Биоорганическая химия : учебник / Н. А. Тюкавкина, Ю. И. Бауков, С. Э. Зурабян. - 2010. - 416 с.
|
|
|
|
ГЛАВА 3. РЕАКЦИОННАЯ СПОСОБНОСТЬ УГЛЕВОДОРОДОВ
3.1. Общая характеристика реакций органических соединений
Знание теоретических закономерностей реакций составляет фундамент для обобщения экспериментальных данных, позволяет выявить сходство и различие между разнообразными химическими и биохимическими реакциями и, наконец, помогает управлять ходом того или иного процесса.
3.1.1. Типы реакций и реагентов
Химические реакции представляют собой процессы, сопровождающиеся изменением распределения электронов внешних оболо- чек атомов реагирующих веществ. Движущей силой химических реакций является стремление к образованию новых, обладающих меньшей свободной энергией, и, следовательно, более стабильных систем. В большинстве случаев органические реакции проходят в виде нескольких последовательных стадий. Детальное описание совокупности всех стадий химического процесса называют механизмом реакции.
Способность вещества вступать в ту или иную химическую реакцию и реагировать с меньшей или большей скоростью называют его реакционной способностью.
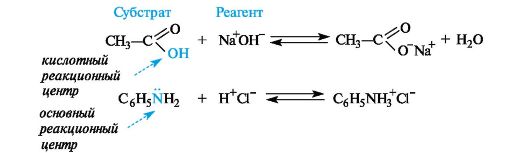
Реакционную способность органического вещества всегда нужно рассматривать только по отношению к конкретному реакционному партнеру. Само вещество при этом называют субстратом, а действующее на него вещество (реакционную частицу) - реагентом. Субстратом, как правило, называют то вещество, в котором у атома углерода происходит разрыв старой и образование новой связи.
В ходе химической реакции обычно затрагивается не вся молекула, а только ее реакционный центр, т. е. атом или группа атомов, непосредственно участвующие в данной реакции.
Органические реакции классифицируют по типу реагента и характеру изменений связей в субстрате и в соответствии с направлением (конечным результатом).
Типы реагентов и характер изменения связей в субстрате. Реагенты подразделяют на приведенные ниже основные типы.
Радикальные реагенты (радикалы) - свободные атомы или частицы с неспаренным электроном. Примеры радикальных реагентов: гидроксильные НО, гидропероксильные HOO, алкильные R* радикалы, атомы галогенов СГ, Br*.
Электрофильные реагенты (электрофилы) - частицы, образующие новую ковалентную связь за счет электронной пары реакционного партнера. Электрофильные частицы обозначаются символом Е+ или Е. Они могут нести положительный заряд - протон Н+, карбокатионы R3C+, ацилий-катионы R-C=O - или быть электронейтральны- ми, например триоксид серы SO3.
Кислотные реагенты (кислоты) - полностью или частично ионизированные в водных растворах нейтральные молекулы (CH3COOH, HCl) либо положительно заряженные частицы (катионы аммония NH4+, гидроксония H3O+), способные быть донором протона для реакционного партнера.
Нуклеофильные реагенты (нуклеофилы) - частицы, образующие новую ковалентную связь с реакционным партнером, предоставляя для этого свою электронную пару. Нуклеофильные частицы обозначаются символами Nu или Nu и могут быть отрицательно заряженными - гидрид-ион Н , гидроксид-ион НО , алкоксид-ион RO , карбанион R3C , хлорид-ион Cl - или быть электронейтральными. В этом случае их нуклеофильность обусловлена n- или π-электронами (NH3, H2O, CH2=CH2, C6H6).
Термин «нуклеофил» применяется к частице, реагирующей с любым электрофильным реакционным партнером за исключением протона Н+.
Основные реагенты (основания) - отрицательно заряженные частицы (НО , RO ) или нейтральные молекулы (NH3, H2O), способные оторвать протон от кислотного реакционного центра. Основные реагенты обозначаются символами B- или B.
Окислители - нейтральные молекулы или ионы (O2, Fe3+, органические восстановители), принимающие электроны или атомы водорода от органического субстрата; обозначаются символом [O].
Восстановители - нейтральные молекулы или ионы (H2, Fe2+, H-, органические восстановители), отдающие электроны или атомы водорода органическому субстрату; обозначаются символом [H].
В соответствии с характером разрыва связи в субстрате и природой реагента различают реакции радикальные и ионные.
В радикальных, или гомолитических, реакциях (символ R) участвуют радикальные реагенты и происходит гомолитический разрыв ковалентной связи в субстрате. При гомолитическом, или свободнорадикальном, разрыве ковалентной связи (гомолизе) у каждого из ранее связанных атомов остается по одному электрону. В результате в качестве промежуточных частиц образуются радикальные реагенты, как показано ниже для молекулы X-Y (где X и Y обозначают ковалентно связанные атомы или группы атомов).
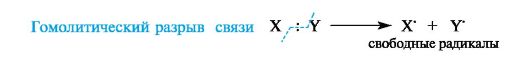
Ионные, или гетеролитические, реакции сопровождаются гетеролитическим разрывом связи в субстрате. При таком разрыве (гетеролизе) ковалентной связи электронная пара, связывающая атомы, остается с одним из партнеров по связи. При этом образуются электрофильная и нуклеофильная частицы.
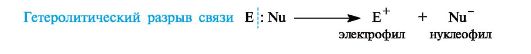
В качестве промежуточных частиц в гетеролитических реакциях принимают участие карбокатионы R3C+ и карбанионы R3C-.
Направление реакции. По направлению (конечному результату) органические реакции делят на несколько основных типов:
• реакции замещения (символ S). Для насыщенных углеводородов характерны процессы радикального замещения SR (см. 3.2.1), для ароматических соединений - реакции электрофильного
замещения SE (см. 3.2.3), для спиртов - реакции нуклеофильного замещения SN у sp3-гибридизованного атома углерода (см. 4.3); для карбоновых кислот - реакции нуклеофильного замещения SN у sp2-гибридизованного атома углерода (см. 6.1.4);
• реакции присоединения (символ A). Эти реакции типичны для соединений, содержащих кратные связи. Для ненасыщенных углеводородов характерны реакции электрофильного присоединения AE (см. 3.2.2), для альдегидов и кетонов - реакции нуклеофильного присоединения AN (см. 5.3);
• реакции отщепления, или элиминирования (символ E). Эти реакции по направлению обратны реакциям присоединения.
Символы реакций и реагентов представляют собой начальные буквы английских терминов: A - addition (присоединение); E - elimination (отщепление); S - substitution (замещение); R - radical (радикал); N - nucleophile (нуклеофил); E - electrophile (электрофил).
• окислительно-восстановительные реакции. Процесс окисления включает переход электронов от органического субстрата к реагенту-окислителю, а процесс восстановления - передачу электронов от реагента к органическому субстрату.
В органической химии более распространен иной подход к трактовке реакций окисления и восстановления. Под окислением пони- мают введение атома кислорода в молекулу субстрата или удаление двух атомов водорода, как, например, в следующих рядах:
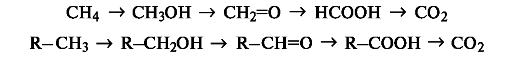
При таком подходе восстановление представляет собой обратный процесс, т. е. удаление атома кислорода или введение двух атомов водорода:
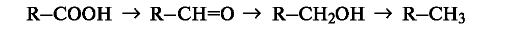
В окислительно-восстановительных реакциях органических соединений, безусловно, меняется степень окисления атома углерода, являющегося реакционным центром. Учет изменения степени окисления, однако, может потребоваться только при необходимости расстановки коэффициентов в уравнении реакции. В то же время многие процессы, протекающие с изменением степени окисления атома углерода, такие, как дегидрирование (-СН2СН2- -- -СН=СН-) или галогенирование (-СН3 - -СН2О), классифицируются не как реакции окисления.
3.1.2. Факторы, определяющие реакционную способность
Многостадийные процессы обычно включают стадии образования промежуточных нестабильных интермедиатов, обладающих высокой реакционной способностью. Во многих случаях можно предположить образование не одного, а нескольких интермедиатов. Реакция будет проходить предпочтительно через стадию образования относительно более устойчивого (обладающего меньшей энергией) интермедиата. В свою очередь, относительная устойчивость интермедиатов, часто представляющих собой высокореакционноспособные промежуточ- ные частицы - свободные радикалы, карбокатионы или карбанионы, определяется возможностью делокализации электронной плотности в этих частицах.
Свободные радикалы. Входящий в состав свободнорадикальных частиц атом углерода с неспаренным электроном находится в состоянии sр2-гибридизации и три его валентные связи лежат в одной плоскости. Неспаренный электрон занимает негибридизованную р-АО, расположенную перпендикулярно плоскости σ-связей (рис. 3.1).
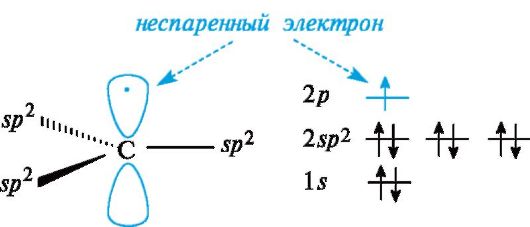
Рис. 3.1. Распределение электронов по орбиталям в свободном радикале
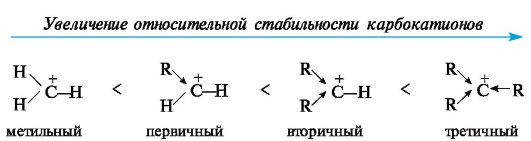
Высокая реакционная способность свободных радикалов объясняется их стремлением достроить внешний электронный уровень до устойчивого октета. Алкильные радикалы - короткоживущие частицы. Их относительная устойчивость соответствует ряду: третичный > вторичный > первичный.

Это связывают с энергией разрыва соответствующей связи С-Н, которая составляет 414 кДж/моль в этане, 396 кДж/моль для группы в пропане и 376 кДж/моль для группы СН в 2-метилпропане.
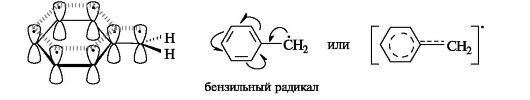
Стабильность свободных радикалов существенно возрастает, когда имеется возможность делокализации неспаренного электрона за счет участия π-электронов соседней двойной связи или бензольного кольца. Для систем с открытой цепью сопряжения наиболее типичным примером является аллильный радикал (см. 2.3.1), а для систем с ароматическим циклом - бензильный радикал (одноэлектронные смещения обозначены стрелкой с одним острием).
Карбокатионы. К ним принадлежат ионы с положительным зарядом на атоме углерода, обладающем в состоянии sp2-гибридизации вакантной орбиталью. Три его гибридные орбитали расположены в одной плоскости, а негибридизованная вакантная орбиталь - перпендикулярно этой плоскости (рис. 3.2).
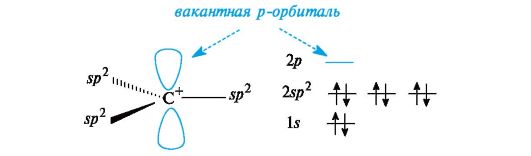
Рис. 3.2. Распределение электронов по орбиталям в карбокатионе
В алкильных карбокатионах алкильные группы, обладающие +/-эффектом, способны понижать положительный заряд у соседнего атома углерода. В связи с этим третичные карбокатионы стабильнее вторичных, а вторичные стабильнее первичных.
Метильный и этильный карбокатионы вследствие крайней неустойчивости являются короткоживущими частицами и их не удается зафиксировать в растворах существующими методами. Среди простых алкильных карбокатионов наиболее устойчив трет-бутилкатион (CH3)3C+.
Возможность делокализации заряда увеличивается, когда положительно заряженный атом углерода находится в сопряжении с двойной связью. Простейшим примером такого карбокатиона является рассмотренный выше аллил-катион СН2=СН-СН2+ (см. 2.3.1).
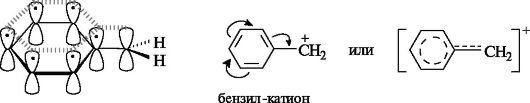
Устойчивость карбокатионов особенно повышается, если возможно сопряжение вакантной орбитали атома углерода с π-электронами ароматического кольца. Например, делокализация положительного заряда в бензил-катионе обусловливает его высокую относительную стабильность.
Карбанионы. В анионах этого типа отрицательно заряжен атом углерода. Простейшие алкильные карбанионы чрезвычайно реакционноспособны в роли основных или нуклеофильных частиц.
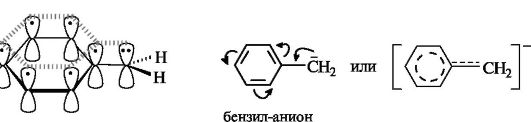
Более устойчивы по сравнению с алкильными карбанионы, у которых неподеленная пара электронов может вступать в сопряжение с двойной связью, находящейся в соседнем положении по отношению к карбанионному центру. Примерами служат карбанионы аллильного СН2=СН-СН2 (см. 2.3.1) и бензильного типов. В том и другом случае осуществляется ρ,π-сопряжение.
3.2. Наиболее важные реакции углеводородов
Углеводороды имеют наиболее простой состав среди органических соединений. Реакционная способность углеводородов зависит от степени их насыщенности. Для них наиболее характерны следующие реакции:
• для насыщенных - реакции радикального замещения Sr;
• для ненасыщенных - реакции электрофильного присоединения АЕ;
• для ароматических - реакции электрофильного замещения SE Эти же реакции свойственны производным углеводородов, т. е.
соединениям других классов, содержащим углеводородные фрагменты указанных типов.
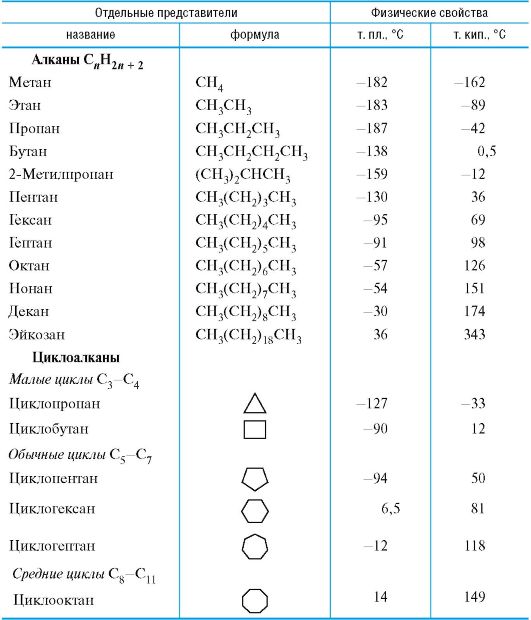
3.2.1. Реакционная способность насыщенных углеводородов
В насыщенных углеводородах - алканах и циклоалканах - имеются только sp3-гибридизованные атомы углерода. Представители алканов и циклоалканов приведены в табл. 3.1. Для этих углеводородов характерны неполярные Csn3-Csn3 и практически неполярные Csp3-H σ-связи,
sp sp sp
обладающие достаточной прочностью (см. табл. 2.1), что объясняет инертность алканов в большинстве гетеролитических реакций.
Алканам свойственны радикальные реакции замещения SR. Аналогично ведут себя обычные циклоалканы, содержащие 5-7 атомов углерода в цикле.
Циклопропан по реакционной способности напоминает ненасыщенные соединения. Так, он реагирует с галогеноводородами с расщеплением трехчленного цикла.
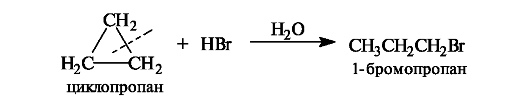
Циклобутан гораздо устойчивее циклопропана, но и для него известны реакции с раскрытием цикла.
В условиях организма важнейшими реакциями по связи С-Н являются химические процессы с участием окислителей (см. 3.1.1).
Взаимодействие органических соединений с молекулярным кислородом служит типичным примером радикальных процессов.
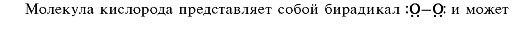
Таблица 3.1. Насыщенные углеводороды
реагировать с соединениями, содержащими связи С-Н, по радикальному механизму с образованием гидропероксидов или продуктов их дальнейших превращений.
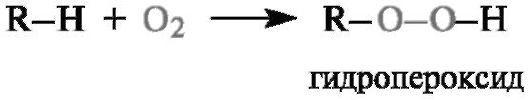
Производные пероксида водорода, в котором один или два атома водорода замещены органическим радикалом, называют гидропе- роксидами и пероксидами соответственно.

В целом насыщенные углеводороды являются наиболее трудно окисляющимися органическими соединениями. Для их окисления необходимы жесткие условия (например, горячая хромовая смесь); более мягкие окислители на них не действуют. Однако в условиях организма окисление с участием связи С-Н является составной частью ряда химических процессов, например, пероксидного окисления липидов (см. Приложение 10-2). Для осуществления этих реакций важно начальное появление в системе свободных радикалов, которые могут возникать в результате взаимодействия ионов металлов переменной валентности (Fe2+ и др.) либо с молекулярным кислородом, либо с гидропероксидами.
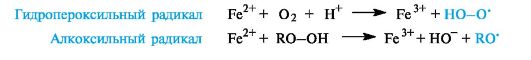
С участием образовавшихся радикальных частиц (обозначим их Х') начинаются и развиваются обычные стадии цепного процесса
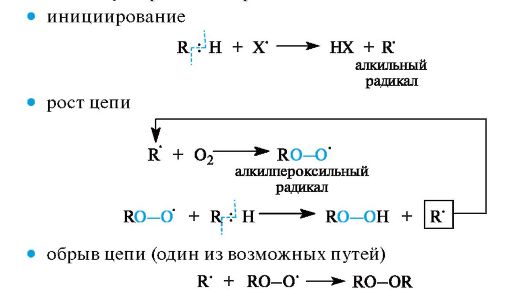
Способность связи С-Н к окислению увеличивается, если она расположена рядом с двойной связью или ароматическим кольцом. В реакциях с кислородом наиболее активны связи С-Н у третичного атома углерода и особенно у атома углерода, находящегося в соседнем положении с двойной связью в аллильном или бензильном фрагментах соответствующих соединений.
Образующиеся in vivo алкилпероксильные радикалы RO-O. сравнительно малоактивны, поэтому они довольно избирательно вступают в последующие реакции, атакуя, например, лишь связи С-Н в аллильном фрагменте (если он имеется в молекуле) и связи О-Н в некоторых фенолах. В случае фенолов образуются еще менее активные арилоксильные радикалы ArO., уже не способные реагировать с новой молекулой субстрата RH, и соответственно на этом прерывается развитие цепного процесса. В таком случае говорят об антиоксидантном действии фенолов (см. 15.5).

Окисление под действием атмосферного кислорода называется автоокислением. Следствием автоокисления часто бывает порча пищевых продуктов при хранении.
Примером автоокисления служит образование гидропероксидов при стоянии на свету и воздухе широко используемого в медицин- ской практике диэтилового эфира. Кислород атакует в молекуле диэтилового эфира прежде всего связи С-Н, находящиеся в α-положении к эфирному атому кислорода.
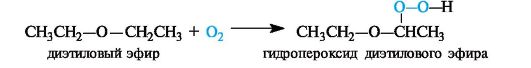
Гидропероксиды и пероксиды легко взрываются, поэтому перед употреблением эфира следует убедиться в отсутствии в нем гидро- пероксидов. Для этого проводят пробу с раствором иодида калия в разбавленной уксусной кислоте. Появление желтой окраски свидетельствует о присутствии гидропероксидов.
3.2.2. Реакционная способность ненасыщенных алифатических углеводородов
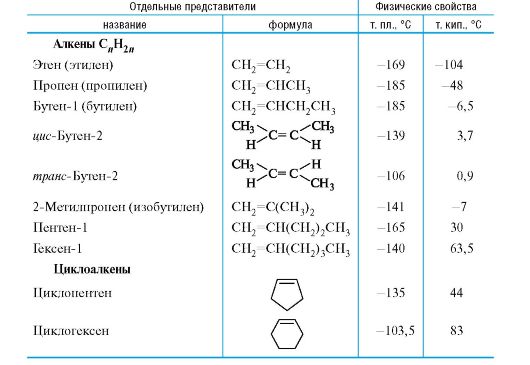
Ненасыщенные углеводороды - алкены и циклоалкены (табл. 3.2) - содержат двойные связи и проявляют способность к реакциям присоединения по этим связям.
Таблица 3.2. Ненасыщенные углеводороды
Типичные примеры реакций присоединения к алкенам приведены на схеме 3.1.
Благодаря электронам π-связи в молекулах алкенов имеется область повышенной электронной плотности (см. 2.1), поэтому они склонны подвергаться атаке электрофильными реагентами (рис. 3.3).
Схема 3.1. Некоторые реакции присоединения к алкенам
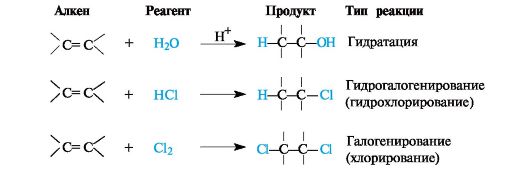
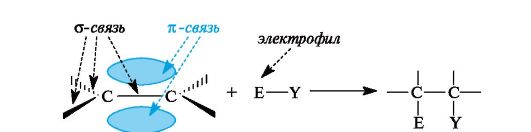
Рис. 3.3. Электрофильное присоединение с участием π-связи
Относительно плоскости π-связи одинаковые заместители у двух атомов углерода могут располагаться по одну (цис) или по разные (транс) стороны. Это приводит к существованию в ряду алкенов пространственных изомеров (стереоизомеров), известных под названием цис- и транс-изомеров (см. 7.1.3). Основная причина существования цис- и транс-изомеров заключается в невозможности вращения вокруг π-связи без ее нарушения.
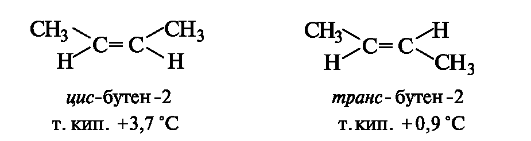
В цис- и транс-изомерах имеется одинаковая последовательность связывания атомов, но они отличаются друг от друга пространственным расположением заместителей и обладают разными свойствами.
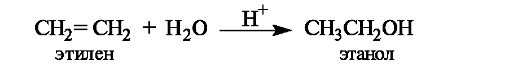
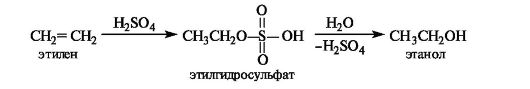
Гидратация. Присоединение воды к алкенам осуществляется только в присутствии катализатора, как правило, серной кислоты. Это известный способ получения спиртов.
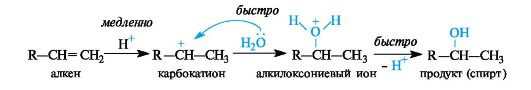
Общее описание механизма реакции электрофильного присоединения AE. Присоединение к алкенам электрофильных реагентов НХ (Н2О, ННа1 и т.п.) протекает по гетеролитическому механизму. Электрофильной частицей в данном процессе служит простейший электрофил - протон.
В реакции выделяют две основные стадии:
• атаку алкена протоном с образованием карбокатиона (медленная стадия, определяющая скорость процесса в целом);
• атаку образовавшегося карбокатиона нуклеофилом; в реакции гидратации это молекула Н2О (быстрая стадия).
При гидратации алкенов в условиях кислотного катализа за двумя этими стадиями следует третья - быстрая стадия отщепления протона (возврат катализатора).
Несимметричные алкены образуют, как правило, один из воз можных структурных изомеров. В случае преимущественного обра зования одного из нескольких возможных структурных изомеров различным положением вводимой функциональной группы говоря о высокой региоселективности реакции.
Региоселективность - предпочтительное протекание реакции п одному из нескольких реакционных центров одинаковой химиче ской природы.
В.В. Марковников (1869) сформулировал закономерность, опре деляющую направление присоединения, которая вошла в мировую химию под названием правила Марковникова.
При взаимодействии реагентов типа НХ с несимметричными алке нами атом водорода присоединяется к атому углерода, связанном с максимальным числом атомов водорода, т. е. к наиболее «гид рогенизированному» атому углерода двойной связи.
Так, при гидратации пропена (несимметричного алкена) в соот ветствии с правилом Марковникова преимущественным продукто реакции является пропанол-2:
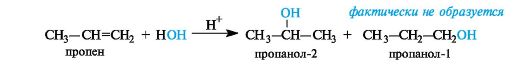
Такое направление реакции объясняется совокупностью двух факторов. В статическом, т. е. нереагирующем, состоянии в несимметричных алкенах электронная плотность π-связи смещена под влиянием заместителя. Возникшие частичные заряды определяют место будущей атаки протоном. В пропене таким местом будет атом С-1 с частичным отрицательным зарядом (как следствие +/-эффекта метильной группы). Таким образом, статический фактор благоприятствует элек-
трофильной атаке по группе СН2 (путь а), что приводит к вторичному карбокатиону (I). При атаке по атому С-2 должен был бы образоваться менее стабильный первичный карбокатион (II) (путь б).
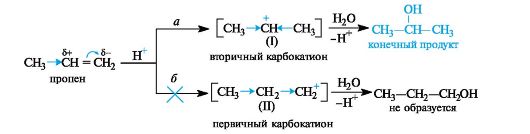
В динамическом состоянии, т. е. в ходе реакции, из двух возможных карбокатионов (I) или (II) будет образовываться более устойчивый. Во вторичном карбокатионе положительно заряженный атом угле- рода связан с двумя электронодонорными алкильными группами, в первичном - с одной. В результате во вторичном карбокатионе за счет +/-эффекта двух алкильных групп осуществляется более эффективное уменьшение положительного заряда. Таким образом, качественная оценка относительной устойчивости промежуточных частиц также говорит в пользу образования вторичного карбокатиона (путь а).
В рассмотренном примере оба фактора, статический и динамический, действуют согласованно. При несогласованном действии статического и динамического факторов последний играет решающую роль. В современной интерпретации правило Марковникова, определяющее региоселективность реакций ненасыщенных соединений, формулируется следующим образом.
Направление присоединения реагентов типа НХ к несимметричным алкенам определяется относительной устойчивостью проме- жуточно образующихся карбокатионов.
Правило Марковникова применяется без оговорок только к алкенам. Однако ненасыщенные соединения часто содержат при двойной связи электроноакцепторные группы (карбоксильную, альдегидную и др.). Учитывая поляризацию связи С=С под влиянием заместителя (статический фактор), можно предсказать иной характер присоединения, а именно, против правила Марковникова. Например, при гидратации α,β-ненасыщенных карбоновых кислот в сильно кислой среде образуются β-гидроксикарбоновые кислоты.
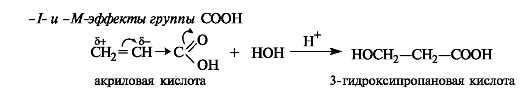
Такой результат можно объяснить и сравнением стабильности промежуточно образующихся карбокатионов (динамический фактор). Катион (I) с одноименными зарядами на соседних атомах углерода менее стабилен, чем катион (II), в котором заряды разделены.
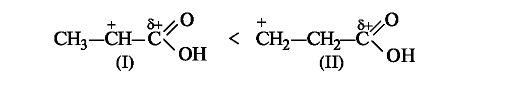
Присоединение галогеноводородов и серной кислоты. Алкены и циклоалкены в достаточно мягких условиях взаимодействуют с галогеноводородами, серной и другими сильными кислотами, способными к диссоциации с образованием протона.
В результате присоединения галогеноводородов образуются галогенопроизводные алканов и циклоалканов.
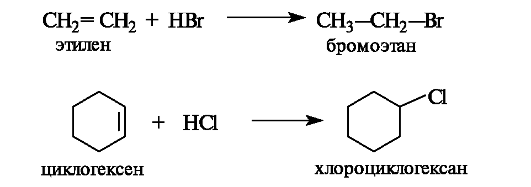
В реакции с концентрированной серной кислотой алкены образую гидросульфаты, в результате гидролиза которых получаются спирты.
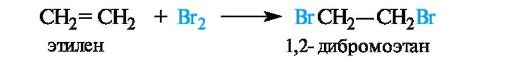
Галогенирование. Алкены в обычных условиях легко присоединяют галогены. Так, быстрое обесцвечивание бромной воды без выделения бромоводорода служит качественной пробой на двойную связь. Еще легче проходит присоединение хлора.
Восстановление и окисление связей C=C. Восстановление двойных связей алкенов осуществляется каталитическим гидрированием - присоединением водорода в присутствии металлов (никеля - при нагревании, платины или палладия - при обычной температуре). Результатом является образование насыщенного продукта - алкана (отсюда, кстати, и возникли термины «насыщенные» и «ненасыщенные» соединения).
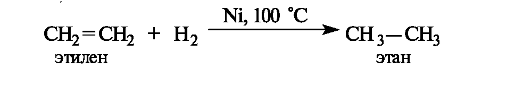
Окисление двойных углерод-углеродных связей в зависимости от условий может приводить к эпоксидам, 1,2-диолам (гликолям) или карбонильным соединениям - продуктам расщепления двойной связи.
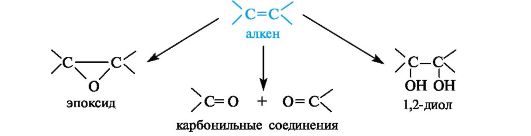
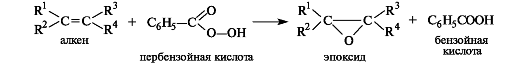
Эпоксиды образуются при обработке алкенов пероксидными соеди- нениями, например пербензойной кислотой.
1,2-Диолы (гликоли) могут быть получены в результате гидролиза эпоксидов в кислой или щелочной среде.
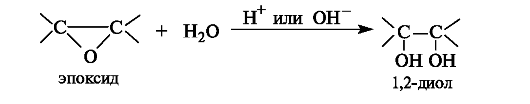
Непосредственно из алкенов 1,2-диолы образуются под действием водного раствора перманганата калия на холоду (реакция Вагнера).
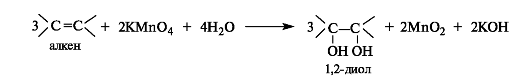
Внешние проявления реакции - исчезновение пурпурной окраски перманганата калия и образование коричневого осадка оксида марганца(IV). Эту реакцию можно использовать как качественную для обнаружения алкенов, а также для их отличия от спиртов, поскольку спирты в таких условиях не окисляются.
В более жестких условиях расщепляется углерод-углеродная связь и образуются кетоны и/или карбоновые кислоты.
3.2.3. Реакционная способность ароматических углеводородов
Ароматическим углеводородам бензольного ряда (аренов) (табл. 3.3) свойственны реакции, не приводящие к нарушению ароматической системы, т. е. реакции замещения. Арены не склонны вступать в реакции присоединения или окисления, ведущие к нарушению ароматичности.
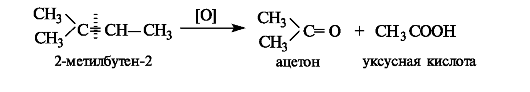
По этой причине при окислении гомологов бензола и других ароматических соединений в жестких условиях (нагревание с пер- манганатом калия или дихроматом калия в кислой среде) окисляются только боковые углеводородные радикалы. Отметим, что более низкая токсичность толуола по сравнению с бензолом объясняется именно легкостью его окисления in vivo в бензойную кислоту.
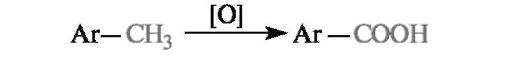
Окисление непосредственно бензольного цикла с его раскрытием и получением малеинового ангидрида осуществляется в чрезвычайно жестких условиях (достаточно обратить внимание на температуру!).
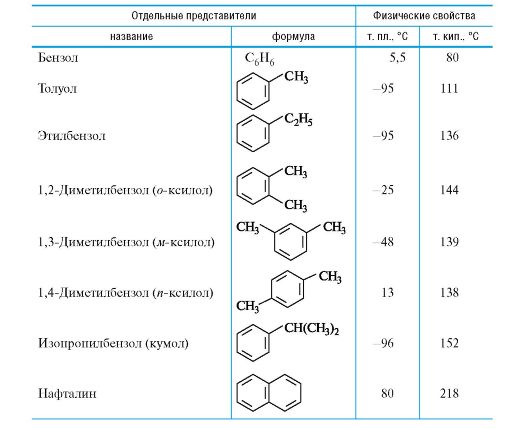
Таблица 3.3. Ароматические соединения бензольного ряда
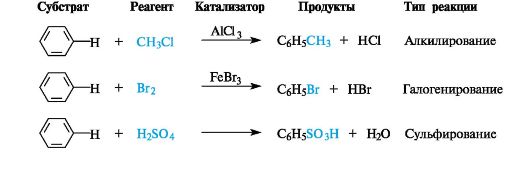
Наиболее типичные для ароматических соединений реакции электрофильного замещения SE приведены на примере бензола на схеме 3.2.
Схема 3.2. Реакции электрофильного замещения в бензоле
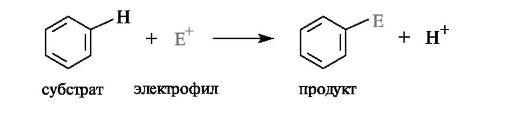
Общее описание механизма реакций электрофильного замещения SE.
Наличие π-электронной плотности с двух сторон плоского ароматического цикла (см. 2.3.2) ведет к тому, что бензольное кольцо является нуклеофилом и в связи с этим склонно подвергаться электрофильной атаке. В общем виде реакции замещения протона в бензольном коль- це на другие электрофилы можно представить следующим образом:
Механизм таких реакций включает ряд общих стадий. Первичной стадией является генерирование электрофильной частицы. Она обычно образуется путем взаимодействия реагента EY с катализатором и может представлять собой либо электронодефицитную часть поляризованной молекулы реагента Εδ+-Υδ-, либо частицу Е+ с полным положительным зарядом (после гетеролитического разрыва связи).
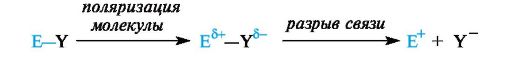
Предполагают, что электрофильная частица, атакуя ароматический субстрат, сначала образует нестойкий π-комплекс, в котором она одновременно связана со всеми π-электронами ароматической системы.
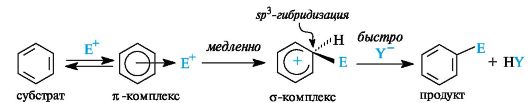
Наиболее важна стадия образования σ-комплекса (самая медленная стадия реакции). Электрофил «забирает» два электрона π-системы и образует σ-связь с одним из атомов углерода бензольного кольца. В σ-комплексе ароматичность нарушена, поскольку один из атомов углерода кольца перешел в sp3 -гибридизованное состояние. Некоторые σ-комплексы удалось выделить в индивидуальном виде.
На последней (быстрой) стадии реакции происходит отщепление протона от σ-комплекса. Ароматическая система восстанавливается (недостающая до секстета пара электронов возвращается в бензо-
льное ядро). Отщепляющийся протон связывается с нуклеофильной частью реагента.
Алкилирование. Арены вступают в реакции алкилирования при участии комплексов алкилгалогенида с галогенидами металлов - AlCl3, FeCl3, ZnCl2 и др.
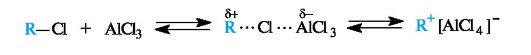
Реакция алкилирования представляет собой общий способ получения гомологов бензола - алкилбензолов.
Алкилирование часто не останавливается на стадии монозамещения и протекает дальше, поскольку введение первой алкильной группы в молекулу бензола активирует ее в реакциях электрофильного замещения.
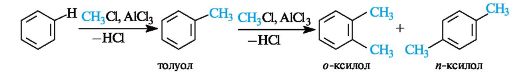
В реакциях алкилирования помимо алкилгалогенидов могут быть использованы и другие источники карбокатионов - алкены и спирты (в кислой среде).
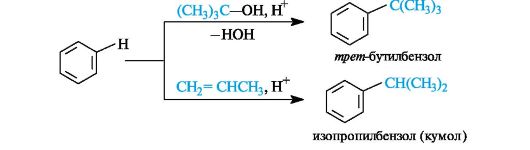
Галогенирование. Замещение свободным хлором или бромом в бензоле непосредственно не происходит, например, бензол не обесцвечивает бромную воду. Реакция проходит только в присутствии катализаторов, в частности FeCl3.
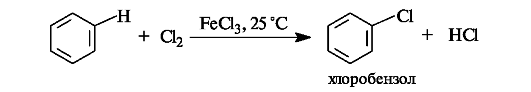
В качестве электрофила выступает либо комплекс галогена с FeCl3, в котором связь между атомами галогена сильно поляризована,
либо катион галогена, образовавшийся в результате диссоциации этого комплекса.

Сульфирование. Бензол сульфируют дымящей серной кислотой, содержащей избыток растворенного в ней оксида серы(VI).
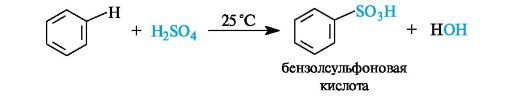
Сульфогруппа может легко удаляться из бензольного ядра, что используется в синтезе бактерицидных препаратов. Так, наиболее общий способ получения фенолов основан на сплавлении ароматических сульфоновых кислот со щелочами.
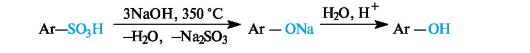
Реакции замещения в производных бензола. При взаимодействии бензола с электрофилом атакуется любой из шести равноценных атомов углерода ароматического кольца, что всегда приводит к единственному монозамещенному продукту. В случае монозамещенного бензола возможно образование трех изомеров - продуктов орто-, мета- и пара-замещения. Соотношение между ними, а также реакционная способность монозамещенного бензола по сравнению с незамещенным определяются природой имеющегося заместителя.
Ориентирующее действие заместителей в бензольном ядре. По влиянию на реакции электрофильного замещения заместители делятся на две группы. Одну из них представляют заместители - ориентанты I рода. К ним относятся алкильные группы, проявляющие +I-эффект по отношению к кольцу, и проявляющие +M-эффект группы OH, OR, NH2, NR2. Все эти заместители являются электронодонорами по отношению к бензольному ядру.
Ориентанты I рода облегчают электрофильное замещение по сравнению с незамещенным бензолом, т. е. являются активирующими, и направляют входящую группу в орто- и пара-положения.
Для соединений, содержащих заместители I рода (XI), реакции электрофильного замещения могут быть представлены в следующем виде:
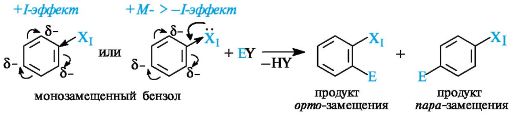
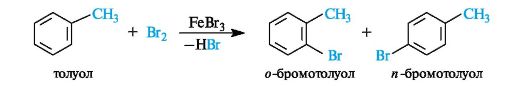
Так, толуол бромируется с образованием смеси продуктов орто- и пара-замещения в соотношении 1:2.
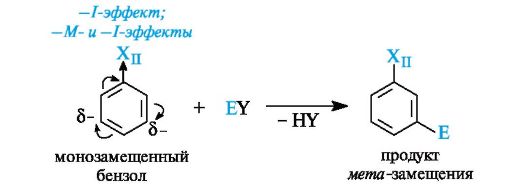
Вторая группа заместителей - это ориентанты II рода. К ним относятся группы NH3+, NR3+, SO3H, CH=O, COOH, являющиеся по отношению к бензольному ядру электроноакцепторами в результате отрицательных индуктивного и/или мезомерного эффектов.
Ориентанты II рода затрудняют реакции электрофильного замещения по сравнению с незамещенным бензолом, т. е. являются дезактивирующими. Если в более жестких условиях реакция все же проходит, то входящая группа вступает в мета-положение.
Для соединений, содержащих заместители II рода (Хц), реакции электрофильного замещения могут быть представлены в следующем виде:
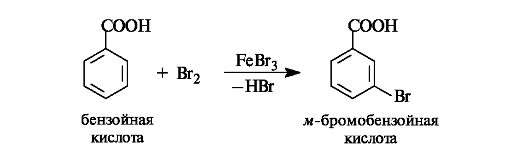
Например, при бромировании бензойной кислоты образуется мета-замещенная кислота.
Правила ориентации не абсолютны; речь идет о предпочтительном направлении реакции.