Биоорганическая химия : учебник / Н. А. Тюкавкина, Ю. И. Бауков, С. Э. Зурабян. - 2010. - 416 с.
|
|
|
|
ПРИЛОЖЕНИЕ
01-1. Префиксы и суффиксы, применяемые для обозначения важнейших характеристических групп (в порядке падения стар- шинства), и примеры построения названий по заместительной номенклатуре ИЮПАК.
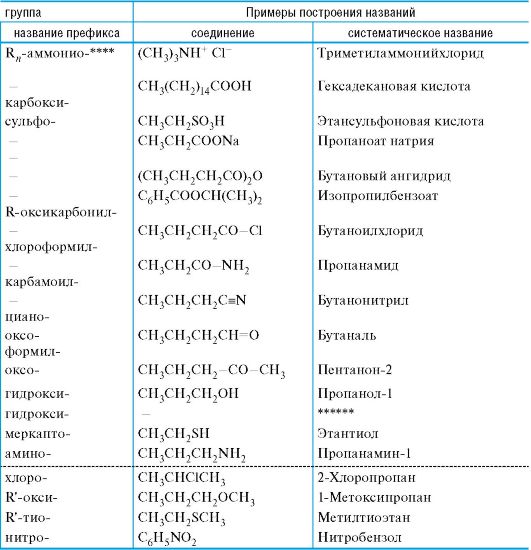
02-1. Атомные орбитали и их заполнение электронами.
03-1. Активные формы кислорода, образующиеся в организме.
03-2. Особенности реакций электрофильного присоединения к сопряженным диенам.
04-1. Количественная оценка кислотности и основности.
04-2. Значения pKa в воде для различного типа кислот.
04-3. Значения рКвн+ в воде для некоторых оснований Брёнстеда.
04-4. Галогенопроизводные углеводородов.
06-1. Реакции конденсации с участием ацетилкофермента А.
10-1. транс-Изомеры высших жирных кислот.
10-2. β-Окисление насыщенных высших жирных кислот.
11-1. Семейство D-альдоз.
11-2. Сиаловые кислоты.
11-3. Участие фосфатов моносахаридов в биохимических процессах. 11-4. Медико-биологическое значение лактозы.
11-5. Полисахариды: декстраны, хитин, пектиновые вещества, альги-
новые кислоты. 11-6. Гликоконъюгаты: протеогликаны и гликопротеины. 12-1. α-Аминокислоты D-стереохимического ряда. 12-2. Расщепление рацематов аминокислот ферментативным путем. 12-3. Метод расчета значений изоэлектрической точки. 12-4. Некоторые превращения α-аминокислот in vivo c участием
кофермента пиридоксальфосфата. 12-5. Некоторые биологически активные пептиды - гормоны, ней-
ропептиды, токсины. 13-1. Тетрапиррольные соединения - порфирины. 13-2. Каталитическое действие имидазольного фрагмента в реакциях
ферментативного гидролиза.
13-3. Гетероциклические производные сульфаниламидов. 13-4. Диазепины - семичленные гетероциклы, содержащие два атома азота.
13-5. Производные птеридина - структурные фрагменты фолиевой
кислоты и рибофлавина. 14-1. Нарушение комплементарных взаимодействий в молекуле ДНК
как причина возникновения мутаций. 14-2. Участие АТФ в переносе ацильных групп.
14-3. Флавинадениндинуклеотид - кофермент окислительно-восстановительных процессов. 15-1. Систематическая номенклатура стероидов. 15-2. Фитоэкдистероиды. 15-3. Пути биосинтеза терпенов и стероидов. 15-4. Основные группы семейства флавоноидов. 15-5. Эйкозаноиды.
15-6. Антибиотики пептидной, аминогликозидной и нуклеозидной природы.
01-1
Префиксы и суффиксы, применяемые для обозначения старшинства*), и примеры построения названий
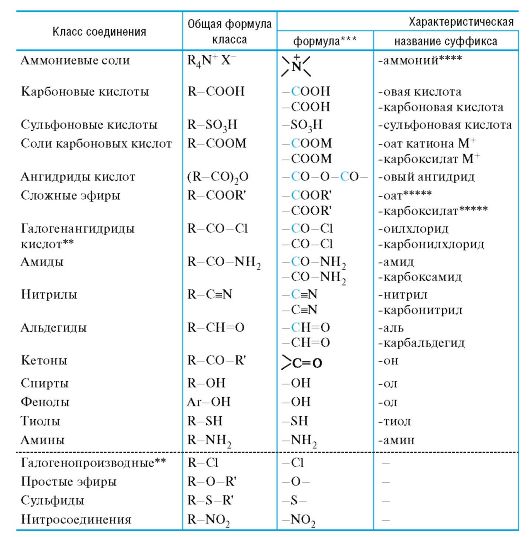
Классы, помещенные ниже пунктирной черты, называются только с использованием префиксов.
* Только для названий, использующих суффикс. ** На примере хлоридов.
*** Атомы, показанные цветом, включаются в родоначальную структуру. **** С перечислением радикалов R и добавлением названия аниона Х-. ***** Перед корнем ставится название радикала R'. ****** Большинство фенолов имеет тривиальные названия.
важнейших характеристических групп (в порядке падения по заместительной номенклатуре ИЮПАК
Атомные орбитали и их заполнение электронами*
Электрон обладает свойствами одновременно волны и частицы. Для описания его движения вокруг ядра используется волновая функция ψ(χ, y, z), где x, y, z - пространственные координаты. Квадрат модуля функции [ψ]2 определяет вероятность нахождения электрона в элементарном объеме, а сама функция описывает орбиталь.
Положение атомных орбиталей (АО) и занимающих их электронов определяется квантовыми числами. Главное квантовое число n характеризует основной уровень энергии орбитали. Побочное (орби- тальное) квантовое число l определяет форму орбитали. При l = 0 АО имеет сферическую форму и называется s-орбиталью (рис. 1). При l = 1 АО имеет форму объемной «восьмерки» (два одинаковых лепестка) и называется р-орбиталью. Она имеет одну узловую плоскость, вероятность нахождения электрона в которой равна нулю. р-АО может иметь различную ориентацию в пространстве (рис. 2).
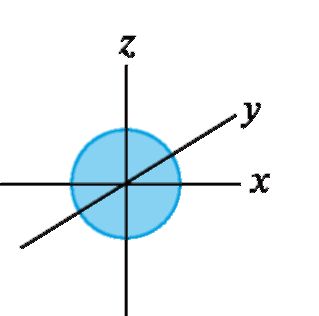
Рис. 1. Атомная s- орбиталь

Рис. 2. Атомные р-орбитали (показаны узловые плоскости)
Общее число электронов, способных заполнить орбитали, на примере двух энергетических уровней показано в табл. 1. При заполнении АО электронами соблюдаются принцип устойчивости, принцип Паули, правило Гунда.
* Подробнее см.: Попков В.А., Пузаков С.А. Общая химия. - М.: ГЭОТАР-Медиа, 2007. - Гл. 1.
Таблица 1. Заполнение энергетических уровней

В соответствии с принципом устойчивости АО заполняются электронами в порядке повышения их энергетических уровней: Is < 2s < 3s < 3p < 4s < 3d, т. е. сначала заполняются орбитали с меньшей энергией.
По принципу Паули на одной АО может находиться не более двух электронов с противоположными спинами.
По правилу Гунда электроны располагаются на АО так, чтобы сохранялось наибольшее число электронов с параллельными спинами, т. е. на АО с одинаковой энергией, так называемых вырожденных орбиталях, электроны стремятся расположиться поодиночке.
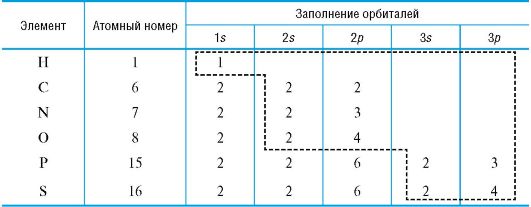
Атом углерода и важнейшие элементы-органогены в основном находятся во втором (C, N, O) и третьем (P, S) периодах Периодической системы. В химических превращениях принимают участие электроны внешнего уровня - валентные электроны (табл. 2).
Таблица 2. Электронная конфигурация элементов-органогенов (пунктирной
рамкой выделены валентные электроны)
03-1
Активные формы кислорода, образующиеся в организме


03-2
Особенности реакций электрофильного присоединения к сопряженным диенам
Для диенов, как и для алкенов, характерны реакции электрофильного присоединения. Изолированные диены по химическому поведению принципиально не отличаются от алкенов. Однако аналогичные реакции в ряду сопряженных диенов имеют особенности, связанные с присутствием в них делокализованного π-электронного облака (сопряжения).
Так, в реакциях с равным молярным количеством реагента обычно получаются два продукта. Один из них образуется за счет участия
только одной из двойных связей, и такой продукт является результатом 1,2-присоединения. В этом случае диен ведет себя подобно алкену. Другой продукт получается в результате 1,4-присоединения, при этом молекула проявляет свойства сопряженной системы, и присоединение идет по ее «концам». В образующемся продукте между атомами С-2 и С-3 возникает двойная связь, как показано на примере гидробромирования бутадиена-1,3.
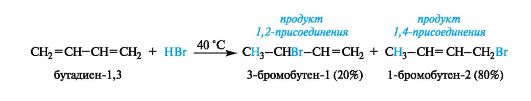
Соотношение между ними в значительной степени зависит от условий эксперимента (главным образом от температуры). Повышенные температуры способствуют 1,4-присоединению. Реакция, осуществленная при -80 ?C, приводит к такому же (4:1) преобладанию продукта 1,2-присоединения. Эта закономерность наблюдается и в ряде других реакций присоединения.
Особенность гетеролитического ступенчатого механизма этих реакций - образование достаточно стабильных карбокатионов, имеющих мезомерное строение (см. 2.3.1). Например, в реакции с участием хлороводорода атака протоном по атому С-1 приводит к карбокатиону аллильного типа, положительный заряд в котором рассредоточен между двумя атомами углерода из-за участия π-электронов соседней двойной связи (ρ,π-сопряжение).
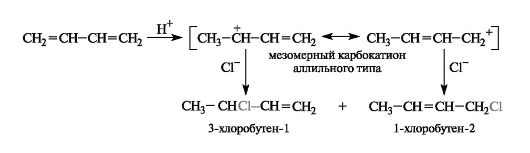
В результате последующей атаки хлорид-ионом мезомерного карбокатиона в соответствии с двумя его граничными структурами получаются два изомерных продукта.
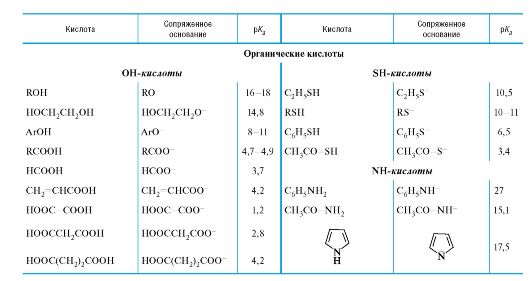
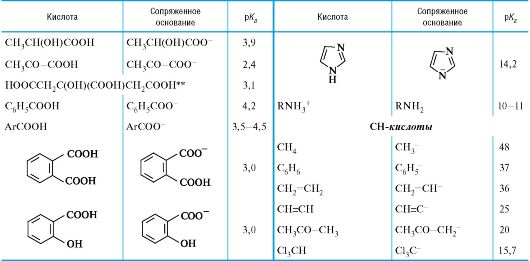
Количественная оценка кислотности и основности


04-2
Значения рК в воде для различного типа кислот*
Продолжение табл.
Окончание табл.
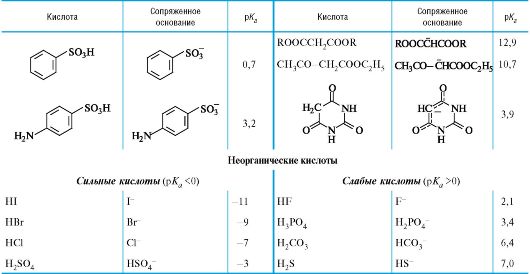
* Значения выше 16 и ниже -2 являются ориентировочными.
** Лимонная кислота, приведена 1-я константа диссоциации; то же для многоосновных неорганических кислот.
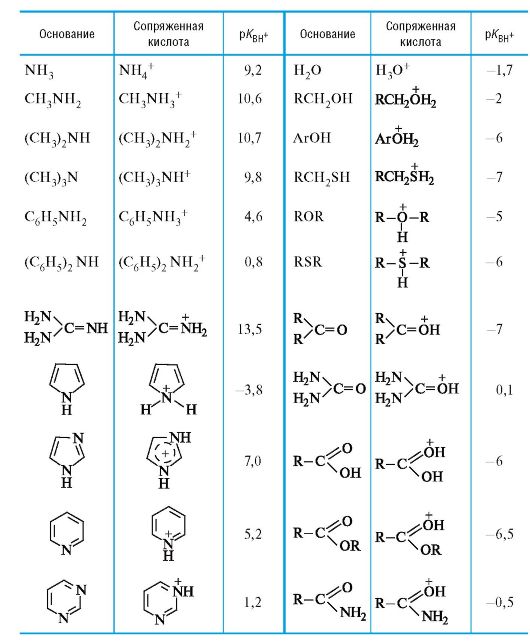
04-3
Значения pKBH+ в воде для некоторых оснований Брёнстеда*
* Значения ниже -3 являются ориентировочными.
Галогенопроизводные углеводородов

Алифатические галогенопроизводные в зависимости от строения углеводородного радикала могут быть первичными, вторичными и третичными. В отличие от углеводородов их молекулы полярны: величина дипольного момента галогенопроизводных составляет 1,25-1,45 D, что обусловлено более высокой электроотрицательностью атомов галогенов по сравнению с атомом углерода (см. 2.2.1). В силу высокой электроотрицательности атомы галогенов в галогенопроизводных прочно удерживают свои n-электроны и не проявляют основных свойств и склонности к образованию межмолекулярных водородных связей. В результате галогеноуглеводороды практически нерастворимы в воде.
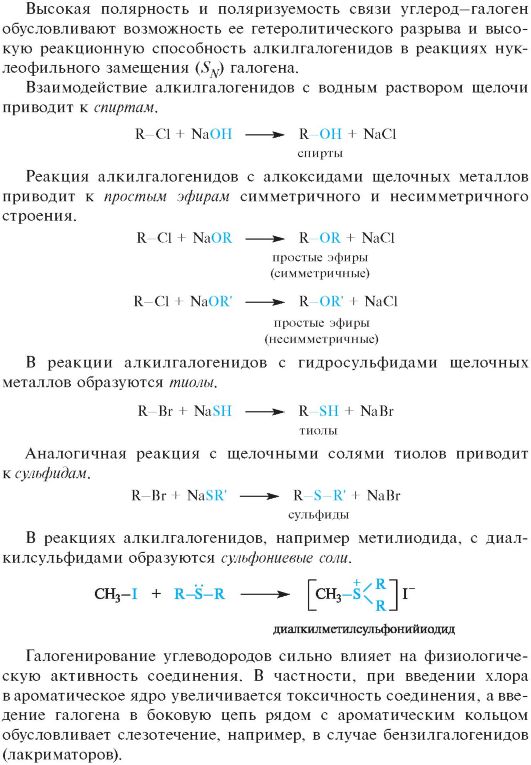

06-1
Реакции конденсации с участием ацетилкофермента А
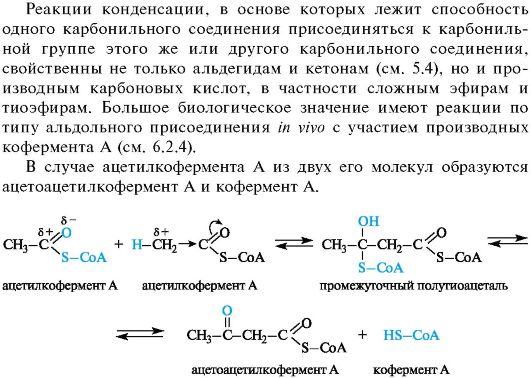
Приведенная схема реакции включает первоначальное образование нестойкого полутиоацеталя, который далее претерпевает распад
до карбонильного соединения и тиола, так как присоединение тиолов к группе С=О, как и аналогичная реакция спиртов с альдегидами и кетонами (см. 5.3), обратимо. Весь процесс в целом можно рассматривать как нуклеофильное замещение у карбонильного атома углерода по тетраэдрическому механизму.
Ацильные производные кофермента А не случайно выбраны природой в качестве реагентов в реакциях по типу альдольного присо- единения в организме, а также переносчиков ацильных групп. Уже отмечалось, что в тиоэфирах по сравнению с обычными сложными эфирами вследствие большего эффективного положительного заряда на карбонильном атоме углерода нуклеофильное замещение у карбонильного атома осуществляется легче. По той же причине α-атомы водорода в тиоэфирах более подвижны и более активны в реакциях конденсации.
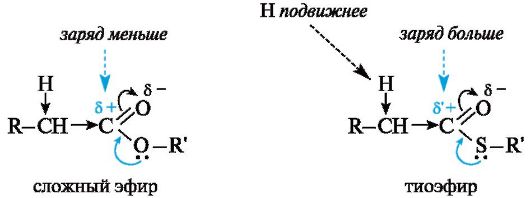
Это обусловлено меньшим (по сравнению с группой OR') +M- эффектом группы SR'.
Взаимодействие между вакантной 2р-АО углерода и Зр-ЛО серы менее эффективно, чем соответствующее взаимодействие с 2р-электронами атома кислорода, находящимися на том же электронном уровне и поэтому более близкими по энергии. В результате частичный положительный заряд на карбонильном атоме углерода в тиоэфирах выше, чем в сложных эфирах (δ'+ > δ+), и соответственно из-за большего -/-эффекта группы -COSR' по сравнению с -/-эффектом группы -COOR' α-атомы водорода в тиоэфирах более подвижны.
Другая важная реакция с участием ацетилкофермента А - взаимодействие с α-оксокислотами, приводящее к образованию β-гидроксикислот. В этом случае реакция аналогична смешанному варианту альдольной конденсации (см. 5.4).
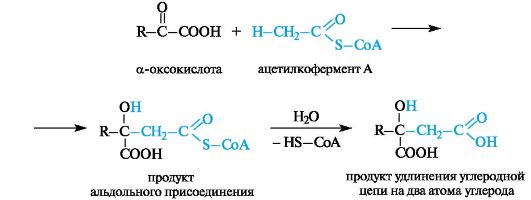
Последующий гидролиз тиоэфирной функции обеспечивает необратимость процесса в целом. Так происходит образование лимонной кислоты в цикле трикарбоновых кислот (см. 9.2.3).
Таким образом, важной функцией тиоэфиров в организме является их использование для создания новых углерод-углеродных связей.
10-1
транс-Изомеры высших жирных кислот
В последнее время большое внимание уделяется обнаруженным в природных пищевых продуктах транс-изомерам высших жирных кислот, имеющим в своем составе хотя бы одну двойную связь с транс-конфигурацией (транс-ВЖК). Обычное содержание транс- ВЖК составляет в сливочном масле 4-11%, в молоке (в пересчете на жир) 2-9%, в мясе жвачных животных 4-11%. В натуральных (недезодорированных) растительных маслах (оливковом, подсолнечном и др.) транс-изомеры ВЖК практически отсутствуют.
Интерес к транс-ВЖК возник в связи с обнаружением взаимосвязи между их употреблением и возрастанием риска сердечно-сосудистых заболеваний. транс-ВЖК могут выступать в роли конкурентных ингибиторов назаменимых ВЖК и, встраиваясь в фосфолипиды клеточных мембран, изменять их физические свойства, что может служить причиной различных метаболических и функциональных расстройств.
транс-ВЖК образуются в небольшом количестве в качестве промежуточных продуктов на начальных стадиях процесса биогидрирования
ненасыщенных высших жирных кислот (линолевой, линоленовой и др.). Например, при биогидрировании линолевой кислоты сначала происходит катализируемая анаэробными бактериями ее изомеризация в цис,транс-форму с расположением двойных связей между С-9-С-10 и С-11-С-12 соответственно. При таком расположении двойных связей в углеродной цепи появляется сопряженный диеновый фрагмент. Этот изомер линолевой кислоты называют «конъюгированной линолевой кислотой». Она может также образовываться путем свободнорадикального автоокисления линолевой кислоты. На дальнейших стадиях процесса биогидрирования образуется смесь вакценовой и элаидиновой кислот. Вакценовая кислота (С18:1) является основной транс-ВЖК, содержащейся в молочных и мясных продуктах. В ее молекуле двойная связь находится между атомами С-11 и С-12.
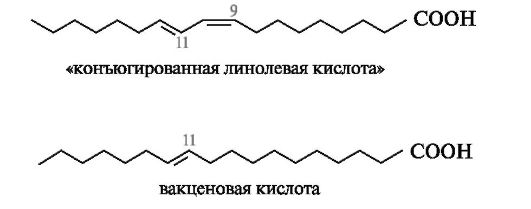
Оказалось, что осуществляемое в широком промышленном масштабе гидрирование масел растительного и морского происхождения с целью получения твердых пищевых жиров (маргаринов) сопряжено с цис-транс-изомеризацией ВЖК. «Легкие» маргарины (с содержанием жира менее 40%) содержат 5-10% транс-ВЖК. Однако в дешевых сор- тах маргаринов, сырье для получения которых значительно обогащено маслами морепродуктов, количество транс-ВЖК увеличено до 50%.
10-2
β-Окисление насыщенных высших жирных кислот
Основным топливом для организма служат углеводы. Ферментативное окисление жирных кислот также является существенным источником энергии. Некоторые детали процесса окисления насыщенных кислот, каждая отдельная стадия которого катализируется соответствующим ферментом, представлены на схеме (см. ниже). Начальная стадия процесса, в котором кислота участвует в активиро-
ванной форме (I), т. е. в виде производного кофермента А, представляет собой α, β-дегидрирование. Последующие стадии - гидратации и затем окисления - приводят к β-кетотиоэфиру (II), поэтому весь процесс в целом рассматривается как β-окисление.
Далее происходит альдольное расщепление (см. 5.4) β-кетотиоэфира (II) под действием коферемента А с образованием ацетилкофермента А и тиоэфира насыщенной кислоты (III), содержащей на два атома углерода меньше по сравнению с исходной, который подвергается новому циклу β-окисления.
Схема. β-Окисление насыщенных жирных кислот в виде производных кофер- мента А
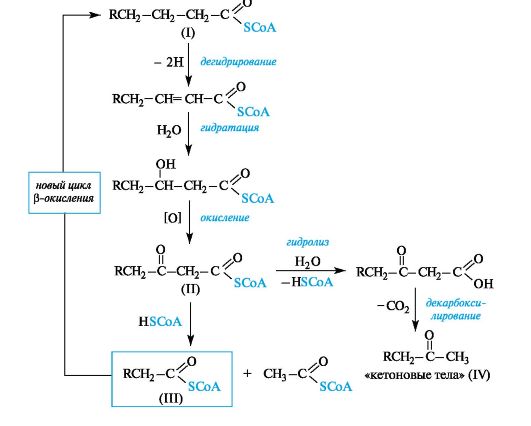
На конечной стадии процесса в случае ацетоацетилкофермента А (II, R = H) при недостаточной активности фермента, ответственного за его альдольное расщепление (см. 5.4), происходит гидролиз аце- тоацетилкофермента А с образованием так называемых «кетоновых тел» (см. 9.2.4) - ацетоуксусного эфира и продукта его декарбоксилирования - ацетона (IV, R = H), что может привести к метаболическому ацидозу.
11-1
Семейство D-альдоз
Семейство альдоз формально можно произвести от родоначального соединения - глицеринового альдегида - путем последовательного наращивания цепи на один атом углерода со стороны альдегидной группы. Так, введение звена СНОН в молекулу D-глицеринового альдегида порождает две D-тетрозы, имеющие тривиальные названия эритроза и треоза (схема). Отсюда, кстати, произошли названия конфигураций эритро- и трео-, означающих соответственно одинаковую и противоположную конфигурации двух хиральных центров. Аналогично из каждой тетрозы можно образовать по две пентозы (всего четыре D-альдопентозы), а из каждой пентозы - по две гексозы (всего восемь D-альдогексоз).
Все сказанное выше относится и к альдозам L-ряда, структуры которых не приводятся, но их легко представить, так как они являются зеркальными отражениями (энантиомерами) альдоз D-ряда.
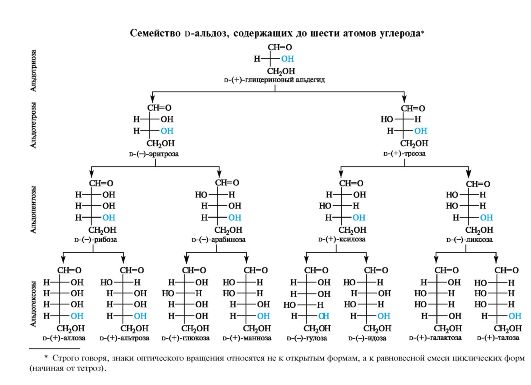
Сиаловые кислоты
Девятиуглеродные моносахариды (кетозы), называемые сиаловыми кислотами, являются уникальными поли- и гетерофункциональными соединениями. Их можно отнести и к альдоновым кислотам, и к дезокси- и аминосахарам. Наиболее значимые из них - нейраминовая кислота и ее производные: N-ацетил- и N-гликолилнейрами- новая кислоты, в которых аминогруппа ацилирована соответственно уксусной и гликолевой (гидроксиуксусной) кислотами.
Сиаловые кислоты распространены в природе как в свободном, так и в связанном виде. Они входят в состав групповых веществ крови, олигосахаридов молока, а также ганглиозидов мозга, участвующих в проведении нервных импульсов.
11-3
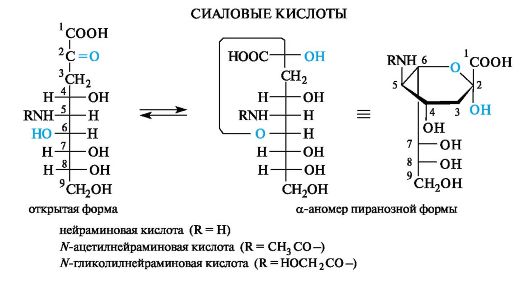
Участие фосфатов моносахаридов в биохимических процессах
Один из важных метаболических процессов - гликолиз - начинается с реакции фосфорилирования глюкозы с помощью АТФ в присутствии фермента глюкокиназы, обеспечивающего избирательное
взаимодействие с участием только первичноспиртовой группы. При этом происходит нуклеофильное замещение у атома фосфора с образованием хорошо уходящей группы в виде молекулы АДФ.
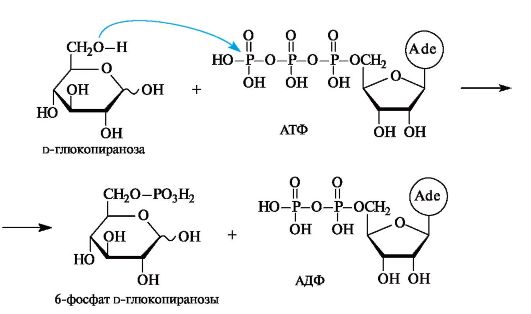
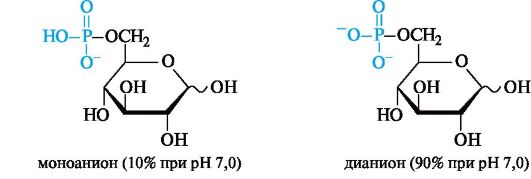
В приведенной выше полностью протонированной форме 6-фос- фат D-глюкозы может существовать в средах с рН ниже 6,0 (его рКа1 0,94, рКа2 6,11). В организме, где в живых клетках рН поддерживается равным 7,0, 6-фосфат D-глюкопиранозы находится преимущественно в форме дианиона.
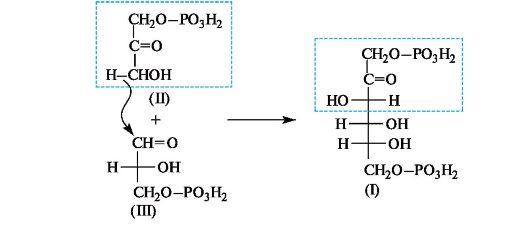
В животных организмах 1,6-дифосфат D-фруктозы (I) подвергается альдольному расщеплению, т. е. реакции, обратной альдольной конденсации. В результате образуются фосфаты двух триоз - дигидроксиацетона (II) и D-глицеринового альдегида (III), которые вовлекаются в дальнейшие превращения.
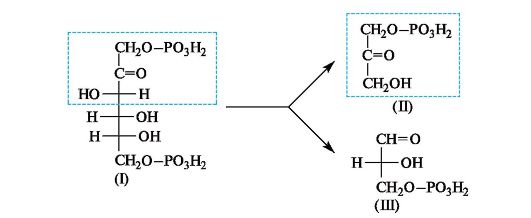
Интересно, что в растениях дифосфат фруктозы (I) образуется на одной из стадий фотосинтеза в результате реакции альдольной конденсации фосфата дигидроксиацетона (II) и фосфата глицеринового альдегида (III).
11-4
Медико-биологическое значение лактозы
Лактоза применяется в фармацевтической практике при изготовлении порошков и таблеток, так как она менее гигроскопична, чем сахар, а также как питательное средство для грудных детей. Лактоза имеет в 4-5 раз менее сладкий вкус, чем сахароза.
Содержание лактозы в женском молоке достигает 8%. Из женского молока выделено более 10 олигосахаридов, структурным фрагментом которых служит лактоза. Эти олигосахариды делят на две группы.
К первой принадлежат три-, тетра- и пентасахариды, соединенные с остатком сиаловой кислоты. Как показано ниже на простейшем примере, остаток сиаловой кислоты присоединен к лактозе в поло- жении 6 галактозного остатка.
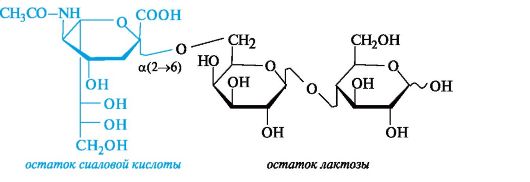
Ко второй группе относят олигосахариды, содержащие остаток L-фукозы (6-дезокси-L-галактозы), присоединенный к лакто- зе. Простейшим примером служит трисахарид, в котором остаток L-фукозы образует гликозидную связь с гидроксильной группой в положении 2 галактозного остатка лактозы.
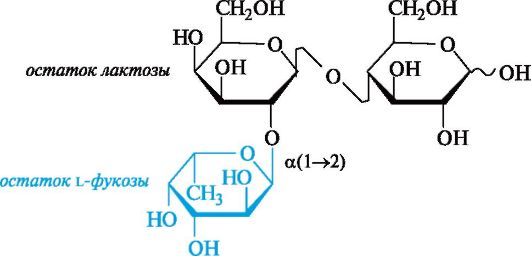
Эти олигосахариды имеют большое значение для формирования кишечной флоры новорожденных. Некоторые из них подавляют рост кишечных болезнетворных бактерий, с их действием связывают целебные свойства грудного молока. Сиалосодержащие олигосахариды активны против возбудителей столбняка и холеры.
11-5
Полисахариды: декстраны, хитин, пектиновые вещества, альгиновые кислоты
Декстраны
Эти полисахариды имеют бактериальное происхождение. В промышленности их получают микробиологическим путем при действии микроорганизмов Leuconostoc mesenteroides на растворы сахарозы.
Декстраны - разветвленные полисахариды, построенные из остатков a-D-глюкопиранозы. Основным типом связи являются a(1-6)-, а в местах разветвления - α(1-4)-, α(1-3)- и реже а(1-2)-гликозидные связи.
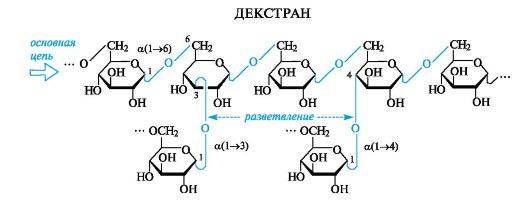
Декстраны используются как заменители плазмы крови, но большая молекулярная масса природных декстранов (несколько милли- онов) делает их непригодными для приготовления инъекционных растворов вследствие плохой растворимости. В связи с этим молекулярную массу снижают до 50-100 тыс. с помощью кислотного гидролиза или ультразвука и получают «клинические декстраны», например препарат полиглюкин. Декстраны обладают антигенными свойствами. Декстраны, синтезируемые обитающими на поверхности зубов бактериями, являются компонентами налета на зубах.
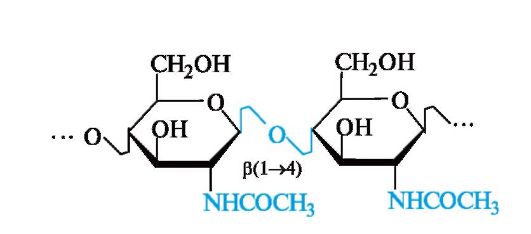
Хитин
Подобно целлюлозе в растениях, хитин выполняет опорные и механические функции в животных организмах (роговые оболочки насекомых, ракообразных и т. п.). Его пространственная упаковка имеет много общего с целлюлозой.
Хитин - линейный полисахарид, в котором остатки Ж-ацетил-D- глюкозамина связаны а(1^4)-гликозидными связями.
Пектиновые вещества
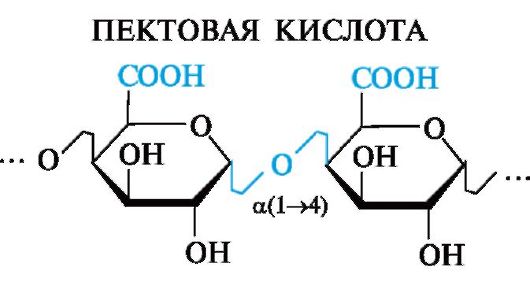
Пектиновые вещества содержатся в плодах и овощах, образуют гель в присутствии органических кислот, что используется в пищевой промышленности (желе, мармелад).
В основе пектиновых веществ лежит пектовая кислота, являющаяся полигалактуроновой кислотой.
Пектовая кислота - полисахарид, в котором остатки D-галактуро- новой кислоты связаны а(1^4)-гликозидными связями
Некоторые пектиновые вещества оказывают противоязвенное действие и являются основой ряда препаратов. Примером может служить плантаглюцид из подорожника.
Альгиновые кислоты
Альгиновыми кислотами называют неразветвленные полисахариды, в которых остатки D-маннуроновой кислоты и ее эпимера по С-5 - L-гулуроновой кислоты - связаны (1^4)-связями.
Альгиновые кислоты как гелеобразователи используются в пищевой промышленности. Источником альгиновых кислот служат бурые водоросли.
Из морских водорослей выделяют многие полисахариды. Например, широко применяемый в биохимических исследованиях агар представляет собой гетерополисахарид, содержащий большое число сульфат- ных групп. Агар состоит из смеси агарозы и агаропектина. В полисахаридной цепи агарозы чередуются остатки D-галактозы и L-галактозы.
11-6
Гликоконъюгаты: протеогликаны и гликопротеины
Многие структурные компоненты клеток представляют собой гликоконъюгаты - биополимеры, включающие углеводы, белки и липиды, причем существенное значение имеет доля того или иного компонента. Гликоконъюгаты с преобладанием полисахаридной части относятся к пептидогликанам и протеогликанам, полипептидной части - к гликопротеинам, липидной - к гликолипидам.
Протеогликаны
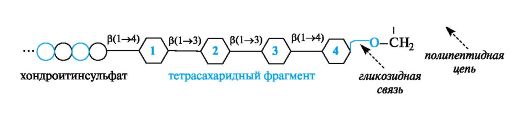
К протеогликанам относят группу углевод-белковых биополимеров, в которых преобладает углеводный компонент. Свойства протеогликанов определяются главным образом полисахаридными составляющими. Типичными представителями этой группы смешанных биополимеров являются протеогликаны соединительной ткани. Полисахаридная и полипептидная цепи соединены гликозидной связью. Полипептидная цепь выступает в роли агликона, поставляющего гидроксильную группу бокового радикала серина для образования гликозидной связи.
Связующим «мостиком» между хондроитинсульфатной цепью и полипептидом служит тетрасахаридный фрагмент. Он состоит последовательно из остатка D-глюкуроновой кислоты (1), двух остатков D-галактопиранозы (2 и 3) и остатка D-ксилопиранозы (4). Остаток D-ксилозы участвует в образовании гликозидной связи с остатком серина, входящего в состав полипептидной цепи. Поскольку остатки серина повторяются многократно, в целом к полипептидной цепи присоединяется много хондроитинсульфатных цепей.
Гиалуроновая кислота и хондроитинсульфатные протеогликановые субъединицы участвуют в образовании более высокоорга- низованных протеогликановых комплексов (агрегатов). С помощью электронографии установлена структура агрегатов хрящевой ткани. По внешнему виду она напоминает ерш для мытья бутылок, отсюда происходит ее название «щеточная структура». Центральную вытянутую часть агрегата (как бы проволочную основу ерша) составляет макромолекула гиалуроновой кислоты. Равномерно вдоль всей цепи гиалуроновой кислоты с интервалом, равным 10 моносахаридным звеньям, т. е. через каждые 30-50 нм, перпендикулярно основной оси располагаются хондроитинсульфатные протеогликановые субъединицы (щетина ерша).
В целом комплексы протеогликановой природы представляют собой поливалентные анионы, способные связывать катионы калия, натрия, кальция и таким образом участвовать в солевом обмене. Важную роль играют протеогликаны, содержащие гепарин.
Гликопротеины
В гликопротеинах с белковыми цепями ковалентно связаны олигосахаридные цепи (от одной до нескольких сотен на одну белковую цепь). Среди гликопротеинов известны ферменты, гормоны, компоненты плазмы крови, защитные белки (иммуноглобулины), муцины (слюна, секреты кишечника, бронхов).
В большинстве случаев олигосахаридная и белковая цепи связаны N-гликозидными связями, образуемыми концевыми остатками N-ацетилглюкозамина (со стороны олигосахарида) и амидной группой аспарагина (в составе белковой цепи).
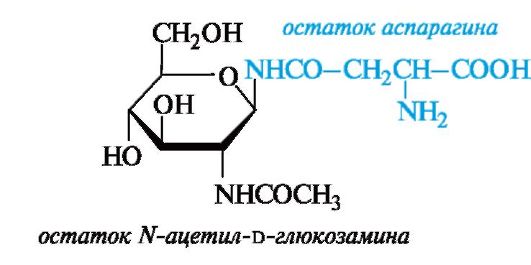
К гликопротеинам принадлежат вещества, определяющие групповую специфичность крови. Структурную основу этих веществ составляет полипептидная цепь, к которой присоединено до 55 олигосахаридных цепей, состоящих в среднем из 21-23 моносахаридных
остатков. Углеводная и пептидная части связываются между собой гликозидными связями с участием гидроксильных групп остатков серина или треонина.
Последовательности из трех-пяти моносахаридных звеньев на невосстанавливающих концах олигосахаридных цепей, называемые детерминантами, определяют групповую специфичность крови. Например, детерминантные олигосахариды групп крови А и В в системе АВ0(Н) различаются тем, что в детерминанте А концевой моносахарид - Ж-ацетил^-галактозамин, а в детерминанте В - D-галактоза. В олигосахариде группы 0(Н) нет ни того, ни другого. С изменением детерминанта меняется группа крови. Так, в 1970-х годах в эксперименте in vitro было показано, что в результате обработки эритроцитов с детерминантом В ферментом галактозидазой отщепляется остаток галактозы и детерминант В превращается в детерминант 0(Н), т. е. из эритроцитов III группы были получены эритроциты I группы крови.
Антигенные детерминанты могут входить в состав не только гликопротеинов, но и гликолипидов. Это свидетельствует о важной роли углеводов в проявлении защитных функций организма (иммунитета).
12-1
α-Аминокислоты D-стереохимического ряда
Использование для построения белков человеческого организма только одного вида стереоизомеров α-аминокислот, а именно энантиомеров L-ряда, имеет важнейшее значение для формирования про- странственной структуры белков. С этим непосредственно связана стереоспецифичность действия ферментов. Макромолекулы ферментов, построенные из α-аминокислот, т. е. хирального материала, в целом являются хиральными и поэтому вступают во взаимодействие только с теми субстратами, которые также имеют определенную конфигурацию.
α-Аминокислоты D-ряда называют иногда «неприродными», так как они не используются для построения белков человеческого организма. В то же время D-аминокислоты встречаются в пептидах, про- дуцируемых микроорганизмами, например в антибиотиках (грамицидин, актиномицин, полимиксин), а также в составе биополимеров
клеточной стенки бактерий, например остаток D-глутаминовой кислоты - в оболочке бактерий сибирской язвы. Против этого вида бактерий бессильны расщепляющие ферменты человека и животных.
Аминокислоты, относящиеся к разным стереохимическим рядам, различаются по вкусу. Так, D-глутаминовая кислота безвкусна, а L-глутаминовая кислота имеет вкус мяса. L-Глутаминовую кислоту, получаемую путем гидролиза клейковины пшеницы, применяют в виде глутамата натрия в качестве вкусовой добавки к пищевым концентратам. Сладкий вкус имеют, как правило, аминокислоты D-ряда: валин, лейцин, треонин, метионин, аспарагиновая кислота, тирозин, триптофан, гистидин. Однако их энантиомеры либо безвкусные, либо горькие. Из изомеров L-ряда сладким вкусом обладают лишь аланин, серин и пролин. В этом отношении аминокислоты привлекают внимание как возможные заменители сладких веществ углеводной природы для больных диабетом.
12-2
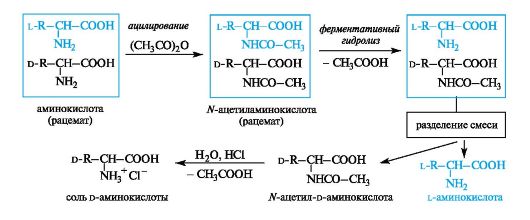
Расщепление рацематов аминокислот ферментативным путем
Рацемическую аминокислоту ацетилируют уксусным ангидридом и получают смесь N-ацетилированных аминокислот, которую обрабатывают ферментом, специфически гидролизующим производные только L-ряда. Свободная аминокислота способна растворяться как в кислотах, так и щелочах, а Ж-ацетиламинокислота - только в щелочах. После отделения L-изомера можно получить D-амино- кислоту с помощью гидролиза оставшегося ацетилированного про- изводного.
Расчет значений изоэлектрической точки

12-4
Некоторые превращения α-аминокислот in vivo c участием кофермента пиридоксальфосфата
Ряд важных химических превращений аминокислот, осуществляемых в организме под действием различных ферментов, имеет общий механизм, обусловленный участием одного и того же кофермента - пиридоксальфосфата (см. 13.4.1).
В результате взаимодействия между альдегидной группой пиридоксальфосфата и аминогруппой α-аминокислоты образуется имин, для которого в биохимической литературе принято название альдимин I (в условиях организма альдимин I протонирован по пиридиновому атому азота).
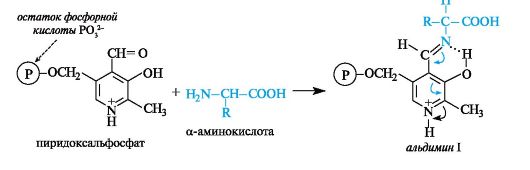
В альдимине I электронная плотность сопряженной системы смещена к протонированному пиридиновому атому азота, в результате чего происходит сильная поляризация связей α-атома углерода аминокислоты. В зависимости от того, какая из этих связей будет принимать участие в дальнейшей реакции (что определяется природой фермента) с аминокислотой, могут осуществляться процессы трансаминирования, декарбоксилирования и др. Эти существенно различающиеся по конечному результату процессы реализуются через общую стадию образования альдимина I.
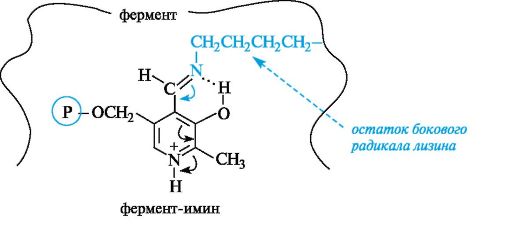
В действительности аминокислота взаимодействует с комплексом ковалентно связанных между собой кофермента и фермента. Пиридоксальфосфат за счет альдегидной группы образует альдиминную связь с аминогруппой бокового радикала лизинового остатка в активном центре фермента. Кроме того, кофермент связан с ферментом и другими связями (фосфатная группа, протонированный пиридиновый атом азота и др.).
Образовавшийся комплекс, назовем его для краткости «фермент-имин», обладает довольно высокой реакционной способностью по отношению к аминокислоте, что связано с большей электрофильностью атома углерода в составе иминной группы по сравнению с альдегидной. Это объясняется более выраженной способностью атома азота иминной группы к протонированию по сравнению с атомом кислорода альдегидной группы.
Трансаминирование
Процесс происходит с участием кофермента пиридоксальфосфата, который выполняет функцию переносчика аминогруппы от донор- ной аминокислоты к акцепторной оксокислоте с промежуточным переходом в форму пиридоксаминфосфата, т. е. пиридоксальфосфат ведет себя как акцептор, а пиридоксаминфосфат - как донор аминогруппы.
Процесс трансаминирования происходит путем последующего превращения альдимина I с участием полярной связи между α-атомом углерода и атомом водорода. СН-кислотный центр и соответственно подвижный атом водорода создают условия для ряда прототропных таутомерных превращений. Альдимин I, отщепляя протон, переходит в промежуточную «хиноидную» форму, в которой путем присоединения протона по двойной связи восстанавливается ароматичность и образуется кетимин. При гидролизе кетимина получаются пиридоксаминфосфат и α-оксокислота.
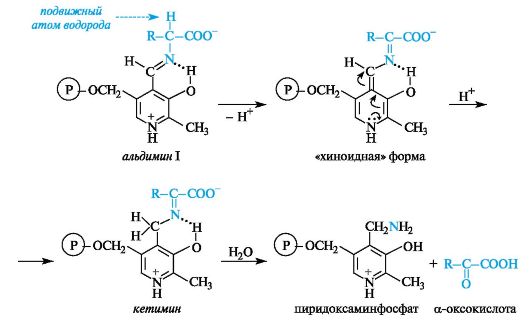
Пиридоксаминфосфат способен взаимодействовать в обратном направлении с акцепторной α-оксокислотой, в результате чего получается аминокислота и «возвращается» пиридоксальфосфат.
Реакция трансаминирования является связующим звеном между процессами метаболизма белков (аминокислоты) и углеводов (α-оксокислоты). С помощью этой реакции устраняется избыток отдельных аминокислот и таким образом регулируется содержание аминокислот в клетках.
Декарбоксилирование
Процесс происходит с участием ферментов декарбоксилаз и кофермента пиридоксальфосфата. Эта реакция осуществляется путем
разрыва в альдимине I полярной связи между α-атомом углерода и карбоксилатной группой. Промежуточная «хиноидная» форма в результате присоединения протона превращается в альдимин Ia, после гидролиза которого получаются пиридоксальфосфат и амин.
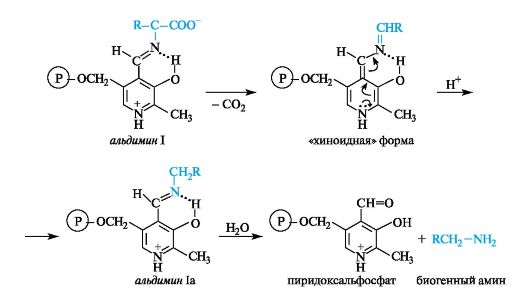
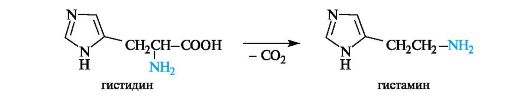
Биогенные амины в организме выполняют важные биологические функции. Например, получающийся при декарбоксилировании гистидина биогенный амин гистамин оказывает разностороннее биологическое действие и участвует в регуляции жизненно важных функций организма.
γ-Аминомасляная кислота (ГАМК), образующаяся при декарбоксилировании глутаминовой кислоты, является нейромедиатором. Большое биологическое значение имеет декарбоксилирование мно- гих природных аминокислот - серина, цистеина, лизина, триптофана, аспарагиновой кислоты и др.
12-5
Некоторые биологически активные пептиды - гормоны, нейропептиды, токсины
Дипептиды
Представителями самых коротких пептидов являются содержащиеся в мышцах животных и человека карнозин (β-аланил-L-гисти- дин) и ансерин β-аланил-1-метил-L-гистидин). В их состав входит остаток необычной аминокислоты - β-аланина (структурный изомер аланина).

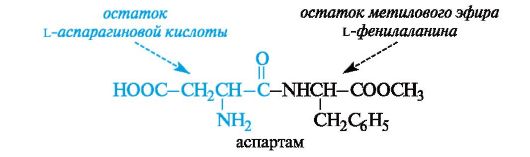
В настоящее время в промышленном масштабе выпускается дипептид аспартам, почти в 200 раз более сладкий, чем сахароза. Аспартам состоит из остатков L-аспарагиновой кислоты и метилового эфира L-фенилаланина.
Пептидные гормоны
Большое значение имеет группа пептидов, проявляющих гормональные свойства, т. е. регулирующих химические реакции в организме. Многие из них детально изучены, некоторые открыты совсем недавно.
В
(3) и лейцина (8) в окситоцине в вазопрессине содержатся остатки фенилаланина (3) и аргинина (8). Оба пептида содержат одну дисульфидную связь, а на конце вместо свободной группы СООН - амидную CONH2.
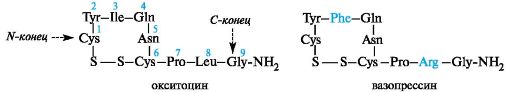
Небольшого различия в аминокислотной последовательности оказывается достаточно для обеспечения специфичности биологического действия каждого из этих гормонов. Окситоцин встречается только у женских особей. Он вызывает сокращение гладкой мускулатуры, особенно мускулатуры матки, и применяется в гинекологии и акушерстве в ветеринарии. Вазопрессин содержится и в женском, и в мужском организме. Он регулирует минеральный обмен и баланс жидкости (антидиуретический гормон). Вазопрессин относится к числу мощных стимуляторов запоминания.
Ответственный за контроль метаболизма углеводов, жиров и белков гормон инсулин вырабатывается
поджелудочной железой. С недостатком инсулина в организме связаны
серьезные нарушения углеводного обмена (сахарный диабет). Молекулярная
масса инсулина 5727, моле- кулярная формула C259H377N65O75S6. Для того чтобы эту молекулярную формулу «превратить» в формулу строения, потребовались годы напряженного труда: в
Инсулин состоит из двух пептидных цепей - А и Б. Цепь А содержит 21, а цепь Б - 30 аминокислотных остатков. Эти цепи соединены двумя дисульфидными мостиками. Кроме того, в цепи А имеется дисульфидная связь между Cys-6 и Cys-11, вызывающая образование петли.
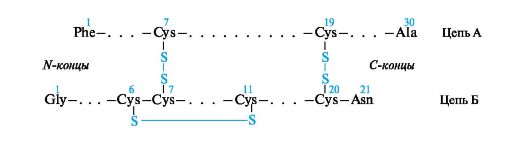
Видовая специфичность инсулинов связана с изменениями на участке 8-10 цепи А. Например, в инсулине человека на этом участке имеется последовательность аминокислотных остатков Thr-Ser-Ile, в инсулине быка - Ala-Ser-Val, барана - Ala-Gly-Val, лошади - Thr-Gly-Ile.
Нейропептиды
К ним относятся пептиды, содержащиеся в головном мозге. Первые два представителя нейропептидов, названные энкефали- нами, были выделены из мозга животных в

Пептидные токсины
Пептидно-белковую природу имеют многие токсичные вещества, например токсины ядовитых грибов, пчел, змей, скорпионов.
Пептид апамин является токсичным компонентом яда пчел и сильно воздействует на ЦНС. Апамин содержит 18 аминокислотных остатков. Это один из низкомолекулярных нейротоксинов. Наименьшую молекулярную массу имеют нейротоксины из морского моллюска - конотоксины, состоящие из 13-15 аминокислотных остатков. Конотоксины очень токсичны.
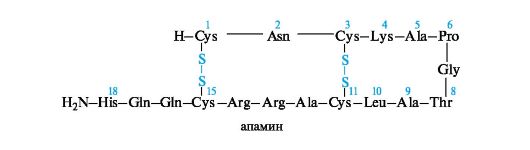
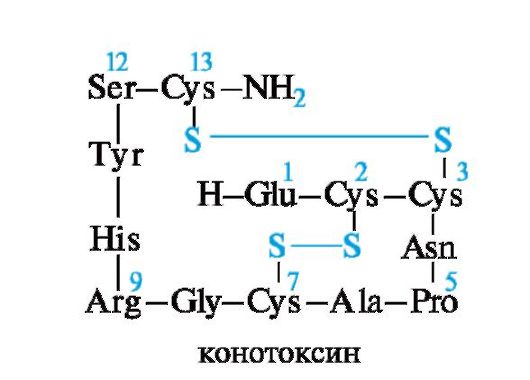
Изучение строения и физиологического действия токсинов представляет интерес не только для их обезвреживания, но и для моде- лирования аналоговых лекарственных средств.
13-1
Тетрапиррольные соединения - порфирины
Плоский
макроцикл порфина представляет собой сопряженную ароматическую систему
из 26 π-электронов (11 двойных связей и две неподеленные пары
электронов пиррольных атомов азота). Замещенные порфины называют порфиринами, один из которых - протопорфирин - показан ниже. О высокой термодинамической стабильности порфиринов говорит тот факт, что они устойчивы до температуры около
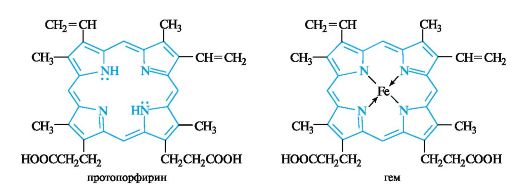
Порфирины в природе находятся в виде комплексов с ионами металлов. Комплекс протопорфирина с ионом Fe2+ служит простетической группой гемопротеинов, к которым относятся кислородпе- реносящие белки, в частности гемоглобин, цитохромы и некоторые ферменты. Производные порфиринов, содержащие ион железа(11),
называют гемами. Примером может служить протогем (чаще называемый гемом) - простетическая группа гемоглобина. Четыре атома азота в геме образуют плоский квадрат с атомом железа в центре.
Комплексы порфиринов с ионом магния лежат в основе структуры хлорофиллов.
13-2
Каталитическое действие имидазольного фрагмента в реакциях ферментативного гидролиза
Особенности строения имидазольного кольца объясняют важную роль гистидина в некоторых ферментативных реакциях, в частности его способность осуществлять кислотный (за счет пиррольной группы NH) и основный (за счет пиридинового атома азота) катализ.
Являясь одновременно и донорами, и акцепторами протонов, имидазол и его производные обладают уникальной способностью катализировать реакции нуклеофильного замещения в функциональных производных карбоновых кислот. В приведенном примере гидролиза сложного эфира имидазол, образуя водородную связь с молекулой воды, повышает ее нуклеофильную активность. Кроме того, переход протона в тетраэдрическом интермедиате от имидазолий-катиона к атому кислорода спиртового остатка способствует отщеплению хорошо уходящей группы - молекулы спирта.
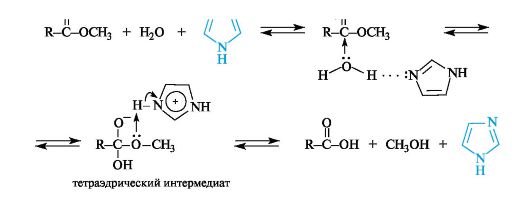
Это свойство имидазола играет важную роль в механизме действия гидролитических ферментов, расщепляющих пептидные связи в белках. В активном центре таких ферментов содержатся остатки аминокислоты гистидина.
Гетероциклические производные сульфаниламидов
Более чем за 70 лет существования сульфаниламидных препаратов было синтезировано около 10 тыс. производных, различающихся главным образом заместителем R в сульфонамидной группе. Реже модификации структуры затрагивали ароматическую аминогруппу. Несколько десятков сульфаниламидов применяют в медицине, причем наибольшую активность проявляют те производные, у которых радикал R имеет гетероциклическую природу.
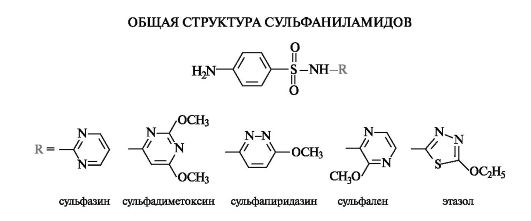
Антибактериальное действие сульфаниламидов основано на том, что они являются антиметаболитами по отношению к n-амино- бензойной кислоте, участвующей в биосинтезе фолиевой кислоты (см. Приложение 13-5) в микроорганизмах. Амид сульфаниловой кислоты имеет структурное сходство с n-аминобензойной кислотой.
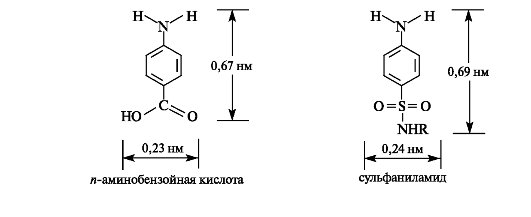
О механизме антибактериального действия сульфаниламидов. Сульфаниламиды конкурируют с и-аминобензойной кислотой на стадии образования птероевой кислоты и связываются с птеридиновым фрагментом. Сульфамидная группа препятствует дальнейшему взаимодействию с глутаминовой кислотой, и биосинтез фолиевой кислоты прекращается, что ведет к гибели бактерий. Избирательность антибактериального действия сульфаниламидов основана на том, что фолиевая кислота в человеческом организме не синтезируется. Таким образом, сульфаниламиды блокируют метаболические реакции, существенные для определенных бактерий (пневмококки, стрептококки и др.), и в то же время не влияют на организм человека.
13-4
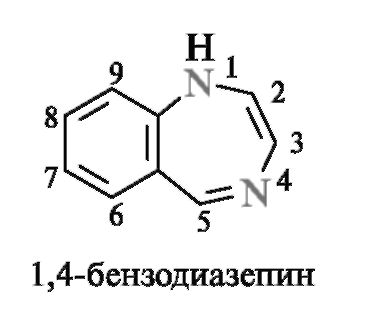
Диазепины - семичленные гетероциклы, содержащие два атома азота
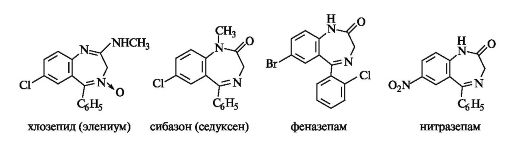
Диазепинами называют семичленные гетеро- циклы с двумя атомами азота. Наиболее изучены из них конденсированные системы - бензодиазеиины. Первым представителем группы диазепинов был хлозепид (элениум), применяемый до настоящего времени в качестве анксиолитического средства (транквилизатора).
За последние 2-3 десятилетия синтезированы тысячи соединений диазепинового ряда, многие из которых нашли практическое применение. К ним относятся транквилизаторы сибазон (известный еще как седуксен), феназепам и снотворное средство нитразепам.
13-5
Производные птеридина - структурные фрагменты фолиевой кислоты и рибофлавина
Бициклическая структура птеридина образована конденсирован- ными ядрами пиримидина и пиразина. Эта система ароматична, устойчива к действию окислителей, проявляет слабые основные свойства.
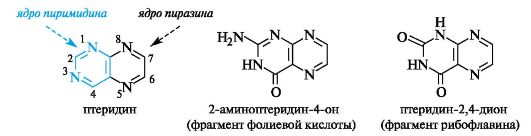
В природе довольно широко распространены оксо- и аминоптеридины. В частности, фрагменты оксопроизводных присутствуют в фолиевой кислоте и рибофлавине (витамин В2, см. Приложение 14-3), являющемся фактором роста живых организмов.
Фолиевая кислота (витамин Вс) помимо птеридинового фрагмента включает остатки L-глутаминовой и п-аминобензойной кислот (ПАБК). Обе функциональные группы ПАБК участвуют в образовании связей с двумя другими компонентами.
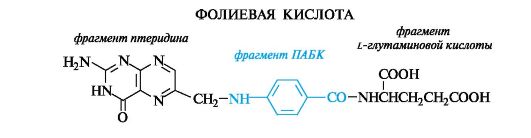
Фолиевая кислота играет важную роль в метаболизме нуклеиновых кислот и белков (см. также Приложение 13-3).
14-1
Нарушение комплементарных взаимодействий
в молекуле ДНК как причина возникновения мутаций
Комплементарность цепей составляет химическую основу важнейшей функции ДНК - хранения и передачи наследственных признаков. Сохранность нуклеотидной последовательности является залогом безошибочной передачи генетической информации. Однако нуклеотидная последовательность ДНК под действием различных факторов может подвергаться изменениям, которые назы- вают мутациями.
Мутации вызывает, в частности, воздействие химических факторов, а также различных видов излучения. Например, если на аденозин подействовать азотистой кислотой, то в результате известной реакции дезаминирования аминогруппа превратится в гидроксильную (см. 4.3), вследствие чего из аденозина получается другой нуклеозид - инозин, содержащий гипоксантин.
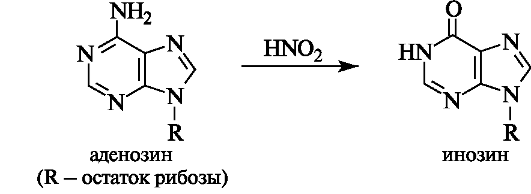
Это может привести к замене в ДНК комплементарной пары оснований, так как аденин комплементарен тимину, а инозин может образовывать комплементарную пару только с цитидином.
14-2
Участие АТФ в переносе ацильных групп
Перевод карбоновых кислот в ангидридную форму составляет химическую основу активации жирных кислот, аминокислот, желчных кислот, необходимой для их участия в последующих превращениях. При этом в состав образующихся ангидридов со стороны АТФ может входить либо остаток фосфорной кислоты (ацилфосфаты), либо остаток АМФ (замещенные ацилфосфаты - ациладенилаты).
Например, в процессе превращения глутаминовой кислоты в глутамин, играющий важную роль в обмене азота в организме, одной из промежуточных стадий является образование ацилфосфата. Непосредственного взаимодействия глутаминовой кислоты с аммиаком не происходит из-за слабой электрофильности атома углерода карбоксильной группы. Однако эта реакция может осуществляться в организме с участием АТФ (в присутствии фермента глутаминсин- тетазы). Глутаминовая кислота при этом образует ангидрид с остатком фосфорной кислоты и таким образом становится более активной на последующей стадии ацилирования аммиака.
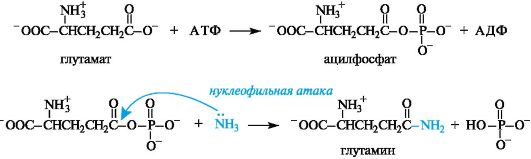
В организме активация карбоксилсодержащих соединений осуществляется также в результате образования ациладенилатов - смешанных ангидридов карбоновых кислот и АТФ.
Например, активация жирных кислот в процессе их β-окисления (см. Приложение 10-2) заключается в образовании ими тиоэфиров. Суммарно этот процесс можно представить в следующем виде:
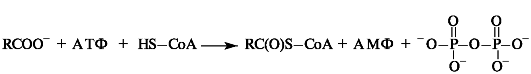
Процесс протекает через промежуточное образование ациладенилата путем взаимодействия жирной кислоты с АТФ.
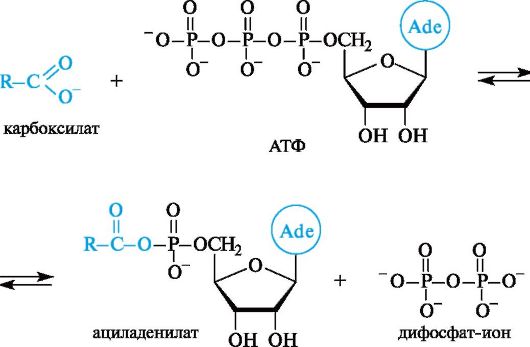
Примером фундаментального процесса жизнедеятельности, в котором происходит образование ациладенилатов, служит биосинтез белка. Сначала α-аминокислота, участвующая в синтезе белка, под- вергается активации с помощью АТФ.
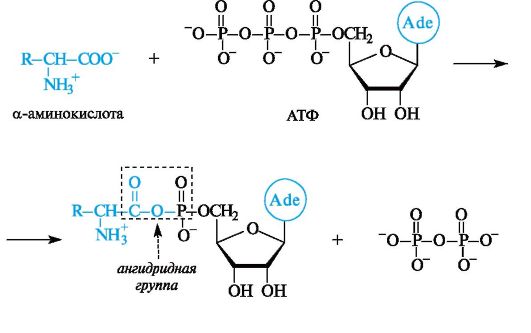
Активированная таким образом α-аминокислота далее взаимодействует с соответствующей ей транспортной РНК (тРНК). Химическая основа этого взаимодействия состоит в ацилировании З'-ОН-группы остатка адениловой кислоты, находящегося на З'-конце тРНК, которая транспортирует связанную с ней α-аминокислоту в рибосому, т. е. к месту синтеза белка.
14-3
Флавинадениндинуклеотид - кофермент окислительно-восстановительных процессов
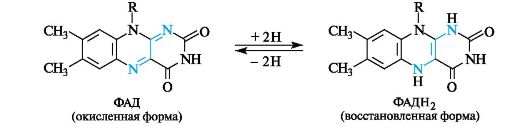
Флавинадениндинуклеотид (FAD, или ФАД) выполняет роль окислителя в некоторых окислительно-восстановительных процессах.
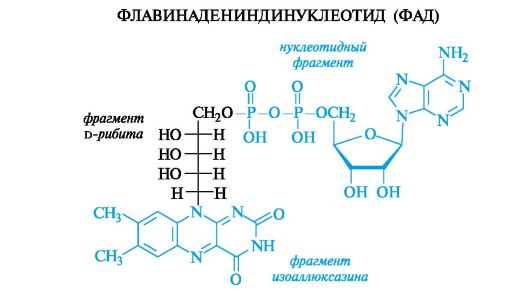
Кроме того, ФАД является метаболически активной формой рибофлавина (витамин В2). В структуру рибофлавина входят остаток D-рибита и гетероциклическая система изоаллоксазина, включающая фрагмент 2,4-диоксоптеридина (см. также Приложение 13-5).
Изоаллоксазин, имеющий ярко-желтый цвет, получил название флавин (от. лат. flavus - желтый) и соответственно витамин В2 - название рибофлавин. За окислительно-восстановительный процесс ответственна изоаллоксазиновая система, способная присоединять два атома водорода (2Н) с образованием восстановленной формы ФАДН2. Процесс обратим.
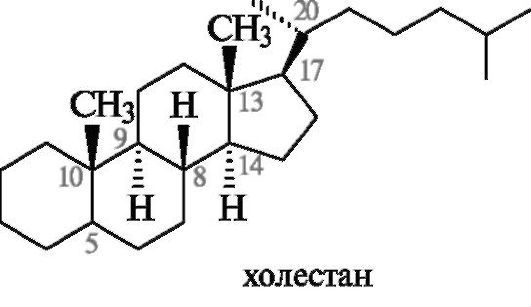
Систематическая номенклатура стероидов
В основе систематических названий стероидов лежат стереородоначальные структуры гомологов гонана: эстран, андростан и др., приведенные в табл. 15.1. В них, как показано на примере холестана, заложена не только структура, но и транс-сочленения колец В/С и С/D, а также R-конфигурация атома С-20. Тип сочленения колец А и В следует при необходимости указывать симво-
лами 5α или 5β (см. рис. 15.1).
Наличие двойных связей и функциональных групп отражают в названии с помощью суффиксов и префиксов по общим правилам заместительной номенклатуры. Если введение заместителя приводит к возникновению нового хирального центра, то локант заместителя дополняют символом α или β в зависимости от его расположения относительно условной плоскости цикла. В качестве примера приведено систематическое название гормона эстриола.
Следует обратить внимание на необычное обозначение бензольного кольца как триеновой системы, в которой локант 5(10) означает двойную связь между атомами С-5 и С-10 (а не С-5 и С-6, как в обычном обозначении ненасыщенности).
Далее приведены систематические названия большинства стероидных соединений, упомянутых в главе 15:
холестерин - холестен-5-ол-3β
копростанол - 5β-холестанол-3;
эргостерин - эргостатриен-5,7,22-ол-3β (от углеводорода эргостана - 24-метилхолестана);
холевая кислота - 3α,7α,12α-тригидрокси-5β-холан-24-овая кислота;
альдостерон - 11β,21-дигидрокси-3,20-диоксопрегнен-4-аль-18; кортикостерон -11β,21-дигидроксипрегнен-4-дион-3,20;
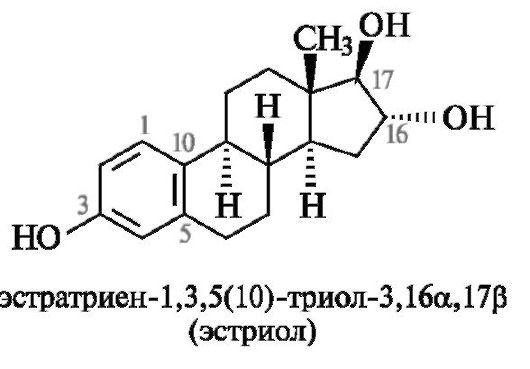
преднизолон - 1β,17,21-тригидроксипрегнадиен-1,4-дион-3,20;
прогестерон - прегнен-4-дион-3,20;
эстрон - 3-гидроксиэстратриен-1,3,5(10)-он-17;
эстрадиол - эстратриен-1,3,5(10)-диол-3,17;
андростерон - 3α-гидрокси-5α-андростанон-17;
тестостерон - ^-гидроксиандростен-4-он-3.
15-2
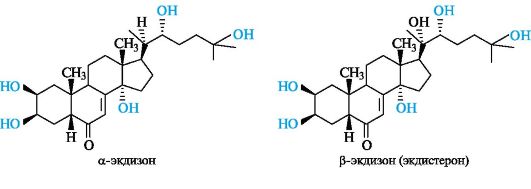
Фитоэкдистероиды
В
Экдистероиды широко распространены в растительном мире. Некоторые растения содержат довольно много (до 2% и более) экдистероидов, например левзея сафлоровидная (маралий корень). Содержащийся в корнях и корневищах этого растения β-экдизон, называемый также экдистероном, тонизирует организм и повышает физическую работоспособность.
В настоящее время идентифицировано более 250 фитоэкдистероидов. Возрастающий интерес к ним обусловлен широким спектром их биологической активности, особенно иммуностимулирующим и адаптогенным действием.
Экдистероиды имеют следующие общие структурные признаки в скелете холестана:
• цис-сочленение колец А/В;
• двойная связь между атомами С-7 и С-8;
• кетонная группа в положении 6;
• гидроксильные группы в положениях 2, 3 и 14, реже в положениях 1, 5 и 11;
• алкильная боковая цепь у атома С-17, как правило, с гидроксильной группой у атома С-22.
Экдистероиды содержат от 5 до 8 гидроксильных групп. Наиболее известными представителями фитоэкдистероидов являются α- и
β-экдизоны. α-Экдизон представляет собой 2β,3β,14 α,(22R),25-пента- гидрокси-5β-холестен-7-он-6. β-Экдизон (экдистерон) по сравнению с α-экдизоном содержит дополнительную группу ОН в положении 20 и является (20^)-гидроксиэкдизоном (2β,3β,14 α,(20R),(22R),25-гекса- гидрокси-5β -холестен-7-он-6).
15-3
Пути биосинтеза терпенов и стероидов
Родство терпенов и стероидов подтверждается возможностью их синтеза в живых организмах из единого предшественника - типич- ного изопреноида сквалена. В качестве исходного вещества используется уксусная кислота. В форме ацетилкофермента А она конденсируется сначала в ацетоацетилкофермент А, который далее по типу альдольной конденсации присоединяет по карбонильной группе новую молекулу ацетилкофермента А с образованием монотиоэфира 3-гидрокси-3-метилпентандиовой кислоты (I).
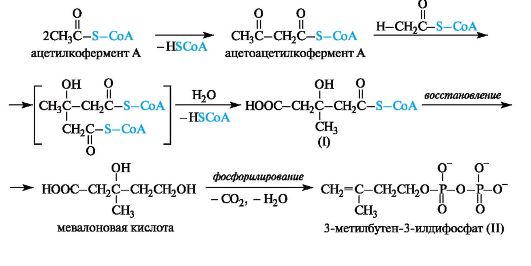
Ферментативное восстановление этой кислоты приводит к мевалоновой кислоте. Последняя с потерей СО2 и Н2О фосфорилируется в 3-метилбутен-3-илдифосфат (II), служащий строительным блоком в синтезе изопреноидов.
3-Метил-3-бутенилдифосфат (II) находится в равновесии с изомерным 3-метилбутен-2-илдифосфатом (III), который может легко ионизироваться, образуя достаточно стабильные дифосфат-ион и катион аллильного типа (IV), и, таким образом, играть роль алкилирующего реагента.
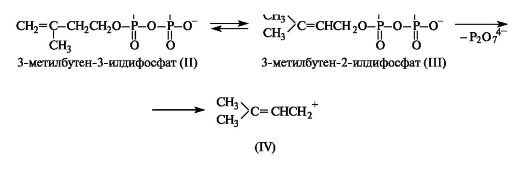
Этот реагент служит источником электрофила (IV) в живых организмах при алкилировании производных одно- и двухатомных фенолов, которое приводит к образованию некоторых коферментов и жирорастворимых витаминов.
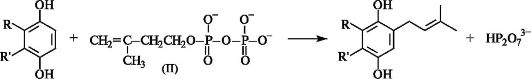
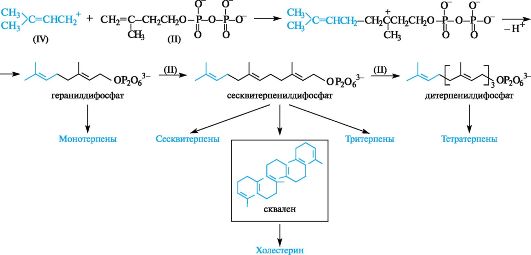
В биосинтезе терпенов и стероидов (схема - см. ниже) последовательное алкилирование катионом (IV) 3-метилбутен-3-илдифосфата (II) ведет к монотерпену геранилдифосфату, сесквитерпенилдифосфату и далее к дитерпенилдифосфату. Последние два соединения затем либо превращаются в соответствующие терпены, либо димеризуются в три- и тетратерпены. При этом из сесквитерпенилдифосфата образуется сквален, который и служит предшественником холестерина и, следовательно, остальных стероидов в организме.
Биосинтез терпенов и стероидов
Основные группы семейства флавоноидов
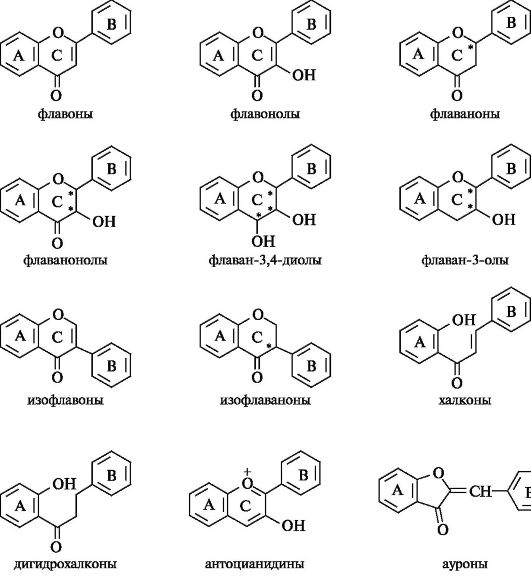
Флавоны, флаваноны
Флавоны, флаваноны и их разнообразные производные составляют самую многочисленную группу в семействе флавоноидов. Общим структурным признаком является наличие в базовой структуре оксогруппы в положении 4; отличительный признак состоит в наличии
двойной связи между атомами С-2 и С-3 у флавонов и отсутствии таковой у флаванонов. Традиционно принято отражать гидроксильную группу в положении 3 (если она имеется) путем добавления суффикса -ол, например флавонолы, флаванонолы (или дигидрофлавонолы).
Базовой структурой флавонов является конденсированная система бензольного кольца А и γ-пирона (477-пиранона-4), т. е. система 4H-1-бензопиранона-4, содержащая фенильный заместитель в положении 2 (см. 15.5)
Базовой структурой флаванонов является конденсированная система бензольного кольца А и 2,3-дигидро-γ-пирона (2,3-дигидро- 4H-пиранона-4), т. е. система 2,3-дигидро-47
H-1-бензопиранона-4, содержащая фенильный заместитель в положении 2 (см. 15.5)
Множественность флавоноидных соединений обеспечивается многообразием моделей замещения в базовой структуре. Варианты замещения различаются числом, местоположением, природой и сочетанием разных заместителей. Всем флавоноидам присущи гидроксильные заместители. Основной моделью гидроксизамещения в кольце А флавонов и флаванонов является наличие гидроксильных групп в положениях 5 и 7.
В наибольшей степени многочисленность флавоноидных соединений обусловлена их способностью образовывать гликозилированные производные - О-гликозиды. Каждое флавоноидное соединение, выступая в роли агликона, может стать прародителем множества своих гликозилированных форм - моногликозидов, дигликозидов, биозидов.
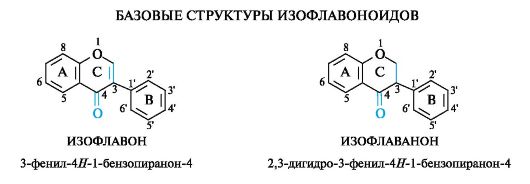
Изофлавоноиды
Структурное отличие изофлавоноидов состоит в перемещении фенильного заместителя В к атому С-3 кольца С.
Базовой структурой изофлавонов является конденсированная система бензольного кольца А и γ-пирона (4H-пиранона-4), т. е. система 4/ -1-бензопиранона-4, содержащая фенильный заместитель в положении 3 гетероциклического кольца С.
Базовой структурой изофлаванонов является конденсированная система бензольного кольца А и 2,3-дигидроγ-пирона (2,3-дигид- ро-4H-пиранона-4), т. е. система 3-дигидро-4H-1-бензопиранона-4, содержащая фенильный заместитель в положении 3 гетероциклического кольца С.
Антоцианидины составляют относительно немногочисленную группу среди других флавоноидных соединений.
Базовой структурой антоцианидинов является конденсированная система бензольного кольца с пирилий-катионом, т. е. система бензопирилий-катиона, содержащая фенильный заместитель в положении 2.
БАЗОВАЯ СТРУКТУРА АНТОЦИАНИДИНОВ
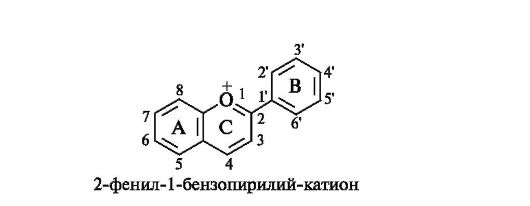
Наиболее известными являются шесть антоцианидинов (агликонов), имеющих одинаковую модель гидроксизамещения:
• в положениях 5 и 7 кольца А;
• в положении 4' кольца В;
• в положении 3 кольца С.
В растениях антоцианидины почти всегда находятся в виде гликозидов. Гликозиды антоцианидинов называют антоцианами. Антоцианы, как правило, бывают 3-моногликозидами или 3,5-диглико- зидами.
Флаван-3-олы
Важнейшими среди флаван-3-олов являются соединения, называемые зачастую катехинами.
Базовой структурой катехинов является конденсированная система бензольного и пиранового колец, т. е. система 3,4-дигидро-2Н- 1-бензопирана, содержащая фенильный заместитель в положении 2, имеющая название флаван.
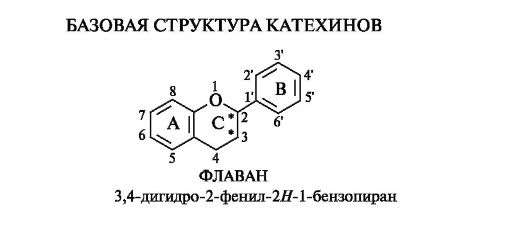
В катехинах в положении 3 гетероциклического кольца С всегда содержится гидроксильная группа. Отсюда их название - флаван- 3-олы. Типичным для флаван-3-олов является 5,7-дигидроксиза- мещение кольца А; в кольце В гидроксильные группы занимают положения 3',4' или 3',4',5'.
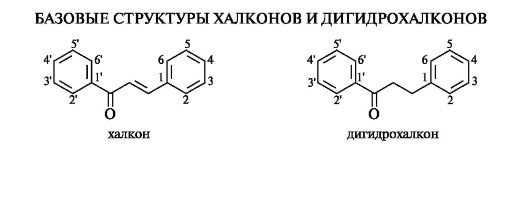
Халконы
Халконы и родственные им дигидрохалконы с позиций химического строения составляют обособленную группу в семействе флавоноидов, поскольку в своей базовой структуре они не содержат гетероциклического кольца С.
Эйкозаноиды
К эйкозаноидам относятся кислородсодержащие производные эйкозаполиеновых кислот, т. е. неразветвленных карбоновых кислот С20 с двумя двойными связями и более, разделенными метиленовой группой.
Эйкозаноиды подразделяются на:
• простаноиды - простагландины (ПГ), простациклины (ПГ-I), тромбоксаны (ТО);
• лейкотриены (ЛТ).
К настоящему времени простагландины являются наиболее изученным классом эйкозаноидов. Они обладают чрезвычайно высокой биологической активностью и широким спектром действия. Единственным местом их образования первоначально считали предстательную железу (простату) - отсюда они и получили свое название. В настоящее время простагландины в малых количествах найдены в большинстве тканей млекопитающих.
С химической точки зрения простагландины - функционально замещенные жирные кислоты С20, которые можно рассматривать как производные не найденной в природе, но полученной синтетическим путем простановой кислоты.
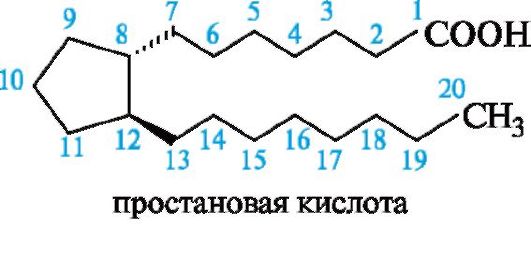
Скелет простановой кислоты в простагландинах включает одну, две или три двойные связи, одну или две гидроксильные группы, а также может содержать карбонильную группу.
В зависимости от природы и положения заместителей в циклопентановом кольце простагландины обозначают буквами A, B, C, D, E и F.
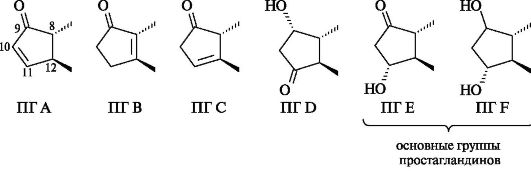
Каждая группа простагландинов далее по числу двойных связей в боковых цепях делится на три серии, как показано ниже на примере простагландинов группы Е.
• ПГ Ej - одна двойная связь транс-конфигурации;
• ПГ Е2 - дополнительно цис-С-5-С-6 двойная связь, т. е. всего две двойные связи;
• ПГ Е3 - дополнительно к ПГ Е2 цис-С-17-С-18 двойная связь, т. е. всего три двойные связи в боковой цепи.
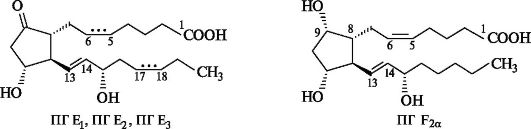
Обозначения α или β после цифрового индекса в простагландинах типов D и F показывают ориентацию гидроксильной группы у атома С-9. Так, α указывает на цис-, а β - на транс-конфигурации этой группы и углеродной цепи при С-8. Стабильные природные простагландины относятся к типам Е и F.
Простагландины являются сильнодействующими биологически активными веществами. Например, их содержание в сперме человека (основном их источнике) составляет 10-6 моль/л, а действие на глад- кую мышцу проявляется уже при концентрациях около 10-9 моль/л. Спектр их биологического действия весьма широк. Они расширяют кровеносные сосуды, ингибируют свертывание крови и выделение желудочного сока, стимулируют работу кишечника, легких и бронхов, активируют синтез гликогена в печени. Отмечено их влияние
на процессы нервного возбуждения, половой цикл у женщин. Так как простагландины вызывают сокращение матки, их можно использовать для стимуляции родовой деятельности или предотвращения беременности.
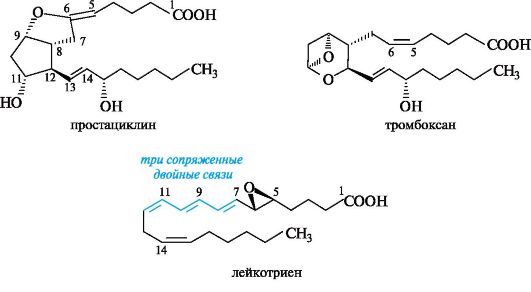
Помимо простагландинов, в настоящее время интенсивно исследуются и другие эйкозаноиды:
• простациклины - вещества, предупреждающие образование тромбов и способствующие расширению сосудов;
• тромбоксаны - нестойкие, но весьма активные вещества, образующиеся в тромбоцитах и ответственные за инициирование сложного механизма, приводящего к формированию тромба;
• лейкотриены - синтезируемые в лейкоцитах активаторы иммунных ответов.
Типичные представители этих соединений приведены ниже.
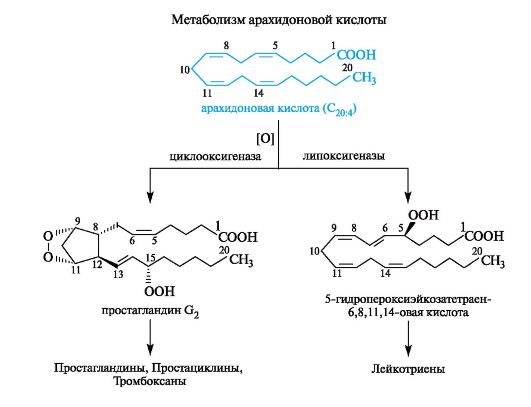
На приведенной ниже схеме в упрощенном виде представлены превращения арахидоновой кислоты, в которой указаны два главных пути биосинтеза различных простаноидов. Первый катализируется ферментом циклооксигеназой и приводит к простагландину G2 и затем к ряду других простагландинов, а также простациклинам и тромбоксанам. Второй путь, катализируемый ферментом липоксигеназой, включает первоначальное образование 5-гидро- пероксиэйкозатетраен-6,8,11,14-овой кислоты и ее последующее пре- вращение в различные лейкотриены.
15-6
Антибиотики пептидной, аминогликозидной и нуклеозидной природы
Пептидные антибиотики. Некоторые пептиды оказывают антибактериальное действие и используются как лекарственные средства. Так, грамицидин С - цик- лический декапептид, действующий на стрептококки, пневмококки и другие мик- роорганизмы, продуцируется споровой палочкой Bacillus brevis. Грамицидин С был выделен Г.Ф. Гаузе и М.Г. Бражниковой (1942). К циклопептидам относятся антибиотики полимиксины. В состав грамицидина С наряду с ранее известными α-аминокислотами входит L-орнитин H2N(CH2)3CH(NH2)COOH, который в орга-
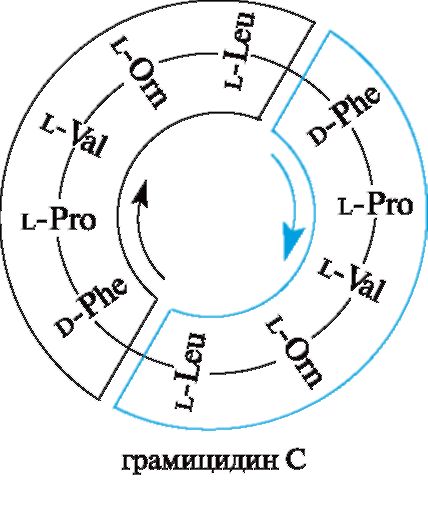
низме человека образуется из аргинина в метаболическом цикле мочевины, но в составе белков человеческого организма не содержится.
Грамицидин С способен быть ионофором, т. е. переносчиком ионов через мембраны. В частности, с его помощью через мембрану переносятся ионы K+, а также Na+ и другие одновалентные катионы.
Ионофором является и другой циклический пептид - валиномицин, способный специфически связывать и переносить ионы калия. В валиномицине наряду с пептидными содержатся и сложноэфирные группы, в образовании которых участвуют α-гидроксикислоты - молочная (2-гидроксипропановая) и α-гидроксиизовалериановая (2-гидрокси-3 -метилбутановая).
Циклическая молекула валиномицина построена из трех идентичных фрагментов. В состав каждого из них последовательно входят остатки D-валина, L-молочной кислоты, L-валина и D-гидроксиизо- валериановой кислоты.
Конформация валиномицина напоминает браслет, внутренний радиус которого точно соответствует ионному радиусу иона K+, который таким образом оказывается «окутанным» гидрофобной оболочкой валиномицина и легко переносится через мембраны. С ионами Na+ валиномицин практически не взаимодействует. Ведется поиск синтетических ионофоров, среди которых наибольшего внимания заслуживают краун-эфиры (см. 8.2).
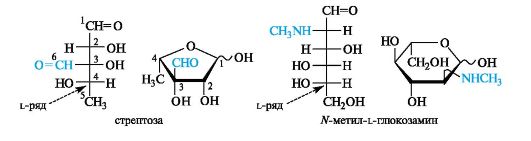
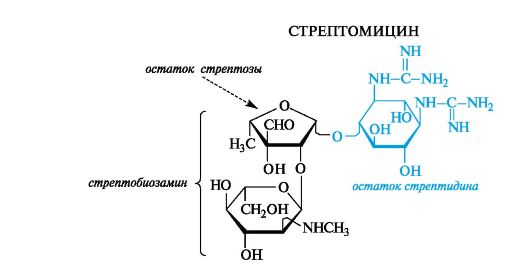
Антибиотики-аминогликозиды. Этот вид антибиотиков продуцируют бактерии. В состав антибиотиков входят углеводные фрагменты, обязательно включающие аминосахара. Типичный представитель - стрептомицин - является гликозидом стрептобиозамина. Указанный дисахарид состоит из остатков двух необычных моносахаридов - стрептозы, имеющей дополнительную альдегидную группу, и алкилированного по аминогруппе N-метил-L-глюкозамина. Оба компонента относятся к L-ряду, что свойственно моносахаридам бактериального происхождения.
Агликоном стрептомицина служит дигуанидиновое производное инозита - стрептидин. Гуанидиновые фрагменты обусловлива- ют основные свойства, и в медицинской практике стрептомицин используется в виде сульфата.
Группа аминогликозидных антибиотиков включает в себя более 100 природных соединений. В качестве агликона все они содержат аминопроизводные инозита (см. 7.2.2). Относящиеся к этой группе известные антибиотики неомицин, канамицин, сизомицин обладают широким спектром действия.
Антибиотики-нуклеозиды. В клетках в свободном состоянии содержатся некоторые нуклеозиды, не являющиеся компонентами нуклеиновых кислот. Эти нуклеозиды обладают антибиотической активностью и приобретают все большее значение при лечении злокачественных новообразований. Известно несколько десятков таких нуклеозидов, выделенных из микроорганизмов, а также из растительных и животных тканей.
Нуклеозиды-антибиотики отличаются от обычных нуклеозидов некоторыми деталями строения либо углеводной части, либо гетероциклического основания. По-видимому, это позволяет им выступать в роли антиметаболитов. Нуклеозидные антибиотики пиримидинового ряда часто подобны цитидину, пуринового ряда - аденозину.
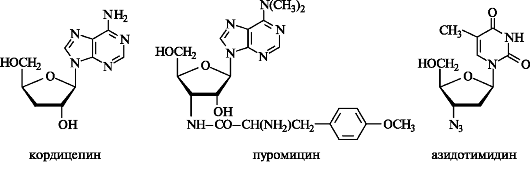
Например, выделенный из грибницы Cordyceps militaries антибиотик кордицепин отличается от аденозина только отсутствием в угле-
водном остатке З'-ОН-группы. Сильными антибиотическими свойствами обладает пуромицин, выделенный из культуральной жидкости Streptomyces alboniger.
Пуромицин представляет собой З'-амино-З'-дезокси-N,N-диметил- аденозин, ацилированный по 3'-аминогруппе остатком О-метилтирозина. Он является ингибитором рибосомального синтеза белка.
Азидотимидин подавляет размножение ВИЧ.
Некоторые микроорганизмы выделяют вещества нуклеозидной природы, в состав которых вместо рибозы входит ее эпимер по атому С-2 - D-арабиноза. Например, сильными антивирусными и антигрибковыми свойствами обладают арабинозилцитозин и арабинозиладенин.
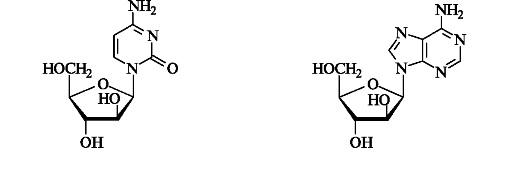
Как видно из приведенных выше примеров, «небольшой» разницы в строении или конфигурации одного атома углерода (С-2) в углеводном остатке достаточно, чтобы вещество выполняло роль ингибитора биосинтеза ДНК. Этот принцип используется при создании новых лекарственных средств методом молекулярной модификации природных моделей.