция жабр. И лишь в конце метаморфоза, на пике секреции тиреоидных гормонов, происходит резорбция хвоста (рис. 1-14). После завершения метаморфоза устанавливается система отрицательной обратной связи в секреции тиреоидных гормонов, и их уровень в крови снижается. Упорядоченность этих процессов во времени в значительной мере определяется особенностями локальной экспрессии йодтирониндейодиназ (изозима D2, превращающего малоактивный T4 в активный гормон T3, и изозима D3, инактивирующего оба гормона). Введение головастикам T4 ускоряет эти процессы, не влияя на их последовательность, а введение T3 индуцирует перечисленные изменения одновременно. Наибольшая чувствительность зачатков конечностей и клеток нервной системы к тиреоидным гормонам, по-видимому, связана с конститутивной экспрессией фермента D2, обеспечивающей уровень T3, достаточный для индукции роста. Последующая индукция экспрессии D2 в кишечнике служит фактором запуска программы укорочения кишечника. В жабрах фермент D2 практически не экспрессируется, и их резорбция в середине периода метаморфоза вызывается возрастающим уровнем T3 и T4 в крови. Хвост остается резистентным к тиреоидным гормонам на протяжении большей части периода метаморфоза по двум причинам: из-за высокой экспрессии изозима D3, которая падает лишь непосредственно перед резорбцией,
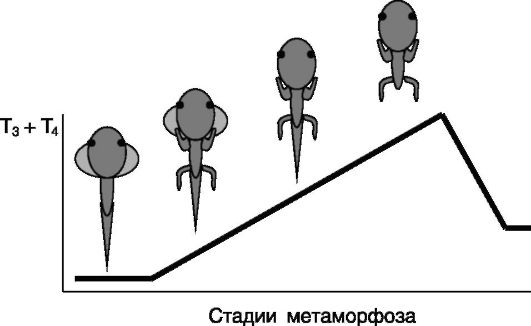 Рис. 1-14. Соответствие морфологических изменений при метаморфозе бесхвостых амфибий уровню тиреоидных гормонов
Рис. 1-14. Соответствие морфологических изменений при метаморфозе бесхвостых амфибий уровню тиреоидных гормонов
и из-за слабой экспрессии изозима D2, индуцируемой лишь высокими концентрациями тиреоидных гормонов в конце периода метаморфоза. Пиковые концентрации тиреоидных гормонов обусловливают также индукцию изозима D2 в тиреотрофах гипофиза, в результате чего образующийся локально T3 подавляет секрецию ТТГ и соответственно секреторную активность фолликулов щитовидной железы.
Тиреоидные гормоны
в развитии мозга млекопитающих
Контроль гормонами щитовидной железы развития ЦНС у млекопитающих затрагивает процессы пролиферации, миграции и дифференцировки нейрональных и глиальных клеток, в частности роста и миелинизации аксонов, ветвления дендритов, формирования межнейронных контактов (рис. 1-15).
Недостаточность йода и/или гормонов щитовидной железы в период формирования структуры ЦНС приводят к необратимым аномалиям развития у человека, проявляющимся в форме кретинизма. У человека «критический период» действия тиреоидных гормонов на ЦНС, по-видимому, включает эмбриональный и плодный периоды, причем в первой четверти беременности созревание головного мозга плода происходит под контролем тиреоидных гормонов матери, а после 12-й недели - щитовидной железы самого плода. У грызунов «критический период» приходится на последнюю треть беременности - первые 15 дней постнатальной жизни. При гипотиреозе экспрессия ряда зависимых от гормонов щитовидной железы генов происходит с отставанием во времени, хотя уровень экспрессии таких генов после завершения критического периода часто не отличается от нормального (рис. 1-16).
Механизмы данного гормонального импринтинга наиболее подробно исследовались на примере формирования мозжечка у грызунов, гипотиреоз у которых может быть вызван скармливанием беременным самкам ингибитора (6-пропил-2-тиоурацила) превращения тироксина в гормонально более активный трийодтиронин. Индуцированное таким образом недоразвитие мозжечка сопровождается нарушениями координации движений и пространственной ориентации. Исследование состава мРНК мозжечка с помощью микроматриц показало, что гипотиреоз вызывает изменения экспрессии
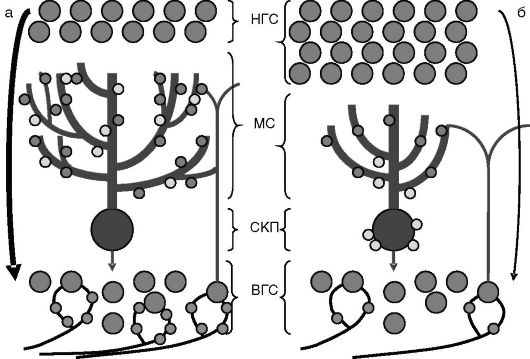 Рис. 1-15. Влияние тиреоидных гормонов в перинатальный период на морфогенез коры мозжечка:
Рис. 1-15. Влияние тиреоидных гормонов в перинатальный период на морфогенез коры мозжечка:
а - эутиреоз, б - гипотиреоз. В отсутствие тиреоидных гормонов подавлены рост и ветвление дендритов клеток Пуркинье, снижен синаптогенез между аксонами гранулярных клеток внутреннего слоя (ВГС) и клетками Пуркинье, замедлена пролиферация и миграция гранулярных клеток наружного слоя (НГС) во внутренний слой, замедлено исчезновение аксосоматических синапсов (светлые кружочки) между извитыми волокнами и клетками Пуркинье, снижено количество синаптических контактов между мшистыми волокнами и гранулярными клетками внутреннего слоя. МС - молекулярный слой; СКП - слой клеток Пуркинье
приблизительно 200 генов, белковые продукты которых участвуют в самых разнообразных процессах, происходящих в клетке, таких, как транскрипционные факторы, белки, регулирующие пролиферацию и апоптоз клеток, белки, участвующие в синаптогенезе и синаптической передаче, и т.д. Последовательность событий, индуцируемых тиреоидными гормонами, остается, однако, неясной.
Ряд зависимых от тиреоидных гормонов генов содержит соответствующие ТЧЭ, однако неизвестно, все ли они функционируют in vivo. Например, хотя in vitro тиреоидные гормоны способны ингибировать активность промотора гена Gai1 (субъединицы ai1 тримерного G- белка), in vivo тиреоидные гормоны, напротив, индуцируют экспрессию
гена Gai1, возможно, через повышение активности транскрипционного фактора C/EBPβ, взаимодействующего с промотором гена Gai1.
Противоречия также касаются типа Т-Рц (Т-Рца или Т-Рцβ), обеспечивающего гормональное действие на мозжечок в критический период. С одной стороны, появление чувствительности мозжечка к действию тиреоидных гормонов по времени совпадает с появлением в клетках Т-Рцβ. Более того, только трансфекция нейрональных клеток в культуре вектором Т-Рцβ, но не вектором Т-Рца обеспечивала индуцирующее действие тиреоидного гормона на BTEB, регулирующий, в частности, количество и длину нейритов. С другой стороны, клетки Пуркинье от мышей с нокаутом гена Т-Рцβ реагируют на тиреоидный гормон интенсивным ростом и ветвлением дендритов. Селективный агонист Т-Рцβ, GC-1, при введении гипотиреоидным животным лишь частично корректировал дифференцировку клеток Пуркинье и был неэффективен в отношении миграции гранулярных клеток мозжечка, но воспроизводил действие природного гормона на клетки коры мозга. Наиболее адекватно отражающей полученные к настоящему времени экспериментальные данные концепцией является предположение о ведущей роли Т-Рца, причем в отсутствие гормона данный рецептор оказывает репрессорное действие на развитие мозжечка, а гормон снимает этот эффект рецептора. Таким образом, тиреоидные гормоны преимущественно выступают в роли не индукторов, а пермиссивных факторов развития мозжечка.
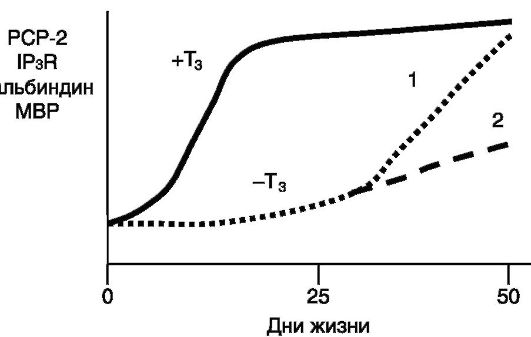 Рис. 1-16. Влияние тиреоидных гормонов на динамику экспрессии белков в мозжечке крысы:
Рис. 1-16. Влияние тиреоидных гормонов на динамику экспрессии белков в мозжечке крысы:
PCP-2 (белок 2 клеток Пуркинье) - фактор обмена нуклеотидов в Ga белках; IP3R - рецептор инозитол-1,4,5-трифосфата; MBP - основной белок миелина
Учитывая генерализованный характер воздействия тиреоидных гормонов, можно предположить, что этот эффект опосредован одним или несколькими транскрипционными факторами. Действительно, давно известна мутация гена сиротского ядерного рецептора RORa (линия staggerer - шатающихся мышей), сопровождающаяся фенотипом, близким к фенотипу врожденного гипотиреоза. Более того, установлено, что тиреоидные гормоны индуцируют экспрессию RORa, а сам RORa способен регулировать активность промоторов некоторых чувствительных к тиреоидным гормонам генов в клетках мозжечка. Аналогичную, но с противоположным знаком функцию, возможно, выполняет транскрипционный репрессор REST (RE1-silencing transcription factor, транскрипционный фактор, подавляющий нейрональные гены в ненейрональных клетках). Показано, что тиреоидные гормоны подавляют экспрессию корепрессора Rcor этого репрессора, что может вести к созданию пермиссивной среды для транскрипции необходимых для данного этапа развития генов. Допускается, что изменения в чувствительности клеточных структур мозжечка к действию тиреоидных гормонов в онтогенезе могут быть связаны с изменениями в экспрессии коактиваторов Т-Рц. Определенную роль во временнóй и пространственной организации действия тиреоидных гормонов на развитие мозжечка могут играть изменения в экспрессии изоформ дейодиназ, активирующих и инактивирующих тиреоидные гормоны. Например, выявлено исчезновение изоформы фермента D2 (активирующей) из клеток Пуркинье после созревания структур мозжечка у цыплят.
Объектами действия тиреоидных гормонов в ЦНС служат как нейроны, так и глиальные клетки. Функции астроцитов в морфогенетическом действии тиреоидных гормонов включают, в частности, превращение малоактивного тироксина в активный трийодтиронин (под действием дейодиназы 2), модификацию белков внеклеточного матрикса (повышение секреции ламинина и фибронектина), секрецию факторов роста (ЭФР, оФРФ), стимулирующих пролиферацию и миграцию нейронов.
Рекомендуемая литература
Bras M., Queenan В., Susin S.A. Programmed cell death via mitochondria: different modes of dying. Biochemistry (Mosc). 2005;70(2):231 - 239.
Brown D.D. The role of deiodinases in amphibian metamorphosis. Thyroid. 2005;15(8):815 - 821.
Fariss M.W., Chan C.B., Patel M. et al. Role of mitochondria in toxic oxidative stress. Mol Interv. 2005;5(2):94 - 111.
Hu Y., Benedict M.A., Ding L., Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999; 18(13):3586 - 3595.
Koibuchi N., Chin W.W. Thyroid hormone action and brain development. Trends Endocrinol Metab. 2000;11(4):123 - 128.
Le Roith D., Bondy C., Yakar S. et al. The somatomedin hypothesis: 2001. Endocr Rev. 2001;22(1):53 - 74.
Solomon A.M., Bouloux P.M. Modifying muscle mass - the endocrine perspective. J Endocrinol. 2006;191(2):349 - 360.
Veldhuis J.D., Roemmich J.N. et al. Endocrine control of body composition in infancy, childhood, and puberty. Endocr Rev. 2005;26(1):114 - 146.