Эндокринная регуляция. Биохимические и физиологические аспекты : учеб. пособие / А.Н. Смирнов ; под ред. В.А. Ткачука - 2009. - 368 с.
|
|
|
|
ГЛАВА 9. ОБМЕН УГЛЕВОДОВ
К углеводам относятся соединения с общей химической формулой [C(H2O)]n. Различают моносахариды (гептозы, гексозы, пентозы, тетрозы, триозы), дисахариды и полисахариды (рис. 9-1). В сбалансированной диете углеводы покрывают приблизительно 50% энергетических потребностей организма.
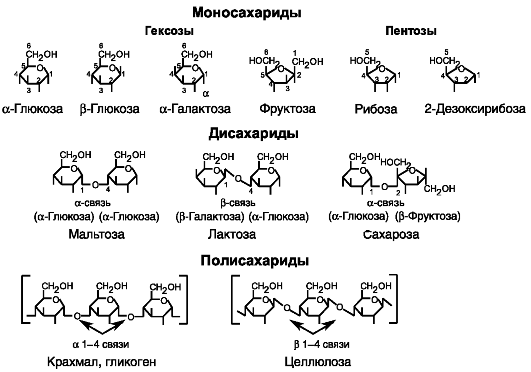 Рис. 9-1. Основные углеводы
Рис. 9-1. Основные углеводы
ПЕРЕВАРИВАНИЕ, ВСАСЫВАНИЕ
Поступающие с пищей полисахариды (растительный крахмал и животный гликоген) гидролизуются слюнной и панкреатической амилазами до дисахаридов мальтозы и изомальтозы, состоящих из
двух остатков глюкозы, соединенных 1,4 и 1,6 гликозидными связями соответственно. Основная часть мальтозы и изомальтозы расщепляется ферментом сахаразой-изомальтазой, заякоренной на плазматической мембране щеточной каемки слизистой кишечника. Этот фермент обеспечивает также гидролиз сахарозы. Часть мальтозы гидролизуется родственным ферментом, мальтазой-глюкоамилазой, который может также использовать в качестве субстратов амилозу и амилопектин. Другие дисахариды, поступающие с пищей, гидролизуются до моносахаридов с помощью ряда специфичных дисахаридаз. В клинической практике широкое применение нашли ингибиторы гидролиза дисахаридов, оказывающие антидиабетическое действие за счет замедления образования и соответственно всасывания моносахаридов.
Секреция панкреатической амилазы стимулируется приемом пищи. Парентеральное кормление ведет к атрофии поджелудочной железы. Стимулирующее действие пищи осуществляется с участием сенсорных и эфферентных нейронов блуждающего нерва и холецистокинина. Стимулирующее действие на секрецию амилазы оказывают также грелин, инсулин, дофамин, бомбезин, секретин, ВИП, вещество P, гастрин. Панкреатический полипептид, соматостатин, пептид, связанный с геном кальцитонина, напротив, подавляют секрецию панкреатической амилазы.
Моносахариды всасываются клетками кишечного эпителия и переносятся в кровоток с помощью транспортеров двух типов: зависимых от натрия (SGLT) котранспортеров Na+/сахар, осуществляющих энергозависимый транспорт, и независимых от натрия транспортеров группы GLUT, обеспечивающих облегченный перенос сахаров через плазматическую мембрану по градиенту концентрации. Известны 3 зависимых от Na+ транспортера (SGLT1-3), осуществляющих активный транспорт глюкозы, и не менее 14 независимых от Na+ транспортеров (GLUT), осуществляющих облегченную диффузию глюкозы.
Однотипно построенные SGLT включают 14 трансмембранных доменов (рис. 9-2). Функционируют как сахар/Na+-котранспортеры. SGLT1 отличается от SGLT2 и 3 тем, что функционирует как высокоаффинный транспортер с низкой емкостью; переносит и глюкозу, и галактозу, тогда как SGLT2 и 3 эффективно транспортируют только глюкозу, обладая низким сродством к субстрату, но высокой емкостью. SGLT экспрессируются в клетках, обеспечивающих поступление сахаров против концентрационного градиента, прежде всего на апикальной поверхности клеток слизистой тонкой кишки и прокси-
мальных канальцев почек. Обнаружены и в некоторых других типах клеток (кардиомиоциты, холангиоциты). В почках основная часть глюкозы реабсорбируется из мочи с помощью низкоаффинных SGLT 2 и 3 в проксимальных извитых канальцах, а остальная часть глюкозы извлекается из мочи за счет работы SGLT1 в нисходящем проксимальном канальце. В кишечнике SGLT1 и 2, по-видимому, могут совместно экспрессироваться в одной клетке. Система трансцеллюлярного транспорта сахаров в почках и кишечнике дополняется экспрессией на базолатеральной мембране клеток транспортеров группы GLUT (12 трансмембранных доменов; см. рис. 9-2), обеспечивающих выход сахара из клетки по градиенту концентрации. Транспортеры GLUT (5 и 7) могут также локализоваться на апикальной поверхности клеток кишечника, обеспечивая часть трансмембранного переноса
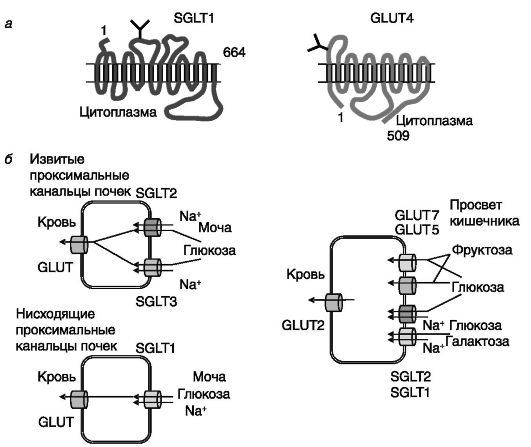 Рис. 9-2. Топология
Na-зависимого транспортера сахаров SGLT1 и Na- независимого,
регулируемого инсулином транспортера глюкозы GLUT4 (а) и локализация
транспортеров сахаров в клетках почек и кишечника (б)
Рис. 9-2. Топология
Na-зависимого транспортера сахаров SGLT1 и Na- независимого,
регулируемого инсулином транспортера глюкозы GLUT4 (а) и локализация
транспортеров сахаров в клетках почек и кишечника (б)
глюкозы и полностью - транспорт фруктозы. Интересно, что в условиях избытка углеводов в пище в апикальную мембрану встраивается также GLUT2, обычно локализующийся в базолатеральной мембране. Поступление глюкозы в клетки большинства органов также осуществляется с участием транспортеров GLUT. Экспрессия генов транспортеров сахаров и их встраивание в плазматическую мембрану находятся под множественным гормональным контролем.
МЕЖУТОЧНЫЙ ОБМЕН
Поступающая в клетки глюкоза быстро подвергается фосфорилированию в положении 6, и образующийся глюкозо-6-фосфат (Г-6-Ф) распределяется между следующими путями метаболизма: гликолизом (цепью анаэробных превращений с образованием триоз и извлечением небольшого количества энергии), пентозо-фосфатным, или фосфоглюконатным путем (цепью необратимых анаэробных превращений с образованием пентоз и - главное - восстановленного НАДФ) и запасанием в форме гликогена, гликогенеза (рис. 9-3). Соотношение
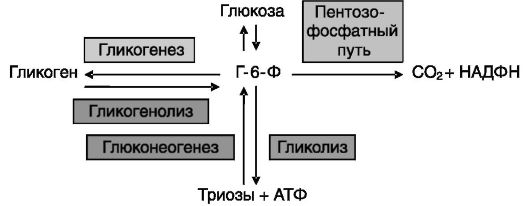 Рис. 9-3. Распределение глюкозы между путями обмена
Рис. 9-3. Распределение глюкозы между путями обмена
этих путей зависит от типа клетки, режима питания и энергозатрат и контролируется рядом гормонов. Гликолиз и гликогенез обратимы (соответствующие цепи реакций носят название глюконеогенез и гликогенолиз).
Ферменты гликолиза
Гликолиз представляет собой последовательную цепь превращений глюкозы до пирувата (или лактата) и предназначен для извлече-
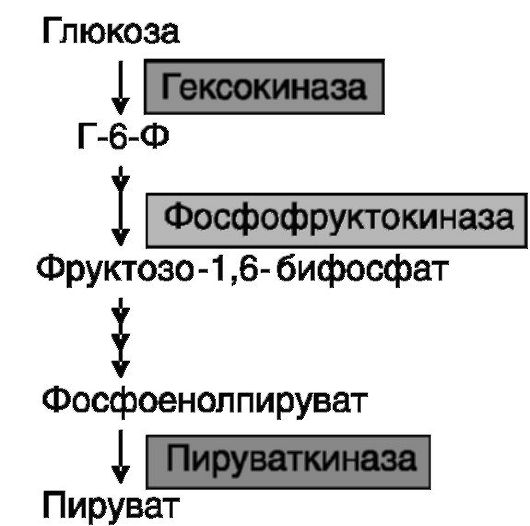 Рис. 9-4. Лимитирующие ферменты гликолиза
Рис. 9-4. Лимитирующие ферменты гликолиза
ния энергии и получения продуктов, используемых для процессов синтеза (липидов, аминокислот). Гликолиз протекает в растворимой фракции клетки и включает 9 (или 10) стадий, часть которых необратима и катализируется лимитирующими ферментами (рис. 9-4), активность которых контролируется гормонами и рядом продуктов обмена. Как правило, гипергликемические гормоны (катехоламины, глюкагон, глюкокортикоиды) подавляют экспрессию ферментов гликолиза, а инсулин - стимулирует. Гексокиназа. Фосфорилирование поступающей в клетки глюкозы с образованием Г-6-Ф катализируется гексокиназой. Известно 4 изозима (I-IV) фермента, различающихся по каталитическим свойствам, регуляции, внутриклеточной локализации и тканевой специфичности экспрессии. Фермент функционирует в форме мономера, но изозимы I-III представляют собой полипептиды, построенные из двух гомологичных половинок, соединенных по типу «голова-хвост», а изозим IV, называемый глюкокиназой , содержит лишь одну такую половинку. В «сдвоенных» изозимах I-III C-концевая половинка выполняет ка-
талитическую функцию, а N-концевая - регуляторную (ингибирование продуктом, регуляция неорганическим фосфатом; рис. 9-5).
Изозимы I и II включают гидрофобную N-концевую область, обеспечивающую заякоривание фермента на наружной мембране митохондрий в области пор, осуществляющих обмен АТФ/АДФ между
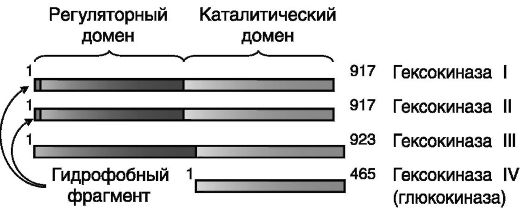 Рис. 9-5. Доменная организация гексокиназ
Рис. 9-5. Доменная организация гексокиназ
митохондриями и цитоплазмой. Полагают, что подобная локализация фермента обеспечивает сопряжение между скоростью фосфорилирования глюкозы и скоростью окислительного фосфорилирования в митохондриях. Изозимы гексокиназы могут экспрессироваться в виде нескольких сплайсинговых вариантов, управляемых разными промоторами. Регуляторная роль гексокиназы проявляется, например, при переходе мышцы из состояния покоя к работе. В покоящейся скелетной мышце лимитирующим фактором потребления глюкозы служит ее транспорт через плазматическую мембрану. При работе и гиперинсулинемии основным барьером становится фосфорилирование глюкозы, катализируемое в этой ткани преимущественно гексокиназой II. Жирная пища подавляет стимулирующее действие инсулина и тренировки на захват глюкозы, возможно, на уровне фосфорилирования.
Промоторы изозимов гексокиназы включают элементы, обеспечивающие регуляторное действие ряда гормональных факторов и уровня питательных веществ. Одним из транскрипционных факторов, опосредующим чувствительность генов гексокиназы к регуляторным воздействиям, служит белок 1, связывающий стеролчувствительный элемент (SREBP-1). Активация изоформ данного белка стимулируется, например, инсулином и подавляется гормонами, оказывающими гипергликемическое действие, с участием цАМФ (например, адреналином, глюкагоном), а также жирными кислотами. Дополнительным путем действия гормонов через цАМФ-зависимые механизмы на экспрессию гексокиназы является регуляция активности группы транскрипционных факторов, таких, как CREB и C/EBP, взаимодействующих цАМФ-чувствительным элементом (CRE) промоторов генов фермента (рис. 9-6).
Глюкокиназа. Глюкокиназа обеспечивает фосфорилирование поступающей в клетки глюкозы для последующего использования по
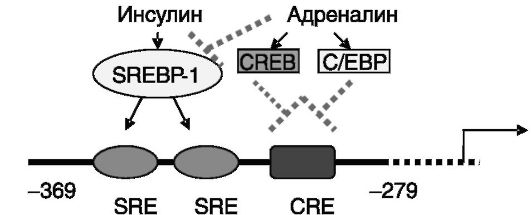 Рис. 9-6. Фрагмент промоторной области гена гексокиназы II человека: SRE - стеролчувствительный элемент
Рис. 9-6. Фрагмент промоторной области гена гексокиназы II человека: SRE - стеролчувствительный элемент
любому из метаболических путей. В отличие от гексокиназы Km глюкокиназы превышает концентрацию глюкозы в крови, и за счет этого фермент обеспечивает практически пропорциональное концентрации глюкозы фосфорилирование, что особенно важно для глюкостатной функции печени. Глюкокиназа экспрессируется в печени, β-клетках поджелудочной железы, нейроэндокринных клетках мозга, в кишечнике. Экспрессия фермента контролируется двумя промоторами: проксимальным в печени (управление инсулином) и дистальным в β-клетках (управление глюкозой). В печени уровень глюкокиназы контролируется режимом питания: при голодании активность фермента снижается, после приема пищи - возрастает. Адаптация активности фермента в значительной мере определяется сравнительно низкой стабильностью его мРНК (t1/2 = 45 мин). Посредниками этих эффектов, по меньшей мере отчасти, служат соответственно глюкагон и инсулин. Механизм стимулирующего действия инсулина включает индукцию экспрессии транскрипционного фактора SREBP-1c (sterol regulatory element-binding proteinic), взаимодействующего с двумя тандемно расположенными стеролчувствительными элементами проксимального промотора гена глюкокиназы. Параллельно инсулин через PI3K и ПК-B стимулирует взаимодействие с промотором транскрипционных факторов HIF-1 (hypoxia-inducible factor-1, индуцируемый гипоксией фактор 1) и HNF-4a (hepatocyte nuclear factor-4a, ядерный фактор гепатоцитов) с участием коактиватора p300 (рис. 9-7). Стимулирующее действие пищи на экспрессию глюкокиназы может также опосредоваться фруктозо-2,6- бифосфатом, действующим также через ПК-B.
Активность глюкокиназы регулируется также на посттрансляционном уровне. В β-клетках поджелудочной железы и печени выявлен белок (GKRP), регулирующий глюкокиназу путем ее обратимой инактивации и транспортировки в клеточное ядро. Взаимодействие двух белков ингибируется высоким уровнем глюкозы. Соответственно при углеводной нагрузке резервированная глюкокиназа высвобождается из комплекса, транспортируется в цитоплазму и обеспечивает усиленное фосфорилирование глюкозы (см. рис. 9-7). Мыши с нокаутированным геном белка, регулирующего глюкокиназу, значительно хуже, чем нормальные животные, справляются с нагрузкой глюкозой.
Фосфофруктокиназа (PFK1). Фосфофруктокиназа направляет обмен глюкозы на образование трехуглеродных фрагментов, что снижает доступность субстрата для биосинтеза гликогена и пенто-
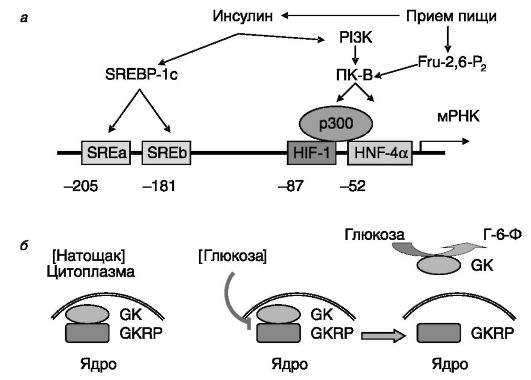 Рис. 9-7. Регуляция активности глюкокиназы (GK) на транскрипционном (а) и посттрансляционном (б) уровнях
Рис. 9-7. Регуляция активности глюкокиназы (GK) на транскрипционном (а) и посттрансляционном (б) уровнях
Инсулин стимулирует активность ряда транскрипционных факторов, взаимодействующих с проксимальным промотором гена глюкокиназы.
зо-фосфатного пути. Фермент функционирует в форме тетрамеров, образуемых из гомологичных полипептидов трех типов - мышечного (M), печеночного (L) и тромбоцитарного (P). В скелетной мышце преобладающая форма фермента представлена гомотетрамером M4, в печени - гомотетрамером L4, в эритроцитах выявлены все возможные сочетания М-и L-субъединиц. Регуляция активности фермента на уровне экспрессии генов субъединиц практически не исследована. Вместе с тем установлен механизм влияния углеводов и гормонов на активность фермента на посттрансляционном уровне. Оказалось, что активность фосфофруктокиназы повышается минорным продуктом обмена глюкозы, фруктозо-2,6-бифосфатом (Fru-2,6-P2), действующим как позитивный аллостерический регулятор. Это же соединение служит негативным алостерическим регулятором фруктозо- 1,6-бифосфатазы, участвующей в глюконеогенезе. Поэтому уровень Fru-2,6-P2 играет важную роль в направленности и интенсивности обмена углеводов в клетке и служит объектом действия ряда регуляторных механизмов.
Фруктозо-2,6-бифосфат образуется из фруктозо-6-фосфата и разрушается до фруктозо-6-фосфата бифункциональным ферментом, 6-фосфофрукто-2-киназой/фруктозо-2,6-бифосфатазой-2 - PFK2/ FBPase2 (рис. 9-8).
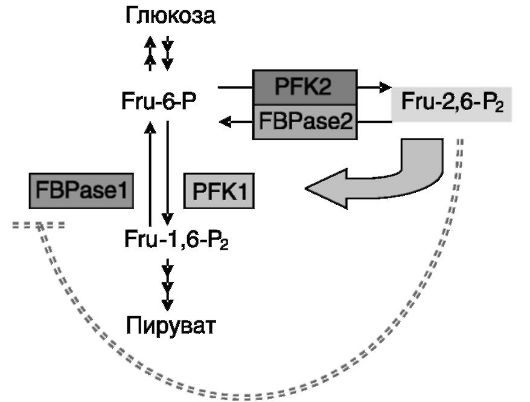 Рис. 9-8. Бифункциональный фермент PFK2/FBPase2 определяет уровень минорного метаболита Fru-2,6-P2, активирующего PFK1 и ингибирующего фруктозобифосфатазу (PBPase1)
Рис. 9-8. Бифункциональный фермент PFK2/FBPase2 определяет уровень минорного метаболита Fru-2,6-P2, активирующего PFK1 и ингибирующего фруктозобифосфатазу (PBPase1)
Полагают, что фермент возник на стадии одноклеточных эукариот в результате слияния генов двух ферментов - 6-фосфофрукто-2-киназы и фруктозо-2,6-бифосфатазы. У млекопитающих фермент функционирует в форме гомодимеров, формируемых из субъединиц четырех типов - изозимов печеночного, сердечного, мозгового (плацентного) и семенникового типов. Изозимы различаются по соотношению киназной и фосфатазной активностей и по наличию фосфорилируемых аминокислотных остатков в N-концевой (киназной) и C-концевой (фосфатазной) половинах субъединиц фермента (рис. 9-9).
Соответственно регуляция активности фермента, уровня Fru- 2,6-P2 и - в конечном итоге - характера обмена глюкозы является тканеспецифичной. Дополнительный уровень тканеспецифичности регуляции определяется использованием альтернативных промоторов при экспрессии генов изозимов. Так, ген изозима печеночного типа (PFKFB1) может давать изоформы белка L и M, различающиеся по N-концевым последовательностям, включая наличие (в L) или отсутствие (в M) одного из фосфорилируемых остатков Ser. В результате активность изоформы L фермента, экспрессируемой в печени,
регулируется ПК-A, а изоформы M, характерной для скелетной мышцы, - нет. Один из путей влияния пищевого режима и гормонов на уровень Fru-2,6-P2 в клетке заключается в регуляции экспрессии генов изозимов PFK2/FBPase2. Наиболее изученным в данном отношении является упоминавшийся ген PFKFB1. Активность специфичного для печени и жировой ткани промотора L данного гена усиливается глюкокортикоидами, глюкозой и митогенными факторами и ингибируется инсулином. Связывание ГК-Рц с глюкокортикоидчувствительной единицей (GRU) интрона 1 гена индуцирует связывание с ДНК другого транскрипционного фактора - ядерного фактора 3 гепатоцитов (HNF-3), действующего кооперативно с ГК-Рц. Инсулин (вероятно, с участием протеинкиназ JNK и SAPK) стимулирует фосфорилирование лигандсвязывающего домена ГК-Рц, подавляя тем самым транскрипционную активность ГК-Рц. Глюкоза увеличивает уровень вариантов F и L мРНК PFK2/FBPase2 в клетках печени через превращение в ксилулозо-5-фосфат (Xu-5-P) и активацию протеинфосфатазы, видимо, дефосфорилирующей и тем самым активирующей транскрипционный фактор ChREBP, способный взаимодейство-
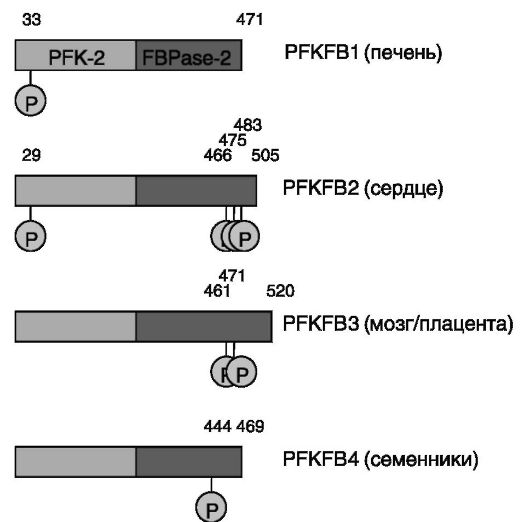 Рис. 9-9. Доменная организация и фосфорилируемые остатки изизимов PFK2/FBPase2 человека
Рис. 9-9. Доменная организация и фосфорилируемые остатки изизимов PFK2/FBPase2 человека
вать с глюкозочувствительным элементом гена. Фосфорилирование (и инактивация) этого фактора происходят под действием ПК-A, активируемой глюкагоном при голодании.
Фосфорилирование печеночного изозима PFK2/FBPase2 по Ser32 ПК-A ведет к ингибированию киназной активности фермента и стимуляции фосфатазной активности фермента. Предполагается, что фосфорилирование домена PFK2 снимает его тормозное действие на домен FBPase2 (рис. 9-10).
В случае сердечного изозима PFK2/FBPase2 фосфорилирование его фосфатазного домена (Ser466 и Ser483 в ферменте быка) ПК-A, активируемой АМФ протеинкиназой (AMPK) или стимулируемыми инсулином или ИФР-I протеинкиназами (ПК-B) ведет к активации PFK2, но не влияет на активность домена FBPase2 (рис. 9-11, а). Предполагается, что фосфорилирование снимает ингибирующее влияние фосфатазного домена на киназный домен.
Повсеместно экспрессируемый ген PFKFB3 стимулируется прогестинами и инсулином. Специфичный для семенников ген PFKFB4, экспрессируемый также в ряде опухолевых клеток, стимулируется гипоксией через HIF1a, взаимодействующий с чувствительным к гипоксии элементом (-422/-429 п.н.) промотора гена (рис. 9-11, б). Индукция гипоксией характерна и для других генов PFKFB, что отражает адаптивную роль фермента при недостатке кислорода.
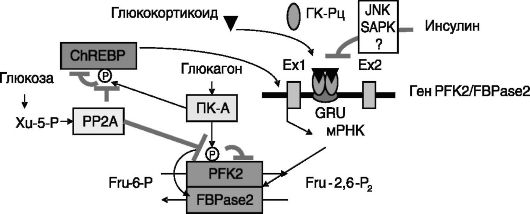 Рис. 9-10. Регуляция уровня Fru-2,6-P2 в гепатоцитах на уровне экспрессии гена и дифференциальной активности PFK2/FBPase2:
Рис. 9-10. Регуляция уровня Fru-2,6-P2 в гепатоцитах на уровне экспрессии гена и дифференциальной активности PFK2/FBPase2:
Ex1(2) - экзон 1(2); P - фосфат; PP2A - протеинфосфатаза 2A; ChREBP - белок, связывающий углеводчувствительный элемент; стрелки - стимуляция; тупые стрелки - ингибирование
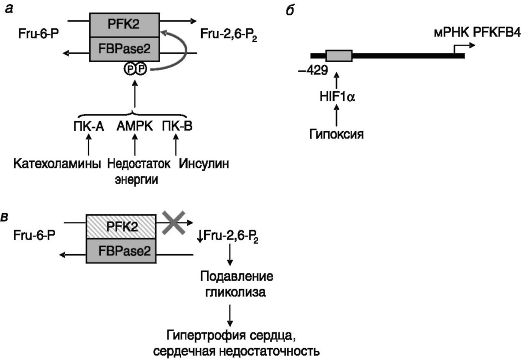 Рис. 9-11. Регуляция и значение изоформ 6-фосфофрукто-2-киназы/фрукто- зо-2,6-бифосфатазы-2:
Рис. 9-11. Регуляция и значение изоформ 6-фосфофрукто-2-киназы/фрукто- зо-2,6-бифосфатазы-2:
а - регуляция активности сердечной изоформы фосфорилированием; б - регуляция экспрессии семенниковой изоформы гипоксией; в - блокада активности сердечной изоформы 6-фосфофрукто-2-киназы ведет к сердечной недостаточности из-за подавления гликолиза
Функциональная значимость поддержания уровня Fru-2,6-P2 для нормальной работы клеток сердца показана с помощью гиперэкспрессии в кардиомиоцитах мутантной PFK2/FBPase2, лишенной киназной активности. В результате в сердце снижался уровень Fru-2,6-P2, снижалось содержание Fru-1,6-P2, но возрастал уровень его предшественников и увеличивалось отложение гликогена. Развивались гипертрофия сердца и сердечная недостаточность. Чувствительность метаболизма к стимулирующему действию инсулина значительно снижалась (рис. 9-11, в).
Пируваткиназа. Пируваткиназа (PK) функционирует в форме гомотетрамеров, субъединицы которых представлены 4 изозимами: L (печеночного типа), R (эритроцитарного типа), M1 и M2 (мышечного типа). Изозимы L и R возникают в результате альтернативного сплайсинга продукта транскрипции одного гена, изозимы M1 и
M2 - другого гена. Установлена регуляция активности фермента L-типа (LPK) глюкозой, инсулином (стимуляция), жирными кислотами и глюкагоном (ингибирование) на транскрипционном уровне. Промотор гена LPK включает углеводчувствительный элемент (ChoRE), обеспечивающий адаптацию активности фермента к условиям питания (рис. 9-12).
Транскрипционная активность связывающего ChoRE белка, ChREBP, действующего в форме гетеродимера с белком Mlx, значительно потенцируется ядерным фактором 4 гепатоцитов, HNF4 , сайт связывания которого на ДНК примыкает к ChoRE. Активность ChREBP регулируется его фосфорилированием/дефосфорилирова- нием, влияющим на транслокацию белка из цитоплазмы в ядро и сродство к ДНК (см. рис. 9-12). Ингибирующее действие глюкагона на экспрессию LPK включает фосфорилирование ChREBP ПК-A в областях, соседствующих с сигналом ядерной локализации и ДНКсвязывающим доменом, что соответственно блокирует транспорт ChREBP в клеточное ядро и взаимодействие с ДНК. Жирные кислоты ингибируют ДНК-связывающую активность ChREBP посредством активации AMPK. Предполагается, что на АТФ-зависимое образование тиоэфиров избытка жирных кислот с коэнзимом A (стадия активации жирных кислот) расходуется значительная часть АТФ клетки, и накапливающийся в результате АМФ активирует AMPK. Не исключено также, что жирные кислоты или их тиоэфиры, будучи лигандами
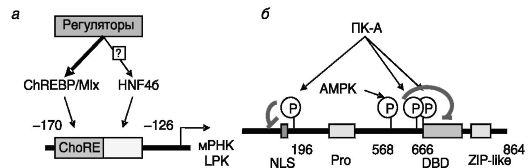 Рис. 9-12. Фрагмент
регуляторной области гена пируваткиназы печеночного типа (LPK),
включающий сайты связывания транскрипционных факторов ChREBP и HNF4α
(а); локализация сайтов фосфорилирования в молекуле ChREBP (б):
Рис. 9-12. Фрагмент
регуляторной области гена пируваткиназы печеночного типа (LPK),
включающий сайты связывания транскрипционных факторов ChREBP и HNF4α
(а); локализация сайтов фосфорилирования в молекуле ChREBP (б):
NLS - сигнал ядерной локализации; DBD-ДНК-связывающий домен; ZIP - лейциновая «застежка-молния»; AMPK - АМФ-зависимая протеинкиназа
HNF4a, могут влиять на способность данного фактора потенцировать действие ChREBP (см. рис. 9-12).
Стимулирующее действие глюкозы на экспрессию LPK включает образование минорного метаболита глюкозы (Xu-5-P), являющегося аллостерическим активатором протеинфосфатаз семейства 2A, которые за счет дефосфорилирования способствуют активации ChREBP. Механизм стимулирующего влияния инсулина на экспрессию LPK остается неясным. Возможно, данный эффект является следствием увеличения образования Xu-5-P из-за стимуляции инсулином пентозофосфатного пути. Не исключено также, что эффект инсулина включает инактивирующее фосфорилирование AMPK или стимуляцию экспрессии ChREBP (рис. 9-13).
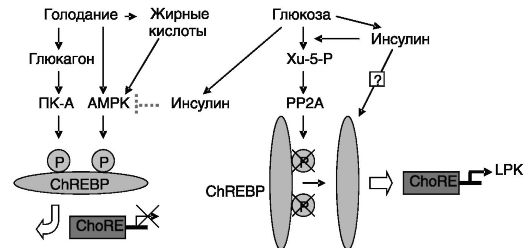 Рис. 9-13. Механизмы
ингибирования (левая часть рисунка) и стимуляции (правая часть рисунка)
экспрессии пируваткиназы печеночного типа (LPK)
Рис. 9-13. Механизмы
ингибирования (левая часть рисунка) и стимуляции (правая часть рисунка)
экспрессии пируваткиназы печеночного типа (LPK)
Пируватдегидрогеназа. Пируватдегидрогеназа (РДН) - гетеротетрамер, построенный из двух α- и двух β-субъединиц, входит в состав мультимерного пируватдегидрогеназного комплекса (PDC), локализованного в матриксе митохондрий. Фермент катализирует необратимую реакцию превращения пирувата в ацетил-КоА и тем самым блокирует возможность использования пирувата для глюконеогенеза. Активность PDH ингибируется фосфорилированием a-субъединицы по трем сайтам под действием киназ пируватдегидрогеназы (PDKs, всего 4 изозима, различающихся тканевой специфичностью экспрессии и кинетикой фосфорилирования трех сайтов PDH). PDK2 и PDK4
индуцируются голоданием и при диабете. Сохраняемый за счет этого глюконеогенез в первом случае обеспечивает эугликемию, а во втором - усугубляет гипергликемию.
В клетках печени экспрессия PDK4 стимулируется лигандами PPARa, включая жирные кислоты, и глюкокортикоидами, а инсулин блокирует эти эффекты. Инсулин снижает также экспрессию PDK2. Таким образом, при голодании и диабете причиной снижения активности PDH может служить повышение экспрессии PDKs из-за сниженного уровня инсулина и повышенного уровня свободных жирных кислот и глюкокортикоидов (рис. 9-14).
Предполагается, ГК-Рц, связываясь с ГКЧЭ промотора гена PDK4, кооперативно с транскрипционными факторами группы FOXO, взаимодействующими с расположенными рядом инсулинчувствительными последовательностями (ИЧП), рекрутирует коактиваторный комплекс p300/CBP, который обеспечивает формирование инициа-
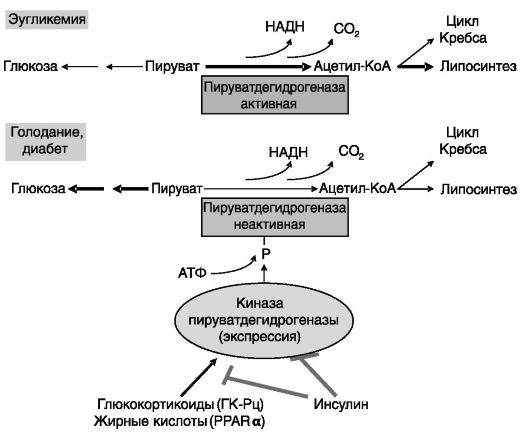 Рис. 9-14. Регуляция активности РDН фосфорилированием
Рис. 9-14. Регуляция активности РDН фосфорилированием
тивного комплекса транскрипции (рис. 9-15, а). Инсулин блокирует действие глюкокортикоидов, индуцируя инактивирующее фосфорилирование факторов FOXO. Последовательность событий при действии инсулина: инсулин → активация PI3K → аккумуляция PI-3,4-P2 → → активация ПК-B → фосфорилирование FOXO → снижение активности FOXO (выход из ядра в цитоплазму, снижение связывания с ДНК и коактиваторами) → утрата кооперативности в действии ГК-Рц и FOXO на транскрипцию (рис. 9-15, б).
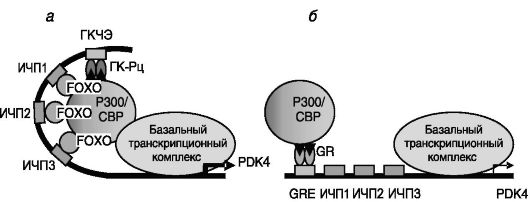 Рис. 9-15. Модель гормональной регуляции экспрессии гена PDK4 человека: а - в присутствии глюкокортикоидов; б - в присутствии глюкокортикоидов и инсулина
Рис. 9-15. Модель гормональной регуляции экспрессии гена PDK4 человека: а - в присутствии глюкокортикоидов; б - в присутствии глюкокортикоидов и инсулина
Множественность сайтов фосфорилирования PDH киназами играет определенную роль в тканевой специфичности регуляции активности фермента, поскольку зависимое от типа изозима PDK фосфорилирование по сайтам 2 и 3 снижает доступность фермента для митохондриальной фосфатазы 2c пируватдегидрогеназы (PDP2c), что имеет особо важное значение при голодании и диабете, при которых происходит фосфорилирование PDH по этим сайтам. Для фосфатазы PDP1c это не является препятствием.
Ферменты глюконеогенеза
Под термином глюконеогенез понимают цепь реакций превращения триоз в глюкозу (рис. 9-16). Нередко этим термином обозначают также образование глюкозы из аминокислот в результате дезаминирования последних.
Лимитирующие глюконеогенез ферменты, катализирующие необратимые реакции на пути пируват - глюкоза, кроме клеток-постав-
 Рис. 9-16. Лимитирующие глюконеогенез ферменты
Рис. 9-16. Лимитирующие глюконеогенез ферменты
щиков глюкозы (гепатоциты, клетки почек, энтероциты) экспрессируются во многих других клетках и выполняют в них иные функции. Соответственно спектры регуляторов активности этих ферментов, механизмы и даже направленность действия регуляторов в разных типах клеток могут различаться. В типичном случае (в печени) ферменты глюконеогенеза регулируются на транскрипционном уровне и индуцируются катехоламинами (работа, стресс), глюкагоном (голодание), глюкокортикоидами (стресс) и ингибируются инсулином (прием пищи). Гормональная регуляция корректируется уровнем энергообеспеченности клетки, доступностью субстратов и рядом других факторов. Соответствующие сигналы могут восприниматься специальными сенсорами клетки, прямо или опосредованно замыкающимися на промоторах генов рассматриваемой группы ферментов.
Пируваткарбоксилаза. Пируваткарбоксилаза катализирует образование оксалоацетата из пирувата и CO2. Функционирует в форме гомотетрамера, локализованного в матриксе митохондрий. Во всех клетках оксалоацетат является элементом цикла трикарбоновых кислот, обеспечивающим энергетические потребности клетки. В клетках, участвующих в глюконеогенезе (клетки печени, почек), оксалоацетат служит субстратом для образования фосфоенолпирувата и далее - глюкозы. В жировой ткани и печени оксалоацетат служит переносчиком ацетил-CoA из митохондрий в цитоплазму в форме цитрата для последующего биосинтеза жирных кислот, а также является предшественником малата - важного поставщика НАДФН, необходимого
для липосинтеза. В нервных клетках оксалоацетат служит биосинтетическим предшественником нейромедиаторов глутамата и ГАМК. В β-клетках поджелудочной железы оксалоацетат неустановленным образом способствует секреции инсулина. Таким образом, функции пируваткарбоксилазы весьма разнообразны и тканеспецифичны. Как и ряд других ферментов глюконеогенеза, пируваткарбоксилаза индуцируется глюкокортикоидами, глюкагоном, катехоламинами и ингибируется инсулином (рис. 9-17, а). По меньшей мере отчасти эти эффекты реализуются на транскрипционном уровне, однако механизмы гормонального действия изучены слабо. Показано, что в органах глюконеогенеза и липосинтеза активен проксимальный промотор гена фермента, а в β-клетках поджелудочной железы - дистальный. Образующиеся мРНК различаются по 5'-нетранслируемой области, что может сказываться на эффективности последующей трансляции. Проксимальный промотор в клетках бурого жира участвует в холодовой адаптации мышей: его активность стимулируется взаимодействи-
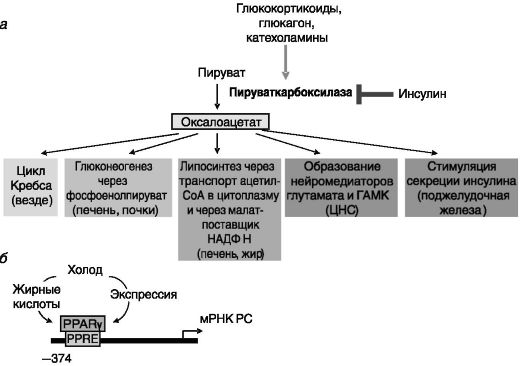 Рис. 9-17. Пируваткарбоксилаза (PC):
Рис. 9-17. Пируваткарбоксилаза (PC):
а - тканеспецифичные функции продукта PC оксалоацетата; б - регуляция экспрессии PC в буром жире при холодовой адаптации
ем с рецептором γ2 активаторов пролиферации пероксисом (PPARγ2), экспрессия которого индуцируется холодом, а активность стимулируется связыванием жирных кислот (рис. 9-17, б).
Фосфоенолпируваткарбоксикиназа (PEPCK). PEPCK - один из лимитирующих ферментов глюконеогенеза и глицеронеогенеза. Кодируется двумя генами. Митохондриальный фермент экспрессируется конституитивно. Цитозольный изозим, о котором пойдет речь ниже, регулируется гормонами. В печени активация экспрессии гена PEPCK под действием гипергликемических факторов (катехоламины, глюкагон, глюкокортикоиды) и торможение под действием инсулина происходит через проксимальную регуляторную последовательность, называемую GRU, в которую входят сайты связывания ГК-Рц, прямой повтор DR1 (gAF1/PCK1), служащий сайтом связывания ряда ядерных рецепторов (HNF4, транскрипционных факторов дальнего промотора овальбумина кур - COUP-TFs, PPARs, рецепторов ретиноевой кислоты - RARs), действующих самостоятельно или в виде гетеродимеров с рецепторами ретиноидов X (RXRs), инсулинчувствительный элемент (рис. 9-18).
В адипоцитах последние два фермента глюконеогенеза (фрукто- зо-1,6-дифосфатаза и глюкозо-6-фосфатаза) не экспрессируются, и
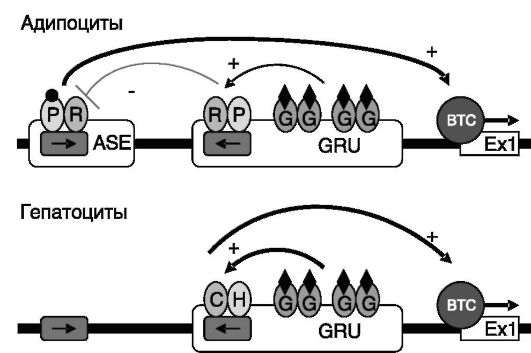 Рис. 9-18. Предполагаемый механизм тканеспецифичности действия глюкокортикоидов на транскрипцию PEPCK:
Рис. 9-18. Предполагаемый механизм тканеспецифичности действия глюкокортикоидов на транскрипцию PEPCK:
P - PPARγ; R - RXR; G - ГК-Рц; C - COUP-TF; H - HNF4; ASE - специфичный для адипоцитов энхансер; BTC - базальный транскрипционный комплекс; Ex1 - экзон 1 PEPCK. Стрелками указаны направления прямых повторов PPRE
PEPCK служит стимулятором глицеронеогенеза через 3-глицеро- фосфат. Этот путь ограничивает выход свободных жирных кислот из адипоцитов и развитие кетоацидоза. Глюкокортикоиды при стрессе снимают это ограничение отчасти за счет торможения экспрессии PEPCK. Таким образом, на один и тот же ген в печени и адипоцитах глюкокортикоиды влияют противоположным образом, но при этом достигается общая конечная цель: повышение в крови энергетических продуктов - глюкозы и жирных кислот.
В адипоцитах экспрессия гена PEPCK, по-видимому, находится под контролем жирных кислот, которые служат агонистами PPARγ стимулирующего экспрессию через дистальный PPAR-чувствитель- ный элемент, PPRE (PCK2), который входит в состав специфичного для адипоцитов энхансера ASE (рис. 9-18). В результате повышение уровня свободных жирных кислот в адипоцитах ведет к аккумуляции связывающего их 3-глицерофосфата, и уровень свободных жирных кислот снижается при росте жировых запасов.
Экспрессия PPARγ (точнее - его сплайсингового варианта PPARγ2) в адипоцитах коррелирует с запасами жира и синергично стимулируется глюкокортикоидами и инсулином. Действие глюкокортикоидов на экспрессию PPARγ2 включает индукцию экспрессии белка δ, связывающего CCAAT/энхансер (C/EBPδ). C/EBPδ, в свою очередь, узнает тандем сайтов в промоторе PPARγ2 и стимулирует активность гена. В печени глюкокортикоиды и жирные кислоты индуцируют экспрессию изоформы α PPAR.
Фруктозо-1,6-бифосфатаза. Выявлено 2 изозима, кодируемых разными генами и специфичных для печени и мышцы. Фермент функционирует в форме гомотетрамеров. Синергично ингибируется АМФ (сигнализирующем о недостаточном энергообеспечении клетки) и фруктозо-2,6-бифосфатом (отражающим уровень глюкозы), причем изозим мышечного типа более чувствителен к ингибированию. Функции мышечного и печеночного изозимов различны: обеспечение синтеза гликогена и секреции глюкозы соответственно. Гормональная регуляция активности фермента изучена слабо. Установлено, что факторы, стимулирующие аккумуляцию цАМФ (глюкагон, катехоламины), повышают экспрессию гена фруктозо-1,6-бифосфатазы за счет наличия в промоторной области цАМФ-чувствительных элементов. Небольшой стимулирующий эффект на экспрессию оказывают также глюкокортикоиды, тогда как инсулин оказывает ингибирующее действие. Повышение активности фермента может также происходить
под действием цАМФ за счет фосфорилирования фермента ПК-А, что ведет к снижению чувствительности к ингибирующему действию АМФ (рис. 9-19). Избыток глюкозы (после фосфорилирования гексокиназой) ускоряет деградацию фермента.
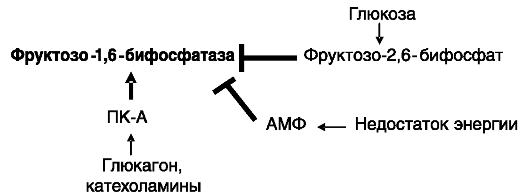 Рис. 9-19. Оперативная регуляция активности фруктозо-1,6-бифосфатазы
Рис. 9-19. Оперативная регуляция активности фруктозо-1,6-бифосфатазы
Глюкозо-6-фосфатаза. Глюкозо-6-фосфатаза является последним лимитирующим ферментом в цепи глюконеогенеза и обеспечивает поступление глюкозы в кровоток из клеток печени, почек, кишечника для поддержания эуглигемии.
В поджелудочной железе и мозгу выявлены белки, сходные с глюкозо-6-фосфатазой. Фермент представляет собой полипептид с 9 трансмембранными доменами, встроенный в мембрану эндоплазматического ретикулума, работающий в тесной кооперации с транспортерами субстрата (Г-6-Ф) и продуктов (глюкоза и неорганический фосфат) через мембрану эндоплазматического ретикулума. Мутации гена фермента являются одной из причин болезни Гирке - формы гликогеноза, для которой характерны гипогликемия и гепатомегалия вследствие избыточной аккумуляции гликогена. Сходные симптомы наблюдаются при мутациях гена транспортера Г-6-Ф, G6PT.
Стратегическое положение глюкозо-6-фосфатазы в глюконеогенезе является причиной того, что активность данного фермента служит объектом регуляции со стороны гормональных факторов, контролирующих обменные процессы. Регуляция осуществляется преимущественно на уровне транскрипции гена глюкозо-6-фосфатазы благодаря наличию в промоторной области гена ряда регуляторных элементов, чувствительных к действию гормонов. К таким элементам относятся ГКЧЭ, CREs и ИЧП, причем каждый из указанных элементов встречается в промоторе 2 раза и более. Эти элементы могут находиться рядом или частично перекрываться и соседствуют со вспомогательными
элементами, усиливающими (или ослабляющими) действие гормончувствительных элементов благодаря взаимодействию со вспомогательными транскрипционными факторами. Элементы CRE и ИЧП способны взаимодействовать с несколькими транскрипционными факторами (например, CREB и C/EBP; HNF3 и FKHR/Foxo соответственно), что обеспечивает возможность проведения гормональных сигналов несколькими путями. Инсулин, как гипогликемический фактор, ингибирует экспрессию гена глюкозо-6-фосфатазы, а глюкокортикоиды и гипергликемические гормоны, действующие через цАМФ, стимулируют экспрессию и препятствуют действию инсулина. Наиболее изученные пути действия гормонов представлены на рис. 9-20.
Помимо гормонов на экспрессию глюкозо-6-фосфатазы могут влиять многие другие факторы. Недостаточность энергообеспечения клеток при голодании приводит к аккумуляции АМФ, который является стимулятором AMPK. Одним из субстратов AMPK служит адаптор-
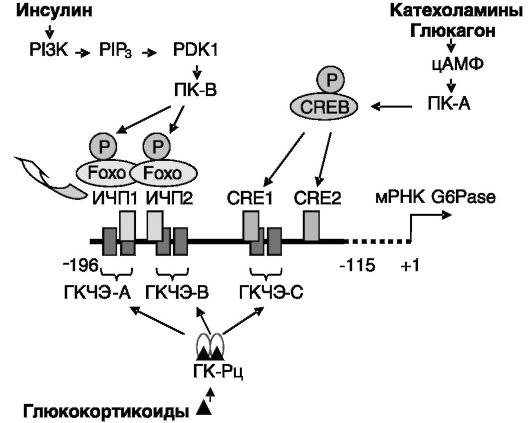 Рис. 9-20. Пути гормональной регуляции экспрессии гена глюкозо-6-фосфа- тазы (G6Pase):
Рис. 9-20. Пути гормональной регуляции экспрессии гена глюкозо-6-фосфа- тазы (G6Pase):
PI3K - фосфатидилинозитид-3-киназа; PIP3 - фосфатидилинозитид-3,4,5- трифосфат; PDK1 - PIP3-зависимая киназа 1; Foxo - транскрипционный фактор, содержащий вилкоголовый блок O (Forkhead box O); CREB - белок, связывающий CRE; P - фосфат; числами отмечены положения нуклеотидов относительно старта транскрипции
ный белок, субстрат 1 рецептора инсулина (IRS-1), обеспечивающий активирующее действие инсулина на PI3K (см. рис. 9-20). В результате при голодании возрастает эффективность проведения сигнала инсулина, в том числе - ингибирования экспрессии глюкозо-6-фосфа- тазы. Другим типом субстратов AMPK являются транскрипционные факторы группы Foxo, фосфорилирование которых приводит к их инактивации (см. рис. 9-20). Гормон адипонектин, секреция которого адипоцитами увеличивается при голодании, вызывает активацию AMPK в печени, обеспечивая тем самым одну из форм связи липидного и углеводного обмена. Другой тип связи выявлен при ингибирующем действии желчных кислот на экспрессию глюкозо-6-фосфатазы. Желчные кислоты через свой сенсор, рецептор X фарнезоидов (FXR), увеличивают экспрессию малого партнера гетеродимеризации (SHP), который конкурентно ингибирует взаимодействие Foxo с коактиватором, белком, связывающим CREB (CBP), в результате чего блокируется трансактивационная эффективность Foxo.
Кроме прямого действия через ПК-A/CREB глюкагон и ряд других гипергликемических гормонов могут стимулировать экспрессию глю- козо-6-фосфатазы и другими путями. Например, аккумуляция цАМФ индуцирует экспрессию коактиватора 1 PPAR γ, PGC-1, который за счет взаимодействия с транскрипционными факторами (например, ГК-Рц, HNF-4, Foxo) усиливает их транскрипционную активность. Инсулин, напротив, подавляет экспрессию PGC-1. Некоторые из перечисленных путей регуляции экспрессии глюкозо-6-фосфатазы представлены на следующем рис. 9-21.
Интересна тканевая специфичность регуляции экспрессии глюко- зо-6-фосфатазы. Фермент быстро индуцируется голоданием в печени и медленно - в кишечнике. Это, видимо, отражает изменения формы участия кишечника в глюконеогенезе при голодании: в начальный период кишечник использует глутамин и отчасти глицерин для образования аланина и лактата, направляемых в печень для синтеза глюкозы. После более длительного голодания (48-72 часа) кишечник начинает сам секретировать глюкозу благодаря индукции глюкозо-6- фосфатазы.
Пентозофосфатный путь
Пентозофосфатный путь (рис. 9-22) служит одним из источников НАДФН, используемого для липосинтеза, а также обеспечивает синтез пентоз, входящих в состав нуклеиновых кислот. Активен в
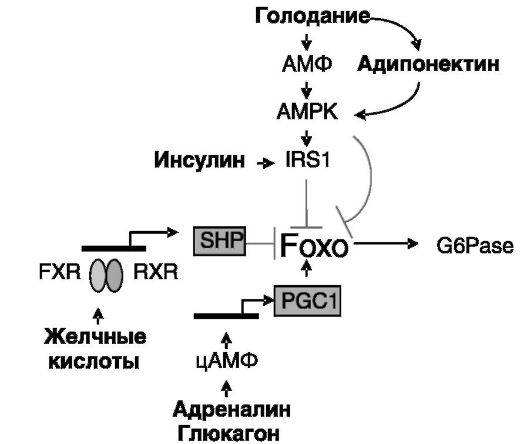 Рис. 9-21. Дополнительные пути регуляции экспрессии глюкозо-6-фосфатазы
Рис. 9-21. Дополнительные пути регуляции экспрессии глюкозо-6-фосфатазы
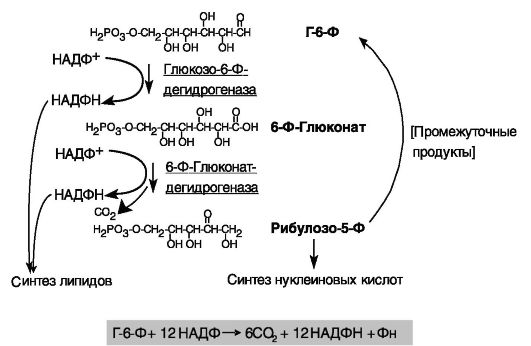 Рис. 9-22. Пентозофосфатный путь
Рис. 9-22. Пентозофосфатный путь
тканях с интенсивным биосинтезом липидов (печень, жировая ткань, гонады). На этом пути образуется также минорный метаболит Xu-5-P,
служащий важным регулятором ферментов липосинтеза и гликолиза. Ключевыми ферментами данного пути служат глюкозо-6-фосфат- дегидрогеназа (G6PDH) и 6-фосфоглюконатдегидрогеназа (6PGDH). Активность данных ферментов адаптивно подавляется голоданием (и, вероятно, глюкагоном) и индуцируется приемом пищи, а также инсулином, эффект которого потенцируется глюкокортикоидами. Регуляция происходит преимущественно на транскрипционном уровне. Действие инсулина включает активацию (через PI3K) белка 1c, связывающего стеролчувствительный элемент (SREBP1c), в регуляторных областях (G6PDH и 6PGDH). SREBP служит посредником действия инсулина на экспрессию многих ферментов липосинтеза.
Гликогенез и гликогенолиз
Гликоген представляет собой быстро мобилизуемую резервированную форму глюкозы. Гликоген печени, составляющий порядка 5% массы органа, что у человека эквивалентно 70-90 г глюкозы, обеспечивает поддержание эугликемии в промежутках между приемами пищи (до 12-24 ч). Функция гликогена скелетной мышцы (порядка 1% по массе) иная - обеспечение работоспособности самой мышцы даже в анаэробных условиях. Непосредственным субстратом для синтеза гликогена служит УДФ-глюкоза, образующаяся из глюкозо- 6-фосфата (через глюкозо-1-фосфат) и УТФ, т.е. гликогенез идет с затратой энергии макроэрга. В соответствии со структурой гликогена (линейные цепи из остатков глюкозы, соединенных 1-4 связями, ветвящиеся с помощью 1-6 связей; рис. 9-23) синтез и распад гликогена катализируется парами ферментов: гликогенсинтаза + ветвящий фермент и гликогенфосфорилаза + 6-глюканогидролаза.
Гликогенсинтаза. Различают 2 изозима гликогенсинтазы - мышечного и печеночного типа. Активность обеих изоформ ингибируется фосфорилированием (в частности, активируемой глюкагоном и катехоламинами ПК-А) и стимулируется аллостерически Г-6-Ф. Считается, что в активации печеночного изозима более важную роль играет уровень глюкозы (через Г-6-Ф), а мышечного изозима - инсулин (подавляющий фосфорилирование фермента; рис. 9-24). Важную роль в физиологической регуляции активности гликогенсинтазы играет киназа 3 гликогенсинтазы.
Киназа 3 гликогенсинтазы (GSK3). Известно два изозима, GSK3a и GSK3β, кодируемых разными генами. Одной из особенностей GSK3 является предпочтительная (100-1000-кратная) каталитическая
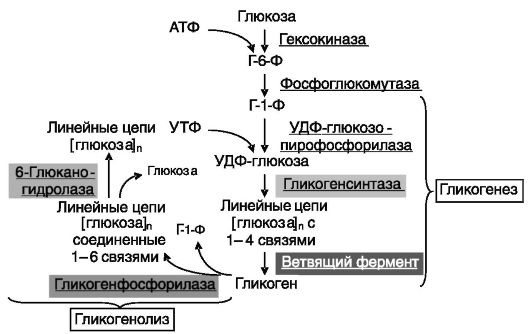 Рис. 9-23. Гликогенез и гликогенолиз
Рис. 9-23. Гликогенез и гликогенолиз
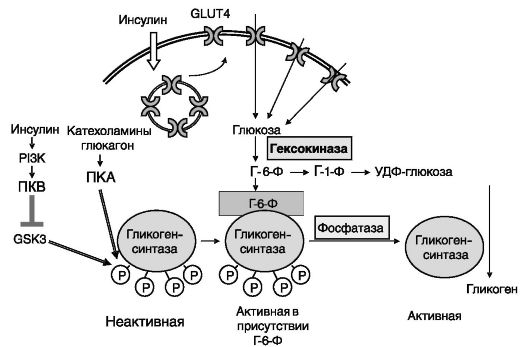 Рис. 9-24. Стимуляция инсулином синтеза гликогена в мышце
Рис. 9-24. Стимуляция инсулином синтеза гликогена в мышце
активность в отношении большинства субстратов, уже фосфорилированных другими протеинкиназами (консенсус Ser/Thr-X-X-X-Ser/
Thr-P, выделена мишень для GSK3). Полагают, что предварительное фосфорилирование субстрата обеспечивает его правильную укладку в каталитическую бороздку фермента. В субстратах, не нуждающихся в предварительном фосфорилировании, заякоривающую роль Ser/ Thr-P выполняет отрицательно заряженный остаток аминокислоты. Фосфорилирование самой GSK3 по остатку тирозина повышает ее активность за счет облегчения доступа субстрата к сайту его связывания в ферменте. Напротив, фосфорилирование GSK3 по остатку серина (Ser21 в GSK3a, Ser9 в GSK3β), индуцируемое инсулином с участием фосфатидилинозитид-3-киназы (PI3K) и ПК-B, ведет к инактивации фермента. В результате начинает преобладать дефосфорилирование субстратов GSK3, в частности гликогенсинтазы, которая при этом активируется и обеспечивает усиление синтеза гликогена. Механизм инактивации GSK3 при ее фосфорилировании по остатку серина связан с возникновением псевдосубстрата, оккупирующего каталитическую бороздку фермента.
Гликогенфосфорилаза. Гликогенфосфорилаза представлена тремя изоформами (мышечного, печеночного и мозгового типа) со сходным типом регуляции. Фермент аллостерически активируется АМФ (сигнал недостаточного энергообеспечения), ингибируется АТФ, АДФ и глюкозо-6-фосфатом (сигналы обеспеченности энергией и глюкозой) и активируется фосфорилированием по N-концу (Ser14). Фосфорилирование ведет к формированию из неактивного гомодимера фосфорилазы в активного гомотетрамера а. Фосфорилирование фосфорилазы катализируется чувствительной к действию гормонов киназой фосфорилазы (рис. 9-25, а).
Киназа фосфорилазы. Киназа фосфорилазы b (Phk) - один из наиболее крупных (молекулярная масса 1,3 МД) и сложноорганизованных ферментов. Основная функция Phk заключается в обеспечении влияния разнообразных стимулов на гидролиз гликогена под действием гликогенфосфорилазы с высвобождением Г-6-Ф, который может быть затем утилизирован посредством гликолиза (в мышце) или экспортирован из клетки в виде глюкозы (в печени).
Phk построена из трех регуляторных (α, β и δ) и одной каталитической (γ) субъединиц, причем каждая из них в ферменте представлена четырьмя молекулами, в результате чего общая организация Phk напоминает бабочку (рис. 9-25, б).
В отсутствие стимулов α- и β-субъединицы ингибируют активность каталитической субъединицы γ. Фосфорилирование a- и β-
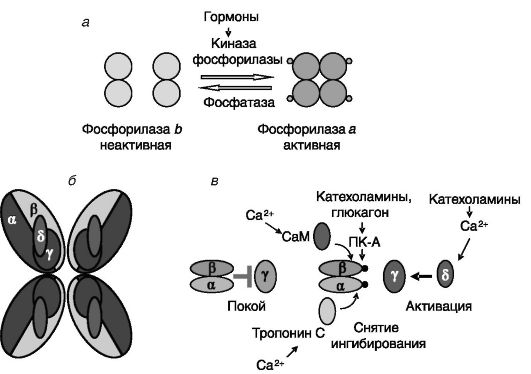 Рис. 9-25. Регуляция
активности гликогенфосфорилазы (а), структурная организация киназы
гликогенфосфорилазы (б) и регуляция ее активности (в)
Рис. 9-25. Регуляция
активности гликогенфосфорилазы (а), структурная организация киназы
гликогенфосфорилазы (б) и регуляция ее активности (в)
субъединиц ПК-А снимает данное ингибирование. Субъединица δ представляет собой конститутивно ассоциированный с ферментом кальмодулин. В присутствии Ca2+ субъединица δ повышает ферментативную активность Phk синергично с фосфорилированием α- и β-субъединиц (см. рис. 25, в). Каждая из субъединиц α, β, и γ Phk представлена несколькими изоформами. В результате даже в пределах одной клетки может иметься более одной изоформы Phk с разными регуляторными свойствами.
γ-Субъединица включает N-концевой каталитический домен и C-концевой регуляторный домен, который в отсутствие стимулов оказывает ингибирующее действие на каталитический домен. В регуляторном домене локализованы два сайта связывания кальмодулина. Предполагается, что именно наличие двух сайтов обеспечивает ассоциацию с кальмодулином даже при низкой концентрации Ca2+. Субъединицы α и β дополнительно усиливают это взаимодействие.
α- и β-Субъединицы гомологичны между собой. В C-концевой области α-субъединицы находится небольшой домен мультифосфо-
рилирования. Один из семи фосфорилируемых остатков серина, входящих в этот домен, фосфорилируется ПК-A. Этот и еще три остатка могут фосфорилироваться самой Phk в результате аутокатализа. Сайт фосфорилирования ПК-A в β-субъединице локализован на N-конце. Этот и два других сайта могут фосфорилироваться также в результате аутокатализа.
Рядом с сайтами фосфорилирования ПК-A в субъединицах α и β идентифицированы участки зависимого от кальция связывания «внешнего», т.е. не ассоциированного конститутивно кальмодулина. Через эти участки в скелетной мышце активность фермента может регулироваться белком, родственным кальмодулину, - тропонином C. Активация фермента «внешним» кальмодулином происходит только в отсутствие фосфорилирования протеинкиназой A. По-видимому, фосфорилирование α- и β-субъединиц и связывание ими «внешнего» кальмодулина сходным образом снимает их ингибирующее действие на γ-субъединицу.
Интересно отметить, что данный путь регуляции (через кальмодулин или тропонин C) не функционирует в случае изоформы Phk сердца (из-за делеции в α'-субъединице, экспрессируемой в сердце, фрагмента, необходимого для взаимодействия с кальмодулином). Различия в аминокислотных последовательностях между изоформами субъединиц (в частности, в областях сайтов фосфорилирования) могут служить причиной и других тканевых особенностей регуляции активности фермента. Следует иметь в виду, что процессы фосфори- лирования/дефосфорилирования по разным сайтам в мультимерном белке происходят не одновременно, что обеспечивает наличие множества промежуточных форм фермента с различающимися свойствами.
Помимо внешних стимулов, на активность Phk может влиять обеспеченность клетки энергией. Данный эффект реализуется аллостерической активацией фермента под действием АДФ и наблюдается лишь в случае отсутствия внешних стимулов. Связывание АДФ стимулирует аутофосфорилирование Phk.
Мутации генов, кодирующих субъединицы Phk, служат наиболее частой причиной наследственных нарушений обмена гликогена - гликогенозов (~1 на 100 000 новорожденных). В зависимости от типа мутаций и генов может наблюдаться полная или частичная недостаточность фермента в том или ином органе. Важным результатом исследования больных явился вывод о координированности экспрессии субъединиц Phk.
АУТОРЕГУЛЯЦИЯ ОБМЕНА ГЛЮКОЗЫ
Диета с высоким содержанием углеводов вызывает быструю активацию ферментов, стимулирующих утилизацию глюкозы посредством гликолиза и липосинтеза в печени. Механизм индукции этих ферментов включает метаболизм глюкозы: неметаболизируемый аналог глюкозы неэффективен в индукции, блокада экспрессии глюкокиназы, превращающей глюкозу в Г-6-Ф, блокирует и индуцирующее действие глюкозы на ферменты. Одним из наиболее важных посредников действия глюкозы считается Xu-5-P (рис. 9-26).
Основным источником Xu-5 -P служит пентозофосфатный шунт, но он может образовываться также из фруктозо-6-фосфата и глицеральде- гид-3-фосфата под действием транскетолазы. Xu-5-P является аллостерическим активатором протеинфосфатазы PP2A. Субстратами PP2A служат белок, связывающий углеводчувствительный элемент (ChREBP) компетентных генов и бифункциональный фермент,
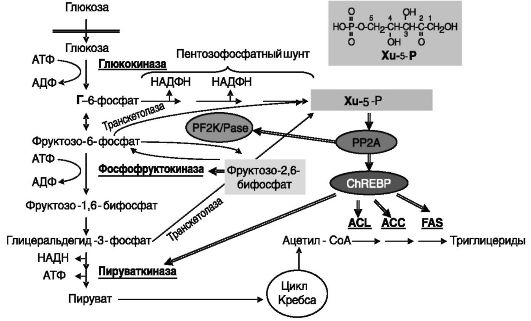 Рис. 9-26. Xu-5-P в действии глюкозы на углеводный и липидный обмен в печени:
Рис. 9-26. Xu-5-P в действии глюкозы на углеводный и липидный обмен в печени:
ACL - АТФ-цитратлиаза; ACC - ацетил-КоА-карбоксилаза; FAS - синтаза жирных кислот; PP2A - протеинфосфатаза 2A; PF2K/Pase - 6-фосфофрук- то-2-киназа/фруктозо-2,6-бифосфатфосфатаза; простые стрелки - пути метаболизма; двойные стрелки - стимуляция
6-фосфофрукто-2-киназа/фруктозо-2,6-бифосфатбифосфатаза (PF2K/ Pase), катализирующий образование и распад минорного метаболита глюкозы, фруктозо-2,6-бифосфата. Данный метаболит служит аллостерическим активатором фосфофруктокиназы - одного из лимитирующих ферментов гликолиза. Активность PF2K/Pase ингибируется ее фосфорилированием ПК-A, активируемой, в свою очередь, такими гормонами, как глюкагон и адреналин. В результате активации PP2A под действием Xu-5-P происходит дефосфорилирование PF2K/Pase, ее активность возрастает, увеличивается концентрация фруктозо-2,6- бифосфата и соответственно возрастают активность фосфофруктокиназы и интенсивность гликолиза.
Транскрипционный фактор ChREBP служит вторым известным субстратом активируемой Xu-5-P PP2A. Фосфорилирование ChREBP в положении Ser-196 ПК-A ведет к ингибированию транслокации ChREBP из цитоплазмы в клеточное ядро. Указанный остаток Ser входит в состав сигнала ядерной локализации (NLS) ChREBP (см. рис. 9-11). Фосфорилирование ChREBP по остатку Thr-666 в клеточном ядре ПК-A, а также по остатку Ser-568 AMPK ингибирует связывание ChREBP с ДНК компетентных генов. (AMPK активируется высоким содержанием жиров в пище.) Оба указанных остатка локализованы в области ДНК-связывающего домена ChREBP. Дефосфорилирование ChREBP активированной Xu-5-P PP2A ведет к транслокации ChREBP в клеточное ядро и повышению его связывания с ДНК, в результате чего интенсивность транскрипции чувствительных генов возрастает. Среди регулируемых ChREBP генов находятся гены печеночной пируваткиназы (одного из лимитирующих ферментов гликолиза) и гены ферментов синтеза жирных кислот: АТФ-цитратлиазы, ацетилСоА-карбоксилазы и синтазы жирных кислот. В результате высокий уровень глюкозы стимулирует собственную утилизацию, координированно усиливая гликолиз и липосинтез в печени.
ChREBP экспрессируется повсеместно, но наиболее интенсивно - в печени, буром и белом жире, тонкой кишке, почках и мышце. Экспрессия ChREBP в адипоцитах стимулируется инсулином, глюкозой и подавляется свободными жирными кислотами. В печени экспрессия ChREBP также стимулируется глюкозой. Нокаут гена ChREBP у мыши ведет к гипергликемии, гиполипидемии, снижению толерантности к глюкозе, повышению отложения гликогена в печени и снижению - в мышце, утрате способности утилизировать фруктозу.
ОБМЕН ГЛЮКОЗЫ
Глюкоза служит основным субстратом для энергообмена в нервной ткани. В условиях низкой или умеренной физической нагрузки основным источником энергии для сердечной и скелетной мышц и, вероятно, многих других органов служат свободные жирные кислоты. Так, при покое у человека (в условиях измерения основного обмена) величина дыхательного коэффициента (отношения выделенного CO2 к потребленному O2) равна ~0,76, что соответствует окислению более 2/3 липидов и менее 1/3 углеводов.
Распределение глюкозы в организме. В крови человека глюкоза находится в плазме и эритроцитах в приблизительно равной концентрации. Скорость поступления глюкозы в эритроциты приблизительно в 1000 раз превышает скорость ее утилизации этими клетками. Свободная глюкоза и гликоген эритроцитов служат буфером для глюкозы плазмы. В условиях гипогликемии и под действием β-адренергических агонистов гликоген эритроцитов может гидролизоваться до глюкозы с выходом последней в плазму крови. В связи с этим эритроциты называют «циркулирующими гепатоцитами».
Захват глюкозы клетками-потребителями осуществляется путем облегченной диффузии с помощью белков-транспортеров семейства GLUT. Неясно, однако, каким образом глюкоза проходит через эндотелий капилляров в интерстициальную жидкость. Это может быть трансцеллюлярный или парацеллюлярный путь диффузии без специальных транспортеров. Во внеклеточной жидкости концентрация глюкозы составляет в среднем 50% от ее концентрации в крови, однако этот показатель существенно варьирует в зависимости от типа ткани и ее физиологической активности. Исключая клеткипроизводители глюкозы (клетки печени, почек, кишечника, эритроциты), концентрация глюкозы в клетках близка к нулю благодаря ее фосфорилированию сразу после поступления в клетку.
Содержание глюкозы в артериальной крови здорового человека в состоянии покоя составляет 5,0±0,05 мМ, однако на протяжении суток эта величина может колебаться от 3 до 9 мМ. По данным артериовенозной разности в концентрации глюкозы покоящаяся скелетная мышца потребляет лишь 3-4% поступающей глюкозы. Под действием инсулина, интенсивной работы или гипоксии до-ля потребляемой мышцей глюкозы может возрастать до 27% от поступления.
Транспортеры глюкозы. Исключая клетки кишечника и почек, в которые глюкоза закачивается против градиента концентрации с
помощью энергозависимых транспортеров группы SGLT, поступление глюкозы в большинство клеток организма происходит по градиенту концентрации путем диффузии, облегчаемой транспортерами группы GLUT.
Транспортеры GLUT (известно 14 изоформ) различаются по сродству к глюкозе, эффективности переноса глюкозы, клеточной локализации и регуляции. В транспорте глюкозы в нейроны мозга участвует GLUT-1 (наиболее распространенный транспортер) хороидного сплетения и GLUT-3 нейронов. Поступление глюкозы в β-клетки поджелудочной железы человека происходит посредством GLUT-1, а кролика - GLUT-2. GLUT-2 служит также для абсорбции глюкозы в кишечнике и почках и переноса глюкозы в гепатоциты и из них. GLUT-4 является преобладающим транспортером глюкозы в клетках, отвечающих на инсулин повышением потребления глюкозы, таких, как адипоциты и мышечные клетки. GLUT-7 обнаружен в эндоплазматическом ретикулуме гепатоцитов и, вероятно, участвует в транспорте новосинтезированной глюкозы из эндоплазматического ретикулума в цитоплазму.
Наибольшим сродством к глюкозе обладает GLUT-3 мозга (Km= 1 - 5 мМ), что соответствует данным о том, что изменения концентрации глюкозы в крови не оказывают влияния на поглощение глюкозы мозгом. Наименьшим сродством к глюкозе обладает GLUT-2 (Km= 20 - 50 мМ), что, по-видимому, связано с тем, что в экспрессирующих GLUT-2 клетках (энтероциты, клетки почек и гепатоциты) концентрация глюкозы может превышать ее уровень в крови. GLUT-1 и GLUT-4 по сродству к глюкозе занимают промежуточное положение, соответствующее нормальным концентрациям глюкозы в плазме крови.
При низкой потребности в транспорте глюкозы оба типа транспортеров (SGLT и GLUT) локализуются преимущественно во внутриклеточных везикулах. По мере роста потребности в транспорте глюкозы данные везикулы встраиваются в плазматическую мембрану.
Регуляция обмена глюкозы режимом питания. Натощак или в начале периода голодания при покое наблюдается квазистабильное состояние. У человека уровень СТГ и инсулина минимален. Уровень глюкозы крови снижается со скоростью менее 1% за 1 ч. Потребление глюкозы мозгом составляет 62% от ее общего потребления организмом, скелетными мышцами - 17%, сердцем - 12%. 40-60% глюкозы, потребленной скелетными мышцами предплечья, расходуется на образование лактата (продукта анаэробного обмена), который может быть утилизирован сердцем или использован для глюконеогенеза печенью. В результате
лишь 16% потребляемого скелетной мышцей O2 расходуется на окисление глюкозы, а 84% - на окисление свободных жирных кислот.
После приема пищи возрастает поступление глюкозы из кишечника в кровь за счет встраивания SGLT в апикальную мембрану и GLUT-2 в люминальную мембрану энтероцитов, а также благодаря активации парацеллюлярного пути (через плотные контакты между энтероцитами). При хроническом потреблении углеводной пищи происходит также индукция общего уровня GLUT-2 в клетках. Повышенный уровень глюкозы в крови снижается за счет ее поступления в клетки с помощью двух механизмов - зависимого и независимого от инсулина. Независимый от инсулина механизм обеспечивается ненасыщенностью большинства GLUT, в результате чего осуществляемый ими транспорт глюкозы возрастает с ростом концентрации глюкозы. Исключение составляют мозг и скелетная мышца. В мозгу GLUT-3 из-за высокого сродства к глюкозе насыщен уже при нормальном уровне глюкозы. В мышце базальная концентрация GLUT слишком низка для обеспечения заметного увеличения транспорта глюкозы при росте ее концентрации.
Зависимый от инсулина механизм повышения потребления глюкозы клетками включает транспортировку и встраивание в плазматическую мембрану GLUT. Стимулами секреции инсулина служат сама глюкоза и ряд пептидов, так называемых инкретинов, к которым относятся глюкагоноподобный пептид 1 (GLP-1), глюкагон, ВИП, пептид, активирующий аденилатциклазу гипофиза (PACAP). Эти пептиды поступают в кровь в ответ на поступление в кишечник пищи, преимущественно углеводов и длинноцепочечных жирных кислот. Уровень инсулина достигает максимума через 30 - 60 мин после приема пищи. Параллельно с ростом концентрации инсулина снижается уровень СТГ в крови. После снижения уровня глюкозы ниже ~3,3 мМ начинают секретироваться антагонисты инсулина: СТГ, глюкагон, β-адренергические агонисты и кортизол.
Влияние инсулина на GLUT тканеспецифично. Инсулин стимулирует встраивание в плазматическую мембрану адипоцитов и GLUT-1, и GLUT-4, а в скелетной мышце - лишь GLUT-4. Эффективность GLUT в качестве транспортеров глюкозы также возрастает. Доля определяемого скелетными мышцами зависимого от инсулина потребления глюкозы в организме при эугликемии составляет около 75%. При гипергликемии эта доля возрастает до 95%. При этом основная часть (≥70%) потребляемой мышцей глюкозы направляется на синтез гликогена.
Помимо инсулина на активность GLUT могут влиять и другие гуморальные факторы. Так, повышенная потребность в глюкозе
растущих гематопоэтических клеток удовлетворяется за счет стимулирующего действия ряда цитокинов (например, ИЛ-3, ИЛ-5) на экспрессию в этих клетках GLUT1, усиления его встраивания в плазматическую мембрану и торможения его эндоцитоза. Как и в случае действия инсулина на GLUT4, влияние цитокинов на GLUT1 в качестве промежуточных этапов включает активацию PI3K и ПК-B.
Свободные жирные кислоты подавляют метаболизм глюкозы. Предполагается, что в результате повышенного метаболизма жирных кислот возрастает продукция ацетил-CoA и цитрата, которые соответственно ингибируют активность пируватдегидрогеназы (направляющей глюкозу на окисление) и фосфофруктокиназы (стимулирующей гликолиз). Эти эффекты пропорциональны концентрации свободных жирных кислот в крови. Жирные кислоты снижают также эффективность GLUT. Кроме опосредованного метаболитами действия на активность ферментов обмена углеводов жирные кислоты могут оказывать влияние на обмен глюкозы через сенсоры жирных кислот, включая PPAR и мембранные рецепторы, сопряженные с G-белками.
Помимо влияния на транспорт глюкозы инсулин подавляет продукцию глюкозы в печени и почках. Прямой механизм включает, в частности, стимуляцию активности гликогенсинтазы и ингибирование активности гликогенфосфорилазы. Непрямой механизм обеспечивается снижением уровня свободных жирных кислот в крови за счет антилиполитического действия инсулина.
Регуляция обмена глюкозы физической нагрузкой. При кратковременной (порядка 20 мин) физической нагрузке малой или средней тяжести потребление глюкозы мышцей возрастает при сохранении уровня глюкозы в крови. В этот период секреция инсулина снижается, а глюкагона и катехоламинов - возрастает, что обеспечивает повышение продукции глюкозы в печени. Параллельно с повышением потребления мышцей глюкозы растет и потребление жирных кислот, в результате чего дыхательный коэффициент мышцы, как и всего организма, практически не меняется. Уровень свободных жирных кислот в крови при физической нагрузке растет благодаря описанным изменениям в секреции гормонов. При интенсивной физической нагрузке глюкоза становится основным поставщиком энергии, а ее важным источником - гликоген мышц. Повышение транспорта глюкозы в мышцы обеспечивается встраиванием GLUT-4 в плазматическую мембрану и повышением его транспортной эффективности. Предполагается, что физическая нагрузка стимулирует GLUT-4 из пула, отличного от пула, активируемого инсулином (эффекты физической нагрузки и
инсулина почти аддидивны). При регулярных тренировках возрастает экспрессия GLUT-4 в мышцах и жировой ткани.
Механизм повышения утилизации глюкозы и жирных кислот мышцами при физической нагрузке (в кратко- и долгосрочном плане) включает сенсоры энергообеспечения, одним из которых служит AMPK.
AMPK построена из трех субъединиц (каталитической α-субъединицы и двух регуляторных - β и γ). Каждая из субъединиц представлена 2 - 3 изоформами, различающимися особенностями тканевой экспрессии и чувствительностью к аллостерическим регуляторам. Активность AMPK регулируется фосфорилированием ее α-субъединицы и аллостерическими регуляторами, АМФ (позитивный регулятор) и креатинфосфатом и АТФ (негативные регуляторы). Накапливающийся при работе мышцы АМФ действует на AMPK несколькими путями: прямо повышает активность AMPK, увеличивает доступность AMPK в качестве субстрата для киназы AMPK (AMPKK), снижает доступность AMPK в качестве субстрата для протеинфосфатазы 2C и аллостерически увеличивает активность AMPKK. Негативное влияние креатинфосфата, по-видимому, не оказывающего влияния на фосфорилирование AMPK, снижается по мере расхода при работе мышцы (рис. 9-27).
Активность AMPK в мышце возрастает не только при выполнении работы, но и под действием ряда других факторов, снижающих уровень энергообеспечения: гипоксии, рассопряжения окисления и фосфорилирования, ингибирования дыхательного комплекса I. Это объясняет тот известный факт, что гипоксия стимулирует захват глюкозы скелетной мышцей сходно с действием физической нагрузки.
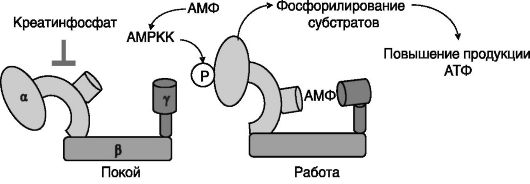 Рис. 9-27. Тримерная
AMPK активируется фосфорилированием AMPKK и образующимся при работе
АМФ, действующим прямо и через активацию AMPKK. Параллельно снимается
ингибирующее действие креатинфосфата, расходуемого при работе на
образование АТФ
Рис. 9-27. Тримерная
AMPK активируется фосфорилированием AMPKK и образующимся при работе
АМФ, действующим прямо и через активацию AMPKK. Параллельно снимается
ингибирующее действие креатинфосфата, расходуемого при работе на
образование АТФ
Эффекторами AMPK служат ферменты и транскрипционные факторы, участвующие в краткосрочной и долговременной стимуляции извлечения энергии из углеводных и липидных субстратов. Одним из объектов действия AMPK является ацетил-CoA-карбоксилаза (ACC), катализирующая превращение ацетил-CoA в малонил-CoA. Фосфорилирование ACC ведет к ее инактивации и соответственно снижению уровня малонил-CoA, являющегося ингибитором фермента, обеспечивающего поступление жирных кислот в митохондрии - карнитинпальмитоилтрансферазы-1 (CPT-1). В результате сокращение мышцы повышает доступность жирных кислот для окисления. Дополнительным механизмом инактивации ACC при ее фосфорилировании AMPK является снижение чувствительности ACC к активирующему действию цитрата. Влияние AMPK на уровень малонил- CoA дополняется стимуляцией (прямой или опосредованной) активирующего фосфорилирования фермента, катализирующего деградацию малонил-CoA - малонил-CoA-декарбоксилазы - MCD (рис. 9-28).
Потенциальным эффектором AMPK может служить NOS, являющаяся субстратом для AMPK in vitro. Известно, что активация NOS через образование NO активирует растворимую гуанилатциклазу, продукт активности которой, цГМФ, способен индуцировать перемещение транспортера глюкозы GLUT-4 из внутриклеточных везикул в плазматическую мембрану. AMPK может также опосредовать долгосрочное действие физических тренировок на способность
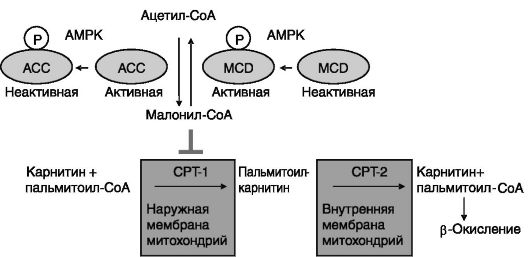 Рис. 9-28. За
счет подавления биосинтеза и ускорения деградации малонил- CoA AMPK
снимает его ингибирующее действие на транспорт жирных кислот в
митохондрии
Рис. 9-28. За
счет подавления биосинтеза и ускорения деградации малонил- CoA AMPK
снимает его ингибирующее действие на транспорт жирных кислот в
митохондрии
мышцы утилизировать глюкозу. Показано, что, как и регулярные физические тренировки, хроническая активация AMPK сопровождается усилением транскрипции гена GLUT-4, увеличением содержания белка GLUT-4 в клетках и повышением эффективности действия инсулина на захват глюкозы клетками. Сходным образом и регулярные физические тренировки, и хроническая активация AMPK повышают экспрессию мРНК и активность гексокиназы в мышечных клетках. AMPK может быть также посредником стимулирующего действия регулярных тренировок на экспрессию и активность ферментов цикла лимонной кислоты и окисления жирных кислот, белков цепи транспорта электронов в митохондриях. Предполагается, что AMPK может прямо или опосредованно влиять на активность транскрипционных факторов, участвующих в регуляции экспрессии указанных белков.
Рекомендуемая литература
Brushia R.J., Walsh D.A. Phosphorylase kinase: the complexity of its regulation is reflected in the complexity of its structure. Front Biosci. 1999;4:D618 - 641.
DobleB.W., Woodgett J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116(Pt 7):1175 - 1186.
DrozdowskiL.A., ThomsonA.B. Intestinal sugar transport. World J Gastroenterol. 2006;12(11):1657 - 1670.
Fujii N, Jessen N, Goodyear L.J. AMP-activated protein kinase and the regulation of glucose transport. Am J Physiol Endocrinol Metab. 2006;291(5): E867 - 877.
Greenberg C.C., Jurczak M.J., Danos A.M., Brady M.J. Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab. 2006;291(1):E1 - 8.
Kabashima T, Kawaguchi T, Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A. 2003;100(9):5107 - 5112.
Rider M.H., Bertrand L, Vertommen D. et al. 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381(Pt 3):561 - 579.
Winder W.W. Energy-sensing and signaling by AMP-activated protein kinase
in skeletal muscle. J Appl Physiol. 2001;91(3):1017 - 1028.
Wilson J.E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206(Pt 12):2049 - 2057. Zierler K. Whole body glucose metabolism. Am J Physiol. 1999;276(3 Pt 1):
E409 - 426.