Клиническая иммунология : учебник / под ред. А.М. Земскова. - 2008. - 432 с.
|
|
|
|
ГЛАВА 7. ИММУНОПРОЛИФЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ
Группа этих заболеваний объединяет патологические иммунопролиферативные процессы, которые исходят из клеток иммунной системы. Патология включает широкий спектр состояний от доброкачественных инфекций (инфекционный мононуклеоз) до нарушений злокачественного характера. Среди иммунопролиферативных состояний можно выделить ситуации с выраженным клеточным полиморфизмом или преобладанием однотипных клеточных форм.
7.1. ЛИМФОГРАНУЛЕМАТОЗ (БОЛЕЗНЬ ХОДЖКИНА)
Характеризуется поражением лимфоидной ткани. Клинически проявляется общей слабостью, субфебрилитетом, потливостью, кожным зудом, потерей массы тела. Иногда первым симптомом является увеличение лимфатических узлов (нижнечелюстных, подмышечных, средостения, паховых). Позднее гипертрофируется печень и селезенка. Как правило, дальнейшее течение заболевания отягощается инфекционными осложнениями. У больных, не получивших лечения, продолжительность заболевания составляет 1-2 года с последующим летальным исходом, хотя иногда возможны длительные спонтанные ремиссии.
Отмечено 2 возрастных пика ЛГМ: ранний взрослый и старше 50 лет. Среди вирусных поражений первое место занимают herpes simplex с хроническим или генерализованным течением, часто заболевание осложняется туберкулезом, бруцеллезом, грибковыми инфекциями. Среди протозойных следует отметить Pn. carinii, токсоплазмоз. В терминальных стадиях - инфекции, вызванные стафилококками, кишечной палочкой, псевдомонадами.
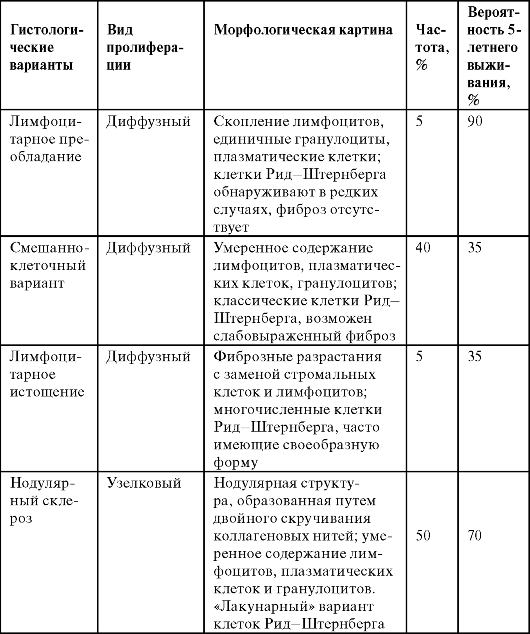
Диагностика включает в первую очередь биопсию и гистологическое исследование периферических лимфатических узлов.
Таблица 5. Морфологическая характеристика болезни Ходжкина (Л. Йегер,
1986)
 Кроме
этого, у больных снижается выраженность клеточного иммунитета - кожные
пробы, например, на динитрохлорбензол бывают отрицательными коррелятивно
со стадией заболевания.
Кроме
этого, у больных снижается выраженность клеточного иммунитета - кожные
пробы, например, на динитрохлорбензол бывают отрицательными коррелятивно
со стадией заболевания.
Пр одукция АТ, кроме терминальной стадии, обычно не страдает. Однако исчезает качественная и количественная разница между первичным и повторным иммунным ответом (эффективность второго резко падает). Иногда в терминальной стадии несколько возрастает концентрация γ-глобулинов в крови. В то же время уровень IgM всегда снижен, даже в начале заболевания.Резко тормозится функциональная активность лимфоцитов, что оценивается РТБЛ. В основном подавляется ответ на Т-митогены (Кон-А и ФГА), реакция на В-митоген (лаконоса) более сохранена.
В периферической крови, как правило, обнаруживается умеренная лимфопения, в терминальной стадии она существенно увеличена. Падает также содержание лимфоцитов в лимфатических узлах (этот критерий диагностически значим).
Значительно уменьшается число Е-РОК CD3 -лимфоцитов. Уровень В-клеток может быть нормальным, но впоследствии он снижается.
Возрастает содержание недифференцированных нулевых лимфоцитов.
Активность К-клеток блокируется, особенно при прогрессировании заболевания.
В 30-60% случаев обнаруживаются антилимфоцитарные АТ.
Образуются неспецифические ингибиторы, например, β-липопротеи-ды, к ним более чувствительны CD3(Т)-клетки больных ЛГМ, чем здоровых лиц.
Возрастает активность супрессорных клеток.
Нарушается хемотаксис фагоцитарных клеток, снижается их метаболическая активность.
В 50-80% случаев увеличивается концентрация ЦИК, но четкой их корреляции с заболеванием нет.
Выделены четыре стадии болезни Ходжкина. Первая характеризуется поражением одного лимфоузла. Вторая - поражением двух и более групп лимфоузлов, расположенных по одну сторону диафрагмы. Третья - поражением двух групп лимфоузлов по обе стороны диафрагмы, возможно, и селезенки. Четвертая - диффузным или диссеминированным вовлечением одного или более экстранодальных органов или печени с наличием или отсутствием поражения лимфоузлов.
Этиология заболевания - вирусная (избирательное поражение CD3(Т)-клеток), опухолевая, возможно, существует генетическая предрасположенность.
Лечение включает лучевую и химиотерапию, экстирпацию пораженных узлов.
7.2. НЕХОДЖКИНСКИЕ ЛИМФОМЫ
Лимфомы образуются при иммунодефицитных состояниях (атаксии-телеангиэктазии, синдром Вискотта-Олдрича); при приеме супрессорных средств у реципиентов с трансплантированными органами и соответствующим лечением вероятность формирования лимфом возрастает в 200 раз; при сопутствующих заболеваниях: СКВ, дерматомиозите, болезни Шегрена. Не исключена и вирусная этиология индукции заболевания.
Неходжкинские лимфомы составляют около 2% всех случаев злокачественных опухолей человека с частотой 2,6-5,8 на 100000 населения.
Клиника лимфом различна. На первом месте стоит поражение лимфоидных тканей, возможны экстранодулярные опухоли в костном мозге, желудочно-кишечном тракте, печени. Заболевание может иметь злокачественный характер с продолжительностью жизни в несколько месяцев, однако встречаются случаи выживания без лечения до 10 лет.
Обычно для лечения используют лучевую и химиотерапию. В последние годы разрабатываются иммунотерапевтические подходы:
- неспецифическая стимуляция, например, вакциной БЦЖ или бактериальными Аг;
- левамизол;
- антилимфоцитарная гетерологичная сыворотка.
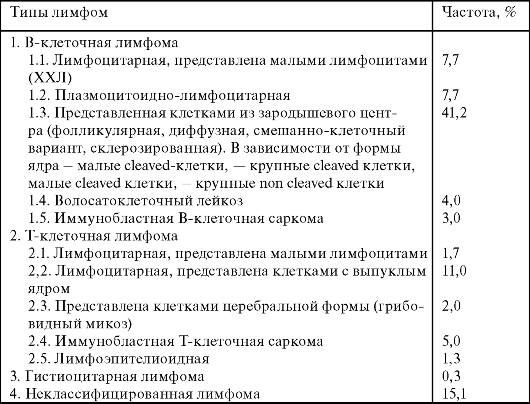
Клинические формы неходжкинских лимфом
Хронический лимфолейкоз чаще поражает пациентов в возрасте 40-70 лет. Это наиболее частый вариант лейкемических форм (25%). Соотношение заболеваемости у мужчин и женщин составляет 2-3:1. Доказана его связь с иммунными дефицитами. У большинства пациентов имеется спленомегалия, увеличение (генерализованное) лимфоузлов. Из экстранодулярных локализаций следует отметить кожу (эритродермии, инфильтрации), печень, желудочно-кишечный тракт и урогенитальный тракт. Содержание лейкоцитов составляет от 20000 до 100000/мкл.
Таблица 6. Классификация неходжкинских лимфом (по Lukes и Collins)
 У больных обычно подавлен клеточный иммунитет:
У больных обычно подавлен клеточный иммунитет:
- снижен ответ (CD3-лимфоцитов) на ФГА, но сохранен на РWМ;
- кожные реакции на динитрохлорбензол и др. снижены;
- подавлена способность к антителообразованию;
- снижен уровень иммуноглобулинов, особенно уменьшен уровень IgM;
- заторможены CD4-лимфоциты;
- угнетены реакции фагоцитоза.
Иммунные расстройства вызывают инфекционные осложнения, обусловленные в первую очередь стафило- и пневмококками, реже вирусным и грибковым агентами ( ветряная оспа, герпес). Чаще всего наблюдаются пневмонии. Второе место занимают бактериальные инфекции кожи.
Принято считать, что ХЛЛ является вариантом новообразований.
Т-клеточный лейкоз - довольно редкая форма хронического лимфолейкоза (5-7% случаев).
У больных подавлен пролиферативный ответ лимфоцитов на ФГА в среднем на 30%, на митоген лаконоса - на 25%.
При Т-клеточном лейкозе часто обнаруживают значительную полиморфную картину с постепенными переходами лейкозных клеток в форму малых клеток Сезари. Клиническая картина заболевания проявляется опухолевым процессом в селезенке при значительно меньшей инфильтрации в коже и нейтропенией. Прогноз относительно благополучен.
Пролимфоцитарный лейкоз также является особой формой хронического лимфолейкоза. Болезнь обычно поражает лиц преклонного возраста. При выраженной гепатоспленомегалии лимфатические узлы обычно не увеличены. Уровень лейкоцитов повышен. Клетки преимущественно лимфоидного типа, с крупным в форме пузырька ядрышком, относительно грубой сетью хроматина и широким ободком цитоплазмы. В более чем 90% случаев пролиферирующие клетки идентифицируются как CD19(B)-, значительно реже как CD3(T)- клетки.
Синдром Сезари - сочетание кожного зуда с эксфолиативной эритродермией: процессом инфильтрации кожи лимфоидными клетками и лейкемизацией периферической крови аналогичными клетками. До 90% клеток белой крови могут составлять клетки Сезари. Их размер 14-20 мкм, они обладают крупным дольчатым ядром.
Волосатоклеточный лейкоз представляет опухолевый процесс в лимфатической системе. Клинические симптомы проявляются постепенно, признаками спленомегалии; в лимфатических узлах, напротив, отмечается снижение клеточной массы. В селезенке происходит выраженная инфильтрация красной пульпы, возможна цитопения. Течение заболевания хроническое, с продолжительностью от нескольких месяцев до 6-8 лет. Характерными признаками заболевания являются особый вид клеток с многочисленными выростами цитоплазмы различной длины, определяемые в периферической крови и костном мозге. Это атипичные CD19(В)-клетки.
Острый лимфолейкоз - заболевание преимущественно детского возраста. У взрослых ОЛЛ развивается обычно после ХЛЛ (бластный криз). Около 80% всех случаев острого лейкоза у детей имеют лимфоцитарную природу. Известны несколько вариантов ОЛЛ:
- Т-клеточный, который развивается в основном у мальчиков. Клиническими проявлениями являются: гепатоспленомегалия, опухоль средостения. Значительно выражен лейкоцитоз;
- В-клеточный, встречается очень редко (3-5%). На мембранах лимфоцитов обнаруживаются глобулины, рецепторы к Fc- фрагменту IgG и С3-компоненту комплемента;
- Ни Т-, ни В-тип - наиболее часто встречаемая форма с благоприятным исходом.
Высокая степень инфицированности при ОЛЛ связана с дефицитом уровня гранулоцитов (концентрация IgG может быть нормальной), уменьшением количества циркулирующих СD3-клеток, снижением их функциональной активности. Обнаружен сывороточный фактор, угнетающий ответ лимфоцитов на ФГА.
Иммунобластная лимфоаденопатия (типа В, ангиобластная лимфоаденопатия с гиперглобулинемией, лимфогранулематоз). Морфологическая картина заболевания сходна с лимфогранулематозом. Характерны следующие признаки: иммунобластная пролиферация с плазмоцитоидными иммунобластами и плазматическими клетками: пролиферация в малых разветвленных сосудах, прежде всего посткапиллярных венулах, с ШИК-положительными клетками, отложением аморфной, ацидофильной субстанции в интерстициальной ткани. Заболевание развивается обычно в зрелом возрасте и характеризуется генерализованным увеличением лимфатических узлов (100% случаев), нередко с гепато- и спленомегалией (60%), повышением температуры тела (70%), потливостью, потерей массы тела (45%), кожным зудом (65%), макулопапулезными поражениями кожи (40%). Эозинофилию периферической крови регистрируют в 20%. Обычно содержание CD3-клеток уменьшено, а CD19-лимфоцитов увеличено. Часто наблюдается образование аутоантител против эритроцитов, лимфоцитов, Аг гладкой мускулатуры, резус-фактора. Средняя продолжительность жизни от момента диагностирования составляет 15 мес, но могут быть спонтанные ремиссии.
Терапия состоит в основном в назначении кортикостероидов, реже - цитостатиков.
Крупнофолликулярная лимфобластома (болезнь Брилла-Симмерса). Относительно редкое заболевание, составляющее 5-10% от всех случаев злокачественных лимфом, поражающее лиц разного возраста. Главным клиническим симптомом является генерализованное или локальное увеличение лимфатических узлов с постепенным развитием патологического процесса. У 20-60% больных регистрируется увеличение селезенки, часто поражается слизистая оболочка желу-
дочно-кишечного, реже - генитального трактов и органов дыхания. Летальный исход обычно наступает через несколько лет.
Гистиоцитарная лимфома (ретикулосаркома). Клиническая картина данного заболевания может развиваться при хроническом лимфолейкозе, болезни Вальденстрема, при других фолликулярных лимфомах. Пролиферация имеет В-клеточную природу, сохраняя вид «гистиоцитарного» процесса. Иногда выявляются клетки плазматического ряда. Крайне редко пролиферирующие клетки имеют Т-маркер.
Лимфоматоидный гранулематоз характеризуется полиморфной инфильтрацией различных органов. У больных регистрируется поражение легких, кожи, почек, центральной нервной системы и печени. Этиология заболевания не выяснена, прогноз неблагоприятный, летальный исход составляет 65-90%. При переходе данного процесса в лимфому рекомендуется активное использование цитостатиков. Примерно в половине случаев возможна ремиссия.
Периферическая Т-клеточная лимфома характеризуется генерализованной лимфоаденопатией, потерей массы тела и часто встречающимся патологическим процессом в легких. Средняя продолжительность заболевания составляет 9 мес.
Гистиоцитоз Х объединяет редкую группу заболевания: болезнь Леттера-Сиве и эозинофильную гранулему. Разные клинические проявления характеризуются общими признаками - образование гранулемы с гистиоцитарной инфильтрацией и пролиферацией. Полагают, что данное заболевание имеет в своей основе комбинированный иммунодефицит и РТПХ.
7.3. ЛИМФОПРОЛИФЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ, ОБУСЛОВЛЕННЫЕ ВИРУСОМ ЭПСТАЙНА-БАРР
Этот вирус широко распространен. У 90% новорожденных в крови обнаруживаются АТ (материнские) против ЭБ. К 5 годам дети приобретают собственный иммунитет против вируса. Вообще же вирусом ЭБ инфицировано 70-90% всего населения, в большинстве случаев - это латентное вирусоносительство, а реактивация вируса сдерживается нормально работающей иммунной системой, и инфекция течет субклинически, характеризуясь только положительными серологическими реакциями. При возникновении дефектов иммунной системы развивается инфекция, поскольку ее вирусный контроль нарушен.
Чаще инфицирование происходит в возрасте 4-20 лет, передача вируса осуществляется воздушно-капельным и бытовым путем через
рук и, предметы обихода. Предполагают трансмиссивную передачу (москиты), передачу с кровью, половой и трансплацентарный путь.
Вирус является В-лимфотропным и инфицирует долгоживущие CD19-клетки памяти IgD-субтипа, трансформирует зрелые и незрелые CD19-лимфоциты. Однако доказано инфицирование им эпителиальных клеток носо- и ротоглотки, канальцев слюнных желез и тимуса, моноцитов и макрофагов, нейтрофилов, фолликулярных дендритных клеток, Т-лимфоцитов и НК. Вирус ЭБ вызывает различные иммунопролиферативные заболевания.
Лимфома Беркитта - локальная опухоль лимфоидной ткани с тенденцией к быстрому росту. Преимущественно поражается верхняя челюсть в области моляров и премоляров. Другие локализации - брюшная полость, забрюшинный отдел, яичники, печень, позвоночник, череп. В основном болеют дети в возрасте 4-7 (3-12 лет), мальчики в 3 раза чаще, чем девочки. Лимфома распространена в определенных районах Африки, в странах с отсутствием тропического климата встречается редко.
Лечение включает хирургическое удаление опухоли, можно использовать лучевую и химиотерапию. При признаках поражения ЦНС препараты необходимо вводить в спинномозговой канал.
Рецидивы наблюдаются в двух формах. Ранние - в первые 3 мес, связаны с первичной локализацией, как правило, не купируются. При поздних рецидивах обнаруживаются новые локализации, более благоприятные к терапии. Прогноз лечения зависит от размеров опухоли. Доказана прямая связь вируса ЭБ с возникновением рака молочной железы, легких и желудка, карциномы тимуса и кожной Т-клеточной лимфомы, болезни Ходжкина и неходжкинских лимфом, недифференцированного рака носоглоточного типа.
Инфекционный мононуклеоз - острое доброкачественное пролиферативное заболевание лимфатической системы. Поражаются в основном лица молодого возраста. Классическая форма начинается с острой фазы, наступающей после 20-50-дневного инкубационного периода. Основные симптомы: повышение температуры тела, увеличение лимфатических узлов (преимущественно шейных), фарингит, ангина. Лимфаденит может быть генерализован. В крови увеличивается содержание лейкоцитов до 15000-20000. Более 60% из них - мононуклеары (лимфо- и моноциты), снижается содержание сегментоядерных лейкоцитов. Диагностически значимым является определение в крови вируса Эпстайна-Барр. В острой фазе снижается выраженность
кожной реакции на туберкулин, РБТЛФГА.
Несмотря на то, что инфекционный мононуклеоз является вирусным заболеванием, он одновременно имеет признаки лимфопролиферативного процесса. Связи с хроническими или острыми лейкозами или лимфогранулематозом не обнаружены. После этого заболевания прививки противопоказаны.
7.4. САРКОИДОЗ
Заболевание с невыясненной этиологией, составляет 20-100 случаев на 100000 населения.
Заболевание протекает часто бессимптомно. Возможны общие клинические признаки в виде слабости, потери массы тела, субфебрильной температуры. В первую очередь отмечаются локальные симптомы, относящиеся к пораженному органу (легкие и лимфоузлы грудной полости - в 90% случаев, печень - в 60, глаза - в 40, сердце - в 20).
Диагноз саркоидоза иногда ставят случайно при оценке состояния кожи или периферических лимфоузлов, вовлекаемых в патологический процесс. Порой выраженный на рентгеновском снимке процесс, как правило, не соответствует относительно слабым жалобам больного.
Главным диагностическим приемом является определение локализации гранулемы, чаще с вовлечением в процесс регионарных лимфатических узлов.
У больных наблюдается массивная реакция на БЦЖ (61%), высокие концентрации γ-глобулина (47%), щелочной фосфатазы (35%), лейкопения (31%), анемия (31%), эозинофилия (25%), гиперкальциемия (17%), гиперкальциурия (30%).
Одновременно отмечается:
- снижение способности CD3 (Т)-клеток к иммунному реагированию;
- нормальная функция CD19 (В)-клеток;
- лимфопения за счет CD3 (Т)-клеток;
- количество моноцитов повышено;
- состояние сенсибилизации у больных укорочено;
- снижена цитотоксичность CD3 (Т)-клеток;
- увеличено количество CD8 (Т)-лимфоцитов;
- повышено количество АТ (к микобактериям, к вирусам кори, герпеса, краснухи, парагриппа);
- концентрация IgG, IgA, IgM повышена.
Лечение включает кортикостероиды, иногда применяют комбинированную иммуносупрессорную терапию (азатиоприн, хлорбутин, метотрексат).
Эффективность лечения приводит в 35-40% случаев к полной ремиссии, у 15-20% больных сохраняются функциональные нарушения. Летальность составляет 4-10% (это расстройства дыхания, сердечно-сосудистая, почечная недостаточность, последствия вторичной инфекции).
7.5. ЗАБОЛЕВАНИЯ, ОБУСЛОВЛЕННЫЕ ПРОЛИФЕРАЦИЕЙ ПЛАЗМАТИЧЕСКИХ КЛЕТОК
Общим критерием заболевания является продукция М-протеина (моноклональный белок). В основе патологии лежит пролиферация плазматических клеток в результате реактивного процесса при хронических инфекциях и опухолях или лейкемизации.
Доброкачественные моноклональные гаммапатии. В основе лежат хронические инфекции (холецистит, туберкулез, сифилис, гепатит, остеомиелит, пиелонефрит, малярия и др.) или процессы, связанные с опухолями в толстой кишке, полости рта, предстательной железе. М-протеин может быть обнаружен при болезнях лимфоретикулярной системы (лимфогранулематоз, лимфосаркома), что несколько размывает понятие доброкачественности. Иногда патология сопровождается индукцией аутоиммунных реакций и пр.
Как правило, в лечении нет необходимости, желателен длительный врачебный контроль, особенно при снижении концентрации иммунных глобулинов. У части больных в последующий период (иногда через 10 лет) развивается картина плазмоцитомы, что объясняется либо переходом от реактивной пролиферации к автономной опухоли, либо скрытой формой опухоли.
Плазмоцитома (множественная миелома). В основе заболевания лежит образование большого количества однородных по структуре моноклональных иммуноглобулинов. Поражает чаще мужчин. Обычно регистрируется у лиц старше 25 лет, пик приходится на возраст после 60 лет. Заболевают обычно 1-3 человека на 100000 населения.
Клиническая картина характеризуется тем, что после латентного периода, иногда более 10 лет, обнаруживается протеинурия, увеличенная СОЭ. Симптомы определяются двумя факторами: процессом непосредственного заселения ткани быстрорастущими плазматическими клетками и свойствами вырабатываемых ими веществ белковой
природы. Происходит замещение клеточного состава в костном мозгу, что вызывает анемию (нормохромную). В периферической крови обнаруживаются плазматические клетки (плазмоклеточный лейкоз).
Случаются проявления остеопороза - гиперкальциемия - патологические переломы костей. В 40-90% случаев обнаруживаются внекостные очаги в органах, богатых лимфоидной тканью, - в респираторном и кишечном трактах, печени, почках, надпочечниках, селезенке, поджелудочной железе. В некоторых случаях заболевание принимает генерализованный характер, иногда в лейкозной форме.
При плазмоцитоме, как правило, синтезируется единственный тип глобулина с идентичной первичной, вторичной, третичной структурами белка (М-протеин). Наряду с ним в сыворотке содержатся нормальные ИГГ в сниженном или стандартном количестве. Однако суммарная концентрация общего белка за счет М-протеина существенно увеличена до 100 г/л. В моче определяются так называемые белки Бенс-Джонса (парапротеин, состоящий из L-цепей), что случается в результате избыточного увеличения их продукции. Протеинурия белков Бенс-Джонса может быть и при других клинических формах лимфопролиферативных процессов.
Парапротеины (М-белки) могут нарушить функцию почек, откладываясь в просвете почечных канальцев и вызывая почечную недостаточность. В 8-10% парапротеин откладывается в тканях, что клинически проявляется амилоидозом.
При плазмоцитоме отмечается снижение уровня CD19(В)-клеток за счет увеличения количества плазматических клеток. При прогрессировании заболевания клеточный иммунитет может быть сохранен или снижен. Образование АТ нарушается, и это сопровождается бактериальными инфекциями (пневмонии, пиелонефриты, абсцессы, сепсис). В то же время туберкулез, грибковая и вирусные инфекции протекают нормально, только ветряная оспа и простой герпес могут принимать злокачественные формы.
Довольно часто данная патология сопровождается формированием других злокачественных новообразований, особенно в желудочнокишечном тракте, включая желчные пути, а также в грудной области (15-19% случаев). Кроме этого, М-протеины обнаруживаются при первичных иммунодефицитах (атаксии-телеангиэктазии, синдроме Вискотта-Олдрича), а также при тимоме.
Лечение. На первом месте стоит лучевая терапия. Способы лечения вариабельны, зависят от степени зрелости клеток, индиви-
дуа л ьной чувствительности. Полезна химиотерапия. Используют цитостатики (сарколизин, мельфалан, циклофосфан), а также преднизолон. Ремиссии наступают в 60-70% случаев, могут продолжаться десятки лет. Правильное использование медикаментов увеличивает жизнь больного с 17 до 31-50 мес. Вполне реальна опасность лейкоза. Позитивным признаком лечения является снижение уровня М-белков.
Макроглобулинемия существует в двух формах: макроглобулинемия вследствие определенных инфекций (трипаносомиаза), которая проявляется в увеличении продукции макроглобулинов в результате интенсификации работы этих белков под воздействием поликлональной активации, и в виде идиопатической формы, характеризующейся продукцией однородного по структуре макроглобулина (моноклональная макроглобулинемия).
Последнюю форму можно отнести к IgM-плазмоцитоме, но с образованием преимущественно незрелых IgM.
Болезнь поражает в основном мужчин 60-70 лет. Симптомы: головная боль, анемия (80%) из-за нарушения эритропоэза, гемолиза, аутогемагглютинации. Иногда в крови обнаруживаются атипичные плазматические клетки. Количество тромбоцитов нормально или снижено. У 60% больных регистрируется геморрагический диатез, предрасположенность к кровотечениям, кровоподтекам на слизистой оболочке, коже. В половине случаев отмечается увеличение периферических лимфатических узлов, селезенки, печени. Содержание лимфоцитов в крови резко увеличено.
Этиология заболевания не выяснена. Течет относительно доброкачественно, в основе лежит переключение синтеза IgG на IgM при отсутствии обратной связи.
Для диагностики используют реакцию, основанную на неспособности макроглобулина растворяться в дистиллированной воде. Для этого каплю сыворотки больного вносят в пробирку с дистиллированной водой, в результате образуется мутный преципитат. Реакция положительна в 50-90% случаев, но может наблюдаться и при системной красной волчанке, хронических инфекциях (туберкулезе, малярии).
Лечение в ряде случаев симптоматическое, направленное против пролиферации плазматических клеток (переливание крови, плазмаферез [при синдроме гипервязкости крови], кортикостероиды). Продолжительность ремиссии измеряется годами.
Поликлональная гипергаммаглобулинемия (болезнь Вальденстрема) характеризуется гипергаммаглобулинемей в сочетании с пурпурой. Часто заболевают молодые женщины 20-30 лет. Среди клинических проявлений отмечают пурпуру, которая развивается в результате нарушения кровообращения. Ее наблюдают при длительном вертикальном положении тела (поражаются ноги в области лодыжек, голени, иногда бедра). Часто пурпуре предшествуют ощущения ожога и зуда. Наиболее известным лабораторным методом является увеличение продукции IgG на БЦЖ. Лечение включает использование кортикостероидов, плазмафереза.
7.6. ЛЕЧЕНИЕ ИММУНОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ ТРАНСПЛАНТАЦИЕЙ КОСТНОГО МОЗГА
Аллогенная трансплантация костного мозга с успехом используется при лечении острых лейкозов, апластической анемии, тяжелой формы талассемии, пароксизмальной ночной гемоглобинурии, иммунодефицитов. В США ежегодно делается до 2000 алломиелотрансплантаций. Кроме этого, разработаны методы поддерживающей терапии больных, лишенных иммунной защиты и собственного кроветворения, - содержание в асептических палатах, применение массивной терапии компонентами крови, например, тромбоцитами, кондиционирование больных, применение химиопрепаратов в качестве противоопухолевого и супрессорного факторов.
Методически в начале осуществляется супрессия собственных иммунных механизмов. Для этого используется циклофосфамид по 120 мг/кг с последующим гиперфракционным тотальным облучением тела по 2 Гр 2 раза в день, 3 дня. Иногда за 4 дня до трансплантации назначается комбинация миелосана 16 мг/кг с циклофосфамидом в указанной дозе.
Проводилось титрование Аг НLА традиционным методом и типирование Аг эритроцитов пар «донор-реципиент» для прогнозирования приживления алломиелотрансплантата.
Трансплантация аллогенного костного мозга сопровождается серьезными изменениями иммунных показателей реципиентов. Со 2-3 нед у реципиентов появляются эритроциты донорского фенотипа. Позднее этот химеризм часто переходил в почти полное замещение эритроцитов реципиента на донорские. Есть факты смены группы крови с А(П) до 0(I) на протяжении 7 лет и В(Ш) на 0(I). Необходимо отметить, что индукция не свойственных реципиенту изогемагглю-
тининов не достигает обычной их активности (1:16-1:64), а составляет разведение 1:2. Однако, несмотря на жесткую цитостатическую и иммуносупрессорную терапию, реципиент все же сохраняет способность продуцировать собственные изогемагглютинирующие АТ.