Клиническая иммунология : учебник / под ред. А.М. Земскова. - 2008. - 432 с.
|
|
|
|
ГЛАВА 4. ИММУНОДЕФИЦИТЫ /ИММУННАЯ НЕДОСТАТОЧНОСТЬ
Иммунодефицитами обозначают устойчивые изменения иммунного статуса, обусловленные дефектами одного или нескольких механизмов иммунного ответа. Различают несколько вариантов иммунодефицитов: врожденные (первичные), возрастные (физиологические) - в раннем детстве, старческом возрасте, приобретенные (вторичные), инфекционные (вирусиндуцированные).
4.1. КЛАССИФИКАЦИЯ ПЕРВИЧНЫХ ИММУНОДЕФИЦИТОВ
В основе первичных иммунодефицитов, или первичной иммунной недостаточности лежит генетически обусловленная неспособность организма реализовать какие-либо звенья иммунного ответа. Они проявляются на ранних этапах постнатального периода, наследуются по аутосомно-рецессивному типу.
В тех случаях, когда дефекты затрагивают специфические механизмы иммунитета (антителообразование и клеточные реакции), их называют специфическими. При поражении фагоцитоза, системы комплемента, речь идет о неспецифических иммунодефицитах. Возможно повреждение Т- и В-клеток и системы фагоцитов или их комбинации. Mногообразие форм иммунодефицитов разделяют на три группы.
1. Комбинированные, с поражением клеточного и гуморального звеньев.
2. С преимущественным дефектом Т-зависимых иммунных реакций.
3. С нарушением продукции антител.
Классификация (по Mеждународной классификации болезней, десятый пересмотр)
D80 - ИMMУНОДЕФИЦИТ С ПРЕОБЛАДАНИЕM ДЕФЕКТОВ АНТИТЕЛ
D80.0 - наследственная гипогаммаглобулинемия Аутосомно-рецессивный тип агаммаглобулинемии (швейцарский тип).
Сцепленная с Х-хромосомой агаммаглобулинемия с дефицитом гормона роста (болезнь Брутона). D80.1 - НЕСЕMЕЙНАЯ ГИПОГАMMАГЛОБУЛИНЕMИЯ D80.2 - Селективный иммунодефицит IgA
D80.3 - Селективный иммунодефицит IgG D80.4 - Селективный иммунодефицит IgM D80.5 - Иммунодефицит с повышенным уровнем IgM D80.7 - Транзиторная гипогаммаглобулинемия детского возраста D81 - КОМБИНИРОВАННЫЕ ИMMУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
D81.1 - Тяжелый иммунодефицит с пониженным количеством Т- и В-клеток
D81.2 - Тяжелый иммунодефицит с пониженным и нормальным количеством В-клеток
D81.3 - Недостаточность аденозиндезаминазы
D81.4 - Синдром Незелофа (французский тип иммунодефицита,
алимфоцитоз)
D81.5 - Недостаточность пуриннуклеозиддифосфорилазы D81.6 - Дефицит Аг главного комплекса гистосовместимости класса II и I. Синдром «голых» лимфоцитов
D81.8 - Другие виды комбинированных иммунодефицитных состояний
D82 - ИMMУНОДЕФИЦИТ В СОЧЕТАНИИ С ДРУГИMИ ЗНАЧИТЕЛЬНЫМИ ДЕФЕКТАМИ
D82.0 - Синдром Вискотта-Олдрича
D82.1 - Синдром Ди-Джорджи
D82.2 - Иммунодефицит с укорочением конечностей D82.4 - Синдром гипергаммаглобулинемии
D82.8 - Иммунодефицит в сочетании со значительными уточненными дефектами
D84.1 - ДЕФЕКТЫ В СИСТЕMЕ КОMПЛЕMЕНТА
УРОВНИ ПЕРВИЧНЫХ ИММУНОДЕФИЦИТОВ (по Е.И. Змушко, Е.С. Белозерову и Ю.А. Mитину, 2001) А. Дефициты специфического звена
1. Дефициты АТ и их компонентов:
- селективный дефицит IgA и его субклассов,
- дефицит IgM и IgA,
- дефицит IgG и IgA,
- агаммаглобулинемия (болезнь Брутона),
- транзиторная младенческая гипогаммаглобулинемия,
- кишечная лимфаангиэктазия.
2. Т-иммунодефициты:
- синдром Ди-Джорджи,
- хронический слизисто-кожный кандидоз,
- тяжелый комбинированный иммунодефицит (ТКИД),
- синдром Незелофа,
- синдром Луи-Барра,
- синдром Вискотта-Олдрича,
- ретикулярная дисгенезия,
- вариабельные иммунодефициты.
В. Дефициты неспецифического звена
1. Дефициты системы комплемента:
- дефицит С1-компонента,
- дефицит С2-компонента,
- дефицит С3-компонента,
- дефицит С4-компонента,
- дефицит поздних компонентов комплемента,
- дефицит ингибиторов комплемента.
2. Дефициты фагоцитоза:
- дефициты хемотаксиса (синдром «ленивых лейкоцитов»),
- дефицит факторов киллинга,
- синдром Чедиака-Хигаси,
- синдром Джоба (Йова),
- наследственный хронический агранулоцитоз,
- периодическая циклическая нейтропения.
4.2. КЛАССИФИКАЦИЯ ВТОРИЧНЫХ ИММУНОДЕФИЦИТОВ
Приобретенные иммунодефициты формируются под действием окружающей среды на уровне фенотипа, в то время как первичная иммунная недостаточность генетически обусловлена и проявляется на уровне генотипа. Не у всех первичных иммунодефицитов есть характерные маркеры, в ряде случаев приобретенная иммунная недостаточность индуцируется латентно протекающей вирусной инфекцией. В последнем случае трудно решить, обусловлен ли данный иммунодефицит генетически или он возникает в организме после рождения. Вторичные иммунодефициты встречаются гораздо чаще, чем первичные.
Вторичная иммунная недостаточность формируется у контингентов с исходно нормальной иммунной системой. У них могут поражаться Т-, В- и фагоцитарные звенья иммунитета, возможны их сочетания, что приводит к снижению количества, функции и взаимодействия лимфоцитов и фагоцитов.
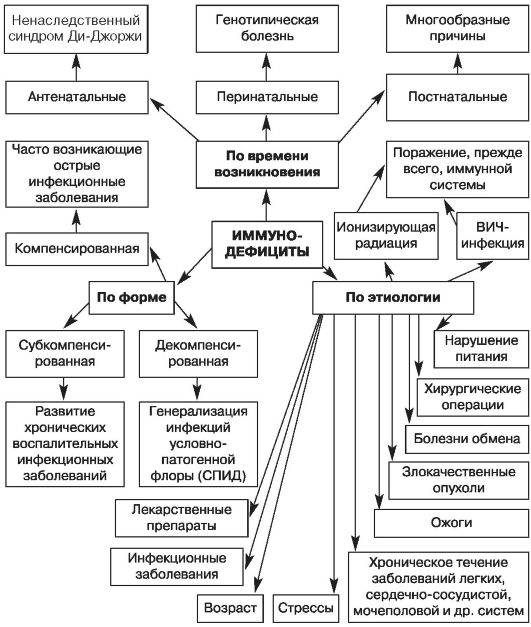 Рис. 2. Классификация вторичных иммунодефицитов по времени возникновения, этиологии, форме (по Белозерову Е.С. и др., 1992).
Рис. 2. Классификация вторичных иммунодефицитов по времени возникновения, этиологии, форме (по Белозерову Е.С. и др., 1992).
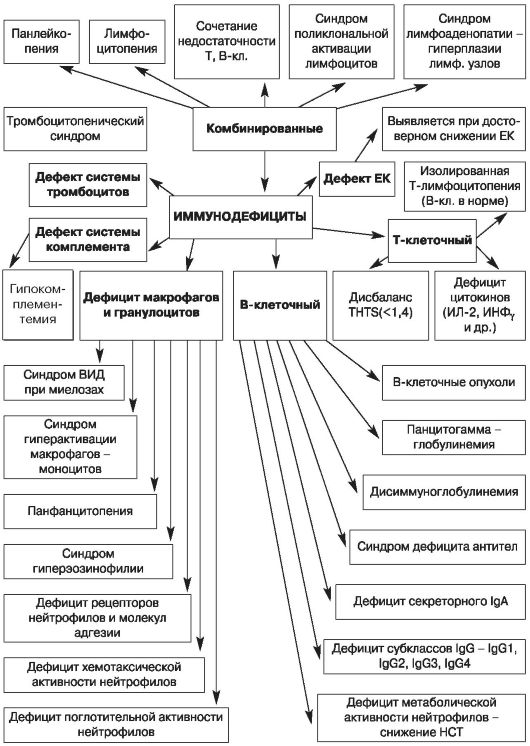 Рис. 3. Классификация вторичных иммунодефицитов по патогенезу (по Новикову Д.К. и Новиковой В.И., 1994)
Рис. 3. Классификация вторичных иммунодефицитов по патогенезу (по Новикову Д.К. и Новиковой В.И., 1994)
4.3. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ИММУНОДЕФИЦИТОВ
Злокачественные новообразования
Смертность от рака у пациентов с иммунодефицитами в 100- 200 раз выше, чем у прочих контингентов. В 65-70% всех случаев встречаются лимфопролиферативные заболевания (лимфомы, лимфосаркомы, лимфогранулематоз, лимфолейкоз, саркома Капоши). Эпителиальные опухоли встречаются реже.
Аллергические заболевания
У больных с первичным иммунодефицитами проявляются кожные поражения по типу упорного экссудативного диатеза, атопического дерматита, экземы, нейродермита.
Аутоиммунные заболевания
У больных часто формируется ревматоидный артрит, системная красная волчанка (СКВ), склеродермия, системные васкулиты, тиреоидит, рассеянный склероз, хроническая почечная недостаточность, сахарный диабет 1 типа.
Другие заболевания
В основном иммунодефициты связываются с характерными изменениями крови: нейтропении, эозинофилии, анемии, тромбоцитопении. Существует сочетание с другими пороками развития: гипоплазией клеточных элементов хрящей, волос, эктодермальной дисплазией, пороками сердца и крупных сосудов.
Дефицит гуморального иммунитета
Иммуноглобулины играют ведущую роль в уничтожении бактерий и других инфекционных агентов. Они также способствуют реализации опсонизирующего эффекта.
Дефицит иммуноглобулинов проявляется рецидивирующими и хроническими бактериальными инфекциями, в том числе вызванными слабыми невирулентными возбудителями. Преимущественно поражаются органы дыхания (бронхоэктазы, фиброз легких), желудочно-кишечный тракт (с диареями, нарушенным всасыванием), придаточные пазухи носа, мозговые оболочки. Инфекции протекают с тяжелыми интоксикациями, часто осложняются септицемией.
Дефицит иммуноглобулинов может протекать в форме тотальной гипогаммаглобулинемии или в виде вариантов со снижением уровня одного класса или подкласса специфических белков.
При дефиците IgM у больных увеличивается риск развития тяжелых менингококковых менингитов, осложняющихся септицемией, повторными респираторными инфекциями с формированием бронхоэктазов. Особенно тяжело протекают инфекции, вызванные высоковирулентными штаммами, поскольку первичный иммунный ответ в виде образования тяжелых иммунных глобулинов у данных пациентов отсутствует.
Дефицит класса IgG, а также пангипоиммуноглобулинемия (агаммаглобулинемия) обозначены как недостаточность образования соответствующих классов иммуноглобулинов. Указанное состояние является преимущественно врожденным, хотя возможны и вторичные пангипогаммаглобулинемии.
Дефицит IgA часто протекает бессимптомно, поскольку он перекрывается образованием иммунных глобулинов классов M и G. Примерно третья часть клеток, синтезирующих IgA, расположена в слизистых оболочках. Иногда дефицит продуцентов IgA в слизистых замещается клетками, образующими IgM, также соединенных с секреторным компонентом. Недостаточность белков может сочетаться с увеличением заболеваний органов дыхания, несколько реже - пищеварительного тракта.
Селективный дефицит IgA или его субклассов довольно часто встречается у лиц обоих полов. Возможно несколько вариантов клиниколабораторного дефицита IgA. Так, транзиторный дефицит IgA или его субклассов наблюдается у детей раннего возраста, чаще у мальчиков. У новорожденных следовые концентрации IgA - обычное явление. Отсутствие IgA у новорожденных говорит или о незрелости иммунной системы, или о вероятности формирования селективного дефицита IgA. Концентрация IgA выше 0,1 г/л у новорожденных свидетельствует о возможности бактериальной инфекции на слизистых. Если IgA не определяется после 9-10-месячного возраста, то при наличии клинических проявлений не вызывает сомнений диагноз селективного дефицита IgA. Если концентрация IgA к 1-2 годам не достигает уровня более 0,5 г/л, то у детей, как правило, имеются признаки дефицита.
Транзиторный дефицит IgA развивается обычно с прекращением вскармливания грудным молоком. Клинически проявляется в виде: а) частых респираторных инфекций, гнойных бактериальных процессов на коже и слизистых конъюнктивы и полости рта, фебрильной судороги, целиакии от всасывания глютена; б) атопии в виде астматического бронхита, бронхиальной астмы, диффузного нейродермита и
пищ е в ой аллергии; в) смешанной формы с гнойно-бактериальными, вирусными, грибковыми инфекциями на фоне поливалентной аллергии, часто встречается дисбактериоз, а также диффузные болезни соединительной ткани.
Селективный дефицит IgA или его субклассов у детей старше 2 лет и у взрослых может носить как транзиторный (IgA не отсутствует, а отмечается снижение его концентрации), так и стойкий характер. В последнем варианте чаще IgA снижен, реже отсутствует. Варианты клинических проявлений те же, но с увеличением продолжительности дефицита больше полиморфизм клинических проявлений. Дефицит IgA может быть вторичным после инфекций, интоксикаций, простагландин-опосредованной супрессии, стволовой ваготомии, гастроэнтеростомии.
Вариантом снижения гуморального иммунитета является синдром отсутствия АТ, когда на фоне нормального содержания иммуноглобулинов в серологических реакциях не обнаруживаются специфические АТ против конкретных возбудителей, что может быть связано со специфической супрессией или генетически обусловленной неспособностью реагировать на определенные Аг. Дефициты АТ - нередкое явление при гипергаммаглобулинемии, поликлональной активации В-клеток, лимфопролиферативном синдроме.
Дефицит клеточного иммунитета
Т-иммунодефициты. Дефициты клеточного иммунитета проявляются развитием инфекций с внутриклеточным паразитированием возбудителя (туберкулез, лепра, бруцеллез, вирусные инфекции, микозы). При менее грубых дефицитах клеточного иммунитета чаще развиваются персистирующие или рецидивирующие вирусные инфекции. Более грубые дефекты манифестируются микозами. Именно на иммунодефицитный фон наслаиваются такие инфекции, как туберкулез, бруцеллез.
Дефицитом клеточного иммунитета объясняется развитие заболеваний, вызванных простейшими. При этом инвазии могут не отражаться существенно на состоянии больных (лямблиоз, трихомониаз) или наслаиваться только на выраженные дефициты клеточного иммунитета (токсоплазмоз, пневмоцистоз). Большинство простейших, гельминтов и прочих инвазирующих агентов сами обладают иммуносупрессорными влияниями.
Кожные поражения при Т-иммунодефиците проявляются герпесом, псориазом, а поражение слизистых - катаральным, пленчатым,
язвенным конъюнктивитом и повреждением ротовой полости и слизистых конъюнктив грибками, особенно часто вирусными афтозными и язвенными стоматитами. Бронхиты характеризуются при клеточном иммунодефиците упорным течением, кашлем без гнойной мокроты, атрофией слизистой (при бронхоскопии) и эффективностью ингаляций интерферона, подтверждающих вирусную природу заболевания. В тяжелых случаях, особенно на фоне неоправданного применения антибиотиков, возможно развитие кандидоза бронхов. Поражение легких может быть в виде фиброза и пневмоцистоза. Со стороны желудочно-кишечного тракта возможно развитие энтеритов и энтероколитов, болезни Крона и кандидоза, лямблиоза. В последующем характерно развитие злокачественных новообразований. Для Т-иммунодефицитов нетипично поражение ЛОР-органов, костей, суставов. Также нехарактерно развитие сепсиса, гнойного менингита. Типично развитие гипоплазии лимфоузлов, миндалин.
Инфекции, вызывающие поликлональную активацию В-клеток (ВИЧ-инфекция), приводят к развитию лимфоаденопатии. Нетипичны аллергия и аутоиммунные заболевания.
Т-иммунодефициты могут быть изолированными, но, учитывая, что к Т-лимфоцитам относятся разнообразные регуляторные клетки, а центральный орган клеточного иммунитета - тимус оказывает влияние на другие системы иммунитета, развитие Т-иммунодефицита приводит к нарушению функционирования других систем иммунитета с формированием комбинированных иммунодефицитов.
Т-иммунодефициты могут быть первичными (врожденными), которые проявляются на первом (реже на третьем) месяце жизни, и вторичными (приобретенными), развивающимися в любом возрасте. Т- иммунодефициты наблюдаются при дефицитах тимуса, в частности, гипоплазии и аплазии, тимомегалии, снижении выработки гормонов вилочкой железы. Они могут быть обусловлены количественным или функциональным дефицитом со стороны СD4+-клеток, Т-контрсупрессоров, Т-киллеров, часто в сочетании с дефектами, идущими от других цитотоксических клеток, что клинически выявляется как Т-иммунодефицит. Лабораторно комбинированный характер иммунодефицита может быть установлен повышением функции специфических и неспецифических Т-супрессоров, недостаточностью аденозиндезаминазы и нуклеозидфосфорилазы.
Клинические проявления комбинированных иммунодефицитов (КИД) характеризуются сочетаниями клиники гуморального и кле-
точ ного дефицита. Такие комбинации чаще всего приводят к смерти уже на первом году жизни ребенка. Для них типичны сочетания пневмонии с инфекциями желудочно-кишечного тракта и кожи, вызванными бактериями, вирусами, грибами. Очень часто развиваются злокачественные новообразования. Инфекции протекают тяжело, слабо поддаются лечению. Пациенты часто погибают от септицемии или злокачественных новообразований.
Следует признать, что наряду с классическими формами комбиниро-ванных иммунодефицитов существуют их более стертые легкие варианты с лучшим прогнозом для жизни и более поддающиеся лечебным мероприятиям.
Дефицит фагоцитарного иммунитета
Дефекты фагоцитоза. Дефекты фагоцитоза развиваются из-за уменьшения количества фагоцитов, что проявляется в виде синдрома нейтропении или вследствие повреждений, которые делятся на нарушения двигательной функции клеток и дефекты киллинга.
Дефект хемотаксиса. К нему можно отнести синдром ленивых лейкоцитов, клинически проявляющийся у детей в виде тяжелых повторных инфекций, особенно в виде микроабсцессов. Представляет собой комбинированный дефект спонтанной миграции и хемотаксиса фагоцитов, сопровождается тяжелой нейтропенией.
Синдром дисфункции актина. Характеризуется подавлением хемотаксиса и фагоцитоза в результате дефекта полимеризации мономерного G-актина в полимерный F-актин. Клетки слабо распластываются (прилипают к поверхности, сильно уплощаясь на площади, превышающей первоначальный размер клетки), но усиленно выделяют лизосомальные ферменты. У больных наблюдаются частые рецидивирующие инфекции, вызванные различными возбудителями, подавление воспалительной клеточной реакции.
Гипериммуноглобулинемия. Обусловлена IgE. У больных подавляется хемотаксис за счет его клеточных дефектов и образования в сыворотке ингибиторов хемотаксиса.
Синдром Йова. При гипериммуноглобулинемии Е (IgE) отмечается клеточный дефект хемотаксиса, «холодные» абсцессы в подкожной клетчатке различной локализации, тяжелый атопический дерматит с гнойничковыми поражениями кожи, циклическая нейтропения с лихорадкой.
Хронический кожно-слизистый кандидоз нередко сочетается с гипер- IgE. Характеризуется выраженным дефектом хемотаксиса фагоцитов
и подавлением их киллинга за счет дефекта дегрануляции. Пациенты страдают бактериальными инфекциями.
Воспалительная кишечная болезнь Крона. При ней отмечается подавление хемотаксиса.
Аномалия Пелгер-Хюэта. Заболевание с аутосомно-доминантным типом наследования, резким нарушением хемотаксиса фагоцитов, неполной сегментацией их ядра.
Ихтиоз сочетается с дефектом хемотаксиса, генерализованной инфекцией трихофитоном.
Существенное снижение хемотаксиса отмечается также при различных аутоиммунных заболеваниях (ревматоидный артрит, СКВ), периодонтальной болезни, бактериальных, вирусных инфекциях, ожогах и т.д.
Дефект киллинга. Прежде всего отмечается при хронической гранулематозной болезни, являющейся первичным иммунодефицитом, который передается либо как аутосомно-рецессивный признак или как заболевание, связанное с X-хромосомой. Фагоцитарные клетки оказываются дефицитными по НАДФН- и НАДН-оксидазам, глутатионпероксидазе, глутатион-редуктазе и глюкозо-6-фосфатдегидрогеназе. В первые дни и недели жизни у больных развивается пиодермия, гнойные лимфадениты, требующие хирургического вмешательства, причем чаще всего поражаются шейные и паховые лимфоузлы. Также развиваются пневмонии с обширным поражением легких, вовлечением в патологический процесс плевры, высокой лихорадкой, лейкоцитозом, повышением СОЭ.
Синдром Чедиака-Хигаси. Комбинированный дефект (носит аутосомно-рецессивный характер) с нарушением хемотаксиса, дегрануляции, дефектом лизосомальных мембран и замедлением внутриклеточного киллинга бактерий.
Недостаточность миелопероксидазы. Наследственное заболевание, передается как аутосомно-рецессивный признак. Выраженный дефект миелопероксидазы в фагоцитах сопровождается дефектом киллинга.
Дефицит фосфоглицераткиназы. Характеризуется дефектом киллинга фагоцитов.
ЛАД-дефициты. Это врожденные дефекты экспрессии молекул адгезии клеток, сопровождающиеся глубокими нарушениями функций лейкоцитов. Например, больные, имеющие дефекты экспрессии на мембране клеток интегринов (LFA-1, Мас-1, р 150,95), характеризуются замедленным отделением пуповины, тяжелыми рецидивиру-
ющи ми бактериальными инфекциями, неспособностью к формированию гноя.
Дефицит компонентов системы комплемента
Система комплемента. Система комплемента входит в группу 4 активаторных каскадных систем плазмы. К этой группе относятся также система кининов, свертывающая система и система фибринолиза. Система комплемента и система кининов тесно связаны с иммунной системой.
Клиника дефицита комплемента характеризуется рецидивирующими или хроническими бактериальными инфекциями органов дыхания, мочевыводящих путей, энтероколитами, воспалением среднего уха, мастоидитами, менингитом, гнойными поражениями кожи и подкожной клетчатки. Заболевания протекают с массивной интоксикацией, склонностью к септицемии. При некоторых вариантах, например, дефиците компонента С6, отмечается относительно изолированная склонность к нейссериальной инфекции (менингококки, гонококки) с менингитами, гонококковыми артритами, септицемией. У некоторых с дефектами системы комплемента инфекционные заболевания протекают без лейкоцитоза.
У больных с дефицитом комплемента возможно снижение противовирусной защиты, так как комплемент-опосредованный лизис необходим для предупреждения распространения инфекции через циркулирующую кровь.
4.4. ПРИНЦИПЫ ЛЕЧЕНИЯ ИММУНОДЕФИЦИТОВ
Первичные иммунодефициты
Проводится адекватное общее лечение, соблюдение соответствующих гигиенических и асептических мер. Рекомендуется диета с исключением углеводов, растительного белка. Необходимо исключить введение живых вакцин и гемотрансфузии.
При расстройствах аденозиндезаминазы и пуриннуклеозидфосфорилазы положительные результаты дает введение больным замороженных или облученных эритроцитов.
При дефекте В-звена иммунитета больным вливают недостающие иммуноглобулины в дозе 25-50 мг/кг в нед.
Эффективность применения иммуномодуляторов не выражена.
При дефектах клеточного иммунитета апробированы следующие виды лечения: пересадка лимфоидных органов от иммунологически зре-
лых доноров, тимуса от несовместимого плода или взрослого донора, цельного костного мозга от совместимого донора, фракций стволовых клеток родителей, одновременный перенос тимуса и грудины от мертворожденного ребенка.
К сожалению, попытки преодолеть несовместимость оказались несостоятельными. Нужно использовать следующие методические подходы:
- подбор совместимых пар (донор-реципиент) по главным Аг HLA;
- уничтожение клеток костного мозга реципиента различными способами;
- обработка клеток донора до трансплантации моноклональными АТ для уничтожения киллеров;
- назначение реципиенту селективных иммунодепрессоров (циклоспорин А).
Вторичные иммунодефициты
(Подробно принципы лечения вторичных иммунодефицитов рассмотрены в гл. 9).
4.5. ВИЧ-ИНФЕКЦИЯ
Среди всех иммунных заболеваний ВИЧ-инфекция занимает особое место. Вирус иммунодефицита человека вызывает инфекционное заболевание, опосредованное первичным вирусным поражением иммунной системы. Это ведет к развитию ярко выраженного вторичного иммунодефицита, который и обусловливает различные инфекционные процессы, индуцированные непатогенными для здорового человека микроорганизмами, и другие патологические состояния.
Возбудителями болезни является ретровирус человека - Т-лимфотропный вирус (HTLV-III). Семейство ретровирусов состоит из двух подсемейств - онко- и ленти (медленных) вирусов. ВИЧ содержит особый фермент - обратную транскриптазу (ревертазу), обусловливающую транскрипцию генетической информации с вирусной РНК на ДНК, а далее - классическим путем через иРНК на белок. Вирус, в отличие от своих соседей по семейству, обладает высокой изменчивостью: 1000 мутаций на 1 ген, (для сравнения - у вируса гриппа, по данным разных авторов, в 30-1 млн раз меньше (!)). Геном ВИЧ включает 7 регуляторных генов, на 4 больше, чем у остальных представителей семейства ретровирусов. Сумма генов обеспечивает ВИЧ-образование вирусных-капсульных белков, ревертазы, белков,
управляющих активностью возбудителя, его инфекционностью и т.д.
Известен ВИЧ-1 с локализацией в Северной Америке,ряде стран Западной Европы, Центральной Африке и ВИЧ-2 - в Западной Африке, Португалии, Франции, Германии.
ВИЧ имеет выраженный тропизм к лимфоидной ткани, в первую очередь, к Т-клеткам, имеющим рецептор CD4. Обычно, проникнув в клетку, вирус остается в латентном состоянии, иногда годами, пока какая-нибудь вторичная инфекция не приведет к стимуляции зараженных Т-лимфоцитов. Тогда наступает активация ВИЧ с бурным синтезом вирусных частиц и гибелью Т-лимфоцитов, несущих маркер CD4. Тем не менее инфицированность типов клеток ВИЧ в организме значительно шире, как и способы проникновения вирусов в клетку. Кроме взаимодействия ВИЧ с рецептором CD4, он может: проникать через Fc γ-рецептор клеток, будучи связанным со специфическими противовирусными АТ и даже с неспецифическими ауто- и аллоантителами к тканевым Аг человека, аминокислотные последовательности которых обнаруживаются в наружном оболочечном белке ВИЧ; попадать в разные клетки-мишени в виде псевдовируса, содержащего геном ВИЧ, а оболочку какого-либо другого вируса, например, герпеса, аденовирусов, паповавирусов и т.д.; проникать в клетки, не имеющие рецептор CD4, например, В-лимфоциты, нейтрофилы (считают возможным взаимодействие пептидов гипервариабельной петли белка gp120 ВИЧ с еще не идентифицированными структурами Т-лимфоцитов и других клеток хозяина); проникать в клетку в виде комплекса ВИЧ с γ-интерфероном через интерфероновый рецептор. В последние годы установлено, что определенный вклад во взаимодействие ВИЧ с Т-лимфоцитами и моноцитами вносят молекулы адгезии, в частности, интегрин LFA-1, который может выполнять роль корецепторной молекулы в дополнение к CD4, взаимодействующей со своими лигандами ICAM-1, -2,-3. Это было подтверждено с помощью моноклональных анти-LFA-1 АТ, блокирующих инфицирование клеток ВИЧ, как и ассоциацией АТ против всех трех лигандов ICAM.
Возбудитель СПИДа обнаруживается не только в Т-лимфоцитах, но и других лимфоидных и нелимфоидных клетках (клетки Лангерганса, макрофаги/моноциты, эозинофилы, мегакариоциты, нейроны, микроглия, астроциты, эндотелиоциты, олигодендроциты, клетки кишечного эпителия и т.д.). Он обнаружен в сперме, крови, слюне, слезной жидкости, грудном молоке кормящих матерей.
Среди заболевших 70-80% составляют гомосексуалисты и бисексу-
алы, 12-18% - наркоманы, 7% - женщины - партнеры больных ВИЧинфекцией, 1,1% - дети больных, 0,6% - больные гемофилией, в 6-7% пути передачи инфекции пока не выяснены.
Главным путем заражения ВИЧ являются половые контакты с больными, переливание инфицированной крови и ее препаратов, зараженные шприцы и иглы, передача с молоком матери, трансплантация органов и тканей. Существует профессиональное заражение стоматологов, хирургов (в 1,5-2% случаев). Передача возбудителя через слюну и кусающими насекомыми до сих пор исключается, однако в организме постельных клопов вирус сохраняет свою жизнеспособность в течение 60 мин (!).
Инкубационный период составляет от 4-6 мес до 3-5 и более лет. Однако у некоторых больных первые признаки ВИЧ-инфекции в форме увеличения лимфатических узлов и повышения температуры развиваются через 4-6 нед. При трансплантационной передаче скрытый период составляет обычно 4-8 мес. Средний период между появлением специфических АТ в крови (наличие инфекции!) и развитием симптомов СПИД составляет 7-11 лет. У 5% инфицированных заболевание развивается в первые 3 года. В последующие 8 лет ВИЧ-инфекция развивается со скоростью 3-7% лиц ежегодно из числа зараженных. У 65% инфицированных лиц клинические проявления СПИД развиваются на протяжении 16 лет (Хаитов Р.М., Игнатьева Г.А., 1992).
На основании нарушения вирусом иммунитета возникает различная патология.
Различают 3 больших и 6 малых симптомов заболевания. Большие - потеря массы на 10 и более процентов, продолжительная лихорадка и хроническая диарея более 1 мес. Малые - постоянный кашель более 1 мес, генерализованный зудящий дерматит, повторный опоясывающий лишай, генерализованная лимфоаденопатия.
В.В. Покровским и О.В. Юриным разработана следующая клиническая классификация ВИЧ-инфекции.
I. Стадия инкубации.
II. Стадия первичных проявлений а/ острая лихорадочная форма
б/ бессимптомная фаза
в/ генерализованная лимфоаденопатия.
III. Стадия вторичных заболеваний
а/ потеря массы тела менее 10%, поверхностные грибковые, бактериальные, вирусные поражения кожи и слизистых оболочек, опоя-
сывающий лишай, повторные фарингиты, синуситы; б/ прогрессирующая потеря массы тела на 10%, необъяснимая лихорадка или диарея более 1 мес, «волосистая» лейкопения, туберкулез легких, повторные или стойкие бактериальные, грибковые, вирусные, протозойные поражения внутренних органов (без диссеминации) или глубокие поражения кожи и слизистых оболочек, повторный или диссеминированный опоясывающий лишай, локализованная саркома Капоши;
в/ генерализованная бактериальная, вирусная, грибковая, протозойная пневмоцистная пневмония, кандидоз, атипичные микобактериозы, внелегочный туберкулез, кахексия, диссеминированная саркома Капоши, поражения ЦНС различной этиологии. IV. Терминальная стадия.
У части больных заболевание протекает с преимущественным поражением желудочно-кишечного тракта, воспалительный процесс от гиперемии до некроза распространяется на всю кишечную трубку. Выявляются упорная диарея, признаки стоматита, эзофагита, проктита. Воспаления вызывают грибки кандида, вирусы, микробактерии, криптококки.
Иногда заболевание приобретает генерализованный характер, в воспалительный процесс вовлекаются легкие, желудочно-кишечный тракт, кожа, нервная система, а возбудителями являются цитомегаловирус, паповавирус, токсоплазмы, грибки кандида, вирусы, микробактерии, криптококки.
Кроме того, заболевание может протекать с преимущественным поражением глаз в виде конъюнктивита, кератита, ретинита, ретинального перифлебита, кровоизлияний в клетчатку, появлением белого пятна со снижением зрения.
Поражение центральной нервной системы может быть самостоятельным проявлением клиники СПИДа или сочетаться с поражениями других органов и систем. Больных беспокоит головная боль, нарушение равновесия, прогрессирует деменция. Причиной неврологических нарушений может быть непосредственно действие вируса на нервную ткань, а также герпес вирусов, например, цитомегаловируса, вируса Эпстайна-Барр, вследствие сформировавшегося у больного иммунодефицита.
Наряду с активацией условно-патогенной флоры у больных СПИДом клинические проявления могут быть обусловлены развитием опухолевого процесса, особенно часто в виде саркомы Капоши.
Действие ВИЧ на иммунную систему характеризуется:
- снижением общего количества Т-лимфоцитов за счет субпопуляции CD4-лимфоцитов;
- снижением функции Т-лимфоцитов за счет резкого нарушения функции CD4-лимфоцитов;
- повышением функциональной активности В-лимфоцитов, изза чего увеличивается спонтанная продукция иммунных глобулинов и рост их концентрации в сыворотке крови;
- в результате вторичных иммунных нарушений снижением способности В-клеток отвечать продукцией иммуноглобулинов на новый Аг, одновременно увеличивается количество ЦИК;
- снижением цитотоксической активности натуральных киллеров и клеточно-опосредованной цитотоксичности;
- нарушением функции клеток моноцитарного ряда (в частности, снижается хемотаксис, цитотоксичность, продукция ИЛ-1). Одновременно увеличивается содержание α-интерферона, появляются антилимфоциторные АТ, супрессорные факторы, снижается уровень тимозина в сыворотке крови и увеличивается β-2-микроглобулин и α-1-тимозин. У части больных также снижается абсолютное число и функциональная активность естественных киллеров, пролиферативный ответ на митогены и Аг, кожные реакции на туберкулин, кандидин, трихофитин, столбнячный, дифтерийный и другие Аг. Выявляется гипергаммаглобулинемия.
Специфическая диагностика включает несколько уровней.
1. Выявление антител против вируса иммунодефицита человека
Выявление АТ против ВИЧ выполняется с помощью методов иммуноферментного (ИФА), радиоиммунного, иммунометрического анализов, когда в используемых тест-системах применяют вирусные лизаты, рекомбинантные вирусные белки или синтетические пептидные вирусные Аг. Кроме них, применяют метод иммунного блотинга, или Вестерн блота, суть которого состоит в том, что после разгонки в электрофорезе белков ВИЧ их отдельные фракции переносят на нитроцеллюлозную (или иную) мембрану, вырезают соответствующую полоску, содержащую конкретный пептид ВИЧ, и инкубируют ее с материалом, заключающим в себе предполагаемые АТ к ВИЧ. Визуализируют реакцию с помощью той же ИФА.
Используют также реакцию агглютинации (латексную, желатиновую, эритроцитарную) и радиоиммунопреципитацию (метаболи-
ческ и меченный изотопами биоматериал разрушают для выделения меченых антигенов, которые затем связываются с антителами и полученные комплексы АГ-АТ выделяются и анализируются на радиоспектрометрах).
2. Обнаружение вирусных Аг
Выполняется с помощью тех же методов ИФА, РИА, радиоиммунопреципитации, в которых применяются, как правило, моноклональные стандартные АТ против Аг ВИЧ.
3. Обнаружение клеток, экспрессирующих белки ВИЧ
Для этой цели применяются методы - микроскопические или проточной лазерной цитофлуориметрии, выявляющие клетки, содержащие вирусные Аг, которые маркированы обработкой специфическими мечеными АТ (ФИТЦ, фикоэритрином, стрептавидином); иммуногистохимические методы (те же принципы, но АТ конъюгируются с ферментами, в последующем вызывающими цветную реакцию при взаимодействии с субстратами, которая выявляется микроскопически или колориметрически); методы электронной микроскопии (АТ метят флуорохромами, ферментами, коллоидным золотом); метод твердофазного ИФА, в котором применяются зараженные ВИЧ клетки, адгезированные на поверхности полистирола.
4. Обнаружение в клетках человека провирусной ДНК или РНК ВИЧ Анализирует клетки периферической крови и клетки, полученные
при биопсии лимфоузлов, миндалин и аденоидов (они содержат ВИЧ в 10-15 раз больше, чем клетки крови!). Выполняется методами гибридизации нуклеиновых кислот и полимеразной цепной реакции (ПЦР). Высокая целесообразность этих методов обусловливается тем, что определение специфических анти-ВИЧ АТ «пропускает» каждого десятого инфицированного. Это вызывает заражение ВИЧ реципиентов при трансфузии им проверенной крови инфицированных лиц. По этой причине в настоящее время любая донорская кровь, даже тестированная на АТ и Аг ВИЧ, считается источником инфекции ВИЧ и может быть применена лишь по жизненным показаниям. Дополнительной гарантией точной ВИЧ-диагностики больного, выявляющей инфицированность вирусом до того, как образуются специфические АТ или клетки, являются поименные методы. Они позволяют обнаружить в инфицированных клетках интегрированную в их геном противовирусную ДНК и саму вирусную РНК.
В первом методе в реакционную среду, содержащую клетки периферической крови (или из других источников) инфицированного
ВИЧ больного, вносят специфические нуклеотидные зонды из генома ВИЧ, которые создают специфический гибрид с ДНК провируса и геномной и информационной РНК ВИЧ, содержащихся в зараженных клетках. Зонд метится либо сульфоновыми группами, которые выявляют ИФА в клетках моноклональными АТ, либо флуорохромами, радиоизотопами (выявляют радиоавтографией, флуориметрией и проч.). Метод высокочувствителен, реагирует при бессимптомной и серонегативной инфекции геном ВИЧ в каждой из десяти мононуклеарных клеток.
При использовании метода ПЦР, в котором применяют специфические олигонуклеотидные «затравки», позволяющие вызвать умножение искомого гена ВИЧ и накопление его в количестве, достаточном для выявления этим методом, удается определить даже один вирусный геном в геноме одной зараженной клетки, находящейся среди миллиона незараженных клеток или клеток, инфицированных какими-либо другими (не ВИЧ) вирусами. Добавляемая нуклеотидная затравка («праймер») соответствует консервативным и уникальным отрезкам генома ВИЧ, которых нет в ДНК клеток хозяина, она многократно копируется с помощью ДНК-полимеразы, накапливаясь в достаточных для анализа количествах. Далее специальными методами она «спаривается» (образует гибридные молекулы) с искомым геном ВИЧ в клетках, если, конечно, клетки им поражены, и вирусный ген выявляется во взятом от больного материале. Обнаружения одной вирусной РНК бывает недостаточно, т.к. позволяет установить лишь 65% носителей провирусной ДНК.
5. Обнаружение зрелых вирионов
Выполняется различными способами. Прежде всего заражением культур клеток in vitro, в которых «размножившиеся» ВИЧ обнаруживаются с помощью АТ, анализа нуклеиновых кислот и обратной транскриптазы. Данный фермент не специфичен для ВИЧ, но обнаружен только в ретровирусах, что позволяет выполнить ориентировочный анализ при подозрении на ВИЧ-инфекцию. В клеточный супернатант, содержащий предполагаемый ВИЧ, добавляют специфическую затравку ДНК (и 3Н-тимидин трифосфат) и обратную транскриптазу, которая синтезирует с нее ДНК на матрице вирусной РНК. Активность фермента определяют по суммарной радиоактивности препарата.
Осуществляется также заражение лабораторных животных (кроликов, мышей линии SCID-hu, обезьян) инфицированным материалом, а далее инфекция выявляется клинически и лабораторно
опи санными методами. Наконец, вирионы исследуются электронномикроскопически, методами гистохимии, флуоресценции, а также по способности индуцировать образование синцития в чувствительных клеточных культурах MOLT-4, CEM-SS, VB, C8166 и др.
В нашей стране достаточно широко используется определение анти-ВИЧ АТ. Отмечают, что если ИФА оказывается дважды положительным, сыворотки далее проверяют в иммуноблотинге (Вестерн блот). Если не обнаруживается АТ к гликопротеидам gp41, 120, 160, а они направлены против других Аг, реакция считается сомнительной и через 3,6,12 мес повторяется. При положительной реакции с Аг р24 (24 кД) сыворотка исследуется на наличие Аг ВИЧ-2. Если и через год реакция с gp120, 41, 160 отрицательна, пациент снимается с учета, но пожизненно освобождается от донорства.
Нередко клиницисты ставят предположительный диагноз СПИДа без лабораторного подтверждения - при кандидозах пищевода, трахеи, бронхов или легких; внелегочном криптоспоридиозе; цитомегаловирусном поражении других органов (кроме печени) длительностью более 1 мес; саркоме Капоши у лиц моложе 60 лет; первичной лимфоме мозга; пневмоцистной пневмонии; прогрессивной многоочаговой лейкоэнцефалопатии; токсоплазмозе мозга у пациентов старше 1 мес; при наличии двух больших и одного малого симптома.
Для лечения ВИЧ-инфекции применяют препараты, препятствующие адсорбции вируса на клетках-мишенях: гепарин, декстрансульфат, троловол, анти-CD4 АТ.
Используют средства, препятствующие размножению ВИЧ в клетках: азидотимидин (зидовудин, ретровир), 2'3'-дидезоксиинозин (видекс) и 2'3'-дидезоксицитидин (диданозин) и их комбинации с азидотимидином, сурамин, влияющий на ревертазу, антисмысловые олигонуклеотиды, ингибиторы вирусных протеаз ( препарат U-75875) и гликозилирования (N-бутил-дезоксиноиримицин, растительный алкалоид - кастаноспермин), производное диазепина - TIBO (R82150, R82913), мурамилдипептид, рибавирин, папаверин, антибиотики фторхинолоны (офлоксацин, ципрофлоксацин, норфлоксацин, эноксацин), препарат из корня солодки - ниглизин, пептид трихосантин из корней китайского огурца, препараты РС6 и РС7, выделенные из шишек белой японской сосны, и др.
Перспективно применение АТ против вирусных белков. Используют гемо-сорбцию, иммуносупрессоры, интерферон, ИЛ и препараты тимуса (хотя о последних мнение не однозначно и ряд исследователей не реко-
мендует их применять), иммунозаместительную терапию, реализуют введение зрелых донорских лимфоидных клеток, имутиола и амплигена, проводят нагревание тела до 420С, используют препараты из поджелудочных желез убойного скота, подавляющие репродукцию вируса; аверол, выделенный из морских губок, желчные кислоты, мембранотропные липиды (12-метокси-ди-деканоат, амфотерицин В), антиоксиданты (дитиокарб, бутилированный гидрокси-анизол), зверобой и т.д.
Однако эффективность описанных методов лечения инфекции невелика, речь может идти только о ремиссии заболевания. Они позволяют продлить жизнь при клинически выраженном СПИДе до 2 лет (без лечения - 6 мес).
Истекшие годы исследований показали, что приготовленные традиционными способами ВИЧ-вакцины не приемлемы. Определенные надежды возлагаются на рекомбинантные препараты генно-инженерной технологии: коревую вакцину с генами ВИЧ, контролирующими образование иммуногенных гликопротеидов, оспенную вакцину, в которую вводят гены, ответственные за синтез gp32, gp120, gp160, и т.д. Разрабатывается идея создания пептидных вакцин, состоящих из синтетических пептидов, аналогичных антигенным эпитопам ВИЧ. Такой подход может привести к синтезу только протективных АТ, исключая усиливающие АТ и аутоантитела. Применение отдельных пептидов позволяет получить АТ к ним, тогда как они не образуются на эти же последовательности, входящие в состав большой естественной пептидной молекулы вируса. Наконец, пептидные вакцины позволят создать полипептидные препараты, состоящие из набора разных вариантов пептидов, соответствующих пептидам циркулирующих в настоящее время ВИЧ и даже тем их вариантам, которые могут появиться в будущем. В Институте иммунологии удалось создать конъюгаты пептида из gp41 с синтетическими иммуноадъювантами (сополимер N-винилпирролидона с N-винил-1,2,4-триазолом), с синтетическими микросферами из N-винилпирролидона, ассоциированного с малеиновым ангидридом, и комплексом пальмитиновой кислоты и хемоаттрактантом. Все они индуцировали синтез специфических АТ, подавляющих образование синцития и некроз в ВИЧинфицированных клетках.