Медицинская и клиническая генетика для стоматологов: учебник для вузов / Под ред. О.О. Янушевича., - 2009. - 400 с.
|
|
|
|
ГЛАВА 7. ВРОЖДЕННЫЕ И НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ ЗУБОВ
Петрин Александр Николаевич РАЗДЕЛ 1
ОБЩАЯ ХАРАКТЕРИСТИКА СТРОЕНИЯ ЗУБОВ
Зубы обеспечивают пережевывание пищи, участвуют в артикуляции и выполняют эстетическую функцию; располагаются в полости рта и занимают примерно 20% ее поверхности.
У человека две генерации зубов: временные (молочные) и постоянные. Во временном прикусе имеется 20 зубов, в постоянном - 32, которые подразделяют на 4 группы: резцы, клыки, малые коренные (премоляры), большие коренные (моляры) (рис. 1.1).
В зависимости от строения корня выделяют одно- и многокорневые зубы. Основу зуба составляет дентин. Снаружи зуб покрыт двумя твердыми обызвествленными тканями: эмалью и цементом.
Анатомически в каждом зубе выделяют коронку, шейку, корень.
Терминология, используемая для идентификации различных поверхностей и структурных компонентов зубов, представлена на рис. 1.2
Коронки представляют собой поверхностную часть зубов, защищенную эмалью. Коронка имеет различную форму, обусловленную функцией зуба. В зависимости от функции коронка снабжена режущим краем или жевательными бугорками.
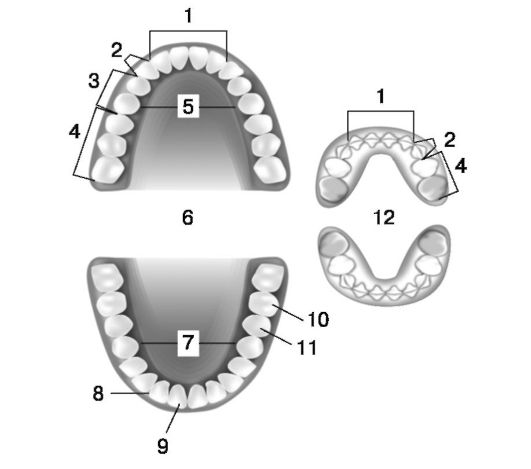 Рис. 1.1. Ориентация зубов и зубные дуги.
Рис. 1.1. Ориентация зубов и зубные дуги.
1. Резцы. 2. Клыки. 3. Премоляры. 4. Моляры. 5. Нёбная поверхность. 6. Постоянные зубы. 7. Язычная поверхность. 8. Резцовая поверхность. 9. Губная поверхность. 10. Щечная поверхность. 11. Поверхность прикуса. 12. Молочные зубы
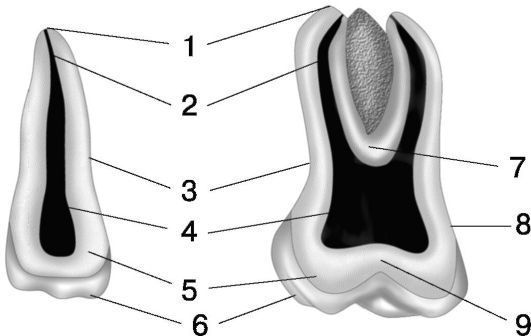 Рис. 1.2. Составные части зубов.
Рис. 1.2. Составные части зубов.
1. Верхушка. 2. Корневой канал. 3. Цемент. 4. Камера пульпы. 5. Дентин. 6. Эмаль. 7. Дно пульпы. 8. Эмалево-дентинная граница. 9. Крыша пульпы
Корень имеет конусовидную форму. К своему свободному концу он сужается и оканчивается верхушкой, имеющей одно или несколько апикальных отверстий, ведущих в полость зуба. Корень погружен в особое углубление челюсти, которое называется зубной альвеолой. Поддерживающий аппарат зуба (парадонт) обеспечивает прикрепление зуба к зубной альвеоле. В его состав входят: цемент, периодонт (периодонтальная связка), стенка зубной альвеолы и десна.
Шейка зуба - участок соединения эмали с цементом, в области которого коронка переходит в корень.
Пульпарная камера вытянута в направлении продольной оси зуба. В ней различают коронковый и корневой отделы. Коронковый отдел составляет основную часть объема пульпарной камеры. Корневой отдел представлен суживающимися в апикальном направлении корневым каналом. Объем пульпарной камеры с возрастом уменьшается вследствие непрерывного отложения дентина на ее стенках.
1.1. ГЕНЕТИЧЕСКИЙ КОНТРОЛЬ НОРМАЛЬНОГО РАЗВИТИЯ И ФОРМИРОВАНИЯ ТКАНЕЙ ЗУБОВ
Развитие и прорезывание зубов оказывает большое влияние на полость рта и соседние органы. Уже на первом году жизни вследствие развития альвеолярных отростков увеличивается высота верхней и нижней челюстей, происходит рост верхнечелюстных пазух. Это приводит к возрастанию вертикального размера полости рта и всего лица. Развитие постоянных зубов способствует росту челюстей и лица в сагитальном направлении, благодаря чему формируется лицевой профиль. После 15 лет, когда завершается в основном прорезывание постоянных зубов, рост лица в сагиттальном направлении и в высоту значительно уменьшается.
Развитие зубов протекает в несколько стадий, которые легко распознаются на микроскопическом уровне, поэтому стадии одонтогенеза традиционно описываются в классических терминах гистологической картины зубов. Эти стадии развития зубов (от ранних до поздних) именуются как пластинка, почка, мешочек (чашечка) и колокольчик. Недавние успехи, достигнутые в понимании молекулярных механизмов, управляющих ростом зубов, привели к появлению новой терминологии, которая описывает четыре стадии развития зубов: инициация, морфогенез, клеточная или цитодифференциация и аппозиция матрикса (рис. 1.3).
Зубная пластинка представляет собой первый морфологический знак начала развития зубов и становится заметной примерно на 5-й неделе внутриутробного развития человека. На этой стадии клетки дентального эпителия и подлежащей эктомезенхимы делятся с разной скоростью (последние быстрее). Индуктивное влияние зубной пластинки, предопределяющее судьбу подлежащей эктомезенхимы, было подтверждено несколькими исследователями.
Стадия почки характеризуется непрерывным ростом клеток зубной пластинки и эктомезенхимы. Эктомезенхима уплотняется и формирует зубной бугорок (сосочек). На этой стадии индуктивный или зубообразующий потенциал передается с зубного эпителия на зубной бугорок. Переход из стадии почки в стадию мешочка представляет собой важный этап в развитии зубов, поскольку именно на этом этапе начинает формироваться коронка. Зубная почка принимает форму мешочка (чашечки), окруженного зубным бугорком. Эктодермальный компартмент будущего зуба называется дентальным или эмалевым органом. Эмалевый орган и зубной бугорок инкапсулируются в другом слое мезенхимных клеток, так называемом зубном фолликуле, который отделяет их от других соединительных тканей в челюсти. Кластер клеток, называемый эмалевым узелком, представляет собой важный организующий центр в пределах зубного органа, который особенно важен для формирования бугорков коронки зуба. Эмалевый узелок экспрессирует уникальный набор сигнальных молекул, определяющих форму коронки и дальнейшее развитие зубного бугорка. Аналогично судьбе сигнальных центров в других организующих тканях, например в почках конечностей, эмалевый бугорок подвергается запрограммированной гибели (апоптоз) после того как завершится формирование бугорков коронки на раннем этапе колокольчика.
Позднее зубной орган принимает форму колокольчика, поскольку клетки продолжают делиться, но с разной скоростью. Единственный слой кубовидных клеток, называемых внешним или наружным зубным эпителием, выстилает периферию зубного органа, а клетки, граничащие с зубным бугорком, которые имеют вид столбиков (призм), формируют внутренний зубной эпителий. Внутренний зубной эпителий дает начало так называемым амелобластам, клеткам, ответственным за образование эмали. Клетки, расположенные в центре зубного органа, вырабатывают довольно много гликозаминогликанов, которые обладают способностью обособлять жидкости
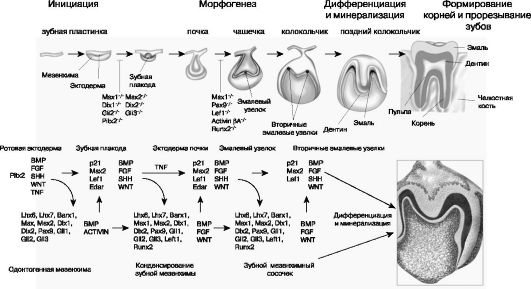 Рис. 1.3. Сигнальные
события при развитии зуба. Схематическое описание диффузионных сигналов
и факторов транскрипции, вовлеченных во взаимодействия между эпителием и
мезенхимой во время развития зубов у мыши. Факторы роста и морфогены,
задействованные в этих процессах, это костные морфогенетические протеины
(BMP) , факторы роста фибробластов (FGF), акустический еж (SHH) и белок
Wingless (WNT). В онтогенезе задействованы многие виды молекул. У
человека мутации в генах PITX2, SHH, MSX1 и PAX9 приводят к
развитию синдрома Ригера , единственного центрального резца верхней
челюсти, агенезии премоляров/третьего моляра и молярной олигодентии
соответственно
Рис. 1.3. Сигнальные
события при развитии зуба. Схематическое описание диффузионных сигналов
и факторов транскрипции, вовлеченных во взаимодействия между эпителием и
мезенхимой во время развития зубов у мыши. Факторы роста и морфогены,
задействованные в этих процессах, это костные морфогенетические протеины
(BMP) , факторы роста фибробластов (FGF), акустический еж (SHH) и белок
Wingless (WNT). В онтогенезе задействованы многие виды молекул. У
человека мутации в генах PITX2, SHH, MSX1 и PAX9 приводят к
развитию синдрома Ригера , единственного центрального резца верхней
челюсти, агенезии премоляров/третьего моляра и молярной олигодентии
соответственно
и факторы роста, ведущие к его экспансии. Эта сеть звездообразных клеток называется звездчатым ретикулумом. Между звездчатым ретикулумом и внутренним зубным эпителием расположен тонкий слой уплощенных клеток, носящий название промежуточного слоя и экспрессирующий высокий уровень щелочной фосфатазы. Считается, что этот промежуточный слой влияет на биологическую минерализацию эмали. В области апикального конца зубного органа внутренний и наружный слои зубного эпителия сливаются, образуя соединение, которое называется цервикальной петлей.
На ранней стадии колокольчика каждый слой зубного органа, повидимому, выполняет специальные функции и осуществляет обмен молекулярной информации, что ведет к дифференциации клеток на поздней стадии колокольчика. Зубная пластинка, соединяющая зубной орган с оральным эпителием, на поздней стадии колокольчика постепенно распадается. В области будущего острия коронки зуба клетки внутреннего зубного эпителия перестают делиться и принимают столбчатую (призматическую) форму. Наиболее периферийные клетки зубного бугорка организуются вдоль базальной мембраны и дифференцируются в одонтобласты, т.е. в дентинообразующие клетки. В это время зубной бугорок становится пульпой зуба. После того как будет накоплен первый слой предентинового матрикса, клетки внутреннего зубного эпителия дифференцируются в амелобласты или эмалеобразующие клетки. По мере накопления эмали над дентиновым матриксом амелобласты отступают к наружной поверхности коронки и, по-видимому, подвергаются запрограммированному апоптозу. В отличие от этого одонтобласты выстилают внутреннюю поверхность дентина и сохраняют метаболическую активность на всем протяжении жизни зуба. Затем происходит образование корня, связанное с тем, что клетки эпителия пролиферируют в апикальном направлении и оказывают влияние на дифференциацию одонтобластов из зубного бугорка и цементобластов из фолликулярной мезенхимы. Это ведет к накоплению корневого дентина и цемента соответственно. В зубах с несколькими корнями одни части оболочки корней растут быстрее других, что приводит к образованию языкоподобных выростов. Когда выросты (два в двухкорневых и три в трехкорневых зубах) входят в соприкосновение друг с другом, устанавливается положение развилки корня. Зубной фолликул, дающий начало компонентам периодонта (периодонтальные связующие фибробласты, альвеолярный костный отросток зубной впадины и цемент), также играет важную роль в прорезывании зубов, обозначающем последнюю стадию одонтогенеза.
Резюмируя, можно сказать, что развитие зубов регулируется ограниченными во времени и пространстве реципрокными взаимодействиями между эпителиальным и мезенхимным компартментами, и потенциал, доминирующий в развитии зубов, перемещается то из эпителия в мезенхиму, то из мезенхимы в эпителий.
В последние годы достигнут большой прогресс в понимании молекулярных механизмов, определяющих место инициации зуба.
Рисунке 1.3 показано, что в мезенхиме первой жаберной дуги экспрессируется много факторов транскрипции, сигнальных молекул (факторов роста и их рецепторов), а также молекул внеклеточного матрикса. Разные линии данных свидетельствуют о том, что в развитии зубов рекурсивно используются синергические и антагонистические взаимодействия сигнальных молекул.
Эмаль. Эмаль - это самая твердая обызвествленная структура в организме позвоночных, покрывающая коронки зубов. Толщина эмали варьирует от 2 до 3 мм в самых массивных частях бугорков коронки зуба до ножевого острия на границе с дентином. Поскольку эмаль это бесклеточная структура, она безжизненна, если не принимать во внимание поверхностную реминерализацию. Поверхностная реминерализация представляет собой результат ионообмена минералов на поверхности эмали. Цвет эмали меняется в диапазоне от прозрачного до желтовато-серого, но в большинстве случаев визуально воспринимаемая окраска покрытых эмалью коронок определяется дентином, просвечивающим через эмаль. Зубы с тонким слоем эмали выглядят желтоватыми, что отражает цвет дентина. Такое явление более характерно для лиц азиатского происхождения.
Процесс формирования эмали концептуально можно описать тремя фазами. Первая, секреторная, фаза характеризуется накоплением органического матрикса в амелобластах. На этой стадии такие клетки называются пресекреторными и секреторными амелобластами. Это вытянутые в длину клетки, в которых можно увидеть полярность ядер и секреторные органеллы. Во второй фазе (фазе минерализации) происходит так называемая нуклеация или образование кристаллов. По мере роста кристаллов процесс переходит в третью и последнюю стадию - созревание матрикса эмали. На этой стадии органический матрикс (особенно амелогенины) разрушается протеазами. Продукты распада амелогенинов реабсорбируются амелобластами.
Основные классы белков в матриксе эмали это амелогенин, амелобластин, эмаль, туфтелин, металлопротеиназа под названием энамелисин (MMP-20), сериновая протеиназа под названием сериновая протеиназа 1 матрикса эмали (EMPSP1) и следовые количества дентинового сиалофосфопротеина (DSPP). Пороки развития эмали часто связаны с мутациями генов, кодирующих эти матричные белки (по крайней мере, некоторые из них).
Основные компоненты эмали - это призмы, оболочки призм и межпризменное вещество. Призмы зубной эмали состоят из органических частиц, вокруг которых нарастают кристаллы апатитов. Каждая призма окружена оболочкой, которая обызвествляется в меньшей степени, чем собственно призма. Природа межпризменной структуры не очень понятна, хотя хорошо известно, что этот компонент эмали облегчает распространение кариеса на дентин.
Под эмалью находится толстый слой дентина и мягкий центральный стержень зуба - камера пульпы.
Дентин. Дентин составляет большую часть массы зуба. Это живая ткань, обладающая многими физическими и химическими свойствами кости. Дентин имеет желтый цвет и гораздо более хрупок по сравнению с эмалью. Процесс образования дентина по существу не отличается от процессов образования других твердых соединительных тканей в организме, например зубного цемента и кости. Основным условием для этого является наличие высокоспециализированных клеток, синтезирующих и секретирующих высокоспециализированный органический матрикс, способный интегрировать биологический апатит и другие минералы. Еще одним условием формирования дентина является хорошее кровоснабжение и высокий уровень фермента щелочной фосфатазы. Дентинообразующие клетки, или одонтобласты, начинают секретировать внеклеточный матрикс (ECM) предентина. Они отступают в направлении пульпы, но сохраняют связь с матриксом, который формируется клеточными расширениями, так называемыми одонтобластными отростками. Органический матрикс предентина превращается в минерализованный слой дентина в результате очень сложного процесса, который начинается на некотором расстоянии от тел одонтобластных клеток. Самый наружный слой дентина, образующийся первым, представляет собой зрелый дентин, а остальная его часть называется околопульпарным дентином.
Органическая часть дентина представлена белками, протеогликанами, липидами, различными факторами роста и водой. В составе белков преобладает коллаген, который придает фиброзному матриксу способность накапливать кристаллы углекислого апатита. Коллаген, присутствующий в дентине, это прежде всего коллаген I типа. Кроме того, здесь можно обнаружить следовые количества коллагена V типа и небольшое количество тримера коллагена I типа. Важность коллагена I типа как ключевого структурного компонента
дентинового матрикса иллюстрируется наследственным заболеванием дентина, так называемым несовершенным дентиногенезом (DGI), который будет подробно обсужден далее.
Важный класс белков дентинового матрикса представляют неколлагеновые протеины (NCP). Дентиноспецифичные NCP - это дентиновые фосфопротеины (DPP), или фосфофорины, и дентиновый сиалопротеин (DSP). После коллагена I типа DPP это следующий по содержанию в дентиновом матриксе белок, на долю которого приходится почти 50% ECM дентина. Типичный DPP представляет собой полиионную макромолекулу с большим содержанием фосфосерина и аспарагиновой кислоты. Высокое сродство с коллагеном I типа и кальцием делает его несомненным кандидатом на роль пускового фактора в минерализации дентина. Содержание DSP в дентиновом матриксе составляет 5-8% и он относительно богат сиаловой кислотой и углеводами. Его роль в минерализации дентина в настоящее время не вполне ясна. Ранее считалось, что DSP и DPP - это два независимых белка, кодируемых разными генами. Сейчас установлено, что эти белки представляют собой специфические продукты расщепления более крупного протеина предшественника, который транслируется из единого большого транскрипта. Этот общий ген, кодирующий как DSP, так и DPP, получил название гена дентинового сиалофосфопротеина (DSPP).
Значение DSPP в образовании дентина было недавно подчеркнуто тем открытием, что мутации этого гена ответственны за основные дефекты дентина, наблюдаемые при несовершенном дентиногенезе (DGI). Локус DGI картирован в регионе q13-21 четвертой хромосомы человека, там же, где и несколько других генов ECM дентина. Вторая категория NCP со способностью связывать кальций классифицирована как минерализующие белки с тканевой специфичностью, поскольку они обнаруживаются во всех обызвествленных соединительных тканях, т.е. в дентине, кости и зубном цементе. К этой группе белков относятся остеокальцин (OC) и костный сиалопротеин (BSP). Богатый серином фосфопротеин, так называемый протеин 1 дентинового матрикса (Dmp-1), экспрессия которого изначально считалась ограниченной только одонтобластами; позднее был обнаружен также в остеобластах и цементобластах, а также в клетках головного мозга. Другими NCP этой группы являются остеопонтин (OP) и остеонектин (SPARC). Четвертая категория NCP дентина не экспрессируется в одонтобластах, а синтезируется, прежде всего в печени, откуда и
попадает в системный кровоток. Примером сывороточного протеина является α2HS-гликопротеин. Диффузионные факторы роста, которые выглядят изолированными в дентиновом матриксе, составляют пятую группу NCP дентина. Эта группа включает костные морфогенетические протеины (BMP), инсулиноподобные факторы роста (IGF) и трансформирующий β-фактор роста (TGF-β).
Цемент. Цемент по строению сходен с костной тканью и является наименее менерализованной твердой тканью зуба. Это обызвествленная ткань мезодермального происхождения. Цемент, покрывающий апикальную треть корня, содержит живые клетки (цементоциты), в то время как остальные две трети цемента бесклеточны. Цемент подвержен рассасыванию в меньшей степени, чем кость, и этот факт, несомненно, связан с его ролью в фиксации зубов и с их способностью прорезываться через кость.
Пульпа. Пульпа зуба заполняет пульпарную камеру в ее коронковом и корневом отделах. Пульпа осуществляет питание дентина, обеспечивает чувствительность зуба, выполняет защитные функции. Резцовая, или окклюзионная, поверхность камеры пульпы является ее крышей. Выступы медиальной и дистальной частей пульпы называются рогами пульпы. Апикальная поверхность камеры пульпы называется ее дном и служит зоной прохода нервов и кровеносных сосудов в корневые каналы. В пульпе можно найти кровеносные и лимфатические сосуды, а также сенсорные и моторные нервы. Единственная сенсорная функция пульпы - это способность передавать боль. Масса пульпы представляет собой рыхлую соединительную ткань, содержащую многие типы клеток, в том числе фибробласты и одонтобласты. Соматические стволовые клетки из пульпы молочных моляров после трансплантации in vivo способны регенерировать разные ткани.
1.2. ГЕНЕТИЧЕСКИЕ ФАКТОРЫ ФОРМИРОВАНИЯ АНОМАЛИЙ ЗУБОВ
Под аномалией в биологии и медицине понимают морфологические или функциональные изменения, возникающие вследствие нарушения развития органов и систем. Аномалии развития зубов и зубочелюстной системы, как и другие аномалии развития, подразделяют на большие, или врожденные, пороки развития и малые аномалии развития. Морфологические нарушения, появившиеся под воздействием неблагоприятных внешних факторов после рождения ребенка,
обозначаются термином «деформация». Врожденные пороки развития - это стойкие морфологические изменения органа или всего организма, выходящие за пределы вариаций границ нормального строения и сопровождающиеся нарушением функции. К ним относятся, например, анэнцефалия, гидроцефалия, агирия, расщелины губы и нёба, редукционные пороки конечностей, атрезия кишечника, удвоение мочевыводящих путей, пороки сердца и др. Малые аномалии развития зубов и зубочелюстной области, в отличие от врожденных пороков развития, не сопровождаются существенными нарушениями функции, не угрожают жизни пациента, но имеют большое эстетическое значение и требуют специфического, иногда длительного и многоэтапного лечения. Для обозначения морфологических вариантов отклонений от нормального строения используются различные термины: «дисгенезии», «дисгенетические признаки», «дисплазии», «стигмы», «микродегенеративные признаки», «дизэмбриогенетические стигмы», «микроформы», «микропроявления», «микросимптомы» и т.д. В генетической литературе наиболее общепризнанным является термин «малые аномалии развития», или «микропризнаки». Анализ малых аномалий развития зубов и зубочелюстной системы имеет большое дифференциально-диагностическое значение как в стоматологии, так при диагностике разнообразных наследственных и врожденных состояний, имеющих сложную этиологию. Аномалии развития фронтальных зубов верхней челюсти, асимметрия носовой перегородки, рубец на верхней губе и альвеолярном отростке, высокое нёбо, короткое мягкое нёбо, видимые на рентгенограмме костные изменения нёбного сегмента, подслизистая расщелина язычка, гипертелоризм, несимметричное расположение крыльев носа, раздвоение язычка, готическое нёбо, атипичная форма верхних боковых резцов, аномалии резцов и клыков, прогения, прямой и перекрестный прикус часто являются маркерами наследственных заболеваний и синдромов.
Наличие у пациента определенных аномалий развития и их сочетание имеет большую диагностическую значимость. Например, частичная или полная олигодентия указывают на синдромы эктодермальных дисплазий, крыловидные складки, нарушение прикуса - на синдром Тернера и синдром Нунан, постаксиальная полидактилия - на синдром Лоуренса-Муна-Барде-Бидля, гипоили аплазия соска на одной стороне - на синдром Поланда, вертикальные насечки на мочке уха, висцеромегалия, большой язык - на синдром Беквита-Видемана и др.).
Диагностическая значимость аномалий развития определяется спектром поражения и спецификой их сочетания у каждого пациента. Например, при синдроме Секкеля, наряду с низким ростом, умственной отсталостью, микроцефалией и своеобразным строением головы и лица, большую роль играют такие признаки, как микрогнатия, высокое нёбо, частичная анадентия, гипоплазия эмали и др. При синдроме Ваарденбурга сочетаются телекант, гетерохромия радужки, сросшиеся брови, белая прядь волос надо лбом; при синдроме Дауна - монголоидный разрез глаз, эпикант, поперечная ладонная складка, брахидактилия, клинодактилия, сандалевидная щель, микродентия, нарушение прикуса и т.д.
Различные врожденные пороки развития зубов и зубочелюстной области встречаются при хромосомных синдромах, моногенных наследственных заболеваниях и при врожденной патологии мультифакториального происхождения.
Выяснение этиологии этих состояний является трудной диагностической задачей и требует консультативного участия представителей разных медицинских специальностей (генетиков, стоматологов, невропатологов, дерматологов, ортопедов, окулистов и др.)
Основными причинами генетически детерминированных аномалий и пороков развития являются разнообразные мутации, регистрируемые на хромосомном, геномном и генном уровнях организации наследственного материала.
Патогенез наследственных и врожденных аномалий развития зубов можно лучше понять, рассматривая их развитие на стадиях инициации и пролиферации, морфогенеза и аппозиции матрикса. Развитие зубов инициирует зубная пластинка. Если эта пластинка не сформирована или ее ранняя организация аномальна, инициация не происходит, и зубы вообще не развиваются (адентия). Если по тем или иным причинам физически разрушена только часть пластинки, инициация отсутствует именно в этой части, что приводит к частичному отсутствию зубов (гиподентия). В некоторых случаях дизруптивные факторы могут придавать пластинке избыточную активность, что приводит к гипердонтии (избыточным зубам). После инициации отделенные друг от друга почки зубов пролиферируют на своих предопределенных местах. Нарушения на стадии пролиферации могут также приводить к такому пороку развития, как гиподентия.
За начальными этапами пролиферации следует гистодифференциация. На этапе гистодифференциации устанавливаются типы
клеток (например, амелобласты и одонтобласты). Если внутренний зубной эпителий дифференцируется неправильно, становится невозможным образование одонтобластов, что, в свою очередь, приостанавливает развитие зубов. Если одонтобласты не могут правильно дифференцироваться, они теряют способность стимулировать образование амелобластов, и соответственно нарушаются процессы формирования эмали. Результатом нарушений дифференциации является аномальная структура зубов с явными изменениями их организации и формы.
Дифференциальный рост частей зубного органа (морфодифференциация) определяет базальный размер и форму зубов. Аномальная морфодифференциация часто приводит к микродентии, макродонтии, глободонтии, добавочным бугоркам на коронках зубов и другим аномалиям.
Аппозиция подразумевает накопление матрикса дентина и эмали. После того как клетки внутреннего зубного эпителия стимулируют подлежащую мезенхиму к образованию одонтобластов, одонтобласты создают слой предентина. После достаточного накопления предентина начинается процесс минерализация. Различные типы дисплазии дентина, по-видимому, отражают процессы дефектов накопления предентина. После формирования небольшого количества предентина амелобласты начинают секретировать матрикс эмали, что продолжается вплоть до достижения конечного (детерминированного) размера коронки. Существенный недостаток матрикса эмали приводит к гипопластическим вариантам дисплазии эмали. После закладки матрикса эмали происходит его минерализация, и любые нарушения на этой стадии развития приводят к дисплазиям эмали с недостаточностью кальцификации. Созревание твердого матрикса идет вслед за аппозиционным ростом. Нарушение созревания ведет к таким формам патологии, как дисплазии эмали с недозрелостью.
1.3. КЛАССИФИКАЦИЯ АНОМАЛИЙ РАЗВИТИЯ ЗУБОВ И ЗУБОЧЕЛЮСТНОЙ ОБЛАСТИ
Имеется множество аномалий, которые имеют схожую клиническую картину, встречаются как изолированные признаки, являются следствием воздействия неблагоприятных факторов внешней среды или общего заболевания. Кроме того, аномалии, пороки
развития зубов и зубочелюстной системы могут иметь сложную генетическую этиологию. Основными этиологическими факторами наследственных аномалий и пороков развития зубов и зубочелюстной системы являются изменения (мутации) генов и хромосом, а также взаимодействие внешнесредовых и наследственных факторов (мультифакториальные заболевания). Проблемы классификации являются достаточно сложными. В свое время были предложены различные варианты классификации зубочелюстных аномалий. Этот вопрос анализируется в специальных руководствах и монографиях (Персин Л.С., 2006). В настоящий момент предложены различные варианты рабочих классификаций, позволяющих систематизировать аномалии развития зубов и зубочелюстной системы, однако единой классификации, учитывающей все этиологические факторы и проявления патологии, в настоящее время еще не существует.
1.3.1. Международная классификация МКБ-10
По международной классификации стоматологических болезней на основе МКБ-10 (третье издание), в рубрике «болезни пищеварения» (класс XI), предложенной Всемирной организацией здравоохранения, выделяют следующие основные категории аномалий развития зубов и зубочелюстной системы.
К00 Нарушение развития и прорезывания зубов
К00.0 Адентия
• Частичная адентия (гиподентия) (олигодентия)
• Полная адентия
• Адентия неуточненная К00.1 Сверхкомплектные зубы
• Областей резца и клыка, мезиодентия (срединный зуб)
• Области премоляров
• Области моляров, дистомолярный зуб, четвертый моляр, парамолярный зуб
• Сверхкомплектные зубы неуточненнные К00.2 Аномалии размеров и формы зубов
• Макродентия
• Микродентия
• Сращение
• Слияние и раздвоение, раздвоение (шизодентия), слияние (синодентия)
• Выпячивание зубов (добавочные окклюзионные бугорки)
• Инвагинированный зуб («зуб в зубе»), (дилатированная одонтома) и аномалии резца, нёбная борозда, копьевидные (конические резцы), лопатообразные резцы, Т-образные резцы
• Премоляризация
• Аномальные бугорки и эмалевые жемчужины (адамантома)
• «Бычий зуб» (тауродонтизм)
• Другие и неуточненные аномалии размеров и формы зубов К00.3 Крапчатые зубы
• Эндемическая (флюорозная) крапчатость эмали
• Неэндемическая крапчатость эмали (нефлюорозное помутнение эмали)
• Крапчатые зубы неуточненные К00.4 Нарушения формирования зубов
• Гипоплазия эмали
• Пренатальная гипоплазия эмали
• Неонатальная гипоплазия эмали
• Аплазия и гипоплазия цемента
• Дилацерация (трещины эмали)
• Одонтодисплазия (региональная одонтодисплазия)
• Зуб Тернера
• Другие уточненные нарушения формирования зубов
• Нарушения формирования зубов неуточненные
К00.5 Наследственные нарушения структуры зуба, не классифицированные в других рубриках
• Незавершенный амелогенез
• Несовершенный дентиногенез, изменения в зубах при незавершенном остеогенезе
• Незавершенный одонтогенез
• Другие наследственные нарушения структуры зуба, дисплазия дентина, раковинные зубы
• Наследственные нарушения структуры зуба неуточненные К00.6 Нарушения прорезывания зубов
• Натальные (прорезавшиеся к моменту рождения) зубы
• Неонатальные (у новорожденного, прорезавшиеся преждевременно) зубы
• Задержка (персистентная) смены первичных (временных) зубов
• Позднее прорезывание
• Преждевременное выпадение первичных (временных) зубов
• Другие уточненные нарушения прорезывания зубов
• Нарушение прорезывания зубов неуточненное К00.7 Синдром прорезывания зубов
К00.8 Другие нарушения прорезывания зубов
• Изменение цвета зубов в процессе формирования вследствие несовместимости групп крови
• Изменение цвета зубов в процессе формирования вследствие врожденного порока билиарной системы
• Изменение цвета зубов в процессе формирования вследствие Порфирии
• Изменение цвета зубов в процессе формирования вследствие применения тетрациклина
• Другие уточненные нарушения развития зубов К00.9 Нарушение развития зубов неуточненное К01 Ретинированные и импактные зубы
К01.0 Ретинированный зуб - зуб, изменивший свое положение при прорезывании без препятствия со стороны соседнего зуба К01.1 Импактные зубы
Импактный зуб - зуб, изменивший свое положение при прорезывании из-за препятствия со стороны соседнего зуба
• Резец верхней челюсти
• Резец нижней челюсти
• Клык верхней челюсти
• Клык нижней челюсти
• Премоляр верхней челюсти
• Премоляр нижней челюсти
• Моляр верхней челюсти
• Моляр нижней челюсти
• Сверхкомплектный зуб
• Импактный зуб неуточненнный К02 Кариес зубов
• Кариес эмали
Стадия «белого (мелового) пятна» (начальный кариес)
• Кариес дентина
• Кариес цемента
• Приостановившийся кариес зубов
• Однотоклазия
• Детская меланодентия
• Меланодентоклазия Другой уточненный кариес зубов Кариес зубов неуточненный
К03 Другие болезни твердых тканей зубов К03.0 Повышенное стирание зубов
• Окклюзионное
• Апроксимальное
• Другое уточненное стирание зубов
• Стирание зубов неуточненное Сошлифовывание (абразивный износ) зубов К03.2 Эрозия зубов
Патологическая резорбция зубов
К03.4 Гиперцементоз
К03.5 Анкилоз зубов
К03.6 Отложения и наросты на зубах
К03.7 Изменения цвета твердых тканей зубов после прорезывания
К06.0 Рецессия десны К06.1 Гипертрофия десны
К06.2 Поражения десны и беззубого альвеолярного края обусловленные травмой
К06.8 Другие уточненные изменения десны и беззубого альвеолярного края
К06.9 Изменение десны и беззубого альвеолярного края неуточненное
К07 Челюстно-лицевые аномалии (включая аномалии прикуса) К07.1 Аномалии челюстно-черепных соотношений К07.2 Аномалии соотношения зубных дуг К07.3 Аномалии положения зубов К07.4 Аномалия прикуса неуточненная.
Как полагают, вышеизложенная классификация слишком громоздка и неудобна для практического применения (Персин Л.С., 2006). В связи с этим кафедрой одонтии и детского протезирования МГМСУ была предложена другая, более совершенная классификация.
1.3.2. Классификация аномалий зубов и челюстей кафедры ортодонтии и детского протезирования МГМСУ (1990)
1. АНОМАЛИИ ЗУБОВ
1.1. Аномалии формы зуба
1.2. Аномалии структуры твердых тканей зуба
1.3. Аномалии цвета зуба
1.4. Аномалии размера зуба (высоты, ширины,толщины)
1.4.1. Макродентия
1.4.2. Микродентия
1.5. Аномалии количества зубов
1.5.1. Гиперодонтия (при наличии сверхкомплектных зубов)
1.5.2. Гиподентия (адентия зубов-полная или частичная)
1.6. Аномалии прорезывания зубов
1.6.1. Раннее прорезывание
1.6.2. Задержка прорезывания (ретенция)
1.7. Аномалии положения зубов (в одном, двух, трех направлениях)
1.7.1. Вестибулярное
1.7.2. Оральное
1.7.3. Мезиальное
1.7.4. Дистальное
1.7.5. Супраположение
1.7.6. Инфраположение
1.7.7. Поворот по оси (тортоаномалия)
1.7.8. Транспозиция
2. АНОМАЛИИ ЗУБНОГО РЯДА
2.1. Нарушение формы
2.2. Нарушение размера
2.2.1. В трансверсальном направлении (сужение, расширение)
2.2.2. В сагиттальном направлении (удлинение, укорочение)
2.3. Нарушение последовательности расположения зубов
2.4. Нарушение симметричности положения зубов
2.5. Нарушение контактов между смежными зубами (скученное или редкое положение)
3. АНОМАЛИИ ЧЕЛЮСТЕЙ И ИХ ОТДЕЛЬНЫХ АНАТОМИЧЕСКИХ ЧАСТЕЙ
3.1. Нарушение формы
3.2. Нарушение размера
3.2.1. В сагиттальном направлении (удлинение, укорочение)
3.2.2. В трансверсальном направлении (сужение, расширение)
3.2.3. В вертикальном направлении (увеличение,уменьшение высоты)
3.2.4. Сочетанные по 2 и 3 направлениям
3.3. Нарушение взаиморасположения челюстей
3.4. Нарушение положения челюстных костей
4. АНОМАЛИИ ОККЛЮЗИИ ЗУБНЫХ РЯДОВ
4.1. Аномальная окклюзия зубных рядов в сагиттальном направлении
Боковой сегмент
4.1.1. Дистальная
4.1.2. Мезиальная Передний сегмент
4.1.3. Сагиттальная резцовая дизокклюзия
4.1.4. Обратная резцовая окклюзия
4.1.5. Обратная резцовая дизокклюзия
4.2. Аномальная окклюзия зубных рядов в вертикальном направлении
Боковой сегмент
4.2.1. Дизокклюзия
Передний сегмент
4.2.2. Вертикальная резцовая дизокклюзия
4.2.3. Глубокая резцовая окклюзия
4.2.4. Глубокая резцовая дизокклюзия
4.2.5. Глубокая резцовая окклюзия
4.3. Аномальная окклюзия зубных рядов в трансверсальном направлении.
Боковой сегмент
4.3.1. Вестибулоокклюзия
4.3.2. Палатоокклюзия
4.3.3. Лингвоокклюзия
Передний сегмент
4.3.4. Трансверсальная резцовая окклюзия
4.3.5. Трансверсальная резцовая дизокклюзия
5. АНОМАЛИИ ОККЛЮЗИИ ПАР ЗУБОВ-АНТОГОНИСТОВ
5.1. По саггитали
5.2. По вертикали
5.3. По трансверсали
Эта классификация выдержана в одном ключе: аномалии смыкания зубных рядов в сагиттальном, вертикальной, трансверсальной плоскостях характеризуется в зависимости от вида смыкания.
Данная классификация, обладая рядом преимуществ, тем не менее не вполне соответствует принятой в клинической генетике системе описания аномальных признаков, используемых при диагностике наследственных заболеваний и синдромов. В современных поисководиагностических системах принята более упрощенная классификация аномалий развития полости рта, зубов и зубочелюстной системы. Эта система предполагает унифицированный методический прием использования стандартного набора диагностических признаков.
1.3.3. Аномальные признаки патологии полости рта, зубов, зубочелюстной системы, используемые для диагностики наследственных заболеваний и синдромов
I. ПОЛОСТЬ РТА-ЗУБЫ
• аномалии зубного ряда;
• аномалии формы зубной коронки;
• тауродонтия/элонгация пульпы моляров;
• макродентия;
• микродентия;
• нарушение формирования эмали;
• нарушение формирования дентина;
• изменения цвета зубов;
• нарушение позиции зубов/аномалия прикуса/открытый прикус;
• позднее прорезывание зубов;
• отсутствие части постоянных зубов;
• адентия/олигодентия;
• наличие зубов у плодов и новорожденных;
• избыточное количество зубов (сверхкомплектные зубы);
• преждевременное прорезывание зубов;
• зубной кариес;
• преждевременная потеря зубов;
• зубные кисты/опухоли;
• другие аномалии зубов.
II. ПОЛОСТЬ РТА-НЁБО/АЛЬВЕОЛЯРНЫЙ ОТРОСТОК
• расщелина твердого нёба;
• расщелина мягкого нёба/раздвоение язычка, подслизистая расщелина;
• короткое нёбо;
• неподвижность мягкого нёба;
• высокое арковидное и узкое нёбо;
• широкий альвеолярный край;
• другие аномалии нёба/альвеолярного отростка.
III. ПОЛОСТЬ РТА-ЯЗЫК/ДЕСНЫ/СЛИЗИСТАЯ
• аномальный язык/десны/слизстая рта;
• глоссоптоз/гипоплазия/атрофия языка;
• выступающий изо рта язык;
• большой язык (макроглоссия);
• бороздчатый язык;
• расщелина/зазубренность языка;
• утолщенные десны;
• гамартомы/другие опухоли языка;
• пигментированные десны/слизистые мембраны;
• синехии/патологические уздечки;
• аномалии слюнных желез;
• другие аномалии языка/десен/слизистой.
РАЗДЕЛ 2
АНОМАЛИИ РАЗМЕРОВ И ФОРМЫ ЗУБОВ
Для объективной оценки аномалий размеров и формы зубов проводят сопоставление измерений отдельных зубов с таблицами их нормальных размеров (по Вайсу или Устименко). Более полное представление о величине зубов может дать показатель модуля коронки. Он определяется как сумма вестибулоязычного и мезиодистального размера деленная на два.
Размеры зубов как антропологическая характеристика могут варьировать. У представителей южной европеоидной расы зубы относительно меньшего размера, чем у людей монголоидной расы. Особенно это касается формы и размера верхних резцов. Известно также, что антропологический тип негроидной расы, особенно представители ее восточной ветви, характеризуются более крупными размерами зубов. Поэтому при оценке размера зубов следует также принимать во внимание этническую и популяционную принадлеж-
Таблица 1.1. Величина коронок зубов
Группы зубов | По Вайсу (1965) | По Устименко (1984) | |||
высота | ширина | высота | ширина | ||
Центральные резцы | Верхние | 8 5-14,0 | 7,0-10,0 | 8,2-9,7 | 8,0-9,0 |
Нижние | 7,5-10,0 | 4,0-6,6 | 7,0-8,6 | 4,9-5,6 | |
Боковые резцы | Верхние | 8,0-11,0 | 5,0-8,0 | 7,1-8,5 | 6,0-7,1 |
Нижние | 8,8-11,3 | 5,2-7,2 | 7,2-8,7 | 5,6-6,4 | |
Клыки | Верхние | 9,5-10,5 | 6,5-8,0 | 8,0-9,6 | 7,1-8,1 |
Нижние | 9,0-14,0 | 5,7-7,2 | 8,5-10,2 | 6,3-7,2 | |
Первые премоляры | Верхние | 7,0-10,8 | 5,0-7,0 | 6,6-8,0 | 6,2-7,2 |
Нижние | 7,5-11,0 | 5,0-9,0 | 7,2-8,5 | 6,4-7,3 | |
Вторые премоляры | Верхние | 6,2-10,2 | 6,0-8,0 | 5,3-6,9 | 6,0-7,0 |
Нижние | 6,9-10,0 | 6,0-8,0 | 6,0-7,8 | 6,5-7,4 | |
Первые моляры | Верхние | 6,8-9,0 | 7,8-11,2 | 4,5-5,9 | 8,7-10,0 |
Нижние | 10,0 | 10,0-12,0 | 4,4-6,1 | 10,3-11,7 | |
Вторые моляры | Верхние | 5,0-7,0 | 9,0-11,0 | 4,5-5,0 | 8,7-10,0 |
Нижние | 10,0 | 9,0-11,0 | 4,5-5,0 | 9,6-10,8 | |
ность индивида. Аномалии величины и формы зубов весьма разнообразны. Неправильную форму могут иметь коронка и корень зуба. В соответствие с МКБ-10 выделяют следующие основные аномалии размеров и формы зубов.
• Макродентия.
• Микродентия.
• Сращение.
• Слияние (синодентия) и раздвоение (шизодентия).
• Выпячивание зубов (добавочные окклюзионные бугорки).
• Инвагинированный зуб («зуб в зубе»), (дилатированная одонтома) и аномалии резца, нёбная борозда, копьевидные (конические резцы), лопатообразные резцы, Т-образные резцы.
• Премоляризация.
• Аномальные бугорки и эмалевые жемчужины (адамантома).
• «Бычий зуб» (тауродонтизм).
• Другие и неуточненные аномалии размеров и формы зубов.
2.1. МАКРОДЕНТИЯ
Макродентия - (син. макродонтия, мегалодонтия)- чрезмерно большие размеры одного или нескольких зубов. Макродентию подразделяют на три типа: 1 - генерализованная макродентия (размеры большинства зубов значительно больших размеров по сравнению с нормой); 2 - относительно генерализованная макродентия (некоторые зубы лишь незначительно превышают нормальные размеры); 3 - изолированная макродентия (только единичный зуб увеличен по своим размерам). Диагностика макродентиии основана на визуальном исследовании и методе сопоставления размеров зуба с его стандартными (средними) популяционными размерами.
Макродентия может встречаться как изолированный признак, как признак множественных аномалий развития зубов или входить в состав наследственных заболеваний и синдромов моногенной и хромосомной этиологии.
Увеличенный размер коронки зуба может быть следствием таких аномалий развития, как слияние и удвоение зуба.
2.2. МИКРОДЕНТИЯ
Микродентия (син. микродентизм) - малые размеры коронки зуба по сравнению со средним размером коронок той же группы зубов. Различают три вида микродентии.
• Генерализованная микродентия - все зубы нормально сформированы, но их размер значительно меньше, чем в норме. Генерализованная микродентия встречается как отдельная аномалия развития, так и в составе некоторых наследственных заболеваний и синдромов, например при гипофизарной карликовости .
• Относительно генерализованная микродентия - имеются зубы нормального и уменьшенного размера, причем на нижней челюсти зубов уменьшенного размера больше, чем нормальных.
• Изолированная микродентия - при изолированной микродентии поражен обычно только один зуб; чаще пораженным зубом являются латеральные резцы и третьи моляры верхней челюсти.
2.3. ЗУБЫ СЛИВШИЕСЯ
Зубы слившиеся - увеличенный горизонтальный размер коронки зуба, сочетающийся в некоторых случаях с наличием добавочного
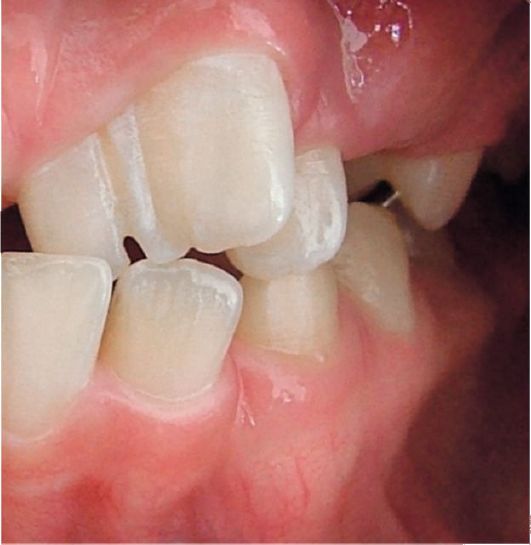 Рис. 1.4. Слияние зубов
Рис. 1.4. Слияние зубов
корня (корней). Эта аномалия развития фенотипически проявляется в виде увеличенного горизонтального размера коронки и является следствием слияния двух (иногда более) зачатков зубов.
При этом отмечается отсутствие латерального резца вследствие слияния с центральным резцом. Эта аномалия развития встречается с частотой примерно 0,5%. В большинстве случаев клинически проявляется как наличие широкой сдвоенной коронки с одной пульпарной камерой.
2.4. УДВОЕНИЕ
Удвоение (germination) - аномалия, обозначаемая как удвоение зуба, наблюдается в том случае, если из одного зубного зачатка формируется как бы два зуба.
По форме зуба иногда сложно различить между собой слияние или удвоение. Для того чтобы отдифференцировать эти состояния, необходимо подсчитать количество зубов. Если при подсчете количество зубов меньше нормы, то это слияние двух зубов; если их число не отличается от нормального количества - удвоение. Частота аномалии в популяции 0,5%.
2.5. ИНВАГИНАЦИЯ ЗУБОВ («ЗУБ В ЗУБЕ»)
Инвагинация встречается либо как изолированный порок развития, либо в сочетании с другими аномалиями в составе некоторых наследственных синдромов. Полагают, что этиология изолированной инвагинации зубов - мультфакториальная. Эта аномалия чаще встречается у китайцев, японцев, американских аборигенов и эскимосов по сравнению с европейцами и выходцами с африканского континента. Встречается изолированная доминантная форма этого состояния. Средняя частота доминантной аномалии аномалия «зуб в зубе», по данным Grahnen et al. (1959), составляет 3%. В настоящее время в мировой литературе имеется описание двух наследственных моногенных заболеваний, сопровождающихся данной аномалией развития. Первое заболевание: тауродонтизм, микродентия и инвагинация зубов (ОМИМ 313490). При данном заболевании, наследующемся по Х-сцепленному рецессивному типу, отмечается генерализованная микродентия, тауродонтизм первых постоянных
моляров и множественные инвагинации зубов. Другое заболевание, при котором описана инвагинация зубов, наследуется по аутосомно-доминантному типу (ОМИМ 125300). Этот синдром так и называется «зуб в зубе и нёбные инвагинации» (DENS IN DENTE AND PALATAL INVAGINATIONS).
2.6. АНОМАЛЬНЫЕ БУГОРКИ И ЭМАЛЕВЫЕ ЖЕМЧУЖИНЫ (АДАМАНТОМА)
«Эмалевые жемчужины , эмалевые капли» - шарообразные образования эмали, прикрепленные к зубу диаметром от 1 до 4 мм, наблюдаются у 1,5% пациентов и располагаются в области шейки зуба на границе эмали и цемента, иногда в зоне бифуркации (трифуркации) корней. «Эмалевые капли» состоят из дентина, покрытого эмалью, внутри них часто имеется полость, заполненная пульпой. Данная аномалия часто не вызывает каких-либо функциональных нарушений.
2.7. ТАУРОДЕНТИЗМ (БЫЧИЙ ЗУБ)
Тауродентизм - аномалия развития, характеризующаяся большой пульповой камерой (рис. 1.5 а, б).
По частоте тауродонтизма наблюдаются межэтнические различия
(табл. 1.2).
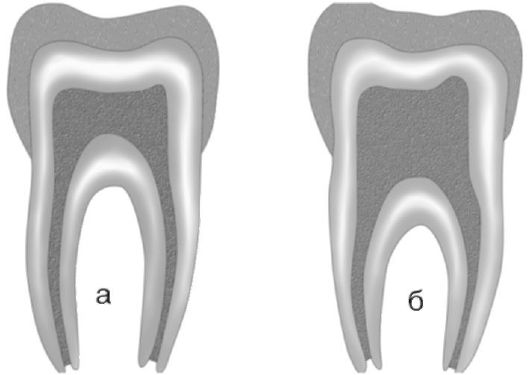 Рис. 1.5 а, б. Тауродонтизм:
Рис. 1.5 а, б. Тауродонтизм:
а - нормальная пульповая камера; б - тауродонтия
Таблица 1.2. Тауродонтия в разных популяциях
Популяция | Частота (%) |
Европейцы | 0,6-3,2 |
Японцы | 0,5 |
Африканцы | 4,4 |
Эта аномалия наиболее часто встречается у зубов-моляров. Клинические варианты тауродонтизма включают в себя гипотауродонтизм, мезотауродонтизм, гипертауродонтизм.
Эти состояния могут быть зарегистрированы как изолированные признаки (аутосомно-доминантные), так и как составная часть хромосомных болезней, моногенных наследственных заболеваний и синдромов, например таких, как трихо-денто-костный синдром, отодентальная дисплазия, эктодермальная дисплазия, зубо-ногтевой синдром, несовершенный эмалегенез, синдром Клайнфельтера, синдром Дауна, гипогидротическая эктодермальная дисплазия и др.
2.8. ДРУГИЕ АНОМАЛИИ РАЗМЕРОВ И ФОРМЫ ЗУБОВ
Зуб Гетчинсона - верхний центральный резец с отверткообразной формой коронки, полулунной выемкой на режущем крае. Наиболее широкий поперечный размер по середине коронки; такую форму зубной коронки иногда обозначают как «отверткообразная» форма зубной коронки.
Зуб Пфлюзера - моляр с наибольшей шириной у шейки, а наименьшей - у жевательной поверхности.
«Зуб рыбий» - клык, похожий по форме на резец.
Зуб Фурнье - первые большие коренные зубы с укороченными коронками и гипоплазией эмали на жевательной поверхности.
Зубы бугорчатые - корень конический, а коронка состоит из ряда бугорков и ямок (зубы в виде плодов шелковицы).
В этом случае форма коронки состоит из ряда бугорков и ямок. Аномалии развития, получившие в стоматологической литературе, обозначение как резцы в виде «отвертки» и моляры в форме «ягод шелковицы», чаще описываются как врожденные состояния вследс-
твие внутриутробного сифилитического поражения. Однако известны и моногенные состояния, сопровождающиеся такими аномалиями развития, в частности так называемый катаракто-дентальный синдром (катаракта, Х-сцепленная с зубами Гетчинсона, мезиоденскатаракта-синдром).
Зубы конусовидные - коронки зубов имеют форму шипа или клина; резцы центральные шиповидные - суженные в диаметре зубы на уровне режущего края.
Гиперцементоз
Гиперцементоз - избыточное отложение цемента, при этом отмечается деформация корня зуба в виде выступов на его поверхности, образовавшихся в результате чрезмерного отложения цемента.
Гиперцементоз может быть локальным, диффузным и генерализованным.
Локальный гиперцементоз проявляется формированием округлых узелков или шипов из цемента на латеральной или межкорневой поверхностях зуба. Наиболее часто это происходит в результате прикрепления к поверхности цемента и погружения в него цементеклей - сферических телец диаметром 0,1-0,4 мм, состоящих из цемента и первоначально расположенных среди пучков волокон периодонтальной связки. Причиной формирования цементиклей служит смещение цементобластов, а ядром, индуцирующим их образование, - эпителиальные остатки Малассе. Цементикли могут расти, сливаясь друг с другом и прикрепляясь к цементу. На их поверхности выявляются цементобласты, образующие новые слои цемента. Локальный гиперцементоз иногда наблюдается в участках, где на поверхности дентина образовались так называемые эмалевые жемчужины. «Эмалевые капли» диаметром от 1 до 4 мм наблюдаются у 1,5% пациентов и располагаются в области шейки зуба на границе эмали и цемента, иногда в зоне бифуркации (трифуркации) корней.
Диффузный гиперцементоз характеризуется усиленным отложением цемента на всей поверхности корня, нередко в связи с хроническим периапикальным инфекционным процессом. В некоторых случаях он приводит к сращению корня со стенкой костной альвеолы. Диффузный гиперцементоз встречается в 2,5 раза чаще в зубах нижней челюсти, особенно в премолярах и молярах.
Генерализованный гиперцементоз - избыточное отложение цемента во всех зубах.
Часто гиперцементоз встречается при болезни Педжета. Болезнь названа по имени английского хирурга Педжета Д. (1814-1899), характеризуется системным поражением скелета с усиленной резорбцией костной ткани, что сопровождается утолщением костной ткани и деформациями скелета. Иногда в процесс вовлечены кости лицевого скелета с признаками гиперцементоза.
Дилацерация (dilaceration) (смещение) - аномалия зубов, при которой корень зуба и его коронка начинают расти под углом друг к другу. В этом случае отмечается девиация (отклонение от нормального развития корня зуба). Наиболее часто такая аномалия встречается при травматическом повреждении формирующегося в процессе роста корня зуба. Данная аномалия встречается также и при наследственных заболеваниях, в частности при врожденном ихтиозе, окулофацио-кардиодентальном синдроме. Наряду с аномалиями развития зубов, при данном заболевании отмечается нейросенсорная глухота, глазные аномалии (микрокорнеа, врожденная катаракта, вторичная глаукома, птоз, блефарофимоз и др.). Пороки сердечно-сосудистой системы и умственная отсталость.
Конкресценция (обширное срастание) - отмечается сращение цемента соседних зубов. Данная аномалия крайне затрудняет экстракцию зубов и требует предварительного рентгеновского исследования для планирования тактики операции.
РАЗДЕЛ 3
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ С АНОМАЛИЯМИ РАЗМЕРОВ И ФОРМЫ ЗУБОВ
3.1. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ
И СИНДРОМЫ С МАКРОДЕНТИЕЙ
В табл. 1.3 приведены данные о некоторых наследственных моногенных заболеваниях и синдромах, где в качестве одного и дифференциально-диагностических признаков фигурирует макродентия.
В настоящее время лишь для относительно небольшого количества заболеваний установлена локализация генов, вызывающих заболевания и синдромы, в состав которых входит макродентия. Как
Таблица 1.3. Наследственные заболевания и синдромы с макродентией
Синдром | Тип наследования | ОМИМ | Локус |
Аарскога синдром | Х-сц. | 305400 | Хр11.21 |
Коффина-Лоури синдром | Х-сц. | 303600 | Хр22.2-р22.1 |
Коэна синдром | АР | 26550 | 8q22-q23 |
Брахман-де-Ланге подобный синдром | АД | 112370 | |
Гемигипертрофия | АР | 235000 | |
KBG-синдром | АД | 148050 | |
Офтальмо-акромиелический синдром (анофтальмия с аномалиями конечностей) | АР | 205920 | |
РЕНО-синдром | АР | 260565 | |
Синдром постаксиальной полидактилии с аномалией зубов и ребер | АР | 263540 | |
Синдром альфа-талассемия/умственная отсталость | Х-сц. | 301040 | Xql3 |
и многие другие, эти заболевания характеризуются широкой клинической вариабельностью.
Аарскога синдром (Аарскога-Скотта). Синдром выделен в самостоятельную нозологическую единицу в 1970 г. Минимальные диагностические признаки: гипертелоризм, брахидактилия, низкий рост. Отмечаются широкая переносица, короткий нос с вывернутыми ноздрями, антимоноголоидный разрез глаз, а также разболтанность суставов, клинодактилия V пальцев, неполная кожная синдактилия рук, переразгибание в межфаланговых суставах. Характерны аномалии гениталий: шалевидная мошонка, крипрорхизм, фемоз, паховые грыжи. Умеренная умственная отсталость отмечается у 14% больных.
К стоматологическим проявлениям синдрома относят: гипоплазию верхней челюсти, относительную прогению, макродентию, аномалии прикуса, олиго-, адентию. Описаны случаи расщелины губы и нёба. Заболевание наследуется Х-сцепленно рецессивно, вызвано мутацией в гене фациогенитальной дисплазии (FGD1). Ген локализован в Xp11.21.
Синдром Коффина-Лоури. Впервые синдром описан в 1966 г. Тип наследования: Х-сцепленный доминантный. Минимальными диаг-ностическими признаками являются: антимоноголоидный разрез глаз, луковицеобразный нос, низкий рост, конусовидные пальцы, умственная отсталость. Данный синдром характеризуется специфическими черепно-лицевыми признаками: микроцефалия, выступающие надбровные дуги, гипертелоризм, антимонголоидный разрез глаз, открытые вперед ноздри, переорбитальная полнота тканей, широкая спинка носа, оттопыренные уши. Характерны также множественные признаки патологии соединительной ткани и скелета (гипермобильность суставов кистей и стоп, деформация грудной клетки, сколиоз, кифоз и др). К стоматологическим проявлениям синдрома относятся: большой приоткрытый рот, узкое, высокое нёбо, гиподентия, нарушение прикуса, широко расставленные зубы, большие срединные резцы (макродентия) (рис. 1.6 а, б). Наряду с умственной отсталостью, характерна мышечная гипотония. При данном заболевании описано также наличие эпиприступов и расширенные желудочки головного мозга. Ген локализован в
районе Xp22.2-p22.1.
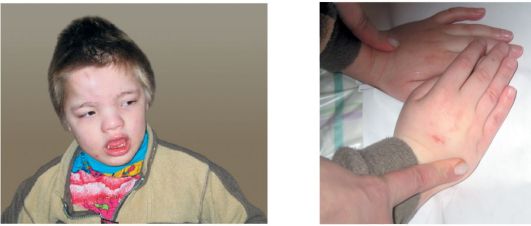 Рис. 1.6. Синдром Коффина-Лоури:
Рис. 1.6. Синдром Коффина-Лоури:
а - антимонголоидный разрез глаз, широкая переносица, оттопыренные уши, приоткрытый рот; б - конусовидные пальцы
Синдром Коэна (гипотония, ожирение, выступающие центральные резцы) описан впервые в 1973 г. Наследуется по аутосомнорецессивному типу. Характерными признаками синдрома являются: мышечная гипотония, ожирение, выступающие центральные резцы. Для данного синдрома характерна широкая вариабельность клинической картины, включающая в себя низкий рост и вес при рождении, наличие микроцефалии, лицевые аномалии в виде гипоплазии верхней челюсти, мягко выраженной микрогнатии, антимонголоидного разреза глаз, миопии, иногда встречаются частичная атрофия зрительных нервов, страбизм, лейкопения. Кисти рук узкие с длинными тонкими пальцами и укороченными метакарпальными и метатарзальными костями. Для синдрома характерна умственная отсталость. К стоматологическим проявлениям, наряду с гипоплазией верхней челюсти и микрогнатией, можно отнести приоткрытый рот, высокое узкое нёбо, выступающие вперед крупные центральные резцы. Чаще, чем в других популяциях, синдром выявляется у евреев - ашкенази и у жителей Финляндии. Заболевание обусловлено мутацией в гене COH1, локализованном в области 8q22-q23.
KBG-синдром. Впервые заболевание описано в 1975 г. Herrmann et al. Основными признаками синдрома являются низкий рост, характерное лицо, макродентия, умственная отсталость и скелетные аномалии (рис. 1.7). Тип наследования: аутосомно-доминант-
ный. Микроцефалия. В детстве округлая форма лица, длинный фильтр, большие оттопыренные уши, гипертелоризм, телекант. Гипопластичные, открытые вперед ноздри. Различные скелетные аномалии: кифоз, аномалии кистей (маленькие кисти, клинодактилия, синдактилия и др.). Типичными и обязательными признаками данного заболевания являются стоматологические аномалии в виде макродентии, крупных верхних центральных резцов, слияния резцов, иногда олигодентия. Заболевание наследуется по аутосомнодоминантному типу. Соотношение пораженных мужского и женского пола 21:8.
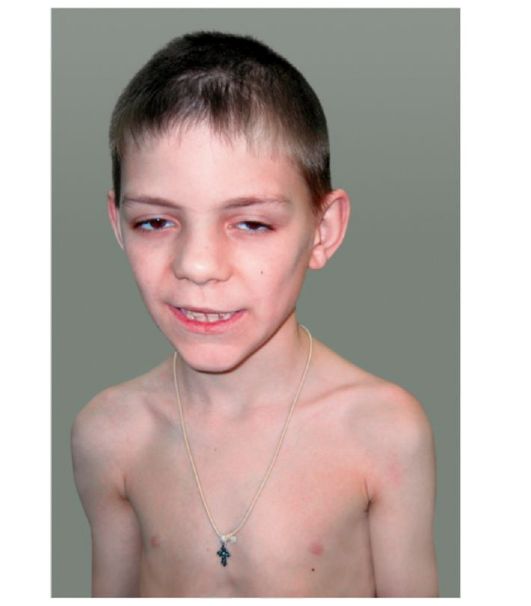 Рис. 1.7 KBG-синдром. Гипертелоризм, эпикант, большие, оттопыренные уши, макродонтия, высокое нёбо
Рис. 1.7 KBG-синдром. Гипертелоризм, эпикант, большие, оттопыренные уши, макродонтия, высокое нёбо
Гемигипертрофия (гемигиперплазия). Впервые заболевание описано Fraumeni et al (1967) у родных брата и сестры. Этиология до конца не установлена. Полагают, что наследование соответствует аутосомно-рецессивному типу, хотя не исключена возможность мультифакториального наследования. Martin et al (2005) полагают, что заболевание связано с мутацией генов LIT1 и H19, расположенных на коротком плече хромосомы 11. Основными признаками патологии являются: гемигипертрофия, гемигиперстезия, гемиарефлексия. Сколиоз и миеломенингоцеле. При данном заболева-
нии в качестве одного из признаков патологии отмечается наличие макродентии.
Брахман-де-Ланге подобный синдром. Заболевание описано в 1992 г. Halal and Silver. Для синдрома характерны: отставание в росте и психическом развитии, умственная отсталость, микроцефалия, мягко выраженные лицевые аномалии. К стоматологическим проявлениям синдрома относятся: микрогнатия и другие пороки развития нижней челюсти, а также макродентия. Заболевание наследуется по аутосомно-доминантному типу.
РЕНО синдром (прогрессирующая энцефалопатия с отеками, гипсаритмией и атрофией зрительных нервов. Тяжелое, редкое, прогрессирующее, аутосомно-рецессивное наследственное заболевание. Описано впервые в 1991 г. у 14 пациентов из 11 семей. Основными признаками данного заболевания являются: прогрессирующая микроцефалия, периферические отеки, неврологические нарушения в виде резкого отставания в развитии. Мышечная гипотония, судороги, изменения кардиограммы (гипсаритмия), прогрессирующая атрофия головного мозга и зрительных нервов и др. К лицевым аномалиям относят: эпикант, открытый рот, макродентию, высокое узкое нёбо.
Полидактилия постаксиальная с аномалиями зубов и ребер.
Заболевание было описано в 1977 г. у трех больных с очень сходной картиной поражения, включавшей в себя постаксиальную полидактилию кистей и стоп (широкие пальцы ног, синдактилию 2-3), гипоплазию и сращение ребер, а также множественные аномалии зубов (сращение зубов, макродентия, гиподентия, укорочение корней зубов и т.д.). У некоторых больных данные аномалии могут сочетаться с пороками сердца.
Синдром α-талласемии, умственной отсталости, не делеционный тип. Заболевание с Х-сцепленным доминантным типом наследования, обусловлено мутацией в ATRX-гене. Ген локализован в районе Хq13. В самостоятельную нозологическую единицу синдром выделен в 1981 г. Наряду с гематологическими отклонениями, характерными для гемоглобинопатий, отмечаются признаки поражения ЦНС в виде умственной отсталости, спастических поражений, судорог, атрофии мозга. При описании синдрома некоторые авторы отмечают наличие характерных лицевых дизморфий и стоматологических аномалий (гипоплазия средней части лица, маленькие, низко посаженные уши, телекант, вывернутые вперед ноздри, широко расставленные резцы, высокое нёбо и макродентия, иногда расщелина нёба).
Микроцефалия, страбизм, макродентия, широко расставленные верхние резцы.
Клайнфельтера синдром (хромосом XXY синдром). Минимальными диагностическими признаками синдрома являются: гипогенетализм, гипогонадизм, высокий рост, непропорционально длинные конечности. Вторичные половые признаки развиты слабо, отмечается оволосение по женскому типу. Популяционная частота 1:1000 мальчиков, коэффициент интеллекта ниже 80%.
3.2. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ С МИКРОДЕНТИЕЙ
Микродентия регистрируется как изолированный признак, встречается в сочетании с другими аномалиями зубов, входит в состав многих наследственных заболеваний и синдромов моногенного и хромосомного генеза. Частота изолированной микродентии 1-2% в европейской популяции и в несколько раз выше (6,2%) у жителей японских островов. Наиболее вероятно, что изолированная микродентия связана с мутаций генов PAX9 и MSXI.
В табл. 1.4 приведен список некоторых моногенных заболеваний с микродентией, наследующихся по аутосомно-рецессивному и аутосомно-доминантному и Х-сцепленному рецессивному типу.
3.2.1. Х-сцепленные наследственные синдромы с микродентией
Тауродонтизм , микродентия , и инвагинация зубов. У пораженных отмечается тауродонтизм, микродентия и множественные зубы с инвагинациями. Характер родословной позволяет предполагать Х-сцепленный рецессивный тип наследования. Локализация гена неизвестна.
Эктодермальная дисплазия 1, ангидротическая. Генетически гетерогенное заболевание. Одна из форм - Х-сцепленной рецессивной эктодермальной дисплазии. Для данного вида патологии, наряду с ярко выраженными признаками эктодермальной дисплазии, характерны аномалии зубов в виде тауродонтизма. У гетерозиготных женщин встречаются варьирующие по своей экспрессивности признаки эктодермальной дисплазии и аномалий зубов. Ген, вызывающий ED1, локализован в районе Xq12-13.1
Таблица 1.4. Наследственные заболевания и синдромы с микродентией
Синдром | Тип наследования | ? ОМИМ | Локус |
Тауродонтизм, микродентия и инвагинация зубов | Х-сц | 313490 | |
Амелогенез несовершенный, гипопластический тип (микродентия генерализованная) | АД | 104530 | |
Симфалангизм дистальный с микродентией | АД | 606895 | |
Спондилоэпифизарная дисплазия. Оманский тип | АР | 608637 | 10q22.1 |
Иммуноссеозная дисплазия, тип Шимке | АР | 242900 | 2q34-q36 |
Роззели-Джулиенетти синдром | АР | 225000 | 11q23-q24 |
Ригера, тип 1 синдром | АД | 180500 | 4q25-q26 |
Ригера, тип 2 синдром | 601499 | 13q14 | |
Эктодермальная дисплазия 1, ангидротическая | Х-сц | 305100 | Xq12-q13.1 |
Аткин(а) синдром | Х-сц | 300431 | |
Ротмунда-Томсона синдром | АР | 268400 | 8q24.3 |
Горлина-Чаудри-Мосса синдром | АР | 233500 | |
Эктодермальная дисплазия ангидротическая | АР | 224900 | 1q42.2-q43, 2q11-q13 |
Вильямса-Бьюрена синдром | АД | 194050 | 7q11.2 |
Эктродактилии, эктодермальной дисплазии и расщелины губы/нёба синдром | АД | 129900 | 7q11.2-q21.3 |
Эктодермальная дисплазия 3, ангидротическая | АД | 129490 | 2q11-q13 |
Микроцефалия, карликовость остеодиспластическая с аномалиями зубов | АР | 607561 | |
Лакримо-аурикуло-денто-дигитальный синдром | АД | 149730 | |
Эктодермальная дисплазия, трихо-адонтоонихиальный тип | АД | 129510 |
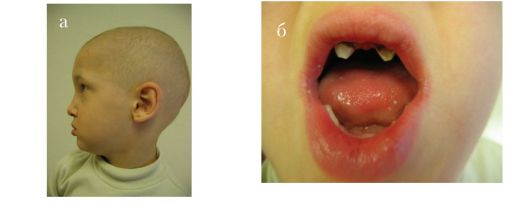
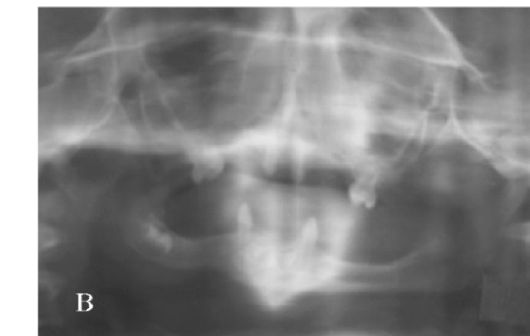 Рис. 1.8. Эктодермальная дисплазия 1, ангидротическая. а - редкие волосы, брови, ресницы; б - олиго- и микродентия, в - рентгенограмма
Рис. 1.8. Эктодермальная дисплазия 1, ангидротическая. а - редкие волосы, брови, ресницы; б - олиго- и микродентия, в - рентгенограмма
Аткина синдром - Х-сцепленное доминантное заболевание. Характеризуется низким ростом, ожирением, макроцефалией, маркорхидизмом, сколиозом, деформацией коленных суставов, укорочением кистей и стоп; умственная отсталость, судороги. Расхождение центральных резцов, микродентия. Ген не идентифицирован.
3.2.2. Аутосомно-доминантные заболевания и синдромы с микродентией
Амелогенез несовершенный, гипопластический тип (микродентия генерализованная)
Для данного заболевания также не известна локализация патологического гена. Тип наследования: аутосомно-доминантный. Это наследственное заболевание имеет несколько клинических и генетических вариантов. Данная форма характеризуется гипопластическим типом несовершенного амелогенеза и генерализованной формой микродентии.
Симфалангизм дистальный с микродентией, минерализованным дентином в пульпе. Аутосомно-доминантное заболевание, характеризущееся аномалиями развития пальцев кистей и стоп (дистальный симфалангизм, брахидактилия, аплазия/гипоплазия средних фаланг, отсутствие или гипоплазия ногтей). К стоматологическим аномалиям относятся: микродентия, минерализация дентина, сужение скуловой дуги. Ген не локализован.
Необычные волосы, катаракта, пигментная дистрофия, зубные аномалии и брахидактилия. Редкий аутосомно-доминантный наследственный синдром, характеризующийся необычной структурой волос, гипотрихозом, признаками эктодермальной дисплазии, юношеской катарактой, пигментной дистрофией сетчатки. Отмечаются олигодентия, добавочный латеральный резец, микродентия.
Эктродактилия, эктодермальная дисплазия и расщелины губы/ нёба синдром, 1. ЕЕС 1. Гетерогенное аутосомно-доминантное заболевание, связанное в большинстве случаев с мутацией гена ТР63 в локусе 7q11.2-q21.3. Стоматологическими признаками синдрома являются: частичная анодентия, микродентия, кариес, слабо выраженная гипоплазия моляров. Лицевые аномалии: гипоплазия верхней челюсти, низко расположенные уши. Главные аномалии: голубые радужки, фотофобия, блефарофимоз, склонность к блефаритам и дакриоциститам, аномалии слезно-носового канала. При данном заболевании описаны расщелина губы, нёба, ксеростома, атрезия хоан. Патология наружных половых органов у мальчиков; микропения и крипторхизм; патология почек и мочевыводящих путей. Отмечаются также ра зличные скелетные аномалии в виде эктродактилии и синдактилии. Могут встречатся признаки патологии кожи, ногтей, волос. Патология эндокринной системы в виде гипогонадотропного гипогонадизма.
Вильямса-Бьюрена синдром. Аутосомно-доминантное заболевание. Вызвано мутацией в эластиновом гене, локализованном в районе 7q11.2. Частота синдрома примерно 1 на 10 000. Низкий рост, лицевые дизморфии, аномалии и пороки развития сердечно-сосудистой системы, деформация грудной клетки, кифосколиоз, почечная патология, умственная отсталость. Зубы: микродентия, гиподентия.
Эктодермальная дисплазия 3 ангидротическая. Аутосомно-доминантное заболевание, вызванное мутацией в эктодисплазин - ангидротическом рецепторном гене (EDAR), локализованном в 2q11-q13.
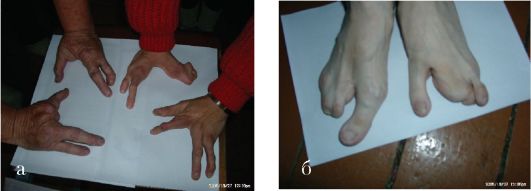 Рис. 1.9 а, б. Эктродактилия кистей (а) и стоп (б)
Рис. 1.9 а, б. Эктродактилия кистей (а) и стоп (б)
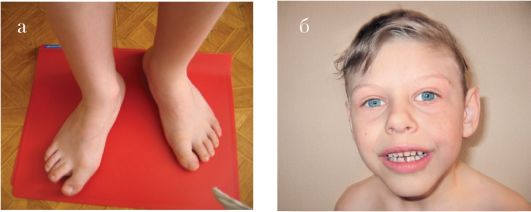
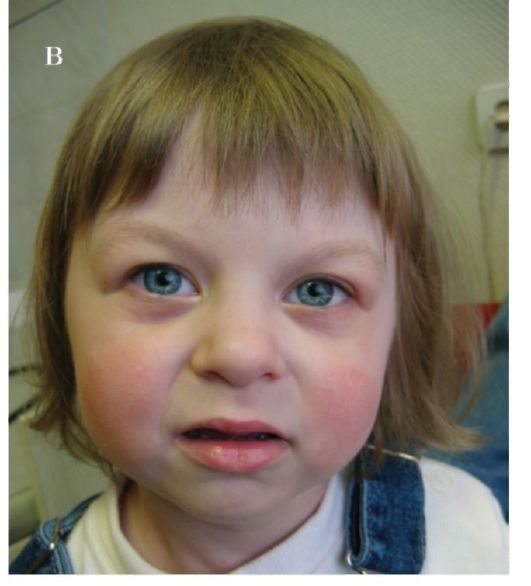 Рис. 1.10 а-в. Синдром ВильямсаБьюрена
Рис. 1.10 а-в. Синдром ВильямсаБьюрена
Характерные признаки: гипогидроз, тонкая, сухая кожа, гипотрихоз, волосы тонкие, ломкие. Гиподентия, анодентия, микродентия.
Синдром Ригера тип II
Аутосомно-доминантное заболевание, ген которого локализован в области 13q14. Характеризуется аномалиями развития глаз (глаукома, микрокорнеа, дисплазия радужки, гипертелоризм, эпикант). Гипоплазия верхней и нижней челюсти, врожденные пороки сердца, атрезия ануса, пупочные грыжи, гипоспадия, крипторхизм. Аномалии зубов в виде микродентии, гиподентии, коническая форма зубных коронок.
Лакримо-арикуло-денто-дигитальный синдром. Синдром обусловлен гетерозиготной мутацией в тирозин-киназном домене, кодирующем рецепторный фактор роста фибробластов 2 (FGFR2). Локализация гена 5p13-p12, 4p16.3. Характерны смешанная (кондуктивная-нейросенсорная), форма тугоухости, патология глаз в виде алакримии, аплазии/гипоплазии слезных желез, телекант, дакриоциститы. Отсутствие слюнных желез, поражение почек, скелетные аномалии, преаксиальная полидактилия, другие аномалии развития пальцев стоп и кистей. Гиподентия, коническая форма резцов, гипоплазия эмали, задержка прорезывания молочных зубов, кариес.
3.2.3. Аутосомно-рецессивные заболевания и синдромы с микродентией
Спондилоэпифизарная дисплазия, оманский тип. Аутосомнорецессивное заболевание, вызвано мутацией в гене CHST3, локализованном в области 10q22.1. Дети рождаются с нормальной длиной тела, затем резкое отставание в росте (взрослые 110-130 см.). Интеллект нормальный. Множественные пороки развития скелета, кистей и стоп. Микродентия, широко расставленные зубы.
Микроцефалия, остеодиспластическая карликовость с зубными аномалиями. Аутосомно-рецессивное заболевание с признаками поражения скелета, низким ростом, брахимезафалангией пальцев кистей и стоп, микроцефалией с нормальным интеллектом. Частичная микродентия, опалесцирущий оттенок эмали, гипоплазия корней моляров, аномалии премоляров верхней челюсти.
Иммунооссеозная дисплазия тип Шимке . Заболевание впервые описано в 1974 г. Шимке как хондроитин-6-сульфатазный мукопо-
лисахаридоз. В последующем отсутствие у больных мукополисахаридурии позволило исключить мукополисахаридоз. Заболевание характеризуется комбинацией признаков спондилоэпифизарной дисплазии и клиническими признаками прогрессирующего иммунодифицита. Больные, как правило, погибают в первые 8 лет жизни. Заболевание вызвано мутацией в гене SMARCAL1, локализованном в районе 2q34-q36. Диспропорциональная карликовость, миопия, астигматизм, почечная гипертензия, поражение почек, спондилоэпифизарная дисплазия, тонкие, ломкие волосы, снижение клеточного иммунитета, аномальный уровень иммуноглобулинов, протеинурия. Микродентия.
Роззелли-Джулиенетти синдром. Аутосомно-рецессивное заболевание с локализацией гена в области 11q23-q24. Впервые заболевание описано в 1961 г. у 4 пациентов с ангидрозом, гипотрихозом, микродентией, дисплазией ногтей, расщелиной губы/нёба, деформацией пальцев и пороками развития мочеполовой системы.
Синдром Ротмунда-Томсона (рис. 1.11 а, б). Аутосомно-рецессивное заболевание, вызванное мутацией гена (REQL4), локализованного в районе 8q24.3. Низкий рост, микрофтальмия, микрокорнеа, страбизм, глаукома; небольшие кисти и стопы, остеопороз. Поражение кожи (эритроматоз, пойкилодерма, телеангиэктазия, участки атрофии кожи) дистрофия ногтей, частичная алопеция, умственная отсталость у 15-30% пациентов. Множественные аномалии зубов (микродентия, сверхкомплектные зубы, аномалии зубных коронок).
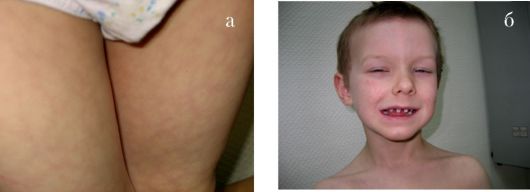 Рис. 1.11 а, б. Синдром Родмунда-Томсона
Рис. 1.11 а, б. Синдром Родмунда-Томсона
Синдром Горлина-Чаудри-Мосса . Аутосомно-рецессивное заболевание. Характеризуется низким ростом, брахицефалией, гипоплазией срединной части лица, кондуктивной тугоухостью, патологией органа зрения (микрофтальмия, гиперопия, гипертелоризм, птоз). Узкое, арковидное нёбо. Скелетные аномалии: краниосиностоз, гипоплазия верхней челюсти и костей носа. Гипертрихоз. Зубные аномалии: аномалии прикуса, гиподентия, микродентия.
Эктодермальная дисплазия, ангидротическая. Аутосомно-рецессивное заболевание, вызванное мутацией в эктодисплазиновом ангидротическом рецепторном гене (EDAR), локализованном в районе 1q42.2-q43, 2q11-q13. Клинические признаки эктодермальной дисплазии: гипогидроз, ангидроз, гипотрихоз. Зубные аномалии в виде гиподентии, анодентии, микродентии.
Эктодермальная дисплазия, трихо-одонто-онихиальный тип. Аутосомно-доминантное заболевание. Одна из форм эктодермальной дисплазии с нормальным потоотделением. Отмечаются гипотрихоз,
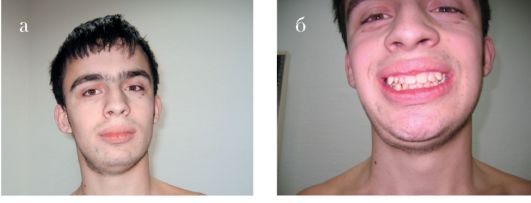
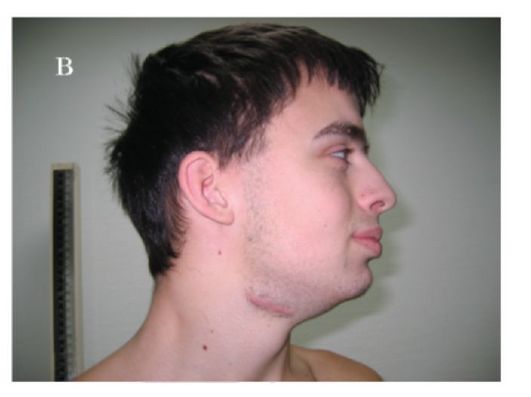 Рис. 1.12 а-в. Синдром Горлина
Рис. 1.12 а-в. Синдром Горлина
гипоплазия ногтей, гиподентия, отсутствие молочных желез (амастия), нейросенсорная тугоухость средней степени.
3.3. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ С ТАУРОДЕНТИЕЙ
Моногенные наследственные заболевания и синдромы, в состав которых тауродентия входит как один из признаков патологии представлены в табл. 1.5. Для некоторых заболеваний установлена локализация патологического гена (синдромы Репп-Ходжкина, эктодермальная дисплазия 1 АД, катаракта-зубной, окуло-дентодигитальная дисплазия, трихо-денто-костный синдром). Тип наследования для большинства этих заболеваний твердо установлен.
Отодентальная дисплазия. Аутосомно-доминантное заболевание, локализация гена неизвестна. Данный наследственный синдром был впервые описан в шести поколениях обширной итальянской семьи. Для синдрома характерно сочетание патологии слуха (нейросенсорная тугоухость) с различными аномалиями зубов. Чаще всего зубные аномалии представлены тауродентией, гиподентией, встречаются первые моляры с двойной пульповой камерой, большие, грушевидные коронки первых и вторых моляров.
Зубы, врожденное отсутствие с тауродонтией и редкими волосами. Заболевание впервые выявлено и описано в 1972 г. Стенвик и соавторы сообщили о четырех пораженных сибсах в норвежской семье. У этих и в дальнейшем описанных больных патология зубов сочеталась с признаками эктодермальной дисплазии. Аутосомнорецессивное заболевание, сочетающее в себе тауродентию, олигодентию, кожу с нормальным потоотделением и редким и волосами.
Пьера-Робена аномалад с аномалиями лица и пальцев - Х-сцепленное заболевание, характеризующееся аномалиями черепнолицевой области и аномалиями пальцев. Для данного синдрома характерны гипоплазия нижней челюсти, расщелина нёба, глоссоптоз. У некоторых больных аномалад Пьера-Робина сочетается с пороками сердца, аномалиями ушей, скелета, глаз, иногда встречается умственная отсталость.
Краниоэктодермальная дисплазия - заболевание наследуется по аутосомно-рецессивному типу. Описано впервые в 1977 г. Характеризуется черепно-лицевыми аномалиями (долихоцефалия, синостоз сагитальных швов черепа) и признаками эктодермальной
Таблица 1.5. Наследственные синдромы с тауродонтией
Синдром | Тип наследования | ? ОМИМ | Локус |
Пьера Робена аномалад с лицевыми и пальцевыми аномалиями | Х-сц | 311895 | |
Краниоэктодермальная дисплазия | АР | 218330 | |
Аккермана синдром | АР | 200970 | |
Рэпп-Ходжкина синдром; RHS | АД | 129400 | 3q27 |
Эктодермальная дисплазия 1, ангидротическая; ED | Х-сц. | 305100 | Xql2-ql3.1 |
Макросомия, ожирение, макроцефалия и глазные аномалии (МОМО-синдром) | АД | 157980 | |
Катаракта-зубной синдром, Нэнс-Хоран синдром; NHS | Х-сц. | 302350 | Хр22.13 |
Окуло-денто-дигитальная дисплазия (ODDD) | АД | 164200 | 6q21-q23.2 |
MOHR-синдром, орофациодигитальный синдром II; OFD2 | АР | 252100 | |
Отодентальная дисплазия | АД | 166750 | |
Зубы, врожденное отсутствие с тауродонтией и редкими волосами | АР | 272980 | |
Трихо-денто-костный синдром | АД | 190320 | 17q21.3-q22 |
Тауродонтизм | АР | 272700 |
дисплазии (редкие, медленно растущие волосы, гиподентия). Зубные аномалии, кроме гиподентии, предствлены тауродонтизмом, сросшимися зубами, дисплазией эмали. Наряду с черепно-лицевыми аномалиями, встречается патология глаз виде миопии, нистагма, гиперопии, патология скелета (брахидактилия, клинодактилия, синдактилия, укорочение нижних конечностей).
Синдром Аккермана (зубо-глазо-кожный синдром). Аутосомнорецессивное заболевание. Названо по имени автора, описавшего это заболевание в 1973 г. Тауродентизм. Корни моляров пирамидальной формы, юношеская глаукома.
Эктодермальная дисплазия 1, ангидротическая. Генетически гетерогенное заболевание. Одна из форм - Х-сцепленная рецессивная эктодермальная дисплазия - уже упоминалась при описании синдромов и заболеваний, сопровождающихся признаками анодентии. Для данного вида патологии, наряду с ярко выраженными признаками эктодермальной дисплазии, характерны аномалии зубов в виде тауродентизма. У гетерозиготных женщин встречаются варьирующие по своей экспрессивности признаки эктодермальной дисплазии и аномалий развития зубов. Ген, вызывающий ED1, локализован в районе Xq12-13.1.
Синдром МОМО (макростомия, ожирение, микроцефалия и аномалии органа зрения). Редкий наследственный синдром, вероятнее всего, обусловленный мутацией de novo. Для данного заболевания характерны большой вес и рост при рождении, ожирение, макрокрания, аномалии органа зрения (колобома сетчатки, нистагм), умственная отсталость. Аномалии зубов в виде нарушения прикуса, тауродонтизма, задержки прорезывания зубов.
Трихо-денто-костный синдром (TDO-синдром). Отмечаются различные признаки патологии скелета в виде укороченной грудины, отставания в костном возрасте, макрокрания, большие кисти и стопы. Нередко умственная отсталость. Аутосомно-доминантное заболевание. Ген локализован в районе 17q21.3-q22. Предполагается, что заболевание может быть вызвано мутацией в гомеобоксном гене DLX3(600525). Долихоцефальная форма черепа, скелетные аномалии и патология зубов в виде истонченной эмали, гипоплазия эмали в виде ямок, тауродентизма; мелкие, широко расставленные зубы, периапикальные абсцессы.
РАЗДЕЛ 4 АНОМАЛИИ КОЛИЧЕСТВА ЗУБОВ
Аномалии развития количества зубов включают в себя как меньшее (агенезия, олигодентия, гиподентия, адентия), так и большее (гипердентия врожденная; син. полиодентия, гиперодентия, сверхкомплектные зубы) по сравнению с нормой количество зубов.
4.1. АГЕНЕЗИЯ ЗУБОВ
Агенезия зубов (олигодентия, гиподентия, адентия) - это врожденное отсутствие одного или более молочных или постоянных зубов. Агенезия отдельных зубов представляет собой одну из самых распространенных аномалий развития у человека. Например, врожденное отсутствие третьих моляров встречается настолько часто, что во многих обзорах по агенезиям зубов вообще выносится за рамки обсуждения.
Частота агенезии временных зубов составляет в среднем 1,4%, агенезия постоянных зубов встречается с частотой 1,6-10%. Многие авторы обращают внимание на межпопуляционные различия в частоте встречаемости данной аномалии. Частота аномалии в азиатских популяциях более высокая, чем у европейцев. Наиболее низкая частота данной аномалии выявлена в африканских популяциях. Соотношение полов (М/Ж) 2:3. Семейные формы патологии 35%. Конкордантность монозиготных близнецов составляет 89%.
Зуб считается врожденно отсутствующим, если он не обнаруживается клинически (как прорезавшийся) или рентгенологически (как непрорезавшийся) в том возрасте, когда должен был появиться. Масштабы, в которых проявляется агенезия зубов, широко вариабельны. Эта аномалия может быть единственным фенотипическим признаком (изолированный порок развития) или входить в комплекс множественных врожденных пороков развития (МВПР) как часть генетического синдрома. Для описания агенезии зубов в литературе используются разные термины, основанные на числе вовлеченных в патологию зубов. Олигодентия - это агенезия шести и более постоянных зубов. Адентией называется полное отсутствие всех молочных и постоянных зубов. В некоторых редких случаях встречается агенезия только части зуба.
4.1.1. Генетические факторы агенезии зубов
Агенезия чаще затрагивает латеральные резцы, вторые премоляры и третьи моляры. Частота этой патологии, по некоторым оценкам, достигает 10-25%. Семейная агенезия зубов наследуется по аутосомно-доминантному, аутосомно-рецессивному и X-сцепленному типу, но может и не проявлять четкого характера наследования, соответствующего основным типам менделевского распределения признаков. У пораженных родственников в пределах семьи нередко наблюдается значительная внутрисемейная вариабельность по локализации, симметрии и числу пораженных зубов. Сохранившиеся зубы могут быть разными по размеру, форме и степени развития. Постоянные зубы страдают в большей степени, чем молочные. Данные, полученные на животных моделях, позволяют предположить, что в развитии отдельных групп зубов участвуют специфические (разные) генетические факторы. У мышей с заданной нулевой мутацией генов гомеобокса Dlx-1 и Dlx-2 не развиваются моляры верхней челюсти, но при этом сохраняются нормальные резцы и нижние моляры. В отличие от этого у мышей с мутантным активином βA (член суперсемейства трансформирующего фактора роста [TGF]β) резцы и нижние моляры не развиваются дальше рудиментарной почки, а верхние моляры развиваются вполне нормально. У таких мышей отсутствуют усы и наблюдаются дефекты вторичного нёба, включая расщелину нёба. У человека с агенезией зубов ассоциированы мутации MSX1 и PAX9, которые встречаются довольно редко. Однако именно эти мутации дают очень четкую картину агенезии зубов: мутация PAX9 приводит к преимущественной агенезии моляров, а мутация MSX1 чаще вызывает агенезию вторых премоляров и третьих моляров. В большинстве случаев агенезия зубов, вероятно, имеет мультифакториальное происхождение и связана с действием многих генов.
В клинической практике агенезия зубов, выраженная в разной степени, часто сочетается с поражением других органов и систем. Эта сочетанная патология, имеющая характерный фенотип, обозначается в медицинской литературе как синдром.
4.1.2. Наследственные болезни и синдромы с анодентией, олиго- и гиподентией
Не редко отсутствие одного или нескольких зубов сочетается с аномалиями других органов и систем. Генетическая природа этих
заболеваний часто связана со специфическими хромосомными или генными мутациями. Например, такое заболевание, как деформация грудной клетки с асфиксией и олигодентией определяется делецией короткого плеча хромосомы 12. Другие заболевания связаны со специфическими доминантными мутациями генов (гиподентия и контрактура Дюпиютрена; гиподентия и колоректальный рак и др.) либо с мутациями аутосомно-рецессивного типа (патология почек и анодентия постоянных зубов; олигодентия и нейросенсорная глухота; олигодентия и поликистоз почек). Одним из наиболее известных наследственных синдромов, сопровождающихся недостаточным количеством зубов, является эктодермальная дисплазия. В настоящее время известно о более чем 150 вариантах этого заболевания, отличающегося широкой клинической вариабельностью и генетической гетерогенностью (известны варианты, наследующиеся как Х-сцепленные, аутосомно-доминантные и аутосомно-рецессивные состояния). При данном заболевании в патологический процесс вовлечены производные эктодермы (волосы, потовые железы, ногти, зубы и др.). Волосы на голове, брови, ресницы, ногти с признаками дистрофии сочетаются либо с олигодентией, либо с анодентией. Сниженная функция потовых желез ведет к гипертермии, особенно в условиях повышения температуры внешней среды.
Отмечается выраженная олигодентия и коническая форма коронок зубов.
У больного редкие волосы, брови и ресницы (Х-сцепленные заболевания у девочек могут иметь более мягкую клиническую картину, что связанно с эффектом лайонизации Х-хромосомы).
4.1.3. Аутосомно-рецессивные болезни и синдромы с недостаточным количеством зубов
В табл. 1.6 представлены аутосомно-рецессивные заболевания и синдромы с анодентией/гиподентией/олигодентией.
Брахиметаподия-анадентия-гипотрихоз-альбинизм. Редкий синдром с аутосомно-рецессивным типом наследования. Описан в 1968 г. у трех сибсов, родившихся в финской семье. У двух сестер и их брата отмечались очень схожие по своим клиническим проявлениям симптомы заболевания. Характерными признаками синдрома являются: врожденная анодентия, гипотрихоз, гипопигментация кожи, поражение органа зрения в виде страбизма, миопии, катаракты, нис-
Таблица 1.6. Наследственные аутосомно-рецессивные заболевания и синдромы с олиго/гипо/адентией
Синдром | Тип наследования | ? ОМИМ | Локус |
Брахиметаподия-анодентия-гипотрихоз-альбинизм | АР | 211370 | |
Полная анодентия постоянных зубов | АР | 206780 | |
Трихо-одонто-онихиальная дисплазия | АР | 275450 | |
Эктродактилия, эктодермальная дисплазия и расщелина губы/ нёба синдром 3; EEC 3 | 604292 | 3q27 | |
Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии рук | АР | 264475 | |
Халлерманна-Штрайфа синдром | АР | 234100 | |
Джапо (GAPO) синдром | АР | 230740 | |
MOHR-синдром, орофациодигитальный синдром II, рото-лицепальцевой синдром, тип II | АР | 252100 |
тагма, отмечаются гипоплазия верхней челюсти, прогнатизм нижней челюсти, умственная отсталость, брахидактилия, укорочение метакарпальных и метатарзальных костей
Анодентия постоянных зубов полная. Тип наследования: аутосомно-рецессивный. Впервые заболевание описано в 1931 г. В дальнейшем стало известно еще о нескольких десятках пациентов с данной патологией. Популяционная частота данного синдрома неизвестна. Подтверждением аутосомно-рецессивного наследования послужили данные о рождении нескольких пораженных детей от здоровых родителей в кровнородственном браке. Наряду с анодентией, у больных имелись пороки развития челюстно-лицевой области.
Трихо-одонто-онихиальная дисплазия. Заболевание описано как новая форма эктодермальной дисплазии. В бразильской семье заболевание было выявлено у четырех родных сестер. Характерными признаками данного заболевания являются: гипоплазия эмали, вторичная анодентия. Признаки эктодермальной дисплазии: дополнительные соски, пигментные невусы, гипотрихоз, дистрофия ногтей.
Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии рук . Синдром как самостоятельная нозологическая единица внесен в каталог Маккюсика на основании описания больных в двух семьях (описано четверо больных). Для этого синдрома характерны следующие стоматологические симптомы: малоккюзия зубов, темный цвет эмали (красно-коричневые зубы), частичная анодентия, иногда сверхкомплектные зубы, задержка прорезывания зубов. Высокое, арковидное нёбо. Отмечались низкий вес и рост, микрогнатия, низкопосаженные уши, атрезия среднего уха, смешанная форма глухоты. Аномалии органа зрения, множествнные аномалии скелета в виде деформации грудной клетки, позвоночника, кифосколиоз. Отмечалось наличие гипоплазии тенара и кератоза ладоней.
Халлермана-Штрайфа синдром. Для данного синдрома характерными признаками являются: частичная анодентия, наличие так называемых неонатальных зубов, малокклюзия, иногда наличие свехкомплектных зубов. При исследовании полости рта также отмечается наличие высокого, узкого, арковидного нёба, микростомия, тонкие губы и оттопыренная нижняя губа. Характерен комплекс пороков развития. Отмечается низкий вес при рождении, отставание в росте и физическом развитии. Брахицефальная форма черепа, низко посаженные уши, микрофтальмия, катаракты, нистагм, колобомы радужки и диска зрительного нерва, множественные скелетные
 Рис. 1.13. Синдром Халлерманна-Штрайфа
Рис. 1.13. Синдром Халлерманна-Штрайфа
аномалии. Отмечается атрофия кожи, наличие телеангиоэктазов, признаков ксероза. Волосы тонкие. У 15% больных отмечаются признаки умственной отсталости разной степени выраженности.
GAPO-синдром (отставание в росте, алопеция, псевдоадентия, атрофия зрительных нервов). При данном заболевании часто отмечается псевдоанодентия (преимущественно вследствие выпадения зубов), толстые, полные губы, низкий рост, патология органа зрения.
Расщелина губы/нёба - эктодермальная дисплазия. Синдром CLPED1, Розелли-Джулинетти синдром. Аутосомно-рецессивное заболевание. Ген локализован в 11q23-q24. Является аллельным вариантом эктодермальной дисплазии острова Маргариты тип (ОМИМ 225060). Характерными признаками синдрома являются: гиподентия, анодентия, микродентия, расщелина губы/нёба. Синдактилии пальцев кистей и стоп, ладонно-подошвенный гиперкератоз, прогрессирующий гипотрихоз, ониходисплазия. Заболевание обусловлено мутацией в гене PVRL 1, локализованном в районе 11q23-q24. Впервые заболевание описано Розелли и Джулинетти в 1961 г. Для этого синдрома характерными стоматологическими признаками являются: гиподентия, анодентия, микродентия в сочетании с ращелиной губы/нёба. Кроме того, отмечается кожная синдактилия на пальцах кистей и стоп, гиперкератоз ладоней и подошв, ониходистрофия, редкие брови, ресницы, редкие волосы на голове.
MOHR-синдром, оро-фацио-дигитальный синдром II, ротолице-пальцевой синдром, тип II. Впервые заболевание описано в 1941 г. генетиками Отто Морхом и Яном Морхом. Это заболевание с аутосомно-рецессивным типом наследования. Ген не локализован. Интеллект в большинстве случаев нормальный. Характеризуется кондуктивной глухотой, аномалиями развития скелета (сколиоз, пре-/постаксиальная полидактилия, короткие кисти, брахидактилия, синдактилия), пороками развития полости рта (срединная расщелина язычка, высокое арковидное нёбо, встречаются расщелины нёба и губы). Характерным признаком является отсутствие центрального резца.
4.1.4. Аутосомно-доминатные болезни и синдромы с недостаточным количеством зубов
Некоторые аутосомно-доминантные заболевания и синдромы с анодентией (олигодентией) гиподентией представлены в табл. 1.7.
Гиподентия аутосомно-доминантная (отсутствие вторых премоляров и третьих моляров; семейная агенезия зубов; агенезия отдельных зубов; частичная анодентия; гиподентия с орофациальными расщелинами). Заболевание связывают с мутацией в МSХ1-гене, локализованном в 14q12-q13, 4p16.1 Впервые заболевание описано Erwin and Cockern в 1949 г. как частичная анодентия и гиподентия третьих моляров. В последующем были описаны семьи, где гиподентия сочеталась с расщелиной губы и нёба.
Эктродактилия, эктодермальная дисплазия и расщелины губы/ нёба синдром 1. ЕЕС 1. Генетически гетерогенное заболевание, связанное в большинстве случаев с мутацией гена ТР63 в локусе 7q11.2- q21.3. Стоматологическими признаками синдрома являются: частичная анодентия, микродентия, кариес, слабо выраженная гипоплазия моляров. Лицевые аномалии: гипоплазия верхней челюсти, низко расположенные уши. Главные аномалии: голубые радужки, фотофобия, блефарофимоз, склонность к блефаритам и дакриоциститам, аномалии слезноносового канала. При данном заболевании описаны расщелина губы, нёба, ксеростома, атрезия хоан. Патология наружных половых органов у мальчиков - микропения и крипторхизм. Патология почек и мочевыводящих путей. Отмечаются также различные скелетные аномалии в виде эктродактилии и синдактилии. Могут встречаться признаки патологии кожи, ногтей, волос.
Таблица 1.7. Аутосомно-доминантные заболевания и синдромы с анодентией/олигодентией/гиподентией
Синдром | Тип наследования | ? ОМИМ | Локус |
Гиподетия, аутосомно-доминантная;НYD1 (гиподентия с орофациальными расщелинами) | АД | 106660 | 14ql2-ql3, 4р16.1 |
Эктродактилия, эктодермальная дисплазия и расщелины губы/ нёба, синдром 1; EEC 1 | АД | 129900 | 7qll.2-q21.3 |
Эктродактилия, эктодермальная дисплазия и расщелина губы/ нёба, синдром 3; EEC 3 | 604292 | 3q27 | |
Окулодентодигитальная дисплазия (ODDD) | АД | 164200 | 6q21-q23.2 |
Латеральный ихтиоз, отсутствие смены зубов, агенезия | АД | 150400 | |
Эктодермальная дисплазия тип 3, ангидротическая; ED3 | АД | 129490 | 2qll-ql3 |
Анкило-блефарон-эктодермальный дефект - расщелина губы/ нёба | АД | 106260 | 3q27 |
Ригера синдром, тип I; RIEG1 | АД | 180500 | 4q25-q26 |
Ригера синдром тип II | АД | 601499 | |
Репп-Ходжкина синдром; эктодермальная дисплазия ангидротическая с расщелиной губы/нёба | АД | 129400 | 3q27 |
Патология эндокринной системы в виде гипогонадотропного гипогонадизма.
Синдром эктродактилия, эктодермальная дисплазия, расщелина губы/нёба 3; ЕЕС3. Аутосомно-доминантное заболевание. Ген локализован в области 3q27. Является генетическим вариантом синдрома ЕЕС 1. Клинические проявления варьируют и характеризуются гипоплазией верхней челюсти и моляров. Отмечается частичная анодентия, микродентия, кариес, расщелина губы/нёба, эктродактилия кистей и стоп, выраженные признаки эктодермальной дисплазии.
Синдром Рэпп-Ходжкина. Rapp and Hodgkin (1968) описали заболевание у матери и двух ее детей в виде ангидротической эктодермальной дисплазии с расщелиной губы и нёба. В последующем было описано еще несколько семей с подобным заболеванием. Методами молекулярной генетики установлена локализация гена, вызывающего данное заболевание. Для данного синдрома характерно множественное поражение эктодермы (гипогидроз, прогрессирующая алопеция, истонченные редкие волосы, истонченная кожа). При осмотре области рта отмечается расщелина губы/нёба, расщелина язычка, гиподентия и маленькие, конические зубы.
Глазо-зубо-пальцевая дисплазия, окуло-денто-оссеозная дисплазия , ODDD-синдром (рис. 1.14 а, б). Генетически гетерогенное заболевание. Известен также аутосомно-рецессивный вариант патологии. Ген локализован в районе 6q21-q23.2. Заболевание обусловлено мутацией в гене коннексина-43 (GJA1); характерными признаками являются: микроцефалия, поражение глаз (микрокорнеа, глаукома, катаракта, аномалии радужки), различные скелетные аномалии и аномалии рук (синдактилия IV и V пальца, укорочение средней фаланги V пальца, камптодактилия V пальца, межфалангеальная гипоплазия. На стопах синдактилия III-IV). Умственная отсталость, дизартрия. Аномалии зубов в виде гипоплазии эмали, частичной анодентии, микродентия, преждевременная потеря зубов, кариес.
Анкило-блефарон-эктодермальный дефект - расщелина губы/ нёба. Ген заболевания локализован в области 3q27. Стоматологическими проявлениями синдрома являются: гиподентия, зубы конической формы, широко расставленные зубы, частичная анодентия. Характерными признаками являются также поражения глаз (коньюктивиты, блефариты, атрезия слезно-носового канала и др.). Встречаются расщелины губы/нёба. Кожные проявления эктодермальной дисплазии (гипогидроз, гиперкератоз, гиперпиг-
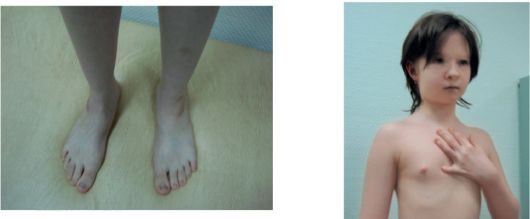 Рис. 1.14 а, б. Глазо-зубо-пальцевая дисплазия
Рис. 1.14 а, б. Глазо-зубо-пальцевая дисплазия
ментация и др.). Редкие волосы, частичная алопеция. Характерными признаком синдрома является микроцефалия.
Синдром Ригера тип I. Генетически гетерогенное заболевание с аутосомно-доминантным типом наследования. Ген локализован в области (4q25-q26). Характерный внешний вид (гипоплазия верхней челюсти, короткий фильтр, дисплазия радужки, аниридия, страбизм, неперфорированный анус, глаукома. Гиподентия.
Синдром Ригера тип II. Аутосомно-доминантное заболевание, ген которого локализован в области 13q14, характеризуется аномалиями развития глаз (глаукома, микрокорнеа, дисплазия радужки, гипертелоризм, эпикант). Гипоплазия верхней и нижней челюсти, врожденные пороки сердца, атрезия ануса, пупочные грыжи, гипоспадия, крипторхизм. Аномалии зубов в виде микродентии, гиподентии, коническая форма зубных коронок.
4.1.5. Х-сцепленные болезни и синдромы с недостаточным количеством зубов
Х-сцепленные заболевания и синдромы с ано-/гипо-/олигоденти- ей представлены в табл. 1.8. Для большинства из этих заболеваний известен хромосомный район локализации патологического гена.
Ото-палато-дигитальный синдром, тип 1. OPD1. Ген заболевания локализован в Xq28. Х-сцепленный доминантный тип наследования. Заболевание характеризуется низким ростом, черепно-лицевыми аномалиями, признаками кондуктивной глухоты. Микростомия,
Таблица 1.8. Х-сцепленные заболевания c ано/гипо/олигодентией
Синдром | Тип наследования | ? ОМИМ | Локус |
Отопалатодигитальный синдром тип I (OPD1) | Х-сц. | 311300 | Xq28 |
Фронтометафизарная дисплазия (FMD) | Х-сц | 305620 | |
Эктодермальная дисплазия 1, ангидротическая: ED1 | Х-сц. | 305100 | Xq12-q13.1 |
Недержания пигмента синдром; IP | Х-сц. | 308300 | Xq28 |
Аарскога синдром | Х-сц. | 305400 | Xq11.21 |
Коффина-Лоури | Х-сц. | 303600 | Xq22.2-p22.1 |
Фокальная дермальная гипоплазия | Х-сц. | 305600 | |
Недержание пигмента | Х-сц. | 308300 | Xq28 |
расщелина нёба, частичная анодентия. Разнообразные скелетные аномалии, гипоплазия ногтей, короткие широкие дистальные фаланги пальцев, средняя степень умственной отсталости.
Фронтометафизарная дисплазия. Заболевание обусловлено мутацией, проводящей к новой функции белка (gainof-function) - мутация в гене филамина А (FLNA;300017). Заболевание впервые описано в 1969 г.
Эктодермальная дисплазия 1, ангидротическая; ED1. Ген заболевания, кодирующий эктодисплазин А, локализован в районе Xq12- q13.1. В настоящее время описано около 150 различных клинических вариантов эктодермальных дисплазий. Большинство этих заболеваний встречается очень редко. Клинические различия определяются различиями в сочетании и частоте встречаемости отдельных симптомов характерных для эктодермальной дисплазии. Известны аутосомно-доминантые, аутосомно-рецессивные и Х-сцепленные формы эктодермальных дисплазий. При Х-сцепленной эктодермальной дисплазии отмечаются олигодентия, микродентия, конические зубы и тауродонтизм, в частности, при Х-сц. Гипогидротической
эктодермальной дисплазией, наряду с зубами, поражаются и другие производные эктодермы: волосы, ресницы, брови (редкие, тонкие), ногти - диспластичные, гипоплазия потовых желез. Больные часто страдают от гипертермии, особенно в жарком климате. Признаком поражения зубов при данной форме эктодермальной дисплазии является наличие латеральных резцов конической формы.
Фокальная дермальная гипоплазия (синдром Гольтца ). Фокальная дермальная гипоплазия наследуется как Х-сцепленное доминантное состояние с летальностью плодов мужского пола. Основными признаками являются атрофия и линейная пигментация кожи (рис. 1.15 а, б), множественные папилломы, слизистые мембраны, аномалии пальцев (синдактилии, полидактилия, каптодактилия и другие аномалии). Аномалии глаз (колобома радужки, стабизм, микрофтальмия). Иногда умственная отсталость. Папилломы на губах, гипоплазия зубов (олигодентия, гиподентия, гипоплазия эмали, задержка прорезывания зубов, отсутствие резцов, аномалии прикуса).
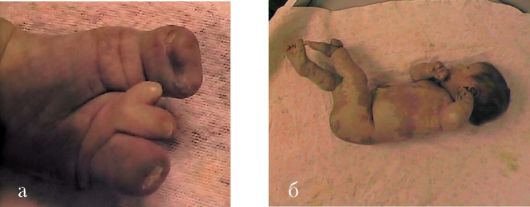 Рис. 1-15 а, б. Синдром Гольца
Рис. 1-15 а, б. Синдром Гольца
Недержание пигмента (синдром Блоха-Сульцбергера). Семейный генодерматоз - недержание пигмента (IP) наследуется как Х-сцепленное доминантное заболевание c летальностью мужских плодов. Заболевание связано с мутацией в IKK-гамма гене (также его обозначают как NEMO), ген локализован в Xq28. Заболевание встречается только у девочек и характеризуется широкой вариабельность клинической картины. Поражаются кожа, волосы, ногти, зубы, глаза и центральная нервная система.
К признакам поражения зубов относятся: гиподентия, задержка прорезывания зубов, коническая форма зубной коронки.
Некоторые выше перечисленные синдромы и заболевания характеризуются полиморфной и сочетанной картиной поражения зубов. Наряду с олиго-, гипоили анодентией, отмечаются признаки аномалий развития формы зубной коронки, например в виде микродентии или других аномалий. Это обстоятельство позволяет в равной степени рассматривать эти заболевания в других разделах.
4.2. ИЗБЫТОЧНОЕ КОЛИЧЕСТВО ЗУБОВ (ГИПЕРДЕНТИЯ, СВЕРХКОМПЛЕКТНЫЕ ЗУБЫ)
Гипердентия - дополнительные зубы. Гипердентия может быть единичной (изолированной) и множественной. Сверхкомплектные зубы часто имеют неправильную форму. Они могут находиться в зубном ряду, либо располагаться вне его. Чаще сверхкомплектные зубы прорезываются во фронтальном участке верхней челюсти, а иногда позади верхних третьих моляров. Исключительно редки случаи прорезывания нескольких сверхкомплектных зубов, создающих как бы второй зубной ряд. Понятие «сверхкоплектные зубы» относится как к временным, так и к постоянным зубам. Частота временных сверхкомплектных зубов 0,1%, постоянных - 1-5%. В разных популяциях соотношениение пораженных мужчин и женщин различно. Так, например, у европейцев соотношение пораженных мужчин и женщин равно 2:1, в популяциях африканского происхождения 4:1, у японцев 5,5:1, у китайцев 6,5:1. Убедительных объяснений данному феномену не найдено, однако можно предположить, что факторы генетического контроля за развитем количества зубов связаны с генами половых хромосом. Основная масса случаев, связанных с наличием сверхкомплектных зубов - спорадические. Семейные случаи составляют 30%. При множественной гипердентии количество зубов значительно отклоняется от нормальных показателей. Такое состояние встречается у больных с клейдокраниальной дисплазией. Примером заболевания, при котором также встречаются сверхкомплектные зубы, является синдром Гарднера. Для этого синдрома кроме сверхкомплектных зубов характерно наличие полипоза кишечника и остеом.
4.2.1. Наследственные заболевания и синдромы с избыточным количеством зубов
В табл. 1.9. представлены наследственные заболевания и синдромы с свехкомплектными зубами. Для большинства заболеваний, представленных в табл. 1.9, наряду с сверхкомплектностью зубов характерно наличие сочетанных зубных аномалий.
4.2.2. Аутосомно-доминантные заболевания и синдромы с сверхкомплектными зубами
В настоящее время известно относительно немного аутосомно-доминантных синдромов с сверхкомплектными зубами (зубы сверхкомплектные, клейдокраниальная дисплазия; аденоматозный полипоз толстого кишечника; трихо-рино-фалангеальный синдром). Для большинства синдромов наряду со сверхкомплектностью отмечается дополнительные признаки поражения зубов и патология других органов и систем. Описания некоторых аутосомно-доминантных заболеваний, сопровождающихся симптомами сверхкомпектных зубов представлены ниже.
Клейдокраниальная дисплазия. Аутосомно-доминантное заболевание. Ген локализован в районе 6p21. Ген заболевания расположен на 6-й хромосоме и кодирует белок CBFA1 Этот белок участвует в процессах формирования скелета, однако его роль в формировании ткани зубов еще недостаточно изучена. Фенотип заболевания формируется у гетерозигот по мутации в гене CBFA1 Гомозиготность по данной мутации летальна. Основными признаками заболевания являются: комплекс множественных аномалий развития, сопровождающихся глухотой, черепно-лицевыми аномалиями, скелетными нарушениями в виде гипо/аплазии ключиц, сколиоза, гипоплазии/ аплазии лобного синуса и др. Зубные аномалии: задержка прорезывания молочных и постоянных зубов, сверхкомплектные зубы, гипоплазия эмали (рис. 1.16).
Аденоматозный полипоз толстой кишки. Ген локализован (рис. 1.16) в области 5q21-q22. Аутосомно-доминантное заболевание, проявляется преимущественно во второй половине жизни. Аденоматозные полипы, помимо толстого кишечника, могут встречаться в других отделах желудочно-кишечного тракта, а также давать очаги малигнизации в головном мозгу и щитовидной железе. При данном заболе-
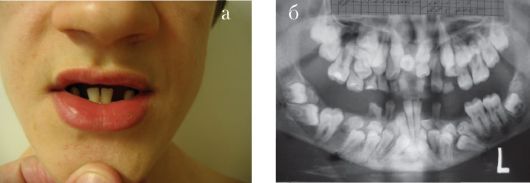
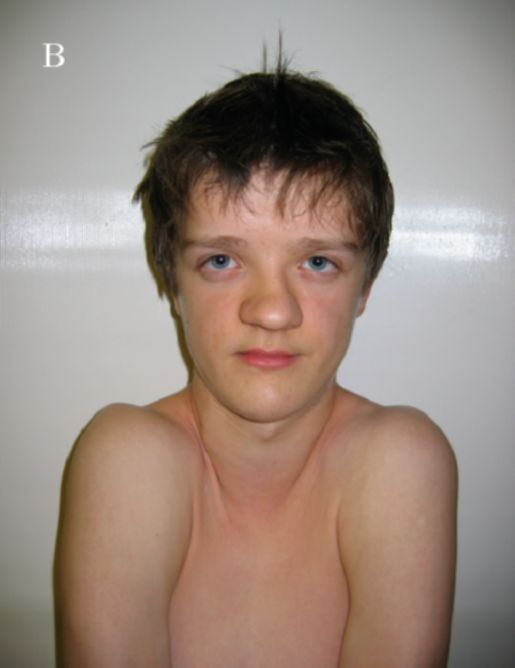 Рис. 1.16 а-в. Клейдокраниальная дисплазия: а - гипоплазия ключиц; б - рентгенограмма зубочелюстной области, общее количество зубов свыше 50.
Рис. 1.16 а-в. Клейдокраниальная дисплазия: а - гипоплазия ключиц; б - рентгенограмма зубочелюстной области, общее количество зубов свыше 50.
вании обнаруживаются зубные аномалии в виде сверхкомплектных зубов, непрорезывания зубов, кариеса, наличия одонтом.
4.2.3. Аутосомно-рецессивные заболевания и синдромы с сверхкомплектными зубами
К аутосомно-рецессивным заболеваниям с сверхкомплектными зубоми относятся: синдром Ротмунда-Томсона; псевдопаппилодерма, глазной гипертелоризм, блефарофимоз и аномалия кистей; синдром Халлермана-Штрайфа. Синдромы Ротмунда-Томсона и ХаллерманаШтрайфа характеризуются множественными аномалиями зубов, где, наряду с сверкомплектными зубами, отмечаются аномалии зубной коронки в виде микродентии. Описание этих заболеваний помещено в раздел «Наследственные синдромы с микродентией».
Таблица 1.9. Наследственные синдромы с сверхкомплектными зубами
Синдром | Тип наследования | ? ОМИМ | Локус |
Зубы сверхкомплектные | АД | 187100 | |
Катаракто-дентальный синдром | Х-сц.д | 302350 | |
Окуло-фацио-кардио-дентальный синдром | Х-сц.д | 300166 | |
Клейдокраниальная дисплазия | АД | 119600 | 6р21 |
Аденоматозный полипоз толстого кишечника | АД | 175100 | 5q21-q22 |
Халлермана-Штрайфа синдром | АР | 234100 | |
Орофациодигитальный синдром | Х-сц.д | 311200 | Хр22.3-р22.2 |
Ротмунда-Томсона синдром | АР | 268400 | 8q24.3 |
Псевдопаппилодерма, глазной гипертелоризм, блефарофимоз и аномалия кистей | АР | 264475 | |
Трихоринофалангеальный синдром, тип III | АД | 190350 | 8q24.12 |
4.2.4. Х-сцепленные заболевания и синдромы со сверхкомплектными зубами
Катаракто-дентальный синдром (катаракта, Х-сцепленная с зубами Гетчинсона, мезиоденс-катаракта-синдром) - Х-сцепленное доминантное заболевание. Ген локализован в районе Xp22.13. Впервые синдром описан в 1974 г. В качестве ведущих стоматологических признаков синдрома фигурируют вывернутые резцы с узкой, коронкой, сверхкомплектные резцы верхней челюсти, заостренные премоляры и моляры, диастема. Из других аномалий: широкие короткие пальцы рук, умственная отсталость средней степени, аутизм. Обязательно встречаются симптомы поражения глаз: двустороння врожденная катаракта, снижение остроты зрения, нистагм, микрофтальмия. У 50% больных отмечается глаукома.
Оро-фацио-дигитальный синдром I (OFD1). Заболевание обусловлено мутацией в гене CXORF5, локализованном в Xp22.3-p22.2. Характеризуется пороками развития лица, полости рта (срединная расщелина верхней губы, расщелина нёба, высокое нёбо, гамартома язычка и др.). Примерно в 40% случаев отмечаются признаки поражения центральной нервной системы (умственная отсталость, агенезия мозолистого тела, гидроцефалия, судороги, гамартома гипоталамуса и др.), пальцев (клинодактилия, синдактилия, брахидактилия). Аномалии зубов выражаются в отсутствии латеральных резцов, гипоплазия эмали, наличии сверхкомплектных зубов, раннего распространенного кариеса.
РАЗДЕЛ 5
НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ ФОРМИРОВАНИЯ
СТРУКТУРЫ ЗУБОВ
В соответствие с классификацией ВОЗ выделяют следующие категории нарушения формирования зубов.
• Несовершенный дентиногенез, изменения в зубах при незавершенном остеогенезе.
• Другие наследственные нарушения структуры зуба, дисплазия дентина, раковинные зубы.
• Наследственные нарушения структуры зуба (неуточненные).
• Гипоплазия эмали.
• Пренатальная гипоплазия эмали.
• Неонатальная гипоплазия эмали.
• Аплазия и гипоплазия цемента.
• Дилацерация (трещины эмали).
• Одонтодисплазия (региональная одонтодисплазия).
• Зуб Тернера.
• Другие уточненные нарушения формирования зубов.
• Незавершенный амелогенез.
5.1. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И СИНДРОМЫ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ
ФОРМИРОВАНИЯ ДЕНТИНА
Аномалии развития дентина могут проявляться как изолированная патология или входить в состав мультифакториальных, внешнесредовых и генетических болезней и синдромов. Одной из наиболее известных форм наследственных аномалий формирования дентина является несовершенный дентиногенез. Это относительно частое заболевание, которое встречается в популяции с частотой примерно 1:8000 человек. Данная форма патологии чаще всего связана с мутацией в дентинсиалопротеиновом гене, расположенном в 4q21. Продуктом гена является дентин специфический матричный белок, состоящий из дентинсиалопротеина (DSP) и дентинфосфопротеина (DFP).
Различают несколько типов несовершенного дентиногенеза
(табл. 1.10).
Таблица 1.10. Классификация несовершенного дентиногенеза
Тип | Заболевание | Генетический дефект |
IA | Несовершенный дентиногенез Шилдса II | Дентин-сиало-фосфопротеин (DSPP) |
IБ | Несовершенный дентиногенез Шилдса III | Дентиновый матрикс фосфопротеин I (DMPI) |
II | Дисплазия дентина I | Не известен |
III | Дисплазия дентина II | Дентин-сиало-фосфопротеин (DSPP) |
IV | Фиброзная дисплазия дентина | Не известен |
Тип IA (несовершенный денитиногенез Шилдса II) - зубы желтокоричневого, серо-голубоватого оттенка, луковицеобразная форма зубной коронки, облитерация атипичным цементом пульповых камер некоторых временных и всех постоянных зубов. Процесс облитерации носит прогрессирующий характер, начинается еще до прорезывания зубов, отмечается искривление и истончение корней зубов. Тип IБ (несовершенный дентиногенез Шилдса III) - как и при типе IA, цвет зубов желто-коричневый или серо-голубоватый. Корни зубов практически те же, что при типе IA, но пульповые камеры, прежде чем подвергнутся облитерации, значительно больше, чем при IA. Количество дентина в зубе значительно снижено и внутренняя часть зуба заполнена преимущественно грубыми коллагеновыми волокнами.
Несовершенный дентиногенез тип I представлен в настоящее время исключительно семейными случаями, когда заболевание удается проследить на протяжении нескольких поколений. На этом основании сделано заключение об аутосомно-доминантном наследовании данного типа несовершенного дентиногенеза.
Тип IA наследуется аутосомно-доминантно с полной пенетрантностью и с незначительно варьирующей экспрессивностью гена.
Тип IB - редкое заболевание, которое обнаружено в изолированных популяциях США и среди евреев-ашкенази.
Тип II (дисплазия дентина I) - более редкое, чем тип I, заболевание. Примерная частота 1:100 000. При данном заболевании коронки зубов нормального размера, цвет практически не изменен. Пульповая камера облитерирована. Клинический диагноз данного типа несовершенного дентиногенеза основан на рентгенологическом исследовании. Корни зубов практически отсутствуют, что является основной причиной ранней утраты зубов.
Тип III (дисплазия дентина II) - наиболее редкое (наследуемое по аутосомно-доминантному типу) заболевание, где встречаются пациенты с аномальной минерализацией дентина временных зубов. Клинически проявляется как дисплазия корней временных зубов, но корни постоянных зубов практически всегда сформированы правильно. Коронки зубов нормального цвета и размера.
Тип IV (фиброзная дисплазия дентина) - наиболее редкое заболевание. К настоящему времени в мировой литературе имеется сообщение только об одной семье, где зарегистрирован данный тип несовершенного дентиногенеза. Коронки зубов нормального цвета и
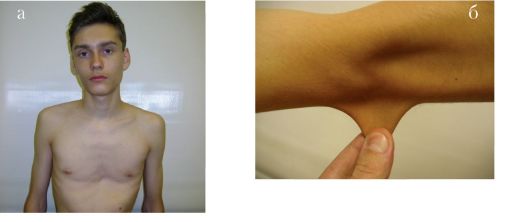
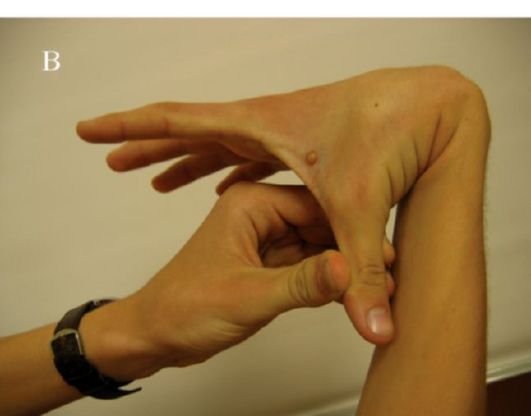 Рис. 1.17 а-в. Синдром ЭлерсаДанло
Рис. 1.17 а-в. Синдром ЭлерсаДанло
размера. Нормального размера пульповая камера, однако она заполнена атипичным дентином, что выявляется при гистологическом исследовании. Генетическая этиология некоторых форм несовершенного дентиногенеза пока еще не установлена. В то же время на основании данных анализа сцепления генов предполагают, что разные типы несовершенного дентиногенеза являются аллельными
состояниями и связаны с мутациями двух тесно сцепленных генов DSPP и DMPI. Как синдромальные состояния, патология дентина описана при синдромах Элерса-Данло тип VIB; дисплазии дентина со склерозом костей; гипофосфатазии; несовершенном остеогенезе, тип III-IV; спондило-метафизарной дисплазии с несовершенным дентиногезом (синдром Голдблатта) и др.
Синдром Элерса-Данло тип VIB. Синдром Элерса-Данло тип VIB, известный также как синдром хрупкой роговицы, характери-
зуется голубыми склерами, разрывами радужки при минимальном травматическом повреждении, кератоконусом, гиперэластичностью кожи, гипермобильностью суставов. Заболевание вызвано мутацией в гене лизил гидроксилазы (PLOD1). Наряду с этим отмечаются макроцефалия, пролапс митрального клапана, сколиоз, спондилолистез, миопия, врожденный вывих бедра. Поражение зубов выражается в признаках несовершенного дентиногнеза. Наследуется аутосомнорецессивно.
Синдром дисплазии дентина со склерозом костей. Аутосомнодоминантное заболевание, сочетающее в себе признаки дисплазии дентина и склероз кортикального слоя трубчатых костей.
Гипофосфатазия детская. Аутосомно-рецессивное заболевание, вызванное мутацией алкалин-фосфатазного гена (ALPL). Тяжелое наследственное поражение с признаками укорочения конечностей. Голубые склеры, дисплазия дентина и полностью несформированные зубы, обширные скелетные аномалии, нефрокальциноз, краниосиностоз, эпиприступы, гипотония. Изменения лабораторных показателей: гиперкальцимия, гиперкацинурия, фосфоэтаноламинурия. Как правило, летальная форма заболевания.
Несовершенный остеогенез, тип III. Заболевание обусловлено мутацией гена коллагена 1 или 2 (COL1A1; COL1A2), локализованного в 17q21, 31-q22, 7q22.1. Генетически гетергенное заболевание наследуется по аутосомно-рецессивному и аутосомно-доминантному типу. Характеризуется резким снижением длины тела (взрослые 92-108 см) и множественными скелетными аномалиями: микрогнатия, голубые склеры, несовершенный дентиногенез.
Несовершенный остеогенез, тип IV. Еще одна генетическая форма несовершенного остеогенеза, вызванная мутацией в тех же генах, что и предыдущее заболевание. Локализовано в области 17q21.31-q22. В настоящее время клинически выделяют несколько форм несовершенного остеогенеза:
• тип I - с голубыми склерами;
• тип II - с внутриутробной летальностью или так называемый врожденный несовершенный остеогенез;
• тип III - прогрессирующая деформирующая форма с нормальными склерами;
• тип IV - с нормальными склерами.
Предполагается существование подтипов А и В в зависимости от наличия или отсутствия признаков несовершенного дентиногенеза.
Несовершенный остеогенез, тип IV - аутосомно-доминантное заболевание. Отмечается некоторое уменьшение длины тела (в среднем на 5% от нормы). Нормально окрашенные склеры, несовершенный дентиногенез, небольшие скелетные деформации, кифосколиоз, снижение слуха, отосклероз.
 Рис. 1.18. Несовершенный остеогенез
Рис. 1.18. Несовершенный остеогенез
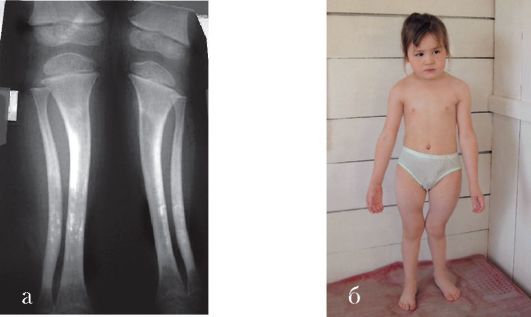 Рис. 1.19 а, б. Спондилоэпифизарная дисплазия
Рис. 1.19 а, б. Спондилоэпифизарная дисплазия
Спондило-метафизарная дисплазия с несовершенным дентиногезом, синдром Голдблатта. Данная клиническая форма спондилоэпифизарной дисплазии наследуется по аутосомно-доминантному типу, сопровождается снижением роста, гипермобильностью суставов, остеопорозом, сколиозом, деформацией коленных суставов, укорочением конечностей. Постоянным признаком заболевания является несовершенный дентиногенез. При лабораторном исследовании отмечается аномальная электрофоретическая подвижность фракции коллагена II типа.
Дисплазия дентина тип II. Аутосомно-доминантное заболевание. Множественные поражения зубов с изменением цвета (янтарный цвет зубов). Облитерация пульповых камер молочных зубов и постоянных зубов, нормальная форма корней молочных и постоянных зубов. Заболевание вызвано мутацией в DSPP-гене. Признаки несовершенного амелогенеза.
Практически постоянным фенотипическим признаком несовершенного дентино- и амелогенеза является изменение цвета зубов. Наряду с наследственными факторами причиной аномалий цвета зубов могут являться различные неблагоприятные факторы внешней и внутренней среды.
Наследственные болезни и синдромы, сопровождающиеся нарушением формирования эмали
Эмаль - наиболее твердая ткань тела человека (твердость эмали 7 по таблице Моосса). Гипоплазия эмали оценивается как порок ее развития, наступающий в результате нарушения метаболических процессов в развивающихся зубах и проявляющийся в качественном и количественном аномальном нарушении эмали зубов. Некоторые исследователи считают, что при гипоплазии нарушается формирование зубных тканей за счет изменения в обра зующих эма ль к летка х- энамелобластах. Другие авторы рассматривают гипоплазию эмали как дефект ее минерализации при нормальном формировании зубных тканей. Считается, что практически невозможно разделение этих двух взаимосвязанных процессов. Полагают, что гипоплазия твердых тканей зубов возникает в результате нарушения как формирования эмали энамелабластами, так и ослабления процесса минерализации эмалевых призм. При гипоплазии нарушены не только процессы минерализации, но в первую очередь построение белковой матрицы эмали зуба в результате недостаточной или замедленной
функции энамелобластов. Системная гипоплазия тканей зуба характеризуется нарушением строения эмали всех или только той группы зубов, которая формируется в один и тот же промежуток времени. Эта форма встречается у 2-14% детей. Причины такого патологического состояния чрезвычайно многобразны. Это и внешнесредовые воздействия (болезни матери во время беременности, плохое питание, прием некоторых лекарственных препаратов во время беременности или препараты, вводимые в организм ребенка, например тетрациклины). Иногда в качестве причины патологии рассматривают недоношенность, гемолитическую желтуху, аллергические состояния и др.
На постоянных зубах гипоплазия эмали развивается под влиянием различных заболеваний, возникших у детей в период формирования и минерализации зубов. Признаки гипоплазии обнаруживают у детей, перенесших рахит, острые инфекционные заболевания, болезни желудочно-кишечного тракта, токсическую диспепсию, алиментарную дистрофию. Системная гипоплазия постоянных зубов встречается также у детей с заболеваниями эндокринной системы, врожденным сифилисом, мозговыми нарушениями. 60% гипопластических дефектов постоянных зубов развивается в первые 9 мес жизни. Элементы гипоплазий чаще определяются в области режущего края резцов, режущего бугра.
Наследственные дефекты эмали, не связанные с общими нарушениями, считаются разновидностями несовершенного амелогенеза. В целом среди населения несовершенный амелогенез всех типов встречается с частотой 1:14 000. Это генетически гетерогенное заболевание.
5.2. ГЕНЕТИЧЕСКИЕ ФАКТОРЫ АНОМАЛИЙ ФОРМИРОВАНИЯ ЭМАЛИ И КЛАССИФИКАЦИЯ
Этиология несовершенного эмалегенеза связана с двумя генами, участвующими в патогенезе. Первая генетическая ассоциация определяется открытием амелогенинового гена, локализованного в регионе р21.1-22.3 на хромосоме Х. Второй ген, участвующий в формировании картины заболевания, - эмалиновый ген на хромосоме 4 (4q21). В настоящее время нет единой классификации несовершенного амелогенеза. Существующие классификации основаны на клинических данных или на данных, сочетающих в себе информацию о клинических проявлениях заболевания и генетической этиологии.
Наиболее распространенный тип несовершенного амелогенеза - аутосомно-доминантно наследуемая гипокальцификация эмали, которая встречается с частотой около 1:20 000. На основе клинической классификации выделяют 4 типа несовершенного амелогенеза:
1) гипопластический;
2) гипоматурационный;
3) гипокальцификационный;
4) гипоматурационный-гипопластический.
Эта классификация соответствует нарушениям, которые могут происходить в разные стадии формирования эмали (секреция органического матрикса, минерализация матрикса, матурация, или созревание эмали). В зависимости от фенотипических вариантов патологии и данных о характере наследования этого признака классификация, основанная на клинико-генетическом принципе, предполагает выделение 15 типов несовершенного амелогенеза (табл. 1.11).
Некоторые мутации, вызывающие изменение структуры или химического состава эмали, проявляются либо как изолированные признаки, либо являются составной частью разнообразных наследственных заболеваний и синдромов, при этом нередко поражаются как временные, так и постоянные зубы, отмечается задержка прорезывания зубов, аномалии прикуса и цвета зубов.
Как следствие мутаций могут наблюдаться: недостаточное образование эмали (гипоплазия), недостаточность обызвествления органической матрицы (гипокальцификация), дефекты образования кристаллов апатита в различных компонентах эмалевых призм (гипосозревание), отложение экзогенного материала, приводящее к аномалиям пигментации, а также комбинация этих нарушений.
При ямочной гипопластический форме амелогенеза эмаль как временных, так и постоянных зубов обычно имеет нормальную толщину, но на ее поверхности беспорядочно расположены ямки небольших размеров. Эмаль прорезавшихся зубов - твердая, желто-белого цвета. Ямки захватывают губные поверхности в большей степени, чем язычные. Отмечается тенденция к расположению ямок вертикальными столбиками. Ямочный гипопластический неполноценный амелогенез чаще наследуется как аутосомно-доминантный признак.
Дефекты зубной эмали выявляются более чем при 140 наследственных заболеваниях и синдромах, соответствующих менделевскому характеру наследования. Некоторые из этих заболеваний приведены в табл. 1.12.
Таблица 1.11. Клинико-генетическая классификация несовершенного амелогенеза
Тип | Вариант несовершенного амелогенеза | Специфические признаки поражения эмали | Тип наследования | Генетический дефект |
IA | Гипопластический | Генерализованный ямчатый дефект | АД | Не известен |
IB | Гипопластический | Локализованный ямчатый дефект | АД (104500) | Анемелин (ΕΝΑΜ) |
IС | Гипопластический | Локализованный ямчатый дефект | АР (204650) | Не известен |
ID | Гипопластический | Гладкая эмаль | АД (104500) | Анемелин (ΕΝΑΜ) |
IE | Гипопластический | Гладкая эмаль | Х-сц.Д | Не известен |
IF | Гипопластический | Грубая, шероховатая эмаль | АД | Не известен |
IG | Гипопластический | Агенезия эмали | АР | Не известен |
IIА | Гипоматурационный | Диффузная пигментация | АР (204700) | Амелогенин (АМЕГХ) |
IIВ | Гипоматурационный | Диффузное поражение | Х-сц. Ρ | Не известен |
IIС | Гипоматурационный | «Снежный» налет | Х-сц | Не известен |
IID | Гипоматурационный | «Снежный» налет | АД | Не известен |
ΠΙΑ | Гипокальцификационный | Диффузное поражение | АД | Не известен |
ΙΙΙΒ | Гипокальцификационный | Диффузное поражение | АР | Не известен |
IVA | Гипоматурационный-гипопластический | Тауродонтизм | АД (104510) | Не известен |
IVB | Гипопластический-гипоматурационный | Тауродонтизм | АД | Не известен |
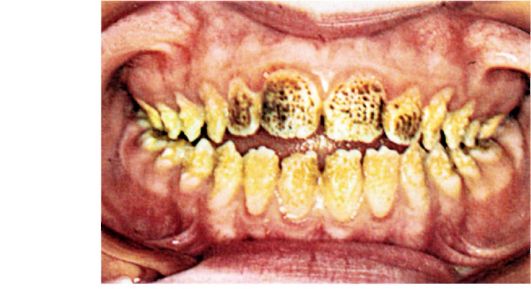 Рис 1.20. Несовершенный амелогенез гипопластический
Рис 1.20. Несовершенный амелогенез гипопластический
5.3. АУТОСОМНО-ДОМИНАНТНЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ С НАРУШЕНИЕМ ФОРМИРОВАНИЯ ЭМАЛИ
Наследственная остеодистрофия Альбрехта. Это гетерогенное по своим проявлениям наследственное аутосомно-доминантное заболевание характеризуется низким ростом, ожирением, катарактой, брахидактилией, умственной отсталостью. Отмечаются гипофосфатемия, гипокальцемия, различные эндокринные аномалии. К стоматологическим признакам относятся: врожденная гипоплазия эмали, задержка прорезывания зубов, ано-/олигодентия. Ген заболевания локализован в районе 20q13.2.
Амело-онихо-гипогидротический синдром. Редкое аутосомнодоминантное заболевание. Описано впервые в 1975 г. как сочетанное поражение, включающее в себя гипокальцифицирующую/гипоплас- тичную эмаль зубов, изменение цвета зубов, онихолизис, гиперкератоз и гипогидроз. Характерными признаками также являются: желто-коричневая окраска зубов, задержка прорезывания постоянных зубов. Себорейный дерматит кожи головы, гипогидроз, ксероз, онихолизиз. Волосы нормальные.
Таблица 1.12. Наследственные заболевания и синдромы с признаками аномального развития эмали
Синдром | Тип наследования | ? ОМИМ | Локус |
Акроренальный дефект, эктодермальная дисплазия, липоатрофический диабет | АР | 207780 | |
Аланинурия с микроцефалией, карликовость, гипоплазия эмали, сахарный диабет | АР | 202900 | |
Фронтоназальная дисплазия с расщелиной крыльев носа | АР | 203000 | |
Наследственная остеодистрофия Альбрехта; псевдопаратиреоидизм тип IA | АД | 103580 | 20ql3.2 |
Алопеция конрактуры, карликовость, умственная отсталость | АР | 203550 | |
Эмало-онихо-гипогидротический синдром | АД | 104570 | |
Эмалегенез несовершенный и нефрокальциноз | АР | 204690 | |
Аутоиммунная полиэндокринопатия, тип I | АР | 240300 | 21q22.3 |
Дистрофия колбочек-палочек и несовершенный эмалегенез | АР | 217080 | |
Краниоэктодермальная дисплазия | АР | 218330 | |
Нейросенсорная тугоухость, гипоплазия эмали, дефекты ногтей | АР | 234580 | |
Несовершенный остеогенез с опалесцирующими зубами, голубыми склерами и вормиевыми костями, но без переломов | АД | 166230 | |
Артрогрипоз и эктодермальная дисплазия | АР | 601701 | |
Буллезный эпидермолиз с умственной отсталостью | 226440 | ||
Эпилепсия и желтые зубы | АР | 226750 | |
Лакримо-аурикуло-денто-дигитальный синдром | АД | 149730 |
Продолжение табл. 1-12
Ленца-Маджевски гиперостатическая карликовость | АД | 151050 | |
Микроцефалия первичная, карликовость, тип Ториелло | АР | 251190 | |
Мукополисахаридоз тип IVA | АР | 253000 | 16q24.3 16q24.3 |
Мукополисахаридоз тип IVB | 253010 | ||
Окулоцеребеллярный синдром LOWE (OCRL) | Х-сц | 309000 | Xq26.1 |
Окулодентодигитальная дисплазия (ODDD) | АД | 164200 | 6q21- q23.2 |
SCARF-синдром (скелетные аномалии, CUTIS LAXA, краниостеноз, аномалии наружных половых органов и лицевые аномалии) | Х-сц. | 312830 | |
Секкеля синдром I | АР | 210600 | 3q22-q24 |
Трихо-адонто-онихиальная дисплазия | АР | 275450 | |
Амелогенез несовершенный 2, гипопластический локальный, аутосомно-доминантный (AIH2) | АД | 104500 | 4q21, 4qll-q21 |
Амелогенез несовершенный 1, гипопластический тип | Х-сц. | 301200 | Xp22.3- p22.1 |
Амелогенез несовершенный, гипопластический, с открытым прикусом, аутосомно-рецессивный | АР | 600653 | 4q21 |
Дистрофия колбочек и палочек, несовершенный остеогенез | АР | 217080 | |
Амелогенез несовершенный с тауродонтизмом | АД | 104510 | 17q21.3- q22 |
Амелогенез несовершенный 3, гипопластический тип (АIHЗ) | Х-сц. | 301201 | Xq22- q28 |
Продолжение табл. 1-12
Амелогенез несовершенный, гипопластический тип, микродентия | АД | 104530 | |
Амелогенез несовершенный, локальный гипопластический тип, рецессивный | АР | 204650 | |
Амелогенез несовершенный, неразвитая пигментация | АР | 204700 | |
Амелогенез несовершенный, неразвитый тип | Х-сц. | 301100 | |
Амелогенез несовершенный, гипопластический/гипоматурацион- ный (незрелый), Х-сцепленный 2 | Х-сц. | 301201 | Xq22- q28 |
Гипоплазия эмали, наследственная | АД | 130900 | |
Ихтиоз-склерозирующий холангит, синдром | АР | 607626 | 3q28- q29, 3q27-q28 |
Скрученные волосы (PILI TORTI) | АР | 261900 | |
Рахит Д-резистентный, тип II | АР | 277000 | 12ql4, 12ql3.1- ql3.3 |
Рахит Д-резистентный, тип II | АР | 277440 | 12ql2- ql4 |
Холла-Риггса умственная отсталость, синдром | АР | 234259 | |
Буллезный эпидермолиз и поясно-конечностная мышечная дистрофия | АР | 226670 |
Окончание табл. 1-12
Буллезный эпидермолиз генерализованный, атрофический, доброкачественный | АР | 226650 | 18qll.2, lq32, 17qll- qter, lq25- q31, 10q24.3 |
Амелогенез несовершенный, гипопластический тип (микродентия генерализованная). Несовершенный эмалегенез клинически и генетически гетерогенное состояние и может проявляться в виде различных клинических вариантов даже в пределах аутосомнодоминантно наследующихся форм. Поводом выделения в самостоятельную нозологическую единицу является стойкое сочетание признаков несовершенного гипопластического амелогенеза с генерализованной микродентией.
Амелогенез несовершенный 2, гипопластический, локальный, аутосомно-доминантный. Данное заболевание представляет собой еще один генетический вариант несовершенного амелогенеза. Заболевание вызвано мутацией в эмалиновом гене (ENAM), локализованном в длинном плече хромосомы 4 (4q21). Наследуется по аутосомно-доминантному типу. Описаны различные клинические варианты аутосомно-доминантного несовершенного амелогенеза.
1. Твердая эмаль, но ее количество значительно уменьшено по сравнению с нормой.
2. Гипокальцифицированная форма, при которой зубная эмаль мягкая; при гистологическом исследовании выявляется нормальное ее количество.
Гипокальцификацированный тип амелогенеза встречается в популяции с частотой примерно 1 на 20 000. Полагают, что клинические, радиографические, гистологические признаки заболевания более ярко выражены при рецессивных формах, чем при доминантных состояниях. Дефект может проявляться как на временных, так и на постоянных зубах. Могут быть поражены все зубы, но в пределах одной семьи обычно доминируют вариации в количестве пораженных зубов и степени поражения ткани. Образование дефекта не соответствует какому-либо специфическому периоду в развитии зубов.
Зубы имеют гладкую блестящую поверхность. Цвет прорезавшихся зубов может варьировать от матово-белого до полупрозрачного коричневого. Толщина эмали составляет приблизительно 1/4-1/3 от нормальной. Боковые контакты зубов отсутствуют. Некоторые участки эмали могут отсутствовать, особенно по режущему краю и на жевательных поверхностях. Иногда эмаль с грубой, гранулообразной поверхностью.
Гиперостатическая карликовость Ленца-Маджевски. В разные годы (1969, 1974, 1977) в специальной литературе были даны описания сходных по клинической картине пациентов. Характерны
множественные пороки развития, затрагивающие черепно-лицевую область, скелет, кожу, нервную систему и др. Характерными признаками являются: микрогнатия, непропорционально большая голова, гипертелоризм, атрезия и стеноз хоан и слезно-носового канала, маленький язык, контрактуры конечностей. Тонкая гиперэластичная кожа с очагами гипотрофии. Неврологические нарушения: умственная отсталость, гипотония, агенезия мозолистого тела. При исследовании зубов отмечается дисплазия эмали.
Несовершенный остеогенез с опалесцирующими зубами, голубыми склерами и вормиевыми костями, но без переломов
Заболевание известно с 1981 г., когда Beigton (1981) сообщил о 20 членах одной семьи, где по меньшей мере в трех поколениях отмечалось стойкое сочетание гипопластичная опалесцирующая эмаль, голубые склеры, вормиевые кости, у 6 членов родословной были выявлены признаки остеопороза.
Лакримо-ауриколо-денто-дигитальный синдром (Леви-Холлистера)
Холлистер в 1973 г., Леви в 1969 г. описали заболевание, впоследствии названное их именами. Характерная однотипная патология была описана у 5 детей (4 девочки и 1 мальчик). У этих пациентов отмечались гипоплазия/аплазия слезной точки, заращение слезноносового канала, аномалии ушей и снижение слуха, гипертелоризм, телекант, склонность к дакриоциститам. Наряду с этим отмечается отсутствие околоушных желез и слюнных протоков. Зубные аномалии: гиподентия, коническая форма коронки резцов, задержка прорезывания постоянных зубов, гипоплазия эмали и склонность к кариесу.
Окуло-денто-дигитальная дисплазия, окуло-денто-оссеозная дисплазия, ODDD-синдром.
Генетически гетерогенное заболевание. Описано Gillespie (1964) как наличие специфических аномалий у брата и сестры в виде двусторонней микрофтальмии, маленького носа, гипотрихоза, зубных аномалий, камптодактилии V пальца, синдактилии IV и V пальцев (III тип синдактилии). Известен также аутосомно-рецессивный вариант патологии. Ген локализован в районе 6q21-q23.2. Заболевание обусловлено мутацией в гене коннексина-43 (GJA1). Характерными признаками являются: микроцефалия, поражение глаз (микрокорнеа, глаукома, катаракта, аномалии радужки), различные скелетные
аномалии и аномалии рук (синдактилия IV и V пальцев, укорочение средней фаланги V пальца, камптодактилия V пальца, межфалангеальная гипоплазия. На стопах синдактилия 3-4). Умственная отсталость, дизартрия. Аномалии зубов в виде гипоплазии эмали, частичной анодентии, микродентии, преждевременной потери зубов, кариеса.
Гипоплазия эмали наследственная. Гипоплазия эмали наследственная описана как изолированный признак, наследующийся по аутосомно-доминантному типу с варьирующей экспрессивностью и неполной пенетрантностью. Не исключено, что этот признак можно рассматривать как локальную форму несовершенного амелогенеза.
5.4. АУТОСОМНО-РЕЦЕССИВНЫЕ НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ С НАРУШЕНИЕМ ФОРМИРОВАНИЯ ЭМАЛИ
Акроренальный дефект, эктодермальная дисплазия, липоатрофический диабет, синдром. Аутосомно-рецессивное заболевание, характеризующееся сочетанием патологических симтомов: липоатрофический диабет, редкие волосы, дистрофия эмали, недостаточное количество молочных зубов, короткая длина тела, сколиоз, аномалия почек, необычное лицо с прогнатией. Множественные аномалии зубов и их сочетание: изменение цвета, олигодентия, аномальное расположение зубов. Встречаются больные с натальными, неонатальными зубами.
Аланинурия с микроцефалией. Характерными признаками являются умственная и физическая отсталость, микроцефалия, микродентия, сахарный диабет и аланинурия с лактозным ацидозом. Кроме того, отмечается врожденный дефект формирования эмали, изменение цвета зубов, аномальное их расположение, малоклюзия.
Фронтоназальная дисплазия с расщелиной крыльев носа.
Аутосомно-рецессивное заболевание. Порок развития лицевой области. Гипертелоризм. Телекант, широкая спинка носа, с колобомами крыльев носа. Встречаются пациенты с расщелиной твердого нёба, врожденный дефект эмали, изменение цвета зубов.
Амелогенез несовершенный и нефрокальционоз. Заболевание известно с 1972 г., когда у родных брата и сестры была обнаружена сходная картина заболевания в виде гипопластического амелогенеза,
патологического разрастания слизистой десен, отмечались явления поражения почек (нефрокальциноз), полиурия. В моче определялся сниженный уровень экскреции кальция и фосфатов.
Дистрофия колбочек-палочек с несовершенным амелогенезом. Ген не идентифицирован, предположительная локализация 2q11. Основными признаками заболевания являются: фотофобия, нистагм, бледные диски зрительных нервов, полная ахроматопсия, прогрессирующее снижение цетрального зрения. Зубы: признаки несовершенного амелогенеза, темно-коричневый цвет зубов, распространенный кариес.
Эпилепсия и желтые зубы. Редкое наследственное заболевание. Локализация гена неизвестна. Тип наследования аутосомно-рецессивный. Отличается внутри, и межсемейным клиническим полиморфизмом. Основными проявлениями явлются симптомы пражения центральной нервной системы (эпилепсия, деменция, умственная отсталость и др.), аномалии зубов: признаки несовершенного амелогенеза временных и постоянных зубов (гипопластический тип), желтый цвет зубов.
Алопеции, контрактур, карликовости и умственной отсталости синдром. Аутосомно-рецессивное заболевание. Характеризуется множественными аномалиями развития скелета (сколиоз, контрактуры, брахидактилия, синдактилии, каптодактилия), аномальной дерматоглификой, алопецией. К зубным аномалиям относят изменение цвета зубов и гипоплазию эмали.
Амелогенез несовершенный, пигментированный гипоматурационный тип. Локализация гена металлопротеиназа-20(ММР20), вызывающего данную форму несовершенного амелогенеза - 19q13.3- q13.4, 11q22.3-q23. Аутосомно-рецессивное заболевание, известное с 1965 г. Ген локализован в 2005 г. Характерно аутосомно-рецессивное наследование, мягкая (рыхлая) эмаль молочных и постоянных зубов. Изменение цвета эмали в виде желто-агарового блестящего оттенка и коричневые пластинчатые включения в эмали. При рентгенографическом исследовании отсутствует рентгеноконтрастная картина между эмалью и дентином. При неполном развитии эмали прорезавшиеся зубы имеют желтый цвет. Поверхность их грубая, гранулообразная. Эмаль почти полностью отсутствует. Зубы расположены редко, прикус открытый. Среди прорезавшихся зубов многие отсутствуют. Поражаются как временные, так и постоянные зубы. Эта форма несовершенного амелогенеза встречается редко.
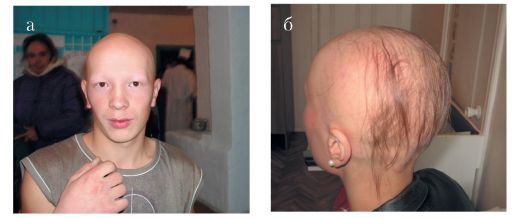
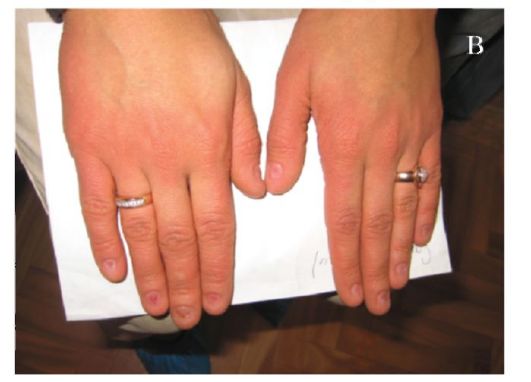 Рис. 1.21 а-в. Алопеция, отсутствие бровей, ресниц, контрактура пальцев
Рис. 1.21 а-в. Алопеция, отсутствие бровей, ресниц, контрактура пальцев
Нейросенсорная тугоухость с гипоплазией эмали и дефектами ногтей. Заболевание впервые описано в 1991 г. У 11-летнего брата и его 9-летней сестры было отмечено двустороннее снижение слуха, развившееся на первом или втором году жизни. У обоих были нормальные молочные зубы, однако постоянные зубы имели генерализованную форму гипоплазии эмали. Этому заболеванию сопутствует гипоплазия ногтей. В последующие годы аналогичное состояние неоднократно описывалось различными авторами, что позволило выделить его в самостоятельную нозологическую форму - синдром Хеймлера с аутосомно-рецессивным типом наследования.
Артрогриппоз и эктодермальная дисплазия. Наследуется аутосомно-рецессивно. Впервые заболевание описано в 1981 г. у 20-лет- ней женщины как необычная форма эктодермальной дисплазии в сочетании с различными пороками развития. Кожные проявления
синдрома: сухая кожа, гиперкератоз, аномальная дерматоглифика, гипогидроз. Дистрофия волос, ногтей, отставание в росте, лицевая дизморфия. Поражения глаз: двустороняя ядерная катаракта, эктропион, трихиаз. Аномалии конечностей, сколиоз. Расщелина губы/ нёба, сахарный диабет. Зубы: олигодентия и аномалии строения эмали.
Ихтиоз-склерозирующий холангит синдром. Аутосомно-рецессивное заболевание, вызванное мутацией в гене CLDN1. Ген локализован в области 3q28-q29, 3q27-q28. Заболевание впервые описано в 2002 г. Включает в себя следующие основные признаки: ихтиоз, гипотрихоз, признаки алопеции, склерозирование желчных протоков и вакуолизацию лейкоцитов. Клинически обширные кожные поражения: распространенный диффузный ихтиоз средней степени выраженности, ксероз, окантоз, папилломатоз и др., поражение печени (гепатомегалия, холестаз). К зубным аномалиям относятся: гиподентия, олигодентия, гипоплазия эмали.
Скелетных аномалий, эластичной кожи (CUTIS LAXA), краниосиностоза, аномального строения гениталий и лицевых аномалий синдром. Синдром описан в 1983 г. у четырех сестер одной семьи. Наследование аутосомно-рецессивное. Для заболевания характерно наличие признаков эктодермальной дисплазии, пегментные невусы, дополнительные соски молочных желез, гипотрихоз, гипоплазия эмали зубов, вторичная анодентия, дистрофия ногтей.
Скрученные волосы (PILI TORTI). Этот наследственный синдром был описан Ronchese в 1932 г. Наследуется аутосомно-рецессивно. Данный признак встречается в составе синдрома Менкеса (ОМИМ 309400), наследующегося как Х-сцепленное рецессивное заболевание. Характерное необычное строение волос (сильно срученные, жесткие, ломкие) и гипоплазия зубной эмали.
Мукополисахаридоз, тип IVA. Наследственное системное заболевание с аутосомно-рецессивным типом наследования. Характеризуется специфическим фенотипом (низкий рост, лицевые аномалии, редкие волосы, широкий рот). Патология зубов: широко расставленные, серый оттенок цвета эмали, обширный кариес.
Мукополисахаридоз, тип IVB. Заболевание связано с дефицитом фермента β-галактозидазы. По клиническим проявлениям и аномалиям зубов сходно с мукополисахаридозом типа IVA.
Микроцефалия, внутриутробное отставание в росте. Тип Ториелло. Заболевание известно с 1983 г. Наследуется аутосомно-
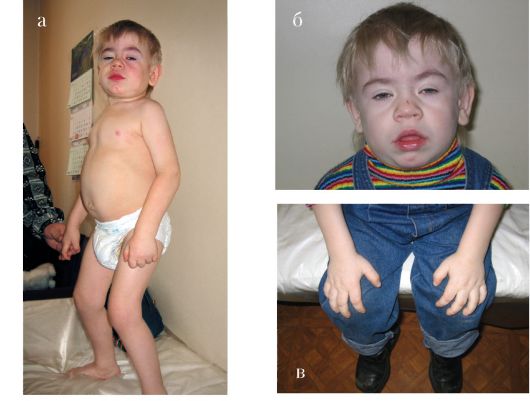 Рис. 1.22 а-в. Мукополисахаридоз
Рис. 1.22 а-в. Мукополисахаридоз
рецессивно. Характерными признаками являются: пропорциональная карликовость, внутриутробная задержка роста, микроцефалия, катаракта, поражение кистей и стоп (короткие кисти и стопы, отставание костного возраста, клинодактилия, укорочение срединных и проксимальных фаланг). Умственная отсталость. Гипоплазия эмали.
Стейва-Видеманна синдром (STUVE-WIEDEMANN). Локализация гена LIFR, в области 5p13.1. Аутосомно-рецессивное заболевание, клиническими признаками которого являются: отставание в росте, гипоплазия срединной области лица, микрогнатия, низко посаженные уши, отсутствие корнеальных рефлексов, частые респираторные заболевания, гипертензия легочной артерии. Диагностическими признаками являются различные скелетные нарушения в виде остеопороза, частых переломов, прогрессирующий сколиоз, камптомиелия, флексорная контрактура пальцев рук, мышечная гипотония
и др. Стоматологические признаки синдрома: нарушения зубного ряда, испещренная эмаль, хронические абсцессы зубов.
Рахит D-резистентный, тип II. Заболевание обусловлено мутацией в гене 25-гидроксивитамин D-3-1-альфа-гидроксилазы CYP27B1. Локализован в 12q14, 12q13.1-q13.3 Сходные по клиническим проявлениям и типу наследования, генетически гетерогенные заболевания с множественными и типичными для рахита признаками. Зубные аномалии выражаются в задержке прорезывания зубов и гипоплазии эмали.
Рахит D-резистентный, тип II. Заболевание обусловлено мутацией в гене рецепторе Д-витамина (VDR). Ген локализован в 12q12-q14.
Холла-Риггса-умственная отсталость синдром. Синдром впервые описан в 1975 г. у детей, рожденных в кровнородственном браке. Наследуется аутосомно-рецессивно. Для синдрома характерны: микроцефалия, умственная отсталость, гипертелоризм, эпикант, широкий, открытый рот. Скелетные аномалии в виде остеопороза, сколиоза, кифоза. Умственная отсталость, эписиндром. Мелкие молочные зубы, гипоплазия эмали.
Буллезный эпидермолиз и поясно-конечностная мышечная дистрофия. Ген заболевания локализован в районе 8q24. Вызвано мутацией в плектиновом гене (PLEC1). Аутосомно-рецессивный тип наследования. Низкий рост, анемия, признаки буллезного эпидермолиза и симптомы мышечной дистрофии. Гипоплазия эмали, распространенный кариес.
Буллезный эпидермолиз генерализованный, атрофический, доброкачественный. Заболевание может вызываться мутациями в нескольких генах: COL17A1, LAMA3, LAMB3, LAMC2. Гены локализованы в районах 18q11.2, 1q32, 17q11-qter, 1q25-q31, 10q24.3. Ведущими клиническими симптомами являются кожные поражения в виде буллезного эпидермолиза. К зубным аномалиям относятся: кариес, гиподентия, ямчатые дефекты эмали.
Краниоэктодермальная дисплазия. Аутосомно-рецессивное заболевание. Характерна долихоцефальная форма черепа, синостоз сагиттальных швов черепа, эпикант, гипетелоризм. Миопия, нистагм. Брахидактилия, клиндактилия, кожная синдактилия. Узкая грудная клетка, гипоплазия эмали.
Секкеля синдром. Клинически гетерогенное аутосомно-рецессивное заболевание. Ген локализован в области 3q22-q24. Характеризуется отставанием в росте, умственной отсталостью, микроцефалией, судо-
рожными приступами, своеобразным строением головы и лица («птицеголовость»). Микрогнатия, низкопосаженные деформированные уши, высокое нёбо, иногда расщелина нёба. Характерны частичная анодентия, гипоплазия эмали, малоклюзия II неправильный рост зубов.
 Рис. 1.23. Синдром Секкеля
Рис. 1.23. Синдром Секкеля
Амелогенез несовершенный, гипопластический, с открытым прикусом, аутосомно-рецессивный. Заболевание обусловлено мутацией в эмалиновом гене (ENAM). Ген локализован в районе 4q21. Rowley et al. (1982), обследовавший 50 больных несовершенным эмалогенезом, обнаружил, что одна из клинических форм данного заболевания всегда ассоциирована с нарушением прикуса, что и позволило выделить данное состояние в отдельную нозологическую единицу. Ретрогнатия верхней челюсти. Вертикальная дисгнатия, сниженная минерализация эмали, желто-коричневая окраска эмали. У гетерозигот ямчатые дефекты эмали.
Ларинго-онихо-кутанный синдром. Заболевание обусловлено мутацией гена LAMA3, локализованном в 18q11.2. Впервые как новый наследственный синдром описан в 1986 г. у 22 больных из 12 семей, проживавших в одной из Пакистанских провинций. Заболевание характеризуется полиморфной клинической картиной поражений кожи (гранулемы, изъязвления кожи), гранулемы голосовых связок,
необычный тембр голоса, аномалии развития ногтей. Зубы: несовершенный амелогенез.
Буллезный эпидермолиз с поздним началом и умственной отсталостью. Аутосомно-рецессивное заболевание. Различные формы буллезного эпидермолиза (БЭ) могут быть подразделены на три основных гистологических группы: эпидермолитический (БЭ), смешанный, дермолитический. В самостоятельную нозологическую единицу заболевание выделено в 1992 г., после того как у болгарских евреев было описано заболевание, характеризующееся проявлениями буллезного эпидермолиза, локализованного на передних поверхностях стоп, признаками дистрофии зубной эмали и ногтей и умственной отсталостью. Кроме того, при данном синдроме иногда встречается подвывих хрусталика, укороченный фильтр, прогнатизм и расщелина нёба.
Аутоиммунной полиэндокринопатии тип 1 синдром (ОМИМ 240300). Ген локализова в районе 21q22.3, мутация в аутоиммунном регуляторном гене (AIRE). Данный синдром характеризуется двумя или тремя основными клиническими симптомами: болезнь Аддисона и/или гипопаротироидизм, а также кандидоз. Часто при этом заболевании присутствует симптом малабсобции и диареи. Заболевание относится к категории «финских болезней» и встречается в Финляндии с достаточно высокой частотой. Наследуется как аутосомно-рецессивно, так и аутосомно-доминантно. Часто сопутствуют кератоконъюнктивиты, хроническая форма гепатита, гипогонадизм, хронический атрофический гастрит, сахарный диабет. На коже часто витилиго, эктодермальная дистрофия, алопеция. В качестве частого признака патологии фигурирует гипоплазия эмали.
5.5. СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ ЗАБОЛЕВАНИЯ И СИНДРОМЫ С НАРУШЕНИЕМ ФОРМИРОВАНИЯ ЭМАЛИ
Амелогенез несовершенный, гипопластический/гипоматураци- онный (незрелый), Х-сцепленный 1 (ОМИМ 301200). Локализация гена, кодирующего амелогенин Хq22.3-p22.1. Х-сцепленное доминантное заболевание. Характеризуется гипопластическим типом амелогенеза. Ген локализован в районе Xp22.3-p22.1. В 1972 г. MacGibbon (1972) сообщил о сестре и брате, у которых отсутствовала зубная эмаль и были признаки нефрокальциноза. Для данного заболева-
ния характерными признаками являются маленький размер зубов, аномальное строение зубной эмали (тонкая, твердая), с шероховатой поверхностью желто-коричневого оттенка. Отмечается иногда непрорезывание зубов и частичная резорбция альвеолярных отростков. Поражаются как временные, так и постоянные зубы. У мужчин и женщин клиническая картина различна. У мужчин постоянные зубы покрыты крапинками и имеют желто-белый цвет, но с возрастом вследствие адсорбции пятен зубы могут темнеть. По толщине эмаль приближается к нормальной. Эмаль мягкая, и кончиком зонда ее можно проткнуть. Несмотря на то что такие зубы более склонны к скалыванию и стиранию, чем здоровые, потеря эмали происходит медленно. У мужчин временные зубы внешне напоминают матовое белое стекло. Иногда отмечается легкое пожелтение временных зубов. Поверхность их относительно гладкая. У женщин как на временных, так и на постоянных зубах видны перемежающиеся вертикальные полосы матово-белой и нормальной полупрозрачной эмали. Эти полосы бывают разной ширины и хаотически распределены по коронке. Симметрия гомологичных зубов справа и слева отсутствует. Как и при других формах неполноценного амелогенеза, часто отмечается открытый прикус.
Амелогенез несовершенный, гипопластический/гипоматураци- онный (незрелый), Х-сцепленный 2. Локализация гена Хq22-q28. По своим клиническим проявлениям напоминает предыдущее заболевание и характеризуется гипопластическим типом амелогенеза. Отличие заключается в районе локализации гена на Х-хромосоме, вызывающего данное заболевание.
Ихтиоз, атрихия и фотофобия. Х-сцепленное рецессивное заболевание. Ведущая клиническая симптоматика этого синдрома с аутосомно-рецессивным типом наследования отражена в самом названии. Заболевание впервые описано в 1909 г. Маклеодом; характерно наличие гипогидроза, фолликулярного ихтиоза, фотофобии. Больные, как правило, низкого роста, с признаками умственной отсталости, подвержены частым респираторным инфекциям. В качестве одного из признаков синдрома отмечается дисплазия эмали.
SCARF-синдром. Редкий наследственный синдром с Х-сцепленным рецессивным типом наследования. К настоящему времени описаны только два пациента с данной патологией. Некоторые признаки синдрома перекрываются с фенотипом синдрома Ленца-Маджевски. К зубным аномалиям относят гипоплазию эмали зубов.
 Рис. 1.24. Ихтиоз Х-сцепленный
Рис. 1.24. Ихтиоз Х-сцепленный
Окуло-церебро-ренальный, синдром Лоу. Х-сцепленное заболевание, обусловлено мутацией в гене OCRL1. Ген локализован в области Xq26.1. Данное заболевание описано в 1958 г. Характерны аномалии глаз (врожденная катаракта, глаукома, микрофтальмия и др.), почетные аномалии, почечный ацидоз. Множественные скелетные аномалии: сколиоз, гипермобильность суставов и искривление конечностей, флексорные контрактуры пальцев, арефлексия, иногда умственная отсталость. Зубные кисты, гипопазия эмали. Таким образом, разнообразные по своим клиническим проявлениям признаки дисплазии эмали могут встречаться как изолированные формы патологии, так и входить в состав разнообразных наследственных синдромов и заболеваний. Клиническая вариабельность этих форм патологии очень широкая, и аномалии развития эмали часто сопряжены с другими аномалиями зубов. Однако отдельные наследственные заболевания могут характеризоваться специфическими особенностями поражения зубной эмали. В табл. 1.13 представлены данные о некоторых наследственных синдромах со специфическими признаками аномалий развития эмали.
Таблица 1-13. Синдромы с признаками дисплазии эмали
Синдром | Тип дисплазии эмали |
Аутосомно-доминантные заболевания | |
Амело-онихо-гипогидротический синдром | Гипокальцификация/гипоплазия |
Окуло-денто-оссеозная дисплазия | Гипоплазия |
Трихо-денто-оссеозный | Гипокальцификация/гипоплазия |
Туберозный склероз | Ямчатая гипоплазия |
ЕЕС-синдром | Гипоплазия |
Эктодермальная дисплазия, тип Базан | Гипокальцификация |
Аутосомно-рецессивные заболевания | |
Синдром Моркио (MPS-IV) | Гипоплазия |
Витамин D-зависимый рахит | Грубая, шероховатая дисплазия |
Синдром Кохлшуттера | Гипоплазия |
Синдром МакГиббона | Аплазия |
Буллезный эпидермолиз (АР тип) | Ямчатая гипоплазия |
Х-сцепленные заболевания | |
Амело-церебро-гидротический синдром | Гипоплазия |
Псевдогипопаратироидизм | Ямчатая гипоплазия |
Фокальная дермальная гипоплазия | Гипоплазия |
Клинический полиморфизм и генетическая гетерогенность данного состояния выражаются в том, что одни и те же признаки встречаются при разных заболеваниях, с разными типами наследования. Так, гипоплазия эмали обнаруживается при окулодентооссеозной дисплазии и ЕЕС-синдроме (аутосомно-доминантный тип наследования), при мукополисахаридозе (синдром Моркио), синдроме Кохлшуттера (аутосомно-рецессивный тип наследования), при амело-церебро-гидротическом синдроме, фокальной дермальной гипоплазии (Х-сцепленный тип наследования).
5.6. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ, СОПРОВОЖДАЮЩИЕСЯ АНОМАЛИЯМИ ЦВЕТА ЗУБОВ
Изменение цвета коронок молочных зубов (желтый, серо-желтый, темно-коричневый, желто-зеленый, коричнево-зеленый, чернокоричневый, серый, серо-синий, зеленый, голубой, лиловый, черный) наблюдается у детей при гемолитическом синдроме и гемолитических желтухах различной этиологии. Образующийся при гемолизе эритроцитов непрямой билирубин откладывается в тканях зуба и обусловливает окрашивание зубов в различные цвета, может привести к недоразвитию эмали или системной гипоплазии эмали. В отличие от системной гипоплазии, вызванной другими заболеваниями, гипоплазия после гемолитической желтухи, вызванной несовместимостью крови матери и ребенка по резус-фактору, обязательно сочетается с изменением окраски коронок молочных зубов. Изменения цвета зубов могут возникать вследствие генетически обусловленных нарушений в тканях зуба или проникновения в них красящего вещества, например при порфирии.
Наиболее обширные сведения о количестве наследственных заболеваний с изменением цвета зубов приведены в базе данных Австралийской компьютерной системы «POSSUM» и в каталоге ОМИМ. При анализе этих данных становится ясным, что количество таких состояний насчитывает несколько десятков нозологических форм. Изменение цвета зубов регистрируют при моногенных, хромосомных и мультфакториальных заболеваниях. Изменение цвета зубов отмечено при таких хромосомных нарушениях, как частичная дупликация длинного плеча хромосомы 1 и 8, интерстициальная делеция длинного плеча хромосом 2 и 4, делеция длинного плеча хромосомы 13 и др.
Практически всегда аномалии цвета зубов встречаются при моногенных заболеваниях, связанных с нарушением формирования эмали и дентина (глава «Наследственные заболевания и синдромы с нарушением формирования эмали и дентина»).
Некоторые моногенные заболевания и синдромы, сопровождающиеся изменением цвета зубов, приведены в табл. 1.14.
Синдром Элерса-Данло, тип VII, аутосомно-рецессивный. В настоящее время известно о нескольких клинических и генетических вариантах индрома Элерса-Данло. Тип VII впервые был описан в 1998 г. Заболевание связано с наличием мутации в гене ADAMTS2. Ген локализован в 5q23 (хромосома 5, длинное плечо, сегмент 23). Ранее было описано 6 основных типов этого синдрома (тип I и II): классический (тип III), гипермобильный, васкулярный тип (IV), с кифосколиозом (тип VI); описаны и некоторые другие варианты этого синдрома как «неспецифические формы». Для данного варианта синдрома характерными признаками являются: микрогнатия, голубые склеры, миопия, гиперплазия слизистой десен. Зубные аномалии: гиподентия, изменение цвета зубов, аномальная морфология моляров молочных зубов, повышенная стираемость эмали молочных зубов. Аномалии скелета: остеопения, укорочение конечностей, короткие пальцы кистей и стоп.
Амело-онихо-гипо-гидротический синдром. Редкий наследственный синдром. Описан в 1975 и 1976 гг. Witkop et al. Для данного синдрома характерно наличие гипокальцифицированной-гипопластичной эмали, изменение цвета зубов (желто-коричневые зубы), задержка прорезывания постоянных зубов. Поражение кожи в виде себореи, гипогидроза, ксероза. Онихолизис и нормальное состояние волос.
Дентиногенез несовершенный, тип Шидлса III. Аутосомно-доминантное заболевание, вызванное мутацией гена DSPP с известной локализацией (4q21.3). Основными признаками этого заболевания являются: поражение молочных и постоянных зубов. Янтарно-опалесцирующая окраска молочных и постоянных зубов, повышенная их стираемость, дефекты эмали, облитерация пульповых камер постоянных зубов, рентгенопрозрачная периапикальная область.
Дентиногенез несовершенный, тип I. Заболевание обусловлено мутацией в дентиновом сиалофосфопротеиновом гене (DSPP). По своим клиническим проявлениям заболевание сходно с другими формами несовершенного дентиногенеза. Тип I отличает его ассоциация с симптомами несовершенного остеогенеза (патологическая
Таблица 1-14. Наследственные заболевания и синдромы, сопровождающиеся аномалиями цвета зубов
Синдром | Тип наследования | ? ОМИМ | Локус |
Синдром Элерса-Данло, тип VII, аутосомно-рецессивный | АР | 225410 | 5q23 |
Синдром Негели | АД | 161000 | 17qll.2-q21 |
Амело-онихо-гипогидротический синдром | АД | 104570 | |
Амелогенез несовершенный, тип Шидлса III | АД | 125500 | 4q21.3 |
Дентиногенез несовершенный, тип I | АД | 125490 | |
Метафизарная дисплазия с гипоплазией верхней челюсти и брахидактилией | АД | 156510 | |
Эпилепсия и желтые зубы, несовершенный амелогенез | АР | 226750 |
ломкость костей). Стоматологическими проявлениями являются: несовершенный, врожденный дентиногенез, коричневый цвет зубов (коричнево-голубой опалесцирующий оттенок), широкие, бульбообразные коронки, узкие корни зубов, корневые каналы узкие, склонные к облитерации, отсутствие пульповых камер. Повышенная стираемость зубов.
Метафизарная дисплазия с гипоплазией верхней челюсти и брахидактилией. Редкий аутосомно-доминантный синдром, описанный впервые в 1982 г. HALLFLet al. (1982). Характерными признаками являются скелетные аномалии (метафизарная дисплазия), низкий рост, гипоплазия верхней челюсти, короткий фильтр, тонкие губы, ранняя потеря зубов, желтый цвет эмали зубов и признаки дистрофии.
Негели синдром (Негели-Франческетти-Ядасона синдром).
Заболевание описано впервые в 1927 г. В 1954 г. Франческетти и Ядасон установили аутосомно-доминантный характер наследования заболевания. Локализация гена известна (17q11.2-q21). Для данного синдрома характерно поражение кожи в виде гипогидроза, сглаженного дерматоглифического рисунка, гиперпигментация периокулярная, периоральная, гиперпигментация груди, шеи, живота. При синдроме Негели патология зубов выражается в изменении их цвета (желтые зубы), сверхкоплектные зубы, аномальная форма зубных коронок, преждевременная потеря зубов, кариес.
РАЗДЕЛ 6 АНОМАЛИИ ПРОРЕЗЫВАНИЯ ЗУБОВ
Прорезывание зубов у человека происходит в определенной последовательности, причем установлены средние сроки прорезывания каждого зуба с учетом их небольших естественных отклонений в ту или другую сторону. Лишь резкие отклонения от естественных сроков прорезывания зубов рассматримают как аномалию. В табл. 1.15 представлена хронология развития и сроки прорезывания временных и постоянных зубов человека.
Первыми в 6-10 мес прорезываются нижние центральные резцы, затем нижние боковые (10-16 мес). Верхние резцы появляются в 8-13 мес; первые моляры - 12-16 мес; клыки - 16-20 мес, вторые моляры - 23-30 мес.
Таблица 1-15. Временная динамика развития молочных зубов
Зубы | Инициация, внутриутробное развитие(нед) | Кальцификация, внутриутробное развитие (нед) | Формирование коронки (мес) | Прорезывание (мес) | Смена (лет) |
Нижний центральный резец | 6-7 | 13-16 | 1-3 | 6-10 | 5-6 |
Нижний латеральный резец | 7 | 14-16 | 2-3 | 10-16 | 6-7 |
Верхние резцы | 7 | 14-16 | 2-3 | 8-13 | 6-7 |
Клыки | 7,5 | 15-18 | 9 | 16-20 | 9-10 |
1-моляры | 8 | 14,5-17 | 6 | 12-16 | 10-11 |
2-моляры | 10 | 16-23 | 10-12 | 23-30 | 12 |
Из аномалий развития наиболее часто встречается задержка прорезывания зубов, или ретенция. Помимо генетических факторов, причиной задержки прорезывания зубов могут являтся различные хронические заболевания, эндокринная патология, неполноценное питание (авитаминоз) и др.
Реже встречается другая аномалия - раннее прорезывание зубов. При такой аномалии дети уже рождаются с зубами или они прорезываются вскоре после рождения. Такие зубы называются натальными или неонатальными. Натальные и неонатальные зубы затрудняют грудное вскармливание и, как правило, подлежат удалению.
Развитие временных и постоянных зубов протекает однотипно, но в разные возрастные периоды. В период, когда временные зубы завершают период своего развития в челюстях, уже имеются закладки постоянных зубов. В ходе прорезывания постоянных зубов происходит выпадение временных. Выпадение временных зубов обычно протекает симметрично на правой и левой сторонах каждой челюсти; у девочек этот процесс происходит раньше, чем у мальчиков. За исключением вторых моляров, зубы нижней челюсти выпадают раньше, чем соответстующие им зубы верхней челюсти.
Исследования, проведенные на монозиготных близнецах, показали, что процесс выпадения временных зубов на 80% определяется генетическими факторами.
Хронология и периоды формирования постоянных зубов представлена в табл. 1.16.
Преждевременное прорезывание зубов является достаточно частым симптомом при многих наследственных моногенных заболеваниях. В настоящее время, по данным каталога МакКюсика, это состояние встречается примерно при 70 формах патологии. Известны заболевания с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным типом наследования и симптомами раннего прорезывания зубов. Поскольку этот признак не является ведущим в клинической картине многих наследственных заболеваний и не всегда учитывается при клиническом описании, можно предположить, что на самом деле количество наследственных состояний, при которых наблюдается преждевременное прорезывание зубов значительно больше. Практически всегда преждевременное прорезывание зубов сопровождается другими аномалиями зубов и пороками развития, затрагивающими многие органы и системы.
Таблица 1-16. Временная динамика развития постоянных зубов
Зубы | Инициация | Начало кальцификации | Формирование коронки (лет) | Прорезывание (лет) |
Нижняя челюсть | ||||
Центральный резец | 20-22 нед ВУ* | 3-4 мес | 4-5 | 6-7 |
Латеральный резец | 21-22 нед ВУ | 3-4 мес | 4-5 | 7-8 |
Клык | 20-26 нед ВУ | 5-5 мес | 6-7 | 9-11 |
Первый премоляр | 38-42 нед ВУ | 1,75-2 года | 5-6 | 10-12 |
Второй премоляр | 7,5-8 мес | 2,25-2,5 лет | 6-7 | 11-12 |
Первый моляр | 15-17 нед ВУ | с рождения | 2,5-3 | 6-7 |
Второй моляр | 8,5-9 | 2,5-3 года | 7-8 | 11-13 |
Третий моляр | 3,5-4 года | 8-10 лет | 12-16 | 17-25 |
Верхняя челюсть | ||||
Центральный резец | 20-22 нед ВУ | 3-4 мес | 4-5 | 7-8 |
Латеральный резец | 21-22 нед ВУ | 3-4 мес | 4-5 | 8-9 |
Клык | 21-26 нед ВУ | 4-5 мес | 6-7 | 11-12 |
Первый премоляр | 38-42 нед ВУ | 1,25-1,75 лет | 5-6 | 10-11 |
Второй премоляр | 7,25-8 мес | 2-2,5 лет | 6-7 | 10-12 |
Первый моляр | 3,5-4 нед ВУ | с рождения | 2,5-3 | 6-7 |
Второй моляр | 8,5-9 мес | 2,5-3 лет | 7-8 | 12-13 |
Третий моляр | 3,5-4 лет | 7-9 лет | 12-16 | 17-25 |
Примечание. * ВУ - внутриутробное развитие.
К аномалиям прорезывания зубов относятся:
• натальные (прорезавшиеся к моменту рождения) зубы;
• неонатальные (у новорожденного, прорезавшиеся преждевременно);
• преждевременное прорезывание (раннее прорезывание);
• задержка (персистентная) смены (первичных) (временных) зубов;
• позднее прорезывание;
• преждевременное выпадение первичных (временных) зубов.
6.1. НАТАЛЬНЫЕ, НЕОНАТАЛЬНЫЕ ЗУБЫ
Впервые как доминантное состояние описано в 1893 г. (Limrick, 1893). По данным разных авторов, частота натальных/неонатальных зубов у новорожденных 1:1000-3000. По имеющимся данным, некоторые известные исторические личности рождались уже с зубами (король Франции Луи XIV, Ричард III, Ганнибал, Ришелье и др.).
6.1.1. Наследственные заболевания и синдромы с натальными/неонатальными зубами
В табл. 1.17 приведен список некоторых известных в настоящее время моногенных заболеваний, сопровождающихся наличием натальных/неонатальных зубов.
Зубы, имеющиеся при рождении. Аутосомно-доминантное состояние, когда прослеживается характерное наследование признака без дополнительных аномалий развития.
Стеатоцитома множественная с натальными зубами. Кинг и Ли в 1987 г. описали китайскую семью, где в 5 поколениях по меньшей мере у 21 человека отмечались множественные стеатоцитомы в сочетании с неонатальными зубами.
Эктодермальная дисплазия с неонатальными зубами. В 1994 г. Turnpenny et al. (1994) описали шотландскую семью с доминантно наследуемой эктодермальной дисплазией и признаками поражения зубов в виде олигодентии, хотя некоторые пораженные имели неонатальные зубы.
Пахионихия врожденная, тип 1. Онихогриппоз, гиперкератоз, ороговение участков кожи, гипергидроз ладоней и подошв, стеатоцитома. У больных отмечается умственная отсталость различной сте-
Таблица 1-17. Синдромы с натальными/неонатальными зубами
Синдром | Тип наследования | ? ОМИМ | Локус |
Зубы, имеющиеся с рождения | АД | 187050 | |
Стеатоцитома множественная, с натальными зубами | АД | 184510 | |
Пахионихия врожденная, тип 1 | АД | 167200 | 17ql2-q21,12ql3 |
Пахионихия врожденная, тип 2 | АД | 167210 | 17ql2-q21,12ql3 |
Эллиса-ван-Кревельда синдром | АР | 225500 | 4р16, 4р16 |
Одонто-трихо-ногте-пальце-ладонный синдром | АД | 601957 | - |
Гиперкератоз контрактуры синдром | АР | 275210 | 1р34 |
Синдром Паллистера-Холла | АД | 146510 | 7р13 |
Врожденные контрактуры, кривошея и злокачественная гипертермия | АР | 217150 | - |
Синдром Меккеля тип 1 | АР | 249000 | 17р23 |
пени выраженности, аномалия волос в виде их истончения, алопеция и др., натальные/неонатальные зубы.
Пахионихия врожденная, тип 2. По своим клиническим проявлениям очень похожа на синдром врожденной пахионихии тип 1. Отличием является отсутствие очагов лейкоплакии.
Эллиса-ван-Кревельда синдром. Ген заболевания локализован на 4-й хромосоме в районе 4р16. Предположительно заболевание связано с мутацией в гене EVC. Заболевание с аутосомно-рецессивным типом наследования. Характеризуется скелетными дисплазиями, укорочением конечностей, постаксиальной полидактилией, дисплазией ногтей и зубов. Иногда (60%) встречаются сердечно-сосудистые аномалии. Из неврологических проявлений отмечается умственная отсталость и аномалия Денди-Уокера. Среди стоматологических аномалий: неонатальные зубы, задержка прорезывания зубов, частичная олигодентия.
Одонто-трихо-ногте-дигитально-ладонный синдром. Mendoza and Valiente (1997) описали предположительно «новый» доминантный синдром эктодермальной дисплазии у двух братьев и их матери. Авторы наблюдали у этих больных натальные зубы, дистрофию волос, патологию пальцев и ладоней, отметили в фенотипе толстые губы, неонатальные зубы, маллоклюзию зубов, аномалии развития кистей и стоп (метакарпальная дисплазия, брахидактилия и др.), дистрофические изменения ногтей и волос.
Гиперкератоз контрактуры синдром. Аутосомно-рецессивное заболевание. Локализация гена известна (1p34). Полагают, что заболевание обусловлено мутацией в генах LMNA или ZMPSTE24. Для данного заболевания характерно наличие неонатальных зубов, внутриутробное отставание массы тела, микрогнатия, расщелина губы/ нёба, низко посаженные уши, гипертелоризм, эктропион, атрезия хоан, пороки развития сердечно-сосудистой и мочеполовой системы, скелета, гипоплазия легких, поражение кожи в виде утолщения, гиперкератоза и др.
Паллистера-Холла синдром. Аутосомно-доминантное заболевание, вызванное мутацией гена GLI3. Ген расположен в 7p13. Большинство случаев спорадические. Для данного наследственного заболевания характерно наличие неонатальных зубов. Множественные аномалии развития с поражением ушей (микротия, отсутстве наружного слухового прохода и др.), глаз (микрофтальмия). В ротовой полости отмечаются уздечки слизистой щек, мик-
роглоссия, расщелина губы/нёба. Пороки сердечно-сосудистой системы, скелета, желудочно-кишечного тракта (неперфорированный анус), мочевой системы (дисплазия и эктопия почек). Пороки развития кистей и стоп (полидактилия, синдактилия, олигодактилия).
Врожденные контрактуры . кривошея и злокачественная гипертермия. Редкое наследственное заболевание с аутосомно-рецессивным типом наследования. Проявляется с рождения в виде артрогриппоза, расщелин нёба, наличия зубов при рождении. Для данного заболевания характерным является злокачественная гипертермия в ответ на анастезию. Этот признак меет большое значение в клинической практике. При использовании анестетиков во время стоматологического лечения не исключены осложнения с тяжелыми последствиями.
Мекеля синдром, тип 1. Данный наследственный синдром (синоним синдром Грубера, Мекеля-Грубера) - аутосомно-рецессивное заболевание. Локализация гена МКS1в районе 17q23. Это широко варьирующее по клиническим проявлениям заболевание включает в себя почечную патологию с пороками развития центральной нервной системы (энцефалоцеле), патологию печени и полидактилию. Характерны микроцефалия, микрогнатия, микрофтальмия, гипо- и гипертелоризм. К стоматологическим признакам относятся: макростомия, расщелина губы/нёба, натальные зубы. Пороки развития внутренних органов (сердца, печени, селезенки). Пороки развития скелета, половых органов и нервной системы (Арнольда-Киари аномалия, Денди-Уокера аномалия, аплазия мозжечка и др.).
РАЗДЕЛ 7 ЗАДЕРЖКА ПРОРЕЗЫВАНИЯ ЗУБОВ
Эта аномалия наблюдается в отношении как временных, так и постоянных зубов и встречается в популяции с частотой (1,5%). В большинстве случаев задержка прорезывания связана с неправильным положением зачатков зубов (ретенция). Чаще всего наблюдается в отношении нижних третьих моляров и верхних клыков. Наряду с генетическими факторами причинами ретенции могут являться разнообразные заболевания (рахит, туберкулез, болезни эндокринной системы, аномалии расположения зачатков зубов, аномалии развития челюстей и др.). Эта аномалия является характерной для многих наследственных заболеваний хромосомной и моногенной этиологии.
В табл. 1.18 представлены данные о некоторых моногенных заболеваниях и синдромах с задержкой прорезывания зубов.
Зубы непрорезавшиеся, с гипоплазией верхней челюсти и вальгусной деформацией коленей. Аутосомно-рецессивное заболевание, характеризующееся аномалиями развития черепа (гипоплазия верхней челюсти и недоразвитие синусов верхней челюсти), деформация коленных суставов, ушных раковин. Зубы: задержка прорезывания временных и постоянных зубов, гипоплазия альвеолярной области.
Клейдокраниальная дисплазия. Это заболевание с аутосомнодоминантным типом наследования и с множественными аномалиями скелета, уже рассматривалось ранее в разделе, посвященном наличию сверхкомплектных зубов. Наряду со сверхкомплектными зубами это заболевание характеризуется также задержкой прорезывания временных и постоянных зубов и признаками гипоплазии эмали.
Пикнодизостоз. Аутосомно-рецессивное заболевание. Характеризуется низким ростом (взрослые меньше 150 см). Отмечаются микрогнатия, узкое нёбо, задержка прорезывания постоянных и молочных зубов, гиподентия, кариес, скелетные аномалии (аплазия, гипоплазия ключиц, остеосклероз, сколиоз, спондилолистез, брахидактилия, акроостеолиз), дистрофия ногтей (ногти плоские, бороздчатые).
Мелника-Нидлса синдром (Мелника-Нидлса остеодисплазия). Х-сцепленное доминантное заболевание, вызвано мутацией гена, кодирующего филамин А (FLNA). Локализация гена Xq28. Заболевание характеризуется задержкой моторного развития, аномалиями лицевой области, микрогнатией, выступающим лбом с низким ростом волос, большими ушами. Часто повторные отиты среднего уха, экзофтальм, гипертелоризм, страбизм. Характерны задержка прорезывания зубов, смещенные зубы. Пороки развития сердечно-сосудистой системы: легочная гипертензия, деформация грудной клетки, укорочение ключиц и лопаток. Скелетные аномалии, аномалии кистей и стоп. Явления акроостеолиза. Патология мочевой системы (гидронефроз, стеноз мочеточников). Нарушение походки.
Рутхерфурда синдром. Аутосомно-доминантное заболевание, сопровождающееся патологией глаз (дистрофия радужки) и признаками задержки прорезывания временных и постоянных зубов.
Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии кистей. Аутосомно-рецессивное заболевание, характеризующееся множественными аномалиями развития (низкий рост,
Таблица 1-18. Наследственные заболевания и синдромы с задержкой прорезывания зубов
Синдром | Тип наследования | ? ОМИМ | Локус |
Зубы непрорезавшиеся с гипоплазией верхней челюсти и вальгусной деформацией коленей | АР | 273050 | |
Клейдокраниальная дисплазия | АД | 119600 | 6р21 |
Пикнодизостоз | АР | 265800 | lq21 |
Синдром Мельника-Нидлса | Х-сц. | 309350 | Xq28 |
Синдром Рутхерфурда | АД | 180900 | |
Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии кистей | АР | 264475 | |
Фиброматоз десен с характерным лицом | АР | 228560 | |
SHORT-синдром | АР | 269880 | |
Эллис-ван-Кревельда синдром | АР | 225500 | 4р16,4р16 |
Витамин D-зависимый рахит, тип II | АР | 277440 | 12ql2-ql4 |
Синдром Коккейна, тип А | АР | 2161400 | 5ql2 |
Синдром Коккейна, тип В | АД | 133540 | lOqll |
Брахидактилия,тип В1 | АД | 113000 | 9q22 |
Лакримоаурикулодентодигитальный синдром | АД | 149730 | 5pl3-pl2,4pl6.3 |
Синдром Дубовица | АР | 223370 | |
Синдром Ротмунда-Томсона | АР | 268400 | 8q24.3 |
Окончание табл. 1-18
Витамин D-зависимый рахит, тип 1 | АР | 264700 | 12ql4, 12ql3.1- ql3.13 |
Синдром ДЖАПО | АР | 230740 | |
Синдром МОМО | АД | 157980 | |
Синдром Винчестера | АР | 277950 | 16ql3 |
Краниометафизарная дисплазия аутосомно-рецессивная | АР | 218400 | 6q21-q22 |
Гемифациальная атрофия прогрессирующая | АД | 141300 | |
Недостаточная адгезия лейкоцитов, тип I | АД | 116920 | 21q22.3 |
Эктодермальная дисплазия с цистами надпочечников | АД | 129550 | |
Фокальная дермальная гипоплазия | Х-сц. | 305600 | |
Синдром камптодактилии | АР | 211910 | |
Наследственная остеодистрофия Альбрехта | АД | 103580 | 20ql3.2 |
Акродизостоз | АД | 101800 | |
Синдром Аперта | АД | 101200 | 10q26 |
Синдром Сотоса | АД | 117550 | 5q35 |
микрогнатия, низкопосаженные уши, атрезия наружного слухового прохода, гипотелоризм, эпикант, псевдопаппилодема, блефарофимоз, высокое узкое нёбо, патология скелета - кифосколиоз, деформации пальцев рук и ног). Для данного заболевания также характерны множественные аномалии развития зубов (неправильный прикус, темный оттенок зубной эмали, анодентия, иногда сверхкомплектные зубы, задержка прорезывания зубов).
Фиброматоз десен с характерным лицом. Синдром с аутосомнорецессивным типом наследования. Для данного синдрома характерно наличие макроцефалии, гипертелоризм, синофриз, гипоплазия крыльев носа, фиброматоз десен, задержка прорезывания и отсутствие некоторых временных зубов, задержка прорезывания постоянных зубов.
Коккейна синдром, тип А. Тяжелое аутосомно-рецессивное заболевание, вызванное гомозиготным состоянием по мутации гена ERCC8. Ген локализован в 5q12. Синдром Коккейна сопровождается нарушениями со стороны центральной и переферической нервной системы (умственная отсталость, деменция, кальцификация базальных ганглиев, дистрофия структур головного мозга, судороги, дисмиелонизация, атаксия, тремор, периферическая нейропатия) и эндокринной систем (гипогонадизм). Патология органа зрения и слуха: пигментная ретинопатия, атрофия зрительных нервов, страбизм, нистагм, катаракта, микрофтальмия, гипоплазия радужки, микрокорнеа, нейросенсорная глухота. Аномалии зубов: кариес, задержка прорезывания, малокклюзия, отсутствие/гипоплазия зубов.
Лакримо-ауриколо-денто-дигитальный синдром. Аутосомнодоминантный синдром. Ген локализован в 5p13-p12,4p16.3. Как следует из его названия, основными составляющими этого синдрома являются: патология слезной системы (алакримия, непроходмость слезно-носового канала, аплазия/гипоплазия слезной точки, апла- зия/гипоплазия слезных желез, дакриоциститы), патология слуха (смешанная кондуктивная и нейросенсорная тугоухость), патология зубов (гиподентия, коническая форма зубной коронки, гипоплазия эмали, задержка прорезывания временных зубов).
Гиперплазия гипофиза, инсулин-резистентный сахарный диабет и соматические аномалии. Аутосомно-рецессивное заболевание, обусловленное гомозиготным состоянием мутации в гене инсулинового рецептора (INSR), для которого характерна патология эндокринной системы (сахарный диабет, диабетический кетоацидоз),
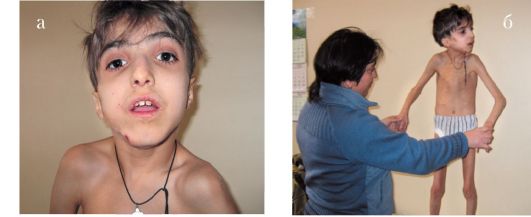 Рис. 1.25 а, б. Синдром Коккейна
Рис. 1.25 а, б. Синдром Коккейна
увеличение гениталий, темная окраска кожи, гипертрихоз. При данной патологии встречаются признаки дисплазии зубов и задержка прорезывания временных зубов.
Брахидактилия, тип В1. Аутосомно-доминатное заболевание. Ген идентифицирован (ROR2). Клиническая картина полиморфна. Одно из редких доминантных заболеваний, когда описаны случаи гомозиготного состояния по доминтной мутации. Ведущей симптоматикой являются признаки поражения скелета: сколиоз, патология ребер, конечностей, кистей (гипоплазия/аплазия фаланг пальцев, гипопла- зия/отсутствие метакарпальных костей, симфалангизм, синдактилия, камптодактилия и др.) и стоп (гипоплазия/аплазия дистальных фаланг). Характерным признаком является задержка прорезывания постоянных зубов.
Дубовица синдром . Аутосомно-рецессивный синдром, для которого характерными признаками являются: низкий рост (в среднем 44 см) и вес (в среднем 2,3 кг) при рождении, с последующим отставанием в темпах развития. Микроцефалия, маленькое лицо, ассиметрия лица, микрогнатия, оттопыренные, низко расположенные уши. Глазные аномалии: телекант, птоз, блефарофимоз, страбизм, микрофтальмия, тапеторетинальная дегенерация сетчатки, гипоплазия и колобомы радужки. Патология скелета, кожи (экзема и др.). У 10-15% пациентов отмечается умственная отсталость. Иногда лимфомы, нейробластомы, апластическая анемия. Склонность к частым хроническим инфекционным заболеваниям, что связано с
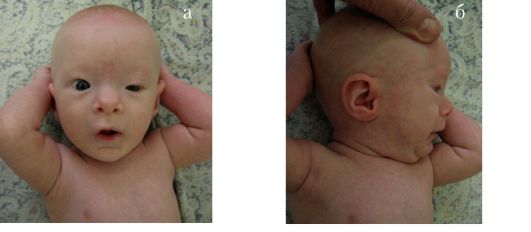 Рис. 1.26 а, б. Синдром Дубовица
Рис. 1.26 а, б. Синдром Дубовица
иммунологическими проблемами в виде гипогаммаглобулинемии и снижения уровня IgA. Отмечаются задержка прорезывания зубов, неправильный их рост, кариес.
Мукополисахаридоз, тип II. Х-сцепленное рецессивное заболевание, обусловленое мутацией в идуронат-2-сульфатазном гене. Ген локализован в районе Xq28. Для данного заболевания характерными признаками являются: низкий рост (взрослые 120-150 см), макроцефалия, снижение слуха, склонность к хроническим отитам, пигментный ретинит, птоз, макроглоссия, гепатомегалия, пупочные и паховые грыжи, кифоз, гипертрихоз, гидроцефалия, иногда судорожные припадки. Зубы: широко расставленные, поздно прорезывающиеся.
Краниометафизарная дисплазия, аутосомно-рецессивная. Макроцефалия, тугоухость, атрофия зрительных нервов, множественные скелетные аномалии (гиперостоз швов черепа, небольшая нижняя челюсть, гипероостоз лицевых костей черепа, облитерация паранозальных синусов и мастоидиты, деформация конечностей), склероз метакарпальных костей и фаланг. Зубы: задержка прорезывания постоянных зубов.
Гемифациальная атрофия прогрессирующая. Семейных случаев не описано. Все известные случаи являются изолированными. Характеризуется односторонним поражением лица, с липоатрофией, мышечной атрофией на пораженной стороне лица, одностороннее поражение органа зрения и слуха. Зубы: задержка прорезывания на одной стороне, нарушение прикуса. Чаще в процесс вовлечена левая половина лица.
Амело-онихо-гипо-гидротический синдром. Аутосомно-доминантное заболевание. Основными признаками синдрома являются: поражение кожи (себорейный дерматит, гипогидроз, ксероз), поражение ногтей (онихолизис) и зубов (гипокальцифицированная-гипопластичная эмаль), желто-коричневый цвет зубов, задержка прорезывания постоянных зубов.
SHORT-синдром. Название синдрома - акроним английских слов, описывающих основные клинические проявления заболевания (Short stature - низкий рост, Hyperextesibility - гиперподвижность суставов/грыжи, Ocular depression - снижение остроты зрения, Rieger anomaly - аномалия Ригера, Teething delay - прорезывание зубов). Аутосомно-доминантноезаболеваниесвышеперечисленнымисимптомами, липоатрофией лица, нормальным интеллектом, инсулин-резистентным диабетом, гипергликемией, нейросенсорной тугоухостью. Патология глаз: миопия, катаракта, глаукома, мегалокорнеа, телекант. Скелетные аномалии: гипермобильность суставов. Отставание костного возраста, большие эпифизы, тонкие диафизы, клинодактилия пальцев рук. Разнообразные варианты патологии зубов: гиподентия, нарушение прикуса, задержка прорезывания зубов.
Рахит витамин D-зависимый, тип II. Аутосомно-рецессивное заболевание, обусловленное мутацией витамин D рецепторном гене (VDR). Заболевание с ранним началом (начинается в первые 6 мес. жизни). Некоторые пациенты могут быть вылечены с помощью больших доз витамина D и кальция. Характерными признаками являются: отставание в росте, признаки поражения скелета, ребер «четки». В крови снижение паратиреоидного гормона. Задержка прорезывания зубов, гипоплазия эмали, ранний кариес (с 2 года жизни).
Витамин D-зависимый рахит, тип I. Заболевание вызвано мутацией в 25-гидроксивитамин D-3-1-альфа - гидроксилазном гене (CYP27B1). Тип наследования: аутосомно-рецессивный. Характерная клиническая картина формируется на втором году жизни. Помимо типичных фенотипических проявлений, лабораторным подтверждением заболевания является определение в сыворотке крови уровня 1,25-дигидроксивитамина D-3. диагностическими лабораторными показателями являются также гипокальцемия, гипофосфатемия, снижение уровня паратиреоидных гормонов, генерализованная аминоацидурия. Частота этого наследственного заболевания очень высокая (носительство гена 1 на 26) среди канадских французов, проживающих в одном из округов Квебека. Хороший терапевтический
эффект оказывает применение 1,25-дигидроксивитамина D-3 или 1-альфагидроксивитамина D-3.
DGAPO-синдром. Название синдрома - акроним английских обозначений ведущих клинических признаков (Growth retardation - отставание в росте, Alopecia -алопеция, Pseudoanodontia - псевдоанадентия, Optic atrophy - атрофия зрительных нервов). При данном заболевании псевдоадентия сочетается с задержкой прорезывания зубов.
Эктодермальная дисплазия с кистами надпочечников. Аплазии участков кожи, гипогидроз, ониходисплазия. Эндокринопатия: кисты надпочечников, алактия. Гипоплазия молочных желез и сосков. Зубные аномалии: задержка прорезывания зубов, малые аномалии развития зубов.
Паллистера-Киллиана синдром. Этиология заболевания: мозаицизм соматических клеток. При лабораторном исследовании обнаруживается мозаицизм (тетрасомия 12р) в фибробластах кожи. При исследовании лимфоцитов обнаруживаются изохромосомные варианты. Тяжелая патология, вызывающая часто внутриутробную гибель плода. Множественные аномалии развития лица, ушей, глаз, полости рта (макроглоссия, расщелина нёба и др.), сердечно-сосудистой системы, центральной нервной системы, скелета. Стоматологические проблемы выражаются в задержке прорезывания зубов.
Миллера-Дикера лиссэнцефалия синдром. Аутосомно-доминанное заболевание, вызванное мутацией в лиссэнцефальном гене 1 (LIS1). При лабораторном исследовании обнаруживается микроделеция хромосомы 17 (район р13.3). Основным способом лабораторной диагностики является флюоресцентная гибридизация in situ критического района хромосомы 17. Для заболевания характерными признаками являются: интра- и постнатальная задержка развития, тяжелая неврологическая сиптоматика (пороки развития мозга: агирия, отсутствие/гипоплазия мозолистого тела, кальцификаты в головном мозгу, ранняя гипотония и позже мышечный гипертонус, отставание в моторном и психическом развитии, умственная отсталость, прогрессирующая спастическая параплегия, судороги и др.). Наряду с неврологической сиптоматикой отмечаются множественные аномалии кистей (полидактилия, клинодактилия, камптодактилия и др.). Характерна задержка прорезывания зубов.
Винчестера синдром. Аутосомно-рецессивное заболевание, вызвано мутацией в матриксе металлопротеиназы-2 гене (ММР2).
Проявляется в раннем возрасте. Признаки заболевания: низкий рост, гипертрофия десен, контрактуры суставов, артралгии, признаки остеопороза, кифосколиоз, компрессия тел позвонков, патология кистей (остеолизис карпальных костей), стоп (остеолизиз тарсальных костей, анкилоз мелких суставов), гипертрихоз. Задержка прорезывания зубов.
Камптодактилии синдром, Гвадалахара тип 1. Аутосомно-рецессивное заболевание, интра- и постнатальное отставание в весе и росте. Микроцефалия, маленькие, низко посаженные, гипопластичные уши. Телекант, микрокорнеа, синофриз, гипертелоризм. Длинная шея. Задержка костного возраста и другие аномалии скелета (деформация грудной клетки, конечностей, брахидактилия, камтодактилия 2-5 пальцев), пигментные невусы, судорожные припадки. К аномалиям развития зубов относятся: олигодентия, шиповидная форма зубной коронки, задержка прорезывания зубов.
МОМО-синдром (макростомия, ожирение, микроцефалия, и аномалии органа зрения). Редкий наследственный синдром, вероятнее всего, обусловленный мутацией de novo. Предположительный тип наследования: аутосомно-доминантный. Для данного синдрома характерны большой вес и рост при рождении, ожирение, макрокрания, аномалии органа зрения (колобома сетчатки, нистагм), умственная отсталость. Аномалии зубов в виде нарушения прикуса, тауродонтизма, задержки прорезывания зубов.
Сотоса синдром. Точный тип наследования не установлен. Практически все случаи изолированные (известно только несколько семейных случаев). Характеризуется увеличенной длиной и весом тела при рождении (средняя длина 55,2 см, вес 3900 гр). Во взрослом состоянии вес и длина тела чаще нормальные (рост мужчины в среднем 184,3 см, женщины - 172,2 см). Макроцефалия, долихоцефалия. Нарушение слуха (кондуктивная тугоухость), патология глаз (нистагм, страбизм, гиперопия), высокое арковидное нёбо. Встречается патология сердечно-сосудистой системы, скелета (разболтанность суставов, вальгусная деформация, большие кисти и стопы). Патология центральной нервной системы (задержка развития, вариабельная степень умственной отсталости, неонатальная гипотония, гиперрефлексия, судороги, поведенческие проблемы, выраженная задержка развития речи, частичная или полная агенезия мозолистого тела, расширенные желудочки головного мозга). Задержка прорезывания временных зубов.
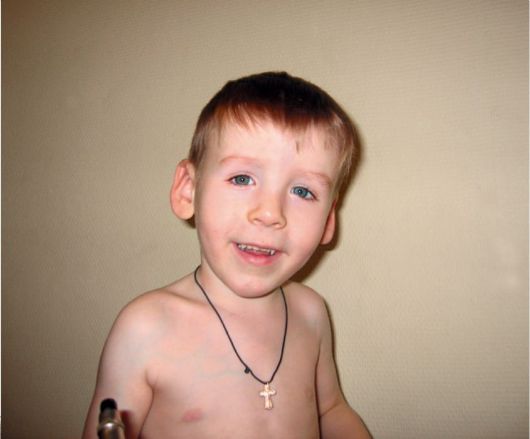 Рис. 1.27. Синдром Сотоса
Рис. 1.27. Синдром Сотоса
Наследственная остеодистрофия Альбрехта. Это гетерогенное по своим проявлениям наследственное аутосомно-доминантное заболевание характеризуется низким ростом, ожирением, катарактой, брахидактилией, умственной отсталостью. Отмечаются гипофосфатемия, гипокальцемия, различные эндокринные аномалии. К стоматологическим признакам относятся: врожденная гипоплазия эмали, задержка прорезывания зубов, ана-/олигодентия. Ген заболевания локализован в районе 20q13.2.
Акродизостоз. Аутосомно-доминантное заболевание. Низкий рост, укорочение конечностей за счет дистальных отделов. Брахицефалия, гипоплазия верхней челюсти, прогнатизм. Запавшая переносица, короткий маленький нос с открытыми вперед ноздрями, снижение слуха, гипертелоризм, страбизм, голубой цвет радужек, множественные скелетные нарушения: деформации плечевой, лучевой, локтевой костей. Кисти широкие, с укороченными пальцами. Иногда гипогинетализм, гипогонадизм. В 90% умственная отсталость (коэффициент интеллекта 24-85). Задержка прорезывания зубов.
Апера синдром. Аутосомно-доминантное заболевание, вызванное мутацией в гене рецепторе фактора роста фибробластов-2 (FGFR2).
Нормальные длина и вес при рождении. Характерные аномалии черепно-лицевой области (краниосиностоз, акробрахицефалия, большой поздно закрывающийся родничок, высокий лоб, прогнатизм, гипертелоризм, опущенные вниз глазные щели, атрезия, стеноз хоан, узкое нёбо, иногда расщелина нёба). Аномалии развития скелета, аномалии почек, патология центральной нервной системы (различная степень умственной отсталости, агенезия мозолистого тела, вентрикуломегалия, аномалии лимбической системы). Зубные аномалии: аномальный прикус, задержка прорезывания зубов.
 Рис. 1.28 а, б. Синдром Апера
Рис. 1.28 а, б. Синдром Апера
РАЗДЕЛ 8
НАСЛЕДСТВЕННЫЕ АНОМАЛИИ НАРУШЕНИЯ
ПРИКУСА
Нарушение прикуса может наблюдаться как изолированная аномалия, являться следствием мультифакториальной патологии или внешнесредовых воздействий, либо входить в состав некоторых наследственных заболеваний и синдромов. Как отдельный фенотипический признак аномалии прикуса встречаются в популяции с высокой частотой (табл. 1-19).
Таблица 1-19. Популяционные частоты аномалий прикуса (Дистель В.А., 2001)
Аномалии прикуса | Частота% |
Прогнатия | 10,0 |
Прогения | 3,2 |
Глубокий | 10,7 |
Косой | 3,2 |
Наследственные заболевания и синдромы с нарушением прикуса представлены в табл. 1-20.
В настоящее время как составляющая часть наследственного моногенного синдрома нарушения прикуса фигурируют более чем при 10 формах аутосомно-рецессивных заболеваний, при 12 формах заболеваний с аутосомно-доминантным типом наследования и при 5 формах Х-сцепленных заболеваний.
8.1. АУТОСОМНО-ДОМИНАНТНЫЕ СИНДРОМЫ
С НАРУШЕНИЕМ ПРИКУСА
Нарушение прикуса из-за выступающих вперед верхних передних зубов . Заболевание описано у 19 человек в 8 ядерных семьях у трех поколений. Наблюдается нарушение прикуса, выступающие вперед верхние передние зубы. Тип наследования: аутосомно-доминантный.
Максилло-назальная дисплазия тип Биндера (Биндера синдром). Синдром описан как самостоятельная нозологическая единица в 1962 г. Binder (1962) описал заболевание как аплазия верхней челюсти, короткий нос с широкой переносицей, аномалия прикуса. Характерной чертой синдрома является гипоплазия срединной части лица. За 20-летний период в Мельбурне (Австралия) было зарегитрировано более 100 пациентов с этим заболеванием. Иногда отмечается гипоплазия терминальных фаланг пальцев, искривление и расщепление ребер. Тип наследования: предположительно аутосомно-доминантный, но не исключена мультифакториальная природа заболевания.
Таблица 1-20. Наследственные заболевания и синдромы с нарушением прикуса
Синдром | Тип наследования | ? ОМИМ | Локус |
Нарушение прикуса из-за выступающих вперед верхних передних зубов | АД | 154300 | |
Нарушение прикуса и низкий рост | АР | 248350 | |
Максилло-назальная дисплазия тип Биндера (синдром Биндера) | АД | 155050 | |
Миастенический синдром врожденный, ассоциированный с недостаточностью ацетилхолинового рецептора | АР | 608931 | 17р12-р11, 17р13-р12, 11р11.2-р11.1 |
Эктопия хрусталика, краниофациальный дизморфизм | АР | 601552 | |
Аурикуло-кондилярный (мыщелковый) синдром | АД | 602483 | |
Синдром Гаррода | АР | 601095 | |
Остеопетроз с почечным канальцевым ацидозом | АР | 259730 | 8q22 |
Синдром камптодактилии, тип I Гвадалахара | АР | 211910 | |
Синдром Секкеля 1 | АР | 210600 | 3q22-q24 |
Гипоспадия, гипертелоризм, колобома верхнего века и смешанный тип тугоухости | АР | 603463 | |
Одонто-трихо-ногте-пальце-ладонный синдром (OTUDP- синдром) | АД | 601957 | |
Симпсона-Голаби-Бехмеля синдром, тип 1 (SGBS-синдром) | Хсц.р | 312870 | Xq26 |
Фронтометафизарная дисплазия | Хсц.р | 305620 | Хр22.2-р22.1 |
Продолжение табл. 1-20
Фокальная дермальная дисплазия | Хсц.р | 305600 | |
Синдром Коффина-Лоури | Х-сц.д | 303600 | Хр22.2-р22.1 |
Микрофтальмия синдромальная 2 (окуло-фацио-кардиодентальный синдром) | Х-сц.д | 300166 | Хр11.4 |
Синдром SHORT | АД | 269880 | |
Склеростеоз (кортикальный гиперостеоз с синдактилией) | АР | 269500 | 17ql2-q21 |
Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии кистей (акроотоокулярный синдром) | АР | 264475 | |
Умственная отсталость, Буэнос-Айрес тип (Мутчник, синдром) | АР | 249630 | |
Франк-Тер-Хаара синдром (синдром Мелника-Нидлса) | АР | 249420 | |
Синдром Халлерманна-Штайфа | АР | 234100 | |
Синдром Горлина-Чаудри-Мосса | АР | 233500 | |
Фациодинитогенитальный синдром, рецессивный | АР | 227330 | |
Синдром Коккейна, тип А | АР | 216400 | |
Трихо-рино-фалангеальный синдром | АД | 190350 | |
Синдром Шпринтзен-Гольдберг краниосиностоз (краниосиностоз с арахнодактилией и грыжами) | АД | 182212 | 15q21.1 |
Синдром Нунан 1 | АД | 163950 | 12q24.1 |
Окончание табл. 1-20
Синдром МОМО | |||
Гипоспадия, гипертелоризм, колобома верхнего века и смешанный тип тугоухости | АР | 603463 | |
Синдром Коккейна, тип Б | АД | 135540 | lOqll |
Акроостеолиз с остеопорозом и аномалиями черепа и нижней челюсти | АД | 102500 | |
Акродизостоз | АД | 101800 | |
Синдром Апера | АД | 101200 | 10q26 |
Аурикуло-кондилярный (мыщелковый) синдром. Uuspaa (1978) сообщил о заболевании у матери и двух ее сыновей. Патология выражалась в пороках развития наружного уха и гипоплазии нижней челюсти. В дальнейшем появилась информация о клинической вариабельности данного синдрома (без существенного нарушения органа слуха). У 25% пациентов отмечается макроцефалия. Практически всегда микрогнатия, микростомия (52%), глоссоптоз (46%), аномалия нёба (63%), аномалии зубной коронки, нарушение прикуса. Изза орофациальных аномалий: нарушение процесса дыхания, апноэ. Заболевание наследуется по аутосомно-доминатному типу.
Одонто-трихо-ногте-пальце-ладонный синдром (OTUDP-син- дром). Тип наследования: аутосомно-доминантный. Как «новый» вариант эктодермальной дисплазии предствлен Mendoza and Valiente (1997). Заболевание было описано у двух братьев их матери и у 18 родственников в пяти поколениях. Авторы наблюдали натальные (при рождении) зубы, дистрофию волос, нарушение прорезывания постоянных зубов, дистрофия ногтей, деформация пальцев, брахидактилию, укорочение метакарпальных и метафизарный костей. Прогнатизм, нарушение прикуса.
SHORT-синдром. Название синдрома - акроним английских слов, описывающих основные клинические проявления заболевания (Short stature - низкий рост, Hyperextesibility - гиперподвижность суставов/грыжи, Ocular depression - снижение остроты зрения, Rieger anomaly - аномалия Ригера, Teething delay - прорезывание зубов). Аутосомно-доминантное заболевание с вышеперечисленными симптомами, липоатрофией лица, нормальным интеллектом, инсулин-резистентным диабетом, гипергликемией, нейросенсорной тугоухостью. Патология глаз: миопия, катаракта, глаукома, мегалокорнеа, телекант. Скелетные аномалии: гипермобильность суставов. Отставание костного возраста, большие эпифизы, тонкие диафизы, клинодактилия пальцев рук. Разнообразные варианты патологии зубов: гиподентия, нарушение прикуса, задержка прорезывания зубов.
Шпринтзен-Гольдберга краниосиностоз (краниосиностоз с арахнодактилией и грыжами). Заболевание вызвано мутацией в фибриллиновом 1 гене FBN1, локализованном в 15q21.1, наблюдается как «новый» наследственный синдром, описано в 1982 г. Shprintzen and Goldberg. Наряду с проявлениями краниосиностоза (микроцефалия, долихоцефалия) отмечаются экзофтальм, телекант, гипертелоризм,
страбизм, миопия, птоз, гипоплазия верхней и нижней челюсти, гипертрофия мягких тканей, низко посаженные уши, множественные грыжи, арахнодактилия, камптодактилия. В детстве: гипотония, отставание в развитии, умственная отсталость, псевдорасщелина нёба. Нарушение прикуса.
Нунан 1 синдром. Заболевание связывают с наличием мутации в 11 гене протеин-тирозин-фосфотаза нерецепторный тип (PTPN11), локализованном в 12.q24.1. Частота заболевания 1:1000-2500 новорожденных. Тип наследования: аутосомно-доминантный. Для данного заболевания характерными признаками являются: гипертелоризм, опущенные вниз наружные углы глаз, низкий рост, короткая шея, широкая плоская шея и грудная клетка, аномалии сердечнососудистой системы, снижение слуха, моторная неловкость, аномалии пальцев, гипогонадизм, крипторхизм и др. К патологии рта и зубов относятся: высокое, арковидное нёбо, микрогнатия, нарушение прикуса.
Акроостеолиз с остеопорозом и аномалиями черепа и нижней челюсти. Аутосомно-доминантный тип наследования. Низкий рост, низко посаженные уши, кондуктивная тугоухость, синофриз, эпикант, телекант, широкий нос, короткая шея, пупочные грыжи, гипоспадия, крипторхизм. Остеопения, патологические переломы, маленькая нижняя челюсть. Гирсутизм, акроостеолизис на кистях и стопах, гидроцефалия. Ранняя потеря зубов, нарушение прикуса.
 Рис. 1.29 а, б. Синдром Нунан
Рис. 1.29 а, б. Синдром Нунан
Трихо-рино-фалангеальный синдром. Заболевание вызвано доминантной мутацией в гене TRPS1. Клинические проявления: грушевидный нос, редкие волосы, конические эпифизы. Низкий рост, микрогнатия, большие оттопыренные уши, деформация проксимальных межфаланговых суставов, укорочение метакарпальных и метатарзальных костей, преждевременный синостоз ростковых зон. Зубные аномалии: маленькие кариозные зубы, нарушение прикуса, задержка прорезывания зубов.
 Рис. 1.30. Трихо-рино-фалангеальный синдром
Рис. 1.30. Трихо-рино-фалангеальный синдром
Коккейна синдром, тип В. Заболевание вызвано мутацией гена (ERCC6), локализованного в 10q11. Клинически сходное заболевание с синдромом Коккейна, тип А. Однако синдром Коккейна, тип А, обусловлен мутацией другого гена (ERCC6), локализованного в 5q11. К зубным аномалиям синдрома относят: кариес, задержку прорезывания и формирования зубов, отсутствие и гипоплазию зубов, аномалии прикуса.
Акродизостоз. Заболевание было впервые описано Maroteaux and Malamut (1968). Авторы отметили у пациентов признаки акродизостоза (специфическое лицо с коротким носом, открытым ртом и прогнатизмом). Данные аномалии сочетались с маленькими кистями и стопами. Аутосомно-доминантное заболевание. Низкий рост, укорочение конечностей за счет дистальных отделов. Брахицефалия, гипоплазия верхней челюсти, прогнатизм. Запавшая переносица, короткий маленький нос с открытыми вперед ноздрями, снижение слуха, гипертелоризм, страбизм, голубой цвет радужек, множественные скелетные нарушения: деформации плечевой, лучевой, локтевой костей. Кисти широкие с укороченными пальцами. Иногда гипогинетализм, гипогонадизм. В 90% умственная отсталость (коэффициент инеллекта 24-85). Аномалии зубо-челюстной системы выражались в виде нарушения прикуса, задержки прорезывания зубов, гиподентии.
Апера синдром (акроцефалосиндактилия, тип 1). Аутосомнодоминантное заболевание, вызванное мутацией в гене рецептора фактора роста фибробластов-2 (FGFR2). Ген локализован в районе 10q26. Впервые заболевание описано в 1906 г. Примерная частота 1:160 000. Исследования, выполненные в Италии, Дании Испании ив4 штатах США, показали, что частота синдрома Апера 15,5: 100 тыс. новорожденных, в Венгрии - 9,9:100 тыс. Средняя частота мутирования гена 7,8Х10-6 за поколоние. Характерными признаками являются нормальная длина и вес при рождении. Специфические аномалии черепно-лицевой области (краниосиностоз, акробрахицефалия, большой, поздно закрывающийся родничок, высокий лоб, прогнатизм, гипертелоризм, опущенные вниз глазные щели, атрезия, стеноз хоан, узкое нёбо, иногда расщелина нёба). Среди всех случаев краниосиностоза синдром Апера составляет 4,5%. Аномалии развития скелета, аномалии почек, патология центральной нервной системы (различная степень умственной отсталости, агенезия мозолистого тела, вентрикуломегалия, аномалии лимбической системы). Зубные аномалии: аномальный прикус, задержка прорезывания зубов. Среди всех случаев заболевания средний возраст матерей составлял 28,9 лет, отцов - 34,1. В 20% случаев рождения детей с синдромом Апера возраст отцов был старше 35 лет. Соотношение пораженных мужчин и женщин 1:1.
8.2. АУТОСОМНО-РЕЦЕССИВНЫЕ СИНДРОМЫ С НАРУШЕНИЕМ ПРИКУСА
Нарушение прикуса и низкий рост. Say et al. (1973) описали родных брата и сестру, рожденных в кровнородственном браке. У пациентов отмечались треугольная форма лица, нарушение прикуса и короткий рост (девочка 14 лет была ростом 146 см, мальчик 6 лет - 107 см). Тип наследования: аутосомно-рецессивный.
Миастенический синдром врожденный, ассоциированный с недостаточностью ацетилхолинового рецептора. Ген локализован в районе 17p12-p11, 17p13-p12, 11p11.2-p11.1. Тип наследования: аутосомно-рецессивный. Полагают, что заболеваение обусловлено мутацией в эпсилон (CHRNE) и бета CHRNB1 субединиц ацетилхолинового рецептора (AchR). Заболевание может также вызываться мутацией в гене RAPSN. Врожденный миастенический синдром, нейромышечное заболевание. Заболевание и типы мутаций были установлены при исследовавнии цыганских семей, проживавших в Индии и Пакистане. Частота носительства мутантного гена в этой этнической группе составляет 3,7%. Заболевание практически не прогрессирует, характерно выраженное поражение периферической нервной системы. Гипотония, дизартрия. Специфическими фенотипическими признаками синдрома являются: удлиненное лицо, прогнатизм, птоз, офтальмопарез, страбизм, высокое арковидное нёбо, нарушение прикуса, нарушение дыхания из-за слабости дыхательной мускулатуры. У некоторых больных признаки множественного артрогрипоза. При мышечной биопсии отмечается атрофия мышечных волокон 2-го типа. В качестве одного из постоянных признаков является аномалия прикуса.
Эктопия хрусталика, краниофациальный дизморфизм. Аутосомно-рецессивное заболевание. До настоящего времени оно описано в нескольких ливанских семьях. Характерные признаки патологии выявляются в виде краниофациальных аномалий, антимонголоидного разреза глаз, нарушения прикуса, эктопии хрусталика, частичной атрофии радужек. Ген не идентифицирован, его локализация неизвестна.
Гаррода синдром. Harrod et al. (1977) сообщили о двух братьях с необычным синдромальным поражением в виде умственной отсталости, характерными чертами лица, с большими оттопыренными ушами, гипотелоризмом, длинным носом, маленьким ртом, арахнодактилией, гипогенетализмом, наличием микрокист в корковом слое почек и другими аномалиями развития. Информация о клинических
проявлениях синдрома была дополнена описанием 46-летнего пациента с аналогичными признаками и патологией соединительной ткани (мегаколон, варикозное расширение вен). Высокое нёбо, нарушение прикуса. Тип наследования: предположительно аутосомнорецессивный.
Остеопетроз с почечным канальцевым ацидозом. Локализация гена 8q22. Sly et al. (1972) описали трех сестер 22, 17 и 15 лет, рожденных от нормальных неродственных между собой родителей. Заболевание характеризуется низким ростом, гепатоспленомегалией, остесклерозом, гипертелоризмом, нарушениями прикуса, анемией. Интеллект нормальный, иногда умственная отсталость. При лабораторных исследованиях отмечается почечный канальцевый ацидоз, периодически гипокалемия.
Камптодактилии синдром, тип I, Гвадалахара. Тип наследования: аутосомно-рецессивный. Низкий рост, задержка внутриутробного развития, микроцефалия, брахицефалия, плоское лицо, гипоплазия срединной части лица, эпикант, телекант, микрокорнеа, гипертелоризм, синофриз. Короткий нос, ноздри повернуты вперед, маленький рот с опущенными вниз углами рта, высокое арковидное нёбо, длинная шея. Деформация грудной клетки и другие аномалии скелета. Брахидактилия, каптодактилия, синдактилия. Судорожные приступы, умственная отсталость. Задержка прорезывания зубов, аномалии прикуса.
Секкеля синдром. Клинически гетерогенное аутосомнорецессивное заболевание. Ген локализован в области 3q22-q24. Характеризуется отставанием в росте, умственной отсталостью, микроцефалией, судорожными приступами, своеобразным строением головы и лица («птицеголовостью»). Микрогнатия, низкопосаженные деформированные уши, высокое нёбо, иногда ресщелина нёба. Характерна частичная анодентия, гипоплазия эмали, малоклюзия II неправильный рост зубов.
Гипоспадия, гипертелоризм, колобома верхнего века и смешанный тип тугоухости. Редкое аутосомно-рецессивное заболевание, выявлено у двух потомков, рожденных в двоюродном браке. Характеризуется низким ростом, прогнатизмом, гипоплазией средней части лица, гипертелоризмом; смешанный тип тугоухости, колобома верхнего века. Гипоспадия, нарушение прикуса.
Склеростеоз (кортикальный гиперостеоз с синдактилией). Заболевание вызвано мутацией в гене, кодирующем склерозин
(SOST). Ген локализован в 17q12-q21. Аутосомно-рецессивный тип наследования. Характерны большой рост, прогнатизм, гипоплазия срединной части лица, вторичная глухота (из-за гиперостоза черепа), гипертелоризм, птоз, страбизм, нистагм, атрофия зрительных нервов. Скелетные аномалии как следствие гиперостоза. Нарушение прикуса.
Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии кистей (акро-ото-окулярный синдром). Тип наследования аутосомно-рецессивный. К настоящему времени имеется описание только четырех пациентов с данным заболеванием. Первая клиническая характеристика заболевания получена при исследовании трех пациентов из двух инбридных бразильских семей. Последнее описание подобного заболевания было в 1997 г. Низкий рост, низкий вес при рождении, микрогнатия, низкопосаженные уши и смешанная форма тугоухости. Псевдопапилледема, гипертелоризм, эпикант, блефарофимоз. Высокое узкое нёбо. Скелетные аномалии: кифосколиоз, аномалии кистей, гипоплазия тенара, гипотенара, кератоз ладоней, укороченные пальцы, частичная синдактилия, укорочение метакарпальных костей. Аномалии стоп: сандалевидная щель, укорочение III-IV пальцев. Множественные пигментные невусы. Зубные аномалии: нарушение прикуса, темноокрашенные зубы, анодентия, сверхкомплектные зубы, аномалии прорезывания зубов.
Умственная отсталость, Буэнос-Айрес тип (Мутчник синдром). Аутосомно-рецессивное заболевание. Mutchinick (1972) описал двух пациентов с умственной и физической отсталостью, характерным лицом, пороками развития сердца и почек. В последующем в медицинской литературе появились описания нескольких случаев подобных заболеваний в Японии и Германии. Характерна микроцефалия, прогнатизм, низко посаженные уши, страбизм, миопия, гипертелоризм, птоз. Большой рот, тонкая верхняя губа, высокое арковидное нёбо, нарушение прикуса, кариес.
Франк-Тер-Хаара синдром (синдром Мелника-Нидлса). Впервые заболевание было описано у 18-летней арабской девушки, рожденой в кровнородственном браке (1973). Клиническая картина складывается из следующих аномалий: генерализованная костная дисплазия, аномалии скелета (аномалии кистей и стоп, грудной клетки и др.). Характерны: большие глаза, экзофтальм, глаукома, гипертелоризм, микрогнатия, нарушение прикуса, склероз сосцевидного отростка, врожденный порок сердца.
Фацио-дигито-генитальный синдром, рецессивный. Аутосомнорецессивное заболевание. Пророрционально низкий рост, брахицефалия, гипертелоризм, выступающая переносица, повернутые вперед ноздри, широкий рот, высокое узкое нёбо, нарушение прикуса. Деформация грудной клетки, маленькие широкие кисти, клинодактилия V пальцев.
Халлермана-Штрайфа синдром. Частичная анодентия, наличие неонатальных зубов, малокклюзия, иногда наличие сверхкомплектных зубов. При исследовании полости рта высокое узкое арковидное нёбо, микростомия, тонкие губы и оттопыренная нижняя губа. Характерен комплекс пороков развития. Отмечается низкий вес при рождении, отставание в росте и физическом развитии. Брахицефальная форма черепа, низко посаженные уши, микрофтальмия, катаракты, нистагм, колобомы радужки и диска зрительного нерва, множественные скелетные аномалии. Атрофия кожи, наличие телеангиоэктазов, признаков ксероза. Волосы тонкие, светлые. У 15% больных отмечаются признаки умственной отсталости разной степени выраженности.
Горлина-Чаудри-Мосса синдром. Аутосомно-рецессивное заболевание. Характеризуется низким ростом, брахицефалией, гипоплазией срединной части лица, кондуктивной тугоухостью, патологией органа зрения (микрофтальмия, гиперопия, гипертелоризм, птоз). Узкое арковидное нёбо. Скелетные аномалии: краниосиностоз, гипоплазия верхней челюсти и костей носа. Гипертрихоз, гипоплазия дистальных фаланг кистей и стоп. Зубные аномалии: аномалии прикуса, гиподентия, микродентия.
Коккейна синдром, тип А. Аутосомно-рецессивное заболевание, вызванное гомозиготным состоянием мутантного гена ERCC8. Ген локализован в 5q12. Характерны нарушения со стороны центральной и периферической нервной системы (умственная отсталость, деменция, кальцификация базальных ганглиев, дистрофия структур головного мозга, судороги, дисмиелонизация, атаксия, тремор, периферическая нейропатия); эндокринной системы (гипогонадизм). Патология органа зрения и слуха (пигментная ретинопатия, атрофия зрительных нервов, страбизм, нистагм, катаракта, микрофтальмия, гипоплазия радужки, микрокорнеа, нейросенсорная глухота). Аномалии зубов: кариес, задержка прорезывания, малокклюзия, отсутствие/гипоплазия зубов.
8.3. НАСЛЕДСТВЕННЫЕ СИНДРОМЫ С НАРУШЕНИЕМ ПРИКУСА, Х-СЦЕПЛЕННЫЕ
Симпсона-Голаби-Бехмеля синдром, тип 1 (SGBS-синдром).
Локализация гена Xq26. Заболевание обусловлено мутацией гена GPC3. Характеризуется пре- и постнатнальным большим весом, широким лицом, макроцефалией, врожденным пороком сердца и другими врождеными аномалиями. По своим клиническим проявлениям напоминает синдром Беквита-Видеманна (имеются преаурикулярные ямки, насечки на мочках ушей, гепатомегалия спленомегалия и др.). Снижение слуха. Диафрагмальные и пупочные грыжи. Скелетные нарушения (сколиоз, брахидактилия, синдактилия и др.). Макростомия, макроглоссия, подслизистая расщелина губы, расщелина нёба, нарушение прикуса.
Фронтометафизарная дисплазия. Х-сцепленный рецессивный тип наследования. Ген локализован в районе Xp22.2-p22.1. Наследственный синдром с множественными аномалиями развития зубов (частичная анодентия, нарушение прорезывания зубов, нарушение прикуса). Множественные аномалии скелета, патология кистей и стоп.
Фокальная дермальная дисплазия. Х-сцепленный доминантный тип наследования. Множественные аномалии развития: микроцефалия, ассиметрия лица, оттопыреные, низкопосаженные уши, снижение слуха, страбизм, колобомы радужки, микрофтальмия (15%), анофтальмия, эктопия хрусталиков (6%), аниридия (3%), атрофия зрительных нервов, нистагм. Аномалии развития скелета, урогенитальной системы кожи (очаги атрофии, телеангиоэктазы, ленейная гиперпигментацияи др.),умственнаяотсталость(15%),гидроцефалия, агенезия мозолистого тела, аномалия Арнольда-Киари. Большинство случаев спорадические (95%). Все пораженные - лица мужского пола, как результат вновь возникшей мутации. Множественные аномалии развития зубов: гиподентия, олигодентия, гипоплазия эмали, нарушение прорезывания зубов, нарушение прикуса.
Коффина-Лоури синдром. Впервые синдром описан в 1966 г. Тип наследования: Х-сцепленный доминантный. Минимальными диагностическими признаками являются: антимоноголоидный разрез глаз, луковицеобразный нос, низкий рост, конусовидные пальцы, умственная отсталость. Синдром характеризуется специфическими черепно-лицевыми признаками: микроцефалия, выступающие надбровные дуги, гипертелоризм, антимонголоидный разрез глаз,
открытые вперед ноздри, переорбитальная полнота тканей, широкая спинка носа, оттопыренные уши. Множественные признаки патологии соединительной ткани и скелета (гипермобильность суставов кистей и стоп, деформация грудной клетки, сколиоз, кифоз и др.). К стоматологическим проявлениям синдрома относятся: большой приоткрытый рот, узкое высокое нёбо, гиподентия, нарушение прикуса, широко расставленные зубы, большие срединные резцы (макродентия).
Микрофтальмия синдромальная 2 (окуло-фацио-кардиодентальный синдром). Заболевание вызвано мутацией в BCL6 корепрессорном гене, локализованном в Xp11.4. Тип наследования: Х-сцепленный доминантный. Впервые описание заболевания выявлено в 1993 г. В последующем было описано еще несколько случаев патологии, включавшей в себя характерные лицевые аномалии (узкое длинное лицо), нейросенсорную глухоту, глазные аномалии (микрокорнеа, врожденная катаракта, вторичная глаукома, птоз, блефарофимоз и др.). Пороки сердечно-сосудистой системы, умственная отсталость средней степени. Множественные аномалии развития зубов (нарушение прорезывания, персистенция временных зубов, олигодентия сверхкомплектные зубы, сращение зубов, дилацерация корней зубов, нарушение прикуса).
8.4. ПРОБЛЕМЫ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ И ЛЕЧЕНИЯ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ В СТОМАТОЛОГИИ
Аномалии развития зубов, неблагоприятно отражаясь на эмоциональном состоянии людей, тем не менее, не входят в круг угрожающих жизни заболеваний и существенно не влияют на репродуктивное поведение. Благодаря этому можно наблюдать значительное количество семейных случаев стоматологических наследственных заболеваний, где наличие патологии регистрируется на протяжении нескольких поколений. Это создает благоприятные условия для изучения генетической этиологии таких болезней. Знание нормы и патологии очень важны для разработки новых подходов в лечении стоматологических заболеваний, в частности на основе развивающихся исследований в области клеточных технологий. Уже в ближайшее время можно рассчитывать на применение этих научных достижений в стоматологической практике. Вся логика исследований в этой
области говорит о том, что перспектива использования современных восстановительных медико-генетических технологий реальна не только на экспериментальных животных, но и у человека. В частности, очень перспективным направлением является использования для этих целей стволовых клеток. Как известно, эти мультипотентные клетки можно получать из стромы костного мозга, а также из тканей эмбриональных зубов или интактных зачатков зубов. Кроме того, было показано, что популяции стволовых клеток, существующие в молочных зубах и со временем утрачивающиеся, обладают потенциалом образовывать минерализованные зубные структуры, в частности дентин и кость, а также регенерировать нервную ткань.
Следует учитывать, что до тех пор, пока агенезия одного или более зубов не является составной частью наследственного синдрома или тяжелого заболевания, генетический и медицинский прогноз для пациента и его семьи относительно благоприятен, даже несмотря на то, что отдельные наследственные формы патологии зубов иногда с вовлечением многих зубов приводят к значительным эстетическим, эмоциональным и финансовым проблемам.
Часто врожденные аномалии зубов сопряжены с дополнительными стоматологическими проблемами в виде нарушения прикуса изза неправильного расположения зубов, слабого роста альвеолярных отростков, отсутствия зубов и наличия пустого пространства в пределах зубных дуг. Хотя клиницисты давно знакомы с гиподентией и олигодентией, возможности ранней диагностики, профилактики и лечения этих состояний всегда были крайне ограничены. Стратегия лечения в каждом случае определяется числом и типом отсутствующих зубов, индивидуальными пропорциями скелета и эстетическими соображениями. Терапия, как правило, включает с в себя много этапов, каждый из которых является часто сложным и длительным и требует участия по меньшей мере двух-трех специалистов разного стоматологического профиля.
Ортодонтические подходы наиболее адекватны для восстановления позиции существующих зубов и консолидации пространства для постоянного или съемного протеза. Однако при утрате нескольких зубов, наблюдаемой при олигодентии моляров, ортодонтическая коррекция дополняется процедурами наращивания кости, направленными на увеличение костной массы и проводимыми еще до установки имплантата. Олигодентия моляров сопровождается самыми тяжелыми последствиями для зубов, если не считать синдромных
форм адонтии. Надежных мер профилактики агенезии зубов нет, а ее пренатальная диагностика невозможна или рассматривается многими специалистами как необоснованная. Теоретически многие аномалии зубов можно выявлять пренатально при помощи ультразвука. Но представляется весьма сомнительным, чтобы супружеская пара решилась на радикальные меры в отношении беременности при изолированных пороках развития зубов, а это делает профилактический подход для части стоматологических заболеваний спорным. Что касается тяжелой, сочетанной патологии, когда поражения зубов составляют часть синдрома, то вопрос о целесообразности пренатальной диагностики и последующего решения относительно пролонгирования беременности должен решаться индивидуально исходя из тяжести поражения, перспектив и эффективности терапевтического или хирургического лечения.