Медицинская и клиническая генетика для стоматологов: учебник для вузов / Под ред. О.О. Янушевича., - 2009. - 400 с.
|
|
|
|
ГЛАВА 2. СЕМИОТИКА НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ И ПРИНЦИПЫ КЛИНИЧЕСКОЙ ДИАГНОСТИКИ
Захарова Ольга Михайловна
2.1. ОБЩАЯ И ЧАСТНАЯ СЕМИОТИКА НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
В энциклопедическом словаре медицинских терминов семиотика определяется как учение о признаках (симптомах) болезней и патологических состояниях. Под общей семиотикой наследственных болезней мы подразумеваем общие признаки, характерные для всех болезней, объединенных в группу наследственных. И несмотря на то что наследственных болезней к настоящему времени описано более 7 тыс. (хромосомные, моногенные, митохондриальные, соматических клеток и т.д.) и проявления их крайне разнообразны, все же необходимо знать общехарактерные черты анамнеза, клиники, течения, исходов, позволяющие отличать наследственные и ненаследственные формы патологии. В клинической практике врачу-генетику нередко приходится сталкиваться с врожденными патологическими состояниями, которые по фенотипическим проявлениям сходны с наследственными формами, но могут быть обусловлены воздействием внешнесредовых факторов. Дифференциальная диагностика в данном случае и правильное установление этиологии - залог патогенетически обоснованного лечения, адекватных мер реабилитации и семейной профилактики.
Для наследственных болезней общими признаками являются: «необычность» клинических проявлений, хронический рецидивирующий прогредиентный характер течения, устойчивость к обычным терапевтическим мероприятиям, поражение нескольких органов и систем (системный характер поражения), повышенная частота семейных случаев.
2.2. МОРФОГЕНЕТИЧЕСКИЕ ВАРИАНТЫ РАЗВИТИЯ И ИХ ЗНАЧЕНИЕ В ДИАГНОСТИКЕ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Диагностика наследственных заболеваний представляет большие трудности, в том числе из-за низкой частоты встречаемости в популяции. Нередко наследственная патология проявляется своеобразным внешним видом пациентов. В связи с этим важно учитывать особенности фенотипа, так называемые микропризнаки, которые реально не влияют на продолжительность жизни, функциональное состояние или иммунный статус пациента, но являются диагностически значимыми. Разрез глаз, расстояние между зрачками, гипертрихоз, длинные пальцы, особенности кожного рисунка ладоней, широкое расстояние между сосками, наличие «кофейных» пятен на коже, низкопосаженные ушные раковины, «насечки» на мочках уха, низкий рост волос на затылке, диастема, высокое нёбо - вот далеко не полный перечень внешних признаков, без тщательного анализа и учета которых невозможно установление правильного диагноза.
Так,патогномоничными признакамидлясиндрома РубинштейнаТейби являются широкие дистальные фаланги I пальцев кистей и стоп, для синдрома Корнелии де Ланге - сросшиеся брови, для синдрома Варденбурга - депигментированная («седая») прядь волос у лба.
2.3. АНТРОПОМЕТРИЯ
Антропометрия как совокупность методов и приемов измерения человеческого тела имеет важное значение в клиническом обследовании пациентов с подозрением на наследственную патологию. В клинике внутренних болезней под антропометрией понимают цифровую оценку трех внешних морфологических признаков: роста, массы тела и окружности груди.
В практике медико-генетического консультирования при необходимости добавляют измерение окружности головы, окружности живота, длины конечностей и их отдельных частей, расстояния между зрачками (или внутренними краями орбит), размера глазной щели, площади кожных пятен, длины носа, объема яичек, длины полового члена, толщины кожной складки, ширины таза и т.д. Границы антро-
пометрического метода могут быть достаточно широкими и включать любые количественные исследования, проводимые в клинике.
Так, например, оценить расстояние между внутренними краями глазниц (увеличенное - гипертелоризм, уменьшенное - гипотелоризм) можно по индексу межорбитальной окружности (ИМО), который высчитывается по формуле:
 Если
ИМО >6,8, то у пациента гипертелоризм, если ИМО <3,8, -
наблюдается гипотелоризм, хотя в практике медико-генетического
консультирования эти признаки нередко оцениваются без столь долгих
вычислений, визуально.
Если
ИМО >6,8, то у пациента гипертелоризм, если ИМО <3,8, -
наблюдается гипотелоризм, хотя в практике медико-генетического
консультирования эти признаки нередко оцениваются без столь долгих
вычислений, визуально.
2.4. ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ
Тератология (от греч. teras, teratos - урод, чудовище) - дословно наука об уродствах, а фактически об этиологии, патогенезе и клинике врожденных пороков развития. К врожденным порокам развития (ВПР) относят(ся) стойкие морфологические изменения, повлекшие за собой грубые нарушения функции органа, ткани или всего организма. В качестве синонимов используются термины «пороки развития» и «врожденные пороки».
Г.И. Лазюк (1983) считает необходимым выделять следующие, сходные с ВПР, понятия.
Врожденные аномалии - пороки развития, не сопровождающиеся нарушением функции органа, например деформации ушных раковин, не обезображивающие лицо и не влияющие на слух.
Ассоциации - устойчивые сочетания врожденных пороков, если есть основания предполагать несколько механизмов возникновения такого комплекса. Например, VATER-ассоциация (V - vertebral, дефекты позвоночника; А - anal atresia, атрезия ануса; ТЕ - tracheooesophageal, трахео-пищеводная фистула; R - radial and renal, дисплазия луча и почек), этиология и патогенез которой неясны, а вклад генетических факторов представляется сомнительным.
Деформация - изменение структуры первоначально правильно сформированного органа. Например, культя конечности после ампутации амниогенными перетяжками.
Аномалад - комплекс нарушений, возникающих в результате одной ошибки морфогенеза, т.е. один первичный порок и цепь его последствий. Например, аномалад Поттер - агенезия почек, маловодие, характерные изменения лица, врожденная косолапость и гипоплазия легких.
Дисплазия - порок развития определенного органа или ткани (лица, соединительной ткани).
Формирование вышеуказанных нарушений происходит во внутриутробном периоде, они становятся очевидными при рождении или проведении пренатальной диагностики.
Формирование морфологических структур эмбриона - так называемый эмбриональный морфогенез - осуществляется при тесном взаимодействии генома зародыша и организма матери, особенно ее гормональной и иммунной систем, и связан с процессами размножения, роста, миграции, дифференциации и отмирания клеток. В основе этих процессов лежит последовательное декодирование морфогенетической информации зародыша, обеспечивающее дифференциальную активность генов. Нарушение любого звена морфогенеза под воздействием генетических или средовых факторов может реализоваться во врожденный порок. Специализацию клеток определяет в основном дифференциальная активность генов, а также состояние клеток-мишеней. Нарушения развития в связи с этим возможны при отсутствии или недостаточном функционировании генов.
Врожденные пороки развития многообразны, количество их велико, они отличаются по этиологии, времени возникновения, клинической картине.
ВПР могут возникать в результате возникновения нарушений на разных этапах развития организма. По времени возникновения все ВПР делятся на: гаметопатии, бластопатии, эмбриопатии, фетопатии.
Под гаметопатиями понимают повреждение половых клеток, чаще всего в результате мутаций, приводящее к наследственным ВПР.
Бластопатии (бластозы) - нарушения в бластоцисте, т.е. в зародыше в первые 15 дней после оплодотворения (до окончания процесса дробления). Следствием бластопатий, возможно, являются нераз-
деленные двойни («сиамские близнецы»), циклопия, сиреномелия, а также мозаичные формы хромосомных нарушений. Выраженные тератогенные воздействия в этот период развития чаще всего приводят к гибели зародыша до установления факта беременности. Если же зародыш выживает, то органоспецифические аномалии не развиваются, так как замещение поврежденных клеток обеспечивает дальнейшее нормальное развитие. Действует принцип «все или ничего». Поэтому пока женщина не знает о своей беременности, природа как бы прощает ей прием лекарств, алкоголя, рентгеновское обследование и т.п. Но как только очередной «критический» день не наступил, с нее будет строго взыскано за каждую сигарету, таблетку, порцию алкоголя, контакт с ионизирующим излучением, полями СВЧ и т.д.
Эмбриопатии - пороки, возникшие в результате повреждения эмбриона, т.е. от 16-го дня после оплодотворения до конца 10 нед беременности. Это период органогенеза и максимальной чувствительности к тератогенам. Большинство ВПР формируются именно в этот период, так как происходит основная закладка всех органов и тканей (диабетическая, талидомидная эмбриопатия, эмбриопатия вследствие поражения вирусом краснухи). Именно в этот период беременной женщине надо быть особенно осторожной при контакте с любыми факторами - возможными или установленными тератогенами.
Фетопатии - повреждения плода, возникают от 11 нед беременности до родов. Этот период характеризуется в основном ростом и увеличением размеров органов, кроме головного мозга, и половых желез. Тератогены в этот период, как правило, не приводят к выраженным порокам развитя. Примером может служить диабетическая фетопатия.
По этиологии все ВПР можно отнести к одной из трех групп: 1) наследственные; 2) экзогенно обусловленные; 3) мультифакториальные.
Наследственные ВПР возникают в результате мутаций (генных, хромосомных, геномных), чаще всего на уровне гамет, реже в зиготе.
Экзогенно обусловленные ВПР возникают в результате воздействия тератогенных факторов во время беременности на эмбрион, плод.
Мультифакториальные ВПР являются результатом совместного действия наследственных и экзогенных факторов, причем в равной степени.
Из общего числа ВПР наследственные занимают примерно 20-30%, экзогенные - 2-5%, мультифакториальные - 30-40%, в остальных 25-50% причина не установлена.
Тератогенные факторы по аналогии с мутагенными (в зависимости от источника воздействия) подразделяют на 1) физические, 2) химические и 3) биологические. Они не вызывают стойких изменений генетического аппарата.
Периоды, когда плод наиболее подвержен повреждающему действию тератогенных факторов, называют критическими. Они совпадают с периодами плацентации и имплантации. Первый критический период у человека приходится на конец 1-й-начало 2-й недели беременности. Исходом повреждающего воздействия в этот период чаще всего является гибель зародыша. Ко второму периоду относятся 3-6 нед беременности. В этот период такие же факторы чаще обусловливают врожденные пороки. Критические периоды связаны с наиболее интенсивным формированием органов в это время.
Широко используется понятие о тератогенном терминационном периоде (ТТП) , т.е. предельном сроке, в течение которого тератогенный фактор может вызвать пороки развития. Если повреждающий фактор действует после окончания формирования органа, он не может быть причиной этого порока. Каждый орган имеет свой ТТП. Так, например, ТТП расщелин губы - до конца 7-й недели, нёба - до 8-й недели, срединной расщелины нижней губы и нижней челюсти - до 5-й недели, срединной расщелины лица - до начала 6-й недели беременности.
К физическим факторам тератогенеза можно отнести ионизирующую радиацию, воздействие электромагнитных полей, полей СВЧ, повышенную температуру беременной, механические факторы (например, сдавление). Некоторые из перечисленных факторов имеют слабый тератогенный эффект (эффект, полученный в эксперименте на животных, и не описанный у человека), однако осторожность при контакте с этими факторами во время беременности считается оправданной.
К химическим факторам тератогенеза относят лекарственные препараты, средства бытовой и промышленной химии, гипоксию, неполноценное или несбалансированное питание.
Большая РоссийскаяЭнциклопедия (БРЭ) лекарственных средств все лекарственные препараты по степени риска развития эмбриотоксического и тератогенного эффекта подразделяет на три группы: 1) высокая; 2) значительная; 3) умеренная степень риска.
К препаратам 1-й группы относятся:
• цитостатические средства (метотрексат, циклофосфамид, винкристин, фторурацил); нарушая обмен фолиевой кислоты, они оказывают эмбриотоксическое или тератогенное действие универсального характера (деформация лицевой части черепа, нарушение его окостенения и т.д.), обладают эмбриолетальным и фетотоксическим действием;
• противогрибковые и противоопухолевые антибиотики (даунорубицин);
• иммунодепрессанты (азатиоприн), которые влияют также и на половые клетки (т.е. действуют и до зачатия).
Действие указанных препаратов сохраняется до 3 мес у мужчин и до 6-12 мес у женщин.
При невозможности отказа от приема препаратов первой группы показано прерывание беременности.
К препаратам 2-й группы относятся:
• антибиотики (аминогликозиды, тетрациклины, рифампицин);
• противопротозойные средства - производные аминохинолина (гидроксихлорохин), препараты хинина;
• противосудорожные средства (фенитоин-гидантоин, депакин, триметин);
• противопаркинсонические средства;
• соли лития;
• глюкокортикостероиды;
• нестероидные противовоспалительные препараты;
• пероральные гипогликемические средства;
• нейролептические средства;
• этиловый спирт;
• антикоагулянты непрямого действия (варфарин);
• антитиреоидные средства (тиамазол; йодиды).
Применение препаратов второй группы в первые 3-10 нед беременности может стать причиной гибели эмбриона и/или самопроизвольного выкидыша.
К препаратам 3-й группы относятся:
• противомикробные сульфаниламидные препараты;
• метронидазол;
• транквилизаторы;
• половые гормоны (эстрогены).
Из противомикробных препаратов с осторожностью следует относиться к применению во время беременности тетрациклинов - из-за риска острой желтой дистрофии печени, а также окрашивания в желтый цвет эмали зубов, их гипоплазии и даже замедления развития костной системы у ребенка. Стрептомицин может оказывать ототоксическое действие, а также вызывать нарушения в строении скелета. Применение во время беременности антибиотиков канамицина и гентамицина чревато ототоксическим эффектом. В последнем триместре беременности противопоказано применение левомицетина из-за опасности развития серого синдрома у новорожденного (вследствие незрелости печени), а также нарушений в системе гемопоэза.
Несмотря на отсутствие указаний о тератогенности, в ранние сроки беременности необходимо воздерживаться от назначения фузидина из-за его высокой эмбриолетальности. В последнем триместре беременности следует избегать применения сульфаниламидных препаратов из-за возможного риска развития желтухи новорожденных и гемолитической анемии у детей с дефицитом глюкозо-6-фос- фатдегидрогеназы.
Тератогенный эффект хлоридина, связанный с влиянием на обмен фолиевой кислоты, проявляется в множественных нарушениях (мозга, глаз, скелета и т.д.) и позволяет считать применение этого препарата в I триместре беременности противопоказанным.
Тератогенное действие хинина связано с повреждением ЦНС и является противопоказанием для применения в период эмбриогенеза. Определенный риск развития тератогенного и эмбриолетального эффекта существует при применении наркотических анальгетиков. Экспериментально установлено эмбриолетальное, тератогенное и фетотоксическое дествие барбитуратов, а у новорожденных наблюдается синдром отмены при приеме высоких доз препаратов этой группы в III триместре беременности. С другой стороны, использование фенобарбитала показано в качестве профилактики гипербилирубинемии при резус-конфликте, а также как противогипоксического средства.
Из группы транквилизаторов выраженным тератогенным эффектом обладает препарат талидомид, который в нашей стране никогда не применялся. В спектре пороков талидомидной эмбриопатии отмечаются недоразвитие конечностей, пороки развития глаз, органа слуха, внутренних органов. Высокая токсичность и возможные
тератогенные эффекты исключают применение психостимуляторов (амфетамин) у беременных.
Высокий потенциальный риск тератогенного эффекта антидепрессантов не позволяет применять их во время беременности (амитриптилин может вызвать пороки мышечной системы, препараты солей лития - врожденные пороки сердца).
Установленная тератогенная активность некоторых противосудорожных препаратов (гидантоина, депакина, триметина) является противопоказанием для применения их во время беременности. «Гидантоиновый» синдром включает пренатальную гипоплазию, умственную отсталость, лицевые дизморфии с расщелиной губы и нёба, врожденные пороки сердца, гипоплазию пальцев и ногтей. «Триметиновый» синдром характеризуется черепно-лицевыми дизморфиями, пороками сердца, задержкой психического и физического развития, изменениями скелета. В состав фетального синдрома, связанного с приемом депакина, могут входить черепно-лицевые дизморфии, задержка психического и физического развития, spina bifida и менингоцеле.
Наркотические анальгетики (героин, метадон) относятся к «поведенческим» тератогенам, так как вызывают у новорожденных абстинентный синдром, отсутствие сосательного рефлекса, нарушения сна, гипервозбудимость, а в дальнейшем - трудности обучения, нарушения памяти, внимания. Можно считать доказанным тератогенный эффект средств, используемых для ингаляционного наркоза. У женщин-хирургов, анестезиологов, операционных сестер в 2-4 раза повышен риск спонтанных абортов и часто встречающихся пороков.
Противопоказано использование в акушерской практике таких антиаритмических средств, как хинидина сульфат (возможны тромбоцитопения, неврит зрительного нерва, миастения у новорожденных), дизопирамид (стимулирующее влияние на сократительную функцию матки), также требует осторожности совместное применение пропроналола и верапамила (нарушение сердечной деятельности у новорожденного). Из обширной группы гипотензивных средств противопоказаны или должны применяться с осторожностью у беременных β-адреноблокаторы, октадин, нитропруссид натрия, гиперстат, каптоприл, папаверина гидрохлорид.
Синтетические антикоагулянты (варфарин) обладают тератогенным (точечная хондродисплазия, седловидный нос, широкие и
короткие пальцы, а также пороки глаз) и фетотоксическим действием (геморрагический синдром).
Противопоказано назначение во время беременности ртутных диуретиков, а также этакриновой кислоты (нарушение слуха у новорожденных) и тиазидов (развитие у новорожденных гипонатриемии, желтухи и тромбоцитопении); нежелательно применение хлорида аммония, верошпирона и диакарба (некомпенсированные изменения КЩС новорожденного).
Использование синтетических прогестинов (оксипрогестерона капронат, этистерон и др.) во время беременности противопоказано из-за указаний на формирование псевдогермафродитизма у девочек, преждевременного полового созревания у мальчиков. Диэтилстильбэстрол также обладает тератогенным действием, вызывая новообразования во влагалище и матке у девочек, патологию яичек у мальчиков.
Гипотетический риск от приема гормональных контрацептивов крайне мал, хотя описаны некоторые тератогенные эффекты. С осторожностью следует принимать андрогенные препараты из-за риска псавдогермафродитизма у девочек, а также антиандрогенные препараты из-за возможной феминизации у мальчиков. Применение пероральных противодиабетических средств (бигуаниды, карбутамид) противопоказано во время беременности из-за тератогенного эффекта. Несколько увеличивет риск патологии у плода передозировка витаминов А, Д, В2, С. Неблагоприятно воздействует дефицит витаминов Е, Д, а также фолиевой кислоты.
Пенициллины и цефалоспорины, проходя плацентарный барьер и достигая в тканях плода терапевтической концентрации, как правило, тератогенным и токсическим действием не обладают. При высокой токсичности хлорамфеникола его эмбриотоксическое действие не доказано. Отсутствуют экспериментальные сведения об эмбриотоксическом и тератогенном действии метронидазола, но предпочтительно не применять его в I триместре беременности. Не обнаружено тератогенное действие эритромицина, цефалоспоринов, нитрофуранов, этамбутола, изониазида, ПАСК, нистатина, клотримазола. При аутоиммунных заболеваниях у беременных возможно применение таких иммунодепрессантов, как плаквенил и хингамин. Левамизол как иммуностимулирующий препарат используют во время родов при тяжелых гнойно-септических осложнениях. Из седативных средств у беременных возможно осторожное примене-
ние магнезии, брома и валерианы как препаратов без тератогенного эффекта.
Большинство клинических наблюдений свидетельствуют об отсутствии нарушений морфогенеза при приеме беременными транквилизаторов - производных бензодиазепина (диазепама, хлордиазепоксида и др.) и мепробамата. Клинические наблюдения показали отсутствие тератогенного эффекта при приеме беременными нейролептиков - производных фенотиазина (аминазин), производных бутирофенона (галоперидол), производных индола (резерпин), что не исключает возможность фетотоксического эффекта (развитие респираторного дистресс-синдрома и нарушение терморегуляции). Из группы противосудорожных препаратов наиболее предпочтительными для использования во время беременности являются карбамазепин, этосуксемд, бензонал в связи с отсутствием данных о потенциальной тератогенной опасности. Отсутствуют убедительные данные о тератогенном эффекте нестероидных противовоспалительных препаратов, в частности салицилатов. Однако не рекомендуется использование этих препаратов перед родами из-за возможного развития геморрагического синдрома у новорожденного.
В качестве противорвотного средства у беременных возможно применение церукала. Препаратами без тератогенного эффекта считаются холинэргические препараты, широко использующиеся в акушерской практике (холиномиметики - прозерин, холинолитики - атропин, ганглиоблокаторы - пентамин, курареподобные препараты - дитилин). Из адренергических средств в акушерской практике используют β-адреноблокаторы, симпатолитик октадин. Клинические данные о других препаратах данной группы отсутствуют.
Тератогенные свойства антигистаминных препаратов не выявлены, однако применение у беременных циметидина противопоказано вследствие возможного развития гинекомастии у новорожденных. Местноанестезирующие средства (новокаин, лидокаин и др.) не обладают тератогенным эффектом, но их нельзя считать абсолютно безопасными для плода из-за указаний на токсическое действие при ацидотических сдвигах (гипоксии).
Широко использующиеся в акушерской практике средства, влияющие на сократительную активность матки (простагландины, окситоцин, эргометрин и др., а также партусистен, коринфар, изоптин и др.) не обладают тератогенными свойствами. Применение сердеч-
ных гликозидов (дигоксин, изоланид, коргликон и пр.) и новокаинамида как антиаритмического средства у беременных также безопасно для будущего потомства.
Нижеперечисленные гипотензивные и сосудорасширяющие средства не вызывают побочных эффектов: α-метилдопа, лабеталол, клофелин, апрессин, но-шпа, эуфиллин, курантил, интенкордин, компламин. Не обладает тератогенным действием антикоагулянт гепарин. Мочегонные препараты дихлотиазид и фуросемид не обладают тератогенным действием. При наличии показаний беременным можно подобрать нетератогенные слабительные (фенолфталеин, бисакодил, листья сенны) и антацидные средства. Гормональные препараты передней (АКТГ, СТГ, ТТГ, ЛГ, ФСГ) и заднй долей (окситоцин, вазопрессин) гипофиза не проникают через плацентарный барьер и не оказывают тератогенного действия. При лечении гипотиреоза показано назначение тиреоидных гормонов (тироксин) еще до зачатия и на протяжении всей беременности.
Антитиреоидные препараты (мерказолил) должны применяться осторожно. Широко используются в акушерской практике безопасные для плода глюкокортикоиды (преднизолон) и минералокортикоиды (дезоксикортикостерон), а также небольшие дозы эстрогенов (сигетин), прогестерон. Лечение сахарного диабета во время беременности проводится с помощью препаратов инсулина, которые не обладают тератогенным действием.
К факторам химического тератогенеза относится также воздействие этилового спирта на плод, что может приводить к развитию фетального алкогольного синдрома. Потомство у женщин, на протяжении всей беременности употребляющих крепкие спиртные напитки, часто имеет пренатальную гипоплазию, лицевые дизморфии, недоразвитие мозга и, как следствие, умственную отсталость и различные нарушения со стороны ЦНС, пороки сердца и других внутренних органов. У новорожденных нередко приходится купировать абстинентный синдром.
Такая все более распространяющаяся в настоящее время вредная привычка, как табакокурение, безусловно, вызывает множество проблем со здоровьем матери и ее ребенка. Курение, в том числе пассивное, во время беременности вызывает пренатальную гипоплазию, гипоксию плода. Есть сообщения о повышенной частоте расщелин губы/нёба у детей курящих женщин. Установлен цитотоксический эффект никотина на развивающиеся половые клетки женщин.
Отмечен повышенный риск бесплодия, спонтанных абортов, мертворождений и преждевременных родов у курящих женщин. Дети курящих матерей часто подвержены «синдрому внезапной смерти», чаще болеют, имеют расстройства со стороны нервной, сердечно-сосудистой, дыхательной, эндокринной систем в перинатальном периоде.
Широко применяемые в промышленности, сельском хозяйстве и быту бензин, бензол, фенолы, формальдегид, ядохимикаты, свинец, пары ртути, различные краски обладают эмбриотоксическими свойствами. Их воздействие может вызвать самопроизвольный выкидыш, внутриутробную гибель плода.
Гипоксия как фактор развития пороков у новорожденных возникает из-за табакокурения, сердечно-сосудистых заболеваний (пороки сердца, анемии) беременных и может вызывать нарушение плацентации, недоразвитие плода, его гибель.
Неполноценное питание во время беременности как причина дефицита микроэлементов цинка, марганца, магния может привести к развитию пороков ЦНС.
К биологическим факторам тератогенеза следует отнести некоторые перенесенные во время беременности инфекции. Первое место по значимости среди них занимает краснуха. Перенесенная в I триместре беременности краснуха может привести к формированию фетального краснушного синдрома (синдром Грега), в состав которого входят пороки сердца, ЦНС, скелета, глаз (чаще в виде катаракты), глухота. Тяжесть поражения диктует необходимость прерывания беременности, если заражение произошло в самые ранние сроки беременности (в первые 4 нед). Характерными признаками перенесенной внутриутробно цитомегаловирусной инфекции являются микроцефалия, хориоретинит, глухота, гепатоспленомегалия, тромбоцитопеническая пурпура. Из других вирусных инфекций определенную роль в развитии некоторых пороков у новорожденных играют грипп, герпес, гепатит, хотя имеется гораздо меньше доказательств их тератогенности.
Нежелательно применение живых вирусных вакцин во время беременности, хотя сведения о развившихся в результате этого пороках немногочисленны. Токсоплазмоз - одна из наиболее распространенных латентных инфекций у человека, вызываемая простейшими. Первичное внутриутробное заражение может привести к поражению ЦНС с внутримозговыми петрификатами, хориоретиниту, гепатоспленомегалии, тромбоцитопенической пурпуре. Бледная спирохета,
вызывающая сифилис, при внутриутробном поражении провоцирует развитие аномалий лицевого черепа и других костей, а также кератита, поражения печени, кожи и слизистых. Поражение может иметь характер латентной инфекции с поздним проявлением (после двух лет).
При оценке тератогенных возможностей различных повреждающих факторов необходимо учитывать дозу (например, радиации, химического вещества, инфекционного агента), интенсивность, продолжительность и время воздействия, а также индивидуальную чувствительность, обусловленную генетическими факторами.
2.5. СЕМЕЙНЫЙ ПОДХОД В ДИАГНОСТИКЕ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Нередко в практике медико-генетического консультирования врач имеет дело с наследственным заболеванием, которое является следствием новой хромосомной, геномной или генной мутации (доминантной в аутосоме или Х-хромосоме, или рецессивной в Х-хромосоме), а также следствием «огомозигочивания» дефектных рецессивных генов. В таком случае другие родственники здоровы. Но для того чтобы убедиться в этом, необходимо тщательное обследование семьи, особенно близких родственников. Если же в родословной большое количество пораженных родственников, это так называемые семейные случаи. Надо помнить, что патология у родственников не обязательно связана с наследственностью. Так, возможны семейные случаи профессиональных заболеваний, воздействие экзогенных эндемических факторов или инфекционного агента.
Классический вариант наследственных болезней предполагает все же наличие семейных форм. Показательная родословная при моногенном заболевании позволяет определить тип наследования и, таким образом, сузить «диагностический поиск». В случае мультифакториальной патологии оценка семейной ситуации помогает правильно рассчитать генетический риск, наметить пути профилактики. При наличии в родословной кровнородственных браков необходимо начинать «тестирование» с аутосомно-рецессивного типа наследования.
Так, при онкологических заболеваниях около 18% здоровых лиц имеют 2 и более родственников, пораженных раком. Однако не во всех таких семьях опухоли наследственные. В некоторых семьях рак может «скапливаться» и случайно. Наследственные формы рака, рас-
пространенные в популяции значительно реже, составляют 5-10% от всех случаев рака конкретных локализаций. Большое количество онкологических больных в семьях определяется рабочим термином «раковые семьи», хотя возможны большие вариации в проявлении опухолевого фенотипа даже среди членов одной семьи.
Идеально, если большинство родственников в сложных случаях диагностики обследуются лично врачом-генетиком или, по его направлению, врачами других специальностей. Если обследование родственников невозможно, необходимо запросить в лечебных учреждениях диагнозы, выписки из историй их болезней, амбулаторных карт, протоколы патанатомического вскрытия или другие важные для диагностики медицинские документы. Иногда большую помощь могут оказать семейные фотоальбомы.
При расчете риска при расщелине губы/нёба важно знать не только, сколько родственников имели этот порок, но и тяжесть поражения, и степень родства с консультируемым; также важны указания на наличие у здоровых родственников гнусавого голоса или фенотипически разнообразных микропризнаков расщелин.
Диагностика наследственных заболеваний, лечение и действенные меры профилактики часто невозможны без понимания семейной ситуации.
2.6. КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОД ДИАГНОСТИКИ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Клинико-генеалогический метод является важнейшим и наиболее разработанным методом диагностики наследственных болезней, основанным на анализе характера передачи различных признаков и заболеваний в отдельной семье с указанием родственных связей между членами родословной. Из названия метода понятно, что его составляющими частями являются две большие компоненты: клиническая и генеалогическая. В узком смысле слова генеалогия (греч. genealogia; от genea - рождение, происхождение, поколение + logos - слово, изложение, наука) - наука о родословных, а в широком смысле - установление родственных связей между индивидуумами в пределах одного поколения или в ряду поколений. Суть генеалогического метода - составление генеалогического древа и проведение генеалогического анализа, т.е. составление родословной и ее анализ.
При медико-генетическом консультировании человек, от которого начинается составление родословной, называется пробандом. На родословной он обозначается стрелочкой. Пробандом может быть больной человек, обратившийся по поводу своего заболевания, или по поводу его заболевания (если речь идет об умершем) обратились ближайшие родственники. Пробандом может быть здоровый человек, обратившийся по поводу «возрастного» или кровнородственного брака, заболеваний в семье, воздействия повреждающих факторов во время беременности или при ее планировании. В подавляющем большинстве случаев медико-генетического консультирования пробандом является больной (или умерший) ребенок. Основное отличие деятельности врача-генетика от врачей-клиницистов других специальностей состоит в том, что генетик имеет дело с семьей, а не только с пациентом (педиатры, правда, тоже общаются с кем-то из взрослых родственников ребенка, но не обследуют его). В медицинской генетике объектом исследования является «ядерная семья» , в которую чаще всего входят больной ребенок-пробанд, его родители и сибсы (т.е. братья и сестры или дети одной супружеской пары). Полусибсы имеют только одного общего родителя; сводные сибсы не являются биологическими родственниками, хотя могут проживать вместе и иметь одну фамилию. Медико-генетическая консультация это, по сути, амбулаторный прием. Медицинским документом по аналогии с амбулаторной картой является генетическая карта. Специализированных отделений для детей с наследственной патологией в педиатрических стационарах не так много, и в этих отделениях оформляется история болезни. Эти главные медицинские документы построены по единому принципу, равно как и клинический метод является универсальным для врача любой специальности и состоит из определенных последовательных этапов. Следует остановиться на особенностях применения клинического метода в практике медикогенетического консультирования.
Поскольку обследованию подвергается семья, то оформляется одна генетическая карта, в которой присутствуют необходимые сведения о всех родственниках. Наиболее подробная информация содержится о пробанде.
Справочная часть, или, в узком смысле, паспортные данные
1. Несмотря на то что пробандом может быть маленький ребенок, его фамилия, имя, отчество записываются полностью. В паспортных
данных женщины отмечают ее девичью фамилию, так как возможные совпадения с фамилией супруга могут свидетельствовать об отдаленном родстве. Иногда фамилия ядерной семьи отношения к «родовым» фамилиям мужа и жены не имеет (смена неблагозвучной фамилии).
2. Пол. Особенно важно указать для тех родственников, по фамилии, имени и отчеству которых невозможно установить половую принадлежность. У лиц с нарушением психосексуальной ориентации записывается паспортный пол.
3. Как правило, в генетической карте отмечают не возраст, а дату рождения, что является более точным показателем.
4. Если пробанд - ребенок, то в карте фиксируют не только место настоящего (если было, то предыдущего) жительства, но и место рождения. Наряду с датой рождения это позволяет оценить воздействие каких-либо неблагоприятных эндемических, экологических и т.п. факторов.
Место жительства предков важно знать для выявления «эффекта родоначальника», особенно при редких рецессивных заболеваниях. В настоящее время этот эффект может наблюдаться чаще в относительных изолятах сельской местности.
В генетической карте записывается почтовый адрес семьи, телефоны (домашний, рабочий, мобильный), так как бывает необходимость в пересылке по почте заключений, результатов анализов, дополнительных запросов в различные лечебные учреждения на других родственников и т.д. Телефон - для удобства оперативной связи с семьей.
5. Сведения о национальности должны быть достаточно подробными. Не секрет, что иногда у всех ближайших родственников в официальных документах значатся разные национальности. Если у родителей пробанда разные национальности, необходимо записать национальность обоих родителей. Эта информация помогает при оценке фенотипических проявлений (пигментация, гипертрихоз, синофриз и т.п.), при оценке вероятности патологии, преимущественно поражающей лиц определенной национальности или этнической группы. К примеру, болезни Тея-Сакса, Нимана-Пика, Гоше чаще встречаются у евреев. Причем популяционная частота для болезни Тея-Сакса у евреев-ашкенази составляет 1:3000, а для других национальностей - от 1:300 000 до 1:3 000 000. Непереносимость конских бобов - фавизм, встречается в основном у азербайджанцев,
что связано с дефектом фермента глюкозо-6-фосфатдегидрогеназы. Негры - гетерозиготы по гену серповидно-клеточной анемии не поражаются малярийным плазмодиумом. В каталоге наследственных болезней встречаются нозологические формы, в названии которых отражается национальная принадлежность. Например, семейная еврейская дизавтономия, периодическая болезнь армян.
6. Важно учитывать характер профессиональной деятельности, наличие производственных вредностей, время и интенсивность их воздействия, полученную дозу. Иногда контакт с особыми повреждающими факторами происходит во время службы в армии. Подробности должны быть зафиксированы в генетической карте.
7. Образование,специальность,квалификация,должность.Иногда по этому показателю косвенно можно судить об интеллектуальном развитии обратившихся и их психологических характеристиках. Эти знания помогают в общении с пациентами, семьей, в выборе адекватной формы изложения генетической информации, которая нередко бывает трудна для понимания людьми с низким образовательным уровнем.
8. Предварительный диагноз или повод для обращения.
9. Цель консультации. Чаще всего это уточнение диагноза, прогноз здоровья будущего потомства, расчет генетического риска для здоровых родственников и проведение среди них профилактических мероприятий, а в последнее время и назначение патогенетического лечения.
10. Дата консультации.
11. Фамилия врача-консультанта.
12. Серия, номер дата выдачи полиса обязательного медицинского страхования, название страховой компании.
13. Если есть, номер и дата выдачи инвалидного удостоверения.
Жалобы
Этот раздел относится к сфере субъективности, ощущений. Жалобы бывают очень разнообразными, порой к изучаемой патологии никакого отношения они не имеют. В то же время иногда пациенты (или родители ребенка) обращают внимание врача на узкий круг жалоб, упуская из виду диагностически значимые, поэтому задачей врача является получение максимальной информации обо всех обращающих на себя особенностях, выходящих за рамки нормы. В отдельных семьях явно патологические признаки рассматриваются как
нормальные, потому что они имеются у многих членов родословной. В данной ситуации необходимо активное выяснение подробностей симптоматики. В некоторых случаях вместо жалоб пациенты предъявляют готовые диагнозы и заключения других врачей-специалистов или результаты параклинических обследований.
Анамнез
Как и в клинической практике, анамнез складывается из двух компонентов: из анамнеза настоящего заболевания (anamnesis morbi) и анамнеза жизни (anamnesis vitae). В анамнез болезни включают все сведения о ее начале, предшествовавшем фоне, развитии клинической симптоматики, эффективности различных видов лечения, ремиссиях-обострениях, провоцирующих факторах, длительности течения.
Анамнез жизни начинают с раннего развития, семейных, бытовых, жилищных, гигиенических, социальных, материальных условий, характере питания. Включают сведения о перенесенных заболеваниях, вредных привычках, половой жизни, а у женщин - акушерско-гинекологический анамнез, методы контрацепции. Далее следует семейный анамнез.
Если пробанд - маленький ребенок, то удобно анамнез записывать единым текстом. Трудно рассматривать анамнез жизни в отрыве от анамнеза болезни, если она проявляется с первых дней (недель, месяцев, лет) жизни. Расспрос начинается с момента зачатия (на каком фоне оно произошло, планированная беременность или «случайная», желанная, нежеланная; проходил ли в это время кто-либо из супругов (или оба супруга) лечение по поводу бесплодия, и, если проходили, то какое; является ли зачатие следствием обращения к новым репродуктивным технологиям - искусственной инсеминации, ЭКО, ИКСИ и т.д.). Беременность описывается так же подробно, как у акушеров-гинекологов, так как первые признаки грядущего неблагополучия могут появляться до рождения ребенка.
Обращаем внимание на токсикозы (гестозы) беременных. Они могут быть тяжелыми, легкими или вообще отсутствовать. Ранние токсикозы (слюнотечение, тошнота, рвота и т.д.), даже субъективно тяжело переживаемые женщинами, реально вряд ли могут повредить развитию ребенка и обычно прекращаются к 12 нед беременности. Поздние токсикозы отмечаются в последнем триместре беременности. Природа и проявления их совсем другие (отеки, повышенное
артериальное давление, белок в моче и т.д.), нередко токсикозам предшествуют хронические заболевания. Даже объективно тяжелые поздние токсикозы могут субъективно переноситься женщинами хорошо. Они не обращаются за помощью, отказываются от госпитализации. Исходом могут быть преэклампсия, эклампсия, преждевременные роды, смерть в родах женщины, гипотрофия и функциональная незрелость плода, высокая перинатальная заболеваемость и летальность.
Угроза выкидыша - в каком сроке беременности отмечалась и в чем выражалась (боли внизу живота, тянущие боли в пояснице, «мажущие» выделения из половых органов или кровотечение). Чем в более ранние сроки и более тяжелой форме проявляется угроза выкидыша, тем выше вероятность каких-либо повреждений плода. Такая беременность может привести к рождению больного ребенка. С другой стороны, угроза выкидыша может быть следствием эндокринных, иммунологических, анатомических, внешнеповреждающих и других причин. В этом случае устранение этиологического фактора или адекватная коррекция позволят произвести на свет полноценное потомство.
Время ощущения первого шевеления и его характер. В норме при первой беременности шевеление ощущается на 20-й неделе, при последующих - несколько раньше - на 18-й, 16-й и даже 14-й неделе. Уже при первой беременности можно оценить характер шевелений. Слабое шевеление с трудом фиксируется женщиной в течение дня. При сильном - живот «ходит ходуном». Гиперактивное шевеление свидетельствует не о хорошем физическом, функциональном и т.д. развитии плода, но о его гипоксии. Слабое шевеление может быть первым признаком какой-либо наследственной патологии. Так могут проявляться амиотрофия Верднига-Гоффмана, синдромы Прадера-Вилли, Дауна (с выраженной мышечной гипотонией).
Перенесенные заболевания. Указывается, какое острое заболевание перенесла женщина, с какими клиническими проявлениями, в каком сроке беременности, количество и дозы лекарственных препаратов или других медикаментозных средств. Обязательно отмечается наличие хронических заболеваний и их терапия.
Прибавка в весе. Нормальной считается прибавка 8-10 кг за весь период беременности. Иногда женщина прибавляет в весе гораздо меньше, но рожает нормального по физическому развитию ребенка. В этом случае, возможно, она теряет в весе, имея его излишки до
беременности. Прибавка более 10 кг бывает тоже нормальной при изначальном дефиците массы или активной потере веса при ранних токсикозах. Избыточное нарастание веса беременной может свидетельствовать о скрытых отеках, что требует незамедлительной коррекции. Прибавка более 15 кг нередко сопровождает многоплодную беременность или указывает на многоводие. Многоплодная беременность считается патологией из-за частых осложнений беременности и родов; по сравнению с одноплодной беременностью при ней увеличена частота перинатальной заболеваемости и смертности близнецов.
Количество околоплодных вод. Многоводие, как и маловодие, часто сочетается с пороками развития плода или даже провоцирует их развитие. По другой теории, пороки развития плода ведут к аномальному образованию околоплодных вод.
Пренатальная диагностика. В каком сроке беременности, какие виды пренатальной диагностики назначались и проводились и каковы их результаты.
Вредные факторы и дополнительные сведения. Необходимо выясняснить о злоупотреблении алкоголем, курении, использовании психоактинвых препаратов, воздействии внешнеповреждающих факторов, стрессах, особенностях питания, труда и быта.
Роды. Нормальная продолжительность беременности - 40 нед, или 10 лунных месяцев, или 280 дней. Нормой считается диапазон в пределах 38-42 нед. Рожденный до 38 нед ребенок считается недоношенным, после 42 нед - переношенным. По сравнению с детьми, рожденными в срок, у этого контингента высок процент случаев врожденной и наследственной патологии, перинатальной смертности, причем чем больше отклонение от нормальной продолжительности беременности, тем выше вероятность патологии. Необходимо помнить, что продолжительность беременности бывает обусловлена генетическими особенностями. В семьях по женской линии может наблюдаться тенденция к невынашиванию или перенашиванию. В этих случаях дети появляются на свет без признаков патологии. Причинами недонашивания также могут быть факторы, связанные с плацентой (низкое прикрепление, отслойка и т.д.), инфекционные и неинфекционные заболевания матери, иммунологическая несовместимость мать-плод, травмы (падение, поднятие тяжестей и т.д.), анатомические особенности/пороки развития матки, поздние токсикозы беременных, многоводие и т.д. Недонашивание чревато незре-
лостью плода, «неготовностью» его к внеутробному существованию, физическим недоразвитием. Большие нагрузки начинают испытывать нервная, эндокринная, пищеварительная, сердечно-сосудистая и другие системы ребенка. Адаптационные возможности недоношенных детей индивидуальны и зависят в том числе и от наследственных факторов. Известны случаи выживания детей, появившихся на свет в 25 нед беременности с массой 500 г.
При перенашивании увеличивается риск травматизации в родах матери и ребенка, так как чаще всего ребенок очень крупный, кости его черепа более плотные, окружность головы больше, чем у рожденного в 40-42 нед ребенка. Одним из диагностических критериев перенашивания является наличие мекониальных околоплодных вод, что может свидетельствовать об асфиксии плода.
В настоящее время роды рассматривают как процесс, активными участниками которого являются и мать, и ребенок. Ребенка не только рожают, но он сам рождается. При слабости родовой деятельности, перенашивании и т.д. можно думать и о внутриутробной патологии плода, которая не позволяет ребенку вовремя и без травм появиться на свет.
Продолжительность родов. Еще недавно считалось, что «солнце над головой роженицы не должно взойти дважды», т.е. продолжительность родов не должна превышать 2 сут. В настоящее время, используя весь арсенал медикаментозных средств, женщинам часто помогают родить быстрее. Однако родить быстро - не значит хорошо. Прохождение плода по родовым путям иногда сравнивают с открыванием головой восьми дверей. Состояние головного мозга родившегося ребенка зависит от того, «открывались ли эти двери» медленно, с постепенным увеличением давления или «вышибались», нанося иногда непоправимую черепно-мозговую травму. Роды длительностью 2 ч считаются стремительными. Они неизбежно ведут к кровоизлияниям в мозг, что требует серьезного и длительного неврологического лечения и реабилитации с непредсказуемыми последствиями. Роды за 5-6 ч считаются быстрыми. Травма головного мозга менее выражена, но диспансерное наблюдение и лечение у невропатолога все равно требуются; прогноз более благоприятный. Обычно продолжительность вторых и последующих родов короче, чем первых. Продолжительность 10-12 ч для первых родов, видимо, маловата, хотя это зависит от разных факторов (срок беременности, наследственность, размеры плода, медикаментозное участие). При
сборе анамнеза необходимо обращать внимание женщины на то, что продолжительность родов считается не от момента поступления в роддом, а с первых признаков родовой деятельности (частое мочеиспускание - подтекание околоплодных вод, периодические болевые ощущения внизу живота - схватки).
Оперативное родоразрешение. Операция кесарева сечения, как и другие операции, проводится строго по показаниям. Плановое кесарево сечение назначается женщинам с психосоматической патологией, анатомическими особенностями или пороками развития половых органов, препятствующими самостоятельному родоразрешению, а также при первых родах у немолодой женщины и т.д. Экстренное кесарево сечение выполняется при клинически узком тазе (невозможности прохождения по родовым путям крупного плода), нарастающей гипоксии плода, эмболии околоплодными водами, слабости родовой деятельности, поперечном положении плода, отслойке плаценты и т.д. Проведение операции по желанию женщины нецелесообразно, даже если она очень боится боли в родах, имеет низкий порог болевой чувствительности. Для ребенка более физиологично появиться на свет в самопроизвольных родах. Любое грамотно проведенное кесарево сечение чревато осложнениями от наркоза, баротравмы (для мозга), травматизации шейного отдела позвоночника и т.д.
Дополнительные сведения. В каком предлежании и виде проходили роды, были ли разрывы внутренние и наружные, проводили ли разрез промежности для облегчения родов, ручное обследование матки и т.д.
После рождения ребенка анамнез собирается так же подробно, как у педиатров.
Закричал ли ребенок сразу после рождения (а также характер крика - громкий, слабый, «кошачий» и т.д.) или после похлопываний, отсасывание слизи из верхних дыхательных путей или проведение реанимационных мероприятий.
Антропометрические данные. Нормальной массой тела родившего ребенка считается 3000 г, рост - 50 см, окружность головы для мальчиков - 36 см, для девочек - 35 см.
Следует подробно описать жизнь ребенка на первом году жизни.
Физическое развитие. На каком вскармливании ребенок и как он прибавлял в весе, росте, как увеличивалась окружность головы. Когда и какой вводили докорм, прикорм. Реакции на пищу (аллергия,
непереносимость и т.д.). Когда появились первые зубы и какое количество их прорезалось к первому году, двум годам; когда молочные зубы начали заменяться на коренные.
Моторное развитие. Когда ребенок стал держать голову, переворачиваться, садиться, сидеть, вставать, ползать, ходить, бегать, прыгать.
Психоречевое развитие. Когда ребенок стал фиксировать взгляд, улыбаться, смеяться, гулить, произносить первые односложные слова, появились первичная и развернутая фразовая речь, когда ребенок стал опрятным (проситься на горшок).
Речевое развитие часто обусловлено генетическими факторами, описаны семейные случаи позднего речевого развития. Нередко ребенку-левше труднее заговорить наравне со сверстниками, так как развитие речи тесно коррелирует с развитием руки, а центр речи находится в левом полушарии головного мозга.
Поведение ребенка дома, вне дома, с родителями и другими родственниками, сверстниками, чужими людьми, в детском дошкольном учреждении, с домашними животными. Обращается внимание на элементы агрессии (в том числе аутоагрессии), аутизма, гиперактивности, расторможенности, а также на чрезмерную общительность и т.д.
Болезни. Какие болезни перенес ребенок, в каком возрасте, клиническая симптоматика, как они повлияли на физическое, психомоторное, речевое развитие, поведение; эффективность лечения, ремиссии, рецидивы.
Прививки. В каком возрасте и какие прививки делали ребенку, реакции на них. Если был отвод от прививок, то от врача какой специальности и с каким диагнозом.
Школа. Успеваемость, поведение. Не всегда школьная неуспеваемость синоним умственной отсталости. Гиперактивный ребенок чаще других подвергается школьным наказаниям, редко любим учителями. Плохие отметки отражают незаинтересованность ребенка в достижении лучших результатов, потере мотивации к учебе. С другой стороны, не секрет, что массовую школу могут окончить люди с легкой степенью умственной отсталости за счет своей усидчивости, неконфликтности, помощи родителей, «хорошего» поведения.
Половое развитие. Половое созревание - период, в течение которого появляются вторичные половые признаки. Преждевременным половое созревание считается при появлении его первых признаков у девочек до 8 лет, у мальчиков - до 10 лет. Отсутствие признаков
полового созревания в возрасте 13-14 лет считается его задержкой. Если к 18 годам признаки полового созревания не появились, то вряд ли они вообще появятся. Задержка полового созревания, равно как и преждевременное его развитие, могут быть обусловлены генетическими факторами (при адреногенитальном синдроме - ускоренное половое развитие у мальчиков; при синдроме ШерешевскогоТернера - отставание или отсутствие полового созревания).
Если пробандом является взрослый человек, не стоит начинать сбор анамнеза с момента зачатия. Пробанд этого не знает, да и вряд ли эта информация необходима врачу-генетику. Анамнез болезни и жизни заполняется так же, как в клинике внутренних болезней. Заинтересовать должны события жизни, состояние здоровья, предшествовавшие визиту в медико-генетическую консультацию.
После сбора анамнеза приступаем к осмотру, т.е. к объективному исследованию пробанда и его родственников. Детальный осмотр происходит с описанием фенотипических проявлений заболевания. Клинический осмотр больных с наследственной патологией имеет большую разрешающую способность. Он приобретает особое значение, поскольку правильный диагноз часто может быть установлен лишь при учете всех особенностей внешнего вида.
Часть наследственных заболеваний диагностируется исключительно на основании осмотра, по сочетанию всех видимых пороков и особенностей строения. Необходим учет всех дисплазий и пороков развития, так как высокая степень генетической предрасположенности проявляется в виде микроаномалий развития, поэтому очень важно выявить эти признаки у фенотипически здоровых родственников. В генетической практике осмотр пробанда и его родственников это фактически фенотипический анализ. Таким образом, диагностика наследственных заболеваний основывается на данных детального обследования больного и его родственников, максимально полном раскрытии фенотипических изменений.
Для того чтобы описание фенотипа пробанда дало по возможности исчерпывающие данные, необходимо применять этот метод по определенной схеме. Наружный осмотр может быть разделен на общий, который касается больного в целом и производится в самом начале, и на осмотр специальный или детальный, относящийся к отдельным частям тела, органам или системам.
Общий осмотр больного нередко дает врачу много ценных для диаг-ноза признаков. Следует оценить внешний вид больного, поло-
жение тела, которое может быть естественным, свободным или вынужденным, конституционные особенности, общее состояние пробанда. Нередко при наследственных и врожденных болезнях общий осмотр пробанда позволяет уверенно поставить диагноз по внешнему виду (что не исключает подтверждающей лабораторной диагностики). Это справедливо при многих хромосомных синдромах (в частности, при синдроме Дауна), моногенных болезнях (ахондроплазия, акроцефалополисиндактилия, мукополисахаридозы), экзогенных поражениях плода (фетальный алкогольный синдром).
Если позволяет температура воздуха в кабинете, ребеночка необходимо раздеть догола и описывать фенотип с «макушки до пят». Предварительно измеряется рост, вес, окружность головы (если необходимо, и груди, живота). В медико-генетических консультациях практикуется фотографирование детей в полный рост на фоне ростомера. Взрослые пробанды и родители больных детей осматриваются с постепенным обнажением различных участков тела (с учетом принципов врачебной этики и деонтологии).
В отличие от клиники внутренних болезней, когда объективное исследование проводится по органам и системам, для диагностики многих наследственных и врожденных болезней удобнее описывать внешние признаки по определенному плану, которые фиксируются в карте фенотипа.
Соответствие внешнего вида паспортному возрасту
Уровень психомоторного, физического, речевого развития детей и интеллектуального развития взрослых, состояние сознания.
Телосложение: правильное, пропорциональное, степень упитанности, отложения жира.
Кожа: цвет, гипертрихоз, истончение, гиперкератоз, ангиомы, аденомы сальных желез, «бабочка» на коже лица, гипер- и гипопигментация, фибромы, липомы, тургор, влажность, потовые железы, стрии, эластичность, участки «лимонной корки», послеоперационные и другие рубцы, «избыточная кожа», веснушки, невусы, папилломы, птеригиумы подколенные, межпальцевые, множественные атрофии, ихтиоз, кисты и пр.
Волосы: алопеция; низкий рост волос на лбу, шее; «мыс вдовы»; волосы редкие, ломкие, пушковые, жесткие; две макушки; неправильный рост; участки депигментации - «седая прядь», раннее поседение, раннее мужское облысение и др.
Мышечная ткань: гипо- и гипертрофии, атрофии, спастические состояния, парезы и параличи, уплотнения, припухлости, болезненность и др.
Череп: макро- и микроцефалия, гидроцефалия, оксицефалия, брахицефалия, акроцефалия, скафоцефалия, тригоноцефалия, выступающий скошенный лоб, деформации, асимметрия, состояние швов, родничков, скошенный затылок и др.
Ушные раковины: состояние слухового анализатора; анотия; макротия; микротия; асимметричные, деформированные, оттопыренные, низкопосаженные; предушные папилломы и фистулы; атрезия наружного слухового прохода; гипертрихоз наружного слухового прохода; бороздки - «насечки» на мочке; «уши Сатира»; дарвиновский узелок и др.
Лицо: грубые черты, «кукольное», «птичье», отечное лицо, «лицо плода», асимметричное, с пухлыми щеками, плоское, круглое, треугольное и др.
Нос: аносмия, короткий, длинный, клювовидный, грушевидный нос; нос со вздернутым кончиком; переносица седловидная, широкая, плоская, высокая, выступающая; гипоплазия; асимметрия; колобома крыльев носа; атрезия хоан; искривление носовой перегородки, раздвоенный кончик и др.
Область глаз:
- цвет, гетерохромия радужек, пятна Брушфильда, кольцо Кайзера-Флейшера, косоглазие, нистагм, микрофтальм, анофтальм, буфтальм, криптофтальм, птоз, асимметрия, эпибульбарный дермоид, слезные железы, дистопия «слезной точки», блефарофимоз и др.;
- брови: сросшиеся, редкие, густые, выступающие надбровные дуги;
- ресницы: двойной, тройной ряд ресниц, густые, длинные загнутые, их отсутствие;
- монголоидный и антимонголоидный разрез глаз, эпикант, телекант, гипо- и гипертелоризм, иридодонез;
- острота зрения, цветовое, сумеречное зрение, поле зрения, амблиопия, нарушения рефракции, «голубые» склеры, телеангиэктазии на склерах, помутнение и аномалии размеров роговицы и хрусталика, аниридия, колобомы века, радужки, сетчатки, глаукома, дистрофии сетчатки, стекловидного тела, атрофия зрительного нерва, симптом «вишневой косточки» и др.
Челюсти: прогения, макро- и микрогения, микро- и макрогнатия, нарушения прикуса, расщелины прямые и косые, асимметрия, аномалад Робена и др.
Область рта: губы тонкие, толстые; расщелина губы; плоское, высокое, готическое нёбо, расщелины нёба; фильтр длинный, короткий, широкий; «карпий рот»; опущенные углы рта; оттопыренная нижняя губа; «лицо свистящего человека»; невозможность открыть широко рот; выемка красной каймы губ; пигментные пятна типа веснушек на губах и около рта; микростомия; макростомия; фиброматоз десен; макроглоссия; географический язык; лобуляция языка; дополнительные уздечки верхней губы, языка; расщепление язычка; ямки или фистулы на нижней губе; аномалии слюнных желез; афония; дисфагия; расстройства вкуса и др.
Зубы: неправильная форма, расположение, олигодентия, сверхкомплектные зубы, двойной ряд зубов; дефекты эмали, дентина; диастемы; тремы; парадонтит; кариес; флуороз; врожденные, молочные зубы; нарушение прорезывания и др.
Шея: короткая, длинная, кривая; крыловидные складки.
Грудная клетка, позвоночник, передняя брюшная стенка: лордоз; кифоз; сколиоз; платиспондилия; пилонидальные ямки; spina bifida; гипертелоризм сосков, дополнительные соски; полимастия; гипоплазия; аплазия большой грудной мышцы с отсутствием соска; грудная клетка бочкообразная, широкая, воронкообразная, килевидная; долихостеномелия; отсутствие ключиц; грыжи; «лягушачий» живот, вздутый и др.
Конечности: асимметрия, изменение длины; деформация костей, суставов; вывихи, подвывихи; дисплазии суставов; синостозы; экзостозы; гипермобильность суставов и ограничение их подвижности; лимфатический отек кистей и стоп; акромикрия кистей и стоп; большой размер кистей и стоп; пре- и постаксиальная полидактилия, олигодактилия, брахидактилия, арахнодактилия, эктродактилия, синдактилия (полная/частичная, кожная/костная), клинодактилия, камптодактилия; недоразвитие и изменение количества и формы фаланг; изменение характера складок ладоней и стоп; косолапость, плоскостопие; сандалевидная щель; врожденные ампутации и некоторые другие редукционные дефекты; артрогрипоз и др.
Ногти: анонихия, изменение формы и структуры.
Мочеполовая система: гипоспадия, крипторхизм, изменения структуры мошонки и клитора, гипоплазия яичек и полового члена,
макроорхидизм, нарушение сроков полового созревания, гипоплазия больших половых губ, сросшиеся большие половые губы, двойственное строение наружных гениталий и др.
Кроме оценки внешних признаков, необходимо получить данные обследования внутренних органов пациентов. Для этого проводят консультации врачей смежных специальностей, дополнительные параклинические методы исследования.
Для уточнения состояния внутренних органов используются рентгенорадиографические (рентгенография, рентгенконтрастные исследования, компьютерная томография, магнитно-резонансная томография), электрофизиологические (ЭКГ, ЭЭГ), ультразвуковые, гематологические, эндокринологические, иммунологические, эндоскопические, гистологические, биохимические и др. методы исследования.
Если предполагается диагноз конкретного наследственного заболевания, то его уточнение проводится с помощью специализированных лабораторно-генетических методов: цитогенетического, биохимического, молекулярно-генетического.
В типичных случаях часто встречающихся наследственных болезней фенотипический анализ и дополнительные клинические методы (иногда и без них) позволяют поставить клинический диагноз. Однако если существует возможность подтверждения его специализированными генетическими обследованиями, не стоит пренебрегать такой возможностью хотя бы из-за существования гено- и фенокопий.
Если пробандом является умерший ребенок или мертворожденный, то фенотипический анализ проводится при патологоанатомическом вскрытии. В этом случае необходимо запросить протокол вскрытия.
2.7. КЛИНИЧЕСКИЕ ОСОБЕННОСТИ ПРОЯВЛЕНИЯ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Почти при любых генетических нарушениях можно выделить общие клинические признаки, к которым относятся.
• Отягощенный семейный анамнез. Несколько случаев одной и той же наследственной патологии в семье. Нередко поводом для обращения к врачу-генетику является первый и единственный случай в родословной, что не исключает наследственного
характера заболевания. Не все семейные заболевания относятся к наследственным; такие факторы окружающей среды, как инфекции и тератогены, могут симулировать наследственные заболевания; иногда могут быть поражены два ребенка и более.
• Специфические симптомы. К ним относятся редко встречающиеся в популяции признаки: «седая» прядь волос, грубые черты лица у ребенка, непропорциональное телосложение, необычный запах, своеобразное поведение, непереносимость отдельных пищевых продуктов, полное отсутствие зубов и т.д. Иногда достаточно одного специфического симптома для установления (предположения) диагноза определенного наследственного синдрома. Гемангиома, занимающая половину лица, - синдром Штурге-Вебера; стереотипные характерные движения рук, напоминающие выжимание, хлопки, мытье и т.д. у ребенка, с грубой задержкой психомоторного развития - синдром Ретта и т.д.
• Генетическая гетерогенность - заключается в том, что развитие сходной клинической картины может быть обусловлено мутацией разных генов, т.е. при клинически одном и том же заболевании могут быть разные типы наследования. Например, мукополисахаридозы, одним из клинических проявлений является гаргоилизм - грубые, гротескные черты лица, а типы наследования разные (аутосомно-рецессивный при типе I, Х-сцепленный рецессивный при типе II). В Лондонской базе данных врожденных и наследственных заболеваний и признаков расщелина губы/нёба отмечена при 847 наследственных синдромах с различными типами наследования.
• Клинический полиморфизм - заключается в различиях клинической картины при одном и том же заболевании: время манифестации, набор, степень выраженности клинических признаков и т.д. На клинический полиморфизм влияют как генетические, так и средовые факторы. Генетические причины связаны с влиянием на проявление патологического гена других генов (супрессоров, модификаторов и т.д.). К средовым причинам относятся условия и образ жизни, климат, проводимое лечение и т.д. Примером может служить аутосомно-доминантный синдром Ван дер Вуд. У одного из родителей ребенка с расщелиной нёба при тщательном осмотре выявлены только «ямки» на нижней губе.
• Хронический, рецидивирующий, прогредиентный характер течения - постоянное прогрессирование патологического процесса, которое развивается в результате постоянного действия мутантного гена. На рецидивы влияют как генетические, так и средовые факторы (погрешности в диете, присоединившиеся инфекции, травмы, стрессы). Необходимо помнить, что существуют наследственные нарушения, не сопровождающиеся прогрессированием клинической симптоматики (односторонняя расщелина губы).
• Плейотропное действие гена - выражается в множественном поражении органов и систем, которое определяется единичной мутацией. Так, при ангидротической эктодермальной дисплазии анодентия, гипоплазия или аплазия потовых, слезных, слюнных желез, гипотрихоз, дефекты ногтей обусловлены действием одного мутантного гена (рецессивного, локализованного в Х-хромосоме). Иногда выделяют первичную плейотропию и вторичную. Для приведенного выше примера эффектами вторичной плейотропии будут нарушения терморегуляции, пищеварения, склонность к конъюнктивитам.
• Взаимодействие аллельных генов - рецессивные гены не проявляются в присутствии доминантных, а только в гомозиготном состоянии. Если патология определяется рецессивными генами, то можно говорить об ее отсутствии у гомозигот по доминантному гену и гетерозигот. В настоящее время тщательное обследование выявляет фенотипические отличия в сходных, на первый взгляд, контингентах. Лабораторные тесты позволяют диагностировать гетерозиготное носительство при фенилкетонурии, муковисцидозе, синдроме ТеяСакса и др.
• Взаимодействие неаллельных генов (полимерия) - заключается в однонаправленном действии нескольких неаллельных генов - полигенов, от суммарного действия которых зависит количественный результирующий признак: рост, масса тела и т.д. или при мультифакториальных болезнях - выраженность патологических проявлений (простая односторонняя расщелина губы - двусторонняя расщелина губы с затрагиванием альвеолярного отростка верхней челюсти, крыльев носа; готическое нёбо - расщелина мягкого нёба, расщелина твердого нёба).
Эпистаз - подавление действие одного гена другим, неаллельным.
Комплементарность - появление нового признака при наличии двух доминантных неаллельных генов (нормальный слух у детей глухих родителей - рецессивных гомозигот по неаллельным генам).
• Устойчивость к стандартным методам терапии. Толерантность к терапевтическим дозам антибиотиков при бронхопневмониях у больных муковисцидозом (эффект наступает при субтоксических дозах), использование сверхбольших доз витамина D при витамин-D-резистентном рахите. Установление патогенетических механизмов наследственных болезней помогает в адекватном и эффективном лечении пациентов (диетотерапия при фенилкетонурии, галактоземии, целиакии; гормонотерапия при адреногенитальном синдроме и т.д.).
• Врожденный характер заболевания - не абсолютный признак наследственной патологии. Комплекс патологических признаков, присутствующий у ребенка при рождении, свидетельствует лишь о том, что дефекты формировались внутриутробно. Большинство хромосомных синдромов носят врожденный характер. Многие моногенные синдромы и мультифакториальные пороки развития - тоже врожденные. Если причиной формирования аномалий были внешние факторы, то диагностируются врожденные краснушный, алкогольный, талидомидный, варфариновый, никотиновый и т.п. синдромы, не являющиеся по сути наследственными. Если при имеющейся патологии диагноз устанавливается при рождении, говорят о сигнальном фенотипе. К сигнальным фенотипам относятся синдромы Дауна, Патау, ахондроплазия и др. Известно большое количество наследственных болезней, при которых патологическое действие мутантного гена начинает реализовываться с возрастом (хорея Гентингтона, поликистоз почек взрослого типа, отосклероз, некоторые виды дегенерации сетчатки и т.д.).
После фенотипического анализа можно приступить к составлению и анализу родословной.
Сбор генеалогической информации о наличии среди родственников больного тех или иных заболеваний может проводиться разными методами: путем опроса на основе личного обследования членов семьи и анкетирования - очного и заочного.
Составление родословной затруднено при следующих обстоятельствах.
1. Недостаточный объем полученной информации. Многие не знают состояния здоровья и причин смерти даже у ближайших родственников, тем более родственников по линии мужа или жены.
2. Пробанд - сирота.
3. Заведомо ложные сведения о состоянии здоровья родственников (психические, венерические и др. заболевания).
4. Неправильно поставленные диагнозы заболевания у больных родственников.
5. Малочисленность семей (пробанд, его мать, отец - единственные дети у своих родителей).
2.8. ГРАФИЧЕСКОЕ ИЗОБРАЖЕНИЕ РОДОСЛОВНОЙ
В родословную заносятся краткие сведения о родственниках в определенном порядке.
1. Данные о пробанде, его возрасте, заболеваниях.
2. Данные о родителях, их возрасте, состоянии здоровья.
3. Данные о сибсах пробанда с указанием порядкового номера рождения, возраста, состояния здоровья.
4. Ближайшие родственники. Сначала следует изобразить родственников по женской линии (дяди, тети, двоюродные сибсы, бабушка, дедушка, их братья и сестры с детьми и внуками, прабабушки и прадедушки), потом то же самое по мужской линии с указанием порядкового номера, возраста, состояния здоровья. Если кого-то из родственников уже нет в живых, то нужно отметить возраст на момент смерти и причину смерти. Как правило, для генеалогического анализа достаточно составления родословной до 4-го колена, т.е. до прабабушек и прадедушек, к тому же очень часто пациенты и сами не знают более далеких предков.
При составлении родословной используются общепринятые условные обозначения (рис. 2.1).
Заболевания в родословной обозначаются произвольно, чаще всего соответствующий символ полностью затушевывается; разные заболевания обозначаются по-разному. В некоторых случаях в одном символе можно отметить несколько заболеваний. Поколения в родословной нумеруются римскими цифрами, члены одного поколения - арабскими.
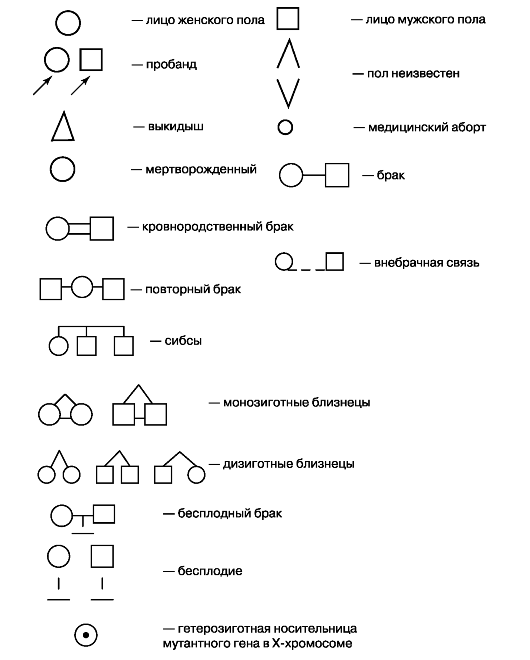 Рис. 2.1. Символы, используемые при составлении родословной
Рис. 2.1. Символы, используемые при составлении родословной
При сборе генеалогической информации желательно стремиться к наиболее полному составлению родословных по восходящему, нисходящему и боковым направлениям и получить сведения о максимальном количестве родственников 3-4-го поколений. Необходимо собрать сведения, касающиеся не только конкретного заболевания в семье, но и информацию обо всех болезнях, встречающихся среди членов семьи. Грубой ошибкой является искусственное укорочение звеньев родословной в связи с трудностями обследования родственников II и III степеней родства.
2.9. АНАЛИЗ РОДОСЛОВНОЙ
Тщательно собрав данные о родословной, уточнив необходимые сведения о больных и обследовав нужных членов семьи, можно приступить к ее генетическому анализу. Генеалогический анализ дает возможность при подробном изучении семейной схемы выяснить, к какому из известных типов наследования она относится, и установить ход наследственной передачи.
При этом необходимо:
1. Установить, является ли данный признак или заболевание единичным в семье или имеется несколько случаев данной патологии (семейный характер).
2. Выделить лиц, подозрительных в отношении данного заболевания, и составить план их обследования и уточнения их диагноза.
3. Определить тип наследования и выяснить, по какой линии - материнской или отцовской - идет передача заболевания.
4. Выявить лиц, нуждающихся в медико-генетическом консультировании, определить клинический прогноз для пробанда и его больных родственников с учетом особенностей заболевания и его генетической характеристики.
5. Разобрать план лечения и профилактики, принимая во внимание индивидуальные и семейные особенности заболевания.
При анализе родословной врач может встретится с генными и хромосомными заболеваниями, с болезнями, в развитии которых принимают участие как генетические, так и средовые факторы, с «неизвестными» заболеваниями.
2.10. ГЕНЕАЛОГИЧЕСКИЙ АНАЛИЗ ПРИ МОНОГЕННЫХ ЗАБОЛЕВАНИЯХ
В настоящее время в большинстве случаев врачу приходится сталкиваться с уже известными моногенными заболеваниями, клинические особенности и тип наследования которых указаны во многих медицинских справочниках. Хотя на практике врачу гораздо чаще приходиться иметь дело с малыми семьями, он должен уметь ориентировочно определять тип наследования генных заболеваний, так как в ряде случаев от этого зависит, правильно ли будет поставлен диагноз. Например, гомоцистинурия и синдром Марфана (подробнее о нем будет сказано ниже) имеют много общих клинических проявлений, а наследуются по-разному. Синдром Марфана - аутосомно-доминантно, а гомоцистинурия - аутосомно-рецессивно.
Рассмотрим основные типы передачи моногенных заболеваний: аутосомно-доминантный, аутосомно-рецессивный и сцепленный с Х-хромосомой.
2.10.1. Аутосомно-доминантный тип наследования
В связи с тем, что доминантные гены, детерминирующие развитие заболевания, в гомозиготном состоянии являются, как правило, летальными, все браки между больными и здоровыми членами семьи относятся в типу Аа и аа, где А - доминантный ген, определяющий развитие наследственного заболевания, а - рецессивный ген.
Родословная имеет следующие характерные признаки.
1. Каждый больной член семьи обычно имеет больного родителя.
2. Заболевание передается из поколения в поколение. Больные встречаются в каждом поколении.
3. У здоровых родителей дети будут здоровы.
4. Заболеть могут и мужчины, и женщины в равной степени, поскольку ген локализуется в аутосоме.
5. Вероятность рождения больного ребенка, если болен один из родителей, равна 50%.
6. От двух больных родителей могут рождаться гомозиготы с более тяжелой формой заболевания, чем у гетерозигот.
7. Число спорадических случаев, вызванных новыми мутациями, зависит от тяжести поражения органов репродукции.
При некоторых аутосомно-доминантных заболеваниях наблюдается неполная пенетрантность гена.
Пенетрантность - вероятность проявления гена, выражается в процентах заболевших от числа носителей. Так, если доминантный ген проявляется в фенотипе всех его носителей, его пенетрантность равна 100%. Если среди носителей патологического доминантного гена болеет только половина, пенетрантность равна 50%, если четверть - 25%.
Носительство доминантного гена без фенотипического проявления можно заподозрить у одного из родителей, если среди его потомков появились больные соответствующей доминантной болезнью. Когда у здоровых родителей появился больной ребенок и в родословной есть еще другие случаи этой болезни, можно предполагать, что у одного из родителей больного был патологический ген, который не пенетрировал, но был передан потомку.
Доминантные гены обладают и различной экспрессивностью. Понятие «экспрессивности» аналогично понятию «тяжести заболевания». При очень низкой экспрессивности гена создается впечатление, что человек здоров, при высокой - развивается тяжелая форма заболевания. Иногда при тщательном исследовании всеми доступными клиническими и параклиническими методами удается поставить диагноз стертой формы доминантного заболевания, и он правомочен только тогда, когда в данной семье встречаются выраженные клинические формы этого заболевания. Крайне вариабельной экспрессивностью обладает аутосомно-доминантный синдром Марфана. Можно встретить очень тяжелые формы заболевания с типичной для синдрома триадой симптомов:
а) поражение костной системы: сколиоз, кифосколиоз, деформация грудины, арахнодактилия, астеническое телосложение, высокий рост;
б) нарушение зрения: двусторонний вывих хрусталика;
в) патология со стороны сердечно-сосудистой системы: аневризма аорты.
Существуют и стертые клинические формы заболевания, которые не диагностируются (астеническое телосложение, сколиоз I степени, арахнодактилия, небольшая миопия, расширение аорты).
2.10.2. Аутосомно-рецессивный тип наследования
Основной особенностью рецессивного гена является то, что он проявляет свое действие только в гомозиготном состоянии, поэтому в гетерозиготном состоянии он может существовать во многих поколениях, никак не проявляясь фенотипически. В результате первый больной рецессивной болезнью появляется через многие поколения после возникновения мутации.
Аутосомно-рецессивное наследование имеет следующие отличительные черты.
1. У здоровых родителей рождаются больные дети. Наиболее частый тип браков - это брак между гетерозигтными носителями (Аа х Аа), когда оба родителя здоровы, но у них могут рождаться дети с гомозиготным генотипом.
2. У больного родителя рождаются здоровые дети. При вступлении в брак больного рецессивной болезнью со здоровым (тип брака, обычно - АА х аа) все дети будут здоровы.
3. Болеют в основном сибсы (братья, сестры), а не родители - дети, как при доминантном типе наследования.
4. Отмечается более высокий процент кровнородственных браков.
5. Все дети больных родителей являются гетерозиготными носителями патологического гена.
6. Одинаково часто болеют мужчины и женщины.
7. У гетерозиготных носителей соотношение больных и здоровых детей 1:3. Вероятность рождения больного ребенка равна 25% для каждого последующего ребенка.
Как и при доминантном типе наследования, это соотношение применимо к семьям с большим количеством детей или к сумме детей из многих семей с одной и той же рецессивной болезнью. Частота возникновения аутосомно-рецессивного заболевания находится в прямой зависимости от степени распространения мутантного гена. Обычно повышается частота рецессивных наследственных заболеваний в изолятах и популяциях, где существует высокий процент кровнородственных браков.
Исходя из того, что общность генов у родителей и детей, братьев и сестер (кроме монозиготных близнецов), т.е. у родственников I степени родства, равна 50% (1/2), можно составить схему общности генов у родственников разных степеней.
Таблица 2.1.
Степень родства | Показатель общности генов |
Монозиготные близнецы | 100% |
I степень родства (родители-дети, сибсы) | 50% (1/2) |
II степень родства (дядя, тетя-племянники, дедушка, бабушка-внуки) | 25% (1/4) |
III степень родства (двоюродные сибсы) | 12,5% (1/8) |
IV степень родства (троюродные сибсы) | 3,125% (1/32) |
Таким образом, вероятность рождения больного ребенка в семьях с кровнородственным браком, отягощенных рецессивным геном, значительно выше, чем в неродственных браках, так как концентрация гетерозиготного носительства в них выше, чем в общей популяции. Чем реже распространен рецессивный ген, тем чаще соответствующая рецессивная болезнь встречается среди детей из кровнородственных браков. Об отрицательном влиянии родственных браков свидетельствует и тот факт, что умственная отсталость среди детей в них в 4 раза выше, чем в семьях неродственных браков, и составляет
16%.
Наиболее часто рецессивные болезни встречаются в семьях спорадически. В такой ситуации появление больного ребенка может быть или результатом первого в семье брака между гетерозиготными родителями, или произойти в браке гетерозиготного носителя со здоровым, в половой клетке которого произошла первичная мутация. Чтобы правильно оценить спорадический случай рецессивной болезни для определения риска рождения других больных детей, необходимо определить гетерозиготное носительство. В настоящее время для многих рецессивных заболеваний разработаны тесты, которые позволяют выявить тонкие фенотипические различия гетерозиготных носителей и здоровых лиц.
2.10.3. Х-сцепленный тип наследования
Гены, локализованные в Х-хромосоме, так же как и при аутосомном наследовании, могут быть доминантными и рецессивными. Главной особенностью Х-сцепленного наследования является отсутствие
передачи соответствующего гена отца сыну, так как мужчины, будучи гемизиготными (имеют только одну Х-хромосому), передают свою Х-хромосому только дочерям.
Если в Х-хромосоме локализуется доминантный ген, такой тип наследования называется Х-сцепленным доминантным. Для него характерны следующие признаки.
1. Если болен отец, то все дочери будут больны, а все сыновья здоровы.
2. Больные дети появляются только в том случае, если болен один из родителей.
3. У здоровых родителей все дети будут здоровы.
4. Заболевание прослеживается в каждом поколении.
5. Если мать больна, то вероятность рождения больного ребенка равна 50%, независимо от пола.
6. Болеют как мужчины, так и женщины, но в целом больных женщин в семье в 2 раза больше, чем больных мужчин.
При локализации в Х-хромосоме рецессивного гена тип наследования называется Х-сцепленным рецессивным. Женщины почти всегда фенотипически здоровы (носители), т.е. гетерозиготы. Тяжесть болезни зависит от степени поражения репродуктивной системы. Для этого типа наследования характерно следующее:
1. Заболеванием поражаются преимущественно лица мужского пола.
2. Заболевание наблюдается у мужских родственников пробанда по материнской линии.
3. Сын никогда не наследует заболевание отца.
4. Если пробанд - больная женщина, ее отец обязательно болен, а также поражены все ее сыновья.
5. В браке между больными мужчинами и здоровыми гомозиготными женщинами все дети будут здоровы, но у дочерей могут быть больные сыновья.
6. В браке больного мужчины и женщины-носительницы дочери: 50% - больные, 50% - носительницы; сыновья: 50% - больные, 50% - здоровые.
7. В браке между здоровым мужчиной и гетерозиготной женщиной вероятность рождения больного ребенка составит: 50% - для мальчиков и 0% - для девочек.
8. Сестры-носительницы имеют 50% больных сыновей и 50% дочерей-носительниц.
2.10.4. Y-сцепленный тип наследования
Признак передается по мужской линии. Доказано, что в Y-хро- мосоме имеются гены, отвечающие за оволосение ушной раковины, сперматогенез (азооспермия), интенсивность роста тела, конечностей, зубов.
2.11. МИТОХОНДРИАЛЬНАЯ НАСЛЕДСТВЕННОСТЬ
1. Передается только по материнской линии.
2. Больные отцы не передают болезнь своим детям.
3. Болеют мальчики и девочки.
Передаются митохондрии только с цитоплазмой яйцеклеток.
2.12. ГЕНЕАЛОГИЧЕСКИЙ АНАЛИЗ
ПРИ МУЛЬТИФАКТОРИАЛЬНЫХ ЗАБОЛЕВАНИЯХ
Анализ родословных при мультифакториальных заболеваниях основан не на законах Менделя, как при моногенных признаках, а на эмпирически полученных данных. В результате многолетних наблюдений были выявлены следующие особенности, характерные для этой формы патологии.
1. Вероятность появления заболевания зависит от степени родства с пораженным членом семьи, так как это определяет количество общих генов (см. табл. 2.1).
2. Количество больных родственников определяет прогноз для пробанда. Например, при сахарном диабете риск для сибсов пробанда в зависимости от числа больных родственников будет следующим:
а) если родители здоровы, риск равен 5-10%;
б) если болен один из родителей, риск равен 10-20%;
в) если больны оба родителя, риск возрастет до 40%.
3. Генетически прогноз зависит от степени тяжести заболевания пораженного родственника, так как степень тяжести при мультифакториальных заболеваниях определяется суммарным действием нескольких генов. Так, человек, получивший 4 гена, от которых зависит артериальная гипертензия, будет иметь более тяжелую форму заболевания и, конечно, большую вероятность передачи патологических генов потомству.
4. Степень наследственной отягощенности для пробанда увеличивается, если его больной родитель относится к редко поражаемому полу. Так, в случае язвенной болезни желудка и двенадцатиперстной кишки в каждой семье, отягощенной этим заболеванием, вероятность передачи генов потомству одинакова. Однако вероятность заболевания всегда больше для лиц мужского пола и в том случае, если поражены женские родственники пробанда.
2.13. ГРУППЫ РИСКА В ЗАВИСИМОСТИ ОТ ВИДА ВОЗМОЖНОЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
В настоящее время, используя клинико-генеалогический метод, можно выделить родственников пробанда, нуждающихся в лечебно-профилактических мероприятиях. Особое внимание следует обратить на профилактику, которая должна проводится у лиц, еще не имеющих клинических проявлений болезни, но «угрожаемых» по данному заболеванию, например при обнаружении основных «факторов риска» ишемической болезни сердца у родственников I-II степени родства пробанда с данной патологией; у лиц с высоким уровнем мочевой кислоты в семьях, где есть больной подагрой, и т.д. Выявление «угрожаемых» по различным наследственным болезням лиц проводится при профилактических осмотрах населения, популяционных исследованиях в результате тотального и селективного скрининга. Однако использование клинико-генеалогического метода позволит решить эту задачу более эффективно, при этом целесообразно в зависимости от вида наследственной патологии относить людей к определенным группам риска (по моногенным и хромосомным болезням, заболеваниям с наследственной предрасположенностью).
2.13.1. Группа риска при хромосомной патологии
В группу риска по хромосомным болезням ребенок попадает в тех случаях, когда:
1) возраст матери больше 35 лет; у нее риск рождения ребенка с синдромом Дауна возрастает почти в 40 раз по сравнению с 20-летней женщиной;
2) в семье имеются дети с хромосомными болезнями;
3) у матери отмечается отягощенный акушерский и семейный анамнез (выкидыши, мертворождения, дети с множественными пороками развития, неустановленным диагнозом, особенно если у матери есть микроаномалии или врожденные пороки развития, которые могут быть признаком мозаицизма или хромосомной аберрации);
4) при наличии у матери (отца) хромосомного мозаицизма или хромосомной аберрации, установленной ранее;
5) если родители контактировали с мутагенными факторами.
2.13.2. Группа риска при моногенных болезнях
Ребенок относится к группе риска по генной патологии, если у родителей, сибсов или других родственников диагностировано наследственное заболевание.
В группу риска входят лица, которые в связи с близким родством с пробандом имеют повышенный риск гетерозиготного носительства мутантного гена. Так, гетерозиготными будут родители и дети гомозигот по рецессивному гену. Например, при фенилкетонурии родители больного и его будущие дети являются гетерозиготными носителями данного рецессивного гена. При доминантных заболеваниях с неполной пенетрантностью носителями патологического гена являются все лица, имеющие больных детей и больных родителей одновременно. При сцепленных с Х-хромосомой заболеваниях гетерозиготными (кондукторами) становятся все дочери больного, например гемофилией, и все матери больных гемофилией. В случае определения гетерозиготности клинико-генеалогическим методом другие методы не используются, а носители патологического гена должны быть поставлены на учет.
Если гетерозиготные носители вступают в брак, то следует определить вероятность гетерозиготности будущего супруга и информировать семью о результатах расчета генетического риска. Также рекомендуется возможным гетерозиготным носителям избегать родственных браков, так как в этом случае увеличивается риск рождения больного ребенка.
2.13.3. Группа риска при мультифакториальных заболеваниях
При мультифакториальных заболеваниях к группе повышенного риска должны быть отнесены лица с учетом величины наследственной отягощенности, которая зависит от тяжести заболевания, степени родства с больными и количества больных в семье.
Выявление группы риска с помощью генеалогического метода позволяет эффективно осуществлять ранние лечебно-профилактические мероприятия для лиц, генетически предрасположенных к различным заболеваниям. Так, при наличии гипертонической болезни у одного из родителей необходимо контролировать уровень артериального давления у ребенка, рекомендовать щадящий режим. В этих семьях необходимо пропагандировать ранние и постоянные занятия спортом, дисциплину труда и отдыха, ограниченное потребление соли. Если такие привычки развиваются с детства, они могут оказать профилактический эффект.
Особое внимание должно уделяться семьям, предрасположенным к таким наследственным заболеваниям, как сахарный диабет, эпилепсия, шизофрения, язвенная болезнь, гипертоническая болезнь и др. Так, в случае если один из родителей болен, риск рождения ребенка с сахарным диабетом равен 10%. Следовательно, существует реальная опасность развития данного заболевания. Такие семьи необходимо поставить на учет, периодически проводить профилактические осмотры с применением дополнительных методов исследования.
Лечебно-профилактические мероприятия могут быть разделены на две группы.
1. Профилактика лиц без клинических проявлений болезни, но с генетическими факторами риска, направленная на предупреждение развития патологии (группы риска на основании семейного фона).
2. Генетическая профилактика - предупреждение повторных заболеваний в семьях.
Генетическая профилактика осуществляется врачами медикогенетической консультации и направлена на предупреждение рождения больного ребенка. Наиболее эффективным методом генетической профилактики является пренатальная диагностика наследственных болезней.