Биоорганическая химия : учебник / Н. А. Тюкавкина, Ю. И. Бауков, С. Э. Зурабян. - 2010. - 416 с.
|
|
|
|
ГЛАВА 6. РЕАКЦИОННАЯ СПОСОБНОСТЬ КАРБОНОВЫХ КИСЛОТ И ИХ ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
6.1. Карбоновые кислоты
6.1.1. Общая характеристика
Карбоновыми кислотами называют соединения, функциональной группой в которых является карбоксильная группа -СООН.
В зависимости от природы органического радикала карбоновые кислоты могут быть алифатическими (насыщенными или ненасыщенными) RCOOH и ароматическими ArCOOH (табл. 6.1). По числу карбоксильных групп они подразделяются на монокарбоновые, дикарбоновые и трикарбоновые. В настоящей главе рассматриваются только монокарбоновые кислоты.

Систематическая номенклатура кислот рассмотрена выше (см. 1.2.1). Для многих кислот используются их тривиальные названия (см. табл. 6.1), которые часто предпочтительнее систематических.
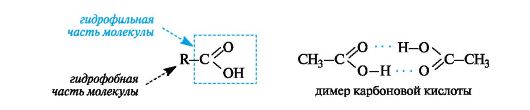
Карбоновые кислоты благодаря карбоксильной группе полярны и могут участвовать в образовании межмолекулярных водородных связей (см. 2.2.3). Такими связями с молекулами воды объясняется неограниченная растворимость низших кислот (C1-C4). В молекулах карбоновых кислот можно выделить гидрофильную часть (карбоксильную группу СООН) и гидрофобную часть (органический радикал R). По мере возрастания доли гидрофобной части снижается растворимость в воде. Высшие карбоновые кислоты алифатического ряда (начиная с С10) в воде практически нерастворимы. Для карбоновых кислот характерна межмолекулярная ассоциация. Так, жидкие карбоновые кислоты, например уксусная кислота, существуют в виде димеров. В водных растворах димеры распадаются на мономеры.
Таблица 6.1. Монокарбоновые кислоты
Увеличение способности к ассоциации при переходе от альдегидов к спиртам и далее кислотам отражается на изменении температур кипения соединений этих классов с близкой молекулярной массой.
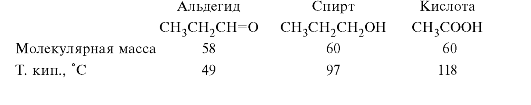
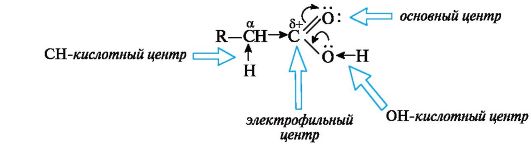
6.1.2. Реакционные центры в карбоновых кислотах
Химические свойства карбоновых кислот обусловлены прежде всего карбоксильной группой, которая в отличие от изученных ранее функциональных групп (спиртовой, карбонильной) имеет более сложное строение. Внутри самой группы имеется р,л-сопряжение в результате взаимодействия р-орбитали атома кислорода группы ОН с π-связью группы С=О (см. также 2.3.1).
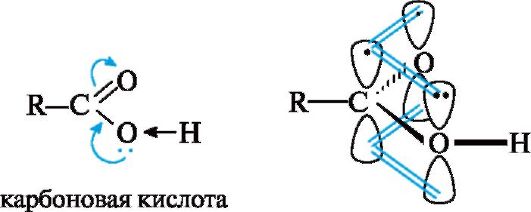
Карбонильная группа по отношению к группе ОН выступает в роли электроноакцептора, а гидроксильная группа за счет +М-эффек- та - в роли электронодонора, подающего электронную плотность на карбонильную группу. Особенности электронного строения карбоновых кислот обусловливают существование нескольких реакционных центров (схема 6.1):
• ОН-кислотный центр, обусловленный сильной поляризацией связи О-Н;
• электрофильный центр - атом углерода карбоксильной группы;
• n- основный центр - атом кислорода карбонильной группы с неподеленной парой электронов;
• слабый СН-кислотный центр, проявляющийся только в производных кислот, так как в самих кислотах имеется несравненно более сильный ОН-кислотный центр.
Схема 6.1. Реакционные центры в молекуле карбоновых кислот
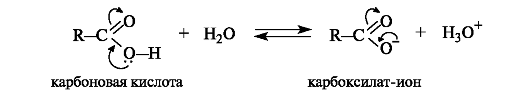
6.1.3. Кислотные свойства
Кислотные свойства карбоновых кислот проявляются в их способности отщеплять протон. Повышенная подвижность водорода обусловлена полярностью связи О-Н за счетр,п-сопряжения (см. схему 6.1). Сила карбоновых кислот зависит от стабильности карбоксилат-иона RCOO , образующегося в результате отрыва протона. В свою очередь, стабильность аниона определяется прежде всего степенью делокализации в нем отрицательного заряда: чем лучше делокализован заряд в анионе, тем он стабильнее (см. 4.2.1). В карбоксилат-ионе заряд делокализуется по р,π-сопряженной системе с участием двух атомов кислорода и распределен поровну между ними
(см. 2.3.1).
Для карбоновых кислот значения рла лежат в интервале 4,2-4,9. Эти кислоты обладают существенно более высокой кислотностью, чем спирты (рКа 16-18), фенолы (рКа ~10) и тиолы (рКа 11-12) (см. табл. 4.5).
Длина и разветвленность насыщенного алкильного радикала не оказывает существенного влияния на кислотные свойства карбоновых кислот. В целом алифатические монокарбоновые кислоты обладают практически одинаковой кислотностью (pKa 4,8-5,0), за исключением муравьиной кислоты, у которой кислотность на порядок выше.
Объяснить более высокую кислотность муравьиной кислоты можно с привлечением еще одного фактора, влияющего на стабильность аниона, а именно сольвата- ции. В водной среде заряд в небольшом по размеру формиат-ионе НСОО лучше делокализован с участием полярных молекул растворителя, чем в более крупных карбоксилат-ионах.
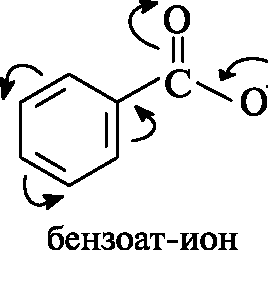
Надо отметить, что ароматические кислоты незначительно превышают алифатические по кислотности (pKa бензойной кислоты 4,2). В делокализации заряда в бензоат-ионе бензольное кольцо выступает как слабый электроноакцептор, не принимая участия в сопряжении с электронами, обусловливающими отрицательный заряд.
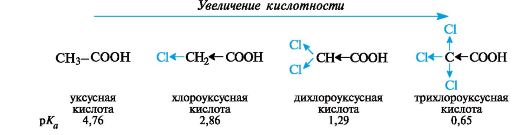
На кислотность карбоновых кислот значительно влияют заместители, введенные в углеводородный радикал. Независимо от механизма
передачи электронного влияния заместителя в радикале (индуктивного или мезомерного), электроноакцепторные заместители способствуют делокализации отрицательного заряда, стабилизируют анионы и тем самым увеличивают кислотность. Электронодонорные заместители, напротив, ее понижают.
В водных растворах карбоновые кислоты слабо диссоциированы.
Кислотные свойства проявляются при взаимодействии карбоновых кислот со щелочами, карбонатами и гидрокарбонатами. Образующиеся при этом соли в заметной степени гидролизованы, поэтому их растворы имеют щелочную реакцию.
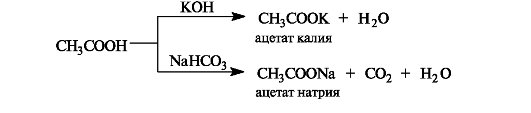
6.1.4. Нуклеофильное замещение
Нуклеофильное замещение у sp2-гибридизованного атома углерода карбоксильной группы представляет наиболее важную группу реакций карбоновых кислот.
Атом углерода карбоксильной группы несет частичный положительный заряд, т. е. является электрофильным центром (см. схему 6.1). Он может быть атакован нуклеофильными реагентами, в результате чего происходит замещение группы ОН на другую нуклеофильную частицу.
Гидроксид-ион является плохой уходящей группой, поэтому реакции нуклеофильного замещения в карбоксильной группе проводятся в присутствии кислотных катализаторов, особенно когда используются слабые нуклеофильные реагенты, такие, как спирты.
Наиболее важные реакции монокарбоновых кислот приведены на схеме 6.2.
Схема 6.2. Некоторые реакции нуклеофильного замещения в карбоновых кислотах
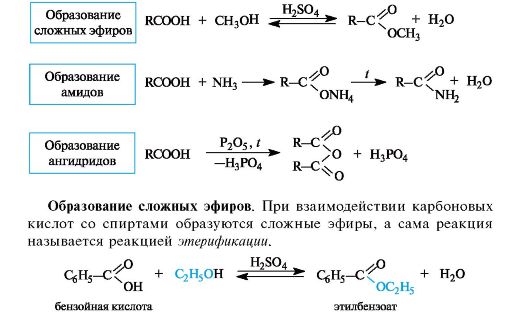
Реакция этерификации катализируется сильными кислотами.
Механизм реакции этерификации. Каталитическое действие серной кислоты состоит в том, что она активирует молекулу карбоновой кислоты, которая протонируется по основному центру - атому кислорода карбонильной группы (см. схему 6.1). Протонирование приводит к увеличению электрофильности атома углерода. Мезомерные структуры показывают делокализацию положительного заряда в об- разовавшемся катионе (I).
Далее молекула спирта за счет неподеленной пары электронов атома кислорода присоединяется к активированной молекуле кис- лоты. Последующая миграция протона приводит к формированию хорошей уходящей группы - молекулы воды. На последней стадии отщепляется молекула воды с одновременным выбросом протона (возврат катализатора).
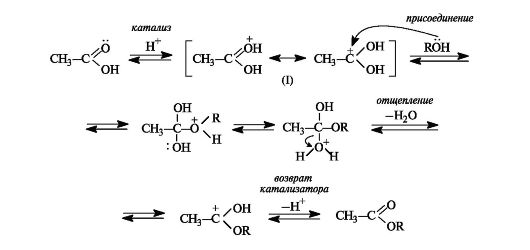
Этерификация - обратимая реакция. Смещение равновесия вправо возможно отгонкой из реакционной смеси образующегося эфира, отгонкой или связыванием воды, либо использованием избытка одного из реагентов. Реакция, обратная этерификации, приводит к гидролизу сложного эфира с образованием карбоновой кислоты и спирта.
Образование амидов. При действии на карбоновые кислоты аммиака (газообразного или в растворе) непосредственно замещения группы ОН не происходит, а образуется аммониевая соль. Лишь при значительном нагревании сухие аммониевые соли теряют воду и превращаются в амиды.
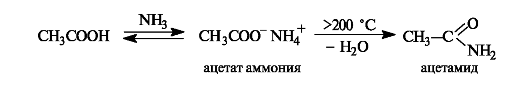
Образование ангидридов кислот. Нагревание карбоновых кислот с оксидом фосфора(V) приводит к образованию ангидридов кислот.
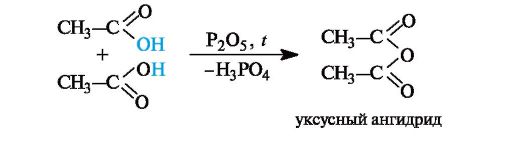
6.2. Функциональные производные карбоновых кислот
6.2.1. Общая характеристика
Функциональные производные карбоновых кислот содержат модифицированную карбоксильную группу, а при гидролизе образуют карбоновую кислоту.
Наиболее важными функциональными производными карбоновых кислот являются соли, сложные эфиры, тиоэфиры, амиды, ангидриды (табл. 6.2). Галогенангидриды кислот - наиболее реакционноспособные производные, имеющие широкое применение в органической химии, однако они не участвуют в биохимических превращениях ввиду их чрезвычайной чувствительности к влаге, т. е. легкости гидролиза.
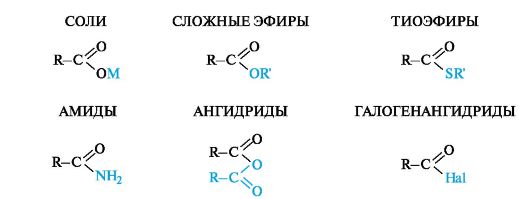
Номенклатура. Названия производных карбоновых кислот строятся с учетом родства их структур со структурой самой карбоновой кислоты, при котором общим фрагментом является ацильный радикал RC(O)-. Эти радикалы называют путем замены сочетания -овая кислота на -оил. Тривиальные названия ацильных радикалов приведены в табл. 6.3.
Соли кислот называют, перечисляя названия аниона кислоты и катиона (в родительном падеже), например, ацетат калия. Названия анионов кислот в свою очередь образуются заменой суффикса -ил в названии ацильного радикала на -ат.
Сложные эфиры называют аналогично солям, только вместо названия катиона употребляют название соответствующего алкила или арила, которое помещают перед названием аниона и пишут слитно
Таблица 6.2. Некоторые функциональные производные карбоновых кислот
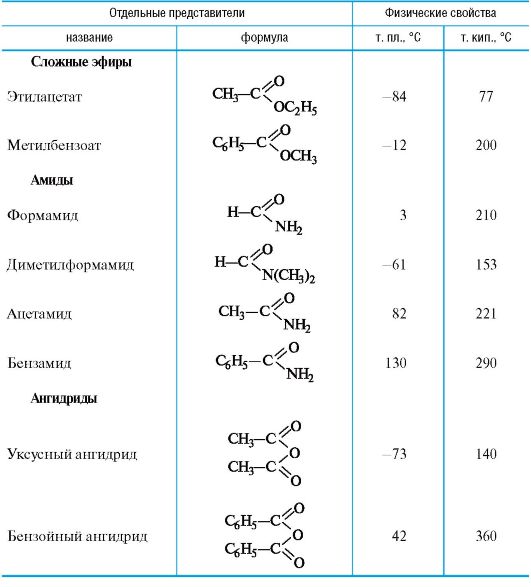
с ним. Сложноэфирную группу COOR можно отразить и описательным способом, например «R-овый эфир такой-то кислоты».

Таблица 6.3. Тривиальные названия ацильных радикалов и производных кислот
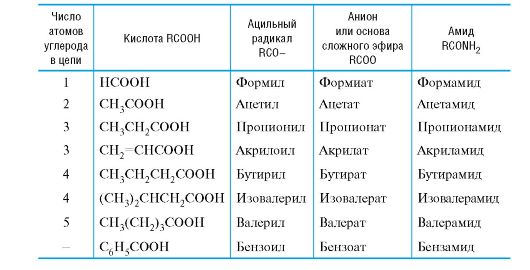
Симметричные ангидриды кислот называют путем замены в названии кислоты слова кислота на ангидрид, например бензойный ангидрид.
Названия амидов с незамещенной группой NH2 производят от названий соответствующих ацильных радикалов заменой суффикса -оил (или -ил) на -амид. В N-замещенных амидах названия радикалов при атоме азота указывают перед названием амида с символом N- (азот).
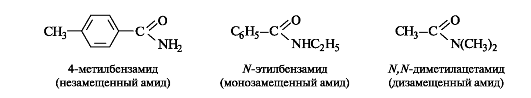
6.2.2. Сравнительная характеристика реакционной способности
Производные карбоновых кислот, как и сами кислоты, способны вступать в реакции нуклеофильного замещения у sp2--гибридизован- ного атома углерода с образованием других функциональных производных. Механизм такого замещения отличается от рассмотренного выше механизма нуклеофильного замещения у sp3--гибридизованного атома углерода в галогеноалканах и спиртах (см. 4.3).
Тетраэдрический механизм нуклеофильного замещения. Сначала нуклеофил присоединяется к атому углерода группы С=О с образованием нестабильного промежуточного аниона (интермедиата). Механизм реакции называют тетраэдрическим, так как атом углерода при этом переходит из sp2- в sр3-гибридное состояние и принимает тетраэдрическую конфигурацию.
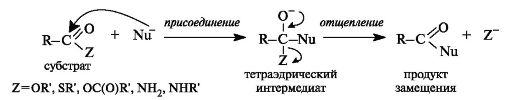
На второй стадии от интермедиата отщепляется частица Z и атом углерода вновь становится sp2-гибридизованным. Таким образом, эта реакция замещения включает стадии присоединения и отщепления.
По такому механизму реакция протекает при наличии достаточно сильного нуклеофила и хорошей уходящей группы Z, напри- мер, в случае щелочного гидролиза сложных эфиров и других функциональных производных карбоновых кислот. Легкость нуклеофильной атаки зависит от величины частичного положительного заряда δ+ на атоме углерода карбонильной группы. В функциональных производных карбоновых кислот он увеличивается с ростом -I-эффекта заместителя Z и уменьшается с увеличением его M-эффекта. В результате этих эффектов величина заряда и, следовательно, способность подвергаться нуклеофильной атаке в рассматриваемых соединениях уменьшаются в приведенной ниже последовательности. К этому же выводу приводит и анализ стабильности уходящих групп Z-, которые выделены цветом (см. 4.2.1).
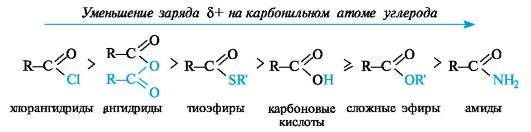
Производные карбоновых кислот по сравнению с альдегидами и кетонами труднее подвергаются нуклеофильной атаке, так как электрофильность карбонильного атома углерода обычно снижается
за счет +M-эффекта заместителя Z. По этой причине в нуклеофильных реакциях функциональных производных карбоновых кислот часто оказывается необходимым кислотный катализ путем протонирования атома кислорода карбонильной группы. Примером такой активации служит уже рассмотренная реакция этерификации (см. 6.1.3).
В результате взаимодействия карбоновых кислот и их функциональных производных со спиртами или аминами в молекулы этих соединений вводится ацильный остаток. По отношению к таким реакциям используют общее название - реакции ацилирования. С этой позиции реакцию этерификации можно рассматривать как ацилирование молекулы спирта.
Функциональные производные кислот обладают разной реакционной способностью в реакциях ацилирования. Наиболее активны хлорангидриды и ангидриды; из них можно получать практически любые производные кислот. Сами кислоты и сложные эфиры (с остатками алифатических спиртов) - значительно менее активные ацилирующие агенты. Реакции замещения с их участием проводятся в присутствии катализаторов. Амиды вступают в реакции ацилирования еще труднее, чем кислоты и сложные эфиры.
Соли карбоновых кислот ацилирующей способностью не обладают, поскольку анион карбоновой кислоты не может быть атакован отрицательно заряженным нуклеофилом или молекулой с неподеленной парой электронов.
6.2.3. Сложные эфиры
Сложные эфиры - широко распространенные в природе производные кислот. Многие лекарственные средства содержат в своей структуре сложноэфирные группировки.
Помимо реакции этерификации, сложные эфиры образуются, причем значительно легче, при ацилировании спиртов или фенолов ангидридами кислот.
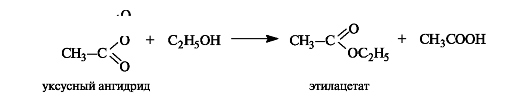
Некоторые реакции сложных эфиров приведены на схеме 6.3.
Схема 6.3. Реакции сложных эфиров
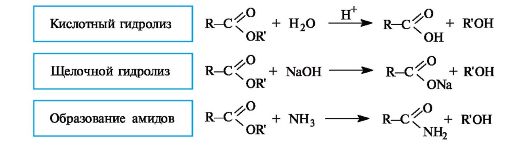
Сложные эфиры способны гидролизоваться и в кислой, и в щелочной среде. Как уже упоминалось (см. 6.1.3), кислотный гидро- лиз сложных эфиров - реакция, обратная реакции этерификации. Несмотря на обратимость этой реакции, кислотный гидролиз легко сделать необратимым при использовании большого избытка воды.
При щелочном гидролизе сложных эфиров щелочь выступает как реагент (на 1 моль сложного эфира расходуется 1 моль щелочи).
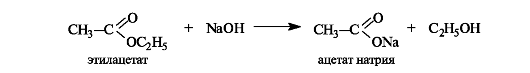
Щелочной гидролиз эфиров - необратимая реакция, поскольку образующийся карбоксилат-ион не способен взаимодействовать с алкоксид-ионом (частицы с одноименными зарядами). Такой гидролиз называют также омылением сложных эфиров. Этот термин связан с тем, что соли высших кислот, образующиеся при щелочном гидролизе жиров, называются мылами.
6.2.4. Тиоэфиры
Тиоэфиры - серные аналоги сложных эфиров - находят весьма ограниченное применение в классической органической химии, но играют важную роль в организме. Известно, что для проявления каталитической активности большинству ферментов, имеющих белковую природу, необходимо соучастие коферментов, которыми служат разнообразные по строению низкомолекулярные органические соединения небелковой природы. Одну из групп коферментов составляют
ацилкоферменты, выполняющие функцию переносчиков ацильных групп. Из них наиболее распространен ацетилкофермент А.
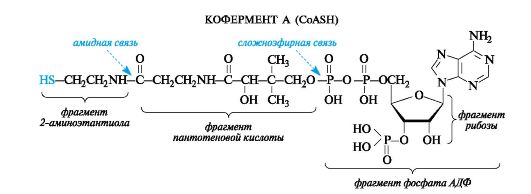
При всей сложности строения молекулы ацетилкофермента А с позиций химического подхода можно определить, что этот кофермент функционирует как тиоэфир.
В качестве тиола, участвующего в его образовании, выступает кофермент А (сокращенно обозначаемый CoASH), молекула которого построена из остатков трех компонентов - 2-аминоэтантиола, пантотеновой кислоты и аденозиндифосфата (дополнительно фосфорилированного по положению 3 в рибозном фрагменте). Аденозиндифосфат (АДФ) рассмотрен в дальнейшем как представитель другой важной группы коферментов - нуклеозидполифосфатов (см. 14.3.1). Пантотеновая кислота образует, с одной стороны, амидную связь с 2-аминоэтанти- олом, а с другой - сложноэфирную связь с остатком АДФ.
По ацилирующей способности все ацилкоферменты А и в том числе ацетилкофермент А, будучи тиоэфирами, занимают «золотую сере- дину» между высокореакционными ангидридами и малоактивными карбоновыми кислотами и сложными эфирами. Их достаточно высокая активность обусловлена, в частности, повышенной стабильностью уходящей группы - аниона CoA-S- - по сравнению с гидроксид- и алкоксид-ионами кислот и сложных эфиров соответственно.
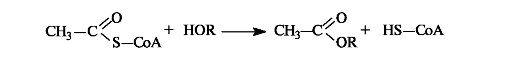
Ацетилкофермент А in vivo является переносчиком ацетильных групп на нуклеофильные субстраты.
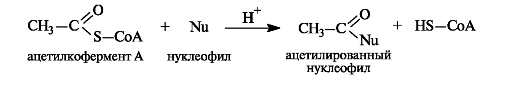
Этим путем, например, осуществляется ацетилирование гидроксилсодержащих соединений.
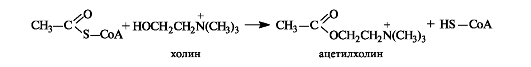
С использованием ацетилкофермента А протекает превращение холина в ацетилхолин, являющегося посредником при передаче нервного возбуждения в нервных тканях (нейромедиатором) (см. 9.2 1).
Кроме этого, можно отметить важное участие в процессах обмена веществ самого кофермента А, функционирующего в качестве тиола. В организме любые карбоновые кислоты активируются путем превращения в реакционноспособные производные - тиоэфиры.
6.2.5. Амиды и гидразиды
Наряду со сложными эфирами важной группой производных кислот являются амиды карбоновых кислот, также широко распростра- ненные в природе. Достаточно упомянуть пептиды и белки, в структуре которых содержатся многочисленные амидные группировки.
В зависимости от степени замещения у атома азота амиды могут быть монозамещенными и дизамещенными (см. 6.2.1).
Амиды образуются при ацилировании аммиака и аминов ангидридами или сложными эфирами.
Амиды обладают самой низкой ацилирующей способностью и гидролизуются намного труднее, чем другие производные кислот. Гидролиз амидов проводится в присутствии кислот или оснований.
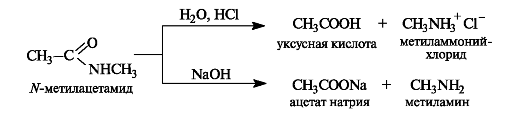
Высокая устойчивость амидов к гидролизу объясняется электронным строением амидной группы, во многом сходным со стро- ением карбоксильной группы. Амидная группа представляет собой р,л-сопряженную систему, в которой неподеленная пара электронов атома азота сопряжена с π-электронами связи С=О. Вследствие сильного +M-эффекта аминогруппы частичный положительный заряд на карбонильном атоме углерода амидов меньше, чем у других функциональных производных кислот. В результате связь углерод-азот в амидах имеет частично двойной характер.
Следствием сопряжения является также чрезвычайно низкая основность атома азота амидной группы. Напротив, у амидов появляются слабые кислотные свойства. Следовательно, амиды обладают амфотерными свойствами.
Амидам родственны гидразиды - производные карбоновых кислот, содержащие остаток гидразина H2NNH2. Немало лекарственных
средств являются по своей природе гидразидами, например, противотуберкулезное средство изониазид (см. 13.4.1). Как и амиды, гидразиды подвергаются гидролизу в достаточно жестких условиях с расщеплением связи C-N.
6.2.6. Ангидриды
Ангидриды кислот чаще встречаются in vivo в виде смешанных ангидридов, включающих ацильные остатки разных кислот, причем одна из кислот - неорганическая (чаще всего фосфорная).
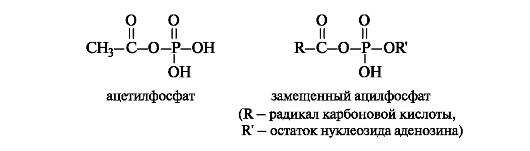
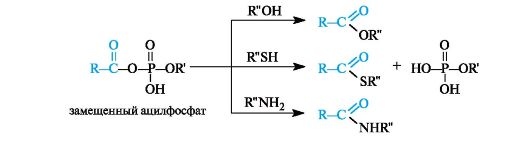
Ацилфосфаты являются хорошими переносчиками ацильных групп, поскольку в реакциях нуклеофильного замещения фосфатные группы представляет собой хорошие уходящие группы.
Замещенные ацилфосфаты - метаболиты, с участием которых в организме осуществляется перенос ацильных остатков к гидроксильным, тиольным группам и аминогруппам различных соединений.
6.3. Сульфоновые кислоты
и их функциональные производные
Сульфоновые кислоты RSO3H можно рассматривать как производные углеводородов, в которых атом водорода замещен сульфогруппой SO3H. Наиболее известны сульфоновые кислоты аромати- ческого ряда; их простейшим представителем является бензолсульфоновая кислота. Подобно серной, сульфоновые кислоты обладают высокой кислотностью.
Сульфоновые кислоты, как и карбоновые кислоты, образуют функциональные производные - соли, эфиры, амиды и т. д.
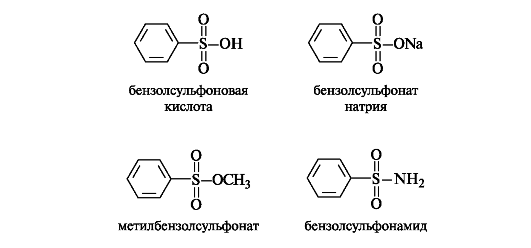
Большое значение в медицинской практике приобрели N-заме- щенные амиды сульфаниловой (n-аминобензолсульфоновой) кислоты - сульфаниламидные средства (см. 9.3).